

Epigenetic and transcriptomic profiling of developing human retina and retinal organoids revealed regulators in human retinal development.
Abstract
Retinal organoids (ROs) derived from human induced pluripotent stem cells (hiPSCs) provide potential opportunities for studying human retinal development and disorders; however, to what extent ROs recapitulate the epigenetic features of human retinal development is unknown. In this study, we systematically profiled chromatin accessibility and transcriptional dynamics over long-term human retinal and RO development. Our results showed that ROs recapitulated the human retinogenesis to a great extent, but divergent chromatin features were also discovered. We further reconstructed the transcriptional regulatory network governing human and RO retinogenesis in vivo. Notably, NFIB and THRA were identified as regulators in human retinal development. The chromatin modifications between developing human and mouse retina were also cross-analyzed. Notably, we revealed an enriched bivalent modification of H3K4me3 and H3K27me3 in human but not in murine retinogenesis, suggesting a more dedicated epigenetic regulation on human genome.
INTRODUCTION
The process of vision starts from the retina, a part of the central nervous system (CNS) that processes both image- and non–image-forming visual information (1, 2). The retina, composed of multiple types of neurons (photoreceptors, horizontal cells, bipolar cells, amacrine cells, and retinal ganglion cells) and a single type of glial cells (Müller cells) differentiated from retinal progenitor cells (RPCs), is an excellent system for studying the regulation of neurogenesis in the CNS (3, 4). Tremendous progress has been made in deciphering the complex molecular mechanisms underlying retinal neurogenesis in rodents (5–7). In contrast, knowledge regarding the molecular mechanisms underlying human retinogenesis remains scarce. Recent advances in human retinal studies provide valuable gene expression and epigenetic profiles of the developing human retina (8, 9). However, the transcriptional regulatory network, which can provide insight into the regulation of interactional transcription factors (TFs), remains poorly understood during human retinal development. The assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) has emerged as a sensitive and robust method for open chromatin assays, nucleosome site mapping, and TF occupancy analysis (10, 11). Moreover, ATAC-seq is also applicable for establishing the transcriptional regulatory network during development, as integration of known TF motifs with chromatin accessibility data from ATAC-seq can predict a genome-wide regulatory network (10). Therefore, systematic ATAC-seq analysis will be a powerful tool to decipher the epigenetic features and transcriptional regulatory network during human retinal development.
Studies on the underlying regulatory mechanisms of human retinal development have been hampered by the impossibility of direct molecular and genetic manipulation of human retinae in vivo. Retinal organoids (ROs) derived from human induced pluripotent stem cells (hiPSCs) are three-dimensional retinal-like structures grown in vitro, which contain the main cell types and proper apical-basal polarity of retinae (12). Thus, ROs provide an opportunity to study human retinal development and disorders with additional flexibility for molecular and genetic manipulations. Previous studies have applied ROs to mimic disease processes and cell transplantation for retinal regeneration (13–15). However, advances in clinical studies using ROs have been hindered by our limited understanding of the molecular and functional differences between the developing human retina and ROs. Furthermore, it is unclear to what extent ROs recapitulate the development of the human retinae in epigenetic modifications. Therefore, it is critical to establish the epigenetic correlations between the developing human retina and ROs.
The dynamics of chromatin accessibility play an important role in regulating human development, including cell fate determination, cell differentiation, and diseases occurrence (16, 17). Recent studies have shown that rod and cone photoreceptors display distinct chromatin accessibility landscapes during fate determination in mice, suggesting that cellular epigenomic states are crucial for retinal neurogenesis (18). In this study, using ATAC-seq and RNA sequencing (RNA-seq) analysis, we explored the chromatin accessibility and transcriptional changes in human retinae and ROs over long-term retinal development. Our results showed that the developing human retina exhibited a complex pattern of chromatin dynamics accompanying retinogenesis. Further analysis indicated that ROs recapitulated the human retinogenesis to a great extent, but divergent epigenetic signatures were found. Moreover, we identified two TFs [nuclear factor I B (NFIB) and thyroid hormone receptor alpha (THRA)] as essential regulators in human retinal development and validated their functions via gene manipulation in ROs. The transcriptional regulatory networks were reconstructed in human and RO, and signaling pathways were analyzed in human and murine retinal development, providing an invaluable data source for future molecular mechanism studies. The chromatin modifications during human and murine retinal development were cross-analyzed and revealed that a bivalent domain of H3K4me3 and H3K27me3 modifications enriched in human only, suggesting a unique and more dedicated epigenetic regulation on human genome. Together, our systematic profiling and integrative analyses of epigenetic and transcriptional changes provide a comprehensive view of the chromatin landscapes that accompany the murine, human retinal, and RO development; establish a developmental temporal-correlation roadmap between the human retinae and ROs; and present a data source for modifying RO culture under the guidance of in vivo human retinal development.
RESULTS
Landscape of chromatin accessibility in developing human retina and ROs
To determine the chromatin accessibility in developing human retina and ROs, the developing human retina from gestational week 6 (GW6) to GW25 at nine time points (GW6, GW10, GW11, GW12, GW14, GW15, GW20, GW24, and GW25; two biological replicates at GW11, GW15, GW20, GW24, and GW25; n = 1 at GW6, GW10, GW12, and GW14), which spanned the key human retinal developmental stages (8), and hiPSC-derived ROs from week 0 to week 30 (w0, w2, w4, w6, w10, w15, w23, and w30; two biological replicates at all stages) were collected for ATAC-seq analysis (Fig. 1A). Each ATAC-seq library was sequenced to obtain, on average, more than 50 million total raw reads per sample. ROs were differentiated as per previous protocols (15). We also conducted RNA-seq for w0, w2, w6, w10, w15, and w23 ROs (n = 1 at all stages). RNA-seq data of the developing human retina were obtained from previous study (8). Here, we stained the developmentally regulated gene RECOVERIN (RCVRN) and nuclear receptor subfamily 2 group E member 3 (NR2E3) in developing human retina and ROs as indicators of differentiation from RPCs to photoreceptors (Fig. 1, B and C, and fig. S1, D and E). Our data revealed that RCVRN protein expression emerged at GW14 and w10 in the human retinae and ROs, respectively, with the expression sustained to GW25 and w30. Like RCVRN, the rod photoreceptor marker NR2E3 was shown at GW20 and w15, respectively. These similar expression trends of the photoreceptor markers indicated that RO culture exhibited progressive retinal neurogenesis, as found in the human retinae.
Fig. 1. Landscapes of chromatin accessibility in the developing human retina and RO.
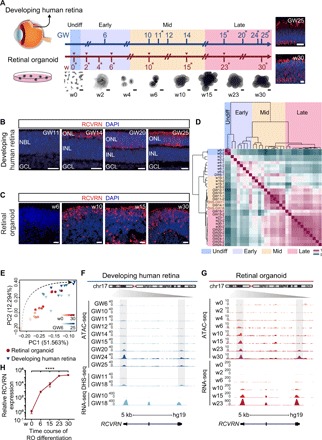
(A) Schematic illustration of the overall experimental designs. Whole human neural retinae and ROs were collected for ATAC-seq (two replicates labeled with asterisk) and RNA-seq (inverted triangle). Development of human retinae and ROs was grouped into early, middle, and late stages and color coded. Immunostaining of GNAT1 in human retinae GW25 and ROs w30 is shown. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Undiff, undifferentiated; Early, early developmental stage; Mid, middle developmental stage; Late, late developmental stage. Scale bars, 500 μm (bright-field images) and 10 μm (fluorescence images). PC1, principal component 1; PC2, principal component 2. (B and C) Immunostaining of RCVRN in human retinae (B) and ROs (C). Nuclei were stained with DAPI. NBL, neuroblastic layer; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bars, 20 μm. (D) Heat map of Pearson correlations across all samples using all ATAC-seq peak signals. Relevant developmental stages are labeled with distinct colors as in (A). (E) PCA of chromatin accessibility during human retinal (blue) and RO (red) development in two dimensions. The black dotted arrow indicates the development process of retinogenesis. (F and G) Normalized epigenetic and expression profiles at the RCVRN loci during human retinal development (F) and RO differentiation (G). All signals were obtained from the University of California, Santa Cruz (UCSC) genome browser. (H) qRT-PCR analysis of the expression level of RCVRN (n = 3) during RO differentiation. Data are means ± SEM. One-way analysis of variance (ANOVA) was performed. ****P < 0.0001.
The ATAC-seq data were analyzed using the ATAC-pipe (19) to obtain the chromatin accessible sites in the developing human retina and RO. Transcription starting site (TSS) enrichment from all samples, aligned fragment length distribution of all samples, and correlation analysis of all the replicate samples indicated high-quality data and excellent reproducibility between replicates (fig. S1, A to C). We also performed correlation analysis of the ATAC-seq peak intensities to define the similarities in chromatin accessibility between the human retinae and ROs (Fig. 1D). The sample replicates were strongly clustered with each other, confirming the high reproducibility of the experiments. Here, except w0 (the undifferentiated hiPSCs), the entire retinal development process can be grouped into three time periods, that is, the early (GW6; w2 to w6), middle (GW10 to GW14; w10 to w15), and late (GW15 to GW25; w23 to w30) stages (color coded in Fig. 1, A and D, and fig. S1F), indicating that human retinae and ROs were developmentally correlated in chromatin accessibility. In addition, the ATAC-seq peaks were also clustered with deoxyribonuclease I (DNase I) hypersensitive site sequencing (DHS-seq) data in day 74 (D74) (DHS-GW11) and D125 (DHS-GW18) retinae produced by the encyclopedia of DNA elements (ENCODE, www.encodeproject.org/) project (Fig. 1D). Principal components analysis (PCA) revealed that the development trajectories of the human retinae and ROs were temporally related in two dimensions. Similar results were found by uniform manifold approximation and projection (UMAP) analysis (Fig. 1E and fig. S1F). Together, these findings suggested the developmental relevance between human retinae and ROs. However, note that although ROs and human retinae were clustered together in the middle stage, the two groups were split apart (fig. S1F, light yellow coded with white dot line), suggesting that the epigenetic signatures might be slightly different between human retinae and ROs at this stage.
