

Abstract
Chlamydia trachomatis are obligate intracellular bacteria that undergo dynamic morphologic and physiologic conversions upon gaining access to a eukaryotic cell. These conversions likely require the detection of key environmental conditions and regulation of metabolic activity. Chlamydia encodes homologs to proteins in the Rsb phosphoregulatory partner-switch pathway, best described in Bacillus subtilis. ORF CT588 has strong sequence similarity to RsbU cytoplasmic phosphatase domain but also contains a unique periplasmic sensor domain that is expected to control phosphatase activity. A 1.7 Å crystal structure of the periplasmic domain of the RsbU protein from C. trachomatis (PDB 6MAB) displays close structural similarity to DctB from Vibrio and Sinorhizobium. DctB has been shown both structurally and functionally to specifically bind to the TCA cycle intermediate succinate. Surface plasmon resonance and differential scanning fluorimetry of TCA intermediates and potential metabolites from a virtual screen of RsbU revealed that alpha-ketoglutarate, malate, and oxaloacetate bound to the RsbU periplasmic domain. Substitutions in the putative binding site resulted in reduced binding capabilities. A RsbU-null mutant showed severe growth defects which could be restored through genetic complementation. Chemical inhibition of ATP synthesis by oxidative phosphorylation phenocopied the growth defect observed in the RsbU null strain. Altogether, these data support a model with the Rsb system responding differentially to TCA cycle intermediates to regulate metabolism and key differentiation processes.
Graphical Abstract
Introduction
Bacteria possess the ability to sense changes in environmental conditions and adjust biologic activities through diverse regulatory components and mechanisms [1]. Often times these reactions are in responses to general environmental stresses as is the case with the Regulator of Sigma B or Rsb system. This phosphoregulatory partner switching system is found mainly in Firmicutes and is most thoroughly described in Bacillus subtilis [2]. A central regulatory component in this system is a phosphatase termed RsbU. Under stressful conditions, such as nutrient depletion, RsbU dephosphorylates a serine on an intermediate protein partner, RsbV. This allows another protein partner and kinase, RsbW, to ‘switch’ from association with sigma B to rephosphorylate RsbV. Ultimately, this enables the alternative sigma factor to freely diffuse and form an RNA holoenzyme polymerase, which activates the transcription of over a hundred genes that assist with the response to environmental stress [3–10]. While the Rsb system is typically associated with general stress responses in Firmicutes, it has also been associated with regulating diverse processes in other bacteria phyla including biofilm formation, type III secretion, and swarming motility [11, 12].
Chlamydia undergo dynamic morphologic and physiologic conversions upon gaining access to a eukaryotic cell. These conversions occur as Chlamydia grows and propagates through a phylum-defining biphasic developmental cycle. The initial phase of the chlamydial developmental cycle is conversion from an infectious, non-replicative and metabolically inert form known as an elementary body (EB) to a non-infectious, metabolically active and replicative form, known as a reticulate body (RB). This conversion occurs upon gaining access to the cell and establishing the intracellular vacuole termed the inclusion [13]. Many ATP requiring processes occur during the EB to RB conversion including protein secretion and de novo transcription and translational activity. RBs also need to acquire most macromolecules from the host cell, including glucose-6-phosphate, nucleotides, amino acids, lipids, and other metabolic precursors for growth and multiple rounds of replication [14]. The second phase is the asynchronous RB to EB conversion which occurs later in the developmental cycle through unknown signals and poorly understood mechanisms. This conversion also requires coordinated events that includes membrane remodeling and infectious capability preparation while metabolic processes, including transcription and translation, are silenced [15, 16]. Chlamydia also organize the escape from the infected host cell through either cell lysis or extruding vacuoles which enables the infection of new cells and possible a new host.
Chlamydia appear to acquire ATP from the host cell using ATP translocases and can also generate ATP through unique substrate-level and oxidative phosphorylation processes. Interestingly, these processes appear to be functional at different developmental stages. ATP stored in EBs may allow for initial protein secretion and transcription and translation processes to occur until RB conversion. After the initial entry into the host cell, ATP translocases are utilized to obtain ATP from the host cell [17]. During RB replication and midcycle growth stage, Liang et al. (2018) demonstrated that Chlamydia can also generate ATP using a sodium-ion gradient to drive the ATP-synthase [17]. Critical for this process is the TCA cycle [17]. Chlamydia spp. lack three canonical TCA enzymes: citrate synthase (gltA), aconitase (acn), and isocitrate dehydrogenase (icd) [18, 19]. Due to the absence of these enzymes, Chlamydia possess a truncated TCA cycle that starts with alpha-ketoglutarate and ends with oxaloacetate that can then be shuttled to other metabolic pathways [14]. This truncated TCA cycle does enable the chlamydial RBs to generate NADH, which drives the sodium-dependent NADH dehydrogenase and, subsequently, ATP generation [17]. However, because of the incomplete TCA cycle, Chlamydia must scavenge dicarboxylate intermediates, such as glutarate and alpha-ketoglutarate, from the host cell [18–20]. Consequently, there are substantial interactions between the parasitic chlamydial cells and the infected host cell. The intimate association between the host and chlamydial metabolisms suggests that signaling pathways in Chlamydia responding to the host’s metabolic milieu play critical roles in development and pathogenesis. Despite the likely importance of these signals, much of the basic biology of these pathways remains poorly understood, including the signal for the EB-to-RB conversion, mechanisms for sensing environmental stimuli, and the differential regulation of ATP acquisition.
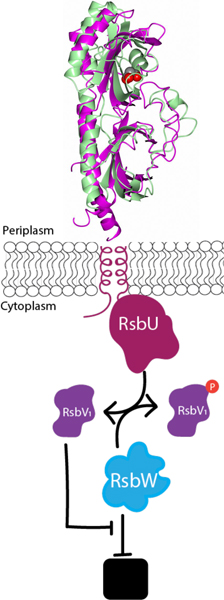
A partner-switching pathway with similarities to the Rsb regulatory pathway could be a primary mechanism for Chlamydia to sense environmental conditions and regulate metabolic activity [19]. The chlamydial genome encodes genes for the production of RsbU (CT588), RsbV1 (CT424), RsbV2 (CT765), and RsbW (CT549) proteins (Figure 1) [19]. However, there are distinct differences from the canonical Rsb system in B. subtilis. For one, chlamydial RsbU is a transmembrane protein situated in the inner membrane with a periplasmic sensor domain attached to a cytoplasmic phosphatase domain (Figure 2) [21–23]. In contrast, RsbU in B. subtilis are strictly cytoplasmic and do not contain a sensor domain, only possessing a phosphatase domain. It is expected that the chlamydial RsbU sensor domain is critical for controlling the phosphatase activitiy and downstream regulatory processes. Chlamydial RsbU has been shown to de-phosphorylate RsbV1 but not RsbV2 [22]. Importantly, multiple studies on the biologic and functional outcomes of the terminal component and kinase, RsbW, support binding and inhibiting the primary sigma factor, σ66 [21–23]. The expected result of this activity would be a global shutdown of most transcription in Chlamydia.
Figure 1. The current model of the Rsb phospho-switching pathway in Chlamydia.

RsbW binds and inhibits the activity of a target protein (black box). However, when RsbV1 is dephosphorylated, RsbW will release its target protein to act as a kinase to phosphorylate RsbV1 (Hua, 2006; Thompson, 2015). RsbU acts as an antagonist of RsbW by dephosphorylating RsbV1 in response to binding a ligand in the periplasm (Thompson, 2015). Ultimately, the binding of the ligand to the RsbU protein leads to the release of the target protein.
Figure 2. Domain organization of RsbU from C. trachomatis and homologs from other bacteria.

RsbU from C. trachomatis bears sequence similarity to RsbU from B. subtilis in the cytoplasmic domain, both containing PP2C domains. RsbU in B. subtilis, however, does not contain any transmembrane helices, nor a periplasmic portion. Structural comparison of the periplasmic portion of RsbU reveals similarity to the periplasmic domain of DctB proteins in V. cholerae and S. meliloti. Amino acids are numbered at the beginning and end of domains. TM denotes transmembrane helices.
To discover the potential binding ligands and response regulatory role of the Rsb system in Chlamydia, a crystal structure of the RsbU periplasmic domain was determined. This structure was used to identify structurally similar proteins for putative function predictions as well as direct virtual and experimental ligand-binding analyses. Growth phenotypes of RsbU null mutant strains and in the presence of chemical inhibitors of key ATP generating functions were evaluated. Together, these observations support that RsbU is binding to TCA cycle intermediates and may play a role in global gene regulation in Chlamydia.
Results
CT588 has a unique domain organization with conserved cytoplasmic RsbU phosphatase domain and an uncharacterized domain predicted to localize to the periplasm.
CT588 is a 650-residue protein with a carboxyl-terminal domain (269–645) that has high sequence similarity to the RsbU superfamily phosphatases (Figure 2). This cytoplasmic domain contains HAMP (residues 338–385) and PP2C serine phosphatase (residues 422–625) subdomains with Smart E-values of 4.35 × 10−5 and 3.09 × 10−72, respectively [24]. The HAMP and PP2C domains together comprise a conserved RsbU family domain (residues 269–645, E-value 5.23 ×10−99) [24] in support of original protein annotation. HAMP domains function as linker regions in order to modulate the transduction between sensor and effector domains [25]. This transduction can occur with cytosolic as well as membrane associated proteins [26]. PP2C domains are metal-dependent protein phosphatases (PPMs), which catalyze the dephosphorylation of either a serine or threonine residue [27].
BLAST search using the N-terminus of CT588 (1–315) revealed sequence similarity to proteins only encoded by Chlamydia; however, the predicted function of these orthologs were unknown. In contrast to other RsbU family members, CT588 has two transmembrane helices that flank residues 40 through 315, which implies that this domain is localized to the periplasm. Based on sequence similarities, it is expected that a periplasmic signal is transduced by the HAMP domain to regulate the PP2C phosphatase activity of CT588. However, while this domain organization of CT588 appears to be unique among bacteria, this protein is widely conserved among Chlamydiaceae family.
I-TASSER was used to model protein structures for the N-terminus RsbU [28]. Four structural models with relatively poor C-scores (range from −3.19 to −3.84) were generated reflecting the absence of sequence similarity to PDB templates. These models predict two protein domains that are tethered to a single alpha helix, which extends the length of these domains (Figure S1). Using these models, DALI searches of the Protein Data Base (PDB) were performed to identify potential structural homologs and associated functional information. This search revealed that the top matches (Z-score > 15; range 16–21) were all periplasmic-localized chemoreceptors with PAS-like domains attached to kinase or methyl-accepting chemotaxis-like domains by linker regions such as HAMP or HisKA domains from diverse bacteria (Table S1). Many of these I-TASSER model structural homologs also had identified ligands that include L-Arginine, C4 dicarboxylates, and asparagine.
A 1.7 Å crystal structure of the RsbU periplasmic domain reveals similarity to periplasmic domains of dicarboxylate binding sensor proteins.
