

We report a massive expansion in RNA-encoded bacterial viral genomes, which have been overlooked within environments.
Abstract
The first sequenced genome was that of the 3569-nucleotide single-stranded RNA (ssRNA) bacteriophage MS2. Despite the recent accumulation of vast amounts of DNA and RNA sequence data, only 12 representative ssRNA phage genome sequences are available from the NCBI Genome database (June 2019). The difficulty in detecting RNA phages in metagenomic datasets raises questions as to their abundance, taxonomic structure, and ecological importance. In this study, we iteratively applied profile hidden Markov models to detect conserved ssRNA phage proteins in 82 publicly available metatranscriptomic datasets generated from activated sludge and aquatic environments. We identified 15,611 nonredundant ssRNA phage sequences, including 1015 near-complete genomes. This expansion in the number of known sequences enabled us to complete a phylogenetic assessment of both sequences identified in this study and known ssRNA phage genomes. Our expansion of these viruses from two environments suggests that they have been overlooked within microbiome studies.
INTRODUCTION
Viruses, particularly bacteriophages targeting prokaryotes, are the most diverse biological entities in the biosphere (1, 2). Currently, there are 11,489 genome sequences available in the NCBI (National Center for Biotechnology Information) Viral RefSeq database (version 94). The vast majority of known phage have a double-stranded DNA (dsDNA) genome (3, 4). Recent metagenomic analysis of 145 marine virome sampling sites identified 195,728 DNA viral populations, highlighting that only a fraction of Earth’s viral diversity has been characterized (5). An additional expansion of known phage populations by Roux et al. (6) revealed that not only dsDNA phages but also single-stranded DNA Inoviridae are far more diverse than previously considered. The rapid expansion in viral discovery through metagenomics is enabling a greater understanding of their roles within environments and their evolutionary relationships, which is subsequently causing a revolution in phage taxonomy (7).
Despite the identification of single-stranded RNA (ssRNA) phages over 50 years ago (8), there are few representative sequences available. The International Committee on Taxonomy of Viruses (ICTV) has currently categorized approximately 5500 viruses (9). Yet, their classification only applies to 25 ssRNA phage sequences (complete or partial) across two genera, Levivirus and Allolevivirus, and an additional 32 sequences unclassified below a family taxonomic rank (10). Historically, methods for classifying Leviviridae depended on molecular weight, density, sedimentation, and serological cross-reactivity (11). A subsequent classification method separated the two genera, with the Alloleviviruses containing a fourth unique gene predicted to encode a lysin (12). Recently, an analysis of the evolution origin of all currently known RNA viruses by Wolf et al. (13) suggested that ssRNA phages may actually be two distinct lineages, which they termed Leviviridae and “Levi-like” viruses.
The ssRNA phage MS2 is a non-enveloped virus with a positive-sense monopartite genome of 3569 nt and was the first biological entity to have its entire genome sequenced (14). MS2 and its relatives were assigned to the family Leviviridae and were generally isolated against Proteobacteria. With additional studies, we can anticipate that ssRNA phages will be found, which target additional bacterial phyla. Genomes of ssRNA phages encode a maturation protein (MP) responsible for host recognition, a coat protein (CP) for genome encapsulation, and an RNA-dependent RNA polymerase (RdRp) required for viral replication. During the phage replication process, there is a negative-sense template produced for genome replication, although it does not persist and no negative-sense ssRNA phages have been isolated or characterized to date (15).
An analysis of the evolution of all RNA viruses recently proposed their primordial origin from reverse transcriptases. ICTV has recently established a new viral realm, Ribovira, to incorporate all known RNA viruses, as they all encode an RdRp for replication (16). The origin of ssRNA phages followed the acquisition of a CP, potentially allowing them to survive ex vivo and prey on the first cellular microbes (13). Despite their small genome size (encoding only three or four genes), ssRNA phages have served as models for understanding some of nature’s most widespread fundamental processes, including genome secondary structure to mechanisms of controlling gene expression and genome replication (17, 18).
Identification of phages was traditionally dependent on culture-based methods (19). In recent years, there has been a shift to culture-independent metagenomic approaches that aim to capture all microbial genomes within a given environment (20). An analysis by Krishnamurthy et al. (21) identified 158 ssRNA phage sequences (complete and partial), remarkably expanding the previously recognized diversity of this group. A more recent study by Starr et al. (22) demonstrated that metatranscriptomics will advance ssRNA phage discovery, with 1338 ssRNA phage RdRp sequences detected in soil. Metatranscriptomics is indeed well suited to capturing ssRNA phage sequences in complex biological samples, given that their genomes resemble the mRNA transcripts that are targeted by this method.
The actual abundance and diversity of ssRNA phages have remained unknown despite recent advancements to better study the phage populations of different environments. Databases are dominated by DNA phage genomes, and novel ssRNA phages may not be recognized. Isolation and purification techniques for phages, such as caesium chloride (CsCl) gradient purification and polyethylene glycol, are biased toward isolating specific phage types (23). Even accepting that specific metatranscriptomic approaches will introduce their own biases in the process of removing ribosomal RNA (24), it is likely to be more representative of the RNA composition of a specific microbiome, including the RNA viral contingents.
RNA phages have served as key models in understanding some of biology’s most intricate pathways such as gene regulation. These phages also offer a potential option in terms of phage therapy, as they have been isolated against many pathogenic bacteria including Acinetobacter and Pseudomonas. Fundamentally, the expansion in ssRNA phage genomes reported here demonstrates that their contributions to the diversity of ecological niches and their impacts on their associated hosts may have been underestimated. Given that we are just starting to explore Earth’s “viral dark matter” through metagenomics, it seems fitting that a portion of this unexplored viral diversity is represented by phages that are not encoded by DNA.
