

SUMMARY
Variants in the ANK3 gene encoding ankyrin-G are associated with neurodevelopmental disorders, including intellectual disability, autism, schizophrenia, and bipolar disorder. However, no upstream regulators of ankyrin-G at synapses are known. Here, we show that ankyrin-G interacts with Usp9X, a neurodevelopmental disorder-associated deubiquitinase (DUB). Usp9X phosphorylation enhances their interaction, decreases ankyrin-G polyubiquitination, and stabilizes ankyrin-G to maintain dendritic spine development. In forebrain-specific Usp9X knockout mice (Usp9X−/Y), ankyrin-G as well as multiple ankyrin-repeat domain (ANKRD)-containing proteins are transiently reduced at two, but recovered at 12 weeks, postnatally. However, reduced cortical spine density in knockouts persists into adulthood. Usp9X−/Y mice display increase of ankyrin-G ubiquitination and aggregation and hyperactivity. USP9X mutations in patients with intellectual disability and autism ablate its catalytic activity or ankyrin-G interaction. Our data reveal a DUB-dependent mechanism of ANKRD protein homeostasis, impairment of which only transiently affects ANKRD protein levels, but leads to persistent neuronal, behavioral, and clinical abnormalities.
Keywords: deubiquitinase, intellectual disability, proximity ligation assay, structured illumination microscopy, ankyrin-G, ANK, SHANK
Graphical Abstract
eTOC
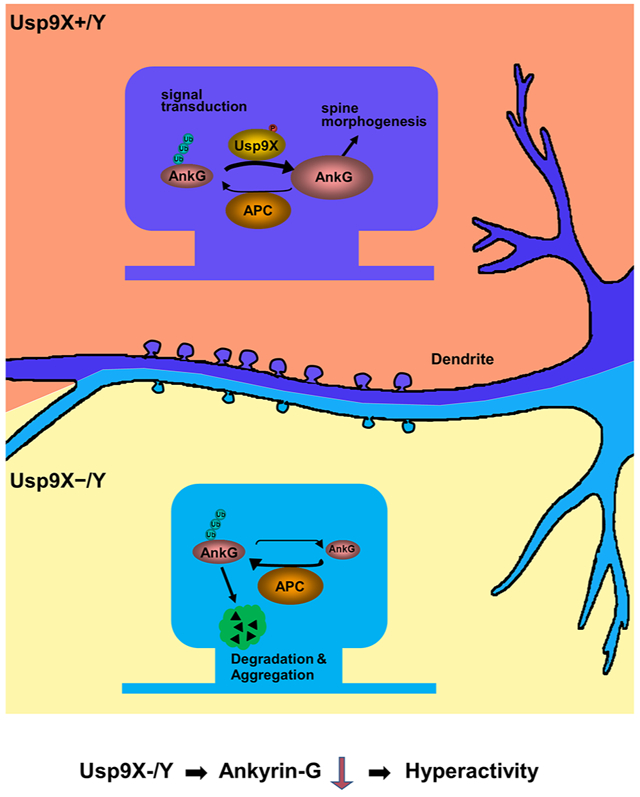
Yoon et al. show that deubiquitination of proteins containing ankyrin-repeat domains is essential for the correct developmental trajectory of cortical synapses, with disruption of the deubiquitinase USP9X resulting in deficient synaptic structural plasticity as well as behavioral and clinical abnormalities.
INTRODUCTION
Rare and common variants in the ANK3 gene have been identified in patients with neurodevelopmental disorders including intellectual disability/attention deficit and hyperactivity disorder (ID/ADHD) (Iqbal et al., 2013) and autism spectrum disorder (ASD) (Sanders et al., 2012), and they are among the most strongly associated risk factors for bipolar disorder (BD) in genome-wide association studies (GWAS) (Ferreira et al., 2008; Schulze et al., 2009). ANK3 encodes ankyrin-G, which acts as a scaffold, linking plasma membrane proteins to the actin/β-spectrin cytoskeleton (Bennett and Healy, 2008). Ankyrin-G plays an important role in multiple neurobiological processes, including synaptogenesis, synaptic plasticity, action potential generation and transmission, and ion channel regulation (Jenkins et al., 2015; Smith et al., 2014; Tseng et al., 2015). Multiple isoforms of ankyrin-G are expressed in the brain, of which the 190 kDa form is enriched in dendrites and postsynaptic densities (PSDs) (Jordan et al., 2004) and regulates dendritic spine structure (Smith et al., 2014). However, few upstream regulators of ankyrin-G at synapses are known.
Ankyrin-G contains 24 ankyrin-repeat domains (ANKRD). The ANKRD has an important role in protein-protein interactions through variations in adaptive surface residues (Mosavi et al., 2004). Remarkably, ANKRDs are among the most common protein-protein interaction domains, present in 270 human proteins with diverse functions (Bork, 1993). By comparison, PDZ domains are present in 154 proteins. The ANKRD family includes prominent synaptic proteins associated with neurodevelopmental disorders, such as ankyrin-G and –B (encoded by ANK2/3) and SHANK2/3 and TRANK1 (De Rubeis et al., 2014; Fromer et al., 2014; Hou et al., 2016). Like ankyrin-G, several ANKRD family members have been shown to regulate dendritic spine structural plasticity (Peca et al., 2011; Schmeisser et al., 2012; Smith et al., 2014).
Structural plasticity of spiny excitatory synapses contributes to brain circuit development, and its abnormalities contribute to neurodevelopmental disorders (Forrest et al., 2018; Penzes et al., 2011). Accordingly, dendritic spine alterations have been observed in postmortem studies of patients with schizophrenia (SZ), BD, ASDs, and ID (Glantz and Lewis, 2000; Hutsler and Zhang, 2010; Irwin et al., 2000; Konopaske et al., 2014). Moreover, genomic studies support a key role for postsynaptic proteins in the pathogenesis of these disorders (Gilman et al., 2011).
Despite their importance, little is known about how the levels of ankyrin-G and other ANKRD proteins, in particular at synapses, are maintained in their normal physiological range. Protein homeostasis (proteostasis) is a mechanism whereby cells maintain physiological levels of proteins, and abnormal neuronal proteostasis leads to disorders associated with abnormal protein levels and to excessive protein aggregation (Galves et al., 2019).
One major mechanism for proteostasis is through protein degradation by the ubiquitin-proteasome system (UPS) (van Roessel et al., 2004), which is also crucial for synaptic remodeling (Ehlers, 2003) and is implicated in neurodevelopmental disorders (Bousman et al., 2010; Glessner et al., 2009). Proteins are targeted for degradation by the UPS through the addition of polyubiquitin chains to their lysine residues. Ubiquitination is performed in a three-tiered enzymatic cascade by ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3), respectively. Of these, the E3 ubiquitin ligase cell division cycle 20/anaphase-promoting complex (Cdc20/APC) controls dendrite morphogenesis and synapse size in the developing brain (Kim et al., 2009).
DUBs oppose this process by removing ubiquitin from proteins, and deubiquitylation is crucial for proteostasis regulation (Bett, 2016). Activation or inhibition of DUBs has been shown to induce synaptic plasticity (Cartier et al., 2009). Recent studies support a role for dysregulation of the UPS pathway in SZ, BD (Ehlers, 2003), ID (Hollstein et al., 2015), and ASD (Glessner et al., 2009), suggesting that protein turnover may play an important role in neurodevelopmental disorders. However, as compared to ubiquitylation, little is known about the synaptic targets of DUBs and about how deubiquitination of PSD proteins impacts spine plasticity and contributes to disease pathogenesis.
To identify novel upstream regulators of ankyrin-G at synapses, here we used an unbiased yeast-2-hybrid screen and found that it interacts with Usp9X, a DUB mutated in patients with neurodevelopmental disability. Phosphorylation enhances Usp9X/ankyrin-G interaction, decreases ankyrin-G poly-ubiquitination, and stabilizes ankyrin-G, to maintain dendritic spines. In situ proximity ligation combined with structured-illumination super-resolution microscopy reveals the postsynaptic spatial organization of this pathway. In forebrain-specific Usp9X conditional knockout (Usp9X−/Y) mice, levels of multiple ANKRD proteins, including key autism risk factors, are transiently reduced at two, but recover at 12 weeks postnatally. However, Usp9X−/Y mice show reduced cortical spine density, which surprisingly persists into adulthood, when it co-occurs with elevated ankyrin-G ubiquitination and aggregation. Consistent with this mechanism, mutations in the USP9X gene in patients with ID and ASD ablate either its catalytic activity or its interaction with ankyrin-G. Our data provide insight into the regulation of homeostasis of ANKRD-containing proteins at synapses as well as the biology of synaptic DUBs. Impairment of this process, while it only transiently affects ANKRD protein levels, leads to persistent neuronal and clinical abnormalities.
RESULTS
Ankyrin-G Stability is Regulated by Ubiquitination, and its ANKRD Interacts with Usp9X
To identify regulators of ankyrin-G, we performed a yeast-2-hybrid screen using its ankyrin repeats 1-12 (aa35-423) as bait. We identified the peptidase domain (aa1719-1842) of the Usp9X deubiquitinase, encoded by USP9X, a gene mutated in ID and epilepsy (Reijnders et al., 2016) (Fig. 1A). We confirmed their interaction in vivo by coimmunoprecipitation from mouse cortex (Fig. 1B) and mapped ankyrin-G’s ANKRD and regulatory domains as interaction sites with Usp9X’s peptidase domain (Fig. 1C).
Figure 1.

Ankyrin-G stability is regulated through ubiquitination and Usp9X-dependent deubiquitination. (A) Ankyrin-G ANKRD interacts with the peptidase domain of Usp9X in a yeast 2-hybrid screen; red lines: interaction domains. (B) Co-immunoprecipitation of ankyrin-G with Usp9X from mouse cortex. (C) Domain-mapping of the ankyrin-G-Usp9X interaction from HEK293T cells. (D) Schematic of ankyrin-G with D- and KEN-boxes. (E) Homology model of ankyrin-G based on ankyrin-B (PDB-4RLV). D-boxes and ubiquitinated lysines are shown. (F) Ankyrin-G levels after MG132 treatment (10 μM). Cell lysates were prepared from primary cultured cortical neurons. (G) Mapping of ubiquitinated domains of ankyrin-G from HEK293T cells. (H) D-box-dependent ubiquitination of HA-ankyrin-G1-807 from HEK293T cells. (I) Kinetics of D-box-dependent degradation of ankyrin-G. Time-lapse of GFP fluorescence after cycloheximide (20 μg/ml) treatment of GFP-ankyrin-G1-807 or GFP-ankyrin-G1-807-Muta;b-expressing neurons (n = 7 cells per each group). Scale bar, 10 μm. Right: Quantification of GFP fluorescence intensity over time (3h, **p < 0.01; 5h, *p < 0.05). Repeated measures of two-way ANOVA were followed by Bonferroni post-tests. Data are represented as mean ± SEM. See also Figure S1.
