

Abstract
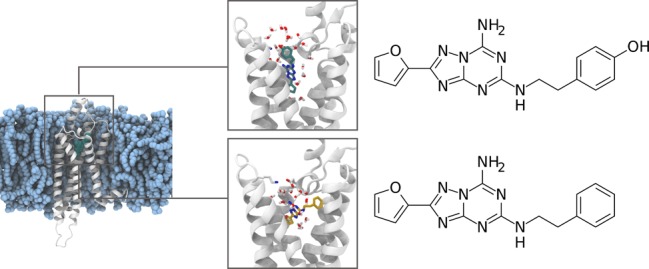
Adenosine receptors are involved in many pathological conditions and are thus promising drug targets. However, developing drugs that target this GPCR subfamily is a challenging task. A number of drug candidates fail due to lack of selectivity which results in unwanted side effects. The extensive structural similarity of adenosine receptors complicates the design of selective ligands. The problem of selective targeting is a general concern in GPCRs, and in this respect adenosine receptors are a prototypical example. Here we use enhanced sampling simulations to decipher the determinants of selectivity of ligands in A2a and A1 adenosine receptors. Our model shows how small differences in the binding pocket and in the water network around the ligand can be leveraged to achieve selectivity.
Introduction
Adenosine receptors are Class A G-protein-coupled receptors (GPCRs) that are widely expressed in neurons, immune system cells, and vascular system.1 The adenosine signaling has long been shown to regulate cytoprotective and immune response in tissues.2 Of the four subtypes, A1R and A2aR have received most attention from the scientific community, mainly due to their therapeutic potential for the treatment of reperfusion injury and neuropathic pain (A1R), cancer, attention deficit hyperactivity disorder (ADHD), and Parkinson’s Disease (PD) (A2aR).3,4 Notwithstanding significant efforts, the success in developing molecules with high potency and selectivity has been limited.5 The proteins have a sequence identity of 37%,6 with limited structural differences and remarkably similar orthosteric binding pockets7−9 (Figure 1), as all key pocket-lining residues are conserved with the exception of the position 7.35 (Ballesteros-Weinstein numbering scheme,10 where the first number refers to the transmembrane helix and the second to the position relative to the most conserved residue of the helix, which is arbitrarily set to 50). The lack of differences in the pockets of the two receptors poses a real challenge for the design of selective ligands.5
Figure 1.
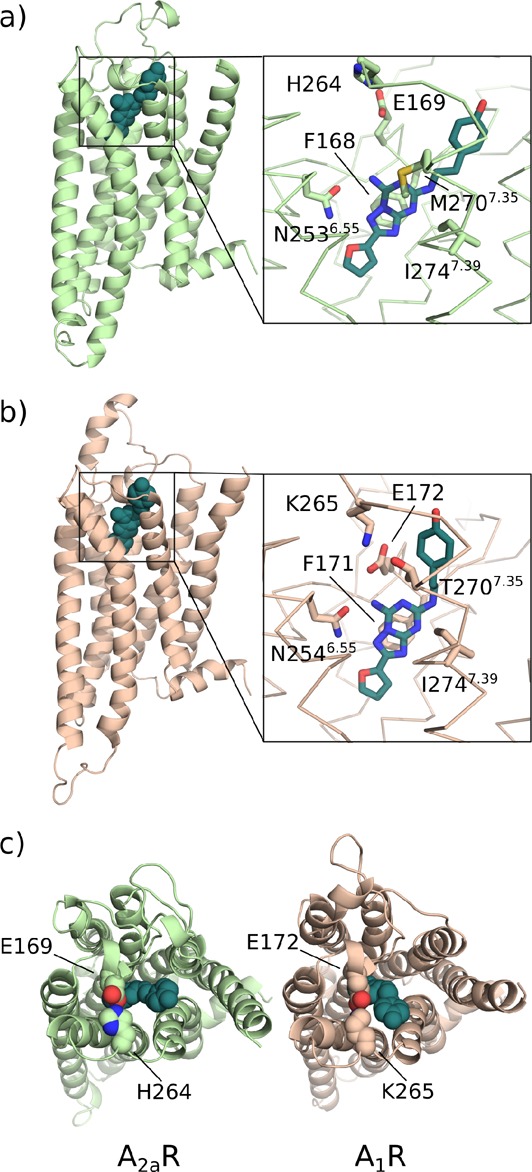
ZM241385 bound to the binding sites of (a) A2aR and (b) A1R in the enhanced-sampling simulations. (c) The salt bridge hindering the pocket entrance is shown as spheres. A zoomed view of the ligands in the binding sites and the key residues is presented in the boxes.