In this study, we report the identification of 15,611 near-complete and partial ssRNA phage sequences. Of these, 1015 were defined as near complete in that they encode all three MP, CP, and RdRp genes that form the recognized ssRNA phage core genome. The identification of ssRNA phage sequences was performed by iteratively developing and applying hidden Markov models (HMMs) based on conserved ssRNA phage proteins. We applied these HMMs to ever-increasing samples from 70 activated sludge and 12 aquatic environments. This expansion in the number of ssRNA phage genomes enabled us to examine the phylogenetic relationships between sequences identified in this study and known sequences and perform a preliminary investigation of phage-host interactions.
RESULTS AND DISCUSSION
Expansion of known ssRNA phage sequences
We collected 193 identifiable unique partial ssRNA phage genome sequences from publicly available databases and relevant studies (fig. S1). An additional 67 Levi-like sequences, described by Shi et al. (25), were used to validate the identification of ssRNA phages from an RNA viral database (see Materials and Methods). We predicted the encoded proteins of the 193 ssRNA phage genomes and used a graph-based clustering method to build a database of HMM sequence profiles representative of their protein sequences (see fig. S2 and Supplementary Text). Four subsequent HMM iterations were built, each using the previous HMM output, and were applied to a final total of 82 publicly available environmental metatranscriptome samples generated from globally sourced activated sludge and aquatic samples. A final manually curated HMM, designated 5-MC, was developed by removing all partial protein sequences.
In total, we identified 15,611 ssRNA phage genomes or partial sequences (Fig. 1B). This represents an approximately 60-fold increase in the number of partial genome sequences. Of the 15,611 identified sequences, there were 5387 ssRNA phage sequences, which had a minimum length of 750 base pairs (bp) and included at least one core gene (MP, CP, or RdRp), 2987 included two core genes, and 1848 had sequences from all three core genes. Of these, 1015 are predicted to encode full-length core genes (see Supplementary Text). Only 29 of the currently publicly identifiable 193 ssRNA phage sequences meet this same criterion (fig. S1D).
Fig. 1. Identification of ssRNA phages in metatranscriptome samples.
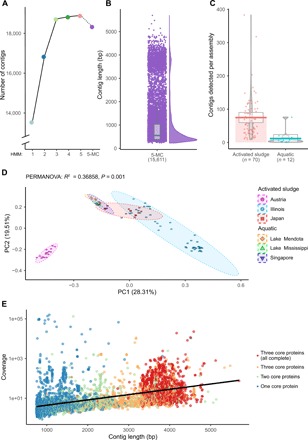
(A) The total number of redundant contigs detected per HMM search. (B) The manually curated HMM 5-MC detected 15,611 nonredundant ssRNA phage sequences. Boxplot displays the median value within the 25th and 75th quartiles, with whiskers representing the interquartile range of ±1.5. (C) The number of contigs (near complete or partial) detected per assembly in activated sludge and aquatic samples. Boxplot horizontal lines indicate the mean, while the gray boxes represent 95% highest-density intervals. (D) Two-dimensional ordination of ssRNA compositional abundance across different geographical locations using the Bray-Curtis Dissimilarity index. The colors and shapes of individual samples differentiate study location and environment, respectively. PC, principle component. (E) Linear model of metatranscriptome sequencing coverage and contig length. Contigs included are of minimum length of 750 bp, and the number of core proteins encoded is indicated.
Significantly more ssRNA phage sequences were detected in activated sludge than in aquatic samples (Kruskal-Wallis, P = 1.847 × 10−6; Fig. 1C). It is possible that activated sludge provides an environment in which proteobacteria, the only known hosts for ssRNA phages, can grow and support phage enrichment. The higher levels of detection could also be due to a variety of technical factors such as increased sequencing depth, microbiome complexity, and metatranscriptome sampling protocols. Our ability to detect longer ssRNA phage sequences correlates with metatranscriptome sequencing depth (Fig. 1E).
Examination of genome-associated proteins and architecture
The 15,611 ssRNA phage sequences encoded 24,419 proteins that could be grouped into three MP, eight CP, and two RdRp clusters (Fig. 2A and fig. S2). It is evident that the RdRp is the most conserved protein, forming only two clusters, whereas the CP is the most diverse of the ssRNA phage–associated core protein, splitting into eight clusters. We next examined all 2987 ssRNA sequences encoding at least two core proteins, which revealed two highly distinct groups (Fig. 2B). Only 5 of the almost 3000 assembled sequences bridge the two groups, and these were investigated further (see Supplementary Text). Briefly, the five outliers only encode partial rather than complete proteins, and their relatedness to a specific protein cluster may be driven by local rather than global sequence similarity.
Fig. 2. Examination of ssRNA phage proteins.
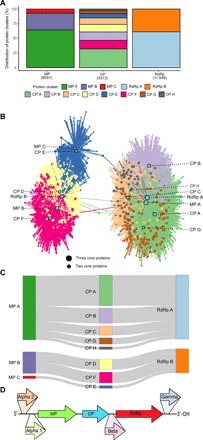
(A) Distribution of protein hits (in parentheses) across MP, CP, and RdRp clusters was identified using HMM 5-MC. (B) Bipartite connection network of contigs (circles) with proteins (squares). Colors are based on the associated CP from (A). (C) Protein cluster co-occurring profiles of ssRNA phages having all three full-length core proteins and (D) the frequently observed positions of hypothetical proteins (genes not drawn to scale).
We analyzed all 1015 near-complete ssRNA phage genomes and observed strictly conserved protein associations (Fig. 2C). In contrast to other viruses, there are no obvious instances of homologous recombination and mosaicism among the identified ssRNA phages. Both mosaicism and horizontal gene transfer are well noted for dsDNA phages, with single genes and whole modules exchanged (26, 27). Recombination frequencies of RNA viruses are reported to vary markedly during coinfection, influenced by various factors such as sequence identity, kinetics of transcription, and RNA genome secondary structure (28). We only recorded eight protein connection profiles between the three MP, eight CP, and two RdRp protein clusters of ssRNA phages. If their genomes underwent extensive recombination events, then it would be expected that the number of core-protein connection profiles would be closer to the theoretical maximum of 48 (3 MP × 8 CP × 2 RdRp). However, as our ssRNA phage discovery pipeline is restricted to finding viruses encoding core proteins similar to those previously identified, future studies with less stringent search criteria may uncover additional unexplored biodiversity.
