

Abstract
It is generally assumed that tethering enhances rates of electron harvesting and delivery to active sites in multidomain enzymes by proximity and sampling mechanisms. Here, we explore this idea in a tethered 3-domain, trimeric copper-containing nitrite reductase. By reverse engineering, we find that tethering does not enhance the rate of electron delivery from its pendant cytochrome c to the catalytic copper-containing core. Using a linker that harbors a gatekeeper tyrosine in a nitrite access channel, the tethered haem domain enables catalysis by other mechanisms. Tethering communicates the redox state of the haem to the distant T2Cu center that helps initiate substrate binding for catalysis. It also tunes copper reduction potentials, suppresses reductive enzyme inactivation, enhances enzyme affinity for substrate, and promotes intercopper electron transfer. Tethering has multiple unanticipated beneficial roles, the combination of which fine-tunes function beyond simplistic mechanisms expected from proximity and restrictive sampling models.
Keywords: copper nitrite reductase, tethering, enzyme catalysis, electron transfer, modular enzyme architecture, intraprotein electron transfer, interprotein electron transfer, protein dynamics
Introduction
Transient protein–protein interactions are central to cellular function and are often linked with interprotein electron transfer events.1,2 While protein–protein complexes are ubiquitous in biology, there are a number of cases where redox partner proteins that are known to interact in a transient manner are found tethered in a single polypeptide chain.3,4 Understanding how and why redox partner proteins are tethered in nature and how tethering impacts on enzyme catalysis is of fundamental importance, as well as being key for the development of novel tethered enzymes for use in biocatalytic and synthetic biology applications.5,6 With an increased number of studies on the few known naturally occurring tethered systems (e.g., P450 BM37 and nitric oxide synthase8,9) in recent years, flexible domain tethering has been shown to play a role in reducing the conformational search space and increasing the population of productive electron transfer configurations. However, as synthetic tethered systems are challenging to design and are often inefficient,6 there is a need to understand why and how nature uses tethered modular enzyme architectures for biological catalysis.
Copper-containing nitrite reductases (CuNiRs) are a highly conserved group of enzymes that are naturally found both tethered and separated from their redox partner proteins.4,10,11 They are highly suited for systematic study to establish the role of redox partner tethering (Figure 1A,B). CuNiRs use electrons generated through denitrification to catalyze one-electron reduction of nitrite (NO2–) to form nitric oxide (NO): NO2– + 2H+ + e– ↔ NO + H2O.12 For nearly 30 years, 2-domain copper-containing nitrite reductases have been extensively studied by computational, structural, and biophysical methods.13−19 Irrespective of organism, these CuNiRs are trimeric enzymes, with each monomer containing two copper centers housed within discrete β-sandwich cupredoxin domains, with the catalytic copper, T2Cu, at the dimer interface (Figure 1A). From a mechanistic viewpoint, 2-domain CuNiRs catalyze the reduction of NO2– to NO by transferring electrons that originate from the partner proteins (pseudo)azurin (PAz/Az) or cytochrome c (cyt c), through a T1Cu center to a catalytic T2Cu. T1Cu to T2Cu electron transfer occurs by proton coupled electron transfer (PCET).13,20,21 Like in many redox proteins,22−24 the interaction between 2-domain CuNiRs and their partner proteins is transient, and conformational search mechanisms involving electrostatic steering are required for successful electron exchange.25
Figure 1.

Rationale and strategy for studying the role of redox partner tethering in copper containing nitrite reductase. (A) Structure (in complex with Ax cytochrome c551; PDB ID 2ZON; top) and proposed mechanism (bottom) of prototypic 2-domain AxNiR. (B) Structure (PDB ID: 3ZIY; top) and proposed mechanism (bottom) of the 3-domain copper containing nitrite reductase, RpNiR, used in this study. In (A) and (B), the three monomers in the structure of the trimeric CuNiRs are shown as green, magenta, and cyan. The isolated cyt c551 protein is shown in yellow. (C) Strategy of dissecting the 3-domain cytochrome c-tethered Ralstonia pickettii copper nitrite reductase into the component domains. In the schematic shown in (C), the three monomers in the structure of RpNiR are shown as green, magenta, and cyan. The isolated RpNiR cyt c protein is shown in yellow.
In recent years, a newly discovered class of 3-domain CuNiRs has been described and structurally characterized (Figure 1B).4,10,11 These 3-domain CuNiRs are widespread in nature and have very similar core structures to the 2-domain CuNiRs but are found with either an Az or a cyt c partner protein fused at the N- or C-terminus of the enzyme, respectively. We have reported a 1.01 Å resolution structure of a 3-domain CuNiR from the denitrifying bacteria Ralstonia pickettii (RpNiR),10 which contains a C-terminal cyt c domain fused to the CuNiR core portion (Figure 1B). This domain proximity in RpNiR might enhance electron transfer between the fused portions of the enzyme. The 3-domain RpNiR has a number of differences to its 2-domain counterparts. These include the presence of Tyr323 (conserved among cyt c tethered 3-domain NiRs), which is located on the linker connecting the haem domain to the core domain, blocking the nitrite binding site, and hydrogen bonding to Asp97, an essential residue in the catalytic pocket. The T2Cu site has two water molecules, one of which is ligated to the T2Cu and another that is hydrogen bonded to this water and also Tyr323. The protein has a single water channel between two monomers, which is blocked by a His residue in 2-domain NiRs.
We have now synthetically deconstructed RpNiR into its constituent domains, namely the core CuNiR portion and cytochrome c (Figure 1C). We have also exchanged Tyr323 for Ala, Glu, and Phe residues in full-length RpNiR to identify the role of Tyr323 and the linker region in substrate access and binding. The deconstructed proteins were studied using biophysical and structural methods alongside the prototypic and well-characterized 2-domain CuNiR (Alcaligenes xylosoxidans NiR; AxNiR) and full-length RpNiR to identify the significance of tethering to RpNiR catalysis. Surprisingly, our data show that tethering does not enhance the rate of electron delivery from haem to T1Cu, a property that was expected from in silico models based on the full-length RpNiR crystal structure and the binary complex of a 2-domain AxNIR with its cognate partner cyt c551.10,25 Here, we show the unexpected and combined importance of the tethered haem domain and linker that harbors the Tyr323 gatekeeper residue to RpNiR catalysis. The effects of tethering are multiple, impacting on mechanisms of long-range redox communication, redox potential modulation, suppression of enzyme inactivation by reductant, electron transfer, activation of the catalytic site, and overall catalysis. Our approach provides a general methodology to discover functional advantages of tethering in other multidomain redox enzyme systems.
Results and Discussion
Reverse Engineering of RpNiR into its Functional Constituent Domains
RpNiR was deconstructed into its constituent catalytic core and cytochrome portions by carefully examining its sequence and structure in comparison with 2-domain CuNiRs (Figure S1). With these constituent proteins and their biophysical characterization (Figure S2), we set out to investigate the functional consequences of tethering in RpNiR catalysis.
First, to assess the impact of removal of the cytochrome c domain on the structure of the RpNiR-core, we solved the structure of the “as-isolated” and “nitrite-bound” RpNiR-core protein at 2.25 and 1.89 Å resolutions, respectively (Table 1). A comparison with the full-length RpNiR structure reveals a number of interesting features. Specifically, the linker region, which connects the core portion to the cytochrome c domain is now unravelled, making a β-strand adjacent to the surface strand of the neighboring molecule, facilitated by formation of a salt bridge between Tyr323 and Glu44 from the adjacent monomer (Figure 2A–C). This new location of the linker region shows a high degree of similarity to other 2-domain CuNiR reductases, which lack Tyr323. Moreover, structures of the RpNiR-core protein show a more compact trimer than the full-length RpNiR (Figure S3), a feature that is attributed to the altered conformation of Ile245 (Figure 2D,E), which interrupts solvent entry via the RpNiR channel. The general tightness of the deconstructed core is visible from an ∼1.6 Å inward movement of the β-strands to the center of the trimeric unit when compared to the full-length protein (Figure S3). Repositioning of Tyr323 and the linker opens up the blocked substrate access channel and changes the conformation of second sphere residues around the T2Cu site similar to 2-domain NiRs. These changes in conformation of the proton channel residues in the RpNiR-core protein resemble those observed in the only other structurally determined cytochrome-fused CuNiR, the enzyme from Pseudoalteromanas haloplanktis (PhNiR) (Figure 2F).26 We infer, therefore that the full-length structures of RpNiR and PhNiR represent two distinct conformations of these tethered complexes.