Proteins are targeted for degradation by UPS through the addition of polyubiquitin chains to their lysines, by a cascade including ubiquitin ligases (E3). Ankyrin-G contains seven “destruction box" (D-box), E3 ligase recognition motifs—two within the ANKRD (aa140-143, 282-285), which are highly conserved, and two KEN boxes (Fig. 1D; Table S1). In addition, three lysines are located within the ANKRD (K39; K260; K268) (Fig. S1B). All seven D-boxes were previously shown to be ubiquitinated (Hornbeck et al., 2015). Threading of ankyrin-G’s ANKRD onto the ankyrin-B structure (Fig. 1E) revealed that these conserved lysines are solvent-exposed, further supporting their availability for ubiquitination. Indeed, treatment of cultured cortical neurons with the proteasome inhibitor MG132 led to increased ankyrin-G levels, demonstrating that ankyrin-G is degraded by the proteasome (Fig. 1F). Because the ANKRD was highly polyubiquitinated, we focused further on this domain (Fig. 1G).
The D-box is a recognition motif for the major neuronal E3 ligase Cdc20/APC, which regulates dendrite morphogenesis (Kim et al., 2009). Mutation of one or both D-boxes (Fig. 1H) or of the lysines (Fig. S1C) within ANKRD attenuated the effect of MG132 on ANKRD polyubiquitination in HEK293T cells. We compared the degradation kinetics in neurons of wild-type GFP-ankyrin-G1-807 with its D-box-mutated version by time-lapse imaging in the presence of the protein synthesis inhibitor cycloheximide. The D-box mutant exhibited slower degradation than wild -type GFP-ankyrin-G1-807 (Fig. 1I), suggesting that ankyrin-G stability is regulated by Cdc20/APC-dependent ubiquitination of its ANKRD.
Super-resolution Imaging Reveals the Relationship between Ankyrin-G and Usp9X Spatial Organization and Spine Architecture
Usp9X was most highly colocalized with ankyrin-G in the somatodendritic compartment (Fig. S2C). Because of the limited resolution of confocal microscopy, we employed structured illumination microscopy (SIM) to analyze the precise spatial organization of ankyrin-G and Usp9X (Fig. 2A). Frequently, multiple ankyrin-G nanodomains were adjacent to or overlapping with Usp9X puncta in perisynaptic areas (Fig. 2B). Their relative localization was substantiated using ratiometric images, colocalization highlighter, linescans, and 3D reconstructions (Fig. 2B-C). Most spines exhibited two or more ankyrin-G nanodomains surrounding Usp9X (Fig. 2D).
Figure 2.

Super-resolution imaging of spatial organization of ankyrin-G and Usp9X relationship with spine architecture. (A) SIM image of ankyrin-G and Usp9X immunofluorescence in a dendritic region outlined by tRFP expression; scale bar, 2 μm. (B) High-resolution image of boxed spine in (A). Lower panels: ratiometric images and colocalization (white). Scale bar, 1 μm. (C) Line scan through spine head. (D) Representative spatial relationships between ankyrin-G and Usp9X within individual spine heads. Scale bar = 0.25 μm. Quantification shows the percentage of spines with each distribution. (E-H) The fraction of spines having Usp9X or ankyrin-G nanodomains in the head or neck. Spine head size related to the presence or absence of Usp9X or ankyrin-G nanodomains in the head or neck. Total 198 spines were analyzed from 6 neurons. ***p < 0.001; One-way ANOVA was followed by Bonferroni post-tests. Data are represented as mean ± SEM. (I-L) Correlation plots of spine head areas, number and size of ankyrin-G nanodomains/spine with number and area of Usp9X nanodomains/spine from (E-H). Two-tailed spearman tests were performed. See also Figure S2.
To understand how the localization of ankyrin-G and Usp9X nanodomains correlates with spine morphology, we analyzed their subcellular distribution by SIM images. Interestingly, Usp9X was observed in only 9.1% of spine necks, despite the presence of Usp9X within 46.0% of the spine heads (Fig. 2E). Furthermore, the presence of Usp9X in spine heads was significantly correlated with larger spine head sizes, but no correlation was observed between its presence in the spine neck and head size (Fig. 2F). Ankyrin-G nanodomains were present in 89.4% of spine heads and 52.0% of spine necks (Fig. 2G), and the spine head size correlated to the presence of ankyrin-G in the spine head and neck (Fig. 2H). The number of Usp9X nanodomains per spine correlated to the spine head area (Fig. 2I), while the total Usp9X nanodomain area per spine correlated to spine head size and to the number and size of ankyrin-G nanodomains within the spine head (Fig. 2J-L).
Usp9X Modulates Spine Morphology through Ankyrin-G Stabilization
Recent studies have suggested that Usp9X plays a role in neuronal development (Oishi et al., 2016; Stegeman et al., 2013). Therefore, we examined the impact of manipulating Usp9X levels on dendritic spine morphology in mature cultured cortical neurons. Usp9X knockdown caused a significant reduction of spine head size and density (Fig. 3A-C). Conversely, overexpression of its peptidase domain resulted in spine head enlargement and rescued the effect of Usp9X knockdown on spine density (Fig. 3E-F). Moreover, overexpression of the peptidase domain of Usp9X attenuated the increase in ubiquitinated ankyrin-G induced by MG132 (Fig. 3G). Furthermore, expression of ankyrin-G with two mutated D-boxes increased spine size in control neurons and rescued spine density in shUsp9X-expressing neurons (Fig. 3H-J). Taken together, these data suggest that Usp9X-mediated deubiquitination stabilizes ankyrin-G levels and maintains spine architecture.
Figure 3.

Usp9X regulates spine morphology by deubiquitination of ankyrin-G. (A) Confocal images of neurons expressing scramble (control RNAi) or Usp9X knockdown (shUsp9X) construct. Scale bar = 40 μm for the left panel and 5 μm for the right panel. (B, C) Bar graph showing shUsp9X causes a decrease in spine head size and density compared with control. Control from 9 cells and shUsp9X from 10 cells were analyzed; **p = 0.0025, *p = 0.0114. Two-tailed unpaired t-test was performed. (D) Confocal images of control RNAi or shUsp9X transfecting neurons co-expressing GFP-Usp9X1555-1958 construct. Scale bar, 5 μm. (E, F) Bar graph of spine head size and density showing co-expression of GFP-Usp9X1555-1958 with control or shUsp9X. 20 neurons were analyzed from each group. ***p < 0.001, †p < 0.05; Two-way ANOVA was followed by Bonferroni post-tests. (G) The peptidase domain of Usp9X (Flag-Usp9X1555-1958) promotes ankyrin-G deubiquitination. Cell lysates were analyzed from HEK293T cells. (H) Confocal images of neurons expressing control RNAi or shUsp9X, co-expressing GFP-ankyrin-G or GFP-ankyrin-G-Muta;b. Scale bar, 5 μm. (I, J) Quantification of the effects on spine size or density. 20 neurons were analyzed per each group. **p < 0.01, ***p < 0.001; One-way ANOVA was followed by Bonferroni post-tests. Data are represented as mean ± SEM. See also Figure S3.
Phosphorylation Enhances the Interaction of Usp9X with Ankyrin-G
Among post-translational modifications (PTMs), phosphorylation of binding hot spots in hydrolases tends to modulate the strength of interaction to target substrates directly (Nishi et al., 2011). Three S/T phosphorylation consensus sites were identified in Usp9X’s peptidase domain (Ser1593/1600/1609) (Hornbeck et al., 2015), we hypothesized that phosphorylation of Usp9X may alter protein-protein interaction. To investigate the role of phosphorylation in regulating the Usp9X and ankyrin-G interaction, we predicted possible models based on the crystal structure of Usp9X. Structural reports have identified the inner groove of the ANKRD as a promiscuous protein-protein interaction site (Al-Khodor et al., 2010). Homology modeling of ankyrin-G and the peptidase domain of Usp9X suggests that lysine deubiquitination would result in extensive contacts between the ankyrin-G scaffolding site and Usp9X (Fig. 4A; Fig. S4). Multiple alignments of Usp9X with related DUBs, including USP7, reveals that these phosphorylation sites lie within a disordered region of low homology that may form an ankyrin-G interaction site-specific to Usp9X within the DUB family (Fig. 4B). Phosphorylation on S1593 and S1609 may alter the Usp9X-ankyrin-G interaction through charge-charge interactions with exposed basic residues present on the inner groove of the ankyrin-G ANKRD. In order to assess whether these sites are subject to PTM, mutagenesis of the peptidase domain of Usp9X (Usp9X1555-1958-S1593A-S1600A-S1609A; Usp9XS3A) was carried out. Usp9X1555-1958 (Usp9XWt) was immunoprecipitated by phosphoserine antibody, but not by Usp9XS3A, suggesting that those serine sites in Usp9X might have critical role for phosphorylation (Fig. S5A).
Figure 4.

Spatial organization of ankyrin-G and phosphomimetic Usp9X interaction and relationship with spine architecture. (A) Schematic of Usp9X-mediated Ub-ankyrin-G DUB activity. Homology model of ankyrin-G ANKRD showing the predicted position of ubiquitinated K260. A potential orientation for Usp9X homology model is indicated based on the position of K260-ubiquitin and minimal steric clash. (B) Alignment of Usp7 and Usp9X peptidase domains reveals three regions of low homology. Red crosses indicate S1593, S1600, S1608 phosphorylation sites. (C) In situ PLA measurement of the interaction between HA-ankyrin-G1-807 and Flag-Usp9X1555-1958 Wt or Flag-Usp9X1555-1958-S1593A-S1600A-S1609A (S3A) in HEK293T cells (Usp9XWt control, n = 16; Usp9XS3A control, n = 9. *p = 0.042). Two-tailed unpaired t-test was performed. (D) In situ PLA measurement of the interaction between HA-ankyrin-G1-807 and phosphomimetic mutants of Flag-Usp9X1555-1958 in HEK293T cells. Bar graph of PLA signal (n = 20 from each condition). *p < 0.05; ***p < 0.001; One-way ANOVA was followed by Bonferroni post-tests. (E-F) The ability of His-Usp9X1547-1962 S3A or His-Usp9X1547-1962-S1593D-S1600D-S1609D (S3D) or each single phosphomimetic mutation to catalyze the cleavage of the fluorogenic substrate was compared to the wild type peptidase domain (WT) and an active site mutant (H1878A). (G) The rate constants of each mutant tested (E, F) were calculated as change in fluorescence intensity per unit time and as a ratio to the WT protein. (H-J) Replacement of RNAi-resistant GFP-Usp9X1555-1958 Wt but not GFP-Usp9X1555-1958 S3A restores spine morphogenesis, as depicted in bar graphs. Scale bar, 5 μm. 20-22 neurons were analyzed from each condition. ***p<0.001; One-way ANOVA was followed by Bonferroni post-tests. (K) Confocal images of primary cortical neurons for detection of interaction between endogenous ankyrin-G and transfecting Flag-Usp9X1555-1958 Wt or S3D or S3A with PLA. Scale bar, 5 μm. Bar graph of PLA signal (n = 16 cells from each condition). *P = 0.011; One-way ANOVA followed by Bonferroni post-tests was performed. (L) Confocal images of primary cortical neurons for detection of interaction between HA-ankyrin-G and Flag-Usp9X1555-1958 Wt or S3D with PLA. Scale bar, 20 μm. Bar graph of PLA signal (Wt: n = 15, S3D: n = 16; **p = 0.005). Two-tailed unpaired t-test was performed. (M) In situ PLA-SIM images of dendritic regions in neurons from (I). The dendritic shapes were outlined by using BFP cell-fill as a guide. Scale bar, 5 μm. (N-Q) Quantification of ratio of spines, spine density and head area expressing HA-ankyrin-G and PLA signal (n= 13 neurons from each condition). **p = 0.002, ***p < 0.001; Two-tailed unpaired t-test was performed. All data are represented as mean ± SEM. See also Figure S4 and S5.