Thus, understanding how ZM241385, an A2aR-selective inverse agonist, achieves its selectivity is of great importance. To this aim we have recently solved and analyzed a new crystal structure of the complex.11 However, the highly dynamical nature of the receptors is not fully captured by the crystal structures,11−14 leaving a number of questions unanswered. For instance, while ZM241385 shows greatly reduced affinity for A1R, its dehydroxy derivative LUF545215 binds with the same affinity to the two proteins (Table 1). The two ligands are very similar (Figure 2), and the crystallographic binding mode of ZM241385 to A2aR shows that the only part of the ligand differing to LUF5452 does not form significant direct interactions with the receptor.11
Table 1. Experimental and Calculated Binding Free Energy of ZM241385 and LUF5452 to A1R and A2aRa.
| A1R |
A2aR |
|||
|---|---|---|---|---|
| ΔGexp | ΔGcalc | ΔGexp | ΔGcalc | |
| ZM241385 | –9.05a,–7.81b | –8.23 ± 0.73 | –12.01a,–12.90b | –11.87 ± 0.43 |
| LUF5452 | –11.19a | –10.34 ± 0.27 | –11.35a | –10.57 ± 0.53 |
Figure 2.
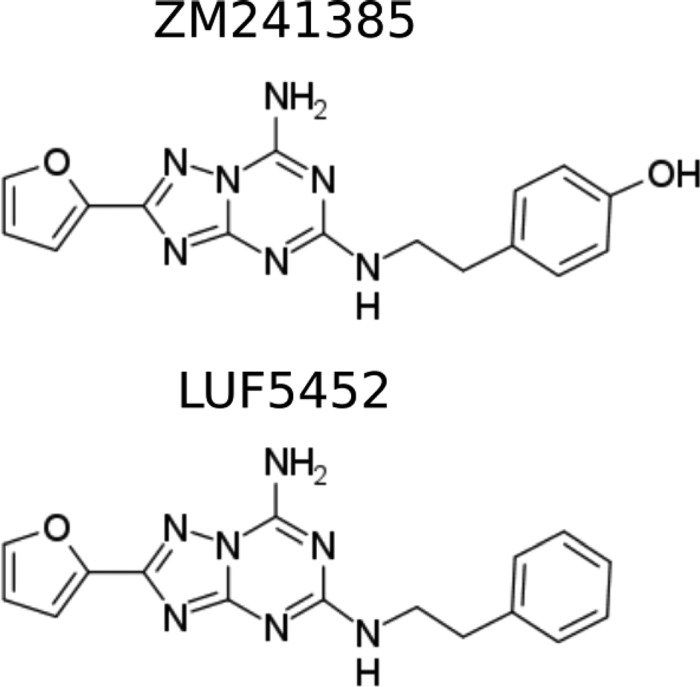
Ligands in this study, ZM241385 and LUF5452.
