

Abstract
Solid tumors frequently metastasize to bone and induce bone destruction leading to severe pain, fractures, and other skeletal-related events (SREs). Osteoclast inhibitors such as bisphosphonates delay SREs but do not prevent skeletal complications or improve overall survival. Because bisphosphonates can cause adverse side effects and are contraindicated for some patients, we sought an alternative therapy to reduce tumor-associated bone destruction. Our previous studies identified the transcription factor Gli2 as a key regulator of parathyroid hormone-related protein (PTHrP), which is produced by bone metastatic tumor cells to promote osteoclast-mediated bone destruction. In this study, we tested the treatment effect of a Gli antagonist GANT58, which inhibits Gli2 nuclear translocation and PTHrP expression in tumor cells. In initial testing, GANT58 did not have efficacy in vivo due to its low water solubility and poor bioavailability. We therefore developed a micellar nanoparticle (NP) to encapsulate and colloidally stabilize GANT58, providing a fully aqueous, intravenously injectable formulation based on the polymer poly(propylene sulfide)135-b-poly[(oligoethylene glycol)9 methyl ether acrylate]17 (PPS135-b-POEGA17). POEGA forms the hydrophilic NP surface while PPS forms the hydrophobic NP core that sequesters GANT58. In response to reactive oxygen species (ROS), PPS becomes hydrophilic and degrades to enable drug release. In an intratibial model of breast cancer bone metastasis, treatment with GANT58-NPs decreased bone lesion area by 49% (p < 0.01) and lesion number by 38% (p < 0.05) and resulted in a 2.5-fold increase in trabecular bone volume (p < 0.001). Similar results were observed in intracardiac and intratibial models of breast and lung cancer bone metastasis, respectively. Importantly, GANT58-NPs reduced tumor cell proliferation but did not alter mesenchymal stem cell proliferation or osteoblast mineralization in vitro, nor was there evidence of cytotoxicity after repeated in vivo treatment. Thus, inhibition of Gli2 using GANT58-NPs is a potential therapy to reduce bone destruction that should be considered for further testing and development toward clinical translation.
Keywords: Bone metastasis, Breast cancer, Nanoparticle, Gli2, PTHrP, GANT58
Graphical abstract:
Introduction
Despite advances in early screening and adjuvant therapy, metastatic disease remains a leading cause of cancer patient morbidity and mortality. One of the most common metastatic sites is the skeleton. Approximately 70–80% of patients with breast or prostate cancer and 30–40% of patients with lung or renal cancer who die from disease develop bone metastases [1, 2]. Patients with bone metastases experience complications including severe bone pain, pathological fractures, and other skeletal-related events (SREs) that significantly reduce their quality of life [2, 3].
Once established in bone, cancer cells disrupt normal bone remodeling to initiate a vicious cycle of tumor-induced bone disease (TIBD) [4]. Specifically, tumor cells in the bone microenvironment secrete parathyroid hormone-related protein (PTHrP), which increases osteoblast expression of receptor activator of nuclear factor kappa-B ligand (RANKL) and consequent osteoclast-mediated bone resorption [5, 6]. Current treatment strategies for TIBD include the anti-resorptive therapies denosumab (RANKL inhibitor) and bisphosphonates (osteoclast inhibitor) [1, 2, 7]. However, these drugs are associated with increased risk of atypical femoral fractures and osteonecrosis of the jaw [8–12]. Conventional chemotherapy and radiation therapies suppress bone marrow cells, resulting in adverse hematologic events and poor bone quality [13, 14]. Tumor-targeted therapies that reduce SREs while minimizing damage to the bone are currently unavailable.
The Hedgehog (Hh) signaling pathway is a key regulator of embryonic development with essential roles in cell differentiation and proliferation, tissue polarity, and stem cell maintenance [15, 16] but has limited activity in the healthy adult skeleton. Aberrant Hh signaling in adults has been implicated in the development and progression of breast, prostate, and lung cancer [17–20]. However, pharmacological inhibitors of the Hh receptor Smoothened (Smo) failed to show significant clinical benefit in patients with advanced solid tumors [21–23] due to acquired mutations in Smo and/or non-canonical Hh pathway activation [24–27]. Small molecule inhibitors that target the Hh transcription factor Gli2 downstream of Smo circumvent these resistance mechanisms [28, 29]. Our previous studies demonstrated that Gli2 stimulates PTHrP expression in bone-destructive tumor cells [30, 31] and that genetic inhibition of Gli2 attenuates the ability of cancer cells to colonize bone and induce osteolysis in vivo [26]. The small molecule Gli-antagonists GANT58 and GANT61 have shown promising anti-tumor effects in vitro and in xenograft models [28, 32–34], but their low water solubility and poor pharmacokinetics (PK) has limited their testing to less translationally-relevant studies using direct injection into subcutaneous tumors. Thus, while Gli2 is a promising therapeutic target for TIBD, small molecule Gli inhibitors have not been tested using systemic delivery nor in models of bone metastasis.
Nanoparticle (NP) drug delivery systems have emerged in recent years as a promising approach to overcome PK and toxicity limitations of otherwise promising drug candidates. Cancer nanomedicines have been widely reported as carriers for chemotherapeutics [35, 36]. However, molecularly targeted agents (MTAs) offer multiple benefits over conventional chemotherapies [37–39]. Most notably, their selectivity reduces normal tissue toxicity, thereby improving the therapeutic index [40]. GANT58 is a therapeutically promising MTA with limited bioavailability, and thus is an excellent candidate for development in a nanoparticle formulation to improve its PK in vivo. Polymer-based micellar nanoparticles have been shown to enhance the solubility and systemic PK of hydrophobic compounds in vivo and have also produced clinical success in cancer patients [41–46]. Further, advances in polymer science toward environmentally-responsive, “smart” polymer formulations have improved target-specific drug delivery [47–50].
Herein, we employ polymeric nanoparticles to encapsulate GANT58 (GANT58-NPs) to enable systemic delivery with distribution to breast and lung cancer bone metastases. Following NP characterization, evaluation of in vitro toxicity, and in vivo biodistribution and PK studies, we tested GANT58-NPs in mouse models of bone metastasis in order to investigate its therapeutic efficacy and safety. We hypothesized that GANT58-NPs delivered intravenously (i.v.) in a fully-aqueous formulation would reduce tumor-induced bone destruction with minimal effects on bone marrow progenitors.
Materials and methods
Cell lines and reagents
Bone-metastatic variants of the human breast cancer cell line MDA-MB-231 and human squamous non-small cell lung carcinoma cell line RWGT2 were generated in our laboratory as previously published [26, 27, 30, 51]. MDA-MB-231-bone and RWGT2-bone clones were maintained in DMEM (Cellgro) and α-MEM (Cellgro) respectively, supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories) and 1% penicillin/streptomycin (P/S; Mediatech). Human mesenchymal stem cells (hMSCs; Extem Biosciences) were maintained in Mesenchymal Stem Cell Growth Medium 2 (PromoCell). GANT58 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). All other reagents were purchased from Sigma Aldrich (St. Louis, MO, USA) unless otherwise specified.
Immunohistologic staining of patient tumor samples
Bone metastatic (n = 17) and soft tissue (n = 3) tumor biopsies were obtained from the Cooperative Human Tissue Network (CHTN) Western Division in accordance with our Institutional Review Board (IRB #151700)-approved protocol and upon informed consent from patients undergoing surgical resection. All patient information was deidentified prior to receipt by investigators to protect subject privacy. The clinical features of the patient samples are summarized in Table S1. Briefly, fresh tissue samples were fixed in 10% formalin (Fisher Scientific) for 48 hr and stored in 70% ethanol at 4°C before being processed and embedded in paraffin. Serial sections (5-μm thickness) were placed on slides, deparaffinized in xylene, and rehydrated with graded alcohol solutions, followed by antigen retrieval in 10 mM sodium citrate buffer at 80°C for 30 min. Sections were then blocked with 5% goat serum in phosphate-buffered saline (PBS)/0.1% Tween-20 for 30 min and incubated with rabbit polyclonal anti-Gli2 primary antibody (1:500, Novus Biologicals) overnight at 4°C. The VECTASTAIN Elite ABC HRP Kit (Vector Laboratories) with biotinylated goat anti-rabbit secondary antibody and the ImmPACT NovaRED Peroxidase Substrate Kit (Vector Laboratories) were both used according to the manufacturer’s protocol. After counterstaining with hematoxylin, tumor sections were mounted with Cytoseal XYL (Thermo Scientific) and imaged using an Olympus BX41 microscope equipped with an Olympus DP71 camera. Gli2-positive staining was quantified using Metamorph software (Molecular Devices, Inc.).
In silico analyses of patient tumors
The Gene Expression Omnibus (GEO) database was queried on 11 October 2018 for the GEO accession number GSE14017 to obtain the gene expression profiling dataset of 29 human breast cancer metastases in different organs, which was generated by X.H. Zhang, et al. [52]. For the purposes of their study, the authors used the Affymetrix Human Genome U133 Plus 2.0 Array (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14017) [52]. To determine Gli2 expression in bone, brain, and lung cancer metastases of breast cancer patients, the identifiers corresponding to Gli2 (207034_s_at, 228537_at, 208057_s_at) were located on the platform record, and gene expression values across dataset samples were retrieved. One brain metastasis (GSM352120) and one lung metastasis (GSM352127) sample from the microarray dataset were omitted from final statistical analyses after performing the Grubb’s test for outliers (GraphPad Software, Inc.).
Polymer synthesis and characterization
The diblock copolymer poly(propylene sulfide)135-b-poly[(oligoethylene glycol)9 methyl ether acrylate]17 (PPS135-b-POEGA17) was synthesized via a combination of anionic and reversible addition-fragmentation chain transfer (RAFT) polymerization as previously published [41, 42]. Briefly, the PPS block was synthesized via anionic polymerization to a degree of polymerization of approximately 135 (10 kDa), conjugated to the RAFT macro-chain transfer agent, and chain-extended with POEGA to a degree of polymerization of approximately 17 (8 kDa). Polymer structure was confirmed by 1H-NMR. Fluorophore-grafted polymers were synthesized by adding a small amount of the amine-reactive intermediate pentafluorophenyl acrylate (PFPA) into the reaction mixture with POEGA at a 1:1 mole ratio (PFPAPPS-ECT). The reaction was allowed to proceed for 24 h at 70°C. The reaction was dialyzed against methanol and dried under vacuum. The resulting polymer (0.021 mmol) was reacted with Cy7-amine or Cy5-amine fluorophores (0.042 mmol) in DMSO for 24 hr at 50°C. The reaction mixture was again dialyzed against methanol and dried under vacuum.
