

Abstract
The aggregation of α-synuclein, a protein involved in neurotransmitter release at presynaptic terminals, is associated with a range of highly debilitating neurodegenerative conditions, most notably Parkinson’s disease. Intraneuronal inclusion bodies, primarily composed of α-synuclein fibrils, are the major histopathological hallmarks of these disorders, although small oligomeric assemblies are believed to play a crucial role in neuronal impairment. We have probed the mechanism of neurotoxicity of α-synuclein oligomers isolated in vitro using antibodies targeting the N-terminal region of the protein and found that the presence of the antibody resulted in a substantial reduction of the damage induced by the aggregates when incubated with primary cortical neurons and neuroblastoma cells. We observed a similar behavior in vivo using a strain of C. elegans overexpressing α-synuclein, where the aggregation process itself is also partially inhibited as a result of incubation with the antibodies. The similar effects of the antibodies in reducing the toxicity of the aggregated species formed in vitro and in vivo provide evidence for a common origin of cellular impairment induced by α-synuclein aggregates.
α-Synuclein (αS) is an intrinsically disordered protein with a molecular weight of 14 kDa whose aggregation and conversion into amyloid fibrils is associated with a range of highly debilitating neurodegenerative disorders, including Parkinson’s disease (PD), Parkinson’s disease with dementia (PDD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA).1−3 Fibrillar aggregates of αS have been identified as the major constituents of the proteinacious inclusions known as Lewy bodies that form inside the neurons of patients suffering from these conditions,4,5 and a number of missense mutations, as well as duplications and triplications of the gene encoding αS, are associated with familial forms of early onset PD.6−8 Because of the link between αS aggregation and PD, intensive efforts have been expended to characterize the structural properties of its fibrillar form,9−16 although it has become evident both in vitro(6,17−22) and in vivo(23,24) that smaller αS oligomers are likely to be the crucial species associated with the underlying mechanism of neurotoxicity. A number of αS oligomeric species have been described so far, providing evidence of the toxicity of these highly heterogeneous species.25−27
The general mechanism of aggregation of αS has been characterized in vitro,28 although the detailed description of the oligomeric species populated during the aggregation process has proved to be challenging, primarily because of the difficulties in studying the structural properties of the often short-lived and highly heterogeneous oligomeric intermediates that are populated prior to the formation of well-defined amyloid fibrils.29 This objective is, however, of vital importance in the quest to define the underlying molecular origins of neurodegenerative conditions associated with αS aggregation. Several lines of evidence have indicated that interactions of αS oligomers with biological membranes, resulting in the loss of cellular or subcellular integrity, are key elements that promote neuronal toxicity.20,23,30−34 In addition, we have observed that natural aminosterol molecules that disrupt binding of αS to cellular membranes, namely, squalamine and trodusquemine, reduce its toxicity in both cells and in a C. elegans model of αS toxicity in vivo.35,36
In the present study, we used an antibody that targets the N-terminal region of αS to probe the mechanism of toxicity of its oligomers when incubated with primary cortical neurons and cultured neuroblastoma cells. The presence of the antibody induced a reduction of the toxicity of the αS oligomers, thereby supporting the role of the N-terminal region of the protein in the mechanism of membrane disruption that generates cellular toxicity.17 We also probed the effect of the antibody in C. elegans animals overexpressing αS, showing a reduction of the aggregation of αS and locomotor impairment in the worms. The finding that the nature of the reduction in toxicity of αS oligomers by this antibody in vitro is similar in the C. elegans model provides evidence for a common mechanism of toxicity induced by αS aggregates, whether these are oligomers isolated in vitro or are the species formed in vivo by aggregation in C. elegans.
Results
Effects of Primary Antibodies on the Membrane Disruption by αS Oligomers
Toxic αS oligomers were formed as previously reported19 and are defined as type B* oligomers.17 According to previous findings,17,20−22 incubation of the type B* αS oligomers with healthy neuronal cells was found to induce cellular damage, resembling the pathophysiological effects observed in pluripotent stem cell-derived neurons from a PD patient with triplication of the αS gene.37 These effects included significant increases in the level of basal intracellular Ca2+ and of intracellular reactive oxygen species (ROS), as well as damage to mitochondrial function and disruption of cellular membranes.17 We identified in a previous study two key elements responsible for the generation of neuronal damage by toxic αS oligomers; these are an exposed highly lipophilic element, found to be the disordered N-terminal region of the protein (residues 1–25), and a highly structured core, identified as residues 70–87, that is rich in β-sheet structure.17 The oligomers interact with lipid bilayers such that the otherwise disordered N-terminal region of the protein adopts an amphipathic helical conformation that anchors the αS oligomers to the membrane surfaces, while the highly structured regions of the assembly insert into the interior of the bilayers thereby disrupting their integrity (Figure 1A). In addition to these structural features that are characteristic of the toxic oligomeric species of αS, the oligomers expose the negatively charged and disordered C-terminus of the protein (residues 100–140), which does not, however, play a role in the mechanism of toxicity by these oligomers.17
Figure 1.
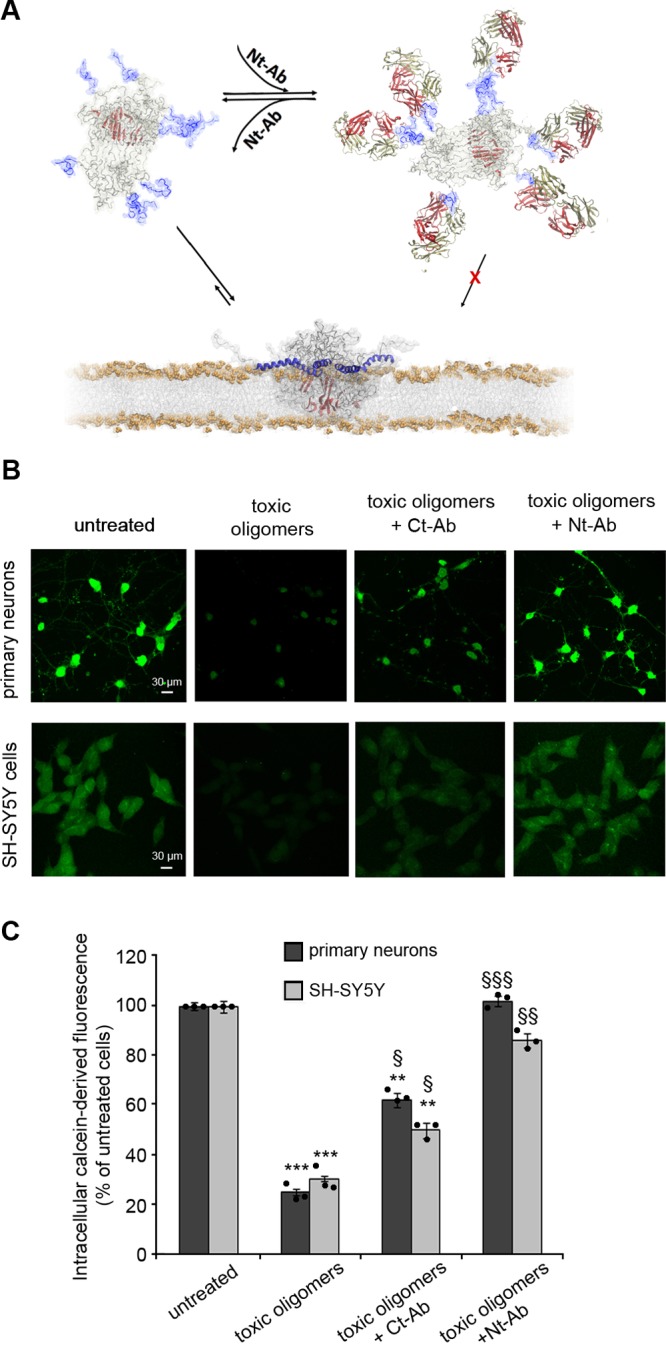
Membrane disruption by αS oligomers and its suppression by Nt-Ab. (A) Model of toxic αS oligomers,17 showing the lipophilic exposed N-terminal region of the protein (blue), and a core region that is rich in β-sheet structure (red). These elements have a role in the mechanisms by which toxic αS oligomers disrupt biological membranes. In particular, upon membrane binding, the N-terminal region of αS adopts an amphipathic α-helical conformation that anchors the oligomers to the membrane surface whereas the structured core inserts into the interior of the lipid bilayer and disrupts its integrity. The scheme shows how binding of Nt-Ab (red and yellow chains) to the N-terminal region of αS inhibits the key initial step promoting membrane binding and disruption. (B) Representative confocal scanning microscopy images of primary cortical neurons (upper panels) and SH-SY5Y cells (lower panels), illustrating the degree of intracellular calcein-induced fluorescence in the two types of cells upon incubation with 0.3 μM (monomer equivalents) αS oligomers in the absence or presence of 1:1 molar ratio of Ct-Ab or Nt-Ab. (C) Quantification of the green fluorescent signal arising from the calcein probe. Error bars indicate the SEM; ** and *** indicate p values ≤ 0.01 and ≤ 0.001 calculated with respect to the data measured on untreated cells (n = 6 per group); §, §§, and §§§ indicate respectively p ≤ 0.05, p ≤ 0.01, and p ≤ 0.001 calculated with respect to the data measured on cells treated with toxic oligomers (n = 6 per group). Samples were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test relative to untreated cells. A total of 80–120 cells were analyzed per condition in total in three independent experiments.