The challenges of explaining the differential binding affinities of ligands based on static poses hints at an important role of the binding mechanisms and perhaps different flexibility of the receptors. Thus, to fully understand the determinants of selectivity, the crystal structures need to be complemented by a method that is able to reconstruct the binding process in atomistic detail and provide information on the different dynamical properties of the receptors. To this end, here we use molecular dynamics simulations (MD) combined with enhanced sampling algorithms, that have been successfully used to model ligand binding in GPCRs.11,16,17
Results and Discussion
Interaction of the Ligands with the Binding Sites
To elucidate the binding pose and the role of the interfacial water, 1 μs-long unbiased MD were run for human A1R and A2aR in complex with either ZM241385 or LUF5452 starting with the ligand bound to the binding site. An important difference in the binding sites of the two receptors is constituted by the 7.35 position, T2707.35 in A1R and M2707.35 in A2aR. Its role in determining the selectivity of ligands with bulky substituents has been proposed in previous studies.9 Indeed, in our simulations we observe a poorer stacking of the ligand cores in A1R due to the smaller side chain of the threonine residue with respect to the methionine of A2aR. Thus, while in A2aR the ligands remained close to the initial pose (with two relatively similar conformations), in A1R a significant reorientation of the (4-hydroxy)phenyl-ethyl tail was observed.
Of the two conformations adopted by the ligands in A2aR, one is close to the starting crystallographic structure (PDB 5IU4(11)), with the (4-hydroxy)phenyl-ethyl tail pointing toward the extracellular side of the protein, and one is very similar to the crystallographic structure of the thermostabilized receptor (PDB 3PWH),13 with the tail pointing toward TM1. The various conformations can be monitored using the RMSD of the common heavy atoms of the two ligands (Figure 3). While ZM241385 preferentially adopts the upright (5IU4-like) position in the simulations, LUF5452 tends to explore 3PWH-like conformations. In this conformation, ZM241385 is able to form polar interactions with Y91.35, E131.39, and A632.61. The interconversion of the poses has significant effect on the hydration of the tail, with partial desolvation happening for 3PWH-like states and allowing LUF5452 to minimize unfavorable interactions between the solvent and the phenyl ring, as seen in Figure 4. A hydrogen bond network involving the ligands and water molecules stabilizes the molecules in the pocket of A2aR (Figure 3, conformations a1 and b1), as is also evident from crystallographic structures.11,12 Although the cavity is wide enough for observing much movement of solvent molecules, there is a tendency to adopt ordered patterns of interaction, that are disrupted in 3PWH-like states. The desolvation of the phenyl ring leads LUF5452 to frequently adopt such conformations and leads to a poor arrangement of water molecules, contributing to the lower affinity for the receptor with respect to ZM241385.
Figure 3.

Analysis of the conformations adopted by ZM241385 and LUF5452 in the binding pocket of A2aR and A1R over the unbiased MD simulation. Probability over the space defined by the RMSD of the common heavy atoms of the ligands to the conformation adopted in PDB 5IU4 or 3PWH, after alignment to the backbone of the proteins. TM7 not shown for clarity.