We next investigated whether chromatin accessibility was related to gene expression changes. As a positive control, we found elevated enrichment of the ATAC-seq and DHS-seq signals at putative promoters and enhancers at the RCVRN gene locus, consistent with the stages when the gene was expressed (Fig. 1, F and G). Moreover, quantitative real-time polymerase chain reaction (qRT-PCR) quantified the expression level of RCVRN in ROs during the differentiation process (Fig. 1H), confirming consistency between the enrichment of RCVRN expression and chromatin dynamics obtained from the ATAC-seq data. In addition, NR2E3 also showed consistency in chromatin accessibility and gene expression dynamics during human retinal and RO development (fig. S1, G and H). Collectively, our data suggested that RO differentiation recapitulated human retinal development to a great extent. On the basis of the chromatin accessibility profile of developing human retina and ROs, we established the maps of the temporal correlation between the human retinae and ROs.
Developmental transitions in human retinae and ROs reflected by chromatin accessibility
To delineate how epigenomic dynamics governs human retinal development, we applied pairwise comparisons of the ATAC-seq signals of human retinae and ROs at different developmental time points. We discovered 10,563 differential DNA accessible sites across the genome (8805 elements from human retinae and 10,160 elements from ROs) and identified five distinct regulatory element clusters (C1 to C5) via unsupervised hierarchical clustering (Fig. 2A). To understand the functions of these notable differential peaks, we applied Gene Ontology (GO) term enrichment analysis using GREAT v3.0.0 (20). GO analysis of the C1 to C5 cluster peaks revealed three main functional groups for the differential accessible sites: The first functional group included C1 and C2, which were composed of 1636 and 2759 elements, respectively. These peaks were highly accessible in the beginning (GW6 and w0 to w6) but progressively declined with human retinal and RO development. GO analysis identified that these peaks were associated with early retinal development, such as neural tube formation (P < 1 × 10–5), neural tube closure (P < 1 × 10–5), regulation of neuron differentiation (P < 1 × 10–7), and neural precursor cell proliferation (P < 1 × 10–6) (fig. S2, A and B). Because C3 consisted of only 478 peaks and showed no enriched GO terms, peaks in C3 were not further analyzed. The second major functional group was C4, which was composed of 3065 peaks. These C4 peaks were accessible from the middle developmental stage (GW10) and sustained to the late stage (GW25) in the human retinae. Strikingly, the C4 peaks were accessible only in the late RO developmental stage (w23 to w30). GO analysis revealed that the peaks in C4 were strongly enriched in nervous system development, including neurogenesis (P < 1 × 10–60) and neuron differentiation (P < 1 × 10–42) (Fig. 2B), suggesting their key roles in retinal neurogenesis. The third functional group was C5, which included peaks that were not accessible in the beginning but were gradually established during the late developmental stage of both human retinae and ROs (GW15 to GW25 and w10 to w30). The C5 group included 2624 peaks enriched in sensory perception of light stimulus (P < 1 × 10–8), visual perception (P < 1 × 10–7), and photoreceptor cell differentiation (P < 1 × 10–6), which represented the functional maturation of the human retinae, especially the photoreceptors (Fig. 2C). Thus, the GO terms from these three functional groups represented the sequential retinogenesis in human retinae, and the classification of chromatin accessibility provided the possibility to define the timing of key developmental events during human retinal and RO development. From the chromatin accessibility data, we observed that in vitro RO differentiation recapitulated the in vivo human retinal development to a great extent. However, note that the peaks in C4 opened later in RO differentiation than those in human retinal development. It is likely that the distinct pattern of C4 provided possible clues to direct RO differentiation closer to human retinae by genetic manipulation of the regulators related to the C4 peaks.
Fig. 2. Epigenetic signatures and expression map of developing human retina and RO.
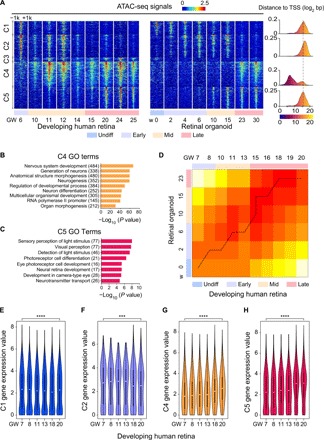
(A) Heat map of 10,563 differential regulatory elements during human retinal and RO development. Each column is a sample, and each row is a peak. Color scale shows the relative ATAC-seq peak intensity centered at the summit of each peak. Distance of cluster peaks to their nearest gene promoters is shown on the right. (B and C) Significant GO terms enriched in C4 (B) and C5 (C) cluster peaks using GREAT v3.0.0. The number of genes enriched in GO terms is shown in the parentheses. (D) Comparison of open-ended DTW analysis between human retinae and ROs. There were 3235 DEGs in humans. Gene expression data were subjected to open-ended DTW analysis, with results plotted as a heat map. (E to H) Violin plot representing the expression level of genes closest to the top 1000 peaks in C1 (E), C2 (F), C4 (G), and C5 (H) during human retinal development showing a variable but positive correlation between chromatin accessibility and gene expression. GREAT was used to annotate peaks to genes. Statistical significance was analyzed with one-way ANOVA. ***P < 0.001, ****P < 0.0001.
We also performed pairwise comparisons of RNA-seq analysis of developing human retina and ROs (fig. S2C). We identified distinct G1 to G4 clusters in the RNA-seq data. Genes in the G1 cluster were associated with early developmental processes, such as cell division, DNA replication, and mitotic cell cycle. G2 genes were associated with axon guidance and regulation of neuron projection development. Genes in G3 were related to visual perception and phototransduction. G4 genes exhibited nonspecific biological processes with retinal development; thus, G4 was not used for further analysis. Therefore, the GO terms (G1 to G3) of RNA-seq data revealed similar sequential retinal development between human retinae and ROs. To further compare the human retinal and RO transcriptome during retinal development, we performed open-ended dynamic time-warping (OE-DTW) analysis (21) of 3235 differentially expressed genes (DEGs) from human retinae (Fig. 2D). We observed a tight temporal correlation between human retinae (GW7 to GW20) and ROs (w0 to w23), confirming that human retinae and ROs shared considerable similarities in gene expression changes. We next examined whether the chromatin signatures in different clusters (C1 to C5) were correlated with the corresponding gene expressions. We chose the top 1000 peaks in each cluster and then applied GREAT to obtain a list of genes regulated by the ATAC-seq peaks and correlated their expression values. By combining the ATAC-seq profiles with the RNA-seq data during retinal development, genes near the loci that gained chromatin accessibility showed significant increases in gene expression levels, whereas genes near the loci that lost chromatin accessibility exhibited decreased expression (Fig. 2, E to H, and fig. S2, D to G), indicating a high correlation between epigenetic and RNA profiling. The correlation between epigenetic and RNA profiling was further analyzed on cell lineage markers, such as PAX6 (retinal progenitor marker), GNAT1 (rod marker), GNGT2 (cone marker), GLUL (Müller cell marker), PROX1 (horizontal cell marker), TFAP2A (amacrine cell marker), and VSX1 (bipolar cell marker) (fig. S3, A to G). Most of the markers showed the similar trends in chromatin accessibility and gene expression during retinal development, suggesting that the chromatin accessibility may govern the gene expression. Together, we observed sequential chromatin changes associated with retinogenesis and correlated with gene transcription; thus, the developmental transitions during retinogenesis can be reflected in the epigenome dynamics.
Identification of potential TFs involved in human retinal development
To identify potential TFs involved in human retinal development, we searched for TFs enriched at accessible sites in C1, C2, C4, and C5 using HOMER v4.8. As accessible DNA sites are often obligated if TFs bind to their cognate DNA motifs, the integration of TF motifs and DNA accessibility data from ATAC-seq can predict TF occupancy on chromatin and thus create regulatory networks (16, 17, 22, 23). Our data revealed distinct patterns of TFs in different clusters. The TFs enriched in the C1 and C2 peaks were identified as potential regulators of early retinal development (fig. S4, A and B). For example, PAX6 is a key regulator for maintaining the multipotency of RPCs (24). SOX3 and RUNX are well known for self-renewal maintenance and morphogenesis (25, 26).
The TFs enriched from C4 and C5 ATAC-seq peaks were identified as critical regulators for neuronal and photoreceptor differentiation, respectively (Fig. 3, A and B). For instance, cluster C4 was enriched with VSX2, SMAD2, and NEUROD1, which are important for retinal neurogenesis (27–29). C5 was enriched with OTX2, CRX, and NR2E3, which are key regulators of photoreceptor differentiation. OTX2 is required for RPC differentiation and cell fate determination (30, 31). CRX is a key regulator for the survival and differentiation of photoreceptors (32). NR2E3 is a direct target of NRL involved in rod and cone photoreceptor differentiation in rodents (33). Therefore, the TFs predicted from the ATAC-seq data were highly associated with retinogenesis and differentiation.
Fig. 3. TF occupancy in developing human retina and RO.
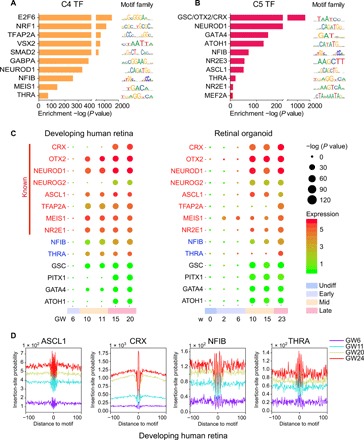
(A and B) TF motifs enriched in C4 (A) and C5 (B) peaks, with P values estimated from HOMER v4.8. (C) Predictions of TFs that may regulate human retinal development (left) and RO differentiation (right). TFs known to be involved in regulating retinal development are shown on top (red). The color of each circle represents expression level of genes that encode corresponding TFs, and the size of the circle represents the enrichment of the motifs. Relevant developmental stages are labeled with distinct colors as in Fig. 1A. (D) Visualization of ATAC-seq footprint for motifs of ASCL1, CRX, NFIB, and THRA in four developmental stages of human retinae. ATAC-seq signals across all motif binding sites in the C4 and C5 genome regions were aligned on the motifs and averaged.
