

Abstract
Understanding the intricacies of how the body regulates pain is fundamental to develop rational strategies to combat the growing prevalence of chronic pain states, opioid dependency, and the resulting increased financial burden to the medical care system. Pain is the most prominent reason why Americans seek medical attention and extensive literature has identified the importance of the endocannabinoid pathway in controlling pain. Endocannabinoid signaling machinery operates in a synapse-specific manner, and its modulation offers new therapeutic opportunities for the selective control of excessive neuronal activity in several pain conditions (acute, inflammatory, chronic, and neuropathic). Cannabinoids have a long history of medicinal use and their analgesic properties are well documented; however, there are major impediments to understanding cannabinoid pain modulation. One major issue is the presence of psychotropic side effects associated with Δ9-tetrahydrocannabinoil (THC) or synthetic derivatives, which puts an emphatic brake on their use. This dose-limiting effect lends to the idea that the appropriate degree of analgesia cannot be reached before the presence of severe side effects. Animal studies have shown that the psychotropic effects are mediated via brain cannabinoid type 1 (CB1) receptors, while analgesic activity in chronic pain states may be mediated via CB1R action in the spinal cord, brainstem, peripheral sensory neurons, or immune cells. The development of appropriate therapies is incumbent on our understanding of the role of peripheral versus central endocannabinoid-driven analgesia. Recent physiological, pharmacological, and anatomical studies provide evidence that one of the main roles of the endocannabinoid system is the regulation of gamma-aminobutyric acid (GABA) and/or glutamate release. This article will review this evidence in the context of its implications for pain. We first provide a brief overview of CB1R’s role in the regulation of nociception, followed by a review of the evidence that the peripheral endocannabinoid system modulates nociception. We then look in detail at regulation of central-mediated analgesia, followed up with evidence that cannabinoid-mediated modulation of pain involves modulation of GABAergic and glutamatergic neurotransmission in key brain regions. Finally, we discuss cannabinoid action on non-neuronal cells in the context of inflammation and direct modulation of neurons. This work stands to reveal long-standing controversies in the cannabinoid analgesia area that have had an impact on failed clinical trials and implementation of therapeutics targeting this system.
Keywords: Cannabinoid, Chronic Pain, Analgesia, Opiate Alternative, Pain Processing, CB1R
Introduction
Chronic pain is a pressing medical issue that costs the United States over $600 billion annually and impacts over 100 million Americans (Dzau & Pizzo, 2014). Despite this growing public health problem, limited therapeutic resources exist to combat this issue. Current resources such as prescription opioids, come with a myriad of negative health consequences that in turn cause their own public health crisis, with opioid overdosing recently becoming one of the leading causes of death in the United States (Chen, Hedegaard, & Warner, 2014; Paulozzi, Budnitz, & Xi, 2006; Webster et al., 2011). Physicians attempting to treat chronic pain are now having to address the dual disorders of chronic pain and addiction that results from the use of current opioid-based analgesics. There is a huge demand to address these public health concerns and find alternatives to the current treatments for pain. Enhanced focus has recently been placed on the endocannabinoid system to investigate its prospective therapeutic use for chronic pain and its potential to combat the current opioid epidemic. With increasing political attention surrounding personal and medical use of cannabis, it has become more imperative than ever to understand the endocannabinoid system and its involvement in pain modulation.
The neurological processing of pain involves a highly complex, multi-level biological system with many components (Woolf, 2013). To further complicate the study of the relationship between the endocannabinoid system and pain processing, the primary sites of action of cannabinoids, cannabinoid receptors (CBRs), are ubiquitously expressed throughout the nervous system at every level (Mackie, 2005). With such wide spread concentrations of these receptors in critical pain processing regions, further described in detail throughout this review, it has long been postulated that cannabinoids directly modulate this system. While there is much research needed to better characterize the analgesic effects of cannabinoids, a clear relationship between the endocannabinoid system and pain modulation has been established.
Cannabis has been used medicinally for millennia to treat numerous illnesses, including pain conditions (Hudson & Puvanenthirarajah, 2018). Its analgesic effects have been demonstrated empirically in animal models and human subjects alike (Skaper & Di Marzo, 2012). These studies have continuously revealed the ability of cannabinoid agonists to inhibit responses to noxious stimuli in different pain states, including mechanical allodynia, thermal hypersensitivity, inflammatory nociception, and neuropathic pain (Amaya et al., 2006). Additionally, cannabinoid antagonists exacerbate nociceptive responses in the same pain modalities, further demonstrating the importance of cannabinoid receptor activation in anti-nociception and analgesia (Bishay et al., 2013).
While there are clearly analgesic aspects of cannabis, there are also many negative effects of the drug, such as anxiety, paranoia, impaired memory and motor functions, and addictive potential (Hill, Palastro, Johnson, & Ditre, 2017). It would be ideal to separate the analgesic effects from the negative experiences associated with marijuana use for chronic pain therapeutics but with the plant containing over 70 lipophilic cannabinoids that activate receptors on a wide number of cells throughout the central and peripheral nervous system, isolating the benefits of cannabinoids will require a more robust understanding of how these molecules function, individually and collectively, in the human body.
There is currently heavy debate on how this issue can be addressed with two conflicting perspectives prevailing. The first argues that cannabinoid modulation of pain occurs through inhibition at the level of the dorsal root ganglia, thus inhibiting ascending nociceptive signals (Agarwal et al., 2007). The second argues that modulation occurs through activity at the level of the brainstem, inhibiting pain signals through descending suppression of nociceptive signals (Meng & Johansen, 2004). Research has yet to delineate the necessity and sufficiency of these two processes in cannabinoid analgesia, and there is evidence in support of both theories. This review summarizes the most recent studies that address cannabinoid modulation in these two systems and aims to provide a comprehensive overview of the different models between peripheral nervous system (PNS) and central nervous system (CNS) modulation.
Cellular Mechanisms of the Endocannabinoid System
Endocannabinoid system receptors are widely expressed throughout the body, including the gastrointestinal system, the endocrine system, the immune system, and the nervous system. Endogenous cannabinoids are lipophilic and therefore synthesized on demand. Two receptor subtypes have been identified as primary cites of action of the endocannabinoid system, cannabinoid receptor 1 (CB1R) and cannabinoid receptor 2 (CB2R). CB1R is prolific throughout the body, with moderate to high concentrations in pain modulating areas, such as the dorsal root ganglia of the spinal cord (DRG), the periaqueductal gray (PAG) and rostral ventral medulla (RVM) in the brainstem, as well as higher cortical brain regions including the limbic system (Ahluwalia, Urban, Capogna, Bevan, & Nagy, 2000). CB2R is also widespread throughout the body but in highest concentrations in the periphery and on immune cells (Gutierrez, Farthing, Zvonok, Makriyannis, & Hohmann, 2007; Pertwee, 1997). These are G-protein coupled receptors that influence firing of neurons through regulation of neurotransmitter release at neuronal synapses. While there is evidence that both receptor sub-types play a role in cannabinoid analgesia, CB1R is believed to have the most direct influence on neuronal pain processing pathways and will be the focus of this review (Seltzman et al., 2016).
CB1 receptors belong to a family of rhodopsin-like G protein-coupled receptors (GPCRs) and influence Gi/o receptor intracellular messenger cascades (Wang et al., 2016). They are negatively-coupled to adenylyl cyclase and promote activation of mitogen-activated protein kinases (MAPK) (Dalton, Bass, Van Horn, & Howlett, 2009). This reduces the excitability of neurons by modulating inward-rectifying K+ channels and Ca2+ channels (McAllister, Griffin, Satin, & Abood, 1999). At the level of singular circuitry, the net modulatory impact of CB1 receptors are highly reliant upon the nature of the presynaptic cell’s intrinsic excitatory (glutamatergic) or inhibitory (GABAergic) properties (Katona & Freund, 2008). This allows CB1 receptors to function as both a modulator and a gate to noxious afferent input from peripheral nociceptors (Stander, Schmelz, Metze, Luger, & Rukwied, 2005). These receptors are known to signal through retrograde transmission, regulating neurotransmitter release post-synaptically (Vaughan & Christie, 2005).
