

Summary
R loops arising during transcription induce genomic instability, but how cells respond to the R loop-associated genomic stress is still poorly understood. Here, we show that cells harboring high levels of R loops rely on the ATR kinase for survival. In response to aberrant R loop accumulation, the ATR-Chk1 pathway is activated by R loop-induced reversed replication forks. In contrast to the activation of ATR by replication inhibitors, R loop-induced ATR activation requires the MUS81 endonuclease. ATR protects the genome from R loops by suppressing transcription-replication collisions, promoting replication fork recovery, and enforcing a G2/M cell-cycle arrest. Furthermore, ATR prevents excessive cleavage of reversed forks by MUS81, revealing a MUS81-triggered and ATR-mediated feedback loop that fine tunes MUS81 activity at replication forks. These results suggest that ATR is a key sensor and suppressor of R loop-induced genomic instability, uncovering a signaling circuitry that safeguards the genome against R loops.
Graphical Abstract
eTOC Blurb
R loops cause genomic instability, but how cells respond to R loop-associated genomic stress is poorly understood. Matos et al. show that the ATR-Chk1 pathway senses R loop-impeded replication forks through a MUS81-mediated mechanism and protects replication forks from excessive MUS81 cleavage, revealing a MUS81-triggered and ATR-mediated feedback loop safeguarding the genome against R loops.
Introduction
The integrity of the genome is constantly challenged by various intrinsic cellular processes, one of which is transcription (Aguilera and Garcia-Muse, 2012; Hamperl and Cimprich, 2016; Santos-Pereira and Aguilera, 2015). During transcription, the nascent RNA generated by RNA polymerases could hybridize with the DNA template, giving rise to a three-stranded structure called an R loop. R loops, which contain DNA:RNA hybrids and displaced single-stranded DNA (ssDNA), arise from many different causes, including high GC skew in DNA sequences, topoisomerase defects, compromised RNA processing and others. R loops are important for a number of cellular processes, such as mitochondrial replication, class-switch recombination, transcription initiation and termination, and DNA repair. However, when R loops accumulate at excessive levels or wrong places in the genome, they become a threat to genomic stability. Therefore, cells have to tightly control the level and distribution of R loops in the genome to maintain genomic integrity. When R loops are dysregulated, cells need to sense and resolve aberrant R loops to survive. Elucidating the mechanisms by which cells detect and respond to aberrant R loops will help explain how the genome is protected from one of its main functions - transcription.
The levels of R loops are regulated by multiple mechanisms in cells. During transcription, a number of factors that function in a transcription-coupled manner are important for preventing R loop formation. These factors include topoisomerases, RNA processing factors, chromatin modulators, and others (Santos-Pereira and Aguilera, 2015). For example, loss of the RNA splicing factor SRSF1 (also known as ASF/SF2) increases R loop formation and genomic instability (Gan et al., 2011; Li et al., 2007). R loops are also suppressed by a second group of proteins that remove DNA:RNA hybrids enzymatically. Proteins such as SETX and AQR may act as helicases to unwind DNA:RNA hybrids (Skourti-Stathaki et al., 2011; Sollier et al., 2014), whereas RNaseH1 and RNaseH2 degrade the RNA hybridized with DNA (Chon et al., 2013; Cornelio et al., 2017; Lima et al., 2016; Nguyen et al., 2017; Parajuli et al., 2017). In addition, several DNA repair factors, including BRCA1, BRCA2, and Fanconi Anemia proteins, are also implicated in R loop suppression (Bhatia et al., 2014; Hatchi et al., 2015; Schwab et al., 2015; Shivji et al., 2018; Zhang et al., 2017). Upon the loss of an R loop suppressor, aberrant R loops may accumulate in the genomic regions where the suppressor functions. For example, loss of SETX leads to R loop accumulation at transcription termination sites (Hatchi et al., 2015; Skourti-Stathaki et al., 2011). Although aberrant R loops arise from a variety of sources, they are commonly associated with genomic instability.
The effects of R loops on genomic stability are complex (Sollier and Cimprich, 2015). The ssDNA exposed at R loops can be targeted by cytosine deaminases, leading to DNA nicks and breaks (Zarrin et al., 2004). Perhaps due to their structural resemblance with DNA repair intermediates, R loops can be cleaved by XPG and XPF, two DNA structure-specific endonucleases involved in the transcription-coupled nucleotide excision repair (Sollier et al., 2014). XPG depletion increases R loops in cells, suggesting that XPG-mediated R loop cleavage is important for suppressing R loop levels (Sollier et al., 2014). The most detrimental effect of R loops on the genome is probably caused by collisions of R loops with DNA replication forks. In replicating cells, R loops impede the progression of replication forks and give rise to DNA double-stranded breaks (DSBs) (Gan et al., 2011; Gomez-Gonzalez et al., 2011). Although the effects of R loop-replication collisions are evident, how DSBs are generated during this process remains unclear.
The ATR kinase is a master regulator of DNA damage and replication stress responses in human cells (Saldivar et al., 2017; Yazinski and Zou, 2016). In response to a broad spectrum of DNA replication problems, ATR is recruited to and activated on RPA-coated ssDNA by the concerted actions of a number of regulators (Marechal and Zou, 2013). When ATR is activated, it functions with the effector kinase Chk1 to protect the genome. Induction of head-on collisions of R loops with replication forks on episomes leads to phosphorylation of several ATR substrates (Hamperl et al., 2017). Nonetheless, how ATR is activated by R loops, and how it functions in the R loop response is still unknown.
In this study, we investigated how ATR is activated by aberrant R loops using cells lacking SRSF1 or treated with an RNA splicing inhibitor. We find that aberrant R loop accumulation activates the ATR-Chk1 pathway in a replication-dependent manner. Surprisingly, in contrast to ATR activation by hydroxyurea (HU), a replication inhibitor, R loop-induced ATR activation requires the MUS81 endonuclease. Furthermore, the proteins promoting replication fork reversal are needed for efficient ATR activation at R loops, suggesting that ATR is activated by R loops through the action of MUS81 on reversed forks. Once activated, ATR protects the genome against R loop-associated DNA damage through several mechanisms: ATR suppresses transcription-replication collisions, promotes replication fork recovery, and enforces a G2/M checkpoint arrest. Importantly, ATR prevents excessive cleavage of reversed forks by MUS81, revealing a MUS81-triggered and ATR-mediated feedback loop that fine tunes MUS81 activity at R loop-impeded replication forks. Together, these results reveal how ATR is activated by aberrant R loops and how it functions in the R loop response, establishing ATR as a key sensor and suppressor of transcription-derived genomic instability.
Results
ATR is activated by aberrant R loop accumulation
To investigate the role of the ATR-Chk1 pathway in the R loop response, we used two independent siRNAs to knock down SRSF1, a suppressor of R loops, in HeLa cells (Gan et al., 2011; Li et al., 2007). Consistent with the effects of AQR depletion (Sollier et al., 2014), knockdown of SRSF1 increased Chk1 phosphorylation (p-Chk1 S317 and S345) (Fig. 1A, S1A). Similarly, knockdown of SRSF1 in HCT116 cells also increased p-Chk1 (Fig. S1B), showing that the induction of p-Chk1 by SRSF1 loss is not cell line-specific. The p-Chk1 in SRSF1 knockdown cells was rapidly abolished by the ATR inhibitor (ATRi) VE-821 but not the ATM inhibitor (ATMi) KU55933 (Figs. 1B, S1C, S1D), suggesting that Chk1 is phosphorylated by ATR. The p-Chk1 levels in SRSF1 knockdown cells were similar to those induced by 32-64 μM HU (Fig. 1C), suggesting that the ATR-Chk1 pathway is modestly activated. Consistent with the interference of DNA replication by R loops, DNA synthesis was reduced in SRSF1 knockdown cells compared with control cells (Fig. S1E). Depletion of SETX, another R loop suppressor, also induced p-Chk1 in an ATR-dependent manner (Fig. S1F), suggesting that loss of R loop suppressors is commonly associated with ATR activation. We recently showed that inhibition of RNA splicing with pladienolide B (Plad-B) increases R loop levels (Nguyen et al., 2017). As detected by immunostaining, Plad-B also induced p-Chk1 (Fig. S1G). Together, these results show that ATR is activated when R loops arise through different mechanisms.
Figure 1. Activation of the ATR-Chk1 pathway by aberrant R loop accumulation.

