

Abstract
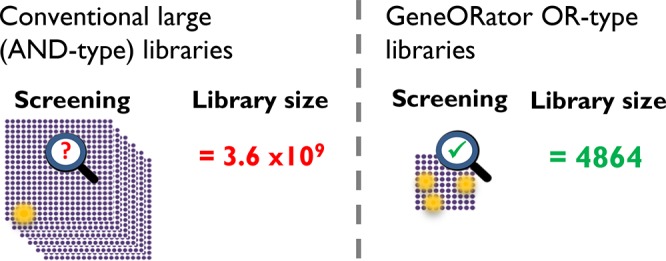
Directed evolution requires the creation of genetic diversity and subsequent screening or selection for improved variants. For DNA mutagenesis, conventional site-directed methods implicitly utilize the Boolean AND operator (creating all mutations simultaneously), producing a combinatorial explosion in the number of genetic variants as the number of mutations increases. We introduce GeneORator, a novel strategy for creating DNA libraries based on the Boolean logical OR operator. Here, a single library is divided into many subsets, each containing different combinations of the desired mutations. Consequently, the effect of adding more mutations on the number of genetic combinations is additive (Boolean OR logic) and not exponential (AND logic). We demonstrate this strategy with large-scale mutagenesis studies, using monoamine oxidase-N (Aspergillus niger) as the exemplar target. First, we mutated every residue in the secondary structure-containing regions (276 out of a total 495 amino acids) to screen for improvements in kcat. Second, combinatorial OR-type libraries permitted screening of diverse mutation combinations in the enzyme active site to detect activity toward novel substrates. In both examples, OR-type libraries effectively reduced the number of variants searched up to 1010-fold, dramatically reducing the screening effort required to discover variants with improved and/or novel activity. Importantly, this approach enables the screening of a greater diversity of mutation combinations, accessing a larger area of a protein’s sequence space. OR-type libraries can be applied to any biological engineering objective requiring DNA mutagenesis, and the approach has wide ranging applications in, for example, enzyme engineering, antibody engineering, and synthetic biology.
Keywords: mutagenesis, directed evolution, protein engineering, synthetic biology, biocatalysis
Natural evolution is based on the random creation of genetic diversity and subsequent selection of a desired fitness.1 Directed evolution attempts to improve on and speed up this process in the laboratory. Genetic diversity is generated for a target gene, enabling the discovery, selection and isolation of variants encoding an improvement in the desired fitness (e.g., increased activity). This process can then be repeated iteratively to improve the properties of the target molecule until an adequate fitness is achieved.2−11 This general process is widely used for the engineering of biocatalysts, enabling the development of enzymes for applications in industrial biotechnology.10,12−15
Since Sewall Wright’s original conception,16 the relationship between a protein’s sequence and its function(s) is often referred to as a “fitness landscape”.17−21 It is conventionally visualized with the position in the sequence space represented via Cartesian x- and y-coordinates and with fitness as a “height”. Proteins are known to exhibit rugged landscapes, where a variety of constraints22 means that sequences of high fitness are surrounded by areas of lower or even negligible activity or fitness.23−27 The objective of directed evolution is therefore to navigate this landscape to discover the best fitnesses possible.10,28 Unfortunately, the size of sequence space (the total number of possible sequences) is vast and impossible to test exhaustively, even for short peptides (full randomization of just 50 amino acids would produce a library of 2050, ∼1 × 1065). As with any combinatorial search problem,29−32 the experimenter therefore needs to devise a strategy that can search efficiently for improved variants while at the same time making libraries of a size that can be screened or selected for in the laboratory.
Some approaches have utilized reduced33 or “smart” library strategies,34−38 which decrease the redundancy of the mixed-base codons used and hence the level of diversity at individual residues. However, even with these methods, the library size quickly becomes too large to test experimentally when looking to mutate multiple amino acids (typically four or more). This problem arises because simultaneous mutation of multiple amino acids leads to a combinatorial explosion, as the size of the search space increases exponentially as the number of residues increases.10,32,39 This is equivalent to the AND operator of Boolean logic (e.g., mutate {residue 1} AND {residue 2} AND {residue 3}, etc.).
Here, we provide an experimental implementation of the Boolean OR function for library generation for directed evolution (e.g., mutate {residue 1} OR {residue 2} OR {residue 3}, etc.). The effect on library size of mutating multiple residues in this way is therefore additive and not multiplicative/exponential. We demonstrate that this strategy can be employed to reduce the library size significantly (often by many orders of magnitude), as well as decreasing its complexity, enabling the mutation of a larger number of regions in the same library. This has the highly desirable effect of significantly reducing the overall size of the library, while still testing all the desired codons and mutations.
We demonstrate the benefit of OR-type libraries through two approaches using monoamine oxidase-N (MAO-N) as the exemplar enzyme target. First, a large-scale mutagenesis approach was adopted, mutating 276 amino acids of MAO-N (of a total of 495 amino acids); these account for every residue known to exhibit a secondary structure. Our approach permitted several (typically up to 12) amino acids to be mutated in a single library without the combinatorial explosion that would occur when using AND-type libraries. We identified multiple variants with increased kcat toward both native and non-native amine substrates, including novel activity for new substrates. Second, we created combinatorial OR-type libraries for a Combinatorial Active-Site Saturation Test (CASTing40,41). Using this approach, 10 active-site residues were mutated simultaneously, such that many different combinations of two-residue mutations were tested in one library. These combinatorial mutations reduced the library size ∼4.4 × 1010-fold compared to simultaneous randomization of all residues (AND mutations). This enabled the screening of a library with more diverse mutations compared to conventional methods, and the rapid discovery of a new variant exhibiting activity toward two novel substrates.
Results and Discussion
2.1. Asymmetric PCR for the Generation of OR-Type Libraries
Numerous studies have utilized asymmetric PCR for the purposes of site-directed mutagenesis.39−43 In this two-step process, the first step consists of an asymmetric PCR that generates a single-stranded DNA (ssDNA) product, created by using an unequal concentration of DNA oligonucleotide primers. The lower concentration (limiting) primer encoding the mutations (termed “mutagenic primer”, MP) becomes depleted during the early cycles of the PCR, after which the corresponding high concentration (excess) primer continues to amplify the amplicon. This generates a ssDNA product encoding all the mutations encoded by the mutagenic primers. Following purification, this product is then used as a “megaprimer” in a second PCR to amplify the full-length gene encoding the library (Supporting Information S1).
