

Paclitaxel failure promotes collateral resistance to EGFR-targeted therapy by adaptive rewiring of the cellular stress response.
Abstract
Secondary drug resistance stems from dynamic clonal evolution during the development of a prior primary resistance. This collateral type of resistance is often a characteristic of cancer recurrence. Yet, mechanisms that drive this collateral resistance and their drug-specific trajectories are still poorly understood. Using resistance selection and small-scale pharmacological screens, we find that cancer cells with primary acquired resistance to the microtubule-stabilizing drug paclitaxel often develop tolerance to epidermal growth factor receptor–tyrosine kinase inhibitors (EGFR-TKIs), leading to formation of more stable resistant cell populations. We show that paclitaxel-resistant cancer cells follow distinct selection paths under EGFR-TKIs by enriching the stemness program, developing a highly glycolytic adaptive stress response, and rewiring an apoptosis control pathway. Collectively, our work demonstrates the alterations in cellular state stemming from paclitaxel failure that result in collateral resistance to EGFR-TKIs and points to new exploitable vulnerabilities during resistance evolution in the second-line treatment setting.
INTRODUCTION
Profuse development of collateral resistance (or cross-resistance) to various drugs defines multidrug resistance (1). The multiplicity and spread of this resistance reduce or even entirely eliminate the utility of available drugs used in chemotherapy, targeted therapy, and their combination regimens (2). This results in frequent, if not inevitable, relapse of cancers despite the initial response of patients to these drugs (3, 4). Cancer relapse due to the development of multidrug resistance reflects the cumulative effects of diverse genetic and nongenetic phenomena, including primary and secondary mutations (5, 6), intratumoral heterogeneity (7, 8), nonmutational rare cell variability (9), and residual cancer persistence (10–12). While the evolution and fitness landscape of multidrug resistance present a trade-off of costs, where the development of resistance toward a drug or drugs also develops a targetable vulnerability [termed “collateral sensitivity” (13)], little is known about the logic of expansion of collateral resistance following acquired resistance to the primary drug. This limits the design of more effective combination therapy and other drug regimens.
In the clinic, multiple chemotherapy cycles have been shown to amplify the selection of innately resistant tumor cells or cells that have acquired resistance over the course of a treatment (2, 14, 15). This presents a unique challenge in designing a drug scheduling approach to preventing relapse following single-drug, sequential, or drug combination modalities. Optimal scheduling of drugs, in the context of combination treatments, has proven to affect the efficacy of cancer therapies albeit with little clinical evidence (16–18). Regardless, current treatment modalities use second-line therapies after adequate failure of patients to first-line therapy regimens (19, 20). The success of this strategy is arguably difficult to predict since collateral sensitivity and resistance are not considered, and biomarkers that can represent specific collateral trajectories of drug-resistant relapsed cancers are yet to be found. It is therefore crucial to understand the nature of the evolutionary routes of primary drug resistance to maximize the treatment success of second-line and combination therapies.
Recent focus on studying resistance mechanisms has shifted from finding mechanisms of resistance at the end of drug selection experiments to the characterization of the dynamic process of tumor evolution, including the understanding of intratumoral heterogeneity and clonal selection during therapy (13). This shift has proven highly informative in exploring the emergence and spread of multidrug resistance, as this has led to the proposal of several alternative routes for acquiring a stable resistance. One emerging evidence implicates that a drug-tolerant persister state (10) feeds this transition to a more stable resistance in a heterogeneous fashion by diversifying genetic mechanisms of drug resistance (10, 12, 21). By entering into a persister state, cancer cells can survive prolonged drug exposure while being dormant, thus predisposing these residual small subpopulations to expand and become stably resistant (12, 22). Although mechanisms of this bottleneck evolution from a persister population have been suggested (23), little is known whether stable drug-resistant tumor cells can reenter a persister state when challenged with chemically different agent/s and influence collateral resistance. Given that collateral resistance trajectories following failure to first-line therapies are poorly understood, uncovering the possibility of reentry to persistence and onset of resistance stability in first-line therapy-resistant cancers may improve the unpredictability of these trajectories, resulting in better management of relapsed cancers.
Using resistance selection experiments and small-scale pharmacological screens in concert with public pharmacogenomic databases, we identified that cancer cells resistant to microtubule-targeting chemotherapy agents such as paclitaxel, docetaxel, epothilone B, and vinorelbine display collateral resistance to epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitors (TKIs) such as gefitinib, erlotinib, and afatinib, among others. With acquired paclitaxel resistance as a first-line therapy failure model, we found that a surviving fraction of resistant cells could undergo persistence to EGFR-TKIs and develop routes to a more stable resistance. We identified that once in the EGFR-TKI persister state, paclitaxel-resistant cells enrich their stemness profile and rewire their glucose metabolism and an apoptotic control pathway to collectively compromise an adaptive stress response during the second-line of therapeutic pressure. Unexpectedly, however, transition from persistence to a stable resistance to EGFR-TKIs following paclitaxel resistance does not seem to require known resistance-promoting mechanisms such as MET amplification, KRAS G12 missense mutation, and the function of adenosine triphosphate (ATP)–binding cassette (ABC) transporters. Together, our findings demonstrate that failure to first-line paclitaxel chemotherapy relays substantial collateral resistance to EGFR-TKIs by following an adaptive logic of reentry to persistence.
RESULTS
Coresistance network across wide array of drugs in the Genomics of Drug Sensitivity in Cancer dataset
We inferred drug responses across thousands of human cancer cell lines previously profiled in pharmacogenomics datasets currently available as a cancer research resource (24). To investigate events of collateral resistance in a large pool of clinically relevant drugs, we used the Genomics of Drug Sensitivity in Cancer (GDSC), a publicly available dataset containing 1001 molecularly annotated human cancer cell lines and their individual drug response to 265 drugs. Mining the preprocessed dataset of binarized median inhibitory concentration (IC50) values (sensitive or resistant), we cross-matched drug profiles and identified substantial coresistance signatures across classes of drugs (grouped in 21 targeted processes or pathways) as analyzed in the GDSC. We visualized these signatures as a network and semiquantitatively sorted coresistance occurrence as drugs with similar “resistant” denomination in the same cell line, evaluating all tested cell lines processed according to GDSC classification (Fig. 1A). We subsequently narrowed down these interactions within classes of drugs that contain >60% of coresistance signatures. Among the >21 drug classes analyzed, antimitotic drugs and EGFR-TKIs represent two chemically different drug classes that have the highest coresistance frequency. Looking into individual cases, an example of noteworthy coresistance identified occurred between paclitaxel and inhibitors of EGFR signaling (gefitinib, erlotinib, afatinib, lapatinib, and cetuximab). The coresistance frequency of these EGFR-TKIs in cancer cell lines classified as paclitaxel resistant is 84 to 92% (Fig. 1B) and was among the high-ranking, chemically different drugs to have coresistance with paclitaxel. Paclitaxel had the lowest discretization threshold among all antimitotic drugs with >80% coresistance match to gefitinib (11 drugs; fig. S1A). Clustering these matched coresistance cases according to cancer types, lung and pancreatic cancer cell lines denote cells to have most likely co-occurring resistance between paclitaxel and EGFR-TKIs (fig. S1B). Identification of vulnerabilities that drive the intermediate stages of this coresistance is important, given that the sequence of paclitaxel-based chemotherapy and EGFR-TKIs and their combinations are actively being considered in the clinic.
Fig. 1. Paclitaxel resistance primes selection trajectories of cancer cells to EGFR-targeted therapies.
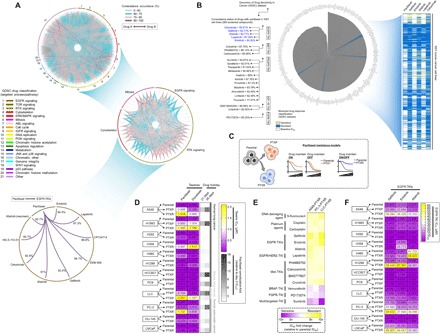
(A) Circos plot visualization of coresistance signatures of 265 drugs screened in 1001 human cancer cell lines processed from the publicly available database of the GDSC. Drugs were classified according to their main target mode of action. Note that coresistance is viewed as either co-occurring bidirectional or one-directional event as identified in the preprocessed data files of binarized response of cell lines to drugs in the GDSC. RTK, receptor tyrosine kinase; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; IGFR, insulin-like growth factor receptor. (B) Radial histogram plot visualization of correlated drugs ranked on the basis of their coresistance classification with paclitaxel resistance derived from their sensitivity response in the panel of cell lines tested in the GDSC datase (left). Discretization threshold of binarized drug response (sensitive or resistant) of paclitaxel and EGFR-TKIs gefitinib, erlotinib, afatinib, lapatinib, and cetuximab (right). (C) Cellular models of paclitaxel resistance used in this study. (D) Characterization of acquired primary paclitaxel and collateral docetaxel resistance in a panel of PTXP (6 cell lines), PTXR (10 cell lines), and their parental lines derived from human and mouse lung and human pancreatic cancers. Stability of resistance was assayed using 21- and 35-day drug holidays. Cells were treated with or without drugs for 72 hours with a concentration dilution series and were assayed for sulforhodamine B (SRB). Representative of three independent experiments. (E) Validation screening of collateral resistance evaluated in a panel of PTXR cell lines with selected drugs representing chemically different classes (selected from a pool of drugs with >80% coresistance classification with paclitaxel) in two PTXR cell lines with the highest resistance (A549-PTXR and PC-3–PTXR). Resistance was assessed on the basis of IC50 fold change relative to parental cell lines assayed as in (D). (F) Characterization of collateral EGFR-TKI (gefitinib, erlotinib, and afatinib) resistance in the same panel of cell lines and assay as in (D). Representative of three independent experiments.
First-line therapy failure to paclitaxel imparts secondary resistance to EGFR-TKIs
Using cancer cell lines derived from human and mouse lung cancers (nine) and human pancreatic cancers (three), we developed an experimental strategy to select which cell lines are suitable to use as sequential resistance models, where first-line therapy resistance (antimitotic drugs) relays drug-specific trajectory to a secondary resistance (EGFR-TKIs). By subjecting a panel of 12 cell lines with diverse oncogenic driver profiles (i.e., KRAS G12 missense mutations, MET amplification, and EGFR exon 19 deletion, among others) to two protocols of short-term (~19 days) drug selection [one expanded from a single formed colony after selection and the other expanded from multiple colonies (admixture) to infer to what intensity heterogeneity influences resistance potential capacity regardless of genetic drivers (fig. S2A and see Materials and Methods for details)], we were able to characterize which cell lines are suitable for further resistance selection experiments and discriminate cell populations that are able to expand after increasing drug-induced pressures to avoid choosing cell lines that become highly senescent during initial selection. A549, H1993, LLC (Lewis lung carcinoma), and PC-3 cells appeared to have high population expansion than any other cell lines upon selection to microtubule-targeting drugs paclitaxel, docetaxel, and epothilone B (fig. S2B). Among the six antimitotic drugs with highest coresistance ranks to EGFR-TKIs, GSK269962A (Rho-associated protein kinase inhibitor) and PF-562271 (FAK inhibitor) prevented further selection of most populations, while paclitaxel and epothilone B enabled most populations to expand. We next inferred from cells expanded in paclitaxel which cell lines are mostly capable of generating tolerant populations under a gefitinib pressure (fig. S2C). The same cell lines that have the highest surviving fraction following paclitaxel selection displayed high gefitinib resistance potential (fig. S2D). Meanwhile, expansion from admixture of multiple single colonies widely altered the resistance potential of most cells upon drug selection (protocol 2 results).
To experimentally explore the coresistance between paclitaxel and EGFR-TKIs, we generated 16 cell lines with primary acquired resistance to paclitaxel (both stable and transient models) by an in vitro stepwise clonal evolution protocol (Fig. 1C, fig. S3A, and see Materials and Methods for details). Most of these stable (PTXR) and transient (PTXP) paclitaxel-resistant cell lines display high collateral resistance to docetaxel, a semisynthetic analog of paclitaxel (Fig. 1D). Across these cell lines, paclitaxel resistance is associated with promotion of G1-S phase cell cycle transition (fig. S3B), indicative of loss of cell growth control. These PTXP and PTXR cell lines have also developed collateral resistance to other microtubule-targeting drugs epothilone B and vinorelbine but not to vinblastine (fig. S3, C to E), recognizing previous reports that looked into the collateral resistance cases of various paclitaxel-resistant cancer models (25–27). As a preliminary validation screen, we characterized three PTXR cell lines (A549-PTXR, LLC-PTXR, and PC-3–PTXR) displaying stably high paclitaxel resistance for collateral sensitivity or resistance to 14 chemotherapy and targeted therapy drugs (Fig. 1E). Consistent with our GDSC data analysis, EGFR-TKIs gefitinib, erlotinib, and afatinib are collaterally resistant with paclitaxel with >5-fold IC50 increase in PTXR cells compared to parental cells. Exemplifying this validation, we observed similar collateral resistance signature across our PTXP and PTXR cell lines based on fold increase in IC50 values relative to their parental counterparts (Fig. 1F and fig. S4A) and colony formation (fig. S4B). Substantiating these results in vivo, A549-derived paclitaxel-resistant xenograft tumors notably displayed collateral resistance to gefitinib even at a high dose (fig. S5). Meanwhile, there is good agreement with GDSC datasets that resistance to other antimitotic drugs such as docetaxel, epothilone B, and vinorelbine may have coresistance to EGFR-TKIs (fig. S6A). To functionally validate this correlation, we established transient acquired resistance to epothilone B in two lung cancer cell lines (A549-EBR and H1993-EBR cells; fig. S6B). Both EBR lines displayed marked collateral resistance signature to gefitinib (fig. S6, C and D), supporting the idea that resistance to microtubule-targeting drugs besides paclitaxel may convey secondary resistance to EGFR-TKIs, regardless of the genomic background of cancer cells.