(A) HeLa cells were transfected with control and two independent SRSF1 siRNAs. Three days after transfection, levels of p-Chk1 were analyzed with phospho-specific antibodies to S317 and S345. (B) HeLa cells were mock transfected or transfected with SRSF1-1 siRNA, and treated with ATRi (VE-821, 10 μM) for the indicated lengths of time. Levels of p-Chk1 and other proteins were analyzed by western blot. (C) HeLa cells were transfected with control or SRSF1-1 siRNA. The cells transfected with control siRNA were treated with increasing concentrations of HU for 1 hr. Levels of HU-induced p-Chk1 S345 were compared to the p-Chk1 detected in SRSF1 knockdown cells. (D) HeLa cells were transfected with control or SRSF1-1 siRNA. Levels of DNA:RNA hybrids in total DNA were analyzed by S9.6 dot blot and quantified. The specificity of S9.6 antibody for DNA:RNA hybrids was confirmed by RNaseH treatment. Levels of input DNA were analyzed with anti-dsDNA antibody. The level of DNA:RNA hybrids in each sample was normalized to the level of input DNA, and relative levels of DNA:RNA hybrids were compared among samples. Error bars, SD (n=4, four technical replicates). (E-F) HeLa cells were transfected with control or SRSF1-1 siRNA and a plasmid expressing RNaseH1-GFP. Cells expressing RNaseH1-GFP were identified by GFP immunofluorescence. Levels of p-Chk1 in individual cells were analyzed by immunostaining and quantified. Representative images are shown in (E). Quantified p-Chk1 S317 signals in the GFP-positive and GFP-negative cell populations are shown in (F). The mean p-Chk1 intensity in each population was determined from > 65 cells (n>65) and shown as a red line. Error bars, SD. ***p < 0.0001, Unpaired Student’s t test.
We next sought to verify whether ATR is indeed activated by R loops in cells lacking R loop suppressors or compromised for RNA splicing. Dot blot analysis of total DNA with the S9.6 monoclonal antibody, which recognizes DNA:RNA hybrids, confirmed that R loop levels were elevated by SRSF1 knockdown (Fig. 1D). The S9.6 signals were abolished by RNase H (Fig. 1D), which cleaves the RNA in DNA:RNA hybrids. To determine whether R loops are required for ATR activation in SRSF1 knockdown cells, we transiently expressed GFP-tagged RNaseH1 and analyzed its effects (Fig. 1E-F). Both SRSF1 knockdown and control cells were divided into GFP− and GFP+ populations. When the GFP− SRSF1 knockdown and control cells were compared (Fig. 1F lanes 1 and 3), knockdown of SRSF1 increased p-Chk1. When the GFP− and GFP+ SRSF1 knockdown cells were compared (Fig. 1E lanes 3 and 4), expression of GFP-RNaseH1 significantly reduced p-Chk1. Similarly, expression of GFP-RNaseH1 also reduced p-Chk1 in Plad-B-treated cells (Fig. S1G). These results demonstrate that ATR is activated in an R loop-dependent manner in cells harboring high levels of R loops.
ATR protects the genome against aberrant R loops
The activation of ATR by aberrant R loops raises the question as to whether ATR is functionally important for the R loop response. To address this, we treated cells with SRSF1 siRNA for two days to induce R loops and then inhibited ATR with ATRi for 24 hours (Fig. 2A). ATRi reduced p-Chk1 in SRSF1 knockdown cells as expected, but increased the levels of γH2AX and phosphorylated KAP1 (p-KAP1 S824), indicating a surge of DSBs. Indeed, neutral comet assays detected an increase of DSBs in SRSF1 knockdown cells after ATRi treatment (Fig. S2). Consistent with the induction of DSBs, SRSF1 knockdown cells were more sensitive to ATRi than control cells (Fig. 2B). In SETX knockdown cells, ATRi rapidly abolished p-Chk1 and reduced γH2AX (Fig. 2C), showing that ATR contributes to these phosphorylation events. Notably, γH2AX reaccumulated in SETX knockdown cells after 4 hours of ATRi treatment and soon exceeded the level prior to ATR inhibition (Fig. 2C), indicating that ATRi induced additional DSBs. Similar to SRSF1 depletion, SETX knockdown also increased cellular sensitivity to ATRi (Fig. 2D). Together, these results suggest that ATR plays an important role in protecting the genome against aberrant R loops.
Figure 2. ATR protects the genome against aberrant R loops.

(A) HeLa cells were transfected with control or SRSF1-1 siRNA and treated with or without ATRi (VE-821, 10 μM) for 24 hr. Levels of p-Chk1, γH2AX, and p-KAP1 were analyzed with phospho-specific antibodies. (B) HeLa cells were transfected with control or SRSF1-1 siRNA. Two days after transfection, cells were treated with increasing concentrations of ATRi (0, 1.25, 2.5, 5, and 10 μM of VE-821) and kept in ATRi for 3 days. Cell viability was determined using Alamar Blue. The mean cell viability at each condition was determined from 3-6 technical replicates (n≥3). Error bars, SD. The relative viability of ATRi-treated cells was normalized to the viability of untreated cells. (C) HeLa cells were mock transfected or transfected with SETX siRNA, and treated with ATRi (VE-821, 10 μM) for the indicated lengths of time. Levels of p-Chk1 and γH2AX were analyzed with phospho-specific antibodies. (D) HeLa cells were transfected twice with control or SETX siRNA on day 0 and day 4. One day after the first transfection, cells were treated with increasing concentrations of ATRi (0, 1.25, 2.5, 5, and 10 μM of VE-821) and kept in ATRi for 8 days. Cell viability was analyzed as in (B) and normalized to untreated control cells. Error bars, SD, (n≥3).
R loops activate the ATR-Chk1 pathway during DNA replication
The importance of ATR in protecting the genome against R loops led us to further investigate how ATR is activated by R loops. To test whether DNA replication contributes to R loop-induced ATR activation, we first sought to reduce replicating cells by inhibiting CDKs. We treated cells with SRSF1 siRNA for two days to induce R loops and then exposed them to roscovitine, a pan CDK inhibitor (CDKi), for 10 hours (Fig. 3A). The roscovitine treatment led to a drastic reduction in DNA synthesis and accumulation of cells in G1 (Fig. S3A-B). The p-Chk1 in SRSF1 knockdown cells was reduced by roscovitine (Fig. 3A), suggesting that the ATR-Chk1 pathway is not efficiently activated by R loops when cells are unable to enter and go through S phase.
Figure 3. R loops activate the ATR-Chk1 pathway in replicating cells.

(A) HeLa cells were transfected with control or SRSF1-1 siRNA, and treated with or without CDKi (roscovitine, 25 μM) for 10 hr. Levels of p-Chk1 and other proteins were analyzed by western blot. (B-C) HeLa cells transfected with control or SRSF1-1 siRNA were analyzed by immunostaining with Cyclin A and p-Chk1 S317 antibodies. Representative images are shown in (B). Levels of p-Chk1 in individual cells were quantified in the Cyclin A-positive and Cyclin A-negative cell populations (C). The mean p-Chk1 intensity in each population was determined from 130 cells (n=130) and shown as a red line. Error bars, SD. ***p < 0.0001, Unpaired Student’s t test. (D-E) HeLa cells were transfected with control or SRSF1-1 siRNA. Newly synthesized DNA was pulse labeled with EdU in control (15 min) and SRSF1 knockdown (30 min) cells. Representative images of p-Chk1 and EdU staining are shown in (D). The intensities of EdU and p-Chk1 staining in >800 individual cells in each cell population were quantified and plotted in 2D (E). Linear regression lines of SRSF1 knockdown and control cell populations are shown. (F) HeLa cells were transfected with control or SRSF1-1 siRNA, and treated with or without CDC7i (XL413, 5 μM) for 4 hr. Levels of p-Chk1 and other proteins was analyzed by western blot.
To further assess the role for DNA replication in R loop-induced ATR activation, we analyzed SRSF1 knockdown and control cells by immunofluorescence of Cyclin A and p-Chk1 (Fig. 3B-C). Cyclin A is primarily expressed in S and G2 cells. In Cyclin A+ cells, knockdown of SRSF1 increased p-Chk1 (Fig. 3C lanes 2 and 4). When Cyclin A+ and Cyclin A− SRSF1 knockdown cells were compared, p-Chk1 levels were significantly higher in Cyclin A+ cells (Fig. 3C lanes 3 and 4). These results suggest that ATR is primarily activated by R loops in S and/or G2 cells. We also analyzed SRSF1 knockdown and control cells by EdU pulse labeling and p-Chk1 immunofluorescence (Fig. 3D-E). The control cells in S phase displayed a range of EdU incorporation but very little p-Chk1. In contrast, although the SRSF1 knockdown cells in S phase incorporated EdU less efficiently than control cells, they displayed significant p-Chk1 signals. Importantly, in S-phase SRSF1 knockdown cells, the levels of EdU incorporation positively correlated with p-Chk1 levels (Fig. 3E), strongly suggesting that the activation of ATR by R loops is driven by DNA replication.