An important advantage of using asymmetric PCR is that, given that the mutagenic primers are depleted, it ensures that all mutations encoded by the primers are present in the final library. We exploit this by using multiple different mutagenic primers in a single reaction to create mutations at different positions in the DNA sequence. If these primers anneal to the same position in the DNA, the final library will conform to Boolean OR logic (i.e., each DNA strand encoding a mutation from {MP1} OR {MP2}). All such primers binding the same position on the DNA template are herein referred to as a “set”. For example, for a simple “set” containing three mutagenic primers, the library is therefore composed of DNA strands with mutations from either MP1 OR MP2 OR MP3 (Supporting Information S1). Consequently, upon transformation into cells, each clone from this library will encode a protein variant with these OR-type mutations which can then be screened (Figure 1). Another benefit is that OR-type libraries simplify the generation of multiple variant libraries by condensing the number of samples required for synthesis, for example, combining three different libraries into one tube (following the example in Figure 1). Extending this approach to include multiple sets creates more complex combinatorial OR-type mutations, discussed further below (section 2.3).
Figure 1.
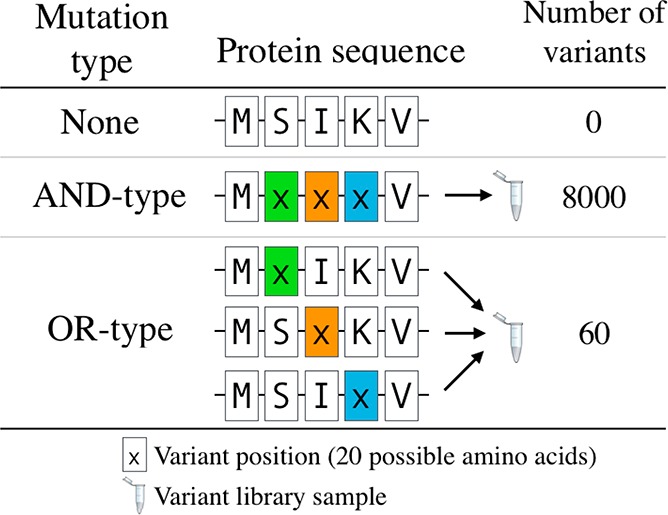
An example of OR-type mutations. When randomizing multiple amino acids (here residues “SIK”, 20 possible amino acids for each position), conventional approaches mutated each residue simultaneously. This “AND-type” mutagenesis approach creates large numbers of variants, as the impact of each position is multiplicative (20 × 20 × 20 = 8000). In contrast, OR-type libraries can randomize any one of these same amino acids, but not all together. In this example the impact of each position is additive (20 + 20 + 20 = 60), thus significantly reducing the size and complexity of the variant library. From another perspective, this OR-type approach is simplifying the generation of multiple libraries by synthesizing three different randomized libraries in one tube.
2.2.1. OR-Type Libraries for Large-Scale Mutagenesis of Monoamine Oxidase-N
MAO-N is an important industrial biocatalyst that oxidizes a variety of primary, secondary, and tertiary amines.42−49 Wild type MAO-N (uniprot: P46882) exhibits strong activity toward primary amine substrates (see section 2.2.3) that are believed to be similar to the native substrates (rates referred to as “wild type speed”). In contrast, the wild type enzyme exhibits very low activity (kcat = 0.17 min–1) toward the primary amine α-methylbenzylamine (α-MBA, chebi:CHEBI:670); however, previous directed evolution studies have generated a variant (I246M/N336S/M348 K/T384N/D385S termed D542,44,46) with a kcat of 154 min–1 for α-MBA. Hence we devised a strategy to seek variants with a “wild type speed” kcat toward the non-native substrate α-MBA.
Our large-scale mutagenesis strategy is guided by the understanding that amino acids throughout the protein structure, often distal to the active site, have a significant effect on the efficiency of catalysis (kcat/Km or kcat).10 Hence, creating mutations throughout the protein structure will enable us to detect those variants with significantly increased kcat for a panel of native and non-native amine substrates.
Given that protein secondary structure often follows a regular binary pattern of polar (P) and nonpolar (NP) residues (e.g., amphiphilic helices can follow a P–NP–P–P–NP–NP–P pattern50,51) one strategy to ensure that the majority of the searched sequence space encodes proteins with similar secondary structure is to follow this semiconservative binary pattern,52 such that the tertiary structure is more-or-less conserved in order to maximize the likelihood of preserving function. Hence, we devised a novel codon mutagenesis approach to increase the proportion of functional protein variants by binary patterning (Supporting Information S2). For example, when Leu is the starting amino acid, we mutated it using the NTN codon (N = A, T, G, or C) to encode Phe, Leu, Ile, Met, or Val. Similarly, small side-chain amino acids were mutated to others with small side-chains (Ala, Gly, Val) and polar residues with H-bonding potential were mutated to other similar residues (Ser, Tyr, Cys, Thr). Secondary structure predictions support the hypothesis that our variants maintain the α-helical and β-sheet content of the native protein, significantly more when compared to full amino acid randomization (Figure 2). Consequently, our strategy is calculated to search a more “functional sequence space”.
Figure 2.
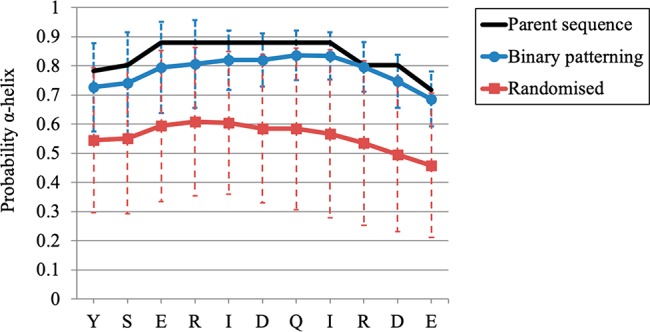
Secondary structure predictions of a binary pattern library. Using α-helix [188]–[198] as an example, every variant encoded by our mutagenesis strategy was calculated for its probability of forming an α-helix (using NetSurfP). For each amino acid the calculated probability is shown (mean and standard deviation), comparing our approach to full randomization using the NNK codon. Our variants are predicted to exhibit similar levels of secondary structure compared to the parent sequence, significantly higher than using full randomization, thus supporting our strategy for maintaining secondary structure in our variants.