GANT58-NP preparation and characterization
GANT58-loaded PPS 135-b-POEGA17 micellar nanoparticles (GANT58-NPs) were formed by the bulk solvent evaporation method. PPS135-b-POEGA17 (10 mg) was co-dissolved with GANT58 (2.5 mg) in chloroform (0.1 mL) and added dropwise to a vial containing vigorously-stirring PBS (1 mL). The oil-in-water biphasic solution was left stirring overnight to evaporate the chloroform and allow for micelle formation. The resulting micelle solution was passed through a 0.45 μm syringe filter producing the final GANT58-NP formulation. The same technique, excluding addition of GANT58, was used to create empty PPS135-b-POEGA17 micellar nanoparticles (Empty-NPs). Dynamic light scattering (DLS) was used to measure the hydrodynamic diameter (Dh) and zeta potential (ζ) of the GANT58-NPs and Empty-NPs via a Malvern Zetasizer Nano-ZS (Malvern Instruments Ltd., Worcestershire, UK) equipped with a 4 mW He-Ne laser operating at λ = 632.8 nm. Transmission electron microscopy (TEM) samples were prepared as previously described [42]. Briefly, 5 μL of GANT58-NPs were added to a pure carbon TEM grid (Ted Pella, Inc., Redding, CA, USA), blotted dry after 60 s, and counterstained with 3% uranyl acetate for 20 s. After vacuum drying, the grids were imaged on an FEI Tecnai Osiris microscope operating at 200 kV for TEM. GANT58 loading was quantified utilizing the fluorescence properties of GANT58. Aliquots of GANT58-NPs in PBS (50 μL) were added in triplicate to a 96-well plate and dissolved by adding an equal volume of DMF. On the same plate, a standard of GANT58 in the same solvent (1:1 DMF:PBS) was prepared. Fluorescence intensity of GANT58 (ex. 485 nm, em. 590 nm) was measured on a micro-plate reader (Tecan Infinite 500, Tecan Group Ltd., Mannedorf, Switzerland) and GANT58 concentration was calculated from the standard curve.
Critical micelle concentration (CMC) determination
The CMC was determined as previously described [41, 42]. Nile Red (NR) was substituted for GANT58 as the loaded species due to its unique fluorescence properties and its similar molecular weight to GANT58. NR is highly fluorescent in hydrophobic environments yet non-fluorescent in polar, aqueous environments, making it useful for characterizing self-assembly properties. Briefly, NR-loaded PPS135-b-POEGA17 micelles (NR-NPs) were prepared by the described bulk solvent evaporation method. NR loading was measured as described for GANT58. Different dilutions of the NR-NPs were prepared in PBS, and NR fluorescence (ex. 535 nm, em. 612 nm) was read on a micro-plate reader (Tecan Infinite 500, Tecan Group Ltd., Mannedorf, Switzerland). The CMC was defined as the intersection point on the plot of NR fluorescence versus polymer concentration as previously described [41, 42].
Hydrogen peroxide (H2O2)-dependent drug release
The ROS-responsive behavior of the PPS135-b-POEGA17 NPs was assessed as described previously, using H2O2 as the ROS-species [42]. Briefly, NR-NPs prepared as described were exposed to a range of concentrations (0–1.5 M) of H2O2. Fluorescence intensity of NR was monitored in a 96-well plate using a micro plate reader (Tecan Infinite 500, Tecan Group Ltd., Mannedorf, Switzerland). Release of the NR due to NP oxidation and destabilization was assessed over time based on disappearance of NR fluorescence. The loss of fluorescence for each sample at each time point was determined by subtracting the fluorescent value from that of the sample prior to H2O2 addition, and the percent fluorescence remaining was determined by normalization to the same value (before addition of H2O2). This value for percent fluorescence remaining was subtracted from 100% and expressed as a percent release for each sample at each time point.
Quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from MDA-MB-231 bone cells after 48 hr GANT58 (0–20 μM) treatment with the RNeasy Mini Kit (Qiagen) as per manufacturer’s instructions. Complimentary DNA (cDNA) was synthesized from 1 μg RNA using the qScript cDNA SuperMix (Quanta Biosciences) and serially diluted to create a standard curve. qPCR was performed on a 7500 Real-Time PCR System (Applied Biosciences) using TaqMan Universal PCR Master Mix (Applied Biosystems) and the TaqMan primers for eukaryotic 18S rRNA (4352930E, Applied Biosystems) or human PTHLH (Hs00174969_m1, Thermo Fisher) under the following conditions: 10 min at 95°C, (15 s at 95°C, 1 min at 60°C) × 40 cycles. Three technical replicates were run for each biological replicate. Quantification was performed using the absolute quantitative method using 18S as an internal control.
Western blotting
Nuclear and cytoplasmic protein lysates were isolated from MDA-MB-231 bone cells after 72 hr treatment with GANT58 (0–20 μM) using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Protein concentrations were quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific). Protein samples (20 μg/well) were separated on a 4–20% Mini-PROTEAN TGX polyacrylamide gel (Bio-Rad) by SDS-PAGE prior to being transferred to a nitrocellulose membrane (Bio-Rad) with the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were then blocked for 1 hr in 1X TBS containing 0.1% Tween-20 and 5% w/v BSA and incubated with the following primary antibodies at 4°C overnight: anti-Gli2 (1:500, Novus Biologicals), anti-TATA binding protein (TBP) (1:1000, Cell Signaling), or anti-Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:5000, Cell Signaling). Following incubation with anti-rabbit IgG HRP-linked secondary antibody (Cell Signaling) at room temperature for 1 hr, protein bands were developed by Western Lightning Plus-ECL (Perkin Elmer) and imaged on a ChemiDoc MP Imaging System (Bio-Rad). The intensity of each band was determined by densitometry using ImageJ software.
Tumor cell proliferation assay
MDA-MB-231-bone cells were seeded in a 96-well plate at 2,000 cells/well in quadruplicate. Vehicle (DMSO), free GANT58, Empty-NPs, or GANT58-NPs were introduced to wells after 24 hr. Cell proliferation was determined after 24 hr treatment by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega) per the manufacturer’s instructions. Absorbance values were measured on a plate reader at optical density (OD) 450 nm.
hMSC proliferation assay
hMSCs were seeded in a 96-well plate at 50,000 cells/mL (5,000 cells/well). Vehicle (DMSO), free GANT58, Empty-NPs, or GANT58-NPs were introduced to the wells after 24 hr. As described, cell proliferation was determined using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega) per manufacturer’s instructions. Absorbance values were measured at OD 490 nm.
Fluorescence microscopy
MDA-MB-231-bone cells were seeded in a 4-chamber well slide at 20,000 cells/well. After 24 hr, media was replaced with media containing treatments. For NP uptake experiments, GANT58-Cy5NPs were added and incubated for 12 hr after treatment. Cells were then stained with DAPI and viewed on a Nikon Eclipse TI confocal microscope for Cy5 and DAPI fluorescence. For Gli2 immunofluorescence, cells were incubated with 20 μM GANT58, 20 μM GANT58-NP, Empty-NP, or DMSO control for 72 hr, fixed with 4% neutral-buffered formalin, and washed with Tris-buffered saline (TBS). The cells were then blocked and permeabilized with TBS containing 2.5% BSA, 0.1% Tween, and 0.2% Triton-X for 5 min. Gli2 antibody (1:500, Novus Biologicals) was then incubated in permeabilization buffer overnight at 4°C. Cells were then washed again with TBS and incubated with secondary Alexa-488 anti-rabbit antibody (1:2000, Thermo Fisher) for 90 min. The cells were then stained with DAPI for 10 min, washed, and mounted with a cover slip. Cells were imaged using an Olympus BX60 fluorescence microscope for DAPI and Alexa-488 fluorescence. ImageJ was used to calculate the Manders coefficient to quantify Gli2 nuclear colocalization.
Cell viability, cytotoxicity, and apoptosis
MDA-MB-231 bone cells were seeded in a 96-well plate (5,000 cells/well) and treated with GANT58 (0–80 μM) for 24 hr and viability, cytotoxicity, and apoptosis were measured using the ApoTox-Glo Triplex Assay (Promega) according to the manufacturer’s instructions. Briefly, 20 μL of Viability/Cytotoxicity Reagent was added to 100 μL of media in each well and incubated for 30 min at 37°C. Fluorescence intensity was then measured for viability (ex. 400 nm, em. 505 nm) and cytotoxicity (ex. 485 nm, em. 520 nm). Luminescence was measured for apoptosis (caspase 3/7 activation) following the addition of 100 μL of Caspase-Glo 3/7 Reagent and 30 min incubation at room temperature.
Mineralization assay
hMSCs were seeded at 25,000 cells/mL (50,000 cells/well) in a 24-well plate and allowed to grow to confluency for 48 hr in MSC Growth Medium 2 (PromoCell) as we have described previously [53]. Cells were then induced with MSC Osteogenic Differentiation Medium (PromoCell) treated with vehicle (DMSO), free GANT58, Empty-NPs, or GANT58-NPs. Cells were cultured for 14 days and media was changed every third day. A cohort of wells remained in MSC Growth Media and served as a control. Cells were then washed with PBS and fixed in 10% formalin for 45 min, and stained with Alizarin Red S (80 mM) for 30 min. After staining, cells were washed five times with deionized (DI) water and imaged under an inverted microscope. Alizarin dye was then extracted with 5% SDS for 1 hr. Absorbance of the extracted dye was read on a plate reader at OD 405 nm.
Osteoclastogenesis assay
Mouse bone marrow-derived stromal cells were obtained from C57BL/6J mice and used in an osteoclastogenesis coculture assay as previously described [54, 55]. Briefly, femora and tibiae were dissected, and both ends cut off. Bone marrow cells were flushed out and collected via centrifugation, suspended in α-MEM with 10% FBS, and plated in 100 mm culture dishes. After 2 hr, nonadherent cells were harvested and pelleted. The non-adherent bone marrow cells (500,000 cells/well) and MDA-MB-231-bone cells supplemented with 10 ng/mL TGF-P (1000 cells/well) were plated in 48-well plates in 300 μL α-MEM (day 1). Treatments with Free GANT58, Empty-NPs, and GANT58-NPs started on day 1. On day 2, 300 μL of fresh α-MEM supplemented with treatments was added to each well, and 300 μL was replaced with fresh, treatment supplemented media each subsequent day until fixation. On day 6, cells were fixed and stained for TRAP using a TRAP kit (Sigma) and counterstained with hematoxylin. TRAP-positive cells with 3 or more nuclei were counted as osteoclasts. Vitamin D3 was used as a positive control for osteoclast formation, whereas absence of MDA-MB-231-bone cells and vitamin D3 was used as a negative control.
NP cargo biodistribution
Athymic female nude mice (4–6 weeks old, Envigo) were intratibially inoculated with GFP-expressing MDA-MB-231-bone tumor cells and given 14 days to progress into tumors. Two independent experiments were performed to assess the biodistribution of the NP cargo and of the NP polymer. For biodistribution of the NP cargo, the near-IR fluorophore Cy7 was loaded into the NP formulation via the bulk solvent evaporation method to serve as the surrogate fluorescent loading species. Cy7-loaded NPs were then injected via tail vein injection (1 mg/kg Cy7). Mice were then imaged on a Pearl Near-IR imager (Licor) immediately, 2 hr, 6 hr, and 24 hr after injection and the images analyzed using region of interest (ROI) analysis in the Licor software. For biodistribution of the NP polymer, Cy7-grafted GANT58-NPs were injected via tail vein injection (8 mg/kg GANT58). Mice were then sacrificed at 1, 4, and 24 hr post NP-injection. Organs and long bones were imaged on a Pearl Near-IR imager (Licor) and the images analyzed using ROI analysis in the Licor software.