Peripheral Nervous System Analgesia
The dorsal horn of the spinal cord consolidates nociceptive and sensory afferent information and accounts for all ascending pathways to higher order cortical structures (Seltzman et al., 2016). In addition to nociceptors in the periphery, many glutamatergic sensory afferents and GABAergic interneurons of the dorsal spinal cord express CB1 (Ware et al., 2010). It is purported that endocannabinoids are produced after stimulation of glutamatergic nociceptive, small- and medium-diameter C-fibers and activate CB1 receptors expressed on inhibitory interneurons within the dorsal horn of the spinal cord. Based on the mechanics of retrograde spinal circuitry, this reduces GABAergic signaling and increases nociceptor excitability leading to maladaptive nociception (Finnerup et al., 2015). New research shows that a small population of astrocytes in the spinal cord express CB1 receptors and activation of these receptors on astrocytes leads to transient Ca2+ currents that stimulate the production of 2-arachidonoylglycerol (2-AG), a potent endogenous CB1 agonist (Herkendell, Tel-Vered, & Stemmer, 2017). These findings would suggest that CB1 receptors expressed in the dorsal horn of the spinal cord are responsible for mediating the effects of chronic, neuropathic pain.
Pre-clinical studies utilizing animal models have demonstrated CB1R modulation at the level of the peripheral nervous system, specifically in nociceptors in the DRG. This is particularly important in cannabinoid research because determining how peripheral pain modulation occurs, peripheral sites can be identified and targeted with therapeutic cannabinoid drugs that specifically produce analgesia, but do not cause adverse psychotropic effects. Because there is such a huge push for non-psychoactive analgesics, most of the research examining cannabinoid modulation of pain processing has focused on peripheral mediation. Functional aspects of the endocannabinoid system have been discovered and suggest a role in peripheral mediation of pain processing. Firstly, CB1 receptors are typically expressed on the terminals of sensory afferent fibers and are found on over 75% of nociceptive neurons in the DRG (Mitrirattanakul et al., 2006). These neurons send CB1R out to the peripheral nerve terminals in response to noxious stimuli, further suggesting a mediating role of endogenous cannabinoids and their receptors on nociception. Additionally, experimental administration of various endogenous and exogenous cannabinoid ligands mediate the activity of spinal neurons (Hohmann & Herkenham, 1999; Khasabova et al., 2013; Mitrirattanakul et al., 2006).
Intrathecal (i.t.) injection of anandamide, an endogenous cannabinoid ligand, was found to inhibit A-delta and C-fiber neuronal responses to inflammatory pain (Harris, Drew, & Chapman, 2000). This was also demonstrated with (i.t.) HU-210, an exogenous cannabinoid ligand, with the most robust findings showing inhibition of hyper-excitability of C-fibers after repeated firing to inflammatory pain. Co-administration of selective CB1R antagonist S141716 (i.t.) blocked the analgesic effects of anandamide and HU-210, indicating a critical role of CB1R activation. Follow-up studies demonstrated that i.t.injection of HU-210 blocked neuronal nociceptive responses, as well as peak action potentials in the DRG (Millns, Chapman, & Kendall, 2001). The inhibitory action of HU-210 was again blocked by intrathecal co-administration of S141716. These results were also seen with WIN 55-212-2, another CB1R agonist (Fox et al., 2001). Systemically administered WIN 55-212-2 inhibited hyper-responsivity of neurons in the DRG to noxious pressure but did not alter non-nociceptive neurons (Hohmann, Martin, Tsou, & Walker, 1995). These finding provide strong support to suggest a specific role of CB1R in anti-nociception and acute analgesia in the peripheral nervous system.
There is also considerable evidence for cannabinoid-induced analgesia in animal behavioral models of chronic pain. In a commonly used neuropathic pain paradigm of sciatic nerve injury, intrathecal injection of WIN 55-212-2 into the spinal cord at the L4 and L5 vertebrate had a strong analgesic effect on thermal and mechanical nociception (Fox et al., 2001). Intraplantar administration of anandamide, an endogenous CBR ligand, but not intraperitoneal administration, inhibited thermal inflammatory pain and edema. These analgesic effects were blocked by intraplantar administration of S141716, a specific CB1R antagonist, suggesting a critical and specific role of CB1R activation for the analgesic effects of locally administered anandamide. This was the first study to demonstrate the role of local CB1R modulation of inflammatory pain in the peripheral nervous system (Richardson, Kilo, & Hargreaves, 1998).
Many studies following have also shown analgesic effects of local administration of cannabinoids in several rodent models. Intraplantar administration of Acrachidonyl-2’chloroethylamide (ACEA) inhibited acute nociception after complete freund’s adjuvant (CFA) an immune-activation agent, injection into the hind paw and strongly attenuated chronic pain 2 days after CFA injection (Amaya et al., 2006). Further, the ratio of neurons expressing CB1R mRNA began to increase 1 day after CFA injection, peaked on day 2, increasing by almost double, and then gradually declined to normal levels 7 days after injection. Further, many nerve endings in the dermis stained strongly positive for CB1R mRNA following CFA injection. Local administration of ACEA also inhibited nociceptive responses to carrageen-induced inflammation in the paw, an effect that was blocked by co-administration with S141716 (Gutierrez et al., 2007).
This anti-nociceptive activity was also shown with the endogenous CB1R ligand anandamide, and synthetic cannabinoid ligands WIN 55-212-2, and CP 55-940 (Calignano, La Rana, Giuffrida, & Piomelli, 1998). Anandamide blocked acute pain and WIN 55-212-2 and CP 55-940 blocked both acute and chronic pain when simultaneously injected with formalin. The anti-nociceptive effects of these agonists were blocked with systemic injection of CB1R antagonist, SR141716A. Local administration of WIN 55-212-2 also attenuated inflammatory nociception after hind-paw injection with carrageenan, reducing mechanical hyperalgesia and allodynia that resulted after carrageenan injection. Further, WIN 55-212-2 suppressed carrageenan-induced up-regulation of Fos protein expression in DRG neurons, suggesting a multi-faceted role pain modulation in the periphery (Nackley, Suplita, & Hohmann, 2003).
Peripherally restricted CB1R agonists have been investigated in pre-clinical pain models. Indole and indene compounds were developed with high affinity for CB1R but are unable to cross the blood brain barrier. These agonists target the peripheral nervous system and do not engage the central nervous system (Seltzman et al., 2016). Intraperitoneal and oral administration of these drugs suppressed sciatic nerve entrapment induced mechanical allodynia, with no behavioral signs of CNS activation, suggesting sufficiency of cannabinoid modulation in the peripheral nervous system of analgesia. URB937 is another peripherally restricted CB1R agonist that has been shown to inhibit nociceptive responses to peripheral nerve injury and inflammation. URB937 acts by inhibiting fatty acid amide hydrolase (FAAH), increasing anandamide concentrations and the duration of ligand action (Clapper et al., 2010). The CB1R allosteric modulator, ZCZ011 also exhibited analgesia after chronic constriction injury but had no central nervous system action as observed with no differences in conditioned place preference, catalepsy, and locomotor activity, indicating possible peripheral site of action of pain modulation (Ignatowska-Jankowska et al., 2015). ZCZ011 is brain permeable however, and the actual specificity of these effects need to be further tested.
Transgenic animal models have been extremely vital to the research of endocannabinoid receptors in the processing and modulation of pain. Utilizing a cre-lox transgenic model, Agarwal, et. al., 2007 was able to create a mouse line of selective CB1R ablation from peripheral sensory neurons only, but left CB1R intact throughout the rest of the body, including in the spinal cord, brain, and other organs (Agarwal et al., 2007). These sensory neuron CB1R knockout mice had significantly increased mechanical allodynia compared to WT after intraplanar CFA, capsaicin, and formalin injections, when tested independently. Importantly, after CB1R agonist, WIN 55-212-2 administration, the knockout mice showed only 17% analgesic responses compared to WT counterparts that showed approximately over 50% pain attenuation. Additionally, mechanical hyperalgesia and thermal allodynia were nearly fully reversed 17 hours after intraperitoneal injection of WIN 55-212-2 in wild-type animals, it was only slightly reduced in the selective knockout mice. This study garnered a lot of attention, as it was the first study to demonstrate the necessity of CB1R activation in peripheral sensory neurons for analgesia (Agarwal et al., 2007).