To test whether ongoing DNA synthesis is needed for maintaining ATR activation, we sought to suppress replication origin firing by inhibiting CDC7, a kinase required for replication initiation. This strategy is expected to gradually reduce the number of replication forks without interfering with fork progression. We treated SRSF1 knockdown cells with the CDC7 inhibitor (CDC7i) XL413 for 4 hours. This treatment reduced DNA synthesis but did not cause a significant cell cycle arrest (Fig. S3A-B). In this context, the p-Chk1 in SRSF1 knockdown cells was only slightly reduced (Fig. 3F). Thus, although the activation of ATR by R loops is driven by DNA replication, the ATR-activating DNA structures induced by R loops during replication may persist for some time after ongoing DNA synthesis is diminished.
R loops activate the ATR-Chk1 pathway through a MUS81-mediated mechanism
The activation of the ATR-Chk1 pathway by R loops in replicating cells suggests that both R loops and replication contribute to ATR activation. Collisions between R loops and replication forks could potentially activate ATR through different mechanisms. For example, collisions may generate stalled replication forks to activate ATR. Alternatively, collisions may activate ATR by stimulating the nucleolytic cleavage of R loops. To test these possibilities, we first knocked down XPG, an endonuclease involved in R loop cleavage (Sollier et al., 2014). Loss of XPG in SRSF1 knockdown cells did not reduce p-Chk1 (Fig. 4A), suggesting that XPG-mediated R loop cleavage is not responsible for ATR activation. Notably, p-Chk1 levels were higher in XPG, SRSF1 double-depleted cells than in cells lacking SRSF1 alone (Fig. 4A lanes 2 and 4). This result is consistent with the role for XPG in suppressing R loops (Sollier et al., 2014), indicating that loss of XPG leads to more R loops, which consequently increase collisions between R loops and replication forks.
Figure 4. R loops induce ATR activation through a MUS81-mediated pathway.

(A) HeLa cells were transfected with control, SRSF1-1, XPG, and MUS81-1 siRNA as indicated. Three days after transfection, levels of p-Chk1 and other proteins were analyzed by western blot. (B) HeLa cells were transfected with control, SRSF1-1, MUS81-1 and MUS81-2 siRNA as indicated. Levels of p-Chk1 and other proteins were analyzed by western blot. (C) HeLa cells were transfected with control, SRSF1-1, and MUS81-1 siRNA as shown. Where indicated, cells were treated with 32 μM of HU for 1 hr. Levels of p-Chk1 and other proteins were analyzed by western blot. (D) HeLa cells were transfected with control, SRSF1-1, EME1, and EME2 siRNA as indicated. The effectiveness of EME2 siRNA was confirmed in Fig. S4A. Levels of p-Chk1 and other proteins were analyzed by western blot. (E) HeLa cells were transfected with control, SRSF-1, and SLX4-1 siRNA as indicated (lower panel). Knockdown of SLX4 was confirmed by SLX4 immunoprecipitation and western blot (upper panel). Levels of p-Chk1 and other proteins were analyzed by western blot. (F) HeLa cells were transfected with control, SRSF1-1, and MUS81 siRNA as shown. Where indicated, cells were infected with lentiviruses expressing siRNA-resistant MUS81WT or MUS81CD. Levels of p-Chk1 S317 were analyzed by immunostaining and quantified. The mean p-Chk1 intensity in each cell population was determined from >1,000 cells and shown as a red line (n>1,000). Error bars, SD. ***p < 0.0005.
While analyzing the effects of XPG on ATR activation, we depleted several other DNA structure-specific endonucleases as controls. Unexpectedly, knockdown of MUS81 by two independent siRNAs reduced p-Chk1 in SRSF1-depleted cells (Fig. 4A-B). Even in XPG, SRSF1 double-depleted cells, MUS81 knockdown diminished p-Chk1 (Fig. 4A). Because MUS81 has not been implicated in ATR activation before, we asked whether MUS81 is also required for HU-induced ATR activation. We treated cells with 32 μM HU, which induces similar levels of p-Chk1 as in SRSF1-depleted cells (Fig. 1C). Strikingly, MUS81 knockdown did not reduce p-Chk1 in HU-treated cells as in SRSF1-depleted cells (Fig. 4C). These results suggest that ATR is activated by R loops and HU through distinct mechanisms.
MUS81 is present in several complexes during the cell cycle. In S phase, MUS81 forms complexes with EME1 or EME2 (Pepe and West, 2014a; Wyatt et al., 2017). When cells approach G2/M, MUS81-EME1 is incorporated into the large SLX4-MUS81-XPF (SMX) complex (Wyatt et al., 2017). To determine which MUS81 complex is involved in R loop-induced ATR activation, we knocked down EME1, EME2, or SLX4 in SRSF1-depleted cells (Fig. 4D-E). Loss of EME1 or EME2 decreased p-Chk1 without reducing the S-phase cell populations (Fig. 4D, S4A-B). Notably, depletion of EME1 but not EME2 significantly reduced MUS81 levels (Fig. 4D) (Forment et al., 2011; Pepe and West, 2014a), leaving the possibility that the effects of EME1 loss are indirect. Depletion of SLX4 by two distinct siRNAs did not reduce p-Chk1 in SRSF1 knockdown cells (Fig. 4E, S4C). Thus, MUS81-EME2 but not SMX is implicated in the R loop-induced ATR activation during S phase.
To test whether MUS81 functions at replication forks to activate ATR, we analyzed the recruitment of MUS81 to replication forks using proximity ligase assay (PLA) with antibodies recognizing MUS81 and PCNA, a core component of the replisome. Knockdown of SRSF1 increased PLA foci (Fig. S4D), suggesting that MUS81 is recruited to replication forks upon collision with R loops. To address whether the nuclease activity of MUS81 is required for R loop-induced ATR activation, we generated cell lines that inducibly express HA-tagged, MUS81 siRNA-resistant wild-type MUS81 (MUS81WT) or the catalytically inactive MUS81 D338A/D339A mutant (MUS81CD) (Fig. S4E) (Fu et al., 2015). While the level of MUS81WT was ~3 folds higher than endogenous MUS81, MUS81CD was expressed at the endogenous MUS81 level (Fig. S4F). After depletion of endogenous MUS81, only MUS81WT but not MUS81CD rescued p-Chk1 levels in SRSF1 knockdown cells (Fig. 4F). Given the caveat that MUS81WT was slightly overexpressed, these results suggest that MUS81 promotes R loop-induced ATR activation by acting as a nuclease at replication forks.
Replication fork reversal is required for R loop-induced ATR activation
In vitro, MUS81-containing complexes cleave a number of fork-like DNA structures, including Holliday junctions (HJs) or reversed forks (Amangyeld et al., 2014; Pepe and West, 2014b), raising the possibility that MUS81 may act on reversed forks to activate ATR at R loops. To test this idea, we knocked down SMARCAL1 or ZRANB3, two factors required for fork reversal, in SRSF1-depleted cells (Fig. 5A) (Betous et al., 2012; Ciccia et al., 2012; Kolinjivadi et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017). Loss of SMARCAL1 or ZRANB3 did not reduce S-phase cells, but decreased p-Chk1 (Fig. 5A, S5A). Depletion of HLTF, another fork reversal factor (Achar et al., 2015), also reduced p-Chk1 in SRSF1 knockdown cells (Fig. S5B). In contrast, loss of RECQ1, which antagonizes fork reversal (Berti et al., 2013), increased p-Chk1 (Fig. S5C). Thus, fork reversal, like MUS81 activity, is an important determinant for R loop-induced ATR activation.
Figure 5. R loop-induced ATR activation involves replication fork reversal and MUS81-mediated ssDNA formation.

(A) HeLa cells were transfected with control, SRSF1-1, SMARCAL1, and ZRANB3 siRNA as shown. Levels of p-Chk1 and other proteins were analyzed by western blot (left panel). The levels of p-Chk1 from three independent experiments were quantified (n=3) (right panel). Error bars, SD. *p < 0.05, Unpaired Student’s t test. (B) Hela cells were transfected with control or MUS81 siRNA for 48 hr. Cells were then treated with Plad-B (1 μM) for 4 hr. Individual cells were analyzed by immunostaining using RPA32 and S9.6 antibodies. Intensities of S9.6 and RPA staining of individual cells were analyzed and plotted in 2D. (C-D) HeLa cells were transfected with control, SRSF1-1, TopBP1 (C), and RAD17 (D) siRNA as shown. Where indicated, cells were treated with 32 or 64 μM of HU for 1 hr. Levels of p-Chk1 and other proteins were analyzed by western blot.
