

Abstract
Raf kinases are important cancer drug targets. Paradoxically, many B-Raf inhibitors induce the activation of Raf kinases. Cryo-EM structural analysis of a phosphorylated B-Raf kinase domain dimer in complex with dimeric 14-3-3, at a resolution of ~3.9 Å, shows an asymmetric arrangement in which one kinase is in a canonical “active” conformation. The distal segment of the C-terminal tail of this kinase interacts with, and blocks, the active site of the cognate kinase in this asymmetric arrangement. Deletion of the C-terminal segment reduces Raf activity. The unexpected asymmetric quaternary architecture illustrates how the paradoxical activation of Raf by kinase inhibitors reflects an innate mechanism, with 14-3-3 facilitating inhibition of one kinase while maintaining activity of the other. Conformational modulation of these contacts may provide new opportunities for Raf inhibitor development.
One Sentence Summary:
A B-Raf dimer bound to 14-3-3 has one active kinase inactivating the other, analogous to the paradoxical activation of Raf by kinase inhibitors.
Raf proteins are the first kinases to be activated in the Ras/Raf/Mek/Erk kinase pathway, one of the key conduits for the transmission of extracellular signals to the nucleus in animal cells (1). The activation of Raf is initiated by the release of autoinhibition, through the binding of Ras•GTP to the Ras-Binding Domain of Raf (2–5). The phosphorylation of the C-terminal tail of Raf then results in its association with the scaffold protein 14-3-3, which promotes dimerization of the Raf kinase domains (6–8). B-Raf is the most studied of the three human Raf kinases, because more than 50% of human melanomas contain an activating mutation (Val600→Glu or V600E) in B-Raf (9). A breakthrough in the treatment of melanoma occurred with the development of small-molecule inhibitors that are effective against the V600E variant of B-Raf (10, 11). Unexpectedly, these inhibitors were found to activate signaling through activation of wild-type Raf isoforms (12–14). This paradoxical activation of Raf by a wide range of inhibitors remains puzzling, and has restricted the full potential of small-molecule therapy targeting B-Raf.
The paradoxical activating effects of Raf kinase inhibitors have their origin in the homo- and hetero-dimerization of Raf isoforms. Drug binding to only one of the Raf proteins in the dimer results in activation of the other (12–16). Active Raf kinase domains adopt a canonical structure, defined by an open conformation of the activation loop and inward rotation of helix αC (identified in Fig. 1). We refer to the active conformation as the “ON-state”, and it is stabilized by side-to-side dimerization of the kinase domains (16, 17). Inactive Raf kinase domains do not dimerize, either because of increased dynamics, or due to adoption of a catalytically unproductive “OFF-state” conformation, common to many kinases and referred to as the Cdk/Src OFF-state conformation (16, 18, 19). Many B-Raf inhibitors destabilize this OFF-state and, surprisingly, promote activation of the partner kinase in a dimer. At sub-saturating concentrations of these inhibitors, inhibitor-bound kinase domains can dimerize with partner kinases that do not have inhibitor bound, stabilizing the ON-state in the inhibitor-free partner (12–14). In contrast, certain sulfonamide inhibitors that favor the adoption of the OFF-state conformation result in destabilization of the side-to-side Raf dimers, and do not exhibit paradoxical kinase activity (16).
Figure 1. Structure of the B-Raf:14-3-3 complex.