Table 1. Data Collection and Refinement Statistics.
| RpNiR-core “as-isolated” | RpNiR-core NO2– | Y323A-“as-isolated” | Y323A-NO2– | Y323E-“as-isolated” | Y323E-NO2– | Y323F-“as-isolated” | Y323F-NO2– | |
|---|---|---|---|---|---|---|---|---|
| space group | C2221 | C2221 | H3 | H3 | H3 | H3 | H3 | I213 |
| unit-cell parameter | ||||||||
| a (Å) | 165.88 | 165.91 | 128.23 | 128.50 | 127.55 | 127.65 | 128.29 | 180.97 |
| b (Å) | 165.88 | 165.91 | 128.23 | 128.50 | 127.55 | 127.65 | 128.29 | 180.97 |
| c (Å) | 143.98 | 143.97 | 172.65 | 172.47 | 172.68 | 86.56 | 86.18 | 180.97 |
| α, β, γ (deg) | 90,90,90 | 90,90,90 | 90,90,120 | 90,90,120 | 90,90,120 | 90,90,120 | 90,90,120 | 90,90,90 |
| resolution (Å) | 45.6–2.25 (2.3–2.25) | 83–1.89 (1.92–1.89) | 42.83–1.50 (1.53–1.50) | 29.32–1.70 (1.73–1.70) | 46.64–1.45 (1.49–1.45) | 46.6–1.40 (1.42–1.40) | 64.14–1.61 (1.64–1.61) | 73.88–2.55 (2.66–2.55) |
| no. of unique reflections | 84912 | 156911 | 149421 | 111132 | 118437 | 97927 | 50094 | 26486 |
| Rmerge | 0.11(0.75) | 0.21(1.25) | 0.13(1.21) | 0.12(1.08) | 0.054 (1.17) | 0.070(1.097) | 0.113(1.14) | 0.17(0.92) |
| Rp.i.m. | 0.076(0.53) | 0.09(0.52) | 0.065(0.65) | 0.091(0.79) | 0.050 (0.905) | 0.059(0.99) | 0.055(0.558) | 0.12(0.66) |
| <I/σ(I)> | 9.3(1.4) | 5.3(0.9) | 8.31(1.03) | 8.6(1.5) | 8.8(0.8) | 6.8(0.8) | 9.3(1.4) | 4.9(1.3) |
| CC1/2* | 0.994(0.54) | 0.99(0.485) | 0.99(0.51) | 0.998(0.62) | 0.99 (0.428) | 0.996(0.3) | 0.997(0.522) | 0.97(0.4) |
| completeness (%) | 94.4(75.4) | 99.9(99.7) | 99.94(99.9) | 99.7(100.0) | 98.5(95.2) | 99.7(99.6) | 100.0(62.5) | 93.9(96.5) |
| redundancy | 3.8(3.4) | 6.75(6.57) | 4.84(4.46) | 5.0(5.0) | 2.8(2.5) | 2.8(2.8) | 5.1(5.2) | 2.9(2.8) |
| Wilson B-factor (Å2) | 25.5 | 34.11 | 24.19 | 17.42 | 24.19 | 13.4 | 19.2 | 37.2 |
| refinement | ||||||||
| no monomers in A.U. | 6 | 6 | 2 | 2 | 2 | 1 | 1 | 1 |
| resolution (Å) | 42.86–2.25 | 50–1.9 | 42.83–1.6 | 23.33–1.70 | 46.64–1.65 | 46.6–1.4 | 64.4–1.75 | 73.99–2.6 |
| Rwork/Rfree (%) | 17.7/23.6 | 17.8/22.4 | 16.3/19.5 | 17.3/20.3 | 15.8/17.8 | 11.35/14.6 | 13.6/16.3 | 15.15/18.75 |
| no. atoms | ||||||||
| protein | 14630 | 14672 | 6968 | 6987 | 7052 | 3525 | 3553 | 3434 |
| ligand/ion | 28/18 | 37/12 | 68/4 | 92/4 | 92/4 | 52/2 | 49/2 | 46/2 |
| waters | 1068 | 1859 | 1308 | 1196 | 1193 | 519 | 537 | 225 |
| B-factors (Å2) | ||||||||
| protein | 32.42 | 28.2 | 17.3 | 20.73 | 20.99 | 19.68 | 20.47 | 43.48 |
| Cu | 35.71 | 13.38 | 12.6 | 15.69 | 16.46 | 14.53/18.26 | 17.02 | 33.95 |
| NO2– | - | 39.97 | - | 29.34 | - | 28.00 | - | 53.68 |
| waters | 34.7 | 36.99 | 33.44 | 36.16 | 36.18 | 41.1 | 37.7 | 38.46 |
| r.m.s deviations | ||||||||
| bond length (Å) | 0.007 | 0.006 | 0.010 | 0.009 | 0.012 | 0.012 | 0.012 | 0.010 |
| bond angles (deg) | 1.505 | 1.880 | 1.784 | 1.509 | 1.682 | 1.670 | 1.668 | 1.765 |
| PDB access code | 6QPU | 6QPT | 6QPV | 6QPX | 6QPZ | 6QQ0 | 6QQ1 | 6QQ2 |
Figure 2.

Structural organization of the RpNiR-core trimer. (A) Cartoon representation of the trimeric RpNiR-core protein (viewed along the 3-fold axis). (B) A close-up view (bottom) of the RpNiR-core protein showing details of the Tyr323 site. In (A) and (B), the monomeric units of the RpNiR-core protein are colored in green, magenta, and cyan. Copper ions are shown as dark blue spheres. (C) Comparison between the RpNiR-core protein (lilac) and the full-length native RpNiR (salmon), showing details of the differences in the linker region (residues 315–333). (D) A close-up view of the T2Cu site and residue Ile245, found in the channel from the T2Cu site toward the surface of the protein in the RpNiR-core protein (lilac and gray). (E) Alignment of the RpNiR-core trimer (lilac and gray) with the full-length RpNiR (PDB ID: 3ZIY; salmon). (F) Alignment of the RpNiR-core trimer (lilac and gray) with PhNiR (PDB ID: 2ZOO; yellow).
Contrary to full-length RpNiR crystals, we were able to soak nitrite into the RpNiR-core crystals to obtain a nitrite-bound structure.27 The “nitrite-bound” RpNiR-core X-ray structure contains 2 trimers in the asymmetric unit and shows different ligands at the T2Cu sites. Sites A and F have two waters bound to T2Cu similar to full-length RpNiR (Figure 3A); four other sites have nitrite bound in two conformations, a “top-hat” (sites C and B) (Figure 3B and Figure S4) and a “side-on” conformation (sites D and F) (Figure 3C and Figure S4). This variation in nitrite binding from “top-hat” to “side-on” coincides with rearrangement of the solvent above the T2Cu site (Figure 3B,C) and stronger hydrogen bonding of the nitrite and water ligands to both His240 and Asp97. Similar conformations of nitrite were observed recently in Achromobacter cycloclases 2-domain CuNiR (AcNiR) structures during in-crystallo enzyme-catalysis studied by serial crystallography.28
Figure 3.

Details of the T2Cu sites of the RpNiR-core proteins and the full-length RpNiR variant proteins in “as-isolated” and “nitrite-bound” form. (A) Water ligands, W1 and W2, bound to T2Cu in the “as-isolated” RpNiR-core protein. (B) Nitrite bound to T2Cu in the “top-hat” and (C) “side-on” conformations in the RpNiR-core protein (see Figure S4 for the stereo view). At the T2Cu site, W1 is coordinated to the T2Cu and W2 is hydrogen bonded to W1. (D), (E), and (F) show structures of “as-isolated” full-length RpNiR (PDB ID: 3ZIY), Y323A, and Y323E variants, respectively. (G), (H), and (I) show the “nitrite-bound” forms of Y323F, Y323A, and Y323E variants, respectively (see Figure S4 for the stereo view). Hydrogen bonds are shown as black dotted lines, coordination bonds are in red, copper ions are shown as blue spheres, and water molecules are small red spheres. A 2Fo-Fc electron density map is contoured at the 1.0σ level for the T1Cu and T2Cu sites and shown as light-gray mesh.
Tether Residue Tyr 323 is a Gatekeeper for Nitrite Binding
We were unable to obtain the structure of the RpNiR-core protein with Tyr323 locked in the active site in a manner similar to that observed in the full-length RpNiR.10,27 We were also unable to obtain a structure of full-length RpNiR with nitrite bound. Consequently, three variants of full-length RpNiR were generated (Y323A/F/E) to investigate the role of residue Tyr323 for nitrite access to and binding at the T2Cu site (Table 1). These variants have specific activities for nitrite reduction that are similar (∼90%) to wild type full-length protein. We solved crystal structures of these variants in “as-isolated” and “nitrite-bound” forms. In all of these structures, the main chain position of residue 323 does not change in comparison with native RpNiR. In the “as-isolated” Y323A/E variant structures, water W1 is coordinated by T2Cu and hydrogen-bonded to W2. Both waters are found in a similar position as those of native RpNiR (Figure 3D–F). Water W1 is also within hydrogen bond distance to both Asp97 and His240. For variants Y323A and Y323E, the water molecules occupying free space above T2Cu are connected by strong hydrogen bonds, while the channel space, opposite to the RpNiR-core, is open and contains full occupancy waters (Figure 3E,F), one of which, W3, ligates to Asp97. The Y323F variant has a single water, W1, bound to T2Cu site. The absence of a second water molecule creates space available above W1 despite the presence of Phe323 in a position similar to Tyr323 of the native protein (Figure S5). In crystals treated with nitrite, we observed nitrite replacing waters W1 and W2 in the Y323A and Y323E variants and water W1 in the Y323F variant (Figure 3G–I). Interestingly, the Y323A variant shows a previously unobserved “inverse-hat” binding mode for nitrite, while both Y323F and Y323E variants have nitrite bound in a “side-on” conformation (Figure S4). The ability of crystals of these variants to bind nitrite despite retention of the linker loop in a “locked-down” position27 similar to that was previously observed in the full-length protein demonstrates the importance of both the nature of residue at position 323 (Tyr) and the position of the linker in controlling substrate access and binding.