Since C4 and C5 peaks were associated with the middle and late stages of retinal development, which were important for neurogenesis and phototransduction, we focused on C4 and C5 to search for previously unknown neurogenesis regulators. One caveat of only using motif analysis for TF prediction is that TFs or TF families can share the same motif; therefore, we integrated motif enrichment analysis by ATAC-seq data and gene expression profiles from the RNA-seq data to better predict the TF occupancy on accessible sites of C4 and C5. At each time point, we plotted the expression value and motif enrichment score on the same figure (Fig. 3C), which showed that many well-known photoreceptor development TFs were highly expressed, and their motifs were enriched at the middle and late stages (GW10 to GW20 and w10 to w23), including CRX, OTX2, ASCL1, and NR2E1. We found TFs NFIB and THRA, which showed similar high expression and motif enrichment at the middle and late retinal developmental stages. NFIB and THRA have not been reported in photoreceptor differentiation. Therefore, to evaluate their involvement in retinal development, we studied all differentially expressed downstream genes containing the binding motifs of NFIB and THRA together with two well-known retinal development regulators (ASCL1 and CRX) as the control (fig. S4C). GO analysis indicated that up-regulated downstream targets of all four factors were involved in retinal development, including visual perception and phototransduction. Thus, NFIB and THRA may participate in the regulation of retinal development.
To further refine our prediction of the potential regulators of retinal development, TF footprint analysis of the ATAC-seq data, which provides evidence of direct occupancy of TF candidates on genomic DNA, was performed. DNA sequences directly occupied by DNA binding proteins are protected from transposition during library construction in ATAC-seq, and therefore, the resulting sequence “footprint” reveals the presence of a DNA binding protein at its binding sites, analogous to DNase digestion footprints. We illustrated the footprints of two known regulators, ASCL1 and CRX, and observed deeper footprints and higher DNA accessibility flanking their motifs in the late stage compared with the early stage of human retinal and RO development (Fig. 3D and fig. S4D). Notably, the footprints of NFIB and THRA were also deeper and more accessible at the late stage, suggesting that the motifs of these two TFs were not only enriched at stage-specific peaks but also most likely physically bonded to the chromatin accessible sites, indicating that they were possible functional regulators of human retinal and RO development. Collectively, the orthogonal footprint results were consistent with the motif enrichment results, indicating that NFIB and THRA were potential previously unidentified regulators of retinal development.
The modulation of retinogenesis-related gene expression by NFIB and THRA
As ROs were similar to human retinae in gene expression and chromatin accessibility, we used ROs as a model to investigate the potential role of NFIB and THRA during retinal development. We established an electroporation method to efficiently overexpress or knock down target genes in ROs. The outer regions of bright neuroretinal epithelium in ROs were cut into ~500-μm (diameter) pieces and placed into cuvettes for electroporation (Fig. 4A). Electroporated RO samples were collected for qRT-PCR or RNA-seq analysis on D10 after electroporation (Fig. 4B). We investigated the function of three genes, CRX, NFIB, and THRA, in retinal development. As a positive control and to test our electroporation system, CRX was knocked down in ROs around w14 and overexpressed around w7. The qRT-PCR results indicated that CRX knockdown (CRX_KD) reduced the expressions of NRL and RAX2, which were targets of CRX related to photoreceptor differentiation (fig. S5A). Conversely, overexpression of CRX (CRX_OE) markedly elevated the expressions of NRL, ARR3, and OPN1SW (fig. S5, B and C). Both CRX_KD and CRX_OE experiments suggested that our system can successfully manipulate gene expression in RO for studying retinal development. GO analysis of down- and up-regulated DEGs of CRX_OE samples suggested that CRX is involved in visual perception (fig. S5, D and E). Thus, these results indicated that we established a reliable gene manipulation system in ROs. Next, specific short hairpin RNA (shRNA) vectors for NFIB or THRA knockdown were electroporated into ROs at ~w14, a time point when NFIB and THRA were expressed. We revealed significantly decreased expression of NFIB and THRA by qRT-PCR or RNA-seq analysis (Fig. 4C and fig. S5, F and G), respectively. To validate the functional knockdown of these two TFs, we next analyzed the expression level of EZH2, a known target of NFIB, and ARNTL, a potential target of THRA. Results revealed that the expressions of EZH2 and ARNTL decreased significantly due to loss of NFIB and THRA, respectively (Fig. 4C and fig. S5G). Notably, we found that a set of photoreceptor-associated genes were down-regulated, including CRX, RHO, and GNAT1, under knockdown of NFIB and THRA, suggesting that NFIB and THRA may be involved in regulating photoreceptor differentiation (Fig. 4, C and D, and fig. S5G). NFIB is highly expressed in fetal cerebral cortex neural progenitor and glial cells and is required for neuronal and glial differentiation in the fetal cerebral cortex (34). Considering that neurogenesis regulation in the CNS is conserved, we selected NFIB for further functional studies. The RNA-seq of NFIB knockdown (NFIB_KD) ROs revealed many down-regulated retinogenesis genes, including GNAT1, NR2E3, and GNGT2 (Fig. 4D). GO analysis of down- and up-regulated genes in NFIB_KD RNA-seq strongly suggested that NFIB was required for retinal development, especially for photoreceptor differentiation (Fig. 4, E and F). In addition, we further used immunohistochemistry to detect the NFIB_KD effect on highly expressed photoreceptor-related protein, RCVRN, between w14 and w15 (Fig. 4G). The quantification results of the relative intensity of RCVRN implied that NFIB_KD reduced the protein expression of RCVRN (Fig. 4H). Similarly, the percentage of RCVRN-positive cells also reduced accordingly in NFIB_KD ROs (Fig. 4I). Together, these data demonstrated that NFIB and THRA were involved in human retinal and RO development. It is possible that NFIB and THRA affected the self-renewal and differentiation ability of RPC to photoreceptors and Müller cells. However, the hypothesis needs to be further investigated.
Fig. 4. NFIB and THRA are necessary for retinal differentiation.
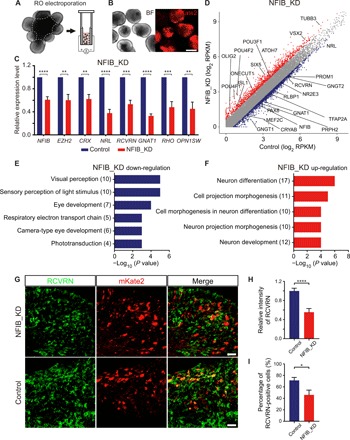
(A) Schematic illustration of ROs split into small sheets for electroporation. Dotted line represents clipping path. (B) Representative images of RO sheets transfected with reported plasmids 10 days after electroporation. Scale bar, 500 μm. (C) qRT-PCR analysis of expression levels of genes after knockdown of NFIB (n = 5). Data are means ± SEM. (D) Plot representing DEGs between control and NFIB_KD groups. Significantly up- and down-regulated genes (fold change >1.5) are highlighted in red and blue, respectively. (E and F) Significant GO terms enriched in down- (E) and (F) up-regulated genes, respectively, in the NFIB_KD experiment. The number of genes enriched in GO terms is shown in the parentheses. (G) Immunostaining of RCVRN in NFIB_KD ROs and control ROs, respectively. Scale bars, 20 μm. (H) Relative intensity of RCVRN signals in the control (n = 366 cells from five independent ROs) and NFIB_KD (n = 135 cells from four independent ROs) groups. (I) Percentage of RCVRN-positive cells in the control (n = 5 independent ROs) and NFIB_KD (n = 4 independent ROs) groups. All statistics by two-tailed Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To further understand the molecular mechanism underlying NFIB-regulated retinal development, we analyzed the potential NFIB-regulated pathways and related functions by ingenuity pathway analysis (IPA) via comparing the gene expression between NFIB_KD ROs and their control (fig. S6A). The results revealed that phototransduction-associated pathways/functions were enriched in down-regulation genes, further confirming the regulation of NFIB of photoreceptors. Moreover, we examined the potential binding of NFIB to retinal development–related genes. We selected the genes enriched in the highlighted pathways in fig. S6A. We then searched for the binding sites of NFIB in the peak region around these genes. To better determine the possibility of these genes bonded by NFIB motifs, we then define their binding affinities using their signals of footprint flank divided by footprint depth signaling (fig. S6B). The results showed that PROM1 and NR2E3 had high affinity with NFIB motifs. NFIB motifs occupying the gene PROM1 and NR2E3 loci also showed opening chromatin states (fig. S6, C and D); therefore, PROM1 and NR2E3 were potential target genes of NFIB. Overall, NFIB is highly possibly involved in retinal development by regulating photoreceptor-related targets.
Prediction of TF regulatory networks during human retinal development
TFs often work in a network by cross-talking with each other to regulate gene transcriptions. To establish the potential connection of enriched TFs, we reconstructed a global picture of the TF regulatory networks during human retinal and RO development. First, we used HOMER v4.8 to identify the enriched TFs bound to the C1 to C5 peaks (P < 1 × 10−20). The connections (edges) between TFs were defined as follows: If TF-X’s motif were on the promoter of TF-Y, then TF-X regulated TF-Y, thereby drawing an arrow from TF-X to TF-Y. Here, only TFs distinctly expressed at this time point were considered. On the basis of this rule, we constructed the transcriptional regulatory networks of human retinae (GW6, GW10, and GW20) and ROs (w6, w10, and w23) at the early, middle, and late stages, respectively (Fig. 5, A and B, and fig. S7, A to D). The regulatory networks at the different time points were highly dynamic. For example, the TFs in the GW6 network, including LHX2 and ISL1, which are required for early retinal development (35–37), were initially highly expressed. However, during development, the expression and enrichment levels of these TFs were reduced. In contrast, in the GW20 network, many known TFs, such as CRX, NR2E3, and VSX2, were increasingly enriched, confirming their important roles in photoreceptor maturation at the late stage. NFIB and THRA were also enriched in the TF network in the late stage and had connections with other TFs involved in retinal development (Fig. 5, A and B). Since TFs interact with different specific TFs to expand their regulatory repertoire and perform regulatory functions, the edge (connection) counts of each TF may represent its importance in regulating retinal development. To better represent the importance of TFs in a network, we defined the connection score of each node in the network as its edge counts multiplied by the SD of its expression (Fig. 5, C and D, and fig. S7, E to H). For example, TFs with the top connection score, such as VSX2, NR2E3, and CRX, are well-known regulators in retinal development. NFIB and THRA were also observed with high connection score, suggesting that NFIB and THRA are also important in retinal development. The TF networks from the human retinae and ROs were highly correlated from early to late developmental time points (Fig. 5E). However, the TF networks in the middle stage showed relatively lower correlations, which may be due to the distinct C4 chromatin accessibility (Fig. 2A) between human retinae and ROs.