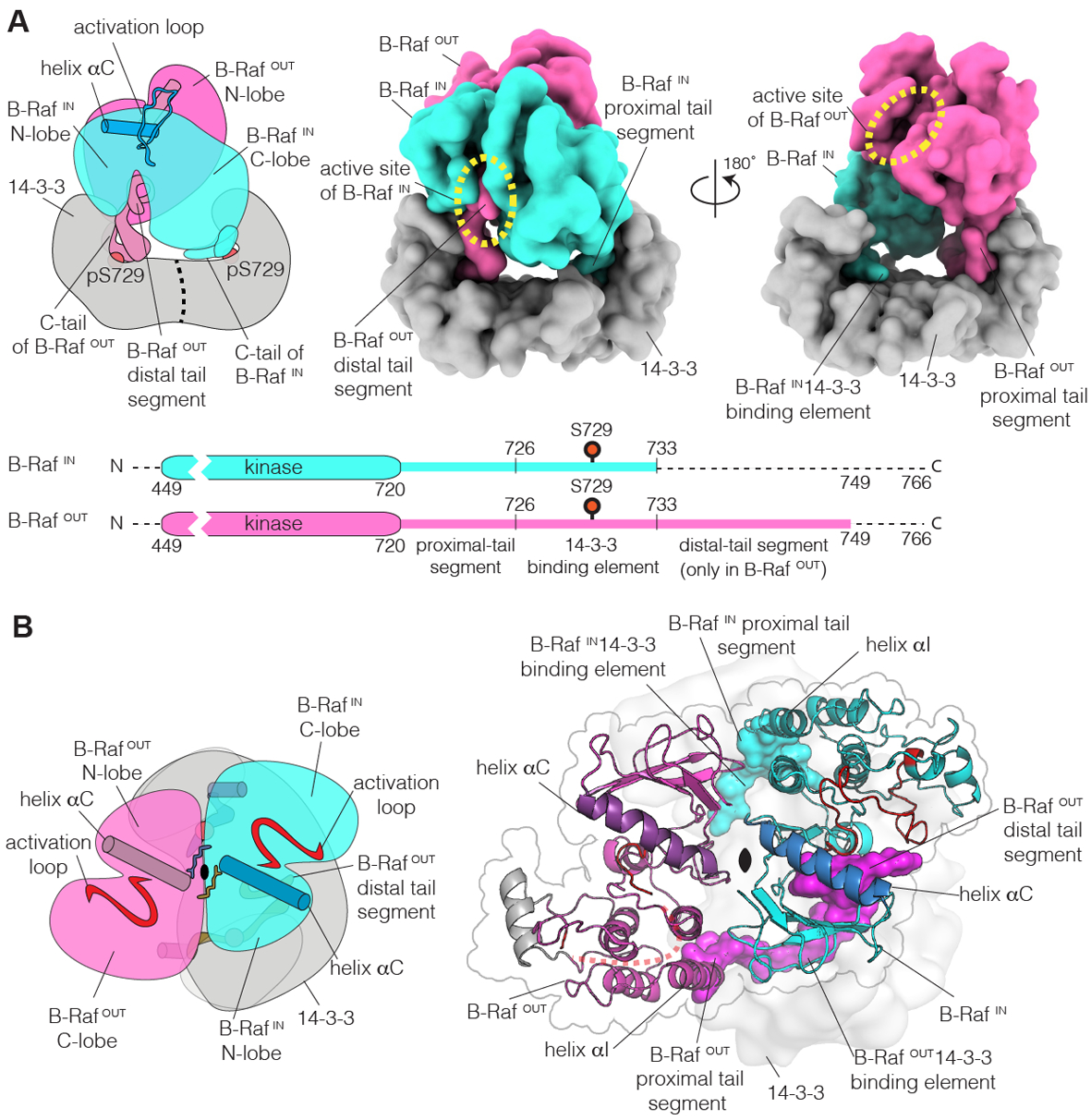
(A) Left, schematic diagram of the structure. Middle and right, two orthogonal views of the molecular surface of the cryo-EM model. The two B-Raf kinases in the dimer are shown in cyan and magenta, respectively, and the 14-3-3 dimer is shown in gray. B-RafIN (cyan) is positioned closer to 14-3-3 than is B-RafOUT (magenta). The schematic diagrams at the bottom of the panel denote the boundaries of the kinase domains and the C-tails of B-Raf that are included in the structural model, and the terms used to identify segments of the C-tails. The kinase domains and the C-tails are not to scale, and that is indicated by the breaks. Dashed lines indicate regions for which there is no interpretable density. (B) A view of the B-Raf:14-3-3 complex, looking down the 2-fold symmetry axis of the B-Raf kinase dimer. The B-Raf kinase dimer seen in the cryo-EM structure closely resembles the dimeric structure of ON-state B-Raf bound to MEK (PDB ID: 4MNE) (22) (Fig S4). Helix αG of B-RafOUT has weaker density compared to the rest of the complex, and is shown in gray.