A crystal structure of the CT588 (RsbU) periplasmic domain was solved in order to better understand its function. A construct comprised of residues 45–313 (RsbU45–313) was recombinantly expressed and purified via affinity and size exclusion chromatography (Figure S2) and then used to screen for crystallization conditions which led to a 1.7 Å resolution crystal structure.
RsbU45–313 adopts a mixed α/β fold with two similar PAS subdomains, each containing antiparallel β-strands flanked by pairs of α-helices (Figure 3). The proximal domain contains five long and two short (two residues) β-strands and the distal domain is composed of five long and one short strand. The secondary structure elements for RsbU were calculated using DSSP [29]. Interestingly, the proximal and distal subdomains exhibit a high degree of structural similarity, reflected by a root mean square deviation (RMSD) value of 1.37 Å for 47 aligned Cα atoms (Z=7.73) [30, 31]. Additionally, the total accessible surface area of the subdomains, calculated using Areaimol via CCP4 [32], are similar with 5,021.6 Å for the distal (K114-D192) and 5,880.8 Å for the proximal subdomain (K210-E302). Another interesting feature in the RsbU structure is that helix α1 is kinked near I54/T55 (Figure 3A, right panel). The angle between the two portions of this helix defined by Q45-S53 (α1) and S56-T72 (α1’) was found to be 36.7° as calculated using least-squares fitting of Cα atoms with Pymol.
Figure 3. Ribbon model of RsbU periplasmic domain (residues 43–313) crystal structure (PDB 6MAB).

A) Tertiary structure showing helices (magenta) and b-sheets (green). The middle and right panels are views rotated 90° and 180° about the vertical direction. The two regions of the kinked helix are denoted as α1 and α 1’. B) Secondary structure annotation relative to the RsbU sequence.
A DALI search comparing the RsbU45–313 crystal structure to all Protein Database (PDB) entries identified 14 non-redundant matches based on global structural similarity (Z-score >3.0; Table S2). The top two hits (Z-scores >15.5) were of the sensor domain from the histidine kinase DctB in Vibrio cholerae and Sinorhizobium meliloti (3BY9 and 3E4O, respectively), which binds to C4 di-carboxylates (e.g. succinate). Looking broadly at the domain organization of RsbU from C. trachomatis compared to DctB in V. cholerae and S. meliloti, these proteins do appear similar in respect to the length of the periplasmic domain and the presence of two flanking transmembrane domains (Figure 2). DctB is the membrane-bound sensor histidine kinase of a two-component system in Rhizobia, Vibrio, Escherichia, and other bacteria that sense extracellular C4-dicarboxylates and reactively regulate their TCA cycle, one of the central metabolic processes [33]. C4-dicarboxylates are four-carbon small molecules, such as the TCA cycle intermediates malate and oxaloacetate. Once DctB senses its ligand, it phosphorylates and thereby activates the system’s response regulator DctD. Activated DctD then activates the expression of the σ54-dependent promoter of a C4-dicarboxylate:cation symporter, DctA [34–36]. Protein homologs of DctB, each of which are membrane bound kinases with periplasmic sensor domains, regulate a variety of responses beyond the TCA cycle as well [37–39].
Nine of the other matches from the DALI search were structures also co-crystalized with ligands. These ligands range from amino acids and other carboxylates to nucleic acids. The remaining three protein matches have no ligands solved in their binding site. These 14 structural matches all are predicted to be membrane-bound proteins with PAS-like domains attached to kinase or methyl-accepting chemotaxis-like domains by linker regions such as HAMP or HisKA domains [24]. They regulate a variety of downstream processes such as chemotaxis, sporulation, and differential metabolite utilization [37, 38, 40–42].
Residues in the DctB binding pocket are not conserved in the putative binding pocket of RsbU45–313.
As noted above, RsbU shares the highest degree of structural similarity with DctB. Superposition of DctB from V. cholera (3BY9) and S. meliloti (3E4O) with RsbU using Gesamt [43] yielded RMSD deviations of 2.58 Å and 3.38 Å between Cα atoms for 205 and 196 residues aligned, respectively (Figure 4A and B). The RMSD deviation between Cα atoms for RsbU and apo DctB (3E4Q) is 3.45 Å (196 residues). Given the structural similarity with DctB, we set out to determine if a similar ligand binding site was present in RsbU. DctB crystal structures from V. cholerae and S. meliloti have both been obtained with succinate bound in the ligand-binding pocket [34, 37]. Additionally, a structure of S. meliloti DctB has also been obtained as a complex with malonate. Although the structural similarity between RsbU45–313 and DctB from V. cholerae is greater, the availability of both apo and ligand-bound structures for DctB from S. meliloti allowed for a more in-depth comparison with RsbU45–313 using these structures. Zhou et al. [34] describe DctB as having an opened apo/C3−dicarboxylate (malonate) bound structure form, and a closed form when bound to a C4-dicarboxylate. The superposition of RsbU45–313 and apo and succinate-bound DctB is depicted in Figure 4C which highlights the ligand binding region. Specifically, when DctB binds to a C4-dicarboxylate, residues 136–153 and 169–175 close around the ligand. For DctB the binding of succinate leads to a 2.2Å movement in residues 169–175 towards the ligand [34]. However, for RsbU these loop regions, which correspond to 132–153 and 159–167, are in a more open position suggesting that a conformational change may occur upon ligand binding.
Figure 4. Superposition of RsbU (6MAB, magenta) with A) Apo (3E4Q, cyan) and B) succinate-bound (3E4O, green) DctB structure.

The succinate molecule is rendered as spheres to highlight the ligand binding region. C) Zoomed in view of the ligand binding pocket with succinate rendered as cylinders. There are evident differences between RsbU and DctB, which is more enclosed. However, structural comparison between the apo and succinate-bound forms of DctB from S. meliloti reveal that the linker between strands 3 and 4 (residues 169–175) shift in distance of 2.2 Å towards the binding pocket when succinate is bound, thereby facilitating pocket enclosure (Zhou et al).
Despite the structural similarity, there is less than 20% sequence identity between RsbU and DctB sensor domains, particularly around the binding site as a BLAST search yielded no significant conservation of this region. Additionally, the ligand binding pocket of DctB contains a large patch of positively charged residues whereas RsbU has both positive and negatively charged regions (Figure 5). Relative to the superimposed structures, S161 and S163 of RsbU are similarly located relative to T171 and S173 of DctB. Additionally, Y142 of RsbU is positioned in a similar location relative to Y149 of DctB. The position of several charged residues within the binding site differ between DctB and RsbU, as highlighted in Figure 5. In RsbU there are no positively-charged residues in the corresponding location of R152 which interacts with succinate (Figure 5C). Instead there is a negatively-charged residue, E145, located in a similar region. Additionally, K197 of DctB, which is located on the middle β-strand of the binding site β-sheet, forms a salt bridge with the dicarboxylate ligand. While there is not a positively-charged residue in the corresponding location on the same β-strand, K114 of RsbU is located on a neighboring β-strand (Figure 5D). Overall, RsbU contains eight charged residues at or around the putative ligand binding site. These include negatively-charged E145, E115, E250 and positively-charged residues K114, R134, K140, and R248 lining the perimeter of the pocket.
Figure 5. Residues in the ligand binding pocket of DctB (3E4O) and putative site of RsbU.

The succinate molecule is rendered as gray cylinders. A) Electrostatic surface for DctB and B) RsbU. C) Charged residues for DctB showing hydrogen bond interactions with succinate (dashed lines). D) Charged residues in the putative ligand binding pocket of RsbU.
I-TASSER ab initio model of CT588 closely matches crystal structure.
A pairwise structure comparison of the I-TASSER model and the RsbU1–315 structure reveals a robust Z-score of 14.4 and RMSD of 4.0 Å (Figure S1). These data support that, despite extremely low sequence similarity between RsbU1–315 and other proteins (maximum sequence similarity below 2% of the templates), I-TASSER was effective in accurately modeling this protein. This ab initio protein modeling is particularly challenging, especially for proteins over 200 amino acids [44]. Additionally, comparison between DALI searches of I-TASSER and crystal structure highlights that more than half of the top 15 proteins with structural similarity are shared, including DctB (Table S1).
Virtual screen of human metabolite and chlamydial metabolite libraries yielded a small list of potential ligands for further testing.
While the structure of RsbU is most similar to DctB, it shares the same fold as several other proteins that bind ligands other than dicarboxylates, such as amino acids, nitrogenous bases, and pyruvate (Table S3). Preliminary docking studies indicated that the C4-dicarboxylate succinate could interact with the positively charged sidechains of K140 and R134, however, these residues are located on the outer edge of the binding pocket, distal to where a tight-binding ligand would be expected to bind. In order to rationally select additional compounds for testing, a virtual screen was performed against compounds that are more likely to interact with by RsbU; namely human metabolites and metabolites associated with Chlamydia trachomatis.
Over 100,000 compounds were computationally screened, and a final library of 26 potential ligands was selected for further testing. This library was composed of the top scoring compounds from the human and chlamydial metabolite libraries supplemented with TCA cycle intermediates or derivatives present in Chlamydia (Table S3). The addition of the TCA cycle intermediates or derivatives was included in order to fully investigate the possibility of the chlamydial RsbU protein binding to a molecule of similar structure and function as the DctB ligand.
Binding studies support TCA cycle intermediates as RsbU ligands.
Surface plasmon resonance (SPR) was selected as a method of screening the library of potential ligands for binding to the RsbU45–313 periplasmic domain due to its sensitivity and ability to determine estimates of binding kinetics [45]. In initial screening with the 26 potential ligands at 100 μM and 1 mM concentration, binding was only observed for alpha-ketoglutarate, malate, and oxaloacetate (Figure S3). Subsequently, dose-dependent binding studies of these three potential ligands were performed. KD values were estimated to be 419 ± 76 µM, 459 ± 91 µM, and 396 ± 69 µM for alpha-ketoglutarate, malate, and oxaloacetate, respectively (Table 2).
Table 2.
Binding kinetic estimations (μM) for RsbU proteins
| Alpha-ketoglutarate |
Malate |
Oxaloacetate |
||||
|---|---|---|---|---|---|---|
| Protein | KD | SD | KD | SD | KD | SD |
| WT RsbU45–313 | 419 | 76 | 459 | 91 | 396 | 69 |
| R134A | 526 | 84 | 601* | 62 | 379 | 57 |
| Q137A | 548 | 112 | 666 | 233 | 459 | 96 |
| K140A | 597* | 120 | 836* | 54 | 569* | 144 |
| K140A/R134A | 558* | 68 | 741* | 49 | 580* | 97 |
SD = Standard deviation
p-value < 0.05 when compared to wild-type protein binding by a two-tailed student’s t-test.
Docking with the alpha-ketoglutarate, malate, and oxaloacetate to RsbU identified specific residues at the putative binding site that could be coordinating ligand binding (Figure 6). Residues R134, Q137, and K140 were predicted to interact with alpha-ketoglutarate, malate, and oxaloacetate. R248 is also in proximity to the ligands. Based on these predicted residue interactions, individual alanine substitutions in the RsbU45–313 protein were created for R134, Q137, and K140, as well as a double-substitution with R134 and K140. SPR was performed with the alanine-substituted proteins compared to the wild-type protein. Table 2 shows the average estimated KD values for the three TCA cycle intermediates with each of the protein variants. Both the signal-substitution variant, K140A, and the double-substitution variant, K140A/R134A, showed statistically-significant, albeit limited, decreases in binding affinity for the three TCA cycle intermediates. The R134A variant displayed a statistically-significant decrease in binding affinity for malate as well as lower binding capabilities to alpha-ketoglutarate (p-value 0.082). Similarly, the Q137A single-substitution also had decreases in binding affinity for alpha-ketoglutarate and malate (p-value <0.1).