Figure 4.
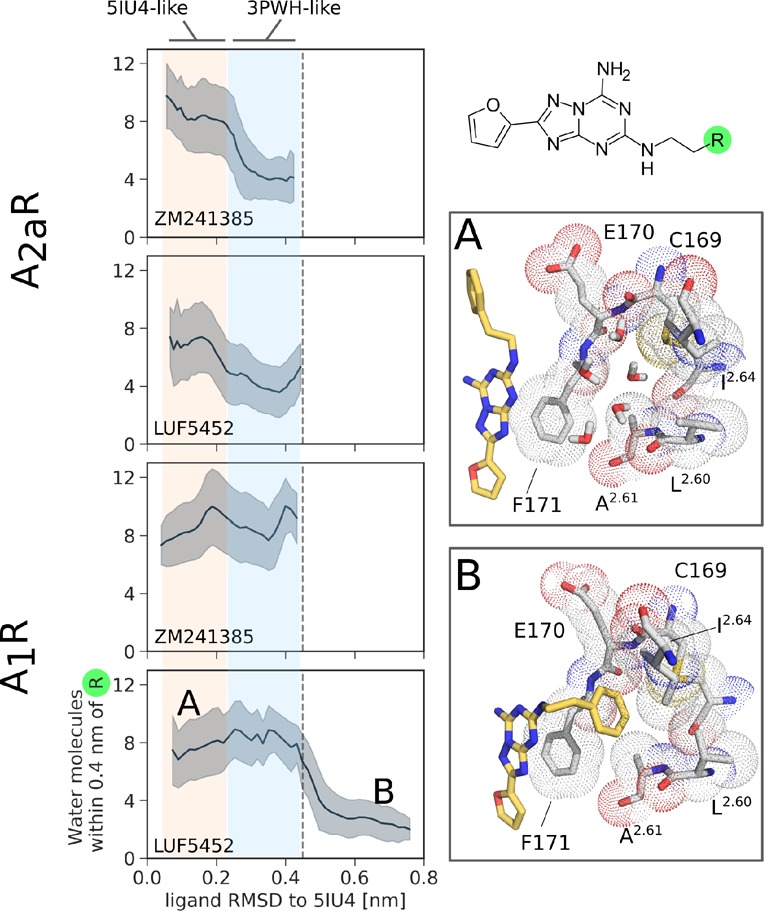
Solvation of the 4-hydroxyphenyl or phenyl groups of the ligands over the unbiased MD simulations, represented by the number of water oxygen atoms within 4 Å from the moieties’ heavy atoms. The plots illustrate the average solvation and its standard deviation as a function of the RMSD of the ligand to an upright, 5IU4-like conformation (see Figure 3). Conformations A and B show the displacement of water molecules in the hydrophobic ECV pocket of A1R by LUF5452.
In A1R, the ligands are much more mobile. ZM241385 was observed rotating the tail to a 3PWH-like conformation (Figure 3, conformation c2) and temporarily interacting with an hydrophobic cavity of the extracellular vestibule (ECV) formed by ECL2 and the ends of the transmembrane helices (TM) 2 and 3 (See Figure S1a). LUF5452 instead undergoes a more significant conformational rearrangement, rotating its core away from the upright orientation and projecting the tail toward the same pocket without reverting to the starting pose (Figure 3, conformation d2). In this conformation the hydrogen bond between the exocyclic primary amine of the ligand and N2546.55 is broken to allow the phenyl ring of the molecule to interact with the pocket. The distance between the amine nitrogen of LUF5452 and the carbonyl oxygen of N2546.55 increases from 3 to 6 Å. This results in a significant rearrangement of the interfacial water molecules (see Figure S2). Similar to A2aR, adopting a 3PWH-like conformation results in a partial desolvation (0.30 > ligand RMSD to 5IU4 > 0.45 nm in Figure 4). Moreover, the wide volume of the pocket of A1R allows for the presence of a large number of solvent molecules. While in this receptor the interactions of the tail of ZM241385 with the solvent are stabilized by the hydroxy group, LUF5452 is instead able to bury the phenyl ring in the hydrophobic ECV pocket, leading to more effective desolvation than in A2aR (Figure 4B).
Binding Poses of the Ligands
To better quantify the relative free energy of the different poses, as well as to compute the full free energy landscape associated with the binding of the ligands, we run parallel tempering metadynamics simulations (PT-metaD)19,20 on the four complexes. For A2aR, the chosen collective variables (CVs) were the projection of the vector connecting the binding pocket and the ligand onto the Z-axis (Z-projection) and onto the XY-plane (XY-projection) parallel to the membrane. In A1R, extensive tests showed that including the distance between the salt bridge-forming residues, E172 and K265 speeds the convergence of the free energy. Thus, for ZM241385, we used the distance and the Z-projection, while for LUF5452 all three CVs were used (see the Supporting Information).
The deepest free energy minimum for the two ligands corresponds to the crystallographic binding pose of ZM24138511,12 in A2aR and closely resemble it in A1R (Figure 5). As expected, the presence of the same key residues in the pocket allows the molecules to form analogous interactions and therefore adopt similar binding poses with respect to A2aR. The binding free energy associated with the minima in all four systems agrees well with experimental values (Table 1).
Figure 5.

