

Abstract
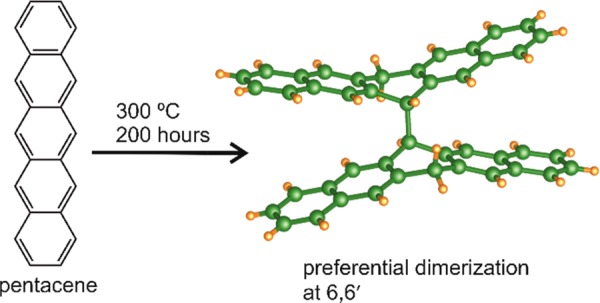
6,6′,13,13′-Tetrahydro-6,6′-bipentacene (HBP), the intermediate molecule connecting pentacene to previously observed peripentacene and extended pentacene oligomers through the formation of a carbon–carbon bond, is synthesized and crystallographically characterized. Heating pentacene to 300 °C under vacuum for 200 h results in pale golden crystals of HBP and amorphous material containing pentacene oligomers, offering experimental evidence that pentacene preferentially dimerizes at the 6,6′-position. Continued heating of HBP results in co-crystals of 6,13-dihydrogenated pentacene and pentacene and further amorphous pentacene oligomers. The amorphous material consists of layered carbonaceous species with a graphenic nature, as determined by Raman spectroscopy and electron microscopy, and suggests HBP as an intermediate to hydrogenated pentacene species and pentacene oligomers, such as peripentacene, of interest in organic electronics.
Introduction
Conjugated small organic molecules have been the focus of intense research for use in organic electronic devices due to high charge mobility, mechanical flexibility, and low cost (compared to typical inorganic compounds).1,2 Pentacene, consisting of five linearly fused aromatic rings (P, Figure 1a), has generated more interest than any other conjugated molecule for use in these devices, thanks to its particularly high conductivity and the availability of a range of deposition techniques leading to uniform highly ordered thin films.1,3 When making thin films, the most widely employed technique is vacuum deposition, not least because it can be considered a simultaneous purification step.3 This technique typically involves high temperature and low pressure to increase the P partial pressure. Roberson et al.4 showed that under an inert gas flow at 320 °C, in competition with its sublimation, P disproportionates to give a 2:1 co-crystal of 6,13-dihydrogenated pentacene (HP, Figure 1a) and P,5 and peripentacene (PP, Figure 1a), detected by mass spectrometry. PP can be considered as two pentacene molecules completely fused along one long axis and has been described as a graphene fragment.6,7 The mechanism for this transformation in the solid state is a matter of some debate,4,8 and since the initial report of PP, considerable effort has gone into developing a rational route to its synthesis.6,9 Thermolysis of a variety of other acenes has been demonstrated to drive both intramolecular C–C bonding10,11 and intermolecular addition reactions to form oligomeric12 and graphitic species.13 Recently, Rogers et al.6 succeeded in preparing single molecules of PP on an Au surface via the dehydrogenation of 6,6′-bipentacene (BP, itself chemically synthesized in bulk from functionalized pentacene derivatives6), which ab initio calculation4 proposes as an intermediate in PP formation in the solid state. Subsequent calculations8 showed that the mechanism is likely to be initiated when a 6-hydropentacenyl radical (HPR, Figure 1a), formed by the hydrogen atom transfer from catalytic amounts of HP to P, dimerizes to give the intermediate species 6,6′,13,13′-tetrahydro-6,6′-bipentacene (HBP, Figure 1a) or adds to a P molecule to form a dimer radical. Either HBP or the dimer radical can be transformed to PP by a series of H abstractions by P or HPR species to generate significant quantities of HP. To date, no experimental reports of the formation of any intermediate dimers (BP or HBP) through C–C bond formation upon heating solid pentacene have been published, and their status as intermediates in the thermal formation of PP remains unclear. The evidence for the existence of PP is mass spectrometry, which may be identifying only the most volatile components of the bulk material, and Raman spectroscopy,14 for which the features are common to all sp2 carbon phases. By heating at the slightly lower reaction temperature of 300 °C under vacuum and studying the resulting materials with a range of bulk characterization methods, we demonstrate the transformation of as-received pentacene to a mixture of single crystalline HBP and a carbon-rich amorphous phase (Figure 1c), likely to be composed of oligomerized pentacene. This represents experimental evidence for preferential intermolecular C–C bond formation at the 6,6′ positions in the thermal oligomerization of pentacene, yielding a crystallographically characterized pentacene oligomer by solid-state thermal transformation. HBP is shown to be unstable, and upon further heating it is further transformed to give the previously reported 2HP/P co-crystals and additional amorphous oligomerized pentacene as formed in the first heating step. The transformation of HBP to 2HP/P co-crystal and amorphous material demonstrates the importance of HBP as an intermediate compound in the thermally driven formation of pentacene-based oligomers and hydrogenated pentacene. The amorphous material has been shown to consist of thin, extended sheets, which can be readily exfoliated, suggesting that this material may be “graphenic” in nature.
Figure 1.
