

Abstract
The structure of free cysteine makes it vulnerable to oxidation by molecular oxygen; consequently, organisms that live in oxic habitats have acquired the ability to import cystine as a sulfur source. We show that cystine imported into E. coli can transfer disulfide bonds to cytoplasmic proteins. To minimize this problem, the imported cystine is rapidly reduced. However, this conversion of cystine to cysteine precludes product inhibition of the importer, so cystine import continues into cells that are already sated with cysteine. The burgeoning cysteine pool is itself hazardous, as cysteine promotes the formation of reactive oxygen species, triggers sulfide production, and competitively inhibits a key enzyme in the isoleucine biosynthetic pathway. The Lrp transcription factor senses the excess cysteine and induces AlaE, an export protein that pumps cysteine back out of the cell until transcriptional controls succeed in lowering the amount of transporter. While it lasts, the overall phenomenon roughly doubles the NADPH demand of the cell. It comprises another example of the incompatibility of the reduced cytoplasms of microbes with the oxic world in which they dwell. It also reveals one natural source of cytoplasmic disulfide stress and sheds light on a role for broad-spectrum amino-acid exporters.
Keywords: Disulfide stress, TcyP, AlaE, oxidative stress, enzyme promiscuity
Graphical Abstract
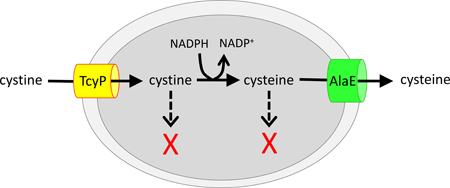
When E. coli encounters environmental cystine, it rapidly imports it, reduces it to cysteine, and then exports most of the cysteine. This odd behavior is ultimately an awkward way to minimize the disulfide stress that results from the import of cystine.
Introduction.
The chemically unique properties of sulfur are exploited through its service in the methionine and cysteine residues of proteins and its incorporation into numerous enzyme cofactors. The atomic concentration of sulfur inside E. coli is approximately 70 mM; thus, the growth of organisms requires the constant acquisition of large amounts of sulfur compounds (Imlay et al., 2015).
In the anoxic environment in which life evolved, sulfur was readily available in the form of hydrogen sulfide. Its assimilation into biomolecules was straightforward, as the sulfur atom was maintained in the same (−2) redox state. However, the appearance of photosystem II two billion years later, and its consequent release of molecular oxygen, eliminated reduced sulfur from air-saturated environments (Anbar, 2008). Hydrogen sulfide was chemically oxidized to sulfate. One upshot is that organisms must expend substantial reducing equivalents and energy to accomplish the eight-electron reduction of sulfate to assimilable sulfide.
This expenditure can be minimized if organisms such as E. coli can access sulfur in the form of organic molecules that have been released from dead cells. The most economical of these is cysteine itself, as it sits at a metabolic hub from which radiate the pathways that synthesize all sulfur-containing biomolecules. Indeed, while E. coli can import and derive sulfur atoms from a range of organic and inorganic molecules, it prioritizes cysteine as its sulfur source. When cysteine is available, E. coli represses the synthesis of the transporters and enzymes that belong to all other sulfur-acquisition pathways (Kredich, 1992).
However, in oxic environments, bacteria are more likely to encounter cysteine in its oxidized disulfide form: as cystine. E. coli expresses two cystine importers—the ion-driven TcyP, and the ATP-driven TcyJLN system (Imlay et al., 2015). One or both of these transporters are apparent in most microbial genomes. When cells import cystine, glutathione-dependent processes in the cytoplasm immediately reduce it to usable cysteine. Glutaredoxins are competent at cystine reduction (Imlay et al., 2015).
Strikingly, when E. coli moves from cystine-free to cystine-containing habitats, it initially over-imports cystine, due to the high titers of TcyP that are induced in low-sulfur environments. The excess cystine is reduced and then exported back into the environment, at an obvious cost and no apparent benefit to the cell. Measurements of cysteine excretion reveal that its import occurs at up to 50 times the rate at which sulfur is actually needed (Imlay et al., 2015).
The purpose of this study was to explore the reasons that the cell behaves in such a wasteful manner. We learned that the absolute sulfur requirement of the cell, coupled to the riskiness of importing a disulfide compound into the reduced cytoplasm, leads almost inevitably to the incongruous futile cycle of cystine import and cysteine export.
Results.
Molecular oxygen rapidly converts cysteine to cystine.
The chemical instability of cysteine is unique among the proteinogenic amino acids. In oxic solutions molecular oxygen oxidizes cysteine, forming the disulfide species cystine plus hydrogen peroxide (Bagiyan et al., 2003). Redox-active metals such as ferric iron are believed to initiate the process by abstracting an electron from the deprotonated thiolate:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
When we incubated cysteine in a simple Tris buffer at neutral pH—without the deliberate provision of metals—the halftime of cysteine oxidation was 0.1 hr (Fig. 1). The reaction was suppressed by the presence of metal chelators, confirming that the reaction was catalyzed by trace metals that contaminate the buffer.
Figure 1. The proximal thiol and amine moieties of cysteine facilitate its oxidation to cystine.
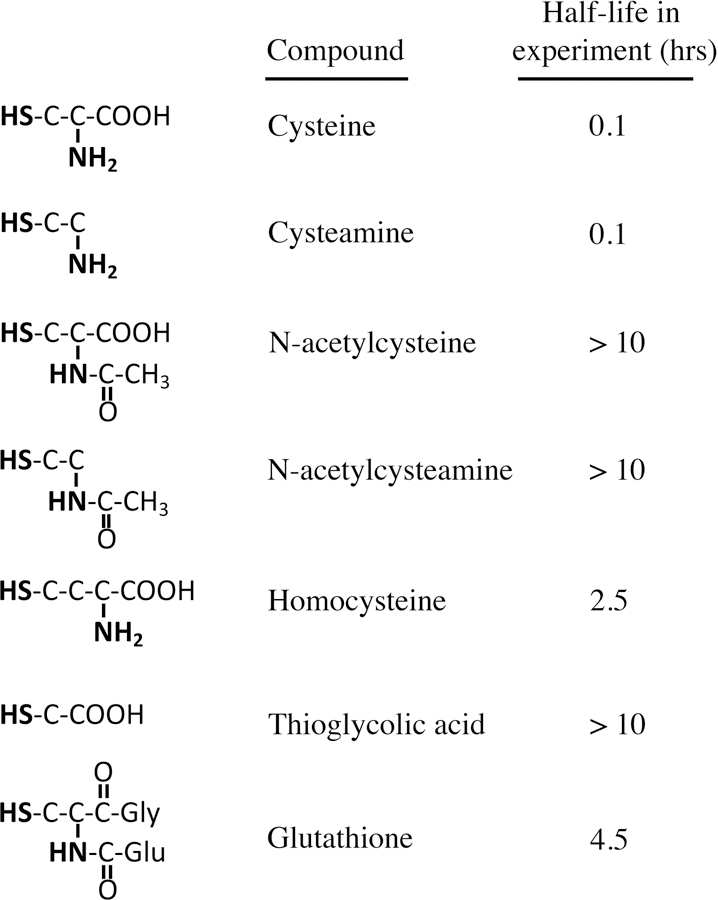
The content of reduced thiol was tracked by DTNB assay; see Experimental Procedures for details. In the presence of 0.1 mM EDTA, the half-life of cysteine was > 10 hrs.
In contrast, our experience is that cysteine residues that are incorporated into proteins do not quickly autoxidize, with the exception of those that are associated with catalytic metals. Therefore we suspected that the facility with which free cysteine autoxidizes is due to the proximity of potent metal-binding groups in its structure. A series of analogs was examined to identify the features that support autoxidation (Fig. 1). Strikingly, the absence of the carboxylate moiety (in cysteamine) did not slow the oxidation rate, but the absence of the amine moiety (in thioglycolic acid) was strongly stabilizing. Derivatization of the amine moiety (in glutathione, N-acetylcysteine, and N-acetylcysteamine) similarly slowed autoxidation. The amide linkage in the latter compounds delocalizes the nitrogen lone pair electrons, making them unavailable for metal binding. The distance between the amine and sulfhydryl groups in homocysteine is two carbon atoms instead of one, and this molecule was significantly more stable than cysteine and cysteamine. Collectively, the data indicate that the proximity of the primary amine to the thiolate promotes its oxidation, probably through bidentate coordination of the catalytic metal through the thiolate anion and the lone pair on the amine. This result fits the fact that in crystals cysteine and penicillamine coordinate cobalt through these same moieties, with the carboxylate uninvolved (Freeman et al., 1978).
These data may explain why E. coli employs the more-stable glutathione, rather than cysteine, in millimolar concentrations as its cellular thiol buffer. Similarly, the autoxidation of cysteine residues in proteins is presumably suppressed because the associated amine group is derivatized by a peptide bond, making it unavailable for metal coordination.
Thus when cysteine is released into oxic environments, it will rapidly convert to the cystine disulfide form. Indeed, most bacteria express transporters that scavenge environmental cystine as a sulfur source (Imlay et al., 2015). Conversely, although E. coli can also import glutathione (Suzuki et al., 2005), it has the capacity to transport only the reduced form (unpublished observations). Perhaps the oxidized form is rarely encountered.
The over-import of cystine creates intracellular disulfide stress.
Cystine is a preferred sulfur source of E. coli, presumably because a single reduction reaction is sufficient to convert it to cysteine. When E. coli is cultured with any other sulfur source, its CysB transcription factor senses a diminished pool of cysteine and activates the synthesis of transporters and enzymes needed for the assimilation of a range of sulfur compounds (Kredich, 1996). These include the two cystine-import systems, TcyP and TcyJLN, which are induced to high titers and allow the cell to bind and import nanomolar cystine from the environment (Imlay et al., 2015). If even low-micromolar cystine then becomes available, it is imported primarily by TcyP and then released as cysteine back into the growth medium (Fig. 2A)(Imlay et al., 2015). This behavior was replicated by Salmonella typhimurium and, to a lesser degree, Bacillus subtilis, but not by P. aeruginosa or yeast (Fig. 2B). Other amino acids can also be over-imported and excreted by E. coli (Fig. 2C), but the magnitude of overshooting of cystine is exceptional. In these experiments the rate of cystine import by TcyP exceeded the sulfur demand of the cell by as much as 80-fold (Fig. 2D). The apparent KM of TcyP is only 2 micromolar (Imlay et al., 2015), so low cystine concentrations are sufficient to produce this effect (Fig. S1). We wished to understand the underlying rationale and mechanism.
Figure 2. Cystine addition triggers profuse cysteine excretion.