Drug-tolerant persistence to EGFR-TKIs reveals unique selection trajectories to a stable resistance in paclitaxel-resistant cancer cells
We next sought to establish an experimental model of minimal residual drug-tolerant persisters derived from parental, PTXP, and PTXR cell lines that can survive cytotoxic exposure to EGFR-TKIs with a reversible resistance capacity (herein referred to as “EGFR-TKI persisters”). Although the reversible, nonmutational mechanisms of this persister model is still poorly understood (10, 22), a “convertible resistance” model whereby drug tolerance can shift from and to a sensitive state is beneficial to uncover trajectories to a more stable resistant state. Defining the state of persistence in cancer remains problematic as it differs from other mechanisms of drug sensitivity and its heritability as a phenotype is still unclear. The terms “drug tolerance” and “drug persistence” are used interchangeably in many instances, but these two phenotypes differ in cases of resilience during drug treatment at the whole and subpopulation levels, respectively (28). Throughout this paper, we reserved to describe persistence as a capacity of an expanded subpopulation of cancer cells from drug-induced selections to regain sensitivity after drug removal that can be reverted back to a drug-tolerant persistence mode. In deriving EGFR-TKI persisters, we primarily chose the cell lines that had considerable tolerability to gefitinib following paclitaxel exposure (rationale of cell line choice based from fig. S2). These cell lines are A549, H1993, and PC-3, which have variable intermediate sensitivities to EGFR-TKIs (gefitinib, erlotinib, and afatinib) at the parental state (IC50 values, 1 to 18 μM). As a supplement to these models, we also derived gefitinib persistence from known EGFR-TKI hypersensitive cell lines PC9 and HCC827, both of which harbor EGFR exon 19 deletion mutation. Modifying a previously established protocol on EGFR-TKI persisters (12), we derived slow-doubling fractions of surviving persisters that emerged from all cell lines (parental, PTXP, and PTXR), which, upon further maintenance with low drug concentrations, can maintain persistence while continuing population expansion in culture. We observed patterns of negligible growth before colony-derived expansions of these EGFR-TKI persisters, which has also been observed in other persister models (12). Drug holidays, short or long term, sensitize the sustained drug tolerance of these persisters, but selection to the same drug allows these persisters to reacquire sensitivity to gefitinib or erlotinib in varying proportions. This reversibility of resistance indicates nonmutational resistance mechanism, as previously observed in other persister models derived from lung adenocarcinoma, melanoma, and breast cancer (10, 12, 22). Persistence to gefitinib or erlotinib is relatively higher in PTXP and PTXR cells and takes a relatively longer drug holiday to be sensitized and their tolerance to be rederived than persistence developed in parental cells (Fig. 2A and fig. S8, A and B). These differences in growth response to gefitinib or erlotinib were coordinated by variable rates of DNA synthesis during persistence selection (fig. S7). This induced persistence by gefitinib or erlotinib also displayed variable collateral persistence folds to other EGFR-TKIs afatinib and lapatinib (Fig. 2B), although it should be noted that collateral persistence to afatinib in all gefitinib or erlotinib persisters was barely rederived after a long-term drug holiday. Expansion of persistence-derived colonies in increasing concentrations of gefitinib generated higher drug tolerance capacities especially in PTXP- and PTXR-derived cells (Fig. 2C and fig. S8C). Next, we sought to determine whether prior paclitaxel selection might influence resistance stability projections of these persisters through a stepwise drug selection approach to gefitinib or erlotinib (Fig. 2D and fig. S8D). As expected, paclitaxel resistance predisposed (PTXP and PTXR) cells to acquire a more stable resistance signature to gefitinib and erlotinib derived from persisters compared to parental-derived cells (Fig. 2, E and F, and fig. S8, E and F). While there is a marked widespread selection for a stable collateral resistance to EGFR-TKIs in all PTXP and PTXR cells upon entry to a persister state, we cannot disregard the fact that most of the parental cells from which these paclitaxel-resistant lines were derived have variable, intermediate sensitivity profiles to EGFR-TKIs, with the exception of EGFR-mutant cell lines. Clinical interpretation of the drug concentrations reflected in our assays can be accurately amended by the use of patient-derived cells and mouse models to which we can only argue as venue for a separate study. Although further work will be needed to confound off-target mechanisms, our data strongly suggest that paclitaxel resistance is a common ground in many cell lines we tested for successful selection in subsequent EGFR-TKI treatment.
Fig. 2. Experimental model of drug persistence reveals varying resistance stabilities to EGFR-TKIs.
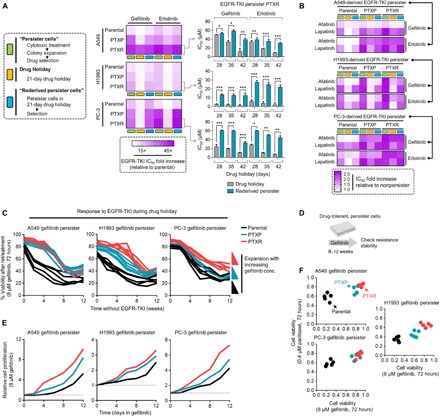
(A) Generation and characterization of reversible drug-tolerant, slow-growing persister A549-, H1993-, and PC-3–derived cells in response to indicated EGFR-TKI–induced selection, expansion, and drug holiday schedules in culture (assayed by SRB). Stability and reversibility of resistance were evaluated using long-term drug holiday schedules (right, bar graph; means ± SD of three biological replicates; *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test). See also Materials and Methods. (B) Characterization of collateral persistence to afatinib and lapatinib in A549-, H1993-, and PC-3–derived gefitinib or erlotinib persisters. Cells were treated with or without drugs for 72 hours with a concentration dilution series and were assayed for SRB. Representative of two independent experiments. (C) Evolution of established A549-, H1993-, and PC-3–derived persisters to gefitinib during a long-term drug holiday. Cells were grown in drug-free media and periodically retested over ~12 weeks for sensitization to EGFR-TKIs (retesting regime: 8 μM gefitinib, 72 hours, assayed by SRB). Representative of two independent experiments. (D and E) Long-term growth of indicated GPs after over ~2 months of stepwise selection to gefitinib to stabilize resistance. Cells were then retested upon treatment in 8 μM gefitinib at indicated times and were assayed by SRB. Values are relative to nontreated. Representative of two independent experiments. (F) Resistance status to both paclitaxel and gefitinib of A549-, H1993-, and PC-3–derived persister pools generated as in (D) and expanded under increasing concentrations of gefitinib. Cells were treated with or indicated concentration of drugs for 72 hours and were assayed by SRB.
Entry to EGFR-TKI persistence following paclitaxel resistance is functionally coupled with an enriched stemness profile
We set out to explore what mechanisms drive the trajectory to an EGFR inhibitor–specific persistence following paclitaxel resistance. The cancer stem cell (CSC) phenotype has been substantially implicated in the reversible drug tolerance to EGFR-TKIs (10). Acquisition and maintenance of this phenotype are closely linked to a concurrent mesenchymal plasticity. Given that most of our cell line panels are epithelial, we first examined whether paclitaxel resistance induces a transition to a mesenchymal state (EMT) using a panoply of known EMT phenotypic assays. The EMT-inducing transcription factor (EMT-TF) Snail1 was generally amplified across our panel of 6 PTXP and 10 PTXR cell lines (Fig. 3A). PTXP and PTXR lines derived from A549, H1993, and PC-3 cells displayed highly mesenchymal profiles compared to their parental lines that resemble primary epithelial tumor characteristics, evidenced by unanimous up-regulation of five EMT markers (N-cadherin, vimentin, fibronectin, Slug, and β-catenin) and down-regulation of the epithelial marker E-cadherin (Fig. 3A). Along with these, extracellular transforming growth factor–β (TGF-β) antagonists bone morphogenetic protein (BMP) ligands exhibited a collective down-regulated expression signature in PTXP and PTXR cells relative to parental cells (fig. S9A) while the opposite for BMP antagonists Gremlin (GREM1) and Chordin-like 2 (CHRDL2) (fig. S9B), indicating that TGF-β is a potent inducer of EMT in our paclitaxel-resistant models. We then examined whether this mesenchymal profile is consequential to acquiring EGFR-TKI persistence. Pharmacological inhibition of TGF-β1 by LY364947 complemented these observations with suppression of both mesenchymal phenotype and secondary resistance to gefitinib and erlotinib, while reversible sensitization was observed upon treatment with recombinant TGF-β1 in PTXP and PTXR cells (fig. S9, C and D). To examine whether EMT is a crucial factor in the transition from persistence to a stable secondary resistance, we used an EMT inhibition and rescue experiment in EGFR-TKI persisters. PTXP- and PTXR-derived EGFR-TKI persisters displayed an impaired growth rate upon EMT inhibition but was recovered upon recombinant TGF-β1 treatment, all during cytotoxic exposure to either gefitinib or erlotinib (fig. S9, E and F). Supporting these signatures, nuclear translocation and accumulation of β-catenin displayed an association with paclitaxel resistance and gefitinib persistence derived from PTXP and PTXR cells (fig. S10, A to C), indicative of stimulated EMT-TFs (29). These results indicate that these persisters might exploit EMT to develop collateral resistance to EGFR-TKI following paclitaxel resistance.
Fig. 3. Paclitaxel resistance–induced EMT enriches and facilitates stemness to consequentially drive EGFR-TKI persistence.
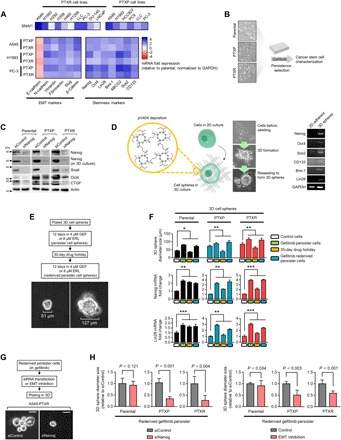
(A) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of SNAI1 expression in our panel of 6 PTXP and 10 PTXR cell lines and expression of other indicated EMT and stemness markers in indicated A549-, H1993-, and PC-3–derived cells. Values are relative to parental and were normalized to glyceraldehyde phosphate dehydrogenase (GAPDH) levels (means ± SD of three biological replicates). Representative of three independent experiments. (B) Schematic of cancer stemness characterization of GPs derived from parental, PTXP, and PTXR lines. (C) Western blot analysis of expression of indicated stemness markers in A549-derived parental, PTXP, and PTXR cells upon NANOG RNA interference (RNAi) for 48 hours. Actin was used as a loading control. Representative of two independent experiments. (D) Schematic of 3D spheroid-like cell formation (herein referred to as “3D cell spheres”) in our in-house built polymeric thin-film 3D culture plates [on the surface of functionalized poly(2,4,6,8-tetravinyl-2,4,6,8-tetramethl cyclotetrasiloxane) (pV4D4)]. 3D sphere formation is characterized with higher stemness as shown in RT-PCR analysis of gene transcripts in A549 cells. (E) Schematic of GP development in A549-derived 3D cell spheres used in (F). GEF, gefitinib; ERL, erlotinib. (F) 3D sphere diameter measurements of indicated A549-derived GPs, grown for 4 days before phase-contrast imaging (top). qRT-PCR analysis of Nanog and Lin28 expressions in the same panel of 3D cell spheres as in the top panel (middle and bottom, respectively). Values are relative to parental control (means ± SD of a representative experiment performed in triplicate; *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test). (G) Schematic of Nanog RNAi and EMT inhibition in 3D cell spheres of A549 rederived GPs used in (H). (H) 3D cell sphere diameter measurements of indicated A549 rederived GP spheres upon Nanog RNAi or EMT inhibition for 3 days (left) or treatment with or without 10 μM LY364947 for 12 hours (right). Values are relative to RNAi or dimethyl sulfoxide control (means ± SD of a representative experiment performed in triplicate; P values by Student’s t test).
We then sought to investigate whether our observed paclitaxel resistance–specific EMT program permits the acquisition of the CSC phenotype (Fig. 3B). Gene expression of seven putative stemness markers (Nanog, Oct4, Lin28, Bmi-1, ABCG2, Sox2, and CD133) demonstrated a close correlation with the stability of paclitaxel resistance in PTXP and PTXR cells (Fig. 3A). Using Nanog as a proxy master regulator of this stemness, we used an RNA interference (RNAi) and multiple three-dimensional (3D) cell culture–based assays to examine the regulation of this phenotype. Nanog RNAi suppressed stemness signals, including connective tissue growth factor (CTGF), a regulator of mesenchymal plasticity (Fig. 3C). Incorporating our in-house built polymeric thin film–based 3D culture system that allows for a relatively faster formation of tumor spheroids (30) (see Materials and Methods), we established a mesenchymal-enriching 3D sphere formation assay that mimics a physiological dimension important for tumor formation (Fig. 3D). We then generated 3D sphere-forming gefitinib persisters (GPs) that are associated with Nanog expression (Fig. 3E and fig. S10D). The diameter size of these 3D spheres correlated with the drug response of GPs derived from parental, PTXP, and PTXR cells (Fig. 3F). We found that Nanog and Lin28 expressions are correlated with reversible persistence specific to PTXP- and PTXR-derived GP spheres but not to parental-derived persister spheres (Fig. 4F). In 3D, CSC-like cells form invadopodia, invasive protrusions important for the dissemination of tumor cells in a constricted physiological space. To explore whether the CSC phenotype of PTXP- and PTXR-derived GPs is associated with invadopodia formation, we cultured the cells in stacked 3D rat-tail collagen gels supplemented with EGF and observed invadopodia-like structures in all derived persisters (fig. S10E). Since invadopodia are retractile structures, we hypothesized that if these structures are associated with gefitinib persistence, then rescue from persistence aversion (after a long-term drug holiday; “rederived persisters”) can induce evident morphological difference among persisters derived from parental, PTXP, and PTXR lines. Rederived GPs from PTXP and PTXR cells exhibited longer protrusions compared to those of their parental origins (fig. S10E). Distribution and accumulation of TKS5, an invadopodia marker, validated the formation of these structures, at least those formed in rederived GPs (fig. S10F). Supporting the hypothesis that Nanog-directed stemness might be responsible for this phenotype, Nanog RNAi resulted in the inhibition of this protrusion in PTXR rederived GPs (fig. S10G), similar to effects observed with the abolished sphere formation capacity of 3D rederived GPs (Fig. 3, G and H). Together, these results support the idea that paclitaxel resistance uses EMT to enrich stemness in developing persistence to EGFR-TKIs.