Pharmacokinetics
Rag 2−/− mice were injected with Cy5-grafted GANT58-NPs via retroorbital injection (8 mg/kg GANT58, 100 μL injection) under isofurane anesthesia. Immediately post-injection, 15 min, 30 min, 1 hr, 2 hr, 4 hr, 12 hr, and 24 hr, a small volume of blood (< 5 μL) was drawn via tail-nick into a heparinized capillary tube and deposited into PCR tubes. PCR tubes containing whole blood samples were then frozen until analysis. Blood samples were thawed at time of analysis and diluted 40x in PBS. Samples were then read on a Take3 microvolume plate (Biotek) and a Synergy H1 fluorescence plate reader (Biotek) and background fluorescence was subtracted. NP concentration was calculated via a standard curve made by doping Cy5-grafted NPs into fresh mouse blood that was then frozen until time of analysis, at which time it was also diluted 40x [56].
Free GANT58 intratibial mouse model of late bone metastasis
Athymic female nude mice (4–6 weeks old, Harlan) were injected with 2.5 × 105 GFP-expressing MDA-MB-231-bone cells in 10 μL PBS into the left tibia under isoflurane anesthesia as previously described [57]. As a control, the contralateral limb was injected with 10 μL PBS. GANT58 was reconstituted in a 4:1 Cremophor EL:ethanol solution to solubilize the drug. GANT58 treatments started 4 days post-tumor inoculation to allow for tumor establishment (n = 8, GANT58; n = 8, vehicle). Mice were then treated 3 days/week with 50 mg/kg GANT58 via subcutaneous, 100 μL injection. Mice were imaged weekly to track tumor progression and sacrificed at 4 weeks.
GANT58-NP intratibial mouse model of late bone metastasis
Athymic female nude mice (4–6 weeks old, Envigo) were injected with 2.5 × 105 GFP-expressing MDA-MB-231-bone or RWGT2-bone cells in 10 μL PBS into the left tibia under isoflurane anesthesia as previously described [57]. As a control, the contralateral limb was injected with 10 μL PBS. GANT58-NP and Empty-NP treatment started 4 days post-tumor inoculation to allow for tumor establishment (n = 12, GANT58-NP; n = 12, Empty-NP). Mice were then treated 5 days/week with 8 mg/kg GANT58 in the GANT58-NP formulation or an equivalent polymer dose of Empty-NPs in 100 μL of PBS via tail vein injection. Mice were imaged weekly to track tumor progression and sacrificed at 4 weeks or 6 weeks post-tumor injection for the MDA-MB-231 and RWGT2 experiments respectively.
GANT58-NP intracardiac mouse model of early bone metastasis
Athymic female nude mice (4–6 weeks old, Envigo) were anesthetized by continuous isoflurane and inoculated with 1 × 105 GFP-expressing MDA-MB-231-bone cells resuspended in PBS via intracardiac injection into the left cardiac ventricle using a 27-gauge needle, as previously described [26, 30, 57]. Mice were then divided into two cohorts: one group which was treated immediately post-tumor inoculation with GANT58-NPs (8 mg/kg, n = 12) or Empty-NPs (equivalent polymer dose, n = 12), and the remaining group started the same treatment 7 days post-tumor inoculation. Mice were imaged weekly to track tumor progression and sacrificed at 4 weeks.
Radiographic imaging
Mice were radiographically imaged weekly beginning 1-week post-tumor cell inoculation using a Faxitron LX-60. Specifically, mice were anesthetized with isoflurane and laid in a prone position on the imaging platform. Images were acquired at 35 kVp for 8 seconds. Using a freehand selection tool, osteolytic lesions in each image were manually outlined and total lesion area and number were measured using the quantitative image analysis software Metamorph (Molecular Devices, Inc.). All data are represented as mean lesion area and number per mouse.
Micro-computed tomography (μCT)
Tibiae and femora were analyzed using a high-resolution benchtop μCT 40 system (Scanco Medical). Tomographic images were acquired at 70 kVp with an isotropic voxel size of 12 μm and at an integration time of 300 ms. Scans were acquired with hindlimbs in 70% ethanol. μCT images were reconstructed, filtered (σ = 0.2, support = 1.0) and thresholded at 230. Tibiae and femora were then contoured using the Scanco software algorithm starting 10 slices below the growth plate and continued 100 slices in the distal direction. Images were then reconstructed using the Scanco Medical Imaging software. The software was also used to calculate the bone morphometric parameters bone volume/total volume (BV/TV, ratio of segmented bone volume to the total volume of interest), trabecular separation (Tb.Sp., mean distance between trabeculae), trabecular thickness (Tb.Th., mean thickness of trabeculae), and trabecular number (Tb.N., measure of the average number of trabeculae per unit length) of the segmented bone.
Histology/histomorphometry
Tibiae and femora were removed during autopsy and fixed in 10% neutral-buffered formalin (Fisher Scientific) for 48 hr at room temperature after which they were stored at 4°C in 70% ethanol. Bone specimens were decalcified in 10% EDTA for 2 weeks at 4°C and embedded in paraffin wax. Bone sections (5-μm thickness) were stained with hematoxylin & eosin (H&E), orange G, and phloxine. Tumor burden was examined under a microscope and quantified using Metamorph software (Molecular Devices, Inc.). Specifically, tumors were manually outlined as ROIs using a freehand selection tool and the total tumor area was measured as a percentage of the total bone marrow area.
The rabbit anti-PTHrP antibody (1:2500, R87, generated against PTHrP (amino acids 1–34)) was a gift from Drs. T.J. Martin (St. Vincent’s Institute of Medical Research, Australia) and Natalie Sims (The University of Melbourne, Australia). Immunohistochemistry (IHC) was carried out on decalcified paraffin-embedded tibial and femoral sections as previously described [58, 59]. PTHrP-positive staining was quantified using Metamorph software (Molecular Devices, Inc.).
Ethics statement
All animal protocols were approved by Vanderbilt University Institutional Animal Care and Use Committee (IACUC) and were conducted according to National Institutes of Health (NIH) guidelines for care and use of laboratory animals.
Statistical analyses
All statistical analyses were performed using Prism version 7 (GraphPad Software, Inc.). Values are presented as mean ± SEM and p-values determined using one-way ANOVA unless otherwise specified where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Gli2 is overexpressed in bone metastatic patient tumors
Our previous data have established that Gli2 regulates PTHrP expression and is strongly correlated with bone-destructive tumor cells [26, 30]. To test our hypothesis that Gli2 is overexpressed in patients with bone metastases, we collected 20 human biopsies through the CHTN (Table S1) and evaluated Gli2 expression in bone-metastatic tumors from a mix of primary origins. Three of the 20 biopsies were from non-metastatic soft tissue sarcomas and were tested as controls. Patient bone metastases expressed significantly higher levels of Gli2 by IHC compared to soft tissue sarcomas (6-fold increase) (Fig. 1A–B). To further show that Gli2 is overexpressed in bone metastases, we queried the GEO database to obtain Gli2 expression values among 29 human breast cancer metastases in different organs [52]. Gli2 gene expression was significantly higher in bone metastases compared to brain and lung metastases from breast cancer patients (Fig. 1C). Together, these observations suggest that the expression of Gli2 correlates with tumors that reside in bone, suggesting that Gli2 inhibitors may be a strategy to reduce TIBD.
Fig. 1.

Gli2 is overexpressed in patient bone-metastatic tumors. (A) Representative images of Gli2 IHC for patient soft tissue sarcomas (n = 3) and bone-metastatic tumors (n = 17) from different primary origins. Scale bar: 50 μm. (B) Quantification of Gli2-stained patient tumor samples. Groups were statistically compared using student’s unpaired t-test. (C) Gli2 gene expression in human breast cancer metastases from bone (n = 10), brain (n = 14), and lung (n = 3) [52]. Minima = 25th percentile, maxima = 75th percentile, centers = median.
Free GANT58 does not reduce tumor growth or bone destruction
Since previous studies using subcutaneous tumors showed efficacy of GANT58 for reducing prostate tumor growth [28], we tested the efficacy of this approach in models of TIBD. Based on this study and other studies using poorly-water soluble drugs such as paclitaxel [60], we used a 4:1 Cremophor EL:ethanol mix for delivery. Athymic nude mice were inoculated with MDA-MB-231 cells into the left tibiae and were then treated 3 days/week with 50 mg/kg GANT58 or vehicle control via subcutaneous injection (Fig. 2A).
Fig. 2.

GANT58 treatment does not block tumor-induced osteolysis when delivered via Cremophor EL vehicle. (A) Tumor inoculation and subcutaneous treatment scheme for intratibial model of bone metastasis. (B) Representative radiographic images of vehicle (control) and GANT58 treated mice at 4-weeks post-tumor (MDA-MB-231) inoculation. Osteolytic bone lesions are outlined in white. (C) Lesion area and (D) lesion number as assessed by radiographic analysis exhibit no significant difference between vehicle and treatment groups. (E & F) μCT analysis of tibiae bone volume fraction (BV/TV) and trabecular separation (Tb. Sp.) showed no significant difference in mice treated with GANT58 versus those treated with Cremophor EL vehicle. (G) Representative μCT reconstructions of vehicle-treated, GANT58-treated, and contralateral, non-tumor bearing tibiae. (H) Representative H&E stained sections and (I) histomorphometric analysis of tumor area in vehicle-treated and GANT58-treated mice inoculated with MDA-MB-231 cells. Growth plate and remaining trabecular bone outlined in black. Scale bar: 200 μm.
Radiographic imaging of the mice over the course of the study showed that by week 3 both the GANT58 and vehicle-treated groups exhibited severe osteolytic lesions, which significantly worsened by week 4, at which time the mice were sacrificed. Further, the mice showed an adverse response to the injections, presenting with necrosis at the drug injection site as early as week 2 (Division of Animal Care was consulted and necessary treatment provided). Radiographic analysis showed that there was no significant difference between GANT58 and control mice in lesion area or lesion number, with both groups exhibiting numerous lesions in the tumor-bearing tibia (Fig. 2B–D). To further investigate the bone outcomes in these mice, μCT was conducted on the tibiae after sacrifice. BV/TV was significantly reduced in all tumor bearing animals and was comparable for vehicle- and GANT58-treated tumor-bearing tibiae (Fig. 2E). Similarly, Tb.Sp. was comparable for vehicle-and GANT58-treated tumor-bearing tibiae and both were significantly higher than the non-tumor controls (Fig. 2F–G). Histological analysis showed that in both treated and control mice, the average tumor area (%) measured by histomorphometry was 60–70%, indicating that tumor had effectively invaded the entire marrow space of the tibia (Fig. 2H–I). These findings demonstrate that delivery of free GANT58 at high doses using standard clinical formulation techniques is not well-tolerated by the animals and is not effective at blocking TIBD, highlighting the need for delivery strategies that solubilize the drug and improve its PK profile to enable bone tumor delivery.
