

Abstract
Neuroinflammation plays a vital role in early brain injury (EBI) following subarachnoid hemorrhage (SAH). The hypothesis of this study was that activation of melanocortin 1 receptor (MC1R) with BMS-470539 attenuates EBI by suppression of neuroinflammation after SAH. We utilized BMS-470539, MSG-606, and MRT-68601 to verify the neuroprotective effects of MC1R. We evaluated brain water content, short-term and long-term neurobehavior after SAH. Western blotting and immunofluorescence staining were utilized to assess the changes of protein levels. The results of western blotting suggested that the expressions of MC1R, phosphorylated-adenosine monophosphate-activated protein kinase (p-AMPK), and phosphorylated-TANK binding kinase 1 (p-TBK1) were increased and reached their peak points at 24 h following SAH. Moreover, BMS-470539 treatment notably attenuated neurological deficits caused by SAH, and also notably improved long-term spatial learning and memory abilities after SAH. The underlying mechanisms of the neuroprotection of BMS-470539 involved the suppression of microglia activation, promotion of CD206+ microglia transformation and reduction of neutrophil infiltration by increasing the levels of p-AMPK and p-TBK1 while decreasing the levels of NF-κB, IL-1β, and TNFα. The neuroprotective effects of BMS-470539 were significantly abolished by MSG-606 and MRT-68601. The activation of MC1R with BMS-470539 notably attenuates EBI after SAH by suppression of microglial activation and neutrophil infiltration via the AMPK/TBK1/NF-κB signaling pathway.
Electronic supplementary material
The online version of this article (10.1007/s13311-019-00772-x) contains supplementary material, which is available to authorized users.
Keywords: Early brain injury, Subarachnoid hemorrhage, Melanocortin 1 Receptor, TBK1, Neuroinflammation
Introduction
SAH is a devastating disease, characterized by high rate of disability and mortality [1]. Recent studies suggested that the poor prognosis of SAH mainly results from early brain injury (EBI), which appears within the first 3 days following SAH [2, 3]. Although inflammation is one of the key mechanisms for tissue repair, overactivation of inflammatory responses after SAH is one of the main pathophysiological processes underlying EBI, which could further amplify the brain injury and cause permanent neurological and cognitive deficits [4]. Given the extensive damage induced by neuroinflammation, suppression of inflammatory responses could be an effective tactic to reduce brain injury after SAH.
Microglia, the resident immune cells of the central nervous system (CNS), are responsible for the inflammatory response [5]. Frequently, microglia are redeemed to exist in a resting status (M0). However, upon activation, they are transformed into two different phenotypes (M1 and M2) with distinct physiological functions [6, 7]. M1 phenotype microglia are pro-inflammatory as they secrete inflammatory molecules, including IL-1β, IL-6, and TNF-α, which are greatly detrimental to the brain tissues [8]. In contrast with the M1 phenotype, M2 microglia are thought to be neuroprotective through the promotion of neurogenesis, axonal regeneration, angiogenesis, and re-myelination due to their anti-inflammatory properties [9]. Therefore, intentional promotion of microglia from M1 phenotype to M2 phenotype could be a promising therapeutic strategy in treating SAH.
The melanocortin receptor (MCR) family, class A (rhodopsin-like) family of G-protein coupled receptors (GPCRs), harbors five members (MCRs 1-5) [10]. Among all, MC1R, MC3R and MC4R have been reported to be highly expressed in the central nervous system (CNS) [11]. Recently, MC1R has been demonstrated on microglia/macrophages and suggested to be involved in immunomodulatory processes [12, 13]. For example, Li et al. reported increased phosphorylated NF-κB levels in the absence of MC1R, indicating that MC1R exerted its anti-inflammatory effects at least partly via the suppression of NF-κB activation [14]. Indeed, several previous studies have demonstrated that NF-κB can promote the transformation of microglia from M0 to M1 phenotype, which in turn increases the secretion of pro-inflammatory cytokines, including IL-1β and TNF-α while decreasing the level of anti-inflammatory cytokines such as IL-10 [15]. Thus, MC1R-mediated anti-inflammatory effects have been consistently suggested in the recent literature [12]. However, the role of MC1R in hemorrhagic stroke has yet to be explored. Although the neuroprotective effect of melanocortin was previously evaluated through the use of α-melanocyte stimulating hormone (α-MSH) analogues in experimental SAH [16], the role of particular activation of MC1R remains largely unknown.
Although the mechanisms underlying the MC1R-mediated suppression of NF-κB activation is not well understood, modulation of the AMPK/TBK1 signaling pathway is one of the potential ways. AMPK is an energy sensor with essential roles in the regulation of inflammation [17], which contributes to the regulation of pro-opiomelanocortin-α (POMC), the precursor protein of endogenous ligand (α-melanocortin) of MC1R [18]. AMPK has been reported to phosphorylate and activate TANK-binding kinase 1 (TBK1) [19], a member of the inhibitor kappa kinase (IKK) family, leading to the alleviation of inflammation via the suppression of NF-κB [20]. However, the role of TBK1 has never been studied in hemorrhagic stroke. BMS-470539, a strong and selective agonist of MC1R, plays essential roles in the suppression of inflammatory responses [21, 22].
Based on the evidence mentioned above, we hypothesized that activation of MC1R by BMS-470539 would reduce pro-inflammatory molecules, promote the transformation of microglia to the M2 phenotype, and alleviate the neurological and cognitive deficits via the AMPK/TBK1/NF-κB signaling pathway in a rat model of SAH.
Methods
Animals
We did the experiments based on the protocols proposed by the Institutional Animal Care and Use Committee (IACUC) at Loma Linda University. The procedures were performed based on NIH guidelines. Male Sprague–Dawley (SD) rats (280-330g, Indianapolis, IN) were used in the present study. Animals were kept at room temperature (22 ± 1°C) and in a 12 h day/night cycle (humidity: 60 ± 5%). Food and water were ad libitum.
Experimental Design
This study was completed in five separate experiments, as shown in Fig. 1. The final number of rats used in this study was 194, including deceased and excluded animals in each group (additional file).
Fig. 1.

Experimental design and animal groups
Experiment 1
We determined the expression of MC1R, AMPK and TBK1 protein levels in the sham group and each group after SAH. Rats were randomly divided into sham and SAH groups with different time-points (3 h, 6 h, 12 h, 24 h, 72 h). The ipsilateral/left cerebral cortex of six rats from each group was collected for western blotting. Additionally, the cellular location of MC1R was evaluated using double immunofluorescence staining in sham and SAH (24 h) groups.