Despite the numerous robust findings in the pre-clinical setting that support the idea that cannabinoid modulation occurs in the peripheral nervous system (Agarwal et al., 2007; Seltzman et al., 2016), these findings have not been replicated in humans. Based on the aforementioned pre-clinical work, clinical researchers were eager to test peripherally restricted cannabinoid agonists, as it could revolutionize therapies for chronic pain. However, human clinical trials using the peripherally restricted synthetic cannabinoids, AZD1940 and AZD1704 have failed to produce any analgesic effect (Kalliomaki et al., 2013) (Pacher & Kunos, 2013). Further, an overwhelming majority of peripherally acting cannabinoid agonists that make it to clinical trials fail for many reasons: lack of efficacy compared to placebo, acute tissue damage to kidneys (Murphy et al., 2013), and a dearth of evidence for the safety of long-term cannabinoid use for the treatment of chronic pain (Finnerup et al., 2015; Kunos, Osei-Hyiaman, Bátkai, Sharkey, & Makriyannis, 2009; Kunos & Tam, 2011; Pacher & Kunos, 2013). It is unclear if these results suggest a failure of peripheral CB1R to inhibit ascending pain signaling or if the cannabis derivatives do not contain the necessary biochemical compounds to achieve optimal activation of CB1R to produce analgesia. Further, these trials utilized capsaicin injection to induce pain, a method that does not reflect many pathological and chronic pain states. Despite these criticisms, these clinical trials debunk the results of the preclinical work summarized above.
Central Nervous System Analgesia
Central nervous system action of cannabinoids has been clearly demonstrated and is historically established. Cannabis has been used throughout history and across various cultures to treat numerous ailments, including pain states (Hudson & Puvanenthirarajah, 2018). CB1 receptors are ubiquitously expressed within the CNS, with the highest aggregates in higher order brain structures like the hippocampus and cerebral cortex, as well as in the periaqueductal gray (PAG) and rostral ventral medulla (RVM) of the brainstem (Seltzman et al., 2016). There are many studies indicating the various and multifaceted CNS effects of cannabinoids on behavior and cognition, including motor impairments, anxiety, memory and learning deficits, as well as addictive potential of cannabis use (Hill et al., 2017). In regards to pain processing, an important distinction between peripheral and central CB1R action is that within the CNS, CB1 receptors may modulate descending pain information as well as regulating the affective or emotional components of pain (Bajic et al., 2018). Direct administration of CB1R agonists on the thalamus, PAG, RVM, and dorsal raphe nucleus all produce anti-nociceptive effects (Martin, Patrick, Coffin, Tsou, & Walker, 1995).
One of the most well studied and well-understood pain modulating areas in the central nervous system is the periaqueductal gray in the brainstem. This region is prominent in opioid analgesia and is likely a critical component of the endocannabinoid system as it is densely saturated with CB1 receptors. This area releases endogenous anandamide in response to noxious stimuli, indicating a pivotal region of endocannabinoid action (Walker, Huang, Strangman, Tsou, & Sanudo-Pena, 1999). Electrical stimulation of the PAG has shown analgesic effects after intradermal formalin injection in rats that are associated to increased anandamide release in the PAG. These analgesic effects are attenuated after injection of the CB1R antagonist, S141716, into the PAG, suggesting a critical role of CB1R in this brain region for pain modulation (Walker et al., 1999). Further, injections of WIN 55-212-2 directly into the dorsolateral PAG inhibited nociceptive responses to noxious heat (Martin et al., 1995).
Early phase algesia in the hind paw after formalin injection was not affected by HU-210 injection directly into the dorsal PAG but late phase algesia was significantly reduced, suggesting an important role of cannabinoids in the PAG in chronic pain inhibition. Further, late phase analgesia produced by HU-210 was blocked by S141716, although injection of S141716 alone did not increase algesic effects of formalin injection acutely or over time. Additionally, PAG administration of HU210 significantly attenuated formalin-evoked increases in c-Fos expression in the caudal lateral PAG (Finn et al., 2003).
Cellular examination of the PAG revealed WIN 55-212-5 inhibits glutamatergic transmission, an effect that was blocked by S141716 but not the opioid antagonist, naloxone (Vaughan, Connor, Bagley, & Christie, 2000). This study reveals critical cellular mechanisms by which cannabinoids may be analgesic via CB1R in the PAG. WIN 55-212-2 did not lead to any changes in membrane current of potassium or calcium, nor did S141716. This indicates analgesic action occurs by a separate mechanism than opioids and other GABAergic agonists.
Another brainstem region that is implicated in cannabinoid descending pain modulation is the RVM. Injection of HU-210 and WIN 55-212-2 into this region strongly increased tail-flick latencies in rats. Co-administration of S141716 blocked these nociceptive effects, suggesting specific CB1R modulation of this analgesic effect (Martin, Tsou, & Walker, 1998). These results are not limited to brainstem nuclei and were also demonstrated in higher cortical regions, including the amygdala and the lateral posterior and submedius regions of the thalamus (Martin et al., 1999). Systematic injection of WIN 55-212-2 into these neurological regions increased latencies in tail-flick responses to noxious heat. This study shows modulation of cannabinoid anti-nociception occurs centrally and at higher order cortical regions than only at well-known pain processing nuclei of the brainstem.
Much of the research investigating cannabinoid modulation of pain in the central nervous system has focused on the differential effects of cannabinoids on different cell types in the brainstem (Bajic et al., 2018). The encoding of pain in the brainstem involves differential neuron firing. It has been shown that some neurons are active just prior to pain response in animals and at the same time, other neurons are inhibited just prior to pain responses. These neurons are referred to as “ON-cells” and “OFF-cells,” respectively. Cannabinoids have been shown to affect these neurons after the induction of noxious stimulus, inhibiting ON-cells and exciting OFF-cells. Meng & Johansen (2004) found that WIN 55-212-2 reduced firing of ON-cell bursts related to tail-flick activity and increased firing of OFF-cells in the rostral ventral medial nucleus of the brainstem. This suggests potential multi-action modulation of cannabinoids through inhibition of excitatory cells and disinhibition of inhibitory cells, specifically in pain pathways of the medullary brainstem (Meng & Johansen, 2004). Further, WIN induced a rapid increase in functioning of OFF-cells with ON-cell inhibition more delayed. The authors argue that these findings suggest that increased activity of OFF-cells may be the most critical to produce anti-nociception. This is likely a fundamental function by which cannabinoids induce analgesia and a potential therapeutic target of chronic pain medication.
Unlike the peripherally restricted cannabinoid clinical trials described above, there have been many promising clinical trials utilizing non-restricted and centrally acting cannabinoids. Both low (3.5% THC) and high (7% THC) doses of cannabis cigarettes produced analgesia in chronic neuropathic pain patients with no significant mood alterations and only limited neurocognitive side effects, including impaired memory and learning. These doses were generally well tolerated by patients who reported a feeling of high but with a “good drug effect” (Wilsey et al., 2008). These analgesic results were supported in patients with HIV (Ellis et al., 2009), multiple sclerosis (MS) (Rog, Nurmikko, Friede, & Young, 2005), and diabetic neuropathy (Wallace, Marcotte, Umlauf, Gouaux, & Atkinson, 2015).
These studies suggest effective uses for centrally acting cannabinoids on neuropathic pain specifically, however, cannabinoid-induced analgesia of other types of pain, such as acute and thermal, have had less robust and more conflicting results. Because cannabis is composed of so many different types of cannabinoids and they act individually on the entire cannabinoid system, often in opposite and competing ways, specific targets of action in the human central nervous system has not yet been determined.
