

Abstract
Chromatin is critical for genome compaction and gene expression. On a coarse scale, the genome is divided into euchromatin, which harbors the majority of genes and is enriched in active chromatin marks, and heterochromatin, which is gene-poor but repeat-rich. The conserved molecular hallmark of heterochromatin is the H3K9me3 modification, which is associated with gene silencing. We found that in Drosophila deposition of most of the H3K9me3 mark depends on SUMO and the SUMO-ligase Su(var)2–10, which recruits the histone methyltransferase complex SetDB1/Wde. In addition to repressing repeats, H3K9me3 also influences expression of both hetero- and euchromatic host genes. High H3K9me3 levels in heterochromatin are required to suppress spurious transcription and ensure proper gene expression. In euchromatin, a set of conserved genes is repressed by Su(var)2–10/SetDB1-induced H3K9 trimethylation ensuring tissue-specific gene expression. Several components of heterochromatin are themselves repressed by this pathway providing a negative feedback mechanism to ensure chromatin homeostasis.
Keywords: chromatin, heterochromatin, epigenetics, gene regulation, transcriptional repression, transposons, germline, cell fate maintenance
eTOC Blurb
The hallmark of heterochromatin is the H3K9me3 modification. Ninova et al. found that H3K9me3 ensures proper gene expression by suppressing spurious transcription in heterochromatin, by ensuring tissue-specific expression of developmentaly regulated genes and by participating in a negative feedback mechanism to ensure chromatin homeostasis.
Graphical Abstract
Introduction
Eukaryotic DNA is packaged in nucleosomes comprised of DNA and histones to form chromatin. Chromatin has historically been divided into euchromatin and heterochromatin based on density reflected by differential staining (Heitz, 1928). Euchromatin is relatively uncondensed, ‘open’ chromatin where transcription is active, whereas heterochromatin is compact and usually transcriptionally silenced. Heterochromatin includes relatively gene-poor and repeat-rich regions near telomeres and centromeres that are condensed in all cell types (constitutive heterochromatin), regions that are silenced in a cell-type specific manner (facultative heterochromatin), as well as transposable elements (TEs) – parasitic DNA elements capable of copying/cutting and integrating their sequences within host genomes (Allshire and Madhani, 2018; Henikoff, 2000). Heterochromatin is essential for genome stability, as it prevents unequal recombination between repetitive regions and confers TE repression, as well as for cell differentiation and llineage commitment (Becker et al., 2016; Janssen et al., 2018; Peng and Karpen, 2009). Although considered transcriptionally silenced, certain regions of heterochromatin host active genes, and at least in Drosophila some heterochromatin-residing genes in fact require this environment to be properly expressed (Eberl et al., 1993; Gatti and Pimpinelli, 1992; Lu et al., 1996, 2000; Wakimoto et al., 1990; Yasuhara and Wakimoto, 2006), although the molecular mechanism of this phenomenon remains elusive.
On the molecular level, chromatin compaction is regulated by post-translational modifactions of histone proteins and associated factors (reviewed in Bannister and Kouzarides, 2011). The best studied histone modifications associated with heterochromatin are histone 3 lysine 9 trimethylation (H3K9me3) and H3 lysine 27 trimethylation (H3K27me3). H3K27me3 is deposited by the polycomb repressive complex 2 (PRC2) and is primarily involved in cell-specific gene repression such as that of the HOX complex (Margueron and Reinberg, 2011; Schuettengruber et al., 2017). H3K9me3 is a conserved hallmark of constitutive heterochromatin, and also present on TEs spread across the genome (Karimi et al., 2011; LeThomas et al., 2013; Martens et al., 2005; Matsui et al., 2010; Mikkelsen et al., 2007; Pezic et al., 2014; Rozhkov et al., 2013; Sienski et al., 2012). H3K9me3 is deposited by H3K9-specific histone methyltransferase enzymes (HMTs) from the SETDB1 and SUV39 families (Nakayama, 2001; Rea et al., 2000; Schotta et al., 2002; Schultz et al., 2002). H3K9me3 provides a high-affinity binding site for members of the conserved heterochromatin protein 1 (HP1) family which mediate chromatin compaction, and can further recruit additional H3K9 HMT complexes enabling heterochromatin spreading (Bannister et al., 2001; Canzio et al., 2011; Hiragami-Hamada et al., 2016; Jacobs et al., 2001; Lachner et al., 2001). H3K9 HMTs can be recruited to target genomic loci by different pathways that employ a diversity of guides such as DNA binding proteins and non-coding RNAs. For example, a vertebrate-specific Krüppel-associated box-containing zinc finger proteins (KRAB-ZFPs) are known for their role to recognize specific DNA motifs of endogenous retrovirus and to recruit SETDB1 resulting in H3K9me3 deposition and silencing of endogenous retroviruses in mammals (Wolf et al., 2015). In fission yeast, small interfering RNAs (siRNAs) and Argonaute proteins recruit the silencing machinery to pericentric heterochromatin and the silent mating type locus; DNA-binding factors that recognize specific regulatory elements can also nucleate heterochromatin formation at some regions (Hall, 2002; Jia, 2004; Kanoh et al., 2005; Verdel et al., 2004; Volpe et al., 2002). A dedicated class of Argonaute proteins (Piwi-s) and associated piRNAs recognize and establish H3K9me3-dependent silencing at TEs in Drosophila and mammalian germline, as well as in somatic cells of the fly ovary (LeThomas et al., 2013; Pezic et al., 2014; Rozhkov et al., 2013; Sienski et al., 2012). A comprehensive picture of the different mechanisms tha regulate H3K9 deposition at different genomic targets is however yet to be established.
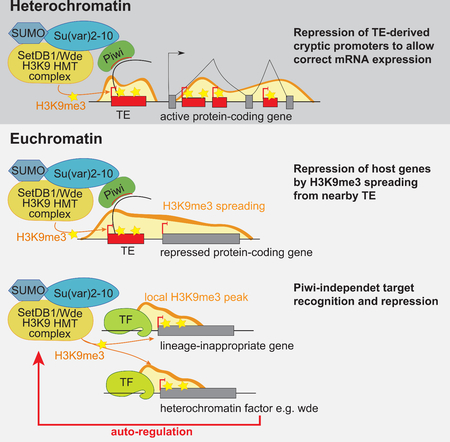
Su(var)2–10/dPIAS encodes a conserved protein from the PIAS family of SUMO E3 ligases, which was previously identified as a suppressor of positional effect variegation and a factor required for chromosomal stability, suggesting a role in heterochromatin formation (Hari et al., 2001; Reuter and Wolff, 1981). We found that Su(var)2–10 plays and important role in genome-wide H3K9me3 deposition and transcriptional repression of TEs in Drosophila female germ cells. Su(var)2–10 interacts with and acts downstream of the piRNA-Piwi complex, where it mediates SUMO-dependent recruitment of the SetDB1/Wde H3K9-HMT complex to TE targets (Ninova et al., accompanying manuscript). We observed that Su(var)2–10 depletion affected the majority of H3K9me3-rich domains, and the expression of multiple host genes. Here, we present a comprehensive analysis of the effect of H3K9me3 depletion resulting from Su(var)2–10 knockdown on the host transcriptome that reveals several distinct mechanisms through which H3K9me3 is involved in gene regulation. First, we show that host gene expression can be altered as a result of H3K9me3 spreading from adjacent TEs, demonstrating that novel TE integrations are an important source of epigenetic diversity. Second, we show that H3K9me3 and Su(var)2–10 are required for the activity of genes residing in heterochromatin and for repression of cryptic promoters upstream of or within host gene introns, thereby suppressing aberrant gene isoforms. Finally, we show that local Su(var)2–10-dependent H3K9me3 peaks are associated with repression of a subset of genes normally expressed in other tissues, highlighting a role for H3K9me3 in cell lineage commitment and facultative heterochromatin – a function usually associated with H3K27me3/PRC2. Such peaks are not associated with TEs and are conserved in other species, pointing to the existance of a piRNA-independent conserved mechanism that guides the H3K9 HMT complex to suppress lineage-inappropriate genes. Remarkably, several genes that encode proteins required for heterochromatin formation are also targets of H3K9me3-mediated repression, pointing to a negative feedback mechanism of heterochromatin control.
Results
Su(var)2–10 controls the expression of host genes
We have shown that Su(var)2–10 represses TEs recognized by the Piwi/piRNA complex (Ninova et al., accompanying manuscript). However, in contrast to Piwi and piRNA, which are predominantly expressed in the germline and required for fertility but not somatic development, Su(var)2–10 is expressed ubiquitously and null-mutants are lethal, suggesting that Su(var)2–10 has other, piRNA-independent, targets and functions. To identify genes that are regulated by Su(var)2–10, we inhibited expression of Su(var)2–10 in ovarian germ cells (GLKD) using small hairpin RNA (shRNA) driven by the maternal tubulin GAL4 driver (mtGAL4) and analyzed changes in steady state RNA levels in shSu(var)2–10-expressing (shSv210) and control shWhite lines (shW). Differential gene expression analysis of RNA-seq libraries performed in duplicates showed that 662 and 417 (389 protein-coding) genes were more than two-fold up- and down-regulated, respectively, upon depletion of Su(var)2–10 (FDR 5%, DESeq2; Fig. 1A, S1). Thus, Su(var)2–10 regulates the expression of a subset of host genes in addition to its function in TE silencing (Ninova et al., accompanying manuscript). Su(var)2–10-dependent TE repression is associated with the repressive H3K9me3 mark, and recruitment of Su(var)2–10 to chromatin leads to H3K9me3 accumulation at the target locus (accompanying manuscript). We therefore analyzed the gene targets of Su(var)2–10 in the context of their vicinity to TEs and H3K9me3 enrichment (ChIP-seq) (Fig. 1B). A subset of Su(var)2–10-regulated genes do not have H3K9me3 peaks or evidence of adjacent transpons (annotated or non-reference, see Methods), and likely represent secondary targets. However, a substantial number of Su(var)2–10 targets overlap with H3K9me3-rich regions that generally lose H3K9me3 signal upon Su(var)2–10 GLKD (Fig. 1B,C). Interestingly, Su(var)2–10 targets that have H3K9me3 peaks include genes both with and without adjacent TE insertions, and might be both up- or down-regulated upon Su(var)2–10 KD. In order to understand how loss of the typically repressive H3K9me3 mark results in such opposing effects in transcriptional activity, we performed detailed analysis of different Su(var)2–10 target categories, which revealed distinct, context-dependent mechanisms by which Su(var)2–10 and H3K9 methylation affect the transcriptome.