To understand how MUS81 regulates ATR activation at reversed forks, we tested whether MUS81 regulates the formation of single-stranded DNA (ssDNA), the key DNA structure triggering ATR activation (Zou and Elledge, 2003). We performed immunostaining with S9.6 and RPA32 antibodies to monitor the levels of R loops and chromatin-bound RPA, a marker of ssDNA. Treatment of cells with Plad-B increased both R loops and ssDNA (Fig. 5B). Knockdown of MUS81 in Plad-B-treated cells reduced ssDNA without altering R loops substantially, suggesting that MUS81 promotes R loop-induced ssDNA formation. Depletion of MUS81 in SRSF1 knockdown cells also decreased ssDNA levels (data not shown). RPA-coated ssDNA is important for the functions of several key ATR regulators, including RAD17 and TopBP1 (Ellison and Stillman, 2003; Lee and Dunphy, 2010; Lee et al., 2007; Zou et al., 2003). Knockdown of TopBP1, an activator of ATR (Kumagai et al., 2006), reduced p-Chk1 in SRSF-depleted cells and HU-treated cells (Fig. 5C). Knockdown of RAD17, a regulator of TopBP1 (Delacroix et al., 2007; Lee and Dunphy, 2010; Lee et al., 2007), also reduced p-Chk1 in SRSF1-depleted cells and HU-treated cells (Fig. 5D). These results suggest that both HU and R loops activate the ATR-Chk1 pathway through the RAD17-TopBP1 circuitry, but the ATR activation by R loops uniquely requires the action of MUS81 at reversed forks.
ATR suppresses transcription-replication collisions
Along with investigating how the ATR-Chk1 pathway is activated by R loops, we sought to determine how ATR protects the genome against R loops. Since collisions between R loops and replication forks may give rise to DNA damage, we asked whether ATR affects transcription-replication collisions. To measure the collision between transcription and replication machineries in SRSF1 knockdown cells, we performed PLA using antibodies to the elongating form of RNA polymerase II (p-RNAPII S2) and PCNA (Fig. 6A) (Hamperl et al., 2017). PLA foci were readily detected in SRSF1 knockdown cells when both p-RNAPII and PCNA antibodies were applied, but not when either antibody was used individually (Fig. 6B, S6A). Furthermore, the PLA foci were reduced by the CDC7i, which suppresses replication origin firing, and by DRB, which inhibits RNAPII (Fig. 6B-C). These results validate the use of PLA to monitor the collision of transcription and replication machineries in SRSF1 knockdown cells.
Figure 6. ATR suppresses transcription-replication collisions and promotes a G2/M arrest.

(A) A schema of the PLA analysis to monitor transcription-replication collisions. (B-C) HeLa cells transfected with SRSF1-1 siRNA were analyzed by PLA using antibodies to PCNA and p-RNAPII S2. PCNA and p-RNAPII S2 antibodies were used individually as negative controls for PLA. Where indicated, cells were treated with CDC7i (XL413, 5 μM) for 4 hr or RNAPIIi (DRB, 80 μM) for 2 hr. Representative images of PCNA and PLA staining are shown in (B). The mean number of PLA foci in each cell population was determined from 900 cells (n=900) and shown as a read line (C). Error bars: SD. (D-E) HeLa cells transfected with SRSF1-1 siRNA were treated with or without ATRi (VE-821, 10 μM) for 24 hr, and analyzed with PLA as in (B). Representative images of PCNA and PLA staining are shown in (D). The mean number of PLA foci in each cell population was determined from >1,200 cells (n > 1,200) and shown as a red line (C). Error bars: SD. (F) HeLa cells were treated with Plad-B (1 μM) for 8 hr, and with CDC7i (XL413, 5 μM) during the last 45 minutes of this treatment. Cells were then washed and released into media containing CDC7i (XL413, 5 μM) and ATRi (VE-821, 10 μM) as indicated. Cells were labeled with EdU for 30 min before and after Plad-B treatment, or for the indicated lengths of time after Plad-B release.
To test whether R loops are involved in transcription-replication collisions, we transfected SRSF1 knockdown cells with an RNaseH1-expressing plasmid. RNaseH1 reduced PLA foci in SRSF1 knockdown cells (Fig. S6B). Similarly, Plad-B also induced the PLA foci of p-RNAPII and PCNA, which were suppressed by RNaseH1 expression (Fig. S6C). These results confirm that R loops indeed contribute to transcription-replication collisions.
Using the PLA of p-RNAPII and PCNA, we tested whether ATR inhibition affects transcription-replication collisions. In the presence of ATRi, the levels of PLA foci were elevated in SRSF1-depleted cells (Fig. 6D-E). ATRi is known to increase replication origin firing (Buisson et al., 2015; Shechter et al., 2004), which drives transcription-replication collisions (Fig. 6B-C). As quantified by S9.6 dot blots, the overall R loop levels in SRSF1 knockdown cells were not significantly altered by ATRi (Fig. S6D). Thus, in SRSF1-depleted cells, ATR likely reduces transcription-replication collisions by suppressing origin firing. The increase of PLA foci may also reflect a defect in resolving collided transcription-replication complexes, which may generate PLA signals persistently at sites of collisions.
ATR promotes DNA synthesis and a G2/M arrest in response to R loops
While analyzing the effects of SRSF1 knockdown on the cell cycle, we noted that SRSF1 loss not only reduced DNA synthesis in S phase but also increased the cells at G2/M (Fig. S6E). This result suggests that the genomic instability in SRSF1 knockdown cells triggers a G2/M checkpoint arrest. Indeed, ATRi treatment of SRSF1 knockdown cells significantly reduced the G2/M cell population (Fig. S6E). Consistent with the role for ATR in suppressing origin firing, ATRi increased DNA synthesis in cells transfected with control siRNA (Fig. S6E). However, ATRi nearly eliminated DNA synthesis in SRSF1 knockdown cells (Fig. S6E), indicating that replication forks fail to recover at R loops in the absence of ATR activity. To test the role for ATR in fork recovery more directly, we used Plad-B to induce R loops and stall replication forks (Fig. 6F). An 8-hour Plad-B treatment of cells largely abolished DNA synthesis. After cells were released from Plad-B in CDC7i (to prevent firing of new origins), substantial DNA synthesis was detected in 60 minutes, indicating the recovery of stalled forks. In the presence of both ATRi and CDC7i, fork restart was significantly reduced and delayed, showing that ATR is required for the efficient recovery of R loop-stalled replication forks.
Knockdown of MUS81 also reduced DNA synthesis in S-phase cells (Fig. S6F), supporting the function of MUS81-EME2 in fork restart. Consistent with the role for MUS81 in ATR activation, depletion of MUS81 in SRSF1 knockdown cells led to a bypass of the G2/M arrest (Fig. S6F). Thus, MUS81 and ATR likely act in concert to promote DNA synthesis in response to R loops, allowing forks to recover from R loop impediments. In cells with high levels of R loops, MUS81 and ATR promote a G2/M arrest, which provides more time for fork recovery and helps cells avoid irreversible damage in mitosis.
ATR suppresses R loop-induced and MUS81-generated DNA damage
We next investigated how DSBs are generated in SRSF1 knockdown cells after ATR inhibition. In the presence of ATRi, knockdown of XPG increased γH2AX in SRSF1-depleted cells (Fig. 7A, lanes 2 and 4), suggesting that XPG suppresses R loops but is not responsible for ATRi-induced DSBs. In contrast, knockdown of MUS81 reduced γH2AX even when XPG was depleted (Fig. 7A-B, compare lanes 2 and 3 of 7A, lanes 4 and 5 of 7A, and lanes 5 and 6 of 7B), suggesting that ATRi-induced DSBs arise from MUS81-mediated DNA cleavage. Consistently, ATRi also induced γH2AX in Plad-B treated cells, and this induction of γH2AX was suppressed by MUS81 knockdown (Fig. S7A). Notably, only MUS81WT but not MUS81CD restored the ATRi-induced γH2AX formation in SRSF1, MUS81 double-knockdown cells (Fig. 7C), showing that the nuclease activity of MUS81 is required for ATRi-induced DSB formation.
Figure 7. ATR suppresses R loop-induced and MUS81-generated DNA damage.