Haem to T1Cu Electron Transfer is Limited by Haem Domain Conformational Sampling
To investigate the consequences of tethering in RpNiR, we measured the kinetics and thermodynamics of the haem to T1Cu electron transfer reaction. This was accomplished by measuring the redox potentials of the haem and copper centers (Table 2 and Figures S6–S9) and the haem to T1Cu electron transfer rates in RpNiR and its constituent domains (Figure S10–11).
Table 2. Redox Potentials of T1Cu, T2Cu, and Haem Centers in the Various CuNiR Proteinsa.
| construct | Haem (mV) vs SHE | T1Cu (mV) vs SHE | T2Cu (mV) vs SHE |
|---|---|---|---|
| full-length RpNiR | 290 ± 1 | 266 ± 2 | 255 ± 4 |
| RpNiR cyt c | 290 ± 1 | n/a | n/a |
| RpNiR-core | n/a | 331 ± 5 | 243 ± 3 |
| Y323F RpNiR-core | n/a | 338 ± 7 | 257 ± 8 |
| AxNiR | n/a | 255 ± 3b | 244 ± 18b |
| Axcyt c551 | 233 ± 1 | n/a | n/a |
Data are shown ± error of fit.
Data for AxNiR T1Cu and T2Cu centers are taken from Leferink et al.13
The midpoint reduction potentials of the haem and T1Cu in full-length RpNiR are similar (290 and 266 mV, respectively). The haem to T1Cu edge-to-edge distance in the X-ray crystal structure of full-length RpNiR is 10.1 Å (Figure 4A). In this situation, electron transfer rates would be fast (ca. 109 s–1 for nonadiabatic electron transfer) unless limited by other factors, such as conformational search mechanisms.29 Conformational search mechanisms are often used to find optimal geometries for electron transfer in biological systems, especially large multidomain redox systems.30,31 We therefore set out to establish if such mechanisms might limit the observed rate of haem to T1Cu electron transfer in RpNiR.
Figure 4.

Domain conformational dynamics limit haem to T1Cu electron transfer in RpNiR. (A) Cartoon representation of the RpNiR structure showing a close-up of the haem and T1Cu centers. (B) Example laser flash photolysis transient for intraprotein electron transfer reaction from the haem to the T1Cu in nitrite-free RpNiR. The red transient shows the background reaction associated with NADH photoexcitation in the presence of the mediator, N-methyl nicotinamide (NMN); the black transient shows the reaction with the RpNiR protein present in the reaction mixture; and the blue transient is the deconvoluted trace, corresponding to the haem to T1Cu electron transfer step in RpNiR. (C) Comparison of experimental small-angle X-ray scattering (SAXS) from full-length wild-type RpNiR33 with that calculated from the crystal structure10 (PDB ID: 4AX3), depicted as inset, χ2 = 21.0. (D) Comparison of experimental SAXS data33 with that calculated for a model, shown as inset, of RpNiR created using the linker conformation of the RpNiR-core structure and refining the position of conformationally plastic cyt c domains, χ2 = 1.5. (E) A comparison of SAXS distance distribution functions (P(R))33 for compact and elongated RpNiR. (F) The rate of interprotein electron transfer from the isolated cytochrome c protein to the core CuNiR proteins.
We used a flash photolysis method to observe electron transfer from reduced haem to the T1Cu center in RpNiR (Figure 4B). In this method, electrons are rapidly injected into the haem center following laser excitation of NADH20,32 (the electron donor). The subsequent electron transfer reaction from haem to T1Cu is slow (16.8 ± 2.0 s–1). Moreover, only a small fraction (<5%; expected to be 38% based on midpoint reduction potential) of electrons delivered to the haem center are transferred to the T1Cu center. This indicates that the thermodynamic equilibrium predicted from the measured reduction potentials of the T1Cu and haem centers is not attained, indicating the possibility of conformational differences between the tethered domains.
Domain motions have been inferred to facilitate electron transfer from tethered domains to the core CuNiR portion in 3-domain CuNiRs.10,11,33 Slow rates of electron transfer from haem to T1Cu in RpNiR are consistent with localized and rate-limiting searches of conformational space in the tethered protein, required to optimize electronic coupling between the haem and T1Cu centers. This sampling of multiple conformations to find reactive geometries is consistent with published X-ray scattering data for RpNiR,33 which had placed the tethered domains far away from the core 2-domain assembly. The structural rearrangement of the Tyr323 linker observed in the high-resolution crystallographic structure of the RpNiR-core has enabled construction of a model of full-length RpNiR that provides a much-improved simulation of the SAXS data. It provides a more reliable model of an extended structure utilizing the position of the linker region in the RpNiR-core protein, placing the three cytochrome domains with three haem to T1Cu separations of 28, 38, and 52 Å. This new model of RpNiR shows that the cyt c domains are highly solvent exposed and distant from the core structure in an extended conformation. We would expect an ensemble of heam-T1Cu distances, which vary around an average of 40 Å as the domains move (Figure 4C–E and Figure S12). The model shown in Figure 4D is however an exceptionally good fit to the experimental data, suggesting that this extended conformation (with small variation) is where the molecule resides most of the time in solution. These observations emphasize the importance of large-scale domain dynamics in RpNiR catalysis.
To gain further knowledge of the potential benefits of tethering, we compared rates of intraprotein electron transfer from haem to T1Cu in full-length RpNiR with those from the untethered haem domain to the RpNiR-core protein. Electron transfer rates were also investigated in the naturally occurring 2-domain AxNiR. For the genetically deconstructed and natural 2-domain NiRs, we used a stopped-flow method in which excess 2-domain CuNiR was rapidly mixed with reduced cyt c protein; the reaction progress was monitored using the haem Soret band (Figure S10). Observed rates of reaction were dependent linearly on RpNiR-core protein concentration, indicating interprotein electron transfer occurs through transient formation of a protein–protein complex (Figure 4F). Also, the natural 2-component AxNiR–cyt c551 system transfers electrons ca 3-fold more effectively than the genetically deconstructed 2-component system.
Redox driving forces for haem to copper electron transfer in the natural 2-domain AxNiR–cyt c551 and genetically deconstructed RpNiR proteins are similar (Table 2). Modest differences in second-order rate constants for genetically deconstructed RpNiR and the natural AxNiR–cyt c551 complexes are likely to be attributed to altered reactive geometries of the respective protein–protein complexes or alternative positioning of the linker region. To ascertain if tethering in RpNiR improves the rate of electron transfer compared to 2-domain AxNiR, rate constants for electron transfer were calculated for 2-domain AxNiR at protein concentrations that represent the average concentration of proteins found in R. pickettii or A. xylosoxidans. This was accomplished using the protein abundance database tool PaxDb.34 At these average protein concentrations, we found that tethering would provide little benefit over the 2-component AxNiR system for haem to T1Cu electron transfer (Table 3). There are differences in redox potential for T1Cu (e.g., full-length RpNiR compared to the constituent RpNiR domains) that will alter the overall redox driving force for electron transfer (Table 2). However, as these reactions are limited by conformational sampling (tethered RpNiR system) or collision and interacting-surface rolling (2-component systems), small differences in midpoint redox potential should not affect observed rates of haem to T1Cu electron transfer.
Table 3. Tabulated Rate Constants for Haem to T1Cu Electron Transfer in Native RpNiR, the Natural 2-Component AxNiR-Axcytc551, and the Reverse Engineered Constituent RpNiR Proteinsa.
| CuNiR construct | k (mM–1 s–1) | kobs (s–1) |
|---|---|---|
| WT RpNiR | n/a | 16.8 ± 0.2 |
| WT AxNiR and WT Axcyt c551 | 2.6 ± 0.1 | 0.26–26b |
| RpNiR-core and RpNiR cyt c | 0.9 ± 0.1 | 0.09–9b |
Data are shown ± error of fit.
kobs was calculated for the 2-component systems by multiplying the second-order rate constants (k) by the average protein concentrations found within Gram-negative bacteria (0.1–10 μM).