The striking ability of kinase inhibitors to activate Raf leads to questions of fundamental interest in cell signaling and the development of effective cancer therapeutics. To help answer these questions, we have determined a structure of Raf in an activated state in complex with 14-3-3, without kinase inhibitors bound to it. We purified full-length human B-Raf in complex with the ε and ζ isoforms of insect cell 14-3-3 (see Methods section for details). The B-Raf protein is produced by intein ligation of the N-terminal regulatory segments of B-Raf, expressed in bacteria, to the C-terminal segment, produced in insect cells. The purified B-Raf protein is phosphorylated at multiple sites, including the activation loop and the C-terminal 14-3-3 binding element (Table S1). The N-terminal segment of B-Raf contains an autoinhibitory phosphorylation site (Ser365) which is unphosphorylated in the B-Raf samples, because the N-terminal segment is produced in bacteria. Thus, the complex we have produced appears to have bypassed the necessity of release of autoinhibition by interaction with Ras•GTP, is active, and can phosphorylate MEK1 (Fig. S1). In the cryo-EM structure of the complex that we determined at an overall resolution of 3.9 Å (Fig. 1A, S2, and S3), the B-Raf kinase domain dimer arrangement corresponds closely to the ON-state dimer seen in many crystal structures of isolated Raf kinase domains (Fig. 1B and S4) (20, 21). The N-terminal regulatory segments are not visualized in the cryo-EM map, presumably because the release of autoinhibition leads to disorder in these segments.
Both of the kinase domains in the dimer are in the ON-state conformation, as shown by comparison to the structure of B-Raf in complex with MEK (Fig S4) (22) but, strikingly, the active site of one kinase is blocked by insertion of the C-terminal tail (C-tail) of the other (Fig. 