To confirm the prediction of our homology modeling, we used in situ Proximity Ligation Assay (PLA), which labels protein-protein interactions within a 16 nm distance (Trifilieff et al., 2011). In basal conditions, PLA signals indicate the interaction of ankyrin-G1-807 and Usp9X1555-1958, but it was absent upon co-expression of phospho-null Usp9XS3A (Fig. 4C) without different expression level of each construct in HEK293T cells (Fig. S5C). Conversely, individual phosphomimetic mutations of each serine (S1598D, S1600D, S1608D) were tested, and S1608D showed significant increase in interaction with ankyrin-G. Moreover, mutagenesis of all three serine sites (Usp9XS3D) showed the most robust enhancement (Fig. 4D; Fig. S5B).
To test whether mutation on these residues alters the activity of Usp9X, we developed an in vitro DUB assay using recombinantly expressed mouse Usp9X1547-1962 alongside Usp9XS3A and a catalytically null construct, Usp9XH1878A. Despite the robust alterations in interaction observed (Fig 4C, D), no difference in Usp9X activity was observed between WT and phospho-null proteins, indicating that mutagenesis does not alter the DUB activity of Usp9X, nor the structural stability of the domain (Fig. 4E). Also, although each Usp9X phosphomimetic mutant showed enhanced interaction with ankyrin-G, their catalytic activity was unaltered in vitro (Fig. 4F).
To investigate the role of phosphorylation of Usp9X in spines, Usp9XWt or Usp9XS3A was co-transfected into cortical cultured neurons with control RNAi or shUsp9X construct. Overexpression of GFP-Usp9XS3A significantly decreased spine size and density, while overexpression of wild-type Usp9X1555-1958 rescued the effect of shUsp9X on spines (Fig. 4H-J). These data suggest that phosphorylation of Usp9X’s peptidase domain regulates spine head size and density.
The PLA method allows interacting proteins to be spatially and quantitatively visualized. To observe interaction patterns of endogenous ankyrin-G and mutated Usp9X in neurons, we overexpressed Flag-Usp9X with either Wt or S3D or S3A. Confocal images showed PLA signals increased in the dendrites and spines with Usp9XS3D (Fig. 4K). To overcome limited access of in situ PLA analysis because of a small number of puncta, HA-ankyrin-G and Flag-Usp9XS3D were co-transfected in primary cultured cortical neurons to investigate the effect of phosphorylated-three serine sites in the peptidase domain of Usp9X (Usp9XS3D) on ankyrin-G and spines. Likewise, overexpression in HEK293T cells, PLA signals increased in Usp9XS3D (Fig. 4L). Because in situ PLA allows interacting proteins to be spatially and quantitatively visualized, we developed an approach combining PLA with SIM (PLA-SIM) to investigate the spatial organization of Usp9X-ankyrin-G interaction within spines and its relationship to spine size. Interestingly, Usp9XS3D increased the ratio of ankyrin-G expressing spines co-incident with an increase in PLA positive spines (Fig. 4N) and it prompted increase of spine density (Fig. 4O). Spine heads expressing ankyrin-G are significantly larger than spine heads without ankyrin-G (Fig. 4P-Q), as previously reported (Smith et al., 2014). These changes were enhanced in the presence of the Usp9XS3D, suggesting that phosphorylation enhanced the interaction between ankyrin-G and Usp9X to regulate spine morphogenesis.
Usp9X Stabilizes Multiple ANKRD Proteins in vivo during Development
We examined the physiology of the ankyrin-G-Ups9x interaction in the context of native mouse brain, in vivo. To identify ankyrin-G-Usp9X co-expression patterns in the forebrain, we examined their immunofluorescence in the mouse primary somatosensory cortex (Bregma, 0.50 mm) (Fig. 5A). The overlap between ankyrin-G and Usp9X was strongest in dendritic areas, rather than the axon initial segment (AIS). Ankyrin-G and Usp9X co-expressed in cortical layers II-III (40.2 ± 2.5%), layer IV (33.2 ± 4.9%), and layer V (30.0 ± 0.0%) (Fig. 5B). In addition, both proteins exhibited overlapping expression pattern in the CA3 of the hippocampal region (46.5 ± 4.2%) (Fig. S6A-B).
Figure 5.

Impaired Usp9X function destabilizes ANKRD proteins. (A) Staining in the primary somatosensory cortex of 12-week old mouse brain; magnified layer II-III is zoomed in to cell level. Scale bar, 200 μm (left); 20 μm (middle); 5 μm (right). (B) The fraction of ankyrin-G or Usp9X-positive cells and quantification (n = 3) in each layer of the cortex. The graph is shown with mean values. (C) Protein levels of ankyrin-G and Usp9X in mouse cortex throughout lifespan. Representative Western blots in mouse cortex at various time periods (n = 3 for each group). (D) Levels of ANKRD and other synaptic proteins in cortex of 2- and 12-week old Usp9X+/Y or Usp9X−/Y mice. (E, F) Relative abundance of proteins in (D) (Usp9X+/Y, n = 4 and Usp9X−/Y, n = 3 in 2 weeks old; Usp9X+/Y, n = 4 and Usp9X−/Y, n = 4 in 12 weeks old). *p < 0.05; **p < 0.01; Two-tailed unpaired t-test was performed. (G) Enrichment of BD risk factors identified through GWAS (top left), de novo ASD/SZ risk factors (top right) and PSD proteins (bottom) among ANKRD proteins (n = 270); statistical significance is in red. (H) Diagram of neuropsychiatric risk genes encoding ANKRD proteins; underlined genes contain Ub-lysine sites within ANKRD. All data are represented as mean ± SEM. See also Figure S6.
Analysis of ankyrin-G and Usp9X protein levels in the mouse cortex throughout the lifespan revealed opposite directionality: while ankyrin-G expression increased throughout postnatal development, Usp9X was highly expressed early, but decreased after 2 weeks (Fig. 5C), suggesting the role of their interaction may be different early in postnatal development versus later.
To assess Usp9X’s role in maintaining ANKRD protein homeostasis, we analyzed cortical lysates from 2- and 12-week old EmxΔUsp9X conditional knockout mice lacking Usp9X in the dorsal telencephalon (Usp9X−/Y) (Fig. 5D-F). Western blotting revealed significantly reduced levels of ankyrin-G at 2 weeks, as compared to WT mice (Usp9X+/Y). Because ANKRD is a common domain among many proteins, we wondered whether the levels of other ANKRD proteins were also affected. Remarkably, we found that ankyrin-B, Shank3, and TNKS2 levels were also significantly reduced in Usp9X−/Y mice, indicating that Usp9X stabilizes multiple ANKRD proteins in vivo.
Surprisingly, however, ANKRD protein levels fully recovered in 12-week old mice. These alterations were likely specific to Usp9X substrates for two reasons: (1) a known Usp9X substrate, β-catenin (Taya et al., 1999), was also reduced, (2) GIT1 and NOTCH1/3, which lack putative ubiquitinated lysines in ANKRD, as well as GluN1, GluN2B, and GSK3β, lacking ANKRD, were not altered.
To estimate whether deubiquitination could potentially play a broader role in regulating the proteostasis of synaptic ANKRD-containing proteins, especially among those implicated in neurodevelopmental disorders, we performed bioinformatics analyses. First, to determine whether ANKRD proteins are globally enriched among neuropsychiatric risk factors, we performed gene set analysis on all ANKRD proteins in Uniprot and neuropsychiatric risk gene datasets (Table S2). We found a significant enrichment of ANKRD protein-encoding genes in genome-wide significant loci from BD GWAS and in de novo variants associated with ASD and SZ, indicating a broader role in neuropsychiatric diseases (Fig. 5G). Notably, ANKRD proteins, especially those associated with neuropsychiatric disorders, were also significantly enriched in the PSD. Among the 59 ANKRD proteins implicated in neuropsychiatric disorders, 20 have been reported to contain ubiquitinated lysine residues within the ANKRD (Hornbeck et al., 2015) (Fig. 5H; Table S3), suggesting that their proteostasis may be regulated through the UPS and DUBs.
Absence of Usp9X Causes Persistent Reduction in Dendritic Spine Density and Increase of Ankyrin-G Ubiquitination and Aggregation in Mouse Cortex
Previous studies have identified reduced spine density in Golgi-impregnated pyramidal neurons within cortex layer III of SZ and BD patients (Glantz and Lewis, 2000; Konopaske et al., 2014), as well as in patients with ID (Irwin et al., 2000). Consistent with the reduced ANKRD protein levels, spine density was significantly reduced in layer III of the frontal cortex of Usp9X−/Y mice 2 weeks postnatally as compared to Usp9X+/Y (Fig. 6A). Surprisingly, spine density remained reduced in 12-week old Usp9X−/Y mice despite recovered Usp9X-substrate protein levels. To assess ubiquitination levels of ankyrin-G across developmental periods and genotypes, we used anti-ubiquitin antibody to immunoprecipitate ubiquitinated proteins from mouse cortex homogenates and detected ankyrin-G in the immunoprecipitates by Western blotting. At 2 weeks of age, ankyrin-G ubiquitination was similar in wild-type and Usp9X−/Y mice (Fig. 6B), however, the normalized ratio of immunoprecipitated ankyrin-G by the innate expression (Fig. 5D) showed significant increase of ubiquitinated ankyrin-G in Usp9X−/Y mice (Fig. 6B). At 12 weeks, ankyrin-G ubiquitination was significantly higher in Usp9X−/Y cortex than in wild-type. On the other hand, no changes in GSK3β ubiquitination were detected, suggesting specificity. To investigate the apparent contradiction between increased levels of ankyrin-G ubiquitination in Usp9X−/Y cortex at 12 weeks, we examine the distribution of ankyrin-G. Because it has been reported that the lower level of Usp9X induced the accumulation and aggregation of ubiquitinated target substrate (Rott et al., 2011). We examined ankyrin-G distribution in the frontal cortex of Usp9X−/Y and Usp9X+/Y mice (Fig. 6C). At 2 weeks, ankyrin G immunofluorescence was reduced in Usp9X−/Y mice as compared to Usp9X+/Y mice. Surprisingly, however, at 12 weeks ankyrin-G was dramatically redistributed into large (0.1-0.2 μm2) aggregates in Usp9X−/Y mice (Fig. 6D-F), indicating that while the overall ankyrin-G levels recovered, it was mostly sequestered in large aggregates, likely due to hyper-ubiquitination. These data indicate Usp9X plays a critical role in regulating ANKRD protein homeostasis and dendritic spine morphogenesis during postnatal development, with effects persisting through adulthood.
Figure 6.