In the present study, we made use of a primary antibody, obtained by rabbit immunization against a peptide encompassing the 25 N-terminal residues of αS (designated as Nt-Ab, see Methods). We found that its addition to solutions containing the αS oligomers rescued the disruption of cellular viability caused by these aggregates, as probed using primary rat cortical neurons and human SH-SY5Y neuroblastoma cells. We first monitored the effects of the presence of Nt-Ab on the ability of the toxic αS oligomers to disrupt the plasma membrane of both neuronal cell types, using calcein-loaded cells and monitoring the efflux of the fluorescent calcein dye from the cytosol to the external medium (Figure 1B, C). Following incubation of the cells with αS oligomers at a concentration of 0.3 μM (monomer equivalents) in the absence of Nt-Ab for 1 h, a reduction of the calcein-derived fluorescence of ca. 70% was observed in both the primary cortical neurons and the neuroblastoma cells, indicating in both cases that the oligomers induce significant disruption of cellular membranes (Figure 1B, C). In the presence of Nt-Ab at a 1:1 molar ratio of αS:Nt-Ab, however, such disruption was almost completely inhibited, as the intracellular calcein-associated fluorescence was found to be restored almost to that of untreated cells (Figure 1B, C). By contrast, analogous experiments carried out in the presence of a primary antibody targeting the C-terminal region, spanning residues 126 to 140 of αS and designated as Ct-Ab (see Methods), resulted in moderate changes in the degree of membrane disruption (ca. 30% and 50% for SH-SY5Y and primary neurons, respectively, Figure 1B,C). This observation is likely to be attributable to steric effects on the interaction of the oligomers with the cell membrane resulting from the binding of Ct-Ab to the C-terminal region of the protein.
Effects of Primary Antibodies on the Interaction of αS Oligomers with Membranes
We then examined whether or not the addition of Nt-Ab to the toxic αS oligomers resulted in the reduction of the binding affinity of the oligomers to cellular membranes. Confocal images of primary cortical neurons and neuroblastoma cells following incubation for 15 min with the toxic αS oligomers showed strong colocalization of the aggregates with the plasma membrane of both types of cells (Figure 2A, B). When the αS oligomers were incubated with the two types of cells in the presence of Nt-Ab, however, the colocalization with the plasma membrane was essentially completely abolished (Figure 2A, B). By contrast, the addition of Ct-Ab reduced to a lower extent (ca. 60% and 50% for primary neurons and SH-SY5Y cells, respectively) the ability of the toxic αS oligomers to colocalize with cellular membranes (Figure 2A, B). The affinity of αS oligomers for cellular membranes was also probed by using a range of concentrations of Nt-Ab (0.03 μM to 0.3 μM) and a constant level of αS oligomers (0.3 μM; Figure 2C, D). The experiments showed a significant reduction in membrane binding by the oligomers even at an αS:Nt-Ab molar ratio of 1:0.5, with only marginal effects found at lower Nt-Ab concentrations (Figure 2C, D). The inhibition of oligomer binding by Nt-Ab was also confirmed with neuroblastoma cells by using oligomers formed by 10% of αS labeled with AF488 dye at the residue 122 (Figure 2E, F).
Figure 2.

Membrane binding by αS oligomers. (A) Representative confocal scanning microscopy images of primary cortical neurons (upper panels) and SH-SY5Y cells (lower panels) under the experimental conditions indicated. Red and green fluorescence indicates the cell membranes and αS oligomers, respectively. (B) Degree of membrane binding by αS oligomers measured following their incubation with primary cortical neurons and human SH-SY5Y neuroblastoma cells using 0.3 μM (monomer equivalents) αS oligomers in the absence or presence of 1:1 molar ratios of Ct-Ab or Nt-Ab. (C, D) Representative confocal scanning microscopy images (C) and quantification analysis (D) of SH-SY5Y cells treated with 0.3 μM (monomer equivalents) αS oligomers in the absence or presence of Nt-Ab at αS:Ab molar ratios of 1:0.1, 1:0.25, 1:0.5, and 1:1, where molar ratios always refer to monomer equivalents. In panel A, αS oligomers alone and in the presence of Ct-Ab were detected using 1:250 diluted rabbit polyclonal anti-αS antibodies, whereas αS oligomers in the presence of Nt-Ab were detected with 1:250 diluted mouse monoclonal anti-αS IgG1. In panel C, αS oligomers alone and in the presence of Nt-Ab were detected using 1:250 diluted mouse monoclonal anti-αS IgG1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). (E,F) Representative confocal scanning microscopy images (E) and quantification analysis (F) of primary rat cortical neurons treated with 0.3 μM (monomer equivalents) AF488 αS oligomers in the absence or presence of 1:1 molar ratios of Ct-Ab or Nt-Ab. In panels B, D, and F, the error bars indicate the SEM, ** and *** indicate p values ≤0.01 and ≤0.001 calculated with respect to the data for untreated cells (n = 6 per group), § and §§§ indicate respectively p ≤ 0.05 and p ≤ 0.001 calculated with respect to the data for cells treated with toxic oligomers (n = 6 per group). Samples were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test relative to untreated cells. A total of 80–120 cells were analyzed per condition in total in three independent experiments.
Taken together, these data indicate that by targeting the N-terminal region of αS using antibodies, it is possible to suppress almost completely the interaction of toxic aggregates with cellular membranes and hence to avoid the consequent disruption of the membrane integrity. This result is consistent with our previous mutational analysis, which showed that the toxicity of αS oligomers can be reduced significantly by modifying the sequence of the N-terminal region of αS.17
Effects of Primary Antibodies on Mitochondrial Activity
We then assessed if the presence of Nt-Ab can reduce the downstream effects arising from the incubation of neuronal cells with toxic αS oligomers, including impairment of mitochondrial activity. The results showed that the αS oligomers (0.3 μM) generated in vitro reduced substantially the mitochondrial activity of both primary cortical neurons (Figure 3A) and neuroblastoma cells (Figure 3B), as probed by the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). MTT reduction was almost fully restored at a 1:1 molar ratio of αS:Nt-Ab, both in primary neurons (Figure 3A) and in neuroblastoma cells (Figure 3B), with very minor effects observed at 1:0.5 or lower molar ratios. By contrast, incubation of the oligomers with different concentrations of Ct-Ab resulted in very modest improvements of the mitochondrial activity in both cell lines even at a 1:1 molar ratio (Figure 3A,B).
Figure 3.