GABAergic vs Glutamatergic
When investigating central cannabinoid modulation, it is necessary to consider the functional influence of cannabinoid action on both GABAergic and glutamatergic neurons in the brain. CB1R is expressed on both of these cell types throughout the central nervous system, including the spinal cord. There is evidence in support of CB1R mediated increases, as well as decreases, in activity of both of these neuronal cell types, which leads to major questions in the functional mechanisms of CB1R modulation of pain. Given that increasing GABAergic activity is systemically inhibitory and increasing glutamatergic activity is systemically excitatory, it is unclear how CB1R functions in these counteracting cell types and if it has a higher affinity for one over the other. Recent studies have sought to characterize the functional and biological mechanisms of central CB1R in the context of both glutamatergic and GABAergic signaling (Bajic et al., 2018) in order elucidate the underlying mechanisms of cannabinoid action and develop more specific and effective therapeutic targets.
Cannabinoid action of glutamatergic neurons in pain modulation has been supported with experiments utilizing both CB1R and glutamatergic receptor agonist. Formalin injection into the hind-paw increased firing bursts of pain responsive neurons (ON-cells) and paused firing of other neighboring neurons (OFF-cells). Low doses of WIN 55-212-2 prevented these changes in neuronal firing patterns in the RVM after formalin injection. At higher doses, WIN 55-212-2 had the opposite effect of formalin in the RVM, decreasing the spontaneous activity of ON-cells and increasing the spontaneous activity of OFF-cells. It also delayed the burst firing of ON-cells in response to nociceptive stimulus and delayed the pause in activity of OFF-cells. Importantly, this effect was blocked by a selective metabotropic glutamate receptor 5 (mGluR5) antagonist, suggesting a critical role of glutamatergic neuron activity in the RVM in cannabinoid modulation of pain (de Novellis et al., 2005). This was further supported by Palazzo (2001), who not only confirmed these results, but also showed the importance of glutamatergic NMDA receptors as well (Palazzo, Luongo, Novellis, Rossi, & Maione, 2010).
At the level of the spinal cord, intrathecal injection of WIN 55-212-2 produced an anti-hyperalgesic effect after sciatic nerve injury that required engagement of glutamatergic signaling via mGluR5, as demonstrated with a blockage of analgesic activity when co-administered with a mGluR5 antagonist (Hama & Urban, 2004). There is also evidence to suggest CB1R spinal nociception occurs via activation on GABAergic neurons. Intrathecal injection of the CB1R antagonist, S141716A partially blocked GABA-induced analgesia to formalin inflammatory pain when simultaneously administered with the GABA-B agonist, baclofen (Naderi, Shafaghi, Khodayar, & Zarindast, 2005). This suggests a necessary role of CB1R activation on GABAergic interneurons of the spinal cord for peripheral anti-nociception.
CB1R influences GABAergic and glutamatergic neurons in other brain regions as well, including the thalamus, limbic system regions, and the hypothalamus with evidence suggesting there may be an analgesic role of CB1R in these brain regions (Rea, Roche, & Finn, 2007). These higher cortical areas are postulated to be involved in the affective component of pain although affective pain processing research is still preliminary (Bajic et al., 2018).
The aforementioned studies provide evidence that CB1R modulates both glutamatergic and GABAergic neurons, although the evidence is limited and far from determining which mechanism of action is more critical for analgesia. This is a crucial argument to delineate if cannabinoid therapeutics hope to emerge. It is possible that CB1R is primarily acting through glutamatergic release, increasing firing rates of pain inhibiting neurons, thus inhibiting nociceptive signals. It is also possible that GABAergic modulation is more critical, decreasing neuronal firing rates of pain sensing and amplifying neurons. The opposite could also be true, in which direct CB1R modulation of glutamatergic activity is actually increasing nociceptive signaling and GABAergic action is actually inhibitory, decreasing neurons that predominantly inhibit nociception. Further, with evidence to support analgesic activity of these neurons in the spinal cord, the brain stem, and higher cortical regions, this further complicates the debate between central versus peripheral cannabinoid mediation of pain signals. The various ways in which CB1R modulates these cells may be the underlying mechanism that causes such conflicting clinical results, in which cannabinoids inhibit pain in some cases but exacerbate it in others (Cooper, Comer, & Haney, 2013) (Corey-Bloom et al., 2012) (Wilsey et al., 2013). Further investigation of the cannabinoid modulation on these cell lines is critical to understanding the potential therapeutic uses of cannabinoids for analgesia.
Affective component of pain
The experience of pain has both sensory and affective components that are necessary to define the experience as salient and unpleasant. Pain is intended to act as a signal that physiological damage is possible or has already occurred. By inducing an aversive sensation, pain acts as a negative reinforcer for the behavior that caused it. The negative emotions associated with pain are evolutionarily adaptive and necessary for the avoidance of harmful stimuli in the environment. There are many brain regions involved in the affective component of pain.
The central nervous system tract postulated to be responsible for the affective components of pain is the spinomesencephalic tract, which connects spinal neurons of the dorsal horn at laminae I and V to the PAG. The PAG then projects to both the amygdala (after synapsing to the parabrachial nucleus) and the RVM. These regions process the painful stimuli, alerting of potentially harmful scenarios but also pain compensation and suppression, likely via projections from the RVM back to nociceptive fibers in the dorsal horn (Ottestad, Angst, & anesthesia, 2013). There are high concentrations of CB1R in both of these nuclei, suggesting a potential role of cannabinoid modulation of this system.
The PAG is well established as a major analgesic contributor in the pathways that control both nociception and affective component processing of pain. Interest in the PAG first developed when early studies found that analgesia resulted from direct stimulation of the PAG (Heinricher, Tavares, Leith, & Lumb, 2009). While whole body CB1R knockouts (CB1-KO) show increased sensitivity to pain, they also show more robust anxiety behaviors in response to noxious stimuli. In a model using partial sciatic nerve ligation, CB1-KO mice showed greater anxiety and depression-like behaviors, such as sucrose anhedonia, disturbed home cage behavior, and increased time in the closed portions of the elevated zero-maze test, compared to wild-type controls, despite both groups showing similar mechanical hypersensitivity (Racz, Nent, Erxlebe, & Zimmer, 2015). Another study using CB1-KO mice showed that following a knee osteoarthritis model, CB1-KO mice have greater anxiety-like behavior in the elevated plus maze compared to wild-type controls, both one and three weeks after intra-articular injection with monosodium iodoacetate (La Porta et al., 2015).
In addition to the PAG, the RVM is a major contributor to the affective pain pathway. Just as stimulation of the PAG causes analgesia, stimulation of the RVM has been shown to inhibit the firing of nociceptors within the dorsal horn, specifically laminae I, II, and V (Koch & Fitzgerald, 2014). Additionally, subcutaneous injection of formalin modified the activity of RVM neurons via CB1R activity in the PAG. Injections of WIN55,212-2 (2 nmol/rat) inhibited the RVM activity change and at higher doses (4nmol/rat) increased tail flick latency (de Novellis et al., 2005). This suggests that the PAG is important for controlling/maintaining RVM neuronal activity and plays a major role in controlling both nociception and affective components of pain.
The amygdala has been extensively studied for its role in fear-learning using Pavlovian Conditioning, in which an animal learns to avoid a specific behavior based on its pairing with an unpleasant or painful stimulus. This fear learning pathway is evolutionarily adaptive and an effective tool for the avoidance of damage however, this process can be maladaptive, especially in cases of chronic pain syndromes leading to chronic depression, anxiety, and other affective disorders. A fear-learning study by Kim et al. (Kim et al., 2013) in male rats showed that stimulation of either the PAG or the amygdala causes fear-responses in rats undergoing a fear-conditioning and foraging task; however, the fear-response was blocked when the amygdala was lesioned prior to PAG stimulation. This change in learning behavior was also found by Di Scala, Mana, Jacobs & Phillips, who found that stimulation of the PAG induces changes in behavior via conditioning mechanisms such as fear-learning and other salient events (Di Scala, Mana, Jacobs, & Phillips, 1987).
