

Abstract
Rationale:
Genome-wide association studies have identified over 100 genetic loci for atrial fibrillation (AF); recent work described an association between loss-of-function (LOF) variants in TTN and early-onset AF.
Objective:
We sought to determine the contribution of rare and common genetic variation to AF risk in the general population.
Methods:
The UK Biobank is a population-based study of 500,000 individuals including a subset with genome-wide genotyping and exome sequencing. In this case-control study, we included AF cases and controls of genetically determined white-European ancestry; analyses were performed using a logistic mixed-effects model adjusting for age, sex, the first 4 principal components of ancestry, empirical relationships and case-control imbalance. An exome wide, gene-based burden analysis was performed to examine the relationship between AF and rare, high-confidence LOF variants in genes with ≥ 10 LOF carriers. A polygenic risk score (PRS) for AF was estimated using the LDpred algorithm. We then compared the contribution of AF PRS and LOF variants to AF risk.
Results:
The study included 1,546 AF cases and 41,593 controls. In an analysis of 9,099 genes with sufficient LOF variant carriers, a significant association between AF and rare LOF variants was observed in a single gene, TTN (OR 2.71, P=2.50×10−8). The association with AF was more significant (OR 6.15, P=3.26×10−14) when restricting to LOF variants located in exons highly expressed in cardiac tissue (TTNLOF). Overall, 0.44% of individuals carried TTNLOF variants, of whom 14% had AF. Among individuals in the highest 0.44% of the AF PRS, only 9.3% had AF. In contrast, an AF PRS explained 4.7% of the variance in AF susceptibility, while TTNLOF variants only accounted for 0.2%.
Conclusion:
Both monogenic and polygenic factors contribute to AF risk in the general population. While monogenic TTNLOF variants confer a substantial AF penetrance, polygenic risk explains a larger proportion of genetic susceptibility to AF.
Keywords: Atrial fibrillation, genetics, TTN, polygenic risk score, gene mutation, exome, association study
Subject Terms: Atrial Fibrillation
Graphical Abstract
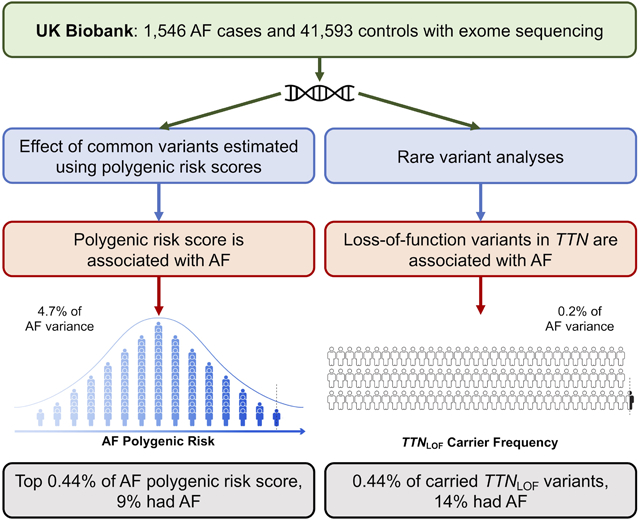
Over the last decade, great progress has been made in defining the genetic basis of AF. Common variants have been identified at more than 100 genetic loci, and rare mutations have implicated many genes in AF. However, the relative contribution of rare and common genetic variants to AF risk remains unclear. The population-based UK Biobank provides a unique opportunity to assess the genetic contributions to AF risk. In an exome wide analysis, we found that LOF mutations in TTN were strongly associated with AF risk and were highly penetrant. A polygenic risk score of common variants explains a great proportion of AF risk than mutations in TTN. Among TTN mutation carriers, it would be interesting in future work to determine if a subtle cardiomyopathy is present by cardiac imaging or to examine the progression to heart failure or other AF co-morbidities.
INTRODUCTION
Atrial fibrillation (AF) is a prevalent cardiac arrhythmia and is associated with an increased risk of stroke, heart failure, dementia, and death1–3. AF currently affects over 3 million Americans and 30 million individuals worldwide4. While cardiometabolic factors play an important role5, a considerable heritable component is thought to contribute to the pathogenesis of AF6, 7. For example, one-fourth of individuals with AF have a first-degree relative affected by the condition8.
Accordingly, genome-wide association studies (GWAS) have successfully established a multitude of genetic loci with common variants predisposing to AF9–12 and subsequent polygenic risk scores (PRS) have identified individuals in the general population who are at high risk of developing the disease13–15. The SNP-heritability for AF has been estimated to be as high as 22%2, and previously reported common variants from GWAS3 explained 5.3% of AF variability2. In contrast, family-based analyses have identified many ‘monogenic’ variants, located mainly in ion-channels16, yet replication for many of these genes is lacking11, 17. We previously demonstrated in a large case-control study that rare loss-of-function variants in the structural sarcomeric gene TTN are associated with a strongly increased risk of early-onset AF18. Despite this observation, the major monogenic contributors to AF risk in the general population remain unclear. Moreover, the relationship between polygenic and monogenic variation to AF susceptibility remains unexplored.
To address these knowledge gaps, we leveraged data from a national biorepository, the UK Biobank. We used a subset of over 40,000 individuals with genome-wide genotyping and whole-exome sequencing (WES)19, 20 to comprehensively assess the respective contributions of common and rare genetic variation to AF risk.
METHODS
Data availability.
Whole exome-sequencing and phenotype data used in this study are available through UK Biobank (www.ukbiobank.ac.uk). Summary level results have been made publicly available at the Cardiovascular Disease Initiative Knowledge Portal and can be accessed at www.broadcvdi.org upon publication.
Study population and phenotypes.
The UK Biobank is a large population-based prospective cohort study from the United Kingdom with deep phenotypic and genetic data on approximately 500,000 individuals aged 40–6919. Phenotypes, including the primary outcome AF, were defined using reports from medical history interviews, ICD-9 and −10 codes, operation codes and death registry records (Online Table I). The UK Biobank resource was approved by the UK Biobank Research Ethics Committee and all participants provided written informed consent to participate. Use of UK Biobank data was performed under application number 17488 and was approved by the local Massachusetts General Hospital institutional review board.
Genotyping, quality control, and variant annotation.
Whole exome sequencing has previously been performed on 50,000 participants from the UK Biobank20. The revised version of the IDT xGen Exome Research Panel v1.0 was used to capture exomes with over 20X coverage at 94.6% of sites20. In the present study, samples were restricted to those of white-European ancestry who also had high-quality genotyping array data available21, 22 (Online Data Supplement). Additional filters were applied to study samples and exome sequence variants: sample call rate (<90%), genotype call rate (<90%) and Hardy-Weinberg equilibrium test (P-value < 1×10−15). Of the 50,000 individuals in the UK Biobank with WES, 40 were removed during initial quality-control, after which we excluded 51 samples who did not have genotyping chip data and 170 samples that failed our additional quality control procedures (Online Data Supplement). Among the remaining 49,739 participants, 43,139 white-European individuals were identified.