Impaired Usp9X function alters cortical spine development. (A) Golgi-Cox staining of layer III pyramidal apical dendrites in 2- and 12-week old Usp9X+/Y and Usp9X−/Y mice. Scale bar, 5 μm. 20 dendrites were analyzed from 3 brains per each group. ***p < 0.001; Two-tailed unpaired t-test was performed. (B) Representative Western blots of co-immunoprecipitation experiments of ankyrin-G or GSK3β with α-Ubiquitin (α-Ub). Intensity of Western blot of immunoprecipitated ankyrin-G or GSK3β by α-Ub (Fig. 6B) was normalized by the expression levels of both proteins (Fig. 5D) (Usp9X+/Y, n = 4 and Usp9X−/Y, n = 3 in 2 weeks old; Usp9X+/Y, n = 4 and Usp9X−/Y, n = 4 in 12 weeks old). *p < 0.05; Two-tailed unpaired t-test was performed. IgG, control IgG; IP, immunoprecipitation; WB, Western blotting. (C, D) Immunofluorescence staining in layer III of the primary somatosensory cortex reveals ankyrin-G aggregates (intensity highlighted in reverse images). Scale bar, 10 μm. (E, F) Distribution of ankyrin-G particle sizes in (B) (n = 4 brains per each group). **p < 0.01; ***p < 0.001; Two-way ANOVA followed by Bonferroni post-tests. All data are represented as mean ± SEM. See also Figure S6 and S7.
Behavioral Analysis of Usp9X−/Y Mice
We have shown that the absence of Usp9X during early development disrupted the stabilization of multiple ANKRD proteins and spine morphology. We wondered whether these biochemical and anatomical alterations were also associated with behavioral phenotypes relevant to neurodevelopmental disorders, and if so, whether they were similar to phenotypes of mice with deletions of genes encoding ANKRD protein. Shared phenotypic domains among such knockout mice include abnormalities in anxiety and locomotor activity levels (Hung et al., 2008; Peca et al., 2011; Schmeisser et al., 2012; Wang et al., 2011; Won et al., 2012; Zhu et al., 2017). We thus investigated these behavioral phenotypes in Usp9X−/y mice. First, Usp9X−/y mice have normal body weight and metabolism (Fig. S8A-C). Usp9X−/y mice showed higher activity levels during testing compared to controls (Fig. 7A). Consistent with hyperactive behavior, Usp9X−/y mice also displayed significant increase of the number of entries in each arm (Fig. 7C) and the number of chamber transitions (Fig. 7E) by the elevated plus maze and light/dark box tests respectively. Less immobilization was also observed in a forced swim test (Fig. 7F).
Figure 7.

Impaired Usp9X function alters behaviors, and occurs in neurodevelopmental disorder patients. (A) Open field test for 10 min. Usp9X+/Y, n = 26; Usp9X−/Y, n = 16. Two-tailed unpaired student’s t-test, t(40) = 5.696, ***p < 0.001. (B-C) Elevated plus maze test for 10 min. Time in open arm (B) and the number of entries in each arm (C) are shown by graphs. Usp9X+/Y, n = 16; Usp9X−/Y, n = 15. Two-tailed unpaired student’s t-test, t(29) = 2.365 for time in open arm; t(29) = 3.855 for the number of entries, *p < 0.05; ***p < 0.001. (D-E) Light/dark box test for 10 min. Time in light space (D) and the number of light chamber transitions are shown by graphs. Usp9X+/Y, n = 16; Usp9X−/Y, n = 15. Two-tailed unpaired student’s t-test, t(29) = 2.855 for time in light space; t(29) = 3.281 for the number of transitions, **p < 0.01. (F) Immobility was assessed in adult mice in the forced swim test for 4 min. Usp9X+/Y, n = 11; Usp9X−/Y, n = 8. Two-tailed unpaired student’s t-test, t(17) = 2.597, *p < 0.05. All data are represented as mean ± SEM. (G) Co-immunoprecipitation of HA-ankyrin-G1-807 with Flag-Usp9X1555-1958 WT, Q1573N, L1693W, G1890E or S3D from HEK293T cells. (H) Cleavage of a fluorogenic substrate by the peptidase domain of wild type, active site mutant His-Usp9X1547-1962 H1878A, and disease-associated USP9X mutants. (I) Homology modeling of human Usp9X. The catalytic pocket (red square) and “thumb” and “finger” domains are indicated. Human mutations were color coded and shown in inset boxes below, with van der Waals radii. Residues likely disrupted by steric hindrance are in blue. Dashed lines indicate predicted steric clash or alterations in hydrogen bonding. See also Figure S8.
Mutations in Humans in the Usp9X Catalytic Domain Impair Its Activity and Interaction with Ankyrin-G
Because mutations in USP9X occur in patients with ID, ASD, and epilepsy (Johnson et al., 2019), we asked whether such mutations in the catalytic domain affected interaction with ankyrin-G and/or DUB activity. We tested recombinant proteins for their ability to interact with ankyrin-G. Mutations Q1573L, detected in a patient with developmental disability (DD) and speech delay (Fig. S8F), and L1693W, detected in a patient with ID, DD, and ASD (Reijnders et al., 2016), displayed normal interaction with ankyrin-G but were catalytically inactivity. Remarkably, the G1890E mutation from two patients with ID, DD, speech delay, and ASD (Fig. S8F) had normal catalytic activity but impaired interaction with ankyrin-G (Fig. 7E-F).
A homology model of the human Usp9X, based on known DUB structures, suggests that Q1573 and L1693 lie within tight internal hydrophobic cores, and mutations in these residues result in steric clashes within the peptidase domain (Fig. 7G). This is consistent with an observed increase in thermodynamic stability (Fig. S8H), suggesting reduced structural flexibility, a finding that likely underlies alterations in catalytic activity. Conversely, the surface exposed mutation, G1890E, does not affect protein structure or catalytic activity in vitro, but disrupts the interaction with ankyrin-G, suggesting that this mutation may produce effects through altered protein-protein interaction.
DISCUSSION
Regulation of ANKRD Proteins in Spines by a DUB
UPS is an important mechanism for activity-dependent PSDs turnover and synaptic remodeling (Amerik and Hochstrasser, 2004). However, target proteins of DUBs, which counterbalance the role for the UPS, are poorly understood in PSDs. Here, we found 20 ANKRD-containing proteins encoded by psychiatric risk genes that contain ubiquitinated lysine residues in their ANKRDs (Fig 5H). Among these, proteins encoded by the ANK1/2/3 and SHANK1/2/3 genes are present in the PSD proteome (Table S3) (Bayes et al., 2012). The proteins encoded by these genes contain D-boxes and E3 ligase recognition motifs. Notably, all of these D-boxes are highly conserved in humans and rodents. Currently, eight DUBs have been identified in the PSD proteome. Of these, Uchl1 and Usp5/Leon have been shown to affect spines (Cartier et al., 2009; Wang et al., 2017). Our findings suggest that it would be worth investigating the synaptic roles of all postsynaptic DUBs.
ANKRD Protein Deubiquitination in Synaptic Structural Plasticity
Deubiquitination is a general mechanism of postsynaptic regulation, alterations of which may cause synaptic pathology. Indeed, we show that deubiquitination by one DUB can control the proteostasis of multiple neurodevelopmental disorder-linked proteins in the PSD that share one of the most common protein-protein interaction domains, the ANKRD. While anecdotal examples implicate genes encoding ANKRD proteins, including ankyrin-G, ankyrin-B, and Shank3, in neurodevelopmental disorders, our bioinformatics analysis shows that ANKRD proteins are enriched as a group among risk factors for neurodevelopmental disorders. Absence of Usp9X in mice drastically reduces the levels of multiple ANKRD proteins, indicating that Usp9X is likely the major DUB regulating their stability during this developmental period. Hence mutations in USP9X could impair the homeostasis of multiple important proteins in convergence among several neurodevelopmental risk factors, whereby one risk factor regulates the stability of many other risk factors.
The relative specificity of Usp9X for its ANKRD targets, in addition to its known target β-catenin, may lie in a structural recognition motif. β-catenin has previously been identified as a substrate for Usp9X, and the Usp9X-binding region was found to lie within the armadillo repeats 3-10 (Taya et al., 1998). An armadillo repeat is about 40 residues in length and forms an α solenoid structure composed of repeating pairs of α-helices. Structural similarities between β-catenin, ankyrin-G and ANKRD proteins, such as the solenoid superstructure and the presence of a positively charged inner groove, may underlie the binding properties of Usp9X to a subset of its substrates. However, this might not extend to all of its substrates because known Usp9X-interacting proteins AF6, MARK4, and NUAK1 do not interact through structural motifs (Al-Hakim et al., 2008; Taya et al., 1999).
The Nanoscale Architecture of a Deubiquitination Pathway in Spines
PLA-SIM provided an unprecedented insight into the nanoscale spatial organization of a deubiquitination pathway in spines, allowing the examination of protein-protein or enzyme-substrate interactions in situ at the nanoscale level. While SIM has been instrumental in showing that ankyrin-G is localized to perisynaptic regions and in spine necks (Smith et al., 2014) here we show that its interaction with Usp9X occurs at spatially restricted sites significantly smaller than their optical colocalization, primarily in a region of the spine head facing the interior of the spine, and not in the neck. PLA-SIM also allowed for the first time an accurate quantitative analysis of the relationships between deubiquitinase-substrate interaction sites and spine architecture.
Phosphorylation of Usp9X Enhances the Interaction with Ankyrin-G
Our results indicate that induced Usp9X phosphorylation within the peptidase domain strengthens its interaction with ankyrin-G. Upregulation of this Usp9X-ankyrin-G interaction appears to facilitate the deubiquitination of ankyrin-G, inhibiting proteasomal degradation and stabilizing ankyrin-G levels within synapses. Phosphorylation is a well-documented mechanism underlying alterations in protein-protein interactions involving ANKRD proteins. It has been reported that phosphorylation motifs of target interactors are important for the modulation of ankyrin-G interaction (Garver et al., 1997; Jenkins et al., 2001; Zhang et al., 1998). In addition, Usp9X is a major regulator of the TGFβ signaling pathway (Dupont et al., 2009; Johnson et al., 2019), controlling neuronal axonal growth and dendritic development in response to TGFβ stimulation (Stegeman et al., 2013). TGFβ receptors are S/T kinase receptors and the activated TGFβ receptor signals through various downstream signaling cascades such as the Smad family, mitogen-activated protein kinase, phosphoinositide 3-kinase, AKT, extracellular signal-regulated kinase, JUN N-terminal kinase, and nuclear factor-κB (Akhurst and Hata, 2012). Phosphorylation of key interacting partners may fine-tune the role of ankyrin-G on synapse formation in response to extracellular signalings, such as TGFβ. The detrimental effect of Usp9X knockout on the expression of a range of ANKRD proteins suggests that this mechanism may be shared with other psychiatric risk factors
Persistent Impact of Usp9X Loss on the Brain
We revealed the regulation of ANKRD protein homeostasis by Usp9X overlapping with synapse maturation, indicating that Usp9X is the major DUB regulating their homeostasis during this period. We observed that Usp9X conditional knockout mice had reduced levels of ANKRD proteins at 2 weeks of age (Fig. 5D-F). However, the persistence of the impact on spines into adulthood indicates that reduced expression level (44.6 ± 1.8 %) of Usp9X in 12 weeks old (Fig. 5C) is sufficient to cause detrimental effects that propagate into adulthood, even when overall protein levels have recovered (Fig. 5D-F).
These deleterious effects in adults, despite restored protein levels, may relate to abnormal protein ubiquitination and aggregate formation, reminiscent of aggresomes in Lewy bodies upon downregulation of Usp9X (Rott et al., 2011). While ubiquitination level of Usp9X-targeted protein returned to normal, these ubiquitinated molecules did not degrade correctly and instead accumulated as aggregates. This model is supported by reports that polyubiquitin chains trigger the formation of fibrillar aggregates (Morimoto et al., 2015). While protein aggregation has been implicated in neurodegenerative disorders (Bousman et al., 2010), its contribution to neurodevelopmental disorders remains to be explored.