Projection of the free energy landscapes onto Z-projection and XY-projection by reweighting.18 Boxes: Representative conformations of the ligands bound to the main binding site of the receptors, LUF5452 interacting with the hydrophobic ECV pocket in A1R, and the two molecules breaking the salt bridge at the entrance of the orthosteric site of the proteins.
LUF5452 Interacts with the Accessory Hydrophobic Site
As observed in the unbiased MD simulations, LUF5452 also shows an equally stable secondary minimum in which it makes significant contacts with the hydrophobic ECV pocket of A1R (Figure 5). The interaction allows the ligand to rotate away from the expected pose, breaking the hydrogen bond between the exocyclic amine and the side chain of N2546.55 (Figure S1a). The region is lined by L652.60, A662.61, I692.64, V833.28, A843.29, C169, E170, and F171. In this conformation, the phenyl group of the ligand is nearly parallel to TM2 and is positioned between the side chains of I692.64 and F171. The ring of F171 is involved in a stacking interaction with the core of the ligand and a T-shaped (quadrupolar) one with its phenyl group. The presence of the aromatic ring of LUF5452 in the pocket displaces a number of water molecules (Figure S1a).
Analysis of the pocket with the GRID software22 confirms the highly hydrophobic nature of its surface, with overlap between the hotspot identified with the C1= (lipophilic, carbon sp2 probe) and the phenyl ring of LUF5452 (Figure S3).
As also observed in the unbiased MD, the hydrophobic nature of the region makes the interaction with the solvent unfavorable and displacing the “unhappy waters” increases the strength of the interaction with the ligand stabilizing the secondary minimum. The importance of exploiting the presence of “unhappy waters” has been previously shown in other GPCRs.23,24
Salt Bridge Influences Binding
In both proteins a salt bridge is present at the entrance of the binding site, hindering the binding and unbinding of the ligands. While in A2aR the interaction is formed between the residues E169 and H264; in A1R a lysine residue (K265) forms a stronger contact with E172. The different nature of the salt bridge in the two receptors results in a much higher stability of the E172-K265 contact in A1R with respect to its counterpart in A2aR (Figure S4a) and was reflected in the need to bias the distance between the side chains in our metadynamics simulations of A1R. We also find that the strong salt bridge in A1R hinders the binding and unbinding of the ligand (Figure 5). On the contrary, the weaker salt bridge in A2aR has a smaller influence on the binding dynamics.
The restriction of the cross-sectional area of the entrance of the binding site determined by the helical turn of ECL2 hosting E169 (A2aR) or E172 (A1R) and ECL3 forces the ligand to approach the salt bridge at a right angle between the common core plane and the bridge axis. Generally, the molecules initially interact with the residues with either the furane ring or with the bicyclic region of the cores. In A2aR, stacking of the core and the side chain of H264 is also observed (Figure 5).
The importance of the salt bridge of A2aR for the binding mechanism is supported by mutagenesis and structural data11,25 and was previously suggested for A1R in the literature.26
Dominant Binding Pathway of the Ligands
The free energy landscapes of the four sets of simulations also indicate a significant role of the extracellular vestibule in driving the binding process. ECL2, in particular, is often the first point of contact between the ligand and the receptors, as reported for β-adrenergic receptors.27 The conformation of ECL2 is different in the two receptors. While a short helical turn is present in both proteins toward the orthosteric binding pocket, hosting F168 and E169 (A2aR) and the equivalent F171 and E172 in A1R, a longer helical segment extending further into the bulk solvent differs in length and conformation (see Figure 1). In the binding path, we observe frequent interactions between the ligands and the loops, especially with A151, A155, A158, and K173 in A1R and K150 or K153 in A2aR. The contact between ECL2 and the ligands likely helps preorient the molecules toward the binding site, forcing the cores of the molecules to lay on the loop surface, and funnelling the ligands to the salt bridge at the entrance of the site (Figure 6), similar to what was observed by Dror et al.27 for β-adrenergic receptors.
Figure 6.

Representative binding or unbinding paths of the ligands, extracted from the PT-metaD simulations. The ligands need to break the salt bridge at the entrance of the binding site in order to unbind. Extensive interactions with ECL2 are found before complete solvation of the ligands in the fully unbound states.
