

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer lacking specific biomarkers that can be correlated to disease onset, promotion and progression. To assess whether tumor cell electrophysiology may serve as a marker for PDAC tumorigenicity, we use multi-frequency impedance cytometry at high throughput (~350 cells/s) to measure the electrical phenotype of single PDAC tumor cells from xenografts, which are derived from primary pancreatic tumors versus those from liver metastases of different patients. A novel phase contrast metric based on variations in the high and low frequency impedance phase responses that is related to electrophysiology of the cell interior is found to be systematically altered as a function of tumorigenicity. PDAC cells of higher tumorigenicity exhibited lowered interior conductivity and enhanced permittivity, which is validated by the dielectrophoresis on the respective cell types. Using genetic analysis, we suggest the role of dysregulated Na+ transport and removal of Ca2+ ions from the cytoplasm on key oncogenic KRAS-driven processes that may be responsible for lowering of the interior cell conductivity. We envision that impedance cytometry can serve as a tool to quantify phenotypic heterogeneity for rapidly stratifying tumorigenicity. It can also aid in protocols for dielectrophoretic isolation of cells with a particular phenotype for prognostic studies on patient survival and to tailor therapy selection to specific patients.
Keywords: Microfluidics, Impedance Cytometry, Dielectrophoresis, Cancer, Pancreatic Ductal Adenocarcinoma
Graphical Abstract
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common malignant cancer of the pancreas and is among the leading causes of cancer-related deaths [1]. The disease initially manifests as an epithelial tumor which arises from the cells lining the ducts of the pancreas, but most fatalities occur due to metastases [2]. Given that the 5-year survival rate for PDAC is 8% [3] and disease incidence is predicted to increase by 55% in the next 20 years [4], PDAC is a growing health concern, especially due to the lack of specific biomarkers which correlate to the tumorigenicity of PDAC cells [5]. While a significant majority of PDAC patients (~95%) exhibit KRAS mutations [6] that activate oncogenic proteins [7], tumorigenicity cannot be assessed solely based on specific mutations. Since the mechanisms by which different KRAS mutations influence the overall cell structure and consequent tumor aggressiveness are poorly understood, it is of great interest to explore phenotypic differences between different PDAC tumor cell types. Using patient-derived PDAC cells of varying tumorigenicity that are expanded in mice as xenografts, we compare PDAC cells obtained from metastatic versus primary tumor sites of KRAS mutant genotype, as well as those from primary tumors of KRAS wild-type genotype, to explore whether subcellular electrophysiology can be used as the phenotype to differentiate between the respective cell types. Metastatic PDAC cells exhibit the greatest level of tumorigenicity, while tumorigenic primary PDAC cells are predominantly of KRAS mutant rather than of KRAS wild-type genotype [6].
Due to the heterogeneity of tumor cells [8] and the lack of surface markers to stratify tumorigenicity [9], there is much interest in biophysical characterization of single tumor cells to yield phenotypic markers that correlate with cancer onset and progression [10], [11]. Label-free methods based on cell size and deformability have been especially useful in this regard [12, 13], but current methods lack the ability for subcellular resolution of the phenotypes, which are altered during tumor development [14]. Cell electrophysiology represents an aggregate of biophysical properties that are influenced by genomic and micro-environmental factors, both of which play critical roles in tumor development. Electrophysiology is not only sensitive to whole-cell characteristics, such as size and shape, but also to subcellular features, such as plasma membrane structure, organelle structure in the cytoplasm and nucleus size. Due to the substantial differences in conductivity and permittivity of these subcellular components, the frequency spectra of impedance of single-cells can yield spatially-resolved information. However, to obtain clinically relevant information, single-cell measurements at truly high throughput levels (i.e., thousands of cells) are necessary for enabling these subtle phenotypic distinctions, while allowing for statistically relevant cytometry of the relative variations within the population of interest.
Single-cell impedance cytometry is a label-free microfluidic technique wherein the electrical impedance of single cells flowing at high throughput (300 – 400 cells/s) past microelectrodes is detected based on the disruption in current flow, under an AC electric field over a range of frequencies (0.5–50 MHz)[15]. While the impedance at low frequencies (~0.5 MHz) is determined by cell volume, the impedance magnitude and phase at successively higher frequencies offer information on cell electrophysiology, such as its membrane capacitance (2–5 MHz) and cytoplasmic conductivity (>10 MHz). Impedance cytometry has been used previously to analyze various cell types, including stem cells [16], waterborne parasites [17] or parasite-infected red blood cells [18]. In prior work, following immuno-capture of circulating tumor cells in blood that originate from PDAC tissues [19],[20], the released tumor cells were distinguished from leukocytes that express similar markers using low frequency (40 kHz) impedance measurements, which are based on size rather than electrophysiology differences. In other work, whole-body impedance measurements have been used to predict disease outcome on pancreatic cancer patients [21]. However, no prior PDAC study has focused on elucidating phenotypic distinctions between tumor cell types, using single-cell methods that are sensitive to heterogeneity. While impedance responses measure alterations in the frequency response of current due to presence of cells between the electrodes, dielectrophoresis (DEP) is based on translation of polarized cells within a spatially non-uniform electric field [22–24], either towards the field by positive DEP (pDEP) or away from the field by negative DEP (nDEP). In prior work, DEP responses have been used to measure differences in tumor cells based on membrane capacitance [25], quantify mitochondrial-phenotype induced cytoplasmic conductivity variations [26] and identify exosomes derived from highly invasive pancreatic tumor cells based on their enhanced conductance levels due to membrane fluidity [27]. Electrophysiology-related differences have also formed the basis for microfluidic selection of tumor cells [28] and bacterial cells [29],[30],[31].
We report on high-throughput multi-frequency single-cell impedance cytometry applied to different patient-derived PDAC tumor cell types, for enabling electrophysiology-based phenotypic distinctions between cells with known differences in tumorigenicity. Single-cell cytometry is needed to obtain sub-cellular sensitivity for quantifying the electrophysiology distinctions, whereas high-throughput measurements (>5000 cells per population) are needed to ensure statistically relevant variations between the respective populations. Based on a normalized impedance phase metric that depends on the cell permittivity to conductivity ratio and its consistence based on DEP spectral measurements, we show systematic differences in electrophysiology of the cell interior as a function of tumorigenicity of the cell population and use genetic analysis to explore some of the molecular processes that likely contribute to the measured subcellular electrophysiological differences. These systematic electrophysiology-based differences could provide a label-free marker for stratification of PDAC cells based on their tumorigenicity and enable strategies for their selective dielectrophoretic isolation from heterogeneous samples, which would eventually aid in prognostic studies on patient survival and therapy selection.