Figure 1. Su(var)2–10 depletion leads to transcriptome-wide changes in gene expression that correlate with changes in chromatin marks.

(A) Differential expression analysis of Su(var)2–10 GLKD and control (shW) ovaries. Data are from two biological replicates. See also Figure S1. (B) Summary of Su(var)2–10 GLKD affected genes by eu- and heterochromatin localization, H3K9me3 peak and adjacent TE (C) RNA and H3K9me3 changes on Su(var)2–10 target genes with H3K9me3 peaks and/or adjacent TEs. Heatmaps show % of repeats in introns and 10Kb-flanking regions. Genes are ordered by RNA-level fold change, presence of H3K9me3 peak and proximal TE, and H3K9me3 levels in control ovaries.
Spreading of the H3K9me3 mark from TE sequences leads to repression of adjacent genes
Silencing of TEs by Su(var)2–10 involves deposition of the H3K9me3 mark by the SetDB1/Wde histone methyltransferase complex (Ninova et al., accompanying manuscript). Repressive chromatin marks can spread from TE sequences up to several kilobases (kb), resulting in local H3K9me3-enriched ‘islands’ within otherwise euchromatic regions, which in turn can influence the expression of adjacent host genes (Karimi et al., 2011; Lee and Karpen, 2017; Pezic et al., 2014; Sentmanat and Elgin, 2012; Sienski et al., 2012). We found that several euchromatic genes regulated by Su(var)2–10 harbor or are adjacent to TE insertions. For example, we observed H3K9me3 spreading from the BARI element at the jheh locus into the downstream jheh3 gene. GLKD of Su(var)2–10 led to reduced H3K9me3 levels and increased expression of jheh3 (Fig. 2A, left). Thus, the BARI insertion represses jheh3 through its effect on the local chromatin state rather than through disruption of gene regulatory elements in the DNA sequence. We found that several host genes are affected by Su(var)2–10 knockdown as a consequence of H3K9me3 deposition on non-reference TEs; an example is the Frl/CG43986 gene (Fig. 2A, right). Thus, spreading of repressive chromatin from TEs can lead to repression of adjacent host genes via epigenetic mechanisms without disrupting cis-regulatory DNA (Fig 2B).
Figure 2. Epigenetic effect of TE-dependent H3K9me3 on adjacent protein-coding gens.

(A) Examples of euchromatic genes adjacent to a reference TE (left) or hosting a non-reference TE (right; red line marks TE integration site). Tracks show RPM-normalized coverage of RNA-seq and ChIP-seq data for indicated marks and conditions. ChIP and Input signals are overlaid. Top panels show the gene structures at corresponding loci. Arrows indicate the direction of transcription. Numbers show RPKM values of exonic regions (RNA-seq, estimated by eXpress) or normalized ChIP/Input signal (ChIP-seq) in manually selected genomic intervals indicated by black bars. (B) Model of piRNA-dependent H3K9me3 deposition at euchromatic TEs and its spreading to adjacent genes.
Su(var)2–10-dependent H3K9me3 deposition ensures proper expression levels and isoform selection of heterochromatic genes
Though new TE insertions often occur in euchromatin, the vast majority of TEs accumulate in heterochromatin. The heterochromatic compartment of D. melanogaster is well characterized cytologically and through genome-wide profiling of chromatin marks and includes nearly the entire chromosomes Y and 4 as well as pericentromeric and telomeric regions of chromosomes X, 2, and 3 (Gatti and Pimpinelli, 1992; Hoskins et al., 2002, 2007; Riddle et al., 2011). Heterochromatin-euchromatin boundaries are similar in different tissues and developmental stages (Riddle et al., 2011; see methods for heterochromatin domain annotation), and our H3K9me3 ChIP-seq profiles from ovaries of control flies (shW) largely overlap with annotated domains (Fig. 3A). Despite its relatively low gene density, several hundred protein-coding genes reside in heterochromatin (Hoskins et al., 2002), and can be actively transcribed. Of the 8255 protein-coding genes expressed in the ovary, 228 reside in heterochromatin including genes that encode conserved proteins with well-established functions such as Parp encoding poly-(ADP-ribose) polymerase and AGO3 required for piRNA repression.
Figure 3. Su(var)2–10 and H3K9me3 are required for heterochromatic gene expression.

(A) Circle plot of the D. melanogaster chromosome arms and heterochromatic scaffolds (dm3). Dots represent positions and RNA fold changes upon Su(var)2–10 GLKD for genes residing within annotated heterochromatin boundaries. Bargraphs show H3K9me3 levels in control ovaries and fold changes upon Su(var)2–10 GLKD for >2-fold H3K9me3-enriched intervals (5Kb genomic intervals, see Methods). (B) Heatmaps of H3K9me3 distribution across heterochromatic Su(var)2–10 targets in control ovaries; side bars show % repeats in indicated regions; barplots show total H3K9me3 enrichment, and H3K9me3 and RNA fold changes upon Su(var)2–10 GLKD. (C) Average profiles of H3K9me3, H3K4me2/3 and RNA Pol II over gene bodies of heterochromatic genes downregulated upon Su(var)2–10 GLKD in indicated conditions. Shaded areas reflect standard errors. See also Figure S2. (D) Tracks show RPM-normalized RNA-seq and ChIP-seq coverage at the nAChRalpha4 locus in indicated conditions. Red bars indicate predicted novel transcripts; asterisks indicate new peaks that appear upon Su(var)2–10 GLKD. Numbers are as in Fig 2. (E) RNA levels from sense exonic and antisense intronic regions of heterochromatic genes in indicated conditions. Data in A-C, E are averages from two biological replicates.
We found that heterochromatic genes are significantly over-represented among Su(var)2–10 targets (86/228, p<0.0001 permutation test). However, in contrast to genes adjacent to TE insertions in euchromatin such as jheh3 and Frl, where loss of Su(var)2–10 led to transcriptional up-regulation, the majority of heterochromatic Su(var)2–10 targets (58 of the 86) were down-regulated upon Su(var)2–10 GLKD (Fig. 3A). Strikingly, almost all genes in this set had high levels of H3K9me3 along their gene bodies (excluding the promoter region) and experienced a decrease in this mark upon Su(var)2–10 GLKD (Fig. 3B, C). Su(var)2–10 loss also led to reduced marks of active transcription (H3K9me2/3 and Pol II), indicating that the observed change in steady-state RNA levels upon Su(var)2–10 depletion and H3K9me3 loss is due to transcriptional down-regulation. Taken together, our results indicate that presence of the “silencing” H3K9me3 mark over gene bodies is not only compatible with transcription, but is also required for the normal expression of gene residing in heterochromatin.
To understand how loss of H3K9me3 interferes with the expression of heterochromatic genes, we carefully examined the RNA-seq profiles and enrichment of RNA polymerase II and H3K4me2/3 at the affected loci. We found that in several cases transcriptional down-regulation upon Su(var)2–10 depletion was associated with the appearance of spurious transcripts from within introns or flanking regions, as illustrated for the nAChRalpha4 gene (Fig. 3D). In wild-type ovaries, RNA-seq reads from nAChRalpha4 mapped predominantly to exons, and few reads were generated from the long introns, which contain repetitive sequences. Upon Su(var)2–10 KD, the signals from exons were reduced, and signals from intronic transcripts, both sense and antisense with respect to the coding gene, were increased (Fig. 3D). The appearance of spurious intronic transcripts was associated with the appearance of H3K4me3 and Pol II peaks (Fig. 3D). Heterochromatic genes that were down-regulated upon germline depletion of Su(var)2–10 displayed a global increase of antisense intronic reads (Fig. 3E). Transcription on either strand of long intronic regions or in flanking sequences of heterochromatic genes might interfere with normal gene expression. Taken together, these results indicate that Su(var)2–10 and H3K9me3 are required for repression of spurious transcription likely initiated from TE promoters located in heterochromatin.