Synthesis of PPS135-b-POEGA17 diblock copolymer
In order to improve drug solubility and potentially improve efficacy of GANT58, a diblock copolymer comprising PPS and POEGA was synthesized via RAFT polymerization (Fig. 3A, Fig. S1A–B). PPS was chosen as the core-forming hydrophobic block primarily due to its promise from other studies in our lab that showed excellent encapsulation and retention of hydrophobic small molecules [42, 50, 61]. Furthermore, the sulfide group in PPS reacts with oxygen radicals to create sulfoxides and sulfones. This reaction causes a hydrophobic to hydrophilic phase transition in the polymer which potentially contributes to both drug release at sites of oxidative stress and ultimate systemic clearance of the polymer through a renal route. Previous studies have also reported that PPS is a potent reactive oxygen species (ROS)-scavenger, which is hypothesized to impart beneficial antioxidant properties to the GANT58-NPs [62, 63]. The POEGA hydrophilic block forms a brush-like conformation of 9 repeating units of ethylene glycol pendant to the hydrocarbon backbone. POEGA was chosen as the NP surface-forming block because this graft polymer architecture has been shown to improve circulation time over linear PEG [64–66].
Fig. 3.

GANT58-NP fabrication and characterization. (A) Synthesis of PPS135-b-POEGA17 polymer and nanoparticle fabrication. (B) GANT58-NPs (2 mg/mL GANT58) are dispersed in PBS while insoluble free GANT58 (2 mg/mL) precipitates from solution. (C) Particle size distribution of GANT58-NPs and Empty-NPs measured by dynamic light scattering (DLS) indicate that both loaded and unloaded formulations have an average diameter < 100 nm A TEM image (right) reveals the spherical morphology of GANT58-NPs. Scale bar: 500 nm. (D) Critical micelle concentration (CMC) determined by Nile red assay. (E) Fluorescence microscopy image of MDA-MB-231-bone cells after 3 hr treatment with Cy5-labeled GANT58-NPs indicating significant NP uptake into the tumor cells.
Nanoparticle Preparation and Characterization
Nanoparticles were formed by the bulk solvent evaporation method to create either GANT58-loaded PPS135-b-POEGA17 nanoparticles (GANT58-NPs) or unloaded PPS135-b-POEGA17 nanoparticles (Empty-NP) (Fig. 3B). GANT58 (Fig. S1C) was introduced into the organic phase resulting in an average loading content of 11.4% (mg drug/mg polymer) and an encapsulation efficiency of 51.8%. The hydrodynamic diameter of both the GANT58-NPs and Empty-NPs were assessed by DLS. The GANT58-NPs have an average hydrodynamic diameter of 93.3 ± 4.1 nm, while the Empty-NPs have a slightly smaller diameter of 73.5 ± 3.4 nm. TEM shows the NPs exhibit a spherical morphology (Fig. 3C). Both the GANT58-NPs and Empty-NPs were monodispersed with an average PDI of 0.22 and 0.18, respectively, and a relatively low ζ-potential (−1.33 mV and −1.20 mV, respectively). The critical micelle concentration (CMC) of the GANT58-NPs was 0.079 mg/mL as determined by the Nile Red (NR) method (Fig. 3D) [41]. This CMC is almost an order of magnitude lower than the initial NP concentration after i.v. administration (~0.5 mg/mL) and remains lower than the NP blood concentration after 12 hr of circulation (~0.15 mg/mL) as indicated by circulation time measurements. Since tumor cells have high levels of ROS [67], this particle chemistry was chosen to be released at higher ROS concentrations (such as the tumor) as previously described [50, 61]. To confirm ROS-responsiveness of the NPs which have previously been characterized by our group [68], we performed in vitro studies using H2O2 as the model ROS species and NR-loaded NPs (Fig. S2). Incubation of Cy5-labeled GANT58-NPs with MDA-MB-231-bone cells revealed that cells readily uptake the NPs. The Cy5-labeled NPs (red) exhibit close proximity to DAPI-stained nuclei (blue), indicating uptake of the NPs within the cells (Fig. 3E).
GANT58-NPs block Gli2 nuclear translocation
The authors of the manuscript on the original drug screen that identified GANT58 hypothesized that it primarily acts at the nuclear level [28], but the molecular mechanism has not been reported. To test the hypothesis that GANT58 inhibits nuclear translocation of Gli2, MDA-MB-231-bone cells were treated with 20 μm GANT58 or GANT58-NP for 72 hr. Immunofluorescent analyses revealed significantly reduced colocalization of Gli2 with DAPI-stained nuclei in cells treated with GANT58 or GANT58-NPs compared to the Empty-NP control (Fig. 4A). Additionally, MDA-MB-231-bone cells treated with increasing concentrations of free GANT58 or GANT58-NPs showed reduced nuclear Gli2 protein levels as shown by immunoblotting (Fig. 4B). Nuclear protein levels of Gli2 in cells treated with 20 μM GANT58 or GANT58-NP was 48% and 57% of the control, respectively. Inhibition of Gli2 nuclear translocation by GANT58 also significantly reduced PTHrP mRNA levels (Fig. 4C). In contrast, Gli2 protein levels in the cytoplasm did not decrease in a dose-dependent manner following treatment with GANT58 or GANT58-NP, but cytoplasmic Gli2 protein levels were reduced (67 – 69% of the control) at the highest GANT58 dose tested (20 μM) (Fig. S3). These findings show that GANT58 blocks translocation of Gli2 into the nucleus and consequent transcription of PTHrP.
Fig. 4.

GANT58-NP treatment inhibits nuclear translocation of Gli2 and tumor cell proliferation. (A) Representative immunofluorescent images of Gli2 nuclear translocation (left) with quantification as measured by the Manders coefficient (right) after treatment with 20 μM GANT58 or GANT58-NP for 72 hr. Scale bar: 50 μm. (B) Nuclear Gli2 protein levels in MDA-MB-231-bone cells at 72 hr. (C) Expression of PTHrP mRNA for MDA-MB-231-bone cells treated with varying concentrations of free GANT58 (red), Empty-NP (blue), or GANT58-NP (green) for 48 hr. Gradient colors indicate varying doses: 1 μM (transparent), 5 μM, 10 μM, to 20 μM (opaque). (D) Tumor cell proliferation as assessed by MTS assay after 24 hr treatment with free GANT58 or GANT58-NPs (same dosage scheme as (C)). (E) Viability, cytotoxicity, and apoptosis of MDA-MB-231-bone cells treated with free GANT58 or GANT58-NPs for 24 hr measured by ApoTox-Glo assay. Four technical replicates from a single experiment are shown in (D) and (E).
GANT58-NPs reduce tumor cell proliferation and induce apoptosis
To determine the effect of GANT58-NPs on tumor cell proliferation and viability, MDA-MB-231-bone cells treated with increasing drug concentrations were cultured for 24 hr. Cell proliferation assessed by MTS assay demonstrated that treatment with GANT58 or GANT58-NP significantly reduced tumor cell growth rate in vitro (Fig. 4D). GANT58 and GANT58-NP also reduced cell viability and increased activation of caspase 3/7 (which is indicative of apoptosis) compared to vehicle-treated cells in the ApoTox-Glo assay (Fig. 4E).
GANT58-NPs do not alter mesenchymal stem cell proliferation
The effect of GANT58-NPs on hMSC proliferation and osteoblast differentiation was assessed to ensure that GANT58-NPs do not negatively impact osteoblast function and behavior. Proliferation (assessed by MTS assay) was not significantly affected by free GANT58, Empty-NPs, or GANT58-NPs at concentrations up to 20 μM (Fig. 5A). Deposition of mineralized matrix by osteoblasts was assessed via Alizarin Red stain after 14 d culture in osteogenic media. Neither the DMSO vehicle nor free GANT58 significantly reduced Alizarin red staining compared to no treatment, and NP treatment had no negative effects on osteoblast mineralization (Fig. 5B). Representative images of the Alizarin Red-stained cells show the deposition of mineralized matrix by the NP-treated osteoblasts (Fig. 5C).
Fig. 5.

GANT58-NP treatment does not inhibit osteoblast function but reduces tumor-mediated osteoclastogenesis in vitro. (A) hMSCs treated with varying concentrations of free GANT58 (red), Empty-NP (blue), or GANT58-NP (green) were cultured for 1, 3, and 5 days (D1, D3, D5) and proliferation assessed by MTS assay. Gradient colors indicate varying doses: 1 μM (transparent), 5 μM, 10 μM, to 20 μM (opaque). (B) hMSCs were cultured in osteogenic medium treated with same dosage scheme described in (A). Deposition of mineralized matrix by osteoblasts was determined by absorbance of extracted Alizarin Red stain after 14 d of culture in osteogenic media. (C) Representative images of Alizarin Red staining of treated osteoblasts after 14 d culture in osteogenic media. Inset image shows hMSCs cultured in non-osteogenic media. Scale bar: 200 μm. (D) Number of osteoclasts (OC)/well in coculture treated with varying doses of GANT58, Empty-NP, and GANT58-NP after 6 days culture with MDA-MB-231 cells. Three technical replicates are shown in (A), (B), and (D) from single experiments. (E) Multinucleated (3 or more nuclei) and TRAP positive cells were counted as OCs. Red arrows: OCs. (F) Representative images of negative control group (no tumor, -vitD), no treatment group, and 20 μM GANT58-NP treatment group. Red arrows: OCs.
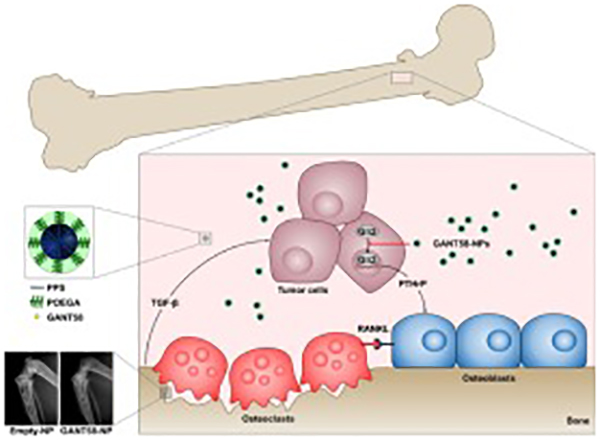
While GANT58 is an antagonist of Gli2 within tumor cells, its functional effect is to reduce secretion of PTHrP protein, which stimulates osteoblasts to express RANKL, a transmembrane protein that stimulates osteoclast differentiation. In order to investigate the effect of GANT58-NPs on osteoclast differentiation, an osteoclastogenesis assay utilizing a coculture of mouse bone marrow-derived stromal cells and MDA-MB-231 cells was performed. Osteoclast number was counted after 7 d culture in the presence of free GANT58, Empty-NP, and GANT58-NP via TRAP stain (Fig. 5D). Osteoclasts were counted at 40x magnification and identified as TRAP+ (red/pink stain), multinucleated (≥ 3 nuclei) cells (Fig. 5E). Osteoclast number showed a GANT58 dose-dependent decrease in osteoclasts for both the free GANT58 and GANT58-NP, while the Empty-NP had no effect on osteoclastogenesis. Representative 10x magnification images of the coculture show very few osteoclasts in the (−) vitamin D negative control and 20 μM GANT58-NP group, whereas the untreated tumor group exhibited extensive osteoclast maturation (Fig. 5F).