Conclusions
Our results suggest that a combination of three structural differences of A1R and A2aR concur to the observed selectivity of ZM241385, while leading to similar binding affinities of LUF5452. First, the salt bridge at the opening of the orthosteric binding site of A1R and A2aR significantly influences the dynamical nature of the interaction between the receptor and ligands. Second, albeit ZM241385 and LUF5452 possess a similar binding mode, the presence of a hydrophobic pocket in the binding site of A1R allows LUF5452 to adopt an alternative pose, displacing weakly interacting water molecules in the region. Third, the desolvation of the 4-hydroxyphenyl and phenyl groups of the two ligands leads to the exploration of alternative conformations at the expense of the water network that stabilizes them in the main binding pose. While in A2aR this results in poor stabilization of LUF5452, the hydrophobic pocket of A1R allows an effective desolvation of the group. Taken together, our findings confirm how the differential behavior of GPCR ligands arises from a complex interplay of small but relevant structural and dynamical differences and from different solvation patterns. The new insights on the molecular determinants of ligand selectivity between A1R and A2aR provide a clear indication for the structure-based rational design of selective drugs for these receptors and possibly for other GPCRs.
Methods
The crystal structures of the human A1 and A2a receptors were downloaded from the Protein Data Bank (A1R: 5N2S,7 A2aR: 5IU4(11)). After correcting mutations and removing the apocytochrome b562 (bRIL) inserted in the ICL3 loop or at the N-terminus, missing residues were added with MODELLER 9.1928 and the proteins were embedded in a pre-equilibrated POPC membrane. The membrane was then aligned to the XY plane, resulting in the main axis of the GPCRs and the Z axis being close to parallel.
The complexes were solvated with TIP3P water, and chloride ions were added to balance the net charge. Ligands were parametrized with the generalized AMBER force field (GAFF)29 and charges were calculated using Gaussian0930 with a 6-31G* basis set at the Hartree–Fock level. Protein, water, and ion parameters were generated with AMBER14SB force field,31,32 and the phospholipid topology was downloaded from LipidBook.33 The simulations were run using GROMACS 5.1.434 with the PLUMED 2.3.1 plugin35 in the NPT ensemble.
The metadynamics simulations were run using a parallel tempering scheme in the 300–310 K temperature range. During the production metadynamics runs, Gaussian hills were deposited every 2 ps in the well-tempered scheme36 with a bias factor of 15. The Gaussian width was set to 0.1 nm for the Z-projection and XY-projection and to 0.033 nm for the salt bridge distance. In the simulations where two CVs were biased, the initial height was 1 kJ/mol, whereas in that biasing three CVs it was set to 1.5 kJ/mol.
The exploration of the bulk water region by the ligand was restrained with the use of a funnel-like restraint in order to aid the convergence of the simulations.17,37
Acknowledgments
F.L.G. acknowledges EPSRC (Grant nos EP/P011306/1 and EP/P022138/1) and the Human Brain Project (Grant SGA2) for financial support. HecBioSim (EPSRC Grant EP/L000253/1), JADE, and PRACE are acknowledged for computer time. The authors thank R. Cooke and S. Acosta-Gutiérrez for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.9b00298.