Nitrite Binding and Enzyme Turnover is Controlled by Haem Reduction and Prevents RpNiR Inactivation
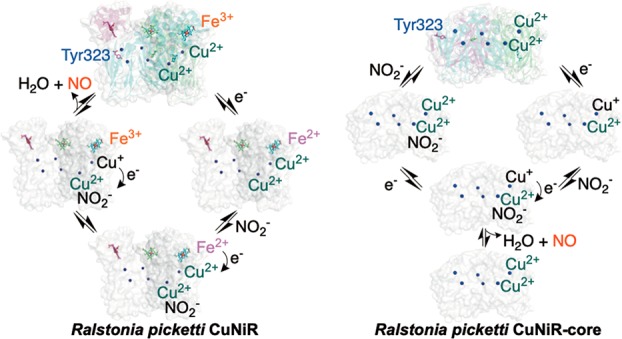
The lack of effect on T2Cu hyperfine feature shows that nitrite does not bind to oxidized RpNiR23 (Figure 5A). It is therefore not possible to determine electron transfer rates from haem to T1Cu in “preloaded” nitrite-bound native RpNiR. We did however investigate electron transfer from reduced haem to the RpNiR copper centers in the presence of nitrite contained in the assay buffer using our flash photolysis assay. These electron transfer reactions were investigated at different concentrations of nitrite contained in the assay solution (Figure S11). With nitrite present, two kinetic phases (compared to the single phase in absence of nitrite) were observed for haem to T1Cu electron transfer. With nitrite, there is also an increase in the number of electrons transferred from the haem to the T1Cu center (Figure S11). This suggests that nitrite facilitates electron flow from the haem domain to the copper centers by increasing the redox potential of the T2Cu center and enabling electron transfer to occur from T1Cu to T2Cu.35 We also found that preincubation of native RpNiR with the strong reductant dithionite reduces the haem domain selectively, but this treatment does not inactivate RpNiR. This contrasts to the well-documented situation with 2-domain NiRs, where dithionite-reduction of the T2Cu in the absence of nitrite leads to enzyme inactivation mediated by dissociation of the water ligated to the T2Cu.36−38 Due to the absence of a lack of dithionite-reduced structure of RpNiR, the precise mechanism responsible for this difference cannot be established, but it is likely to arise from the position of Tyr323 in the catalytic pocket and the associated conformation of the linker.
Figure 5.

Nitrite binding and redox communication between the tethered haem domain and the catalytic T2Cu in RpNiR. (A) EPR spectra of the different RpNiR constructs with and without 5 mM nitrite present. In (A), the WT RpNiR spectra is shown in black and the RpNiR-core is shown in red. (B) Steady state Michaelis–Menten plots of the different CuNiR constructs (WT AxNiR is shown as red circles; WT RpNiR is shown as black circles; RpNiR-core is shown as black squares; and Y323F RpNiR-core is shown as black diamonds). The kinetic parameters obtained are shown in Table 4. (C) UV–vis spectra of oxidized (black) and 1-electron reduced (red) WT RpNiR, showing how 1-electron predominately sits on the haem cofactor. (D) Effect of titrating nitrite into 1-electron reduced WT RpNiR measured by the absorbance of the haem Soret peak. The insert in (D) shows the observed rate constants for the reaction between 1-electron reduced RpNiR and nitrite. The Ks value for nitrite binding measured by UV–vis spectral titration is 3.7 × 10–3 ± 0.3 × 10–3 mM, and the Ks and klim values for the nitrite dependent stopped-flow are 3.5 × 10–3 ± 1.2 × 10–3 mM and 0.34 ± 0.02 s–1, respectively.
We attribute the slow phase observed in flash photolysis studies of RpNiR to the binding of nitrite at the T2Cu site following rapid reduction of the haem by photoactivated NADH. When nitrite is unavailable, RpNiR is protected from inactivation by preventing electron flow from the reduced haem center to T1Cu as noted above (<5% T1Cu is reduced compared to 38% expected on the basis of midpoint reduction potential). The process of substrate binding in RpNiR is an even more highly coordinated sequence of events than 2-domain CuNiRs, protecting the enzyme against reductive inactivation when nitrite is not available.37,38
Inter-Cu Electron Transfer in the Catalytic Core of RpNiR Mimics that of 2-Domain AxNiR
We investigated intercopper electron transfer by monitoring the electron transfer rate from T1Cu to T2Cu and its nitrite-dependence. These studies were performed with the RpNiR-core protein because the dominant haem absorption feature masks the T1Cu features in full-length RpNiR (Figure S2). Laser flash was used to rapidly transfer electrons from NADH to the T1Cu center. Subsequent electron transfer from the T1Cu to the T2Cu as a new equilibrium became established was monitored by absorption spectrophotometry to report on the oxidation state of the T1Cu center (Figure S13).13,20 In the absence of nitrite, T1Cu to T2Cu electron transfer did not occur in the RpNiR-core protein, consistent with measured midpoint reduction potentials of the RpNiR-core protein (+331 mV and +243 mV for T1 and T2Cu centers, respectively; Table 2), which are within the same range as those previously reported for the 2-domain Pseudomonas chlororaphis CuNiR, though different from the AxNiR for the T1Cu site.13,39
Two discrete kinetic phases were observed for RpNiR-core intercopper electron transfer in the presence of the nitrite substrate (Figure S13). The faster of the two phases shows a nitrite dependence that is similar to the native 2-domain AxNiR.20 At low concentrations of nitrite, the rate of electron transfer is fast, but inhibition is observed at higher nitrite concentrations (Figure S13). The slower kinetic phase shows a hyperbolic dependence on nitrite concentration. Kinetic parameters associated with this phase are similar to those determined for steady-state assays of the RpNiR-core protein (Table 4 and 5; Figure 5B and S13). This second phase is likely to be rate-limiting in steady-state turnover of the RpNiR-core protein. In native 2-domain CuNiRs and full-length RpNiR, intercopper electron transfer is not rate limiting in steady-state turnover (Figure 5), but it can become so in selected variant forms (e.g., residues targeted in the substrate access channel).15 The fast phase is not seen in the Y323F variant of the RpNiR-core in the presence of nitrite. In the RpNiR-core protein, the two phases might relate to multiple conformational states. Studies with the RpNiR-core have established rates for intercopper electron transfer in RpNiR and revealed a complex nitrite dependence similar to that of 2 domain AxNiR.20
Table 4. Tabulated Steady-State Kinetic Parameters for RpNiR, the RpNiR-Core Protein, and AxNiRa.
| CuNiR construct | kcat per monomer (s–1) | Km (mM) | Ki (mM) | kcat/Km (mM–1 s–1) |
|---|---|---|---|---|
| WT RpNiR | 1.1 ± 0.03 | 1.6 × 10–3 ± 0.2 × 10–3 | n/a | 6.9 × 102 ± 0.9 × 102 |
| WT Ax NiR | 202.7 ± 0.7 | 2.1 × 10–2 ± 0.2 × 10–2 | n/a | 9.3 × 103 ± 0.9 × 103 |
| RpNiR-core | 2.3 ± 0.2 | 4.1 × 10–1 ± 1.6 × 10–1 | 1.3 × 102 ± 0.8 × 102 | 5.6 ± 2.1 |
| Y323F RpNiR-core | 2.6 ± 0.2 | 2.9 × 10–1 ± 0.8 × 10–1 | 1.6 × 102 ± 0.7 × 102 | 8.8 ± 2.5 |
Data are shown ± error of fit.
Table 5. Tabulated Kinetic Parameters Associated with T1 to T2 Cu Electron Transfer in the RpNiR-Core and Y323F RpNiR-Corea.
| RpNiR construct | klim (s–1) | Ks (mM) | Ki (mM) | klim/Ks (mM–1 s–1) |
|---|---|---|---|---|
| RpNiR-core | 3.5 ± 0.4 | 1.2 ± 0.3 | 1.8 × 101 ± 0.6 × 101 | 2.8 ± 0.8 |
| Y323F RpNiR-core | 2.7 ± 0.4 | 0.8 ± 0.4 | 4.2 × 101 ± 2.9 × 101 | 3.4 ± 1.6 |
Data are shown ± error of fit.
Tethering Influences Overall Steady-state Catalysis in RpNiR
We investigated the effects of tethering using steady-state turnover assays with full-length and genetically deconstructed RpNiR proteins. Comparisons were made also to 2-domain AxNiR (Table 4; Figure 5B). RpNiR shows Michaelis–Menten behavior over a range of nitrite concentrations (up to 50 mM). By contrast, the RpNiR-core protein shows inhibition at high nitrite concentrations (beyond 10 mM). Similar inhibition has been observed in AxNiR variants, where active site residues involved in PCET (e.g., Asn90) were modified by site-directed mutagenesis.13 A consequence of tethering is that this nitrite inhibition is suppressed in full-length RpNiR.
Kinetic parameters are shown in Table 4. Like other 2-domain CuNiRs, AxNiR has a low apparent nitrite affinity (KM = 2.1 × 10–2 ± 0.2 × 10–2 mM) and high turnover (kcat = 202.7 ± 0.7 s–1) value. It is important to compare these with corresponding values for RpNiR and the RpNiR-core proteins. The core protein has a relatively weak affinity for nitrite (4.1 × 10–1 ± 1.6 × 10–1 mM). Conversely, the full-length RpNiR has a higher relative affinity for nitrite (KM = 1.6 × 10–3 ± 0.2 × 10–3 mM). The RpNiR-core protein is an inefficient nitrite reductase (kcat/KM values ca. 1500 times lower than AxNiR). Full-length RpNiR, by contrast, is a relatively efficient enzyme (kcat/KM = 6.9 × 102 ± 0.9 × 102 mM–1 s–1) despite having an ∼200-fold lower kcat value than that of the AxNiR. The difference in Michaelis constant for nitrite observed for full-length and core RpNiR proteins is consistent with a requirement to reduce the RpNiR haem in order to facilitate nitrite binding at the T2Cu center.