The protein consequences of variants were explored using the LOFTEE plug-in implemented in the Variant Effect Predictor23 (https://github.com/konradjk/loftee, Online Data Supplement). The most severe predicted consequences for canonical gene transcripts were ascertained for each variant and used for the primary analysis. All variants in significantly associated genes were re-annotated using LOFTEE to identify additional high-confidence loss-of-function (LOF) variants in other transcripts.
Single variant association analyses.
An exome-wide single variant association analysis for AF was performed using AF cases and controls of white-European ancestry. Variants with minor allele frequency (MAF) ≥ 1% were tested for association with AF assuming an additive genetic model. To correct for the relatedness among participants and the imbalanced case-control ratio, we used a logistic mixed-effects model implemented in SAIGE (https://github.com/weizhouUMICH/SAIGE)24. Age, sex, and the first 4 principal components of ancestry were used as fixed effects. The genetic relatedness matrix was estimated using independent high-quality variants from the genotyping array (N = 93,491, Online Data Supplement). The exome-wide significance threshold was set to alpha=3.1×10−7 (0.05/162,514, Bonferroni correction).
Polygenic risk score estimation.
We closely followed a previously published approach to derive and validate an AF PRS13 as shown in Online Figure I. In short, effect estimates for common variants from a large AF GWAS meta-analysis11 were adjusted to account for linkage-disequilibrium using LDpred (https://github.com/bvilhjal/ldpred)25. Multiple PRSs were constructed using high quality imputed variants (Online Data Supplement) based on 7 different values of ρ (the assumed fraction of variants with nonzero effects) and were applied to the UK Biobank. In this study, we used the LDpred-adjusted effect estimates for each value of ρ and identified the best performing PRS in a validation cohort of unrelated white-European individuals distinct from the exome sequencing cohort (N = 322,161, Online Data Supplement). The performance of PRS was assessed by the area under the receiver operating characteristic curve (AUC). AUC and confidence intervals were calculated using R-package ‘pROC’ version 1.12.126. The best performing PRS was subsequently applied to the imputed genotypes from the exome-sequencing cohort. Effect-estimates and respective confidence intervals by profile likelihood were calculated using Firth’s bias-reduced logistic regression, implemented in R-package ‘logistf’ version 1.23 (https://rdrr.io/cran/EHR/man/Logistf.html)27, 28.
Rare variant burden analyses.
Rare variants (MAF ≤ 1%) that were predicted to be LOF were associated with AF in the exome sequencing cohort using a gene-based burden analysis. LOF variants were collapsed into a single variable (carrier vs. non-carrier) by sample, for each gene. Genes with ≥ 10 LOF variant carriers were analyzed using SAIGE as described, for the single variant analysis above. The exome-wide significance threshold was determined to be 5.04×10−6 by using a Bonferroni correction of 0.05 / 9,909 genes. Odds ratios (OR) and confidence intervals were estimated using Firth’s regression in an unrelated (relatedness estimated to be 3rd degree or closer was removed) subset of the cohort (N = 41,335). Additionally, sensitivity analyses were performed adjusting for the AF PRS.
Upon identifying a significant association between AF and LOF variants in TTN (Results), we analyzed LOF variants located in exons that are highly expressed (percentage splicing index ≥ 90%) in left ventricular tissue29, denoted as TTNLOF variants. From then on, all individuals with a diagnosis of heart failure concurrent or prior to diagnosis of AF were removed for AF analyses, as heart failure is strongly associated with both TTN LOF variants and atrial arrhythmias30, 31. We further compared the prevalence of TTNLOF variants among individuals with AF, heart failure, and nonischemic cardiomyopathy and calculated the penetrance of TTNLOF variants for those diseases in the unrelated population.
Association analyses between TTNLOF and multiple traits.
To identify novel phenotypic associations with TTNLOF variants and to confirm known associations30, 31, we then performed association analyses using a curated set of disease phenotypes and continuous cardiometabolic traits. Association tests were performed for LOF variants in TTN, using a list of curated disease phenotypes and continuous cardiometabolic traits (N = 58, Online Table I). Disease phenotypes with < 50 cases or a prevalence of LOF variant carriers ≤ 0.5% were excluded to avoid spurious associations. Association tests on remaining diseases (N = 31) were carried out using the same logistic mixed-effects models implemented in the rare variant analyses. Age, sex, and first 4 principal components of ancestry were implemented as fixed effects. Effect estimates and confidence intervals were estimated using Firth’s logistic regression in the unrelated subset. In addition, quantitative traits of BMI, blood pressure, and electrocardiogram measurements (Online Table II) were inverse normalized using the “—invNormalize” flag implemented in SAIGE and then associated with LOF variants in TTN using a linear mixed-effects model adjusting for the same covariates. For PR interval, P wave duration, and QRS complex, we additionally adjusted for the RR-interval. A P-value of 6.41 × 10−4 (0.05 / (39 × 2) traits; Bonferroni correction) was considered significant. For significantly associated diseases, a sensitivity analysis was performed where individuals with AF prior to the diagnosis were removed.
Monogenic and polygenic risk.
Within the entire unrelated population (N = 41,212), AF prevalence conferred by high polygenic risk was calculated by comparing increasingly extreme tails of the PRS distribution. Odds ratios conferred by high polygenic risk were estimated by comparing these tails to the remainder of the population, using Firth’s logistic regression adjusting for age, sex, and the first 4 principal components of ancestry. We further identified what increment of PRS in standard deviations (SD) was predicted to be equivalent to the risk conferred by TTNLOF variants. This was based on the assumed linear relationship between the PRS and the log of odds for AF in logistic regression. We then assessed the effect of PRS on AF penetrance among TTNLOF variant carriers. This was done by testing the association between PRS and AF within carriers only, using Firth’s logistic regression adjusted for age, sex, and the first 4 principal components of ancestry. Finally, the variance in AF susceptibility explained by both TTNLOF variants and AF PRS were calculated. This was done by calculating the improvement in R2 on the liability scale, upon adding either predictor to Firth’s regression models which included age, sex and the first 4 principal components of ancestry as covariates. The prevalence of AF (3.3%) was determined in the exome sequencing samples.
RESULTS
Baseline characteristics.
After sample level quality controls, 1,546 AF patients were identified with a mean age at AF onset of 62.6 years and 33.7% of AF cases were female (Table 1). The remaining 41,593 participants were considered controls. A total of 8.7 million distinct genetic variants were available from the exome sequencing data.
Table 1.