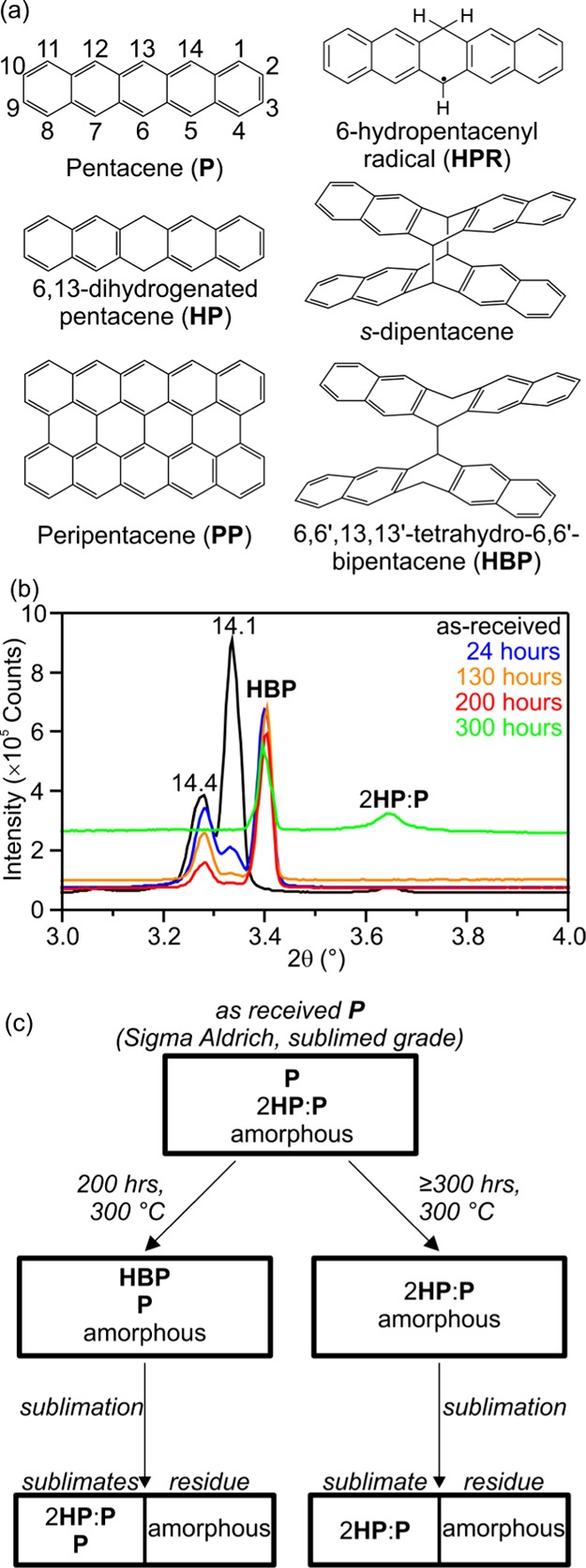
(a) Pentacene (with carbon positions numbered) and related derivatives relevant to its thermal reactivity. (b) Synchrotron powder X-ray diffraction (SXRD) patterns of pentacene heated at 300 °C for 0–300 h, showing the peaks arising from polymorphs of pentacene (so called “14.1 Å” and “14.4 Å” polymorphs), HBP and 2HP/P co-crystals. (c) The scheme of observed products from the heating of as-received pentacene.
Results and Discussion
Figure 2b shows the synchrotron powder X-ray diffraction (SXRD) pattern of as-received pentacene (Sigma-Aldrich, sublimed grade >99.9% trace metals basis). Fitting to this pattern shows that it contains a mixture of the two known bulk-phase polymorphs3,15−18 but also a significant proportion (12.3(2)% by mass, Table S2) of 2HP/P co-crystals, where HP has been previously suggested as a likely catalyst in pentacene disproportionation.8 As-received pentacene powder (50 mg) was pelletized and sealed in a Pyrex tube evacuated to 2 × 10–5 mbar. Upon heating at 300 °C for 24 h pale, golden-colored crystals were observed on the surface of the black pellet. Single-crystal X-ray diffraction confirms the identity of these crystals as the previously undetected HBP (Figures 2a and S3). The unit cell (space group P21/n, Figure S4) contains just one 6′-hydropentacenyl subunit within the asymmetric unit, with a center of inversion at the center of the dimer molecule. The C–C bond between the two 6-hydropentacenyl subunits forms between the 6 and 6′ carbons, yielding the isomer calculated to be the most energetically favored.8 This bond is notably long at 1.601(2) Å (at 150(2) K); ∼0.1 Å longer than a typical C–C bond. This can be thought of as arising from quadrupolar repulsion of the neighboring aromatic systems with C···C distances of as little as 3.166(2) Å between the 5 and 5′ (and 7 and 7′) positions, Figure 2a. Similar bond lengths are also observed in “sandwich photodimers” of acenes,19−21 whereby two new C–C bonds form between two parallel stacked acene subunits across an aromatic ring overlapping π systems. For example, in s-dipentacene21 (Figure 1a), C–C bonds form between the 6:6′ and 13:13′ positions with a mean bond length of 1.611(2) Å, leading to π···π interactions between the closest aromatic rings of a mean distance 3.885(3) Å (as measured from the aromatic centroids), close to the ideal distance for π···π stacking.22 Note that this is significantly longer than the C5–C5′ distance in HBP, due to a larger bond angle around the sp3 carbon atoms in the sandwich dimer (∼112.5°, compared to ∼109° in HBP). In HBP, there is only one C–C bond between the subunits, and the resulting “stepped” configuration minimizes the quadrupolar repulsion within the molecule. Raman spectroscopy of a HBP (point group C2h) single crystal (Figure 3a) shows sharp peaks at 790, 843, 931, 1017, 1387, 1469 cm–1 and is used as a fingerprint for the identification of HBP in the Raman spectra of bulk samples.
Figure 2.