Experiment 2
In order to explore the neuroprotective effects of BMS-470539 (sc-362716A, Santa Cruz), we chose three different dosages of BMS-470539, based on a previous study [22]. Rats were randomly divided into five groups: sham, SAH + vehicle (10 μl sterile saline), SAH + BMS-470539 (50 μg/kg, 10 μl), SAH + BMS-470539 (160 μg/kg, 10 μl), and SAH + BMS-470539 (500 μg/kg, 10 μl). BMS-470539 was administrated intra-nasally (i.n.) 1 h after SAH. Neurobehavior tests (including modified Garcia score and beam balance test) and brain water content analysis were performed at 24 h after SAH.
Experiment 3
The medium dosage (160 μg/kg) was chosen for further study according to the better results obtained from the previous experiment. To study the effects of BMS-470539 (160 μg/kg, 10 μl) on long-term neurological functions, rats were randomly distributed into three groups: sham, SAH + vehicle, and SAH + BMS-470539. Rotarod tests were performed in the first, second and third weeks after SAH. Morris water maze tests were conducted between days 21 and 25. Two rats from each group were used for Nissl staining.
Experiment 4
We used a selective MC1R antagonist (MSG-606, 5954, R&D systems) to study the role of MC1R in BMS-470539 mediated anti-neuroinflammation. Rats were randomly divided into five groups: sham, SAH + vehicle 1(10 μl sterile saline, i.n), SAH + BMS-470539 (160 μg/kg, 10 μl), SAH + BMS-470539 + vehicle 2 (6 μl sterile saline, i.c.v), and SAH + BMS-470539 + MSG-606 (1 nmol/μl, 6 μl). MSG-606 was administered 1 h before SAH via i.c.v. route. Neurological testing was performed at 24 h after SAH. Ipsilateral/left cerebral cortex was sampled for western blotting.
Experiment 5
To assess the role of TBK1 on the anti-neuroinflammation effects of BMS-470539, the selective TBK1 inhibitor MRT-68601 (5067, R&D systems) was administrated through the intracerebroventricular (i.c.v.) route 1 h before SAH. Rats were randomly divided into five groups: sham, SAH + vehicle 1(10 μl sterile saline, i.n), SAH + BMS-470539 (160 μg/kg, 10μl), SAH + BMS-470539 + vehicle 2 (9 μl sterile saline, i.c.v), and SAH + BMS-470539 + MRT-68601 (1.8 nmol/μl, 9 μl). Neurological testing was performed 24 h after SAH. Ipsilateral/left cerebral cortex was sampled at 24 h after induction of SAH for western blotting and immunofluorescence staining.
Animal Model
The SAH model was achieved by endovascular perforation, which has been widely established [23]. Briefly, the rats received anesthetization with 5% isoflurane and then they were positioned on a ventilator. The rats were kept under anesthesia with 3% isoflurane in 65/35% medical air/oxygen during surgery (heart and respiration rates were monitored throughout the procedure). We exposed the carotid artery and its bifurcation. Afterwards, a 4-0 sharpened nylon suture was inserted from the external carotid artery. The suture then went along the internal carotid artery and finally reached the bifurcation of the anterior cerebral artery and middle cerebral artery, where a perforation was executed. After euthanasia, we evaluated the degree of SAH according to a new grading system [24].
Drug Administration and Intracerebroventricular Injection
BMS-470539 was given by intranasal injection with a volume of 10 μl. The dosage of BMS-470539 was determined based on previous literature [22]. MSG-606 (1 nmol/μl, 6 μl) and MRT-68601 (1.8 nmol/μl, 9 μl) were administrated by i.c.v. injection 1 h before SAH, the procedures of which were according to a previous report [25]. Briefly, the rats received anesthetization with 5% isoflurane and kept under anesthesia as described above. Then we made a burr hole with a drill 1 mm posterior to the bregma and 1.5 mm right lateral to the midline, at which point the drug was slowly administrated (3.5 mm in depth, 0.5 μl/min). The needle was kept in place for 5 minutes and then withdrawn after another 5 minutes. Finally, the burr hole and incision were closed with bone wax and sutures, respectively (all surgical procedures were conducted with sterile techniques).
Mortality and Short-Term Neurobehavior Evaluation
We set the timing of the neurobehavior tests at 24 h after SAH, since the protein levels of MC1R, p-AMPK, and p-TBK1 were found to peak at 24 h after the injury. We then calculated the mortality rate and evaluated the neurobehavior at 24 h after SAH. Modified Garcia scoring system [24] and beam balance test [26] were used for neurological assessment. Parameters and modifications of these two methods can be found in Supplemental Tables 1 and 2. The neurological tests were conducted by an independent researcher.
Long-Term Neurological Assessment
Rotarod test was adopted to assess the long-term neurobehavior in the first, second and third weeks following SAH. Additionally, Morris water maze test was applied during days 21 to 25 to evaluate the spatial learning and memory abilities between different groups, the procedures of which were based on a previous report [27]. Detailed description of the Morris water maze test can be found in Supplemental 3.
Brain Water Content Assessment
The wet-dry method was used to assess the brain edema 24h after SAH. Briefly, the rats underwent euthanasia and the brains were quickly collected, which were then separated into right hemisphere, left hemisphere, cerebellum, and brain stem. Afterwards, these four parts of the brain were weighed separately (wet weight) and then put into an oven at a temperature of 105°C for three days. Dry brains were weighed (dry weight) and the brain water content was calculated as follows: [(wet weight − dry weight) / (wet weight)] × 100% [28].
Immunofluorescence Staining
After being anesthetized with 5% isoflurane, the rats received trans-cardiac perfusion (0.1 M PBS) and then 10% formalin. Afterwards, we collected the brains quickly and put them into 10% formalin (4°C, 24 h). Formalin was replaced with 30% sucrose solution for 3 days. The brains were cut into 10-μm sections. The slices were then fixed on slides and used for immunofluorescence staining. The brain slices were incubated at 4°C overnight with the following primary antibodies: MC1R (1:50, ThermoFisher, PA5-75342), IL-1β (1:500, Abcam ab9722), myeloperoxidase (MPO) (1:500, Abcam ab65871), Iba-1 (1:100, Abcam ab48004), glial fibrillary acidic protein (GFAP) (1:100, Abcam ab53554), NeuN (1:100, Abcam ab104224), CD206 (1:200, Santa Cruz sc-34577). Secondary antibodies were then applied at room temperature (21 °C) for 2 h and assessed with a fluorescence microscope (Leica Microsystems, Germany). To evaluate the numbers of Iba-1, MPO, CD206, and IL-1β positive cells, we chose three different fields from each of the five sections per brain and quantified the positive cells using a magnification of 200X.