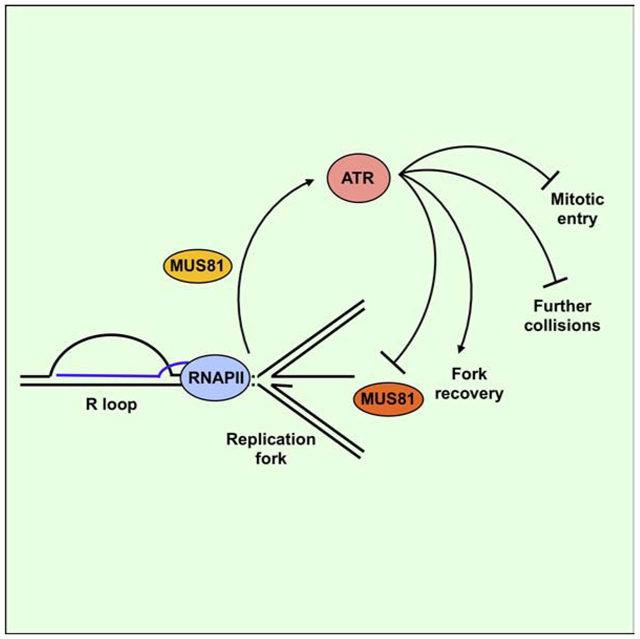
(A) HeLa cells were transfected with control, SRSF1-1, XPG, and MUS81-1 siRNA as indicated, and treated with ATRi (VE-821, 10 μM) for 24 hr. Levels of γH2AX and other proteins were analyzed by western blot. (B) HeLa cells were transfected with control, SRSF1-1, and MUS81-1 siRNA as indicated, and treated with or without ATRi (VE-821, 10 μM) for 24 hr. Levels of γH2AX and other proteins were analyzed by western blot. (C) HeLa cells were transfected with SRSF1 or MUS81 siRNA and treated with ATRi for 12 hr. Where indicated, cells were infected with lentiviruses expressing siRNA-resistant MUS81WT or MUS81CD. Levels of γH2AX were measured by immunostaining in >1,000 cells (n>1,000). The fractions of cells displaying γH2AX signals above the ground were colored in read and quantified. ***p<0.0001, Unpaired Student’s t test. (D) HeLa cells were transfected with control, SRSF1-1, EME1, and EME2 siRNA as indicated, and treated with ATRi as in (A). Levels of γH2AX and other proteins were analyzed by western blot. (E) HeLa cells were transfected with control, SRSF1-1, SMARCAL1, and ZRANB3 siRNA as indicated, and treated with ATR as in (A). (F) A model for an ATR-mediated feedback loop that protects the genome against R loop-associated DNA damage.
Interestingly, while MUS81 promotes R loop-induced ATR activation, the activity of MUS81 is restricted by ATR. These findings led us to hypothesize that ATR protects the genome through a MUS81-triggered feedback loop. To understand which MUS81 complex generates DNA damage at R loops after ATR inhibition, we knocked down SRSF1 with EME1, EME2, or SLX4 and analyzed the induction of γH2AX by ATRi. Knockdown of EME2 did not reduce γH2AX (Fig. 7D), suggesting that MUS81-EME2, which is important for ATR activation, is not involved in ATRi-induced DSB generation. Depletion of SLX4 increased ATRi-induced γH2AX (Fig. S7B, S7C), excluding SMX as the DSB-generating nuclease. Consistently, CDK1i and PLK1i did not reduce ATRi-induced γH2AX (Fig. S7D), ruling out premature CDK1 activation and CDK1/PLK1-dependent SMX activity as the cause of DSBs (Duda et al., 2016). In contrast, loss of EME1 reduced γH2AX (Fig. 7D). Since EME1 is not only important for MUS81 activity but also its stability (Fig. 4D) (Chang et al., 2008), EME1 may contribute to the action of MUS81 both directly and indirectly.
Finally, we asked whether MUS81 acts on reversed forks at R loops to generate ATRi-induced DSBs. Knockdown of SMARCAL1, ZRANB3, or HLTF reduced the ATRi-induced γH2AX in SRSF1-depleted cells (Fig. 7E, S7E). In contrast, depletion of RECQ1 increased ATRi-induced γH2AX (Fig. S7F). These results suggest that R loop-induced reversed forks are not only a trigger for MUS81-mediated ATR activation, but also a target for excessive MUS81 cleavage when ATR activation is compromised.
Discussion
R loops activate the ATR-Chk1 pathway through reversed replication forks
Recent studies have implicated ATR in the R loop response. Chk1 is increasingly phosphorylated in AQR knockdown cells (Sollier et al., 2014). Chk1 phosphorylation is also induced by head-on collisions of R loops and replication forks on episomes (Hamperl et al., 2017). In both budding and fission yeasts, R loop accumulation leads to phosphorylation of the functional counterparts of human Chk1 (Costantino and Koshland, 2018; Zhao et al., 2018). Using ATRi, RNaseH1, and different means to induce R loops, we demonstrate that Chk1 is indeed phosphorylated in an ATR-dependent manner in response to aberrant R loop accumulation. These observations raise the question as to how ATR is activated by R loops.
RPA-coated ssDNA, a nucleoprotein structure critical for ATR activation, is present at both R loops and replication forks (Marechal and Zou, 2015; Nguyen et al., 2017; Zou and Elledge, 2003). We recently suggested that RPA is a sensor of R loops even in non-S-phase cells (Nguyen et al., 2017). A mitosis-specific ATR-Chk1 pathway is locally activated at centromeres by RPA-coated R loops to promote faithful chromosome segregation (Kabeche et al., 2018). In contrast to centromeric R loops, the aberrant R loops resulting from SRSF1 loss induce p-Chk1 in a replication-dependent manner, suggesting that the RPA-ssDNA in R loops is not sufficient for Chk1 activation outside of centromeres. Chk1 localizes to centromeres during mitosis, but is not known to associate with R loops directly. The inability of aberrant R loops to activate Chk1 in the absence of replication may stem from the lack of Chk1 at these sites. Although aberrant R loops do not activate the ATR-Chk1 pathway directly, they may recruit the ATR-ATRIP kinase complex and allow ATR to phosphorylate some of its substrates (Nguyen et al., 2017). Nonetheless, the efficient phosphorylation of Chk1 at aberrant R loops requires the contribution of replication forks.
We also find that the efficient phosphorylation of Chk1 in SRSF1 knockdown cells requires SMARCAL1, ZRANB3, and HLTF, three factors needed for replication fork reversal (Betous et al., 2012; Ciccia et al., 2012; Kolinjivadi et al., 2017; Taglialatela et al., 2017; Vujanovic et al., 2017). RECQ1, which promotes resolution of reversed forks (Berti et al., 2013), suppresses Chk1 phosphorylation in SRSF1 knockdown cells. These findings establish a strong correlation between fork reversal and the activation of ATR by R loops. Although ATR was suggested to restrict SMARCAL1 activity (Couch et al., 2013), SMARCAL1 has not been implicated in ATR activation. Furthermore, while fork reversal is commonly induced by replication inhibitors and genotoxic agents (Zellweger et al., 2015), it has not been linked to ATR activation. Our data suggest that fork reversal is induced by the collision of R loops and replication forks, and that reversed forks are needed for efficient activation of the ATR-Chk1 pathway in this context (Fig. 7F). We do not exclude the possibility that R loops also contribute to Chk1 activation after colliding with replication forks. Head-on collisions of R loops and replication forks may expand and/or stabilize R loops (Hamperl et al., 2017), generating more RPA-ssDNA to facilitate ATR activation. Collisions may also induce remodeling of R loops, giving rise to DNA or DNA:RNA structures contributing to ATR activation.
A unique role for MUS81 in R loop-induced ATR activation
In this study, we also unexpectedly find that MUS81 is required for activation of the ATR-Chk1 pathway in SRSF1 knockdown cells. While MUS81 has been implicated in the recovery of stalled forks (Pepe and West, 2014a), it has not been linked to ATR activation. Indeed, we show that MUS81 is required for ATR activation in SRSF1 knockdown cells but not in HU-treated cells, suggesting that ATR is activated by R loops through a unique pathway. The nuclease activity of MUS81 is needed for the activation of ATR by R loops. EME2 is also required for efficient ATR activation in SRSF1 knockdown cells, implicating the MUS81-EME2 complex in this pathway. Although EME1 depletion also reduces p-Chk1 in SRSF1 knockdown cells, we cannot conclude whether MUS81-EME1 is directly involved in ATR activation because MUS81 levels are decreased in the absence of EME1 (Fig. 7F).