2. Materials and methods
2.1. Microfabrication
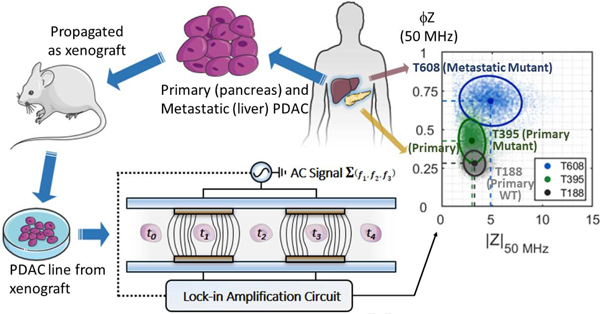
The electrode design was defined on two 4” glass wafers (D263, Schott) using a photomask (Photosciences) to pattern AZ 1505 (Microchem), before a Ti/ Au (20 nm/ 200 nm) layer was deposited by e-beam evaporation (CHA-50, CHA). Upon lift-off in acetone with ultrasonication, SU-8 3050 (Microchem) was spun onto one of the identical substrates to give a film thickness of ~30 μm for use as the microchannel layer. Upon UV exposure and resist development, the two wafers (one containing the SU8 structures) with facing gold microelectrodes ~45 μm wide (with a spacing of ~15 μm) were aligned and brought into contact (EVG 620, EV Group). Thermal bonding of the wafer complex was performed at 180 °C using a bonding pressure of 10 kN for 1 h with vacuum established at 5900 mbar (EVG 510, EV Group). The bonded wafer complex was cut into individual dies using a dicing saw (DAD3220, Disco). Fluidic access holes were drilled using a CO2 laser (50 W at 80% power and 40% speed, VSL 3.5, Versa). A finalized device can be seen in Figure 1a, with key characteristics highlighted. Chips were assembled in a 3D printed holder which housed all fluidic and electrical connections.
Fig 1:

(a) Schematic of the experimental setup and photo of a finalized impedance cytometry device. Some of the key characteristics are highlighted. Insert shows a schematic of the device detection region. A custom 3D printed holder houses the device and all fluidic and electrical connections. The inlet is connected to a syringe pump and the outlet is connected to a waste syringe. An impedance spectroscope (HF2IS, Zurich Instruments) outputs the AC signal and a custom printed circuit board (PCB) connects the output signal to the top detection electrodes. The current flowing through the bottom detection electrodes is transmitted via another custom PCB to the signal inputs of the impedance spectroscope. (b) From PDAC patient to microfluidics via the xenograft model. (1) Tumor samples collected from the pancreas or liver of PDAC patients through surgical resection and (2) implanted and propagated in immunocompromised mice as a xenograft (PDX). (3) PDAC cell lines were then established from surgically resected PDX and (4) maintained in RPMI1640 at 37 °C. (5) PDAC samples were then aspirated, washed and resuspended in impedance cytometry buffer. (c) Schematic representation of the impedance cytometer detection region operation. PDAC tumor cells in PBS flow through a microchannel which includes the two sets of detection electrodes. PDAC tumor cells in PBS flow through a microchannel which includes two sets of facing microelectrodes. Voltages at three discrete frequencies (ƒ1, ƒ2 and ƒ3) are applied to the top electrodes and the differential current at the bottom electrodes goes through a current amplifier. The response at each frequency is demodulated using lock-in amplification to give real and imaginary impedance data at discrete frequencies. The steps of the idealized measurement are annotated t0 to t4.
2.2. Derivation of patient-derived xenografts and cell lines
PDAC tumor samples MAD 12–188, 09–366, 12–395, 14–449, 08–608 and 08–738 were generated from remnant human tumor surgical pathology specimens collected in collaboration with the University of Virginia Biorepository and Tissue Research Facility and with the approval of the University of Virginia Institutional Review Board for Health Sciences Research following written informed consent from each patient. Tumors were propagated orthotopically on the pancreata of immunocompromised mice. Tumor growth characteristics were measured, samples were collected for genotyping, and xenograft lines were established, as previously described [32, 33]. Cells were transduced with firefly luciferase lentivirus (KeraFAST), selected using puromycin and maintained in RPMI1640 with 10% FBS and 2 mM glutamine. Fresh cell aliquots were thawed, propagated, and used for experiments.
2.3. Sample preparation
Confluent cells in Dulbecco’s Modified Eagle Medium (DMEM) [Gibco, USA] were aspirated, washed in PBS and exposed to 0.5% trypsin for 5 mins at 37 °C. Cells were resuspended in 5 mL DMEM and centrifuged at 300 g for 10 mins. DMEM was aspirated, the cell pellet was resuspended in 1x PBS, 500 mM EDTA, and 0.5% Bovine Serum Albumin and filtered through a 100 μm cell strainer. Cells were then counted with a hemocytometer and diluted to a concentration of ~2 x 105 mL−1. Reference particles (7 μm diameter) at a concentration of ~1.2 x 105 were added to the buffer to account for system non-linearities and inter-measurement variability. An aliquot containing cells suspended for 2 mins 1:1 in a solution of trypan blue (Fisher Scientific) and were counted (100 cells) to ensure that sample viability was ≥ 90%. A total of n = 9 samples were measured from three separate batches of each cell type.
2.4. Impedance cytometry
Sample was introduced to the microfluidic channel (~30 μm tall by ~60 μm wide) device (set-up in Fig. 1a) at a flow rate of 100 μL min−1 (neMESYS, Cetoni). Sinusoidal voltages at three discrete frequencies were applied to the top electrodes using a digital impedance spectroscope (HF2IS, Zurich Instruments) – Fig 1. A voltage of 2 Vpp was applied at each signal frequency. The reference frequency was applied at 18.3 MHz, the probe frequency was swept over 24 discrete frequencies between 250 kHz – 50 MHz, and the third frequency was applied at 500 kHz. The current flowing through the bottom electrodes was converted to voltage using a current amplifier (HF2TA, Zurich Instruments) which had a gain factor of 1000. A sample-rate of 115,000 Samples s−1 was used to for data acquisition. Lock-in amplification was used to separate the real and imaginary signal components at each frequency, from which impedance magnitude and phase are derived (see Fig S1).