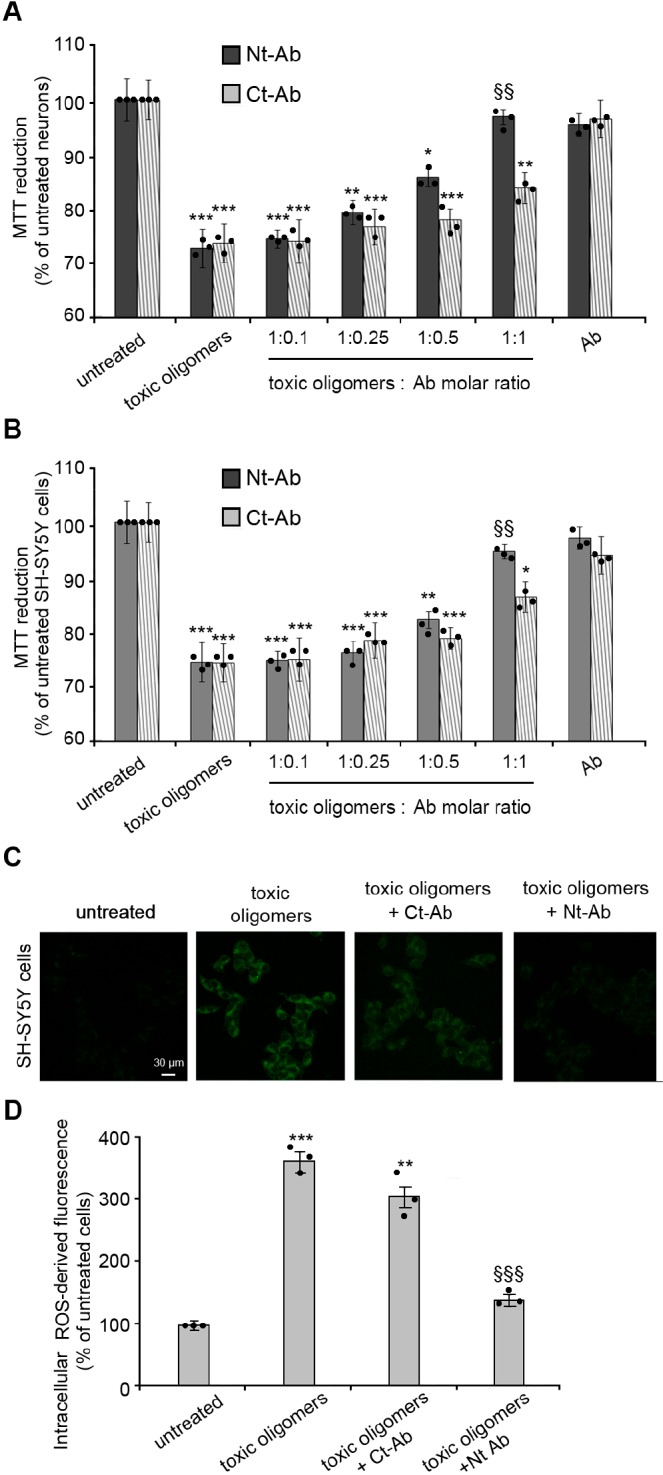
Measurements of the cellular toxicity of αS oligomers. (A,B) Mitochondrial activity monitored by the reduction of MTT, measured for rat primary cortical neurons (A) and human neuroblastoma SH-SY5Y cells (B) upon incubation with 0.3 μm αS oligomers (monomer equivalents), in the absence or presence of Nt-Ab or Ct-Ab at αS/Ab molar ratios of 1:0.1, 1:0.25, 1:0.5 and 1:1, where molar ratios always refer to monomer equivalents. (C) Intracellular ROS production in SH-SY5Y cells resulting from incubation with 0.3 μM (monomer equivalents) αS oligomers in the absence or presence of a 1:1 molar ratio of Ct-Ab or Nt-Ab. The images are representative of those obtained with confocal scanning microscopy under various experimental conditions. The green fluorescence arises from intracellular ROS. In both panels, the error bars indicate the SEM; *, **, and *** indicate p values ≤ 0.05, ≤ 0.01, and ≤ 0.001 calculated with respect to the data measured on untreated cells; §§ and §§§ indicate, respectively, p ≤ 0.01 and p ≤ 0.001 calculated with respect to the data measured on cells treated with toxic oligomers (n = 9 and n = 6 per group for MTT and ROS, respectively). Samples were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison test relative to untreated cells. A total of 80 000–120 000 cells for MTT and 80–120 cells for ROS were analyzed per condition in total in three independent experiments.
Effects of Primary Antibodies on the Generation of Intracellular Reactive Oxygen Species
Since a rise in the levels of oxidative stress has been reported to be an important feature in PD,21 we also analyzed the generation of reactive oxygen species (ROS) in neuroblastoma cells under the various conditions explored for the other examined toxicity readouts. In particular, toxic αS oligomers were found to induce a large increase (nearly 4-fold relative to untreated cells) in intracellular ROS levels (Figure 3C,D).17 A very significant reduction of intracellular ROS generation was, however, observed following incubation of the oligomers with Nt-Ab at a 1:1 molar ratio, with the levels of ROS being restored almost to the values of untreated cells (Figure 3C,D). By contrast, analogous experiments with Ct-Ab resulted in the intracellular production of ROS in neuroblastoma cells remaining very high, indicating that the presence of Ct-Ab generates only a slight reduction of oxidative stress (Figure 3C,D). Taken together, these data indicate that targeting the N-terminal region of αS oligomers by Nt-Ab results in the potent suppression of the neurotoxicity of these aggregates, in contrast to the small effects arising from targeting the C-terminal region with Ct-Ab.
Structural Analysis of Toxic Oligomers in the Presence of N-Terminal Antibody
The evidence described above suggests that Nt-Ab either blocks the interaction between αS oligomers and the cell membrane or promotes a structural reorganization (or disaggregation) of the oligomers by interacting with the exposed N-terminus. Thus, we assessed whether the structural properties of the β-sheet core and solvent-exposed hydrophobicity of the αS oligomers were perturbed by Nt-Ab using Thioflavin T (ThT) fluorescence and 1-anilino-8-naphthalenesulfonate (ANS) fluorescence, respectively. Using ThT staining, we observed that αS oligomers present a rudimentary β-sheet core, as revealed by the weak increase of ThT fluorescence (Figure 4A). An equimolar concentration of free Nt-Ab also increased weakly the fluorescence of ThT, most probably because of its large content of β-sheet structure in the native state (Figure 4A). The oligomers preincubated with Nt-Ab produced a higher increase of ThT fluorescence, as both species contribute to ThT binding (Figure 4A). The obtained blank-subtracted spectrum is similar to that determined from the sum of the blank-subtracted spectra obtained with free Nt-Ab and free oligomers (or slightly lower as the complex formation inevitably masks ThT binding sites).
Figure 4.
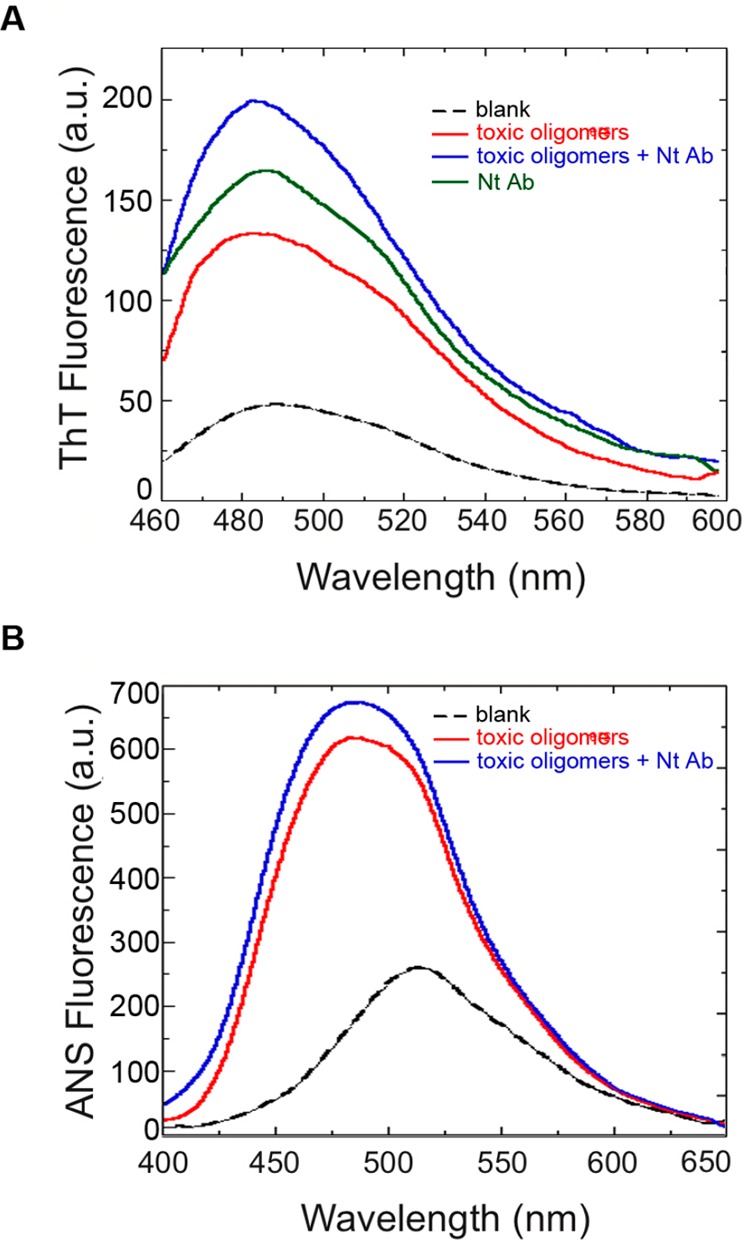
Structural characterization of αS oligomers in the absence and presence of Nt-Ab. (A) ThT fluorescence spectra of αS oligomers (4 μM) incubated in the absence (red) or presence (blue) of a 1:1 molar ratio of Nt-Ab for 15 min before performing the measurement. The ThT fluorescence spectra of free Nt-Ab (green) and without any protein (dashed line) are also shown. Samples were excited at 440 nm, and the emission fluorescence spectra were recorded between 460 and 600 nm. (B) ANS fluorescence spectra of αS oligomers (4 μM) incubated in the absence or presence of a 1:1 molar ratio of Nt-Ab for 15 min before performing the measurement. The ANS fluorescence spectrum without any protein is also shown (dashed line). Samples were excited at 350 nm, and the emission fluorescence spectra were recorded between 400 and 650 nm.