Characteristics of controls, atrial fibrillation cases, and atrial fibrillation cases with LOF carriers
| Controls | AF* cases | AF cases carrying TTNLOF† variants | |
|---|---|---|---|
| Participants, N§ | 41,593 | 1,546 | 34 |
| Female, N (%) | 22,825 (54.88) | 521 (33.7) | 14 (41.18) |
| Age at baseline, Mean (SD||) | 57.2 (7.9) | 62.7 (6) | 62.3 (6) |
| Age at onset, Mean (SD) | - | 62.6 (7.3) | 62.6 (7.6) |
| Hypertension, N (%) | 13,198 (31.73) | 987 (63.84) | 17 (50) |
| Heart Failure, N (%) | 293 (0.7) | 237 (15.4) | 12 (35.29) |
| Myocardial Infarction, N (%) | 1,096 (2.64) | 198 (12.81) | 4 (11.76) |
| Diabetes, N (%) | 2,630 (6.32) | 229 (14.81) | 11 (32.35) |
AF: atrial fibrillation,
TTNLOF: predicted to be high-confidence loss-of-function variant in TTN exons highly expressed in cardiac tissues,
N: the number of samples,
SD: standard deviation.
Polygenic risk scores are strongly associated with AF risk.
Among 7 candidate PRSs, we found that the PRS derived with ρ = 0.003 was the best predictor of AF in the validation cohort (AUC 0.613; 95% CI 0.608–0.618) (Online Table III). In the exome sequencing cohort, the PRS performed slightly better (AUC 0.636, 95% CI 0.622–0.650) than in the validation cohort. The OR for AF per SD increment of PRS was 1.63 (95% CI 1.55–1.71, P-value <1×10−15). Individuals in the top decile of the PRS were at 2.53-fold increased odds of AF compared to the remainder of the population (95% CI 2.21–2.89, P-value <1×10−15, Online Table IV).
Mutations in TTN are associated with AF in the general population.
Associations between AF and 162,514 variants with MAF ≥ 1% were assessed using a logistic mixed-effects model accounting for age, sex, population structure, and sample relatedness. There were no single exonic variants that reached exome-wide significance in the present analysis.
Next, we sought to determine if there were any genes with a burden of LOF mutations that were associated with AF. Among 18,350 protein-coding canonical gene transcripts, 9,099 had ≥ 10 LOF variant carriers and were tested for the association with AF. We found that LOF variants in TTN (N = 259 variants) were significantly associated with AF (OR 2.71, 95% CI 1.97–3.66, P-value = 2.50×10−8, N = 554 carriers, Online Table V). When we performed a sensitivity analysis adjusting for AF PRS (Figure 1A, Online Figure II), the significant association between LOF variants in TTN and AF remained similar (OR 2.70, 95% CI 1.95–3.66, P-value = 3.12×10−8, Online Table V). The prevalence of LOF variants in TTN among AF cases was 3.2% (N = 49 carriers) vs 1.2% (N = 505 carriers) among controls.
Figure 1. High-confidence loss-of-function variants in TTN among atrial fibrillation cases and controls in UK Biobank.
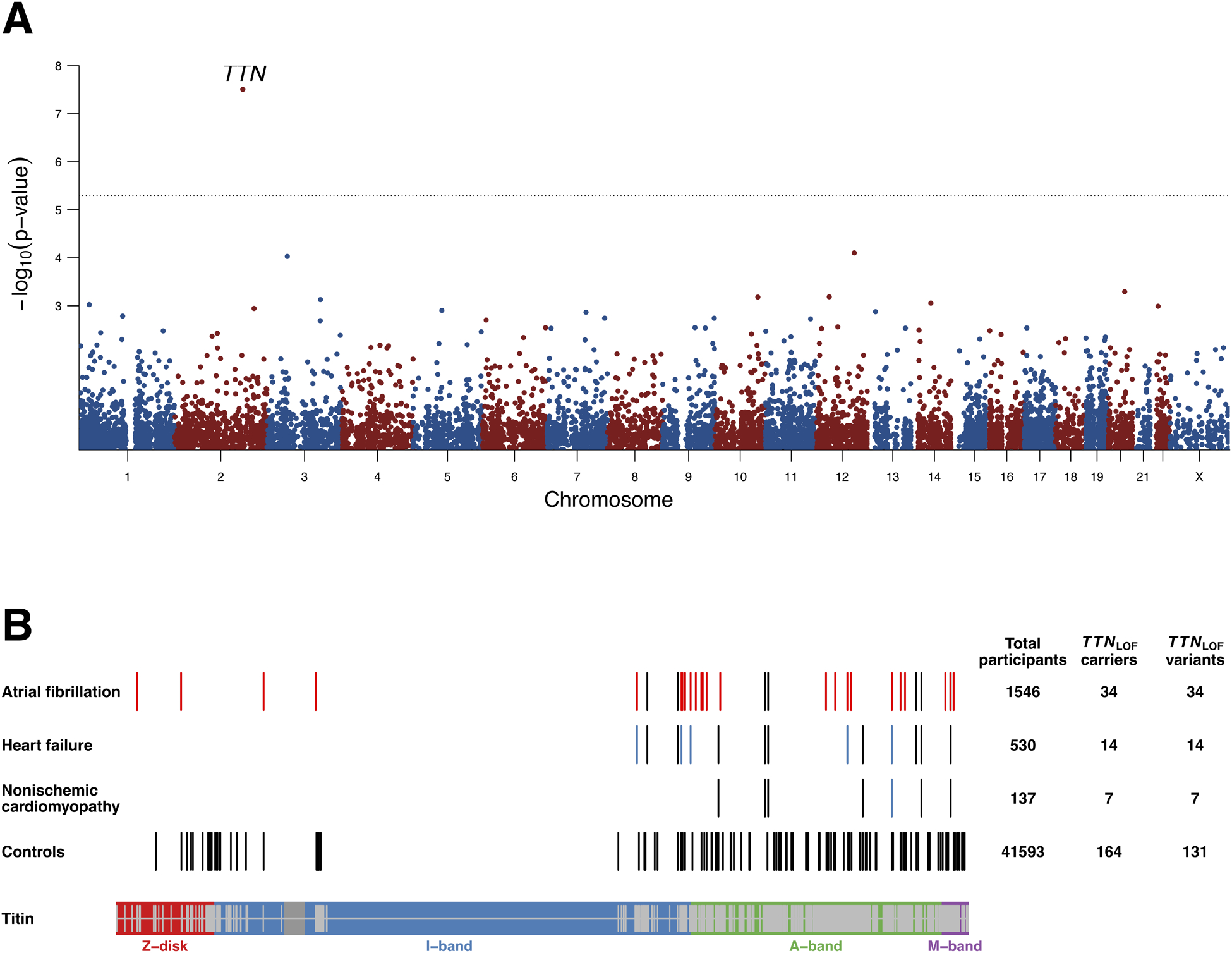
Figure 1A is a Manhattan plot of the gene-based burden analysis for predicted high-confidence loss-of-function (LOF) variants and atrial fibrillation. Grey dotted line represents the exome-wide significance level. Results are based on LOF variants in canonical transcripts only, and are adjusted for sex, age, polygenic risk score and the first four principal components of ancestry. LOF variants in TTN are associated with AF. Figure 1(B) shows the locations of LOF variants in titin (protein encoded by TTN) found in heart failure, non-ischemic cardiomyopathy and atrial fibrillation patients, as well as in controls. Shown variants are restricted to those found in exons highly expressed in cardiac tissue. Red bars (N = 27) in atrial fibrillation cases are LOF variants found among patients who did not have heart failure prior to atrial fibrillation. Blue bars from the second and third rows represent LOF variants identified from patients who had atrial fibrillation prior to heart failure or non-ischemic cardiomyopathy. The bottom of the Figure 1B illustrates different bands of TTN. The TTN exons highly expressed in heart tissue are shown on the inside of the band in grey.