Fig. 5. Dynamics of transcriptional regulatory network in developing human retina and RO.
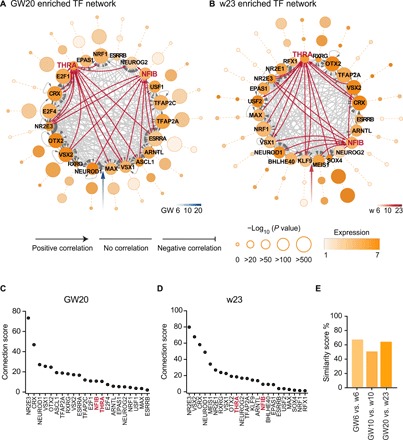
(A and B) Cis-regulatory networks of TFs (nodes) in human retinae GW20 (A) and ROs w23 (B). Circle groups from inner to outer represent different time points. Arrow on edge from node X to node Y indicates that TF-X regulates TF-Y by binding to the promoter site of the latter. Size of each node indicates TF enrichment, and color of each node indicates TF expression levels in that stage. Connection types indicate Pearson correlation between gene expression profiles of connected TFs. (C and D) Ranking of the connection score in human retinae GW20 (C) and ROs w23 (D) networks. The connection score of each node was defined as SD of its expression multiplied by its degree. (E) Similarity of human retinal and RO networks in different developmental stages. We selected GW6/w6, GW10/w10, and GW20/w23 to represent the early, middle, and late stages of retinal development and calculated their similarity score, respectively.
Distinct histone modifications in human and murine retinal development
To determine the distinct epigenetic modifications during human and murine retinal development, mouse DHS-seq data at three time points [embryonic day (E14.5), postnatal day 0 (P0) and P7] were downloaded from ENCODE, which can be clustered into five clusters (MC1 to MC5) (fig. S8A). There was no GO term enriched in MC1. The GO terms of MC2 and MC3 showed that they were involved in stem cell proliferation and regulation of cell development, similar to C1 and C2 in Fig. 2A. MC4 was involved in neurogenesis similar to C4, and MC5 was required for visual perception similar to C5. To decipher the similarity between human and murine retinal development in chromatin accessibility, we chose the top 500 peaks in each cluster and then applied GREAT to obtain a list of genes regulated by these ATAC-seq or DHS-seq peaks. The ratio of overlapping genes from human and mouse clusters was calculated (fig. S8B). The results indicated that genes in C1 and C2 were highly overlapped with MC2 and MC3, and genes in C4 and C5 were overlapped with MC4 and MC5.
Then, we coanalyzed human ATAC-seq data or mouse DHS-seq data with chromatin immunoprecipitation sequencing (ChIP-seq) data in the study by AlDiri et al. (9) during retinal development. Eleven chromatin hidden Markov modeling (chromHMM) states (9) were copy used to systematically annotate the epigenetic states across the C1 to C5 (except C3) and MC2 to MC5 regions during retinogenesis (Fig. 6, A and B). State 1 has active epigenetic marks, states 2 and 3 are predominantly enhancers, and state 4 marks bivalent promoters. State 5 is defined by PolII binding, and states 6 and 7 are consistent with gene bodies (H3K36me3). State 8 is a polycomb-repressed chromatin (H3K27me3) outside of the promoter or enhancers. State 9 is empty chromatin, and state 10 marks the H3K9me3-repressed chromatin. State 11 is marked by the insulator protein CCCTC-Binding Factor (CTCF). The results revealed that in murine retinal development, the chromatin accessible sites were mainly regulated by the active promoter/enhancer marks (state 2), whereas the modifications on human genome were diverse. In human retinal development, the active promoter/enhancer marks (states 2 and 3) were involved in C1 and C2 regulation, which progressively decreased during development. States 1 and 4 highly marked C4, and C5 was mainly marked by state 2. It was likely that active epigenetic states were highly associated with chromatin accessibility during both human and murine retinal development. A similar phenomenon was also detected in cell type markers, such as retinal progenitor marker RAX, rod photoreceptor marker NR2E3, cone photoreceptor marker RXRG, and Müller cell marker GLUL (fig. S8C). The peaks from either ATAC-seq or DHS-seq in these markers were mainly modified by active states, such as states 1 and 2, during both human and murine retinal development.
Fig. 6. Cross epigenetic analysis of human and murine retinal development.
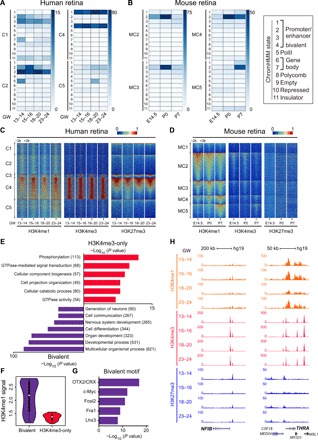
(A and B) Heat map of different ChromHMM state enrichment in each cluster during human (A) and murine (B) retinal development. Each column is a sample, and each row is a ChromHMM state. Color scale shows the relative enrichment. Each state is used to represent the ChromHMM states (rectangle on the right). (C and D) Heat map of H3K4me1, H3K4me3, and H3K27me3 signals for differential regulatory elements in each cluster during human (C) and murine (D) retinal development. Each column is a sample, and each row is a peak region. Color scale shows the relative ChIP-seq peak intensity centered at the summit of each peak. (E) Significant GO terms enriched in bivalent subgroup and H4K4me3-only subgroup peaks using GREAT v3.0.0. The number of genes enriched in GO terms is shown in the parentheses. (F) Violin plot representing H3K4me1 ChIP-seq peak intensity in bivalent subgroup peaks and H4K4me3 subgroup peaks. (G) TF motifs enriched in bivalent subgroup peaks, with P values estimated from HOMER v4.8. (H) Normalized H3K4me3 and H3K27me3 profiles at NFIB and THRA loci during human retinal development. All signals were obtained from the UCSC genome browser.
The bivalent modifications (state 4) marked C4 peak regions specifically in human retinal neurogenesis but not in mouse MC4. To clearly present the dynamic changes of different histone modifications during both human and murine retinal development, we calculated the signals of histone modifications in each cluster (Fig. 6, C and D). The histone modification signals of H3K4me3 and H3K27me3 were enriched in C4 open regions. However, no such notable bivalency modifications were enriched in mouse MC4. These data suggested that the bivalent H3K4me3 and H3K27me3 modifications are distinguished between human and mouse during retinal neurogenesis, which indicate that developing human retina had a more dedicated epigenetic regulation than mouse due to the co-operation of these histone modifications on genome. The bivalent domains were considered to poise the expression of developmental genes, which allowed timely activation while maintaining repression in the absence of differentiation signals (38), matching the critical role of C4 in neurogenesis.
Next, we divided C4 into two subgroups, namely, bivalent subgroup (H3K4me3 and H3K27me3) and H3K4me3-only subgroup, according to the enrichment of different histone modifications in peak regions (Fig. 6E). GO analysis found that the bivalent subgroup was significantly associated with organ development, generation of neurons, and developmental process, suggesting the important role of bivalency in neurogenesis. The H3K4me3-only subgroup was enriched in phosphorylation and guanosine triphosphatase (GTPase)–mediated signal transduction, which were involved in general biological process. As expected, we observed H3K4me3-only enrichment in wildly expressed gene PDK2, which functions as a mechanotransducer that stimulates an increase in intracellular calcium in response to fluid flow (39), whereas the bivalent domain of H3K4me3 and H3K27me3 was detected on the developmental gene BMP8B (fig. S8, D and E), further confirming the important role of bivalent domains in neurogenesis. H3K4me1 is one of the critical modifications for neurogenesis (9). Consistent with this, the C4 bivalent subgroup had more H3K4me1 signals than the C4 H3K4me3-only subgroup (Fig. 6F), further confirming the key role of the C4 bivalent subgroup in neurogenesis. Using motif analysis, we predicted the TFs that regulated the motif with bivalency domains (Fig. 6G). Well-known developmental TFs (OTX2/CRX) and proliferation TFs (c-Myc) were enriched in the bivalent subgroup, which are crucial for various regulations of retinal neurogenesis. Since we determined the role of NFIB and THRA in retinal development, we further investigated the histone modifications around their chromatin regions (Fig. 6H). As expected, NFIB and THRA were bivalently modified with H3K4me3 and H3K27me3, further confirming that these factors were involved in retinal neurogenesis. Together, the bivalent histone modifications in C4 were highly associated human retinal neurogenesis but were relatively weak or missing in mice.
Signaling pathways in human and murine retinal development
We next used GREAT to decipher the key signaling pathways that were differentially activated during human and murine retinal development (fig. S9, A and B). In early human retinal development, the heparan sulfate proteoglycan (HSPG)/fibroblast growth factor (FGF) signaling pathway was enriched. In agreement, the FGF pathway played a number of roles in eye development, including patterning of the optic vesicle, proliferation and differentiation of progenitor cells, and survival of neurons and photoreceptors. Cell surface heparan sulfate (HS) acts as a co-factor for FGF signaling, forming a trimeric complex with FGF and the FGF receptor. It is therefore expected that the HSPG/FGF pathway was discovered in human retinal development. In the early murine development, several known pathways were identified to be involved in retinal development, including Notch, Wnt and E-cadherin pathways. Notch signaling is an important component of RPC maintenance and Müller cell specification during development. Wnt signaling pathway is a known key regulator of optic vesicle establishment, cornea and lens development, and maintenance of retinal stem cell and neuronal specification. Here, histone deacetylase (HDAC) pathway was also emerging as an important regulator for early retinal development in our pathway analysis. Histone acetylation is a posttranslational modification that leads to changes in chromatin structure and transcription repression, which can regulate retinal fate determination. The roles of HDACs in retinal development need further study. In the middle stage of retinal development, pathways including neuronal system, axon guide, and adenylate cyclase activating were enriched. In the late retinal development, we identified a visual transduction pathway, an Na+/Cl−-dependent neurotransmitter transporter pathway, and so on, which related with phototransduction or synaptogenesis. The chromatin accessibilities of the genes involved in different signaling pathways matched with their role in retinal development (fig. S9, C and D). The average expression levels of the genes in each pathway were shown, which matched with their functions in retinal development (fig. S9E). Combined, we were able to generate robust genomic, transcriptomic, and epigenomic datasets, which provided a foundation for future studies for retinal development.