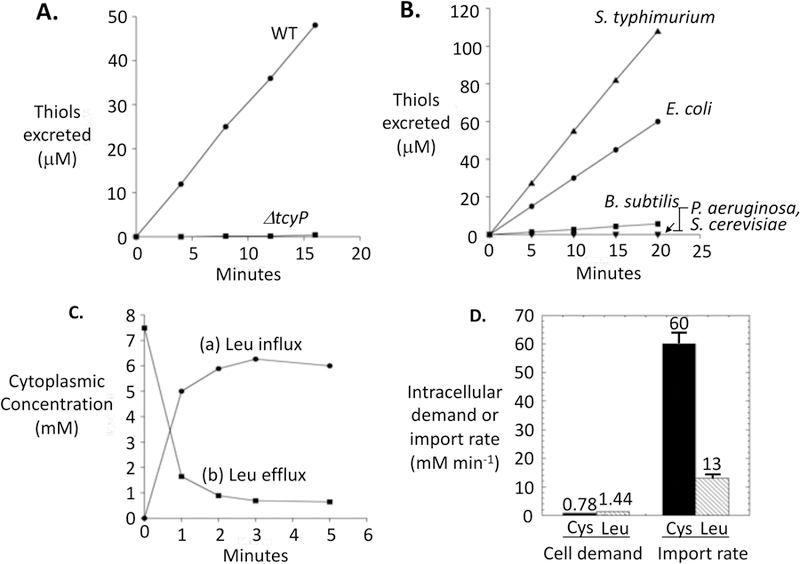
(A) Excretion of thiols upon the addition of cystine (time zero) to wild-type (MG1655) and ΔtcyP (KCI1254) E. coli strains. Cells (0.1 OD600) were growing exponentially in medium containing 18 amino acids (cystine and methionine omitted). (B) Cystine was added to microbes growing in the same media as in panel A, and thiol excretion was monitored. E. coli, S. typhimurium, and B. subtilus contain TcyP homologs, whereas P. aeruginosa and S. cerevisiae do not. (C) 14C-leucine accumulation (curve a) and excretion (curve b) by E. coli. Chloramphenicol was added to cells in amino-acids-free glucose medium, and then 14C-leucine (50 μM) was added at time zero. Intracellular leucine was tracked by filtration (curve a). After 7 min, cells were washed and resuspended in medium containing 1 mM cold leucine, and efflux of 14C-leucine was tracked (curve b). Leucine was selected because E. coli lacks enzymes to catabolize it. (D) Comparison of the cellular demand for leucine and cystine (left bars) with the initial rate of influx when these amino acids are added to cells growing in amino-acid-free medium (right bars). See Experimental Methods for calculations.
We conjectured that most importers become product-inhibited as sufficient levels of their substrates accumulate in the cytoplasm, but the reduction of imported cystine to cysteine renders it unable to effect product inhibition of the importers. This idea is supported by the observation that glutathione-deficient mutants, which are defective in cystine reduction, import cystine much more slowly (Imlay et al., 2015). Indeed, in wild-type cells the reduction process occurred too rapidly for us to reliably detect intracellular cystine using standard extraction methods and mass spectrometry.
The rapid reduction of cystine would be important if cystine would otherwise transfer disulfide bonds to the cysteine residues of cytoplasmic proteins. Proteins in the bacterial cytoplasm are normally devoid of such bonds (Pollitt & Zalkin, 1983, Derman & Beckwith, 1991). The scheme of disulfide transfer is shown in Fig. 3A. The capacity of cystine to participate in such reactions was tested by adding it to reduced alkaline phosphatase (PhoA) in vitro. PhoA is active only when it contains two disulfide bonds. Incubation of fully reduced PhoA with cystine triggered the appearance of substantial activity, confirming that disulfide exchange reactions are facile and can be detected with this enzyme (Fig. 3B). Oxidized glutathione also activated PhoA in vitro.
Figure 3. Cystine can transfer disulfide bonds to proteins in vitro and in vivo.
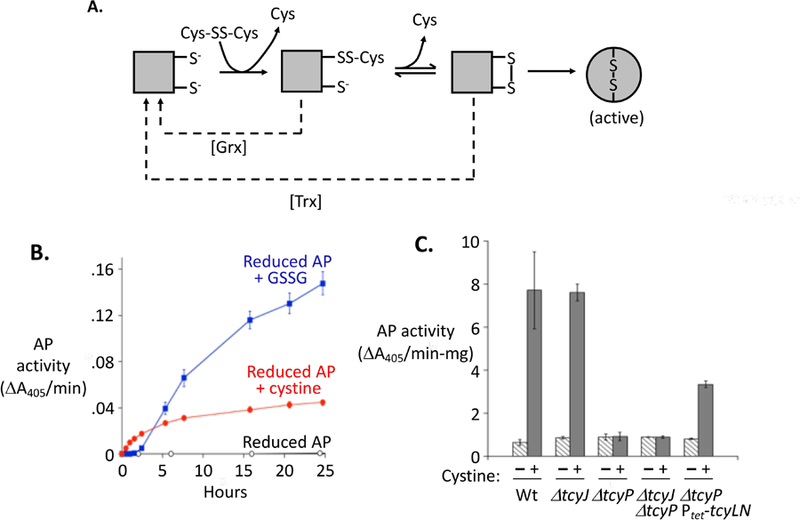
(A) Steps by which low-molecular-weight disulfide compounds can transfer disulfide bonds to alkaline phosphatase (AP), thereby activating it. AP ultimately folds around the disulfide bonds, making activation irreversible (Akiyama & Ito, 1993). For clarity, only one of the two disulfide bonds in AP is represented. Dashed lines: The glutaredoxin (Grx) and thioredoxin (Trx) systems suppress AP activation, likely by reducing mixed and the initial intra-protein disulfide bonds, respectively. (B) Cystine and oxidized glutathione (glutathione disulfide, GSSG) at 0.5 mM activate purified AP in vitro. (C) Cystine import activates cytoplasmic AP in vivo. Cystine was added (or not) to strains containing the pcAP plasmid, and intracellular AP activity was measured 40 min later. Strains bearing the pcAP plasmid are WP551 (WT); KCI1630 (ΔtcyJ); KCI1632 (ΔtcyP); KCI1820 (ΔtcyJ ΔtcyP); and KCI1638 (ΔtcyP Ptet-RAc-tcyLN), which constitutively overexpresses the integral membrane proteins of the TcyJLN cystine importer. Error bars in this and all other figures represent SEM of at least three biological replicates.
To test whether such reactions occur at a significant rate in vivo, we used a modified form of PhoA. Native PhoA is secreted to the periplasm, and during this process the DsbAB system activates it by oxidizing two cysteine pairs to form the necessary disulfide bonds (Collet & Bardwell, 2002). In contrast, a modified PhoA developed by the Beckwith lab lacks its leader sequence and therefore is retained in the cytoplasm (Derman et al., 1993). When expressed in E. coli the leader-less enzyme was inactive. However, the addition of cystine to sulfate-grown cells caused the immediate appearance of PhoA activity (Fig. 3C). This effect depended upon the TcyP cystine importer. No activation was observed when reduced cysteine was imported (Fig. S2). Thus the rapid import of cystine does trigger disulfide formation in cytoplasmic proteins.
This conclusion was further supported by the observation that cystine import activates the OxyR transcription factor. In the presence of hydrogen peroxide, its natural effector, two key cysteine residues on OxyR are oxidized, forming a disulfide bond and activating it as a transcription factor. We observed that some activation also occurred when cystine was added to cells. To ensure that this activation was mediated by disulfide-exchange reactions rather than by the evolution of hydrogen peroxide, this result was repeated under anoxic conditions (Fig. S3).
These effects were also produced when cystine was imported by TcyJLN, but only when the membrane-bound components of this transporter were artificially overproduced (Fig. 3C). That observation fits the fact that cystine influx through TcyJLN is substantially slower than through TcyP. It is typical that ion-driven importers (like TcyP) display higher flux rates, while ATP-driven importers (like TcyJLN) exhibit lower fluxes but higher substrate affinities.
E. coli expresses two thioredoxins and three glutaredoxins that can reduce disulfide bonds (Stewart et al., 1998). The absence of glutathione or glutaredoxins enhanced PhoA activation by cystine (Fig. 4). The glutathione/glutaredoxin systems can reduce cystine, and they are also effective at reducing mixed disulfide bonds, potentially including the cysteinylated PhoA intermediate that precedes formation of the intramolecular disulfide bond (Fig. 3A). Mutants that lack thioredoxin A also suffered a greater activation of PhoA, consistent with its role as the primary reducer of protein disulfide bonds (Fig. 4C). Thus both disulfide reducing systems act to minimize the disulfide stress that follows upon cystine import.
Figure 4. The glutaredoxin and thioredoxin systems diminish disulfide stress arising from imported cystine.
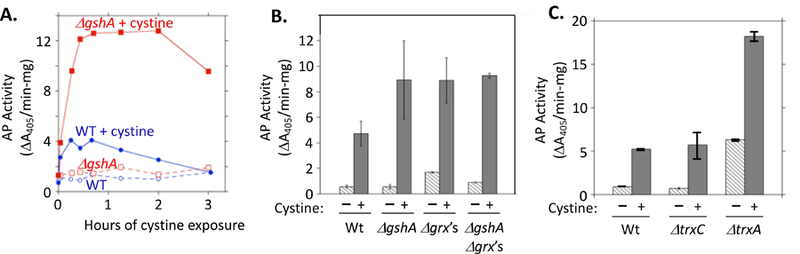
(A) Time course of AP activation inside wild-type (WP551) and ΔgshA (KCI1622) mutants upon treatment with 0.5 mM cystine. Dashed lines: no cystine addition. (B) AP activity 40 min after the addition of cystine to WT (WP551), ΔgshA (KCI1622), ΔgrxA ΔgrxB ΔgrxC (KCI1812, Δgrx’s), and ΔgshA ΔgrxA ΔgrxB ΔgrxC (KCI1846) strains. No single glutaredoxin mutant phenocopied the triple mutant, suggesting significant cross-complementation. (C) AP activity 40 min after the addition of cystine to WT (WP551), ΔtrxC (KCI1824), and ΔtrxA (KCI1810) mutants.
Cysteine oxidation also imposes ROS stress.
Disulfide stress is not the sole adverse effect of cystine import. When cystine-fed cells excreted cysteine into aerobic medium, its consequent chemical oxidation by oxygen generated hydrogen peroxide (Fig. 5A). The H2O2 can flow back into cells, with the potential for the inactivation of iron-containing enzymes. E. coli expresses NADH peroxidase (AhpCF) and catalase to cope with environmental H2O2 (Imlay, 2013), and indeed mutants that lacked these enzymes were unable to tolerate sudden cystine supplementation (Fig. 5B). The growth defect was reversed by the addition of Ile, Leu, and Val, as H2O2 blocks growth by inactivating iron-sulfur cluster-containing dehydratases in the branched-chain biosynthetic pathway (Jang & Imlay, 2007). Notably, however, in scavenging-proficient cells these growth defects were absent.
Figure 5. The autoxidation of exported cysteine creates H2O2 stress.
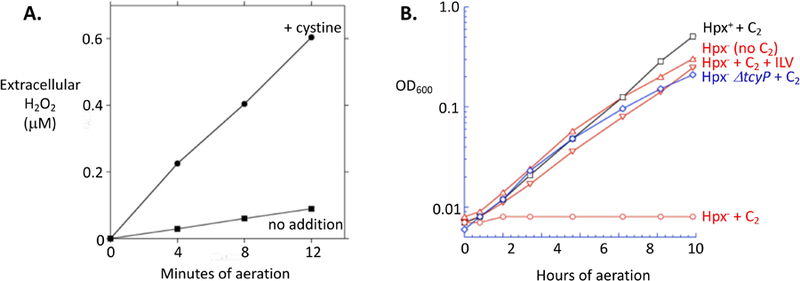
(A) H2O2 efflux into culture medium was measured using a mutant lacking catalases and NADH peroxidase (LC106). Cystine (0.1 mM) was added at time zero. Error bars are obscured by the markers. Cystine addition under anoxic conditions generated no H2O2 signal (< 0.01 μM). (B) The H2O2 generated during cystine uptake is sufficient to poison branched-chain biosynthesis in a cell lacking catalase/peroxidase (Hpx−). C2, cystine added; +ILV, 0.5 mM of isoleucine, leucine, and valine added. Strains were Hpx+ (MG1655), Hpx− (LC106), or Hpx− ΔtcyP (KCI1153).