αS oligomers in the absence or presence of an equimolar ratio of Nt-Ab induced a high ANS fluorescence intensity and a virtually identical blue-shift of the wavelength of maximum ANS emission (Figure 4B). Our data indicate that the tinctorial properties and the degree of hydrophobic-exposed surfaces of αS oligomers are not affected by Nt-Ab in vitro, ruling out a significant structural reorganization or a disassembly of the oligomers by Nt-Ab.
Mechanism of Toxicity in a C. elegans Model of αS-Mediated Dysfunction
Finally, in this study, we extended our analysis of the inhibition of αS aggregation and toxicity by investigating the effects of Nt-Ab and Ct-Ab in a C. elegans model of αS-mediated dysfunction.35 In this model, nematode worms were engineered to overexpress αS, tagged with the yellow fluorescent protein (YFP), in their large muscle cells.38−40 This specific C. elegans model of αS toxicity has been instrumental in identifying genes and pathways connected with the onset and progression of PD38−40 and in characterizing the inhibition by naturally occurring aminosterols of the aggregation and toxicity of αS in vivo.35,36
As a result of the overexpression of YFP-αS, significant quantities of aggregates were observed to be deposited in the worms, as shown by fluorescence microscopy (Figure 5A, see Methods). The extent of YFP-αS aggregation determined from the images was, however, observed to be significantly reduced (by ca. 70%) in YFP-αS worms incubated with 0.4 μM Nt-Ab administered in the worm medium since day 0 of adulthood (Figure 5A,B); by contrast, the reduction of YFP-αS aggregates was marginal (ca. 15%) in YFP-αS C. elegans treated with 0.4 μM Ct-Ab. These data provide strong evidence to support the conclusion that the N-terminal region of αS plays a critical role in defining the underlying mechanism of the formation of aggregates. Indeed, this process has been shown to be promoted by the interaction of αS monomers with membranes.36,41−43 It is also possible, however, that Nt-Ab is also able to inhibit the formation of fibrils by binding to small aggregates, including oligomeric species, of αS and preventing their conversion into fibrillar species that are able to elongate. In addition to reducing the extent of aggregation as a result of the treatment with Nt-Ab, we observed the almost complete suppression of the ability of the aggregates to cause motility impairment in the animals. In particular, untreated YFP-αS animals show a very significant reduction in their motility properties, measured as bends per minute in an automated tracking device (see Methods), compared to control worms overexpressing YFP alone (Figure 5C). By administering 0.4 μM Nt-Ab in the medium, we observed an essentially complete recovery of the motility of the YFP-αS C. elegans to the levels of healthy YFP controls (Figure 5C). By contrast, no significant improvements in motility were detected as a result of the treatment with Ct-Ab (Figure 5C).
Figure 5.

Effect of Nt-Ab and Ct-Ab on the aggregation and toxicity of αS in C. elegans. (A) Representative fluorescence microscopy images of worms at day 11 of adulthood expressing YFP (left) or YFP-αS (right) in the absence (top) or presence of 0.4 μM of Ct-Ab (middle) or Nt-Ab (bottom). The inclusions are indicated by white arrows. (B) Estimates of the number of YFP-αS inclusions in worms in the absence and presence of 0.4 μM Ct-Ab or Nt-Ab; the error bars indicate the SEM. (C) Effects of Ct-Ab and Nt-Ab on the behavior of the worms overexpressing YFP-αS. The motilities measured in bends per minute are reported for control animals overexpressing YFP (dotted lines) and YFP-tagged αS (solid lines). Experiments were performed in the absence (red) or presence of 0.4 μM of Ct-Ab (green) or Nt-Ab (blue), and the error bars indicate the SEM (D) Fingerprint map64 probing different parameters of the fitness of YFP-αS C. elegans in the absence (red) or presence of 0.4 μM of Ct-Ab (green) or Nt-Ab (blue). Experiments performed in panels A–C were performed by direct incubation of Nt-Ab and Ct-Ab in the worm medium, whereas experiments in panel D were performed with the vesicle encapsulation method.44
We also employed a different protocol to administer Nt-Ab and Ct-Ab to YFP-αS C. elegans. This protocol was developed to facilitate the ingestion and cell uptake of proteins and peptides in the worms by encapsulating the biomolecules into cationic lipid vesicles.44 Antibodies administered in this way were shown to colocalize with intracellular protein aggregates formed in YFP-αS C. elegans.44 The results obtained with the vesicle encapsulation protocol confirmed that Nt-Ab and Ct-Ab have different effects on the health status of YFP-αS C. elegans (Figure 5D). In particular, five indicators of worm health showed a significant improvement of the fitness status of the YFP-αS C. elegans upon administration of Nt-Ab, in contrast to very minor effects associated with the utilization of Ct-Ab.
Overall, the antibody appears to have a double action against the effects of overexpression of αS in vivo, as it reduces significantly the extent of aggregation of the protein in C. elegans and also suppresses the ability of the residual aggregates that are formed in the worms to impair the locomotor abilities of the animals. The rescuing effect of Nt-Ab in the C. elegans experiment resembles that obtained with aminosterols35,36 despite a significant difference in their mechanism of action: binding to the membrane with consequent displacement of the oligomers in the case of aminosterols and binding to the oligomers resulting in inhibition of the interaction with the membrane in the case of Nt-Ab. These findings suggest that these two different effects act together to inhibit the key steps for the generation of toxicity following overexpression of αS in C. elegans. Moreover, these data indicate that the biological properties of isolated αS oligomers generated in vitro correlate well with the behavior of the species that are formed in vivo in a living organism.
Discussion
There is increasing evidence that the disruption of cell membranes by misfolded protein aggregates is a key step in the induction of the downstream loss of neuronal viability.17,27−29,45,46 The interaction between membranes and monomeric αS is also crucial for both function47,48 and aggregation.45,49 In this study, we have provided evidence that aggregates formed both in vitro and in vivo by αS are able to induce damage to cells by membrane disruption. In addition, using antibodies we show that the N-terminal region of αS is a crucial structural element in the generation of toxicity, both by stabilized oligomers generated in vitro and through the formation and toxicity of the aggregates generated in vivo in C. elegans. The employment of antibodies to target prefibrillar protein oligomers, including αS species,50,51 has generated encouraging results.52,53
In previous work, we have shown that toxic oligomers of αS can be formed by the structural rearrangement of initially disordered and nontoxic aggregates, resulting in the formation of oligomeric species characterized by a structured core rich in β-sheet conformation and exposed hydrophobic regions.18 Two structural features of the αS toxic oligomers were shown to be necessary to enable them to disrupt biological membranes, namely, the presence of a highly lipophilic N-terminal region of the protein promoting strong interactions with the membrane surface and the existence of a rigid core region rich in β-sheet structure that is able to insert into the lipid bilayer and cause a loss of membrane integrity.17
In the present study, we assessed the origin of the neuronal toxicity generated by αS oligomers isolated in vitro by using Nt-Ab, a specific antibody that interacts with the exposed N-terminal sequence of the protein. The results show that the antibody restores the normal viability of cultured primary cortical neurons or neuronal cells incubated in the presence of the oligomers by inhibiting their interaction with the plasma membranes of the cells, therefore avoiding the disruption of the membranes and/or alteration of the protein components that result in the generation of intracellular ROS and the impairment of mitochondrial activity. In addition, the presence of Nt-Ab was found to reduce significantly the formation of aggregates induced by the overexpression of αS in C. elegans, a process that is itself related to the interaction of monomeric αS with cell membranes. Thus, inhibition of aggregation, in conjunction with suppression of the deleterious effects of any aggregates that do form, restores the behavioral properties of the worms to those characteristic of wild-type controls. The observed reduction in locomotor impairment is particularly significant because the aggregation of αS in a living system such as C. elegans is likely to be associated with a highly heterogeneous ensemble of aggregated states of the protein.1 Yet, the toxicity of these multiple species is suppressed by the antibody as effectively as that of the highly homogeneous oligomers generated in vitro.