Analysis of gene expression upon Su(var)2–10 GLKD revealed additional effects of H3K9me3 loss on heterochromatic genes. Loss of H3K9me3 correlated with activation of a cryptic promoter in an intron of the Nipped-B gene that led to the appearance of a new mRNA isoform (Fig. 4A). This new isoform is missing 14 coding exons of the canonical Nipped-B transcript and has one new exon, which shares homology with the gypsy TE. Expression of the full-length Nipped-B isoform is only moderately affected by Su(var)2–10 depletion, but expression of the truncated protein from the new mRNA isoform might cause dominant negative effects and interfere with proper protein function. In the case of unc-13, Su(var)2–10 GLKD and associated H3K9me3 loss resulted in the up-regulation of a longer isoform from an upstream transcription start site (TSS) adjacent to an Invader TE and in reduced activity of the canonical TSS (Fig. 4B). Ectopic transcriptional activation can affect genes at long distances as evident for the gene CG10417, which encodes a conserved protein phosphatase (Fig. 4C). H3K9me3 loss and transcriptional up-regulation near a TE-rich region about 5 kb upstream of the canonical CG10417 promoter resulted in the emergence of a long fusion transcript between the upstream region and the protein-coding gene. As was observed for unc-13, up-regulation of transcription from the upstream TSS was associated with reduced H3K4me3 at the canonical promoter and with repression of the normal mRNA isoform (Fig. 4C). Collectively, these examples show that loss of H3K9me3 causes activation of TE transcription within introns or in regions adjacent to host genes and interferes with the activity of gene promoters, leading to reduced gene expression and/or appearance of new mRNA isoforms encoding novel proteins (Fig. 4D). Overall, our results indicate that genes positioned in heterochromatin have very different properties than their euchromatin counterparts: they require high levels of H3K9me3 for proper expression and canonical isoform selection.
Figure 4. H3K9me3 loss induces transcriptional interference and aberrant isoform expression.

(A-C) Tracks showing RPM-noramlzied coverage of RNA-seq and ChIP-seq data across three different heterochromatic loci in the indicated conditions; annotations are as in Fig 2. Asterisks mark new peaks appearing upon Su(var)2–10 GLKD. Bottom panels show the relative expression (FPKM) of detected isoforms; asterisks mark differentially expressed isoforms. (D) Summary of different effects of H3K9me3 loss and TE activation on heterochromatic genes (Fig 3D and 4A–C).
Su(var)2–10-dependent H3K9me3 deposition restricts expression of tissue-specific genes
The presence of TE sequences that are targeted by Su(var)2–10 in close proximity to genes explains the effect of Su(var)2–10 on such genes. However, a number of euchromatic genes repressed by Su(var)2–10 do not have proximal TE insertions, yet still display H3K9me3 peaks (Fig. 1B, 5A). While some H3K9me3 peaks may be due to polymorphic TEs that escaped detection, we found that a subset of these genes display H3K9me3 peaks in other fly strains based on ChIP-seq datasets from previous studies (LeThomas et al., 2013; Muerdter et al., 2013; Yu et al., 2015, see Methods) (Fig. 5A). H3K9me3 deposition at TEs in the ovary is mediated by the piRNA pathway. Analysis of small RNA-seq data showed that piRNA-sized reads target regions with TE insertions. In contrast, regions of Su(var)2–10 targets with no TE insertions were devoid of piRNA-sized reads, suggesting piRNA-independent H3K9me3 deposition (Fig. 5B). Finally, we found that the D. virilis homologs of more than half of the conserved D. melanogaster genes suppressed by Su(var)2–10 with H3K9me3 peaks but lacking adjacent TE insertions (32 out of 58) also have H3K9me3 peaks. D. virilis diverged from D. melanogaster more than 45 million years ago (Hedges and Kumar, 2009), thus conserved H3K9me3 signals are indicative of TE-independent deposition. Taken together, these data suggest that Su(var)2–10 is involved in an alternative, piRNA-independent pathway of H3K9me3 deposition that directly targets euchromatic genes for silencing (Fig 5C). Analysis of tissue expression profiles of direct targets of Su(var)2–10/H3K9me3 repression showed biases to specific tissues such as the central nervous system or head, larval imaginal discs and testis (Fig. 5D). These genes typically showed low or no expression in control ovaries, and became ectopically expressed in the female germline upon Su(var)2–10 GLKD and H3K9me3 loss. For example, the testis-specific gene mtsh (encoding male meiosis factor) is repressed in the ovary and marked by a strong H3K9me3 peak at its promoter region that depends on Su(var)2–10 and SUMO, but not on Piwi (Fig. 5E). Su(var)2–10 GLKD led to loss of the H3K9me3 peak and ectopic expression of mtsh. In addition to protein-coding genes, direct targets of Su(var)2–10/H3K9me3 repression include non-coding RNA loci, including well-characterized testis-biased endogenous hp-siRNAs (Wen et al., 2015), as well as the testis-specific microRNA cluster mir-992/991/2498 (Mohammed et al., 2014) (Fig. 5F,G; Fig. S3). Thus, Su(var)2–10 plays a role in maintaining appropriate female-specific expression patterns in germline cells by repressing testis-expressed genes. More generally, Su(var)2–10 prevents ectopic expression of genes that are normally restricted to other cell fates through a mechanism that is independent of TE suppression.
Figure 5. TE-independent H3K9me3 deposition represses lineage-inappropriate genes.

(A) H3K9me3 signal over eurchromatic genes repressed by Su(var)2–10; side heatmaps show % repeats in indicated regions; black marks denote H3K9me3 peaks conserved in other strains (see Methods) and non-reference TEs. (B) Relationship between proximity to TEs and number of piRNA-sized reads that can be aligned to euchromatic Su(var)2–10/H3K9me3 repressed genes (C) Model of TE-independent gene repression by Su(var)2–10. (D) Tissue expression bias (modEncode Anatomy database) of euchromatic, TE-independent targets of Su(var)2–10/H3K9me3 repression. Sidebars indicate fold upregulation upon Su(var)2–10 GLKD and presence of conserved H3K9me3 peaks in D. virilis. (E-G) Examples of RNA-seq and H3K9me3 ChIP-seq tracks at TE-independent testis-specific loci mtsh (protein-coding), CG18854 (endo-siRNA) and mir-991/992/2498 (microRNA cluster, small RNA-seq) in indicated conditions. Numbers are as in Fig 2 (microRNA levels quantified as RPM). See also Figure S3.
Su(var)2–10 represses a set of genes involved in heterochromatin formation
We found that several genes that encode proteins involved in heterochromatin formation and maintenance are repressed by Su(var)2–10 dependent H3K9me3 (Fig. 6A). Su(var)2–10 regulates the expression of wde, a conserved co-factor of SetDB1/Eggless (Koch et al., 2009), the smt3 gene encoding the single copy of SUMO (Small Ubiquitin-like Modifier) in Drosophila and two HP1 interactors, Sov and CG30403. SUMO has been implicated in various aspects of heterochromatin formation and transcriptional repression from yeast to human, and we showed that SUMO is required for H3K9me3 deposition downstream of Su(var)2–10 and for TE repression in the Drosophila ovary (Ninova et al., accompanying manuscript). Sov is a suppressor of positional effect variegation and plays a role in recruitment of HP1 to chromatin (Alekseyenko et al., 2014; Jankovics et al., 2018). To characterize the role of Sov in heterochromatin formation, we tested the effect of sov GLKD on the genome-wide distribution of the H3K9me3 mark in the ovary by ChIP-seq. H3K9me3 levels decreased globally upon depletion of sov (Fig. S4A, B), indicating that Sov is required for H3K9me3 deposition, explaining its role in HP1 recruitment. CG30403 encodes a DNA-binding protein that emerged as an HP1 interactor in a proteomic screen (Alekseyenko et al., 2014). We confirmed that CG30403 interacts with HP1 by co-immunoprecipitation from Drosophila S2 cell lysate (Fig. S4C). We were unable to achieve efficient depletion of CG30403 using an shRNA prohibiting further investigation of its function.
Figure 6. H3K9me3 regulates factors involved in heterochromatin formation.

(A) RNA-seq and ChIP-seq coverage tracks for wde, CG30403, smt3 and sov in indicated conditions and orthologous loci in D. virilis ovaries. Annotations are as in Fig 2. (B) Expression levels of wde and sov nascent transcripts (RT-qPCR using intron-specific primers) in indicated conditions. (C) Expression levels of silencing factors (RT-qPCR) in control (shW) ovaries and upon indicated GLKD. (B,C) Dots correspond to independent biological replicates; bars show the mean and SD. Asterisks indicate significant difference compared to the shW samples, *p<0.05, **p<0.01, ***p<0.001; “n.s.”=not significant (Student’s two-tailed t-test (B) or one-way ANOVA followed by Tukey’s posthoc test (C)). (D) Heatmap shows all-versus-all summaries (data in A-C) of fold changes in silencing factor expression upon GLKD of different factors compared to shW controls. Numbers for RNA-seq based data are from DESeq2 and RT-qPCR based data are ratios of mean values from panels B and C; “n.s.” denotes changes that were not statistically significant. §For sov and wde change upon their corresponding mRNA knockdown, data is for nascent transcript levels measured by intronic primers (see main text and B). (E) Model of how euchromatic sensor genes regulate cellular H3K9me3 levels. See also Figures S4,5.
All four genes, wde, sov, CG30403, and smt3, have H3K9me3 peaks that are deposited in a manner dependent on both Su(var)2–10 and SUMO (Fig. 6A). Loss of the H3K9me3 mark upon depletion of Su(var)2–10 and SUMO correlated with transcriptional up-regulation of each gene as evidenced by RNA-seq and Pol II and H3K4me3 ChIP-seq (Fig. 6A). Notably, H3K9me3 peaks for all four genes are conserved for their D. virilis orthologs (Fig. 6A), and wde, smt3 and CG30403 showed no evidence of local TE insertions (from DNA-seq data and genomic PCR, data not shown) or piRNA targeting, suggesting that these genes are direct, TE-independent targets of H3K9me3 deposition through Su(var)2–10/SUMO.