Comparison of rat and human receptor sequences; details of the setup of the molecular dynamics and parallel-tempering metadynamics simulations; and further information on the hydrophobic pocket of A1R, the role of water in the orthosteric pocket of A1R, and on the results of the metadynamics simulations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sheth S.; Brito R.; Mukherjea D.; Rybak L.; Ramkumar V. Adenosine Receptors: Expression, Function and Regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. 10.3390/ijms15022024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Muller C. E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors-An Update. Pharmacol. Rev. 2011, 63, 1–34. 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z.-G. Adenosine Receptors as Therapeutic Targets. Nat. Rev. Drug Discovery 2006, 5, 247–264. 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero Y.; Gumpert C.; Serlachius E.; Lichtenstein P.; Walum H.; Johansson D.; Anckarsäter H.; Westberg L.; Eriksson E.; Halldner L. A Study of the Possible Association Between Adenosine A2A receptor Gene Polymorphisms and Attention-deficit Hyperactivity Disorder Traits. Genes, Brain Behav. 2013, 12, 305–310. 10.1111/gbb.12015. [DOI] [PubMed] [Google Scholar]
- Chen J.-F.; Eltzschig H. K.; Fredholm B. B. Adenosine Receptors as Drug Targets - What Are the Challenges?. Nat. Rev. Drug Discovery 2013, 12, 265–286. 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piirainen H.; Ashok Y.; Nanekar R. T.; Jaakola V.-P. Structural Features of Adenosine Receptors: From Crystal to Function. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 1233–1244. 10.1016/j.bbamem.2010.05.021. [DOI] [PubMed] [Google Scholar]
- Cheng R. K. Y.; Segala E.; Robertson N.; Deflorian F.; Doré A. S.; Errey J. C.; Fiez-Vandal C.; Marshall F. H.; Cooke R. M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285. 10.1016/j.str.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Khoshouei M.; Thal D. M.; Liang Y.-l.; Nguyen A. T. N.; Furness S. G. B.; Venugopal H.; Baltos J.-a.; Plitzko J. M.; Danev R.; Baumeister W.; May L. T.; Wootten D.; Sexton P. M.; Glukhova A.; Christopoulos A. Structure of the Adenosine-bound Human Adenosine A1 Receptor - GI Complex. Nature 2018, 558, 559–563. 10.1038/s41586-018-0236-6. [DOI] [PubMed] [Google Scholar]
- Glukhova A.; Thal D. M.; Nguyen A. T.; Vecchio E. A.; Jörg M.; Scammells P. J.; May L. T.; Sexton P. M.; Christopoulos A. Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell 2017, 168, 867–877. 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H.. Methods in Neuroscience; Academic Press, 1995; Vol. 25; pp 366–428. [Google Scholar]
- Segala E.; Guo D.; Cheng R. K. Y.; Bortolato A.; Deflorian F.; Doré A. S.; Errey J. C.; Heitman L. H.; Ijzerman A. P.; Marshall F. H.; Cooke R. M. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor Through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- Jaakola V.-P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; IJzerman A. P.; Stevens R. C. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science 2008, 322, 1211–1217. 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré A. S.; Robertson N.; Errey J. C.; Ng I.; Hollenstein K.; Tehan B.; Hurrell E.; Bennett K.; Congreve M.; Magnani F.; Tate C. G.; Weir M.; Marshall F. H. Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 2011, 19, 1283–1293. 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherezov V.; Stevens R. C. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337, 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zwart M.; Vollinga R. C.; Beukers M. W.; Sleegers D. F.; von Frijtag Drabbe Künzel J. K.; de Groote M.; Ijzerman A. P. Potent Antagonists for the Human Adenosine A2b Receptor. Derivatives of the Triazolotriazine Adenosine Receptor Antagonist ZM241385 with High Affinity. Drug Dev. Res. 1999, 48, 95–103. . [DOI] [Google Scholar]
- Saleh N.; Saladino G.; Gervasio F. L.; Haensele E.; Banting L.; Whitley D. C.; Sopkova-de Oliveira Santos J.; Bureau R.; Clark T. A Three-site Mechanism for Agonist/Antagonist Selective Binding to Vasopressin Receptors. Angew. Chem., Int. Ed. 2016, 55, 8008–8012. 10.1002/anie.201602729. [DOI] [PubMed] [Google Scholar]