In conclusion, we have provided experimental evidence for a common molecular mechanism inducing membrane disruption by oligomeric species formed by the aggregation of αS in vitro and in vivo. It is likely that there are other factors that contribute to cellular toxicity, including the interaction with other cellular components,54,55 but the similarity of the behavior of the aggregated species generated under a variety of conditions provides support for the ability of in vitro systems to provide detailed mechanistic information that is relevant for living systems.
Materials and Methods
αS Purification
αS was expressed and purified in E. coli using plasmid pT7-7 encoding for the protein.56 After transforming in BL21 (DE3)-gold cells (Agilent Technologies, Santa Clara, CA USA), αS was obtained by growing the bacteria at 37 °C under constant shaking at 250 rpm in lysogeny broth (LB) medium supplemented with 100 μg·mL–1 of ampicillin to an OD600 of 0.6. Subsequently the expression of the protein was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 37 °C for 4 h, and the cells were harvested by centrifugation at 6200g (Beckman Coulter, Brea, CA USA). The cell pellets were resuspended in lysis buffer (10 mM Tris-HCl pH 8, 1 mM EDTA) and EDTA-free complete protease inhibitor cocktail tablets (Roche, Basel, Switzerland) and lysed by sonication. The cell lysate was centrifuged at 22 000g for 30 min, and the supernatant was heated for 20 min at 70 °C and centrifuged again at 22 000g. Subsequently, streptomycin sulfate was added to the supernatant to a final concentration of 10 mg·mL–1. The mixture was stirred for 15 min at 4 °C followed by further centrifugation at 22 000g. Then, ammonium sulfate was added to the supernatant to a concentration of 360 mg·mL–1 in order to precipitate the protein. The solution was stirred for 30 min at 4 °C and centrifuged again at 22 000g. The resulting pellet was resuspended in 25 mM Tris-HCl, at pH 7.7, and dialyzed against the same buffer to remove salts. The dialyzed solutions were then loaded onto an anion exchange column (26/10 Q Sepharose high performance, GE Healthcare, Little Chalfont, UK), eluted with a 0–1 M NaCl step gradient, and then further purified by loading onto a size exclusion column (Hiload 26/60 Superdex 75 preparation grade, GE Healthcare, Little Chalfont, UK). All fractions containing the monomeric protein were pooled together and concentrated by using Vivaspin filter devices (Sartorius Stedim Biotech, Gottingen, Germany). Protein purity was analyzed by SDS-PAGE, and protein concentrations were determined spectrophotometrically using ε275 = 5600 M–1 cm–1.
Preparation of αS Oligomers
Toxic αS oligomeric samples were prepared as previously described.19 Briefly, 6 mg of lyophilized protein was resuspended in PBS buffer at a pH of 7.4 and at a concentration of 12 mg·mL–1. The solution was passed through a 0.22 μm cutoff filter and subsequently incubated at 37 °C for 24 h in stationary mode and without agitation in order to avoid acceleration of fibril formation.19 Residual fibrillar species were removed by ultracentrifugation for 1 h at 288 000g using a TLA-120.2 Beckman rotor (Beckman Coulter, Brea, CA USA). The excess of αS monomers in the sample was then removed by means of several filtration steps using 100 kDa cutoff membranes, which resulted in the enrichment of the oligomeric αS species. Samples of the toxic αS oligomers prepared in this manner have been found to be stable for many days but in this study were used within 2 days of their production. Fluorescently labeled αS molecules carrying the AF488 dye (Invitrogen, Carlsbad, CA, USA) were obtained by using the N122C mutational variant, allowing the dye molecules to react with the thiol moiety of Cys122.18 The labeled protein was then purified from the excess of free dye by a P10 desalting column with a Sephadex G25 matrix (GE Healthcare, Waukesha, WI, USA) and concentrated using Amicon Ultra Centricons (Merck, Darmstadt, Germany). Fluorescent oligomers were generated by mixing 90% and 10% of unlabeled and labeled αS, respecitvely. The low ratio of labeled to unlabeled monomers and the C-terminal position of Cys122, which is not involved in oligomer formation and membrane interaction,17 ensured the absence of significant modifications to the properties of the oligomers.
Incubation of αS Oligomers with Antibodies
Nt-Ab (128003, Synaptic Systems, Gottingen, Germany) is a polyclonal rabbit antibody generated against a synthetic peptide spanning residues 2 to 25 in human αS. Ct-Ab (128211, Synaptic Systems, Gottingen, Germany) is a monoclonal mouse antibody generated against the synthetic peptide spanning residues 126 to 140 in human αS. All the experiments reported in this study were based on direct incubation of the αS oligomers (0.3 μM monomer equivalents) with an equimolar concentration of the antibodies (either Nt-Ab or Ct-Ab), using rat primary cortical neurons or human neuroblastoma SH-SY5Y cells. In a series of experiments, αS oligomers (0.3 μM monomer equivalents) were added to the cells in the presence of different concentrations of antibodies (from 0.03 μM to 0.3 μM), with αS/Ab molar ratios of 1:0.1, 1:0.25, 1:0.5, and 1:1. We also tested a protocol involving a preincubation of 1 h of the αS oligomers (0.3 μM monomer equivalents) with an equimolar concentration of the antibodies (either Nt-Ab or Ct-Ab) in the cell medium, and subsequent addition of this mixture to the neuronal cell lines, generating results that are undistinguishable from those obtained with the first protocol.
Cell Cultures
Primary rat cortical neurons were obtained from embryonic day (ED)-17 Sprague–Dawley rats (Harlan) as described in ref (57). The experimental procedures were in accordance with the standards defined in the Guide for the Care and Use of Laboratory Animals (published by the National Academy of Sciences, National Academy Press, Washington, DC). Cortical neurons were maintained in neuronal basal medium (NBM) at 37 °C in a 5.0% CO2-humidified atmosphere and analyzed 14 days after plating, as previously described.22,58 Authenticated human neuroblastoma SH-SY5Y cells were purchased from A.T.C.C. (Manassas, VA, USA). The cells were tested to ensure that they were free from mycoplasma contamination and were cultured in Dulbecco’s modified Eagle’s medium (DMEM), F-12 HAM with 25 mM HEPES, and NaHCO3 (1:1) and supplemented with 10% FBS, 1 mM glutamine, and 1.0% antibiotics.57−59 Cell cultures were maintained in a 5% CO2 humidified atmosphere at 37 °C and grown until they reached 80% confluence for a maximum of 20 passages.22,58−61
Cellular Membrane Permealization by Calcein-Derived Fluorescence
Primary rat cortical neurons and SH-SY5Y cells were plated on glass coverslips and treated for 10 min at 37 °C with 1.0 μM calcein-AM (Molecular Probes, Eugene, Oregon) diluted in culture medium, as previously described.22,61,62 The levels of intracellular calcein-derived fluorescence at 488 nm were then measured using confocal microscopy following incubation of the two types of cells with αS oligomers for 1 h at a concentration of 0.3 μM (monomer equivalents) in the absence or presence of an equimolar concentration of either Nt-Ab or Ct-Ab. Cell fluorescence was analyzed by a TCS SP5 scanning confocal microscopy system (Leica Microsystems, Mannheim, Germany) equipped with an argon laser source, using the 488 nm excitation line. A series of 1.0-μm-thick optical sections (1024 × 1024 pixels) was taken through the cells for each sample using a Leica Plan Apo 63× oil immersion objective and then projected as a single composite image by superimposition. The confocal microscope was set at optimal acquisition conditions, e.g., pinhole diameters, detector gain and laser powers. The settings were maintained at constant values for each analysis.