Figure 6. Ligand docking showing the predicted binding modes of A) ketoglutarate, B) malate, and C) oxaloacetate in the binding pocket of RsbU.

All the ligands are rendered as gray cylinders. Interacting residues are annotated in panel A.
Orthogonal analyses using differential scanning fluorimetry (DSF) was also performed with of potential ligands [46]. Significant stabilizing temperature shifts were observed with 5 mM and 10 mM additions of alpha-ketoglutarate and malate (Table S4). Oxaloacetate only showed a significant positive temperature shift at the highest ligand concentration tested and only in one of two biological replicates. Additionally, a single trial of isothermal titration calorimetry (ITC) resulted in binding curves indicating stronger ligand binding with alpha-ketoglutarate, malate, and oxaloacetate (KD values of 25.8 μM, 22.0 μM, 55.5 μM, respectively), but not succinate or malonate (data not shown). Overall, all three of these binding studies support the binding of the RsbU periplasmic domain to TCA cycle intermediates alpha-ketoglutarate, malate, and oxaloacetate.
Nonsense mutation in rsbU gene suggests importance of Rsb pathway in chlamydial growth.
Based on the three ligands and the potential for the regulation of ATP generation (oxidative phosphorylation), it was hypothesized that the absence of this sensing system could be detrimental to the growth of Chlamydia. To evaluate this hypothesis, a C. trachomatis L2 EMS mutant (CTL2M401) was obtained (Dr. R. Valdivia; Duke University) that contained a SNP causing a nonsense mutation at W284 in the ct588 gene coding for the RsbU protein [47, 48]. This nonsense mutation occurs towards the C-terminal end of the periplasmic domain resulting in a truncated protein lacking the cytoplasmic domain. Western blot analysis using antibodies raised against the periplasmic domain supported the absence of the full-length RsbU, as well as any lack of truncated product, in this mutant strain and is deemed a null mutant (RsbU*; Figure 7A). Growth of this RsbU* null mutant strain was assessed with DNA harvested at 0, 12, 24, 36, 48, and 72 hours post-infection. Genome copy numbers were compared between Chlamydia (secY) and host (rpp30) (Figure 7B). Striking differences in the growth pattern of the mutant were observed compared to wild-type Chlamydia, with the mutant strain displaying minimal replication capabilities and generation of detectable infectious progeny (Figure 7B and 7D).
Figure 7. The Rsb pathway affects normal growth of Chlamydia.

(A) Western blot of WT and RsbU* expression of RsbU protein at 24 hours post-infection. No RsbU protein fragment is detected in the RsbU* mutant strain. MOMP is used as a loading control. (B) Chlamydia genome copy numbers (secY) were compared to host cell genome copy numbers (rpp30) over 72 hours after the initial infection. RsbU* appears to begin replicating around 72 hours post-infection (* p-value <0.05 with student’s T-test). (C) Growth curves with chemical inhibitors, HQNO and BKA, show significant differences between WT and WT +BKA after 24 hours, as well as RsbU* and WT +BKA (p-value >0.05). With HQNO added, both WT and RsbU* were not statistically different from RsbU* without any inhibitors. (D) Progeny assay looking at the difference in IFUs produced after 36 hpi compared to the IFUs in the initial infection. The decrease in IFUs produced by the RsbU* strain, as well as WT treated with HQNO, suggest that the chlamydial cells are largely in the RB form at this point in the infection rather than the EB form capable of propagating the infection to new host cells. While all three experimental conditions were significantly different from the WT untreated condition (**p-value <0.001 with student’s T-test), the RsbU* mutant compared to the HQNO treated infection shows no significant difference (p-value = 0.3). (E) The RsbU+ recombinant strain, with WT RsbU expression but retaining the majority of the other EMS-induced SMPs, restores growth to WT levels.
Whole genome sequencing of this RsbU* mutant confirmed the truncating SNP in ct588 (rsbU). Thirty-two additional SNPs were also determined (Table S5). Of these SNPs, seven are silent mutations and four are in intergenic regions. The remaining 21 SNPs were evaluated for their potential effect on their respective coding regions with the majority predicted to have no obvious effect on protein function based on their likelihood to alter secondary structures or active domains as predicted by EMBOSS secondary structure prediction or BLASTp domain predictions. Two SNPs are predicted to alter secondary structures: a G105E mutation in CT259 and a Q204* mutation in CT163. The mutation in CT259 is predicted to form an alpha helix spanning E99 to F113 not predicted in the wild-type CT259 and has been associated with reduced phosphatase activity of the protein [49]. The most significant SNP, outside of rsbU*, is the additional truncation in CT163, a hypothetical protein with no conserved motifs. The CT163 protein is predicted to be a membrane protein with one transmembrane domain in C. trachomatis. The truncation stops translation one third of the way through the large putative extracellular domain, likely altering protein function. It is unclear what effect the truncation of this protein would have on the chlamydial developmental cycle and we cannot rule out the possibility that the SNP is contributing to the growth and morphological defects that have been determined for the RsbU* null mutant.
In order to more confidently attribute the growth defect and phenotype to the RsbU disruption rather than the other SNPs induced by EMS mutagenesis, complementation efforts were pursued. However, because of the extremely poor growth of the RsbU* mutant, standard transformation with a wild-type rsbU gene on a vector plasmid proved unsuccessful. To overcome this limitation, lateral gene transfer was performed between C. trachomatis rsbU* (RifR) and another mutant strain that has a transposon (β-lactamase) inserted in mutL (ct575::Tn bla), which is near the rsbU coding region (CT588). After mixed infection and dual antibiotic selection, this was expected to encourage homologous recombination between the two genomes and restore rsbU coding region (Figure S4). This was expected to also leave the majority of SNPs including the ct163 mutant truncation. Importantly, the transposon mutant strain (ct575::Tn bla) showed growth phenotypes matching wild-type C. trachomatis L2 strain (Figure 7E and Figure S5).
Sequencing of amplicons from various genomic regions revealed a cross-over region in one of the resulting clones obtained following mixed infection and dual selection. Upon whole genome sequencing this complemented strain, the RsbU+ strain was revealed to be a mosaic between the RsbU* and wild-type genomes with a couple different regions of recombination apparent. In addition to a wild-type rsbU gene, the RsbU+ strain also has wild-type versions at 14 of the 32 SNP loci, 11 of which are in coding regions (Figure S4). Because the complemented strain does not retain all of the RsbU* SNPS, it does leave open the possibility that one or more of those SNPs could be playing a role in the growth defect of the null mutant that is restored in the complemented strain. In particular, the SNP in the rpoD gene encoding σ66 could effect on growth of the organism, however, the position of the SNP does not appear like it would have an effect on the structure of the protein, and is a region of the protein that does not appear to interact with the DNA binding [50]. Importantly, however, in the RsbU+ complemented strain, the non-sense mutation in the ct163 gene is maintained, meaning that any growth difference between the mutant and complemented strain is not due to this mutation. Growth curves were done with the parental transposon strain and the RsbU+ complemented strain, revealing that the RsbU+ strain showed a restoration in growth rate (Figure 7E).
We then hypothesized that the binding of TCA cycle intermediates to RsbU could indicate that the Rsb pathway is playing a regulatory role on TCA cycle activation in the chlamydial developmental cycle, leading to the poor growth of the RsbU* mutant. In order to test this hypothesis, we looked into chemical inhibitors targeting Chlamydia’s ability to produce ATP itself, as well as to steal ATP from the host cell using ATP translocases. 2-heptyl-4-hydrosyquinoline N-oxide (HQNO) has been shown to selectively inhibit at low concentrations (1 μM) the sodium-dependent NADH dehydrogenase that Chlamydia utilizes to produce the ion gradient that drives ATP synthesis by the chlamydial ATP synthase [17, 51]. Alternatively, bongkrekic acid (BKA) has been shown to inhibit ATP translocases in Chlamydia, limiting the ability to utilize host ATP [52]. Growth curves were repeated with wild-type Chlamydia and the RsbU* mutant strain with the addition of the chemical inhibitors (Figure 7C). BKA caused a decrease in the growth of wild-type Chlamydia that is statistically significant from wild-type (p-value <0.05) after 24 hours, as well as from the RsbU* mutant after 24 hours. The addition of HQNO to a wild-type infection, however, was not statistically different from the RsbU* mutant growth at any time point.
Additionally, progeny production was assessed for the RsbU* mutant strain, as well as wild-type infections with the BKA and HQNO chemical inhibitors (Figure 7D). This assay revealed that while there is a decrease in IFUs produced in the presence of BKA compared to the untreated WT infection, viable EBs are still being produced. However, in the RsbU* and WT + HQNO conditions, there is a decrease in the number of IFUs produced compared to the initial infection, suggesting that these cells are in the RB non-infectious form rather than converting to the infectious EB form. This is consistent with the growth curves in Figure 7C, where genome copies can be detected for these conditions, but RB-to-EB conversion appear to stalled in the infection.
To further investigate the poor growth by the wild-type Chlamydia in the presence of the sodium-dependent NADH dehydrogenase inhibitor (HQNO) and translocase inhibitor (BKA) as well as by RsbU*, confocal microscopy was carried out to view L929 cells infected with wild-type Chlamydia or RsbU* with and without inhibitors at 24 and 72 hours (Figure 8). Image analysis revealed that the wild-type Chlamydia at 24 hours post-infection in the presence of HQNO inhibitor formed smaller inclusions and appear to contain fewer EBs (puncta), although chlamydial RB cells appear like wild-type. At 72 hours post-infection, Chlamydia infected in the presence of HQNO had inclusions that were grossly under-full compared to wild-type Chlamydia with no inhibitor. No obvious morphological abnormalities were apparent for wild-type Chlamydia in the presence of the BKA inhibitor. RsbU* was shown to have a severe growth defect with no defined development of an inclusion. Additionally, Chlamydia cells appear dispersed in the host cytosol at levels well under that seen by wild-type Chlamydia within inclusions at both 24 and 72 hours post-infection. RsbU* mutant infections at both 24 and 72 hpi do appear to contain both EB and RB Chlamydia cell forms. Addition of the HQNO and BKA inhibitors appeared to have no effect on levels of RsbU* Chlamydia or their dispersion within the host cell.
Figure 8. Immunofluorescent microscopy of Chlamydia with RsbU* disruption and inhibitors of sodium-dependent NADH dehydrogenase and ATP translocase.

L929 cells infected with wild-type C. trachomatis or RsbU* with and without the presence of inhibitor (HQNO or BKA) at 24 and 72 hours post-infection. Blue: DAPI, nucleus; Red: Evan’s Blue, cytoplasm; Green: OmpA, C. trachomatis organisms. Images were acquired by confocal microscopy using a 150X objective and are comprised of 7 compressed Z-stacks (maximum projection) for each field.