We did not observe a significant association between AF and rare variation in 37 genes previously reported as candidate genes for monogenic forms of AF (Online Table VI, Online Figure III).7 Similarly, we tested for an association between LOF variation and the genes at recently described GWAS loci for AF10. At the 94 AF GWAS loci, there were 421 of 1,181 genes that had a sufficient number of loss of function variants to test for an association with AF. Of these 421 genes, only TTN was significantly associated with AF (Online Table VII). Furthermore, LOF variants in TTN were more common than LOF variants in other genes implicated in monogenic forms of cardiovascular disease, such as LDLR, MYBPC3, SCN5A, and KCNQ1 (Online Table VIII).
LOF variants in cardiac exons of TTN are strongly associated with AF.
We then performed a series of post-hoc analyses focused on the association between TTN and AF. First, we identified 20 additional LOF variants in non-canonical sequences, which in aggregate with canonical transcript variants were still significantly associated with AF (OR 2.66, 95% CI 1.94–3.56, P-value = 2.80×10−8, N = 591 carriers, Online Tables IX–X). Second, we restricted our analysis to LOF variants in exons highly expressed in cardiac tissue29 (TTNLOF), which left 198 carriers with 178 distinct LOF variants. Using these variants, the association with AF substantially strengthened (OR 6.15, 95% CI 4.07–9.06, P-value = 3.26×10−14, N = 198 carriers, Online Table X) whereas LOF variants in other exons were not associated with AF (OR 1.26, 95% CI 0.74–2.00, P-value = 0.58, N = 395, Online Table X). Third, there is a well-described relationship between LOF variants in TTN and dilated cardiomyopathy30, 31. Because of this, we excluded AF cases with heart failure concurrent or prior to AF diagnosis. Even after removal of 132 of such cases, the association between TTNLOF variants and AF persisted at exome-wide significance (OR 5.35, 95% CI 3.39–8.13, P-value = 1.12×10−10, Figure 1B, Online Table XI). The prevalence of TTNLOF variants among these AF cases was 1.9% (N = 27 carriers) vs 0.4% (N = 164 carriers) among controls.
TTNLOF variants are more penetrant for AF than for heart failure.
Next, we investigated the frequency and phenotypic presentation of TTNLOF variants with respect to both AF and heart failure phenotypes in unrelated participants. Of the phenotypes, TTNLOF variants were most common among individuals with nonischemic cardiomyopathy (5.4% of cases; N = 7 carriers, Figure 2A). In contrast, TTNLOF variants were more penetrant for AF than for heart failure: 14.4% (N = 26 carriers) of carriers had AF while only 7.4% (N = 14 carriers) had heart failure (Figure 2B).
Figure 2. Prevalence and penetrance of TTNLOF variants with respect to atrial fibrillation and heart failure.
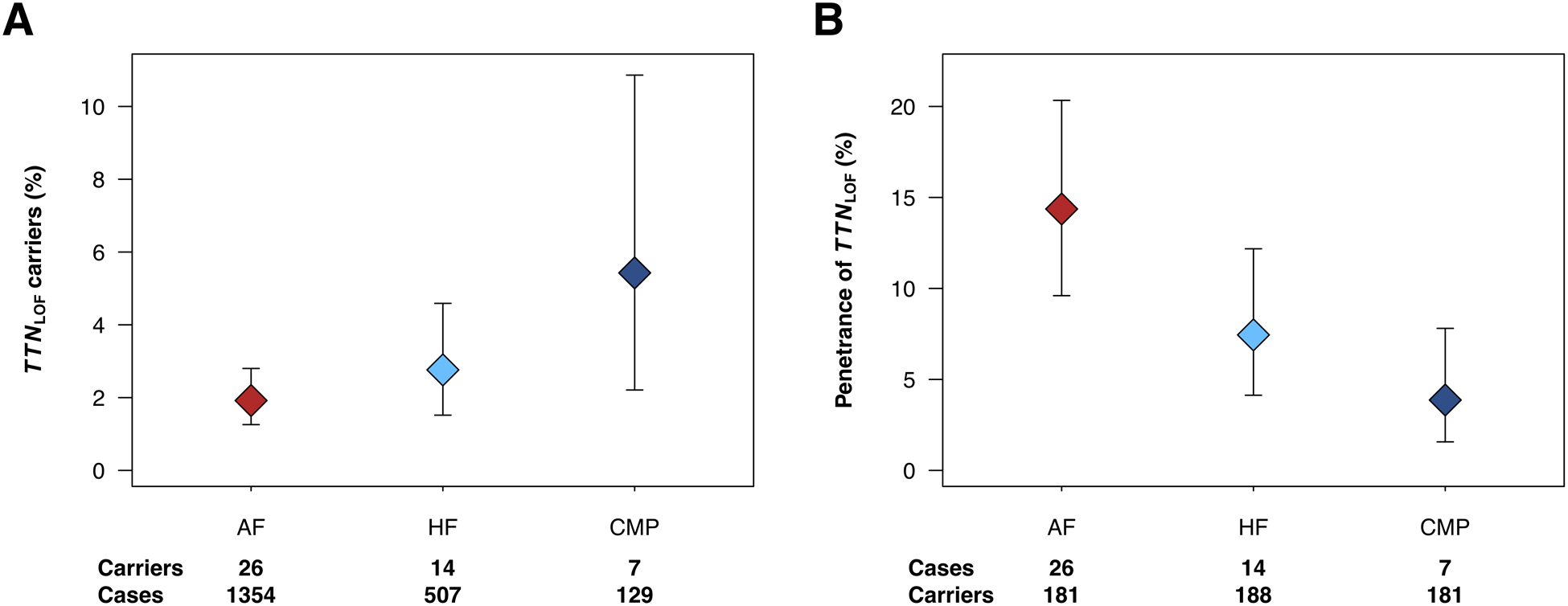
Figure 2A exhibits the proportion of carriers with high confidence loss-of-function variants in cardiac TTN (TTNLOF) and 95% confidence intervals among unrelated atrial fibrillation (AF), heart failure (HF), and nonischemic cardiomyopathy (CMP) cases. Figure 2B shows the penetrance of TTNLOF variants for AF, HF, and CMP. Of the three diseases, TTNLOF variants are most frequent among individuals with non-ischemic cardiomyopathy. All values are calculated from an unrelated subset of the exome sequencing cohort (N = 41,212). AF cases with HF prior to AF are excluded.