With such a tremendous expansion in the quantity of identifiable complete ssRNA phages, we undertook an examination of their genome structure. First, we investigated the specific order of MP, CP, and RdRp core proteins. Notably, on no occasion did we identify the recognizable CP situated either before the MP- or after the RdRp-encoding genes. In all 1015 instances, a CP was situated between the MP and RdRp genes. We noted that hypothetical proteins could exist before the MP, after the CP, or following the RdRp (Fig. 2D). In 20 instances, there were two hypothetical proteins situated before the MP. We termed the locations the alpha position (closest to the 5′ terminus), the beta position (between the CP and RdRp), and the gamma position (closest to the 3′ terminus). We labeled any hypothetical immediately preceding the MP gene as the alpha 1 position, and if a second hypothetical was identified, then it was deemed to occupy the alpha 2 position.
Further investigation revealed that hypothetical genes predicted to follow the RdRp often had weak similarity to the native RdRp termini. Therefore, these proteins annotated as hypothetical could be an artifact of stop codons inadvertently introduced during metatranscriptome assembly, or alternatively, RNA phages are known to bypass stop codons as part of their replication (29). The hypothetical genes located upstream of the MP and between the CP and RdRp were also analyzed. These genes encode proteins with high sequence diversity, and hence, they did not generate clusters. However, several isolated ssRNA phages are known to contain a gene encoding a lysin in these positions (12, 30). These hypothetical genes may encode this and/or other putative functions, which may be revealed in future studies through biochemical analysis.
Phylogenetic assessment of near-complete ssRNA phage genomes
Comparisons of RNA viruses infecting all kingdoms of life have previously been undertaken using the RdRp protein (13). For greater resolution, we estimated the evolutionary relatedness of ssRNA phages using all three core proteins. We included the 29 publicly identifiable complete ssRNA phage sequences with the 1015 identified in this study. Through phylogenetic analysis, we observed the higher-level taxonomy of ssRNA phages that follow the clustering of the RdRp and CP (Fig. 3A). Lower-level taxonomy of ssRNA phages was performed using pairwise identity comparisons (fig. S5). A potential restructuring of ssRNA taxonomy is outlined in (fig. S6).
Fig. 3. Phylogenetic assessment of ssRNA phages.
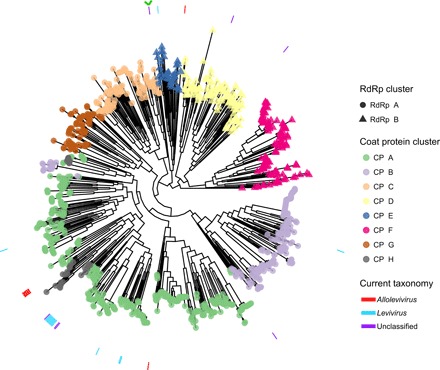
Phylogeny of ssRNA phages using their core protein sequences (MP, CP, and RdRp). The 29 previously characterized and 1015 newly identified phages were included. Branch tip shapes highlight specific RdRp protein clusters, while color indicates CP clustering. The encircling annotation ring depicts current ICTV taxonomy. A green arrowhead represents AVE006, which encodes a unique RdRp and CP association. Bootstrap support values shown are for 100 iterations.
The phylogenetic divergence of ssRNA phages by their core proteins supports the hypothesis of Wolf et al. (13) that the current Leviviridae family is two distinct lineages. However, our analysis further classifies ssRNA phages into eight subfamilies (currently denoted A to H) based on CP clustering. While this suggested that classification system can be applied to previously identified ssRNA phages, it does not support the current Levivirus and Allolevivirus taxonomic division (Fig. 3 and fig. S6).
Correlation analysis between the newly proposed taxa and the source locations identified a possible link. The ssRNA subfamilies were statistically different by geographical location (Kruskal-Wallis test; P < 0.001). This may signify that specific ecological niches are occupied by specific phage taxa. For example, CP A was strongly associated with ssRNA phages identified from the Illinois study site (254 of 1015; 25.0%), whereas it was infrequently observed among Singapore-associated phages (0.5%). A specific global distribution of dsDNA phages was recently detailed for crAssphage (31). However, because of the inherent differences introduced through different study protocols and sequencing methodologies, a single study investigating multiple geographical locations is necessary to confirm the potential global localization of specific ssRNA phage taxa.
Examination of phage-host interactions
In an attempt to further elucidate ssRNA phage interactions with their host bacteria, we examined bacterially encoded CRISPR systems and also examined the phage receptor binding protein, MP. CRISPR systems have recently been identified to target RNA phages (32); however, actively transcribed CRISPR spacers were only found against a handful of viral RefSeq database sequences in this study (see fig. S7B and Supplementary Text). No CRISPR spacers were identified to target ssRNA phage sequences. A similar observation was noted by Silas and colleagues (33). Therefore, advances in alternative techniques may be required to identify ssRNA phage–host partners, as has been demonstrated for dsDNA phages using Hi-C sequencing and single-cell viral tagging (34, 35).
Our expanded number of host-recognizing MP protein sequences allowed for a comparative structural analysis. Focusing on the MP cluster A, we revealed three variable regions associated with the MP β-binding region (fig. S8). Conserved and variable regions of MP proteins were previously highlighted during an analysis of ssRNA phage AP205 (36). Structural analysis of cluster A host-recognizing MP proteins also revealed the association of different sections with various viral components, with the conserved α-helical domain interacting with the CP subunits and the viral genome. The identification of variable ssRNA phage genomic regions through multiple sequence comparisons will further reveal areas under evolutionary selective pressure.