Overall, growth with HQNO causes a marked reduction in growth in wild-type chlamydial infections, but does not have an additive effect on the growth defect observed in the RsbU* mutant. These observations support that the Rsb pathway in Chlamydia is linked to the ability of the bacteria to generate ATP via oxidative phosphorylation.
Transcriptional analysis of TCA cycle-associated and constitutively active genes suggestive of Rsb pathway regulation of σ66 activity.
σ66 is the primary sigma factor of only three sigma factors that Chlamydia sp. possess, and is responsible for transcription of the vast majority of genes throughout the developmental cycle. In order to further explore the proposed link between the Rsb pathway in Chlamydia to the regulation of σ66 [22], transcript levels of σ66-transcribed genes were assessed for differential expression between the RsbU* mutant and WT L2 C. trachomatis (Figure 9). Genes chosen for this analysis included TCA cycle associated genes (gltT, sucA, sdhB, mdhC, pckA), constitutively active “housekeeping” genes, (secY, rpoA, dnaK), and other genes associated with dicarboxylate processing or transport (xasA, ybhI, pdhB) [20]. All σ66-transcribed genes were observed to have lower transcript counts compared to wild-type, while the σ28-transcribed gene, hctB, did not appear to be differentially expression between the two strains. These results suggest that when the Rsb pathway is disrupted, as in the RsbU* mutant strain, there is decrease in transcription of these genes under regulation of σ66 activity.
Figure 9. Differential expression of TCA cycle-associated genes and other sigma-66 transcribed genes in RsbU* mutant compared to WT L2 transcript levels at 24 hpi.

Genomic levels of DNA per infection were used to normalize transcript counts. Sigma-28 transcribed gene, hctB, shows similar transcript levels between the RsbU* mutant and WT L2, while those genes with σ66 promoters all show significant decreases in the level of transcripts (*p-value >0.05; **p-value >0.01 with student’s T-test). Genes selected for this analysis included TCA cycle-associated genes (gltT, sucA, sdhB, mdhC, pckA), constitutively active genes (secY, rpoA, dnaK), and other genes associated with dicarboxylate processing or transport (xasA, ybhI, pdhB), all of which as σ66-transcribed genes.
Discussion
In order to characterize the role of the Rsb phosphoregulatory partner-switching pathway in Chlamydia, we focused on the structure and ligand-binding capabilities of the periplasmic domain of RsbU. A 1.7 Å crystal structure for the periplasmic domain (Figure 3) allowed for structural comparisons to other proteins, leading to the identification of a putative binding pocket, and a possible association to the native ligand.
SPR (Table 2 and Figure S3), DSF (Table S4), and ITC (data not shown) experiments suggest that alpha-ketoglutarate, malate, and oxaloacetate are binding to RsbU45–313. Dose-dependent SPR binding studies allowed for calculation of an estimated KD value of 419, 459, and 396 µM for alpha ketoglutarate, malate, and oxaloacetate, respectively. This is a relatively high KD value, indicative of weak binding; however, the concentrations of alpha-ketoglutarate and malate used were those similar to physiological levels in the cell [53]. Similar proteins including Tlp3 from Campylobacter jejuni and PctA, PctB, and PctC from Psuedomonas aeruginosa have been shown to bind ligands at similar binding affinities [54, 55]. Alternatively, there are several factors that could be having an effect on the RsbU45–313 protein’s ability to bind to the ligand, including the need for dimerization for ligand binding and the lack of the cytoplasmic and transmembrane portions of the protein that my help to stabilize the protein binding [34, 35, 38, 56]. The KD values from the single ITC experiment were about a log lower than those values calculated from SPR, indicating tighter binding affinity. It is possible this discrepancy is due to the difference in the condition of the protein (free in solution with ITC compared to cross-linked to a surface with SPR). The KD values from the ITC experiment are closer to the KD determined for DctB binding to succinate, also determined by ITC [35].
The binding of multiple ligands allows for the possibility of differential responses upon binding. DctB has been shown to bind to both succinate and malonate, with a conformational change and loop closure of 2.2 Å with succinate, but not with malonate binding [34]. The aforementioned structurally similar Tlp3 and Pct proteins also have been shown to bind to multiple ligands and have differential responses based on the identity of the ligand [54, 55].
Determining that RsbU is binding to TCA cycle intermediates lends itself to the question of what role this protein and its related pathway are playing in the chlamydial developmental cycle. To investigate the effect of RsbU on chlamydial growth, an RsbU* mutant showed a severe deficit in growth compared to the wild-type strain supporting that the Rsb pathway plays a role in the normal pattern of chlamydial growth (Figure 7B) [48]. When complementation of the rsbU gene was accomplished through homologous recombination with the ct575::Tn strain, the growth pattern returned to wild-type-like levels (Figure 7E).
Chlamydia has different ways that it can acquire energy. The presence of two ATP-ADP translocases allow for ATP uptake from the host cell appears to be the main source of energy when in the early stages of the developmental cycle, immediately after entry into the cell [57]. Chlamydia is then able to manufacture its own ATP utilizing a sodium-ion gradient to drive its ATP synthase activity during RB replication in midcycle time points as demonstrated by a recent publication by Liang et al. [17]. Wild-type chlamydial growth with HQNO, a sodium-dependent NADH dehydrogenase inhibitor, appears to mimic the growth pattern of the RsbU* mutation, potentially stalling the RB-to-EB conversion reducing the number of infectious progeny in the late stage of the developmental cycle as well (Figure 7C and 7D). While it possible that the loss of the NADH-driven sodium gradient might also impact other processes that utilize the ion gradient, such as amino acid transport, when RsbU* was grown in the presence of HQNO, the growth pattern was similar to that of wild-type with HQNO. These data suggest that the inhibition of the sodium-dependent NADH dehydrogenase in the RsbU* strain does not have an additive effect on the growth defect, and that the Rsb pathway may be playing a role in Chlamydia’s production of ATP through oxidative phosphorylation.
The dynamic energy utilization could account for the non-lethality of the RsbU* mutation. If Chlamydia is able to actively scavenge ATP and other metabolites from the host in its early developmental cycle, then there is the possibility of replication as well, albeit much more slowly. Moreover, there is the possibility for redundant pathways for activation of metabolic and replicative machinery. A second antagonist to the RsbW protein, RsbV2 (CT765), is also present in Chlamydia. Previous studies have shown that RsbU only dephosphorylated RsbV1; while RsbW phosphorylated both RsbV proteins, but has a bias towards RsbV1 [22, 23]. This duality of RsbW antagonists could potentially mean that there is a secondary signal that has a similar, but possibly lesser effect on the repression of RsbW inhibition of the downstream target protein, and thus why the RsbU signaling disruption is not lethal.
The target protein(s) for RsbW in Chlamydia is debatable. Several studies have investigated the potential protein interaction partners of RsbW in order to identify its target protein. Based on the Rsb system in B. subtilis, the target is presumed to be a sigma factor, for which Chlamydia has only three [9]. However, conflicting results have been observed in interaction studies with the primary chlamydial sigma factor, σ66, in addition to the alternative sigma factors, σ28 and σ54. Douglas and Hatch demonstrated that RsbW pulled down with σ28 in vitro, while Hua and colleagues found that RsbW did not interact with any of the chlamydial sigma factors using a yeast two-hybrid system and an in vitro σ28-dependent transcription assay [21, 23]. Most recently, Thompson et al. found using a bacterial two-hybrid system, and validated using surface plasmon resonance experiments, that RsbW binds σ66, but not σ54 or σ28 [22]. All this data combined has led to some uncertainty for any one sigma factor as the target protein, and the possibility for a non-sigma factor target has yet to be fully investigated. While the Rsb pathway described in B. subtilis and other gram-positive bacteria regulates an alternative sigma factor, it is also worth considering that this pathway in Chlamydia may not be regulating such transcriptional machinery. In Bordetella, an RsbU homolog has been shown to be an important regulator of type three effector protein secretion without affecting transcription [11, 58]. Further efforts are being made to more definitively determine the target protein of the Rsb pathway and its specific role in the chlamydial developmental cycle; however, in this study, differential expression of σ66-transcribed genes, TCA cycle-associated and otherwise, was also assessed (Figure 9). Of the genes selected for transcriptional analysis, all σ66-transcribed genes appear to be down-regulated in the RsbU* mutant compared to WT L2 C. trachomatis, in contrast to σ28-transcribed hctB [59]. This differential expression pattern between the RsbU* mutant and wild-type Chlamydia show a correlation between a disruption in the Rsb pathway and a decrease in σ66-transcribed gene transcript levels.
If the Rsb pathway regulates σ66, as the most recent publication and this study suggests [22], the binding of alpha-ketoglutarate seems rational. Chlamydia is known to obtain alpha-ketoglutarate from the host cell as means for fueling its truncated TCA cycle to produce ATP through oxidative phosphorylation [20]. The presence of a pool of alpha-ketoglutarate that Chlamydia can access could be an indicator that the bacteria is inside of the host cell and in a favorable environment for replication, and thus the activation of the primary sigma factor. The regulation of σ66 by the Rsb pathway may also explain the difference in the morphology of the RsbU* mutant compared to the wild-type Chlamydia with the HQNO inhibitor. While the HQNO inhibitor in the wild-type infection does mimic the RsbU* growth pattern, the IFA imaging (Figure 8) is not an exact phenocopy. There is still an obvious inclusion present in the wild-type infection in the presence of HQNO, although the amount of Chlamydia is clearly less, compared to the RsbU* mutant which does not appear to be inside of an inclusion, but instead clustered together in the host cell cytoplasm. The wild-type infection in this case would still have the ability to activate σ66, while the RsbU* would have σ66 repression, thus having a larger pleiotropic effect and be diminished in its ability to transcribe genes for the establishment and maintenance of the inclusion, TCA cycle enzymes, and effective growth and replication of the organism. Liang et al. were also able to show similar growth phenotype when wild-type Chlamydia is in the presence of monensin, a Na+/H+ exchanger that dissociates the Na+ ion gradient driving the chlamydial ATP synthase [17].