Fig. 4. Stemness is functionally associated with a glycolytic metabolism in EGFR-TKI persisters derived from paclitaxel-resistant cancer cells.
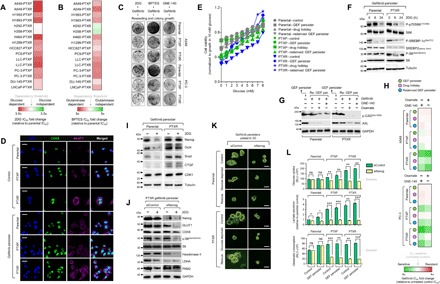
(A and B) Characterization of glycolysis and glutaminolysis dependency of indicated PTXP and PTXR cell lines upon treatment with or without 2DG or BPTES with a concentration dilution series and assayed for SRB (means ± SD of three biological replicates). (C) Colony formation assay of parental, PTXP, and PTXR derived from A549 and PC-3 cells following indicated drug treatment regimes (10 mM 2DG, 10 μM BPTES, or 8 μM GNE-140 for 24 hours, followed by 5 μM gefitinib for 72 hours and then equally reseeded in drug-free media following 8 days of culture). Representative of two independent experiments. (D) Representative images of immunofluorescence of CDK8 (green) and GLUT1 (magenta) in indicated A549-derived control and GPs. Nuclei are shown in blue [4′,6-diamidino-2-phenylindole (DAPI)]. Scale bars, 25 μm. Representative of two independent experiments. (E) Glucose titration curves for indicated A549-derived GPs. Values were normalized to growth in 10 mM glucose-containing media (0 mM glucose represents glucose-deprived media; means ± SD of two biological replicates). (F) Western blot analysis of indicated proteins in the mTOR/AMPK pathway in A549-derived GPs upon treatment with or without 10 mM 2DG for indicated times. Tubulin was used as a loading control. Representative of two independent experiments. (G) Western blot analysis of phospho-CAD (Ser1859) [p-CAD(Ser1859)] and AXL expressions in indicated A549-derived GPs upon treatment with or without LDHA inhibitors GNE-140 (8 μM) or oxamate (40 mM) for 24 hours, followed by treatment with or without 5 μM gefitinib for 24 hours. Tubulin was used as a loading control. Representative of two independent experiments. (H) Sensitivity characterization of indicated A549- and PC-3–derived GPs upon treatment with or without 8 μM GNE-140 or 40 mM oxamate, followed by treatment with gefitinib for 72 hours with a concentration dilution series and assayed for SRB (means ± SD of three biological replicates). (I and J) Western blot analysis of indicated stemness and glycolysis marker expressions in indicated A549-derived GPs upon treatment with or without 10 mM 2DG for 24 hours. A549-PTXR GPs underwent NANOG RNAi for 48 hours, followed by the same 2DG treatment. Tubulin and GAPDH were used as loading controls. Representative of two independent experiments. (K) Phase-contrast images of indicated A549-derived GP 3D cell spheres upon NANOG RNAi for 48 hours, followed by indicated glucose modifications for 12 hours (rescue indicates culture in 10 mM glucose–containing media). Representative of two independent experiments. Scale bars, 70 μm. (L) Glucose uptake, lactate secretion, and glutamate consumption measurements in indicated A549-derived GPs upon NANOG RNAi for 48 hours (RLU, relative luciferase units; means ± SD of three biological replicates; ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test).
Stemness regulates glycolytic metabolism to develop selection routes to EGFR-TKIs in paclitaxel-resistant cancer cells
A requirement for the dissemination of CSCs is an altered metabolic state such as a metabolic switch leading to a glycolytic dependency (31). To examine whether paclitaxel resistance rewires the metabolic demand by enriching stemness to develop EGFR-TKI persistence, we characterized our panel of 16 PTXP and PTXR cell lines for dependency to glycolysis and glutaminolysis. Incorporating the glycolysis inhibitor 2DG (2-deoxy-d-glucose) and glutaminase inhibitor BPTES [bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide], most PTXP and PTXR cell lines displayed sensitization to these inhibitors (Fig. 4, A and B). Treatment with 2DG, BPTES, or GNE-140 [lactate dehydrogenase A (LDHA) inhibitor] obliterated the collateral gefitinib resistance in PTXP and PTXR cells (Fig. 4C). Furthermore, expressions of glucose transporter GLUT1 and glycolytic dependency promoter CDK8 were increased in PTXP and PTXR cells compared to parental cells, and both were generally associated with gefitinib persistence (Fig. 4D). We then examined how paclitaxel resistance might abolish any tolerance to reduced glucose levels in GPs. Consistent with an escalated glycolytic dependency, PTXP- and PTXR-derived GPs have a reduced tolerance to glucose deprivation yet remained responsive to supplementation (Fig. 4E). Next, we inspected the status of the mammalian target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK) pathway, a key regulator of cellular energy production that defines the integrity of glycolytic dependence (32). 2DG, also an inhibitor of signaling to mTOR effectors, abolished the mTORC1-mediated phosphoactivation of S6K1 at 6 hours only in PTXR-derived GPs. Note that at 24 hours, a striking blockade of phosphorylation was observed in both parental- and PTXR-derived GPs. The specificity of regulation in PTXR-derived GPs was also supported by inhibition of phosphoactivation of ribosomal protein S6, persistence of AMPK-mediated Ser372 phosphorylation of sterol regulatory element–binding protein–1c (SREBP-1c), and an inhibited basal level of the active form of SREBP2 upon glycolysis disruption (Fig. 4F). Since EGFR signaling can maintain the de novo pyrimidine biosynthesis pathway in addition to altering glucose metabolism in response to EGFR-TKIs, we determined whether our observed regulation of mTORC1-mediated glycolytic dependence might also hold true in the context of other glycolysis inhibitors (i.e., LDHA inhibitors). We used our rederived GPs derived from both parental and PTXR cells, as they were the most responsive to low concentrations of GNE-140 (fig. S11A). PTXR rederived GPs halted a rather sustained Ser1859 phosphorylation of CAD, a first step requirement in de novo synthesis of pyrimidines, upon treatment with GNE-140 or oxamate compared to those from parental cells. Notably, this PTXR specificity was supported by suppression of the EGFR-diversifying receptor tyrosine kinase AXL in GNE-140– or oxamate-treated PTXR rederived GPs. However, note that in parental rederived GPs, AXL was also suppressed but most likely because of the gefitinib treatment while rederiving persistence, which is apparent in gefitinib-treated only cells (Fig. 4G). On the basis of their subsequent effect on AXL, we further examined whether GNE-140 or oxamate sensitizes EGFR-TKI persisters derived from paclitaxel resistance to gefitinib or erlotinib. Treatment with GNE-140 or oxamate highly sensitized both gefitinib and erlotinib persistence (derived and rederived) in PTXR cells compared to those in parental and PTXP cells (Fig. 4H and fig. S11B), suggesting that high paclitaxel resistance affects a demanding glycolytic process in developing persistence to EGFR-TKIs that can totally be reversed by blocking multiple important nodes in the glycolysis pathway such as blocking the conversion of pyruvate to lactate.
We next asked whether stemness is responsible for maintaining this shift to a more glycolytic dependence. We initially characterized the expression of stemness markers upon glycolysis inhibition. 2DG effectively inhibited stemness gene expression and induced a modest down-regulation of CDK1 in parental-derived GPs but not in PTXR-derived cells where these markers were either unaltered or affected subtly (Fig. 4I). Since Nanog can metabolically reprogram the subset of tumor-initiating CSC-like cells (33), we aimed to further characterize the state of glycolysis in the context of Nanog regulation. Upon Nanog RNAi, treatment with GNE-140 induced both apoptotic morphology (fig. S11C) and complete sensitization during drug persistence maintenance in culture (fig. S11, D and E), while treatment with 2DG suppressed glycolytic proteins and enzymes GLUT1 and hexokinase-II complemented with total inhibition of phosphoactivation of S6 in PTXR-derived GPs (Fig. 4J). This suggests that Nanog controls the glycolytic dependence state in response to 2DG. To functionally confirm whether Nanog is a glycolytic reprogramming factor specific to developing gefitinib persistence derived from PTXR cells, we examined the sphere-forming capacity of these cells upon glucose deprivation and rescue. Nanog RNAi abolished the stemness rescue mechanism in PTXR-derived GPs (Fig. 4K). Next, we checked the functional parameters of glycolysis dependence upon Nanog RNAi. Basal levels of glucose uptake, lactate secretion, and glutamine consumption were higher in PTXP- and PTXR-derived GPs than those derived from parental cells. Silencing Nanog prevented the increase in these parameters only in PTXR-derived GPs but not in parental-derived cells (Fig. 4L and fig. S11F).
Adaptive senescence is a metabolically responsive phenotype exploited by paclitaxel-resistant cancer cells under EGFR-TKI pressure
To link stemness-regulated control of metabolism in GPs derived from paclitaxel resistance to stress adaptation, we focused on a recently described adaptive stress response phenotype exploited by cancer cells to evade killing: a therapy-induced senescence (TIS)–associated stemness reprogramming (34). We tested whether the glycolytic stemness associated with our gefitinib persistence model derived from paclitaxel resistance uses senescence as a stress response during therapy. We first subjected the cells to serial long-term passaging in culture and subsequently starved them of glucose to trigger a glycolytic cell cycle arrest, followed by 1-week expansion in culture. Notably, only GPs derived from PTXP and PTXR, but not from parental, stained positive for a strong senescence-associated β-galactosidase (SA-β-gal) activity (Fig. 5, A and B). However, rescue in high-glucose media for 3 days was not sufficient to escape from senescence, at least in cells derived from PTXR cells (Fig. 5A). We then observed the effects of serial passaging in population expansion upon glucose modification. As expected, only those derived from PTXP and PTXR cells showed an arrested colony growth when compared to parental-derived GPs (Fig. 5C), supporting the active SA-β-gal positivity in PTXP- and PTXR-derived GPs. In addition, elevated levels of genes encoding p16INK4a and the senescence-associated secretory phenotype (SASP) components and reduced expression of Lamin B1 further confirmed this senescence phenotype (Fig. 5D). This induced senescence upon glucose modification was associated with maintained expression of stemness markers in PTXR-derived GPs (Fig. 5E). Because this signature of stemness-associated TIS has previously been observed to be regulated by the activation of a specific Wnt cascade (34) with β-catenin degradation-promoting glycogen synthase kinase 3β (GSK3β) acting as the cell-autonomous driver of this Wnt program (34), we checked the Ser9 phosphorylation activity of GSK3β under our induced senescence condition. Unexpectedly, there was higher phosphorylated-GSK3β activity and a down-regulated signature of active β-catenin (dephosphorylated at Ser37) in senescent PTXR-derived GPs (Fig. 5E). Despite our premature expression data, this signature reflects that induction of senescence in PTXR-derived GPs might be governed by Wnt-directed capability. To test whether regulation of canonical Wnt signaling via GSK3β functionally influences our model, we used the GSK3β inhibitor CHIR99021 (GSK3βi). Examining the exit from senescence to apoptosis, we found that GSK3βi did not significantly affect the activities of caspase 3/7 and caspase 9, with some modest exceptions in senescent PTXR-derived GPs when compared to parental-derived persisters (Fig. 5F). However, Nanog RNAi significantly increased the activities of these caspases and led to a robust suppression of SASP components interleukin-1α and CXCL-1 upon GSK3βi in PTXR-derived GPs, suggesting that inhibition of stemness might be required to exit from senescence and facilitate apoptosis in these cells (Fig. 5G), as previously observed in senescent B cell lymphomas with a reprogrammed stemness profile (34). We next characterized whether stemness-directed migration is still retained in senescent PTXR-derived GPs and whether it is regulated upon GSK3βi. Senescent PTXR-derived GPs continue to migrate despite the active senescence positivity, reflective of an adaptive senescence phenotype previously shown in a melanoma model. GSK3βi alone can abolish this migration but was superimposed upon Nanog RNAi (Fig. 5H). Furthermore, we found a much higher fraction of cells positive for active nuclear β-catenin before senescence induction in PTXR-derived GPs compared to parental-derived cells where active β-catenin is mostly cytoplasmic. Upon senescence induction, there was an evident decrease in active β-catenin, but this was abrogated upon GSK3βi with sparse expression both in the nucleus and cytoplasm. Nanog RNAi reinforced this signal suppression and was able to fully repress nuclear β-catenin (Fig. 5I). Although we cannot completely preclude alternative interpretations, these results favor our hypothesis that gefitinib persistence following paclitaxel resistance uses stemness-associated senescence as an adaptive stress response mechanism during perturbations in glucose metabolism.
Fig. 5. An adaptive-like senescence phenotype is a stemness-directed metabolic stress response of EGFR-TKI persisters derived from paclitaxel-resistant cancer cells.