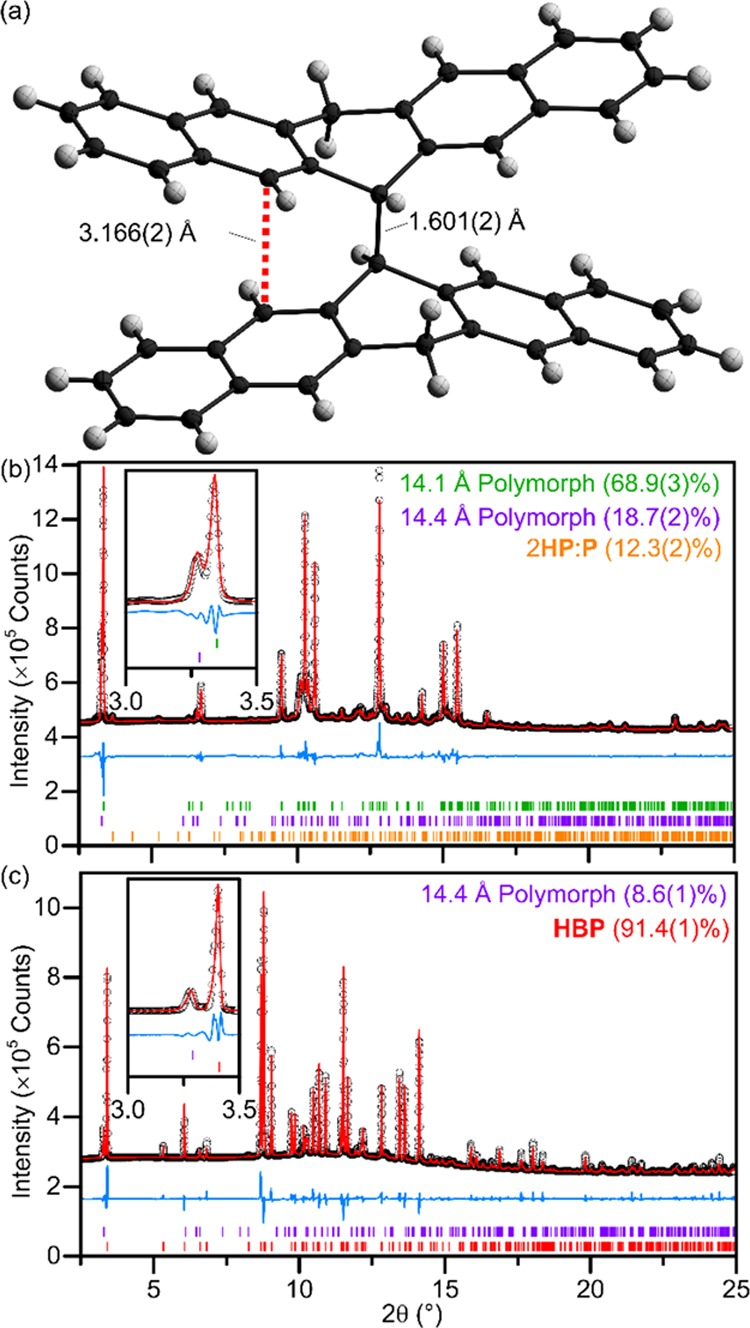
(a) Refined structure of HBP from single-crystal X-ray refinement (data measured at 150(2) K) with thermal ellipsoids at 50% probability. (b) SXRD of as-received Sigma-Aldrich pentacene with the multiphase Rietveld refinement of two pentacene polymorphs3,16 and 2HP/P co-crystals.5Rwp = 5.03%. The inset shows partially resolved (001) peaks of both pentacene polymorphs. (c) SXRD of pentacene after heating in vacuo at 300 °C for 200 h, with the multiphase Rietveld refinement of HBP with a residual amount of pentacene. Rwp = 4.48%. Full details of refinements in Tables S2 and S3.
Figure 3.
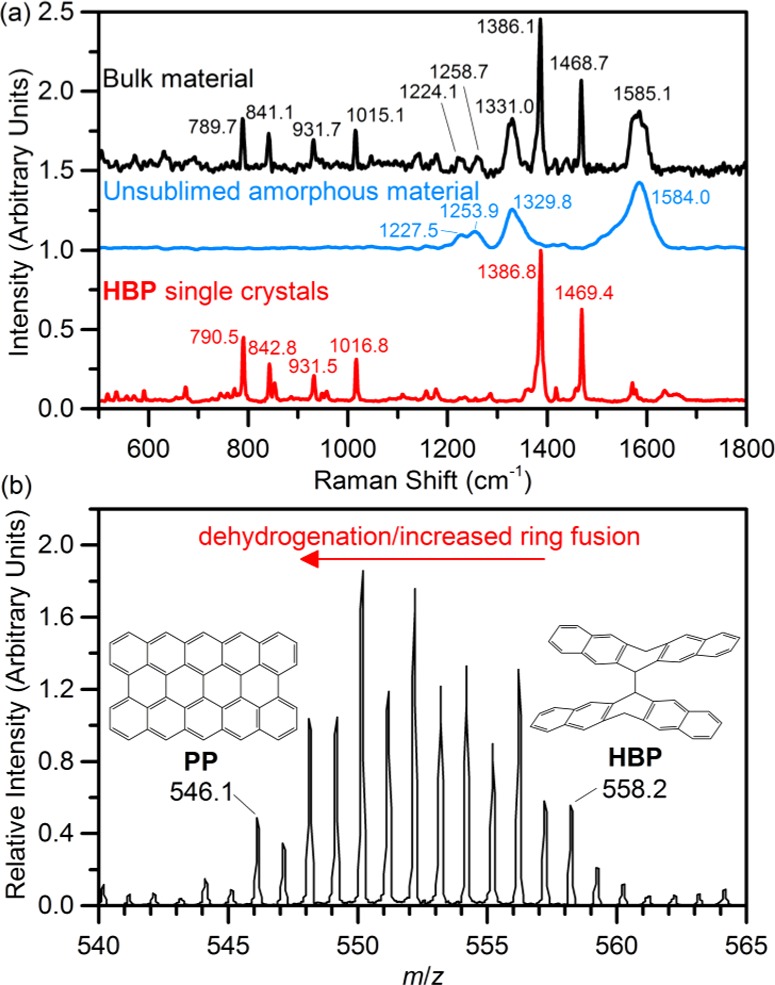
(a) Raman spectra (exciting laser λ = 785 nm) of single crystals of HBP, residual post-sublimed amorphous material, and the bulk mixture of HBP and amorphous material. (b) High-resolution electron ionization-mass spectrometry (EI-MS) data of residual post-sublimed amorphous material. Full MS spectrum (50 ≤ m/z ≤ 900) shown in Figure S9.