Nitrite does not bind to oxidized full-length RpNiR but can bind to 2-domain CuNiRs and RpNiR-core proteins (Figure 5A). In titration experiments, nitrite binds tightly to the one-electron reduced state (Ks = 3.7 × 10–3 ± 0.3 × 10–3 mM) but only when the tethered haem domain is reduced (Figure 5D). We confirmed this finding by performing catalytic turnover of prereduced RpNiR under single turnover conditions in a stopped-flow device. Using dithionite, we titrated a single electron into RpNiR, which remained localized at the haem (Figure 5C). Electron flow from the haem to the copper centers was then triggered by addition of nitrite. This confirms that nitrite binding triggers the flow of electrons to the T2Cu site via the T1Cu and thus facilitates catalysis, consistent with single turnover flash photolysis studies. Stopped-flow measurements were performed at different nitrite concentrations to determine nitrite-binding affinity to 1-electron reduced RpNiR (Figure 5D; Figure S14). The apparent affinity (3.5 × 10–3 ± 1.2 × 10–3 mM; stopped-flow studies) and the apparent Michaelis constant (1.6 × 10–3 ± 0.2 × 10–3 mM; conventional steady-state kinetic assays) are similar to the dissociation constant (3.7 × 10–3 ± 0.3 × 10–3 mM; static titration) for nitrite-bound one-electron-reduced RpNiR. The data indicate that the limiting rate constant (klim) for oxidation of RpNiR with nitrite is three times lower than the recorded steady-state per RpNiR monomer, consistent with only one of the three monomeric units found within the RpNiR protein to be active during turnover or a “one-third site reactivity” mechanism for RpNiR (see the Supporting Information for further discussion).25,40 Altogether, stopped-flow and spectral titration data presented here confirm the importance of haem reduction prior to events that trigger substrate access and binding for catalysis to occur.
Conclusions
On the basis of our findings, we propose a mechanism for RpNiR in which the haem domain and linker region containing Tyr323 are important to RpNiR catalysis. The structure of the genetically obtained core enzyme shows that the linker region containing Tyr323 can unravel dramatically in a manner that maps onto the extended conformation obtained from SAXS of the intact RpNiR molecule placing the tethered domain at >30 Å from T1Cu. One-electron reduction of the cytochrome domain facilitates both nitrite binding and a conformational search that is necessary for the cytochrome domain to interact effectively with the core portion of RpNiR. These structural changes facilitate haem to T1Cu electron transfer.
In principle, electron transfer to the T2Cu could either be concerted or sequential with respect to nitrite binding, but a concerted mechanism, facilitated by a repositioning of the gatekeeper Tyr323 residue, would seem the most plausible mechanism. Repositioning of Tyr323 may in part be aided by the conformational changes accompanying reduction of the haem. This could then unblock the substrate access channel to provide access for nitrite to the T2Cu catalytic site. Unblocking of the substrate access channel can also be accomplished by directed mutagenesis. The structures of variants reported here show that this is accomplished without any rearrangement of the linker in the compact tethered RpNiR.
Premature electron transfer to the T2Cu site is prevented when nitrite is unavailable. This suppresses the reductive inactivation of RpNiR at the T2Cu site by mechanisms that have been documented in the natural 2-domain CuNiRs.35 Genetic deconstruction of RpNiR into its constituent domains has provided additional insights. Electron transfer in the RpNiR-core protein indicates that nitrite can bind to the T2Cu site in the oxidized form of the proteins and that intercopper electron transfer is rate limiting in catalysis. This is fundamentally different to the RpNiR full-length protein, where nitrite does not bind to the oxidized enzyme.
To conclude, our study has revealed a complex and subtle interplay implicating long-range structural transitions facilitated by haem reduction to deliver electrons and nitrite to the T2Cu catalytic site in RpNiR. This coordinated action is only possible because of the tethered structure of RpNiR. These subtle and beneficial effects on catalysis as reflected by very high affinity for nitrite suggest that laboratory-based improvement design through simple domain fusions will not suffice in the development of efficient redox enzymes. Instead, a more intricate design strategy should be used in which the multiple roles of tethering are considered. This would then open up redox partner tethering to improve catalysis through a multitude of previously unforeseen mechanisms.
Materials and Methods
Materials
All reagents were of analytical grade and were purchased from Sigma-Aldrich, apart from NADH, which was purchased from Melford (Ipswich, UK). All spectroscopic measurements were performed in 50 mM potassium phosphate buffer (pH 7.0). For anaerobic measurements, all buffers were degassed with N2 before being introduced into the anaerobic glovebox (<5 ppm of O2). Prior to performing anaerobic spectroscopic measurements, protein samples were taken into the glovebox, oxidized with a few grains of potassium ferricyanide, and passed down a Bio-Rad Econo-Pac 10DG-desalting column equilibrated with anaerobic buffer.
Molecular Biology Methods
Codon optimized nirK genes encoding full-length wild-type (WT) and truncated portions of RpNiR were synthesized by GeneArt (Thermo Fisher Scientific, Paisley, UK). For periplasmic haem c incorporation in both WT and the isolated cyt c portion of RpNiR, the gene encoding the RpNiR N-terminal leader sequence was added at the 5′ end of the NiR genes. Synthesized genes for full-length RpNiR and the cyt c domain were cloned into the pET22b vector between Nde I and Xho I restriction sites, while the genes for the core regions were cloned into the pETM-11 expression vector between the Nco I and the Nde I sites by GeneArt (Thermo Fisher Scientific Paisley, UK). All genes inserted into pET22b included a 3′ sequence encoding for a TEV cleavage site, which was used to remove the C-terminal His-tag following protein purification.
To generate the Y323F RpNiR-core variant, the Q5 Site-Directed Mutagenesis Kit and protocol from New England BioLabs (Hitchin, UK) were used with the forward (5′-TTT CTG GGT GAT CGT GCA GCA C-3′) and reverse (5′-AAC GCT ATC CAG TTC TTT ACC GCT ATA CA-3′) custom primers ordered from Eurofins Genomics (Ebersberg, Germany). All mutations were confirmed by DNA sequencing (Eurofins Genomics, Ebersberg, Germany).
NirK genes encoding full-length Y323A/E/F RpNiR mutants were synthesized and cloned into pET26b vector between Nde I and Xho I restriction sites by GenScript (USA).
Expression and Purification of Proteins
E. coli strain C41(DE3) cells were used to express WT RpNiR. Briefly, the haem maturation plasmid, pEC86, and the pET22b plasmid containing the C-terminal His-tagged RpNiR genes were transformed into C41(DE3) cells and grown in 0.5 L of Terrific Broth media at 30 °C. Once an optical density at 600 nm (OD600) of 0.6 was attained, the culture incubation temperature was dropped to 20 °C and cells were supplemented with 0.1 mM CuSO4, 0.3 mM 5-aminolevulinic acid, and 0.3 mM IPTG. Cultures were incubated overnight and subsequently, cells were harvested by centrifugation. Cell pellets were stored at −20 °C prior to performing purification steps.
WT RpNiR was purified using nickel affinity chromatography. Briefly, cell lysates contained in 40 mM MOPS (pH 7.8), 150 mM NaCl, and 40 mM imidazole buffered solutions were loaded onto a HisTrap HP nickel-charged IMAC column from GE Healthcare (Little Chalfont, U.K.) equilibrated with the same buffer. RpNiR was eluted from the column using 200 mM imidazole. For removal of the C-terminal His-tag, RpNiR was passed down a desalting column equilibrated with 50 mM Tris-HCl (pH 7.8), 200 mM NaCl, 1 mM DTT before being incubated overnight with a His-tagged TEV protease at room temperature. Following proteolytic cleavage, TEV protease was separated from RpNiR by using a reverse nickel affinity purification step and RpNiR proteins were supplemented with copper by incubating protein solutions with 50 mM Tris-HCl, 150 mM NaCl, and 0.1 mM CuSO4 for 2 h at room temperature. Following a final desalting step to remove excess CuSO4, RpNiR was flash-frozen in liquid N2 and stored at −80 °C. Variant forms of RpNiR were purified using a similar procedure.
For RpNiR-core protein expression, a pETM-11 plasmid encoding the N-terminal His-tagged protein was transformed into E. coli strain BL21(DE3) cells and grown in 0.5 L of terrific broth media incubated at 30 °C. After cell cultures attained an OD600 of 0.6, cells were supplemented with 0.1 mM CuSO4 and the incubation temperature was dropped to 20 °C. RpNiR-core protein expression was induced by addition of IPTG (final concentration 0.5 mM). Cultures were incubated overnight and harvested the following morning by centrifugation. Cell pellets were stored at −20 °C. The RpNiR-core proteins were purified using the same protocols described above for the WT RpNiR protein.