In this study, every amino acid in MAO-N D5 exhibiting secondary structure was mutated according to our mutagenesis design (see below), totalling 276 amino acids. Mutagenic primers were limited to mutating three amino acids or less, using our design of ambiguous codons (Supporting Information S2). These primers were used as part of a “set” to mutate single strand α-helices or β-sheets in one library. In one example (Figure 3, Supporting Information S3), four mutagenic primers were created in one set to mutate 11 consecutive codons. Simultaneous mutation of all 11 codons together (the same mutations but using AND-type mutations) would create 5.9 × 1011 genetic combinations, whereas a corresponding OR-type library encodes 5136 combinations, a 1.1 × 108-fold reduction.
Figure 3.

An example of how OR-type libraries were used to mutate the secondary structure of MAO-N in this study. (A) The selected α-helix (residues [188]–[198], 11 amino acids) was divided into four, each mutating 2 or 3 amino acids with a mutagenic primer (MP). The number of variants per residue is highlighted, and these follow our mutagenesis strategy. OR-type libraries produced by this approach encoded 5136 genetic combinations, a 1.1 × 108-fold reduction compared to simultaneous AND-type mutations. (B) The design of DNA oligonucleotides for mutagenesis is shown for this α-helix, aligned to the target amino acid sequence and corresponding DNA sequence.
2.2.2. MAO-N Improved Variants to Non-native Amine Substrates
Using the previously described colony-based screening method to analyze oxidase activity by detection of hydrogen peroxide,42,53 we screened every OR-type library using α-MBA, attempting to improve the kcat toward this non-native substrate. For each library, the top (fastest) colonies were selected and the DNA sequenced. Sequences that showed a clear selection for a new variant (e.g., a mutation selected multiple times) were characterized.
We identified four variants with an elevated kcat compared to that of the D5 variant (Supporting Information S4). One variant, A289V (kcat = 242 min–1), exhibited a 1.6-fold increase to that of its parent D5. We compiled all the screening data to understand the mutability of each mutated amino acid, providing an insight into the in vitro selection of every amino acid mutated in the study (Figure 4). We discovered strong selection for 120 residues, where the amino acid encoded in the parent sequence was invariant. Conversely, many amino acids were tolerant of several different mutations while still maintaining good catalytic activity. In total, of those assessed, 53 residues could encode one other residue, 44 could accommodate two mutations, and 50 could accommodate three or more mutations. High-frequency selection for a new mutation was discovered for nine amino acids and each of these mutations was characterized (above). We also found that a strong selection for native (parent D5) residues was more frequently observed for amino acids closer to the protein core and to the FAD cofactor.
Figure 4.

Enzymatic improvements for selected MAO-N variants. (A) The most significant improved activity to the primary target non-native substrate α-methylbenzylamine was demonstrated by the D5 variant A289V, exhibiting a 1.6-fold increase to that of D5. (B) Every amino acid mutated in this study is shown, with its color denoting whether it (i) showed strong selection for the wild-type amino acid (red); (ii) exhibited robustness, where at least one alternative mutation could be accommodated while still maintaining activity (green); and (iii) exhibited strong selection for a new mutation that increased kcat (blue). (C) Amino acid selection (as in panel B) showing the secondary structure elements. Images generated using PyMol using MAO-N D5 structure (2vvm). (D) Improved activity to three native amine substrates was shown by the D5 variant F128L, with a kcat between 1.6 to 2.25-fold higher than the WT, and 2.2 and 3-fold higher than the parent D5 variant.
2.2.3. MAO-N Activity to Native Primary Amine Substrates
In addition to characterizing MAO-N variants toward α-MBA, we also tested our variants against the native WT substrates, where several variants also exhibited increased activity (Figure 4, Supporting Information S5). Interestingly, the best α-MBA variant (A289V) was not the fastest toward these substrates, but F128L was faster for all three “native” substrates. F128L activity to N-amylamine (AA, chebi:CHEBI:74848, 655 min–1) is the highest kcat published for MAO-N for any substrate to date, 1.7-fold higher than the WT and 3-fold faster than its parent D5 variant.
2.2.4. MAO-N Activity to Novel Substrates
No published MAO-N variants to date (including WT and D5) exhibit detectable activity toward the primary amine cyclohexylamine (CHA, chebi:CHEBI:15773). However, we detected activity (kcat = 17 min–1, Supporting Information S5) for one of our variants (A266V). To improve this activity, we created double mutants combining A266V with other mutations found in this study (both neutral and positive for activity). Interestingly, combining A266V with other mutations known to improve activity for other substrates (e.g., F128L) did not improve activity for CHA (kcat = 15 min–1). However, combining A266V with C50T did improve activity over 2-fold (kcat = 38 min–1). Interestingly, the C50T mutation alone does not improve activity (kcat toward α-MBA, AA, BTA, and BZA is not increased), thus demonstrating an unpredictable epistatic interaction between A266V and C50T. Given that neither C50 nor A266 are positioned in the active site (29 and 16 Å from the FAD amine where catalysis occurs, respectively, Supporting Information S6), such data show that residues distal to the active site also contribute specificity for substrates, and that mutagenesis of these residues can yield variants with activity toward novel substrates.54
2.3. Active Site Mutagenesis Using Combinatorial OR-Type Mutations
We envisaged that the benefit of OR-type mutations becomes more significant when this method is applied to screening multiple combinatorial mutations, given that its additive nature prevents the combinatorial explosion of mutation combinations associated with conventional AND-type libraries. It is worth noting that combinatorial OR-type mutations can also be described as AND–OR mutations. To demonstrate this, we created OR-type combinatorial mutations for 10 amino acids in and around the active site of MAO-N for CASTing. The residues were divided into two sets (each containing five amino acids) and the megaprimers for each set were pooled together in the second PCR step to create combinatorial OR-type mutations (Figure 5, Supporting Information S7). Each amino acid was mutated using the NNK codon (32 possible combinations encoding all 20 amino acids). Consequently, in this CASTing library every amino acid substitution for all five amino acids in set 1 was mutated with every amino acid substitution in set 2. Mutation of all 10 amino acids together (AND-type library) would create ∼1 × 1015 codon combinations (= 3210), whereas our library encodes 25600 combinations, a 4.4 × 1010-fold reduction in DNA library size. Alternatively, to recreate each of these mutation combinations without OR-type libraries would require the synthesis of 25 separate libraries. This demonstrates the benefit of combinatorial OR-type mutations for the screening of many mutation combinations, significantly reducing the experimental effort of creating all the different mutations separately. Effectively this strategy permits the screening of a more diverse number of mutation combinations quickly in the search for improved and novel enzyme function.