Multiple trait analysis confirms known associations between TTNLOF and cardiovascular traits.
We then performed association tests for TTNLOF variants using a curated set of 31 disease phenotypes and continuous cardiometabolic traits. We found that nonischemic cardiomyopathy, heart failure, supraventricular arrhythmia, mitral valve disease, and the RR interval were significantly associated with TTNLOF variants (P < 6.41 × 10−4, Online Figure IV, Online Table XII). After removal of participants who had AF prior to the diagnosis of disease, TTNLOF variants remained significantly associated with nonischemic cardiomyopathy (OR 21.61, 95% CI 8.56–46.02, P= 7.9 × 10−7, Online Figure V).
TTNLOF variants are substantially penetrant for AF while PRS explains more genetic susceptibility.
We then sought to determine the relative contribution of both PRS and TTNLOF variants to the overall risk of AF. We began by comparing the AF prevalence conferred by high PRS to the prevalence conferred by TTNLOF variants. In our study population, 0.44% of individuals carried TTNLOF variants, of whom 14% had AF (Figure 3). In contrast, among individuals in the highest 0.44% of the AF PRS, only 9.3% had AF (Figures 3–4, Online Table IV). Only individuals in the highest 0.10% of the PRS had an AF prevalence comparable to the prevalence observed among TTNLOF variant carriers (Figure 3, Online Table IV). TTNLOF variants were predicted to confer a risk equivalent to a 3.39 SD increment of PRS. Only 0.10% of the cohort had a polygenic score of 3.39 SD from the mean or higher (Online Figure VI). In contrast, TTNLOF variants explained only 0.2% of the variance in AF susceptibility in the study population, and inclusion of LOF variants from additional 13 testable AF genes explained only 0.4% of the variance in AF risk. The AF PRS, on the other hand, explained 4.7% of the variance in AF susceptibility (Online Table XIII).
Figure 3. Prevalence of atrial fibrillation conferred by loss-of-function variants in cardiac TTN compared to polygenic risk in the UK Biobank.
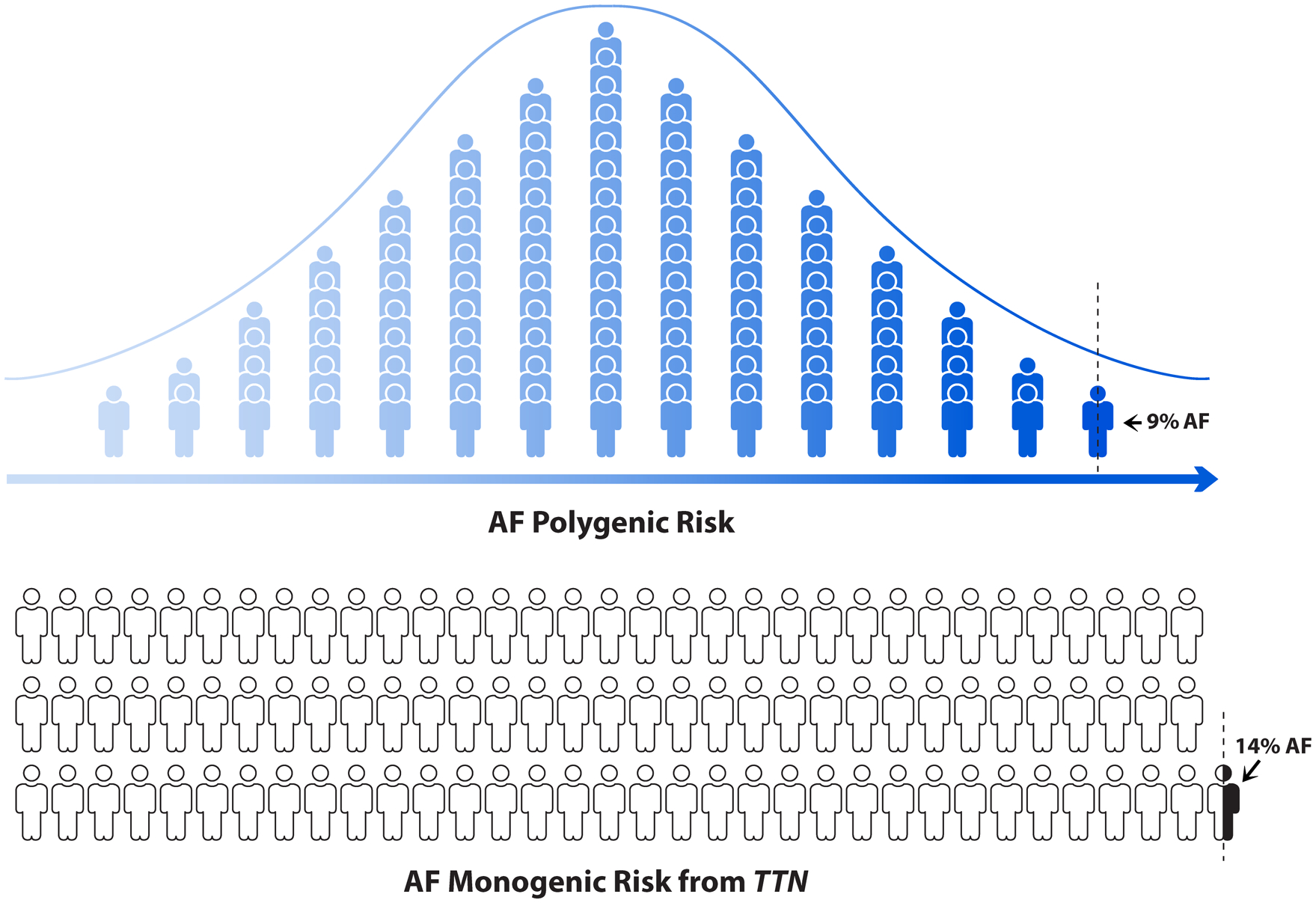
The first figure illustrates the distribution of AF polygenic risk score in the UK Biobank. Each human icon represents 1% of the population and a dotted vertical line exhibits highest 0.44% of AF polygenic risk group. Among this 0.44% group, 9.3% individual had atrial fibrillation. The bottom figure illustrates the carriers with high confidence loss-of-function variants in cardiac TTN (TTNLOF). As shown in the last human icon, 0.44% participants of the UK Biobank carried TTNLOF variants and among those, 14.3% had atrial fibrillation.