2.5. Dielectrophoretic analysis
Confluent cells in Dulbecco’s Modified Eagle Medium (DMEM) [Gibco, USA] were aspirated, washed in PBS and exposed to 0.5% trypsin for 5 mins at 37 °C. Cells were resuspended in 5 mL DMEM and centrifuged at 300 g for 10 mins. DMEM was aspirated, the cell pellet was resuspended in isotonic dielectrophoresis (DEP) buffer (8.8% sucrose, 0.5% BSA solution in DI water), with the conductivity adjusted to 0.15 S m−1 by titrating back in 1x PBS. Cell concentrations were adjusted to 1 x 106 cells mL−1 and counted by hemocytometer to confirm concentration. Cells were analyzed using a 3DEP analyzer (DEPtech, UK) as described previously [26], [34] (see SI Appendix Section B). Data were analyzed using MATLAB (R2018b).
2.6. Flow cytometry
Flow cytometry was carried out at the University of Virginia Flow Cytometry Core Facility using a FACS Calibur flow cytometer (BD Biosciences). Data were exported as standard FCS files and the forward and side scattered light (FSC and SSC) signals were analyzed using MATLAB (R2018b).
2.7. Data analysis
Code was written in MATLAB (R2018b) for data processing and statistical analysis. The impedance signal of each tumor cell was normalized relative to the frequency-independent impedance response of the polystyrene beads. Tumor cell populations were gated from smaller debris and larger reference beads using normalized impedance data gathered at the reference frequency (18.3MHz). The normalized impedance response of gated cells at each frequency was then plotted and analyzed (Fig S1). Statistical analyses (MATLAB R2018b) were performed on processed datasets. Significance level was defined at α < 0.05 for all cases, unless otherwise stated. Independent datasets for each cell line and electrophysiology parameter, i.e., the collections of mean values for electrical diameter, impedance magnitude opacity and impedance phase contrast (n = 9), were tested for normality using a one-sample Kolgomorov-Smirnov test (using MATLAB kstest function). The independent parameter datasets passed normality tests. One-way ANOVA tests were then performed on each group of parameter datasets, i.e. comparing datasets for all cell lines for each electrophysiology parameter, with the null hypothesis being rejected for all cases (p ≪ 0.05). Thus, multiple two sample Students’ t-tests were performed, comparing individual cell lines for each electrophysiology parameter in order to assess statistically significant differences between cell lines parameters. Statistical significance levels were adjusted using Bonferroni correction for the multiple comparisons (n = 15) performed: *p < 0.05 / 15 ≈ 0.003 level; **p < 0.01 / 15 ≈ 0.0007 level; ***p < 0.001 / 15 ≈ 0.00007 level.
2.8. Bioinformatics Analysis
Gene expression profiling was performed on PDX PDAC tumors from all samples. Microarray data were generated using the GeneChip Human Gene 2.0 ST Array Chipset and analysed in an R/Bioconductor programming environment. Raw gene expression data were preprocessed using the pd.hugene.2.0.st the oligo [PMID: 20688976] packages. Differential expression analysis and gene set variability analysis were implemented using the limma [PMID: 25605792] and gsva [PMID: 23323831] packages. A gene set collection of gene ontology terms describing molecular function (GO:MF) were obtained from the Molecular Signature Database (MSigDB) [PMID: 21546393]. Comparisons were made between the four KRAS mutant samples and two KRAS wild-type samples.
3. Results and discussion
3.1. Tumor growth characteristics of patient-derived xenografts
Key clinical and genetic characteristics for the group of six patient-derived xenografts (PDXs) of PDAC tumor cell types used in this study are outlined in Table 1. These include two tumor cell types: T608 and T366, derived originally from liver metastases (so-called metastatic mutant or met mut) and two tumor cell types: T395 and T449, derived originally from primary pancreatic cancers (so-called primary mutant or pri mut). While the aforementioned tumor cell types were identified to incorporate KRAS mutations at codon G12, which occurs in ~98% of all KRAS mutant PDAC cases, the remaining tumor cell types: T188 and T738, were derived from primary stage tumors with no known KRAS mutations (so-called primary wild-type or pri wt). Tumorigenicity in Table 1 is determined based on time to tumor growth in a mouse model and invasiveness is determined by abdominal or liver metastasis in the mouse [32, 33, 35]. Based on these definitions, the tumorigenicity of the T608 and T366 tumors (i.e. met mut) is greater than the T395 tumor (i.e. pri mut), while the ‘tumorigenicity’ is lowest for the T188 and T738 tumors (i.e. pri wt). The T449 tumor (i.e. pri mut) shows rapid growth ability, but limited invasiveness. Since, both the metastatic and primary PDAC cells from the patient were expanded in the spleen of the mouse model, their respective microenvironments are somewhat similar over the several generations of expansion of the PDXs. Hence, the chief difference is the far greater invasiveness of the implanted metastatic PDXs (T608 and T366) versus the primary PDXs of the same genotype (T395 and T449). While differences from the original patient cells are possible, the metastatic cell lines tested here should still present a strong similitude to the metastatic PDAC cells from the tumor site.
Table 1.
Characteristics of PDX PDAC tumors and cells.
| Tumor | Tumorigenicity | Invasiveness | Clinical Stage* | KRAS status† | Codon mutation details‡ | ||
|---|---|---|---|---|---|---|---|
| Time to growtha | Metastatic rateb | G12 | G12 | G12 | |||
| T608 | 2.0 months | 100% | met | mut | aa12(G➔D) | None | None |
| T366 | 2.8 months | 75% | met | mut | aa12(G➔D) | None | None |
| T395 | 4.5 months | 40% | pri | mut | aa12(G➔L) | NT | NT |
| T449 | 2.0 months | 0% | pri | mut | aa12(G➔D) | NT | NT |
| T188 | 6.0 months | 100% | pri | wt | None | NT | NT |
| T738 | 7.2 months | 25% | pri | wt | None | None | None |
- in mouse model
– in abdominal or liver
met - metastatic; pri – primary
mut - mutant; wt - wild-type.
NT - not tested.