Figure 5.
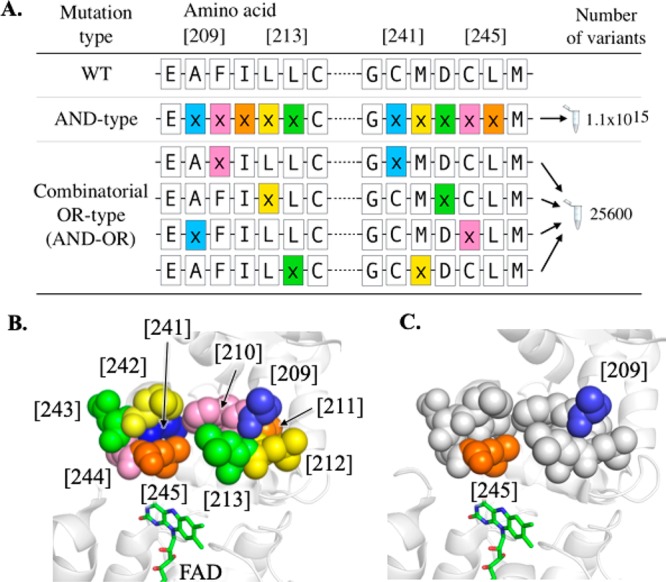
Combinatorial OR-type libraries for CASTing. (A) OR-type mutations at two separate positions in a sequence generates combinatorial OR-type mutations, where all different combinations of mutations from set 1 and set 2 are created, such that optimal paired mutations can be discovered. Simultaneous mutagenesis using the NNK codon using conventional AND-type mutations produces over 1015 genetic combinations, while the corresponding combinatorial OR-type library encoded 25600 combinations, a 4.4 × 1010-fold reduction in library size. (B) Sets 1 (residues [209]–[213]) and 2 ([241]–[245]) were selected as they sit on either side of the MAO-N active site channel. Both sets contained five mutagenic primers, each randomizing one amino acid. The library created every mutation combination between sets 1 and 2, that is, {[MP1] AND [MP1]} OR {[MP1] AND [MP2]}, etc. (C) The “hit” combination, exhibiting novel activity to non-native substrates, encoded mutations at the [1] (A209S) and [5] (L245C) positions.
Screening of the CASTing library identified a new variant (D5 A209S/L245C) with novel activity to two non-native substrates (1-(3-bromophenyl)ethan-1-amine and 1-(3-methoxyphenyl)ethan-1-amine; Supporting Information S8).55 These mutations were encoded in positions [1] and [5] in sets 1 and 2 (respectively, Figure 5), a combination that could not realistically have been predicted by structural or sequence analysis, thus demonstrating the benefit of our approach.
Conclusions
In this study we demonstrate a methodology to create a novel type of variant library, whereby multiple discrete DNA regions can be mutated in an OR-type fashion. The result is that each region contributes an additive effect to the total library size (Boolean OR logic), in contrast to conventional site-directed mutagenesis methods (utilizing AND logic) where multiple mutations create a combinatorial explosion. Boolean logic rules have recently been exploited in different biological applications, most notably in synthetic biology to provide control over cellular systems and pathways. Siuti et al.56 have implemented logic gate functions in E. coli using recombinases, while others utilize small molecules.57,58 However, to our knowledge GeneORator is the first application of Boolean OR logic for the construction of variant protein libraries for directed evolution.
Here, we exploit OR-type libraries to implement a novel mutagenesis scheme based on the binary patterning feature of protein secondary structure. We devised an ambiguous codon design strategy and used this to mutate every amino acid in the secondary structure-containing regions of MAO-N. This design sought to conserve the pattern of polar and nonpolar residues present in the MAO-N sequence, an approach predicted to improve the proportion of variants with the secondary structure required to create the tertiary fold required for catalysis. Taken together, our mutagenesis methodology and library design enabled large-scale mutagenesis studies to improve the search of “functional sequence space”, in a way that is not economic (nor feasible) using existing approaches. Regardless of the codon mutagenesis strategy, we have demonstrated that our experimental approach was efficient at generating the designed OR-type mutations for screening. A similar strategy could be therefore be employed for different enzyme targets for which multiple mutations are to be created and screened.
In this study we discovered several residues distal to the active site that conferred an increase in kcat in a manner that was not predictable from the knowledge of an amino acid sequence, tertiary structure, or catalytic mechanism. Given that these mutations are not predicted to alter the protein’s basic secondary structure, it is expected that these mutations improve activity through the alteration of protein dynamics during catalysis, rather than via major ground-state structural changes (see also refs (59 and 60)). Recently, Curado-Carballada et al.61 described molecular dynamics simulations of MAO-N wild type and D5 variant, describing the presence of previously unknown conformations during catalysis. Interestingly, the F128L variant identified in this study is located close to a β-hairpin loop, predicted to be involved in the recognition of the different substrates.
Given the knowledge of which variants had been screened in each library we obtained sequence-activity data for every library that was screened. Combining these data with that of the mutations that increase kcat provides important information on the selection pressure exerted on every residue in the secondary structure during our screening. Interestingly, combining multiple mutations known to improve activity together did not yield an additive improvement; thus, no double mutants exhibited an increased kcat for α-MBA above the single mutants. Accordingly, the highest activity variant for native substrates (F128L) had a neutral effect on activity for CHA (variant D5 A266V/F128L) while the neutral mutation C50T had an improved effect on CHA activity (variant D5 A266V/C50T). These data serve to illustrate the highly epistatic nature of this protein’s fitness landscape.
There is widespread interest in exploiting in silico learning algorithms for biological applications. Machine learning provides the opportunity to learn complex sequence-activity relationships and to predict variants with improved fitness.17 Principled search algorithms such as “protein sequence activity relationships” (ProSAR) have been used to help engineer enzymes by creating partial least-squares (PLS) regression models, and recent updates may accommodate epistatic interactions between two residues.28,62,63 We envisage that improved technology in DNA library synthesis and “deep mutational scanning”64,65 will empower learning algorithms to predict proteins with improved fitness for a variety of directed evolution applications. Given the complexity of protein sequence–activity relationships, especially the importance of epistasis, learning algorithms require the ability to design specific yet complex DNA libraries for screening. GeneORator is capable of creating these libraries in a way that does not suffer from the combinatorial explosion associated with conventional libraries, and is a powerful tool in the rapid discovery of new biocatalysts with improved and novel activity.