CB1R is present in high concentrations throughout the central nervous system, including higher order regions such as the amygdala (Katona et al., 2001). Utilizing a conditioned place preference (CPP) paradigm in male rats, the CB1R agonist arachidonylcyclopropylamide (ACPA) increased place preference in a dose-dependent manner. ACPA was administered to directly to the central amygdala, providing strong evidence that CB1R plays a role in affective processing and can modulate behavior (Rezayof, Sardari, Zarrindast, & Nayer-Nouri, 2011). Further, the CB1R antagonist, AM251, reversed these effects and even induced conditioned place aversion when injected directly into the amygdala. These results taken together indicate a modulatory role of CB1R in higher order brain regions, above and beyond the level of the brainstem, and may play a critical role in how pain is subjectively experienced.
CB1R Action on Non-neuronal Cells
Peripheral CB1R mediation of pain is likely not specific to only nociceptors and may occur on other non-neuronal cells. There is a wealth of evidence to suggest pain mediation occurs via cellular mechanisms of immune cells, glial cells, cancer cells, epidermal cells, and stem cells (Ji, Chamessian, & Zhang, 2016). For example, CB1R expression has been found on Schwann cells (Freundt-Revilla, Kegler, Baumgärtner, & Tipold, 2017), the peripheral glial cells responsible for myelination of sensory neurons. The idea that these cells may be involved in nociceptive processing is still novel and empirical research assessing the level of that involvement is preliminary. It has been shown that Schwann cells are involved in the propagation and facilitation of neuronal firing through ionic regulation. Further, Schwann cells recruit and activate immune cells via secretion of matrix metalloproteinase 9 (MMP-9 ) (Calvo, Dawes, & Bennett, 2012), potentially influencing inflammatory pain processes. How CB1R agonists act on these cells and in turn, on peripheral inflammatory pain has yet to be determined but is an intriguing possible mediator.
As mentioned earlier, synthetic and endocannabinoids possess the ability to reduce excitability in cells through G-coupled intracellular signaling cascades, however it may be immune cells that possess the ability to sensitize these nociceptors and are critically involved in modulating inflammatory pain. CB1R antagonism exhibits anti-inflammatory effects on immune cells, specifically macrophages (Sugamura et al., 2008). Cytokine-producing macrophages play a pivotal role in the sensitization of peripheral sensory neurons by promoting a pro-inflammatory microenvironment through the production of cytokines like interleukins (IL) - 1-beta (IL-1β) and IL-6 (Ghasemlou, Chiu, Julien, & Woolf, 2015). Inhibition of the ability of immune cells to produce inflammatory cytokines by tight regulation of transcription factors lends itself to reduce excitability in neurons by decreasing inflammatory input. It may be by this mechanism that cannabinoids inhibit inflammatory pain. Cannabinoid agonists have been shown to bind to Peroxisome-proliferator-activated receptors (PPARs) and increase transcriptional activity of specifically PPARγ (Liu, Li, Burstein, Zurier, & Chen, 2003). PPARs are found in a variety of cells but here we will focus specifically on macrophages (Kliewer et al., 1994). PPAR activation in immune cells negatively regulates transcription of pro-inflammatory transcription factors such as AP-1 and NFκB (Berghe et al., 2003). It is a potential that CB1R agonists work to modulate activity of immune cells that play an important role in neuronal sensitization. By decreasing the capabilities of cytokine producing cells by either direct CB1R activation or PPAR these cells in turn, indirectly, decrease activity and sensitization in nociceptive neurons.
Non-neuronal cells may be a missing link to cannabinoid pain mediation in the periphery and may be an alternative therapeutic target in cannabinoid analgesics. The behavior of these cells may also strongly influence the results of clinical studies, leading to conflicting results and the continued debate of the efficacy of cannabinoid analgesics. Future directions in cannabinoid research should address the concept of non-neuronal analgesic activity of CB1R signaling pathways.
Conclusions
While it is clear that peripherally restricted CB1R agonists do not significantly produce analgesia in humans, it is still not yet determined if central acting cannabinoids can produce reliable and consistent analgesic results that are well tolerated in clinical trials with few adverse side effects (Ellis et al., 2009; Wilsey et al., 2008). This paper reviews the analgesic efficacy of cannabinoids that specifically activate CB1R at every level of pain processing, through local administration in the paw, the dorsal root ganglia, the periaqueductal gray, and through higher-level cortices in affective processing. Even though each level of nociceptive processing has been examined, the research has yet to determine the most critical area for pain processing and if there is a most optimal target for therapeutic treatments.
Summary Figure:
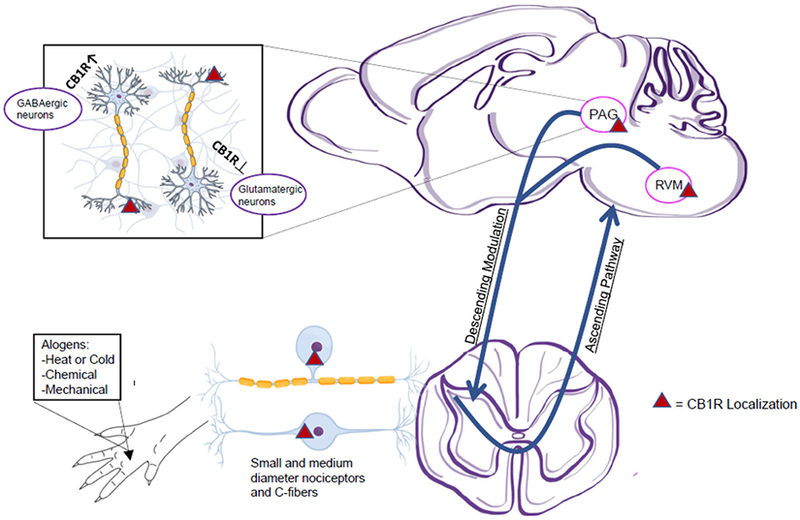
This figure visually summarizes important concepts discussed in the article. Pain inducing stimuli (alogens) is received at the extremities. That information travels to the dorsal root ganglia via small to medium diameter nociceptors and c-fibers. These neurons have high concentrations of CB1R (approx. 70-80%) and are the likely target of peripheral CB1R modulation. These neurons send nociceptive signals to the brainstem and higher level pain processing regions. Nociceptive signals are also modulated through descending spinal tracts, likely from the periaqueductal grey (PAG) and rostral ventral medulla (RVM) of the brainstem, although this theory has not yet been confirmed with concrete evidence. Within the central nervous system, GABAergic and Glutamatergic neurons are modulated by CB1R signaling, although the specific mechanisms by which this occurs have not been determined. Further, we are only able to postulate the role of these neurons in pain processing, as there is much controversy over their action in relation to cannabinoid modulation.
Table 1:
An organized table listing the various drugs used in pre-clinical and clinical studies discussed in the paper.
| Drug Abreviation | Drug Full Name | Derivative | Specificity |
|---|---|---|---|
| AGONISTS | |||
| WIN 55212-2 | WIN 55212-2 | Synthetic | CB1R Full Agonist CB2R Full Agnoist |
| Indole | Indole | Synthetic | Peripherally restricted CB1R agonist |
| Indene | Indene | Synthetic | Peripherally restricted CB1R agonist |
| URB937 | URB937 | Synthetic | Peripherally restricted CB1R agonist |
| ZCZ011 | ZCZ011 | Synthetic | CB1R Allosteric Modulator |
| AZD1940 | AZD1940 | Synthetic | Peripherally restricted CB1R agonist |
| AZD1704 | AZD1704 | Synthetic | Peripherally restricted CB1R agonist |
| CP-55,940 | CP-55,940 | Synthetic | CB1R Full Agonist CB2R Full Agnoist |
| HU-210 | HU-210 | Synthetic | CB1R Full Agonist CB2R Full Agnoist |
| Δ 9-THC | Δ9-tetrahydrocannabinol | Plant-based Derivative | CB1R Full Agonist CB2R Full Agnoist |
| Anandamide | N-arachidonoylethanolamine | Endogenous | CB1R Partial Agonist CB2R Partial Agnoist |
| 2-AG | 2-arachidonoylglycerol | Endogenous | CB1R Full Agonist CB2R Partial Agnoist |
| ACEA | Arachidonyl-2-chloroethylamide | Endogenous | CB1R Full Agonist |
| ANTAGONISTS | |||
| ACPA | Arachidonylcyclopropylamide | Synthetic | CB1R Antagonist |
| SR141716 | [3H]rimonabant | Synthetic | CB1R Antagonist |
| This table is meant to be a reference and assist the reader throughout the article of distingusihing important drug informaiton and how it pertains to the experimental results described. | |||
Note. CB1R = cannabinoid receptor type 1; CB2R = cannabinoid receptor type 2.