The pellet of P treated at 300 °C for 24 h was ground into a fine powder, and SXRD (Figures 1b and S5) shows that a majority of the P is converted to HBP. Optimization of the synthesis time found that heating at 300 °C for 200 h yields a sample where >90% (by mass) of the crystalline component was HBP, with P comprising the remainder of the crystalline portion (Figure 2c, Table S3). Despite this high degree of conversion, the pellet remains a black color, inconsistent with the observed pale color of the single crystals of HBP, suggesting the presence of a secondary amorphous phase. Raman spectroscopy confirms the presence of the secondary amorphous phase by comparison of the bulk material and single crystals of HBP extracted from the surface of the pellet (Figure 3a). The Raman spectra show four broad peaks (ν = 1224, 1259, 1331, and 1585 cm–1) present in the bulk material in addition to peaks assigned to HBP. Elemental analyses of as-received pentacene gave a measured C/H mass ratio of 18.9, and the mixture of HBP and amorphous material produced by the thermal treatment shows the same measured C/H mass ratio (18.8), and since HBP is hydrogenated with respect to pentacene (theoretical C/H mass ratio for HBP is 17.5), the amorphous material is assumed to be carbon rich with respect to the starting material. Quantification of the amorphous component by SXRD using an internal standard (monocrystalline diamond powder, Sigma-Aldrich) shows that the sample contains approximately one-third (by mass) amorphous material (Figure S6, Table S4). This is a larger amorphous contribution than would be expected if PP were the only component of the amorphous phase (∼17%). For comparison, as-received pentacene was found by the same method to contain ∼10% amorphous material. An attempt to separate HBP from the amorphous material by heating the mixture to 300 °C at the end of a sealed evacuated tube with the opposing end of the tube at room temperature (Figure S1) led to the decomposition of HBP, resulting in sublimation of only yellow and purple-red 2HP/P co-crystals and a small quantity of unreacted pentacene. Subsequent powder SXRD of the residual post-sublimed material showed no crystalline peaks, confirming that HBP is unstable over extended heating periods and decomposes to form hydrogenated pentacene species and amorphous material.
Heating pentacene at 300 °C for a longer time of 300 h leads to the formation of a product consisting of 2HP/P co-crystals and amorphous material (Figures 1b and S5) with a small quantity of HBP remaining, suggesting that HBP initially formed in the reaction is transformed over the extended heating period. The transient nature of HBP is the most likely explanation for the difficulty in observing it in earlier experiments.4 Sublimation of the mixture of 2HP/P co-crystal and amorphous material, as described above for the HBP/amorphous mixture, resulted in 2HP/P co-crystals at the cool end of the tube, with a significant proportion of residual post-sublimed black material remaining, which was further characterized to determine the nature of any oligomerized pentacene products potentially formed during the annealing of sublimed pentacene thin films for use in organic electronics, as discussed in the Introduction. The black material is completely amorphous by SXRD (Figure S7), and the Raman spectrum (Figure 3a), an effective probe of carbonaceous structure,23−27 consists of only four peaks at 1228, 1254, 1330, and 1584 cm–1, coinciding with the peaks of the secondary phase observed in the Raman spectrum of the as-made mixture of HBP and amorphous material (Figure 3a), suggesting that this amorphous material is formed concurrently with HBP and is unchanged by further heating. The spectrum agrees broadly with both the calculated Raman spectrum for PP and the observed spectra of pentacene samples heated under vacuum at higher temperatures (425–800 °C).14 However, the two strongest peaks, at 1584 and 1330 cm–1, can also be assigned to the G (resulting from in-plane optic phonons) and D (caused by structural defects) modes of extended sp2 carbon structures, such as graphene and carbon nanotubes, respectively. The high ratio of intensities I(D)/I(G) and the lack of any observed second-order modes (>2000 cm–1, Figure S8) would suggest a high degree of disorder,24,27−29 consistent with a material containing a distribution of pentacene oligomers, including PP, or an extended, partially hydrogenated graphitic material, or graphene nanoribbons, which could be formed by extensive oligomerization. Recently graphene nanoribbons have been shown to exhibit weak phonon modes in the range of 1200–1300 cm–1, which were ascribed to breathing modes of six-atom rings30 and which closely resemble features seen in the Raman spectrum of the amorphous material at 1228 and 1254 cm–1 (Figure 3a). Elemental analysis of the residual post-sublimed amorphous material showed an increased C/H mass ratio (21.9) over the starting pentacene (18.9, consistent with the sublimation of 2HP/P co-crystals) but still significantly lower than expected for pure PP (theoretical C/H mass ratio of 29.1), suggesting only partial fusion or a mixture of products. Sampling by gas chromatography (GC) of the gas in the sealed Pyrex tube after the reaction (Figure S2) shows that no significant quantity of H2 gas is evolved at any stage (corresponding to ≪1% of the total number of H atoms in the starting material; Figure S10, Table S5). This suggests that fusion under vacuum is not generating gaseous H2 as previously proposed14 but is driven by H atom transfers to P or HPR, calculated to be a much lower-energy process.8
In the previous work, mass spectrometry (MS) data have offered the most definitive evidence for the presence of PP.4 High-resolution electron ionization MS (EI-MS) data from the residual post-sublimed amorphous material in the mass range for dimeric pentacenyl species (Figure 3b) show that the sample contains a spectrum of m/z values from 546.1 to 558.2, coinciding with masses of PP and HBP, respectively. A similar distribution of m/z values was seen in the previous work4 and was ascribed by the authors to the partial fusion of the pentacene monomers and/or a misalignment of the pentacene subunits. Based on the computed high favourability of 6,6′-dimerization of HPR over the formation of other adducts8 and our experimental observation of HBP as the only (crystalline) intermediate, we assign this distribution of masses to partial fusion and hydrogenation, rather than significant occurrence of misaligned monomers. There is also another cluster of m/z peaks between 818.2 and 834.4 (Figure S9), corresponding to the mass range of trimeric, partially dehydrogenated pentacene species (the trimeric analogues of PP and HBP have masses of 814.2 and 836.3, respectively), demonstrating the ability of the system to further oligomerize. Higher mass oligomers were not observed, but it is unclear whether this is due to their low abundance or poor volatility.