Materials and Methods
Design of Oligonucleotide Primers for OR-Type Libraries
The MAO-N D5 gene (uniprot:P46882) was designed using GeneGenie66 and synthesized using the SpeedyGenes gene synthesis method, as previously described.67,68 In the design of OR-type libraries, first the number of target regions and the number of codons to be mutated were identified (typically up to four target regions, each containing up to three codon mutations). Flanking sequences to these target regions were selected, such that the annealing temperature (Tm) was predicted to be 60 °C at both the 5′ and 3′ termini. The relevant ambiguous codons were designed by CodonGenie38 then inserted into the oligonucleotide sequence, depending on the amino acids present in the parent D5 sequence. One mutagenic primer was designed for each target region, such that a set of primers encoded the same 5′ and 3′ flanking sequences but each different target region mutations. Corresponding end PCR (nonmutagenic) primers were also designed with a predicted annealing temperature (Tm) of 60 °C for the 5′ and 3′ termini of the gene.
Synthesis of OR-Type Libraries
DNA oligonucleotides were synthesized by Integrated DNA Technologies. For asymmetric PCR, the reaction contained 25 nM mutagenic (limiting, forward read) primer and 500 nM end (excess, reverse read) primer, with 0.5 ng μL–1 template (MAO-N D5), 0.2 mM dNTP mix, Q5 reaction buffer, and 0.02 U μL–1 Q5 hot-start high-fidelity polymerase (New England Biolabs) in 50 μL total volume. The PCR consisted of denaturation at 98 °C for 30 s, then 25 cycles of 98 °C for 20 s, 60 °C for 20 s, and 72 °C for 40 s. PCR products containing ssDNA were purified using a PCR purification kit (Qiagen).
For symmetric PCR to assemble the full gene, the ssDNA PCR product from asymmetric PCR was used as the megaprimer (reverse read) together with the relevant end primer (forward read). The reaction contained 16.5 μL of megaprimer, 500 nM end primer, and other reagents as above. The PCR consisted of denaturation at 98 °C for 30 s, then 25 cycles of 98 °C for 30 s, 60 °C for 20 s, and 72 °C for 40 s. For combinatorial OR-type libraries, megaprimers were created for each set of mutations and pooled together in the in the PCR above (also see ref (69)). PCR products were visualized and purified by gel electrophoresis and gel extraction (Qiagen kit) (Supporting Information S9). Purified libraries were ligated into a linearized expression vector (pET16b, Novagen) using the In-Fusion cloning kit (Clontech), following the manufacturers’ protocol. Quality control of the synthesized libraries was performed using Sanger sequencing (Eurofins) and next-generation DNA sequencing (Supporting Information S10–S11).
Screening for MAO-N Activity
Ligation reactions were transformed into E. coli competent cells (T7 express, New England Biolabs) and spread onto an LB agar with 100 μg mL–1 ampicillin covered with a Hybond-N membrane (Amersham Biosciences). Following incubation overnight at 30 °C, the membrane containing single colonies was transferred to an LB agar plate (100 μg mL–1 ampicillin and 1 mM IPTG) and incubated for 2 h at 30 °C. Oxidase activity was then assayed following the protocol outlined previously.39,53 Briefly, the membrane containing colonies was transferred to a membrane soaked in 0.1 mg mL–1 HRP (Sigma) and 100 mM potassium phosphate pH 7.7 for 30 min (the prescreen). Colonies were then transferred to a membrane soaked in 0.1 mg mL–1 HRP, DAB (Sigma), 2.5 mM α-methylbenzylamine (Sigma), and 100 mM potassium phosphate pH 7.7. Oxidase activity was observed by the formation of a brown DAB precipitate.
Colonies that exhibited the fastest color change were picked and inoculated into LB (100 μg mL–1 ampicillin) and grown overnight (37 °C, 180 rpm), and the plasmids were extracted using a plasmid miniprep kit (Qiagen). The sequencing of variants was performed as above.
Expression and Purification of MAO-N
Selected variants were overexpressed by BL21 (DE3) E. coli strain in 700 mL of LB medium with 100 μg mL–1 ampicillin. A 0.5 mM sample of IPTG was introduced to the culture when OD600 reached 0.6, and the culture was incubated at 25 °C, 180 rpm. Cells were harvested after 16–20 h and purified using 5 mL of Histrap FF crude column (GE Healthcare) with an AKTA Explorer 100 protein purification system as described.48
Liquid Phase Kinetic Assay
Different amine stock solutions including α-methylbenzylamine, N-amylamine, butylamine, benzylamine, or cyclohexylamine (all from Sigma) were prepared in 0.1 M potassium phosphate pH 7.7. The final concentration range of the substrate was between 0.5 mM and 100 mM. A colorimetric assay solution was made up by dissolving Pyrogallol red (0.3 mM final concentration) in 0.1 M potassium phosphate, pH 7.7. The assay was conducted by combining 35 μL of substrate solution, 50 μL of Pyrogallol red solution and 5 μL of horseradish peroxidase (1 mg mL–1) in a flat bottom 96-well plate and started by adding 110 μL of purified MAO-N. Assay progress was monitored at 550 nm at 25 °C in a Molecular Devices Spectramex M2 plate reader. The data were analyzed using Prism7 (GraphPad), which was also used to calculate the kinetic parameters kcat, kM, and vmax.
Data Availability
The data sets generated during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
We acknowledge funding from the Biotechnology and Biological Sciences Research Council (BBSRC) under Grants BB/M017702/1, “Centre for synthetic biology of fine and speciality chemicals (SYNBIOCHEM)” and BB/K00199X/1, as well as a Proof of Concept grant from the BBSRC BIOCATNET NIBB. This is a contribution from the Manchester Centre for Synthetic Biology of Fine and Speciality Chemicals (SYNBIOCHEM) and we gratefully acknowledge the support of the centre for parts of this study.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.9b00063.
Figures and tables: diagrams for mutagenesis, enzyme variant kinetics data and sequencing data (PDF)
Author Present Address
# Dept of Biochemistry, Institute of Integrative Biology, University of Liverpool, Crown Street, Liverpool L69 7ZB, United Kingdom.
The authors declare the following competing financial interest(s): AC is the named inventor on a submitted patent describing the GeneORator method.