3.2. Impedance analysis
The overall device is described in Fig. 1a and the sample collection and preparation steps are described in Fig. 1b. Using single-cell impedance cytometry (Fig 1b), the electrical properties were analyzed to investigate detectable differences in the electrical impedance responses between the respective tumor cell types with known differences in tumorigenicity. The impedance of each detected event was measured simultaneously at three discrete frequencies across three decades, thereby creating multiple single-cell datapoints. All data were normalized using the impedance of co-flowing 7 μm reference particles, since these particles have a tight size distribution and constant impedance across the measurement range [36]. At low frequencies (≤0.5 MHz), the presence of an intact lipid membrane makes biological cells electrically insulating. The resistive maximum measured in the real component of the impedance signal within this frequency range is related to cell size [37]. At intermediate frequencies (2–5 MHz), capacitance of the cell membrane causes a reactance peak in the imaginary component of the signal, which occurs directly before the membrane capacitance is effectively short-circuited [37]. The ratio of impedance magnitude at intermediate to low frequency (|Z|2 MHz / |Z|500 kHz) or the so-called |Z| opacity [37] is inversely related to the cell membrane capacitance. At frequencies above peak reactance (10–50 MHz), dielectric properties of the conducting and capacitive regions at the cell interior dominate the impedance response [37], with the impedance phase determined by the ratio of permittivity to conductivity.
Fig 2a–c show the real and imaginary components of the impedance signal (250 kHz – 50 MHz) for representative cell types of decreasing tumorigenicity: T608 (met mut), T395 (pri mut) and T188 (pri wt). Using the real and imaginary impedance components, impedance magnitude (|Z|) and phase (ϕZ) can be calculated – refer to Supplementary Information (SI) Appendix Section A for a description of the background impedance theory. Based on this, a scatter plot of single-cell data plotted as impedance phase (ϕZ) versus magnitude (|Z|) are shown at three key frequencies (0.5, 2 and 50 MHz) for met mut T608 (Fig. 2d–f), pri mut T395 (Fig. 2g–i) and pri wt T188 (Fig. 2j–l), which were acquired at a throughput of ~350 tumor cells per second. For reference, the impedance data for all the tumor cell types of Table 1 are shown in Fig S3 and Fig S4. As apparent from the low frequency impedance (0.5 MHz) in Fig. 2d, 2g, 2j, no statistically significant difference in size is detected for the PDAC cells used in this work (see Fig S5a and S6). Based on the impedance at intermediate frequency (2 MHz) per Fig 2e, 2h and 2k, the |Z| opacity shows no statistically significant difference between the respective tumor types (Fig 3a). Despite this, peak reactance occurs at slightly different frequencies for each tumor cell type (see imaginary impedance data in Fig S3), demonstrating variance between the cell membrane capacitance values. The largest differences between tumor types are most visible in the phase response at 50 MHz (Fig 3b). Using the ratio of the impedance phase at a high frequency to that at a frequency below the peak reactance (e.g., ϕZ50 MHz / ϕZ500 kHz; Fig 3c), the phase response of the cell interior and cell membrane can be compared to calculate the novel metric that we describe as the ϕZ contrast. This parameter aggregates the low and high frequency phase differences into a contrast term that describes the frequency-dependent electrophysiology of different cell components. By using phase at a low frequency, such as 500 kHz, to effectively normalize phase signals at higher frequencies, it is possible to take into consideration both permittivity and conductivity differences at membrane and interior level between samples.
Fig 2:

Impedance data for representative cell types of decreasing tumorigenicity: T608 (met mut), T395 (pri mut) and T188 (pri wt). Normalized data for the real component (downward-facing arrows) and imaginary component (upward-facing arrows) of the impedance signal for (a) T608 (met mut), (b) T395 (pri mut), and (c) T188 (pri wt) plotted as mean ± SEM; n = 9 for each datapoint. Low frequencies are within the red-shaded frequency decade, intermediate frequencies within the green-shaded decade and high frequencies within the blue-shaded frequency decade. Red, green or blue markers indicate impedance at 500 kHz, 2 MHz or 50 MHz, respectively. Impedance phase (ϕZ) is plotted against magnitude (|Z|) for met mut T608 (d-f), pri mut T395 (g-i) and pri wt T188 (j-l) at 500 kHz, 2 MHz and 50 MHz, respectively. Red circular markers with dashed red lines show population means and annotated confidence ellipses, which contain ~50% of detected events. Each scatter plot shows ~6000 plotted events.
Fig 3.

Electrical properties for PDAC samples calculated using multi-frequency impedance data: a) magnitude opacity and c) phase contrast for PDAC cells. Violin plots show population distribution for each cell line (color-coded) with median values (solid line). Overlaying markers show individual means (n = 9) for all cell lines. Statistical significance (post Bonferroni correction for multiple comparisons): *p ≲ 0.003 level; **p ≲ 0.0007 level; ***p ≲ 0.00007 level. b) Impedance phase (ϕZ) is plotted against magnitude (|Z|) for met mut T608, pri mut T395 and pri wt T188 at 50 MHz. Circular markers (color-coded) with dashed red lines show population means and annotated confidence ellipses, which contain ~50% of detected events. Each scatter plot shows ~6000 plotted events.
The impedance phase contrast term (ϕZprobe frequency / ϕZ500 kHz) for varying probe frequencies (500 kHz – 50 MHz) is plotted in Fig. 4 for each of the investigated tumor types. The ϕZ contrast is greatest at high frequencies, which justifies the use of 50 MHz data for ϕZ contrast calculation (as suggested per Fig 3b and c). A high ϕZ contrast indicates larger disparity between the phase shift at high vs low frequency, for example, due to the presence of capacitive cell components near the cell exterior, the conductive portions of the cell interior, or a combination of both factors. Such high impedance phase differences have been reported for so-called “ghost” red blood cells, known for their lower cytoplasmic conductivity and compromised membrane in comparison to viable red blood cells [38],[39]. Tumor cells derived from liver metastases (T608 and T366) were found to exhibit ϕZ contrast with a minimum of 1.25× greater than primary tumor-derived samples of the same KRAS mutant genotype (T395 and T449) and a minimum of 2× greater than primary tumor-derived samples of KRAS wild-type genotype (T188 and T738; see Table S1). Specifically, T608 (met mut) shows 2.3× larger ϕZ contrast than T188 (pri wt), while T395 (pri mut) shows 1.5× larger ϕZ contrast than T188 (pri wt). Furthermore, there were no statistical differences in ϕZ contrast between tumor cells with same origin and KRAS mutant genotype, i.e., T608 versus T366, T395 versus T449, and T188 versus T738 (Fig 3c; see Table S2). However, when individually comparing met mut tumor cells (T608 or T366) to either pri mut tumor cells (T395 or T449) or pri wt tumor cells (T188 or T738), statistically significant differences were observed, ranging from p ≲ 0.003 to p ≲ 0.00007 levels. Finally, when individually comparing pri mut to pri wt tumor cells, only T188 showed statistically significant differences to either T395 or T449.