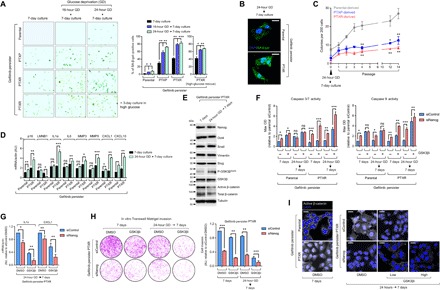
(A) SA-β-gal staining of indicated A549-derived GPs under indicated culture conditions and glucose modification at indicated times, followed by 3-day high-glucose culture for PTXR GPs (bottom). Cells were passaged over ~36 times before experiment. Representative images are shown (left) along with quantified data as percentage (right; means ± SD of three biological replicates; *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test). (B) Representative images of SA-β-gal (green) immunofluorescent labeling using a C12FDG substrate in indicated A549-derived GPs under the indicated culture conditions. Nuclei are shown in blue (DAPI). Scale bars, 20 μm. Representative of two independent experiments. (C) Colony counts of indicated of early serial passages of A549-derived GPs under indicated culture conditions (means ± SD of three biological replicates; *P < 0.05, **P < 0.01, Student’s t test). (D) qRT-PCR analysis of expression of p16INK4a and other indicated SASP components in indicated A549-derived GPs upon indicated culture conditioning. Values were analyzed relative to actin levels to control for complementary DNA (cDNA) quantity (means ± SD of three biological replicates; *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test). AU, arbitrary units. (E) Western blot analysis of indicated stemness marker and Wnt5a pathway marker expressions in A549-PTXR GPs upon indicated culture conditioning. Tubulin was used as a loading control. Representative of two independent experiments. (F) Caspase 3/7 DEVDase and caspase 9 activities of indicated A549-derived GPs upon indicated culture conditioning following NANOG RNAi for 48 hours (means ± SD of three biological replicates; *P < 0.05, **P < 0.01, ***P < 0.005, Student’s t test). OD, optical density. (G) qRT-PCR analysis of interleukin-1α (IL1α) and CXCL1 expressions in A549-PTXR GPs under indicated culture condition upon treatment with or without GSK3βi inhibitor (1 μM) for 48 hours following NANOG RNAi for 48 hours (means ± SD of three biological replicates; *P < 0.05, **P < 0.01, Student’s t test). (H) Transwell Matrigel invasion assay of A549-PTXR GPs under the same condition, treatment, and RNAi as in (G) (means ± SD of three biological replicates; **P < 0.01, ***P < 0.005, Student’s t test). DMSO, dimethyl sulfoxide. (I) Representative images of active β-catenin (gray) in indicated A549-derived GPs under indicated culture conditions upon treatment with or without CHIR99021 (low, 1 μM; high, 5 μM) for 48 hours following NANOG RNAi for 48 hours. Nuclei are shown in blue (DAPI). Scale bars, 25 μm. Representative of two independent experiments.
A rewired apoptosis control pathway supports the development of secondary EGFR-TKI resistance in paclitaxel-resistant cancer cells by regulating stemness-directed fates
Apoptosis is a commonly hijacked program in developing drug resistance (1). To collectively resolve how EGFR-TKI persistence generated from paclitaxel-resistant cancer cells phenotypically couples apoptosis evasion to cellular state rewiring, we studied the potential functional role of FOXO3a, a transcriptional promoter of apoptotic effectors. FOXO3a has been observed to be required for paclitaxel resistance (25, 35), and its loss is associated with an impaired cancer-initiating cell function, at least in leukemia where FOXO3a was needed for maintenance of the CSC phenotype (36, 37) and resistance development (38). Despite these, FOXO3a is a bona fide tumor repressor and a major inactivation target of the Akt pathway in many cancers. A natural idea from this paradox would be that FOXO3a can be programmed to stall apoptosis and promote resistance. To resolve this issue, we used a potpourri of strategies to understand how paclitaxel resistance exploits FOXO3a to influence cell fate during EGFR-TKI treatment. We first examined the correlation between FOXO3a expression and survival in clinical cohorts of patients with breast or lung cancers, two cancer types that represent a large fraction of cell lines concurrently resistant to paclitaxel and EGFR-TKIs in our GDSC analysis. Using publicly available microarray data, we found that although low FOXO3a expression was generally correlated with poor relapse-free survival (RFS) of treatment-naïve patients with breast cancer, primary and adjuvant chemotherapy led to a wider gap between low and high FOXO3a expressions, with high FOXO3a expression significantly correlated with poor RFS (Fig. 6A, top). We then further assessed the prognostic value of FOXO3a for treatment-associated relapse from a smaller cohort of patients with breast cancer that received sequential cycles of multiagent chemotherapy with paclitaxel as the last cycle component. Gene expression and immunohistochemical (IHC) analysis of FOXO3a in formalin-fixed paraffin-embedded (FFPE) tumor tissues from these patients revealed that high FOXO3a expression is strongly correlated with relapse (Fig. 6, B and C, and fig. S12A). Unexpectedly, high FOXO3a IHC positivity is mostly associated with metastasis to the lymph node of these patients with breast cancer (fig. S12B). Intrigued by this correlation, we adopted our LLC-derived cells to establish a murine model of lung metastasis and see whether the metastatic propensity of LLC-PTXR cells is associated with FOXO3a expression (fig. S12C). Consistent with this hypothesis, basal gene expression of FOXO3a and its putative transcription targets insulin receptor substrate 2 (IRS2) and tumor necrosis factor superfamily member 10 (TNFSF10) were up-regulated, while ribosomal protein S6 kinase beta-1 (RPS6KB1), a gene encoding for a downstream protein target of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, was down-regulated in PTXR metastasized tumors from the lung, liver, and spleen (fig. S12D), encouraging the notion that paclitaxel resistance of metastasized tumors uses FOXO3a possibly to promote further resistance development. Likewise, in large cohorts of patients with breast cancer, there is a significant signature of FOXO3a overexpression in patients that underwent chemotherapy alone or in combination regimes with hormone and radiotherapy (fig. S13A). In patients with lung cancer, poor overall and first progression survival probabilities are significantly correlated with high FOXO3a expression (Fig. 6A, bottom).
Fig. 6. Apoptosis evasion by FOXO3a is crucial to initiate and maintain phenotypic transitions to a stable secondary resistance.
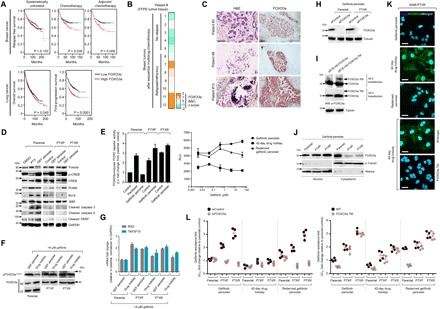
(A) Kaplan-Meier plots of RFS of patients with breast cancer, categorized in cohort groups of systemically untreated, chemotherapy-treated, and adjuvant chemotherapy–treated patients (top) and overall and first progression survival of patients with lung cancer (bottom). Patient survival data were stratified by FOXO3a expression (low or high) in their primary tumors. P values were calculated using a log-rank test. (B) qRT-PCR analysis of FOXO3a expression in FFPE tumor tissue sections from patients with breast cancer who underwent sequential multidrug chemotherapy. Log-transformed gene expression values are relative to the sample with the lowest FOXO3a expression and were normalized to GAPDH levels (means ± SD of two biological replicates). (C) Immunohistochemical analysis of indicated FFPE tumor tissue sections used in (B). Sections were blocked and probed with FOXO3a antibody and detected using a 3,3’-Diaminobenzidine (DAB) chromagen kit (right). Replicate tissue sections were stained with hematoxylin and eosin (H&E) (left). All sections were photographed with an inverted phase-contrast microscope (original magnification, ×200). Scale bars, 150 μm. Representative of two independent experiments. (D) Western blot analysis of indicated apoptotic signal expressions in A549-derived parental, PTXP, PTXR, and indicated GPs. Treatment with 20 nM paclitaxel for 24 hours in parental cells served as a positive control. GAPDH was used as a loading control. Representative of two independent experiments. PARP, poly(adenosine 5´-diphosphate–ribose) polymerase. (E) FOXO reporter activity induced by FOXO3 in the same cells and conditions as in (D). Values represent ratio fold induction normalized reporter activity in the presence of FOXO3 to that in the presence of the negative control expression vector (transfected for 48 hours); all relative to parental control. Reporter activity was measured upon treatment with indicated concentration dilution series of gefitinib for 72 hours in indicated A549-PTXR GPs before plasmid transfections for 48 hours (right; means ± SD of three biological replicates). LU, luciferase units. (F) Western blot analysis of phospho-FOXO3a (T32) expression in indicated A549-derived persisters upon treatment with 8 μM gefitinib for 72 hours. Representative of two independent experiments. SE, short exposure; LE, long exposure. (G) qRT-PCR analysis of expression of FOXO3a target genes IRS2 and TNFSF10 in the same cells and treatment as in (E). Values are relative to parental and were normalized to GAPDH levels (means ± SD of three biological replicates). (H) Western blot analysis of FOXO3a expression in indicated A549-derived GPs upon FOXO3a RNAi for 48 hours. Tubulin was used as a loading control. Representative of two independent experiments. (I) Western blot analysis of active FOXO3a expression in A549-PTXR GPs upon transfection with indicated plasmids for 36 hours to induce transcription of an active mutant form of FOXO3a [hemagglutinin (HA)–tagged FOXO3a.TM]. Tubulin was used as a loading control. Representative of two independent experiments. (J and K) Characterization of FOXO3a translocation in indicated A549-derived GPs by Western blotting (J) (α-tubulin and histone were used as loading controls for nuclear and cytoplasmic lysates, respectively) and immunofluorescence (K) (cells were transfected with or without HA-tagged FOXO3a.TM for 36 hours). Representative of two independent experiments. (L) Gefitinib resistance characterization of indicated A549-derived GPs upon FOXO3a RNAi for 48 hours or transfection with control or HA-tagged FOXO3a.TM plasmid for 36 hours following gefitinib treatment for 72 hours with a concentration dilution series and were assayed for SRB. Values are relative to parental control (means ± SD of four biological replicates).
We have previously shown that high FOXO3a activity can drive multidrug collateral resistance in paclitaxel-resistant cancer cells (25, 35). Expanding on this, an initial analysis of GDSC and Cancer Cell Line Encyclopedia (CCLE) datasets revealed that FOXO3a expression is widely associated with collateral resistance to paclitaxel and EGFR-TKIs in several malignancies (fig. S13B). However, note that these associations appear to be varied because of the aggregate categorization of cell lines per cancer type as processed in an open-tool application used for the analysis. In support of this, FOXO3a expression, when mapped in individual cell lines, is broadly associated with concurrent taxane and EGFR-TKI resistance in a separate pharmacogenomics dataset (fig. S13C). Next, we examined the state of apoptosis in GPs during drug maintenance. We affirmed that FOXO3a expression was retained in both PTXP- and PTXR-derived GPs but was down-regulated in parental-derived persisters. Phosphoactivation of CREB (adenosine 3′,5′-monophosphate response element–binding protein), a prosurvival signal, was induced in all lines of GPs, while expressions of apoptotic markers were subtle in PTXP- and PTXR-derived GPs compared to those derived from parental (Fig. 6D). A luciferase-based reporter activity assay showed that FOXO3a was mostly active in PTXP- and PTXR-derived GPs but was inactive in parental-derived persisters (Fig. 6E, left). This specificity in persisters derived from paclitaxel-resistant cells was supported by the correlation of FOXO3a activity with gefitinib persistence during gefitinib treatment (Fig. 6E, right). Threonine-32 phosphorylation (Thr32) of FOXO3a, a critical posttranslational modification for the inhibition of FOXO3a proapoptotic function, was inhibited, but total FOXO3a was induced upon drug holiday sensitization in parental-derived GPs, but not in PTXP- and PTXR-derived cells in response to gefitinib treatment (Fig. 6F), supporting the idea that stability of FOXO3a activity in the state of gefitinib persistence is relayed by paclitaxel resistance. Upholding this shift of function to an antiapoptotic role of FOXO3a, two well-known FOXO3a targets IRS2 and TNFSF10 were increased in drug holiday–induced sensitization in parental-derived GPs but were decreased in PTXP- and PTXR-derived persisters (Fig. 6G and fig. S14A), suggesting that expression of FOXO3a and its targets is adaptable depending on the persistence state (high in resistant mode and low in sensitive mode) of paclitaxel resistance–derived GPs. Supporting these, gefitinib inhibited the kinase activities of EGFR, AKT, and extracellular signal–regulated kinase (ERK) in parental cells, whereas in PTXP and PTXR cells, only that of EGFR was suppressed with AKT and ERK activities sufficiently maintained, functionally contextualizing the maintained expressions of Thr32-phosphorylated FOXO3a and p70S6K, a downstream target of the PI3K/Akt pathway, in PTXR cells upon gefitinib treatment (fig. S15, A and B). Regardless, gefitinib-induced inhibition of EGFR kinase activity was rescued following GP selection in parental, PTXP, and PTXR cells (fig. S15B). To uncouple these activity correlations and to better understand how FOXO3a-mediated apoptosis is rewired as a result of gefitinib pressure in paclitaxel-resistant cells, we used FOXO3a RNAi and nonphosphorylatable constitutive active mutant FOXO3a.TM (TM is a triple mutant form of FOXO3a in which all three Akt phosphorylation sites are mutated to alanine; T32A/S253A/S315A) transfections in these cells (Fig. 6, H and I, and fig. S14B). Upon sensitization, FOXO3a inhibition led to a sufficient suppression of Tyr1068 phosphorylation of EGFR in PTXR-derived GPs but not those derived from parental (fig. S15C). Gefitinib persistence induced a chronic nuclear accumulation of FOXO3a, which can be translocated to and from the cytoplasm upon drug holiday sensitization and rederivation of persistence, respectively, in PTXR-derived cells, while transduction of FOXO3a.TM led to a strong nuclear signal translocation in sensitized GPs (Fig. 6, J and K, and fig. S14C). To investigate whether FOXO3a is sufficient and/or required to rewire the apoptosis pathway in GPs derived from paclitaxel resistance, we characterized the resistance fold to gefitinib in these cells upon FOXO3a RNAi or FOXO3a.TM transduction. We found that FOXO3a inhibition led to reversal of resistance in PTXP- and PTXR-derived GPs but not those derived from parental, while FOXO3a.TM modestly affected the response to gefitinib in PTXP- and PTXR-derived persisters but consistently inhibited resistance in parental-derived, rederived, and drug holiday–sensitized GPs (Fig. 6L and fig. S14D). Substantiating this resistance inhibition, FOXO3a.TM led to an increased activation of caspase 3/7 and caspase 9 activities in parental-derived GPs but not those derived from PTXP and PTXR (fig. S16A), while inhibition of FOXO3a forced an activation of these caspases in PTXP- and PTXR-derived cells but not in parental-derived GPs upon staurosporine (STS)–induced apoptosis (fig. S16B). Thus, FOXO3a on its own can rewire a caspase-dependent apoptosis in GPs specifically derived from paclitaxel-resistant cells.