CONCLUSION
In summary, we iteratively optimized an HMM-based ssRNA phage discovery pipeline. Through intensive data mining of multiple metatranscriptomic datasets from just two environmental ecosystems, we identified 15,611 near-complete and partial genomes. These samples originated from America, Austria, Japan, and Singapore, highlighting the global distribution of these viruses. This represents an approximate 60-fold expansion of previously known genome sequences. Phylogenetic comparison of 1044 near-complete genomes allowed us to construct a robust, yet elastic, taxonomic scheme that provides a hierarchal foundation, which will accommodate the expected increase in ssRNA phage discoveries. Given the amount of the ssRNA phages identified in this study from two environments, we suspect that their low abundance in metagenomic studies of other ecosystems may be attributed to a variety of factors, including isolation protocols and computational shortcomings.
MATERIALS AND METHODS
Assembly of metatranscriptome samples
The assembly of metatranscriptome samples is portrayed in fig. S2A. Fastq raw reads were downloaded from the NCBI Sequence Read Archive (SRA) database using accession numbers provided in the Supplementary Materials, with files separated into forward and reverse reads using the “--split-files” option. Illumina adapter sequences were removed using Cutadapt [version 1.9.1; (37)]. The overall read quality was improved using Trimmomatic [version 0.32; (38)], pruning sequences where the read quality dropped below a Phred score of 30 for a 4-bp sliding window. Reads less than 70 bp were discarded, with surviving reads assembled using rnaSPAdes [version 3.12.0; (39)]. Only metatranscriptome sample SRR5466337, which generated an error during rnaSPAdes assembly, was assembled differently using MEGAHIT [version 1.1.1-2; (40)]. This one sample, of the total 82 samples, failed to assemble using rnaSPAdes. The reasons were not investigated further. All contig assemblies less than 500 bp were discarded. Only the rnaSPAdes “hard filtered transcript” outputs were examined for the presence of ssRNA phages.
Generation of profile HMMs
The pipeline for generating profile HMMs is depicted in fig. S2B, with the numerical breakdown of the HMM building and testing stages depicted in fig. S2 (D and E), respectively. To generate the first HMM, “HMM 1,” all ssRNA phage near-complete and partial genome sequences were downloaded from the NCBI Taxonomy database (October 2018) and previous published studies (21). The encoded proteins of all identifiable ssRNA phage sequences (n = 193) were predicted using Prodigal with the “-p meta” option enabled for small contigs, and “-n” option was specified to do a full motif scan per nucleotide sequence [version 2.6.3; (41)]. Predicted proteins were clustered using OrthoMCL using a BLASTp all-v-all E value of 1 × 10−5 and default settings [version 2.0; (42)]. Clusters of ssRNA phage proteins with 10 or more sequences were aligned using MUSCLE [version 3.8.31; (43)] and used to generate HMMs via hmmbuild [version 3.1b1; (44)]. Multiple HMMs were combined into a single HMM search tool through hmmpress (version 3.1b1).
The number of samples tested by each HMM iteration is outlined in fig. S2C. HMMs 2 to 5 were built in a similar fashion to HMM 1 with the following alterations. Subsequent to the detection of contigs in metatranscriptome samples encoding two or more functionally distinct ssRNA phage proteins (hmmscan score of 50 or greater), the predicted proteins were combined with those from the initial 193 ssRNA phage sequences, obtained from NCBI and a previous publication (21). Using a BLAST all-v-all approach, the proteins used to generate HMMs 2 to 5 were made nonredundant at 70% amino acid identity, removing the shorter of two protein sequences when the overlap exceeded 70%. Before the generation of HMM 5-MC, proteins were manually curated to remove sequences encoded at the edge of contigs (termed “edge proteins”).
Validating HMM detection of ssRNA phages
The metatranscriptome sample SRR1027978, which was an activated sludge sample previously shown by Krishnamurthy et al. (21) as containing ssRNA phage sequences using a tBLASTn approach, was downloaded as a positive control and examined for the presence of ssRNA phage proteins. Briefly, a random subset of 10 million reads was extracted from the SRA file with the seqtk “sample” command [version 1.0-r31; (45)] using a user-defined seed (“-s13”). Adaptor and read trimming was performed as described above, with surviving reads assembled using MEGAHIT. Proteins were predicted in all contigs greater than 500 bp, using options “-p meta -n”, before scanning with HMM 1.
After manual curation of ssRNA phage hits, it was decided to adopt a conservative approach for the remainder of the study. Only hmmscan hits with a score of 50 or greater were considered during the generation of HMM iterations, with hmmscan scores of 30 further investigated during metatranscriptome sample analyses. Future studies may benefit from less stringent ssRNA phage discovery cutoffs, by lowering the hmmscan score requirements and/or using rnaSPAdes “soft filtered transcripts.” However, results would need to be treated cautiously to avoid false positives.
A comparison between a BLAST and an HMM-based approach to identify ssRNA phages was performed using the complete ssRNA phage proteins, which built the final HMM model 5-MC. The BLAST and HMM approaches were applied to the 2308 unique viral sequences described by Shi and colleagues (25). This database contains 67 ssRNA Levi-like viruses. Using a relaxed BLASTp E value of 1 × 10−5, 78 viral sequences were considered ssRNA phages (11 false positives). However, with a more stringent BLASTp E value of 1 × 10−15, only the expected 67 sequences were returned. Using an HMM scan with a score of 30 identified the 67 Levi-like viruses without any false positives identified.
When the strict BLASTp search approach (E value of 1 × 10−15) was applied to the assembled contigs from the metatranscriptome sample SRR1027978, 12 ssRNA phages were identified. The HMM-based approach identified 13 ssRNA phages. Reducing the BLASTp stringency to 1 × 10−5 did identify 13 putative ssRNA phages. However, because of the false positives noted while using a less strict BLASTp approach against a curated database, only HMM searches were used throughout this study.