Fig 4:

Phase contrast spectra for PDAC samples. For each detected event, phase at the varying probe frequency (500k – 50 MHz) is divided by phase at the low reference frequency (500 kHz) to calculate phase contrast across three frequency decades. The color map shows average phase contrast across the probe frequency range for each sample (n = 9) of the cell lines; 0 (black) = low phase contrast, 3 (blue= high phase contrast.
On average, PDAC cells originally isolated from liver metastases (T366 and T608) had a ∼1.4× greater ϕZ contrast than pancreas-derived samples of the same KRAS mutant genotype (T449 and T395; see Fig 4 and Table S1); with these differences being statistically significant ( p ≲ 0.003; Fig 3c). Since metastaticity is linked to a higher expression of the mesenchymal phenotype genes [40], [41], [42], it would be of interest in future studies to investigate whether impedance phase can provide information on the expression level of this mesenchymal phenotype of tumor cells. Given that four out of five of PDAC patients die due to the development of metastases [2, 43], such an electrical measurement of the phenotype can aid in predicting patient survival in prognostic studies and help in therapy selection.
3.2. Dielectrophoresis analysis
To validate the electrophysiology differences between the PDAC cell types observed by impedance cytometry, we used dielectrophoresis (DEP) analysis to compare the respective cells. Specifically, we compare representative cell types of decreasing tumorigenicity (per Table 1): T608 (met mut), T395 (pri mut) and T188 (pri wt). Using single-shell model fits [23] to the DEP spectra, we quantitatively compare each cell line based on their subcellular electrophysiology (see SI Appendix Section B and Table S3a and S3b for a detailed description of the modelled parameters). Fig. 5 shows the DEP frequency spectra (measured in media of moderate conductivity - 0.15 S m−1) and the optimal single-shell model fitting curves. Cell size estimation from the single shell model (Table S3) is in agreement with cell size estimation from impedance cytometry (Figure S6), with the met mut T608 being the largest of the three analysed cell lines (8.5 μm - DEP and 8.8 μ m – impedance cytometry). Furthermore, the DEP frequency spectra in Fig. 5 show the highest crossover frequencies (fcross-over) for T608 (fcross-over ≈ 1.2 MHz), followed by T395 (fcross-over ≈ 640 kHz) and T188 (fcross-over ≈ 260 kHz). These differences in fcross-over indicate a clear dissimilarity in reactive elements between the cell lines, which are quantified by the estimated membrane capacitance (Cnenbrane) from the single shell model (Table S3). This stratification in membrane properties could be related to known alterations of membrane conformations or intracellular folds observed in multiple cancer cell lines [25]. At frequencies above the DEP crossover frequency, wherein cells are polarized to exhibit positive DEP (pDEP), the met mut T608 tumor cell type exhibits the lowest pDEP levels, followed by those of pri mut T395 tumor cell type of the same mutant genotype, while the pri wt T188 tumor cell type lacking mutations shows the highest pDEP levels. Since electrophysiology of the cell interior dominates the DEP force response beyond fcross-over, fits to the DEP spectra using the single-shell model (Table S3b) show significantly higher interior conductivity for T188 (1.2 S m−1) versus the more invasive T395 and T608 cell types (≈0.4 S m−1). Hence, the lower interior conductivity of the metastatic T608 and the aggressive primary T395 cell types versus that of the T188 cell type are consistent with the higher levels in impedance phase for T608 and T395 cell types, since impedance phase depends inversely on conductivity.
Fig 5:

Dielectrophoretic spectra of: a) metastatic PDAC cells of KRAS mutant genotype (T608 - met mut), b) primary PDAC cells of KRAS mutant genotype (T395 - pri mut) and c) primary PDAC cells of KRAS wild-type genotype (T188 - pri wt). In each case, the dielectric force data are plotted as mean ± SEM for n = 10 samples. Color-coded single-shell model fitting curves (solid line) and estimated crossover frequencies (dashed-lines) are indicated for each sample type.
3.3. Bioinformatics Analysis
To identify the factors contributing to the described electrophysiological differences, gene expression data was gathered for all six PDAC cell lines. Recent genomic analysis [44] showed little difference between the primary versus metastatic cell types used in this work, even though we observed significant phenotypic differences between the respective cell types based on their electrophysiology. Impedance phase enhancement with tumorigenicity that is measured in the current study is consistent with the trend of prior studies showing a greater level of intracellular folds within tumorigenic cells [25], which would increase cell permittivity to enhance impedance phase. Since our impedance and DEP results suggest that the most significant electrophysiology differences occurred between tumor cell types: pri wt vs. mut (including pri and met), we stratified gene expression data along these categories to identify potential mechanisms involved between KRAS wild-type vs. KRAS mutant electrophysical phenotypes.