GANT58 encapsulation using nanoparticles allows for solubilization in a formulation compatible with intravenous delivery for in vivo studies
GANT58-NPs distribute to the tumor bearing bones
After validation of in vitro efficacy, the biodistribution and pharmacokinetics (PK) of the PPS 135-b-POEGA17 NPs and its cargo was evaluated via near-IR fluorescent dye grafting and loading (for NP and cargo tracking, respectively) of the micelles. First, Cy7 fluorophore-loaded PPS 135-b-POEGA17 NPs (Cy7-NPs) were administered via i.v. tail vein injection to tibial tumor-bearing athymic nude mice to evaluate NP cargo biodistribution and PK of in tumor and organs of interest. Cy7 was chosen as the surrogate compound for GANT58 due to its strong near-IR fluorescence and solubility properties similar to that of GANT58 (logP = 3.4 and 3.9, respectively). In vivo biodistribution was assessed in mice over time to observe Cy7 cargo localization in the tumor-bearing tibia site versus control tibia (Fig. 6A). Near-IR in vivo imaging and Pearl image analysis software were used to quantify Cy7 fluorescence intensity in both the tumor and control ROI. Cy7 fluorescence intensity was significantly higher in the tumor-bearing tibia at each time point, indicating that the Cy7 cargo was preferentially localizing in the tumor bearing site.
Fig. 6.

GANT58-NPs accumulate in tumor and demonstrate prolonged blood circulation. (A) Cy7-loaded NPs (Cy7-NPs) time course biodistribution in tumor-bearing mice. Cy7 fluorescence is significantly higher in the tumor-bearing tibia (red arrow) compared to the contralateral control. (B) Cy7-grafted GANT58-NPs biodistribution ex vivo analysis at 24 hr shows significantly higher Cy7 fluorescence in the tumor-bearing hindlimb compared to other long bones. L(R)HL = left (right) hindlimb; L(R)FL = left (right) forelimb. (C) Time-course ex vivo analysis of Cy7-grafted GANT58-NPs shows that NPs increasingly accumulate in tumor over 24 hr period. (D) Representative organ Cy7 fluorescence at 24 hr post-NP injection. (E) GFP imaging (tumor) and Cy7 fluorescence (NP) confirm colocalization of NPs within the tumor site. (F) Circulation time of Cy5-grafted GANT58-NPs as assessed by tail nick method.
Next, Cy7-grafted GANT58-NPs (GANT58-Cy7NPs) were administered via tail vein injection to track GANT58-Cy7NP biodistribution over time. Mice were sacrificed at predetermined time points, and Cy7 fluorescence was measured in organs of interest ex vivo. At 24 hr post-injection, the tumor-bearing left hindlimb (LHL) had significantly increased uptake over the other long bones, while the liver had the highest intensity as expected due to clearance by the mononuclear phagocyte system (MPS). The kidneys had the next highest level of fluorescence, potentially serving as a route for clearance of the Cy7-grafted polymer following NP disassembly (Fig. 6B). Cy7 fluorescence steadily decreased over time in all long bones aside from the tumor-bearing hindlimb, indicating stable accumulation in the bone-tumor site (Fig. 6C). Similarly, there was a steady decrease in Cy7 fluorescence in all organs aside from liver and kidney, where there was an expected increase in intensity as the NPs were cleared from circulation (Fig. 6C–D). To further validate the findings that the NPs are preferentially accumulating in the tumor, GFP intensity from the MDA-MB-231 cells was measured and spatially compared to the Cy7 fluorescence (Fig. 6E). A strong colocalization was observed, suggesting the NP polymer accumulation is primarily within the tumor site.
The PK profile of the GANT58-NPs was next investigated by measuring the blood circulation time of Cy5-grafted GANT58-NPs (GANT58-Cy5NPs). The findings from the biodistribution study suggested a significant concentration of NPs are still in circulation after 12 hr due to the increased NP polymer accumulation in tumor, liver, and kidneys at the 24 hr time point. We designed the circulation time experiment to encompass both short (< 30 min) and long (> 12 hr) time periods for accurate measurement of the α and β phase circulation half-lives. After retroorbital injection of the GANT58-Cy5NPs, a tail nick method was used to draw small volumes (< 5 μL) of blood immediately post-injection (t=0) and at subsequent predetermined intervals up to 24 hr. These small blood volumes were then measured for Cy5 fluorescence intensity on a micro-volume plate using a fluorescence plate reader and NP concentration calculated from a standard curve. A rapid α-phase distributive half-life of 12.3 min was observed, after which there was a more gradual β-phase with a 28 hr half-life (Fig. 6F). Even after 24 hr, roughly 15% of the initial NP dose was still in circulation, which supports the biodistribution findings showing increased uptake in tumor and clearance organs at 24 hr.
GANT58-NPs reduce bone destruction in mouse model of established tumors in bone
The promising in vitro results and preferential uptake in the tumor site presented compelling evidence that GANT58-NPs would successfully block tumor-induced bone destruction in vivo and possibly reduce tumor burden. To test this hypothesis, MDA-MB-231-bone cells were inoculated into the left tibiae of female athymic nude mice. After 4 days to allow tumor establishment, mice were treated daily with GANT58-NPs (8 mg/kg GANT58) or Empty-NPs via tail vein injection (Fig. 7A). Empty-NPs were chosen as the control due to the in vitro results suggesting the positive effect the Empty-NPs may have on osteoblast differentiation.
Fig. 7.

GANT58-NP treatment reduces tumor-induced osteolysis in mouse model of late bone metastasis in multiple tumor cell lines. (A) Tumor-inoculation and treatment scheme for intratibial model of bone metastasis. (B) Representative radiographic images of Empty-NP (control) and GANT58-NP treated mice at 4-weeks post-tumor (MDA-MB-231) inoculation. Osteolytic bone lesions are outlined in white. (C) Lesion area and (D) lesion number as assessed by radiographic analysis are significantly reduced in GANT58-NP treated mice. (E-G) μCT analysis of tibiae bone volume fraction (BV/TV), trabecular separation (Tb. Sp.), and trabecular number (Tb. N.) showed significantly improved bone quality in mice treated with GANT58-NPs. (H) Representative μCT reconstructions of GANT58-NP-treated, Empty-NP-treated, and contralateral, non-tumor bearing tibiae. (I) Representative H&E stained sections and (J) histomorphometric analysis of tumor area (indicated by T) in Empty-NP-treated and GANT58-NP-treated in mice inoculated with MDA-MB-231 cells. Scale bar: 200 μm (K) Representative images and (L) quantification of immunohistochemical staining for PTHrP (red-brown staining) in sections from Empty-NP-treated and GANT58-NP-treated tumor-bearing mice. Scale bar: 200 μm
Substantial osteolytic lesions were observed radiographically in the mice treated only with the Empty-NPs after 4 weeks, while smaller and fewer lesions were visible in mice treated with GANT58-NP (Fig. 7B). Quantitative assessment of both lesion area and number confirmed that the osteolytic lesions in the GANT58-NP-treated mice were significantly smaller and fewer (Fig. 7C–D).
After sacrifice, mouse tibiae were examined by μCT to assess the effects of the drug on bone morphometric properties. Mice treated with the GANT58-NPs had 2.5-fold higher (p < 0.001) BV/TV than those treated with Empty-NPs and 22% lower (p < 0.05) BV/TV than the treated, non-tumor bearing control (Fig. 7E). Tb.Sp. in the GANT58-NP group was 34% lower (p < 0.05) than the Empty-NP group and 48% higher (p < 0.05) than the untreated control (Fig. 7F). Similarly, trabecular number (Tb.N.) in the GANT58-NP group trended higher than the Empty-NP group and was 28% lower than the untreated control (Fig. 7G). 3D μCT renderings of representative tibiae from each group further demonstrate the significant tumor-induced bone destruction in the mice treated with Empty-NPs compared to those treated with GANT58-NPs (Fig. 7H). Histomorphometric analysis of the tibiae showed no significant differences in tumor burden between groups (Fig. 7I–J); however, a significant reduction in PTHrP protein expression was detected by IHC in GANT58-NP-treated mice compared to those treated with Empty-NPs (Fig. 7K–L).
In order to establish the broader generalizability of these findings, another human osteolytic cancer cell line was tested in the same intratibial model. In this experiment, a bone-metastatic variant of a squamous non-small cell lung carcinoma line, RWGT2-bone, was used and the same bone outcomes were assessed. Again, radiograph analysis showed a significant reduction in lesion area and number in GANT58-NP-treated mice (Fig. S4A). BV/TV was also significantly higher in the mice treated with GANT58-NPs than those treated with Empty-NPs (Fig. S4B). Further, there was no significant difference in BV/TV in GANT58-NP-treated mice and the non-tumor-bearing control tibiae (Fig. S4B). Other morphometric parameters, however, were not significantly different between the GANT58-NP and Empty-NP groups (Fig. S4C–E). 3D μCT renderings of representative tibiae from each group again demonstrate the tumor-induced bone destruction in the mice treated with Empty-NPs compared to those treated with GANT58-NPs (Fig. S4F). It is important to note that the RWGT2-bone cell line was not as aggressive as the MDA-MB-231-bone line, as indicated by the higher BV/TV in the RWGT2-bone Empty-NP group compared to the same group in the MDA-MB-231-bone experiment. This lower tumor burden could explain the diminished significance in μCT outcomes for the GANT58-NP treatment group relative to controls for this study relative to the MDA-MB-231 study.
GANT58-NPs reduce TIBD in bone metastatic mouse model
The intratibial model of bone metastasis was informative in showing that GANT58 is effective in blocking bone destruction induced by established tumors, but its effect on tumor metastasis could not be elucidated using this model. Therefore, an intracardiac model of bone metastasis was used in order to investigate the effect of GANT58-NPs on blocking tumor metastasis in addition to bone destruction. In this experiment, mice were injected with MDA-MB-231 cells via intracardiac injection and were subsequently divided into two cohorts. The first cohort, denoted the “immediate” group, was immediately treated via tail vein injection with either GANT58-NPs (treatment) or Empty-NPs (control). The second cohort of mice, denoted the “delayed” group, was allowed 7 days for tumor establishment before starting treatment (Fig. 8A).
Fig. 8.

GANT58-NP treatment reduces tumor-induced osteolysis in early metastasis model of tumor-induced bone disease. (A) Tumor-inoculation and treatment scheme for intracardiac model of bone metastasis. (B) μCT analysis of tibiae bone volume fraction (BV/TV) and trabecular thickness (Tb. Th.) showed significantly improved bone quality in mice treated with GANT58-NPs. (C) Representative H&E stained sections and histomorphometric analysis of tumor area (indicated by T) in Empty-NP-treated and GANT58-NP-treated in mice inoculated with MDA-MB-231 cells. Scale bar: 200 μm (D) Representative images and quantification of immunohistochemical staining for PTHrP (red-brown staining) in femoral sections from Empty-NP-treated and GANT58-NP-treated tumor-bearing mice. Scale bar: 200 μm
Tibiae and femurs were scanned by μCT after sacrifice, and subsequent observation of histological sections showed that tumor presence was more substantial in the femur than in the tibia. Thus, the femur was chosen for thorough μCT and histological analysis. μCT analysis revealed that there was significantly higher BV/TV and Tb. Th. in the mice that were treated with GANT58-NPs, indicating reduced bone destruction and improved bone quality in GANT58-NP-treated mice. However, there was no significant difference between the mice treated immediately and the mice that received delayed treatment (Fig. 8B). Histological analysis showed no significant reduction in tumor burden across all groups (Fig. 8C); however, a significant reduction in PTHrP expression was observed by IHC in tumor-bearing mice that were immediately treated with GANT58-NPs compared to control-treated mice (Fig. 8D). Taken together, these findings are consistent with those of the intratibial model where GANT58-NP treatment significantly improved bone outcomes, but tumor burden was not significantly affected. While more studies are needed to further characterize potential effects on tumor growth, these studies strongly demonstrate that GANT58-NPs reduce tumor-induced bone destruction in two mouse models of bone metastasis.