Imaging and Quantification of αS Oligomers Bound to the Plasma Membrane
Primary cortical neurons or SH-SY5Y cells were seeded on glass coverslips and treated for 15 min with αS oligomers at a concentration of 0.3 μM (monomer equivalents) with or without an equimolar concentration of either Nt-Ab or Ct-Ab. In each series of experiments, αS oligomers were added to SH-SY5Y cells in the absence or presence of different concentrations of Nt-Ab (from 0.03 μM to 0.3 μM). The αS/Ab molar ratios were 1:0.1, 1:0.25, 1:0.5, and 1:1. After incubation, the cells were washed with PBS and counterstained for 15 min using 5.0 μg mL–1 of Alexa Fluor 633-conjugated wheat germ agglutinin in order to label fluorescently the plasma membrane (Life Technologies, Carlsbad CA, USA).17,22,35 After washing with PBS, the cells were fixed in 2% (w/v) buffered paraformaldehyde for 10 min at RT (20 °C). αS oligomers were detected using 1:250 diluted mouse monoclonal anti-αS IgG1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or with 1:250 diluted rabbit polyclonal anti-αS antibodies (Life Technologies, Carlsbad CA, USA) that were coupled with 1:1000 diluted Alexa Fluor 488-conjugated antimouse or antirabbit secondary antibodies (Life Technologies, Carlsbad CA, USA). To detect only the oligomers bound to the cell surface, the cellular membrane was not permeabilized at this stage, thus preventing antibody internalization. Fluorescence emission was detected after double excitation at 633 and 488 nm by the scanning confocal microscopy system described above and a series of 1.0-μm-thick optical sections were projected as a single composite image by superimposition. In another series of experiments, AF488 αS oligomers (0.3 μM monomer equivalents) were also added to the cells with an equimolar concentration of the antibodies (either Nt-Ab or Ct-Ab) for 15 min. The cells were washed with PBS and counterstained with Alexa Fluor 633-conjugated wheat germ agglutinin and analyzed by confocal microscopy as described above.
MTT Reduction Assay
αS oligomers at a concentration of 0.3 μM (monomer equivalents) in the absence or presence of either Nt-Ab or Ct-Ab were added to the cell culture medium of primary rat cortical neurons or SH-SY5Y cells, seeded in 96-well plates, for 24 h. The αS/Ab molar ratios were 1:0.1, 1:0.25, 1:0.5, and 1:1. The MTT reduction assay was performed as previously described.22,58−61
ROS Measurements
αS oligomers at a concentration of 0.3 μM (monomer equivalents) in the absence or presence of an equimolar concentration of either Nt-Ab or Ct-Ab were added to the cell culture medium of primary rat cortical neurons or SH-SY5Y cells and seeded on glass coverslips for 15 min. Cells were loaded with 10 μM 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA, Life Technologies, CA, USA) as previously described.22,59 Cell fluorescence was analyzed by the confocal microscopy system described above.
ThT and ANS Fluorescence
Fluorescence measurements were performed using a 3 × 3 mm quartz cell and a PerkinElmer LS 55 spectrofluorimeter (Waltham, MA, USA) equipped with a thermostated cell holder attached to a ThermoHaake C25P water bath (Karlsruhe, Germany). ThT fluorescence was monitored by exciting the sample at 440 nm and recording the emission fluorescence spectrum between 460 and 600 nm. αS oligomers (4 μM) were incubated in the absence or presence of an equimolar ratio of Nt-Ab for 15 min with 10 μM ThT in PBS, 25 °C, before performing the measurement. ANS fluorescence was monitored by exciting the sample at 350 nm and recording the emission spectrum between 400 to 650 nm. αS oligomers (4 μM) were incubated in the absence or presence of an equimolar ratio of Nt-Ab for 15 min with 150 μM ANS and PBS, 25 °C, before recording the spectra.
C. elegans Cultures
The following C. elegans strains were used: zgIs15 [P(unc-54)::αS::YFP]IV (OW40) and rmIs126 [P(unc-54)Q0::YFP]V (OW450), kindly provided by Prof. Ellen Nollen (University of Groningen, The Netherlands). The animals were synchronized by hypochlorite bleaching, hatched overnight in M9 buffer (3 g/L KH2PO4, 6 g/L Na2HPO4, 5 g/L NaCl, 1 μM MgSO4), and subsequently cultured at 20 °C on nematode growth medium (NGM; 1 mM CaCl2, 1 mM MgSO4, 5 μg mL–1 cholesterol, 250 μM KH2PO4 pH 6, 17 g/L Agar, 3 g/L NaCl, 7.5 g/L casein) in plates seeded with the E. coli strain OP50. Saturated cultures of OP50 were grown by inoculating 50 mL of LB medium (10 g/L tryptone, 10 g/L NaCl, 5g/l yeast extract) with OP50 and incubating the culture for 16 h at 37 °C.38−40 NGM plates were seeded with bacteria by adding 350 μL of saturated OP50 to each plate and leaving the plates at 20 °C for 2–3 days. Worms were transferred from seeded NGM plates on day 3, after synchronization, to the multiwell plates. All the worms were suspended in a solution of S Medium at 75 worms/mL containing 5 mg mL–1 OP50 as a constant food source. On day 4 after synchronization, when the worms had reached the L4 stage, 75 μM 5-fluoro-2′-deoxy-uridine (FUDR) was introduced to prevent the development of future generations. On the same day, Nt-Ab or Ct-Ab was added to the wells at a concentration of 0.4 μM.
Automated C. elegans Motility Assays
Automated motility assays58 in multiwells were performed by placing OP50 worms on a benchtop plate shaker at 750 rpm for 2 min to distribute them evenly. Immediately after shaking, the worms were staged on the platform for imaging, and collection was initiated 60 s after shaking. Up to 200 animals were counted in each experiment. This protocol was adapted from ref (63) and optimized for analysis of motility.
Determination of YFP-αS Inclusions in C. elegans
Individual animals were mounted on 2% agarose pads, containing 40 mM NaN3 as an anesthetic, on glass microscope slides for imaging. Quantification of the number of inclusions in YFP-αS animals was performed only for the frontal region of the worms38 as described previously64 using a Leica MZ16 FA fluorescence dissection stereomicroscope (Leica Microsystems, Wetzlar, Germany) at a nominal magnification of 20×, images were acquired using an Evolve512 Delta EMCCD Camera, with high quantum efficiency (Photometrics, Tucson, AZ, USA). Measurements on inclusions were performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). At least 50 animals were examined for each condition. All experiments were carried out in triplicate, and the data from one representative experiment are shown in the figure.
Acknowledgments
This research was supported by Parkinson’s UK (G-1508 to A.D.S., G.F., M.V., C.M.D.), the UK Medical Research Council (MR/N000676/1 to A.D.S., M.V., C.M.D.), the Agency of Science, Technology and Research of Singapore (to S.W.C.), the Fondi di Ateneo of the University of Florence (to F.C., C.C.), the Ministry of Education, Universities and Research of Italy (Progetto Dipartimento di Eccellenza “Gender Medicine” to C.C.) and the Centre for Misfolding Diseases of the University of Cambridge (C.M.D., M.V.).
Author Contributions
R.C., F.C. and C.C. designed the cell study. M.P., G.F., and A.D.S. designed the C. elegans study. G.F., C.M.D., and A.D.S. identified the strategy to suppress the toxicity of αS oligomers based on structural analysis. R.C. performed the cellular and biopysical experiments and the associated data analysis. M.P. performed the C. elegans experiments and the associated data analysis. S.W.C. and G.F. purified αS and generated its oligomers. R.C., F.C., C.M.D., and A.D.S. drafted the manuscript. All authors contributed to the manuscript revisions.
The authors declare no competing financial interest.
