

Abstract
Background:
Lung cancer kills more people than any other cancer in the United States. In addition to environmental factors, lung cancer has genetic risk factors as well, though the genetic etiology is still not well understood. We have performed whole exome sequencing on 262 individuals from 28 extended families with a family history of lung cancer.
Methods:
Parametric genetic linkage analysis was performed on these samples using two distinct analyses – the lung cancer only (LCO) analysis, where only lung cancer patients were coded as affected, and the all aggregated cancers (AAC) analysis, where other cancers seen in the pedigree were coded as affected.
Results:
The AAC analysis yielded a genome-wide significant result at rs61943670 in POLR3B at 12q23.3. POLR3B has been implicated somatically in lung cancer, but this germline finding is novel and is a significant expression quantitative trait locus in lung tissue. Interesting genome-wide suggestive haplotypes were also found within individual families, particularly near SSPO at 7p36.1 in one family and a large linked haplotype spanning 4q21.3–28.3 in a different family. The 4q haplotype contains potential causal rare variants in DSPP at 4q22.1 and PTPN13 at 4q21.3.
Conclusions:
Regions on 12q, 7p, and 4q are linked to increased cancer risk in highly aggregated lung cancer families, 12q across families and 7p and 4q within a single family. POLR3B, SSPO, DSPP, and PTPN13 are currently the best candidate genes.
Impact:
Functional work on these genes is planned for future studies and if confirmed would lead to potential biomarkers for risk in cancer.
Keywords: Lung cancer, genetic linkage, family studies, whole exome sequencing, cancer risk
Introduction
Lung cancer remains the deadliest cancer in the United States. In 2019, more Americans will die of lung cancer than breast, colon, and prostate cancers combined (https://www.cancer.org/cancer/non-small-cell-lung-cancer/about/key-statistics.html). Lung cancer is caused by a variety of environmental factors; tobacco smoking (1–4) is responsible for 85–90% of all lung cancers (5, 6).
Tobacco smoking does not account for all cases of lung cancer, however. Approximately 10–15% of lung cancers develop in nonsmokers. Passive smoking only accounts for about 16–24% of lung cancer cases in nonsmokers. Even as governments have passed stringent laws against tobacco use for minors and in public spaces, lung cancer frequency in nonsmokers appears to be increasing (7).
There is significant genetic predisposition to lung cancer risk. Tokuhata and Lilienfeld observed familial aggregation of lung cancer in 1963(8, 9). They found relatives of lung cancer patients have a higher risk of developing lung cancer compared to relatives of controls. Further studies confirmed that nonsmoking relatives of lung cancer patients have a higher risk of lung cancer, possibly as high as 2–3% (10–12). Segregation analyses support a codominant Mendelian inheritance of a rare autosomal gene that interacts with smoking (13–15).
Genome-wide association studies (GWAS) became popular in the early 2000s with the advent of commercially produced SNP microarrays. GWAS are designed to identify risk variants that are common, have low penetrance and a moderate/small effect on lung cancer risk. GWAS have identified multiple lung cancer risk variants, including the neuronal acetylcholine receptor cluster subunit genes, CHRNA3, CHRNA5, and CHRNB4, at 15q25 (16–18).
Linkage analyses are an alternative approach to GWAS that use family-based data to trace the co-segregation of a phenotype and a given variant throughout the generations. Linkage studies are better at identifying rare, highly penetrant risk variants that have a large effect on phenotype risk. Variants that are rare in the general population will be enriched in a family that carries it. Further, linkage studies can take advantage of long, linked haplotypes that exist within individual families. The individuals in population studies are unrelated; countless meioses through thousands of generations have broken apart the haplotypes into only variants with the strongest linkage disequilibrium (LD) between them. In family studies, haplotypes are determined by the haplotypes of the founders of each family. Family studies rarely extend beyond five or six generations, so there are only a limited number of recombinations that can break apart the haplotypes resulting in a longer haplotype across chromosomal regions. The longer haplotype in turn increases power to find ungenotyped causal variants along the haplotype. Linkage studies have been used to identify potential risk loci in families with a history of lung cancer, including at 6q23–25 (19), among others (20).
The Genetic Epidemiology of Lung Cancer Consortium (GELCC) has collected highly aggregated lung cancer families for over 20 years at eight sites across the United States. Here, we present the genetic linkage analysis on whole exome sequence (WES) data from 262 patients from 28 extended families with a strong history of familial lung cancer.
Materials and Methods
Patient Recruitment
We recruited probands with a strong familial history of lung cancer, defined as four or more related persons with lung cancer. Samples of blood, saliva, and archival tissue were collected for all participants. Cancer status was verified through medical records, pathology reports, and death certificates for 80% of the lung cancer affecteds. For the 20% missing relevant documentation, at least three family members corroborated cancer diagnoses, with higher weight given to the testimony of first-degree relatives. Previous studies have reported family member reports of cancer diagnoses have a high accuracy rate (21, 22). Further data regarding age at onset and smoking statistics were also collected when possible. This study adhered to the tenets of the Declaration of Helsinki and all participants provided written informed consent. This study was approved by the institutional review boards of the National Human Genome Research Institute and all other participating institutions.