The idea of dynamic energy utility also leads to our proposed model of how the Rsb partner-switching pathway is playing a role in the Chlamydia developmental cycle (Figure 10). When an EB enters the host cell it comes in contact with an increased level of alpha-ketoglutarate, which binds to the RsbU periplasmic domain. In the current model, upon binding to alpha-ketoglutarate, the cytoplasmic effector domain of RsbU performs its phosphatase activity on RsbV1. RsbW then releases its target protein in order to re-phosphorylate the RsbV1 protein. That target protein then affects the activation of the TCA cycle in Chlamydia. This effect could be indirect, being a sigma factor, such as σ66, or through the other transcriptional regulators or machinery that lead to the expression of other proteins involved in the TCA cycle; or direct, through activation of transport proteins for TCA cycle substrates or enzymes in the TCA cycle itself. Then when levels of alpha-ketoglutarate are waning, potentially towards the end of the developmental cycle, RsbU is no longer bound and the target protein is again inhibited by RsbW. Interestingly, temperature-sensitive mutants generated by Brothwell et al. for both sodTi (the putative dicarboxylate transporter) and gltT (the putative glutamate transporter) support that acquisition of alpha-ketoglutarate is important for chlamydial growth [60]. In addition, the levels of malate and/or oxaloacetate in the periplasm could act as an inhibitor for RsbU signaling. Malate or oxaloacetate could build up in the periplasm as it is transported out of the chlamydial cytoplasm by transporter proteins such as SodTi [61]. The phosphoenolpyruvate carboxylkinase (Pck) enzyme catalyzing the conversion of oxaloacetate to phosphoenolpyruvate has been shown to be differentially regulated as a mid-late stage gene, possibly leading to more malate and oxaloacetate being present in the cytoplasm to be exported into the periplasm by SodTi in exchange for alpha-ketoglutarate [62, 63]. Additionally, malate converted to oxaloacetate can also be used to synthesize meso-diaminopimelate (mDAP), a crosslinker in the A1γ-type peptidoglycan Chlamydia synthesizes during growth [64, 65]. Peptidoglycan is only needed during growth of the Chlamydia cell [64, 66], and therefore a buildup of malate could occur as the cell ceases growth in preparation for the conversion to the EB form.
Figure 10. Working model of the Rsb phospho-regulatory pathway integrated with the truncated TCA cycle in Chlamydia.

Alpha-ketoglutarate binding to the periplasmic domain of RsbU, as could be the case when an EB enters the host cell, leads to the activation of the phosphatase function of the cytoplasmic domain. RsbW then releases its target protein (black box), allowing for its normal function to be performed. That target protein then, either directly or indirectly, activates the chlamydial TCA cycle, allowing for alpha-ketoglutarate to be utilized. Chlamydia has been shown to be capable of creating its own ATP during mid-cycle using a truncated TCA cycle to generate electron-carrying molecules (i.e. NADH, FADH2) and a sodium pumping NADH:quinone oxidoreductase (Na+-NQR) (Liang,2018). As malate builds up in the periplasm, through the export by the SodTi protein (Weber, 1995), it acts as an inhibitor as the concentration of alpha-ketoglutarate is depleted. The inhibition of the RsbU protein or the depletion of alpha-ketoglutarate, potentially later in the developmental cycle, could lead to a slowing of the TCA cycle as the Chlamydia cells prepare to convert to the EB form.
Aspects of the Rsb phosphoregulatory partner-switching pathway still remain to be explored. While the transcriptional analysis supports the hypothesis that the target protein of the pathway could be σ66, it does leave open the possibility of RsbW binding to a secondary transcriptional regulator. A phosphoproteomic analysis done in Chlamydia caviae showed that phosphorylated RsbV1 and RsbV2 can be detected in EBs, but not in RBs, rather than the other way around; calling into question the nature of this intermediate connection between RsbU and RsbW, and the mechanism of which these proteins communicate [67]. Furthermore, the true response of RsbU to binding either alpha-ketoglutarate, malate, or oxaloacetate; whether they are activating or inhibiting to the phosphatase activity of the RsbU cytoplasmic domain, still remains to be fully examined.
In this study, we were able to solve a 1.7Å crystal structure for the periplasmic domain of the chlamydial RsbU protein and utilize structural similarities to a dicarboxylate-binding protein to determine alpha-ketoglutarate, malate, and oxaloacetate as binding ligands. Moreover, an RsbU-null mutant was utilized to show the importance of the Rsb pathway in normal chlamydial growth. Finally, we proposed a working model for how this pathway may be sensing the aforementioned ligands to regulate the TCA cycle.
Materials and Methods
Overexpression and purification of recombinant RsbU45–313.
A fragment of ctl0851 encoding residues 45 through 313 of the open reading frame was amplified via polymerase chain reaction (PCR) from C. trachomatis L2 434/Bu (AM884176) genomic DNA. ctl0851 is homologous and 99% identical to CT588 (RsbU) from C. trachomatis D/UW-3. This fragment was inserted into the pTBSG vector in frame and immediately downstream of a sequence encoding an N-terminal hexahistidine tag and TEV protease recognition site. After confirming sequence accuracy, this vector was transformed into BL21 (DE3) E. coli competent cells, which were then grown at 37°C (200 rpm) in Terrific Broth supplemented with 100 μg/mL Carbenicillin to an OD600 of 0.8. Overnight protein expression (15°C, 200 rpm) was induced at this optical density with the addition of IPTG (isopropyl 1-thio-β-D-galactopyranoside) to a final concentration of 1mM. Following E. coli collection by centrifugation (10,000g; 15 minutes), cells were resuspended in lysis buffer [20 mM Tris pH 8.0, 500 mM NaCl, 10 mM imidazole, 1 mM phenylmethane sulfonyl fluoride (PMSF), and 1000U benzonase endonuclease (EMD Millipore) per liter of Terrific broth culture] and lysed by sonication. After centrifugation (23,000g; 30 minutes), the supernatant was clarified through a 0.45 μm filter and purified on a gravity flow column containing 3 mL of HisPur Cobalt Resin (ThermoFisher) per liter of Terrific Broth culture. Following washes with 5 column volumes (CVs) of lysis buffer and then 3 CVs of wash buffer (20 mM Tris pH 8.0, 500 mM NaCl, and 50 mM imidazole), immobilized His6-RsbU45–313 was eluted with 3 CVs of elution buffer ( 20 mM Tris pH 8.0, 500 mM NaCl, and 500 mM imidazole). The eluate was buffer exchanged into Buffer A (20 mM Tris pH 8.0, 500 mM NaCl, and 10 mM imidazole) on a HiPrep 26/10 Desalting column (GE Healthcare) and then incubated overnight at 4°C with 5 mM dithiothreitol (DTT) and recombinant polyhistidine-tagged TEV protease for His6-tag removal. Recombinant TEV protease and cleaved His6-tag were then removed from this mixture via flow over a 5 mL HisTrap HP column (GE Healthcare). Following buffer exchange into Buffer × (10 mM Tris pH 7.5, 50 mM NaCl, and 1mM DTT) as described above, the sample was concentrated to a volume of 1.5 mL with an Amicon-15, Ultracel-10 centrifugal filter (EMD Millipore). Final purification was achieved via size exclusion chromatography using a flow rate of 0.2 mL/minute on a HiPrep 16/60 Sephacryl S-200 HR column (GE Healthcare). Collected fractions containing RsbU45–313 were concentrated to 15.9 mg/mL (by Bradford assay) via ultracentrifugation and stored at 4°C until further use.
Crystallization and data collection.
All crystallization screening was conducted in Compact 300 (Rigaku Reagents) sitting drop vapor diffusion plates at 18°C using equal volumes of protein solution and crystallization solution equilibrated against 75 μL of the latter. Prismatic crystals grew within one day and continued to grow for approximately one week from Wizard 1–2 screen (Rigaku Reagents) condition E10 (1M ammonium phosphate dibasic, 100 mM Tris pH 8.5) and the Crystal Screen HT (Hampton Research) condition D5 [20% (w/v) PEG 4000, 10% (v/v) 2-propanol, 100 mM Hepes pH7.5]. A heavy atom derivative was prepared by soaking a crystal obtained from Wizard 1–2 condition E10 for 22 hours in crystallant containing 5 mM K2PtCl4. Native and heavy atom-soaked crystals were transferred to a fresh drop containing 80% crystallant and 20% ethylene glycol before flash freezing in liquid nitrogen. Data were collected at the Advanced Photon Source IMCA-CAT beamline 17-ID using a Dectris Pilatus 6M pixel array detector.
Structure solution and refinement.
Intensities were integrated using XDS via Autoproc, and the Laue class analysis and data scaling were performed with Aimless [29, 68, 69]. The highest probability Laue class was 4/m, for either space group I4 or I41. The Matthew’s coefficient (Vm) and solvent content were estimated to be Vm = 2.3/47% solvent for 1 molecule in the asymmetric unit [70]. Data for phasing were collected using the platinum-soaked crystals, at the absorption edge λ = 1.0716 Å (11.570 keV) as determined from an X-ray fluorescence scan. Integrated defraction data from two crystals were scaled together with Aimless in order to increase the multiplicity. Structure solution was conducted using the SAD method with Autosolve via the Phenix interface, which yielded a figure of merit of 0.23 and a Bayes-CC of 0.299 [71]. The Autobuild step of Autosolve produced a model containing 188 of the possible 272 residues which converged at R= 0.35, Rfree = 0.44 following refinement. Crystals of native RsbU obtained from the Crystal Screen HT condition D5 yielded the highest resolution diffraction (1.7 Å) and were used from this point forward. The resulting model from Autosolve was used for molecular replacement with Phaser against a native RsbU data set and the top solution was obtained in the space group I4 (TFZ = 45.8, LLG = 1,836) [72]. The model was further improved by automated model building using Arp/wARP and subsequent rounds of structure refinement and manual model building were carried out using Phenix and Coot [73, 74]. Residues P162, L163 and R313 were not modeled due to inadequate electron density. TLS refinement was incorporated in later rounds to model anisotropic atomic displacement parameters [75, 76]. Structure validation was conducted with Molprobity, and relevant crystallographic data are provided in Table 1 [77]. Coordinates and structure factors for RsbU were deposited to the Worldwide Protein Databank (wwPDB) with the accession code 6MAB.
Table 1.
X-ray diffraction data and structure refinement
| RsbU (K2PtCl4) |
RsbU (Native) |
|
|---|---|---|
| Data Collection | ||
| Cell dimentions | ||
| a, b, c (Å) | 96.71, 96.71, 96.49 | 96.71, 96.71, 96.39 |
| a, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 |
| Space group | l4 | l4 |
| Resolution (Å)a | 48.36 – 2.30 (8.91 – 2.30) | 48.36 – 1.70 (1.73 – 1.70) |
| Wavelength (Å) | 1.0716 | 1 |
| Temperature (K) | 100 | 100 |
| Observed reflections | 352,983 (33,886) | 225,529 (11,050) |
| Unique reflections | 13,526 (1,297) | 33,233 (1,758) |
| <l/σ (l)>a | 18.5 (1.7) | 17.9 (1.7) |
| Completences (%)a | 100.0 (100.0) | 100.0 (100.0) |
| Multiplicity | 26.1 (26.1) | 6.8 (6.3) |
| Rmerge (%)a,b | 11.5 (119.6) | 6.1 (111.0) |
| Rmerge (%)a,c | 11.5 (119.6) | 6.6 (121.1) |
| Rpim (%)a,c | 2.3 (23.9) | 2.5 (47.8) |
| Refinement | ||
| Resolution (Å) | 36.07 – 1.70 | |
| Reflections (working/test) | 33,231 (3,266) | |
| Rfactor/Rfree (%)d | 16.36/19.73 | |
| No of atoms | 2,101/1/203 | |
| (protein/ligand/water | ||
| Model Quality | ||
| R.m.s deviations | ||
| Bond length (Å) | 0.008 | |
| Bond angles () | 0.914 | |
| Average B factor (Å) | ||
| All Atoms | 23.11 | |
| Coordinate error, maximum likelihood (Å) | 0.19 | |
| Ramachandran Plot | ||
| Most favored (%) | 98.11 | |
| Additionally allowed (%) | 1.89 |
Values in parentheses are for the highest resolution shell.