Table 2:
A table summarizing appropriate references in pre-clinical research describing peripheral vs. central nervous system action of CB1R.
| Reference | Model | CB Drugs | Mechanism of Action | Results |
|---|---|---|---|---|
| PERIPHERAL NERVOUS SYSTEM ACTION | ||||
| Agarwal, N., et. al. 2007 | CB1R-KO in peripherial nociceptors ONLY | WIN 55212-2 | Agonist | ↑ Mechanical Pain ↑ Cold Allodynia |
| Seltzman, H. H., et. al., 2016 | WT Rats - Peripherally restricted drug | Indole Indene |
Agonist | ↓ Allodynia |
| Mitrirattanakul, S., et. al. 2006 | WT Rats | None | Normal CB1R function | ↑ CB1R in L4 neruons after SNL ↑ Co-localtization with TRPV1 and IB4 ↑ AEA and 2-AG concentrations in spinal cord neurons. |
| Sideris, A., et. al., 2016 | CB1R-KO | None | Loss of CB1R function | ↑ Cold Allodynia ↑ Mechanical Pain but better recovery outcomes 2 weeks after injury |
| Nackley, A., et. al., 2003 | WT Rats | WIN 55-212-2 | Agonist | ↓ Fos up-regulation after injury in DRG neurons |
| Calignano, A., et. al., 1998 | WT Mice | Anandamide WIN55-212 CP-55,940 |
Agonist | ↓ Acute and Chronic inflammatory pain |
| Amaya, F., et. al., 2006 | WT Rats | ACEA | Agonist | ↓ Acute and Chronic inflammatory pain |
| Ignatowska-Jankowska, B., et. al., 2015 | FAAH-KO but CB1R functionally intact | ZCZ011 | Allosteric Modulator | ↓ Mechanical Pain |
| CENTRAL NERVOUS SYSTEM ACTION | ||||
| Zimmer, A., et. al., 1999 | CB1R-KO | Δ9-THC | Agonist | ↓ Algesia |
| Bajic, D., et. al., 2018 | Glu-CB1R-KO GABA-CB1R-KO CaKM-CB1R-KO |
SR141716 | Antagonist | ↓ Thermal Pain ↓ Mechanical Allodynia |
| Fox, A., et. al., 2001 | WT Rats | WIN55-212 CP-55,940 HU-210 |
Agonist | ↓ Thermal Pain ↓ Mechanical Allodynia |
| Finn, D., et. al. 2003 | WT Rats | HU-210 | Agonist | ↓ Late Phase Algesia after PAG injection |
| Meng, I., & Johansen, J., 2004 | WT Rats | WIN 55-212-2 | Agonist | ↑ Increased Tailflick Latencies ↑ Activity of pain inihibiting neurons in RVM ↓ Activity of pain processing neurons in RVM |
| Amaya, F., et. al., 2006 | WT Rats | ACEA | Agonist | ↓ Acute and Chronic inflammatory pain |
| de Novellis, V., et. al. 2005 | WT Rats | WIN 55-212-2 | Agonist | ↑ Increased Tailflick Latencies |
Note. CB1R-KO = cannabinoid receptor type 1 knockout; WT = wild type; SNL = spinal nerve ligation; TRPV1 = transient receptor potential cation channel subfamily V member 1; AEA = N-arachidonoylethanolamine; 2-AG = 2-arachidonoyglycerol; DRG = dorsal root ganglion; ACEA = arachidonyl-2’-choroethylamide; FAAH-KO = fatty acid hydrolase knockout; THC = tetrahydrocannabinol; PAG = periaqueductal gray; RVM = rostral ventromedial medulla.
Acknowledgments
FUNDING
Supported by the National Institute of Neurological Disorders and Stroke (NINDS) grant: K22NS096030 (MDB), Texas Rising Star, (MDB), The American Pain Society Future Leaders Award (MDB), and The Rita Allen Foundation (MDB)
Footnotes
DISCLOSURES
The authors have no conflict of interest or additional support to disclose
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, . . . Kuner R (2007). Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neuroscience, 10(7), 870–879. doi: 10.1038/nn1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, & Nagy I (2000). Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience, 100(4), 685–688. doi: 10.1016/s0306-4522(00)00389-4 [DOI] [PubMed] [Google Scholar]
- Amaya F, Shimosato G, Kawasaki Y, Hashimoto S, Tanaka Y, Ji RR, & Tanaka M (2006). Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain, 124(1-2), 175–183. doi: 10.1016/j.pain.2006.04.001 [DOI] [PubMed] [Google Scholar]
- Bajic D, Monory K, Conrad A, Maul C, Schmid RM, Wotjak CT, & Stein-Thoeringer CK (2018). Cannabinoid receptor type 1 in the brain regulates the affective component of visceral pain in mice. Neuroscience, 384, 397–405. doi: 10.1016/j.neuroscience.2018.05.041 [DOI] [PubMed] [Google Scholar]
- Berghe WV, Vermeulen L, Delerive P, De Bosscher K, Staels B, & Haegeman G (2003). A paradigm for gene regulation: Inflammation, NF-κB and PPAR In Peroxisomal Disorders and Regulation of Genes (pp. 181–196): Springer. [DOI] [PubMed] [Google Scholar]
- Bishay P, Haussler A, Lim HY, Oertel B, Galve-Roperh I, Ferreiros N, & Tegeder I (2013). Anandamide deficiency and heightened neuropathic pain in aged mice. Neuropharmacology, 71, 204–215. doi: 10.1016/j.neuropharm.2013.03.021 [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, & Piomelli D (1998). Control of pain initiation by endogenous cannabinoids. Nature, 394(6690), 277–281. doi: 10.1038/28393 [DOI] [PubMed] [Google Scholar]
- Calvo M, Dawes JM, & Bennett DL (2012). The role of the immune system in the generation of neuropathic pain. The Lancet Neurology, 11(7), 629–642. doi: 10.1016/s1474-4422(12)70134-5 [DOI] [PubMed] [Google Scholar]
- Chen LH, Hedegaard H, & Warner M (2014). Drug-poisoning deaths involving opioid analgesics: United States, 1999-2011. NCHS Data Brief (166), 1–8. [PubMed] [Google Scholar]
- Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, . . . Spradley JM (2010). Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature Neuroscience, 13(10), 1265–1270. doi: 10.1038/nn.2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ZD, Comer SD, & Haney M (2013). Comparison of the analgesic effects of dronabinol and smoked marijuana in daily marijuana smokers. Neuropsychopharmacology, 38(10), 1984–1992. doi: 10.1038/npp.2013.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey-Bloom J, Wolfson T, Gamst A, Jin S, Marcotte TD, Bentley H, & Gouaux B (2012). Smoked cannabis for spasticity in multiple sclerosis: A randomized, placebo-controlled trial. CMAJ, 184(10), 1143–1150. doi: 10.1503/cmaj.110837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Bass CE, Van Horn CG, & Howlett AC (2009). Signal transduction via cannabinoid receptors. CNS & Neurological Disorders - Drug Targets, 8(6), 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Novellis V, Mariani L, Palazzo E, Vita D, Marabese I, Scafuro M, . . . Maione S (2005). Periaqueductal grey CB1 cannabinoid and metabotropic glutamate subtype 5 receptors modulate changes in rostral ventromedial medulla neuronal activities induced by subcutaneous formalin in the rat. Neuroscience, 134(1), 269–281. doi: 10.1016/j.neuroscience.2005.03.014 [DOI] [PubMed] [Google Scholar]
- Di Scala G, Mana MJ, Jacobs WJ, & Phillips AG (1987). Evidence of Pavlovian conditioned fear following electrical stimulation of the periaqueductal grey in the rat. Physiology & Behavior, 40(1), 55–63. doi: 10.1016/0031-9384(87)90185-5 [DOI] [PubMed] [Google Scholar]
- Dzau VJ, & Pizzo PA (2014). Relieving pain in America: Insights from an Institute of Medicine committee. JAMA, 312(15), 1507–1508. doi: 10.1001/jama.2014.12986 [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Toperoff W, Vaida F, van den Brande G, Gonzales J, Gouaux B, . . . Atkinson JH (2009). Smoked medicinal cannabis for neuropathic pain in HIV: A randomized, crossover clinical trial. Neuropsychopharmacology, 34(3), 672–680. doi: 10.1038/npp.2008.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn DP, Jhaveri MD, Beckett SR, Roe CH, Kendall DA, Marsden CA, & Chapman V (2003). Effects of direct periaqueductal grey administration of a cannabinoid receptor agonist on nociceptive and aversive responses in rats. Neuropharmacology, 45(5), 594–604. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, . . . Wallace M (2015). Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurology, 14(2), 162–173. doi: 10.1016/S1474-4422(14)70251-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, & James I (2001). The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain, 92(1-2), 91–100. [DOI] [PubMed] [Google Scholar]