Sequencing and Quality Control
We chose 270 people from the 28 most highly informative families for whole exome sequencing. Informative families were determined by several factors, primarily the number of lung cancer affecteds in the family, the number of lung cancer affecteds with available biospecimens or whose offspring had available biospecimens, and the total other informative (linking) individuals with biospecimens in the pedigree. Sequencing was performed at Washington University in St. Louis, MO, USA, and the National Intramural Sequencing Center (NISC) in Bethesda, MD, USA. Data from both sites was jointly realigned, recalled and cleaned together using Genome Analysis Tool Kit (GATK), following their best practices (23, 24), including removing variants with depth (DP) > 10, genotype quality (GQ) > 10, and GQ/DP > 0.5.
The software package PLINK (25) was used for further QC as follows. All variants that were monomorphic or not genotyped in at least 80% of the data set were removed. Identity-by-descent (IBD) calculations were performed to ensure familial relationships were correct; this resulted in the removal of three individuals. Variants that showed a Mendelian error in a single family were removed in the offending family. Variants that displayed Mendelian errors in more than one family were removed from all families. Another five individuals were removed since they were married into the family but had no children and provided no information to the pedigree. This resulted in 262 sequenced individuals that passed QC, 60 of which were lung cancer cases.
We added 266 ungenotyped individuals that were needed to connect disjointed affecteds in pedigrees. These individuals were known to exist from family history, but for various reasons were unwilling or unable to participate in the study. Genotypes for all variants were set to missing for these individuals. In some cases, the phenotype data on these ungenotyped individuals was known such as individuals with lung cancer but died prior to biosamples being taken. We sampled from surviving, usually unaffected relatives such as children and parents to reconstruct the affected individuals’ genotypes in the linkage analysis via the Elston-Stewart algorithm, which calculates the likelihood of the pedigree across all possible genotypes for ungenotyped individuals incorporating the genotypes of their relatives in this likelihood (26).
The final dataset contained 262 sequenced subjects (60 sequenced lung cancer cases) and 266 unsequenced subjects (81 unsequenced lung cancer cases) for a total of 528 subjects (135 lung cancer cases). The average age of the participants was 58.13 with a standard deviation of 17.52. 52.27% of the participants were female. There was a total of 397781 SNVs and indels across 22 autosomes.
Founder Allele Frequency Estimation
All individuals included were European-Americans. Founder allele frequencies were estimated from the data set using an EM algorithm in sib-pair (https://genepi.qimr.edu.au/staff/davidD/Sib-pair/Documents/sib-pair.html). Estimating allele frequencies directly from the data set in homogeneous populations has been shown to effectively control type I error rates and power (27–29).
Parametric Linkage Analysis
Parametric linkage analysis was performed under two distinct affection classification schemes. Under the first scheme, henceforth referred to as lung cancer only (LCO) analysis, all individuals affected with lung cancer were coded as affected, all other individuals were then coded as unknown. This allowed for the high degree of uncertainty between smoking and lung cancer risk as well as jointly allowing for smoking status; 80% of affected individuals in the pedigrees smoked. It also allowed for the possibility that individuals who are presently unaffected may be carriers of the disease allele who will develop lung cancer later in life. Linkage analysis was carried out using TwoPointLods (http://www-genepi.med.utah.edu/~alun/software/), assuming an autosomal dominant mode of inheritance and 1% disease allele frequency (DAF). Penetrance was set at 80% for carriers and 1% for non-carriers as used in previous analysis (20). LOD scores were then added across families for a cumulative LOD score at each variant and heterogeneity LOD (HLOD) scores were calculated at each variant. HLODs consider potential heterogeneity across the different families by incorporating a measure of the proportion of families that are linked to the variant to the LOD score (30, 31).
The data was reanalyzed under a second affection classification scheme, termed the all aggregated cancers (AAC) analysis, using the identical parameters from the LCO analysis. We decided to use this additional analysis after observing multiple other cancers in these highly aggregated lung cancer families. The specific cancers varied from family to family, but the most common were breast, prostate, skin, and bladder (Table 1). Inherited version of one cancer type can lead to an increased risk in other cancers; this is true of Lynch syndrome. Lynch syndrome sufferers have a significantly increased genetic risk of colorectal cancer, inherited autosomal dominantly (32, 33), but also have an increased risk of other cancer types including pancreatic cancer (34), ovarian, gastric, and possibly breast, among others (35). We hypothesize something similar with the AAC analysis, where the risk variant significantly increases the risk of lung cancer within families but also increases the risk of additional cancers. Anyone with either lung cancer or another cancer that had an affected parent to ensure it was inherited within the same pedigree was coded as affected; all other individuals were coded as unknown. This resulted in the addition of 30 non-lung cancer individuals with a different type of cancer. All parameters used in the LCO analysis were identical in the AAC analysis.
Table 1:
List of Cancers Present in 28 Sequenced Strongly Familial Lung Cancer Families
| Cancer Type | Number of Cases |
|---|---|
| Lung | 135 |
| Breast | 6 |
| Skin | 5 |
| Prostate | 4 |
| Bladder | 3 |
| Cervix | 2 |
| Leukemia | 1 |
| Bone | 1 |
| Colon | 1 |
| Lip | 1 |
| Lymphosarcoma | 1 |
| Pancreas | 1 |
| Pharynx | 1 |
| Stomach | 1 |
| Throat | 1 |
| Unknown | 1 |
Table displaying the different cancer cases within the data set.
Annotation
All variants were annotated via wANNOVAR (36–38) a web-based version of the functional software ANNOVAR, which provided annotation for all sequenced variants such as rsID, allele frequencies from both 1000Genomes and ExAC, and whether a variant was exonic/intronic/intergenic. It also collates functional predictions from popular predictions algorithms like CADD (39), SIFT (40) and PolyPhen2 (41). REVEL (42) was also used for functional annotation, while RegulomeDB (43) was used for regulatory sites. RegulomeDB is an integrated database that collates data from multiple sources including all ENCODE transcription factor data and data from NCBI Sequence Read Archive.