Gene Set Variation Analysis (GSVA) [45] was performed using the Gene Ontology Molecular Function (GO MF) collection [46, 47]. Of the 24 gene sets found to have statistically relevant differences in expression levels in KRAS mutant PDAC cells compared to those for wild-type cells (p < 0.05), six were associated with transmembrane transport of ions/ solutes, with three gene sets related to the upregulation of glutamate-activated ion channels, two to the upregulation of various sodium ion (Na+) cotransporters, and one to adenosine triphosphatase (ATPase)-driven cation transport per Fig 6. Membrane transport proteins can influence cell electrophysiology by allowing the transfer of charge across the membrane or change the interior conductivity through the accumulation or depletion of intracellular ions in the cytoplasm. Contained in Table S4 are details of the dysregulated genes within each gene set associated with ion/ solute transport. Three gene sets associated with glutamate-activated ion transport were found to have upregulated expression in KRAS mutants, which implies that glutamate may be used by KRAS mutant PDAC cells [6]. Specifically, gene sets associated with glutamate receptor activity (p < 0.01), ionotropic glutamate receptor activity (p < 0.01) and metabotropic glutamate GABAB-like receptor activity (p < 0.05) were upregulated. Within the former two gene sets, expression of genes associated with various subunits of each of the three types of ionotropic glutamate receptors were upregulated, i.e., the N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) and kainite receptors [48]. Upon activation by glutamate or glutamate-like agonists, these ion channels have high permeability for Na+ and potassium (K+) ions, plus a lower permeability for calcium ions (Ca2+) [48]. With reference to dysregulated metabotropic glutamate GABAB-like receptor activity gene set, expression of GRM2, GRM4 and GRM6 were upregulated. These genes encode metabotropic glutamate receptors that are believed to reduce NMDA receptor activity [49, 50]. Altered sensitivity to glutamate and glutamate-like agonists in KRAS mutant cells may contribute to the detected electrophysiological changes reported here, by means of significantly altering ion transport patterns. The expression of two gene sets associated with Na+ cotransport was also significantly upregulated in KRAS mutant PDAC cells: solute sodium symporter activity (p < 0.05) and inorganic anion transmembrane transporter activity (p < 0.01). Specifically, genes were upregulated which encode solute symporters or ion cotransporters that are permeable to Na+ and either of glucose, citrate, bicarbonate, phosphate, sulfate and chloride – the latter four of which are anions and could contribute to changes in electrophysiology. Dysregulated transport of Na+ would have a significant role in the induced electrophysiological change detected for KRAS mutant cells. Not only were several genes related to Na+ symporters and cotransporters upregulated, but Na+ is also highly permeable through the glutamate receptors highlighted previously [48]. Since KRAS mutant PDAC cells exhibited a lower level of interior conductivity (Fig 4 and Fig 5), the net direction of ion/ solute transport is likely biased towards cellular extrusion, which is especially significant given the number of genes identified with upregulated expression which are involved in Na+ transport.
Fig 6:
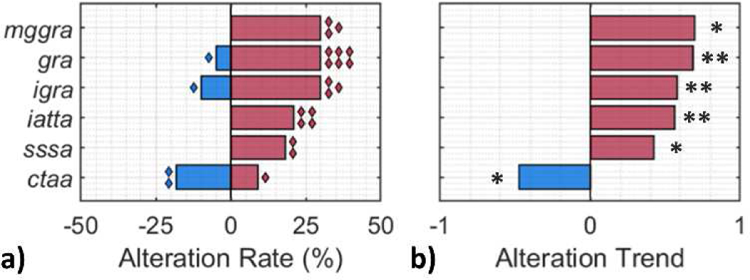
Gene sets associated with ion/ solute transport in PDAC cells were enriched for differential expression when comparing KRAS mutant to KRAS wild-type samples. (a) Proportion of upregulated (red) and downregulated (blue) genes within each gene set from a collection of GO (Gene Ontology) terms describing molecular functions. Diamond markers indicate the number of genes with dysregulated expression using gene set variability analysis. (b) Overall expression dysregulation trend for each gene set. Darker shade indicates larger variation in (a) rate or (b) trend. gra = glutamate receptor activity; igra = ionotropic glutamate receptor activity; mggra = metabotropic glutamate GABAB-like receptor activity; iatta = inorganic anion transmembrane transporter activity; sssa = solute sodium symporter activity; and ctaa = cation transporting ATPase activity. Statistical significance: *p < 0.05 level; **p < 0.01 level.
Another potentially contributing factor that has been discussed in recent literature is the effect that KRAS mutation has on calcium signalling [51, 52], since the KRAS-MAPK effector pathway and Ca2+ pathways interact at many levels [53]. Previously, we showed that combination therapy with the MEK inhibitor trametinib plus the T-type calcium channel inhibitor mibefradil caused significant growth inhibition of KRAS mutant patient-derived PDAC tumors [50]. Moreover, this combination therapy resulted in near complete growth inhibition of PDAC tumors that had acquired resistance to trametinib. Meanwhile, Pierro et al. [51, 52] showed that colorectal cancer cell lines with oncogenic KRAS exhibited Ca2+ release suppression mechanisms, which conferred a survival advantage. A gene set associated with cation transporting ATPase activity was identified to have significantly dysregulated levels of expression in KRAS mutant cells than wild-type cells (p < 0.05) – see Fig 6. Specifically, expression of ATP2A3, which encodes a protein that enables the transfer of Ca2+ from the cytosol to the endoplasmic reticulum [54], was upregulated. The expression of ATP2C1, which encodes a membrane-bound, ATP-powered calcium pump called hSPCA1 [55], was downregulated. Transport of cytosolic Ca2+ to the Golgi apparatus, for intracellular and extracellular distribution, is enabled by hSPCA1. Hence, the lowered interior conductivity of KRAS mutant PDAC cells may be explained by the removal of Ca2+ ions from the cytoplasm and their subsequent stockpiling at the endoplasmic reticulum.
4. Conclusions
Based on high-throughput single-cell impedance cytometry measurements using patient-derived PDAC cell types of varying tumorigenicity, we observed significantly higher levels in impedance phase signal for cell types obtained from metastatic versus from primary tumor sites of the same KRAS mutant genotype, while cells from primary tumors possessing KRAS mutations exhibited higher impedance phase signal versus those lacking KRAS mutations. Since impedance phase varies in inverse proportion to conductivity, these observations were consistent with a lowered level of interior cell conductivity determined based on dielectrophoretic spectra for metastatic versus primary PDAC cell types and for those with versus without KRAS mutations, as apparent from lowered positive dielectrophoresis levels and upshifted dielectrophoretic crossover frequencies. Since the most significant electrophysiological differences were observed between tumor cells obtained from KRAS wild-type versus KRAS mutant genotype (including primary and metastatic tumors), the gene expression data were stratified along these categories to suggest oncogenic KRAS-driven processes that may potentially be responsible for the detected variation in electrophysiological phenotype. Upon validation of the findings using a larger sample pool, we envisage that single-cell impedance cytometry could be used to rapidly provide clinicians with information about the tumorigenicity and/ or metastatic potential of an individual’s PDAC for tailoring therapies and prognoses.
Supplementary Material
Highlights.