- Freundt-Revilla J, Kegler K, Baumgärtner W, & Tipold A (2017). Spatial distribution of cannabinoid receptor type 1 (CB1) in normal canine central and peripheral nervous system. PLoS ONE, 12(7), e0181064. doi: 10.1371/journal.pone.0181064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemlou N, Chiu IM, Julien JP, & Woolf CJ (2015). CD11b+Ly6G− myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America, 112(49), E6808–6817. doi: 10.1073/pnas.1501372112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez T, Farthing JN, Zvonok AM, Makriyannis A, & Hohmann AG (2007). Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: A comparative analysis. British Journal of Pharmacology, 150(2), 153–163. doi: 10.1038/sj.bjp.0706984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama AT, & Urban MO (2004). Antihyperalgesic effect of the cannabinoid agonist WIN55, 212-2 is mediated through an interaction with spinal metabotropic glutamate-5 receptors in rats. Neuroscience Letters, 358(1), 21–24. [DOI] [PubMed] [Google Scholar]
- Harris J, Drew LJ, & Chapman V (2000). Spinal anandamide inhibits nociceptive transmission via cannabinoid receptor activation in vivo. NeuroReport, 11(12), 2817–2819. doi: 10.1097/00001756-200008210-00041 [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, & Lumb BM (2009). Descending control of nociception: Specificity, recruitment and plasticity. Brain Research Reviews, 60(1), 214–225. doi: 10.1016/j.brainresrev.2008.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkendell K, Tel-Vered R, & Stemmer A (2017). Switchable aerobic/anaerobic multi-substrate biofuel cell operating on anodic and cathodic enzymatic cascade assemblies. Nanoscale, 9(37), 14118–14126. doi: 10.1039/c7nr06233h [DOI] [PubMed] [Google Scholar]
- Hill KP, Palastro MD, Johnson B, & Ditre JW (2017). Cannabis and pain: A clinical review. Cannabis & Cannabinoid Research, 2(1), 96–104. doi: 10.1089/can.2017.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, & Herkenham M (1999). Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience, 92(4), 1171–1175. doi: 10.1016/s0306-4522(99)00220-1 [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Martin WJ, Tsou K, & Walker JM (1995). Inhibition of noxious stimulus-evoked activity of spinal cord dorsal horn neurons by the cannabinoid WIN 55,212-2. Life Sciences, 56(23-24), 2111–2118. doi: 10.1016/0024-3205(95)00196-d [DOI] [PubMed] [Google Scholar]
- Hudson R, & Puvanenthirarajah N (2018). Cannabis for pain management: Pariah or panacea? University of Western Ontario Medical Journal, 87(1), 58–61. doi: 10.5206/uwomj.v87i1.1922 [DOI] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, . . . Zanda M (2015). A cannabinoid CB 1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology, 40(13), 2948–2959. doi: 10.1038/npp.2015.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R-R, Chamessian A, & Zhang Y-Q (2016). Pain regulation by non-neuronal cells and inflammation. Science, 354(6312), 572–577. doi: 10.1126/science.aaf8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalliomaki J, Annas P, Huizar K, Clarke C, Zettergren A, Karlsten R, & Segerdahl M (2013). Evaluation of the analgesic efficacy and psychoactive effects of AZD1940, a novel peripherally acting cannabinoid agonist, in human capsaicin-induced pain and hyperalgesia. Clinical & Experimental Pharmacology & Physiology, 40(3), 212–218. doi: 10.1111/1440-1681.12051 [DOI] [PubMed] [Google Scholar]
- Katona I, & Freund TF (2008). Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nature Medicine, 14(9), 923–930. doi: 10.1038/nm.f.1869 [DOI] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, & Freund TF (2001). Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. Journal of Neuroscience, 21(23), 9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Holman M, Morse T, Burlakova N, Coicou L, Harding-Rose C, . . . Seybold VS (2013). Increased anandamide uptake by sensory neurons contributes to hyperalgesia in a model of cancer pain. Neurobiology of Disease, 58, 19–28. doi: 10.1016/j.nbd.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Horovitz O, Pellman BA, Tan LM, Li Q, Richter-Levin G, & Kim JJ (2013). Dorsal periaqueductal gray-amygdala pathway conveys both innate and learned fear responses in rats. Proceedings of the National Academy of Sciences of the United States of America 110(36), 14795–14800. doi: 10.1073/pnas.1310845110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, . . . Evans RM (1994). Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proceedings of the National Academy of Sciences of the United States of America, 91(15), 7355–7359. doi: 10.1073/pnas.91.15.7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SC, & Fitzgerald M (2014). The selectivity of rostroventral medulla descending control of spinal sensory inputs shifts postnatally from A fibre to C fibre evoked activity. Journal of Physiology, 592(7), 1535–1544. doi: 10.1113/jphysiol.2013.267518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunos G, Osei-Hyiaman D, Bátkai S, Sharkey KA, & Makriyannis A (2009). Should peripheral CB1 cannabinoid receptors be selectively targeted for therapeutic gain? Trends in Pharmacological Sciences, 30(1), 1–7. doi: 10.1016/j.tips.2008.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunos G, & Tam J (2011). The case for peripheral CB1 receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. British Journal of Pharmacology, 163(7), 1423–1431. doi: 10.1111/j.1476-5381.2011.01352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Porta C, Bura SA, Llorente-Onaindia J, Pastor A, Navarrete F, Garcia-Gutierrez MS, . . . Maldonado R (2015). Role of the endocannabinoid system in the emotional manifestations of osteoarthritis pain. Pain, 156(10), 2001–2012. doi: 10.1097/j.pain.0000000000000260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Li H, Burstein SH, Zurier RB, & Chen JD (2003). Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Molecular Pharmacology, 63(5), 983–992. doi: 10.1124/mol.63.5.983 [DOI] [PubMed] [Google Scholar]
- Mackie K (2005). Distribution of cannabinoid receptors in the central and peripheral nervous system. Handbook of Experimental Pharmacology, 168, 299–325. doi: 10.1007/3-540-26573-2_10 [DOI] [PubMed] [Google Scholar]
- Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, & Walker JM (1999). Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Research, 822(1-2), 237–242. doi: 10.1016/s0006-8993(98)01368-7 [DOI] [PubMed] [Google Scholar]
- Martin WJ, Patrick SL, Coffin PO, Tsou K, & Walker JM (1995). An examination of the central sites of action of cannabinoid-induced antinociception in the rat. Life Sciences, 56(23-24), 2103–2109. doi: 10.1016/0024-3205(95)00195-c [DOI] [PubMed] [Google Scholar]
- Martin WJ, Tsou K, & Walker JM (1998). Cannabinoid receptor-mediated inhibition of the rat tail-flick reflex after microinjection into the rostral ventromedial medulla. Neuroscience Letters, 242(1), 33–36. doi: 10.1016/s0304-3940(98)00044-5 [DOI] [PubMed] [Google Scholar]