The cyt c portion of RpNiR was expressed and purified using the same protocol described for WT RpNiR except that CuSO4 was excluded during recombinant protein expression and purification. The 2-domain copper-containing nitrite reductase from Alcaligenes xylosoxidans (AxNiR), the cyt c551 protein from Alcaligenes xylosoxidans, and N-terminal His-tag tobacco etch virus (TEV) protease were expressed and purified by following previously published protocols.41,42
For Y323A/E/F full-length RpNiR variant expression, a pET26b plasmid encompassing the DNA of the Y323 variant was transformed into Escherichia coli BL21(DE3) cells containing the pEC86 vector (used for c-type haem maturation). Cells were grown in 1 L of modified Terrific Broth medium, containing 24 g yeast extract, 12 g tryptone, 10 g NaCl, and 2 mL of glycerol at 37 °C. At an OD600 of 0.8, 0.1 mM CuSO4 and 1 mg of Hemin were added to the medium and cultures were left to incubate overnight at 16 °C. Following expression, cells were harvested by centrifugation and stored at −20 °C until purification.
All Y323A/E/F purification steps were performed at 4 °C. Harvested cells were sonicated in 20 mM Tris HCl (pH 8.4) buffer, containing cocktail protease inhibitor (Roche, Hertfordshire, UK) and subsequently centrifuged to remove the insoluble fraction. Cell lysates were applied to a DEAE Sepharose Fast Flow column equilibrated with 20 mM Tris HCl (pH 8.4). The red-colored band, symbolic of the RpNiR protein, was monitored when eluting RpNiR with a 20 mM Tris HCl (pH 8.4) buffer, containing 50 mM NaCl. The eluted protein was passed down a Superdex200 gel filtration column equilibrated with 20 mM Tris-HCl (pH 7.6) and 200 mM NaCl. To monitor the RpNiR elution, an ÄKTA pure system (GE Healthcare, Little Chalford, UK) was used and 280 and 410 nm wavelengths were selected to track the presence of protein and the haem soret band, respectively. Eluted fractions were dialyzed overnight against 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 0.1 mM CuSO4, for reconstitution of T2Cu sites and reapplied onto Superdex200 gel filtration column to attain the RpNiR trimer.
Crystallography
Crystallization
The C2221 crystal form of the RpNiR-core protein was obtained by mixing 2 μL of a 10 mg/mL protein solution present in 50 mM Tris-HCl (pH 7.8), 100 mM NaCl with similar amount of reservoir solution containing 0.2 M MgCl2, and 20% P6000 in MES buffer (pH 6.0). Crystals grew at 4 °C over a 2-day period. For formation of the nitrite complex, crystals were soaked in 100 mM nitrite, which was added to the reservoir solution. Crystals were flash-frozen in the same solution they were grown in which was supplemented with 20% glycerol, as a cryo-protectant.
Crystals for full-length RpNiR-Y323E and Y323A variants were obtained in H3 crystal form, and crystal for Y323F variant appeared in I213 at 4 °C using the hanging drop vapor diffusion method. For crystallization of the RpNiR variants, 2 μL of 10 mg/mL protein solution present in 20 mM Tris-HCl and 200 mM NaCl (pH 7.5) was mixed with an equal volume of reservoir solution containing 0.1 M HEPES (pH 7.50, 20% PEG 3350, 0.2 M sodium citrate) and equilibrated over 500 μL of the reservoir solution. To obtain RpNiR-nitrite complexes in the full-length variants, crystals were incubated in the reservoir solution with 200 mM sodium nitrite. All crystals were cryo-protected in reservoir or soaking solutions containing 15% glycerol and flash cooled in liquid N2.
Data Collection, Structure Solution, and Refinement
For the “as-isolated” RpNiR-core variant, data were collected at Barkla X-ray laboratory a Rigaku FR-E+ Super-Bright rotating anode generator with an EIGER R 4M detector with 1.54 Å wavelength. “Nitrite-bound” RpNiR-core data were collected at IO4–1 beamline (Diamond Synchrotron, UK) using Pilatus 6M-F detector and 0.91587 Å wavelength. Data for Y323A-“as-isolated”, Y323A-NO2–, Y323E-NO2–, and Y323F-NO2– were collected at the I24 Diamond Synchrotron using a Pilatus 6M-F detector and 0.96861 Å wavelength. Data for Y323F-“as-isolated” were collected at the IO3 beamline, Diamond Synchrotron using a Pilatus3–6M detector and 0.9686 Å wavelength. Data for Y323E-“as-isolated” were collected at the PROXIMA1 beamline (SOLEIL synchrotron, France) using a PILATUS 6M detector. All data were collected ca. 100 K. Data were integrated by DIALS43 and scaled by Aimless.44 Structure of RpNiR-core was solved by molecular replacement using MOLREP45 with the core region of full-length wild-type RpNiR (PDB ID: 3ZWI) (residues 4–344) as the starting model. When there was no isomorphous model available, structures of Y323A/E/F RpNiR variants were solved by molecular replacement, using the full-length wild-type RpNiR model (PDB ID: 3ZWI). Where a similar model was available, data were reindexed to match the orientation of the model in CCP446 followed by refinement with REFMAC5.47 Refinement was iterated with manual model building in COOT.48 TLS refinement was implemented at the last stage of the refinement as hydrogen atoms were placed in riding positions. The quality of the model was assessed in PDBDEP. Data collection and refinement statistics are shown in Table 1.
Protein Modeling Based on Small-Angle X-ray Scattering (SAXS)
C-terminal cytochrome domains were appended to the RpNiR-core structure and allowed to move freely throughout several torsion angle molecular dynamics simulations using CNS.49 The core structure was fixed, and the globular cytochrome domain treated as a rigid body. The scattering curves of structures generated throughout these simulations were compared to experimental scattering data for RpNiR collected and published previously33 using FoXS for Mac.50 Distance distribution functions were calculated with ScÅtter (Robert Rambo, DIAMOND, UK).
Nitrite Reductase Activity Measurements
Nitrite Reductase Activity Measurements Using a NO Electrode
The nitrite reductase activity measurements were carried out in an anaerobic glovebox under a nitrogen +1.5% hydrogen gas atmosphere. The reaction was started by adding 1 μL of purified protein at a concentration of 5 mg/mL to a total volume of 3 mL of the assay reaction mixture consisting of 50 mM MES, pH 6.5, 250 μM phenazine methosulfate, 5 mM sodium ascorbate, and 5 mM sodium nitrite in a reaction vessel equipped with a magnetic stirrer at room temperature. The nitric oxide produced from the reaction was detected by a nitric oxide electrode (ISO-NOP World Precision Instruments, Serasota, USA). NO concentrations were determined by comparison with calibration curves of known concentrations of NO.
Nitrite Reductase Activity Measurements Monitoring Dithionite Oxidation
The conversion of nitrite to nitric oxide by NiR was followed using a previously published method by monitoring the oxidation of sodium dithionite.20,33 Briefly, all steady-state activity measurements were carried out at 10 °C in an anaerobic glovebox (<5 ppm of O2) using 0.1 mM phenazine methylsulfate (PMS) reduced by 0.5 mM sodium dithionite as the electron donor. All assays contained catalytic amounts of NiR (10–50 nM). To maintain ionic strength across the nitrite range used in the measurements, KCl was used to ensure [KCl] + [KNO2] = 50 mM.
For each assay, NiR and nitrite was mixed with a reactant solution of PMS and sodium dithionite and the depletion in the sodium dithionite absorbance at 315 nm (Δε315 8 mM–1cm–1) was used to determine the activity of NiR proteins. The linear regions of progress curves were used to obtain initial velocities, which were fitted to the Michaelis–Menten equation after considering the NiR concentration (eq 1) by using OriginPro 9.1 software.
| 1 |
When substrate inhibition was observed, data were fitted to a modified version of the Michaelis–Menten equation (eq 2), where Ki is the dissociation constant for the enzyme–inhibitor complex.
| 2 |
Rapid Mixing Stopped-Flow Spectroscopy
The rate of interprotein electron transfer from Axcyt c551 to AxNiR and from RpNiR-cyt c domain to RpNiR-core was monitored under single-turnover conditions using stopped-flow spectroscopy. All assays were conducted at 10 °C in an Applied Photophysics SC18MV spectrometer housed within an anaerobic glovebox (<5 ppm of O2). To reduce the haem in RpNiR-cyt c for stopped-flow studies, an excess of sodium dithionite was mixed with the protein before the sample was passed down a desalting column to remove surplus reducing agent. For each measurement, 1 μM (final concentration) of the reduced RpNiR-cyt c domain was mixed with a range of NiR or core proteins (final concentration 10–80 μM). Reactions were monitored by recording the change in absorbance at the haem Soret peak (417–420 nm). Exponential decay functions were fitted to reaction transients. Plots of observed rate constant versus concentration were fitted to linear functions to determine second-order rate constants for reactions using OriginPro 9.1.