In vitro, the MUS81-EME2 complex cleaves fork-like structures, including reversed forks (Amangyeld et al., 2014; Pepe and West, 2014b). Since R loop-induced ATR activation requires fork reversal, MUS81-EME2 may cut reversed forks to activate ATR (Fig. 7F). In the absence of MUS81, the ssDNA induced by R loops is much reduced. During ATR activation, the RAD17-RFC complex acts at junctions of ssDNA and double-strand DNA (dsDNA) to load RAD9-RAD1-HUS1 (9-1-1) complexes onto dsDNA, which subsequently enable TopBP1 to stimulate ATR (Delacroix et al., 2007; Ellison and Stillman, 2003; Lee and Dunphy, 2010; Lee et al., 2007; MacDougall et al., 2007; Zou et al., 2002; Zou et al., 2003). We find that both RAD17 and TopBP1 are required for the activation of ATR by R loops. These results suggest that MUS81 may cleave reversed forks to generate ssDNA-dsDNA junctions and extend ssDNA, thereby allowing the RAD17-TopBP1 circuitry to activate ATR (Fig. S7G). Unraveling the DNA structural determinants and protein components of the MUS81-mediated ATR pathway is an important task for future studies.
ATR coordinates multiple cellular responses to suppress R loop-induced DNA damage
Yeast genetic studies suggested that the S-phase checkpoint is required for the survival of THO mutants, which contain high levels of R loops (Gomez-Gonzalez et al., 2009). In fission yeast, both the S-phase checkpoint and Mus81 are critical for cell survival when aberrant R loops accumulate in the absence of RNase H (Zhao et al., 2018). Consistently, we found that human cells lacking SRSF1 or SETX are increasingly sensitive to ATRi. Together, these results suggest that ATR plays a critical role in protecting the genome against R loop-associated genomic instability.
Our analyses of the effects of ATR inhibition in SRSF1 knockdown cells reveal several concerted functions of ATR in the R loop response (Fig. 7F). First, ATR suppresses the collision between transcription and replication complexes. ATR probably carries out this function by suppressing replication origin firing. Although ATRi did not significantly increase R loop levels in SRSF1-depleted cells, we do not exclude the possibility that ATR suppresses R loops in specific regions of the genome. Of note, a recent study showed that the ATR-Chk1 pathway promotes the function of DDX19 in unwinding DNA:RNA hybrids (Hodroj et al., 2017). Second, ATR promotes recovery of stalled replication forks at R loops. ATRi nearly eliminated DNA synthesis in SRSF1 knockdown cells, and it prevented efficient fork restart after Plad-B release. Third, ATR enables an R loop-induced G2/M arrest. This cell cycle arrest may provide more time for fork recovery and prevent detrimental consequences in the ensuing mitosis. Finally, ATR prevents excessive cleavage of DNA by MUS81, possibly by inhibiting MUS81 and/or its functional partners, or by suppressing the formation of MUS81 substrates (Forment et al., 2011; Kai et al., 2005). Collectively, these results suggest that ATR orchestrates the cellular responses to R loops, explaining its critical role in the survival of cells harboring high levels of R loops.
A MUS81-triggered and ATR-mediated feedback loop fine tunes MUS81 activity
Interestingly, we find that MUS81 acts both upstream and downstream of ATR in the R loop response, which suggests that ATR mediates a feedback loop that starts from and ends at MUS81 (Fig. 7F). MUS81 is known to have both pro-repair and pro-damage effects in cells (Abraham et al., 2003; Buisson et al., 2015; Couch et al., 2013; Forment et al., 2011; Munoz-Galvan et al., 2012; Pepe and West, 2014a; Ragland et al., 2013; Wechsler et al., 2011), but how these opposing effects of MUS81 are regulated is not clear. Our results suggest that ATR fine tunes MUS81 activity. During S phase, the DNA structural specificity of MUS81-EME2 or its low abundance may enable it to promote ATR activation and fork recovery without generating high levels of DSBs. Compared with MUS81-EME2, MUS81-EME1 is less active on HJs in vitro and may have a distinct substrate preference (Amangyeld et al., 2014; Pepe and West, 2014b). Although MUS81 complexes have been extensively characterized in vitro, how these complexes cleave replication forks associated with replisomes in cells is still poorly understood. ATR inhibition may alter the activity, specificity, stability, or localization of MUS81-EME1 in cells, allowing it to cleave reversed forks excessively. Loss of ATR activity may also affect the replisomes impeded by R loops or promote altered fork remodeling (Couch et al., 2013; Mutreja et al., 2018; Ragland et al., 2013), generating more preferred substrates for MUS81-EME1. Thus, in the context of R loop-induced fork stalling and reversal, ATR may allow MUS81-EME2 to promote fork restart and prevent the toxic cleavage by MUS81-EME1, thereby suppressing the genomic instability at R loops.
Notably, in contrast to the ATRi-induced DSB formation in SRSF1 knockdown cells, the induction of DSBs by ATRi in HU-treated cells depends on SLX4 but not MUS81 (Couch et al., 2013), suggesting that ATR suppresses DSBs through a distinct mechanism. It is tempting to speculate that the replication forks impeded by R loops undergo reversal in a unique way, which makes the MUS81-triggered and ATR-mediated feedback loop particularly important in this context. Alternatively, this ATR-mediated feedback may have a general role in protecting reversed forks that goes beyond the R loop context. Interestingly, it was recently reported that ATRi reduced the levels of detectable reversed forks in response to DNA interstrand crosslinks (Mutreja et al., 2018), which is consistent with the possibility that reversed forks are increasingly cleaved by nucleases upon ATR inhibition. Future studies are needed to fully understand the physiological and pathological contexts in which reversed fork-triggered and ATR-mediated feedback loops operate to suppress genomic instability.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the lead contact, Dr. Lee Zou (zou.lee@mgh.harvard.edu), Massachusetts General Hospital Cancer Center, Harvard Medical School, Boston, MA, USA
Materials Availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
EXPERIMENTAL MODEL AND SUBJECT DETAILS.
Cell culture
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 4 mM L-glutamine, and 1% peniciliin/streptomycin. HCT116 cells were cultured in DMEM with high glucose, GlutaMAX, and pyruvate supplemented with 10% FBS and 1% penicillin/ streptomycin. To express siMUS81-1-resistant MUS81WT and MUS81CD in HeLa cells, these protein sequences were tagged with HA in a Tet-inducible lentiviral vector using the Gateway cloning system (Life Technologies). Cells were infected with lentiviruses expressing MUS81WT or MUS81CD and selected with 1.8 mg/mL of geneticin.
METHOD DETAILS
RNA interference
siRNA transfections were done by reverse transfection using Lipofectamine RNAiMAX and harvested 72 hr after transfection unless otherwise noted. All siRNAs were used at 5 nM except siHLTF, which was used at 2.5 nM, siSLX4-1, which was used at 15 nM in reverse transfection during seeding, and siRad17 (15 nM), which was used at 5 nM in reverse transfection during seeding followed by forward transfection 24 hours later at 10 nM. The sequences of the siRNAs used are the following:
| List of siRNAs used in this study: | |
|---|---|
| Control siRNA | GGGUAUCGACGAUUACAAA |
| EME1 siRNA | GUAAGUUUCUGACCCACAA |
| EME2 siRNA | CACUGACCCAGAUCUCUGG |
| HLTF siRNA | GGAAUAUAAUGUUAACGAU |
| Rad17 siRNA | CAGACUGGUUGACCCAUC |
| MUS81 siRNA#1 | CAGCCCUGGUGGAUCGAUA |
| MUS81 siRNA #2 | CGCGCUUCGUAUUUCAGAA |
| RECQ1 siRNA | GCAGUUCCCUAACGCAUCA |
| SETX siRNA | GCCAGAUCGUAUACAAUUA |
| SLX4 siRNA #1 | GCUACCCGGACACUUGUCAUUGUUA |
| SLX4 siRNA #2 | AAACGUGAAUGAAGCAGAA |
| SMARCAL1 siRNA | GGCUCUCACUGGAAUCUCU |
| SRSF1 siRNA #1 | GGAUAACACUAAGUUUAGA |
| SRSF1 siRNA #2 | GCAUCUACGUGGGUAACUU |
| TopBP1 siRNA | GUGGUUGUAACAGCGCAUCUU |
| XPG siRNA | GAACGCACCUGCUGCUGUA |
| ZRANB3 siRNA | GAGAUAUCAUCGAUUAUGA |
Plasmid transfection
The plasmid expressing RNaseH1-GFP was provided by Dr. H. D. Nguyen of the Zou laboratory. Two days after siRNA transfection, the RNaseH1-GFP plasmid was transfected using X-tremeGENE HP DNA Transfection Reagent (Sigma-Aldrich) or Lipofectamine 3000 Transfection Reagent (ThermoFisher Scientific) following manufacturer’s instructions for 24 hours. For EME2 experiments, EME2 was tagged with HA and expressed from a CMV promotor in a Gateway destination vector. One day after siRNA transfection, HA-EME2 plasmid was transfected using the Lipofectamine 3000 Transfection Reagent following manufacturer’s instructions for 48 hours.