- McAllister SD, Griffin G, Satin LS, & Abood ME (1999). Cannabinoid receptors can activate and inhibit G protein-coupled inwardly rectifying potassium channels in a xenopus oocyte expression system. The Journal of Pharmacology & Experimental Therapeutics, 291(2), 618–626. [PubMed] [Google Scholar]
- Meng ID, & Johansen JP (2004). Antinociception and modulation of rostral ventromedial medulla neuronal activity by local microinfusion of a cannabinoid receptor agonist. Neuroscience, 124(3), 685–693. doi: 10.1016/j.neuroscience.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Millns PJ, Chapman V, & Kendall DA (2001). Cannabinoid inhibition of the capsaicin-induced calcium response in rat dorsal root ganglion neurones. British Journal of Pharmacology, 132(5), 969–971. doi: 10.1038/sj.bjp.0703919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, . . . Spigelman I (2006). Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain, 126(1-3), 102–114. doi: 10.1016/j.pain.2006.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TD, Weidenbach KN, Van Houten C, Gerona RR, Moran JH, Kirschner RI, . . . Newman A (2013). Acute kidney injury associated with synthetic cannabinoid use—multiple states, 2012. Morbidity and Mortality Weekly Report, 62(6), 93. [PMC free article] [PubMed] [Google Scholar]
- Nackley AG, Suplita RL 2nd, & Hohmann AG (2003). A peripheral cannabinoid mechanism suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience, 117(3), 659–670. doi: 10.1016/s0306-4522(02)00870-9 [DOI] [PubMed] [Google Scholar]
- Naderi N, Shafaghi B, Khodayar M-J, & Zarindast M-R (2005). Interaction between gamma-aminobutyric acid GABAB and cannabinoid CB1 receptors in spinal pain pathways in rat. European Journal of Pharmacology, 514(2-3), 159–164. doi: 10.1016/j.ejphar.2005.03.037 [DOI] [PubMed] [Google Scholar]
- Ottestad E, Angst MJP, & anesthesia, p. f. (2013). Nociceptive physiology. 235–252. [Google Scholar]
- Pacher P, & Kunos G (2013). Modulating the endocannabinoid system in human health and disease–successes and failures. FEBS Journal, 280(9), 1918–1943. doi: 10.1111/febs.12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo E, Luongo L, Novellis V, Rossi F, & Maione S (2010). The role of cannabinoid receptors in the descending modulation of pain. Pharmaceuticals, 3(8), 2661–2673. doi: 10.3390/ph3082661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulozzi LJ, Budnitz DS, & Xi Y (2006). Increasing deaths from opioid analgesics in the United States. Pharmacoepidemiology & Drug Safety, 15(9), 618–627. doi: 10.1002/pds.1276 [DOI] [PubMed] [Google Scholar]
- Pertwee RG (1997). Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacology & Therapeutics, 74(2), 129–180. doi: 10.1016/s0163-7258(97)82001-3 [DOI] [PubMed] [Google Scholar]
- Racz I, Nent E, Erxlebe E, & Zimmer A (2015). CB1 receptors modulate affective behaviour induced by neuropathic pain. Brain Research Bulletin, 114, 42–48. doi: 10.1016/j.brainresbull.2015.03.005 [DOI] [PubMed] [Google Scholar]
- Rea K, Roche M, & Finn DP (2007). Supraspinal modulation of pain by cannabinoids: The role of GABA and glutamate. British Journal of Pharmacology, 152(5), 633–648. doi: 10.1038/sj.bjp.0707440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezayof A, Sardari M, Zarrindast MR, & Nayer-Nouri T (2011). Functional interaction between morphine and central amygdala cannabinoid CB1 receptors in the acquisition and expression of conditioned place preference. Behavioral Brain Research, 220(1), 1–8. doi: 10.1016/j.bbr.2011.01.023 [DOI] [PubMed] [Google Scholar]
- Richardson JD, Kilo S, & Hargreaves KM (1998). Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain, 75(1), 111–119. doi: 10.1016/s0304-3959(97)00213-3 [DOI] [PubMed] [Google Scholar]
- Rog DJ, Nurmikko TJ, Friede T, & Young CA (2005). Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology, 65(6), 812–819. doi: 10.1212/01.wnl.0000176753.45410.8b [DOI] [PubMed] [Google Scholar]
- Seltzman HH, Shiner C, Hirt EE, Gilliam AF, Thomas BF, Maitra R, . . . Spigelman I (2016). Peripherally selective cannabinoid 1 receptor (CB1R) agonists for the treatment of neuropathic pain. Journal of Medicinal Chemistry, 59(16), 7525–7543. doi: 10.1021/acs.jmedchem.6b00516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, & Di Marzo V (2012). Endocannabinoids in nervous system health and disease: The big picture in a nutshell. Philosophical Transactions of the Royal Society B: Biological Sciences, 367(1607), 3193–3200. doi: 10.1098/rstb.2012.0313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stander S, Schmelz M, Metze D, Luger T, & Rukwied R (2005). Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. Journal of Dermatological Science, 38(3), 177–188. doi: 10.1016/j.jdermsci.2005.01.007 [DOI] [PubMed] [Google Scholar]
- Sugamura K, Sugiyama S, Nozaki T, Matsuzawa Y, Izumiya Y, Nakayama M, . . . Ogawa H (2008). Activated endocannabinoid system in coronary artery disease and anti-inflammatory effects of cannabinoid 1 receptor blockade on macrophages. In: Am Heart Assoc. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, & Christie MJ (2005). Retrograde signalling by endocannabinoids. Handbook of Experimental Pharmacology (168), 367–383. doi: 10.1007/3-540-26573-2_12 [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Connor M, Bagley EE, & Christie MJ (2000). Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Molecular Pharmacology, 57(2), 288–295. [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, & Sanudo-Pena MC (1999). Pain modulation by release of the endogenous cannabinoid anandamide. Proceedings of the National Academy of Sciences of the United States of America, 96(21), 12198–12203. doi: 10.1073/pnas.96.21.12198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MS, Marcotte TD, Umlauf A, Gouaux B, & Atkinson JH (2015). Efficacy of inhaled cannabis on painful diabetic neuropathy. Journal of Pain, 16(7), 616–627. doi: 10.1016/j.jpain.2015.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu W, Cai Y, Ma L, Ma C, Luo A, & Huang Y (2016). Glutaminase 1 is a potential biomarker for chronic post-surgical pain in the rat dorsal spinal cord using differential proteomics. Amino Acids, 48(2), 337–348. doi: 10.1007/s00726-015-2085-z [DOI] [PubMed] [Google Scholar]
- Ware MA, Wang T, Shapiro S, Robinson A, Ducruet T, Huynh T, . . . Collet JP (2010). Smoked cannabis for chronic neuropathic pain: A randomized controlled trial. CMAJ, 182(14), E694–701. doi: 10.1503/cmaj.091414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster LR, Cochella S, Dasgupta N, Fakata KL, Fine PG, Fishman SM, . . . Wakeland W (2011). An analysis of the root causes for opioid-related overdose deaths in the United States. Pain Medicine, 12(Suppl 2), S26–35. doi: 10.1111/j.1526-4637.2011.01134.x [DOI] [PubMed] [Google Scholar]
- Wilsey B, Marcotte T, Deutsch R, Gouaux B, Sakai S, & Donaghe H (2013). Low-dose vaporized cannabis significantly improves neuropathic pain. Journal of Pain, 14(2), 136–148. doi: 10.1016/j.jpain.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsey B, Marcotte T, Tsodikov A, Millman J, Bentley H, Gouaux B, & Fishman S (2008). A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. Journal of Pain, 9(6), 506–521. doi: 10.1016/j.jpain.2007.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ (2013). Pain: Morphine, metabolites, mambas, and mutations. Lancet Neurology, 12(1), 18–20. doi: 10.1016/S1474-4422(12)70287-9 [DOI] [PubMed] [Google Scholar]