Nissl Staining
Nissl staining was conducted according to a previous report [29]. Briefly, rats were treated as described above for immunofluorescence staining. The brains were then cut into 16-μm slices. The slices were immersed with 0.5 % cresyl violet (Sigma-Aldrich, St. Louis, MO, USA) solution and dehydrated with 100% alcohol. Next, the sections were washed with xylene and covered with a coverslip. Finally, the sections were observed under a light microscope by a blinded investigator.
Western Blot Analysis
After being anesthetized, the rats received trans-cardiac perfusion (0.1 M PBS). Ipsilateral/left cerebral cortex was then sampled for western blotting, the procedures of which were according to a previous report [30]. In brief, 40 μg protein from each sample were applied for electrophoresis and then the protein was transferred onto nitrocellulose membranes (100 V, 80 minutes). Afterwards, the membranes were incubated with the following primary antibodies overnight (4°C): MC1R (1:500, ThermoFisher, PA5-75342), p-AMPK (1:1000, Cell Signaling, #2535), AMPK (1:2000, Cell Signaling, #5832), p-TBK1 (1:5000, Abcam ab109272), TBK1 (1:5000, Abcam ab40676), p-NFκB (1:500, Biorbyt orb107125), NFκB (1:1000, NOVUS NBP1-87760), IL-1β (1:500, Abcam ab9722), TNFα (1:1000, Abcam ab6671), β-actin (1: 3000, Santa Cruz sc-47778). Secondary antibodies were then applied at room temperature for 1 h. Finally, the bands were probed with ECL Plus chemiluminescence reagent Kit (Amersham Biosciences, Arlington Heights, PA) and visualized with the imaging system (Bio-Rad, Versa Doc, model 4000). Then, Image J software (NIH) was used to measure intensity. The results were displayed as relative density (grayscale value of the target proteins /β-actin or total proteins). For quantification of western blot results, when we detected the proteins, the beta-actin from the same sample on the same membrane corresponding to the proteins was always detected at the same time. That means, all proteins were normalized to their own beta-actin from the same sample, no exceptions. For quantification of phosphorylated and total proteins (p-NFkB/NFkB, p-AMPK/AMPK, p-TBK1/TBK1), we ran the same sample twice, once for phosphorylated protein and once for total protein. We did not strip the membrane during the experiments.
Statistical Analysis
The data were shown as the mean ± standard deviation (SD). SPSS 22.0 software (IBM, USA) was used to perform power analysis based on α = 0.05 and the number of the group, with a power of 0.85 or higher indicating reliable evidence. GraphPad Prism (GraphPad Software, San Diego, CA, USA) was applied to analyze the data. First, we assessed the normality of the data. If the data met the requirement of satisfied normality and homogeneity of variance, one-way analysis of variance (ANOVA) is followed by multiple comparisons between different groups using Tukey’s post hoc test. For the data that failed the normality test, non-parametric statistics was applied. Additionally, two-way repeated measures ANOVA was applied to analyze the data of long-term neurological functions and brain water content. Statistical significance was set at P < 0.05.
Results
Mortality Rates and SAH Grade Score
The physical parameters after surgery are shown in Supplemental Table 4. No significant difference was observed among different groups. Of the 194 rats, 38 were in the sham group and 156 received perforation surgery. The overall mortality of the SAH groups was 11.5% (18/156) (Supplemental Tables 5 and 6). Seven rats were excluded as SAH grade was less than 7 (Supplemental Table 5). After exclusion, we added new animals to the groups with animals excluded to make sure that each group had n=6. Basal cortex of the left hemisphere was sampled for immunofluorescence staining and western blotting (Fig. 3A). There was no significant difference between different groups in SAH grade score (Fig. 3B).
Fig. 3.

Effects of BMS-470539 on short-term neurological functions and brain edema. (A) Representative pictures of brains in the sham and SAH groups. (B) The SAH grades for each group. (C, D) Beam balance and Modified Garcia scores for each group. (E) The quantification of brain water content 24 h after SAH. Data of SAH grade and beam balance scores are shown as the medians with interquartile range, and compared by Kruskal-Wallis test followed by the Dunn post hoc test. Data of brain water content are shown as mean ± SD and compared by two-way ANOVA test. Other data were shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH + vehicle, N = 6 per group.
Temporal Patterns of MC1R, p-AMPK, and p-TBK1 were evaluated after SAH
Western blotting was used to assess the levels of MC1R, p-AMPK, and p-TBK1 at 3, 6, 12, 24, and 72 h after SAH in the left brain. The levels of MC1R and p-AMPK started to increase at 3 h after SAH and peaked at 24 h (Fig. 2A, 2B). Additionally, the level of p-TBK1 increased at 6 h after SAH and reached the highest point at 24 h, which was significantly higher than that of the sham group (P < 0.05; Fig. 2C).
Fig. 2.

Expression of MC1R, p-AMPK and p-TBK1. Representative Western blot images and quantitative analyses of MC1R time course (A), p-AMPK time course (B), and p-TBK1 time course (C) obtained from the left hemisphere after SAH; N=6 for each group. Data of comparison between sham and SAH (24 h) are shown as mean ± SD and compared by using T-test. Other data are shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH (3 h), &p<0.05 versus SAH (6 h), @p<0.05 versus SAH (24 h); (D) Representative microphotographs of immunofluorescence staining showing localization of APJ (green) with NeuN, iba-1 and GFAP (red) 24 h in sham group and SAH (24 h) group. (N=2 for each group). Scale bar=50μm.
Cellular Location of MC1R
The results of double immunofluorescence staining indicated that MC1R was colocalized with neurons (NeuN), microglia (Iba-1), and astrocytes (GFAP) and the number of MC1R positive cells was increased after SAH. Among these three cell types, MC1R was mainly colocalized with neurons and microglia (Fig. 2D).
Administration of BMS-470539 Improved Short-term Neurological Functions and attenuated Brain Edema after SAH
Modified Garcia and beam balance tests were used to evaluate short-term neurological functions. The results showed that both of these scores were notably lower in the SAH + vehicle group compared to the sham group. Moreover, medium (160 μg/kg) and high (500 μg/kg) dosages of BMS-470539 treatment notably alleviated the neurological deficits after SAH (P < 0.05; Fig. 3C and 3D).