Subsequently, we sought to examine whether a rewired FOXO3a function coordinates stemness specific to gefitinib persistence derived from paclitaxel-resistant cells. Co-occurrence of genes annotated to be involved in EMT and cancer stemness was high in patients with lung cancer with an amplified copy number of FOXO3a while low in patients with FOXO3a deletion (fig. S16C). To further pursue this correlation in our model, we analyzed ex vivo the xenografts derived from parental and PTXR cells following gefitinib therapy. Residual tumors that are left behind after the therapy were considered resistant following a 20-day daily gefitinib regimen (fig. S16D). Phosphorylation of EGFR (Tyr1068) was partially inhibited in all of these residual tumors (fig. S16D). Exemplifying our FOXO3a model, residual PTXR-derived tumors expressed a more pronounced activation of FOXO3a compared to parental-derived tumors following high-dose gefitinib therapy (fig. S16D). This activation strongly correlated with high gene and protein expressions of stemness markers (fig. S16, E to G), including CD133, a putative cancer stemness marker previously shown to be highly enriched in drug-tolerant persister cancer cells (10). Supporting this, we observed a reversal of the 3D mesenchymal morphology in PTXR-derived GPs and an inhibited stemness potential in vitro (fig. S16, H and I). Consistent with previous studies showing a functional shift in the role of FOXO3a in drug-resistant cancer cells (38), all of these results argue that FOXO3a is sufficient to direct apoptosis evasion that phenotypically affects the state of stemness in a GP model following prior paclitaxel resistance.
Dispensability of some putative resistance-promoting mechanisms in drug-tolerant cancer cells with various genomic backgrounds
Accumulating evidence suggests that the reversibility of the drug-tolerant persistence phenotype is independent of classic stable resistance-mediating mechanisms. Development of a stable paclitaxel resistance often requires the prominent overexpression of ABC transporters (39). This led us to question the requirement for ABC transporter–driven drug efflux in eliciting (or maintaining) a drug-specific secondary resistance trajectory in our paclitaxel-resistant models. We profiled the gene expression of nine ABC transporters in A549-PTXR cells, the model with the most stable paclitaxel resistance, and found that ABCB1 and ABCC1, genes that encode for P-glycoprotein (P-gp) and multidrug resistance–associated protein 1 (MRP1), respectively, were strongly up-regulated relative to expressions in parental cells (fig. S17A). In the GDSC and CCLE datasets, ABCB1 is more commonly associated with resistance to taxanes and EGFR-TKIs than ABCC1 in cell lines derived from various malignancies (fig. S17B). We then evaluated the gene expression of these two genes in our panel of transient and stable paclitaxel-resistant cell lines. While both of their expression profiles reveal strong association with paclitaxel resistance, ABCC1 shows a more varied expression pattern, whereas ABCB1 is extensively up-regulated across PTXP and PTXR cell lines (fig. S17C). Given that the EGFR-TKIs gefitinib and erlotinib were previously shown to have antagonizing effects on ABC transporters P-gp, MRP1, and breast cancer resistance protein (BCRP) (ABCG2) (40, 41), we assumed that persistence to EGFR-TKIs in paclitaxel-resistant cells precludes the necessity for drug efflux in maintaining a secondary resistance. To test this assumption, we first characterized the effects of gefitinib or erlotinib treatment on expression and adenosine triphosphatase (ATPase) activity of P-gp in comparison with verapamil, a P-gp inhibitor. As expected, expression of P-gp, but not MRP1, was inhibited upon treatment with gefitinib or erlotinib (fig. S17D) along with activation of P-gp ATPase activity (fig. S17E) in A549-PTXP and A549-PTXR cells, suggesting the direct inhibition of P-gp afforded by EGFR-TKIs (41). However, these results do not translate into inhibition of secondary resistance to EGFR-TKIs in A549-PTXP and A549-PTXR cells as observed in colony formation and viability assays (fig. S17, F and G). We next used the A549-derived GPs to further corroborate these results. Verapamil-stimulated ATPase activity was higher in PTXP- and PTXR-derived GPs than those derived from parental (fig. S17H), and drug holiday sensitization led to scant P-gp protein expression in parental-derived persisters but not in PTXP- and PTXR-derived persisters upon gefitinib treatment (fig. S17I), suggesting that gefitinib persistence established in paclitaxel-resistant cells is less likely to be driven by P-gp. Demonstrating this case, gefitinib persistence established in PTXP and PTXR cells, but not in parental cells, abolished verapamil-induced intracellular accumulation of rhodamine-123, a P-gp substrate (fig. S17J). In addition, paclitaxel-resistant A549- and PC-3–derived GPs showed a negligibly inhibited gefitinib resistance fold despite pharmacological inhibition of P-gp at high concentrations of verapamil, further precluding the requirement for P-gp (fig. S17K). Together, these results dispute the necessity for paclitaxel resistance–specific high drug efflux activity of P-gp to maintain persistence or perpetuate secondary resistance to EGFR-TKIs (fig. S17L). Nevertheless, there are previous observations on rapid emergence of persistence mediated by drug efflux upon chemotherapy (42). Whether there is a strict logic of chemical specificity required to exploit drug efflux to expand persister clones and become more stably resistant remains an open question.
We next wondered whether EGFR-TKI persistence derived from our paclitaxel-resistant cells requires some well-known putative genetic drivers of resistance expressed in our model systems. In our H1993-derived persisters, we sought to address the functional implication of MET amplification, an oncogenic background often associated with EGFR-TKI resistance (43), because the H1993 cell line is MET-amplified (44). While MET expression and phosphorylation appeared to be correlated with gefitinib persistence derived from A549 and H1993 parental cells, these MET parameters were variable and anticorrelated in the derivation and rederivation of gefitinib persistence in PTXR-derived persisters (fig. S18A). Nonetheless, MET copy number was highly amplified in H1993-derived PTXR cells than parental cells, but selection for gefitinib persistence resulted in variable changes in the MET copy number in A549- and H1993-derived PTXR cells (fig. S18B). We next evaluated the functional consequences of MET inhibition in H1993-derived GPs using RNAi and PHA665752, a MET-TKI (fig. S18C). MET inhibition retarded the proliferation of parental GPs but had no significant effect on PTXR-derived GPs (fig. S18D). We then sensitized these GPs upon withdrawal of the drug for 35 days in culture to extrapolate the effects of MET on gefitinib-induced killing. PHA665752 inhibited MET kinase activity more effectively in sensitized parental-derived cells upon gefitinib treatment, and this inhibition appeared to be rescued by recombinant hepatocyte growth factor (HGF), MET’s endogenous ligand (fig. S18, E and F). However, this HGF-mediated rescue was not observed in sensitized PTXR-derived GPs. These data argue that MET amplification is not a requirement by paclitaxel-resistant cells to enter into a GP state.
In our A549-derived models, we addressed KRAS mutation. Because the A549 cell line harbors an oncogenic homozygous KRAS G12S mutation (45), we evaluated the functional consequences of loss of KRAS oncogenic addiction. Using an RNAi strategy previously shown to functionally inhibit oncogenic KRAS mRNA in cell lines harboring mutations at codon 12 (G12S and G12C) (45), we first tested whether mutant KRAS depletion could influence any resistance signature in A549-derived cells, all of which harbor a homozygous missense KRAS mutation (fig. S19, A to D). In parental and PTXP cells, mutant KRAS inhibition led to profound sensitization to taxanes and EGFR-TKIs, but not in PTXR cells, suggesting that a stable paclitaxel resistance is required for mutant KRAS-independent mechanisms (fig. S19E). Moreover, mutant KRAS inhibition significantly impeded the tumor growth of parental-derived xenografts but not of PTXR-derived xenografts (fig. S19F). Similarly, this inhibition revealed notable sensitization to gefitinib in parental- and PTXP-derived GPs where the ability to rederive drug tolerance was retarded but not in PTXR-derived GPs (fig. S19, G and H). To identify whether mutant KRAS affects the state of a rewired apoptosis in our PTXR-derived model, we checked the status of FOXO3a and its activity upon KRAS RNAi. There was a robust activation of FOXO3a and its target genes IRS2 and TNFSF10, inhibition of RPS6KB1, and increased FOXO3a-induced FOXO reporter activity in parental-derived GPs following mutant KRAS inhibition and gefitinib treatment, indicating that mutant KRAS remains a bona fide oncogene in this drug-tolerant persister model, but not in the PTXR-derived model, as the gene and protein expression signatures of these targets and FOXO reporter activity remain unaffected (fig. S19, I and J). Together, our data reveal that oncogenic KRAS, at least in mutant PTXR-derived cells, is inconsequential to the development of collateral EGFR-TKI resistance and selection of drug-tolerant persisters following acquisition of a stable paclitaxel resistance.
DISCUSSION
Drug resistance has genetic and nongenetic duality (46). This dynamic view of acquiring resistance has led us to examine whether selective pressures from a “sequential multidrug” treatment engage adaptive mechanisms to achieve stability. Mining the GDSC database revealed many coresistance signatures and trajectories across a wide array of drugs and identified an interesting coresistance between classes of antimitotic drugs and EGFR-TKIs. Notably, by combining in vitro drug resistance selection and pharmacological perturbation experiments, we uncovered that prior acquired resistance to paclitaxel confers persistence to EGFR-TKIs, such as gefitinib and erlotinib, relaying the potential to become a stable resistance. Our analysis implicates that paclitaxel resistance remodels a subset of cancer cell populations using a reversible persistence as a reservoir of diverse adaptive cell states. Our study covers a wide range of phenotypic analysis of known hallmarks of drug resistance: mesenchymal plasticity, metabolic adaptation, and apoptosis evasion (Fig. 7). These reprogrammed mechanisms, enabled by paclitaxel failure, collectively serve as an evolutionary advantage during a selection pressure brought forth by the new drug. Although it might appear that these diverse mechanisms lack a causative relationship among them, our EGFR-TKI persister models allowed us to differentiate paclitaxel-resistant cells from sensitive cells in using these mechanisms to leverage their survival and cope under the pressure of second-line selection to EGFR-TKIs. Ultimately, our work, while mainly a proof-of-principle study, indicates that there is a considerable biological logic for EGFR-TKI cross-resistance evolution upon acquisition of paclitaxel resistance.
Fig. 7. Schematic overview of our study.
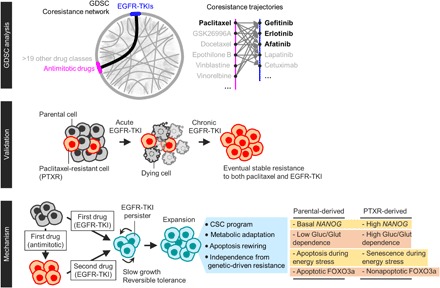
The mechanisms of this EGFR-TKI–specific tolerance in paclitaxel-resistant cells involve distinct phenotypic regulations in stemness, metabolism, and stress response. Cancer cells with prior paclitaxel resistance can enter a unique and evolvable EGFR-TKI–specific persister state by exploiting the acquired EMT phenotype when these cells survived paclitaxel pressure as opposed to sensitive cells that did not evolve under this pressure. This EMT-induced loss of the differentiation status in paclitaxel-resistant cells resulted in an enriched stemness profile. Our subsequent characterization revealed that this stemness is exploited as a mechanism to isolate EGFR-TKI persisters within isogenic populations by promoting an adaptive mode of glycolytic stress response. We assumed that this is a type of a “retractable glycolytic switch” in which during entry into an EGFR-TKI persister state, paclitaxel-resistant cells exhibit an exceptionally high glycolytic activity under a normal glucose condition but when glucose is scarce, these persisters become senescent, a cellular growth arrest program that is able to conserve a high metabolic rate (47). To our surprise, these metabolically responsive senescent cells maintain their enriched stemness and mesenchymal activities. Although the expression of EMT and CSC markers is a frequent hallmark of the reversible persistence phenotype (28), our work is the first to link stemness with a senescence-associated metabolic adaptation strategy during entry into a drug-persistent state. We speculate that this adaptive metabolism is required for slow-doubling resistant cells to enter into dormancy during metabolic stress where there is a scant energy resource.
The ability of paclitaxel resistance–induced selection to relay EGFR-TKI persistence appears to involve a rewired FOXO3a-directed apoptotic control pathway. There was a functional shift in the role of proapoptotic and tumor suppressor FOXO3a in this context, where higher levels of active FOXO3a are important for maintaining the induced EGFR-TKI persistence in paclitaxel-resistant cells. This shows that FOXO3a can evade PI3K/Akt-dependent sequestration and inactivation in drug-resistant cells as previously observed (25, 38) yet it is still unknown as to how this mechanism, which may or may not involve posttranslational modifications, proceeds. This rewired apoptotic response is functionally linked with the state of stemness of an EGFR-TKI persister model derived from paclitaxel-resistant cells. These raise the possibility that FOXO3a is a control point for paclitaxel-resistant cells to remodel their stress response to invoke a survival capacity during EGFR-TKI pressure.