1A). Thus, the dimer is asymmetric, with only one kinase ready for catalysis. The asymmetry in the kinase dimer is due to a close but asymmetric interaction between the B-Raf kinase dimer and the 14-3-3 dimer, mediated by the C-tails of B-Raf (Fig. 2A). The kinase-proximal segment of the C-tail (“proximal tail segment”, see Fig. 1A) makes specific interactions with 14-3-3 (Fig. S5), and it is followed by Ser729, which is phosphorylated and docks on 14-3-3 in the canonical manner (23). These interactions constrain each kinase domain to be near the corresponding 14-3-3 subunit.
Figure 2. Interaction of the distal tail segment of B-RafOUT with the active site of B-RafIN.
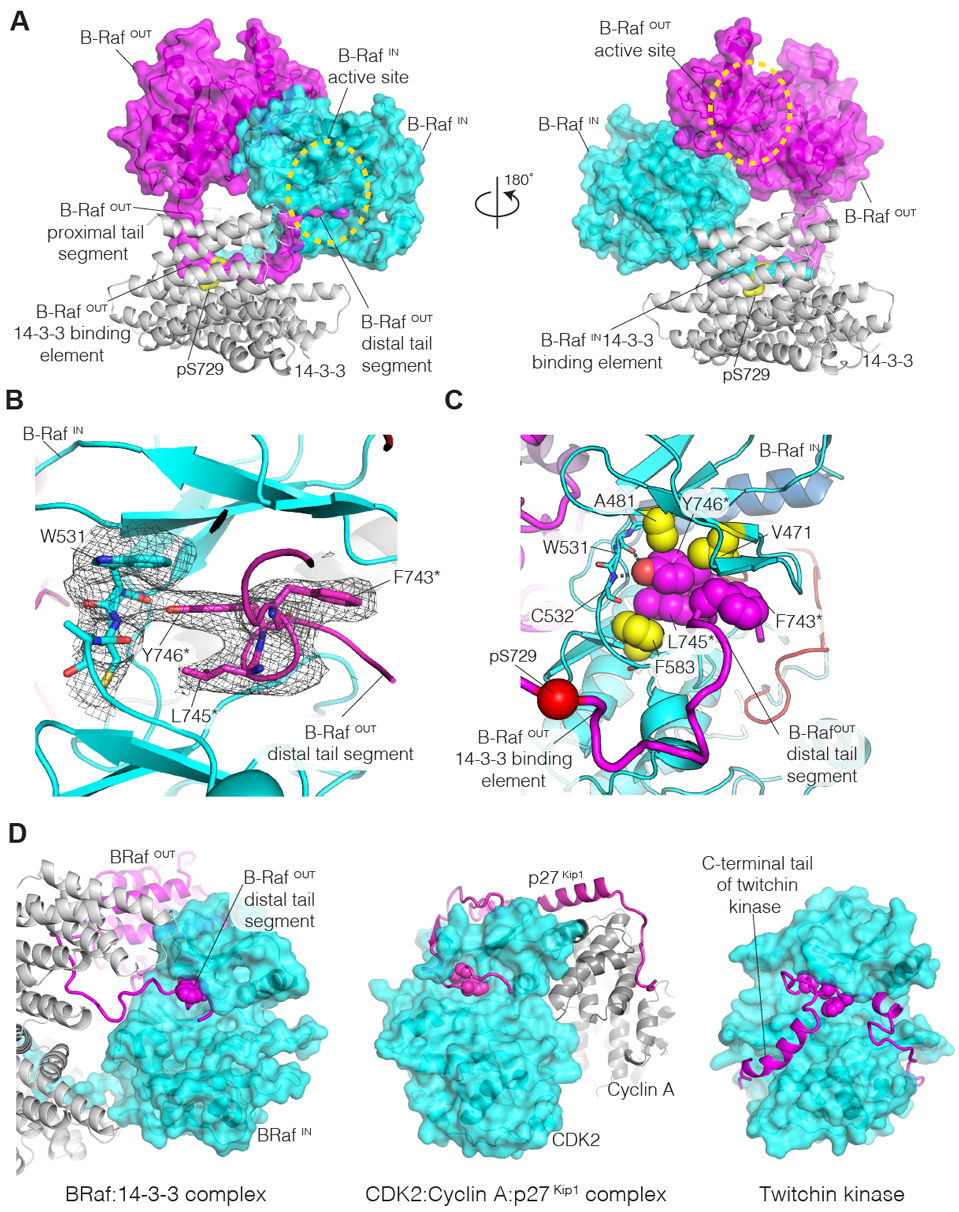
(A) Orthogonal views of the cryo-EM structure of the B-Raf:14-3-3 complex. On the left, the C-tail of B-RafOUT (magenta) is seen bound to 14-3-3 (gray) and the distal tail segment enters the active site of B-RafIN (cyan). (B) View of the ATP binding site of B-RafIN (cyan), with cryo-EM density shown in gray. Residues in the distal tail segment of B-RafOUT (magenta) are identified by asterisks. (C) The hydrophobic sidechains of the C-spine of B-RafIN are shown as yellow spheres, with two sidechains of the B-RafOUT distal tail segment (magenta) completing the C-spine. (D) Comparison of the structure of the B-Raf:14-3-3 complex with that of the CDK2:Cyclin A:p27Kip1 complex (PDB ID: 1JSU) (33), and the autoinhibited form of twitchin kinase (PDB ID: 1KOB) (34). In the B-Raf:14-3-3 complex, the distal tail segment of B-RafOUT (magenta) enters the ATP-binding site of B-RafIN (cyan). In the CDK2:Cyclin A:p27Kip1 complex, the p27Kip1 inhibitor (magenta) enters the ATP-binding site of CDK2 (cyan). In twitchin kinase, the C-terminal tail of the kinase (magenta) enters the ATP-binding site. Selected hydrophobic sidechains in the inhibitory segments are shown as spheres.