Lung eQTL Analyses
The top significant SNVs in regions showing genome-wide and suggestive signals were investigated for expression quantitative trait loci (eQTL) in lung tissues. eQTLs are variants that are responsible for at least part of a gene’s mRNA expression. The lung tissues were from 409 subjects who underwent lung surgery at the Institut universitaire de cardiologie et de pneumologie de Québec – Université Laval, Quebec City, Canada. Details about phenotyping, genotyping, and gene expression profiling were previously described (44, 45). Probe sets located 1 Mb up and downstream of selected SNVs were tested for cis-eQTL effects. The genetic associations between SNVs and gene expression were assessed using quantitative trait association analyses as implemented in PLINK.
Results
Lung Cancer Only Analysis
No genome-wide significant results were observed; 110 genome-wide suggestive results were identified (Supplementary Figure S1 and Supplementary Table S1). Significant is defined as (H)LOD score >= 3.3 while suggestive is defined as (H)LOD score >= 1.9 (46). The highest HLOD score, 2.7, was found at intronic SNV rs28675295 in the SKOR1 gene at 15q23. SNVs in SKOR1 had four of the top seven overall HLOD scores ranging from 2.3–2.7; rs7170185 was nonsynonymous exonic and predicted damaging by SIFT. Suggestive variants were found on 18 autosomes.
The highest individual family LOD scores were in Family 102 (Supplementary Table S2 and Supplementary Figure S2A), a five generational pedigree with 26 individuals, 6 of whom are lung cancer cases. The family had three suggestive variants; two of the suggestive variants were an exonic SNV and an intronic deletion both located in the SSPO gene at 7q36.1 (Figure 1A). The other variant was an intronic SNV at 10p11.22 in ARHGAP12 (Figure 1B). Four additional variants were close to suggestive with LOD = 1.8; two exonic (rs10262505 and rs2079335) and one intronic SNVs were in SSPO and an intronic deletion in KIF5B at 10p11.22.
Figure 1:
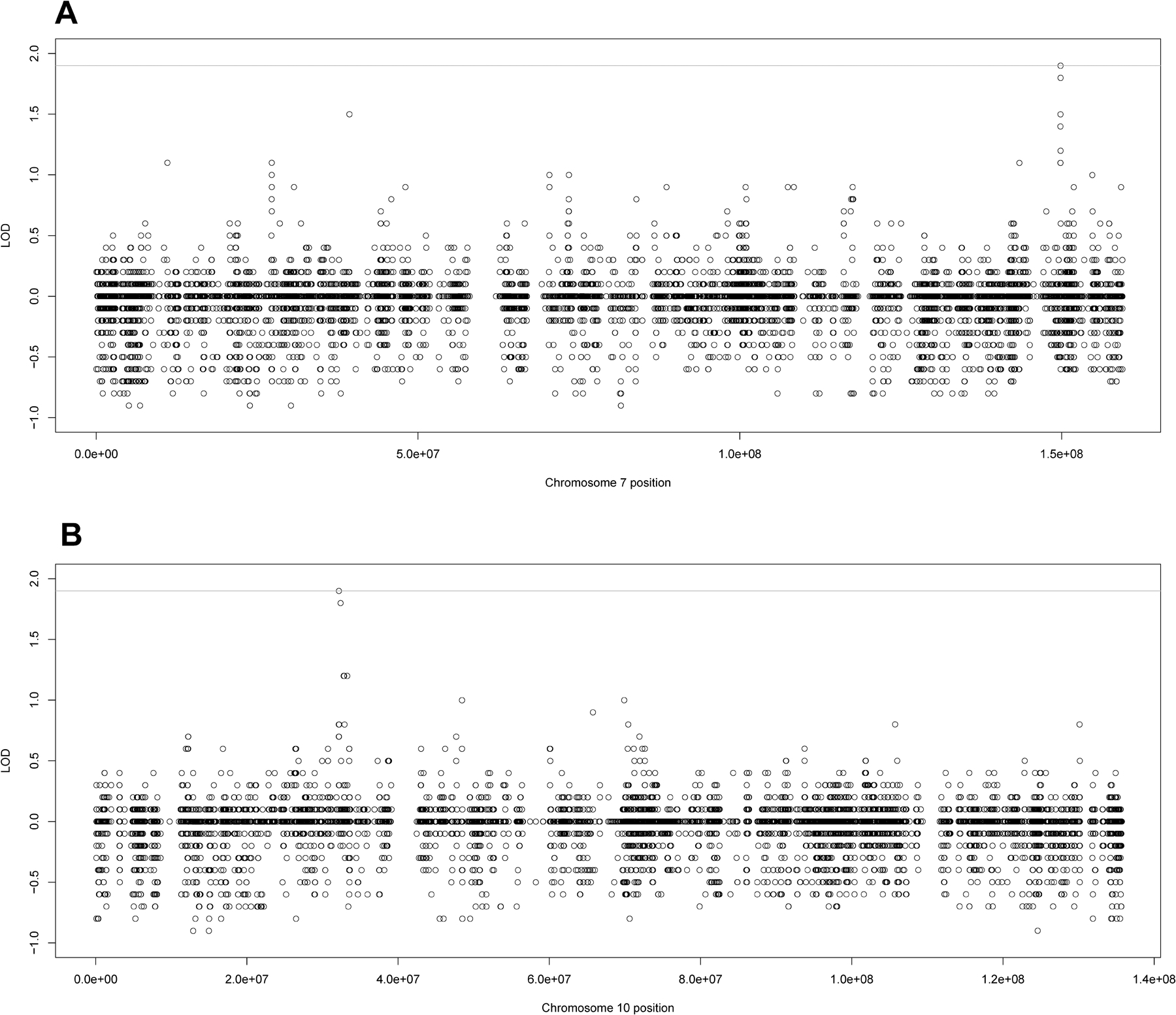
Individual LOD scores for Family 102 for the Lung Cancer Only analysis. A) The individual LOD scores for family 102 for chromosome 7 and B) the individual LOD scores for family 102 for chromosome 10. The line at 1.9 represents the genome-wide suggestive threshold as recommended by Lander and Kruglyak.