In which cases does coevolution of a secondary resistance establish itself during resistance development to first-line (or front-line) therapies? Our results offer differential regulatory modes of developing collateral resistance to EGFR-TKIs during a phenotypic transition from a sensitive to a paclitaxel-resistant state. The diversity of EGFR-TKI persistence mechanisms we observed from our paclitaxel-resistant models suggests that reprogramming of stemness and apoptosis are passageways of persistence that can inform a resistance or a sensitive trajectory to a secondary line of therapy following failure to a first-line therapy. Perhaps what remains complicated is the need for “preexisting” resistance-conferring oncogenic backgrounds in driving an evolutionary selection for a secondary resistance. Despite our preliminary data, we substantially showed that paclitaxel resistance is independent of MET amplification and oncogenic KRAS mutation in driving a selection for a second-line resistance to EGFR-TKIs, especially to gefitinib. Ultimately, capturing the optimal window for a therapeutic intervention before developing this type of collateral resistance will also require knowledge on time window for both collateral resistance and sensitivity, which remains to be challenging as therapeutic vulnerabilities of these resistance nodes are still poorly understood.
In many cases, improvement of cancer patient survival depends on therapeutic opportunities in the second-line setting. While there are successes in using EGFR-TKIs as second-line or maintenance therapy following first-line chemotherapy (48), there is deficiency in the availability of predictive biomarkers that can trace the historical evolution of mechanisms that only confer a collateral resistance, as these can be confused as the same mechanisms that relayed acquired resistance to the first-line therapy. Our study is built on this need. Certainly, acquired drug resistance involves a multitude of distinct molecular mechanisms that can arise independently within the same patient who receive a monotherapy, thereby complicating the evolutionary traces of resistance of tumors that survive the pressures of multiple and sequential therapy cycles. Our work offers an understanding of potential entry points to a drug-specific collateral resistance following failure to a prior therapy. Needless to say, our study has limitations. One would be the potential off-target toxicity in cases where we used concentrations of EGFR-TKIs higher than those relevant to on-target EGFR inhibition in vitro and in vivo. As a result, we caution the possible clinical interpretation of our data. Regardless, the expansion of this work to other working models of resistance will take on increasing importance as sequential, or simultaneous treatment strategies will continue to be adapted to any form of anticancer therapy regimens.
MATERIALS AND METHODS
Data reporting and statistics
No statistical methods were used to predetermine sample size. The experiments were not randomized unless otherwise stated. The investigators were not blinded to allocation during experiments and outcome assessment. Images were processed using Fiji/ImageJ [National Institutes of Health (NIH) software]. All quantitative data are presented as means ± SD unless otherwise specified. Statistical tests were performed using GraphPad Prism 7.01 and SPSS 22 unless otherwise specified. Two-sided Student’s t tests assuming Gaussian distribution were used for comparisons of the means of data between two groups. Data visualization was mostly performed using GraphPad Prism 7.01 and R in conjunction with the ggplot2 software package. Other relevant methods not listed here are detailed in Supplementary Methods.
Human cancer patient samples and ethics statement
All human tissue samples were collected and analyzed with approved protocols in accordance with the ethical requirements and regulations of the Institutional Review Board of Seoul National University Hospital. Informed consent was obtained from all participants. All patients were diagnosed to have invasive breast carcinoma by core needle biopsy and were started on primary systemic therapy (PST). Thirty-three adult patients received adjuvant chemotherapy with four cycles of adriamycin (doxorubicin) and cychlophosphamide regimen, followed by four additional cycles of paclitaxel. Pathological complete response following PST was defined as complete disappearance of all invasive cancer or only residual ductal carcinoma in situ. FFPE paired tissue samples of prechemotherapy biopsy and postchemotherapy surgical resection were collected from each patient. Post- or preoperative radiation therapy was not performed. The tumor differentiation was categorized on the basis of the grading system described by the World Health Organization classification. Clinicopathologic information was obtained by reviewing medical records, pathology reports, and hematoxylin and eosin–stained sections (table S1). Recurrence/relapse, metastasis status, and all patient data were independently reviewed by a breast cancer pathologist (coauthor H.S.R.)
Cell lines
Human A549, H1299, H1993, H292, H358, H460, LNCaP clone FGC, and PC-3 cell lines [American Type Culture Collection (ATCC) nos. CCL-185, CRL-5803, CRL-5909, CRL-1848, CRL-5807, HTB-177, CRL-1740, and CRL-435, respectively; obtained in 2014] were grown under standard conditions in RPMI 1640 (Welgene) supplemented with 10% fetal bovine serum alternative Fetalgro bovine growth serum (RMBIO) or EqualFETAL bovine serum (Atlas Biologicals) and penicillin (100 U/ml)–streptomycin (100 μg/ml; Invitrogen) in a humidified incubator at 37°C and 5% CO2. Human DU-145 cell line (ATCC no. HTB-81; obtained in 2014) was grown in Eagle’s minimum essential medium (Merck) with the same supplementation as mentioned above. Mouse LL/2 (LLC1; ATCC no. CRL-1642; obtained in 2015) cell line was grown in Dulbecco’s modified Eagle’s medium with high glucose content (Invitrogen) with the same supplementation as mentioned above. Human PC9 and HCC827 cell lines expressing the EGFR exon 19 deletion mutation (ΔE746-A750), originally provided by J. K. Rho (Asan Medical Center, University of Ulsan, Seoul, Korea) in 2017, were grown in RPMI 1640 with the same supplementation as mentioned above. A549, PC-3, PC9, and HCC827 cell lines were authenticated at the College of Pharmacy, Seoul National University in 2014, 2016, and 2017, respectively, and the LLC cell line was authenticated at the College of Veterinary Medicine, Seoul National University in 2014, while others were not authenticated in-house, but all cells were tested for mycoplasma contamination before experiment, which all tested negative (Cosmogenetech mycoplasma test service).
Establishing resistant cell lines
Paclitaxel resistance in human A549, DU-145, H1299, H1993, H292, H358, H460, LNCaP clone FGC, and PC-3 cell lines and mouse LLC1 cell line and epothilone B resistance in human A549 and H1993 cell lines were established by first seeding cells at low density, which were continuously exposed to gradually increasing concentrations of the drug for 5 to 13 months for a stable phenotype (PTXR) and 1 to 2 months for a transient phenotype (PTXP). Some cell lines were expanded from colonies considering very long recovery from cytotoxic concentrations. Paclitaxel resistance in human PC9 and HCC827 cell lines was established as mentioned above except that selection lasted for 3 weeks. All resistant cell lines were additionally maintained weekly at low concentrations of the drug (fig. S3A), except for PC9- and HCC827-derived PTXR cell lines in which maximum tolerated concentrations of paclitaxel were used to maintain the cells at least for an additional 1 week before any assay. Cells were passaged every 4 or 5 days with adding fresh drug concentration.
Establishing drug-tolerant persistence
We generated EGFR-TKI (gefitinib or erlotinib)–induced drug-tolerant persisters from A549-, H1993-, PC-3–, PC9-, and HCC827-derived parental and paclitaxel-resistant (PTXP and PTXR) cells in 2D or 3D cultures modified from (12), based on the idea of colony-expanded persister-derived resistance. See Supplementary Methods for details on PC9- and HCC827-derived GPs. In a 2D monolayer, all cells were seeded 150,000 to 250,000 cells in 10-cm plates and allowed to adhere overnight and treated with EGFR-TKIs (7, 2.5, and 8.5 μM gefitinib and 10, 4.5, and 8 μM erlotinib for A549-, H1993-, and PC-3–derived cells, respectively). Preliminary concentrations used were based on 20-concentration range (nanomolar to micromolar) cytotoxicity analysis. The cells were treated for 6 to 18 days (washed and replaced with fresh drug every 3 days) until clearly well-spaced colonies are visible. These colonies were physically picked, placed, and cultured onto wells of a 96-well plate containing standard medium for 36 hours, followed by expansion in very low concentrations of EGFR-TKIs (0.2 to 4 μM gefitinib and 0.09 to 2 μM erlotinib) between 3 and 7 weeks. At weeks 4 and 5, many cells died and were seen floating, but few, slow-doubling cells remained and started to expand again as colonies from weeks 6 to 7. After this selection, cells were transferred onto 24-well plates, allowed to expand with the same drug treatment, transferred onto 6-well plates, and allowed to expand for another 3 weeks with drug maintenance (5, 1, and 4 μM gefitinib and 8, 2, and 6 μM erlotinib for A549-, H1993-, and PC-3–derived cells, respectively, for minimum tolerated concentrations and 10, 5, and 9 μM gefitinib and 15, 12, and 10 μM, respectively, for maximum tolerated concentrations). Cells were only plated onto 10-cm plates before analysis with the same drug maintaining concentrations until cells became confluent (70 to 90%; no colony formation) and then seeded accordingly per experimental protocol. Rederived persisters were generated after subjecting freshly selected persisters to long-term drug holidays (21-, 28-, 35-, or 42-day drug withdrawal as indicated), followed by EGFR-TKI selection for another 2 weeks similar to expansion protocol as above. As a 3D culture proxy model of EGFR-TKI persistence, we tested a simple protocol of drug selection in our in-house built 3D culture plates. Upon forming 3D “spheres” by repeated replating, A549-derived cells were treated with 4 μM gefitinib or 8 μM erlotinib for 12 days (replaced with fresh drug every 3 days). At the end of the treatment, there were a lot of floating cell spheres; after washout, the remaining cell spheres were replated onto a new plate and subjected to a 30-day drug holiday before rederiving persistence by resubjecting the cells to the same EGFR-TKI treatment for 12 days. We confirmed the “sensitization” of this persistence model by measuring the diameter size of the cell spheres. Note that we did not confirm whether these spheres were hollow or filled structures.
Cell proliferation assays
Cell viability and survival were assessed by sulforhodamine B (SRB) and colony formation assays, as we previously described (25). For small-scale screenings, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) colorimetric assay was simultaneously used, as we previously described (25), to validate the range of IC50 values per drug obtained by SRB assay. Briefly, for SRB assay, cells were seeded from 4 × 104 to 8 × 104 cells/ml in 96- or 24-well plates, allowed to adhere overnight, and treated with various concentrations of drugs. After 72 hours (or after indicated time), cells were fixed with 10% trichloroacetic acid solution, incubated at 4°C, stained with 0.038% SRB solution (Sigma-Aldrich), and incubated for 40 min at room temperature, followed by multiple quick rinses in 1% acetic acid to remove unbound dye. One hundred microliters (96-well) or 400 μl (24-well) of tris buffer (0.01 M, pH 10.4) was added per well and shaken gently for 5 min. Optical density was measured at 515 nm with common background subtraction at 650 nm in a microplate reader (VersaMax, Molecular Devices). For primary screening, we used a threshold of 50% cell growth inhibition as a cutoff for cytotoxicity against cell lines. IC50 values were calculated using nonlinear regression analysis (plotted as percent survival versus drug concentration) in TableCurve 2D v5.01 software (Systat). For colony formation, cells were seeded onto 12- or 24-well plates at densities of 700 to 2000 cells per well (depending on derived cell lines). Cells were allowed to adhere for 3 days and treated with indicated drugs (indicated schedules). Cells were additionally cultured for 1 or 2 weeks as indicated, and adherent colonies were fixed with 2% paraformaldehyde, stained with 0.01 to 0.03% crystal violet (in water), washed with water several times, and dried. Colonies with a diameter of >0.20 mm or colony areas were counted/quantified using the ColonyArea plugin in Fiji/ImageJ (NIH software).
3D culture–based assays
Before seeding, cells were collected using a scraper and filtered using a 40 μM pore cell strainer (BD Biosciences). Single cells (0.5 × 104 to 5 × 104) were seeded onto different 3D scaffold environments as indicated. For 3D culture used in Figs. 3 (D to H) and 4K and figs. S10D and S16 (H and I), we used our in-house developed poly(2,4,6,8-tetravinyl-2,4,6,8-tetramethl cyclotetrasiloxane) (pV4D4) polymer thin film–based scaffold culture plates (30). The thickness of pV4D4 films on plates was monitored in situ using a He-Ne laser (JDS Uniphase) interferometer system before use. Growing cancer cells in pV4D4 polymer thin film platform resulted in formation of multicellular spheroids within 72 hours and an observed phenotypic acquisition of stem cell–like properties (30). Rapid formation of these spheroids (~40 mm in diameter) can be observed within 24 hours; however, replatable spheroids (~200 mm in diameter) were observed at least after 36 hours. For A549-derived cells, 72-hour culture in these 3D plates resulted in variable multicellular spheroid formation with “aggregated” spheroids as large as ~410 mm in diameter. To achieve spatially separated individual cell spheres, ~50% confluent 3D cultures were physically replated onto new 3D culture plates and were repeated at least twice until individual 3D cell spheres were observed. Further information about this material is available upon reasonable request.
RNAi and plasmid transfections
pECE-HA-FOXO3a (Addgene plasmid no. 1787), pECE-HA-FOXO3a.TM (Addgene plasmid no. 1788), and the pECE-HA expression plasmids were transfected into cells using Lipofectamine 3000 (Life Technologies) or FuGene 6 transfection reagents (Promega) according to the manufacturer’s instructions and analyzed 24 or 48 hours after transfection. Short interfering RNAs (siRNAs) were delivered with Lipofectamine 3000 (Life Technologies) or DharmaFECT (Dharmacon) reagents at final concentrations of 40 nM (for Nanog RNAi) and 10 nM (for FOXO3a RNAi) according to the manufacturer’s instructions (sequences used were as follows: 5′-UUC UCC GAA CGU GUC ACG U-3′ for siControl, 5′-CUU ACG CUG AGU ACU UCG A-3′ for siLuciferase, 5′-CCA GAC CUG GAA CAA UUC A-3′ for siNANOG, and 5′-AAU GUG ACA UGG AGU CCA UUA-3′ for siFOXO3a). Scrambled were used as nontargeting siRNA controls (siControl). RNA oligonucleotides were purchased from IDT Korea (Integrated DNA Technologies) and Bioneer. Experiments were performed 48 hours after transfection as Western blotting indicated protein depletion to background levels.