References
- Chiti F.; Dobson C. M. (2017) Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Eliezer D. (2009) Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 10, 483–499. 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth E. S.; Stavrovskaya I. G.; Bartels T.; Kristal B. S.; Selkoe D. J. (2014) Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 289, 21490–21507. 10.1074/jbc.M113.545749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel H. A.; Overk C. R.; Oueslati A.; Masliah E. (2013) The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J.; Masliah E. (2015) Neurodegeneration: Aggregates feel the strain. Nature 522, 296–297. 10.1038/nature14526. [DOI] [PubMed] [Google Scholar]
- Roberts H. L.; Brown D. R. (2015) Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules 5, 282–305. 10.3390/biom5020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin M. C.; Kachergus J.; Roumier C.; Mouroux V.; Douay X.; Lincoln S.; Levecque C.; Larvor L.; Andrieux J.; Hulihan M.; Waucquier N.; Defebvre L.; Amouyel P.; Farrer M.; Destée A. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Singleton A. B.; Farrer M.; Johnson J.; Singleton A.; Hague S.; Kachergus J.; Hulihan M.; Peuralinna T.; Dutra A.; Nussbaum R.; Lincoln S.; Crawley A.; Hanson M.; Maraganore D.; Adler C.; Cookson M. R.; Muenter M.; Baptista M.; Miller D.; Blancato J.; Hardy J.; Gwinn-Hardy K. (2003) Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Tuttle M. D.; Comellas G.; Nieuwkoop A. J.; Covell D. J.; Berthold D. A.; Kloepper K. D.; Courtney J. M.; Kim J. K.; Barclay A. M.; Kendall A.; Wan W.; Stubbs G.; Schwieters C. D.; Lee V. M.; George J. M.; Rienstra C. M. (2016) Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat. Struct. Mol. Biol. 23, 409–415. 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J.; McFarlane H. T.; Madsen AØ.; Riekel C.; Eisenberg D. (2007) Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 447, 453–457. 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Vilar M.; Chou H. T.; Lührs T.; Maji S. K.; Riek-Loher D.; Verel R.; Manning G.; Stahlberg H.; Riek R. (2008) The fold of alpha-synuclein fibrils. Proc. Natl. Acad. Sci. U. S. A. 105, 8637–8642. 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J. A.; Ivanova M. I.; Sawaya M. R.; Cascio D.; Reyes F. E.; Shi D.; Sangwan S.; Guenther E. L.; Johnson L. M.; Zhang M.; Jiang L.; Arbing M. A.; Nannenga B. L.; Hattne J.; Whitelegge J.; Brewster A. S.; Messerschmidt M.; Boutet S.; Sauter N. K.; Gonen T.; Eisenberg D. S. (2015) Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 525, 486–490. 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousset L.; Pieri L.; Ruiz-Arlandis G.; Gath J.; Jensen P. H.; Habenstein B.; Madiona K.; Olieric V.; Böckmann A.; Meier B. H.; Melki R. (2013) Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 4, 2575. 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gath J.; Bousset L.; Habenstein B.; Melki R.; Meier B. H.; Bockmann A. (2014) Yet another polymorph of alpha-synuclein: solid-state sequential assignments. Biomol. NMR Assignments 8, 395–404. 10.1007/s12104-013-9526-y. [DOI] [PubMed] [Google Scholar]
- Heise H.; Hoyer W.; Becker S.; Andronesi O. C.; Riedel D.; Baldus M. (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. U. S. A. 102, 15871–15876. 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G.; Lemkau L. R.; Nieuwkoop A. J.; Kloepper K. D.; Ladror D. T.; Ebisu R.; Woods W. S.; Lipton A. S.; George J. M.; Rienstra C. M. (2011) Structured regions of alpha-synuclein fibrils include the early-onset Parkinson’s disease mutation sites. J. Mol. Biol. 411, 881–895. 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G.; Chen S. W.; Williamson P. T. F.; Cascella R.; Perni M.; Jarvis J. A.; Cecchi C.; Vendruscolo M.; Chiti F.; Cremades N.; Ying L.; Dobson C. M.; De Simone A. (2017) Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 358, 1440–1443. 10.1126/science.aan6160. [DOI] [PubMed] [Google Scholar]
- Cremades N.; Cohen S. I.; Deas E.; Abramov A. Y.; Chen A.; Orte A.; Sandal M.; Clarke R. W.; Dunne P.; Aprile F. A.; Bertoncini C. W.; Wood N. W.; Knowles T. P.; Dobson C. M.; Klenerman D. (2012) Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 149, 1048–1059. 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. W.; Drakulic S.; Deas E.; Ouberai M.; Aprile F. A.; Arranz R.; Ness S.; Roodveldt C.; Guilliams T.; De-Genst E. J.; Klenerman D.; Wood N. W.; Knowles T. P.; Alfonso C.; Rivas G.; Abramov A. Y.; Valpuesta J. M.; Dobson C. M.; Cremades N. (2015) Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation. Proc. Natl. Acad. Sci. U. S. A. 112, E1994–2003. 10.1073/pnas.1421204112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova P. R.; Ludtmann M. H.; Horrocks M. H.; Negoda A.; Cremades N.; Klenerman D.; Dobson C. M.; Wood N. W.; Pavlov E. V.; Gandhi S.; Abramov A. Y. (2016) Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. 10.1242/jcs.180737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E.; Cremades N.; Angelova P. R.; Ludtmann M. H.; Yao Z.; Chen S.; Horrocks M. H.; Banushi B.; Little D.; Devine M. J.; Gissen P.; Klenerman D.; Dobson C. M.; Wood N. W.; Gandhi S.; Abramov A. Y. (2016) Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signaling 24, 376–391. 10.1089/ars.2015.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchi C.; Stefani M. (2013) The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 182, 30–43. 10.1016/j.bpc.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Winner B.; Jappelli R.; Maji S. K.; Desplats P. A.; Boyer L.; Aigner S.; Hetzer C.; Loher T.; Vilar M.; Campioni S.; Tzitzilonis C.; Soragni A.; Jessberger S.; Mira H.; Consiglio A.; Pham E.; Masliah E.; Gage F. H.; Riek R. (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U. S. A. 108, 4194–4199. 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan O. W.; Chung K. K. (2012) The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson’s disease. PLoS One 7, e38545. 10.1371/journal.pone.0038545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieri L.; Madiona K.; Melki R. (2016) Structural and functional properties of prefibrillar α-synuclein oligomers. Sci. Rep. 6, 24526. 10.1038/srep24526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M. E.; Greimel S. J.; Amar F.; LaCroix M.; Boyle G.; Sherman M. A.; Schley H.; Miel C.; Schneider J. A.; Kayed R.; Benfenati F.; Lee M. K.; Bennett D. A.; Lesné S. E. (2017) Selective lowering of synapsins induced by oligomeric α-synuclein exacerbates memory deficits. Proc. Natl. Acad. Sci. U. S. A. 114, E4648–E4657. 10.1073/pnas.1704698114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Diggelen F.; Hrle D.; Apetri M.; Christiansen G.; Rammes G.; Tepper A.; Otzen D. E. (2019) Two conformationally distinct α-synuclein oligomers share common epitopes and the ability to impair long-term potentiation. PLoS One 14, e0213663. 10.1371/journal.pone.0213663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell A. K.; Galvagnion C.; Gaspar R.; Sparr E.; Vendruscolo M.; Knowles T. P. J.; Linse S.; Dobson C. M. (2014) Solution conditions determine the relative importance of nucleation and growth processes in alpha-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 111, 7671–7676. 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P.; Vendruscolo M.; Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Grey M.; Linse S.; Nilsson H.; Brundin P.; Sparr E. (2011) Membrane interaction of alpha-synuclein in different aggregation states. J. Parkinsons Dis 1, 359–371. 10.3233/JPD-2011-11067. [DOI] [PubMed] [Google Scholar]
- Lorenzen N.; Lemminger L.; Pedersen J. N.; Nielsen S. B.; Otzen D. E. (2014) The N-terminus of alpha-synuclein is essential for both monomeric and oligomeric interactions with membranes. FEBS Lett. 588, 497–502. 10.1016/j.febslet.2013.12.015. [DOI] [PubMed] [Google Scholar]
- Danzer K. M.; Haasen D.; Karow A. R.; Moussaud S.; Habeck M.; Giese A.; Kretzschmar H.; Hengerer B.; Kostka M. (2007) Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S.; Mannini B.; Zampagni M.; Pensalfini A.; Parrini C.; Evangelisti E.; Relini A.; Stefani M.; Dobson C. M.; Cecchi C.; Chiti F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147. 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Stefani M. (2010) Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 277, 4602–4613. 10.1111/j.1742-4658.2010.07889.x. [DOI] [PubMed] [Google Scholar]