High-throughput (~350 cells/s) impedance cytometry to measure ~10,000 cells/sample
Samples of patient-derived pancreatic tumor xenografts of varying tumorigenicity
Impedance phase due to electrophysiology of cell interior varies with tumorigenicity
Lowered interior conductivity and enhanced permittivity for cells of higher tumorigenicity
Validated based on level of positive dielectrophoresis and its crossover frequency
5. Acknowledgements
Support from NIH Grant 1R21AI130902-01, NIH Grant R01 CA200755, from University of Virginia’s Cancer Center and the Global Infectious Diseases Institute are acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest to report in relation to this manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Research, 2014. 74(11): p. 2913–2921. [DOI] [PubMed] [Google Scholar]
- 2.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, and Lacaine F, A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. New England Journal of Medicine, 2004. 350(12): p. 1200–1210. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2017. CA: A Cancer Journal for Clinicians, 2017. 67(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 4.Smith BD, Smith GL, Hurria A, Hortobagyi GN, and Buchholz TA, Future of cancer incidence in the United States: burdens upon an aging, changing nation. Journal of Clinical Oncology, 2009. 27(17): p. 2758–2765. [DOI] [PubMed] [Google Scholar]
- 5.Swords DS, Firpo MA, Scaife CL, and Mulvihill SJ, Biomarkers in pancreatic adenocarcinoma: current perspectives. OncoTargets and therapy, 2016. 9: p. 7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryant KL, Mancias JD, Kimmelman AC, and Der CJ, KRAS: feeding pancreatic cancer proliferation. Trends in Biochemical Sciences, 2014. 39(2): p. 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu S, Jang H, Nussinov R, and Zhang J, The structural basis of oncogenic mutations G12, G13 and Q61 in small GTPase K-Ras4B. Scientific reports, 2016. 6: p. 21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cros J, Raffenne J, Couvelard A, and Poté N, Tumor heterogeneity in pancreatic adenocarcinoma. Pathobiology, 2018. 85(1–2): p. 64–71. [DOI] [PubMed] [Google Scholar]
- 9.Friedman AA, Letai A, Fisher DE, and Flaherty KT, Precision medicine for cancer with next-generation functional diagnostics. Nature Reviews Cancer, 2015. 15(12): p. 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wlodkowic D and Cooper JM, Microfabricated analytical systems for integrated cancer cytomics. Analytical and bioanalytical chemistry, 2010. 398(1): p. 193–209. [DOI] [PubMed] [Google Scholar]
- 11.Yang R-J, Fu L-M, and Hou H-H, Review and perspectives on microfluidic flow cytometers. Sensors and Actuators B: Chemical, 2018. 266: p. 26–45. [Google Scholar]
- 12.Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M, Lenz D, Erickson HM, Ananthakrishnan R, and Mitchell D, Optical deformability as an inherent cell marker for testing malignant transformation and metastatic competence. Biophysical journal, 2005. 88(5): p. 3689–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraning-Rush CM, Califano JP, and Reinhart-King CA, Cellular traction stresses increase with increasing metastatic potential. PloS one, 2012. 7(2): p. e32572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gascoyne P and Shim S, Isolation of circulating tumor cells by dielectrophoresis. Cancers, 2014. 6(1): p. 545–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer D, Caselli F, Bisegna P, and Morgan H, High accuracy particle analysis using sheathless microfluidic impedance cytometry. Lab on a Chip, 2016. 16(13): p. 2467–2473. [DOI] [PubMed] [Google Scholar]
- 16.Song H, Wang Y, Rosano JM, Prabhakarpandian B, Garson C, Pant K, and Lai E, A microfluidic impedance flow cytometer for identification of differentiation state of stem cells. Lab on a Chip, 2013. 13(12): p. 2300–2310. [DOI] [PubMed] [Google Scholar]
- 17.McGrath J, Honrado C, Spencer D, Horton B, Bridle H, and Morgan H, Analysis of parasitic Protozoa at the single-cell level using microfluidic impedance cytometry. Scientific reports, 2017. 7(1): p. 2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honrado C, Ciuffreda L, Spencer D, Ranford-Cartwright L, and Morgan H, Dielectric characterization of Plasmodium falciparum-infected red blood cells using microfluidic impedance cytometry. Journal of The Royal Society Interface, 2018. 15(147): p. 20180416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamande J, Hupert M, Witek M, Wang H, Torphy R, Dharmasiri U, Njoroge S, Jackson J, Aufforth R, and Snavely A, Modular microsystem for the isolation, enumeration, and phenotyping of circulating tumor cells in patients with pancreatic cancer. Analytical chemistry, 2013. 85(19): p. 9092–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torphy RJ, Tignanelli CJ, Kamande JW, Moffitt RA, Loeza SGH, Soper SA, and Yeh JJ, Circulating tumor cells as a biomarker of response to treatment in patient-derived xenograft mouse models of pancreatic adenocarcinoma. PloS one, 2014. 9(2): p. e89474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta D, Lis CG, Dahlk SL, Vashi PG, Grutsch JF, and Lammersfeld CA, Bioelectrical impedance phase angle as a prognostic indicator in advanced pancreatic cancer. British Journal of Nutrition, 2004. 92(6): p. 957–962. [DOI] [PubMed] [Google Scholar]
- 22.Pohl HA, The motion and precipitation of suspensoids in divergent electric fields. Journal of Applied Physics, 1951. 22(7): p. 869–871. [Google Scholar]
- 23.Pethig R, Dielectrophoresis: Status of the theory, technology, and applications. Biomicrofluidics, 2010. 4(2): p. 022811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan H, Sun T, Holmes D, Gawad S, and Green NG, Single cell dielectric spectroscopy. Journal of Physics D: Applied Physics, 2006. 40(1): p. 61. [Google Scholar]
- 25.Gascoyne PR, Shim S, Noshari J, Becker FF, and Stemke-Hale K, Correlations between the dielectric properties and exterior morphology of cells revealed by dielectrophoretic field-flow fractionation. Electrophoresis, 2013. 34(7): p. 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohani A, Moore JH, Kashatus JA, Sesaki H, Kashatus DF, and Swami NS, Label-free quantification of intracellular mitochondrial dynamics using dielectrophoresis. Analytical chemistry, 2017. 89(11): p. 5757–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore JH, Varhue WB, Su Y-H, Linton SS, Farmehini V, Fox TE, Matters GL, Kester M, and Swami NS, Conductance-Based Biophysical Distinction and Microfluidic Enrichment of Nanovesicles Derived from Pancreatic Tumor Cells of Varying Invasiveness. Analytical chemistry, 2019. 91(16): p. 10424–10431. [DOI] [PubMed] [Google Scholar]