The brain water content in different parts of the brain (right hemisphere, left hemisphere, cerebellum and brain stem) were all significantly increased after SAH. However, medium and high dosages of BMS-470539 notably reduced brain edema in the left and right hemispheres, while no significant difference was noted between these two groups (P < 0.05; Fig. 3E). Based on the results above and priority principle, we elected to proceed with the medium dosage of BMS-470539 for long-term outcome and mechanism studies.
Administration of BMS-470539 Improved Long-term Neurobehaviors after SAH
Rotarod and Morris water maze tests were used to assess the long-term neurobehaviors (Supplemental Table 3). Rotarod test was performed in the first, second and third weeks following SAH. The results showed that rats receiving puncture surgery had shorter falling latency when compared with the rats in the sham group (P < 0.05; Fig. 4A and 4B) in both 5 RPM and 10 RPM. However, administration of BMS-470539 significantly increased the falling latency in the first, second and third weeks (P < 0.05; Fig. 4A and 4B).
Fig. 4.

The effects of BMS-470539 on long-term neurobehavioral outcomes after SAH. (A, B) Rotarod test of 5RPM and 10RPM. (C) Velocity of water maze test. (D, E) Escape latency and swim distance of water maze test, and (F, G) probe quadrant duration and typical traces of water maze test. Data of Rotarod test, escape latency and swim distance are shown as mean ± SD and compared by two-way ANOVA test. Other data are shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH + vehicle; N=9 per group.
In the Morris water maze test, no significant difference of velocity was noted across each group (Fig. 4C). The rats in SAH + vehicle group had poorer escape latency and longer swimming distance to find the platform in block 2, block 3 and block 4 compared to the rats in the sham group. After the use of BMS-470539, the escape latency of the rats was significantly improved in block 2 and block 3, and the swim distance to find the platform was significantly decreased in block 2 (P < 0.05; Fig. 4D and 4E). In the probe trials, the rats in the SAH + vehicle group stayed for a shorter time (24%) in the target quadrant compared to the sham group (38%). BMS-470539 treatment notably increased the duration in the probe quadrant (33%, Fig. 4F, G).
To assess the morphological changes of neurons in the long-term experiment, we performed Nissl staining in both hippocampus and cerebral cortex. As shown in Fig. 5, sharp demarcation and rich cytoplasm were noted in the sham group, while neurons in the SAH + vehicle group were presented as shrunken cell bodies and condensed nuclei. The administration of BMS-470539 significantly attenuated neuronal injury compared to SAH + vehicle group (P < 0.05; Fig. 5A, B, C).
Fig. 5.

Nissl staining of hippocampus and cerebral cortex demonstrating reduced injury in BMS-470539 treated rats. (A) DG, CA1, CA2, and CA3 areas of hippocampus. (B) Cerebral cortex. Scale bar =200 μm (general) and 50 μm (regions). (C) The quantitative analysis of Nissl staining. N = 4 for each group. Data of Nissl staining are shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH + vehicle.
Inhibition of MC1R with MSG-606 Abolished the Anti-inflammatory Effects of BMS-470539
To verify the neuroprotective role of BMS-470539, we used MSG-606 to inhibit the function of MC1R. The rats in the SAH + BMS-470539 group displayed significantly improved neurological functions; however, the use of MSG-606 abolished the neuroprotective effects of BMS-470539 after SAH when compared with SAH + BMS-470539 + vehicle 2 (P < 0.05; Fig. 6A, B). The results of western blotting showed that the levels of p-AMPK and p-TBK1 were significantly increased, while the levels of NFκB, IL-1β, and TNFα were decreased at 24 h after SAH in the SAH + BMS-470539 group when compared to the SAH + vehicle group (P < 0.05). However, inhibition of MC1R with MSG-606 significantly decreased the levels of p-AMPK and p-TBK1, and increased the levels of NFκB, IL-1β, and TNFα at 24 h after SAH (P < 0.05; Fig. 6C, D).
Fig. 6.

Inhibition of MC1R with MSG-606 abolished the neuroprotective effects of BMS-470539 at 24 h after SAH. (A) Representative Western blot images. (B) Quantitative analyses of MC1R, p-AMPK, p-TBK1, NFκB, IL-1β, TNFα. N=6 for each group. Data of beam balance scores, and MC1R/β-actin protein expressions are shown as the medians with interquartile range and compared by Kruskal-Wallis test followed by the Dunn post hoc test. Other data are shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH + vehicle, &p<0.05 versus SAH + BMS-470539.
TBK1 inhibitor, MRT-68601, Abolished the Anti-inflammatory effects of BMS-470539
TBK1 inhibitor, MRT-68601, was used to further verify the neuroprotective pathway of BMS-470539. The use of MRT-68601 significantly reversed the positive effects of BMS-470539 after SAH when compared with SAH + BMS-470539 + vehicle 2 (P < 0.05; Fig. 7A, B). Western blot analysis indicated that MRT-68601 did not influence the levels of MC1R and p-AMPK. However, the level of p-TBK1 was significantly decreased while the levels of NFκB, IL-1β, and TNFα were increased in the SAH + BMS-470539 + MRT-68601 group compared to the SAH + BMS-470539 group (P < 0.05, Fig. 7C, D). In addition, the evaluation of IL-1β with immunofluorescence revealed that BMS-treatment notably reduced the number of IL-1β positive cells, the results of which were abolished by the use of MRT-68601 (P < 0.05; Fig. 8A, B).
Fig. 7.

TBK1 inhibitor, MRT-68601, reversed the anti-inflammatory effects of BMS-470539 at 24 h after SAH. (A) Representative Western blot images. (B) Quantitative analyses of MC1R, p-AMPK, p-TBK1, NFκB, IL-1β, TNFα. N=6 for each group. Data of modified Garcia scores, beam balance scores, IL-1β, and TNFα protein expressions are shown as the median with interquartile range and compared by Kruskal-Wallis test followed by the Dunn post hoc test. Other data were shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH + vehicle, &p<0.05 versus SAH + BMS-470539.
Fig. 8.

BMS-470539 attenuated neuroinflammation, promoted M2 microglia activation, and reduced neutrophil infiltration at 24 h after SAH. Representative images of immunofluorescence staining and quantification of (A, B) IL-1β + cells; (C, D) Iba-1 activation; (E, F) MPO + cells; (G, H) CD206 + cells. N = 3 for each group. Scale bar=50μm. Data are shown as mean ± SD and compared by one-way ANOVA followed by the Tukey post hoc test. *p<0.05 versus sham, #p<0.05 versus SAH + vehicle, &p<0.05 versus SAH + BMS-470539.