These biochemical and structural changes in Usp9X−/y mice likely underlie behavioral alterations. Indeed, here we show that Usp9X KO mice display an abnormality in locomotor activity levels. Usp9X−/y mice displayed hyperactivity in open field, elevated plus maze, light/dark box, and forced swim tests. Likewise, conditional deletion of Ank3 in adult forebrain pyramidal neurons produced hyperactivity (Zhu et al., 2017) consistent with the behavioral phenotypes of Usp9X−/Y mice. Notably, a recent publication has shown cognitive deficits in male Usp9X−/y mice (Johnson et al., 2019). Overall, these data show that Ups9X mice display behavioral phenotypes are consistent with Usp9X and ankyrin-G functioning in the same pathway.
Deubiquitination and Neurodevelopmental Disorders
Mutations in USP9X occur in patients with ID, ASD, and epilepsy, but the effects of these mutations on Ups9X protein function have not yet been evaluated directly. By focusing on mutations within the catalytic domain, here we show that they affected its DUB activity, or, more interestingly, its interaction with ankyrin-G. Remarkably, these mutations were detected in patients with DD, speech delay, and ASD, which overlaps with clinical cases linked to ANKRD protein mutations, such as SHANK2, SHANK3, and ANK2. Abnormal deubiquitination of ANKRD proteins may thus be a pathogenic mechanism for USP9X-associated neurodevelopmental disorders.
Among the eight DUBs identified in the PSD proteome, seven are implicated in neurological disorders: UCHL1 is implicated in Parkinson's disease, Alzheimer's disease, and cerebellar ataxia; mutations in USP14 result in NMJ synaptic defects in ataxia (Nijman et al., 2005); Usp15 is implicated in speech and communication disorders; mutations in USP9X is implicated in ID and seizure disorders (Reijnders et al., 2016); Usp5 is connected to neuropathic pain (Garcia-Caballero et al., 2014); Atxn2 and Atxn10 are implicated in Parkinson's disease (Schule et al., 2017; Zhou et al., 2018). Our data may thus provide a general mechanism of pathogenesis in DUB-associated disorders.
Taken together, our findings demonstrate that deubiquitination during a full life span of synapse maturation is essential for a correct developmental trajectory of cortical synapses, and its alteration results in behavioral, and clinical abnormalities.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Peter Penzes (p-penzes@northwestern.edu).
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Usp9X conditional knockout mice were generated by crossing Emx1-Cre heterozygous males (Iwasato et al., 2000) with Usp9XloxP/loxP female mice. Detailed information for generation of Usp9XloxP/loxP mice was described previously (Stegeman et al., 2013). All mouse breeding was performed under the ethical clearance approved by Griffith University Animal Ethics Committee. As Usp9X is located on the X chromosome, male offspring that inherit the Emx1-Cre allele lacked Usp9X in the telencephalon and derived cortex and hippocampal structures (referred to as Usp9X−/Y). Cre-negative male mice were used as controls (referred to as Usp9X+/Y). Female mice were not analyzed. Potential Usp9X−/Y mice were identified by two PCRs on extracted tail-tip DNA targeting Sry and Cre genes, using REDExtract-N-Ampa Tissue PCR Kit (Sigma; Cat# - XNAT). The mice were housed together with mixed-genotype home cages until testing for all assays. Mice were kept within an SPF barrier area in an air-conditioned room on a 12:12 h light/dark schedule, under constant conditions of temperature and humidity. Food and tap water (membrane filterpurified and autoclaved water) were provided ad libitum. 12-14 weeks old mice were used for behavioral analyses. 2 or 12 weeks old mice were used for immunohistochemistry, brain lysate analyses, and Golgi-Cox staining. All experiments were performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and were carried out in accordance with The University of Queensland Institutional Biosafety committee.
Neuronal cell culture and transfection
Dissociated cultures of primary cortical neurons were prepared from P0 C57BL/6J (The Jackson Laboratories) pups. All procedures were approved by Northwestern University’s Animal Care and Use Committee and were in compliance with the National Institutes of Health standards. Brains were dissected in ice-cold Leibowitz’s L-15 media with penicillin/streptomycin, and cortical tissue isolated, digested with 0.25% trypsin-EDTA solution at 37°C, and mechanically dissociated in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 1.4 mM L-glutamine, and 6.0 g/L glucose. Cortical neurons were plated at 320,000 cells per 18 × 18 mm coverslip or 640,000 cells per six-well plates precoated with 50 μg/ml poly-D-lysine (Sigma) and 2 μg/ml laminin (Sigma). Neuronal cultures were maintained at 37°C in 5% CO2 in Neurobasal media supplemented with B27 and GlutaMax-1 and penicillin/streptomycin. Neurons were transfected at DIV14 with Lipofectamine 2000, providing a transfection efficiency of 4 μg total DNA and Lipofectamine 2000 (Invitrogen) were diluted in DMEM + HEPES (10 mM), mixed thoroughly together, and incubated for 20-30 minutes at 37°C before adding to cultured cells. Following transfection, neurons were supplanted in antibiotic-containing feeding media containing half conditioned and half fresh media and allowed to express constructs for 4 days.
METHOD DETAILS
Plasmids
3XHA-ankyrin-G (EX-Mm25668-M06) and 3XFlag-Usp9X (EX-Mm24322-M12) were purchased from GeneCopoeia. RNAi constructs were purchased from Origene, in the pRFP-C-RS vector with a turboRFP element to enable identification of transfected cells. The target sequences used were AGA AGA CCA TAC ACT GGC AAT CCT CAG TA for Usp9X. Four domain fragments of ankyrin-G (amino acids 1–807, 808–1475, 808–1961, 1476–1961) were amplified from 3XHA-ankyrin-G. Peptidase domain fragment of Usp9X (amino acids 1555-1958) were amplified from 3XFlag-Usp9X. GFP-tagged ankyrin-G and GFP-Usp9X1555-1958 were amplified from 3XHA-ankyrin-G and 3XFlag-Usp9X1555-1958, and subcloned into pEGFP-N2 (#6801-1; Clontech) using the Infusion ligation independent cloning kit (Clontech). His-Usp9X1547-1962 was amplified from 3XFlag-Usp9X and subcloned into SSP1 cut pMCs-G53. 3XHA-ankyrin-G1-807 Muta;b, GFP-ankyrin-G1-807 Muta;b, GFP-ankyrin-G Muta;b, 3XHA-ankyrin-G1-807 K39A, K260A, K268A, K260;268A, K39;260;268A, and 3XFlag-Usp9X1555-1958 S1593;1600;1609A (S3A) and His-Usp9X1547-1962 S3A, S1593D, S1600D, S1609D, S1593;1600;1609D (S3D), Q1573L, L1693W, G1890E, H1878A were generated using QuickChange Site-Directed Mutagenesis Kit (Agilent) as per the manufacturer’s instructions. 3XFlag-Usp9X1555-1958 S1593D, S1600D, S1609D, S3D, Q1573L, L1693W, G1890E was amplified from His-Usp9X1547-1962 mutants (using Usp9X 1555-1958 forward and reverse primers) and subcloned using the Infusion ligation independent cloning kit (Clontech). All constructs were verified by sequencing (Genewiz).
SIM imaging and analysis
Imaging and reconstruction parameters were empirically determined with the assistance of the expertise in the Nikon Imaging Center at Northwestern. The acquisition was set to 10MHz, 14 bit with EM gain and no binning. Auto exposure was kept between 100-300ms and the EM gain multiplier restrained below 300. Conversion gain was held at 1x. Laser power was adjusted to keep LUTs within the first quarter of the scale (<4000). Reconstruction parameters (0.96, 1.19, and 0.17) were kept consistent across experiments and imaging sessions. Resolution of images was validated with full-width half maximum (FWHM) measurements of a small structure within the image. For each spine analyzed, the single-plane in which the spine head was in focus, based on the cell fill, was chosen for analysis. Using Image J software, each spine head was outlined manually in the channel of the cell fill to detect the area. Fluorescence intensities in spines were measured in ImageJ with a linescan across the spine head. Gaussian fits of these intensity profiles were performed in GraphPad Prism. Based on the localization of ankyrin-G within the spine head, dendritic spines were classified into one of three groups. The 1 ANK group included spines with only one punctum of ankyrin-G nearby (adjacent-to or just within the edge of the Usp9X puncta). The 2+ ANK group contained all dendritic spine heads containing two or more ankyrin-G within or near the Usp9X as long as those ankyrin-G puncta within the Usp9X were confined to the edges. The Overlap group contained spines in which ankyrin-G puncta were entirely enveloped within the Usp9X, with close to 100% colocalization. Whether or not ankyrin-G was present within the spine neck was assessed based on the presence or absence of fluorescent signal within the confines of the turboRFP- or BFP-derived spine neck outline. ankyrin-G and Usp9X puncta within the spine head were manually outlined and the area recorded. A 100μm dendritic region was selected and puncta counts were made; puncta smaller than 0.065 μm2 were excluded from analysis. Visual assessment of fluorescence intensity was used to delineate separate or connected puncta. Puncta were considered separate if a region of decreased intensity was readily visible. The number of puncta within the spine head was quantified manually and recorded. Whether or not ankyrin-G was present within the spine neck was assessed based on the presence or absence of fluorescent signal within the confines of the turboRFP- or BFP-derived spine head outline. Colocalization highlighter images and Manders’ colocalization coefficients were determined in ImageJ, after thresholding. Three-dimensional reconstructions of dendritic spines were created in the Nikon Elements Software by merging of the two other channels.
Immunocytochemistry
Cells were fixed for 10 min in 4% formaldehyde in PBS at 4°C. After 3 times PBS washing for 5 min, fixed neurons were permeabilized and blocked simultaneously in PBS containing 1% bovine serum albumin (BSA) and 0.3% Triton-X-100 for 1 hr at room temperature. Primary antibodies were added in PBS containing 1% BSA and 0.3% Triton-X-100 overnight at 4 °C, followed by 3 × 10 min washes in PBS. Secondary antibodies were incubated for 1 hr at room temp, also in 1% BSA and 0.3% Triton-X-100 in PBS. Three further washes (5 min each) were performed before coverslips were mounted using Fluorescent Mounting Medium (Dako).
Confocal microscopy
Confocal images of immunostained neurons were obtained with a Nikon C2+ confocal microscope. Images of neurons were taken using the 63x oil-immersion objective (NA = 1.4) as z-series of 8-10 images, averaged 2 times, taken at 0.4 μm intervals, with 1024×1024 pixel resolution. Detector gain and offset were adjusted in the channel of cell fill (turboRFP or mCherry) to include all spines and enhance edge detection due to variable levels of plasmid expression.