Expression of genes encoding heterochromatin proteins is regulated by a negative feedback loop
The repression of genes encoding heterochromatin proteins by local H3K9me3-rich chromatin islands suggests that their expression might be controlled by a negative feedback loop in which gene activity is repressed by its own product. To test if such negative feedback occurs, we asked if depletion of the gene product affected its own transcription. To this end, we took advantage of the facts that RNAi destroys mRNAs in the cytoplasm causing protein depletion, and that the abundance of nascent (non-spliced) pre-mRNA can by analyzed by RT-PCR to give a proxy of the level of transcription. We found that GLKD of wde and sov led to increases in abundance of their respective pre-mRNAs, confirming that expression of these genes is auto-regulated (Fig. 6B). sov auto-regulation was further evidenced by a decrease in H3K9me3 signal at the sov promoter upon sov GLKD (Fig. 6A). We were unable to test CG30403 and smt3 auto-regulation due to poor knock-down efficiency and lack of utilizable introns, respectively.
Wde, SUMO(smt3), CG30403, and Sov might function independently to mediate heterochromatin formation or may participate in one common pathway that regulates expression of all four genes. To discriminate between these two possibilities, we explored the dependence of these genes on each other as well as on the effector methyltransferase SetDB1. Analysis of RNA-seq data upon GLKD of smt3 showed that wde, CG30403, and sov are up-regulated (Fig. 6A). Expression of CG30403 and wde was increased >2 fold upon GLKD of SetDB1 or wde, and slightly in sov GLKD ovaries (Fig. 6C, D). In contrast, smt3 and sov transcript levels showed modest or no significant increase upon SetDB1 and wde depletion. Taken together, these results indicate that Su(var)2–10, SetDB1/Wde, and Smt3 act in the same pathway to confer H3K9me3 deposition and transcriptional repression at the wde and CG30403 loci. The observation that repression of smt3 and sov is not strongly impacted by depletion of SetDB1 and Wde implies existence of a distinct SUMO/Su(var)2–10/Sov-dependent silencing pathway that does not require SetDB1/Wde.
Discussion
Su(var)2–10 and H3K9me3 play important roles in the regulation of gene expression
H3K9me3 is a conserved hallmark of constitutive heterochromatin from yeast to human and is generally associated with gene repression. We identified Su(var)2–10 as a crucial factor required for deposition of the H3K9me3 mark in large heterochromatic domains of the D. melanogaster genome, such as pericentromeric regions and the 4th chromosome, as well as at discrete islands in euchromatin.
Here we showed that, in addition to effects on TE silencing (Ninova et al., accompanying manuscript), Su(var)2–10 and H3K9me3 influence expression of protein-coding genes. Su(var)2–10-dependent H3K9me3 deposition on TEs impacts expression of genes located in heterochromatin and expression of euchromatic genes adjacent to TE insertions. Su(var)2–10 is also involved in TE-independent H3K9me3 deposition on host genes, which is essential for suppression of ectopic expression of tissue-specific genes, thereby conferring correct cell type identity.
Epigenetic effects of TEs influence host gene expression
About half of the human genome is comprised of TE sequences, and the TE fraction is as high as 90% in several plant species (Lander et al., 2001; Sabot et al., 2005; Schnable et al., 2009). One new TE insertion per generation is estimated to propagate to the offspring (Kazazian, 2004). Somatic TE insertions, although difficult to detect, are likely even more prevalent (Kazazian, 2011). Thus, TE activity is a major source of genetic variation that can occur on a very short time scale. The effects of TEs on the host transcriptome have been the subject of many studies ever since Barbara McClintock identified “control” elements that regulate gene expression before genome compositions were known (McClintock, 1950). TEs can disrupt gene expression by inserting into coding regions or into or close to cis-regulatory sequences (Chuong et al., 2017). TE insertions are not always disruptive: Insertions into non-coding regions can bring new regulatory elements that change gene expression patterns resulting in increased fitness (Feschotte, 2008). Instances of positive selection for TE insertions are well documented in Drosophila (González et al., 2008; Maside et al., 2002; Schlenke and Begun, 2004). TE-derived promoters also drive expression of numerous mouse and human genes, suggesting that TE insertions can be co-opted into gene regulatory pathways (Chuong et al., 2017; Jordan et al., 2003; Nigumann et al., 2002; Peaston et al., 2004).
In addition to changes in the DNA sequence, TE insertions may also introduce local epigenetic effects (Slotkin and Martienssen, 2007). Active TEs are transcriptionally silenced by H3K9 trimethylation and/or DNA methylation. The H3K9me3 mark can spread several kilobases outside the TE region (Lee and Karpen, 2017; Pezic et al., 2014; Sentmanat and Elgin, 2012; Sienski et al., 2012), affecting adjacent cis-regulatory elements of host genes, interfering with their normal expression. TE insertions with high levels of H3K9me3 are strongly selected against, supporting a model that TEs can alter expression of host genes through epigenetic changes (Lee and Karpen, 2017).
The finding that Su(var)2–10 is responsible for deposition of H3K9me3 on TE bodies and flanking sequences allows us to separate the effect of direct damage to cis-regulatory elements from the effect on chromatin. We found evidence that TE insertions can lead to H3K9me3-dependent changes in gene expression, as shown for the jheh3 and frl loci (Fig. 1B). Notably, the BARI insertion at the jheh3 locus was shown to be positively selected in the D. melanogaster population (Gonzalez et al., 2009; González et al., 2008) indicating that Su(var)2–10-dependent epigenetic silencing caused by a TE insertion can be used for beneficial re-wiring of host gene regulatory networks.
Our results suggest that TEs can rewire gene regulatory networks on a short time scale at least in part via effects on chromatin. Euchromatic H3K9me3 peaks due to TE insertions are widespread in Drosophila (Lee, 2015; Lee and Karpen, 2017; Sienski et al., 2012), indicating that TE insertions may be a common cause of gene regulatory variation. New TE insertions during development generate genomic diversity between different cell types in human and mouse with implications for tumorigenesis and brain development (Faulkner and Garcia-Perez, 2017). Future studies are required to elicit the epigenetic effects of somatic TE insertions on gene regulatory networks.
Genes located in heterochromatin require H3K9me3 for proper expression
Heterochromatin domains include nearly 30% of the fly genome (Hoskins et al., 2002, 2007). Although relatively gene poor, heterochromatin hosts several hundred protein-coding genes. Studies of chromosomal rearrangements suggested that heterochromatic localization is required for proper expression of heterochromatic genes (Wakimoto et al., 1990; Yasuhara and Wakimoto, 2006). However, the molecular mechanism of the positive effect of the heterochromatin environment on expression is not fully understood.
Consistent with previous studies (Riddle et al., 2011, 2012), we observed many active genes in H3K9me3-rich heterochromatic regions and found that for many active heterochromatic genes Su(var)2–10-induced H3K9 methylation is not only permissive, but is required for proper expression (Fig. 3, 4).
How can the same chromatin mark lead to repression of genes in euchromatin and activation in heterochromatin? H3K9me3 is present over the gene bodies and regions flanking heterochromatic genes, but depleted at promoters, which instead carry typical active marks such as H3K4me3 and Pol II occupancy. Thus, H3K9me3 over gene bodies appears to be compatible with transcription. H3K9me3 loss upon Su(var)2–10 GLKD correlated with increased levels of intronic RNAs and the appearance of H3K4me2/3 and Pol II signals in introns, indicating up-regulation of spurous transcripts originating from within host-gene introns. One possible source of such transcripts is the activation of TE promoters that are highly abundant within introns and flanking sequences of heterochromatic genes (Fig. 4B, C). We propose that transcription from TE promoters located in introns and flanking sequences interferes with proper gene expression through transcriptional interference (Shearwin et al., 2005; Fig 4D).
H3K9me3 loss also disrupted normal isoform regulation of heterochromatic genes, as we observed both truncated and extended mRNA isoforms with coding potential distinct from the canonical gene mRNA upon depletion of Su(var)2–10 (Fig. 4). Activation of cryptic promoters might disrupt proper gene expression through multiple mechanisms, such as reduction in canonical mRNA output or dominant negative effects of the extended or truncated protein isoforms. Not all heterochromatic genes that lose H3K9me3 upon Su(var)2–10 GLKD show signs of interfering transcripts or cryptic promoters, indicating that H3K9me3 might have other functions in heterochromatic gene activation. For example, compaction of heterochromatin by HP1 might bring distant enhancers of heterochromatic genes into physical proximity of promoters to activate expression. Overall, our results combined with previous studies indicate that genes positioned in heterochromatin require high H3K9me3 levels for proper expression and isoform selection.
H3K9me3 in euchromatin restricts gene expression to correct cell lineages
Discrete Su(var)2–10-dependent H3K9me3 peaks are present at a number of euchromatic genes. Some of these peaks have no TEs in their vicinity and their H3K9me3-based repression is conserved between D. melanogaster and D. virilis - two species that separated more than 45 million years ago and have no common TE insertions (Fig. 5A, B). Expression of many of these TE-independently repressed genes is restricted to specific tissues such testis, digestive system, or central nervous system and loss of H3K9me3 leads to ectopic expression in the female germline. Our finding is in line with a recent report that SetDBI depletion in the female germline was associated with loss of H3K9me3 and mis-expression of male-specific genes (Smolko et al., 2018). H3K9me3, SetDBI and the SUMO pathway were also implicated in lineage-specific gene expression and cell fate commitment in mammals (Becker et al., 2016; Cheloufi et al., 2015; Cossec et al., 2018; Ivanov et al., 2007; Wang et al., 2018; Yang et al., 2015). Collectively, these data suggest that a TE-independent H3K9me3 deposition via the SUMO-SetDB1 pathway plays an evolutionarily conserved role in restricting gene expression to proper cell lineages.