DISCUSSION
Here, we performed a comprehensive assessment of chromatin accessibility and transcriptional changes during human retinogenesis in vivo and in RO differentiation in vitro and revealed stage-specific chromatin dynamics, which regulate human retinogenesis in line with global transcriptional changes. We reconstructed the transcriptional regulatory network and signal pathways regulating human retinogenesis. Notably, we also identified TFs, NFIB, and THRA involved in retinal development, validated by in vitro gene manipulation in the ROs. Therefore, our study provides valuable data for studying human retinal and RO development and a viable framework to optimize in vitro RO differentiation. Moreover, we showed the difference in epigenetic regulation between human and mice. This kind of difference probably contributes gene expression pattern and timing, giving the species difference in retinal development. Therefore, this study gives valuable information to understand species-specific epigenomic regulation.
In our study, we established a temporal-correlation relationship between human retinal and RO development according to epigenetic and transcriptomic profiles (Figs. 1D and 2D). ROs recapitulated the time courses of retinal morphogenesis, retinal neurogenesis, and photoreceptor differentiation of human retinae in the early, middle, and late stages. Hence, our study provided a correlation time frame for the study of stage-specific human retinal development in the RO system. The high transcriptome and chromatic accessibility similarities between human retinae and ROs indicate that ROs are a good model to study human retinal development. Likewise, the human retinal epigenomic and transcriptomic data provide molecular insights for the further improvement of RO differentiation. It is worth noting that the distinct C4 pattern in developing human retina could be related to the complexities of cell types and differential processes in human retinae. Thus, to improve the RO culture, further studies should focus on the DEGs related to C4 particularly.
The transcriptional regulatory networks were reconstructed in both human retinae and ROs. In these networks, we observed many known key TFs for human retinal development, such as OTX2, NR2E3, and ASCL1, which are also critical for retinogenesis in murine. Therefore, the TFs regulating retinal development are conserved among humans, murine, and ROs. We also observed that the TF networks were highly correlated between the developing human retina and ROs, further confirming that the RO system is a good model to study retinal development. Moreover, NFIB and THRA were identified as potential regulators involved in retinogenesis. Thus, the transcriptional regulatory network expands our understanding of molecular regulation during human retinal development. Previous studies have demonstrated that NFIB plays an important role in regulating neural progenitor cell proliferation and differentiation in the cortex (40). Nfib function was very recently verified in regulating cell cycle and the differentiation of late-born retinal progenitors in mice, further supporting our prediction of the function of NFIB, and suggested the reliability of our data analysis (41). In our study, loss of NFIB at the middle retinal differentiation stage reduced the expression of photoreceptor-associated genes. From pathways and motif analysis, we identified that PROM1 and NR2E3 might be the potential targets of NFIB, partially explaining how NFIB regulates human retinogenesis. The thyroid hormone receptors TRɑ1 and TRβ are encoded by the genes THRA and THRB, respectively. THRB plays important roles in cone photoreceptor development (42, 43). However, whether THRA is also involved in human retinal development remains unclear. In our transcriptional regulatory network, we found that THRA may interact with NR2E3, VSX1, and CRX, which are well-known regulators in retinal development. We also demonstrated the role of THRA in retinal development via RO molecular manipulations. The function of THRA in retinal development, which has not been reported previously, may be due to the compensatory effects of THRB. The genesis of Müller cells and photoreceptors in ROs detected by immunostaining started after w14, around w17 (15). Here, we knocked down NFIB and THRA at ~14-week-old ROs, which mainly contained retinal progenitors but limited cell numbers of Müller cells (RLBP+) and photoreceptors (GNAT1+ or RHO+). Therefore, it is highly possible that NFIB and THRA mainly function in regulating retinal progenitor differentiation, thereby affecting both the photoreceptor and Müller differentiation.
Histone modifications are crucial for the control of gene expression, cell fate decisions, and differentiation. Many chromatin regions in embryonic stem cells and early embryonic development harbor a distinctive histone modification signature that combines the active H3K4me3 and the repressive H3K27me3 marks (44). These bivalent domains are considered to poise the expression of developmental genes, allowing timely activation while maintaining repression in the absence of differentiation signals. Here, in human retinal development, C4 is bivalently modified and associated with human retinal neurogenesis only, demonstrating a fine-tuning on the gene expression that associated human neurogenesis. Thus, these bivalent features in C4 may facilitate neurogenesis via timely gene activation and silencing. Moreover, cross-analysis with ATAC-seq, RNA-seq, and pathway analysis highlighted numerous signaling pathways, which seemed to be differentially activated in retinal development. The differential activation of these pathways is consistent with changes in the expression of key genes in long retinal development, providing potential regulators involved in retinal development. Thus, our data provided a large scope of data sources for further molecular study underlying retinal development.
In summary, we provided a comprehensive view of the chromatin landscapes that accompany human retinal and RO development; established a comprehensive resource for temporal and molecular correlations between human retinal and RO development; discovered TFs for human retinal development; and reconstructed the transcription regulatory network and signaling pathways, which greatly expand our understanding of human retinal development and provide a roadmap for further studies.
MATERIALS AND METHODS
Informed consent
Human embryo collection was approved by the Reproductive Study Ethics Committee of Beijing Anzhen Hospital (2014012x). All embryos were obtained with written informed consent signed by the patient who had made the decision to legally terminate her pregnancy. Informed consent confirmed that the patients were voluntarily donating embryos for research on human embryonic development mechanisms with no financial payment. The deidentified fetal retinae were collected with patient informed consent in strict observance of the legal and institutional ethical regulation approved by the Institutional Review Board (ethics committee) at the Institute of Biophysics, Chinese Academy of Sciences. All samples used in these studies had never been involved in previous procedures (drugs or other tests). All protocols followed the Interim Measures for the Administration of Human Genetic Resources administered by the Chinese Ministry of Health.
Tissue sample preparation
Fetal retinae were collected in ice-cold artificial cerebrospinal fluid, which included 125.0 mM NaCl, 26.0 mM NaHCO3, 2.5 mM KCl, 2.0 mM CaCl2, 1.0 mM MgCl2, and 1.25 mM NaH2PO4 (pH 7.4), bubbled with carbogen (95% O2 and 5% CO2). Retinae were gently cut into small pieces, and tissue samples were centrifuged for 5 min at 500g at room temperature (RT). The supernatant was removed followed by the addition of 900 μl of 0.25% trypsin-EDTA (Thermo Fisher Scientific, 25200114) with 5 μl of DNase I (Thermo Fisher Scientific, EN0521) for digestion for 15 min at 37°C. Shaking and gentle pipetting were performed at 5-min intervals. We added 100 μl of fetal bovine serum (FBS; Thermo Fisher Scientific, 10270) to stop digestion. Samples were centrifuged and washed with 1 ml of Dulbecco’s phosphate-buffered saline (DPBS; Thermo Fisher Scientific, 14190250). We collected ~50,000 cells for ATAC-seq and ~1 million cells for RNA-seq.
hiPSC culture
The BC1–eGFP (enhanced green fluorescent protein) (45) hiPSC line was obtained from L. Cheng (Johns Hopkins University, Baltimore, MD, USA), with verified normal karyotype and was contamination free. hiPSCs were maintained on Geltrex-coated plates (Thermo Fisher Scientific, A1413302) with TeSR-E8 medium (STEMCELL Technologies, 05940) in a 37°C, 5% CO2 humidified incubator. Cells were passaged with ACCUTASE (STEMCELL Technologies, 07920) every 4 to 5 days at ~70% confluence, and autodifferentiated cells were marked and mechanically removed before passaging. We added 10 μM Y-27632 (STEMCELL Technologies, 72304) into the TeSR-E8 medium on the first day after passaging.
RO differentiation
The hiPSC line was induced to differentiate into ROs, as described previously (15). Briefly, on D0, hiPSCs were treated with dispase (STEMCELL Technologies, 07923) until the edges of the clones began to curl, after which they were scraped into small pieces and cultured in suspension with mTeSR1 medium (STEMCELL Technologies, 05850) and 10 μM blebbistatin (Sigma-Aldrich, B0560) to induce aggregate formation. Aggregates were gradually transitioned into neural induction medium (NIM) containing Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) (Thermo Fisher Scientific, 11330), 1% N2 supplement (Thermo Fisher Scientific, 17502), 1× MEM Non-Essential Amino Acids Solution (NEAA) (Thermo Fisher Scientific, 11140), heparin (2 μg/ml; Sigma-Aldrich, H3149), and 1% antibiotic-antimycotic (Thermo Fisher Scientific, 15240) by replacing the medium with a 3:1 ratio of mTeSR1/NIM on D1, 1:1 on D2, and 100% NIM on D3. On D7, aggregates (average size of 250 ± 50 μm) were seeded onto Geltrex-coated dishes containing NIM at an approximate density of 10 aggregates/cm2 and switched to retinal differentiation medium (RDM) on D16 containing 70% Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, 11965) and 30% Ham’s F-12 Nutrient Mixture (F-12) (Thermo Fisher Scientific, 11765) supplemented with 2% B27 supplement (Thermo Fisher Scientific, 12587010), 1× NEAA, and 1% antibiotic-antimycotic. On D28, the neural retina domains were manually detached with a sharpened Tungsten needle under an inverted microscope and then collected and cultured in suspension in RDM. On D42, RDM was transitioned into retinal maturation medium (RMM) containing 60% DMEM, 25% F-12, supplemented with 10% FBS (Thermo Fisher Scientific), 100 μM taurine (Sigma-Aldrich, T0625), 2% B27, 1× NEAA, 1× GlutaMAX supplement (Thermo Fisher Scientific, 35050), and 1% antibiotic-antimycotic. We freshly added 1 μM retinoic acid (Sigma-Aldrich, R2625) to the RMM when the medium was changed twice a week.