The impact of H2O2 formation is exacerbated because intracellular cysteine plays a second role in sensitizing cells to H2O2: the tendency of cysteine to reduce ferric iron produces ferrous iron that serves as an electron donor for the Fenton reaction (Park & Imlay, 2003; Imlay et al., 2015):
| (2) |
| (8) |
The combination of these reactions comprises a cycle that continuously generates hydroxyl radicals, which can kill cells by damaging their DNA. Figure S5A shows that cystine addition conferred great sensitivity even to external sources of H2O2. In natural habitats, the excretion of excess cysteine would spare the cell this H2O2 stress by ensuring that cysteine autoxidation occurs externally rather than internally.
In prior work we observed that rapid cystine import causes the evolution of hydrogen sulfide, as the elevated cysteine becomes an adventitious substrate for tryptophanase. This hydrogen sulfide inhibits the main cytochrome bo oxidase of the respiratory chain, and continued respiration depends upon the less energy-conserving cytochrome bd oxidase (Korshunov et al., 2016, Forte et al., 2016).
Excessive intracellular cysteine can disrupt metabolism.
Experiments here and in previous study (Korshunov et al., 2016) have shown that cystine over-import threatens the cell with stress from disulfide bonds, reactive oxygen species, and hydrogen sulfide; however, their impacts upon cell performance are minimized by redoxins, peroxide scavengers, and an alternative cytochrome oxidase. Yet workers have noted that when non-physiological (millimolar) concentrations of cysteine are added to cultures, the pathway of isoleucine synthesis is inhibited (Harris, 1981). High concentrations of cysteine can compete with threonine for the active site of threonine deaminase, the first enzyme in the isoleucine biosynthetic pathway. We observed that the same effect occurred when low-micromolar cystine was supplied to cells (Fig. 6A). This effect again depended upon TcyP (Fig. S4). The growth defect was suppressed by the addition of isoleucine. Interestingly, the growth problem was temporary, suggesting that the cell activates a mechanism that gradually alleviates the problem.
Figure 6. Excess intracellular cysteine impairs isoleucine synthesis.
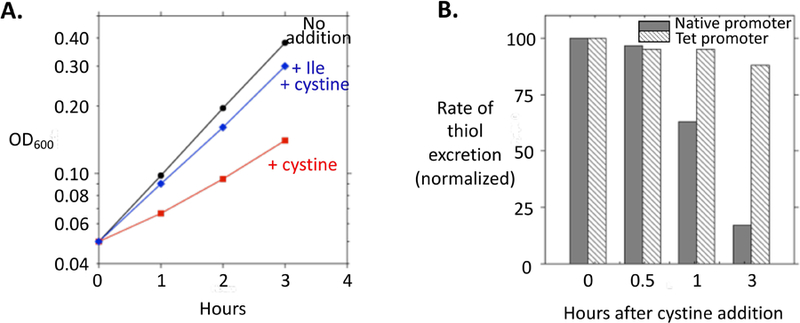
(A) As indicated, at time zero cystine and/or isoleucine were added to log-phase WT cells (MG1655) in minimal glucose medium. (B) Renewed repression of tcyP stops the futile cycle of cystine import and cysteine efflux. The rate of thiol efflux was determined at intervals after the addition of cystine to log-phase cells. Isoleucine was present, and the doubling time was 70 min. Solid bars: WT (MG1655). Hatched bars: TcyP was driven at a constant rate from the Tet promoter (SSK168 with Tet added).
Induction of the AlaE exporter minimizes the adverse effects of cysteine accumulation.
The problems caused by cystine import diminish over time. After cystine was added, the initially high rate of cystine influx and cysteine efflux gradually declined over subsequent generations, because tcyP transcription drops abruptly and extant TcyP is diluted out by cell division (Fig. 6B). However, the relief of the isoleucine auxotrophy occurred prior to much cell division, indicating a rapid adjustment to cysteine stress. We suspected that the cell might induce whichever transporter is involved in pumping cysteine back out of the cell.
To investigate this point, RNA-sequencing analysis was performed. Transcripts were collected both before and 10 minutes after the addition of cystine to sulfate-grown cells. The data are summarized in Table 1 and Table S1, and the full data set is presented in Table S2.
Table 1:
Effects of cystine influx upon transcripts of interest.
| Gene | Fold Induction | FDR | (regulator) |
|---|---|---|---|
| yhaO | 130 | 0 | Cysteine importer, desulfidase (YbaO control). |
| yhaM | 56 | 0 | Cysteine importer, desulfidase (YbaO control). |
| alaE | 44 | 6.6E-154 | Alanine exporter (Lrp). |
| dadA | 17 | 0 | D-amino acid dehydrogenase/alanine racemase (Lrp). |
| dadX | 16 | 0 | D-amino acid dehydrogenase/alanine racemase (Lrp). |
| ilvG | 6.5 | 1.4E-137 | Isoleucine biosynthetic operon (attenuation). |
| ilvM | 10 | 2.0E-106 | Isoleucine biosynthetic operon (attenuation). |
| ilvE | 3.2 | 3.6E-103 | Isoleucine biosynthetic operon (attenuation). |
| ilvD | 4.6 | 2.6E-78 | Isoleucine biosynthetic operon (attenuation). |
| ilvA | 4.7 | 1.3E-148 | Isoleucine biosynthetic operon (attenuation). |
| aaeA | 1.5 | 0.00041 | Efflux pump (Crp). |
| eamA | 1.3 | 4.5E-05 | Putative cysteine efflux pump. |
| eamB | 1.2 | 0.076 | Putative cysteine efflux pump. |
| yjcB | 5.9 | 6.8E-166 | Uncharacterized protein. |
| mdtK | 5.3 | 7.4E-121 | Multidrug efflux pump. |
| mdtM | 5.3 | 5.2E-80 | Multidrug efflux pump (SlyA). |
| yohC | 6 | 2.8E-266 | Inner-membrane protein. |
| tnaC | −11 | 5.1E-29 | leader peptide, tryptophanase |
| tnaA | −9 | 1.5E-113 | leader peptide, tryptophanase |
RNA sequencing analysis was performed upon triplicate samples of log-phase cells, with and without 10 min exposure to 50 μM cystine (Experimental Procedures). Fold induction denotes the level of gene mRNA shortly after cystine addition versus without cystine. Key transcriptional regulatory controls are indicated in parentheses; shaded genes belong to a common operon. See supplementary Table S1 for impact upon the CysB, MetJ, SOS, and OxyR regulons, and Table S2 for the full data set.
More genes were strongly repressed than induced (Table S1). The repressed genes included members of the CysB and MetJ regulons, as these transcription factors respond to the elevation of cytoplasmic cysteine (via N-acetylserine) and S-adenosylhomocysteine, respectively (Ostrowski & Kredich, 1989). The Fur regulon was abruptly repressed; we suspect that the burgeoning cysteine rapidly reduces intracellular iron to the ferrous form (reaction 2), which then metallates the Fur repressor (Troxell & Hassan, 2013). Some members of the OxyR regulon were mildly induced, in keeping with the fusion data (Table S1). Other effects, such as repression of the PurR regulon, do not present an obvious rationale.
The induced genes did not fall as tidily into known regulons. A number of SOS genes (recN, umuCD, sulA) responded, presumably due to a rise in Fenton-driven oxidative DNA damage. The yhaOM operon was strongly induced. It encodes a cysteine import/degradation system that is controlled by the transcription factor DecR (YbaO), which is believed to detect elevated levels of intracellular cysteine (Oguri et al., 2012, Shimada et al., 2016). Our focus, however, was upon the possibility that the cell induces exporters of cysteine. None of the candidates proposed in the literature (eamA, eamB, aaeA (Franke et al., 2003, Yamada et al., 2006, Ohtsu et al., 2010)) were significantly induced. However, the alaE monocistronic mRNA was induced 44-fold. The AlaE protein has been identified as an alanine exporter, because mutants that lack both alaE and alanine-degrading enzymes cannot grow when they are fed Ala-Ala dipeptide (Hori et al., 2011). No phenotype had yet been demonstrated for the single alaE mutant.
We wondered whether AlaE might also export cysteine. Indeed, mutants that lacked alaE exhibited a prolonged growth lag when cystine was added to cells (Fig. 7A). Single mutations in other induced membrane proteins (yohC, yjcB, mdtK, mdtM) and in proposed cysteine exporters did not have any effect.
Figure 7. AlaE exports cysteine.
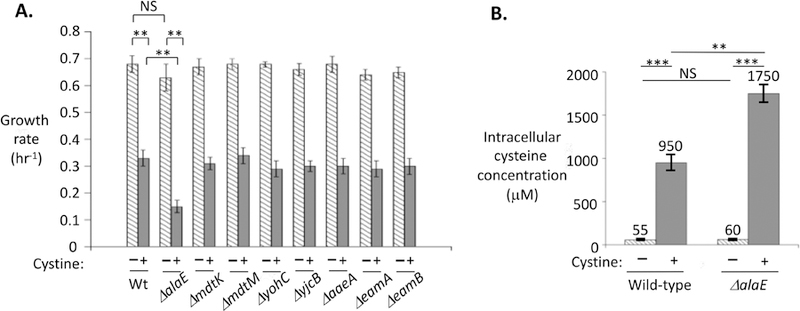
(A) The AlaE exporter partially suppresses the cystine-imposed growth defect. Cystine was added to mutants lacking export proteins, and growth in unsupplemented medium over the next hour was quantified as a net growth rate. (B) LC-MS was used to quantify levels of cysteine inside WT (MG1655) and ΔalaE (SSK250) strains both before and shortly after addition of 0.1 mM cystine. For comparison, the KM values for cysteine of the two primary cysteine-using enzymes—cysteinyl tRNA synthetase and O-succinylhomoserine lyase—are 30 and 50 μM (Holbrook et al., 1990, Zhang et al., 2003). NS: not significant; **: P < 0.01; ***: P < 0.001.
The ability of AlaE to suppress growth problems may be underestimated in this experimental design, because in closed cultures the YhaO importer may have the perverse effect of re-importing the cysteine that AlaE exports. The logic of such an import/export cycle is considered in the Discussion. Indeed, in a strain lacking YhaO, AlaE almost completely eliminated growth inhibition (Fig. 8).
Figure 8. Cysteine export by AlaE ameliorates cysteine toxicity.