Scanning electron microscopy (SEM) of the residual post-sublimed amorphous material suggests that it is composed of thin layers (Figures 4a,b and S11), consistent with an extended sp2 material. A suspension of the material in N-methyl-2-pyrrolidone (NMP), a common solvent used in the exfoliation of graphene sheets,31 was generated by sonication for 35 h. After sonication, the suspensions have a dark-brown/black color, which is retained for (at least) several months. A droplet of the sonicated suspension was deposited onto a glass slide, and the solvent allowed to evaporate. Optical microscopy reveals the presence of sheets up to 200 μm across (Figure 4c), the Raman spectra of which (collected at multiple locations to confirm homogeneity) have modes at the same frequencies, as observed in the bulk material (Figure S12). Transmission electron microscopy (TEM) and SEM (Figures 4 and S13) show the presence of layers in the material, but their thickness was difficult to quantify. For comparison, pristine pentacene in NMP was also sonicated, yielding a suspension, which rapidly (<5 min) settles to give a black powder and yellow solution of 6,13-pentacenequinone formed by oxidation of pentacene.32 Immediately after sonication, a droplet of the suspension was deposited onto a glass slide. After the NMP evaporated, optical microscopy shows only small black crystals of pentacene, and their agglomerates can be observed with no indication of extended sheets forming (Figure S14).
Figure 4.

(a) SEM of the bulk amorphous residue material after sublimation of HBP and 2HP/P, zoomed area shows a single layer exfoliated from the bulk material, (b) SEM of bulk residual post-sublimed amorphous material at high magnification showing the layered structure, (c) optical microscopy and (d) TEM of exfoliated residual post-sublimed amorphous material.
Conclusions
We have shown that the previously unobserved dimer HBP forms readily upon heating samples of pentacene containing trace amounts of HP. The observation of HBP represents the first crystallographic characterization of a C–C bond between pentacene units on the path to extended sp2 C–C-based species. HBP is unstable and upon further heating is easily cracked to give 2HP/P co-crystals and an amorphous byproduct, consisting of oligomerized pentacene species at various stages of fusion. Although the previous work4 has suggested that PP is the main constituent of this amorphous material, high-resolution mass spectrometry data we present suggest that dimeric and higher-order oligomers with various degrees of fusion are also present. Further characterization of this amorphous mixture indicates that partially hydrogenated graphitic or graphene-like constituents may be present, formed by extensive oligomerization of pentacene, which are not detectable by mass spectrometry due to low volatility or fragmentation. No H2 gas is observed in the reaction vessel at any stage, suggesting that fusion occurs by a series of H-transfer steps. The results confirm the theoretical predictions8 that 6,6′ fusion is strongly preferred and that the formation of isolatable HBP is the first step in pentacene oligomerization, providing a starting point for future experimental mechanistic studies. Reliable synthesis of HBP offers a useful starting material in the rational synthesis of PP and nanographene.
Experimental Section
Pentacene Thermolysis
As-received pentacene (50 mg, Sigma-Aldrich, sublimed grade, >99.9% trace metals basis) was pressed into a 5-mm-diameter pellet under a pressure of 1 ton in an Ar-filled glovebox (with measured levels of O2 and H2O < 1 ppm). The pellet was then placed into a Pyrex tube (outer diameter ∼9.5 mm, inner diameter ∼5.5 mm), which was subsequently evacuated to a pressure of 2 × 10–5 mbar. The Pyrex tube was then sealed at a length of 10–12 cm and placed in a fan-assisted oven at room temperature. The oven was then heated to 300 °C at a heating rate of 5 °C min–1. After heating for 24–300 h, the oven was cooled to room temperature at a cooling rate of −5 °C min–1.
Heating at slightly lower temperatures (280–290 °C) yields the same reaction, albeit at a much reduced rate, necessitating heating times of over 1000 h to achieve the maximum conversion from pentacene to HBP. Single crystals of HBP could then be isolated by manual separation (Raman 1469.4, 1386.8, 1016.8, 931.5, 842.8, 790.5 cm–1). Heating pentacene at 320 °C failed to isolate HBP, instead a mixture containing only amorphous material and 2HP/P co-crystals was produced.