Immunohistochemistry
Human retinae were fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich, 16005) for 2 hours at RT, and ROs were fixed in 4% PFA for 30 min at RT. All samples were washed in DPBS (three times for 10 min), dehydrated with a sucrose gradient (15% for 30 min at RT and 30% overnight at 4°C), and embedded in Tissue-Tek OCT Compound (Sakura, 4583) for freezing. Samples were sectioned (10 μm unless otherwise stated), air dried for 1 hour, washed in DPBS (three times for 10 min), blocked in 10% bovine serum albumin (BSA; Sigma-Aldrich, B2064) in DPBS with 0.25% Triton X-100 (Sigma-Aldrich, T9284) for 1 hour at RT, and incubated with a primary antibody in 10% BSA in DPBS with 0.25% Triton X-100 at 4°C overnight. The next day, slides were washed in DPBS (three times for 10 min) and incubated with corresponding species-specific Alexa Fluor 568– or Alexa Fluor 647–conjugated secondary antibodies (1:500; Thermo Fisher Scientific, A-11036 and A-21245, respectively) in DPBS for 2 hours at RT. The slides were incubated in 4′,6-diamidino-2-phenylindole (DAPI) (1:1000; Thermo Fisher Scientific, D1306) in DPBS for 5 min, washed in DPBS (three times for 10 min), and cover slipped. Primary antibodies against the following proteins were used at the indicated dilutions: GNAT1 (1:200; Santa Cruz Biotechnology, sc-389), RCVRN (1:100; Millipore, AB5585), and NR2E3 (1:100; R&D Systems, PP-H7223-00). Fluorescence images were acquired with an LSM 800 confocal microscope (Zeiss).
Plasmid construction and electroporation
shRNA sequences of targeted genes (table S1) were synthesized by Tsingke Biological Technology and cloned into shRNA expression vector pAAV-U6-shRNA-CMV-mKate2-SV40, and pAAV-U6-shRLuc-CMV-mKate2-SV40 vector with luciferase shRNA (gtgcgttgctagtaccaacttcaagagagttggtactagcaacgcactttttt) was used as control. The complementary DNA (cDNA) of the CRX gene for overexpression was cloned into the pEF1α-cDNA-IRES2-mKate2 vector, and the pEF1α-IRES2-mKate2 vector without CRX cDNA was used as control. Primers used are listed in table S1. High-quality vectors were extracted using the NucleoBond Xtra Maxi EF Kit (Macherey-Nagel, 740424.50), with a final concentration of 1 μg/μl used for electroporation. In all experiments, electroporation was performed on both control and experimental groups. ROs for electroporation were manually cut into ~500-μm (diameter) pieces under an inverted microscope, equally distributed to both control and experimental groups, and resuspended in the Human Stem Cell Nucleofector Kit 1 (Lonza, VPH-5012). Ten pulses of electroporation were performed on both sides of the small balls under the following parameters: square wave, 35 V, 1 Hz, and 5% duty.
Quantitative RT-PCR and RNA-seq
Total RNA was isolated from samples with TRIzol (Thermo Fisher Scientific, 15596018) and converted into cDNA with a PrimeScript RT Master Mix (TaKaRa, RR036A). FastStart Essential DNA Green Master (Roche, 06924204001) was then used for qRT-PCR analysis, with RNA-seq performed on an Illumina HiSeqXten-PE150. Primer sequences for qRT-PCR are shown in table S1.
ATAC sequencing
ATAC-seq was performed as described previously (11, 46). Briefly, a total of 50,000 cells were washed twice with 50 μl of cold DPBS and resuspended in 50 μl of lysis buffer [10 mM tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, and 0.1% (v/v) NP-40 substitute (Sigma-Aldrich, 11332473001)]. The suspension of nuclei was then centrifuged for 10 min at 500g at 4°C, followed by the addition of 50 μl of transposition reaction mix (10 μl of 5× TTBL buffer, 4 μl of TTE mix, and 36 μl of nuclease-free H2O) of TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme Biotech, TD501). Samples were then incubated at 37°C for 30 min. DNA was isolated using the QIAquick PCR Purification Kit (QIAGEN, 28106). ATAC-seq libraries were first subjected to five cycles of preamplification using NEBNext High-Fidelity 2X PCR Master Mix (New England Biolabs, M0541S). To determine the suitable number of cycles required for the second round of PCR, the library was assessed by qPCR as described previously (11) using NEBNext High-Fidelity 2X PCR Master Mix with SYBR Green I Nucleic Acid Gel Stain (Thermo Fisher Scientific, S7563) and then PCR amplified for the appropriate number of cycles. Libraries were purified with the QIAquick PCR Purification Kit. Library quality was checked using the High Sensitivity DNA Analysis Kit (Agilent, 5067-4626). Last, 2 × 150 paired-end sequencing was performed on an Illumina HiSeq X-10.
RNA-seq analysis
FASTQ files were evaluated for quality control using FastQC (v0.11.5) (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence alignment was performed using STAR (v2.5.2a) with reference assembly hg19. We estimated gene expression levels using reads per kilobase of transcript per million mapped reads (RPKM) values. DEGs were filtered by an RPKM value of >5 for one stage and an absolute value of log2 fold change >1 between any two groups. GO analysis was performed using David v6.8. The RNA-seq data of the developing human retina were obtained from the previous study (8), and we defined D52/D54, D57, D67, D80, D94, D105, D115, D125, D132, and D136 as GW7, GW8, GW10, GW11, GW13, GW15, GW16, GW18, GW19, and GW20.
ATAC-seq primary data processing
Primary data were processed as described previously (19). In simple terms, we removed adapter sequences and then mapped reads to hg19 using Bowtie2. The PCR duplicates and chromosome M were removed. The uniquely mapped reads were shifted +4/−5 base pair (bp) according to the strand of the read. All mapped reads were then extended to 50 bp centered through the cleavage position. Peak calling was performed using MACS2 with options - f BED -g hs, -q 0.01, --nomodel, --shift 0.
ATAC-seq data quality control
ATAC-seq data quality was comprehensively studied in the previous work (17). Briefly, we used several parameters to evaluate data quality, including number of raw reads, overall alignment rate, final mapped reads, final mapped rate, percentage of reads mapped to chromosome M, percentage of reads mapped to repeat regions (black list), percentage of reads filtered out by low MAPQ score, percentage of PCR duplicates, TSS enrichment score (reads enriched at ±2 kb around TSS versus background), and read length distribution.
ATAC-seq significance analysis
Peak calling was performed using MACS2 from all sample reads. The number of raw reads mapped to each peak at each condition was quantified using the intersectBed function in BedTools. Raw counts in peaks were normalized using the DESeq package in R. Peak intensity was defined as the log2 of the normalized counts. Samples were then grouped into 17 categories (30 samples; RO: w0, w2, w4, w6, w10, w15, w23, and w30; human retina: GW6, GW10, GW11, GW12, GW14, GW15, GW20, GW24, and GW25). First, to remove the genomic regions dominated by hiPSCs, we compared peaks between RO w0 and other time points (w2 to w30) to remove peaks with log2 fold change >1 on w0 and SD <1 on other time points. Significance analysis was then performed by pairwise comparison with eight categories of RO samples and nine categories of human retinal samples using DESeq with P < 0.01, false discovery rate (FDR) < 0.01, log2 fold change >1, and intrinsic analysis with z score >1. We lastly obtained 10,563 differential accessible peaks. We used the long-distance peaks (located 1 kb outside the TSS of the gene) to calculate the correlation between samples. Unsupervised clustering was performed using Cluster 3.0 and visualized in Treeview. GO and other enriched functions of cis-regulatory regions were performed with GREAT.
Intrinsic analysis
Intrinsic analysis followed the steps described in the previous research (10). We defined correlation matrix C, where Cp,q is the Pearson correlation between samples p and q where all peaks were included. Similarly, we defined correlation matrix Ci, where Cip,q is the Pearson correlation between samples p and q where all peaks except for peak i were excluded. We defined delta matrix, deltaCi = C − Ci. We defined wbScorei = average (deltaCireplicates) − average (deltaCinonreplicates). Replicates were defined as samples at the same time point, and nonreplicates otherwise. For peak i, the greater the wbScorei, the less variance the peak intensity was within the replicates and the greater variance within nonreplicates. We then calculated the average and SD of all wbScore (from I = 1 to N). We defined z score = (wbScorei − average (wbScorei=1,N))/SD (wbScorei=1,N).
DNase hypersensitivity analysis
Details of protocols and standards for DHS-seq are described by ENCODE (www.encodeproject.org/). All replicates of DHS-seq samples were combined before analysis for the D74 (DHS-GW11) and D125 (DHS-GW18) time points. The Bowtie2 algorithm was implemented to align the reads to the human (hg19) reference genome (with option --very-sensitive). PCR duplicates, reads mapped to repeated regions, and chromosome M were removed.
Comparison of human and RO transcriptomes
To correlate the developmental stages between human retina and RO, we applied OE-DTW analysis (21). DTW is a popular technique for comparing time series. The rationale behind DTW is that given two time series, they should be stretched or compressed locally to make one resemble the other as much as possible. We used the R package DTW on expression profiles of human retinal development spanning from GW6 to GW25. The analysis was performed on the 3235 genes from the human RNA-seq data to find an optimal alignment between the human retinal and RO development.
TF motif enrichment analysis
The TF motif enrichment analysis was performed using HOMER with options: findmotifs.pl input.fa fasta output. To obtain genes that may be regulated by a certain TF, we overlapped all the binding site of TFs with the open sites. Genes with TF binding sites in the promoter region were then considered to be possible regulated genes.