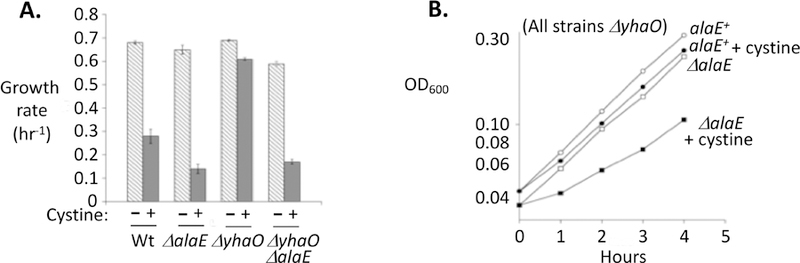
(A) In a ΔyhaO background (right two strains), AlaE almost completely suppresses growth inhibition during cysteine intoxication. (B) Growth curves exhibiting AlaE suppression of growth inhibition. Both strains lacked the cysteine importer YhaO. MG1655 (Wt), SSK226 (ΔalaE yhaO+), SSK209 (alaE+ ΔyhaO), SSK238 (ΔalaE ΔyhaO).
Other measures of cysteine toxicity supported these conclusions. The lethal effect of H2O2 upon cystine-treated cells was profoundly diminished by the expression of alaE (Fig. S5), consistent with the ability of AlaE to reduce the amount of cysteine inside the cell. Finally, mass spectrometric analysis confirmed that cysteine levels are elevated in alaE mutants (Fig. 7B).
Notably, although AlaE diminished the level of cytoplasmic cysteine, it had no impact upon its net efflux rate (Fig. S6A). We inferred that other less-efficient transporters have some capacity to export cysteine, and as cysteine pools quickly rise to very high levels in the alaE mutant, efflux through these high-KM secondary exporters also rises until it matches the rate of cystine import, thereby creating a higher steady-state level of cysteine. Indeed, the growth defect that cystine imposed upon alaE mutants was further enhanced by eliminating secondary exporters (Fig. S6B). Thus both the transcriptomic and functional data indicate that AlaE is the primary exporter of excessive intracellular cysteine.
The induction of alaE by cysteine is mediated by Lrp.
We sought to explain alaE induction. The two transcription factors known to respond to intracellular cysteine are CysB (Kredich, 1996) and DecR (Oguri et al., 2012, Shimada et al., 2016), but mutants lacking these proteins continued to induce alaE during cystine exposure (Fig. S7). Previous work by Ihara et al. demonstrated that the transcription of alaE could be induced by treatment of E. coli with Ala-Ala dipeptide, Ala, and Leu (Ihara et al., 2017). In this situation Leucine-Responsive Protein (Lrp) was necessary for induction. In vitro both Ala and Leu stimulated Lrp binding to a site approximately 100 bp upstream of the alaE transcriptional start site.
We determined that Lrp is also necessary for the alaE induction that occurs during cystine import (Fig. 9A, B). This outcome was somewhat surprising, as cysteine had not been regarded as a likely ligand of Lrp. A survey of amino acids suggested that alaE is induced when cells are exposed to high levels of a range of non-polar linear amino acids. Fig. 10 shows that four amino acids (Ala, Leu, Met, and 2-aminobutyrate) and Ala-Ala dipeptide each induced the alaE-lacZ fusion more than 10-fold; norvaline, cystine, cysteine, homocysteine, and norleucine each had moderate inducing effects (> 4-fold); lysine had minimal impact (ca. 2-fold); and valine and isoleucine were ineffective. In each case, induction did not occur in an lrp mutant.
Figure 9. Excessive intracellular cysteine induces alaE expression by activating Lrp.
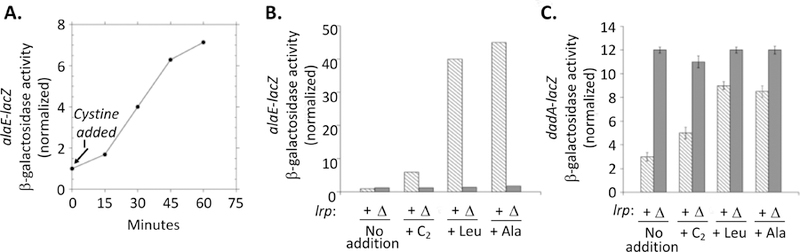
(A) Induction of a chromosomal alaE-lacZ fusion upon cystine addition to cells in unsupplemented medium. (B) The alaE-lacZ fusion was induced upon addition of 0.2 mM cystine, leucine, or alanine. Induction depends upon lrp. Strains: SSK250 and SSK248. (C) Cystine also derepresses expression of a dadA-lacZ fusion for which Lrp acts as a repressor. Strains: SSK252 and SSK253.
Figure 10. AlaE is induced when cells are overloaded with non-polar amino acids.
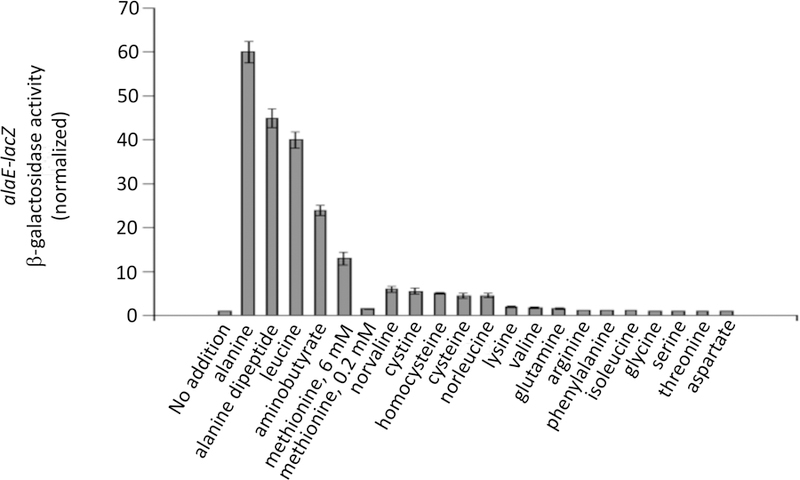
SSK250 cells were exposed to the indicated amino acids for 60 min prior to assay. Concentrations are listed in Experimental Procedures. Minimal glucose medium contained 0.05 mM Ile to circumvent toxicity from valine and cysteine.
The effectiveness of cysteine at affecting gene expression via Lrp was not limited to the alaE promoter. Apo-Lrp represses the dadA gene (Mathew et al., 1996), and repression was relieved when cells were treated with Leu, 2-aminobutyrate, or cystine (Fig. 9C). We considered the possibility that cysteine might be degraded by IscS to alanine in vivo (Mihara et al., 2000), allowing the latter amino acid to be the direct activator of Lrp; however, full alaE-lacZ induction persisted in an iscS mutant (Fig. S7). We infer that Lrp likely forms functionally active complexes with amino acids that have C1-C4 side chains that are linear and hydrophobic.
AlaE is a broad-spectrum exporter.
While AlaE was originally identified as an alanine exporter, its induction by a variety of other amino acids raised the possibility that it might be able to export these other inducers, too. Both norvaline and 2-aminobutyrate are non-productive amino acids that can be generated by the off-target reactions of the branched-chain biosynthetic pathway (Soini et al., 2008). Like cysteine, in excess these compounds can toxify E. coli. We found that norvaline toxicity was unaffected by the presence or absence of AlaE (Fig. S8). In contrast, 2-aminobutyrate toxicity was markedly higher in strains that lacked either alaE or lrp (Fig. 11). Thus the Lrp/AlaE duo exhibit similarly broad recognition of hydrophobic linear amino acids.
Figure 11. AlaE is a broad-spectrum exporter that protects cells against the toxicity of a non-proteinogenic natural amino acid.
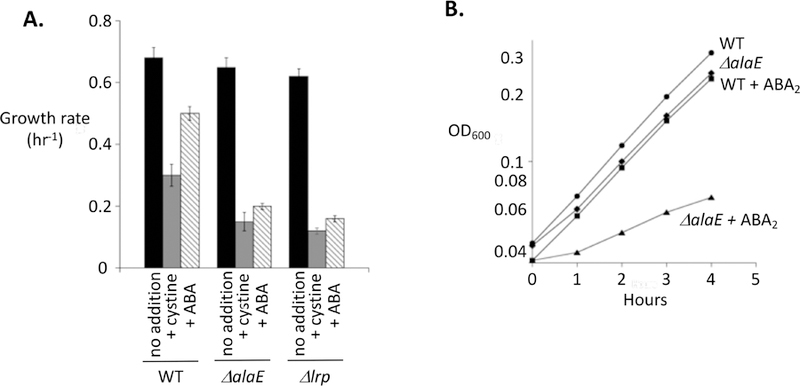
Cystine or α-aminobutyrate (ABA) (A) or its dipeptide (ABA2) (B) were added (0.2 mM). Subsequent growth was especially diminished in ΔalaE and Δlrp mutants that cannot induce the AlaE efflux pump. Strains: MG1655, SSK250, SSK244.
The futile cycle of cystine import and cysteine excretion is costly to the cell.
The import and reduction of cystine, followed by the excretion of cysteine, minimizes the amount of disulfide stress, oxidative damage, and biosynthetic inhibition that would otherwise ensue. However, because the flux rate is so high, the energetic cost is likely to be considerable. By comparing the rate of cysteine efflux to the NADPH demands of biosynthesis, we estimated that the process will consume 10% of ATP production and double the normal NADPH flux of the cell (Imlay et al., 2015). The latter prediction was tested qualitatively by examining whether cystine cycling was problematic for strains that are partially defective in the main routes of NADP+ reduction. NADPH production in E. coli is primarily mediated by the pentose-phosphate pathway and by Pnt, a membrane-bound transhydrogenase (Hanson & Rose, 1980). A strain carrying a null zwf allele (encoding glucose-6-phosphate dehydrogenase) lacks the oxidative pentose phosphate pathway; its growth was suppressed by cystine addition even when isoleucine was supplied (Fig. 12). A strain lacking both zwf and pnt, encoding transhydrogenase, was even more sensitive. These results support the prediction that cystine reduction consumes a substantial fraction of the NADPH pool.
Figure 12. Substantial NADPH is consumed by the futile cycle of cystine import and cysteine export.
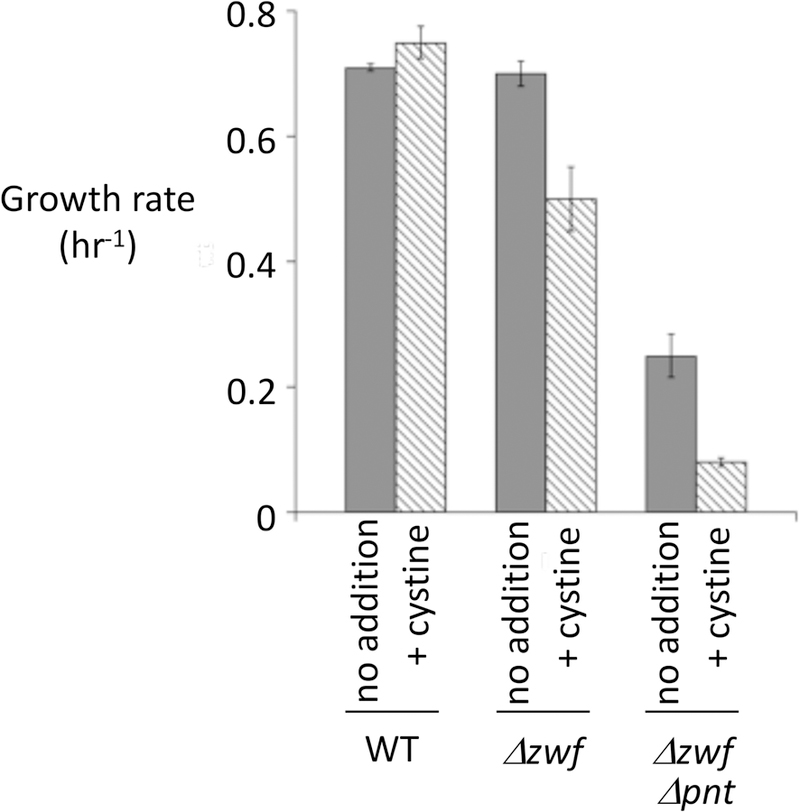
The primary sources of NADPH are the zwf-dependent pentose phosphate pathway and the pnt-dependent transhydrogenase (Hanson & Rose, 1980). Strains: AN387, ALN10, and ALN11. Calculations indicate that cystine reduction doubles the cellular NADPH demand.