While no other family had any suggestive variants, we observed long linked haplotypes within some families. Long haplotypes are expected within a single family, where the haplotypes are determined solely by the family founders and have only a few generations to break apart haplotypes. Thus, if a disease variant exists, one expects to see linkage to other variants around the causal variant that are on the same long haplotype. We observed a long haplotype at 4q21.3–28.3 haplotype in Family 105 (Figure 2) with little to no negative signal beneath it. Family 105 is a four-generation pedigree with 17 individuals containing 6 cases/4 genotyped cases. This haplotype consisted of approximately 70 variants with LOD scores from 1.0 – 1.4 (Supplementary Table S3). Three rare, nonsynonymous exonic variants are particularly interesting - rs148827799 in DSPP at 4q22.1, rs115836094 in PTPN13 at 4q21.3, and rs748116911 in COL25A1 at 4q25. These variants are extremely rare and do not appear in the 1000Genomes European population. rs148827799 does not appear in the ExAC non-Finnish Europeans; rs115836094 and rs748116911 appear with respective frequencies of 0.00003 and 0.00002. rs148827799 and rs748116911 are predicted damaging by PolyPhen2, CADD, and MetaLR. For each of these three extremely rare variants, the minor allele appears within Family 105 five times, in four cases and one unknown individual. It does not appear in any other individual in any family. The cases are two pairs of cousins. The unknown individual is a niece of two of the cousins, and she is only 45 and still at risk of developing lung cancer later in life. The two ungenotyped cases also become obligate carriers of the rare variant by virtue of having children with the variant and the fact that the variant is extremely unlikely to have come from either of the married-in unaffected parents.
Figure 2:

Individual LOD scores for Family 105 for the Lung Cancer Only analysis. A) The genome-wide individual LOD scores for family 105 and B) the chromosome 4 individual LOD scores for family 105 showing a closer look at the linked haplotype at 4q21.3–28.3. The line at 1.9 represents the genome-wide suggestive threshold as recommended by Lander and Kruglyak.
All Aggregated Cancers Analysis
rs61943670 was the only variant to reach genome-wide significance with an HLOD = 3.3. It is located at 12q23.3 and is an intronic variant in POLR3B, a subunit of RNA polymerase III. It has a MAF of 0.22 in 1000Genomes Europeans. RegulomeDB found it likely to affect transcription factor (TF) binding. This variant was found to be a significant lung eQTL (p=4.19E-07, Supplementary Figure S3A) affecting the expression of POLR3B. rs61943670 does not have a particularly high LOD score in any one family; it has LOD scores over 0.2 in 10 families. There are other suggestive signals around this variant, which decreases its chances of being a false positive (Figure 3B). Six families, which had additional cancers besides lung cancer, had increased LOD scores at this variant when compared to the LCO analysis, with those increases ranging from 0.1 – 0.4 (Supplementary Table S4).
Figure 3:

The HLOD scores Across All 28 Families for the All Aggregated Cancers analysis. A) The genome-wide HLOD scores and B) the HLOD scores for chromosome 12. The lines at 3.3 and 1.9 represent the respective genome-wide significant and suggestive thresholds as recommended by Lander and Kruglyak.
There were 204 genome-wide suggestive variants, covering all autosomes except 18. The 6q24.3–27 region had the most suggestive variants with 17, including 8 of the top 20 overall HLOD scores. The most strongly linked variant in this region, rs1062067, had an HLOD= 2.8 (Table 2), is in a non-coding RNA and was also significant in the lung eQTL analysis (p=1.16E-17) with probes mapping to LOC100507557 (Supplementary Figure S3B). Another variant, rs2251666 at 16p13.3 was also strongly linked with an HLOD = 3.0. It is in an intron of UBN1 and was found to be a significant eQTL (p=3.83E-08) in the lung, controlling expression not of UBN1, but the nearby gene SMIM22 (Supplementary Figure S3C). A comparison of the top three variants in both the LCO and AAC analyses can be found in Supplementary Table S4. The top 10 overall variants can be found in Table 2 and all genome-wide significant and suggestive variants can be found in Supplementary Table S5.