Confocal image analysis
Colocalization highlighter images (ankyrin-G and Usp9X) and Manders’ colocalization coefficients were determined in ImageJ, after thresholding, with the use of the MBF set of plugins. Background-subtracted images were thresholded, regions along dendrites were outlined, and total immunofluorescence intensity for each cluster was measured automatically. For dendritic spine quantification, two-dimensional, background-subtracted maximum projection reconstructions of images for dendritic spine morphometric analysis (area), and quantification of spine linear density (# of spines / 10 μm dendritic length) were performed using Image J software. A threshold was applied to the maximum projection images to include all detectable spines, and regions along dendrites containing dendritic spines were manually traced to enclose spines but not the dendritic shaft or other structures. Dendritic spine “objects”, restricted to objects with areas greater than 0.1 μm2, were automatically detected, and the area of each spine head was measured. Two dendritic branches, (approximately 100 μm) of each neuron were analyzed. Only spines on secondary and tertiary apical dendrites were measured to reduce variability. Cultures that were directly compared were stained simultaneously and imaged with the same acquisition parameters.
Immunofluorescence histochemistry
For immunostaining, 12-week old mice were injected with EUTHASOL (Virbac; 710101) of 5 μl/g and were perfused first with filter-sterilized PBS and then with 4% paraformaldehyde in PBS. All procedures were approved by Northwestern University’s Animal Care and Use Committee and were in compliance with the National Institutes of Health standards. The brain was removed, incubated for 4 hours at 4°C in 4% paraformaldehyde, and dehydrated for 48 hours at 4°C in PBS containing 30% sucrose. Free-floating serial sections (60 μm) for immunohistochemical staining of ankyrin-G and Usp9X were prepared with a cryostat (Leica). Sections were washed with PBS three times for 10 min before consecutive incubations at room temperature with PBS containing 0.3% Triton X-100 and 1% BSA for 30 min. The sections were then incubated overnight at 4°C with mouse anti-ankyrin-G (NeuroMab, 1:200) and rabbit anti-Usp9X (abcam, 1:200), washed with PBS, and incubated for 1 hour at room temperature with Alexa Fluor 488-conjugated goat anti-mouse and Alexa Fluor 568–conjugated donkey anti-rabbit secondary antibodies (Invitrogen, 1:2,000), with all antibodies being diluted in PBS containing 1% BSA and 0.3% Triton X-100. After washed with PBS twice, DAPI (0.25 ng/ml) solution applied for nucleic acids staining. The sections were finally mounted in Fluorescent Mounting Medium.
Coimmunoprecipitation assays
Coimmunoprecipitation assays were performed as described in Smith et al., 2014 with modifications. Briefly, 3-week old mouse cortex was dissected and homogenized in pull-down buffer (50 mM HEPES pH 7.5, 1% triton-X-100, 150 mM NaCl, 1 mM EDTA, 1mM AEBSF with protease inhibitor cocktail (Roche) and phosphatase inhibitor (Sigma) and solubilized for 1 hour at 4°C. Solubilized material was centrifuged at 13,200 rpm for 25 min at 4°C and the supernatant was precleared with protein A/G beads for 1 hour. Proteins were immunoprecipitated with 3 μg of antibody overnight at 4°C, followed by 1 hour incubation with protein A/G beads. Beads were then washed extensively and bound complexes were analyzed by SDS-PAGE and western blotting. For assays of overexpressed ankyrin-G and Usp9X constructs, HEK293T cells were transfected with 3XHA-ankyrin-G and 3XFlag-Usp9X using PEI transfection reagent (Sigma) in 100-mm dishes. Cells were treated in lysis buffer (20 mM Tris pH 7.5, 1% triton-X-100, 150 mM NaCl, 1 mM EGTA, 1mM AEBSF with protease inhibitor cocktail) and equal protein aliquots of cell lysates (300 μg) were incubated with 3 μg of antibody overnight at 4 °C followed by the addition of 30 μl of slurry Sepharose A/G for 4 hr at 4°C. The complexes were washed three times with washing buffer. Samples were then denatured (5 min at 95 °C). The immunoprecipitated proteins were separated with a 10% SDS-PAGE and western blotting.
Protein production
All mutants were produced as follows; pMCSG53-USP9Xpeptidase and mutagenized constructs were used to transform BL-21 chemically competent cells (NEB) which were plated on selective plates. Single colonies were used to inoculate overnight starter cultures in LB broth (Thermo-Fisher) supplemented with ampicillin. TB broth (KH2PO4, K2HPO4 pH 7.5, 1.2% (w/v) tryptone, 2.4% (w/v) yeast extract, 0.5% (v/v) glycerol) supplemented with ampicillin, 0.1% antifoam B and 1% (w/v) glucose was inoculated 1:50 with starter culture and grown to OD600 1.2. Cells were chilled on ice and ampicillin was replenished before induction with 0.25 mM IPTG overnight at 25°C. Cells were lysed by pressure-driven homogenization (2 times at 15,000 PSI) in binding buffer (10 mM Tris pH 8.3, 500 mM NaCl, 1 mM BME) supplemented with Complete, EDTA-free Protease Inhibitor Cocktail (Roche Applied Science), and kept at 4°C throughout purification. The lysate was clarified by centrifugation (21,000 × g, 30 mins) and the supernatant was filtered through a 0.45 μm filter. Purification was performed by AKTAxpress on a nickel affinity column charged with Ni2+ and equilibrated in binding buffer. The resin was washed with binding buffer supplemented with 25 mM imidazole. His-tagged protein was eluted with binding buffer containing 500 mM imidazole, followed by size exclusion chromatography on a Superdex 200 (GE Healthcare). Protein containing fractions, indicated by A280, were pooled and buffer exchanged to deubiquitination assay buffer (50 mM Tris pH 7.6, 150 mM NaCl, 1 mM TCEP) and concentrated using a centrifugal filter (30 kDa MWCO). Protein concentration was calculated by absorbance at 280 nm and coomassie gels revealed greater than 95% purity. Proteins were flash-frozen in liquid nitrogen and stored at −80°C until use.
In vitro deubiquitination (DUB) assay
His-Usp9X-peptidase proteins were prepared as a 2x (2 μM) stock solution in DUB assay buffer. All stock solutions were compared by coomassie gel to confirm equal assay concentrations. The fluorogenic substrate, ubiquitin-7-amido-4-methylcoumarin (Ub-AMC, Sigma U2258), was prepared as a 2x (1 μM) stock solution in DUB assay buffer. Protein and substrate were mixed 1:1 in black 96 well assay plates and immediately read by C5 cytation plate reader at 380/460 (ex/em) every two minutes for one hour. Conditions were tested in quadruplicate with 3-7 replicates. Data were normalized to maximum (1 mM Usp9X-peptidase domain) and minimum (no DUB) controls. Data points were fit to a curve and activities calculated from the change in fluorescence intensity between time points 0 and the time point where half-maximal inhibition was observed.
Homology modeling
Human USP9X/1555-1975 was aligned to multiple DUB templates (PDB IDs;5UQX_A, 5UQV_A, 3M99_A, 3MHH_A, 4FIP_A, 4ZUX_U, 5K16_A, 5K1A_A, 5L8H_A, 5K1B_A, 5L8W_A, 5CVM_A) using Maestro multiple sequence viewer (Schrödinger, LLC). A homology model was generated and characterized using Maestro Prime module (Schrödinger) using template-based modeling, with unaligned regions modeled ab initio through energy minimization. AnkG ANKRD homology model was generated as above through alignment with the AnkB ANKRD structure (PDB; 5Y4D). Homology model image generation, mutagenesis, and structural alignments were performed using PyMol V1.8.2.0 (Schrödinger, LLC).
Proximity ligation assay (PLA)
Cortical neurons plated on coverslips were transfected with 3XHA-ankyrin-G and 3XFlag-Usp9X at DIV14. At 4 days post-transfection, the transfected cells were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C. HEK 293T cells were transfected with 3XHA-ankyrin-G1-807 and 3XFlag-Usp9X using PEI transfection reagent. At 24 hr post-transfection, the transfected cells were fixed. All procedures were followed by the manufacturer’s instructions (Sigma). To confirm the transfected cells, mouse 488 conjugated-Flag antibody and rabbit 568 conjugated-HA antibody were used. To detect PLA signal, far-red reagent (Sigma, DUO92013) was used.
Brain lysate analysis
Mice were euthanized by cervical dislocation. Brains were extracted and immediately flashed frozen using liquid nitrogen. Frozen tissue was stored at −80°C until further use. All procedures were approved by the Griffith University Animal Ethics Committee.
Golgi-Cox staining
We used a modified Golgi staining method adapted from the protocols used by Bayram-Weston et al., 2016. Briefly, Usp9X+/Y or Usp9X−/Y mice were sacrificed and then perfused with 4% PFA in PBS. The brain was removed, incubated for 24 hours at 4°C in 4% paraformaldehyde, and dehydrated for 48 hours at 4°C in PBS containing 30% sucrose. Next procedures were followed by the manufacturer’s instructions (FD Rapid GolgiStain Kit). Mounted samples which cut 120 μm thickness sections were imaged on a microscope (LSM 510; Carl Zeiss) using a 63X oil lens with AxioCam. For apical dendritic spine quantification, only the first branch of apical dendrite over 30 μm away from cell body was concerned for the analysis of spine linear density (# of spines/10 μm dendritic length).
Behavior analysis
All statistical analyses for the behavioral experiments were performed using two-tailed unpaired student’s t-test.
Open field test
Mice were placed in an arena measuring 30 cm × 30 cm × 30 cm and recorded using an overhead camera. The analysis was done using Ethovision software (Noldus). The software provides the total distance traveled. The locomotor activity recorded for the first 10 min was analyzed.
Elevated Plus Maze
Mice were recorded for 10 minutes on an elevated platform (1 m from the ground, with 4 arms measuring 30 cm × 5 cm). Two of the platforms were covered with 30 cm walls. The analysis was done using Ethovision software (Noldus).
Light/Dark Box Test
The light/dark emergence test for the Usp9X+/y and Usp9X−/y mice is conducted in four individual activity monitors (27.9 × 27.9 cm) with three 16 beam infrared arrays (MED Associates, Inc., Georgia, VT, USA), each containing a darkened acrylic insert that was penetrable by infrared light and sheltered half the arena. To start the test the mouse is placed into the dark compartment and each mouse was recorded for 30 minutes. The activity was measured as counts of infrared beam breaks as well as the duration spent being mobile or stationary in each compartment. The results are presented in the second 10-20 minutes bin.
Forced Swim Test
The apparatus used in the forced swim test was a clear round container (20 cm high × 14 cm diameter) with a column of water (16 cm deep) maintained at 25 °C. Each mouse was placed in the container for 6 min and recorded using a USB digital camera and recording software. The activity was measured only from 2 min to 6 min and scored using the mobility threshold settings within the Ethovision software by measuring the percentage change in area of the tracked object from one sample to the next. Immobility was defined as less than 5% movement using these settings and was validated by a human observer.
Phenomaster metabolic readings
During the period of recording in the Phenomaster (TSE Systems), several metabolic readings were taken of the mice between the genotypes/individual mice for 24 hours.
Gene set analysis
Ankyrin repeat domain containing proteins were retrieved from Uniprot (http://www.uniprot.org/). A list of genomewide significant BD risk factor was compiled from published GWAS (Hou et al., 2016; Iossifov et al., 2014). de novo variants from exome sequencing studies in ASD and SZ were acquired from published supplementary material (De Rubeis et al., 2014; Fromer et al., 2014; Iossifov et al., 2014). Neuropsychiatric risk gene sets from de novo exome sequencing data were filtered to only include variants predicted to affect protein sequence (i.e. missense, frameshift, nonsense, splice-site, and start lost mutations). A hypergeometric test was used to test for statistical significance of each overlap.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were performed with GraphPad Prism8. A two-sample comparison was performed using unpaired Student’s t-test, and multiple comparisons were made using two-way ANOVA or one-way ANOVA followed by a Bonferroni test. Bar graphs are displayed as mean ± SEM. Significance was shown with an asterisk represents *p<0.05, **p<0.01, ***p<0.001.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study. All data are available from the Lead Contact upon request.