Detecting ssRNA within metatranscriptome samples
After confirming that HMM 1 could detect ssRNA phage proteins in a positive control sample, HMM 1 was implemented against nine previously untested metatranscriptome samples of activated sludge. This environment was chosen as Krishnamurthy et al. (21) demonstrated sewage as a rich source for ssRNA phages (21). These nine SRA files analyzed represent three activated sludge samples from each of the study locations from Austria, Illinois, and Japan (see the Supplementary Materials). The total collection of activated sludge and aquatic samples cumulatively analyzed during this study is outlined in fig. S2C. The remaining samples tested represent 13 activated sludge samples from Austria, 39 activated sludge samples from Illinois, 9 activated sludge samples from Japan, 4 freshwater aquatic samples from Lake Mendota (Wisconsin), 4 aquatic samples from the Mississippi river (Louisiana), and 4 freshwater aquatic samples from Singapore.
Analysis of ssRNA phage proteins
Analyses were conducted using the R programming language (version 3.5.3) implemented through RStudio (46). Images were generated using the “ggplot2” package (47), with additional colors obtained from the “RColorBrewer” (48), the “wesanderson” (49), and the “YaRrr” package (50). The bipartite network of ssRNA phage proteins, for sequences containing two or three core proteins, was generated using the “igraph” package (51). The distance between core proteins (squares) was automatically calculated on the basis of the number of ssRNA sequences (circles) that share similar protein profiles. The ssRNA phage partial genomes are colored on the basis of the associated CP. The Sankey plot demonstrating the connection patterns of ssRNA phage–encoded proteins was illustrated using the R package “networkD3” (52).
Phylogeny of ssRNA phage proteins was performed as follows. Proteins fulfilling the same functions among ssRNA phages were assigned the name of their originating contig and subsequently aligned using MUSCLE. The alignment of the three core proteins were concatenated using MEGA [version 10.0.5; (53)]. After the three proteins were concatenated, the MUSCLE alignment was performed with default settings—no alignment trimming, all positions were retained, and the substitution model was applied to all proteins together. These alignments were imported into R using the “seqinr” package (54, 55) with “ape” package dependencies (56) before conversion to a phyDat format using the “phangorn” package (57). The best evolutionary model was estimated using the phangorn “modelTest” function, with the model yielding the lowest Akaike Information Criterion score selected for maximum likelihood tree construction. Blosum62 was determined as the best amino acid substitution model. Phylogenetic trees were bootstrapped 100 times and saved using the “treeio” package (58), before visualization using “ggtree” (59). The R scripts and input data used to generate this study’s images and infer results are provided in the Supplementary Materials.
Supplementary information
The newly identified unique RNA phage sequences and genomes (n = 15,611 and 1015, respectively) and the final ssRNA phage detection tool (HMM 5-MC) are provided in data S1. All the accession number details, raw data, tables, and R scripts used in the analysis and creation of images are provided in data S2.
Supplementary Material
Acknowledgments
Funding: This publication has emanated from research conducted with the financial support of Science Foundation Ireland under grant number SFI/12/RC/2273. Author contributions: J.C. and S.R.S. conceived the study, performed the analysis, produced the images, and wrote the manuscript. A.S. and L.A.D. helped interpret the results, provided helpful suggestions, and corrected manuscript drafts. R.P.R. and C.H. secured the funding, conceived the study, contributed to data analysis, and assisted in generating the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay5981/DC1
Supplementary Text
Fig. S1. Workflow depiction of known ssRNA phage sequences.
Fig. S2. Workflow depiction of the study pipeline.
Fig. S3. Identification of ssRNA phage contigs within 82 metatranscriptome samples.
Fig. S4. Genome architecture of ssRNA phages.
Fig. S5. Taxonomic cutoff values for ssRNA phage genera and species.
Fig. S6. Potential taxonomic restructuring for ssRNA phages.
Fig. S7. Analysis of microbial community complexity.
Fig. S8. Structural investigation of ssRNA phage–host interactions.
Data S1. ssRNA phage finding hidden Markov model and associated sequences.
Data S2. Bioinformatic scripts used during data analysis.
REFERENCES AND NOTES
- 1.Cobián Güemes A. G., Youle M., Cantú V. A., Felts B., Nulton J., Rohwer F., Viruses as winners in the game of life. Annu. Rev. Virol. 3, 197–214 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Clokie M. R. J., Millard A. D., Letarov A. V., Heaphy S., Phages in nature. Bacteriophage 1, 31–45 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manrique P., Bolduc B., Walk S. T., van der Oost J., de Vos W. M., Young M. J., Healthy human gut phageome. Proc. Natl. Acad. Sci. U.S.A. 113, 10400–10405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Norman J. M., Handley S. A., Baldridge M. T., Droit L., Liu C. Y., Keller B. C., Kambal A., Monaco C. L., Zhao G., Fleshner P., Stappenbeck T. S., McGovern D. P. B., Keshavarzian A., Mutlu E. A., Sauk J., Gevers D., Xavier R. J., Wang D., Parkes M., Virgin H. W., Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregory A. C., Zayed A. A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C., Dimier C., Domínguez-Huerta G., Ferland J., Kandels S., Liu Y., Marec C., Pesant S., Picheral M., Pisarev S., Poulain J., Tremblay J.