Table 2:
Top 10 HLOD Scores from the All Aggregated Cancers Analysis
| CHR | POS | TYPE | rsID | CLOD | HLOD | ALPHA | FUNC | GENE |
| 12 | 106751805 | SNV | rs61943670 | 3.3 | 3.3 | 1.0 | intron | POLR3B |
| 20 | 31660489 | SNV | rs11700200 | 3.2 | 3.2 | 1.0 | intron | BPIFB3 |
| 20 | 31671599 | SNV | rs13036385 | 3.1 | 3.1 | 1.0 | nonsyn exon | BPIFB4 |
| 9 | 5233558 | DEL | N/A | 3.0 | 3.0 | 1.0 | intron | INSL4 |
| 16 | 4923091 | SNV | rs2251666 | 3.0 | 3.0 | 1.0 | intron | UBN1 |
| 20 | 31671209 | SNV | rs4339026 | 2.9 | 2.9 | 1.0 | nonsyn exon | BPIFB4 |
| 3 | 10258762 | SNV | rs2302860 | 2.8 | 2.8 | 1.0 | intron | IRAK2 |
| 20 | 31678534 | SNV | rs2070326 | 2.8 | 2.8 | 1.0 | syn exon | BPIFB4 |
| 6 | 146207563 | SNV | rs1062067 | 2.8 | 2.8 | 1.0 | ncRNA | LOC100507557 |
| 3 | 10261294 | SNV | rs3895947 | 2.8 | 2.8 | 1.0 | intron | IRAK2 |
HLOD scores for Top Ten Variants in the All Aggregated Cancers (AAC) analysis. Headers are as follows: CHR = chromosome, POS = position of the variant in basepairs (hg 19), TYPE = Type of variant; either single nucleotide variant (SNV) or deletion (DEL), rsID = rsID of the variant (if applicable), CLOD = cumulative LOD score of the variant across all 28 families, HLOD = heterogeneity LOD score of the variant across all 28 families, ALPHA = Alpha value of variant used in HLOD score calculation, FUNC = functional description of the variant; nonsynonymous exonic (nonsyn exon), synonymous exonic (syn exon), noncoding RNA (ncRNA), or intronic (intron), GENE = gene location of the variant.
Family 102 had seven genome-wide suggestive LOD scores (Supplemental Figure S2B, Supplementary Table S2), all which were previously identified in the LCO analysis. Five variants were found in SSPO at 7p36.1 (Figure 4A) with MAFs ranging from 0.046–0.049. Three of the variants were synonymous exonic and one was a single nucleotide deletion. One intronic SNV in ARHGAP12 and one single-nucleotide intronic deletion KIF5B were identified at 10p11.22 (Figure 4B). The AAC analysis resulted in the addition of one case with leukemia to family 102 that shared the same haplotypes as the other cases and resulted in the power boost at 7p36.1 and 10p11.22. There were three additional SNVs that were located within SSPO with LOD score above 1.4 in this family.
Figure 4:

Individual LOD scores for Family 102 for the All Aggregated Cancers analysis. A) The individual LOD scores for family 102 for chromosome 7 and B) the individual LOD scores for family 102 for chromosome 10. The line at 1.9 represents the genome-wide suggestive threshold as recommended by Lander and Kruglyak.
No other families contained any suggestive LOD scores. 10 families had no additional cancers; their LOD scores remained identical from the LCO analysis, including the 4q21.3–28.3 haplotype from Family 105.
Discussion
This study identified a genome-wide significant variant at 12q23.3 in highly aggregated lung cancer families when including other cancers reported among family members. The significant variant, rs61943670, is an intronic variant located in POLR3B, subunit B of RNA polymerase III. RegulomeDB found it likely that this variant affects transcription factor binding and it was found to be a significant eQTL in the lung. Somatic mutations in POLR3B have been implicated in lung cancer; it was found to be differentially methylated in stage I lung adenocarcinoma (47) and recurrent mutations in POLR3B were identified in pulmonary carcinoid tumors (48). This is the first time that POLR3B has been implicated as a germline risk variant for lung cancer. Functional studies revealed a truncated form of POLR3B represses the transcriptional activities of p53 and AP-1 and may play a role in tumorigenesis (49). Biologically, POLR3B makes sense as a possible susceptibility gene for cancer.
rs61943670 is intronic and not particularly rare in Europeans - MAF = 0.22. The variant does not exhibit a large effect on any one family with LOD scores ranging from 0.2 – 0.5. If the variant is causal, it likely exhibits a small/moderate effect on cancer risk, possibly through being a TF binding site and affecting POLR3B transcription. It does not seem to cluster in individuals with early onset lung cancer (defined as having lung cancer before age 50); it is prevalent in individuals that developed lung cancer in their 50s and 60s. It is also possible that is variant is not causal, but simply in linkage disequilibrium with a more penetrant rare variant along the haplotype that was not sequenced in this WES study. We also note that since the variant was identified as significant only under the AAC analysis, it may be a risk factor in familial cancers in general as well as a potential risk modifier of common cancer predisposition syndromes.
Two of the highly suggestive variants were significant eQTLs in the lung. rs1062067 is on 6q24.3 located in LOC100507557, a noncoding RNA gene. Its function is unknown, but noncoding RNAs in general have been found to be important in both lung cancer (50) and other cancers (51, 52). rs2251667 on 16p13.3 is in an intron of UBN1 and controls expression of SMIM22. UBN1 is involved in cellular senescence and is a potential tumor suppressor (53) while SMIM22 is differentially expressed in prostate cancer (54).
Lung cancer is almost certainly heterogeneous, so it is likely that the individual families are also harboring unique risk variants, possibly of large effect. Only Family 102 had family-specific genome-wide suggestive variants, one at 7p36.1 and another at 10p11.22. The signals appeared in the LCO analysis and were boosted in the AAC analysis by a single leukemia case that shared the same haplotypes as the lung cancer cases.