Negative feedback regulation of ‘heterochromatin sensors’ ensures chromatin homeostasis
SUMO- and Su(var)2–10-dependent H3K9me3 repression also regulates several factors involved in heterochromatin formation and maintenance, such as SUMO (smt3), Wde, Sov and CG30403. Wde is the homologue of the mammalian MCAF1/ATF7IP, which is required for nuclear localization and stability of SetDB1 (Koch et al., 2009; Timms et al., 2016) and promotes its methyltransferase activity (Wang et al., 2003). Drosophila Wde also associates with SetDB1 and their germline depletion results in similar phenotype, supporting the role of Wde as a conserved SetDB1 co-factor (Koch et al., 2009; Smolko et al., 2018). Our data in Drosophila and studies in mammals suggest that SUMO is involved in SetDB1/Wde recruitment to its targets (Ninova et al., accompanying manuscript, Ivanov et al., 2007). HP1 is a H3K9me3 reader that is responsible for the structural properties of heterochromatin and also serves as a hub for many other heterochromatin proteins (Canzio et al., 2011; Eissenberg and Elgin, 2014; Hiragami-Hamada et al., 2016; Larson et al., 2017; Strom et al., 2017). Both Sov and CG30403 interact with HP1, and Sov is critical for heterochromatin maintenance (Jankovics et al., 2018, Fig. S4).
The genes encoding Wde, SUMO, Sov, and CG30403 reside in euchromatin and are repressed by local H3K9me3. Unlike tissue-restricted genes, which are often completely repressed by Su(var)2–10 in the female germline, these factors are not fully silenced although they are upregulated upon Su(var)2–10 depletion (Fig. 6A). Our results indicate that these four genes are part of a negative feedback mechanism that controls heterochromatin formation. Negative feedback in biological circuits maintains protein levels within a certain range, providing homeostatic regulation. We propose that SUMO-dependent repression of heterochromatin proteins provides such homeostatic regulation to maintain the proper ratio and boundaries of hetero- and euchromatin. According to our model (Fig. 6E), specific genes, such as wde, act as sensors of overall H3K9me3 level. Insufficient level of H3K9 methylation lead to elevated sensor gene expression due to decreased H3K9me3 at their promoters, which in turn enhances H3K9me3 deposition and heterochromatin formation throughout the genome. Concomitant repression of sensor genes ensurs that H3K9me3 is restricted to proper genomic domains and does not spread to euchromatic regions that should remain active. Inspection of ENCODE data showed that the mammalian homolog of wde, ATF7IP, is decorated by H3K9me3 in some human cell lines (Fig. S5), suggesting that this mode of regulation might be deeply conserved.
A reminiscent negative feedback loop was identified in yeast: the single H3K9 methyltransferase clr4 is suppressed by H3K9me3 to restrict ectopic spreading of silencing chromatin (Wang et al., 2015). In mammals, genes encoding proteins from the KRAB-ZFP family of transcriptional repressors reside in H3K9me3- and HPI-enriched loci (Frietze et al., 2010; O’Geen et al., 2007; Vogel et al., 2006). Thus, auto-regulation of heterochromatin effectors is a conserved mode of chromatin regulation, although the genes involved in the feedback mechanism differ between different organisms. In the future, it will be important to dissect the network architecture of heterochromatin regulation. As heterochromatin formation and maintenance was reported to be disrupted in cancer and during aging, this mechanism might be a promising target of therapeutic interventions.
Molecular mechanism of Su(var)2–10 recruitment to genomic targets
H3K9me3 writer enzymes are targeted to genomic loci by different mechanisms. In the case of TE repression in germ cells, piRNAs bound to nuclear Piwi proteins serve as sequence-specific guides that bind complementary nascent transcripts (LeThomas et al., 2013; Rozhkov et al., 2013; Sienski et al., 2012) and recruit Su(var)2–10, which induces H3K9me3 deposition by SetDB1 (Ninova et al., accompanying manuscript). We found that Su(var)2–10 identifies non-TE targets in a piRNA-independent fashion (Fig. 4A), in agreement with a broader function of Su(var)2–10 in development (Hari et al., 2001). The observation that H3K9me3 peaks at homologous euchromatic genes are also present in the distantly related D. virilis points to a conserved mechanism of H3K9me3 deposition in host-gene regulation.
The molecular mechanism of piRNA-independent recruitment of Su(var)2–10 remains to be explored. Su(var)2–10 has a putative DNA binding SAP domain that might be sufficient for its binding to DNA (Aravind and Koonin, 2000). However, motif enrichment analysis failed to identify a common sequence motif among TE-independent Su(var)2–10 targets (MEME-ChIP), suggesting that different partners might recruit Su(var)2–10 to distinct targets. In mammals, a large family of transcription factors, the KRAB-ZFPs, are responsible for SetDB1 recruitment and H3K9me3 deposition on many different targets, primarily endogenous retroviruses (reviewed in Wolf et al., 2015). Individual members of the KRAB-ZFP family influence distinct targets due to differences in DNA-binding specificities of their zinc-finger DNA-binding domains. Notably, SetDB1 recruitment through KRAB-ZFPs occurs through a SUMO-dependent mechanism (Ivanov et al., 2007). The KRAB-ZFP family is vertebrate-specific, and there are no known proteins in D. melanogaster that can recruit H3K9me3 activity. A preliminary search for direct Su(var)2–10 interactors using a yeast two-hybrid screen identified several proteins with putative DNA-binding domains (Ninova et al., manuscript in preparation). Thus, we propose that analogously to the KRAB-ZFP pathway in mammals, Su(var)2–10 may link DNA-binding proteins to the SetDB1 silencing machinery. Future studies are necessary to identify the proteins that guide Su(var)2–10 to target loci and to elucidate TE-independent recruitment mechanisms of the silencing machinery.
STAR METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katalin Fejes Toth (kft@caltech.edu).
Experimental model and subject details
Fly stocks and husbandry
shRNA-mediated knockdown experiments were performed to target the following genes of interest using the listed stocks obtained from the Bloomington Drosophila Stock Center encoding UASp-driven shRNAs: su(var)2–10 (shSv210, BDSC #32956), piwi (shPiwi, BDSC #33724), wde (shWde, BDSC #33339), CG30403 (BDSC #57286), and white (shWhite, BDSC #33623). For UASp-shSmt3, the anti-Smt3 shRNA sequence based on the TRiP line HMS01540 was ligated into the pValium20 vector (Ni et al. 2011) using T4 DNA ligase from NEB, according to the manufacturer’s instructions and was integrated at the attP2 landing site by BestGene. UASp-shSetDB1 stocks were a gift from Julius Brennecke, and UASp-shSov stocks were generated by Miklos Erdelyi (Jankovics et al., 2018). The expression of all constructs was driven by maternal alpha-tubulin67C-Gal4 (MT-Gal4, BDSC #7063), except for the shSUMO H3K9me3 ChIP-seq samples, where the BDSC #7062 stock encoding alpha-tubulin67C-Gal4 driver was used. All flies were maintained on standard medium at 24 °C and were supplemented with yeast for 2 to 3 days before ovary dissections.
Method details
qRT-PCR and data analysis
Ovaries from 2–5 day-old females were hand dissected. Three biological replicates of 10 pairs of ovaries were used for each condition. RNA was extracted using TRIzol reagent (Invitrogen). Approximately 1 pg RNA was treated with DNase I (Invitrogen), and subsequently used as a template for cDNA synthesis using Super Script III Reverse Transcriptase (Invitrogen). SYBR Green qPCR was performed using MyTaq HS Mix (BioLine) with primers listed in the Key Reources Table. CT values were calculated from technical duplicates or triplicates. Relative gene expression was calculated using Rp49/RpL13 as an endogenous reference. Statisical significance of observed differences was estimated by two-tail Student’s t-test or, in the case of multiple comparisons, one-way ANOVA followed by Tukey’s post-hoc test (TukeyHSD R function).