Sublimation
A subsequent sublimation step was used to separate crystalline, hydrogenated products from the amorphous, hydrogen-poor material, which exhibits low volatility. The mixture was ground into a fine powder using a pestle and mortar and loaded into a narrow Pyrex tube (outer diameter ∼5 mm, inner diameter ∼3 mm, length ∼6 cm), which was then itself loaded into a Pyrex tube (outer diameter ∼9.5 mm, inner diameter ∼5.5 mm). The tube was then evacuated to 2 × 10–5 mbar and sealed at a length of ∼30 cm. The tube was then placed in a three-zone tube furnace such that the sample was at the center of the furnace and the opposing end was at room temperature (Figure S1a). The center of the furnace was heated to 300 °C at a heating rate of 5 °C min–1. A thermocouple was used to determine the temperature profile along the length of the tube furnace (Figure S1b), which shows a gradient from 300 °C at the center to 120 °C at the edge of the furnace. After 18 h, the furnace was cooled at a rate of 5 °C min–1 to room temperature. After heating, crystals of P and 2HP/P co-crystals were found to be sublimed at the cool end of the tube (Figure S1c).
X-ray Diffraction
Single-crystal XRD data for HBP were collected using a Rigaku MicroMax-007 HF with a molybdenum rotating anode microfocus source and a Saturn 724+ detector. The structure was solved and refined using SHELX-2013.22 Hydrogen atoms were placed in calculated positions using built-in SHELX riding models and assigned isotropic thermal parameters 1.2 times those of their parent atoms. Full crystallographic details are included in Table S1. The supplementary crystallographic data for HBP can be obtained from CCDC identifier 1811701.
Powder XRD and synchrotron powder X-ray diffraction (SXRD) data were collected at room temperature in transmission mode from 0.7 mm borosilicate capillaries. Laboratory XRD data were collected from a Bruker-AXS D8 Advance diffractometer with a fine focus Mo Kα source (λ = 0.71073 Å), whereas SXRD data were collected at Beamline I11 at Diamond Light Source, U.K. (λ = 0.82602(1) Å) using Mythen PSD detectors.
Rietveld refinements were carried out using TOPAS Academic v5.1 Starting models were either taken from structures reported in the literature or (in the case of the new compound HBP) obtained from single-crystal XRD. For the Rietveld refinements, lattice parameters and a single C constrained thermal displacement parameter were the only structural parameters allowed to refine.
To calculate the amorphous content of HBP/amorphous mixture and pristine pentacene, the sample (20 mg) was ground and thoroughly mixed with a known quantity of monocrystalline diamond powder. A sample of this mixture was then loaded into a 0.7 mm borosilicate capillary for SXRD. Collected SXRD patterns were fitted to multiphase models yielding the ratio of crystalline sample mass/internal standard mass. By comparison to the ratio of total sample mass/internal standard mass, the proportion of the sample that is crystalline (and thus amorphous) is determined (Table S4).
Other Characterization Techniques
Raman spectroscopy was collected using a Renishaw inVia Raman Spectrometer with an excitation laser wavelength of 785 nm.
Elemental analysis for C and H content was performed using a Thermo EA1112 Flash CHNS-O Analyser.
Gas chromatography (GC) to detect any hydrogen emission from the sample was carried out using an Agilent 6890N with He as the carrier gas. After reaction at 300 °C, the circumference of the sealed Pyrex reaction tube was scored at its center, and PTFE washers were placed around the top and bottom. This was then placed in a custom-made break-seal apparatus (Figure S2) adapted from an earlier design by Caldwell et al.,33 inside an Ar-filled glovebox (O2 and H2O levels <1 ppm). The ground glass joint was sealed with a SubaSeal septum. The greaseless stopcock was turned to exert a force on the Pyrex tube at the score mark until it was opened. After 1 min (to allow the gas to equilibrate), a needle was used to collect a 500 μL sample of the gas, which was injected into the GC. The gas evolved from 50 mg of samples of pentacene heated at 300 °C for 200 h (i.e., at maximum conversion to HBP and amorphous oligomerized material) and 450 h (i.e., after the formation and subsequent decomposition of intermediate HBP to 2HP/P co-crystals and further amorphous oligomerized material) was measured. In addition, the H2 levels from an empty tube and 50 mg of picene (an isomer of pentacene consisting of zig-zag (rather than linearly) fused aromatic rings that is not susceptible to reaction), both heated at 300 °C for 200 h were measured. The GC was calibrated to allow quantification using a reference gas of accurately known concentration (500 ppm), which determined a sensitivity of 7(2) × 10–9 mgH per unit area of the H2 peak. The approximation of the total volume of the break-seal apparatus allowed an estimate of total H2 gas inside to be calculated.
Electron ionized mass spectrometry was conducted at the EPSRC UK National Mass Spectrometry Facility using a Thermo Scientific DSQ-II.
Exfoliation of the amorphous material was conducted using sonication. Amorphous material (2 mg) was added to 0.5 mL of N-methyl-2-pyrrolidone (NMP) in a 2 mL vial. The vial was then sonicated for 3 h to form a suspension that remains stable for (at least) several weeks. A drop of this suspension was deposited onto either a glass slide (for optical microscopy) or a bare (i.e., no carbon support) 400 mesh nickel TEM grid (for TEM). The sample was left open to the air for at least an hour to allow solvent evaporation.
Scanning electron microscopy (SEM) was carried out using a Hitachi S-4800 field-emission SEM using an accelerating voltage of either 3 kV (for imaging sonicated material suspended on TEM grid) or 10 kV, to collect images of the bulk amorphous material, for which a small amount of powder was spread onto carbon tape and sputter-coated with gold.