Discussion.
Biological systems exploit many of the singular traits of sulfur atoms: their redox capacity, nucleophilicity, metal affinity, thioester energetics, disulfide strength, and sulfonium and thiyl-radical stabilities. Consequently, all organisms manifest a substantial demand for sulfur species. Access to sulfur atoms was not problematic in the reducing atmosphere in which life evolved. However, things changed when oxygenic photosynthesis filled the atmosphere with oxygen (Anbar, 2008). The predominant environmental form of inorganic sulfur switched from hydrogen sulfide to sulfate—and the primary organic source changed from cysteine to cystine. From a bioenergetic perspective sulfate is a disaster: eight electrons are required to convert it back to hydrogen sulfide, the form in which it is assimilated, at a cost of ~12 ATP equivalents. Cystine is a far more economical choice, as a single divalent reduction generates 2 cysteine molecules at only 1.5 ATP per each. Therefore, most aerobes feature regulatory systems that prioritize cystine as the sulfur source whenever it is available. E. coli strongly induces two cystine transporters not only when sulfur is scarce, but also whenever the cell is relying upon any other sulfur source (Imlay et al., 2015). The hopeful strategy is that even if environmental cystine concentrations are scant, the induced transporters might snag enough to satisfy the cellular sulfur demand.
In this study we have observed a series of calamities that ensue when the cell then encounters micromolar cystine. In the long run the cell adjusts to this sulfur-rich state by diminishing the synthesis of the cystine transporters, as lower titers provide a sufficient influx. However, during the several-generation-long transition, the cell imports cystine at a rate that enormously exceeds its demand and compromises fitness. This bizarre behavior is ultimately a logical consequence of the fact that the cell is importing a disulfide compound into a cell interior that operates as a reducing environment.
The daisy chain of problems, fixes, secondary problems, and secondary fixes can be quickly summarized, with the specific steps enumerated in Fig. 13. The nonnegotiable elemental demand for sulfur requires that the cell populate its surface with sulfur transporters whenever the supply is inadequate; not to do so would paralyze growth. Any subsequent encounter with cystine triggers its import. The introduction of this disulfide compound into the cytoplasm initiates uncontrolled sulfur-exchange reactions, which threaten to inactivate enzymes by derivatizing their cysteine residues. The cell minimizes this problem by using its glutathione pool to rapidly reduce imported cystine molecules. By doing so, however, the cell loses any possibility that the cystine importers can be product-inhibited by cytoplasmic cystine—so cystine import continues even as the cysteine pools swell. The burgeoning pools present a multiplicity of unfortunate consequences: oxidative stress due to autoxidation reactions, accidental production of toxic sulfide by tryptophanase, and competitive inhibition of enzymes that normally bind other amino-acid substrates. If unchecked, the downstream consequences would include poisoning of energetic and biosynthetic pathways and a cessation of growth. Instead, LRP senses the excess cysteine and induces the AlaE exporter, whose action shrinks the intracellular pool to a tolerable level. The net effect: rapid cystine import and cysteine excretion, at a high energetic cost.
Figure 13. Summary of the threats posed by cystine import and of the tactics by which E. coli defends itself.
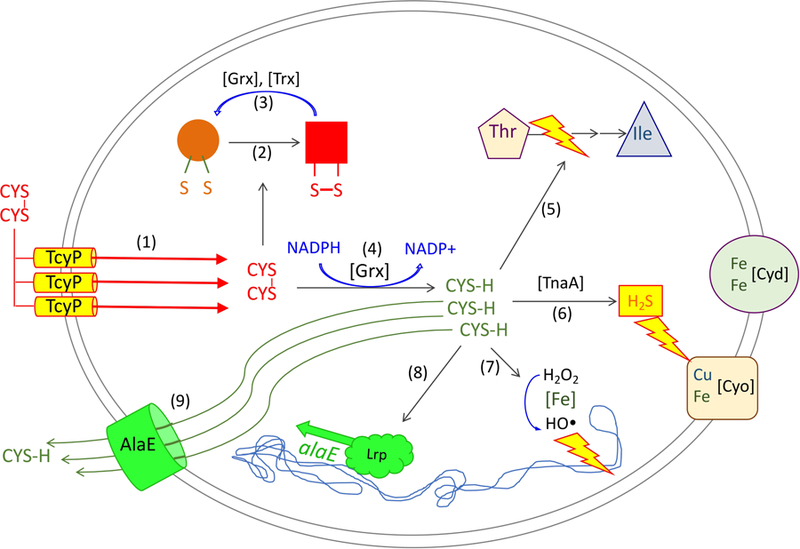
To acquire adequate sulfur, the TcyP cystine importer is 10-fold induced when sulfur sources are scarce (1). Upon a subsequent encounter with low-micromolar cystine, TcyP can import excess cystine into the cytoplasm, where it can transfer disulfide bonds to proteins (2). This effect is countervailed by glutaredoxin- and thioredoxin-dependent protein disulfide reduction processes (3) and by the rapid reduction of cystine to cysteine, which is mediated by NADPH/GOR/GSH/glutaredoxin (4). However, the consumption of cystine precludes product inhibition of TcyP, and cysteine accumulation greatly outstrips cell demand. Elevated cysteine levels are problematic because cysteine can inhibit Ile synthesis (5); act as a pseudosubstrate for tryptophanase, which releases sulfide that inhibits cytochrome bo oxidase (6); and both generates H2O2 by oxidation and mobilizes ferrous iron for the DNA-damaging Fenton reaction (7). The transcription factor Lrp is activated by the high cysteine pools (8), and its induction of AlaE provides an exporter that drives down the level of cysteine and abates these toxic effects (9).
Why doesn’t TcyP stop importing cystine when cysteine pools are adequate?
All these hazards would be avoided were TcyP feedback-inhibited by intracellular cysteine. We have pondered why this does not happen; a couple points may be relevant. First, cysteine does not physically resemble cystine, and so it cannot competitively inhibit TcyP by directly occupying its active site. Second, it appears not to be common for transporters to have allosteric sites. Only a few examples are known—methionine can inhibit its ABC-type importer (Yang & Rees, 2015), and magnesium does so with MgtX (Subramani et al., 2016)—and these appear to be exceptions. Product inhibition, in which the accumulated substrate binds and clogs the importer, is more likely to be the rule.
Third, cysteine-mediated allostery is rare in general. Cysteine has the unusual circumstance of having an isostructural twin, serine, and so protein binding that is dictated by the standard characteristics of metabolite size, shape, and charge will fail to discriminate between the two. Indeed, the mechanism by which cysteine feedback-inhibits its own synthesis is by competing with serine in the active site of O-acetylserine transacetylase (Subramani et al., 2016). Were serine able to bind inappropriately to a notional allosteric site on TcyP, then during sulfur starvation the outcome would be perverse: cysteine deficiency would cause translation to stall and serine (and other amino acids) to accumulate, and the serine would shut down any chance of cystine import. One E. coli enzyme that successfully binds cysteine but excludes serine is the cysteinyl tRNA synthetase; it accomplishes this trick by presenting a prosthetic zinc ion to which the cysteinyl sulfur atom binds far more tightly than does the seryl oxygen atom (Zhang et al., 2003). An analogous zinc atom at an allosteric site on TcyP might work, but perhaps it would bind cysteine with too great an avidity for the rapid ligand exchange that is needed for continuous control.
Notably, the ABC-type TcyJLN transporter does not over-import cystine. The reason is that during sulfur restriction, the cell induces only the binding protein, leaving the membrane proteins at constitutive levels. When cystine is scarce, transporter turnover is dictated by the pace at which the binding proteins grab cystine, so induction of the binding protein is useful and induction of the transmembrane proteins would be unproductive. However, when cystine is abundant and the binding protein (KM = 20 nM) is saturated, then the rate-limiting step is catalyzed by the (non-induced) membrane proteins—so over-import occurs only if these proteins are artificially overproduced (Imlay et al., 2015).
Amino acid efflux is a key mechanism of balancing pools.
Most amino acid importers do not over-import their substrates. TcyP is special, both because the non-negotiable desperation for sulfur warrants high titers of transporters, and because import control via product inhibition is precluded by the immediate conversion of cystine to cysteine. Yet workers have repeatedly observed that bacteria have the capacity to export other amino acids, too. Often these observations have arisen in the context of identifying genes that improve resistance to amino-acid analogs or of enabling the overproduction and excretion of amino acids by engineered strains (Kruse et al., 2002, Kutukova et al., 2005). One natural need for export arises when the cell primarily derives energy by hydrolyzing peptides. E. coli catabolizes many amino acids, but others it cannot degrade, including branched-chain amino acids. When the peptide influx exceeds the biosynthetic demand for these non-degradable amino acids, the excess must be exported. Apparently authentic exporters of lysine and arginine have been identified in E. coli and other bacteria (Nandineni & Gowrishankar, 2004, Pathania & Sardesai, 2015). Other exporters have been found that, when overproduced, can pump artificially elevated levels of amino acids out of the cell (Kutukova et al., 2005, Doroshenko et al., 2007, Park et al., 2007, Liu et al., 2015). It is not yet clear whether amino acids are the natural substrates of these proteins. Thus AlaE is exceptional in that its regulation and native substrates are known and a clear phenotype is apparent under a natural growth circumstance.
When abundant, cysteine can disruptively insert itself into the active sites of tryptophanase and threonine deaminase (Korshunov et al., 2016; Harris, 1981). These problematic events are reminders that the substrate specificities of enzymes are not unlimited. In fact, the breadth of substrate recognition by Lrp and AlaE is useful, as they allow cells to cope, for example, with excessive pools of both cysteine and alanine. They also permit the excretion of unnatural amino acids. This behavior is useful to bioengineers, who have long coaxed bacteria to overproduce amino acids without quite understanding how the products ended up in the culture supernatant. These fermentations can be bottlenecked at the excretion step (Nandineni & Gowrishankar, 2004, Kutukova et al., 2005, Doroshenko et al., 2007, Park et al., 2007, Pathania & Sardesai, 2015, Liu et al., 2015), so they might be assisted by our identification of AlaE as a primary exporter of the linear neutral class of amino acids.