BMS-470539 Treatment Suppressed Microglia Activation and Neutrophil Infiltration
Microglia were notably activated, demonstrating larger cell body and shorter processes, and the number of Iba-1 (Fig. 8C, D) and MPO (Fig. 8E, F) positive cells were significantly increased in the SAH group (P < 0.05). However, BMS-470539 treatment significantly reduced the number of Iba-1 and MPO positive cells and alleviated overactivation of microglia, the results of which were abolished by MRT-68601 (P < 0.05).
The selective polarization of microglia could be beneficial for attenuating neuroinflammation after SAH [9]. Therefore, we evaluated the number of CD206+ cells in different groups. Compared with the sham group, the number of CD206+ cells increased after the induction of SAH. Interestingly, the use of BMS-470539 further increased the number of CD206+ cells, while MRT-68601 significantly reversed these results (P < 0.05, Fig. 8G, H).
Discussion
In this study, we demonstrated a novel mechanism of MC1R-mediated neuroprotection in a rat model of SAH, by activating MC1R with BMS-470539, which then decreased neuroinflammation through the AMPK/TBK1/NF-κB signaling pathway (Fig. 9). The novel findings of this study were: (1) the levels of MC1R, p-AMPK, and p-TBK1 were significantly increased and reached a maximum point at 24 h after SAH; MC1R was extensively expressed in the brain and was colocalized with microglia, neurons, and astrocytes; (2) the use of BMS-470539 significantly improved the neurological functions and alleviated brain edema after SAH; (3) BMS-470539 significantly strengthened learning and memory abilities leading to improved long-term neurobehavior scores after SAH; (4) the activation of MC1R with BMS-470539 significantly suppressed the activation of microglia and neutrophil infiltration, increased the number of CD206+ microglia, and the levels of p-AMPK and p-TBK1, while decreasing the levels of NF-κB, IL-1β, TNF-α; (5) the inhibition of MC1R with MSG-606 significantly abolished the anti-inflammatory effects of BMS-470539 when compared to SAH + BMS-470539 + vehicle 2 group; and (6) the selective TBK1 inhibitor MRT-68601 reversed the neuroprotective effects of BMS-470539/MC1R system at 24 h following SAH. Based on the evidence above, the activation of MC1R showed neuroprotective effects and improved short-term and long-term neurological functions by controlling microglia-mediated neuroinflammation following SAH, and these effects were at least partly mediated by the AMPK/TBK1/NF-κB signaling pathway.
Fig. 9.
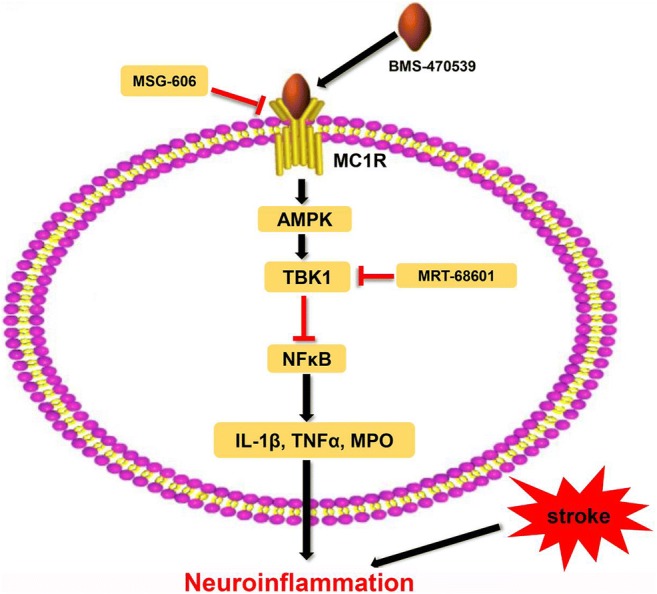
The potential molecular mechanisms of BMS-470539-mediated anti-inflammatory effects via the activation of AMPK/TBK1 and suppression of NFκB
MC1R, a G-protein coupled receptor, has gained attention due to its roles in a number of important physiological processes, including modulation of skin color, defense behavior and inflammatory responses [31, 32]. For example, Johannes et al. found increased protein and mRNA levels of MC1R on murine podocytes which were treated with puromycin [33]. Consistent with the results of the present study, the expression of MC1R in the CNS and its neuroprotective effects have been extensively verified as well. Furthermore, Schaible et al. showed that the activation of MC1R with its endogenous ligand α-MSH significantly attenuated brain injury by reducing neuroinflammation and neuronal apoptosis following a mouse model of experimental traumatic brain injury [34].
In the first part of our study, we evaluated the temporal pattern of MC1R, p-AMPK and p-TBK1, revealing significantly upregulated and peaked levels of these three proteins at 24 h after SAH. The level of MC1R increased and peaked at 24 hours after SAH, which could be explained by the attack of SAH which is conducted inside the cell and increases the genetic expression of MC1R. Besides, it’s reasonable to believe that both the protective and harmful factors would increase after brain injury as the body is trying to keep homeostasis. The induction of SAH would lead to continuous injury to the brain tissues, including brain edema, neuroinflammation, oxidative stress and finally neuronal apoptosis. At the same time, the SAH would also upregulate the genetic expression of protective proteins, such as MC1R. Therefore, the upregulation of MC1R and increased neural injury could be paralleled and is reasonable. In addition, we were able to demonstrate colocalization of MC1R with microglia (Iba-1), neurons (NeuN) as well as astrocytes (GFAP) by double immunofluorescence staining, which was still unclear up to now. Among all, MC1R was mainly expressed in neurons and microglia. The evidence above suggested the potential involvement of MC1R in the regulation of neuroinflammation.
The blood leaking into the subarachnoid space following SAH has the potential to cause severe brain edema and subsequent neurological deficits [35, 36]. In addition, the toxic agents resulting from the breakdown of the blood cells cause further neural death, leading to permanent neurological deficits, including cognition, memory and learning disabilities [37]. In this study, we found that BMS-470539 treatment significantly alleviated neurological deficits and reduced brain edema at 24 h after SAH. Moreover, evaluation of long-term neurological functions with rotarod and Morris water maze tests demonstrated that BMS-470539 treatment administered at the early stage of SAH was able to improve the long-term movement coordination, memory, and spatial learning abilities.