Cellular fractionation and Western blotting
Isolation of nuclear and cytosolic extracts was carried out using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) following the manufacturer’s instructions. Whole-cell pellets were lysed in lysis buffer [150 mM NaCl, 50 mM tris (pH 7.5), 0.2% Triton X-100, 1 mM EDTA, 1 mM dithiothreitol, and 0.1 mM NaVO4] supplemented with a protease inhibitor cocktail (Calbiochem) and phosphatase inhibitor cocktails I and II (AG Scientific). Protein concentrations were determined by Bradford protein assay (Bio-Rad). Samples were boiled for 5 min in Laemmlli buffer. Equivalent amounts of proteins (usually 25 to 40 μg per loading sample) were separated on a SDS–polyacrylamide gel electrophoresis gel (usually on 7.5, 10, and 12% gels). Proteins were transferred onto Immobilon PVDF membranes (Millipore) using an Amersham semidry transfer system. The detection system was Clarity Max Western ECL Substrate (Bio-Rad) and Western Lightning Plus-ECL (PerkinElmer). Secondary antibodies were either goat antibodies to mouse immunoglobulin G–horseradish peroxidase (IgG-HRP) (DACO) or donkey antibodies to rabbit IgG-HRP (GE Healthcare). Primary antibodies used were P-gp (ab129450, Abcam), MRP1 (QCRL-1, Santa Cruz Biotechnology), E-cadherin [no. 3195, Cell Signaling Technology (CST)], fibronectin (ab2413, Abcam), vimentin (no. 3932, CST), Nanog (no. 4903, CST), Snail (no. 3879, CST), Oct4 (no. 2750, CST), CTGF (no. 86641, CST), CDK1 (ab18, Abcam), Glut1 (no. 12939, CST), CDK8 (no. 4106, CST), pS6(Ser240/244) (no. 2215, CST), S6 (no. 2217, CST), Hexokinase II (no. 2867, CST), LDHA (no. 2012, CST), PKM2 (no. 4053, CST), p-p70S6K(Thr389) (no. 9205, CST), S6K (no. 9202, CST), pSREBP-1c(Ser372) (no. 9874, CST), active SREBP2 (1C6, Santa Cruz Biotechnology), pCAD(Ser1859) (no. 12662, CST), Slug (no. 9585, CST), pGSK3β(Ser9) (no. 5558, CST), GSK3β (no. 12456,CST), active β-catenin (05-665, Millipore), β-catenin (610153, BD Biosciences), FOXO3a (no. 2497, CST), active FOXO3a (07-702, Millipore), pCREB (no. 9198, CST), CREB (no. 4820, CST), PUMA (no. 12450, CST), Bcl-6 (no. 5650, CST), BIM (H-5, Santa Cruz Biotechnology), cleaved caspase 3 (no. 9661, CST), cleaved caspase 9 (no. 9501, CST), KRAS (ab81075, Abcam), p70S6K (no. 9202, CST), CD133 (no. 130-090-422, Miltenyi Biotec), ABCG2 (no. 42078, CST), ALDH1A1 (ab134188, Abcam), α-tubulin (no. 2144, CST), histone H3 (no. 9715, CST), β-tubulin (D-10, Santa Cruz Biotechnology), β-actin (no. 4967, CST), and glyceraldehyde phosphate dehydrogenase (GAPDH) (6C5, Santa Cruz Biotechnology).
Reverse transcription polymerase chain reaction and quantitative polymerase chain reaction
Total cellular RNA (1 to 2 μg in a total volume of 10 μl) was extracted using TRIzol (Life Technologies) following the manufacturer’s protocol and was mixed with 1 μl of 10 mM deoxynucleotide triphosphates (2.5 mM of each; LifeTechnologies) and 1 μl of 50 μM random hexaprimers (New England Biolabs). Purified RNA was treated with deoxyribonuclease I (DNase I) (Takara) and reverse-transcribed using RevertAid reverse transcriptase (Fermentas). Complementary DNA (cDNA) was amplified by an SYBR Green PCR master mix (Applied Biosystems). Quantitative polymerase chain reaction (qPCR) was performed using SureCycler 8800 (Agilent) and AriaMx Real-Time PCR System (Agilent). The reaction contained 5 ng of cDNA, 2 μl of 1 μM qPCR primer pair (final concentration of each primer 200 nM), 5 μl 2× Master Mix, and final volume made up to 10 μl with DNase-free water. The specificity of the reaction was verified by melt curve analysis. Each reaction was performed in three replicates. Fold changes were calculated by the ΔΔCt method using GAPDH or β-actin as internal standards and normalized to the experimental control as indicated. For nucleic acids extraction (total RNA and genomic DNA) from FFPE tumor samples, we used the QIAGEN FFPE All-Prep kit and followed the manufacturer’s protocol. Small portion of specimens were prepared from ~80-μm slices of FFPE tumor blocks, followed by dewaxing using Deparaffinization Solution (QIAGEN). Purified RNA was subjected to reverse transcription PCR (RT-PCR) and qPCR as mentioned above. Primers used in this study are provided in table S2.
Immunocytochemistry
Cells were plated onto 0.1% gelatin-coated glass-bottom 30-mm dishes (except for 3D culture) and incubated overnight unless otherwise specified. The cells were fixed with 4% paraformaldehyde [in phosphate-buffered saline (PBS)] for 8 min at room temperature, quenched for 1 min in 10 mM tris (in PBS) at room temperature, and permeabilized in 0.1% Triton X-100 (in PBS), except for cells subjected for P-gp detection, since P-gp is a transmembrane protein. Cells were then blocked in 2% bovine serum albumin (BSA) (in PBS containing 0.01% Triton X-100) for 30 min at room temperature and incubated with primary antibodies diluted in 2% BSA for 2 hours. Alexa Fluor or fluorescein isothiocyanate–conjugated secondary antibodies were used to label primary antibodies. DAPI (4′,6-diamidino-2-phenylindole; 0.35 μg/ml) or Hoechst 33342 was used to counterstain the nuclei. Cells were mounted using VECTASHIELD Mounting Medium (Vector Laboratories). Immunocytochemistry was carried out using a ZEISS LSM 780 ApoTome microscope (Carl Zeiss) using C-Apochromat 40× lens with a numerical aperture of 1.20.
Glycolysis assays
Cells were seeded onto 96- or 24-well plates and cultured for 8 hours. The cells were then depleted in culture medium supplemented with 2 or 5% of in-house dialyzed fetal bovine serum (dialyzed against 0.15 M NaCl until glucose is <5 mg/dl; 10,000 molecular weight cutoff) for an additional 18 hours (unless otherwise indicated following drug treatments). Glucose uptake was analyzed using bioluminescent-based Glucose Uptake-Glo Assay (Promega) according to the manufacturer’s instructions. For measuring lactate secretion, cells were replenished with fresh medium after depletion and cultured 36 hours longer. Lactate levels in conditioned medium were measured using a colorimetric-based Lactate Assay Kit (Cell Biolabs). For measuring glutamine consumption, we used the bioluminescent-based Glutamine/Glutamate-Glo Assay (Promega) following the manufacturer’s protocol. Glutamine uptake was terminated by adding 6 mM glutamine to yield a final concentration of 4 mM. Luminescence was measured using a POLARstar Omega luminometer.
FOXO reporter assay
FOXO reporter activity induced by FOXO3a was monitored using a luminescence-based assay kit (BPS Bioscience) following the manufacturer’s protocol, with slight modifications on transfection time and incubation. The assay is designed to assess the activity of FOXO transcriptional activity triggered by transfection-ready FOXO3 expression vector in cultured cells. The reporter vector is PI3K/Akt pathway responsive, which contains a firefly luciferase gene under the control of multimers of the FOXO responsive element located upstream of a minimal promoter. A constitutively expressing sea pansy (Renilla) luciferase vector served as an internal control for the transfection efficiency, while a noninducible firefly luciferase vector premixed with constitutively expressing Renilla luciferase vector was used as a negative control. Luminescence was read on a POLARstar Omega luminometer.
Bioinformatics
We downloaded and curated pharmacological data from the GDSC. We accessed the preprocessed dataset on drug response and drug sensitivity of cell lines (available as an Excel file TableS5C.xlsx in their database), which contains discretized data for logic modeling of binarized drug response classification as “drug-resistant” (R) or “drug-sensitive” (S) assigned on each drug per cell line. Blank corresponds to baseline IC50. Note that the threshold values used in this process corresponded to 3 SDs above/below the mean of the normalized scores. The output feature was the response of the cancer cell lines to an anticancer drug as measured by the IC50. IC50 values were recorded as the natural logarithm of the half-maximal inhibitory micromolar concentrations. Using Circos, we visualized the coresistance status (that is, two drugs that have the same “R” classification per cell line) of all the drugs screened that are grouped according to their target pathway or process (grouping of drugs as in TableS1F.xlsx GDSC file). We then curated the cell lines identified to have resistance to paclitaxel (or other microtubule-targeting drugs) and individually ranked the coresistance of other drugs and presented as radial plots. Custom R and Python scripts used for plotting are available in https://github.com/borrisHUBO/Aldonza-et-al.-Science-Advances-Paper. In exploring the correlation of specific gene expression (ABCB1 and FOXO3a) with drug sensitivity, we used a previously developed in silico prediction interface using data from the CCLE and GDSC (49). For survival analysis, the overall, first progression, and RFS were queried through Kaplan-Meier plotter for breast and lung cancers in http://kmplot.com/analysis, using the 2017 version of the database. All biased arrays were excluded. The probed cohorts used in the analyses are indicated in datasets with Affymetrix IDs 204132_s_at (breast cancer cohorts) and 217399_s_at (lung cancer cohorts). Median expression of queried genes was used to separate high/low expression patient groups.
Supplementary Material
Acknowledgments
We thank the members of Y.K.’s lab for insightful discussions, S. Y. Bae and J. Song for technical help in establishing some paclitaxel-resistant cell lines, H. Lee for providing the FFPE All-Prep kit and help in clinical sample processing, and H.-K. Chang, M. Y. Kim, W. D. Heo, D. Helfman, and K. J. Jeong for providing some antibodies, fluorescent dyes, and plasmids. We are also grateful to the Department of Biological Sciences and GSMSE of KAIST and College of Pharmacy of Seoul National University for animal, confocal microscopy, and FACS core facilities. Funding: This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2016R1C1B2009886) and by the KAIST Future Systems Healthcare Project (KAISTHEALTHCARE42) funded by the Korean government Ministry of Science and ICT (MSIT). M.B.D.A. was supported by the KAIST long-term Undergraduate Research Participation (URP) program (18-C-01). Dayeon Kim and J.H. were supported by the NRF grant funded by the MSIT (NRF-2019R1C1C1010482). Author contributions: M.B.D.A. conceived the project; designed, performed, and analyzed most of the experiments; and wrote the manuscript. J.K. and M.B.D.A. mined the GDSC database and processed raw data files. J.-Y.H. and S.K.L. supervised M.B.D.A. in developing drug-resistant cell lines and provided quality controls. S.J.Y., M.-S.L., and S.G.I. developed and provided the 3D culture plates. H.S.R. provided, curated, and prepared all human cancer patient specimens. Donghwa Kim, M.C.P., and S.T. assisted in performing experiments required for the revision. Dayeon Kim and J.H. assisted in setting up the animal experiments. Y.K. supervised the project and wrote the manuscript with M.B.D.A. All authors reviewed the manuscript. Competing interests: S.G.I. is a co-inventor of the polymeric thin film–based 3D culture used in this study and is currently filing a patent for it. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the first and/or corresponding author/s.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaav7416/DC1
Supplementary Methods
Fig. S1. GDSC parameters on selected compounds.
Fig. S2. Resistance potential assay on all parental cell lines used in the study.
Fig. S3. Characterization of paclitaxel-resistant cancer cells for cell cycle progression and collateral resistance to other microtubule-targeting drugs.
Fig. S4. Collateral EGFR-TKI resistance in paclitaxel-resistant cancer cells.
Fig. S5. Collateral gefitinib resistance in paclitaxel-resistant xenograft tumors.
Fig. S6. GDSC-based coresistance ranking of some microtubule-targeting drugs with EGFR-TKIs and validation study using epothilone B–resistant cell lines.
Fig. S7. BrdU incorporation assay on all EGFR-TKI persister models used in the study.
Fig. S8. Characterization of GPs derived from EGFR-TKI hypersensitive cell lines.
Fig. S9. Characterization of EMT profile.
Fig. S10. β-Catenin translocation and invadopodia protrusion are correlated with stemness in GPs derived from paclitaxel-resistant cancer cells.
Fig. S11. Characterization of glycolysis parameters in EGFR-TKI persisters.
Fig. S12. FOXO3a activation is associated with the metastatic propensity of paclitaxel-resistant tumors.
Fig. S13. FOXO3a expression is correlated with therapy relapse breast cancer patients and with drug resistance to various chemotherapy and targeted therapy agents in cancer cell lines.
Fig. S14. Consequences of FOXO3a inhibition in GPs derived from transient and stable paclitaxel-resistant cells.
Fig. S15. FOXO3a affects protein kinase activities of EGFR and downstream signaling to facilitate apoptosis rewiring in PTXR-derived GPs.
Fig. S16. Phenotypic consequences of FOXO3a inhibition on the state of apoptosis and stemness.
Fig. S17. Expression and activity of ABC drug efflux pumps are not required for a more stable secondary EGFR-TKI resistance.
Fig. S18. MET amplification is dispensable for entering gefitinib persistence in paclitaxel-resistant cancer cells.
Fig. S19. Mutant KRAS is dispensable for collateral EGFR-TKI persistence development in paclitaxel-resistant cancer cells.
Fig. S20. Calculated IC50 values.
Table S1. Clinicopathologic information of human breast cancer patients.
Table S2. Primer sequences for qRT-PCR.