The benefits of metabolite excretion can be masked by closed culture systems.
The excretion of excess cysteine was markedly more protective in strains in which the cysteine importer YhaO was absent. Why does E. coli excrete something if a competing importer will return it to the cytoplasm? In natural habitats—the gut, or surface waters—compounds that E. coli excretes will largely be diluted and lost to the environment. In a closed flask with high-density cultures, however, the extracellular compounds accumulate locally and reenter the cell. This is an example in which the experimental set-up can give a deceptive outcome.
The same issue pertains to the H2O2 that results from cysteine oxidation. Were this process to occur in the cytoplasm, it would threaten peroxide-sensitive enzymes. When it occurs after cysteine excretion, the resultant H2O2 would be lost to the open environments in which E. coli naturally grows. However, in a lab experiment the H2O2 gradually accumulates and reenters the cell; the AlaE-dependent excretion fails to shield the cell at all from the H2O2. A similar conclusion was drawn from the localization in the periplasm of monoamine oxidase (TynA), the only high-flux enzyme in E. coli that stoichiometrically produces H2O2 (Kumar & Imlay, 2013). This localization would spare the cytoplasm from H2O2 exposure in natural habitats, but in lab cultures every molecule of H2O2 ultimately flows into a cytoplasm, creating as much stress as if it had been generated there.
Conclusion: Disulfide stress is another problem derived from aeration of an anoxic world.
Bacteria rely upon enzyme machinery that is best suited to the anoxic habitats in which life evolved: their cytoplasmic biochemistry relies upon reduced sulfur, ferrous iron, and solvent-exposed flavins. Aeration occurred 2 billion years after the appearance of life, which is long after these features had become hard-wired into the cellular plan. The consequences were dire: flavin oxidation by molecular oxygen begat reactive oxygen species; oxidation of environmental iron made soluble iron vanishingly scarce. Both of these problems have been imperfectly solved by layers of adaptations—the evolution of scavenging and repair enzymes to defray ROS stress (Imlay, 2013), and of complex iron acquisition and storage strategies to cope with iron deficiency (Troxell & Hassan, 2013). We now see that the hazard of disulfide import appears to have elicited a similar string of adjustments.
To date most studies of intracellular disulfide stress have employed artificial stressors—most frequently, diamide, a synthetic compound that was designed for this purpose (Kosower et al., 1969). Workers have speculated that protein disulfide bonds might also derive from natural oxidants—in particular, molecular oxygen and/or hydrogen peroxide. Yet H2O2 is a slow oxidant of typical cysteine residues, with half-times of many days at physiological concentrations (Winterbourn & Metodiewa, 1999, Li & Imlay, 2018). Molecular oxygen is also slow, in part because the peptide linkage prevents the amino moiety from binding iron nearby. Hypochlorous acid (HOCl, a.k.a. bleach), which is generated by neutrophils, is a more avid former of disulfide bonds (Hawkins et al., 2003, Dahl et al., 2015, Ezraty et al., 2017), and this action might be important when bacteria enter regions of host inflammation. But cystine import may comprise the most stressful routine source of disulfide bonds that has been identified thus far.
Experimental Procedures.
Chemicals.
All proteinogenic amino acids (except for glycine), alanine dipeptide, cystine, norvaline, norleucine, 2-aminobutyrate, ampicillin, chloramphenicol, kanamycin, tetracycline, cysteamine, glutathione, homocysteine, N-acetyl-cysteamine, oxidized glutathione, guanidinium hydrochloride, thioglycolic acid, sulfosalicylic acid, hydrogen peroxide, catalase, horseradish peroxidase, p-nitrophenyl phosphate (pNPP), 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB), and o-nitrophenol-β-galactoside (ONPG) were purchased from Sigma. Glycine, potassium mono- and dibasic phosphates, magnesium sulfate, ammonium sulfate, sodium citrate, sodium chloride, and glucose were from Fisher. Coomassie reagent was from Thermo Scientific. 14C-labeled leucine was from PerkinElmer, Amplex Red was from Invitrogen, and X-gal was from RPI. Aminobutyrate dipeptide was synthesized by request in GenScript. Restriction and ligation enzymes were from New England Biolabs, and Qiagen kits were used for genomic and plasmid DNA preparation.
Strains and plasmids.
Strains and plasmids are listed in the supplementary tables 1 and 2. Mutations were moved by P1 transduction (Miller, 1972) with selection for associated drug markers and confirmation by PCR.
Tetracycline-controlled expression of tcyP and tcyLN was achieved by replacing each of the native promoters with the tetRA cassette that was amplified by PCR from the chromosome of strain JRE520; replacement was achieved by a modified lambda red recombinase method (Martin & Imlay, 2011, Merighi et al., 2005). TcyP was induced by addition of tetracycline with subsequent incubation for minimum 2 hours. Without tetracycline the level of TcyP with this construct, as judged by cysteine efflux rates upon cystine addition, was about 12% of the level when wild-type cells are grown in sulfate medium; after tetracycline addition, the TcyP level rose to 2.5 times the level in the wild-type cells. In the case of tcyLN, the insertion of the tetRA cassette involved deletion of the dcyD ORF as well. (See Fig. S9.) In PtetRA-tcyLN the native sequence leading to tcyL is retained, and expression is tetracycline-dependent. In PtetRAC-tcyLN (in KCI1382 and its derivatives) the likely Shine-Dalgarno sequence was altered in order to improve expression; the bases preceding the yecS ATG are no longer ccggaaacgctcATG (native) but rather ccggaggcgctcATG. In addition, a fortuitous G-to-T mutation in tetR created a stop codon 12 amino acids prematurely, disabling the repressor (Orth et al., 2000) and causing transcription to be tetracycline independent. The overall effect, as gauged by cystine import assays, was to increase TcyJLN turnover about five-fold higher than that of the native genes under sulfate growth conditions.
A plasmid overexpressing alaE was constructed. Linear DNA that included the alaE gene and 300 bp upstream of the gene was inserted into the BamHI and HindIII restriction sites of pBR322 using tgtgggatcccgcgactggcgatgccagtcgcgaaaagaa and tgtgaagcttcggcatgtaaaaatccacaatgtacaaaaa primers.
Growth conditions and rate calculations.
E. coli was grown in 37 °C minimal A medium (Miller, 1972) containing 0.2% glucose; this basic medium provides sulfate as the sulfur source. Amino acids were supplemented (0.25–0.5 mM) as specified in individual experiments. Salmonella typhimurium (LT2), Bacillus subtilis (RS24), and Pseudomonas aeruginosa (ATCC10145) were grown in minimal A medium supplemented with glucose and 18 standard amino acids (excluding Cys and Met). Saccharomyces cerevisiae (EG103) was grown in the same medium with the following modifications: sodium citrate was replaced by 0.2% sodium chloride; glucose concentration was 1%; calcium pantothenate, nicotinic acid, thiamine and pyridoxine were added (5 mg/l each); pH was adjusted to 5.0; and the temperature was 30 °C.
Relative growth rates were determined after the addition of potentially inhibitory compounds. Cells were grown in appropriate medium from OD 0.005 to 0.08–0.1 and then appropriate chemicals were added. Optical density was taken at the moment of addition of the agent and one hour later. Growth rates (hr−1) were calculated as ln(A1/A0) where A denotes the optical density of the culture at 600 nm.
Assays of thiol excretion.
Thiols were measured using the DTNB method (Imlay et al., 2015). Cells were grown to mid-exponential phase (0.2 OD) in media specified for each experiment. Cells were then collected by centrifugation and resuspended to OD 0.1 in fresh medium at 37°C (30 °C for S. cerevisae). EDTA (0.1 mM) was added to suppress cysteine oxidation, and 0.1 mM cystine was added. Every 4 minutes 1 ml aliquots were removed and filtered through 0.22 uM syringe filters. Filtrate (0.5 ml) was mixed with 0.5 ml of 0.4 mM DTNB solution in 50 mM KPi (pH 7.1). After 2 min of incubation, absorbance was measured at 412 nm. Cysteine was used to generate a standard curve.
Monitoring of thiol excretion at very low cystine concentrations was done in DU-640 Beckman spectrophotometer. The cell culture was cultured to 0.1 OD in minimal A medium supplemented with 18 amino acids. DTNB (0.2 mM) was added, and then an appropriate amount of cystine was added and the rate of absorbance increase was determined.
To examine the effect of cystine pre-exposure on the thiol efflux, cells were grown in minimal A glucose medium supplemented with 0.25 mM of isoleucine to OD 0.1, then diluted to 0.02 OD. Sub-cultures were grown for three hours, and during this period, cystine (0.6 mM) was added at a defined interval before measurement of thiol efflux. For efflux measurement, chloramphenicol (0.1 mg/ml) was added 2 min before harvesting and was included during all washings and measurements. Cells were centrifuged at 10000 g for 6 min, washed twice with minimal A medium at 4 °C, and then resuspended for thiol-efflux measurements using the DTNB method described above.
Measurements of thiol oxidation in vitro.
The oxidation of thiol compounds in model system was measured in 100 mM Tris buffer (pH 7.5) in 50 ml glass flasks at 37°C in a shaking water bath. All flasks used in a single experiment were rinsed first with distilled/deionized water and then with 30 ml of the buffer. All the buffer used for the flask rinses was collected in a single glass beaker and then was employed for the experiment, ensuring that the trace metal levels were equivalent in all samples. Thiols (0.2 mM final concentration) were added to 10 ml of prewarmed buffer in each flask. At intervals, 0.5 ml aliquots were removed and mixed with 0.5 ml of Tris-DTNB solution, and absorbance was determined at 412 nm.
Amino acid influx and efflux.
Cells were cultured into exponential phase in amino-acid-free minimal A glucose medium. The cells were centrifuged and then suspended to 0.4 OD in the same warm medium containing 50 μg/ml chloramphenicol, and rate of leucine import was quantified upon the addition of 0.03 mM of 14C-leucine (25 mCi/mmol) to 1 ml of cell suspension. Each min 0.1 ml aliquots were filtered through nitrocellulose membranes. Membranes were washed twice with 5 ml of ice-cold KPi buffer (50 mM, pH 7.1) and then dried under a heat lamp. Radioactivity was measured in a scintillation counter.
The rate of leucine export was measured by loading the cell suspension with labeled leucine for 5 minutes as described above and then blocking further influx of 14C-leucine by adding unlabeled leucine to the final concentration of 3 mM (effecting a 100-fold dilution of radioactive leucine). Leucine efflux could then be monitored by the progressive diminution of cell-associated radioactivity. The rate constant for leucine export was determined using the equation
Where C(leu) represents the current concentration of radiolabeled leucine in the bacterial cytoplasm, k is the rate constant of the first-order export reaction, t is time, and B is the concentration of leucine somehow unavailable for export (i.e. membrane bound, incorporated into protein, partially degraded, representing < 15% of the radioactivity). The absolute intracellular free leucine concentration was calculated using the information that 1 liter of 1.0. OD600 E. coli contains 0.5 ml total cytoplasm (Imlay & Fridovich, 1991); B was determined experimentally as the residual value of cytosolic radioactivity after 7 min of export. The rate constant k was estimated using experimental data for the first two minutes of export. At the steady-state (about 7 mM) the rate of leucine export (k × 7 mM) is equal to the rate of the import, allowing calculation of the import rate.