Supplementary Material
References
- Cobb R. E.; Chao R.; Zhao H. (2013) Directed Evolution: Past, Present, and Future. AIChE J. 59 (5), 1432–1440. 10.1002/aic.13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt C.; Kauffman S.; Wang Z.-G. (2001) Rational Evolutionary Design: The Theory of in Vitro Protein Evolution. Adv. Protein Chem. 55, 79–160. 10.1016/S0065-3233(01)55003-2. [DOI] [PubMed] [Google Scholar]
- Arnold F. H. (2006) Fancy Footwork in the Sequence Space Shuffle. Nat. Biotechnol. 24 (3), 328–330. 10.1038/nbt0306-328. [DOI] [PubMed] [Google Scholar]
- Jäckel C.; Kast P.; Hilvert D. (2008) Protein Design by Directed Evolution. Annu. Rev. Biophys. 37 (1), 153–173. 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- Turner N. J. (2009) Directed Evolution Drives the next Generation of Biocatalysts. Nat. Chem. Biol. 5 (8), 567–573. 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Huisman G. W.; Kazlauskas R. J.; Lutz S.; Moore J. C.; Robins K. (2012) Engineering the Third Wave of Biocatalysis. Nature 485 (7397), 185–194. 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- Reetz M. T. (2013) Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J. Am. Chem. Soc. 135 (34), 12480–12496. 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]
- Romero P. A.; Krause A.; Arnold F. H. (2013) Navigating the Protein Fitness Landscape with Gaussian Processes. Proc. Natl. Acad. Sci. U. S. A. 110 (3), E193–E201. 10.1073/pnas.1215251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F.; Zhu L.; Schwaneberg U. (2015) Directed Evolution 2.0: Improving and Deciphering Enzyme Properties. Chem. Commun. 51 (48), 9760–9772. 10.1039/C5CC01594D. [DOI] [PubMed] [Google Scholar]
- Currin A.; Swainston N.; Day P. J.; Kell D. B. (2015) Synthetic Biology for the Directed Evolution of Protein Biocatalysts: Navigating Sequence Space Intelligently. Chem. Soc. Rev. 44 (5), 1172–1239. 10.1039/C4CS00351A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza R. O. M. A.; Miranda L. S. M.; Bornscheuer U. T. (2017) A Retrosynthesis Approach for Biocatalysis in Organic Synthesis. Chem. - Eur. J. 23 (50), 12040–12063. 10.1002/chem.201702235. [DOI] [PubMed] [Google Scholar]
- Dalby P. A. (2011) Strategy and Success for the Directed Evolution of Enzymes. Curr. Opin. Struct. Biol. 21 (4), 473–480. 10.1016/j.sbi.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Cobb R. E.; Si T.; Zhao H. (2012) Directed Evolution: An Evolving and Enabling Synthetic Biology Tool. Curr. Opin. Chem. Biol. 16 (3–4), 285–291. 10.1016/j.cbpa.2012.05.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N. J.; Truppo M. D. (2013) Biocatalysis Enters a New Era. Curr. Opin. Chem. Biol. 17 (2), 212–214. 10.1016/j.cbpa.2013.02.026. [DOI] [PubMed] [Google Scholar]
- Wang J.; Li G.; Reetz M. T. (2017) Enzymatic Site-Selectivity Enabled by Structure-Guided Directed Evolution. Chem. Commun. 53 (28), 3916–3928. 10.1039/C7CC00368D. [DOI] [PubMed] [Google Scholar]
- Wright S. (1932) The Roles of Mutation, Inbreeding, Crossbreeding, and Selection in Evolution. 6th Intl. Congress Gene. 1, 356. [Google Scholar]
- Knight C. G.; Platt M.; Rowe W.; Wedge D. C.; Khan F.; Day P. J. R.; McShea A.; Knowles J.; Kell D. B. (2009) Array-Based Evolution of DNA Aptamers Allows Modelling of an Explicit Sequence-Fitness Landscape. Nucleic Acids Res. 37 (1), e6 10.1093/nar/gkn899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P. A.; Arnold F. H. (2009) Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 10 (12), 866–876. 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Qian W.; Maclean C. J.; Zhang J. (2016) The Fitness Landscape of a TRNA Gene. Science 352 (6287), 837–840. 10.1126/science.aae0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues J. V.; Bershtein S.; Li A.; Lozovsky E. R.; Hartl D. L.; Shakhnovich E. I. (2016) Biophysical Principles Predict Fitness Landscapes of Drug Resistance. Proc. Natl. Acad. Sci. U. S. A. 113 (11), E1470–E1478. 10.1073/pnas.1601441113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisyan K. S.; Bolotin D. A.; Meer M. V.; Usmanova D. R.; Mishin A. S.; Sharonov G. V.; Ivankov D. N.; Bozhanova N. G.; Baranov M. S.; Soylemez O.; et al. (2016) Local Fitness Landscape of the Green Fluorescent Protein. Nature 533 (7603), 397–401. 10.1038/nature17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth C. L.; Gong S.; Blundell T. L. (2009) Structural and Functional Constraints in the Evolution of Protein Families. Nat. Rev. Mol. Cell Biol. 10 (10), 709–720. 10.1038/nrm2762. [DOI] [PubMed] [Google Scholar]
- Kauffman S. A.; Johnsen S. (1991) Coevolution to the Edge of Chaos: Coupled Fitness Landscapes, Poised States, and Coevolutionary Avalanches. J. Theor. Biol. 149 (4), 467–505. 10.1016/S0022-5193(05)80094-3. [DOI] [PubMed] [Google Scholar]
- Kauffman S. A. (1993) The Origins of Order: Self-Organization and Selection in Evolution, Oxford University Press, New York. [Google Scholar]
- Hayashi Y.; Aita T.; Toyota H.; Husimi Y.; Urabe I.; Yomo T. (2006) Experimental Rugged Fitness Landscape in Protein Sequence Space. PLoS One 1 (1), e96 10.1371/journal.pone.0000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S. A.; Weinberger E. D. (1989) The NK Model of Rugged Fitness Landscapes and Its Application to Maturation of the Immune Response. J. Theor. Biol. 141 (2), 211–245. 10.1016/S0022-5193(89)80019-0. [DOI] [PubMed] [Google Scholar]