While both the kinase dimer and the 14-3-3 dimer are two-fold symmetric, their symmetry axes are not aligned, which results in the active site of one B-Raf kinase domain (denoted B-RafIN, and shown in cyan) being closer to 14-3-3 than that of the other (B-RafOUT, in magenta; Fig. 1A and 2A). This unexpected asymmetric disposition of the two kinases with respect to 14-3-3 is a consequence of an important difference in the paths of the B-Raf C-tails as they exit their respective 14-3-3 binding sites. The two B-Raf C-tails run in opposite directions, so that the tail of one kinase domain points towards the other kinase domain (Fig. 1A and 2A). The active site of B-RafIN is positioned close to the 14-3-3 surface, and it captures the kinase-distal segment of the C-tail of B-RafOUT (“distal tail segment”) as it exits from the 14-3-3 binding site (Fig. 1, 2 and S5C). In contrast, the active site of B-RafOUT points away from the 14-3-3 surface, and is too far away for a reciprocal interaction with the distal tail segment of B-RafIN.
For the C-tail of B-RafOUT, continuous density is seen extending from Leu733 in the 14-3-3 binding element, towards the active site of B-RafIN (Fig. S6). Strong density is observed for the backbone of a seven-residue segment of the distal tail segment (residues Asp742-Cys748), within which the sidechains of Phe743, Leu745, and Tyr746 can be resolved clearly (Fig. 2B and S6). The distal tail segment forms a helical turn between residues 743 and 747, thereby exiting the active site of B-RafIN in a direction that is orthogonal to the direction of entry. In this way, the distal tail segment of B-RafOUT makes an L-shaped wedge within the active site of B-RafIN, with marked resemblance to the way in which some kinase inhibitors are bound at this site (Fig. S5D). The hydroxyl group of the sidechain of Tyr746 in the distal tail segment of B-RafOUT forms hydrogen bonds with the backbone of the hinge connecting the N- and C-lobes of the B-RafIN kinase domain (Fig. 2C). The formation of such hydrogen bonds is a characteristic feature of the interaction of small-molecule inhibitors with protein kinases (24). The tyrosine sidechain occupies the space that is normally occupied by the adenine group of ATP in the structures of active kinases (Fig. S5D). The wedge formed by the distal tail segment of B-RafOUT prevents closure of the N-lobe of B-RafIN over the C-lobe, and in this way favors maintenance of the ON-state conformation of B-RafIN (Fig. S7) and, as a consequence, is expected to stabilize the B-Raf kinase dimer.
As a first step towards validating the interactions seen in our model, we utilized Ba/F3 pro B cells that rely on interleukin-3 (IL-3) for growth (25, 26). In agreement with a critical role for IL3/B-Raf signaling, we were unable to expand single Ba/F3 clones in which B-Raf was deleted by CRISPR/Cas9 (data not shown). We therefore applied heterologous expression of B-Raf variants fused to a fluorescent protein (EGFP) coupled to dual parameter flow cytometry, simultaneously measuring levels of phospho-ERK (pERK), a downstream product of Raf activation, and GFP levels in Ba/F3 cells (Fig. S8). Since Ba/F3 cells express endogenous Raf proteins, we restricted the measurement of pERK levels to cells expressing a high level of the EGFP-B-Raf fusion protein. Under these conditions, we assume that B-Raf dimers predominantly contain either two molecules of the heterologous fusion protein, or one molecule each of the fusion protein and endogenous Raf kinases. Heterologous overexpression of wild-type EGFP-B-Raf resulted in similar pERK levels compared to those observed for GFP-negative cells, both at baseline and after IL-3 stimulation, indicating that regulation of B-Raf signaling to ERK is intact (Fig. S9). By contrast, EGFP-B-RafV600E resulted in uniformly spontaneous phospho-ERK induction in non-stimulated cells relative to the untransfected control, as expected (Fig. 3A and B) (20).
Figure 3. Mutational analysis of B-Raf.