REFERENCES AND NOTES
- 1.Gottesman M. M., Lavi O., Hall M. D., Gillet J.-P., Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol. 56, 85–102 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Komarova N. L., Wodarz D., Drug resistance in cancer: Principles of emergence and prevention. Proc. Natl. Acad. Sci. U.S.A. 102, 9714–9719 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garraway L. A., Jänne P. A., Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2, 214–226 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Shekhar M. P. V., Drug resistance: Challenges to effective therapy. Curr. Cancer Drug Targets 11, 613–623 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Pao W., Miller V. A., Politi K. A., Riely G. J., Somwar R., Zakowski M. F., Kris M. G., Varmus H., Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, 225–235 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nazarian R., Shi H., Wang Q., Kong X., Koya R. C., Lee H., Chen Z., Lee M.-K., Attar N., Sazegar H., Chodon T., Nelson S. F., McArthur G., Sosman J. A., Ribas A., Lo R. S., Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M., Rowan A. J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P., Varela I., Phillimore B., Begum S., McDonald N. Q., Butler A., Jones D., Raine K., Latimer C., Santos C. R., Nohadani M., Eklund A. C., Spencer-Dene B., Clark G., Pickering L., Stamp G., Gore M., Szallasi Z., Downward J., Futreal P. A., Swanton C., Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marusyk A., Polyak K., Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 1805, 105–117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaffer S. M., Dunagin M. C., Torborg S. R., Torre E. A., Emert B., Krepler C., Beqiri M., Sproesser K., Brafford P. A., Xiao M., Eggan E., Anastopoulos I. N., Vargas-Garcia C. A., Singh A., Nathanson K. L., Herlyn M., Raj A., Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S. V., Lee D. Y., Li B., Quinlan M. P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M. A., Wong K.-K., Brandstetter K., Wittner B., Ramaswamy S., Classon M., Settleman J., A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tetsu O., Phuchareon J., Eisele D. W., Hangauer M. J., McCormick F., AKT inactivation causes persistent drug tolerance to EGFR inhibitors. Pharmacol. Res. 102, 132–137 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Ramirez M., Rajaram S., Steininger R. J., Osipchuk D., Roth M. A., Morinishi L. S., Evans L., Ji W., Hsu C.-H., Thurley K., Wei S., Zhou A., Koduru P. R., Posner B. A., Wu L. F., Altschuler S. J., Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 7, 106904 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao B., Sedlak J. C., Srinivas R., Creixell P., Pritchard J. R., Tidor B., Lauffenburger D. A., Hemann M. T., Exploiting temporal collateral sensitivity in tumor clonal evolution. Cell 165, 234–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park I. H., Lee K. S., Ro J., Effects of second and subsequent lines of chemotherapy for metastatic breast cancer. Clin. Breast Cancer 15, e55–e62 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Jensen P. B., Holm B., Sorensen M., Christensen I. J., Sehested M., In vitro cross-resistance and collateral sensitivity in seven resistant small-cell lung cancer cell lines: Preclinical identification of suitable drug partners to taxotere, taxol, topotecan and gemcitabin. Br. J. Cancer 75, 869–877 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foo J., Michor F., Evolution of resistance to targeted anti-cancer therapies during continuous and pulsed administration strategies. PLoS Comput. Biol. 5, e1000557 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez J. S., Banerji U., Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 14, 57–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roubaud G., Liaw B. C., Oh W. K., Mulholland D. J., Strategies to avoid treatment-induced lineage crisis in advanced prostate cancer. Nat. Rev. Clin. Oncol. 14, 269–283 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmitt M. W., Loeb L. A., Salk J. J., The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 13, 335–347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonarakis E. S., Armstrong A. J., Evolving standards in the treatment of docetaxel-refractory castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 14, 192–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hata A. N., Niederst M. J., Archibald H. L., Gomez-Caraballo M., Siddiqui F. M., Mulvey H. E., Maruvka Y. E., Ji F., Bhang H.-e. C., Krishnamurthy Radhakrishna V., Siravegna G., Hu H., Raoof S., Lockerman E., Kalsy A., Lee D., Keating C. L., Ruddy D. A., Damon L. J., Crystal A. S., Costa C., Piotrowska Z., Bardelli A., Iafrate A. J., Sadreyev R. I., Stegmeier F., Getz G., Sequist L. V., Faber A. C., Engelman J. A., Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 22, 262–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hangauer M. J., Viswanathan V. S., Ryan M. J., Bole D., Eaton J. K., Matov A., Galeas J., Dhruv H. D., Berens M. E., Schreiber S. L., McCormick F., McManus M. T., Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oxnard G. R., The cellular origins of drug resistance in cancer. Nat. Med. 22, 232–234 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Iorio F., Knijnenburg T. A., Vis D. J., Bignell G. R., Menden M. P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H., Cokelaer T., Greninger P., van Dyk E., Chang H., de Silva H., Heyn H., Deng X., Egan R. K., Liu Q., Mironenko T., Mitropoulos X., Richardson L., Wang J., Zhang T., Moran S., Sayols S., Soleimani M., Tamborero D., Lopez-Bigas N., Ross-Macdonald P., Esteller M., Gray N. S., Haber D. A., Stratton M. R., Benes C. H., Wessels L. F. A., Saez-Rodriguez J., McDermott U., Garnett M. J., A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldonza M. B. D., Hong J.-Y., Alinsug M. V., Song J., Lee S. K., Multiplicity of acquired cross-resistance in paclitaxel-resistant cancer cells is associated with feedback control of TUBB3 via FOXO3a-mediated ABCB1 regulation. Oncotarget 7, 34395–34419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giannakakou P., Gussio R., Nogales E., Downing K. H., Zaharevitz D., Bollbuck B., Poy G., Sackett D., Nicolaou K. C., Fojo T., A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. U.S.A. 97, 2904–2909 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dittrich C., Zifko U., Fazeny B., Fiegl M., Grisold W., Huber H., Vinorelbine after paclitaxel in breast cancer: Cross resistance and cumulative neurotoxicity? Ann. Oncol. 5, 473–474 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Kochanowski K., Morinishi L., Altschuler S. J., Wu L. F., Drug persistence – From antibiotics to cancer therapies. Curr. Opin. Syst. Biol. 10, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vermeulen L., De Sousa E Melo F., van der Heijden M., Cameron K., de Jong J. H., Borovski T., Tuynman J. B., Todaro M., Merz C., Rodermond H., Sprick M. R., Kemper K., Richel D. J., Stassi G., Medema J. P., Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Choi M., Yu S. J., Choi Y., Lee H. R., Lee E., Lee E., Lee Y., Song J., Son J. G., Lee T. G., Kim J. Y., Kang S., Baek J., Lee D., Im S. G., Jon S., Polymer thin film–induced tumor spheroids acquire cancer stem cell–like properties. Cancer Res. 78, 6890–6902 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Sancho P., Barneda D., Heeschen C., Hallmarks of cancer stem cell metabolism. Br. J. Cancer 114, 1305–1312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pusapati R. V., Daemen A., Wilson C., Sandoval W., Gao M., Haley B., Baudy A. R., Hatzivassiliou G., Evangelista M., Settleman J., mTORC1-dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cells. Cancer Cell 29, 548–562 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Chen C.-L., Uthaya Kumar D. B., Punj V., Xu J., Sher L., Tahara S. M., Hess S., Machida K., NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 23, 206–219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milanovic M., Fan D. N. Y., Belenki D., Däbritz J. H. M., Zhao Z., Yu Y., Dörr J. R., Dimitrova L., Lenze D., Monteiro Barbosa I. A., Mendoza-Parra M. A., Kanashova T., Metzner M., Pardon K., Reimann M., Trumpp A., Dörken B., Zuber J., Gronemeyer H., Hummel M., Dittmar G., Lee S., Schmitt C. A., Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Aldonza M. B. D., Hong J.-Y., Lee S. K., Paclitaxel-resistant cancer cell-derived secretomes elicit ABCB1-associated docetaxel cross-resistance and escape from apoptosis through FOXO3a-driven glycolytic regulation. Exp. Mol. Med. 49, e286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naka K., Hoshii T., Muraguchi T., Tadokoro Y., Ooshio T., Kondo Y., Nakao S., Motoyama N., Hirao A., TGF-β–FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463, 676–680 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Pellicano F., Scott M. T., Helgason G. V., Hopcroft L. E. M., Allan E. K., Aspinall-O'Dea M., Copland M., Pierce A., Huntly B. J. P., Whetton A. D., Holyoake T. L., The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells 32, 2324–2337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hui R. C.-Y., Gomes A. R., Constantinidou D., Costa J. R., Karadedou C. T., Fernandez de Mattos S., Wymann M. P., Brosens J. J., Schulze A., Lam E. W.-F., The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol. Cell. Biol. 28, 5886–5898 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottesman M. M., Fojo T., Bates S. E., Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Lainey E., Lainey E., Sébert M., Thépot S., Scoazec M., Bouteloup C., Leroy C., De Botton S., Galluzzi L., Fenaux P., Kroemer G., Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle 11, 4079–4092 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitazaki T., Oka M., Nakamura Y., Tsurutani J., Doi S., Yasunaga M., Takemura M., Yabuuchi H., Soda H., Kohno S., Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 49, 337–343 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Pisco A. O., Brock A., Zhou J., Moor A., Mojtahedi M., Jackson D., Huang S., Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat. Commun. 4, 2467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai G. G. Y., Lim T. H., Lim J., Liew P. J. R., Kwang X. L., Nahar R., Aung Z. W., Takano A., Lee Y. Y., Lau D. P. X., Tan G. S., Tan S. H., Tan W. L., Ang M.-K., Toh C. K., Tan B. S., Devanand A., Too C. W., Gogna A., Ong B. H., Koh T. P. T., Kanesvaran R., Ng Q. S., Jain A., Rajasekaran T., Yuan J., Lim T. K. H., Lim A. S. T., Hillmer A. M., Lim W. T., Iyer N. G., Tam W. L., Zhai W., Tan E.-H., Tan D. S. W., Clonal MET amplification as a determinant of tyrosine kinase inhibitor resistance in epidermal growth factor receptor–mutant non–small-cell lung cancer. J. Clin. Oncol. 37, 876–884 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Kubo T., Yamamoto H., Lockwood W. W., Valencia I., Soh J., Peyton M., Jida M., Otani H., Fujii T., Ouchida M., Takigawa N., Kiura K., Shimizu K., Date H., Minna J. D., Varella-Garcia M., Lam W. L., Gazdar A. F., Toyooka S., MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. Int. J. Cancer 124, 1778–1784 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pecot C. V., Wu S. Y., Bellister S., Filant J., Rupaimoole R., Hisamatsu T., Bhattacharya R., Maharaj A., Azam S., Rodriguez-Aguayo C., Nagaraja A. S., Morelli M. P., Gharpure K. M., Waugh T. A., Gonzalez-Villasana V., Zand B., Dalton H. J., Kopetz S., Lopez-Berestein G., Ellis L. M., Sood A. K., Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol. Cancer Ther. 13, 2876–2885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salgia R., Kulkarni P., The genetic/non-genetic duality of drug ‘resistance’ in cancer. Trends Cancer 4, 110–118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiley C. D., Campisi J., From ancient pathways to aging cells—Connecting metabolism and cellular senescence. Cell Metab. 23, 1013–1021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lazzari C., Bulotta A., Ducceschi M., Viganò M. G., Brioschi E., Corti F., Gianni L., Gregorc V., Historical evolution of second-line therapy in non-small cell lung cancer. Front Med (Lausanne) 4, 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qin Y., Conley A. P., Grimm E. A., Roszik J., A tool for discovering drug sensitivity and gene expression associations in cancer cells. PLOS ONE 12, e0176763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/6/eaav7416/DC1
Supplementary Methods
Fig. S1. GDSC parameters on selected compounds.
Fig. S2. Resistance potential assay on all parental cell lines used in the study.
Fig. S3. Characterization of paclitaxel-resistant cancer cells for cell cycle progression and collateral resistance to other microtubule-targeting drugs.
Fig. S4. Collateral EGFR-TKI resistance in paclitaxel-resistant cancer cells.
Fig. S5. Collateral gefitinib resistance in paclitaxel-resistant xenograft tumors.
Fig. S6. GDSC-based coresistance ranking of some microtubule-targeting drugs with EGFR-TKIs and validation study using epothilone B–resistant cell lines.
Fig. S7. BrdU incorporation assay on all EGFR-TKI persister models used in the study.
Fig. S8. Characterization of GPs derived from EGFR-TKI hypersensitive cell lines.
Fig. S9. Characterization of EMT profile.
Fig. S10. β-Catenin translocation and invadopodia protrusion are correlated with stemness in GPs derived from paclitaxel-resistant cancer cells.
Fig. S11. Characterization of glycolysis parameters in EGFR-TKI persisters.
Fig. S12. FOXO3a activation is associated with the metastatic propensity of paclitaxel-resistant tumors.
Fig. S13. FOXO3a expression is correlated with therapy relapse breast cancer patients and with drug resistance to various chemotherapy and targeted therapy agents in cancer cell lines.
Fig. S14. Consequences of FOXO3a inhibition in GPs derived from transient and stable paclitaxel-resistant cells.
Fig. S15. FOXO3a affects protein kinase activities of EGFR and downstream signaling to facilitate apoptosis rewiring in PTXR-derived GPs.
Fig. S16. Phenotypic consequences of FOXO3a inhibition on the state of apoptosis and stemness.
Fig. S17. Expression and activity of ABC drug efflux pumps are not required for a more stable secondary EGFR-TKI resistance.
Fig. S18. MET amplification is dispensable for entering gefitinib persistence in paclitaxel-resistant cancer cells.
Fig. S19. Mutant KRAS is dispensable for collateral EGFR-TKI persistence development in paclitaxel-resistant cancer cells.
Fig. S20. Calculated IC50 values.
Table S1. Clinicopathologic information of human breast cancer patients.
Table S2. Primer sequences for qRT-PCR.