- Kondrashov D. A.; Kondrashov F. A. (2015) Topological Features of Rugged Fitness Landscapes in Sequence Space. Trends Genet. 31 (1), 24–33. 10.1016/j.tig.2014.09.009. [DOI] [PubMed] [Google Scholar]
- Fox R. J.; Huisman G. W. (2008) Enzyme Optimization: Moving from Blind Evolution to Statistical Exploration of Sequence–Function Space. Trends Biotechnol. 26 (3), 132–138. 10.1016/j.tibtech.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Voigt C. A.; Mayo S. L.; Arnold F. H.; Wang Z.-G. (2001) Computational Method to Reduce the Search Space for Directed Protein Evolution. Proc. Natl. Acad. Sci. U. S. A. 98 (7), 3778–3783. 10.1073/pnas.051614498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corne D., Dorigo M., and Glover F. (1999) New Ideas in Optimization, McGraw-Hill. [Google Scholar]
- Knowles J. (2009) Closed-Loop Evolutionary Multiobjective Optimization. IEEE Comput. Intell. Mag. 4 (3), 77–91. 10.1109/MCI.2009.933095. [DOI] [Google Scholar]
- Kell D. B. (2012) Scientific Discovery as a Combinatorial Optimisation Problem: How Best to Navigate the Landscape of Possible Experiments?. BioEssays 34 (3), 236–244. 10.1002/bies.201100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reetz M. T.; Kahakeaw D.; Lohmer R. (2008) Addressing the Numbers Problem in Directed Evolution. ChemBioChem 9 (11), 1797–1804. 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- Tang L.; Gao H.; Zhu X.; Wang X.; Zhou M.; Jiang R. (2012) Construction of “Small-Intelligent” Focused Mutagenesis Libraries Using Well-Designed Combinatorial Degenerate Primers. BioTechniques 52 (3), 149–158. 10.2144/000113820. [DOI] [PubMed] [Google Scholar]
- Sebestova E.; Bendl J.; Brezovsky J.; Damborsky J. (2014) Computational Tools for Designing Smart Libraries. Methods Mol. Biol. 1179, 291–314. 10.1007/978-1-4939-1053-3_207. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zheng K.; Zheng H.; Nie H.; Yang Z.; Tang L. (2014) DC-Analyzer-Facilitated Combinatorial Strategy for Rapid Directed Evolution of Functional Enzymes with Multiple Mutagenesis Sites. J. Biotechnol. 192A, 102–107. 10.1016/j.jbiotec.2014.10.023. [DOI] [PubMed] [Google Scholar]
- Wang X.; Lin H.; Zheng Y.; Feng J.; Yang Z.; Tang L. (2015) MDC-Analyzer-Facilitated Combinatorial Strategy for Improving the Activity and Stability of Halohydrin Dehalogenase from Agrobacterium Radiobacter AD1. J. Biotechnol. 206, 1–7. 10.1016/j.jbiotec.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Swainston N.; Currin A.; Green L.; Breitling R.; Day P. J.; Kell D. B. (2017) CodonGenie: Optimised Ambiguous Codon Design Tools. PeerJ. Comput. Sci. 3, e120 10.7717/peerj-cs.120. [DOI] [Google Scholar]
- Moore J. C.; Jin H. M.; Kuchner O.; Arnold F. H. (1997) Strategies for the in Vitro Evolution of Protein Function: Enzyme Evolution by Random Recombination of Improved Sequences. J. Mol. Biol. 272 (3), 336–347. 10.1006/jmbi.1997.1252. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Wang L.-W.; Bocola M. (2006) Directed Evolution of Enantioselective Enzymes: Iterative Cycles of CASTing for Probing Protein-Sequence Space. Angew. Chem., Int. Ed. 45 (8), 1236–1241. 10.1002/anie.200502746. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Carballeira J. D.; Peyralans J.; Höbenreich H.; Maichele A.; Vogel A. (2006) Expanding the Substrate Scope of Enzymes: Combining Mutations Obtained by CASTing. Chem. - Eur. J. 12 (23), 6031–6038. 10.1002/chem.200600459. [DOI] [PubMed] [Google Scholar]
- Alexeeva M.; Enright A.; Dawson M. J.; Mahmoudian M.; Turner N. J. (2002) Deracemization of α-Methylbenzylamine Using an Enzyme Obtained by In Vitro Evolution. Angew. Chem., Int. Ed. 41 (17), 3177–3180. . [DOI] [PubMed] [Google Scholar]
- Atkin K. E.; Reiss R.; Koehler V.; Bailey K. R.; Hart S.; Turkenburg J. P.; Turner N. J.; Brzozowski A. M.; Grogan G. (2008) The Structure of Monoamine Oxidase from Aspergillus Niger Provides a Molecular Context for Improvements in Activity Obtained by Directed Evolution. J. Mol. Biol. 384 (5), 1218–1231. 10.1016/j.jmb.2008.09.090. [DOI] [PubMed] [Google Scholar]
- Dunsmore C. J.; Carr R.; Fleming T.; Turner N. J. (2006) A Chemo-Enzymatic Route to Enantiomerically Pure Cyclic Tertiary Amines. J. Am. Chem. Soc. 128 (7), 2224–2225. 10.1021/ja058536d. [DOI] [PubMed] [Google Scholar]
- Bailey K. R.; Ellis A. J.; Reiss R.; Snape T. J.; Turner N. J. (2007) A Template-Based Mnemonic for Monoamine Oxidase (MAO-N) Catalyzed Reactions and Its Application to the Chemo-Enzymatic Deracemisation of the Alkaloid (±)-Crispine A. Chem. Commun. 35, 3640. 10.1039/b710456a. [DOI] [PubMed] [Google Scholar]
- Rowles I.; Malone K. J.; Etchells L. L.; Willies S. C.; Turner N. J. (2012) Directed Evolution of the Enzyme Monoamine Oxidase (MAO-N): Highly Efficient Chemo-Enzymatic Deracemisation of the Alkaloid (±)-Crispine A. ChemCatChem 4 (9), 1259–1261. 10.1002/cctc.201200202. [DOI] [Google Scholar]
- Ghislieri D.; Houghton D.; Green A. P.; Willies S. C.; Turner N. J. (2013) Monoamine Oxidase (MAO-N) Catalyzed Deracemization of Tetrahydro-β-Carbolines: Substrate Dependent Switch in Enantioselectivity. ACS Catal. 3 (12), 2869–2872. 10.1021/cs400724g. [DOI] [Google Scholar]