Dot blot
Following manufacturer’s instructions, total DNA was purified using QIAamp DNA mini kit (Qiagen). DNA concentration was initially estimated using a spectrophotometer. To determine DNA concentration more precisely, DNA was titrated and blotted on a positively charged nylon transfer membrane (Amersham) using a Bio-Dot Apparatus (Bio-Rad) in duplicate. The membrane was UV crosslinked (120 mJ/cm2), blocked with 5% milk in TBST buffer (1xTBS with 0.1% Tween-20), and probed with anti-dsDNA antibody (1:15,000) in blocking solution. Based on the titration of DNA and dsDNA signals, we normalized the amounts of input DNA from different samples for S9.6 dot blots. S9.6 dot blots were performed in at least triplicates with S9.6 antibody (1:15,000) in blocking solution. To test the specificity of S9.6 antibody, samples were also digested with RNase H (NEB). After incubations in dsDNA and S9.6 antibodies, blots were incubated with HRP-conjugated secondary antibodies and dsDNA and S9.6 signals were visualized using a ChemiDoc (Bio-Rad). Images were analyzed using Quantity One software (Bio-Rad).
Immunofluorescence
For experiments involving RPA32 and S9.6, samples were prepared as previously described (Nguyen et al., 2017). For experiments involving EdU labeling, SRSF1 knockdown and control cells were treated with EdU (10 μM) for 30 and 15 minutes, respectively. Cells were fixed in PBS containing 3% paraformaldehyde and 2% sucrose for 15 minutes. After washes with 1xPBS, cells were permeabilized using cold 1xPBS containing 0.5% Triton X-100 for 20 minutes. After 1xPBS washes, the Click-iT EdU Alexa Fluor 488 Imaging Kit was used to visualize EdU incorporation following manufacturer’s instructions. Subsequently, cells were blocked in 1xPBS containing 3% BSA and 0.05% Tween-20 for 1 hour. Primary antibodies, diluted to 1:500 in blocking solution, were then added to cells and incubated overnight at 4°C. After washes with PBST buffer (1xPBS with 0.05% Tween), cells were incubated with secondary antibodies conjugated to Cy3 or Alexa Fluor 488 for 30 minutes at 37°C. Finally, cells were washed three times with PBST, stained with DAPI during the second wash, and mounted on slides with Prolong Gold and sealed with nail polish. Images were captured with a Nikon 90i microscope and quantified using ImageJ.
PLA
Cells were washed once with PBS and then treated with CSK extraction buffer (0.2% Triton X-100, 20 mM HEPES-KOH pH 7.9, 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 1 mM EGTA) on ice for 3 minutes. Cell were then fixed on ice with PFA for 5 minutes followed by ice-cold methanol treatment at −20°C for 20 minutes. Subsequently, cells were permeabilized with 1x PBS containing 0.5% Triton-x100 for 4 minutes and then blocked with 3% BSA in PBST buffer at room temperature for 1 hour. Cells were washed with 1x PBS between each step. Afterwards, cells were incubated with the indicated primary antibodies diluted at 1:500 (PCNA and MUS81) and 1:1000 (p-RNAPII Ser2) at 4°C overnight. After three washes with 1x PBST, cells were incubated with anti-mouse minus and anti-rabbit plus PLA probes (PLA kit from Sigma) at 37°C for 1 hour. Following the manufacturer’s instructions, the PLA reaction was performed with the Duolink In Situ Detection Reagents (PLA kit). After a wash with Buffer B (PLA kit), cells were blocked again with 3% BSA in PBST and incubated with secondary antibody conjugated to fluorescence 488 at room temperature for 1 hour. Finally, cells were washed three times, stained with DAPI during the second wash, and mounted on slides with Prolong Gold and sealed with nail polish. Images were captured with a Nikon 90i microscope and quantified using ImageJ.
Cell viability assay
For measuring the viability of SRSF1 knockdown cells, cells were subjected to reverse transfection with control or SRSF1-1 siRNA (3 nM) in 6-well plates. After an overnight incubation, cells were seeded in a 96-well plate at 2,500 cells/well, and on the following day, treated with increasing concentrations of VE-821. After three days of ATRi treatment, cells were stained with Alamar Blue (Thermo Fisher) and analyzed with the EnVision 2103 multilabel plate reader from PerkinElmer. For measuring the viability of SETX knockdown cells, cells were seeded in a 96-well plate at 500 cells/well and subjected to forward transfection with control or SETX siRNA (5 nM). After an overnight incubation, cells were treated with escalating doses of ATRi. Four days after the initial transfection, cells were re-transfected with the same siRNA and treated with fresh ATRi to ensure continuous SETX knockdown and ATR inhibition. Four days after the second transfection, cell viability was measured with Alamar Blue as above.
FACS
For cell cycle profiling, cells were labeled with 10 μM EdU for 30 minutes and then processed with the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Life technologies) following manufacturer’s instructions. Flow cytometry was done using a BD LSR II or Fortessa apparatus equipped with the FACS Diva software (BD Biosciences).
Immunoprecipitation
Cells were pelleted in Eppendorf tubes and washed twice with 1xPBS. Cells were then lysed using ice-cold NETN lysis buffer [100 mM NaCl, 20 mM Tris-Cl (ph 8.0), 0.5 mM EDTA, 0.5% Nonidet P-40, and 1x Protease Inhibitor Cocktail] and incubated on ice for 30 minutes with occasional mixing. Crude cell lysates were obtained after centrifugation. The lysates (500 μg) were incubated with 3 μg of SLX4 antibody (Bethyl; A302-269A) with rotation at 4°C overnight. Pre-washed Protein G Dynabeads (50 μL; Invitrogen) were added to the samples and incubated at 4°C for 2 hours. Beads were then washed with NETN buffer before being resuspended with 2x SDS sample buffer and heated at 95°C for 5 minutes. The Western blot of SLX4 immunoprecipitates was probed with a second SLX4 antibody (Bethyl; A302-270A).
Neutral comet assay
To visualize double-strand DNA breaks, HeLa cells were transfected with siSRSF1 for 72 hours and treated with 10 μM ATRi during the last 12 hours. Neutral comet assay (also known as single-cell gel electrophoresis assay) were carried out as per the instructions provided by Trevigen (Cat No. 4250-050-K). A least 100 comet images from each condition were scored using OpenComet software (Gyori et al., 2014).
Replication recovery assay
HeLa cells were treated with Pladienolide B (Tocris, Cat No.6070) for 8 hours, and with 5 μM CDC7i (XL413) during the last 45 minutes of this treatment. Then, the cells were wash once with media, and released in 10 μM ATRi (VE-821), 5 μM CDC7i (XL413), and 10 μM EdU for the indicated time. Cell cycle distribution was analyzed as previously mentioned.
QUANTIFICATION AND STATISTICAL ANALYSIS
For S9.6 dot blots, S9.6 staining intensity was quantified from at least three replicates of each sample using the Quantity One software and normalized to dsDNA intensity. For western blots, the Quantity One software was used to analyze band intensity. For SMARCAL1, ZRANB3, HLTF, and RECQ1 western blot analysis, three independent experiments were analyzed. Cell viability was assayed using the Perkin Elmer EnVision Manager software. In cell viability assays, three to six technical replicates were analyzed at each dose of ATRi, and the cell viability at each dose was normalized to the untreated cells. For p-Chk1 S317, EdU, γH2AX, and PLA immunofluorescence intensities, ImageJ was used for quantifications. FIJI was used for foci identification and quantification. In all experiments analyzing cell populations, the numbers of cells analyzed are described in figure legends. Mean signal intensities or foci numbers in cell populations and standard deviations (SD) were determined with Prism or Excel. Unpaired Student’s t test was used to analyze the differences among samples.