Inflammatory responses are suggested to be one of the significant factors contributing to the EBI after SAH. The cellular debris and toxic agents resulting from the decomposition of blood clots activate the resident immune cells (microglia). The increase in the number of M1 phenotype of microglia produces pro-inflammatory cytokines and chemokines, which further disrupts BBB and attracts the accumulation of macrophages and neutrophils into the damaged brain tissue. The effects of this extensive inflammatory cascade can cause devastating and permanent damage to the brain. The anti-inflammatory effect of MC1R has been previously reported. Chen et al. demonstrated that MC1R decreased the levels of pro-inflammatory cytokines (TNF-α, IL-6, etc.) and alleviated inflammation both in vivo and in vitro [38]. Also, the binding of MC1R to its endogenous ligand, α-MSH, significantly suppressed the activation of macrophages and NF-Κb signaling, and these effects were significantly abolished by decreased protein expression of MC1R via the administration of siRNA [14].
Mykicki et al. showed that activation of MC1R with Nle4-D-Phe7-α-melanocyte-stimulating hormone (NDP-MSH) reduced neuroinflammation by restoring the functions of the BBB and regulatory T cells (Treg) in a mice model of autoimmune encephalomyelitis [12]. Moreover, MC1R displayed a strong ability to inhibit neutrophil recruitment following cerebral ischemia-perfusion injury. Consistent with the present study, BMS-470539, the selective agonist of MC1R, was reported to display anti-inflammatory and neuroprotective effects as well [39]. In the present study, the results showed that BMS-470539 treatment significantly improved neurological deficits, attenuated inflammatory responses, and reduced neutrophil infiltration by decreasing the levels of pro-inflammatory molecules (IL-1β, TNF-α, MPO). Additionally, BMS-470539 treatment increased the number of CD206+ microglia. However, the selective inhibition of MC1R with MSG-606 significantly reversed the neuroprotective effects of BMS-470539. Moreover, the administration of BMS-470539 did not increase the level of MC1R, which means that the activation of MC1R by BMS-470539 may lead to the structure changes of MC1R, but not the level.
Next, we explored the molecular basis of MC1R-mediated anti-inflammatory effects after SAH. AMPK, an evolutionarily conserved energy sensor, was mainly regulated by three upstream kinases: liver kinase B (LKB), Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKK-β) and transforming growth factor-β-activated kinase 1 (TAK1) [40]. The activation of AMPK was reported to promote the transformation of microglia/macrophages to M2 phenotype and reduce pro-inflammatory molecules [41, 42]. Moreover, AMPK was also reported to exert its anti-inflammatory effects by activating TBK1 and suppressing NF-κB [19]. In this study, we found that the levels of p-AMPK and p-TBK1 were increased in parallel with MC1R (3 h, 6 h, 12 h, 24 h after SAH) while the level of p-NF-κB was decreased at 24 h after SAH. Thus, the evidence above indicated that AMPK and TBK1 might participate in MC1R-mediated anti-inflammatory effects. Since MC1R is expressed on various cells of the CNS and little is documented about the anti-inflammatory effects of MC1R/AMPK/TBK1/NF-κB pathway in microglia, we utilized MRT-68601 (TBK1 inhibitor) to inhibit the activation of TBK1 selectively. The results suggested that MRT-68601 administration significantly reversed the neuroprotective effects of MC1R and AMPK by decreasing the levels of IL-1β, TNF-α, and MPO. The evidence above confirmed that activation of MC1R with BMS-470539 suppressed inflammation, promoted M2 transformation and reduced neutrophil infiltration, which was, at least partly mediated by the AMPK/TBK1/NF-κB signaling pathway.
Some limitations of this study should not be ignored. First, although MC1R exerts its neuroprotective effects through many different pathways (AMPK, PKA, et al), we only studied the AMPK-dependent pathway. We will keep studying other pathways in the next study. Second, we only evaluated the anti-inflammatory characteristics of MC1R in a rat model of SAH, without further investigation regarding its roles in anti-apoptosis, anti-autophagy, anti-oxidative stress, etc. Third, although we verified a novel mechanism of MC1R-mediated neuroprotection via the AMPK/TBK1/NF-κB signaling pathway, the exact mechanism how MC1R upregulates the level of p-AMPK after SAH is still unclear. Fourth, the inhibitor of MC1R, MSG-606, also has some effects on MC3R, which in some degrees weaken the credibility of the conclusions of this study. Therefore, more specific inhibitors of MC1R is needed to verify these results. Moreover, further studies should also focus on exploring more detailed characteristics of MC1R and the underlying mechanism of how MC1R increases the level of p-AMPK.
Conclusions
Activation of MC1R by BMS-470539 reduces pro-inflammatory molecules, promotes the transformation of microglia to the M2 phenotype, and alleviates the neurological and cognitive deficits via the AMPK/TBK1/NF-κB signaling pathway in a rat model of SAH.
Electronic supplementary material
(PDF 463 kb)
(DOCX 22 kb)
Acknowledgements
This work was supported by the National Institutes of Health (Grants NS081740 and NS082184); National Key Research and Development Program of China (2017YFC1308500), Key Program of Science and Technology Development of Zhejiang (2017C03021); Grant WKJ-ZJ-1615 (2016149634) from a major science and technology project in medical and health of Zhejiang province. Basic Public Interest Research Plan of Zhejiang Province (LY18H090001).
Author Contributions Statement
This study was designed by WX, JM, JT, JZ, and JHZ. The experiments were completed by WX, JM, UO, ZDT, TZ, PW, TL, and YZ. JP, BE and AS performed statistical analysis. WX and JM finished writing the manuscript. JZ and JHZ provided the supervision and final check. All the authors read the final version of this paper and approved it.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Abbreviations
- EBI
Early brain injury
- SAH
Subarachnoid hemorrhage
- MC1R
Melanocortin 1 receptor
- AMPK
Adenosine monophosphate-activated protein kinase
- TBK1
TANK-binding kinase 1
- CNS
Central nervous system
- IKK
IκB kinase β
- SD
Sprague–Dawley
- MPO
Myeloperoxidase
- GFAP
Glial fibrillary acidic protein
- LKB
Liver kinase B
- CaMKK-β
Ca2+/calmodulin-dependent protein kinase kinase-β
- TAK1
Transforming growth factor-β-activated kinase 1
Compliance with Ethical Standards
Ethical Approval and Consent to Participate
All animal experiments were performed according to the Institutional Animal Care and Use Committee (IACUC) at Loma Linda University. The procedures were conducted according to the National Institutes of Health’s Guide for the Care and the Use of Laboratory Animals and the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
Availability of Supporting Data
The datasets analyzed during the present study are available from the corresponding author on reasonable request.