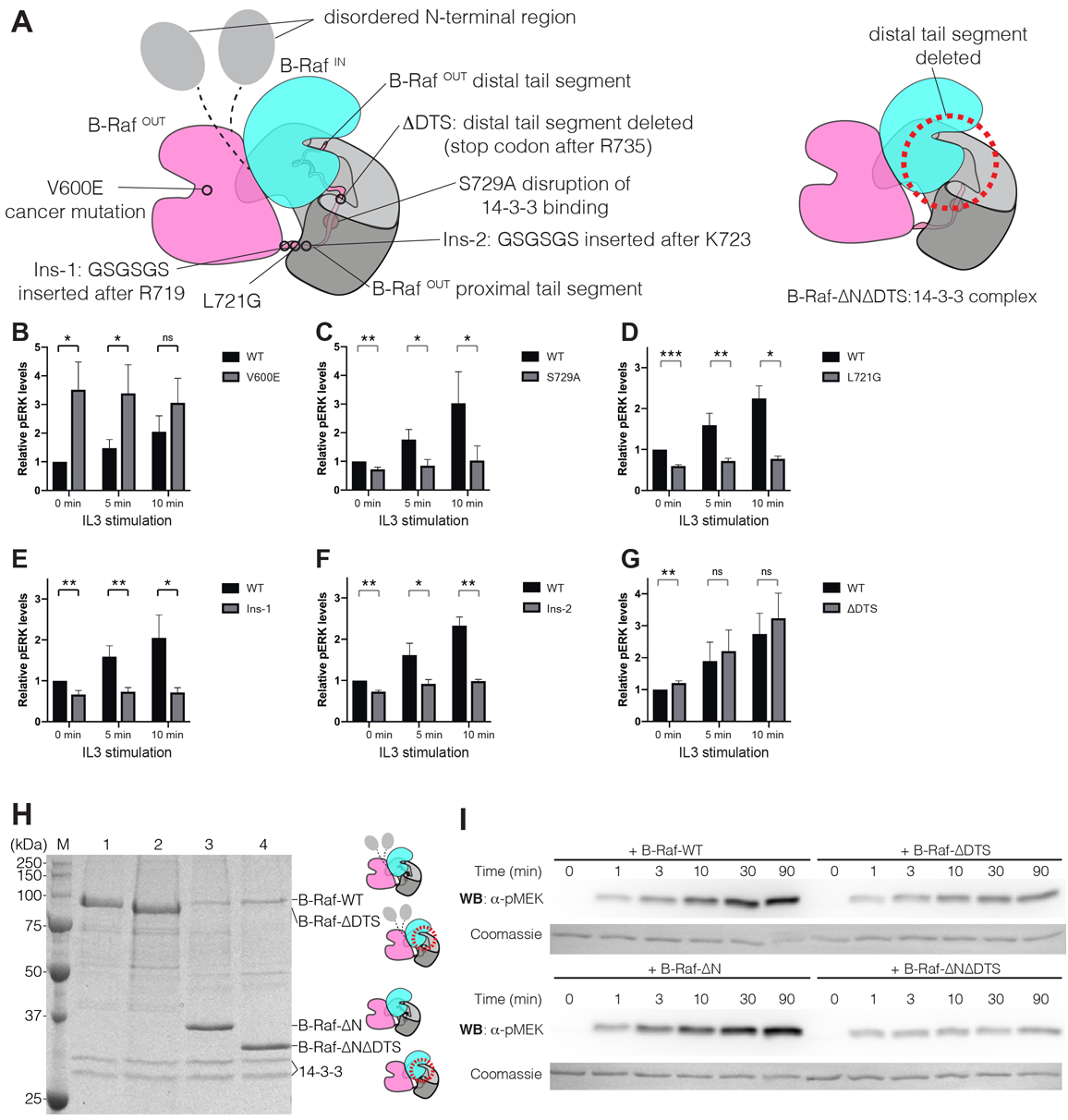
(A) Left, schematic diagram of the cryo-EM structure, indicating the B-Raf variants that were analyzed. Right, schematic diagram of the B-Raf-ΔNΔDTS:14-3-3 complex, which lacks the N-terminal region and the distal tail segment (dotted circle). (B-G) Relative levels of phospho-ERK (pERK) for cells expressing B-Raf variants. Mean values for relative pERK levels and standard deviations were plotted from three flow cytometry experiments (the complete histograms for pERK levels in the experiments are shown in Fig. S9B). For each experiment, the pERK level for unstimulated wild-type EGFP-B-Raf transfected cells at 0 min was set to 1, and all other values were normalized to this. The statistical significance of each measurement is indicated by ns (p value>0.05), * (p value <=0.05), ** (p value<=0.01), *** (p value<= 0.001). (H) SDS-PAGE gel analysis of B-Raf constructs purified from HEK293T cells. M – Precision Plus Protein Unstained Standards (Bio-Rad); 1 – B-Raf-WT; 2 – B-Raf-ΔDTS; 3 – B-Raf-ΔN; 4 – B-Raf-ΔNΔDTS. (I) Western blot analysis of MEK1 phosphorylation by B-Raf constructs with the N-terminal regulatory region present and without the N-terminal regulatory region. Coomassie brilliant blue staining of the membrane shows the total amount of MEK1 protein loaded to each lane on the gel.