Rmege = ShklSi | li(hkl) - <l(hkl)>| / ShklSi li(hkl), where li(hkl) is the intensity measured for the ith reflection and <l(hkl)> is the average intensity of all reflection with indices hkl.
Rmeas = redundance-independent (multiplicity-weignted) Rmerge (Evans, 2011; Diedrivhs, 1997). Rpim = precision-indicating (multiplicity-weighted) Rmerge (Evans, 2006; Weiss, 2001).
Rfactor = Shkl || Fobs (hkl) | - |Fcalc (hkl) || / Shkl | Fobs (hkl)|; Rfree is calculated in an identical manner using 5% of randomly selected reflections that were not included in the refinement
Structural alignments and superimposition.
Structures of DctB were obtained from the PDB. Apo DctB (3E4Q), Malonate-bound DctB (3E4P), and apo RsbU were aligned to beta sheet residues (120–198) of succinate-bound DctB (3E4O) using the Combinatorial Extension alignment method [78]. Alignments were performed using the NCBI Blast webserver [79]. Global alignments were performed using the Needleman-Wunch method, and local alignments were performed using BLAST. Proteins with the same fold were identified by performing a TM-alignment [80] of RsbU against the non-redundant structures from the PDB [28]. Proteins that had a TM-Score of at least 0.5, when normalized against RsbU, were considered to have the same fold [81, 82].
Virtual screen of human metabolite and chlamydial metabolite libraries.
The human metabolites set of compounds was downloaded from the Human Metabolite Database and compounds with molecular weight greater than 300 were discarded [83–85]. The Chlamydia metabolites set of compounds was downloaded from the Chlamydia trachomatis database in BioCyc [86]. Up to 250 conformers were generated using Omega (version 2.5.1.4) by OpenEye (Santa Fe, NM) [87]. The receptor was prepared using APOPDB2RECEPTOR and compounds were docked into using FRED (version 3.2.0.2) at the “Standard” docking resolution (Santa Fe, NM), [88]. Docked models were refined using SZYBKI (version 1.9.0.3) (Sant Fe, NM). Compounds with docking scores above −6 (chosen based on the docking score of succinate), positive interaction energies, and minimized ligand poses that moved more than 1.5 Å were discarded. The remaining compounds were enriched with malate, malonate, alpha-ketoglutarate, succinate, α-D-glucose, fumarate, glutamate, pyruvate, 3-phosphoglyceric acid, oxaloacetate, and aldohexose stereoisomers. Compounds were prepared using LigPrep by Schrodinger using the default settings (New York, NY) to identify the physiologically relevant protonation states. The receptor was prepared using the protein preparation wizard in Schrodinger, which optimizes the hydrogen bonding and protonation state, followed by a constrained minimization. These compounds were then docked into the receptor using Glide (release 2017–3) by Schrodinger. Up to 5 docked poses were generated per compound, using extra precision (XP) settings [89–91]. Docked poses were then refined and free energies of binding were predicted using Prime MM-GBSA, allowing flexibility in residues within 8Å of the ligand [92, 93]. Compounds were selected based on the docking score, MM-GBSA predicted energy, predicted ligand efficiency, and visual inspection of the models.
The docking models of oxaloacetate, alpha-ketoglutarate, and malate to RsbU were generated by docking using Glide XP followed by Prime MM-GBSA refinement, allowing flexibility in residues within 8Å of the ligand.
Surface plasmon resonance.
SPR runs were performed on a Biacore T200 (GE Healthcare Life Sciences) with cell culture grade Phosphate Buffered Saline (Corning). Purified RsbU45–313 protein in PBS was immobilized onto a Series S NTA or CM5 sensor chip (GE Healthcare Life Sciences). All ligands were dissolved in PBS and PBS only was used as a negative control. A flow cell with no protein bound was used as a reference cell for all runs. Ligands were injected over the chip for 30 seconds, with a 60 second dissociation period. Binding affinity was manually estimated using the steady state affinity equation:
where Req is the measured resonance units at steady state binding levels, C is the concentration of the ligand, and Rmax is the maximum binding capacity determined for each respective ligand assuming a 1:1 ratio of binding to protein.
Data was analyzed using Biacore T200 Software (version 3.0).
Differential scanning fluorimetry (DSF).
RsbU45–313 was purified as described above and buffer exchanged into PBS (Corning). DSF were performed with SyproOrange (Invitrogen) in 384-well plate (Roche) format [46]. The following potential ligands were tested: succinate, malonate, glutamate, alpha-ketoglutarate, fumarate, oxaloacetate, malate, 2-phosphoglycerate, glucose, pyruvate, phosphoenolpyruvate, and ATP (Sigma-Aldrich). All ligands were dissolved in PBS. Compounds were added to each well, followed by DSF buffer HEPES-NaOH pH7.5 (100mM), and a 10X SyproOrange dye. Reliable baselines for Tm shifts were established using 10X SyproOrange and 10 μM RsbU45–313. The mixture was heated from 20 to 85 °C. Melting curves were analyzed on Roche Tm Analysis Software.
Isothermal titration calorimetry (ITC).
RsbU45–313 (30 μM) was purified as described above and buffer exchanged into PBS (Corning). Alpha-ketoglutarate, malate, oxaloacetate, malonate, and succinate (Sigma-Aldrich) were dissolved in the same PBS used for the buffer exchange of the RsbU45–313 protein at a concentration of 30 mM. ITC was performed on a MicroCal PEAQ-ITC (Malvern Panalytical and analyzed using MicroCal ITC Analysis Software (version 1.21).
Growth Curves.
An EMS mutant strain of Chlamydia trachomatis L2 was obtained from the Valdivia lab at the Duke University Medical Center [47, 48]. A confluent monolayer of L929 mouse fibroblast cells was infected with an MOI of 0.5 mutant or wild-type chlamydial cells with centrifugation and using Hanks’ Balanced Salt Solution with calcium and magnesium (Corning). After centrifugation, the HBSS was removed from the cells and replaced with RPMI (Corning) supplemented with 5% FBS (Millipore), 10 μg/mL gentamycin, and 1 μg/mL cycloheximide. For the growth curves with the addition of chemical inhibitors, the BKA and HQNO were added into the RPMI at the time of infection. HQNO was added at a final concentration of 1μM. BKA was added at a final concentration of 0.25 μM. The infected cells were incubated at 37°C, 5% CO2 until harvested. Total DNA was harvested from infected cells at 0, 12, 24, 36, 48, and 72 hours post infection. DNA was harvested by adding 200 μl of 5mM DTT, 200 μL of Buffer AL from a Blood and Tissue Kit (Qiagen), and 20 μl of Proteinase K (Qaigen) to each well and incubated at room temperature for 10 minutes. Wells were then scrapped and washed twice with the lysate before being collected. Following harvest, the lysate was heated at 56°C for 10 minutes and then frozen until all time point samples were collected. The remainder of the DNA isolation was performed using the Blood and Tissue Kit (Qiagen).
After DNA isolation was complete, the number of host genome copies and
Chlamydia genome copies was determined by Droplet Digital PCR (ddPCR) [94]. Chlamydia genome copies were assessed by the amplification of secY, and host cell genome copies were assessed by amplification of rpp30. Quantification of copy numbers was determined using Quantasoft software version 1.7 (Bio-Rad).
Progeny assay.
L929 cells were infected with wild-type or RsbU* mutant strains of C. trachomatis L2 with BKA (0.25 μM) and HQNO (1μM) added at the time of infection when indicated. At 36 hpi, cells were either fixed and stained using MicroTrack C. trachomatis culture confirmation test (Syva Co., Palo Alto, CA), or lysed with water and passaged onto a new monolayer of host cells. An additional 36 hours after passaging, the infections were fixed and stained. Fold changes were calculated by counting the IFUs of the infections after the first 36 hours and comparing to the IFU counts after the infections were passaged.
Immunofluorescence microscopy.
L929 cells were grown to confluency in an 8-well ibiTreat µ-Slide (Ibidi, Martinsried, Germany) and were infected with respective wild-type C. trachomatis L2, RsbU* mutant, RsbU+ complemented strain, or ct575::Tn bla strain. Chemical inhibitors (HQNO and BKA) were added to the indicated conditions immediately after infection. At 24 and 72 hpi, infected cells were fixed with 100% methanol for 10 minutes at room temperature. Cells were washed once with HBSS and again with PBS then stained using 180µl of the MicroTrack C. trachomatis culture confirmation test (Syva Co., Palo Alto, CA) diluted 1:40 in PBS 1 hour and 50 minutes at room temperature. 20µl of 1µM 4’, 6-diamidino-2-phenylindole (DAPI) diluted 1:100 in PBS was then added to wells and allowed to stain for 10 minutes, room temperature in the dark. Stain was then removed, and the cells washed with PBS. A final overlay of Vectashield antifade mounting medium (Burlingame, CA) was added and slides were immediately imaged. Cells were visualized on an Olympus IX81/3I spinning disk confocal inverted microscope at 150X magnification and captured on an Andor Zyla 4.2 sCMOS camera (Belfast, Northern Ireland). Microscope and camera were operated using SlideBook 6 software (Intelligent Imaging Innovations, Denver, USA). Exposure time remained consistent for all fields captured, with exposure for DAPI at 2 seconds, OmpA 3 seconds, and cytoplasm 3 seconds. Seven Z-stack images at 0.3µm apart were taken per field imaged. Images were processed in SlideBook 6 and a No Neighbors Deconvolution with a subtraction constant of 0.4 was applied to all images. Images represent a maximum projection over the Z axis of all 7 acquired stacks for each field shown.
Whole genome sequencing.
Chlamydial DNA was extracted from RsbU* EBs. Briefly, 200uL of renografin-purified EBs were pelleted, resuspended in RQ1 DNase buffer, water and RQ1 Dnase, incubated and stopped as per manufacturer’s instructions (Promega, Madison, WI). 2uL DTT was added to the EBs and DNA was extracted using the Qiagen Blood and Tissue DNA Extraction Kit (Qiagen, catalog number 69506) with following steps that optimize for DNA sequencing. Libraries were generated using the NEBNext Ultra II DNA library Prep kit (New England Biolabs, catalog number E7645S). DNA was sequenced by the Illumina Nextseq MO-SR150bp. Over 91 million reads were generated with a mean quality score of 32.78. Approximately 3% of reads were mapped to the Chlamydia trachomatis L2/434 (NC_010287) parent genome through reference-guided assembly using the Geneious assembler with up to 5 iterations. Total average coverage for the RsbU* genome was 400x. Through direct comparison with the reference genome, 33 SNPs were evaluated, including the RsbU* truncation which was confirmed to be a monoclonal polymorphism as 98.6% of reads at that site confirmed the SNP. For the 32 other SNPs discovered in the RsbU* genome, potential effects on secondary structure were analyzed using Geneious secondary structure predictions based on the EMBOSS 6.5.7 tool garnier or signal cleavage site prediction with sigcleav.
Generation of RsbU complemented mutant (RsbU+) by lateral gene transfer.