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Flag (HRP conjugated) | Sigma | Cat#A8592 |
| Rabbit polyclonal anti-GFP | Chen et al., 2016 | |
| IRDye®-conjugated anti-rabbit | LiCor | Cat#925–68071 |
| GFP-Trap® | ChromoTek | Cat#gtma-20 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat#ab8898 |
| Mouse monoclonal anti-RNA Pol II | Abcam | Cat#ab5408 |
| Mouse monoclonal anti-H3K4me2/3 | Abcam | Cat#ab6000 |
| Critical Commercial Assays | ||
| Ribo-Zero™ rRNA Removal Kit | Epicentre/Illumina | Cat#MRZH11124 |
| NEBNext® Ultra™ Directional RNA Library Prep Kit | NEB | Cat#E7760 |
| NEBNext ChIP-Seq Library Prep Master Mix Set | NEB | Cat#E6240 |
| Deposited Data | ||
| shPiwi, shW H3K9me3 ChIP-seq | LeThomas et al. 2013 | |
| Raw data and bigwig files (RNA-seq, ChIP-seq) | This paper | GSE115277, GSE125055 |
| Experimental Models: Organisms/Strains | ||
| maternal alpha-tubulin67C-Gal4 | BDSC #7063 | |
| maternal alpha-tubulin67C-Gal4 | BDSC #7062 | |
| UASp-shSv210–2 | BDSC #32956 | |
| UASp-shSUMO | this study | |
| UASp-shWde | BDSC #33339 | |
| UASp-shWhite | BDSC #33623 | |
| UASp-shSov | Jankovics et al., 2018 | |
| UASp-shSetDB1 | J Brennecke | |
| Oligonucleotides | ||
| Wde (long isoform) F | IDT | AAACGAGTTTGTCGTGTC |
| Wde (long isoform) R | IDT | TGGTCTGGTTTACTCCCATCA |
| Wde (nascent transcript) F | IDT | GAGCAGGGATCCTCATTGAA |
| Wde (nascent transcript) R | IDT | GCTGCCTGTTTCCTCATCTC |
| Sov (all isoforms) F | IDT | AATGTTACCGCCAATGGAAG |
| Sov (all isoforms) R | IDT | TTCCCGTTTTGATTGCTAGG |
| Sov (long isoform) F | IDT | CCTTATGGCCAGCTCCTTCG |
| Sov (long isoform) R | IDT | TTGCCGGCGAATACGATAGA |
| Sov (nascent transcript) F | IDT | GGAATGTGCATCAACGACGA |
| Sov (nascent transcript) R | IDT | AGTCGCAAAAGCTCGTCCAC |
| SUMO (smt3) F | IDT | ACCATCGAGGTTTACCAGCA |
| SUMO (smt3) R | IDT | TGTGTTTTTGCTTTTGTGGTTT |
| CG30403 F | IDT | TGGTTTGCCCCACTTCACG |
| CG30403 R | IDT | GGCGCTTTAGTTGAGGAATTGAT |
| Recombinant DNA | ||
| pValium20 | DRSC/TRiP | |
| Drosophila Gateway Vector collection | DGRC | |
| pENTR™/D-TOPO™ | Invitrogen | Cat #K240020 |
| pActin-HP1-GFP | This study | |
| pActin-Flag-CG30403 | This study | |
| Software and Algorithms | ||
| Bowtie 0.12.17 | Langmead et al., 2009 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.7/ |
| Cutadapt | Marcel 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| UCSC browser | Karolchik et al., 2004 | https://genome.ucsc.edu/ |
| Samtools | Li et al. 2009 | http://samtools.sourceforge.net/ |
| BEDtools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| USCS/BigWig tools | Kent et al., 2010 | http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/ |
| Circos 0.67.7 | Krzywinski et al., 2009 | http://circos.ca/software/ |
| TIDAL | Rahman et al., 2015 | https://github.com/laulabbrandeis/TIDAL |
| eXpress | Roberts and Pachter, 2013) | https://pachterlab.github.io/eXpress/ |
| ballgown | Pertea et al., 2016 | https://github.com/alyssafrazee/ballgown |
| stringtie | Pertea et al., 2016 | https://ccb.jhu.edu/software/stringtie/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| ngs.plot.r | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot |
| HISAT2 | Kim et al., 2015 | https://ccb.jhu.edu/software/hisat2/index.shtml |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| pheatmap | https://cran.r-project.org/web/packages/pheatmap/index.html | |
| Other | ||
| Schneider’s Drosophila Medium | Gibco (Life Technologies) | 21720–024 |
| Fetal Bovine Serum | GEMINI bio-products | 100–106 |
| Penicillin/Streptomycin | Gibco (Life Technologies) | 15140–122 |
Heterochromatic gene definition
Heterochromatic genes were defined as genes residing on chromosome 4, chromosome U, chromosome U extra, chr2RHet, chr2LHet, chr3RHet, chr3LHet, and chrXHet, as well as the cytological borders of heterochromatin on chromosome arms 2R, 2L, 3R, 3L, and X listed in Riddle et al. 2011, dm3 assembly.
RNA-seq and data analysis
RNA was extracted from dissected ovaries of 2–5 day old shW (control), shSu(var)2–10, and shSmt3 flies using TRIzol reagent (Invitrogen). Approximately 1 pg RNA was treated with DNase I (Invitrogen) followed by rRNA depletion using the RiboZero Gold kit (Illumina) supplemented with D. melanogaster rRNA antisense oligonucleotides. rRNA-depleted RNA was used as a starting material for library construction using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs). Two biological replicates per condition were sequenced on Illumina HiSeq 2500 instrument at Millard and Muriel Jacobs Genetics and Genomics Laboratory at Caltech yielding 15–20 million single-end 50-bp reads. Reads were first filtered against D. melanogaster rRNA sequences (Stage and Eickbush, 2007) and 5S rRNA in dm3 annotated by RepeatMasker available via the UCSC Genome Browser, allowing three mismatches using bowtie v0.12.17 (Langmead et al., 2009). Less than 10% of all reads in each condition aligned to rRNA. The remaining reads were then aligned to the D. melanogaster genome (dm3) allowing up to two mismatches and a single mapping position bowtie v0.12.17 (v 2, −m 1).
For differential expression analysis, read counts corresponding to the exonic regions of protein-coding genes were calculated using an in-house python script. Genes with fewer than 10 reads in all samples were excluded from downstream analysis. For TE expression, reads were aligned to the D. melanogaster genome (dm3) allowing 10,000 mapping positions and zero mismatches. TE annotations were obtained from RepeatMasker tables available on the UCSC browser (Karolchik et al., 2004). Read counts for individual TE families were calculated using a custom Python script correcting for multiple mapping positions. The resulting gene and TE count data was analyzed using the DESeq2 R package using default settings (Love et al., 2014). Significantly up- and down-regulated genes were considered to be those displaying >2-fold change between control and Su(var)2–10 GLKD libraries with an adjusted p-value <0.05. We also quantified gene expression by alignment to the reference transcriptome (dm3) followed by read count estimation by eXpress (Roberts and Pachter, 2013). UCSC genome browser tracks were generated from the uniquely mapped read alignments using the UCSC utilities (Kent et al., 2010), using the total numbers of uniquely mapped reads to the genome per million as a normalization factor. For isoform and novel transcript analysis, reads were aligned to the D. melanogaster genome using hisat2 (v2.1.0), and de novo transcript assembly was performed using stringtie (v1.3.4d) according to published guidelines (Pertea et al., 2016). Structure and expression levels of different isoforms were visualized using the ballgown R package (Frazee et al., 2015).
Gene proximity to TE definition
Euchromatic genes were classified as TE-independent if the following criteria were met: no non-reference TE insertion (see below) within 1 kb of gene ends and no more than 10% and 1000 bp of the intronic sequence and 500 bp of the 10-kb flaking regions sharing homology to TEs as determined by RepeatMasker. To test if piRNAs mapped within gene loci, we aligned 23–29-nt reads from small RNA-seq libraries from mtGal4>UASp-shW ovaries to the genome allowing multiple mapping positions and calculated the number of reads that aligned within gene bodies and 1 −kb flanking regions.
ChIP-seq and data analysis
ChIP-seq experiments were performed as described previously (Ninova et al., accompanying manuscript) with the following antibodies: anti-H3K9me3 (Abcam, ab8898), anti-RNA Pol II (Abcam, ab5408), and anti-H3K4me2/3 (Abcam, ab6000). ChIP-seq library construction was performed using the NEBNext ChIP-Seq Library Prep Master Mix Set (NEB). Libraries were sequenced on the Illumina HiSeq 2000/2500 platform, generating single-end 49-bp or 50-bp reads. Reads were aligned to the D. melanogaster genome (dm3) allowing up to two mismatches and single mapping position using bowtie v0.12.17 (−v 2, −m 1). In order to determine H3K9me3-rich regions present across different fly strains, we processed with the same mapping parameteres previously published H3K9me3 data from the ovaries of other D. melanogaster strains including nanos-gal4>dsWhite (GSE71374, Yu et al., 2015), nanos-gal4>dsPanx (GSE71374, Yu et al. 2015), and DGRC#204406 heterozygous strain (GSE46009, Muerdter et al., 2013).
UCSC genome browser tracks were generated as for RNA-seq tracks. H3K9me3 coverage heatmaps and average ChIP profiles on gene bodies were performed using the ngs.plot R package (Shen et al., 2014), using input libraries for normalization. For genome-wide H3K9me3 enrichment, the dm3 genome was partitioned into 5-kb intervals. Interval coverage was calculated as the ratio of RPKM-normalized reads from IP libraries to input libraries using custom python and R scripts. Low coverage intervals with fewer than 1 RPKM in input libraries were excluded from the analysis. H3K9me3-enriched 5-kb genomic intervals were defined as intervals that had H3K9me3 ChIP/Input enrichment ratio >2 in both replicate control ovaries (shW), as well as >1.5 enrichment in control ovaries of the additional analyzed datasets (shW control of shSUMO, nanos-gal4>dsWhite from GSE71374, DGRC#204406 heterozygous from GSE46009). Circular plots were generated using Circos 0.67.7 (Krzywinski et al., 2009). H3K9me3 enrichment over gene bodies was calculated as the ratio of the RPKM-normalized ChIP and input read coverage within annotated gene ends. H3K9me3 peaks were called using macs14 using the broad peak calling protocol with the parameters (Feng et al., 2012) –nomodel, –shiftsize 73, –B, –S, and –pvalue 1e-3 using reads uniquely aligned to the dm3 D. melanogaster genome assembly. Peaks consistent between strains (Fig. 5A) were considered those detected independently in at least five of the seven H3K9me3 ChIP-seq data sets analyzed (four biological replicates of control UASp>shW libraries, two replicates of nanos-gal4>dsWhite from GSE71374, and one replicate of DGRC#204406 from GSE46009).