Supplementary Material
Table S1: Related to Figure 1. The prediction of D-box and KEN box results for the mouse ankyrin-G (National Center for Biotechnology Information accession number NM_146005) are shown.
Table S3: Related to Figure 5. The ubiquitinated lysine sites of ANKRD in ANKRD containing psychiatric risk genes.
Table S4: Related to Figure 1, 3, 4, and 7, Figure S1, S3, S5, and S8. Sequences of oligos used this study.
Table S2: Related to Figure 5. The list of ANKRD genes from Uniprot (Homo sapiens).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-AnkG | NeuroMab Facility | Cat#75-146; RRID: AB 10673030 |
| Goat polyclonal anti-AnkG (P-20) | Santa Cruz Biotechnology | Cat#sc-31778; RRID: AB 2289736 |
| Rabbit polyclonal anti-Usp9X | Abcam | Cat#ab19879; RRID: AB 470300 |
| Goat polyclonal anti-PSD95 | Abcam | Cat#ab12093; RRID: AB 298846 |
| Chicken polyclonal anti-MAP2 | Abcam | Cat#ab15452; RRID: AB 805385 |
| Mouse monoclonal anti-Flag (clone M2) | Sigma-Aldrich | Cat#F1804; RRID: AB _262044 |
| Mouse monoclonal anti-HA (clone HA-7) | Sigma-Aldrich | Cat#H3663; RRID: AB 262051 |
| Rabbit polyclonal anti-HA | Enzo Life Sciences | Cat#ADI-MSA-106; RRID: AB _10615792 |
| Mouse monoclonal anti-β-actin (clone AC-74) | Sigma-Aldrich | Cat#A2228; RRID: AB 476697 |
| Rabbit polyclonal anti-phosphoserine | Abcam | Cat#ab9332; RRID: AB 307184 |
| Chicken polyclonal anti-mCherry | Abcam | Cat#ab205402; RRID: AB 2722769 |
| Goat polyclonal anti-mCherry | SICGEN | Cat#AB0040; RRID: AB_2333092 |
| Mouse monoclonal anti-Flag-488 (clone M2) | Abcam | Cat#ab117505; RRID: AB 10972518 |
| Rabbit polyclonal anti-HA-568 | Synaptic System | Cat#245 003C3; RRID: AB 2619954 |
| Rabbit polyclonal anti-AnkB (H-300) | Santa Cruz | Cat#sc-28560, RRID: AB_2242828 |
| Rabbit polyclonal anti-Shank3 | LSBio | Cat#LS-C290822 |
| Mouse monoclonal anti-pan-Shank | NeuroMab | Cat#N23B/49, RRID: AB_10672418 |
| Rabbit polyclonal anti-TNKS2 | Abcam | Cat#ab155545 |
| Rabbit monoclonal anti-TRPV5 | Abcam | Cat#ab137028 |
| Rabbit polyclonal anti-β-catenin | Cell Signaling | Cat# 9562, RRID: AB_331149 |
| Rabbit polyclonal anti-GIT (CAT1) | Abcam | Cat# ab37588, RRID: AB_2190720 |
| Rabbit polyclonal anti-Notch1 | Abcam | Cat# ab27526, RRID: AB_471013 |
| Rabbit polyclonal anti-Notch3 | Abcam | Cat# ab23426, RRID: AB_776841 |
| Rabbit monoclonal anti-NMDAR1 | Abcam | Cat# ab109182, RRID: AB 10862307 |
| Rabbit monoclonal anti-NMDAR2B | Abcam | Cat#ab183942 |
| Rabbit monoclonal anti-GSK-3β (27C10) | Cell Signaling | Cat# 9315, RRID: AB_490890 |
| Rabbit recombinant anti-Ubiquitin (EPR8830) | Abcam | Cat# ab134953, RRID: AB_2801561 |
| Mouse monoclonal anti-Ubiquitin (Ubi-1) | Abcam | Cat# ab7254, RRID: AB_305802 |
| Mouse monoclonal anti-turboRFP (2D9) | OriGene | Cat# TA150061,RRID: AB_2622262 |
| Chicken polyclonal anti-GFP | Abcam | Cat#ab13970; RRID: AB _300798 |
| Goat anti-chicken 488 | Thermo Fisher Scientific | Cat#A-11039; RRID: AB _2534096 |
| Donkey anti-rabbit 488 | Thermo Fisher Scientific | Cat#A-21206; RRID: AB _2535792 |
| Donkey anti-rabbit 568 | Thermo Fisher Scientific | Cat#A-10042; RRID: AB _2534017 |
| Goat anti-chicken 568 | Thermo Fisher Scientific | Cat#A-11041; RRID: AB _2534098 |
| Goat anti-mouse 568 | Thermo Fisher Scientific | Cat#A-11031; RRID: AB _144696 |
| Donkey anti-goat 647 | Thermo Fisher Scientific | Cat#A-21447; RRID: AB _2535864 |
| Goat anti-chicken 647 | Thermo Fisher Scientific | Cat#A-21449; RRID: AB _2535866 |
| Donkey anti-mouse 647 | Thermo Fisher Scientific | Cat#A-31571; RRID: AB _162542 |
| Goat polyclonal anti-Rabbit IgG, HRP | Thermo Fisher Scientific | Cat# 31460, RRID: AB 228341 |
| Goat polyclonal anti-Mouse IgG, HRP | Millipore | Cat# AP308P, RRID: AB_92635 |
| Bacterial and Virus Strains | ||
| E. coli HST08 | TaKaRa | Cat#636766 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG132 | Sigma-Aldrich | Cat#M7449 |
| Cycloheximide | Sigma-Aldrich | Cat#C4859 |
| Ubiquitin-7-amido-4-methylcoumarin (Ub-AMC) | Sigma-Aldrich | Cat#U2258 |
| Critical Commercial Assays | ||
| Phire Animal Tissue Direct PCR Master Kit | Thermo Fisher Scientific | Cat#F140WH |
| Duolink® In Situ PLA® Probe Anti-Mouse MINUS | Sigma-Aldrich | Cat#DUO92004 |
| Duolink® In Situ PLA® Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat#DUO92002 |
| Duolink® In Situ Detection Reagents FarRed | Sigma-Aldrich | Cat#DUO92013 |
| FD Rapid GolgiStain Kit | FD NeuroTechonologies | Cat#PK401 |
| In-Fusion HD Cloning Kit | Clontech | Cat#638910 |
| QuickChange Site-Directed Mutagenesis Kit | Agilent Technologies | Cat#200516 |
| Experimental Models: Cell Lines | ||
| HEK293T/17 cells | ATCC | Cat#CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | #000664 |
| Mouse: Emx cre | Iwasato et al., 2000 | N/A |
| Mouse: Usp9X loxP | Stegeman et al., 2013 | N/A |
| Oligonucleotides | ||
| Primers for PCR, see Table S4 | This paper | N/A |
| Emx Cre F: CTG ACC GTA CAC CAA AAT TTG CCT G | Iwasato et al., 2000 | N/A |
| Emx Cre R: GAT AAT CGC GAA CAT CTT CAG GTT C | Iwasato et al., 2000 | N/A |
| Usp9X loxP F: GAG GCA CAA GTT GGC CCA GCA G | Stegeman et al., 2013 | N/A |
| Usp9X loxP R: GGT TCC TGT CCC ACT GCA GAA G | Stegeman et al., 2013 | N/A |
| Recombinant DNA | ||
| pEZ-3XHA-AnkG | GeneCopoeia | Cat# EX-Mm25668-M06 |
| pEZ-3XFlag-Usp9X | GeneCopoeia | Cat#EX-Mm24322-M12 |
| pRFP-C-RS | Origene | Cat#TR30014 |
| pRFP-C-RS Control | Origene | Cat#TR30015 |
| pRFP-C-RS shUsp9X | Origene | Cat#TF510959D |
| pmCherry-C 1 | Clontech | Cat#632524 |
| pEBFP2-N1 | Addgene | Cat#54595 |
| pEZ-3XHA-AnkG1-807 | This paper | N/A |
| pEZ-3XHA-AnkG808-1475 | This paper | N/A |
| pEZ-3XHA-AnkG1476-1961 | This paper | N/A |
| pEZ-3XHA-AnkG808-1961 | This paper | N/A |
| pEZ-3XHA-AnkG1-807 Muta | This paper | N/A |
| pEZ-3XHA-AnkG1-807 Mutb | This paper | N/A |
| pEZ-3XHA-AnkG1-807 Muta;b | This paper | N/A |
| pEZ-3XHA-AnkG1-807 K39A | This paper | N/A |
| pEZ-3XHA-AnkG1-807 K260A | This paper | N/A |
| pEZ-3XHA-AnkG1-807 K268A | This paper | N/A |
| pEZ-3XHA-AnkG1-807 K260;268A | This paper | N/A |
| pEZ-3XHA-AnkG1-807 K39;260;268A | This paper | N/A |
| pEGFP-N2-AnkG | This paper | N/A |
| pEGFP-N2-AnkG Muta;b | This paper | N/A |
| pEGFP-N2-AnkG1-807 | This paper | N/A |
| pEGFP-N2-AnkG1-807 Muta;b | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 S3A | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 S1593D | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 S1600D | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 S1609D | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 S3D | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 Q1573L | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 L1693W | This paper | N/A |
| pEZ-3XFlag-Usp9X1555-1958 G1890E | This paper | N/A |
| His-Usp9X1547-1962 | This paper | N/A |
| His-Usp9X1547-1962 H1878A | This paper | N/A |
| His-Usp9X1547-1962 S3A | This paper | N/A |
| His-Usp9X1547-1962 S1593D | This paper | N/A |
| His-Usp9X1547-1962 S1600D | This paper | N/A |
| His-Usp9X1547-1962 S1609D | This paper | N/A |
| His-Usp9X1547-1962 S3D | This paper | N/A |
| His-Usp9X1547-1962 Q1573L | This paper | N/A |
| His-Usp9X1547-1962 L1693W | This paper | N/A |
| His-Usp9X1547-1962 G1890E | This paper | N/A |
| pEGFP-N2-Usp9X1555-1958 | This paper | N/A |
| pEGFP-N2-Usp9X1555-1958 S3A | This paper | N/A |
| Software and Algorithms | ||
| FIJI (ImageJ) | NIH | https://fiji.sc/ |
| NIS-Elements v4.51.00 | Nikon | http://www.nikon.com/products/microscope-solutions/support/index.htm |
| Prism 8 | GraphPad | http://www.graphpad.com/scientificsoftware/prism/ |
| PyMOL V1.8.2.0 | Schrödinger | https://pymol.org/2/ |
| Maestro multiple sequence viewer | Schrödinger | https://www.schrodinger.com/ |
| Maestro Prime module | Schrödinger | https://www.schrodinger.com/ |
| EthoVision XT 11.5 | Noldus | http://www.noldus.com |
| Image Lab 3.0 | Bio-Rad Laboratories | http://www.bio-rad.com/en-us/product/image-lab-software |
HIGHLIGHTS.