Competing Interests
The authors report no conflicts of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Weilin Xu and Jun Mo contributed equally to this work.
Contributor Information
Jianmin Zhang, Email: zjm135@zju.edu.cn.
John H. Zhang, Email: johnzhang3910@yahoo.com
References
- 1.Laiwalla AN, Ooi YC, Liou R, Gonzalez NR. Matched cohort analysis of the effects of limb remote ischemic conditioning in patients with aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2016;7(1):42–8. doi: 10.1007/s12975-015-0437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu W, Gao L, Li T, Zheng J, Shao A, Zhang J. Apelin-13 Alleviates early brain injury after subarachnoid hemorrhage via suppression of endoplasmic reticulum stress-mediated apoptosis and blood-brain barrier disruption: possible involvement of ATF6/CHOP pathway. Neuroscience. 2018;388:284–296. doi: 10.1016/j.neuroscience.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Pang J, Chen Y, Kuai L, Yang P, Peng J, Wu Y, Chen Y, Vitek MP, Chen L, Sun X, Jiang Y. Inhibition of blood-brain barrier disruption by an apolipoprotein E-mimetic peptide ameliorates early brain injury in experimental subarachnoid hemorrhage. Transl Stroke Res. 2017;8(3):257–272. doi: 10.1007/s12975-016-0507-1. [DOI] [PubMed] [Google Scholar]
- 4.Zhou K, Enkhjargal B, Xie Z, Sun C, Wu L, Malaguit J, Chen S, Tang J, Zhang J, Zhang JH. Dihydrolipoic acid inhibits lysosomal rupture and NLRP3 through lysosome-associated membrane protein-1/calcium/calmodulin-dependent protein kinase II/TAK1 pathways after subarachnoid hemorrhage in rat. Stroke. 2018;49:175–183. doi: 10.1161/STROKEAHA.117.018593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang J, Peng J, Matei N, Yang P, Kuai L, Wu Y, Chen L, Vitek MP, Li F, Sun X, Zhang JH, Jiang Y. Apolipoprotein E exerts a whole-brain protective property by promoting M1? Microglia quiescence after experimental subarachnoid hemorrhage in mice. Transl Stroke Res. 2018;9(6):654–668. doi: 10.1007/s12975-018-0665-4. [DOI] [PubMed] [Google Scholar]
- 7.Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Progress in Neurobiology. 2016;142:23–44. doi: 10.1016/j.pneurobio.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Huang XP, Peng JH, Pang JW, Tian XC, Li XS, Wu Y, Li Y, Jiang Y, Sun XC. Peli1 contributions in microglial activation, neuroinflammatory responses and neurological deficits following experimental subarachnoid hemorrhage. Frontiers in Molecular Neuroscience. 2017;10:398. doi: 10.3389/fnmol.2017.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai W, Liu S, Hu M, Sun X, Qiu W, Zheng S, Hu X, Lu Z. Post-stroke DHA treatment protects against acute ischemic brain injury by skewing macrophage polarity woward the M2 phenotype. Transl Stroke Res. 2018;9(6):669–680. doi: 10.1007/s12975-018-0662-7. [DOI] [PubMed] [Google Scholar]
- 10.Wolf Horrell EM, Boulanger MC, D'Orazio JA. Melanocortin 1 Receptor: structure, function, and regulation. Frontiers in Genetics. 2016;7:95. doi: 10.3389/fgene.2016.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catania A. Neuroprotective actions of melanocortins: a therapeutic opportunity. Trends Neurosci. 2008;31(7):353–60. doi: 10.1016/j.tins.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Mykicki N, Herrmann AM, Schwab N, Deenen R, Limmer ST, Wachsmuth LA, Klotz L, Köhrer K, Faber C, Wiendl H, Luger TA, Meuth SG, Loser K. Melanocortin-1 receptor activation is neuroprotective in mouse models of neuroinflammatory disease. Sci. Transl. Med. 2016;8:1–16. doi: 10.1126/scitranslmed.aaf8732. [DOI] [PubMed] [Google Scholar]
- 13.Auriemma M, Brzoska T, Klenner L, Kupas V, Goerge T, Voskort M, Zhao Z, Sparwasser T, Luger TA, Loser K. alpha-MSH-stimulated tolerogenic dendritic cells induce functional regulatory T cells and ameliorate ongoing skin inflammation. The Journal of Investigative Dermatology. 2012;132:1814–1824. doi: 10.1038/jid.2012.59. [DOI] [PubMed] [Google Scholar]
- 14.Li, D. & Taylor, A.W. Diminishment of alpha-MSH anti-inflammatory activity in MC1r siRNA-transfected RAW264.7 macrophages. Journal of Leukocyte Biology. 2008; 84:191-198. [DOI] [PMC free article] [PubMed]
- 15.Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T, Wan J, Zhu W, Wang J. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain, behavior, and immunity. 2017;61:326–339. doi: 10.1016/j.bbi.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gatti S, Lonati C, Acerbi F, Sordi A, Leonardi P, Carlin A, Gaini SM, Catania A. Protective action of NDP-MSH in experimental subarachnoid hemorrhage. Exp Neurol. 2012;234(1):230–8. doi: 10.1016/j.expneurol.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 17.Hall, D.T., Griss, T., Ma, J.F., Sanchez, B.J., Sadek, J., Tremblay, A.M.K., Mubaid, S., Omer, A., Ford, R.J., Bedard, N., Pause, A., Wing, S.S., Di Marco, S., Steinberg, G.R., Jones, R.G. & Gallouzi, I.E. The AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), but not metformin, prevents inflammation-associated cachectic muscle wasting. EMBO Molecular Medicine. 2018;10. [DOI] [PMC free article] [PubMed]
- 18.Oh TS, Cho H, Cho JH, Yu SW, Kim EK. Hypothalamic AMPK-induced autophagy increases food intake by regulating NPY and POMC expression. Autophagy. 2016;12(11):2009–2025. doi: 10.1080/15548627.2016.1215382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172(4):731–743. doi: 10.1016/j.cell.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, Brittain GC, Chang M, Cheng X, Sun SC. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nature Immunology. 2012;13:1101–1109. doi: 10.1038/ni.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang L, McIntyre KW, Gillooly KM, Yang Y, Haycock J, Roberts S, Khanna A, Herpin TF, Yu G, Wu X, Morton GC, Tuerdi H, Koplowitz B, Walker SG, Wardwell-Swanson J, Macor JE, Lawrence RM, Carlson KE. A selective small molecule agonist of the melanocortin-1 receptor inhibits lipopolysaccharide-induced cytokine accumulation and leukocyte infiltration in mice. Journal of Leukocyte Biology. 2006;80:897–904. doi: 10.1189/jlb.1204748. [DOI] [PubMed] [Google Scholar]