- Perni M.; Galvagnion C.; Maltsev A.; Meisl G.; Müller M. B. D.; Challa P. K.; Kirkegaard J. B.; Flagmeier P.; Cohen S. I. A.; Cascella R.; Chen S. W.; Limboker R.; Sormanni P.; Heller G. T.; Aprile F. A.; Cremades N.; Cecchi C.; Chiti F.; Nollen E. A. A.; Knowles T. P. J.; Vendruscolo M.; Bax A.; Zasloff M.; Dobson C. M. (2017) A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. U. S. A. 114, E1009–E1017. 10.1073/pnas.1610586114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perni M.; Flagmeier P.; Limbocker R.; Cascella R.; Aprile F. A.; Galvagnion C.; Heller G. T.; Meisl G.; Chen S. W.; Kumita J. R.; Challa P. K.; Kirkegaard J. B.; Cohen S. I. A.; Mannini B.; Barbut D.; Nollen E. A. A.; Cecchi C.; Cremades N.; Knowles T. P. J.; Chiti F.; Zasloff M.; Vendruscolo M.; Dobson C. M. (2018) Multistep Inhibition of α-Synuclein Aggregation and Toxicity in Vitro and in Vivo by Trodusquemine. ACS Chem. Biol. 13, 2308–2319. 10.1021/acschembio.8b00466. [DOI] [PubMed] [Google Scholar]
- Devine M. J.; Ryten M.; Vodicka P.; Thomson A. J.; Burdon T.; Houlden H.; Cavaleri F.; Nagano M.; Drummond N. J.; Taanman J. W.; Schapira A. H.; Gwinn K.; Hardy J.; Lewis P. A.; Kunath T. (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2, 440. 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham T. J.; Holmberg M. A.; van der Goot A. T.; Teuling E.; Garcia-Arencibia M.; Kim H. E.; Du D.; Thijssen K. L.; Wiersma M.; Burggraaff R.; van Bergeijk P.; van Rheenen J.; Jerre van Veluw G.; Hofstra R. M.; Rubinsztein D. C.; Nollen E. A. (2010) Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612. 10.1016/j.cell.2010.07.020. [DOI] [PubMed] [Google Scholar]
- van Ham T. J.; Thijssen K. L.; Breitling R.; Hofstra R. M.; Plasterk R. H.; Nollen E. A. (2008) C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 4, e1000027. 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot A. T.; Zhu W.; Vázquez-Manrique R. P.; Seinstra R. I.; Dettmer K.; Michels H.; Farina F.; Krijnen J.; Melki R.; Buijsman R. C.; Ruiz Silva M.; Thijssen K. L.; Kema I. P.; Neri C.; Oefner P. J.; Nollen E. A. (2012) Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. U. S. A. 109, 14912–14917. 10.1073/pnas.1203083109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodner C. R.; Maltsev A. S.; Dobson C. M.; Bax A. (2010) Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 49, 862–871. 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G.; De Simone A.; Arosio P.; Vendruscolo M.; Veglia G.; Dobson C. M. (2016) Structural Ensembles of Membrane-bound alpha-Synuclein Reveal the Molecular Determinants of Synaptic Vesicle Affinity. Sci. Rep. 6, 27125. 10.1038/srep27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G.; Pape T.; Stephens A. D.; Mahou P.; Costa A. R.; Kaminski C. F.; Kaminski Schierle G. S.; Vendruscolo M.; Veglia G.; Dobson C. M.; De Simone A. (2016) Structural basis of synaptic vesicle assembly promoted by alpha-synuclein. Nat. Commun. 7, 12563. 10.1038/ncomms12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perni M.; Aprile F. A.; Casford S.; Mannini B.; Sormanni P.; Dobson C. M.; Vendruscolo M. (2017) Delivery of Native Proteins into C. elegans Using a Transduction Protocol Based on Lipid Vesicles. Sci. Rep. 7, 15045. 10.1038/s41598-017-13755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck P. K.; Caraveo G.; Lindquist S. (2010) alpha-Synuclein, membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Soper J. H.; Kehm V.; Burd C. G.; Bankaitis V. A.; Lee V. M. (2011) Aggregation of alpha-synuclein in S. cerevisiae is associated with defects in endosomal trafficking and phospholipid biosynthesis. J. Mol. Neurosci. 43, 391–405. 10.1007/s12031-010-9455-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead D.; Eliezer D. (2014) Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol 23, 292–313. 10.5607/en.2014.23.4.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G.; Sanz-Hernandez M.; De Simone A. (2018) Order and disorder in the physiological membrane binding of α-synuclein. Curr. Opin. Struct. Biol. 48, 49–57. 10.1016/j.sbi.2017.09.004. [DOI] [PubMed] [Google Scholar]
- Galvagnion C.; Buell A. K.; Meisl G.; Michaels T. C.; Vendruscolo M.; Knowles T. P.; Dobson C. M. (2015) Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234. 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson G.; Lindström V.; Rostami J.; Nordström E.; Lannfelt L.; Bergström J.; Ingelsson M.; Erlandsson A. (2017) Alpha-synuclein oligomer-selective antibodies reduce intracellular accumulation and mitochondrial impairment in alpha-synuclein exposed astrocytes. J. Neuroinflammation 14, 241. 10.1186/s12974-017-1018-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emadi S.; Barkhordarian H.; Wang M. S.; Schulz P.; Sierks M. R. (2007) Isolation of a human single chain antibody fragment against oligomeric alpha-synuclein that inhibits aggregation and prevents alpha-synuclein-induced toxicity. J. Mol. Biol. 368, 1132–1144. 10.1016/j.jmb.2007.02.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Canto I.; Breydo L.; Rasool S.; Lukacsovich T.; Wu J.; Albay R. 3rd; Pensalfini A.; Yeung S.; Head E.; Marsh J. L.; Glabe C. (2010) Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57. 10.1186/1750-1326-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Ferreira; Temido-Ferreira M.; Vicente Miranda H.; Batalha V. L.; Coelho J. E.; Szegö ÉM.; Marques-Morgado I.; Vaz S. H.; Rhee J. S.; Schmitz M.; Zerr I.; Lopes L. V.; Outeiro T. F. (2017) α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 20, 1569–79. 10.1038/nn.4648. [DOI] [PubMed] [Google Scholar]
- Diógenes M. J.; Rombo D. M.; Vicente Miranda H.; Maiolino F.; Guerreiro P.; Näsström T.; Franquelim H. G.; Oliveira L. M.; Castanho M. A.; Lannfelt L.; Bergström J.; Ingelsson M.; Quintas A.; Sebastião A. M.; Lopes L. V.; Outeiro T. F.; Dias R. B. (2012) Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 32, 11750–11762. 10.1523/JNEUROSCI.0234-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G.; De Simone A.; Gopinath T.; Vostrikov V.; Vendruscolo M.; Dobson C. M.; Veglia G. (2014) Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat. Commun. 5, 3827. 10.1038/ncomms4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongers G.; Sallmen T.; Passani M. B.; Mariottini C.; Wendelin D.; Lozada A.; van Marle A.; Navis M.; Blandina P.; Bakker R. A.; Panula P.; Leurs R. (2007) The Akt/GSK-3beta axis as a new signaling pathway of the histamine H(3) receptor. J. Neurochem. 103, 248–258. 10.1111/j.1471-4159.2007.04752.x. [DOI] [PubMed] [Google Scholar]
- Cascella R.; Conti S.; Tatini F.; Evangelisti E.; Scartabelli T.; Casamenti F.; Wilson M. R.; Chiti F.; Cecchi C. (2013) Extracellular chaperones prevent Abeta42-induced toxicity in rat brains. Biochim. Biophys. Acta, Mol. Basis Dis. 1832, 1217–1226. 10.1016/j.bbadis.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Capitini C.; Conti S.; Perni M.; Guidi F.; Cascella R.; De Poli A.; Penco A.; Relini A.; Cecchi C.; Chiti F. (2014) TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS One 9, e86720. 10.1371/journal.pone.0086720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelisti E.; Cascella R.; Becatti M.; Marrazza G.; Dobson C. M.; Chiti F.; Stefani M.; Cecchi C. (2016) Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 6, 32721. 10.1038/srep32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella R.; Evangelisti E.; Bigi A.; Becatti M.; Fiorillo C.; Stefani M.; Chiti F.; Cecchi C. (2017) Soluble Oligomers Require a Ganglioside to Trigger Neuronal Calcium Overload. J. Alzheimer's Dis. 60, 923–938. 10.3233/JAD-170340. [DOI] [PubMed] [Google Scholar]
- Zampagni M.; Cascella R.; Casamenti F.; Grossi C.; Evangelisti E.; Wright D.; Becatti M.; Liguri G.; Mannini B.; Campioni S.; Chiti F.; Cecchi C. (2011) A comparison of the biochemical modifications caused by toxic and non-toxic protein oligomers in cells. J. Cell Mol. Med. 15, 2106–2116. 10.1111/j.1582-4934.2010.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis G. M.; Petrascheck M. (2011) Measuring Caenorhabditis elegans life span in 96 well microtiter plates. J. Visualized Exp. 49, 2496. 10.3791/2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perni M.; Challa P. K.; Kirkegaard J. B.; Limbocker R.; Koopman M.; Hardenberg M. C.; Sormanni P.; Müller T.; Saar K. L.; Roode L. W. Y.; Habchi J.; Vecchi G.; Fernando N.; Casford S.; Nollen E. A. A.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. (2018) Massively parallel C. elegans tracking provides multi-dimensional fingerprints for phenotypic discovery. J. Neurosci. Methods 306, 57–67. 10.1016/j.jneumeth.2018.02.005. [DOI] [PubMed] [Google Scholar]