Transmission electron microscopy (TEM) was carried out using a 300 kV JEOL 3010.
Optical microscopy was carried out using a Meiji MT9430 microscope.
Acknowledgments
Verity Piercy and Dr. Alex Cowan are thanked for assistance with GC measurements. Prof Paul Chalker and Prof Laurence Hardwick are thanked for useful discussions on the Raman spectroscopy of carbonaceous materials, Prof Laurence Hardwick is also thanked for the use of the Renishaw inVia Raman Spectrometer. This work was carried out with the support of the Diamond Light Source on Beamline I11. We are grateful to the EPSRC UK National Mass Spectrometry Facility at Swansea University (Dr. Ann Hunter). The work was funded through EPSRC grants EP/K027212/1 and EP/K027255/2.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b00671.
Experimental details: sublimation set up; HBP crystallographic details; powder synchrotron X-ray diffraction data; additional Raman spectroscopy data; additional mass spectrometry data; gas chromatography data; optical and electron microscopy (PDF)
Author Present Address
† University of Manchester, School of Chemistry, Oxford Road, Manchester M13 9PL, U.K. (G.F.S.W.).
The authors declare no competing financial interest.
Notes
Underlying data is available at http://dx.doi.org/10.17638/datacat.liverpool.ac.uk/445.
Supplementary Material
References
- Dimitrakopoulos C. D.; Malenfant P. R. L. Organic Thin Film Transistors for Large Area Electronics. Adv. Mater. 2002, 14, 99–117. . [DOI] [Google Scholar]
- Garnier F. Thin-film transistors based on organic conjugated semiconductors. Chem. Phys. 1998, 227, 253–262. 10.1016/S0301-0104(97)00308-X. [DOI] [Google Scholar]
- Kitamura M.; Arakawa Y. Pentacene-based organic field-effect transistors. J. Phys.: Condens. Matter 2008, 20, 184011 10.1088/0953-8984/20/18/184011. [DOI] [Google Scholar]
- Roberson L. B.; Kowalik J.; Tolbert L. M.; Kloc C.; Zeis R.; Chi X.; Fleming R.; Wilkins C. Pentacene Disproportionation during Sublimation for Field-Effect Transistors. J. Am. Chem. Soc. 2005, 127, 3069–3075. 10.1021/ja044586r. [DOI] [PubMed] [Google Scholar]
- Mattheus C. C.; Baas J.; Meetsma A.; de Boer J. L.; Kloc C.; Siegrist T.; Palstra T. T. M. A 2:1 cocrystal of 6,13-dihydropentacene and pentacene. Acta Crystallogr., Sect. E: Struct. Rep. Online 2002, 58, o1229–o1231. 10.1107/S1600536802018445. [DOI] [Google Scholar]
- Rogers C.; Chen C.; Pedramrazi Z.; Omrani A. A.; Tsai H.-Z.; Jung H. S.; Lin S.; Crommie M. F.; Fischer F. R. Closing the Nanographene Gap: Surface-Assisted Synthesis of Peripentacene from 6,6′-Bipentacene Precursors. Angew. Chem., Int. Ed. 2015, 54, 15143–15146. 10.1002/anie.201507104. [DOI] [PubMed] [Google Scholar]
- Heya A.; Matsuo N.. Guidelines for bottom-up approach of nanocarbon film formation from pentacene using heated tungsten on quartz substrate without metal catalyst Jpn. J. Appl. Phys. 2018, 57, 04FL03. 10.7567/JJAP.57.04FL03. [DOI]
- Northrop B. H.; Norton J. E.; Houk K. N. On the Mechanism of Peripentacene Formation from Pentacene: Computational Studies of a Prototype for Graphene Formation from Smaller Acenes. J. Am. Chem. Soc. 2007, 129, 6536–6546. 10.1021/ja070392a. [DOI] [PubMed] [Google Scholar]
- Zöphel L.; Berger R.; Gao P.; Enkelmann V.; Baumgarten M.; Wagner M.; Müllen K. Toward the peri-Pentacene Framework. Chem. - Eur. J. 2013, 19, 17821–17826. 10.1002/chem.201302859. [DOI] [PubMed] [Google Scholar]
- Sarobe M.; Jenneskens L. W.; Wiersum U. E. Thermolysis of benzo[c]phenanthrene: Conversion of an alternant C18H12 PAH into Non-alternant C18H10 PAHs. Tetrahedron Lett. 1996, 37, 1121–1122. 10.1016/0040-4039(95)02320-8. [DOI] [Google Scholar]
- Zeis R.; Besnard C.; Siegrist T.; Schlockermann C.; Chi X.; Kloc C. Field Effect Studies on Rubrene and Impurities of Rubrene. Chem. Mater. 2006, 18, 244–248. 10.1021/cm0502626. [DOI] [Google Scholar]
- Lewis I. C.; Edstrom T. Thermal Reactivity of Polynuclear Aromatic Hydrocarbons1. J. Org. Chem. 1963, 28, 2050–2057. 10.1021/jo01043a025. [DOI] [Google Scholar]
- Field L. D.; Sternhell S.; Wilton H. V. Mechanochemistry of some hydrocarbons. Tetrahedron 1997, 53, 4051–4062. 10.1016/S0040-4020(97)00017-3. [DOI] [Google Scholar]