Safety profile of GANT58-NPs
In addition to efficacy, successful clinical translation of the GANT58-NPs hinges upon safety of the treatment at therapeutic doses. To test the safety profile of the GANT58-NPs, blood was drawn at time of sacrifice for the cohort of mice in the intratibial study after having received GANT58-NP or Empty-NP treatment 5 days/week at 8 mg/kg GANT58 (or equivalent dose of Empty-NP) for 4 weeks. Biochemical analysis of blood serum markers for liver and kidney toxicity was performed. There was no significant increase in aspartate aminotransferase (AST), alanine aminotransferase (ALT), or blood urea nitrogen (BUN) levels above two standard deviations from average levels reported by the animal supplier (Envigo) (Fig. S5A). Further, histological sections of the liver and kidney of the same mice were observed by a blinded, board certified pathologist and there was no evident toxicity in either group (Fig. S5B).
Discussion
Metastatic disease accounts for over 90% of cancer-related deaths and therefore remains a major clinical concern [69]. In patients with bone metastases, standard-of-care RANKL and osteoclast inhibitors improve quality of life but do not target tumor-specific aberrations in signaling that cause TIBD. We identified Gli2 as a promising therapeutic target for bone metastases based on our previous studies establishing its overexpression in bone-destructive cell lines [26, 30]. While free GANT58 did not inhibit bone resorption in vivo due to its poor solubility and PK, GANT58-NPs significantly reduced bone resorption in intracardiac and intratibial injection models of TIBD using bone-metastatic breast and lung cancer cell lines.
Since RANKL and osteoclast inhibitors target osteoclast function throughout the body, osteoblast activity, which is tightly coupled with and triggered by bone resorption, is also decreased, which severely impairs bone remodeling and fracture healing [70, 71]. Consistent with these findings, other studies have reported dose-dependent inhibitory effects of bisphosphonates on osteoblast proliferation, differentiation, and mineralization [72, 73]. In contrast, the MTA GANT58 did not inhibit osteoblast hMSC proliferation or deposition of mineralized matrix by osteoblasts (Fig. 5A–C). GANT58 blocked nuclear translocation of Gli2 (Fig. 4A–B) and consequent PTHrP expression in vitro (Fig. 4C) and in vivo (Fig. 7K–L, 8D). Considering that PTHrP stimulates expression of RANKL (a stimulator of osteoclast formation) by osteoblasts, we investigated the effects of GANT58 treatment on osteoclastogenesis in vitro. GANT58 treatment of mouse bone marrow-derived cells co-cultured with MDA-MB-231-bone tumor cells significantly inhibited osteoclastogenesis (Fig. 5D–F). These findings suggest that GANT58 blocks tumor-induced stimulation of osteoclastogenesis without impairing osteoblast differentiation.
While the current study establishes the therapeutic potential of Gli2 inhibition to block tumor-mediated bone destruction, other studies have previously utilized nanomedicine strategies for delivery of Hh pathway inhibitors to tumors. Delivery of Hedgehog Pathway Inhibitor-1 (HPI-1) has been investigated for treatment of medulloblastoma and hepatocellular carcinoma [43, 74]. While these studies showed that HPI-1 nanoparticles inhibited systemic metastases in an orthotopic model of human hepatocellular carcinoma, the effects of Hh inhibition on bone metastases were not investigated [43, 74]. Nanomedicine strategies have also been applied to inhibit bone metastases by delivering conventional chemotherapies to reduce bone tumor burden in the bone marrow [75, 76]. These studies found that nano-encapsulated docetaxel and paclitaxel reduced tumor burden, but the effects of the drugs on bone were not investigated extensively in vivo. Other studies have reported that conventional chemotherapies cause DNA damage and apoptotic cell death in human bone marrow cells, and that these treatments negatively affect trabecular bone microarchitecture and mechanical properties [77, 78]. Thus, targeted delivery of chemotherapies to tumors in bone is anticipated to have adverse effects on the bone microenvironment and warrants further investigation. In contrast, nano-encapsulated GANT58 blocked tumor-induced bone destruction in tumor-bearing bones, but the drug did not inhibit mineralization (Fig. 5B–C).
GANT58-NPs inhibited tumor proliferation (Fig. 4D) and viability (Fig. 4E) in vitro, but there were only modest changes in cytotoxicity with increasing GANT58 dose (Fig. 4E). These data suggest that GANT58 slows tumor growth but is not cytotoxic to tumor cells, which is consistent with previous studies reporting that GANT58 and GANT61 inhibit tumor proliferation by inducing cytostasis (cell cycle arrest) and early phase apoptosis rather than direct cytotoxic killing [28, 33, 79]. Our previous study showed that genetic inhibition of Gli2 using an Engrailed repressor construct significantly attenuated the ability of cancer cells to colonize bone (only micrometastases were observed) and induce osteolysis in vivo [26]. However, GANT58-NPs did not significantly reduce tumor burden in either the intratibial or intracardiac models. Micellar nanoparticles are passively targeted to tumor sites via the enhanced permeability and retention (EPR) effect, which requires the tumor vasculature to be permeable or “leaky” in order for nanoparticles to accumulate within the tumoral interstitial space [80]. Though the significant reduction in PTHrP expression confirms on-target delivery of GANT58-NPs into bone-associated tumors, the nanoparticles likely do not sufficiently accumulate at bone-tumor sites until larger, vascularized tumors are established, at which point it is difficult to reduce tumor burden in the short time-line of current animal models. In future studies, we will evaluate the nanoparticle carrier to determine if conferring bone-binding affinity to the polymer chemistry will improve targeted delivery to bone and block initial bone tumor establishment.
There may also be spatial variation in the tumor cell responsiveness to GANT58 within the bone and bone marrow. Matrix rigidity in the bone microenvironment is a key mediator of Gli2 expression in bone-metastatic tumor cells. Gli2 expression was significantly increased in patient bone-metastatic tumors but not soft tissue tumors (Fig. 1A–B) or brain and lung metastases (Fig. 1C). Furthermore, Gli2 is not expressed on collagen-like (30 MPa) substrates but is overexpressed on bone-like (>100 MPa) substrates in vitro [81]. Considering the relatively low elastic modulus of bone marrow (0.25–24.7 kPa) [82], tumor cells growing in the marrow space are anticipated to express low levels of Gli2 and are therefore likely to be less sensitive to GANT58. However, as tumor cells approach the interface with bony trabeculae, they overexpress Gli2 and PTHrP in response to the rigid (>100 MPa) mineralized bone matrix [81, 83]. Thus, GANT58 is anticipated to inhibit tumor cell proliferation, expression of PTHrP, and the transition to the bone-destructive phenotype in tumor cells within close proximity of bone but potentially have less direct impact on tumor cells in the marrow space. In future studies using bone targeted approaches, we will evaluate whether GANT58 can block the transition of tumor cells from micro-metastatic cells in the bone marrow to more aggressive and bone destructive tumors that have invaded the bone.
Previous studies have also implied the importance of tumor-stromal interactions in promoting bone metastasis [6]. Recent work has elucidated new molecular mechanisms and therapeutic targets for prevention of bone metastasis and/or halting the vicious cycle of bone destruction. In addition to Hh signaling, Notch signaling has been implicated in bone metastasis, and the Notch ligand Jagged1 drives tumor progression in bone [84]. A subsequent study developed a Jagged1 antibody that not only reduced incidence of bone metastasis, but also sensitized the tumor to chemotherapy and reduced tumor recurrence [85]. Another recent study showed that crosstalk between ROR1-HER3 and the Hippo-YAP pathway promotes breast cancer bone metastasis and identified multiple new therapeutic targets for inhibition [86]. While these tumor-targeted approaches offer the potential of reducing tumor burden in bone, inter-patient heterogeneity may limit their ability to benefit all bone-metastatic cancer patients [87]. In patients with metastatic cancer, PTHrP is expressed in > 90% of bone-residing tumors compared to < 20% at non-bony sites [88]. Similarly, we show that Gli2 is also overexpressed in bone-metastatic tumors from various primary sites (Fig. 1A–B). The prevalence of Gli2 and PTHrP expression in bone suggests that GANT58 treatment is likely to be effective in a broad spectrum of patients suffering from bone metastases, which is supported by our observations of bone protection by GANT58-NPs across both breast and lung cancer models. Thus, combined delivery of GANT58, which blocks the transition to the bone-destructive phenotype, with targeted therapies or conventional chemotherapeutics that block tumor growth in the bone marrow could potentially slow the progress of TIBD and improve patient quality of life.
Conclusions
Polymeric NP encapsulation of GANT58 provided an injectable aqueous dispersion that significantly decreased bone lesions and increased trabecular bone volume in two breast cancer models of bone metastasis (intratibial and intracardiac). Importantly, GANT58-NPs did not alter hMSC proliferation or osteoblast mineralization, essential processes for bone remodeling and fracture repair in cancer patients. Pharmacokinetic and biodistribution analysis showed the GANT58-NPs exhibit an extensive circulation time, preferentially localize at the tumor site, and show no evidence of cytotoxicity in the liver or kidneys, the major organs through which the GANT58-NPs are cleared. Thus, the efficacy and safety profile of GANT58-NPs provide promising rationale to continue testing GANT58-NPs in pre-clinical models of TIBD to potentially lead to novel therapies for reducing TIBD.