A confluent layer of Vero monkey kidney cells in a T-75 cell culture flask was infected with 100µl of RsbU* lysate in 1X SPG buffer. Briefly, the monolayer was washed once with HBSS, and 10ml of HBSS was added to the culture flask along with RsbU* lysate. Cells were spun at 550XG for 30 minutes at room temperature. Infection material was aspirated from the flask and 15ml of RPMI containing 1µg/ml cyclohexamide was added to the flask. Infected cells were incubated at 37°C for 85 hours post-infection. RsbU* infected cells were then co-infected, as described above, with a C. trachomatis mutant containing a transposon insertion in ct575 (ct575::Tn bla). Co-infected cells were incubated another 48 hours at 37°C. Cells were then lysed by water lysis and transferred to Vero cell monolayers in a 24-well plate with each well containing variable concentrations of rifampicin and ampicillin to facilitate successful lateral gene transfer of the bla resistance marker of the ct575::Tn into the RsbU* mutant clone. After 24hpi, WT-like Chlamydia growth was identified by phase contrast microscopy in a well containing 0.01µg/ml rifampicin and 5 ug/ml ampicillin. After 32hpi, cells in the well containing growth were lysed by water and the lysate then underwent two rounds of limiting dilution in a 96-well plate to isolate a clonal population of RsbU complemented mutant recombinants. Mutants with dual antibiotic resistance to rifampicin and ampicillin were evaluated by PCR amplification and sequencing for the genotype of the rsbU and ct163 genes, followed by the other SNPs present in the EMS mutant genome to determine where the area of homologous recombination occurred.
Transcriptional analysis.
A confluent monolayer of L929 cells were infected with either WT L2 C. trachomatis or the RsbU* mutant strain at an MOI of 1. At 24 hpi, the infections were harvested for RNA using TRIzol (Invitrogen). RNA was purified by phenol/chloroform extraction followed by DNase treatment with TURBO DNase (Invitrogen). A final purification step was performed using the RNeasy Mini Kit (Qiagen) before converting the RNA to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher). DNA contamination was assessed using a no reverse transcriptase control reaction. After gDNA depletion has been confirmed for all RNA samples, transcript counts are quantified using ddPCR (Bio-Rad). gDNA taken from the infections was used to normalized the transcript counts.
Supplementary Material
Figure S1. I-TASSER model of RsbU1-315 shares structural similarity to periplasmic domain RsbU45-313 crystal structure. A) I-TASSER protein structure model with residues 1 through 315 of C. trachomatis RsbU. B) Structure overlay of RsbU45-313 crystal structure (magenta) and I-TASSER model (yellow). Structure comparison has Z-score of 14.4 and a RMSD of 4.0 Å.
Figure S2. SDS-PAGE gel showing purification of RsbU45-313. Lane 1, protein marker; lane 2, lysate supernatant; lane 3, lysate pellet; lane 4, IMAC flow-through; lane 5, IMAC elution; lane 6, post-buffer exchange; lane 7, post-TEV protease treatment; Lane 8, post-reverse nickel column; lane 9, SEC column peak fraction.
Figure S3. SPR dose-dependent binding curves. Alpha-ketoglutarate, malate, and oxaloacetate show dose-dependent binding to RsbU45-313, while succinate and malonate, the ligands found to bind to DctB, do not appear to be binding by SPR.
Figure S4. Chromosome schematic of cross between RsbU* EMS strain and ct575::Tn strain to create the complemented RsbU+ strain that retains the majority of the other SNPs induced by EMS mutagenesis. The ct575::Tn strain contains a beta-lactamase resistance gene in the transposon, while the RsbU* has a SNP in the rpoB gene that incurs rifampicin resistance. Dual selection with ampicillin and rifampicin was utilized to select for recombinants that retained the RsbU* strain backbone but a wild-type version of the rsbU gene. The red region on the RsbU+ chromosome represents the region of recombination between the genomes confirmed by PCR of the SNPs present in the RsbU* strain. In addition to the restoration of the rsbU gene, the RsbU+ strain restores three other SNPs close to the position of the transposon: two in coding regions for secY and polA; and one in an intergenic region (IGR).
Figure S5. Immunofluorescent microscopy of C. trachomatis ct575::Tn parent strain and RsbU+ complemented strain. At 24 and 72 hours post-infection, the RsbU+ complemented strain does not appear to be phenotypically different from the Tn parent strain.
Acknowledgements
University of Kansas Protein Structure Laboratory was supported by the NIH NIGMS (P30 GM110761). Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02–06CH11357. KRS was supported by NIH T32 GM008545 and SDL, ZED and PSH were supported by AI126785 and AI125929. PSH and DKJ had support by P20 GM113117. AD was supported by the Arnold and Mabel Beckman Foundation (University of Kansas).
Footnotes
There are no apparent conflicts of interests.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Zhulin IB, Nikolskaya AN, and Galperin MY, Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. J Bacteriol, 2003. 185(1): p. 285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hecker M, Pane-Farre J, and Volker U, SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol, 2007. 61: p. 215–36. [DOI] [PubMed] [Google Scholar]
- 3.Hughes KT and Mathee K, The anti-sigma factors. Annu Rev Microbiol, 1998. 52: p. 231–86. [DOI] [PubMed] [Google Scholar]
- 4.Voelker U, Dufour A, and Haldenwang WG, The Bacillus subtilis rsbU gene product is necessary for RsbX-dependent regulation of sigma B. J Bacteriol, 1995. 177(1): p. 114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voelker U, et al. , Separate mechanisms activate sigma B of Bacillus subtilis in response to environmental and metabolic stresses. J Bacteriol, 1995. 177(13): p. 3771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang CM, et al. , Homologous pairs of regulatory proteins control activity of Bacillus subtilis transcription factor sigma(b) in response to environmental stress. J Bacteriol, 1996. 178(13): p. 3846–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang CM, Vijay K, and Price CW, Serine kinase activity of a Bacillus subtilis switch protein is required to transduce environmental stress signals but not to activate its target PP2C phosphatase. Mol Microbiol, 1998. 30(1): p. 189–96. [DOI] [PubMed] [Google Scholar]
- 8.Yang X, et al. , Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev, 1996. 10(18): p. 2265–75. [DOI] [PubMed] [Google Scholar]
- 9.Wise AA and Price CW, Four additional genes in the sigB operon of Bacillus subtilis that control activity of the general stress factor sigma B in response to environmental signals. J Bacteriol, 1995. 177(1): p. 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson AK and Haldenwang WG, Regulation of sigma B levels and activity in Bacillus subtilis. J Bacteriol, 1993. 175(8): p. 2347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozak NA, et al. , Interactions between partner switcher orthologs BtrW and BtrV regulate type III secretion in Bordetella. J Bacteriol, 2005. 187(16): p. 5665–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris AR and Visick KL, The response regulator SypE controls biofilm formation and colonization through phosphorylation of the syp-encoded regulator SypA in Vibrio fischeri. Mol Microbiol, 2013. 87(3): p. 509–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elwell C, Mirrashidi K, and Engel J, Chlamydia cell biology and pathogenesis. Nat Rev Microbiol, 2016. 14(6): p. 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehlitz A, et al. , Metabolic adaptation of Chlamydia trachomatis to mammalian host cells. Mol Microbiol, 2017. 103(6): p. 1004–1019. [DOI] [PubMed] [Google Scholar]
- 15.Hatch TP, Allan I, and Pearce JH, Structural and polypeptide differences between envelopes of infective and reproductive life cycle forms of Chlamydia spp. J Bacteriol, 1984. 157(1): p. 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hackstadt T, Todd WJ, and Caldwell HD, Disulfide-mediated interactions of the chlamydial major outer membrane protein: role in the differentiation of chlamydiae? J Bacteriol, 1985. 161(1): p. 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang P, et al. , Dynamic energy dependency of Chlamydia trachomatis on host cell metabolism during intracellular growth: Role of sodium-based energetics in chlamydial ATP generation. J Biol Chem, 2018. 293(2): p. 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliffe-Lee ER and McClarty G, Glucose metabolism in Chlamydia trachomatis: the ‘energy parasite’ hypothesis revisited. Mol Microbiol, 1999. 33(1): p. 177–87. [DOI] [PubMed] [Google Scholar]
- 19.Stephens RS, et al. , Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science, 1998. 282(5389): p. 754–9. [DOI] [PubMed] [Google Scholar]
- 20.Iliffe-Lee ER and McClarty G, Regulation of carbon metabolism in Chlamydia trachomatis. Mol Microbiol, 2000. 38(1): p. 20–30. [DOI] [PubMed] [Google Scholar]
- 21.Douglas AL and Hatch TP, Expression of the transcripts of the sigma factors and putative sigma factor regulators of Chlamydia trachomatis L2. Gene, 2000. 247(1–2): p. 209–14. [DOI] [PubMed] [Google Scholar]
- 22.Thompson CC, et al. , The Rsb Phosphoregulatory Network Controls Availability of the Primary Sigma Factor in Chlamydia trachomatis and Influences the Kinetics of Growth and Development. PLoS Pathog, 2015. 11(8): p. e1005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hua L, et al. , Core of the partner switching signalling mechanism is conserved in the obligate intracellular pathogen Chlamydia trachomatis. Mol Microbiol, 2006. 59(2): p. 623–36. [DOI] [PubMed] [Google Scholar]
- 24.Schultz J, et al. , SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A, 1998. 95(11): p. 5857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hulko M, et al. , The HAMP domain structure implies helix rotation in transmembrane signaling. Cell, 2006. 126(5): p. 929–40. [DOI] [PubMed] [Google Scholar]
- 26.Elliott KT, et al. , Conserved residues in the HAMP domain define a new family of proposed bipartite energy taxis receptors. J Bacteriol, 2009. 191(1): p. 375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Serine/threonine phosphatases: mechanism through structure. Cell, 2009. 139(3): p. 468–84. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, et al. , The I-TASSER Suite: protein structure and function prediction. Nat Methods, 2015. 12(1): p. 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch WS, C., Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 1983. 22(12): p. 2577–637. [DOI] [PubMed] [Google Scholar]
- 30.Bryant SH and Altschul SF, Statistics of sequence-structure threading. Curr Opin Struct Biol, 1995. 5(2): p. 236–44. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen MN, Tan KP, and Madhusudhan MS, CLICK--topology-independent comparison of biomolecular 3D structures. Nucleic Acids Res, 2011. 39(Web Server issue): p. W24–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winn MD, et al. , Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr, 2011. 67(Pt 4): p. 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janausch IG, et al. , C4-dicarboxylate carriers and sensors in bacteria. Biochim Biophys Acta, 2002. 1553(1–2): p. 39–56. [DOI] [PubMed] [Google Scholar]
- 34.Zhou YF, et al. , C4-dicarboxylates sensing mechanism revealed by the crystal structures of DctB sensor domain. J Mol Biol, 2008. 383(1): p. 49–61. [DOI] [PubMed] [Google Scholar]
- 35.Nan B, et al. , From signal perception to signal transduction: ligand-induced dimeric switch of DctB sensory domain in solution. Mol Microbiol, 2010. 75(6): p. 1484–94. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, et al. , Mutational analysis of dimeric linkers in peri- and cytoplasmic domains of histidine kinase DctB reveals their functional roles in signal transduction. Open Biol, 2014. 4(6): p. 140023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheung J and Hendrickson WA, Crystal structures of C4-dicarboxylate ligand complexes with sensor domains of histidine kinases DcuS and DctB. J Biol Chem, 2008. 283(44): p. 30256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu YC, et al. , Structural basis for amino-acid recognition and transmembrane signalling by tandem Per-Arnt-Sim (tandem PAS) chemoreceptor sensory domains. Acta Crystallogr D Biol Crystallogr, 2015. 71(Pt 10): p. 2127–36. [DOI] [PubMed] [Google Scholar]