Data and Code Availability
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CHK1 | Santa Cruz | sc-8408 |
| CHK1 pS317 | Cell Signaling | 2344 |
| CHK1 pS345 | Cell Signaling | 2348 |
| Cyclin A | Cell Signaling | 4656 |
| dsDNA | Abcam | ab27156 |
| EME1 | Santa Cruz | sc-53275 |
| GAPDH | Millipore | ABS16 |
| GFP | Millipore | MAB2510 |
| γH2AX | Millipore | 05-636 |
| HA | Abcam | ab18181 |
| HLTF | Sana Cruz | sc-398357 |
| KAP1 pS824 | Bethyl | A300-767A |
| MUS81 | Abcam | ab14387 |
| PCNA | Abcam | ab18197 |
| PCNA | Santa Cruz | sc-56 |
| Rad17 | Abcam | ab151513 |
| RECQ1 | Bethyl | A300-450A |
| RNAP II pSer2 | Novus Bio | NB100-1805 |
| RPA32 | Abcam | ab109084 |
| S9.6 | Antibodies Incorporated | Protein A purified from hybridoma S9.6 |
| SETX | Bethyl | A301-105A |
| SLX4 | Bethyl | A302-269A |
| SLX4 | Bethyl | A302-270A |
| SMARCAL1 | Santa Cruz | sc-376377 |
| SRSF1 | Bethyl | A302-053A |
| TopBPl | Bethyl | A300-111A |
| XPG | Bethyl | A301-484A |
| ZRANB3 | Bethyl | A303-033A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pladienolide B | Tocris Biosciences | 6070 |
| RNase H | NEB | M0297 |
| Critical Commercial Assays | ||
| alamarBlue™ Cell Viability Reagent | Thermo Fischer | DAL1025 |
| Comet Assay Kit | Trevigen | 4250-050-K |
| Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit | Thermo Fischer | C10337 |
| Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Thermo Fischer | C10634 |
| Duolink™ In Situ Detection Reagents Red | Sigma-Aldrich | DUO92008 |
| Oligonucleotides | ||
| siRNA | This Study | See List in Methods |
| Recombinant DNA | ||
| Plasmid: EME2 | Harvard PlasmID Repository | |
| Software and Algorithms | ||
| GraphPad Prism 5 | NIH | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Fiji | (Schindelin et al., 2012) | https://fiji.sc/ |
| NIS element viewer | Nikon | https://www.nikoninstruments.com/Products/Software/NIS-Elements-Advanced-Research/NIS-Elements-Viewer |
| Quantity One | Bio-rad | http://www.bio-rad.com/en-us/product/quantity-one-1-d-analysis-software?ID=1de9eb3a-1eb5-4edb-82d2-68b91bf360fb |
| Python 3.7 | Python | https://www.python.org/downloads/release/python-370/ |
Highlights.
The ATR-Chk1 pathway is activated by aberrant R loops in S phase.
ATR suppresses R loop-induced DNA double-strand break formation.
R loop-induced ATR activation requires fork reversal and the MUS81 nuclease.
ATR prevents excessive cleavage of reversed forks by MUS81.
Acknowledgements
We thank Dr. J. W. Harper for the MUS81 plasmid, and Drs. N. Dyson, J. Walter, J. K. Joung, A. Elia, L. Lan and members of the Zou and Dyson labs for discussions. D.A.M. was partially supported by an NIH F31 fellowship (1F31CA210311). J.-M.Z. is partially supported by the Massachusetts General Hospital Cancer Center Excellence Award. H.D.N. was supported by an NIH T32 postdoctoral training grant (T32 DK007540) and a grant from the Edward P. Evans Foundation. M.-M.G. was partially supported by a postdoctoral fellowship from Fonds de recherche Santé Québec (FRQS). L.Z. is the James & Patricia Poitras Endowed Chair in Cancer Research, and was supported by a Jim & Ann Orr Massachusetts General Hospital Research Scholar Award. This work is supported by grants from the NIH (GM076388, CA197779, and CA218856) to L.Z.
Footnotes
Declaration of interests
L.Z. has consulted for EMD Serono and received research funding from Calico. All other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham J, Lemmers B, Hande MP, Moynahan ME, Chahwan C, Ciccia A, Essers J, Hanada K, Chahwan R, Khaw AK, et al. (2003). Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J 22, 6137–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achar YJ, Balogh D, Neculai D, Juhasz S, Morocz M, Gali H, Dhe-Paganon S, Venclovas C, and Haracska L (2015). Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res 43, 10277–10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, and Garcia-Muse T (2012). R loops: from transcription byproducts to threats to genome stability. Mol Cell 46, 115–124. [DOI] [PubMed] [Google Scholar]
- Amangyeld T, Shin YK, Lee M, Kwon B, and Seo YS (2014). Human MUS81-EME2 can cleave a variety of DNA structures including intact Holliday junction and nicked duplex. Nucleic Acids Res 42, 5846–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, and Cortez D (2012). SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, and Aguilera A (2014). BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365. [DOI] [PubMed] [Google Scholar]
- Buisson R, Boisvert JL, Benes CH, and Zou L (2015). Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol Cell 59, 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JH, Kim JJ, Choi JM, Lee JH, and Cho Y (2008). Crystal structure of the Mus81-Emel complex. Genes Dev 22, 1093–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, and Cerritelli SM (2013). RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41, 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. (2012). Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelio DA, Sedam HN, Ferrarezi JA, Sampaio NM, and Argueso JL (2017). Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair (Amst) 52, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, and Koshland D (2018). Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Mol Cell 71, 487–497 e483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, and Karnitz LM (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 21, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison V, and Stillman B (2003). Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5' recessed DNA. PLoS Biol 1, E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forment JV, Blasius M, Guerini I, and Jackson SP (2011). Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS One 6, e23517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Martin MM, Regairaz M, Huang L, You Y, Lin CM, Ryan M, Kim R, Shimura T, Pommier Y, et al. (2015). The DNA repair endonuclease Mus81 facilitates fast DNA replication in the absence of exogenous damage. Nat Commun 6, 6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011). R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalez B, Felipe-Abrio I, and Aguilera A (2009). The S-phase checkpoint is required to respond to R-loops accumulated in THO mutants. Mol Cell Biol 29, 5203–5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalez B, Garcia-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marin A, Foiani M, and Aguilera A (2011). Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J 30, 3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786 e719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2016). Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 167, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 57, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, and Maiorano D (2017). An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J 36, 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Boddy MN, Russell P, and Wang TS (2005). Replication checkpoint kinase Cds1 regulates Mus81 to preserve genome integrity during replication stress. Genes Dev 19, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, and Dunphy WG (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. [DOI] [PubMed] [Google Scholar]
- Lee J, and Dunphy WG (2010). Rad17 plays a central role in establishment of the interaction between TopBP1 and the Rad9-Hus1-Rad1 complex at stalled replication forks. Mol Biol Cell 21, 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, and Dunphy WG (2007). The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem 282, 28036–28044. [DOI] [PubMed] [Google Scholar]
- Li X, Niu T, and Manley JL (2007). The RNA binding protein RNPS1 alleviates ASF/SF2 depletion-induced genomic instability. RNA 13, 2108–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima WF, Murray HM, Damle SS, Hart CE, Hung G, De Hoyos CL, Liang XH, and Crooke ST (2016). Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res 44, 5299–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall CA, Byun TS, Van C, Yee MC, and Cimprich KA (2007). The structural determinants of checkpoint activation. Genes Dev 21, 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res 25, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Galvan S, Tous C, Blanco MG, Schwartz EK, Ehmsen KT, West SC, Heyer WD, and Aguilera A (2012). Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol Cell Biol 32, 1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, and Lopes M (2018). ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep 24, 2629–2642 e2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, and Zou L (2017). Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol Cell 65, 832–847 e834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli S, Teasley DC, Murali B, Jackson J, Vindigni A, and Stewart SA (2017). Human ribonuclease H1 resolves R-loops and thereby enables progression of the DNA replication fork. J Biol Chem 292, 15216–15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe A, and West SC (2014a). MUS81-EME2 promotes replication fork restart. Cell Rep 7, 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe A, and West SC (2014b). Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res 42, 3833–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragland RL, Patel S, Rivard RS, Smith K, Peters AA, Bielinsky AK, and Brown EJ (2013). RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev 27, 2259–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira JM, and Aguilera A (2015). R loops: new modulators of genome dynamics and function. Nat Rev Genet 16, 583–597. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, and Niedzwiedz W (2015). The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell 60, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, and Gautier J (2004). ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6, 648–655. [DOI] [PubMed] [Google Scholar]
- Shivji MKK, Renaudin X, Williams CH, and Venkitaraman AR (2018). BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep 22, 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, and Gromak N (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, and Cimprich KA (2015). Breaking bad: R-loops and genome integrity. Trends Cell Biol 25, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, and Cimprich KA (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler T, Newman S, and West SC (2011). Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HD, Laister RC, Martin SR, Arrowsmith CH, and West SC (2017). The SMX DNA Repair Tri-nuclease. Mol Cell 65, 848–860 e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, and Zou L (2016). Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu Rev Genet 50, 155–173. [DOI] [PubMed] [Google Scholar]
- Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, and Tian M (2004). An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol 5, 1275–1281. [DOI] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chiang HC, Wang Y, Zhang C, Smith S, Zhao X, Nair SJ, Michalek J, Jatoi I, Lautner M, et al. (2017). Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat Commun 8, 15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhu M, Limbo O, and Russell P (2018). RNase H eliminates R-loops that disrupt DNA replication but is nonessential for efficient DSB repair. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Cortez D, and Elledge SJ (2002). Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev 16, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
- Zou L, Liu D, and Elledge SJ (2003). Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A 100, 13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.