H3K9me3 peak conservation in D. virilis
H3K9me3 ChIP-seq data from D. virilis ovaries was previously published (LeThomas et al., 2014). Libraries were processed using the same pipeline as for D. melanogaster, using the dvir-r-1.06 genome assembly version from FlyBase (St. Pierre et al., 2014). Only H3K9me3 peaks detectable in both biological replicates were considered. Orthologous genes between D. melanogaster and D. virilis were retrieved from FlyMine (Lyne et al., 2007). Genic H3K9me3 peaks of D. melanogaster genes were considered conserved if present within 2 kb of the corresponding D. virilis homolog.
Non-reference TE insertions
Non-reference insertions were annotated using the TIDAL pipeline (Rahman et al., 2015) with default parameters using merged reads that do not map to the reference genome (allowing two mismatches) from all experiments involving DNA sequencing (DNA from Input and ChIPs) from each strain (mtGAL4>shW and mtGAL4>shSu(var)2–10).
Co-immunoprecipitation
Expression vectors encoding GFP-fusion HP1 protein and Flag-fusion CG30403 proteins under the control of the actin promoter were cloned into entry cDNA clones and then transferred into pAWG or pAFW destination vectors from the Drosophila Gateway Vector collection using the Gateway system. Vectors were co-transfected into S2 cells, cultured in Schneider’s Drosophila Medium containing 10% heat-inactivated FBS and 1X penicillin-streptomycin with the TransIT-LT1 reagent (Mirus). Flag-CG30403 only (no bait) transfected cells were used as negative control. At 24–48 hours post transfection cells were harvested and lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.2% NP-40, 0.2% Triton-X, 5% glycerol) supplemented with protease inhibitor cocktail (Roche). Lysates were incubated with GFP-Trap magnetic agarose beads (Chromotek) for 1–2 hours at 4 °C with end-to-end rotation. After incubation, the beads were washed five times with 500 μl wash buffer (0.1% NP40, 20 mM Tris, pH 7.4, 150 mM NaCl) containing protease inhibitor. Proteins were detected by Western blot using HRP-conjugated mouse anti-FLAG (Sigma, A8592) and rabbit polyclonal anti-GFP (Chen et al., 2016), followed by IRDye anti-rabbit secondary antibody (Li-cor 925–32211).
Data and software availability
H3K9me3 ChIP-seq data from shPiwi and corresponding shW control lines were previously published (LeThomas et al., 2013). All RNA-seq and ChIP-seq data generated for this study and for the accompanying manuscript (Ninova et al, accompanying manuscript) were deposited into the Gene Expression Omnibus (GEO) database under accession GSE115277. H3K9me3 ChIP-seq data for shSov and respective controls was deposited to GEO under accession GSE125055.
Supplementary Material
Highlights.
Heterochromatic gene expression requires H3K9me3 to suppress spurious transcription
Deposition of H3K9me3 via Su(var)2–10 ensures tissue-specific gene expression
Negative feedback regulation controls expression of several heterochromatin proteins
Acknowledgements
We thank members of the Fejes Toth and Aravin labs for discussion. We are grateful to the Bloomington Stock Center and Julius Brennecke for providing flies. We thank Igor Antoshechkin (Caltech) for help with sequencing. This work was supported by grants from the National Institutes of Health (R01 GM097363), the Ministry of Education and Science of Russian Federation (14.W03.31.0007) and by the Packard Fellowship Awards to A.A.A., and the National Institutes of Health (R01GM110217) and Ellison Medical Foundation Awards to K.F.T.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alekseyenko AA, Gorchakov AA, Zee BM, Fuchs SM, Kharchenko PV, and Kuroda MI (2014). Heterochromatin-associated interactions of Drosophila HP1a with dADDI, HIPP1, and repetitive RNAs. Genes Dev. 28, 1445–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, and Madhani HD (2018). Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol 19, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, and Koonin EV (2000). SAP - A putative DNA-binding motif involved in chromosomal organization. Trends Biochem. Sci. 25, 112–114. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, and Kouzarides T (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, and Kouzarides T (2001). Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124. [DOI] [PubMed] [Google Scholar]
- Becker JS, Nicetto D, and Zaret KS (2016). H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 32, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D, Chang EY, Shankar S, Kuchenbecker KM, Simon MD, Madhani HD, Narlikar GJ, and Al-Sady B (2011). Chromodomain-Mediated Oligomerization of HP1 Suggests a Nucleosome-Bridging Mechanism for Heterochromatin Assembly. Mol. Cell 41, 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Elling U, Hopfgartner B, Jung YL, Murn J, Ninova M, Hubmann M, Badeaux AI, Ang CE, Tenen D, et al. (2015). The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-CA, Stuwe E, Luo Y, Ninova M, Le Thomas A, Rozhavskaya E, Li S, Vempati S, Laver JD, Patel DJ, et al. (2016). Cutoff Suppresses RNA Polymerase II Termination to Ensure Expression of piRNA Precursors. Mol. Cell 63, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, and Feschotte C (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet 18, 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossec J-C, Theurillat I, Chica C, Búa Aguín S, Gaume X, Andrieux A, Iturbide A, Jouvion G, Li H, Bossis G, et al. (2018). SUMO Safeguards Somatic and Pluripotent Cell Identities by Enforcing Distinct Chromatin States. Cell Stem Cell 23, 742–757.e8. [DOI] [PubMed] [Google Scholar]
- Eberl DF, Duyf BJ, and Hilliker AJ (1993). The role of heterochromatin in the expression of a heterochromatic gene, the rolled locus of Drosophila melanogaster. Genetics 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg JC, and Elgin SCR (2014). HP1a: a structural chromosomal protein regulating transcription. Trends Genet. 30, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner GJ, and Garcia-Perez JL (2017). L1 Mosaicism in Mammals: Extent, Effects, and Evolution. Trends Genet. 33, 802–816. [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C (2008). Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet 9, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, and Leek JT (2015). Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol 33, 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze S, O’Geen H, Blahnik KR, Jin VX, and Farnham PJ (2010). ZNF274 recruits the histone methyltransferase SETDB1 to the 39 ends of ZNF genes. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti M, and Pimpinelli S (1992). Functional Elements in Drosophila Melanogaster Heterochromatin. Annu. Rev. Genet 26, 239–276. [DOI] [PubMed] [Google Scholar]
- Gonzalez J, Macpherson JM, and Petrov DA (2009). A Recent Adaptive Transposable Element Insertion Near Highly Conserved Developmental Loci in Drosophila melanogaster. Mol. Biol. Evol. 26, 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J, Lenkov K, Lipatov M, Macpherson JM, and Petrov DA (2008). High rate of recent transposable element-induced adaptation in Drosophila melanogaster. PLoS Biol. 6, e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall IM (2002). Establishment and Maintenance of a Heterochromatin Domain. Science 297, 2232–2237. [DOI] [PubMed] [Google Scholar]
- Hari KL, Hari KL, Cook KR, Cook KR, Karpen GH, and Karpen GH (2001). The Drosophila Su(var)2–10 locus regulates chromosome structure and function and encodes a member of the PIAS protein family. Genes Dev. 15, 1334–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, and Kumar S (2009). The timetree of life. Oxford Biol. [Google Scholar]
- Heitz E (1928). Das Heterochromatin der Moose. Jahrb Wiss Bot 762–818. [Google Scholar]
- Henikoff S (2000). Heterochromatin function in complex genomes. Biochim. Biophys. Acta 1470, O1–8. [DOI] [PubMed] [Google Scholar]
- Hiragami-Hamada K, Soeroes S, Nikolov M, Wilkins B, Kreuz S, Chen C, De La Rosa-Velázquez IA, Zenn HM, Kost N, Pohl W, et al. (2016). Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun 7, 11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Smith CD, Carlson JW, Carvalho AB, Halpern A, Kaminker JS, Kennedy C, Mungall CJ, Sullivan BA, Sutton GG, et al. (2002). Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol. 3, RESEARCH0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, et al. (2007). Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science 316, 1625–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, et al. (2007). PHD Domain-Mediated E3 Ligase Activity Directs Intramolecular Sumoylation of an Adjacent Bromodomain Required for Gene Silencing. Mol. Cell 28, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis Cd., and Khorasanizadeh S (2001). Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 20, 5232–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovics F, Bence M, Sinka R, Faragó A, Bodai L, Pettko-Szandtner A, Ibrahim K, Takacs Z, Szarka-Kovacs AB, and Erdelyi M (2018). Drosophila small ovary gene is required for transposon silencing and heterochromatin organisation and ensures germline stem cell maintenance and differentiation. Development 145. [DOI] [PubMed] [Google Scholar]
- Janssen A, Colmenares SU, and Karpen GH (2018). Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol 34, 265–288. [DOI] [PubMed] [Google Scholar]
- Jia S (2004). RNAi-Independent Heterochromatin Nucleation by the Stress-Activated ATF/CREB Family Proteins. Science 304, 1971–1976. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Glazko GV, and Koonin EV (2003). Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 19, 68–72. [DOI] [PubMed] [Google Scholar]
- Kanoh J, Sadaie M, Urano T, and Ishikawa F (2005). Telomere Binding Protein Taz1 Establishes Swi6 Heterochromatin Independently of RNAi at Telomeres. Curr. Biol. 15, 1808–1819. [DOI] [PubMed] [Google Scholar]
- Karimi MMM, Goyal P, Maksakova IAA, Bilenky M, Leung D, Tang JXX, Shinkai Y, Mager DLL, Jones S, Hirst M, et al. (2011). DNA Methylation and SETDB1/H3K9me3 Regulate Predominantly Distinct Sets of Genes, Retroelements, and Chimeric Transcripts in mESCs. Cell Stem Cell 8, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, and Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH (2004). Mobile elements: drivers of genome evolution. Science 303, 1626–1632. [DOI] [PubMed] [Google Scholar]