The 7p36.1 signal was particularly interesting because it was localized to a single gene, SSPO. The SSPO signal consists of seven variants in the gene with LOD scores from 1.34–2.17. SSPO encodes the protein SCO-Spondin, which is involved in the modulation of neuronal aggregation. It is upregulated in brain tissue harboring metastases (55) and somatic mutations are associated with aggressive thyroid microcarcinomas (56). This is the first time that germline mutations have been linked to any type of familial cancers. The exonic variants are synonymous, though possibly affecting TF binding, and the deletion was intronic and only a single nucleotide. All SNVs were moderately rare with MAF = 0.046–0.049 in 1000Genomes Europeans. It is possible these variants are not causal and that an unsequenced or failed variant is in fact the true causal variant and these variants are simply located on the same haplotype. Targeted sequencing of SSPO would capture some of these unsequenced variants and subsequent linkage analysis could reveal any more linked variants in the gene. It is clear however, that even if the causal variant was not found, the SSPO gene is linked to lung cancer risk in this family and should be the focus of further study.
The second suggestive signal in Family 102 was localized to ARHGAP12 and KIF5B at 10p11.22. ARHGAP12 was implicated in early onset colorectal cancer in Finns (57). KIF5B is a driver of lung cancer adenocarcinoma through a fusion with the RET gene (58), though this is not a germline mutation.
Family 105 had an interesting long, linked haplotype from 4q21.3–28.3. Family 105 contained only lung cancer affecteds, so the AAC analysis did not change its results. The haplotype has little to no negative signal underneath it, which is characteristic of a true linked haplotype and not a false positive. The haplotype encompasses several nonsynonymous variants; three SNVs were interesting because they were exceedingly rare; they were not present in 1000 Genomes Europeans and had MAF < 0.00004 in ExAC). The variants only appeared in cases within the family, one unknown individual with the potential to develop lung cancer, and the two ungenotyped cases must be obligate carriers of the variants. Two of the variants are in good candidate genes, DSPP and PTPN13. DSPP is an extracellular matrix glycophosphoprotein that silences tumorigenic activities in oral cancer (59), predicts the transition from oral epithelial dysplasia to oral squamous carcinoma (60) is expressed in prostate cancer (61). Loss of the closely related glycophosphoprotein DMP1 results in lung cancer tumorigenesis (62). PTPN13 is a protein tyrosine phosphatase that is a known tumor suppressor gene in lung cancer (63).
The strength of this study was its family-based nature, which was designed to find potential, possible rare, risk variants for lung cancer and other aggregated cancers in these pedigrees. The linkage analysis also allowed for the utilization of long linked haplotypes within families. We were able to identify one common variant of small/moderate effect across all families and several interesting individual family-specific variants. These individual family-based variants could not have been found in a population-based study and the long, linked haplotype at 4q led to the potential rare causal variants in PTPN13 and/or DSPP. The study is not without weakness, as this study only used WES data and thus could have missed linked noncoding variants. Targeted sequencing is planned to address this issue. We note that although we found significant evidence of linkage across all families under the AAC analysis, we did not find any significant linkage under the LCO analysis. This is most likely due to lack of power under the LCO analysis; 19 of the 28 families had only one or two sequenced lung cancer affected individuals and the genotypes of other affected individuals were imputed from their descendants’ genotypes. A larger number of families and updates to the affection status of individuals in these families will certainly add to the power of this study.
In conclusion, this study identified a significant linkage signal at 12q23.3 centered on POLR3B for general cancer risk in highly aggregated lung cancer families. The risk is cumulative and moderate; the variant is a significant lung eQTL, likely TF binding site, and potentially causal. We also identified highly suggestive variants that were significant eQTLs in the lung at 6q24.3 and 16p13.3 and interesting familywise signals were identified on 7p and 4q. Targeted sequencing is planned for 12q, 7p, and 4q for better coverage of the non-coding regions. Functional analysis, including knock-outs and knock-ins, is planned on POLR3B and family candidate genes SSPO in Family 102 and DSPP and PTPN13 in Family 105 and potentially the other eQTLs. These genes were prioritized because they were either genome-wide significant (POLR3B), had multiple individual family scores that were suggestive (SSPO), or were very rare variants located along a linked haplotype that only appeared in cases within that family (PTPN13 and DSPP). Finally, ongoing follow-up of potential risk allele carriers to identify newly affected individuals in these high-risk families is likely to increase our ability to identify causal variants in the future.