Laser Flash Spectroscopy
A laser flash spectroscopy method was used to monitor intraprotein haem to T1Cu and T1Cu to T2Cu electron transfer in RpNiR and the genetically deconstructed RpNiR, respectively. There is potential complexity in using the laser flash approach to study haem to T1Cu electron transfer, in that solvated electrons generated by laser uncaging of NADH could in principle transfer to all three redox centers of RpNiR. However, the majority of the electrons generated by laser excitation are transferred to the haem cofactor under the conditions used. This is expected given that the haem domain is the most solvent accessible and has a higher redox potential compared to the other copper centers (Table 2).10 Also, we demonstrated that if RpNiR is titrated against sodium dithionite to the level of one reducing equivalent, the electron is located on the haem (Figure 5C). All samples were prepared in an anaerobic glovebox (<5 ppm of O2) under dim red light. Samples containing 200 μM NADH, 50 mM N-methyl nicotinamide (NMN), RpNiR protein, and various concentrations of KNO2 were made up in a quartz cuvette, sealed with a rubber Suba-seal stopper, and taken from the glovebox for laser flash analysis. The ionic strength was maintained constant using additional KCl such that [KCl] + [KNO2] = 50 mM.
Laser flash measurements were performed at 10 °C using methods previously described.20 Briefly, samples were excited at 355 nm using the third harmonic of a Q-switch Nd:YAG laser (Brilliant B, Quantel) and spectral changes associated with the haem c (418 nm; full-length RpNiR) or the T1Cu (600 nm; genetically deconstructed core protein) were used to monitor rates of electron transfer. Acquired transients were fitted to exponential decay functions and plots of the dependence of these observed rate constants on nitrite concentration were analyzed by fitting to a modified version of the hyperbolic functions (eq 3) using OriginPro 9.1. In eq 3, klim is the limiting rate constant, Ks is the saturation constant, krev is the reverse rate of reaction, and Ki is the dissociation constant.
| 3 |
EPR and Redox Potentiometry
The reduction potentials of the haem c, the T1Cu, and the T2Cu in the various NiR/cyc c551 proteins were determined by electrochemical titration. NiR/cyt c protein was titrated against sodium dithionite. To facilitate communication between the electrode and the protein, mediators were used. Between each addition of sodium dithionite the electrode potential was allowed to stabilize. The electrochemical potential of the solution was measured using a Thermo Orion ORP electrode at 25 °C. A factor of +207 mV was used to correct values to the standard hydrogen electrode (SHE). During titration against dithionite, UV–vis absorbance spectra were recorded using a Cary UV-50 Bio UV–visible scanning spectrophotometer and samples (300 μL) were withdrawn for EPR analysis. Samples were placed in 4 mm Suprasil quartz EPR tubes (Wilmad LabGlass) and sealed with a Suba-seal rubber stopper and immediately frozen in liquid N2.. Samples were stored in liquid N2 to prevent reoxidation until they were analyzed. Continuous wave X-band EPR spectra (∼ 9.4 GHz) were recorded using a Bruker ELEXSYS E580 EPR spectrometer (Bruker GmbH, Rheinstetten, Germany). Temperature was maintained using an Oxford Instruments ESR900 helium flow cryostat coupled to an ITC 503 controller from the same manufacturer. EPR experiments were carried out at 20 K and employed 0.5 mW microwave power, 100 kHz modulation frequency, and 5 G (0.5 mT) modulation amplitude.
Redox potentials of the copper and haem centers were determined by fitting data to the Nernst eq (eq 4):
| 4 |
where Eh is the measured potential, Em is the midpoint potential of the cofactor, R is the gas constant (8.31 J K–1 mol–1), T is the temperature in K, n is the number of electrons, F is the Faraday constant (96.5 kJ V–1 mol–1) and [ox] and [red] are the concentrations of oxidized and reduced enzyme, respectively.
Acknowledgments
The work was supported by the UK Biotechnology and Biological Sciences Research Council (BB/N019380/1, BB/N013972/1, and BB/L006960/1). The authors would like to thank Diamond, UK, and SOLEIL, France, Light Sources (MX11740 and 20171212 BAG allocations, respectively) and Instruct for the synchrotron access and support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b01266.
Sequence alignment, biochemical characterization, crystallography, SAXS, redox potentiometric, stopped-flow and laser-flash photolysis spectroscopic data, as well as a kinetic model suggesting “one-third” site reactivity in RpNiR (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Perkins J. R.; Diboun I.; Dessailly B. H.; Lees J. G.; Orengo C. Transient Protein-Protein Interactions: Structural, Functional, and Network Properties. Structure 2010, 18, 1233–1243. 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Nooren I. M. A.; Thornton J. M. Diversity of Protein–Protein Interactions. EMBO J. 2003, 22, 3486–3492. 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. A New Function Evolved from Gene Fusion. Genome Res. 2000, 10, 1655–1657. 10.1101/gr.165700. [DOI] [PubMed] [Google Scholar]
- Ellis M. J.; Grossmann J. G.; Eady R. R.; Hasnain S. S. Genomic Analysis Reveals Widespread Occurrence of New Classes of Copper Nitrite Reductases. JBIC, J. Biol. Inorg. Chem. 2007, 12, 1119–1127. 10.1007/s00775-007-0282-2. [DOI] [PubMed] [Google Scholar]
- Grünberg R.; Serrano L. Strategies for Protein Synthetic Biology. Nucleic Acids Res. 2010, 38, 2663–2675. 10.1093/nar/gkq139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.; Liu C.; Kim B. G.; Lee D. Y. Synthetic Fusion Protein Design and Applications. Biotechnol. Adv. 2015, 33, 155–164. 10.1016/j.biotechadv.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Whitehouse C. J. C.; Bell S. G.; Wong L. L. P450BM3 (CYP102A1): Connecting the Dots. Chem. Soc. Rev. 2012, 41, 1218–1260. 10.1039/C1CS15192D. [DOI] [PubMed] [Google Scholar]
- Hedison T. M.; Hay S.; Scrutton N. S. A Perspective on Conformational Control of Electron Transfer in Nitric Oxide Synthases. Nitric Oxide 2017, 63, 61–67. 10.1016/j.niox.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leferink N. G. H.; Hay S.; Rigby S. E. J.; Scrutton N. S. Towards the Free Energy Landscape for Catalysis in Mammalian Nitric Oxide Synthases. FEBS J. 2015, 282, 3016–3029. 10.1111/febs.13171. [DOI] [PubMed] [Google Scholar]
- Antonyuk S. V.; Han C.; Eady R. R.; Hasnain S. S. Structures of Protein-Protein Complexes Involved in Electron Transfer. Nature 2013, 496, 123–126. 10.1038/nature11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojiri M.; Xie Y.; Inoue T.; Yamamoto T.; Matsumura H.; Kataoka K.; Deligeer; Yamaguchi K.; Kai Y.; Suzuki S. Structure and Function of a Hexameric Copper-Containing Nitrite Reductase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4315–4320. 10.1073/pnas.0609195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumft W. G. Cell Biology and Molecular Basis of Denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leferink N. G. H.; Han C.; Antonyuk S. V.; Heyes D. J.; Rigby S. E. J.; Hough M. A.; Eady R. R.; Scrutton N. S.; Hasnain S. S. Proton-Coupled Electron Transfer in the Catalytic Cycle of Alcaligenes Xylosoxidans Copper-Dependent Nitrite Reductase. Biochemistry 2011, 50, 4121–4131. 10.1021/bi200246f. [DOI] [PubMed] [Google Scholar]
- Lintuluoto M.; Lintuluoto J. M. Intra-Electron Transfer Induced by Protonation in Copper-Containing Nitrite Reductase. Metallomics 2018, 10, 565–578. 10.1039/C7MT00323D. [DOI] [PubMed] [Google Scholar]
- Leferink N. G. H.; Antonyuk S. V.; Houwman J. A.; Scrutton N. S.; Eady R. R.; Hasnain S. S. Impact of Residues Remote from the Catalytic Centre on Enzyme Catalysis of Copper Nitrite Reductase. Nat. Commun. 2014, 5, 4395. 10.1038/ncomms5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godden J. W.; Turley S.; Teller D. C.; Adman E. T.; Liu M. Y.; Payne W. J.; LeGall J. The 2.3 Angstrom X-Ray Structure of Nitrite Reductase from Achromobacter Cycloclastes. Science 1991, 253, 438–442. 10.1126/science.1862344. [DOI] [PubMed] [Google Scholar]