- 39.Chang C, et al. , Extracytoplasmic PAS-like domains are common in signal transduction proteins. J Bacteriol, 2010. 192(4): p. 1156–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z and Hendrickson WA, Structural characterization of the predominant family of histidine kinase sensor domains. J Mol Biol, 2010. 400(3): p. 335–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu R, et al. , Insight into the sporulation phosphorelay: crystal structure of the sensor domain of Bacillus subtilis histidine kinase, KinD. Protein Sci, 2013. 22(5): p. 564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishiyama S, et al. , Identification of a Vibrio cholerae chemoreceptor that senses taurine and amino acids as attractants. Sci Rep, 2016. 6: p. 20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krissinel E, Enhanced fold recognition using efficient short fragment clustering. J Mol Biochem, 2012. 1(2): p. 76–85. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, et al. , Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11. Proteins, 2016. 84 Suppl 1: p. 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jason-Moller L, Murphy M, and Bruno J, Overview of Biacore systems and their applications. Curr Protoc Protein Sci, 2006. Chapter 19: p. Unit 19 13. [DOI] [PubMed]
- 46.Niesen FH, Berglund H, and Vedadi M, The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc, 2007. 2(9): p. 2212–21. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen BD and Valdivia RH, Virulence determinants in the obligate intracellular pathogen Chlamydia trachomatis revealed by forward genetic approaches. Proceedings of the National Academy of Sciences of the United States of America, 2012. 109(4): p. 1263–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kokes M, et al. , Integrating Chemical Mutagenesis and Whole-Genome Sequencing as a Platform for Forward and Reverse Genetic Analysis of Chlamydia. Cell Host & Microbe, 2015. 17(5): p. 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claywell JE, et al. , Inhibition of the Protein Phosphatase CppA Alters Development of Chlamydia trachomatis. J Bacteriol, 2018. 200(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paget MS, Bacterial Sigma Factors and Anti-Sigma Factors: Structure, Function and Distribution. Biomolecules, 2015. 5(3): p. 1245–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuz K, et al. , The Kinetic Reaction Mechanism of the Vibrio cholerae Sodium-dependent NADH Dehydrogenase. J Biol Chem, 2015. 290(33): p. 20009–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winkler HH and Neuhaus HE, Non-mitochondrial ATP transport. Trends Biochem Sci, 1999. 24(2): p. 64–8. [DOI] [PubMed] [Google Scholar]
- 53.Albe KR, Butler MH, and Wright BE, Cellular concentrations of enzymes and their substrates. J Theor Biol, 1990. 143(2): p. 163–95. [DOI] [PubMed] [Google Scholar]
- 54.Rico-Jimenez M, et al. , Paralogous chemoreceptors mediate chemotaxis towards protein amino acids and the non-protein amino acid gamma-aminobutyrate (GABA). Mol Microbiol, 2013. 88(6): p. 1230–43. [DOI] [PubMed] [Google Scholar]
- 55.Rahman H, et al. , Characterisation of a multi-ligand binding chemoreceptor CcmL (Tlp3) of Campylobacter jejuni. PLoS Pathog, 2014. 10(1): p. e1003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Delumeau O, et al. , Functional and structural characterization of RsbU, a stress signaling protein phosphatase 2C. J Biol Chem, 2004. 279(39): p. 40927–37. [DOI] [PubMed] [Google Scholar]
- 57.Tjaden J, et al. , Two nucleotide transport proteins in Chlamydia trachomatis, one for net nucleoside triphosphate uptake and the other for transport of energy. J Bacteriol, 1999. 181(4): p. 1196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattoo S, et al. , Regulation of type III secretion in Bordetella. Mol Microbiol, 2004. 52(4): p. 1201–14. [DOI] [PubMed] [Google Scholar]
- 59.Yu HH and Tan M, Sigma28 RNA polymerase regulates hctB, a late developmental gene in Chlamydia. Mol Microbiol, 2003. 50(2): p. 577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brothwell JA, et al. , Interrogating Genes That Mediate Chlamydia trachomatis Survival in Cell Culture Using Conditional Mutants and Recombination. J Bacteriol, 2016. 198(15): p. 2131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber A, et al. , The 2-oxoglutarate/malate translocator of chloroplast envelope membranes: molecular cloning of a transporter containing a 12-helix motif and expression of the functional protein in yeast cells. Biochemistry, 1995. 34(8): p. 2621–7. [DOI] [PubMed] [Google Scholar]
- 62.Belland RJ, et al. , Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A, 2003. 100(14): p. 8478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicholson TL, et al. , Global stage-specific gene regulation during the developmental cycle of Chlamydia trachomatis. J Bacteriol, 2003. 185(10): p. 3179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Packiam M, et al. , Structural characterization of muropeptides from Chlamydia trachomatis peptidoglycan by mass spectrometry resolves “chlamydial anomaly”. Proc Natl Acad Sci U S A, 2015. 112(37): p. 11660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pilhofer M, et al. , Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ. Nat Commun, 2013. 4: p. 2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liechti GW, et al. , A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature, 2014. 506(7489): p. 507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fisher DJ, Adams NE, and Maurelli AT, Phosphoproteomic analysis of the Chlamydia caviae elementary body and reticulate body forms. Microbiology, 2015. 161(8): p. 1648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans PR, An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr, 2011. 67(Pt 4): p. 282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vonrhein C, et al. , Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr, 2011. 67(Pt 4): p. 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matthews BW, Solvent content of protein crystals. J Mol Biol, 1968. 33(2): p. 491–7. [DOI] [PubMed] [Google Scholar]
- 71.Adams PD, et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr, 2010. 66(Pt 2): p. 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCoy AJ, et al. , Phaser crystallographic software. J Appl Crystallogr, 2007. 40(Pt 4): p. 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Langer G, et al. , Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc, 2008. 3(7): p. 1171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emsley P, et al. , Features and development of Coot. Acta Crystallogr D Biol Crystallogr, 2010. 66(Pt 4): p. 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Painter J and Merritt EA, Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr, 2006. 62(Pt 4): p. 439–50. [DOI] [PubMed] [Google Scholar]
- 76.Winn MD, Isupov MN, and Murshudov GN, Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr, 2001. 57(Pt 1): p. 122–33. [DOI] [PubMed] [Google Scholar]
- 77.Chen VB, et al. , MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr, 2010. 66(Pt 1): p. 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O’Hearn SD, Kusalik AJ, and Angel JF, MolCom: a method to compare protein molecules based on 3-D structural and chemical similarity. Protein Eng, 2003. 16(3): p. 169–78. [DOI] [PubMed] [Google Scholar]
- 79.Coordinators NR, Database resources of the National Center for Biotechnology Information. Nucleic Acids Res, 2017. [DOI] [PMC free article] [PubMed]
- 80.Zhang Y and Skolnick J, TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res, 2005. 33(7): p. 2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y and Skolnick J, Scoring function for automated assessment of protein structure template quality. Proteins, 2004. 57(4): p. 702–10. [DOI] [PubMed] [Google Scholar]
- 82.Xu J and Zhang Y, How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics, 2010. 26(7): p. 889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wishart DS, et al. , HMDB: the Human Metabolome Database. Nucleic Acids Res, 2007. 35(Database issue): p. D521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wishart DS, et al. , HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res, 2009. 37(Database issue): p. D603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wishart DS, et al. , HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res, 2018. 46(D1): p. D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caspi R, et al. , The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res, 2016. 44(D1): p. D471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hawkins PC, et al. , Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model, 2010. 50(4): p. 572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McGann M, FRED pose prediction and virtual screening accuracy. J Chem Inf Model, 2011. 51(3): p. 578–96. [DOI] [PubMed] [Google Scholar]
- 89.Friesner RA, et al. , Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem, 2004. 47(7): p. 1739–49. [DOI] [PubMed] [Google Scholar]
- 90.Friesner RA, et al. , Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem, 2006. 49(21): p. 6177–96. [DOI] [PubMed] [Google Scholar]
- 91.Halgren TA, et al. , Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem, 2004. 47(7): p. 1750–9. [DOI] [PubMed] [Google Scholar]
- 92.Jacobson MP, et al. , On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol, 2002. 320(3): p. 597–608. [DOI] [PubMed] [Google Scholar]
- 93.Jacobson MP, et al. , A hierarchical approach to all-atom protein loop prediction. Proteins, 2004. 55(2): p. 351–67. [DOI] [PubMed] [Google Scholar]
- 94.Hindson BJ, et al. , High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem, 2011. 83(22): p. 8604–10.’ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. I-TASSER model of RsbU1-315 shares structural similarity to periplasmic domain RsbU45-313 crystal structure. A) I-TASSER protein structure model with residues 1 through 315 of C. trachomatis RsbU. B) Structure overlay of RsbU45-313 crystal structure (magenta) and I-TASSER model (yellow). Structure comparison has Z-score of 14.4 and a RMSD of 4.0 Å.
Figure S2. SDS-PAGE gel showing purification of RsbU45-313. Lane 1, protein marker; lane 2, lysate supernatant; lane 3, lysate pellet; lane 4, IMAC flow-through; lane 5, IMAC elution; lane 6, post-buffer exchange; lane 7, post-TEV protease treatment; Lane 8, post-reverse nickel column; lane 9, SEC column peak fraction.
Figure S3. SPR dose-dependent binding curves. Alpha-ketoglutarate, malate, and oxaloacetate show dose-dependent binding to RsbU45-313, while succinate and malonate, the ligands found to bind to DctB, do not appear to be binding by SPR.
Figure S4. Chromosome schematic of cross between RsbU* EMS strain and ct575::Tn strain to create the complemented RsbU+ strain that retains the majority of the other SNPs induced by EMS mutagenesis. The ct575::Tn strain contains a beta-lactamase resistance gene in the transposon, while the RsbU* has a SNP in the rpoB gene that incurs rifampicin resistance. Dual selection with ampicillin and rifampicin was utilized to select for recombinants that retained the RsbU* strain backbone but a wild-type version of the rsbU gene. The red region on the RsbU+ chromosome represents the region of recombination between the genomes confirmed by PCR of the SNPs present in the RsbU* strain. In addition to the restoration of the rsbU gene, the RsbU+ strain restores three other SNPs close to the position of the transposon: two in coding regions for secY and polA; and one in an intergenic region (IGR).
Figure S5. Immunofluorescent microscopy of C. trachomatis ct575::Tn parent strain and RsbU+ complemented strain. At 24 and 72 hours post-infection, the RsbU+ complemented strain does not appear to be phenotypically different from the Tn parent strain.