Supplementary Material
Acknowledgements
The authors wish to acknowledge Joshua Johnson for histological processing and sectioning of femora/tibiae as well as Kelli Boyd, D.V.M. Ph.D., veterinary pathologist, for pathological assessment of liver/kidney tissues. In addition, we thank Professors T.J. Martin and Natalie Sims from the St. Vincent’s Institute of Medical Research and The University of Melbourne for kindly providing the PTHrP antibody used in the immunohistochemistry studies. We also acknowledge the CHTN Western Division supported by the NCI/NIH grant U01CA091664, Translational Pathology Shared Resource (TPSR) core which is supported by the NCI/NIH grant 5P30 CA68485-19, and the μCT core funding 1S10RR027027631. We are also grateful to the Vanderbilt Institute for Nanoscale Science and Engineering (VINSE) for access to the DLS and TEM for nanoparticle characterization. This work was supported by the NIH grant R01CA163499 (S.A.G. and J.A.S.), VA Merit award 1I01BX001957 (J.A.S.), and the DOD CDMRP awards W81XWH-15-1-0627 (C.L.D.) and W81XWH-15-1-0622 (J.A.S). R.W.J. is supported in part by the NIH grant R00CA194198 and DOD CDMRP award W81XWH-18-1-0029.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
References
- [1].Buijs JT, van der Pluijm G, Osteotropic cancers: From primary tumor to bone, Cancer Lett. 273 (2009) 177–193, https://doi.org/10.1016Zj.canlet.2008.05.044. [DOI] [PubMed] [Google Scholar]
- [2].Coleman RE, Clinical features of metastatic bone disease and risk of skeletal morbidity, Clin. Cancer Res 12 (2006) 6243s–6249s, 10.1158/1078-0432.CCR-06-0931. [DOI] [PubMed] [Google Scholar]
- [3].Saad F, Lipton A, Cook R, Chen YM, Smith M, Coleman R, Pathologic fractures correlate with reduced survival in patients with malignant bone disease, Cancer 110 (2007) 1860–1867, 10.1002/cncr.22991. [DOI] [PubMed] [Google Scholar]
- [4].Mundy GR, Mechanisms of bone metastasis, Cancer 80 (1997) 1546–1556, [DOI] [PubMed] [Google Scholar]
- [5].Sterling JA, Edwards JR, Martin TJ, Mundy GR, Advances in the biology of bone metastasis: How the skeleton affects tumor behavior, Bone 48 (2011) 6–15, 10.1016/j.bone.2010.07.015. [DOI] [PubMed] [Google Scholar]
- [6].Weilbaecher KN, Guise TA, McCauley LK, Cancer to bone: A fatal attraction, Nat. Rev. Cancer 11 (2011) 411–425, 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sousa S, Clezardin P, Bone-targeted therapies in cancer-induced bone disease, Calcif. Tissue Int 102 (2018) 227–250, 10.1007/s00223-017-0353-5. [DOI] [PubMed] [Google Scholar]
- [8].Edwards BJ, Sun M, West DP, Guindani M, Lin YH, Lu H, et al. , Incidence of atypical femur fractures in cancer patients: The MD Anderson Cancer Center experience, J. Bone Miner. Res 31 (2016) 1569–1576, 10.1002/jbmr.2818. [DOI] [PubMed] [Google Scholar]
- [9].Yang SP, Kim TW, Boland PJ, Farooki A, Retrospective review of atypical femoral fracture in metastatic bone disease patients receiving denosumab therapy, Oncologist 22 (2017) 438–444, 10.1634/theoncologist.2016-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bamias A, Kastritis E, Bamia C, Moulopoulos LA, Melakopoulos I, Bozas G, et al. , Osteonecrosis of the jaw in cancer after treatment with bisphosphonates: Incidence and risk factors, J. Clin. Oncol 23 (2005) 8580–8587, 10.1200/jco.2005.02.8670. [DOI] [PubMed] [Google Scholar]
- [11].Coleman R, Woodward E, Brown J, Cameron D, Bell R, Dodwell D, et al. , Safety of zoledronic acid and incidence of osteonecrosis of the jaw (ONJ) during adjuvant therapy in a randomised phase III trial (AZURE: BIG 01–04) for women with stage II/III breast cancer, Breast Cancer Res. Treat 127 (2011) 429438, 10.1007/s10549-011-1429-y. [DOI] [PubMed] [Google Scholar]
- [12].Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. , Denosumab and bone - metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial, Lancet 379 (2011) 39–46, 10.1016/s0140-6736(11)61226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fan C, Georgiou KR, Morris HA, McKinnon RA, Keefe DMK, Howe PR, et al. , Combination breast cancer chemotherapy with doxorubicin and cyclophosphamide damages bone and bone marrow in a female rat model, Breast Cancer Res. Treat 165 (2017) 41–51, 10.1007/s10549-017-4308-3. [DOI] [PubMed] [Google Scholar]
- [14].Green DE, Rubin CT, Consequences of irradiation on bone and marrow phenotypes, and its relation to disruption of hematopoietic precursors, Bone 63 (2014) 87–94, 10.1016/j.bone.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ingham PW, McMahon AP, Hedgehog signaling in animal development: Paradigms and principles, Genes Dev. 15 (2001) 3059–3087, 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- [16].Briscoe J, Therond PP, The mechanisms of hedgehog signalling and its roles in development and disease, Nat. Rev. Mol. Cell Biol 14 (2013) 416–429, 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- [17].Pasca di Magliano M, Hebrok M, Hedgehog signalling in cancer formation and maintenance, Nat. Rev. Cancer 3 (2004) 903–911, 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- [18].Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, et al. , Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer, Cancer Res. 64 (2004) 6071–6074, 10.1158/0008-5472.can-04-0416. [DOI] [PubMed] [Google Scholar]
- [19].Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. , Hedgehog signalling in prostate regeneration, neoplasia and metastasis, Nature 431 (2004) 707–712, 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- [20].Yuan Z, Goetz JA, Singh S, Ogden SK, Petty WJ, Black CC, et al. , Frequent requirement of hedgehog signaling in non-small cell lung carcinoma, Oncogene 26 (2006) 1046–1055, 10.1038/sj.onc.1209860. [DOI] [PubMed] [Google Scholar]
- [21].LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. , Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors, Clin. Cancer Res 17 (2011) 2502–2511, 10.1158/1078-0432.ccr-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jimeno A, Weiss GJ, Miller WH Jr., Gettinger S, Eigl BJ, Chang AL, et al. , Phase I study of the hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors, Clin. Cancer Res 19 (2013) 2766–2774, 10.1158/1078-0432.ccr-12-3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wagner AJ, Messersmith WA, Shaik MN, Li S, Zheng X, McLachlan KR, et al. , A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors, Clin. Cancer Res 21 (2014) 1044–1051, 10.1158/1078-0432.ccr-14-1116. [DOI] [PubMed] [Google Scholar]
- [24].Metcalfe C, de Sauvage FJ, Hedgehog fights back: Mechanisms of acquired resistance against smoothened antagonists, Cancer Res. 71 (2011) 5057–5061, 10.1158/0008-5472.can-11-0923. [DOI] [PubMed] [Google Scholar]
- [25].Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. , Smoothened variants explain the majority of drug resistance in basal cell carcinoma, Cancer Cell 27 (2015) 342–353, 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Johnson RW, Nguyen MP, Padalecki SS, Grubbs BG, Merkel AR, Oyajobi BO, et al. , TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical hedgehog signaling, Cancer Res. 71 (2011) 822–831, 10.1158/0008-5472.can-10-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Johnson RW, Merkel AR, Page JM, Ruppender NS, Guelcher SA, Sterling JA, Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer, Clin. Exp. Metastasis 31 (2014) 945–959, 10.1007/s10585-014-9682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lauth M, Bergstrom A, Shimokawa T, Toftgard R, Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists, Proc. Natl. Acad. Sci. USA 104 (2007) 8455–8460, 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, et al. , Small-molecule inhibitors reveal multiple strategies for hedgehog pathway blockade, Proc. Natl. Acad. Sci. USA 106 (2009) 14132–14137, 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sterling JA, Oyajobi BO, Grubbs B, Padalecki SS, Munoz SA, Gupta A, et al. , The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells, Cancer Res. 66 (2006) 7548–7553, 10.1158/0008-5472.can-06-0452. [DOI] [PubMed] [Google Scholar]
- [31].Cannonier SA, Sterling JA, The role of hedgehog signaling in tumor induced bone disease, Cancers (Basel) 7 (2015) 1658–1683, 10.3390/cancers7030856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Neelakantan D, Zhou H, Oliphant MUJ, Zhang X, Simon LM, Henke DM, et al. , EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells, Nat. Commun 8 (2017) 15773, 10.1038/ncomms15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gonnissen A, Isebaert S, McKee CM, Dok R, Haustermans K, Muschel RJ, The hedgehog inhibitor GANT61 sensitizes prostate cancer cells to ionizing radiation both in vitro and in vivo, Oncotarget 7 (2016) 84286–84298, 10.18632/oncotarget.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang L, Walter V, Hayes DN, Onaitis M, Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer, Clin. Cancer Res 20 (2014) 1566–1575, 10.1158/1078-0432.ccr-13-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R, Nanocarriers as an emerging platform for cancer therapy, Nat. Nanotechnol 2 (2007) 751–760, 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- [36].Wicki A, Witzigmann D, Balasubramanian V, Huwyler J, Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications, J. Control Release 200 (2015) 138–157, 10.1016/j.jconrel.2014.12.030. [DOI] [PubMed] [Google Scholar]
- [37].Ashton S, Song YH, Nolan J, Cadogan E, Murray J, Odedra R, et al. , Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo, Sci. Transl. Med 8 (2016) 325ra317, 10.1126/scitranslmed.aad2355. [DOI] [PubMed] [Google Scholar]
- [38].Mizrachi A, Shamay Y, Shah J, Brook S, Soong J, Rajasekhar VK, et al. , Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma, Nat. Commun 8 (2017) 14292, 10.1038/ncomms14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB, et al. , T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity, Nat. Commun 8 (2017) 1747, 10.1038/s41467-017-01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kwak EL, Clark JW, Chabner B, Targeted agents: The rules of combination, Clin. Cancer Res 13 (2007) 5232–5237, 10.1158/1078-0432.ccr-07-1385. [DOI] [PubMed] [Google Scholar]
- [41].Gupta MK, Meyer TA, Nelson CE, Duvall CL, Poly(PS-b-DMA) micelles for reactive oxygen species triggered drug release, J. Control Release 162 (2012) 591–598, 10.1016/j.jconrel.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Uddin MJ, Werfel TA, Crews BC, Gupta MK, Kavanaugh TE, Kingsley PJ, et al. , Fluorocoxib A loaded nanoparticles enable targeted visualization of cyclooxygenase-2 in inflammation and cancer, Biomaterials 92 (2016) 71–80, 10.1016/j.biomaterials.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chenna V, Hu C, Pramanik D, Aftab BT, Karikari C, Campbell NR, et al. , A polymeric nanoparticle encapsulated small-molecule inhibitor of hedgehog signaling (NanoHHI) bypasses secondary mutational resistance to smoothened antagonists, Mol. Cancer Ther 11 (2012) 165–173, 10.1158/1535-7163.mct-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Blanco E, Shen H, Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol 33 (2015) 941–951, 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wei T, Chen C, Liu J, Liu C, Posocco P, Liu X, et al. , Anticancer drug nanomicelles formed by self-assembling amphiphilic dendrimer to combat cancer drug resistance, Proc. Natl. Acad. Sci. USA 112 (2015) 2978–2983, 10.1073/pnas.1418494112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim DW, Kim SY, Kim HK, Kim SW, Shin SW, Kim JS, et al. , Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer, Ann. Oncol 18 (2007) 2009–2014, 10.1093/annonc/mdm374. [DOI] [PubMed] [Google Scholar]