- Ellis M. J.; Dodd F. E.; Sawers G.; Eady R. R.; Hasnain S. S. Atomic Resolution Structures of Native Copper Nitrite Reductase from Alcaligenes Xylosoxidans and the Active Site Mutant Asp92Glu. J. Mol. Biol. 2003, 328, 429–438. 10.1016/S0022-2836(03)00308-5. [DOI] [PubMed] [Google Scholar]
- Antonyuk S. V.; Strange R. W.; Sawers G.; Eady R. R.; Hasnain S. S. Atomic Resolution Structures of Resting-State, Substrate- and Product-Complexed Cu-Nitrite Reductase Provide Insight into Catalytic Mechanism. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12041–12046. 10.1073/pnas.0504207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S.; Yamaguchi K.; Kataoka K.; Kobayashi K.; Tagawa S.; Kohzuma T.; Shidara S.; Iwasaki H.; et al. Spectroscopic Characterization and Intramolecular Electron Transfer Processes of Native and Type 2 Cu-Depleted Nitrite Reductases. JBIC, J. Biol. Inorg. Chem. 1997, 2, 265–274. 10.1007/s007750050132. [DOI] [Google Scholar]
- Brenner S.; Heyes D. J.; Hay S.; Hough M. A.; Eady R. R.; Hasnain S. S.; Scrutton N. S. Demonstration of Proton-Coupled Electron Transfer in the Copper-Containing Nitrite Reductases. J. Biol. Chem. 2009, 284, 25973–25983. 10.1074/jbc.M109.012245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leferink N. G. H.; Eady R. R.; Hasnain S. S.; Scrutton N. S. Laser-Flash Photolysis Indicates That Internal Electron Transfer Is Triggered by Proton Uptake by Alcaligenes Xylosoxidans Copper-Dependent Nitrite Reductase. FEBS J. 2012, 279, 2174–2181. 10.1111/j.1742-4658.2012.08601.x. [DOI] [PubMed] [Google Scholar]
- Bashir Q.; Volkov A. N.; Ullmann G. M.; Ubbink M. Visualization of the Encounter Ensemble of the Transient Electron Transfer Complex of Cytochrome c and Cytochrome c Peroxidase. J. Am. Chem. Soc. 2010, 132, 241–247. 10.1021/ja9064574. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Ubbink M. Close Encounters of the Transient Kind: Protein Interactions in the Photosynthetic Redox Chain Investigated by NMR Spectroscopy. Acc. Chem. Res. 2003, 36, 723–730. 10.1021/ar0200955. [DOI] [PubMed] [Google Scholar]
- Mei H.; Wang K.; Peffer N.; Weatherly G.; Cohen D. S.; Miller M.; Pielak G.; Durham B.; Millett F. Role of Configurational Gating in Intracomplex Electron Transfer from Cytochrome c to the Radical Cation in Cytochrome c Peroxidase. Biochemistry 1999, 38, 6846–6854. 10.1021/bi983002t. [DOI] [PubMed] [Google Scholar]
- Nojiri M.; Koteishi H.; Nakagami T.; Kobayashi K.; Inoue T.; Yamaguchi K.; Suzuki S. Structural Basis of Inter-Protein Electron Transfer for Nitrite Reduction in Denitrification. Nature 2009, 462, 117–120. 10.1038/nature08507. [DOI] [PubMed] [Google Scholar]
- Tsuda A.; Ishikawa R.; Koteishi H.; Tange K.; Fukuda Y.; Kobayashi K.; Inoue T.; Nojiri M. Structural and Mechanistic Insights into the Electron Flow through Protein for Cytochrome c-Tethering Copper Nitrite Reductase. J. Biochem. 2013, 154, 51–60. 10.1093/jb/mvt023. [DOI] [PubMed] [Google Scholar]
- Dong J.; Sasaki D.; Eady R. R.; Antonyuk S. V.; Samar Hasnain S. Identification of a Tyrosine Switch in Copper-Haem Nitrite Reductases. IUCrJ 2018, 5, 510–518. 10.1107/S2052252518008242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrell S.; Antonyuk S. V.; Eady R. R.; Hasnain S. S.; Hough M. A.; Strange R. W. Serial Crystallography Captures Enzyme Catalysis in Copper Nitrite Reductase at Atomic Resolution from One Crystal. IUCrJ 2016, 3, 271–281. 10.1107/S205225251600823X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page C. C.; Moser C. C.; Chen X.; Dutton P. L. Natural Engineering Principles of Electron Tunnelling in Biological Oxidation-Reduction. Nature 1999, 402, 47–52. 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Hedison T. M.; Scrutton N. S. Tripping the Light Fantastic in Membrane Redox Biology: Linking Dynamic Structures to Function in ER Electron Transfer Chains. FEBS J. 2019, 10.1111/febs.14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys D.; Basran J.; Talfournier F.; Sutcliffe M. J.; Scrutton N. S. Extensive Conformational Sampling in a Ternary Electron Transfer Complex. Nat. Struct. Mol. Biol. 2003, 10, 219–225. 10.1038/nsb894. [DOI] [PubMed] [Google Scholar]
- Orii Y. Immediate Reduction of Cytochrome c by Photoexcited NADH: Reaction Mechanism As Revealed by Flow-Flash and Rapid-Scan Studies. Biochemistry 1993, 32, 11910–11914. 10.1021/bi00095a021. [DOI] [PubMed] [Google Scholar]
- Han C.; Wright G. S. A.; Fisher K.; Rigby S. E. J.; Eady R. R.; Samar Hasnain S. Characterization of a Novel Copper-Haem c Dissimilatory Nitrite Reductase from Ralstonia Pickettii. Biochem. J. 2012, 444, 219–226. 10.1042/BJ20111623. [DOI] [PubMed] [Google Scholar]
- Wang M.; Weiss M.; Simonovic M.; Haertinger G.; Schrimpf S. P.; Hengartner M. O.; von Mering C. PaxDb, a Database of Protein Abundance Averages Across All Three Domains of Life. Mol. Cell. Proteomics 2012, 11, 492–500. 10.1074/mcp.O111.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Dey A.; Sun Y.; Scholes C. P.; Solomon E. I. Spectroscopic and Computational Studies of Nitrite Reductase: Proton Induced Electron Transfer and Backbonding Contributions to Reactivity. J. Am. Chem. Soc. 2009, 131, 277–288. 10.1021/ja806873e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijma H. J.; Jeuken L. J. C.; Verbeet M. P.; Armstrong F. A.; Canters G. W. Protein Film Voltammetry of Copper-Containing Nitrite Reductase Reveals Reversible Inactivation. J. Am. Chem. Soc. 2007, 129, 8557–8565. 10.1021/ja071274q. [DOI] [PubMed] [Google Scholar]
- Strange R. W.; Murphy L. M.; Dodd F. E.; Abraham Z. H. L.; Eady R. R.; Smith B. E.; Hasnain S. S. Structural and Kinetic Evidence for an Ordered Mechanism of Copper Nitrite Reductase. J. Mol. Biol. 1999, 287, 1001–1009. 10.1006/jmbi.1999.2648. [DOI] [PubMed] [Google Scholar]
- Murphy M. E. P.; Turley S.; Adman E. T. Structure of Nitrite Bound to Copper-Containing Nitrite Reductase from Alcaligenes Faecalis: Mechanistic Implications. J. Biol. Chem. 1997, 272, 28455–28460. 10.1074/jbc.272.45.28455. [DOI] [PubMed] [Google Scholar]
- Pinho D.; Besson S.; Brondino C. D.; de Castro B.; Moura I. Copper-Containing Nitrite Reductase from Pseudomonas Chlororaphis DSM 50135. Eur. J. Biochem. 2004, 271, 2361–2369. 10.1111/j.1432-1033.2004.04155.x. [DOI] [PubMed] [Google Scholar]
- Krzemiński Ł.; Ndamba L.; Canters G. W.; Aartsma T. J.; Evans S. D.; Jeuken L. J. C. Spectroelectrochemical Investigation of Intramolecular and Interfacial Electron-Transfer Rates Reveals Differences between Nitrite Reductase at Rest and during Turnover. J. Am. Chem. Soc. 2011, 133, 15085–15093. 10.1021/ja204891v. [DOI] [PubMed] [Google Scholar]
- Prudêncio M.; Eady R. R.; Sawers G. The Blue Copper-Containing Nitrite Reductase from Alcaligenes Xylosoxidans: Cloning of the NirA Gene and Characterization of the Recombinant Enzyme. J. Bacteriol. 1999, 181, 2323–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea J. E.; Cherry S.; Waugh D. S. Expression and Purification of Soluble His6-Tagged TEV Protease. Methods Mol. Biol. 2009, 498, 297–307. 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- Winter G.; Waterman D. G.; Parkhurst J. M.; Brewster A. S.; Gildea R. J.; Gerstel M.; Fuentes-Montero L.; Vollmar M.; Michels-Clark T.; Young I. D.; Sauter N. K.; Evans G. DIALS: Implementation and Evaluation of a New Integration Package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. 10.1107/S2059798317017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. 10.1107/S0021889897006766. [DOI] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Brunger A. T. Version 1.2 of the Crystallography and NMR System. Nat. Protoc. 2007, 2, 2728–2733. 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D.; Hammel M.; Tainer J. A.; Sali A. FoXS, FoXSDock and MultiFoXS: Single-State and Multi-State Structural Modeling of Proteins and Their Complexes Based on SAXS Profiles. Nucleic Acids Res. 2016, 44, W424–W429. 10.1093/nar/gkw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.