TF footprint analysis
For footprint, we adjusted the read start sites to represent the center of the transposon’s binding. Previous descriptions of the Tn5 transposase show that the transposon binds as a dimer and inserts two adaptors separated by 9 bp. Therefore, we modified the reads’ aligned file in the SAM format by offsetting +4 bp for all the reads aligned to the forward strand and −5 bp for all the reads aligned to the reverse strand. We then converted a shifted base SAM file to the BAM format and had the BAM file sorted using SAMtools. We overlaid TF binding sites with reads in each sample of the C4 and C5 peak regions. We then averaged the overlaid read counts of a 150-bp genomic region centered by the motif sites for motif footprints.
Binding affinity analysis
To study the regulatory mechanisms of TF NFIB, we predicted the potentiality of NFIB to bind to retinal-related genes. First, we selected a total of 35 genes enriched in the highlighted term in fig. S6A. We expanded each gene with the upstream of TSS by 10 kb and the downstream of gene transcription termination site by 5 kb. Next, we overlapped the binding site of NFIB in the peak regions of our ATAC-seq with extended gene regions. To better compare the possibility of these genes bonded by the NFIB motif, we calculated the flanking accessibility and the footprint depth of the motif binding regions. This is based on previous work (47). We first defined two key relative positions: (i) The footprint base is the region encompassing the very center of the motif and is defined as a motif binding site, and (ii) the footprint flank is the region immediately adjacent to the TF binding site and is defined as the region between the end of the footprint base and 100 bp away from the motif end. The accessibility of the motif region and the flank region was calculated. The higher value for footprint depth and the lower value for flanking accessibility indicate strong factor occupancy. We defined binding potential = flanking accessibility/footprint depth to characterize the binding potential of a gene region.
Reconstruction of transcriptional regulatory network
To map the regulatory network throughout the retinal developmental process, we selected human GW6, GW10, and GW20 and RO w6, w10, and w23 to represent the early, middle, and late stages of retinal development, respectively. According to Fig. 2A, we assigned motifs enriched in the C1 and C2 to the early stage and motifs enriched in C4 and C5 to the late stage. Because the development of ROs in the middle stage appeared to be slower than that of the human retina in C4, we assigned motifs enriched in C2 and C4 to the human middle stage and motifs enriched in C2 to the RO middle stage. First, we used HOMER to find the TFs that were bound to each cluster (P < 1 × 10−20). A TF with a higher gene expression level on a time point than the average expression of all time points was considered as a member of the regulatory network of this time point, and then we constructed a transcriptional regulatory network. We defined the promoter region of TF-Y as going from the upstream 10 kb to the downstream 1 kb of TSS. If TF-X binds to the promoter of TF-Y, then we assumed that TF-X regulates TF-Y. The size of each node indicates the TF enrichment in that stage, and the color of each node indicates the TF expression in that stage. The correlation between TF-X and TF-Y expression (CX,Y) was calculated: CX,Y > 0.5, positive correlation; CX,Y < −0.5, negative correlation; –0.5 < CX,Y < 0.5, no correlation. We defined the connection score of each node in the network as its edge counts multiplied by the SD of its expression. To measure the extent of consistency between human retinae and ROs, we compared the similarities of their regulatory networks. We defined similarity score: S = 1 − D (D = 0.5 × N/M + 0.5 × (I × 0.5)/J), where D values represent differences between human retinae and ROs over the same time period. The differences in regulatory networks come from two aspects. One is that the two regulatory networks share some common TFs, so we used the ratio of the different connections (N) and all connections (M) in the same TF to represent this difference. The other is derived from the different TFs. We divided half the number of different TFs (I) by the number of all TFs (J). We assumed that they may share the same contribution to differences in regulatory networks; therefore, we multiplied the respective coefficients by 0.5.
Epigenetic modification analysis between human and murine retinal development
DHS-seq data for mouse retinae (E14.5, P1, and P7) from ENCODE (www.encodeproject.org/) were analyzed. The Bowtie2 algorithm was implemented to align the reads to the mouse (mm9) reference genome (with option --very-sensitive). PCR duplicates, reads mapped to repeated regions, and chromosome M were removed. After peak calling and DESeq normalization, significance analysis was then performed on adjacent time points with P < 0.01, FDR < 0.01, log2 fold change >2. We lastly obtained 8967 differential peaks. Unsupervised clustering was performed using Cluster 3.0 and visualized in Treeview. GO and other enriched functions of cis-regulatory regions were performed with GREAT. To identify chromatin states, we used the ChromHMM software (v.1.06) according to the previous paper (48). Neighborhood enrichment command was used to calculate the enrichment of different chromatin states in cluster peak regions.
We calculated the signals of H3K4me3 and H3K27me3 modifications in each peak of C4 at GW13 and GW14. We defined average signals of a modification (m) in one peak region (g) as S (mg). If S (mg) > 1, then the peak region g enriched modification m. We divided the peaks of C4 into two subgroups, namely, H3K4me3-only subgroup and bivalent subgroup according to the enrichment of different histone modifications in peak region. If a peak region simultaneously enriched with both modifications of H3K4me3 and H3K27me3, then the peak is in a bivalent state, and if enriched with the H3K4me3 but not H3K27me3 modification, then the peak is H3K4me3-only state.
Statistical relative intensity of RCVRN
We extracted the fluorescence intensity of RCVRN by ImageJ software, following the ImageJ User Guide. Corrected total cell fluorescence (CTCF) of each calculated cell (mKate2+ cells and mKate2-RCVRN+ cells) in the same slice was compared with the mean CTCF of mKate2-RCVRN+ cells to normalize the CTCF between different slices. The normalized CTCF of mKate2+ cells in both the control and NFIB_KD groups was compared with the mean of the normalized CTCF of mKate2+ cells in the control group, and then the relative intensity of the RCVRN in each cell was obtained.
Statistical analysis
Statistical significance was analyzed with unpaired two-tailed t tests or one-way analysis of variance (ANOVA). A value of P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are presented as means ± SEM as indicated in the figure legends. All statistical analyses were performed in GraphPad Prism v7.00.
Supplementary Material
Acknowledgments
We thank L. Cheng for the BC1-eGFP cell line. Funding: This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16020603 to T.X.), the National Key R&D Program of China (2017YFA0102903 to K.Q. and 2016YFA0400900 to T.X.), and the National Natural Science Foundation of China (81925009, 61890953, 91748212, 61890953, 81790644, 31322024, 81371066, and 91432104 to T.X.; 91940306, 91640113, 31970858, and 31771428 to K.Q.; and 81900855 to M.Z.). The study was also supported by the Anhui Provincial Natural Science Foundation (1808085MH289 to M.Z.), and the Fundamental Research Funds for the Central Universities (WK2070000174 to M.Z. and WK2070000158 to K.Q.). We thank the USTC supercomputing center and the School of Life Science Bioinformatics Center for providing supercomputing resources for this project. Author contributions: T.X., K.Q., and M.Z. conceived the project and designed the experiments. H.X. performed the ATAC-seq, RNA-seq, and RO electroporation experiments. W.Z. worked on the data analysis. T.A., Y.L., W.Y., X.S., Z.Z., M.W., X.F., Z.Y., K.D., S.Z., Q.L., Y.S., Q.W., X.W., H.Z., and J.B. worked on sample collection and data analysis. T.X., K.Q., and M.Z. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw and processed sequencing data can be obtained from the Genome Sequence Archive at the BIG Data Center with the accession number CRA0011280 and is available at http://bigd.big.ac.cn/gsa/s/oda82b5P. Additional data related to this paper may be requested from T.X.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay5247/DC1
Fig. S1. Qualification of ATAC-seq data.
Fig. S2. Analysis of differential chromatin elements and RNA-seq data in developing human retina and ROs.
Fig. S3. Epigenetic and expression profiles of retinal cell type markers during retinal development stages.
Fig. S4. Identification of putative TFs involved in retinal differentiation.
Fig. S5. Functional assay of THRA and NFIB using RO electroporation.
Fig. S6. NFIB-regulated signaling pathways in retinal development.
Fig. S7. Transcriptional regulatory network during human retinal and RO development.
Fig. S8. Comparative analyses of human and murine epigenetic data during retinal development.
Fig. S9. Signaling pathways in different stages of retinal development in mouse and human.
Table S1. Primers in all experiments.