Supplementary Material
Acknowledgments:
The authors thank all study participants and their families. This work was funded in part by the National Institutes of Health, National Cancer Institute grants U01CA76293 (S.M. Pinney and M.W. Anderson), U19CA148127 (C.I. Amos and M. You) , P30CA22453 (A.G. Schwartz), R03CA77118 (P. Yang), R01CA80127 (P. Yang), R01CA84354 (P. Yang), National Institutes of Health, National Institute of Environmental Health Sciences P30ES006096 (S.M. Pinney), and Department of Health and Human Services contracts HHSN26820100007C (D. Mandal) and HHSN268201700012C (D. Mandal) C.I. Amos is a Research Scholar of the Cancer Prevention Research Institute of Texas (CPRIT). This research was partially supported by CPRIT grant RR170048. J.E. Bailey-Wilson, A.M. Musolf, B.A. Moiz, and H. Sun were funded in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health. P. Yang and M. de Andrade were funded in part by the Mayo Foundation Fund. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Financial Support: NIH grants U01CA76293, U19CA148127, P30CA22453, P30ES006096, HHSN26820100007C, HHSN268201700012C, R03CA77118, R01CA80127, R01CA84354, Intramural Research Program of the National Human Genome Research Institute, NIH, Mayo Foundation Fund. Christopher Amos is a Research Scholar of the Cancer Prevention Research Institute of Texas (CPRIT); CPRIT grant RR170048 partially supported this work.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest
References
- 1.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. Journal of the National Cancer Institute. 1981;66:1191–308. [PubMed] [Google Scholar]
- 2.Doll R, Peto R, Wheatley K, Gray R, Sutherland I. Mortality in relation to smoking: 40 years’ observations on male British doctors. Bmj. 1994;309:901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carbone D. Smoking and cancer. The American journal of medicine. 1992;93:13S–7S. [DOI] [PubMed] [Google Scholar]
- 4.Burch PR. Smoking and lung cancer. Tests of a causal hypothesis. Journal of chronic diseases. 1980;33:221–38. [DOI] [PubMed] [Google Scholar]
- 5.Mattson ME, Pollack ES, Cullen JW. What are the odds that smoking will kill you? American journal of public health. 1987;77:425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. Bmj. 2000;321:323–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenks S Is Lung Cancer Incidence Increasing in Never-Smokers? Journal of the National Cancer Institute. 2016;108. [DOI] [PubMed] [Google Scholar]
- 8.Tokuhata GK, Lilienfeld AM. Familial aggregation of lung cancer in humans. Journal of the National Cancer Institute. 1963;30:289–312. [PubMed] [Google Scholar]
- 9.Tokuhata GK, Lilienfeld AM. Familial aggregation of lung cancer among hospital patients. Public health reports. 1963;78:277–83. [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon-Albright LA, Thomas A, Goldgar DE, Gholami K, Rowe K, Jacobsen M, et al. Familiality of cancer in Utah. Cancer research. 1994;54:2378–85. [PubMed] [Google Scholar]
- 11.Ooi WL, Elston RC, Chen VW, Bailey-Wilson JE, Rothschild H. Increased familial risk for lung cancer. Journal of the National Cancer Institute. 1986;76:217–22. [PubMed] [Google Scholar]
- 12.Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. Journal of the National Cancer Institute. 1994;86:1600–8. [DOI] [PubMed] [Google Scholar]
- 13.Sellers TA, Bailey-Wilson JE, Elston RC, Wilson AF, Elston GZ, Ooi WL, et al. Evidence for mendelian inheritance in the pathogenesis of lung cancer. Journal of the National Cancer Institute. 1990;82:1272–9. [DOI] [PubMed] [Google Scholar]
- 14.Bailey-Wilson JE, Sellers TA, Elston RC, Evens CC, Rothschild H. Evidence for a major gene effect in early-onset lung cancer. The Journal of the Louisiana State Medical Society : official organ of the Louisiana State Medical Society. 1993;145:157–62. [PubMed] [Google Scholar]
- 15.Sellers TA, Bailey-Wilson JE, Potter JD, Rich SS, Rothschild H, Elston RC. Effect of cohort differences in smoking prevalence on models of lung cancer susceptibility. Genetic epidemiology. 1992;9:261–71. [DOI] [PubMed] [Google Scholar]
- 16.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nature genetics. 2008;40:616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–7. [DOI] [PubMed] [Google Scholar]
- 18.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey-Wilson JE, Amos CI, Pinney SM, Petersen GM, de Andrade M, Wiest JS, et al. A major lung cancer susceptibility locus maps to chromosome 6q23–25. American journal of human genetics. 2004;75:460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Musolf AM, Simpson CL, de Andrade M, Mandal D, Gaba C, Yang P, et al. Parametric Linkage Analysis Identifies Five Novel Genome-Wide Significant Loci for Familial Lung Cancer. Human heredity. 2016;82:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellers TA, Ooi WL, Elston RC, Chen VW, Bailey-Wilson JE, Rothschild H. Increased familial risk for non-lung cancer among relatives of lung cancer patients. American journal of epidemiology. 1987;126:237–46. [DOI] [PubMed] [Google Scholar]
- 22.King TM, Tong L, Pack RJ, Spencer C, Amos CI. Accuracy of family history of cancer as reported by men with prostate cancer. Urology. 2002;59:546–50. [DOI] [PubMed] [Google Scholar]
- 23.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Human heredity. 1971;21:523–42. [DOI] [PubMed] [Google Scholar]
- 27.Mandal DM, Sorant AJ, Atwood LD, Wilson AF, Bailey-Wilson JE. Allele frequency misspecification: effect on power and Type I error of model-dependent linkage analysis of quantitative traits under random ascertainment. BMC genetics. 2006;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandal DM, Wilson AF, Bailey-Wilson JE. Effects of misspecification of allele frequencies on the power of Haseman-Elston sib-pair linkage method for quantitative traits. American journal of medical genetics. 2001;103:308–13. [PubMed] [Google Scholar]
- 29.Mandal DM, Wilson AF, Elston RC, Weissbecker K, Keats BJ, Bailey-Wilson JE. Effects of misspecification of allele frequencies on the type I error rate of model-free linkage analysis. Human heredity. 2000;50:126–32. [DOI] [PubMed] [Google Scholar]