- [47].Ramasamy T, Ruttala HB, Gupta B, Poudel BK, Choi HG, Yong CS, et al. , Smart chemistry-based nanosized drug delivery systems for systemic applications: A comprehensive review, J. Control Release 258 (2017) 226–253, 10.1016/j.jconrel.2017.04.043. [DOI] [PubMed] [Google Scholar]
- [48].Joshi RV, Nelson CE, Poole KM, Skala MC, Duvall CL, Dual pH- and temperature-responsive microparticles for protein delivery to ischemic tissues, Acta Biomater. 9 (2013) 6526–6534, 10.1016/j.actbio.2013.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li H, Miteva M, Kirkbride KC, Cheng MJ, Nelson CE, Simpson EM, et al. , Dual MMP7-proximity-activated and folate receptor-targeted nanoparticles for siRNA delivery, Biomacromolecules 16 (2014) 192–201, 10.1021/bm501394m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Poole KM, Nelson CE, Joshi RV, Martin JR, Gupta MK, Haws SC, et al. , ROS-responsive microspheres for on demand antioxidant therapy in a model of diabetic peripheral arterial disease, Biomaterials 41 (2015) 166–175, 10.1016/j.biomaterials.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guise TA, Yoneda T, Yates AJ, Mundy GR, The combined effect of tumor-produced parathyroid hormone-related protein and transforming growth factor-alpha enhance hypercalcemia in vivo and bone resorption in vitro, J. Clin. Endocrinol. Metab 77 (1993) 40–45, 10.1210/jcem.77.1.8325957. [DOI] [PubMed] [Google Scholar]
- [52].Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. , Latent bone metastasis in breast cancer tied to src-dependent survival signals, Cancer Cell 16 (2009) 67–78, 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Guo R, Lu S, Merkel AR, Sterling JA, Guelcher SA, Substrate modulus regulates osteogenic differentiation of rat mesenchymal stem cells through integrin β1 and BMP receptor type IA, J. Mater. Chem. B Mater. Biol. Med 4 (2016) 3584–3593, 10.1039/c5tb02747k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mbalaviele G, Chen H, Boyce BF, Mundy GR, Yoneda T, The role of cadherin in the generation of multinucleated osteoclasts from mononuclear precursors in murine marrow, J. Clin. Invest 95 (1995) 2757–2765, 10.1172/jci117979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Michigami T, Shimizu N, Williams PJ, Niewolna M, Dallas SL, Mundy GR, et al. , Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast-stimulating activity, Blood 96 (2000) 1953–1960, [PubMed] [Google Scholar]
- [56].Tietjen GT, DiRito J, Pober JS, Saltzman WM, Quantitative microscopy-based measurements of circulating nanoparticle concentration using microliter blood volumes, Nanomedicine 13 (2017) 1863–1867, 10.1016/j.nano.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wright LE, Ottewell PD, Rucci N, Peyruchaud O, Pagnotti GM, Chiechi A, et al. , Murine models of breast cancer bone metastasis, Bonekey Rep. 5 (2016) 804, 10.1038/bonekey.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lam MH, Thomas RJ, Loveland KL, Schilders S, Gu M, Martin TJ, et al. , Nuclear transport of parathyroid hormone (PTH)-related protein is dependent on microtubules, Mol. Endocrinol 16 (2002) 390–401, 10.1210/mend.16.2.0775. [DOI] [PubMed] [Google Scholar]
- [59].Ansari N, Ho PW, Crimeen-Irwin B, Poulton IJ, Brunt AR, Forwood MR, et al. , Autocrine and paracrine regulation of the murine skeleton by osteocyte-derived parathyroid hormone-related protein, J. Bone Miner. Res 33 (2017) 137–153, 10.1002/jbmr.3291. [DOI] [PubMed] [Google Scholar]
- [60].Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, et al. , Cremophor EL-mediated alteration of paclitaxel distribution in human blood: Clinical pharmacokinetic implications, Cancer Res. 59 (1999) 1454–1457, [PubMed] [Google Scholar]
- [61].Gupta MK, Martin JR, Werfel TA, Shen T, Page JM, Duvall CL, Cell protective ABC triblock polymer-based thermoresponsive hydrogels with ROS-triggered degradation and drug release, J. Am. Chem. Soc 136 (2014) 14896–14902, 10.1021/ja507626y. [DOI] [PubMed] [Google Scholar]
- [62].O’Grady KP, Kavanaugh TE, Cho H, Ye H, Gupta MK, Madonna MC, et al. , Drug-free ROS sponge polymeric microspheres reduce tissue damage from ischemic and mechanical injury, ACS Biomater. Sci. Eng 4 (2018) 1251–1264, 10.1021/acsbiomaterials.6b00804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dollinger BR, Gupta MK, Martin JR, Duvall CL, Reactive oxygen species shielding hydrogel for the delivery of adherent and nonadherent therapeutic cell types, Tissue Eng. Part A 23 (2017) 1120–1131, 10.1089/ten.tea.2016.0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Monfardini C, Schiavon O, Caliceti P, Morpurgo M, Harris JM, Veronese FM, A branched monomethoxypoly(ethylene glycol) for protein modification, Bioconjug. Chem 6 (1995) 62–69, [DOI] [PubMed] [Google Scholar]
- [65].Gao W, Liu W, Mackay JA, Zalutsky MR, Toone EJ, Chilkoti A, In situ growth of a stoichiometric PEG-like conjugate at a protein’s N-terminus with significantly improved pharmacokinetics, Proc. Natl. Acad. Sci. USA 106 (2009) 15231–15236, 10.1073/pnas.0904378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gao W, Liu W, Christensen T, Zalutsky MR, Chilkoti A, In situ growth of a PEG-like polymer from the C terminus of an intein fusion protein improves pharmacokinetics and tumor accumulation, Proc. Natl. Acad. Sci. USA 107 (2010) 16432–16437, 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gurer-Orhan H, Ince E, Konyar D, Saso L, Suzen S, The role of oxidative stress modulators in breast cancer, Curr. Med. Chem 25 (2017) 4084–4101, 10.2174/0929867324666170711114336. [DOI] [PubMed] [Google Scholar]
- [68].Werfel TA, Jackson MA, Kavanaugh TE, Kirkbride KC, Miteva M, Giorgio TD, et al. , Combinatorial optimization of PEG architecture and hydrophobic content improves ternary siRNA polyplex stability, pharmacokinetics, and potency in vivo, J. Control Release 255 (2017) 12–26, 10.1016/j.jconrel.2017.03.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chaffer CL, Weinberg RA, A perspective on cancer cell metastasis, Science 331 (2011) 1559–1564, 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- [70].Baron R, Ferrari S, Russell RG, Denosumab and bisphosphonates: Different mechanisms of action and effects, Bone 48 (2011) 677–692, 10.1016/j.bone.2010.11.020. [DOI] [PubMed] [Google Scholar]
- [71].Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, et al. , Atypical subtrochanteric and diaphyseal femoral fractures: Second report of a task force of the American Society for Bone and Mineral Research, J. Bone Miner. Res 29 (2013) 1–23, 10.1002/jbmr.1998. [DOI] [PubMed] [Google Scholar]
- [72].Pozzi S, Vallet S, Mukherjee S, Cirstea D, Vaghela N, Santo L, et al. , High-dose zoledronic acid impacts bone remodeling with effects on osteoblastic lineage and bone mechanical properties, Clin. Cancer Res 15 (2009) 5829–5839, 10.1158/1078-0432.CCR-09-0426. [DOI] [PubMed] [Google Scholar]
- [73].Idris AI, Rojas J, Greig IR, Van’t Hof RJ, Ralston SH, Aminobisphosphonates cause osteoblast apoptosis and inhibit bone nodule formation in vitro, Calcif. Tissue Int 82 (2008) 191–201, 10.1007/s00223-008-9104-y. [DOI] [PubMed] [Google Scholar]
- [74].Xu Y, Chenna V, Hu C, Sun HX, Khan M, Bai H, et al. , Polymeric nanoparticle-encapsulated hedgehog pathway inhibitor HPI-1 (NanoHHI) inhibits systemic metastases in an orthotopic model of human hepatocellular carcinoma, Clin. Cancer Res 18 (2012) 1291–1302, 10.1158/1078-0432.ccr-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ross MH, Esser AK, Fox GC, Schmieder AH, Yang X, Hu G, et al. , Bone-induced expression of integrin beta3 enables targeted nanotherapy of breast cancer metastases, Cancer Res. 77 (2017) 6299–6312, 10.1158/0008-5472.can-17-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Yamashita S, Katsumi H, Hibino N, Isobe Y, Yagi Y, Tanaka Y, et al. , Development of PEGylated aspartic acid-modified liposome as a bone-targeting carrier for the delivery of paclitaxel and treatment of bone metastasis, Biomaterials 154 (2018) 74–85, 10.1016/j.biomaterials.2017.10.053. [DOI] [PubMed] [Google Scholar]
- [77].Hu W, Sung T, Jessen BA, Thibault S, Finkelstein MB, Khan NK, et al. , Mechanistic investigation of bone marrow suppression associated with palbociclib and its differentiation from cytotoxic chemotherapies, Clin. Cancer Res 22 (2016) 2000–2008, 10.1158/1078-0432.ccr-15-1421. [DOI] [PubMed] [Google Scholar]
- [78].Fonseca H, Carvalho A, Esteves J, Esteves VI, Moreira-Goncalves D, Duarte JA, Effects of doxorubicin administration on bone strength and quality in sedentary and physically active Wistar rats, Osteoporos. Int 27 (2016) 3465–3475, 10.1007/s00198-016-3672-x. [DOI] [PubMed] [Google Scholar]
- [79].Mazumdar T, DeVecchio J, Agyeman A, Shi T, Houghton JA, Blocking hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells, Cancer Res. 71 (2011) 5904–5914, 10.1158/0008-5472.can-10-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Torchilin V, Tumor delivery of macromolecular drugs based on the EPR effect, Adv. Drug Deliv. Rev 63 (2011) 131–135, 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- [81].Page JM, Merkel AR, Ruppender NS, Guo R, Dadwal UC, Cannonier S, et al. , Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin beta3 and TGF-beta receptor type II, Biomaterials 64 (2015) 33–44, 10.1016/j.biomaterials.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Jansen LE, Birch NP, Schiffman JD, Crosby AJ, Peyton SR, Mechanics of intact bone marrow, J. Mech. Behav. Biomed. Mater 50 (2015) 299–307, 10.1016/j.jmbbm.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ruppender NS, Merkel AR, Martin TJ, Mundy GR, Sterling JA, Guelcher SA, Matrix rigidity induces osteolytic gene expression of metastatic breast cancer cells, PLoS One 5 (2010) e15451, 10.1371/journal.pone.0015451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sethi N, Dai X, Winter CG, Kang Y, Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells, Cancer Cell 19 (2011) 192–205, https://doi.org/10.1016Zj.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zheng H, Bae Y, Kasimir-Bauer S, Tang R, Chen J, Ren G, et al. , Therapeutic antibody targeting tumor- and osteoblastic niche-derived jagged1 sensitizes bone metastasis to chemotherapy, Cancer Cell 32 (2017) 731–747.e736, 10.1016/j.ccell.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Li C, Wang S, Xing Z, Lin A, Liang K, Song J, et al. , A ROR1-HER3-IncRNA signalling axis modulates the Hippo-YAP pathway to regulate bone metastasis, Nat. Cell Biol 19 (2017) 106–119, 10.1038/ncb3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wan L, Pantel K, Kang Y, Tumor metastasis: Moving new biological insights into the clinic, Nat. Med 19 (2013) 1450–1464, 10.1038/nm.3391. [DOI] [PubMed] [Google Scholar]
- [88].Powell GJ, Southby J, Danks JA, Stillwell RG, Hayman JA, Henderson MA, et al. , Localization of parathyroid hormone-related protein in breast cancer metastases: increased incidence in bone compared with other sites, Cancer Res. 51 (1991) 3059–3061, [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.