-É., Vik D., Acinas S. G., Babin M., Bork P., Boss E., Bowler C., Cochrane G., de Vargas C., Follows M., Gorsky G., Grimsley N., Guidi L., Hingamp P., Iudicone D., Jaillon O., Kandels-Lewis S., Karp-Boss L., Karsenti E., Not F., Ogata H., Pesant S., Poulton N., Raes J., Sardet C., Speich S., Stemmann L., Sullivan M. B., Sunagawa S., Wincker P., Babin M., Bowler C., Culley A. I., de Vargas C., Dutilh B. E., Iudicone D., Karp-Boss L., Roux S., Sunagawa S., Wincker P., Sullivan M. B., Marine DNA viral macro- and microdiversity from Pole to Pole. Cell 177, 1109–1123.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roux S., Krupovic M., Daly R. A., Borges A. L., Nayfach S., Schulz F., Sharrar A., Carnevali P. B. M., Cheng J.-F., Ivanova N. N., Bondy-Denomy J., Wrighton K. C., Woyke T., Visel A., Kyrpides N. C., Eloe-Fadrosh E. A., Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. , 1895–1906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barylski J., Enault F., Dutilh B. E., Schuller M. B. P., Edwards R. A., Gillis A., Klumpp J., Knezevic P., Krupovic M., Kuhn J. H., Lavigne R., Oksanen H. M., Sullivan M. B., Jang H. B., Simmonds P., Aiewsakun P., Wittmann J., Tolstoy I., Brister J. R., Kropinski A. M., Adriaenssens E. M., Analysis of spounaviruses as a case study for the overdue reclassification of tailed phages. Syst. Biol. , syz036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loeb T., Zinder N. D., A Bacteriophage Containing RNA. Proc. Natl. Acad. Sci. U.S.A. 47, 282–289 (1961). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker P. J., Siddell S. G., Lefkowitz E. J., Mushegian A. R., Dempsey D. M., Dutilh B. E., Harrach B., Harrison R. L., Hendrickson R. C., Junglen S., Knowles N. J., Kropinski A. M., Krupovic M., Kuhn J. H., Nibert M., Rubino L., Sabanadzovic S., Simmonds P., Varsani A., Zerbini F. M., Davison A. J., Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019). Arch. Virol. 164, 2417–2429 (2019). [DOI] [PubMed] [Google Scholar]
- 10.R. C. L. Olsthoorn, J. Van Duin, Leviviridae - Positive Sense RNA Viruses - Positive Sense RNA Viruses (2011) - International Committee on Taxonomy of Viruses (ICTV). Int. Comm. Taxon. Viruses ICTV (2017); https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/263/leviviridae.
- 11.Olsthoorn R., van Duin J., Bacteriophages with ssRNA. Encycl. Life Sci. 10.1002/9780470015902.a0000778.pub3 , (2011). [Google Scholar]
- 12.Atkins J. F., Steitz J. A., Anderson C. W., Model P., Binding of mammalian ribosomes to MS2 phage rna reveals an overlapping gene encoding a lysis function. Cell 18, 247–256 (1979). [DOI] [PubMed] [Google Scholar]
- 13.Wolf Y. I., Kazlauskas D., Iranzo J., Lucía-Sanz A., Kuhn J. H., Krupovic M., Dolja V. V., Koonin E. V., Origins and evolution of the global RNA virome. mBio 9, e02329-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiers W., Contreras R., Duerinck F., Haegeman G., Iserentant D., Merregaert J., Jou W. M., Molemans F., Raeymaekers A., den Berghe A. V., Volckaert G., Ysebaert M., Complete nucleotide sequence of bacteriophage MS2 RNA: Primary and secondary structure of the replicase gene. Nature 260, 500–507 (1976). [DOI] [PubMed] [Google Scholar]
- 15.Koonin E. V., Senkevich T. G., Dolja V. V., The ancient virus world and evolution of cells. Biol. Direct 1, 29 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A. Gorbalenya, M. Krupovic, S. Siddell, A. Varsani, J. H. Kuhn, Riboviria: Establishing a single taxon that comprises RNA viruses at the basal rank of virus taxonomy (2017).
- 17.Gytz H., Mohr D., Seweryn P., Yoshimura Y., Kutlubaeva Z., Dolman F., Chelchessa B., Chetverin A. B., Mulder F. A. A., Brodersen D. E., Knudsen C. R., Structural basis for RNA-genome recognition during bacteriophage Qβ replication. Nucleic Acids Res. 43, 10893–10906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lodish H. F., Bacteriophage f2 RNA: Control of translation and gene order. Nature 220, 345–350 (1968). [DOI] [PubMed] [Google Scholar]
- 19.Kannoly S., Shao Y., Wang I.-N., Rethinking the evolution of single-stranded RNA (ssRNA) bacteriophages based on genomic sequences and characterizations of two R-plasmid-dependent ssRNA phages, C-1 and Hgal1. J. Bacteriol. 194, 5073–5079 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dantas G., Sommer M. O. A., Degnan P. H., Goodman A. L., Experimental approaches for defining functional roles of microbes in the human gut. Annu. Rev. Microbiol. 67, 459–475 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnamurthy S. R., Janowski A. B., Zhao G., Barouch D., Wang D., Hyperexpansion of RNA bacteriophage diversity. PLOS Biol. 14, e1002409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starr E. P., Nuccio E. E., Pett-Ridge J., Banfield J. F., Firestone M. K., Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Microbiology 116, 25900–25908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleiner M., Hooper L. V., Duerkop B. A., Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 16, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alberti A., Belser C., Engelen S., Bertrand L., Orvain C., Brinas L., Cruaud C., Giraut L., Da Silva C., Firmo C., Aury J.-M., Wincker P., Comparison of library preparation methods reveals their impact on interpretation of metatranscriptomic data. BMC Genomics 15, 912 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi M., Lin X.-D., Tian J.-H., Chen L.-J., Chen X., Li C.-X., Qin X.-C., Li J., Cao J.-P., Eden J.-S., Buchmann J., Wang W., Xu J., Holmes E. C., Zhang Y.-Z., Redefining the invertebrate RNA virosphere. Nature 540, 539–543 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Hatfull G. F., Hendrix R. W., Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rokyta D. R., Burch C. L., Caudle S. B., Wichman H. A., Horizontal gene transfer and the evolution of microvirid coliphage genomes. J. Bacteriol. 188, 1134–1142 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon-Loriere E., Holmes E. C., Why do RNA viruses recombine? Nat. Rev. Microbiol. 9, 617–626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiner A. M., Weber K., A single UGA codon functions as a natural termination signal in the coliphage Qβ coat protein cistron. J. Mol. Biol. 80, 837–855 (1973). [DOI] [PubMed] [Google Scholar]