Ankyrin-G interaction with Usp9X regulates spine development
Usp9X controls ankyrin-repeat domain protein homeostasis
Usp9X knockout mice show synaptic abnormalities, ankyrin-G aggregates, and hyperactivity
USP9X mutations in patients ablate its catalytic activity or ankyrin-G interaction
ACKNOWLEDGMENTS
This work was supported by R01MH107182 to P.P. We thank NU Nikon Cell Imaging Facility for use of the N-SIM. Protein expression and purification was performed in the Recombinant Protein Production Core Facility at Northwestern University. Special thanks go to Sergii Pshenychnyi for assistance throughout. In vitro analyses were performed in the Analytical BioNanoTechnology Core Facility of the Simpson Querrey Institute at Northwestern University. Northwestern University provided funding to develop this facility and ongoing support is being received from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205). Molecular modeling was performed by the Medicinal and Synthetic Chemistry Core at Northwestern University.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akhurst RJ, and Hata A (2012). Targeting the TGF beta signalling pathway in disease. Nat Rev Drug Discov 11, 790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, and Alessi DR (2008). Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J 411, 249–260. [DOI] [PubMed] [Google Scholar]
- Al-Khodor S, Price CT, Kalia A, and Abu Kwaik Y (2010). Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol 18, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik AY, and Hochstrasser M (2004). Mechanism and function of deubiquitinating enzymes. Bba-Mol Cell Res 1695, 189–207. [DOI] [PubMed] [Google Scholar]
- Bayes A, Collins MO, Croning MDR, van de Lagemaat LN, Choudhary JS, and Grant SGN (2012). Comparative Study of Human and Mouse Postsynaptic Proteomes Finds High Compositional Conservation and Abundance Differences for Key Synaptic Proteins. Plos One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, and Healy J (2008). Organizing the diseases fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol Med 14, 28–36. [DOI] [PubMed] [Google Scholar]
- Bett JS (2016). Proteostasis regulation by the ubiquitin system. Essays Biochem 60, 143–151. [DOI] [PubMed] [Google Scholar]
- Bork P (1993). Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17, 363–374. [DOI] [PubMed] [Google Scholar]
- Bousman CA, Chana G, Glatt SJ, Chandler SD, Lucero GR, Tatro E, May T, Lohr JB, Kremen WS, Tsuang MT, and Everall IP (2010). Preliminary evidence of ubiquitin proteasome system dysregulation in schizophrenia and bipolar disorder: convergent pathway analysis findings from two independent samples. Am J Med Genet B Neuropsychiatr Genet 153B, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier AE, Djakovic SN, Salehi A, Wilson SM, Masliah E, and Patrick GN (2009). Regulation of Synaptic Structure by Ubiquitin C-Terminal Hydrolase L1. Journal of Neuroscience 29, 7857–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–U119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, et al. (2009). FAM/USP9x, a Deubiquitinating Enzyme Essential for TGF beta Signaling, Controls Smad4 Monoubiquitination. Cell 136, 123–135. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6, 231–242. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, O'Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Parnell E, and Penzes P (2018). Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci 19, 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galves M, Rathi R, Prag G, and Ashkenazi A (2019). Ubiquitin Signaling and Degradation of Aggregate-Prone Proteins. Trends Biochem Sci. [DOI] [PubMed] [Google Scholar]
- Garcia-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen LN, Hamid J, Pizzoccaro A, et al. (2014). The Deubiquitinating Enzyme USP5 Modulates Neuropathic and Inflammatory Pain by Enhancing Ca(v)3.2 Channel Activity. Neuron 83, 1144–1158. [DOI] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, and Bennett V (1997). Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol 137, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, and Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. (2009). Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein R, Parry DA, Nalbach L, Logan CV, Strom TM, Hartill VL, Carr IM, Korenke GC, Uppal S, Ahmed M, et al. (2015). HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J Med Genet 52, 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Bergen SE, Akula N, Song J, Hultman CM, Landen M, Adli M, Alda M, Ardau R, Arias B, et al. (2016). Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum Mol Genet 25, 3383–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AY, Futai K, Sala C, Valtschanoff JG, Ryu J, Woodworth MA, Kidd FL, Sung CC, Miyakawa T, Bear MF, et al. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J Neurosci 28, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, and Zhang H (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 1309, 83–94. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–U136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z, Vandeweyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA, Roca LT, Vulto-van Silfhout AT, Nijhof B, Kramer JM, et al. (2013). Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet 22, 1960–1970. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, and Greenough WT (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10, 1038–1044. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, and Itohara S (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406, 726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins PM, Kim N, Jones SL, Tseng WC, Svitkina TM, Yin HH, and Bennett V (2015). Giant ankyrin-G: A critical innovation in vertebrate evolution of fast and integrated neuronal signaling. P Natl Acad Sci USA 112, 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Kizhatil K, Kramarcy NR, Sen A, Sealock R, and Bennett V (2001). FIGQY phosphorylation defines discrete populations of L1 cell adhesion molecules at sites of cell-cell contact and in migrating neurons. J Cell Sci 114, 3823–3835. [DOI] [PubMed] [Google Scholar]
- Johnson BV, Kumar R, Oishi S, Alexander S, Kasherman M, Vega MS, Ivancevic A, Gardner A, Domingo D, Corbett M, et al. (2019). Partial Loss of USP9X Function Leads to a Male Neurodevelopmental and Behavioral Disorder Converging on Transforming Growth Factor beta Signaling. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu CF, Grigorean G, Ziff EB, and Neubert TA (2004). Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics 3, 857–871. [DOI] [PubMed] [Google Scholar]
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, and Bonni A (2009). A Centrosomal Cdc20-APC Pathway Controls Dendrite Morphogenesis in Postmitotic Neurons. Cell 136, 322–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, and Benes FM (2014). Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 71, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto D, Walinda E, Fukada H, Sou YS, Kageyama S, Hoshino M, Fujii T, Tsuchiya H, Saeki Y, Arita K, et al. (2015). The unexpected role of polyubiquitin chains in the formation of fibrillar aggregates. Nat Commun 6, 6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosavi LK, Cammett TJ, Desrosiers DC, and Peng ZY (2004). The ankyrin repeat as molecular architecture for protein recognition. Protein Sci 13, 1435–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SMB, Luna-Vargas MPA, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK, and Bernards R (2005). A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786. [DOI] [PubMed] [Google Scholar]
- Nishi H, Hashimoto K, and Panchenko AR (2011). Phosphorylation in Protein-Protein Binding: Effect on Stability and Function. Structure 19, 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi S, Premarathne S, Harvey TJ, Iyer S, Dixon C, Alexander S, Burne TH, Wood SA, and Piper M (2016). Usp9x-deficiency disrupts the morphological development of the postnatal hippocampal dentate gyrus. Sci Rep 6, 25783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, and Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, and Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijnders MR, Zachariadis V, Latour B, Jolly L, Mancini GM, Pfundt R, Wu KM, van Ravenswaaij-Arts CM, Veenstra-Knol HE, Anderlid BM, et al. (2016). De Novo Loss-of-Function Mutations in USP9X Cause a Female-Specific Recognizable Syndrome with Developmental Delay and Congenital Malformations. Am J Hum Genet 98, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, and Engelender S (2011). alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. P Natl Acad Sci USA 108, 18666–18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–U124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. [DOI] [PubMed] [Google Scholar]
- Schule B, McFarland KN, Lee K, Tsai YC, Nguyen KD, Sun C, Liu M, Byrne C, Gopi R, Huang N, et al. (2017). Parkinson's disease associated with pure ATXN10 repeat expansion. Npj Parkinson Dis 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze TG, Detera-Wadleigh SD, Akula N, Gupta A, Kassem L, Steele J, Pearl J, Strohmaier J, Breuer R, Schwarz M, et al. (2009). Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol Psychiatry 14, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Kopeikina KJ, Fawcett-Patel JM, Leaderbrand K, Gao RQ, Schurmann B, Myczek K, Radulovic J, Swanson GT, and Penzes P (2014). Psychiatric Risk Factor ANK3/Ankyrin-G Nanodomains Regulate the Structure and Function of Glutamatergic Synapses. Neuron 84, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman S, Jolly LA, Premarathne S, Gecz J, Richards LJ, Mackay-Sim A, and Wood SA (2013). Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFbeta-mediated axonogenesis. PLoS One 8, e68287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya S, Yamamoto T, Kanai-Azuma M, Wood SA, and Kaibuchi K (1999). The deubiquitinating enzyme Fam interacts with and stabilizes beta-catenin. Genes Cells 4, 757–767. [DOI] [PubMed] [Google Scholar]
- Taya S, Yamamoto T, Kano K, Kawano Y, Iwamatsu A, Tsuchiya T, Tanaka K, Kanai-Azuma M, Wood SA, Mattick JS, and Kaibuchi K (1998). The Ras target AF-6 is a substrate of the Fam deubiquitinating enzyme. J Cell Biol 142, 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Rives ML, Urizar E, Piskorowski RA, Vishwasrao HD, Castrillon J, Schmauss C, Slattman M, Gullberg M, and Javitch JA (2011). Detection of antigen interactions ex vivo by proximity ligation assay: endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. Biotechniques 51, 111-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng WC, Jenkins PM, Tanaka M, Mooney R, and Bennett V (2015). Giant ankyrin-G stabilizes somatodendritic GABAergic synapses through opposing endocytosis of GABA(A) receptors. P Natl Acad Sci USA 112, 1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roessel P, Elliott DA, Robinson IM, Prokop A, and Brand AH (2004). Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell 119, 707–718. [DOI] [PubMed] [Google Scholar]
- Wang CH, Huang YC, Chen PY, Cheng YJ, Kao HH, Pi HW, and Chien CT (2017). USP5/Leon deubiquitinase confines postsynaptic growth by maintaining ubiquitin homeostasis through Ubiquilin. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet 20, 3093–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. [DOI] [PubMed] [Google Scholar]
- Zhang X, Davis JQ, Carpenter S, and Bennett V (1998). Structural requirements for association of neurofascin with ankyrin. J Biol Chem 273, 30785–30794. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang C, Ding D, Chen Z, Peng Y, Peng H, Hou X, Wang P, Hou X, Ye W, et al. (2018). Analysis of (CAG)(n) expansion in ATXN1, ATXN2 and ATXN3 in Chinese patients with multiple system atrophy. Sci Rep-Uk 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Cordner ZA, Xiong J, Chiu CT, Artola A, Zuo Y, Nelson AD, Kim TY, Zaika N, Woolums BM, et al. (2017). Genetic disruption of ankyrin-G in adult mouse forebrain causes cortical synapse alteration and behavior reminiscent of bipolar disorder. Proc Natl Acad Sci U S A 114, 10479–10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Related to Figure 1. The prediction of D-box and KEN box results for the mouse ankyrin-G (National Center for Biotechnology Information accession number NM_146005) are shown.
Table S3: Related to Figure 5. The ubiquitinated lysine sites of ANKRD in ANKRD containing psychiatric risk genes.
Table S4: Related to Figure 1, 3, 4, and 7, Figure S1, S3, S5, and S8. Sequences of oligos used this study.
Table S2: Related to Figure 5. The list of ANKRD genes from Uniprot (Homo sapiens).
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. All data are available from the Lead Contact upon request.