A previous study determined that 64 micromolar sulfur atoms are needed in growth medium to achieve an optical density of 1.0 (Imlay et al., 2015). This cell mass represents 0.5 microliter of cytoplasm per ml of cells {Imlay, 1991 #1353}, indicating that the total intracellular sulfur-atom concentration is 130 mM. At the measured doubling time of 58 min, k = 0.012 min−1, and k × 130 mM = 1.56 mM-min−1. Because cystine provides two sulfur atoms, the demand for cystine as sole sulfur source is 0.78 mM-min−1. The total leucine content in E. coli is 1.84 times the cysteine + methionine content (Neidhardt & Unbarger, 1996), indicating a leucine demand of 1.44 mM-min−1.
Activation of purified reduced APase by disulfide compounds in vitro.
To prepare E. coli alkaline phosphatase that is reduced and inactive, the purified enzyme (200 U, 4 mg) (Sigma catalog # P5931) was denatured with guanidinium HCl and reduced with β-mercaptoethanol, in 37 °C buffer under aerobic conditions as described (Walker & Gilbert, 1994). AP was then transferred to the anaerobic chamber. Guanidinium HCl and β-mercaptoethanol were removed using Amicon Ultra-0.5 (Millipore) Centrifugal Filter Device: 800 μL of the denatured APase was loaded onto two 30K spin columns, 400μL each. The samples were centrifuged at 14K × g at RT for 15 minutes, leaving 40 μL of sample. The filtrate was discarded, and 400 μL folding buffer was added (1 mM ZnAc2, 1 mM MgCl2, 100 mM Tris, pH 8). Columns were centrifuged as above. For protein collection, the columns were inverted and centrifuged into a fresh tube, 1000 × g for 2 minutes. The volume was adjusted to 400 μL with folding buffer. The remaining reduced, inactive enzyme was kept on ice in the anaerobic chamber to avoid activation in air; it was stable in this form for at least a month.
To probe the ability of disulfide compounds to transfer disulfide bonds to AP, the reduced AP was diluted 50-fold into 1 mL reactions volumes of folding buffer with 0.5 mM GSSG or 0.5 mM cystine added. The reaction was executed at 37°C in an anaerobic chamber, and at intervals aliquots were removed and assayed for AP activity. AP activity was determined at A405 in 1-mL reactions in 1 M Tris, pH 8, and 0.4% p-nitrophenyl phosphate under aerobic conditions (Brickman & Beckwith, 1975). This degree of oxygen exposure did not itself cause detectable activation of AP.
Activation of cAP and OxyR in vivo during cystine import into cells.
Intracellular activation of alkaline phosphatase was monitored using strains bearing a plasmid (pAID135) that encodes a leader-less form of alkaline phosphatase, which is therefore localized in the cytoplasm (cAP) (Derman et al., 1993). For experiments with aerobic cells, strains bearing the cAP-expressing plasmid (WP551) were grown in minimal A glucose medium containing 18 amino acids (no Cys or Met) and 200 μg/mL ampicillin for four generations to OD600 = 0.2. Cells were diluted to OD = 0.05 into fresh medium, and 5 mM IPTG was added. Cultures were grown for two more doublings to OD = 0.2, split to fresh medium with or without 0.5 mM cystine, and grown for 40 minutes. Cultures (15 ml) were then pelleted in ice-chilled tubes, washed in ice cold wash-lysis buffer (20 mM Tris, pH 8, 1 mM EDTA), re-suspended in 1.5 mL ice-cold wash-lysis buffer, and lysed by French press. The extract was cleared of cell debris by centrifugation for 20 minutes at 4 °C in a microcentrifuge. The supernatant was collected and assayed for AP activity as described in the preceding section.
We wished to confirm that oxygen was uninvolved in intracellular AP activation and that cystine, rather than cysteine, drove enzyme activation. To do so, a form of the experiment was repeated with minor changes under anoxic conditions. Strain KCI1842 (as WP551, transduced to Leu+) was grown in anoxic minimal A glucose medium containing 0.5 mM histidine and 200 μg/mL ampicillin for four doublings to OD600= 0.2. Amino acid-free medium was used because branched-chain amino acids impair the import of cysteine; histidine was supplied because MG1655 is a histidine bradytroph under anoxic conditions. The cells were then sub-cultured to OD600 = 0.05 in the same medium supplemented with 5 mM IPTG, to induce expression of cAP from pAID135, for two doublings. The culture was split and, where indicated, supplemented with 0.5 mM cysteine or 0.5 mM cystine. After one doubling cells were washed and harvested as described above, albeit with anoxic buffers, and the supernatant was used for the AP activity assay.
The activation of the OxyR transcription factor was detected by the induction of a katG-lacZ transcriptional fusion (Liu & Imlay, 2013). To avoid any involvement of H2O2, the natural activator of OxyR, the experiments were conducted in an anaerobic chamber. Fifteen-ml cultures were inoculated to 0.003 OD in minimal A glucose supplemented with 18 amino acids. After four doublings, 0.5 mM cystine was added to cells at 0.05 OD. After two more generations of growth, cells were centrifuged, washed with ice-cold 50 mM Tris, pH 8, resuspended in 1.5 ml of the same buffer, and lysed by French press. Lysates were clarified by centrifugation, and β-galactosidase assays were performed by tracking 420 nm in a RT reaction containing Z buffer (Miller, 1972), 0.67 mg/ml ONPG, and extract.
Measurements of hydrogen peroxide production.
The strain LC106, which lacks the H2O2 scavenging catalases and NADH peroxidase (katG katE ahpCF) (Seaver & Imlay, 2004), was grown in minimal A medium with glucose and non-sulfurous amino acids prior to the addition of cystine (see Media). Hydrogen peroxide that is formed either inside or outside these cells accumulates in the growth medium. At timepoints, aliquots were removed, and H2O2 was quantified using the horseradish peroxidase/Amplex UltraRed assay (Messner & Imlay, 2002).
Growth curves that detect H2O2 stress.
Cultures were grown overnight in the anaerobic chamber, 37°C, in minimal A glucose medium containing 0.5 mM histidine, phenylalanine, tyrosine and tryptophan. Pre-cultures were grown anaerobically for 4 doublings to an OD600 = 0.1 in the same medium. Cells were then inoculated into the same base aerobic media to OD600 = 0.007 and grown with shaking, 37°C. Where indicated, the aerobic medium was supplemented with 0.5 mM cystine and/or 0.5 mM isoleucine, leucine, and valine.
RNA sequencing analysis.
Three independent cultures of the wild-type strain MG1655 were grown for > 6 generations to 0.1 OD in minimal A glucose medium supplemented with His, Trp, Tyr, and Phe. The cultures were divided, and 50 μM cystine was added to one flask of each pair. After 10 min the RNA from the triplicate pairs was harvested and sequenced, exactly as described (Imlay et al., 2015), as that reference presents data from an accompanying experiment.
LC-MS determination of cytosolic cysteine.
MG1655 and its ΔalaE derivative (SSK226) were cultured in amino acid-free minimal A glucose medium and suspended at 0.5 OD in the same warm medium. Cystine (20 μM) was added for 3 minutes, and then metabolites were extracted and quantified by LC-MS as described (Imlay et al., 2015).
Cell killing by cystine/H2O2 treatment.
Cells were grown in appropriate medium to OD 0.1 and then diluted to 0.001 OD in 5 ml of the same pre-warmed (37 °C) medium. Where indicated, alanine dipeptide (0.1 mM) was added. After 15 min of incubation 0.1 mM of cystine was added followed 1 min later by 4 mM of hydrogen peroxide. At time points 0.1 ml aliquots were transferred to 0.5 ml of minimal A glucose medium (room temperature) containing 2000 U/ml catalase. After twenty minutes cells were plated in top agar. Both agar and plates were of minimal A glucose basis. In some experiments cells (0.02 OD) were pre-incubated with 0.06 mM cystine for 10 min at 37 °C on a shaker. They were then diluted 1:20 into pre-warmed minimal A glucose medium, and after 1 minute 4 mM H2O2 was added.
Expression of alaE and dadA.
The ability of amino acid supplements to trigger induction of alaE and dadA genes was determined using translational lacZ fusions. Fusions of lacZ to alaE and dadA were made as described (Ellermeier et al., 2002) using the pCE40 vector. JW2645 and JW1178 strains from the Keio collection were used as the sources of the original gene deletions, which were transduced to SJ130, a ΔlacZ derivative of MG1655. The original kanr cassettes then were eliminated using pCP20 plasmid (Datsenko & Wanner, 2000), then the corresponding pCP20-containing strain was transformed by pCE40 plasmid. Kan-resistant colonies were tested on LB plates using X-gal, and the correct insertion was confirmed by PCR.
To test induction of the lacZ fusion, 35 ml of cells were grown in minimal A glucose medium to 0.1 OD, and either 0.2 mM or 6 mM of the tested amino acid was then added. Because of their toxicity, norvaline was added at 1 mM and cysteine at 0.6 mM. In experiments that tested gene induction by cystine and cysteine, 0.05 mM isoleucine was included in the growth medium to circumvent growth inhibition. Norleucine was added at 12 mM because of its apparently poor transport. Dipeptides were added at 0.2 mM and 0.4 mM concentrations; however, in all cases no difference in effect between these concentrations was noticed. After the additions cells were incubated for 60 minutes and then collected by centrifugation. Cells were lysed by French press, and lysates were spun for 10 min at 13000 G to get rid of the debris. B-galactosidase activity was measured using the ONPG-based method (Miller, 1972): 0.1 ml of lysate was added to 1 ml of 100 mM KPi buffer (pH 7.1), and the reaction was started by addition of 0.2 mL of ONPG solution in the same buffer (8 mg/ml). Absorbance was monitored at 420 nm for 10 minutes. Total protein was measured using a Coomassie blue reagent (Protein Assay Reagent) from Thermo-Fisher.
Statistics.
Error bars represent the standard error of the mean from triplicate biological samples. * P < 0.05; ** P < 0.01; *** P < 0.001.
Supplementary Material
Acknowledgments.
We are grateful to Stefano Mancini for his assistance with the RNA seq analysis. This study was funded by grant GM101012 from the National Institutes of Health.
References.