- O’Reilly E.; Iglesias C.; Ghislieri D.; Hopwood J.; Galman J. L.; Lloyd R. C.; Turner N. J. (2014) A Regio- and Stereoselective ω-Transaminase/Monoamine Oxidase Cascade for the Synthesis of Chiral 2,5-Disubstituted Pyrrolidines. Angew. Chem., Int. Ed. 53 (9), 2447–2450. 10.1002/anie.201309208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Yao P.; Gong R.; Li J.; Liu P.; Lonsdale R.; Wu Q.; Lin J.; Zhu D.; Reetz M. T. (2017) Simultaneous Engineering of an Enzyme’s Entrance Tunnel and Active Site: The Case of Monoamine Oxidase MAO-N. Chem. Sci. 8 (5), 4093–4099. 10.1039/C6SC05381E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamtekar S.; Schiffer J. M.; Xiong H.; Babik J. M.; Hecht M. H. (1993) Protein Design by Binary Patterning of Polar and Nonpolar Amino Acids. Science 262 (5140), 1680–1685. 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- West M. W.; Hecht M. H. (1995) Binary Patterning of Polar and Nonpolar Amino Acids in the Sequences and Structures of Native Proteins. Protein Sci. 4 (10), 2032–2039. 10.1002/pro.5560041008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley L. H., Thumfort P. P., and Hecht M. H. (2006) De Novo Proteins from Binary-Patterned Combinatorial Libraries, in Protein Design; Methods in Molecular Biology (Guerois R., and Paz M. L., Eds.) Vol. 340, pp 53–69, Humana Press. [DOI] [PubMed] [Google Scholar]
- Alexeeva M.; Carr R.; Turner N. J. (2003) Directed Evolution of Enzymes: New Biocatalysts for Asymmetric Synthesis. Org. Biomol. Chem. 1 (23), 4133–4137. 10.1039/b311055a. [DOI] [PubMed] [Google Scholar]
- Morley K. L.; Kazlauskas R. J. (2005) Improving Enzyme Properties: When Are Closer Mutations Better?. Trends Biotechnol. 23 (5), 231–237. 10.1016/j.tibtech.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Herter S.; Medina F.; Wagschal S.; Benhaïm C.; Leipold F.; Turner N. J. (2018) Mapping the Substrate Scope of Monoamine Oxidase (MAO-N) as a Synthetic Tool for the Enantioselective Synthesis of Chiral Amines. Bioorg. Med. Chem. 26 (7), 1338–1346. 10.1016/j.bmc.2017.07.023. [DOI] [PubMed] [Google Scholar]
- Siuti P.; Yazbek J.; Lu T. K. (2013) Synthetic Circuits Integrating Logic and Memory in Living Cells. Nat. Biotechnol. 31 (5), 448–452. 10.1038/nbt.2510. [DOI] [PubMed] [Google Scholar]
- Miyamoto T.; Razavi S.; DeRose R.; Inoue T. (2013) Synthesizing Biomolecule-Based Boolean Logic Gates. ACS Synth. Biol. 2 (2), 72–82. 10.1021/sb3001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Qian K.; Huang Y.; Jin N.; Lai H.; Zhang T.; Li C.; Zhang C.; Bi X.; Wu D.; et al. (2015) SynBioLGDB: A Resource for Experimentally Validated Logic Gates in Synthetic Biology. Sci. Rep. 5, 8090. 10.1038/srep08090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Osés G.; Osuna S.; Gao X.; Sawaya M. R.; Gilson L.; Collier S. J.; Huisman G. W.; Yeates T. O.; Tang Y.; Houk K. N. (2014) The Role of Distant Mutations and Allosteric Regulation on LovD Active Site Dynamics. Nat. Chem. Biol. 10 (6), 431–436. 10.1038/nchembio.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Rivera A.; Garcia-Borràs M.; Osuna S. (2017) Computational Tools for the Evaluation of Laboratory-Engineered Biocatalysts. Chem. Commun. 53 (2), 284–297. 10.1039/C6CC06055B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curado-Carballada C.; Feixas F.; Iglesias-Fernández J.; Osuna S. (2019) Hidden Conformations in Aspergillus Niger Monoamine Oxidase Are Key for Catalytic Efficiency. Angew. Chem., Int. Ed. 58 (10), 3097–3101. 10.1002/anie.201812532. [DOI] [PubMed] [Google Scholar]
- Fox R. J.; Davis S. C.; Mundorff E. C.; Newman L. M.; Gavrilovic V.; Ma S. K.; Chung L. M.; Ching C.; Tam S.; Muley S.; et al. (2007) Improving Catalytic Function by ProSAR-Driven Enzyme Evolution. Nat. Biotechnol. 25 (3), 338–344. 10.1038/nbt1286. [DOI] [PubMed] [Google Scholar]
- Berland M.; Offmann B.; André I.; Remaud-Siméon M.; Charton P. (2014) A Web-Based Tool for Rational Screening of Mutants Libraries Using ProSAR. Protein Eng., Des. Sel. 27 (10), 375–381. 10.1093/protein/gzu035. [DOI] [PubMed] [Google Scholar]
- Fowler D. M.; Stephany J. J.; Fields S. (2014) Measuring the Activity of Protein Variants on a Large Scale Using Deep Mutational Scanning. Nat. Protoc. 9 (9), 2267–2284. 10.1038/nprot.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starita L. M.; Fields S. (2015) Deep Mutational Scanning: A Highly Parallel Method to Measure the Effects of Mutation on Protein Function. Cold Spring Harb. Protoc. 2015 (8), pdb.top077503. 10.1101/pdb.top077503. [DOI] [PubMed] [Google Scholar]
- Swainston N.; Currin A.; Day P. J.; Kell D. B. (2014) GeneGenie: Optimized Oligomer Design for Directed Evolution. Nucleic Acids Res. 42 (W1), W395–W400. 10.1093/nar/gku336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currin A.; Swainston N.; Day P. J.; Kell D. B. (2014) SpeedyGenes: An Improved Gene Synthesis Method for the Efficient Production of Error-Corrected, Synthetic Protein Libraries for Directed Evolution. Protein Eng., Des. Sel. 27 (9), 273–280. 10.1093/protein/gzu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currin A.; Swainston N.; Day P. J.; Kell D. B. (2017) SpeedyGenes: Exploiting an Improved Gene Synthesis Method for the Efficient Production of Synthetic Protein Libraries for Directed Evolution. Methods Mol. Biol. 1472, 63–78. 10.1007/978-1-4939-6343-0_5. [DOI] [PubMed] [Google Scholar]
- Sadler J. C.; Green L.; Swainston N.; Kell D. B.; Currin A. (2018) Fast and Flexible Synthesis of Combinatorial Libraries for Directed Evolution. Methods Enzymol. 608, 59. 10.1016/bs.mie.2018.04.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during the current study are available from the corresponding author upon reasonable request.