Figure 4. Prevalence of atrial fibrillation conferred by loss-of-function variants in cardiac TTN compared to polygenic risk in the UK Biobank.

Figure 4 shows the prevalence of atrial fibrillation (AF) conferred by loss-of-function variants in cardiac TTN (TTNLOF) and the prevalence conferred by high AF polygenic risk scores (PRS) in an unrelated subset of the exome sequencing cohort where cases of AF with heart failure prior to AF are excluded (N = 41,212). Increasingly extreme tails of the PRS distribution are shown in blue. TTNLOF variant carriers are shown in red. Approximately 0.44% of the population carried TTNLOF variants, of which 14% had AF. Meanwhile, only 9.3% of individuals in the top 0.44% of AF PRS had AF.
PRS associates with AF penetrance among TTNLOF carriers.
Finally, we investigated whether AF polygenic risk affects the penetrance of TTNLOF variants. The overall prevalence of AF among TTNLOF variants was 14.4% compared to 3.2% among non-carriers (Figures 3–4; Online Figure VI). Within the 181 carriers of TTNLOF variants, the PRS significantly associated with AF (OR per SD 1.79, 95% CI 1.17–2.85, P-value = 0.007). The observed prevalence of AF among TTNLOF variant carriers in the highest tertile of polygenic risk was 21.5% compared to 6.7% in the lowest tertile (Figure 5).
Figure 5. Prevalence of atrial fibrillation stratified by monogenic and polygenic risk in the UK Biobank.
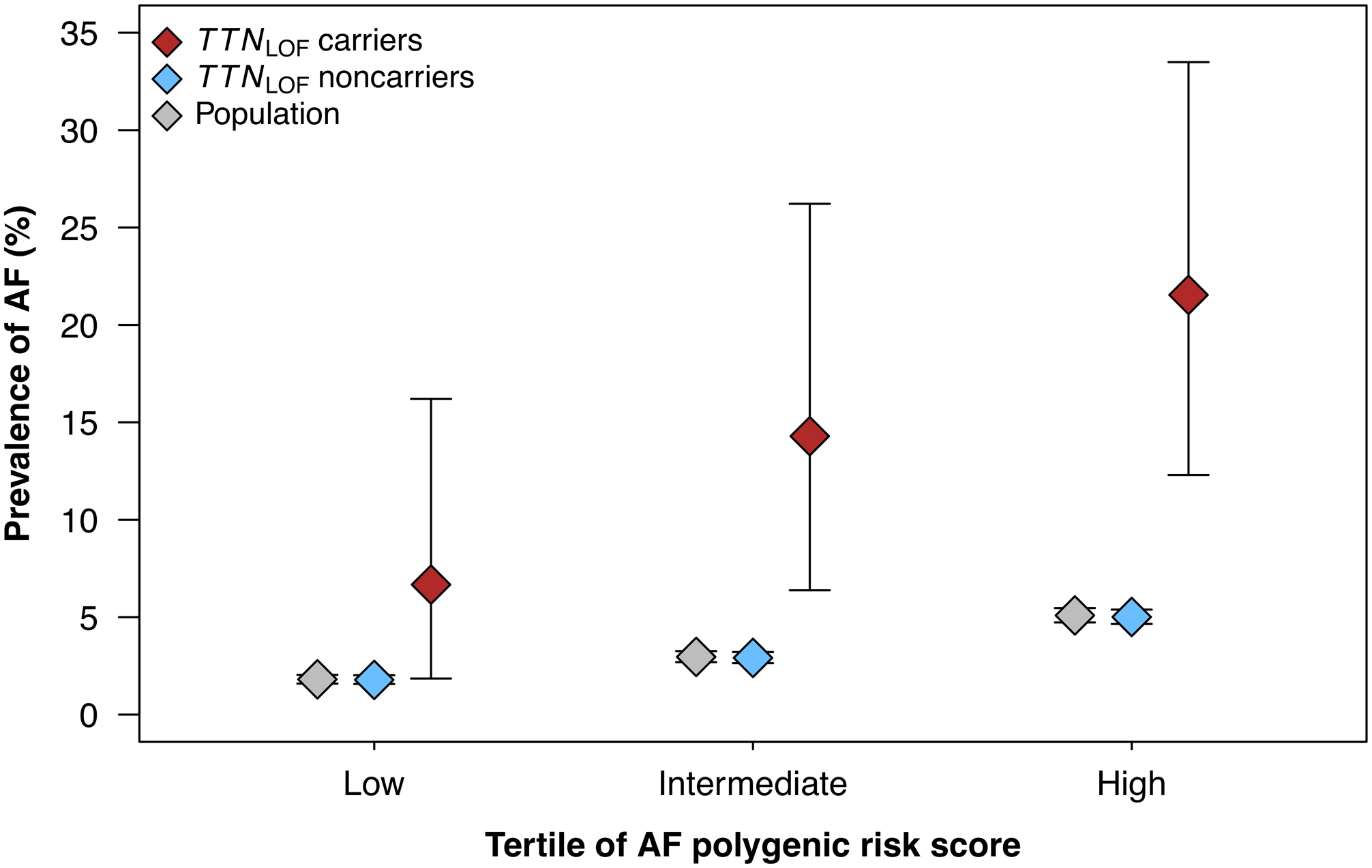
Figure 5 shows the prevalence of atrial fibrillation (AF), stratified by polygenic and monogenic risk in the unrelated subset of the exome sequencing cohort (N = 41,212). In the population, AF prevalence increased with increasing AF polygenic risk score (PRS) and was considerably higher in carriers of loss-of-function variants in cardiac TTN (TTNLOF) which are shown in red. Among TTNLOF carriers, AF PRS associated with AF penetrance: Carriers in the lowest tertile of PRS had an AF prevalence of 6.7% compared to 21.5% in the highest tertile.
DISCUSSION
The availability of both genotyping array and exome sequencing data in over 43,000 individuals from the UK Biobank provided a unique opportunity to explore the contributions of common and rare genetic variation to AF. In the current work, we had four primary observations. First, we found that TTN is the most frequently implicated gene for AF in the general population in terms of LOF variation. Second, LOF variants in the cardiac exons29 of TTN are strongly associated with AF, regardless of a prior history of heart failure. Third, despite the substantial AF penetrance conferred by TTN mutations, the polygenic risk for AF explains a larger proportion of genetic susceptibility in the general population. Finally, the polygenic risk of AF markedly alters the disease penetrance among TTN mutation carriers.