REFERENCES AND NOTES
- 1.Nguyen-Legros J., Functional neuroarchitecture of the retina: Hypothesis on the dysfunction of retinal dopaminergic circuitry in Parkinson’s disease. Surg. Radiol. Anat. 10, 137–144 (1988). [DOI] [PubMed] [Google Scholar]
- 2.Xue T., Do M. T. H., Riccio A., Jiang Z., Hsieh J., Wang H. C., Merbs S. L., Welsbie D. S., Yoshioka T., Weissgerber P., Stolz S., Flockerzi V., Freichel M., Simon M. I., Clapham D. E., Yau K.-W., Melanopsin signalling in mammalian iris and retina. Nature 479, 67–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harada T., Harada C., Parada L. F., Molecular regulation of visual system development: More than meets the eye. Genes Dev. 21, 367–378 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Cepko C. L., Austin C. P., Yang X., Alexiades M., Ezzeddine D., Cell fate determination in the vertebrate retina. Proc. Natl. Acad. Sci. U.S.A. 93, 589–595 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cepko C., Intrinsically different retinal progenitor cells produce specific types of progeny. Nat. Rev. Neurosci. 15, 615–627 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Mears A. J., Kondo M., Swain P. K., Takada Y., Bush R. A., Saunders T. L., Sieving P. A., Swaroop A., Nrl is required for rod photoreceptor development. Nat. Genet. 29, 447–452 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Roger J. E., Hiriyanna A., Gotoh N., Hao H., Cheng D. F., Ratnapriya R., Kautzmann M.-A. I., Chang B., Swaroop A., OTX2 loss causes rod differentiation defect in CRX-associated congenital blindness. J. Clin. Invest. 124, 631–643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshino A., Ratnapriya R., Brooks M. J., Chaitankar V., Wilken M. S., Zhang C., Starostik M. R., Gieser L., La Torre A., Nishio M., Bates O., Walton A., Bermingham-McDonogh O., Glass I. A., Wong R. O. L., Swaroop A., Reh T. A., Molecular anatomy of the developing human retina. Dev. Cell 43, 763–779.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aldiri I., Xu B., Wang L., Chen X., Hiler D., Griffiths L., Valentine M., Shirinifard A., Thiagarajan S., Sablauer A., Barabas M. E., Zhang J., Johnson D., Frase S., Zhou X., Easton J., Zhang J., Mardis E. R., Wilson R. K., Downing J. R., Dyer M. A.; St. Jude Children’s Research Hospital—Washington University Pediatric Cancer Genome Project , The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron 94, 550–568.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu K., Zaba L. C., Giresi P. G., Li R., Longmire M., Kim Y. H., Greenleaf W. J., Chang H. Y., Individuality and variation of personal regulomes in primary human T cells. Cell Syst. 1, 51–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J., ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S., Lowe A., Dharmat R., Lee S., Owen L. A., Wang J., Shakoor A., Li Y., Morgan D. J., Hejazi A. A., Cvekl A., DeAngelis M. M., Zhou Z. J., Chen R., Liu W., Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. U.S.A. 116, 10824–10833 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng W. L., Gao M. L., Lei X. L., Lv J. N., Zhao H., He K. W., Xia X. X., Li L. Y., Chen Y. C., Li Y. P., Pan D., Xue T., Jin Z. B., Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Reports 10, 1267–1281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volkner M., Zschatzsch M., Rostovskaya M., Overall R. W., Busskamp V., Anastassiadis K., Karl M. O., Retinal organoids from pluripotent stem cells efficiently recapitulate retinogenesis. Stem Cell Reports 6, 525–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong X., Gutierrez C., Xue T., Hampton C., Vergara M. N., Cao L.-H., Peters A., Park T. S., Zambidis E. T., Meyer J. S., Gamm D. M., Yau K.-W., Canto-Soler M. V., Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 5, 4047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corces M. R., Granja J. M., Shams S., Louie B. H., Seoane J. A., Zhou W., Silva T. C., Groeneveld C., Wong C. K., Cho S. W., Satpathy A. T., Mumbach M. R., Hoadley K. A., Robertson A. G., Sheffield N. C., Felau I., Castro M. A. A., Berman B. P., Staudt L. M., Zenklusen J. C., Laird P. W., Curtis C.; Cancer Genome Atlas Analysis Network, Greenleaf W. J., Chang H. Y., The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qu K., Zaba L. C., Satpathy A. T., Giresi P. G., Li R., Jin Y., Armstrong R., Jin C., Schmitt N., Rahbar Z., Ueno H., Greenleaf W. J., Kim Y. H., Chang H. Y., Chromatin accessibility landscape of cutaneous T cell lymphoma and dynamic response to HDAC inhibitors. Cancer Cell 32, 27–41.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mo A., Luo C., Davis F. P., Mukamel E. A., Henry G. L., Nery J. R., Urich M. A., Picard S., Lister R., Eddy S. R., Beer M. A., Ecker J. R., Nathans J., Epigenomic landscapes of retinal rods and cones. eLife 5, e11613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuo Z., Jin Y., Zhang W., Lu Y., Li B., Qu K., ATAC-pipe: General analysis of genome-wide chromatin accessibility. Brief. Bioinform. 20, 1934–1943 (2019). [DOI] [PubMed] [Google Scholar]
- 20.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tormene P., Giorgino T., Quaglini S., Stefanelli M., Matching incomplete time series with dynamic time warping: An algorithm and an application to post-stroke rehabilitation. Artif. Intell. Med. 45, 11–34 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Mazumdar C., Shen Y., Xavy S., Zhao F., Reinisch A., Li R., Corces M. R., Flynn R. A., Buenrostro J. D., Chan S. M., Thomas D., Koenig J. L., Hong W. J., Chang H. Y., Majeti R., Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 17, 675–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao X., Rubin A. J., Qu K., Zhang J., Giresi P. G., Chang H. Y., Khavari P. A., A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 16, 284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klimova L., Kozmik Z., Stage-dependent requirement of neuroretinal Pax6 for lens and retina development. Development 141, 1292–1302 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Guo Y., Yin J., Tang M., Yu X., Downregulation of SOX3 leads to the inhibition of the proliferation, migration and invasion of osteosarcoma cells. Int. J. Oncol. 52, 1277–1284 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Anderson G., Mackay N., Gilroy K., Hay J., Borland G., McDonald A., Bell M., Hassanudin S. A., Cameron E., Neil J. C., Kilbey A., RUNX-mediated growth arrest and senescence are attenuated by diverse mechanisms in cells expressing RUNX1 fusion oncoproteins. J. Cell. Biochem. 119, 2750–2762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou C., Levine E. M., Vsx2 controls eye organogenesis and retinal progenitor identity via homeodomain and non-homeodomain residues required for high affinity DNA binding. PLOS Genet. 8, e1002924 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu A. Q., Popova E. Y., Barnstable C. J., Activin signals through SMAD2/3 to increase photoreceptor precursor yield during embryonic stem cell differentiation. Stem Cell Reports 9, 838–852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pennesi M. E., Cho J.-H., Yang Z., Wu S. H., Zhang J., Wu S. M., Tsai M.-J., BETA2/NeuroD1 null mice: A new model for transcription factor-dependent photoreceptor degeneration. J. Neurosci. 23, 453–461 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glubrecht D. D., Kim J.-H., Russell L., Bamforth J. S., Godbout R., Differential CRX and OTX2 expression in human retina and retinoblastoma. J. Neurochem. 111, 250–263 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akagi T., Mandai M., Ooto S., Hirami Y., Osakada F., Kageyama R., Yoshimura N., Takahashi M., Otx2 homeobox gene induces photoreceptor-specific phenotypes in cells derived from adult iris and ciliary tissue. Invest. Ophthalmol. Vis. Sci. 45, 4570–4575 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Furukawa T., Morrow E. M., Cepko C. L., Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91, 531–541 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Oh E. C., Cheng H., Hao H., Jia L., Khan N. W., Swaroop A., Rod differentiation factor NRL activates the expression of nuclear receptor NR2E3 to suppress the development of cone photoreceptors. Brain Res. 1236, 16–29 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betancourt J., Katzman S., Chen B., Nuclear factor one B regulates neural stem cell differentiation and axonal projection of corticofugal neurons. J. Comp. Neurol. 522, 6–35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Melo J., Zibetti C., Clark B. S., Hwang W., Miranda-Angulo A. L., Qian J., Blackshaw S., Lhx2 is an essential factor for retinal gliogenesis and Notch signaling. J. Neurosci. 36, 2391–2405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter F. D., Drago J., Xu Y., Cheema S. S., Wassif C., Huang S. P., Lee E., Grinberg A., Massalas J. S., Bodine D., Alt F., Westphal H., Lhx2, a LIM homeobox gene, is required for eye, forebrain, and definitive erythrocyte development. Development 124, 2935–2944 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Mu X., Fu X., Beremand P. D., Thomas T. L., Klein W. H., Gene regulation logic in retinal ganglion cell development: Isl1 defines a critical branch distinct from but overlapping with Pou4f2. Proc. Natl. Acad. Sci. U.S.A. 105, 6942–6947 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voigt P., Tee W.-W., Reinberg D., A double take on bivalent promoters. Genes Dev. 27, 1318–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nauli S. M., Alenghat F. J., Luo Y., Williams E., Vassilev P., Li X., Elia A. E., Lu W., Brown E. M., Quinn S. J., Ingber D. E., Zhou J., Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129–137 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Fane M. E., Chhabra Y., Hollingsworth D. E. J., Simmons J. L., Spoerri L., Oh T. G., Chauhan J., Chin T., Harris L., Harvey T. J., Muscat G. E. O., Goding C. R., Sturm R. A., Haass N. K., Boyle G. M., Piper M., Smith A. G., NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. EBioMedicine 16, 63–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark B. S., Stein-O’Brien G. L., Shiau F., Cannon G. H., Davis-Marcisak E., Sherman T., Santiago C. P., Hoang T. V., Rajaii F., James-Esposito R. E., Gronostajski R. M., Fertig E. J., Goff L. A., Blackshaw S., Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron 102, 1111–1126.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng L., Hurley J. B., Dierks B., Srinivas M., Salto C., Vennström B., Reh T. A., Forrest D., A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat. Genet. 27, 94–98 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Eldred K. C., Hadyniak S. E., Hussey K. A., Brenerman B., Zhang P. W., Chamling X., Sluch V. M., Welsbie D. S., Hattar S., Taylor J., Wahlin K., Zack D. J., Johnston R. J. Jr., Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science 362, eaau6348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X., Wang C., Liu W., Li J., Li C., Kou X., Chen J., Zhao Y., Gao H., Wang H., Zhang Y., Gao Y., Gao S., Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537, 558–562 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Cheng L., Hansen N. F., Zhao L., Du Y., Zou C., Donovan F. X., Chou B. K., Zhou G., Li S., Dowey S. N., Ye Z.; NISC Comparative Sequencing Program, Chandrasekharappa S. C., Yang H., Mullikin J. C., Liu P. P., Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell 10, 337–344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baek S., Goldstein I., Hager G. L., Bivariate genomic footprinting detects changes in transcription factor activity. Cell Rep. 19, 1710–1722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ernst J., Kellis M., ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaay5247/DC1
Fig. S1. Qualification of ATAC-seq data.
Fig. S2. Analysis of differential chromatin elements and RNA-seq data in developing human retina and ROs.
Fig. S3. Epigenetic and expression profiles of retinal cell type markers during retinal development stages.
Fig. S4. Identification of putative TFs involved in retinal differentiation.
Fig. S5. Functional assay of THRA and NFIB using RO electroporation.
Fig. S6. NFIB-regulated signaling pathways in retinal development.
Fig. S7. Transcriptional regulatory network during human retinal and RO development.
Fig. S8. Comparative analyses of human and murine epigenetic data during retinal development.
Fig. S9. Signaling pathways in different stages of retinal development in mouse and human.
Table S1. Primers in all experiments.