- Ishii Y.; Sakashita T.; Kawasaki S.; Kato H.; Takatori M. Fusing Treatment of Pentacenes: Toward Giant Graphene-Like Molecule. Mater. Express 2011, 1, 36–42. 10.1166/mex.2011.1005. [DOI] [Google Scholar]
- Mattheus C. C.; Dros A. B.; Baas J.; Meetsma A.; de Boer J. L.; Palstra T. T. M. Polymorphism in pentacene. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2001, 57, 939–941. 10.1107/S010827010100703X. [DOI] [PubMed] [Google Scholar]
- Siegrist T.; Besnard C.; Haas S.; Schiltz M.; Pattison P.; Chernyshov D.; Batlogg B.; Kloc C. A Polymorph Lost and Found: The High-Temperature Crystal Structure of Pentacene. Adv. Mater. 2007, 19, 2079–2082. 10.1002/adma.200602072. [DOI] [Google Scholar]
- Siegrist T.; Kloc C.; Schön J. H.; Batlogg B.; Haddon R. C.; Berg S.; Thomas G. A. Enhanced Physical Properties in a Pentacene Polymorph. Angew. Chem., Int. Ed. 2001, 40, 1732–1736. . [DOI] [PubMed] [Google Scholar]
- Holmes D.; Kumaraswamy S.; Matzger A. J.; Vollhardt K. P. C. On the Nature of Nonplanarity in the [N]Phenylenes. Chem. - Eur. J. 1999, 5, 3399–3412. . [DOI] [Google Scholar]
- Ehrenberg M. The crystal structure of di-para-anthracene. Acta Crystallogr. 1966, 20, 177–182. 10.1107/S0365110X66000380. [DOI] [Google Scholar]
- Gaultier J.; Hauw C.; Desvergne J. P.; Lapouyade R. Major tetracene photodimer, C36H24. Cryst. Str. Commun. 1975, 4, 497–500. [Google Scholar]
- Berg O.; Chronister E. L.; Yamashita T.; Scott G. W.; Sweet R. M.; Calabrese J. s-Dipentacene: Structure, Spectroscopy, and Temperature- and Pressure-Dependent Photochemistry. J. Phys. Chem. A 1999, 103, 2451–2459. 10.1021/jp984066g. [DOI] [Google Scholar]
- Sinnokrot M. O.; Sherrill C. D. Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer: Sandwich, T-Shaped, and Parallel-Displaced Configurations. J. Phys. Chem. A 2004, 108, 10200–10207. 10.1021/jp0469517. [DOI] [Google Scholar]
- Nemanich R. J.; Solin S. A. First- and second-order Raman scattering from finite-size crystals of graphite. Phys. Rev. B 1979, 20, 392–401. 10.1103/PhysRevB.20.392. [DOI] [Google Scholar]
- Ferrari A. C.; Robertson J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. 10.1103/PhysRevB.61.14095. [DOI] [Google Scholar]
- Ferrari A. C.; Meyer J. C.; Scardaci V.; Casiraghi C.; Lazzeri M.; Mauri F.; Piscanec S.; Jiang D.; Novoselov K. S.; Roth S.; Geim A. K. Raman Spectrum of Graphene and Graphene Layers. Phys. Rev. Lett. 2006, 97, 187401 10.1103/PhysRevLett.97.187401. [DOI] [PubMed] [Google Scholar]
- Ferrari A. C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. 10.1016/j.ssc.2007.03.052. [DOI] [Google Scholar]
- Malard L. M.; Pimenta M. A.; Dresselhaus G.; Dresselhaus M. S. Raman spectroscopy in graphene. Phys. Rep. 2009, 473, 51–87. 10.1016/j.physrep.2009.02.003. [DOI] [Google Scholar]
- Martins Ferreira E. H.; Moutinho M. V. O.; Stavale F.; Lucchese M. M.; Capaz R. B.; Achete C. A.; Jorio A. Evolution of the Raman spectra from single-, few-, and many-layer graphene with increasing disorder. Phys. Rev. B 2010, 82, 125429 10.1103/PhysRevB.82.125429. [DOI] [Google Scholar]
- Cançado L. G.; Jorio A.; Ferreira E. H. M.; Stavale F.; Achete C. A.; Capaz R. B.; Moutinho M. V. O.; Lombardo A.; Kulmala T. S.; Ferrari A. C. Quantifying Defects in Graphene via Raman Spectroscopy at Different Excitation Energies. Nano Lett. 2011, 11, 3190–3196. 10.1021/nl201432g. [DOI] [PubMed] [Google Scholar]
- Verzhbitskiy I. A.; Corato M. D.; Ruini A.; Molinari E.; Narita A.; Hu Y.; Schwab M. G.; Bruna M.; Yoon D.; Milana S.; Feng X.; Müllen K.; Ferrari A. C.; Casiraghi C.; Prezzi D. Raman Fingerprints of Atomically Precise Graphene Nanoribbons. Nano Lett. 2016, 16, 3442–3447. 10.1021/acs.nanolett.5b04183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M.; Shen Z. A review on mechanical exfoliation for the scalable production of graphene. J. Mater. Chem. A 2015, 3, 11700–11715. 10.1039/C5TA00252D. [DOI] [Google Scholar]
- Nagano M.; Hasegawa T.; Myoujin N.; Yamaguchi J.; Itaka K.; Fukumoto H.; Yamamoto T.; Koinuma H. The First Observation of 1 H-NMR Spectrum of Pentacene. Jpn. J. Appl. Phys. 2004, 43, L315. 10.1143/JJAP.43.L315. [DOI] [Google Scholar]
- Caldwell W. E.; Odom J. D.; Williams D. F. Glass-Sample-Tube Breaker. Anal. Chem. 1983, 55, 1175–1176. 10.1021/ac00258a600. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.