- 22.Leoni G, Voisin MB, Carlson K, Getting S, Nourshargh S, Perretti M. The melanocortin MC1 receptor agonist BMS-470539 inhibits leucocyte trafficking in the inflamed vasculature. British Journal of Pharmacology. 2010;160:171–180. doi: 10.1111/j.1476-5381.2010.00688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke. 2010;41:1783–1790. doi: 10.1161/STROKEAHA.110.586537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. Journal of Neuroscience Methods. 2009;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Z, Huang L, Enkhjargal B, Reis C, Wan W, Tang J, Cheng Y, Zhang JH. Recombinant Netrin-1 binding UNC5B receptor attenuates neuroinflammation and brain injury via PPARgamma/NFkappaB signaling pathway after subarachnoid hemorrhage in rats. Brain, Behavior, and Immunity. 2018;69:190–202. doi: 10.1016/j.bbi.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Fujimoto M, Nakano F, Nishikawa H, Okada T, Kawakita F, Imanaka-Yoshida K, Yoshida T, Suzuki H. Deficiency of tenascin-C alleviates neuronal apoptosis and neuroinflammation after experimental subarachnoid hemorrhage in mice. Mol Neurobiol. 2018;55(11):8346–8354. doi: 10.1007/s12035-018-1006-z. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Zhao L, Sherchan P, Ding Y, Yu J, Nowrangi D, Tang J, Xia Y, Zhang JH. Activation of melanocortin receptor 4 with RO27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. Journal of Neuroinflammation. 2018;15:106. doi: 10.1186/s12974-018-1140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Wu Q, Zhang Q, Lu Y, Liu J, Li W, Lv S, Zhou M, Zhang X, Hang C. Resveratrol attenuates early brain injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Frontiers in Neuroscience. 2017;11:611. doi: 10.3389/fnins.2017.00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang, Z., Zhou, F., Dou, Y., Tian, X., Liu, C., Li, H., Shen, H. & Chen, G. Melatonin alleviates intracerebral hemorrhage-induced secondary brain injury in rats via suppressing apoptosis, inflammation, oxidative stress, DNA damage, and mitochondria injury. Transl Stroke Res. 2018; 9(1):74-91. [DOI] [PMC free article] [PubMed]
- 30.Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotoxicity Research. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- 31.Catania A, Lonati C, Sordi A, Carlin A, Leonardi P, Gatti S. The melanocortin system in control of inflammation. The Scientific World Journal. 2010;10:1840–1853. doi: 10.1100/tsw.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleiner S, Braunstahl GJ, Rudrich U, Gehring M, Eiz-Vesper B, Luger TA, Steelant B, Seys SF, Kapp A, Bohm M, Hellings PW, Raap U. Regulation of melanocortin 1 receptor in allergic rhinitis in vitro and in vivo. Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology. 2016;46:1066–1074. doi: 10.1111/cea.12759. [DOI] [PubMed] [Google Scholar]
- 33.Elvin J, Buvall L, Lindskog Jonsson A, Granqvist A, Lassén E, Bergwall L, Nyström J, Haraldsson B. Melanocortin 1 receptor agonist protects podocytes through catalase and RhoA activation. Am J Physiol Renal Physiol. 2016;310(9):F846–56. doi: 10.1152/ajprenal.00231.2015. [DOI] [PubMed] [Google Scholar]
- 34.Schaible EV, Steinsträßer A, Jahn-Eimermacher A, Luh C, Sebastiani A, Kornes F, Pieter D, Schäfer MK, Engelhard K, Thal SC. Single administration of tripeptide α-MSH (11-13) attenuates brain damage by reduced inflammation and apoptosis after experimental traumatic brain injury in mice. PLoS One. 2013;8(8):e71056. doi: 10.1371/journal.pone.0071056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen T, Wang W, Li JR, Xu HZ, Peng YC, Fan LF, Yan F, Gu C, Wang L, Chen G. PARP inhibition attenuates early brain injury through NF-kappaB/MMP-9 pathway in a rat model of subarachnoid hemorrhage. Brain research. 1644;2016:32–38. doi: 10.1016/j.brainres.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Yan F, Tan X, Wan W, Dixon BJ, Fan R, Enkhjargal B, Li Q, Zhang J, Chen G, Zhang JH. ErbB4 protects against neuronal apoptosis via activation of YAP/PIK3CB signaling pathway in a rat model of subarachnoid hemorrhage. Experimental Neurology. 2017;297:92–100. doi: 10.1016/j.expneurol.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Hamming AM, van der Toorn A, Rudrapatna US, Ma L, van Os HJ, Ferrari MD, van den Maagdenberg AM, van Zwet E, Poinsatte K, Stowe AM, Dijkhuizen RM, Wermer MJ. Valproate reduces delayed brain injury in a rat model of subarachnoid hemorrhage. Stroke. 2017;48:452–458. doi: 10.1161/STROKEAHA.116.014738. [DOI] [PubMed] [Google Scholar]
- 38.Chen W, Li J, Qu H, Song Z, Yang Z, Huo J, Jiang H, Huang Q, Huo M, Liu B, Zhang Q. The melanocortin 1 receptor (MC1R) inhibits the inflammatory response in Raw 264.7 cells and atopic dermatitis (AD) mouse model. Molecular Biology Reports. 2013;40:1987–1996. doi: 10.1007/s11033-012-2256-x. [DOI] [PubMed] [Google Scholar]
- 39.Holloway, P.M., Durrenberger, P.F., Trutschl, M., Cvek, U., Cooper, D., Orr, A.W., Perretti, M., Getting, S.J. & Gavins, F.N. Both MC1 and MC3 receptors provide protection from cerebral ischemia-reperfusion-induced neutrophil recruitment. Arterioscler Thromb Vasc Biol. 2015; 35(9):1936-44. [DOI] [PMC free article] [PubMed]
- 40.Hardie DG. AMPK--sensing energy while talking to other signaling pathways. Cell metabolism. 2014;20:939–952. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu F, Cui WQ, Wei Y, Cui J, Qiu J, Hu LL, Gong WY, Dong JC, Liu BJ. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J Exp Clin Cancer Res. 2018;37(1):207. doi: 10.1186/s13046-018-0878-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181(12):8633–41. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 463 kb)
(DOCX 22 kb)