- Akiyama Y, and Ito K. (1993) Folding and assembly of bacterial alkaline phosphatase in vitro and in vivo. J. Biol. Chem 268: 8146–8150. [PubMed] [Google Scholar]
- Anbar AD. (2008) Elements and evolution. Science 322: 1481–1483. [DOI] [PubMed] [Google Scholar]
- Bagiyan GA, Koroleva IK, Soroka NV, and Ufimtsev AV. (2003) Oxidation of thiol compounds by molecular oxygen in aqueous solutions. Russ. Chem. Bull 52: 1135–1141. [Google Scholar]
- Brickman E, and Beckwith J. (1975) Analysis of the regulation of Escherichia coli alkaline phosphatase synthesis using deletions and phi80 transducing phages. J. Mol. Biol 96: 307–316. [DOI] [PubMed] [Google Scholar]
- Collet JF, and Bardwell JC. (2002) Oxidative protein folding in bacteria. Mol. Microbiol 44: 1–8. [DOI] [PubMed] [Google Scholar]
- Dahl J-U, Gray MJ, and Jakob U. (2015) Protein quality control under oxidative stress conditions. J. Mol. Biol 427: 1549–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman AI, and Beckwith J. (1991) Escherichia coli alkaline phosphatase fails to acquire disulfide bonds when retained in the cytoplasm. J. Bacteriol 173: 7719–7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman AI, Puziss JW, P J Bassford J, and Beckwith J. (1993) A signal sequence is not required for protein export in priA mutants of Escherichia coli. EMBO J 12: 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroshenko V, Airich L, Vitushkina M, Kolokolova A, Livshits V, and Mashko S. (2007) YddG from Escherichia coli promotes export of aromatic amino acids. FEMS Microbiol. Lett 275: 312–318. [DOI] [PubMed] [Google Scholar]
- Ellermeier CD, Janakiraman A, and Slauch JM. (2002) Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290: 153–161. [DOI] [PubMed] [Google Scholar]
- Ezraty B, Gennaris A, Barras F, and Collet J-F. (2017) Oxidative stress, protein damage, and repair in bacteria. Nat. Rev. Microbiol 15: 385–396. [DOI] [PubMed] [Google Scholar]
- Forte E, Borisov VB, Falabella M, Colaco HG, Tinajero-Trejo M, Poole RK, Vicente JB, Sarti P, and Giuffre A. (2016) The terminal oxidase cytochrome bd promotes sulfide-resistant bacterial respiration and growth. Sci. Rep 6: 23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke I, resch A, Dassler T, Maier T, and Bock A. (2003) YfiK from Escherichia coli promotes export of O-acetylserine and cysteine. J. Bacteriol 2003: 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman HC, Moore CJ, Jackson WG, and Sargeson AM. (1978) Synthesis, structure, and steriochemistry of some cysteine- and penicillaminecobalt(III) complexes. Inorg. Chem 17: 3513–3521. [Google Scholar]
- Hanson RL, and Rose C. (1980) Effects of an insertion mutation in a locus affective pyridine nucleotide transhydrogenase (pnt::Tn5) on the growth of Escherichia coli. J. Bacteriol 141: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CL. (1981) Cysteine and growth inhibition of Escherichia coli: threonine deaminase as the target enzyme. J. Bacteriol 145: 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins CL, Pattison DI, and Davies MJ. (2003) Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 25: 259–274. [DOI] [PubMed] [Google Scholar]
- Holbrook EL, Greene RC, and Krueger JH. (1990) Purification and properties of cystathionine gamma-synthase from overproducing strains of Escherichia coli. Biochemistry 29: 435–442. [DOI] [PubMed] [Google Scholar]
- Hori H, Yoneyama H, Tobe R, Ando T, Isogai E, and Katsumata R. (2011) Inducible L-alanine exporter encoded by the novel gene ygaW (alaE) in Escherichia coli. Appl. Environ. Micro 77: 4027–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara K, Sato K, Hori H, Makino Y, Shigenobu S, Ando T, Isogai E, and Yoneyama H. (2017) Expression of the alaE gene is positively regulated by the global regulator Lrp in response to intracellular accumulation of L-alanine in Escherichia coli. J. Biosci. Bioeng 123: 444–450. [DOI] [PubMed] [Google Scholar]
- Imlay JA. (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol 11: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, and Fridovich I. (1991) Assay of metabolic superoxide production in Escherichia coli. J. Biol. Chem 266: 6957–6965. [PubMed] [Google Scholar]
- Imlay KRC, Korshunov S, and Imlay JA. (2015) The physiological roles and adverse effects of the two cystine importers of Escherichia coli. J. Bacteriol 197: 3629–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, and Imlay JA. (2007) Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem 282: 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov S, Imlay KRC, and Imlay JA. (2016) The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol 101: 62–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosower NS, Kosower EM, and Wertheim B. (1969) Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem. Biophys. Res. Commun 37: 593–596. [DOI] [PubMed] [Google Scholar]
- Kredich NM. (1992) The molecular basis for positive regulation of cys promoters in Salmonella typhimurium and Escherichia coli. Molecular Microbiology 6: 2747–2753. [DOI] [PubMed] [Google Scholar]
- Kredich NM, (1996) Biosynthesis of cysteine In: Escherichia coli and Salmonella. Neidhardt FC. (ed). Washington, D.C.: ASM Press, pp. 514–527. [Google Scholar]
- Kruse D, Kramer R, Eggeling L, Rieping M, Pfefferle W, Tchieu JH, Chung YJ, M H Saier J, and Burkovski A. (2002) Influence of threonine exporters on threonine production in Escherichia coli. Appl. Microbiol. Biotechnol 59: 205–210. [DOI] [PubMed] [Google Scholar]
- Kumar SR, and Imlay JA. (2013) How Escherichia coli tolerates profuse hydrogen peroxide formation by a catabolic pathway. J. Bacteriol 195: 4569–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutukova EA, Livshits VA, Altman IP, Ptitsyn LR, Zyiatdinov MH, Tokmakova IL, and Zakataeva NP. (2005) The yeaS (leuE) gene of Escherichia coli encodes an exporter of leucin, and the protein regulates its expression. FEBS Lett 579: 4629–4634. [DOI] [PubMed] [Google Scholar]
- Li X, and Imlay JA. (2018) Improved measurements of scant hydrogen peroxide enable experiments that define its threshold of toxicity for Escherichia coli. Free Rad. Biol. Med 120: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Liang Y, Zhang Y, Shang X, Liu S, WEn J, and Wen T. (2015) YjeH is a novel exporter of L-methionine and branched-chain amino acids in Escherichia coli. Appl. Environ. Microbiol 81: 7753–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, and Imlay JA. (2013) Cell death from antibiotics without the involvement of reactive oxygen species. Science 339: 1210–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JE, and Imlay JA. (2011) The alternative aerobic ribonucleotide reductase of Escherichia coli, NrdEF, is a manganese-dependent enzyme that enables cell replication during periods of iron starvation. Mol. Microbiol 80: 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew E, Zhi J, and Freundlich M. (1996) Lrp is a direct repressor of the dad operon in Escherichia coli. J. Bacteriol 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi M, Ellermeier CD, Slauch JM, and Gunn JS. (2005) Resolvase-in vivo expression technology analysis of the Salmonella enterica serovar Typhimurium PhoP and PmrA regulons in BALB/c mice. J Bacteriol 187: 7407–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner KR, and Imlay JA. (2002) In vitro quantitation of biological superoxide and hydrogen peroxide generation. Meth. Enzymol 349: 354–361. [DOI] [PubMed] [Google Scholar]
- Mihara H, Kurihara T, Yoshimura T, and Esaki N. (2000) Kinetic and mutational studies of three NifS homologs from Escherichia coli: mechanistic difference between L-cysteine desulfurase and L-selenocysteine lyase reactions. J. Biochem 127: 559–567. [DOI] [PubMed] [Google Scholar]
- Miller JH, (1972) Experiments in Molecular Genetics Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. [Google Scholar]
- Nandineni MR, and Gowrishankar J. (2004) Evidence for an arginine exporter encoded by yggA (argO) that is regulated by the LysR-type transcriptional regulator ArgP in Escherichia coli. J. Bacteriol 186: 3539–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, and Unbarger HE. (1996). Chemical composition of Escherichia coli In: Escherichia coli and Salmonella. Edited by Neidhardt FC. et al. ASM Press, Washington, D.C. [Google Scholar]
- Oguri T, Schneider B, and Reitzer L. (2012) Cysteine catabolism and cysteine desulfhydrase (CdsH/STM0458) in Salmonella enterica serovar typhimurium. J. Bacteriol 194: 4366–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu I, Wiiyathanawudhiwong N, Morigasaki S, Nakatani T, Kadokura H, and Takagi H. (2010) The L-cysteine-L-cystine shuttle system provides reducing equivalents to the periplasm in Escherichia coli. J. Biol. Chem 285: 17479–17487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth P, Schnappinger D, Hillen W, Saenger W, and Hinrichs W. (2000) Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system Nat. Struct. Biol 7: 215–219. [DOI] [PubMed] [Google Scholar]
- Ostrowski J, and Kredich NM. (1989) Molecular characterization of the cysJIH promoters of Salmonella typhimurium and Escherichia coli: regulation by CysB protein and N-acetyl-L-serine. J. Bacteriol 171: 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Lee KH, Kim TY, and Lee SY. (2007) Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. USA 104: 7797–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, and Imlay JA. (2003) High levels of intracellular cysteine promote oxidative DNA damage by driving the Fenton reaction. J. Bacteriol 185: 1942–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania A, and Sardesai AA. (2015) Distinct paths for basic amino acid export in Escherichia coli: YbjE (LysO) mediates export of L-lysine. J. Bacteriol 197: 2036–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollitt S, and Zalkin H. (1983) Role of primary structure and disulfide bond formation in b-lactamase secretion. J. Bacteriol 153: 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaver LC, and Imlay JA. (2004) Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J. Biol. Chem 279: 48742–48750. [DOI] [PubMed] [Google Scholar]
- Shimada T, Tanaka K, and Ishihama A. (2016) Transcription factor DecR (YbO) controls detoxification of L-cysteine in Escherichia coli. Microbiology 162: 1698–1707. [DOI] [PubMed] [Google Scholar]
- Soini J, Falschlehner C, Liedert C, Bernhardt J, Vuoristo J, and Neubauer P. (2008) Norvaline is accumulated after a down-shift of oxygen in Escherichia coli W3110. Microb. Cell Fact 7: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EJ, Aslund F, and Beckwith J. (1998) Disulfide bond formation in the Escherichia coli cytoplasm: an in vivo role reversal for the thioredoxins. EMBO J 19: 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S, Perdreau-Dahl H, and Morth JP. (2016) The magnesium transporter A is activated by cardiolipin and is highly sensitive to free magnesium in vitro. Elife 5: e11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Koyanagi T, Izuka S, Onishi A, and Kumagai H. (2005) The yliA, -B, -C, and -D genes of Escherichia coli K-12 encode a novel glutathione importer with an ATP-binding cassette. J. Bacteriol 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troxell B, and Hassan HM. (2013) Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front. Cell Infect. Microbiol 3: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KW, and Gilbert HF. (1994) Effect of redox environment on the in vitro and in vivo folding of RTEM-1 B-lactamase and Escherichia coli alkaline phosphatase. J. Biol. Chem 269: 28487–28493. [PubMed] [Google Scholar]
- Winterbourn CC, and Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Rad. Biol. Med 27: 322–328. [DOI] [PubMed] [Google Scholar]
- Yamada S, Awano N, Inubushi K, Maeda E, Nakamori S, Nishino K, Yamaguchi A, and Takagi H. (2006) Effect of drug transporter genes on cysteine export and overproduction in Escherichia col. Appl. Environ. Microbiol 72: 4735–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JG, and Rees DC. (2015) The allosteric regulatory mechanism of the Escherichia coli MetNI methionine ATP binding cassette (ABC) transporter. J. Biol. Chem 290: 9135–9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CM, Christian T, Newberry KJ, Perona JJ, and Hou YM. (2003) Zinc-mediated amino acid discrimination in cysteinyl-tRNA synthetase. J. Mol. Biol 327: 911–917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.