- 30.Ruokoranta T. M., Grahn A. M., Ravantti J. J., Poranen M. M., Bamford D. H., Complete genome sequence of the broad host range single-stranded RNA phage PRR1 places it in the Levivirus genus with characteristics shared with Alloleviviruses. J. Virol. 80, 9326–9330 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards R. A., Vega A. A., Norman H. M., Ohaeri M., Levi K., Dinsdale E. A., Cinek O., Aziz R. K., McNair K., Barr J. J., Bibby K., Brouns S. J. J., Cazares A., de Jonge P. A., Desnues C., Muñoz S. L. D., Fineran P. C., Kurilshikov A., Lavigne R., Mazankova K., McCarthy D. T., Nobrega F. L., Muñoz A. R., Tapia G., Trefault N., Tyakht A. V., Vinuesa P., Wagemans J., Zhernakova A., Aarestrup F. M., Ahmadov G., Alassaf A., Anton J., Asangba A., Billings E. K., Cantu V. A., Carlton J. M., Cazares D., Cho G.-S., Condeff T., Cortés P., Cranfield M., Cuevas D. A., De la Iglesia R., Decewicz P., Doane M. P., Dominy N. J., Dziewit L., Elwasila B. M., Eren A. M., Franz C., Fu J., Garcia-Aljaro C., Ghedin E., Gulino K. M., Haggerty J. M., Head S. R., Hendriksen R. S., Hill C., Hyöty H., Ilina E. N., Irwin M. T., Jeffries T. C., Jofre J., Junge R. E., Kelley S. T., Mirzaei M. K., Kowalewski M., Kumaresan D., Leigh S. R., Lipson D., Lisitsyna E. S., Llagostera M., Maritz J. M., Marr L. C., McCann A., Molshanski-Mor S., Monteiro S., Moreira-Grez B., Morris M., Mugisha L., Muniesa M., Neve H., Nguyen N., Nigro O. D., Nilsson A. S., O’Connell T., Odeh R., Oliver A., Piuri M., Prussin A. J. II, Qimron U., Quan Z.-X., Rainetova P., Ramírez-Rojas A., Raya R., Reasor K., Rice G. A. O., Rossi A., Santos R., Shimashita J., Stachler E. N., Stene L. C., Strain R., Stumpf R., Torres P. J., Twaddle A., Ibekwe M. U., Villagra N., Wandro S., White B., Whiteley A., Whiteson K. L., Wijmenga C., Zambrano M. M., Zschach H., Dutilh B. E., Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 4, 1727–1736 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strutt S. C., Torrez R. M., Kaya E., Negrete O. A., Doudna J. A., RNA-dependent RNA targeting by CRISPR-Cas9. eLife 7, e32724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silas S., Makarova K. S., Shmakov S., Páez-Espino D., Mohr G., Liu Y., Davison M., Roux S., Krishnamurthy S. R., Fu B. X. H., Hansen L. L., Wang D., Sullivan M. B., Millard A., Clokie M. R., Bhaya D., Lambowitz A. M., Kyrpides N. C., Koonin E. V., Fire A. Z., On the origin of reverse transcriptase-using CRISPR-Cas systems and their hyperdiverse, enigmatic spacer repertoires. mBio 8, e00897-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marbouty M., Baudry L., Cournac A., Koszul R., Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci. Adv. 3, e1602105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Džunková M., Low S. J., Daly J. N., Deng L., Rinke C., Hugenholtz P., Defining the human gut host–phage network through single-cell viral tagging. Nat. Microbiol. 4, 2192–2203 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Klovins J., Overbeek G. P., van den Worm S. H. E., Ackermann H.-W., van Duin J., Nucleotide sequence of a ssRNA phage from Acinetobacter: Kinship to coliphages. J. Gen. Virol. 83, 1523–1533 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 38.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bushmanova E., Antipov D., Lapidus A., Przhibelskiy A. D., rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. bioRxiv 10.1101/420208, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W., MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Hyatt D., Chen G.-L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J., Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer S., Brunk B. P., Chen F., Gao X., Harb O. S., Iodice J. B., Shanmugam D., Roos D. S., Stoeckert C. J., Using OrthoMCL to Assign Proteins to OrthoMCL-DB Groups or to Cluster Proteomes Into New Ortholog Groups.Curr. Protoc. Bioinf., Ed. D. S. Goodsell. 35, 6.12.1–6.12.19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finn R. D., Clements J., Eddy S. R., HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.H. Li, Toolkit for processing sequences in FASTA/Q formats: lh3/seqtk (2019); https://github.com/lh3/seqtk.
- 46.RStudio Team (2015). RStudio: Integrated Development for R. RStudio, Inc., Boston, MA. RStudio Support, www.rstudio.com/.
- 47.H. Wickham, ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, New York, 2009); www.springer.com/gp/book/9780387981413, Use R!
- 48.E. Neuwirth, RColorBrewer: ColorBrewer Palettes (2014); https://CRAN.R-project.org/package=RColorBrewer.
- 49.wesanderson: A Wes Anderson Palette Generator version 0.3.6 from CRAN, https://rdrr.io/cran/wesanderson/.
- 50.N. Phillips, yarrr: A Companion to the e-Book “YaRrr!: The Pirate’s Guide to R” (2017); https://CRAN.R-project.org/package=yarrr.
- 51.G. Csardi, T. Nepusz, The igraph software package for complex network research. (2006); http://igraph.org.
- 52.J. J. Allaire, C. Gandrud, K. Russell, C. Yetman, networkD3: D3 JavaScript Network Graphs from R version 0.4 from CRAN (2017); https://rdrr.io/cran/networkD3/.