- 30.Smith CA. Testing for Heterogeneity of Recombination Fraction Values in Human Genetics. Ann Hum Genet. 1963;27:175–82. [DOI] [PubMed] [Google Scholar]
- 31.Ott J Analysis of Human Genetic Linkage. Baltimore: Johns Hopkins University Press; 1985. [Google Scholar]
- 32.Lynch HT, Kimberling W, Albano WA, Lynch JF, Biscone K, Schuelke GS, et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). I. Clinical description of resource. Cancer. 1985;56:934–8. [DOI] [PubMed] [Google Scholar]
- 33.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clinical genetics. 2009;76:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. Risk of pancreatic cancer in families with Lynch syndrome. Jama. 2009;302:1790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Familial cancer. 2013;12:229–40. [DOI] [PubMed] [Google Scholar]
- 36.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. Journal of medical genetics. 2012;49:433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nature protocols. 2015;10:1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012;40:W452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2 Current protocols in human genetics / editorial board, Jonathan L Haines [ et al. ]. 2013;Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. American journal of human genetics. 2016;99:877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome research. 2012;22:1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao K, Bosse Y, Nickle DC, Pare PD, Postma DS, Laviolette M, et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS genetics. 2012;8:e1003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamontagne M, Berube JC, Obeidat M, Cho MH, Hobbs BD, Sakornsakolpat P, et al. Leveraging lung tissue transcriptome to uncover candidate causal genes in COPD genetic associations. Human molecular genetics. 2018;27:1819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature genetics. 1995;11:241–7. [DOI] [PubMed] [Google Scholar]
- 47.Luo WM, Wang ZY, Zhang X. Identification of four differentially methylated genes as prognostic signatures for stage I lung adenocarcinoma. Cancer cell international. 2018;18:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asiedu MK, Thomas CF Jr., Dong J, Schulte SC, Khadka P, Sun Z, et al. Pathways Impacted by Genomic Alterations in Pulmonary Carcinoid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24:1691–704. [DOI] [PubMed] [Google Scholar]
- 49.Yunlei Z, Zhe C, Yan L, Pengcheng W, Yanbo Z, Le S, et al. INMAP, a novel truncated version of POLR3B, represses AP-1 and p53 transcriptional activity. Molecular and cellular biochemistry. 2013;374:81–9. [DOI] [PubMed] [Google Scholar]
- 50.Jin M, Ren J, Luo M, You Z, Fang Y, Han Y, et al. Long noncoding RNA JPX correlates with poor prognosis and tumor progression in non-small cell lung cancer by interacting with miR-145–5p and CCND2. Carcinogenesis. 2019. [DOI] [PubMed] [Google Scholar]
- 51.Wang C, Yang Y, Zhang G, Li J, Wu X, Ma X, et al. Long noncoding RNA EMS connects c-Myc to cell cycle control and tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong HG, Li H, Xiao Y, Yang QC, Yang LL, Chen L, et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. Journal of experimental & clinical cancer research : CR. 2019;38:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banumathy G, Somaiah N, Zhang R, Tang Y, Hoffmann J, Andrake M, et al. Human UBN1 is an ortholog of yeast Hpc2p and has an essential role in the HIRA/ASF1a chromatin-remodeling pathway in senescent cells. Molecular and cellular biology. 2009;29:758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li F, Ji JP, Xu Y, Liu RL. Identification a novel set of 6 differential expressed genes in prostate cancer that can potentially predict biochemical recurrence after curative surgery. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2019;21:1067–75. [DOI] [PubMed] [Google Scholar]
- 55.Sato R, Nakano T, Hosonaga M, Sampetrean O, Harigai R, Sasaki T, et al. RNA Sequencing Analysis Reveals Interactions between Breast Cancer or Melanoma Cells and the Tissue Microenvironment during Brain Metastasis. BioMed research international. 2017;2017:8032910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song J, Wu S, Xia X, Wang Y, Fan Y, Yang Z. Cell adhesion-related gene somatic mutations are enriched in aggressive papillary thyroid microcarcinomas. Journal of translational medicine. 2018;16:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanskanen T, Gylfe AE, Katainen R, Taipale M, Renkonen-Sinisalo L, Jarvinen H, et al. Systematic search for rare variants in Finnish early-onset colorectal cancer patients. Cancer genetics. 2015;208:35–40. [DOI] [PubMed] [Google Scholar]
- 58.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nature medicine. 2012;18:375–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joshi R, Tawfik A, Edeh N, McCloud V, Looney S, Lewis J, et al. Dentin sialophosphoprotein (DSPP) gene-silencing inhibits key tumorigenic activities in human oral cancer cell line, OSC2. PloS one. 2010;5:e13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogbureke KU, Abdelsayed RA, Kushner H, Li L, Fisher LW. Two members of the SIBLING family of proteins, DSPP and BSP, may predict the transition of oral epithelial dysplasia to oral squamous cell carcinoma. Cancer. 2010;116:1709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaplet M, Waltregny D, Detry C, Fisher LW, Castronovo V, Bellahcene A. Expression of dentin sialophosphoprotein in human prostate cancer and its correlation with tumor aggressiveness. International journal of cancer. 2006;118:850–6. [DOI] [PubMed] [Google Scholar]
- 62.Inoue K, Sugiyama T, Taneja P, Morgan RL, Frazier DP. Emerging roles of DMP1 in lung cancer. Cancer research. 2008;68:4487–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scrima M, De Marco C, De Vita F, Fabiani F, Franco R, Pirozzi G, et al. The nonreceptor-type tyrosine phosphatase PTPN13 is a tumor suppressor gene in non-small cell lung cancer. The American journal of pathology. 2012;180:1202–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.