In an exome wide analysis of the population-based UK Biobank, we identified mutations in a single gene, TTN, that were significantly related to AF. The TTN gene encodes a very large sarcomeric protein, titin, that is crucial for sarcomere assembly, cardiac muscle contraction, and elasticity32. Truncating variants in TTN are a well-known cause of dilated cardiomyopathy30 and have also been identified in other cardiac and skeletal muscle myopathies. Recently, we and others found a significant association between TTN truncating variants in selected individuals with familial AF33 and early-onset AF18. Given that AF is a risk factor for heart failure34, and heart failure is also a risk factor for AF35, it will be interesting to explore the temporal relationship between each of these diseases among TTNLOF mutation carriers. In the current work, we establish a significant association between TTNLOF variants and AF even after removal of any individuals with heart failure prior to the onset of AF (OR 5.35, 95% CI 3.39–8.13, P-value = 1.12×10−10). Notably, we also find that TTNLOF variants are more penetrant for AF than for heart failure in the UK Biobank; while the relative risk of heart failure conferred by TTNLOF variants is higher than the relative risk of AF, the absolute risk of AF is higher among carriers. In future years as exome sequencing data and MRI imaging data become available on more UK Biobank participants, further delineation of the long-term outcomes in TTNLOF mutation carriers will be possible.
TTN is the largest gene in the human genome. As such, more LOF variants are expected to exist in the population for TTN compared to most other genes. Indeed, TTNLOF variants are 5–10 times more common than mutations in other smaller, well-known cardiovascular disease susceptibility genes. For example, LOF mutations in LDLR underlying familial hyperlipidemia occur in only 0.028% individuals in the current dataset. Similarly, LOF mutations in MYBPC3 (hypertrophic cardiomyopathy), KCNQ1 (long-QT syndrome), and SCN5A (Brugada syndrome; conduction disorders) are observed in only 0.044%, 0.046%, and 0.065% of individuals, respectively. In contrast, TTNLOF mutations are relatively common, as we find that 0.44% of individuals harbor a TTNLOF variant.
Our findings also highlight the robust contribution of polygenic risk to AF susceptibility and the complimentary nature of polygenic risk and rare variation. AF polygenic risk explains a considerably larger proportion of AF susceptibility in the general population (4.7% of variance) compared to TTNLOF mutations. Though polygenic risk accounts for a greater proportion of AF risk, by nature it has a lower average degree of penetrance. In fact, only 0.10% of the population has a polygenic score conferring an equivalent AF prevalence to that observed among TTNLOF variant carriers. We also find that AF polygenic risk results in a striking difference in the prevalence of AF among TTNLOF variant carriers (OR per SD 1.79). For example, TTNLOF variant carriers in the lowest tertile of AF PRS have an AF prevalence of only 6.7% compared to 21.5% in the highest tertile. Thus, polygenic risk results in an additive risk that further increases the likelihood of AF in TTNLOF mutation carriers.
With the observation that TTN mutations are associated with AF in families, early-onset cases, and now in a population-based biobank, a number of potential further lines of investigation can be considered. In the future, it will be interesting to determine whether the identification of a TTNLOF mutation in an individual with AF could alter clinical management. For example, one could consider cardiac magnetic resonance imaging to identify subtle structural abnormalities or screening of at-risk relatives. It will also be interesting to determine if TTNLOF variant carriers with AF may benefit from treatment with neurohormonal therapy or may respond differently to standard AF treatments such as antiarrhythmic medications or catheter ablation.
To date, mutations in more than 35 genes including ion channels, gap junction proteins, and transcription factors have been identified in individuals and families with AF; however, we were not able to replicate an association between LOF variants in these genes and AF in our analyses. We also performed gene-based testing accounting for functional classes, but did not observe any significant associations between these genes and AF risk (Online Table XIV). However, since many of these genes had few LOF variants, the power to establish an association between these genes and AF was limited. Furthermore, our analyses did not consider other forms of genetic variation. It is possible that nonsynonymous variation in some of these previously reported genes may contribute to AF risk. For example, the gain-of-function nonsynonymous variants previously described in KCNQ136, 37 would not have been identified in our current approach. Similarly, there have been over 100 GWAS loci reported for AF, yet we did not identify an association between LOF variants in any gene at a GWAS locus other than TTN. Since GWAS loci are typically associated with non-coding variants with small effects, it is possible that LOF variants in nearby genes are only rarely associated with AF. Finally, despite inclusion of over 1,500 AF cases, our power remains modest and future studies with significantly larger sample sizes will be informative. For example, a recent analysis of exome sequencing data for diabetes with over 20,000 cases identified multiple genes implicated in the disease38.
Our study has several other potential limitations. First, we focused on a relatively homogeneous middle aged, white-European population. As such, our findings may not be applicable to other age strata, races or ethnicities. Second, disease status in the UK Biobank relies on self-reports, ICD codes, operation codes, and death registry codes. As a consequence, some misclassification is possible. However, we recently used the same phenotypic definitions in GWAS for AF and heart failure and replicated well-described genetic loci6, 10, 39. In addition, well-known associations between TTNLOF and nonischemic cardiomyopathy were replicated in a PheWAS in the present study. Misclassification of these diseases may therefore be limited. Third, there is the potential for ascertainment bias among participants in the UK Biobank, making it unlikely that the study perfectly reflects the overall UK population. The participants in the UK Biobank are known to be healthier than the overall British population, and individuals with an overt cardiomyopathy due to a TTNLOF mutation may be less likely to participate in this longitudinal study. It is reassuring, however, that we found a similar frequency of TTNLOF variants in the UK Biobank (0.44%) compared to prior reports of 0.5% among controls33 and the general population40.
In conclusion, both polygenic and monogenic factors contribute to AF risk in the general population. While monogenic TTNLOF variants confer a substantial AF penetrance, polygenic risk explains a larger proportion of genetic susceptibility to AF.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Over 100 distinct genetic loci have been identified for atrial fibrillation (AF).
Rare loss-of-function mutations in TTN have been associated with early-onset AF.
The contribution of rare and common genetic variation to AF risk in the general population is not clear.
What New Information Does this Article Contribute?
Loss-of-function (LOF) mutations in TTN are significantly associated with AF in a large population-based study.
TTN mutations associated with AF are rare but have a high penetrance of approximately 14%.
A much larger proportion AF risk in the population is explained by the additive effect of many common variants than by loss-of-function mutations in TTN.
SOURCES OF FUNDING
This work was supported by the Fondation Leducq (14CVD01), and by grants from the National Institutes of Health to Dr. Ellinor (1RO1HL092577, R01HL128914, K24HL105780) and Dr. Lubitz (1R01HL139731). This work was also supported by a grant from the American Heart Association to Dr. Ellinor (18SFRN34110082) and to Dr. Lubitz (18SFRN34250007). Dr. Weng is supported by an American Heart Association Postdoctoral Fellowship Award (17POST33660226). This work was also supported by an American Heart Association Strategically Focused Research Networks (SFRN) postdoctoral fellowship to Drs. Weng and Hall (18SFRN34110082).