- 28.Li M and Anand RK, Integration of marker-free selection of single cells at a wireless electrode array with parallel fluidic isolation and electrical lysis. Chemical science, 2019. 10(5): p. 1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su Y-H, Warren CA, Guerrant RL, and Swami NS, Dielectrophoretic monitoring and interstrain separation of intact Clostridium difficile based on their S (Surface)-layers. Analytical chemistry, 2014. 86(21): p. 10855–10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohani A, Moore JH, Su Y-H, Stagnaro V, Warren C, and Swami NS, Single-cell electro-phenotyping for rapid assessment of Clostridium difficile heterogeneity under vancomycin treatment at sub-MIC (minimum inhibitory concentration) levels. Sensors and Actuators B: Chemical, 2018. 276: p. 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowther CV, Hilton SH, Kemp L, and Hayes MA, Isolation and identification of Listeria monocytogenes utilizing DC insulator-based dielectrophoresis. Analytica chimica acta, 2019. 1068: p. 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stokes JB, Adair SJ, Slack-Davis J, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, and Hwang RF, Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Molecular cancer therapeutics, 2011: p. MCT.0261.2011. [DOI] [PMC free article] [PubMed]
- 33.Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT, and Chopivsky ME, Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia, 2013. 15(2): p. IN8–IN10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su Y-H, Rohani A, Warren CA, and Swami NS, Tracking Inhibitory alterations during interstrain clostridium difficile interactions by monitoring cell envelope capacitance. ACS infectious diseases, 2016. 2(8): p. 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newhook TE, Lindberg JM, Adair SJ, Kim AJ, Stelow EB, Rahma OE, Parsons JT, and Bauer TW, Adjuvant trametinib delays the outgrowth of occult pancreatic cancer in a mouse model of patient-derived liver metastasis. Annals of surgical oncology, 2016. 23(6): p. 1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spencer D, Hollis V, and Morgan H, Microfluidic impedance cytometry of tumour cells in blood. Biomicrofluidics, 2014. 8(6): p. 064124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gawad S, Schild L, and Renaud P, Micromachined impedance spectroscopy flow cytometer for cell analysis and particle sizing. Lab on a Chip, 2001. 1(1): p. 76–82. [DOI] [PubMed] [Google Scholar]
- 38.Küttel C, Nascimento E, Demierre N, Silva T, Braschler T, Renaud P, and Oliva AG, Label-free detection of Babesia bovis infected red blood cells using impedance spectroscopy on a microfabricated flow cytometer. Acta tropica, 2007. 102(1): p. 63–68. [DOI] [PubMed] [Google Scholar]
- 39.Honrado C, L.C., Spencer D, Ranford-Cartwright L, Morgan H, Dielectric characterization of Plasmodium falciparum-infected red blood cells using microfluidic impedance cytometry. J. R. Soc. Interface 2018. 15: p. 20180416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, and Lin M-L, The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature, 2010. 467(7319): p. 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, and Jakkula L, Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature medicine, 2011. 17(4): p. 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Large T, Bijlsma M, Kazemier G, van Laarhoven H, Giovannetti E, and Jimenez C, Key biological processes driving metastatic spread of pancreatic cancer as identified by multi-omics studies. Seminars in cancer biology, 2017. 44: p. 153–169. [DOI] [PubMed] [Google Scholar]
- 43.Herman JM, Swartz MJ, Hsu CC, Winter J, Pawlik TM, Sugar E, Robinson R, Laheru DA, Jaffee E, and Hruban RH, Analysis of fluorouracil-based adjuvant chemotherapy and radiation after pancreaticoduodenectomy for ductal adenocarcinoma of the pancreas: results of a large, prospectively collected database at the Johns Hopkins Hospital. Journal of Clinical Oncology, 2008. 26(21): p. 3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brar G, Blais EM, Bender RJ, Brody JR, Sohal D, Madhavan S, Picozzi VJ, Hendifar AE, Chung VM, and Halverson D, Multi-omic molecular comparison of primary versus metastatic pancreatic tumours. British journal of cancer, 2019: p. 1. [DOI] [PMC free article] [PubMed]
- 45.Hänzelmann S, Castelo R, and Guinney J, GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics, 2013. 14(1): p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, and Eppig JT, Gene ontology: tool for the unification of biology. Nature genetics, 2000. 25(1): p. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Consortium GO, The Gene Ontology (GO) database and informatics resource. Nucleic acids research, 2004. 32(1): p. D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dingledine R, Borges K, Bowie D, and Traynelis SF, The glutamate receptor ion channels. Pharmacological reviews, 1999. 51(1): p. 7–62. [PubMed] [Google Scholar]
- 49.Ambrosini A, Bresciani L, Fracchia S, Brunello N, and Racagni G, Metabotropic glutamate receptors negatively coupled to adenylate cyclase inhibit N-methyl-D-aspartate receptor activity and prevent neurotoxicity in mesencephalic neurons in vitro. Molecular pharmacology, 1995. 47(5): p. 1057–1064. [PubMed] [Google Scholar]
- 50.Sharon D, Vorobiov D, and Dascal N, Positive and negative coupling of the metabotropic glutamate receptors to a G protein–activated K+ channel, GIRK, in Xenopus oocytes. The Journal of general physiology, 1997. 109(4): p. 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pierro C, Cook SJ, Foets TC, Bootman MD, and Roderick HL, Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodeling of IP3Rs isoform composition and ER luminal Ca2+ levels in colorectal cancer cell lines. Journal of Cell Science, 2014: p. jcs-141408. [DOI] [PubMed]
- 52.Pierro C, Zhang X, Kankeu C, Trebak M, Bootman MD, and Roderick HL, Oncogenic KRAS suppresses store-operated Ca2+ entry and ICRAC through ERK pathway-dependent remodelling of STIM expression in colorectal cancer cell lines. Cell calcium, 2018. 72: p. 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monteith GR, Prevarskaya N, and Roberts-Thomson S, The calcium–cancer signalling nexus. Nature Reviews Cancer, 2017. 17(6): p. 367. [DOI] [PubMed] [Google Scholar]
- 54.Korošec B, Glavač D, Volavšek M, and Ravnik-Glavač M, ATP2A3 gene is involved in cancer susceptibility. Cancer genetics and cytogenetics, 2009. 188(2): p. 88–94. [DOI] [PubMed] [Google Scholar]
- 55.Foggia L, Aronchik I, Aberg K, Brown B, Hovnanian A, and Mauro TM, Activity of the hSPCA1 Golgi Ca2+ pump is essential for Ca2+-mediated Ca2+ response and cell viability in Darier disease. Journal of cell science, 2006. 119(4): p. 671–679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.