- Kazazian HH (2011). Mobile DNA transposition in somatic cells. BMC Biol. 9, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CM, Honemann-Capito M, Egger-Adam D, and Wodarz A (2009). Windei, the Drosophila homolog of mAM/MCAF1, is an essential cofactor of the H3K9 methyl transferase dSETDB1/eggless in germ line development. PLoS Genet. 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: An information aesthetic for comparative genomics. Genome Res. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, and Jenuwein T (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DnA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nat. 2017 5477662 547, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YCG (2015). The Role of piRNA-Mediated Epigenetic Silencing in the Population Dynamics of Transposable Elements in Drosophila melanogaster. PLOS Genet. 11, e1005269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YCG, and Karpen GH (2017). Pervasive epigenetic effects of drosophila euchromatic transposable elements impact their evolution. Elife 6, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeThomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, and Tóth KF (2013). Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 27, 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeThomas A, Marinov GK, and Aravin AA (2014). A Transgenerational Process Defines piRNA Biogenesis in Drosophila virilis. Cell Rep. 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu BY, Bishop CP, and Eissenberg JC (1996). Developmental timing and tissue specificity of heterochromatin-mediated silencing. EMbO J. 15, 1323–1332. [PMC free article] [PubMed] [Google Scholar]
- Lu BY, Emtage PCR, Duyf BJ, Hilliker AJ, and Eissenberg JC (2000). Heterochromatin Protein 1 Is Required for the Normal Expression of Two Heterochromatin Genes in Drosophila. Genetics 155, 699 LP–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyne R, Smith R, Rutherford K, Wakeling M, Varley A, Guillier F, Janssens H, Ji W, Mclaren P, North P, et al. (2007). FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biol. 8, R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JHA, O’Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, and Jenuwein T (2005). The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 24, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maside X, Bartolomé C, and Charlesworth B (2002). S-element Insertions Are Associated with the Evolution of the Hsp70 Genes in Drosophila melanogaster. Curr. Biol 12, 1686–1691. [DOI] [PubMed] [Google Scholar]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, and Shinkai Y (2010). Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931. [DOI] [PubMed] [Google Scholar]
- McClintock B (1950). The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. U. S. A 36, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Bortolamiol-Becet D, Flynt AS, Gronau I, Siepel A, and Lai EC (2014). Adaptive evolution of testis-specific, recently evolved, clustered miRNAs in Drosophila. RNA 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Guzzardo PM, Gillis J, Luo Y, Yu Y, Chen C, Fekete R, and Hannon GJ (2013). A genome-wide RNAi screen draws a genetic framework for transposon control and primary piRNA biogenesis in drosophila. Mol. Cell 50, 736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J. -i. (2001). Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Nigumann P, Redik K, Matlik K, and Speek M (2002). Many Human Genes Are Transcribed from the Antisense Promoter of L1 Retrotransposon Genomics 79, 628–634. [DOI] [PubMed] [Google Scholar]
- O’Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, Green R, and Farnham PJ (2007). Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet. 3, 0916–0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, and Knowles BB (2004). Retrotransposons Regulate Host Genes in Mouse Oocytes and Preimplantation Embryos. Dev. Cell 7, 597–606. [DOI] [PubMed] [Google Scholar]
- Peng JC, and Karpen GH (2009). Heterochromatic Genome Stability Requires Regulators of Histone H3 K9 Methylation. PLoS Genet. 5, e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, and Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc 11, 1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezic D, Manakov SA, Sachidanandam R, Castro-diaz N, Ecco G, Coluccio A, and Aravin AA (2014). piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev. 28, 1410–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Pierre SE, Ponting L, Stefancsik R, McQuilton P, and Consortium, the F. (2014). FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42, D780–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman R, Chirn G-W, Kanodia A, Sytnikova YA, Brembs B, Bergman CM, and Lau NC (2015). Unique transposon landscapes are pervasive across Drosophila melanogaster genomes. Nucleic Acids Res. 43, 10655–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun Z-W, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. [DOI] [PubMed] [Google Scholar]
- Reuter G, and Wolff I (1981). Isolation of dominant suppressor mutations for position-effect variegation in Drosophila melanogaster. Mol. Gen. Genet. MGG 182, 516–519. [DOI] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder-Basso D, et al. (2011). Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 21, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Jung YL, Gu T, Alekseyenko AA, Asker D, Gui H, Kharchenko PV, Minoda A, Plachetka A, Schwartz YB, et al. (2012). Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain. PLoS Genet. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, and Pachter L (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 10, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, and Hannon GJ (2013). Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev. 27, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabot F, Guyot R, Wicker T, Chantret N, Laubin B, Chalhoub B, Leroy P, Sourdille P, and Bernard M (2005). Updating of transposable element annotations from large wheat genomic sequences reveals diverse activities and gene associations. Mol. Genet. Genomics 274, 119–130. [DOI] [PubMed] [Google Scholar]
- Schlenke TA, and Begun DJ (2004). Strong selective sweep associated with a transposon insertion in Drosophila simulans. Proc. Natl. Acad. Sci. U. S. A 101, 1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. (2009). The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 326, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, Rea S, Jenuwein T, Dorn R, and Reuter G (2002). Central role of Drosophila SU(VAR)3–9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 21, 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon H-M, Di Croce L, and Cavalli G (2017). Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57. [DOI] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, and Rauscher Iii FJ (2002). SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentmanat MF, and Elgin SCR (2012). Ectopic assembly of heterochromatin in Drosophila melanogaster triggered by transposable elements. Proc. Natl. Acad. Sci. U. S. A 109, 14104–14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearwin KE, Callen BP, and Egan JB (2005). Transcriptional interference – a crash course. Trends Genet. 21, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Donertas D, and Brennecke J (2012). Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151, 964–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, and Martienssen R (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285. [DOI] [PubMed] [Google Scholar]
- Smolko AE, Shapiro-Kulnane L, and Salz HK (2018). The H3K9 methyltransferase SETDB1 maintains female identity in Drosophila germ cells. Nat. Commun 9, 4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stage DE, and Eickbush TH (2007). Sequence variation within the rRNA gene loci of 12 Drosophila species. Genome Res. 17, 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, and Lehner PJ (2016). ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex. Cell Rep. 17, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SIS, and Moazed D (2004). RNAi-Mediated Targeting of Heterochromatin by the RITS Complex. Science 303, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel MJ, Guelen L, de Wit E, Peric-Hupkes D, Loden M, Talhout W, Feenstra M, Abbas B, Classen A-K, and van Steensel B (2006). Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 16, 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SIS, and Martienssen RA (2002). Regulation of Heterochromatic Silencing and Histone H3 Lysine-9 Methylation by RNAi. Science 297, 1833 LP–1837. [DOI] [PubMed] [Google Scholar]
- Wakimoto BT, Hearn MG, Berloco M, Wakimoto BT, Eissenberg JC, Henikoff S, Rosen C, and Dorsett D (1990). The effects of chromosome rearrangements on the expression of heterochromatic genes in chromosome 2L of Drosophila melanogaster. Genetics 125, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu X, Gao Y, Yang L, Li C, Liu W, Chen C, Kou X, Zhao Y, Chen J, et al. (2018). Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 20, 620–631. [DOI] [PubMed] [Google Scholar]
- Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, and Zhang Y (2003). mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 12, 475–487. [DOI] [PubMed] [Google Scholar]
- Wang J, Reddy BD, and Jia S (2015). Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Duan H, Bejarano F, Okamura K, Fabian L, Brill JA, Bortolamiol-Becet D, Martin R, Ruby JG, and Lai EC (2015). Adaptive Regulation of Testis Gene Expression and Control of Male Fertility by the Drosophila Hairpin RNA Pathway. Mol. Cell 57, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Greenberg D, and Macfarlan TS (2015). Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mob. DNA 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang BX, El Farran CA, Guo HC, Yu T, Fang HT, Wang HF, Schlesinger S, Seah YFS, Goh GYL, Neo SP, et al. (2015). Systematic Identification of Factors for Provirus Silencing in Embryonic Stem Cells. Cell 163, 230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara JC, and Wakimoto BT (2006). Oxymoron no more: the expanding world of heterochromatic genes. Trends Genet. 22, 330–338. [DOI] [PubMed] [Google Scholar]
- Yu Y, Gu J, Jin Y, Luo Y, Preall JB, Ma J, Czech B, and Hannon GJ (2015). Panoramix enforces piRNA-dependent cotranscriptional silencing. Science 350, 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137–R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
H3K9me3 ChIP-seq data from shPiwi and corresponding shW control lines were previously published (LeThomas et al., 2013). All RNA-seq and ChIP-seq data generated for this study and for the accompanying manuscript (Ninova et al, accompanying manuscript) were deposited into the Gene Expression Omnibus (GEO) database under accession GSE115277. H3K9me3 ChIP-seq data for shSov and respective controls was deposited to GEO under accession GSE125055.