DISCLOSURES
Dr. Ellinor is supported by a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases. Dr. Ellinor has also served on advisory boards or consulted for Bayer AG, Quest Diagnostics, and Novartis. Dr. Lubitz receives sponsored research support from Bristol Myers Squibb / Pfizer, Bayer AG, and Boehringer Ingelheim, and has consulted for Bristol Myers Squibb / Pfizer and Bayer AG.
Nonstandard Abbreviations and Acronyms:
- AF
Atrial fibrillation
- GWAS
Genome-wide association studies
- PRS
Polygenic risk scores
- WES
Whole-exome sequencing
- MAF
Minor allele frequency
- AUC
Area under the receiver-operating-characteristics curve
- LOF
High-confidence loss-of-function
- TTNLOF
Loss-of-function variants located in TTN exons highly expressed in cardiac tissue
- OR
Odds ratio
- SD
Standard deviation
REFERENCES
- 1.Zoni-Berisso M, Lercari F, Carazza T, Domenicucci S. Epidemiology of atrial fibrillation: European perspective. Clinical Epidemiology. 2014;6:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilke T, Groth A, Mueller S, Pfannkuche M, Verheyen F, Linder R, Maywald U, Bauersachs R, Breithardt G. Incidence and prevalence of atrial fibrillation: An analysis based on 8.3 million patients. Europace. 2013;15:486–493 [DOI] [PubMed] [Google Scholar]
- 3.Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial fibrillation: A global burden of disease 2010 study. Circulation. 2014;129:837–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, Singer DE. Prevalence of diagnosed atrial fibrillation in adults. JAMA : the journal of the American Medical Association. 2001;285:2370. [DOI] [PubMed] [Google Scholar]
- 5.Kumar P, Gehi AK. Atrial fibrillation and metabolic syndrome: Understanding the connection. J Atr Fibrillation. 2012;5:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weng LC, Choi SH, Klarin D, et al. Heritability of atrial fibrillation. Circ Cardiovasc Genet. 2017;10:e001838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christophersen IE, Ellinor PT. Genetics of atrial fibrillation: From families to genomes. Journal of Human Genetics. 2015;61:1–10 [DOI] [PubMed] [Google Scholar]
- 8.Lubitz SA, Yin X, Fontes JD, Magnani JW, Rienstra M, Pai M, Villalon ML, Vasan RS, Pencina MJ, Levy D, Larson MG, Ellinor PT, Benjamin EJ. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA : the journal of the American Medical Association. 2010;304:2263–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen JB, Thorolfsdottir RB, Fritsche LG, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roselli C, Chaffin MD, Weng LC, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christophersen IE, Rienstra M, Roselli C, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JY, Kim TH, Yang PS, Lim HE, Choi EK, Shim J, Shin E, Uhm JS, Kim JS, Joung B, Oh S, Lee MH, Kim YH, Pak HN. Korean atrial fibrillation network genome-wide association study for early-onset atrial fibrillation identifies novel susceptibility loci. Eur Heart J. 2017;38:2586–2594 [DOI] [PubMed] [Google Scholar]
- 13.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weng LC, Preis SR, Hulme OL, Larson MG, Choi SH, Wang B, Trinquart L, McManus DD, Staerk L, Lin H, Lunetta KL, Ellinor PT, Benjamin EJ, Lubitz SA. Genetic predisposition, clinical risk factor burden, and lifetime risk of atrial fibrillation. Circulation. 2018;137:1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khera AV, Chaffin M, Wade KH, et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell. 2019;177:587–596 e589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahida S, Lubitz SA, Rienstra M, Milan DJ, Ellinor PT. Monogenic atrial fibrillation as pathophysiological paradigms. Cardiovasc Res. 2011;89:692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lubitz SA, Brody JA, Bihlmeyer NA, et al. Whole exome sequencing in atrial fibrillation. PLoS Genet. 2016;12:e1006284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi SH, Weng LC, Roselli C, et al. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA : the journal of the American Medical Association. 2018;320:2354–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M. Uk biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hout CVV, Tachmazidou I, Backman JD, Hoffman JX, Ye B, al. e. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the uk biobank. BioRxiv. 2019 [Google Scholar]
- 21.Regier AA, Farjoun Y, Larson DE, et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat Commun. 2018;9:4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bycroft C, Freeman C, Petkova D, et al. The uk biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou W, Nielsen JB, Fritsche LG, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50:1335–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vilhjalmsson BJ, Yang J, Finucane HK, et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet. 2015;97:576–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. Proc: An open-source package for r and s+ to analyze and compare roc curves. BMC Bioinformatics. 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X. Firth logistic regression for rare variant association tests. Front Genet. 2014;5:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Statistics in Medicine. 2002;21:2409–2419 [DOI] [PubMed] [Google Scholar]
- 29.Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lubitz SA, Benjamin EJ, Ellinor PT. Atrial fibrillation in congestive heart failure. Heart Fail Clin. 2010;6:187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng J, Raddatz K, Molkentin JD, Wu Y, Labeit S, Granzier H, Gotthardt M. Cardiac hypertrophy and reduced contractility in hearts deficient in the titin kinase region. Circulation. 2007;115:743–751 [DOI] [PubMed] [Google Scholar]
- 33.Ahlberg G, Refsgaard L, Lundegaard PR, et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun. 2018;9:4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, D’Agostino RB, Murabito JM, Kannel WB, Benjamin EJ. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: The framingham heart study. Circulation. 2003;107:2920–2925 [DOI] [PubMed] [Google Scholar]
- 35.Alonso A, Krijthe BP, Aspelund T, et al. Simple risk model predicts incidence of atrial fibrillation in a racially and geographically diverse population: The charge-af consortium. Journal of the American Heart Association. 2013;2:e000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das S, Makino S, Melman YF, Shea MA, Goyal SB, Rosenzweig A, Macrae CA, Ellinor PT. Mutation in the s3 segment of kcnq1 results in familial lone atrial fibrillation. Heart Rhythm. 2009;6:1146–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y-H, Xu S-J, Bendahhou S, et al. Kcnq1 gain-of-function mutation in familial atrial fibrillation. Science (New York, N.Y.). 2003;299:251–254 [DOI] [PubMed] [Google Scholar]
- 38.Flannick J, Mercader JM, Fuchsberger C, et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. 2019;570:71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aragam KG, Chaffin M, Levinson RT, et al. Phenotypic refinement of heart failure in a national biobank facilitates genetic discovery. Circulation. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole exome-sequencing and phenotype data used in this study are available through UK Biobank (www.ukbiobank.ac.uk). Summary level results have been made publicly available at the Cardiovascular Disease Initiative Knowledge Portal and can be accessed at www.broadcvdi.org upon publication.