- 53.Kumar S., Stecher G., Li M., Knyaz C., Tamura K., MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.D. Charif, O. Clerc, C. Frank, J. R. Lobry, A. Necşulea, L. Palmeira, S. Penel, G. Perrière, seqinr: Biological Sequences Retrieval and Analysis (2017); https://CRAN.R-project.org/package=seqinr.
- 55.D. Charif, J. R. Lobry, in Structural Approaches to Sequence Evolution: Molecules, Networks, Populations, U. Bastolla, M. Porto, H. E. Roman, M. Vendruscolo, Eds. (Springer Berlin Heidelberg, Berlin, Heidelberg, 2007); 10.1007/978-3-540-35306-5_10, Biological and Medical Physics, Biomedical Engineering, pp. 207–232. [DOI]
- 56.Paradis E., Schliep K., ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Schliep K. P., phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.G. Yu, treeio: Base Classes and Functions for Phylogenetic Tree Input and Output version 1.6.2 from Bioconductor (2019); https://rdrr.io/bioc/treeio/.
- 59.Yu G., Smith D. K., Zhu H., Guan Y., Lam T. T.-Y., GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017). [Google Scholar]
- 60.Drake J. W., Charlesworth B., Charlesworth D., Crow J. F., Rates of spontaneous mutation. Genetics 148, 1667–1686 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holland J., Spindler K., Horodyski F., Grabau E., Nichol S., VandePol S., Rapid evolution of RNA genomes. Science 215, 1577–1585 (1982). [DOI] [PubMed] [Google Scholar]
- 62.Gu X., Tay Q. X. M., Te S. H., Saeidi N., Goh S. G., Kushmaro A., Thompson J. R., Gin K. Y.-H., Geospatial distribution of viromes in tropical freshwater ecosystems. Water Res. 137, 220–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Linz A. M., He S., Stevens S. L. R., Anantharaman K., Rohwer R. R., Malmstrom R. R., Bertilsson S., McMahon K. D., Freshwater carbon and nutrient cycles revealed through reconstructed population genomes. PeerJ. 6, e6075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schulz F., Yutin N., Ivanova N. N., Ortega D. R., Lee T. K., Vierheilig J., Daims H., Horn M., Wagner M., Jensen G. J., Kyrpides N. C., Koonin E. V., Woyke T., Giant viruses with an expanded complement of translation system components. Science 356, 82–85 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Mei R., Narihiro T., Nobu M. K., Kuroda K., Liu W.-T., Evaluating digestion efficiency in full-scale anaerobic digesters by identifying active microbial populations through the lens of microbial activity. Sci. Rep. 6, 34090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dolja V. V., Wolf Y. I., Kazlauskas D., Iranzo J., Lucía-Sanz A., Kuhn J. H., Krupovic M., Koonin E. V., Origins and evolution of the global RNA virome. bioRxiv 10.1101/451740, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beekwilder M. J., Nieuwenhuizen R., van Duin J., Secondary structure model of the last two domains of single-stranded RNA phage Qβ. J. Mol. Biol. 247, 903–917 (1995). [DOI] [PubMed] [Google Scholar]
- 68.J. van Duin, in The Bacteriophages (Springer, Boston, MA, 1988); https://link.springer.com/chapter/10.1007/978-1-4684-5424-6_4), The Viruses, pp. 117–167.
- 69.Rumnieks J., Tars K., Diversity of pili-specific bacteriophages: Genome sequence of IncM plasmid-dependent RNA phage M. BMC Microbiol. 12, 277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kazaks A., Voronkova T., Rumnieks J., Dishlers A., Tars K., Genome structure of caulobacter phage phiCb5. J. Virol. 85, 4628–4631 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li W., Jaroszewski L., Godzik A., Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 17, 282–283 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Grazziotin A. L., Koonin E. V., Kristensen D. M., Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 45, D491–D498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zimmermann L., Stephens A., Nam S.-Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A. N., Alva V., A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243 (2018). [DOI] [PubMed] [Google Scholar]
- 74.A. D. Millard, in Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions, M. R. J. Clokie, A. M. Kropinski, Eds. (Humana Press, Totowa, NJ, 2009); 10.1007/978-1-60327-164-6_4, Methods in Molecular BiologyTM, pp. 33–42. [DOI]
- 75.Roux S., Krupovic M., Daly R. A., Borges A. L., Nayfach S., Schulz F., Cheng J.-F., Ivanova N. N., Bondy-Denomy J., Wrighton K. C., Woyke T., Visel A., Kyrpides N., Eloe-Fadrosh E. A., Cryptic inoviruses are pervasive in bacteria and archaea across Earth’s biomes. bioRxiv 2019, 548222 (2019). [Google Scholar]
- 76.Edgar R. C., PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8, 18 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dai X., Li Z., Lai M., Shu S., Du Y., Zhou Z. H., Sun R., In situ structures of the genome and genome-delivery apparatus in an ssRNA virus. Nature 541, 112–116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gorzelnik K. V., Cui Z., Reed C. A., Jakana J., Young R., Zhang J., Asymmetric cryo-EM structure of the canonical Allolevivirus Qβ reveals a single maturation protein and the genomic ssRNA in situ. Proc. Natl. Acad. Sci. U.S.A. 113, 11519–11524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay5981/DC1
Supplementary Text
Fig. S1. Workflow depiction of known ssRNA phage sequences.
Fig. S2. Workflow depiction of the study pipeline.
Fig. S3. Identification of ssRNA phage contigs within 82 metatranscriptome samples.
Fig. S4. Genome architecture of ssRNA phages.
Fig. S5. Taxonomic cutoff values for ssRNA phage genera and species.
Fig. S6. Potential taxonomic restructuring for ssRNA phages.
Fig. S7. Analysis of microbial community complexity.
Fig. S8. Structural investigation of ssRNA phage–host interactions.
Data S1. ssRNA phage finding hidden Markov model and associated sequences.
Data S2. Bioinformatic scripts used during data analysis.