- Saleh N.; Ibrahim P.; Saladino G.; Gervasio F. L.; Clark T. An Efficient Metadynamics-based Protocol to Model the Binding Affinity and the Transition State Ensemble of G-protein-coupled Receptor Ligands. J. Chem. Inf. Model. 2017, 57, 1210–1217. 10.1021/acs.jcim.6b00772. [DOI] [PubMed] [Google Scholar]
- Tiwary P.; Parrinello M. A Time-independent Free Energy Estimator for Metadynamics. J. Phys. Chem. B 2015, 119, 736–742. 10.1021/jp504920s. [DOI] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. Replica-exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. 10.1016/S0009-2614(99)01123-9. [DOI] [Google Scholar]
- Laio A.; Parrinello M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D.; Xia L.; van Veldhoven J. P. D.; Hazeu M.; Mocking T.; Brussee J.; IJzerman A. P.; Heitman L. H. Binding Kinetics of ZM241385 Derivatives at the Human Adenosine A2A Receptor. ChemMedChem 2014, 9, 752–761. 10.1002/cmdc.201300474. [DOI] [PubMed] [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Mason J. S.; Bortolato A.; Congreve M.; Marshall F. H. New Insights from Structural Biology into the Druggability of G Protein-coupled Receptors. Trends Pharmacol. Sci. 2012, 33, 249–260. 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Mason J. S.; Bortolato A.; Weiss D. R.; Deflorian F.; Tehan B.; Marshall F. H. High End GPCR Design: Crafted Ligand Design and Druggability Analysis Using Protein Structure, Lipophilic Hotspots and Explicit Water Networks. In Silico Pharmacol. 2013, 1, 23. 10.1186/2193-9616-1-23. [DOI] [Google Scholar]
- Guo D.; Pan A. C.; Dror R. O.; Mocking T.; Liu R.; Heitman L.; Shaw D. E.; IJzerman A. P. Molecular Basis of Ligand Dissociation from the Adenosine A2A Receptor. Mol. Pharmacol. 2016, 89, 485–491. 10.1124/mol.115.102657. [DOI] [PubMed] [Google Scholar]
- Jespers W.; Schiedel A. C.; Heitman L. H.; Cooke R. M.; Kleene L.; van Westen G. J. P.; Gloriam D. E.; Müller C. E.; Sotelo E.; Gutiérrez-de Terán H. Structural Mapping of Adenosine Receptor Mutations: Ligand Binding and Signaling Mechanisms. Trends Pharmacol. Sci. 2018, 39, 75–89. 10.1016/j.tips.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Pan A. C.; Arlow D. H.; Borhani D. W.; Maragakis P.; Shan Y.; Xu H.; Shaw D. E. Pathway and Mechanism of Drug Binding to G-protein-coupled Receptors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13118–13123. 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M.; Eramian D.; Shen M.-y.; Pieper U.; Sali A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinf. 2006, 15, 5.6.1–5.6.30. 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. a.; Case D. a. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision B.01; 2009.
- Case D. A.; Babin V.; Berryman J. T.; Betz R. M.; Cai Q.; Cerutti D. S.; Cheatham T. E.; Darden T. A.; Duke R. E.; Gohlke H.; Goetz A. W.; Gusarov S.; Homeyer N.; Janowski P.; Kaus J.; Kolossváry I.; Kovalenko A.; Lee T. S.; LeGrand S.; Luchko T.; Luo R.; Madej B.; Merz K. M.; Paesani F.; Roe D. R.; Roitberg A.; Sagui C.; Salomon-Ferrer R.; Seabra G.; Simmerling C. L.; Smith W.; Swails J.; Walker; Wang J.; Wolf R. M.; Wu X.; Kollman P. A.. Amber 14; University of California: San Francisco, 2014. [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domański J.; Stansfeld P. J.; Sansom M. S. P.; Beckstein O. Lipidbook: A Public Repository for Force-Field Parameters Used in Membrane Simulations. J. Membr. Biol. 2010, 236, 255–258. 10.1007/s00232-010-9296-8. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations Through Multi-level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-tempered Metadynamics: A Smoothly Converging and Tunable Free-energy Method. Phys. Rev. Lett. 2008, 100, 1–4. 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Limongelli V.; Bonomi M.; Parrinello M. Funnel Metadynamics as Accurate Binding Free-Energy Method. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 6358–6363. 10.1073/pnas.1303186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.