Using this platform, we first investigated the importance of the positioning of the C-tails of B-Raf onto 14-3-3. Expression of EGFP-B-RafS729A, a variant lacking the phosphorylation site that triggers binding to 14-3-3, resulted in impaired pERK induction upon IL-3 stimulation (Fig. 3C), as expected (27). The cryo-EM structure reveals that Leu721 in both proximal tail segments is sandwiched between the kinase domain and the 14-3-3 surface (Fig. S5) and a L721G mutation is expected to disrupt these interactions. Expression of EGFP-B-RafL721G resulted in lower levels of pERK relative to control (Fig. 3D). We also made two variants of B-Raf in which glycine and serine residues were inserted into the proximal tail segment to increase flexibility, and to alter the length of the proximal tail segment, which is highly conserved in B-Raf (Fig. S10). In one variant, the sequence GSGSGS was inserted after the last residue in the kinase domain (Arg719), and in the second variant the same six residues were inserted in the middle of the proximal tail, between residues Lys723 and Ile724 (denoted Ins-1 and Ins-2, respectively, in Fig. 3A and S5). Expression of the Ins-1 and Ins-2 variants of B-Raf resulted in lower levels of pERK relative to control, at baseline and following IL-3 stimulation (Fig. 3E and F). Thus, precise positioning of the B-Raf dimer onto the 14-3-3 dimer is essential for optimal pERK signaling.
We expressed EGFP-tagged variants of Raf containing mutations that alter or delete the distal tail segment. These mutations did not lead to a reduction in activity in this cellular assay but, instead, led either to no change, or to a slight increase in pERK levels (Fig. 3G and S11A–D). These results suggest that the distal tail segment could also play an autoinhibitory role, as has been proposed for the C-tail of C-Raf (28), and discussed further below. Only one distal tail segment in a dimer (that of B-RafOUT) is required for activation, and endogenous B-Raf can provide this function, prompting our conservative interpretation of the results of these cellular signaling assays.
To further investigate the importance of the distal tail segment for B-Raf activity, we carried out phosphorylation assays with purified proteins. We prepared two sets of N-terminally Flag-tagged B-Raf constructs for mammalian cell expression, one based on full-length B-Raf (B-Raf-WT), and one without the N-terminal regulatory domain, corresponding to the cryo-EM model (B-Raf-ΔN). For these two constructs, we either retained the distal tail segment, or deleted it, generating two additional constructs (B-Raf-ΔDTS and B-Raf-ΔNΔDTS) (Fig. 3A). We purified the B-Raf proteins corresponding to these constructs in complex with endogenous mammalian 14-3-3 proteins (Fig. 3H) and used them to phosphorylate the Raf substrate MEK1, with phospho-MEK1 detected by western blotting (Fig. 3I). Both B-Raf-WT and B-Raf-ΔN phosphorylate MEK1 robustly, and for both, the deletion of the distal tail segment resulted in substantially lower levels of MEK1 phosphorylation, while the binding of ΔDTS mutants to 14-3-3 is unaffected (Fig. 3H, I, S11E and F). The reduction of MEK1 phosphorylation by B-Raf-ΔNΔDTS is a particularly important result, showing that interactions made by the distal tail segment are crucial, even in a stripped-down construct that lacks all of the regulatory domains. The interpretation of these mutational data is complicated, however, by a possible autoinhibitory role for the distal tail segment (see below). In addition, the distal tail segment contains two highly conserved phosphorylation sites (Ser750 and Thr753), whose mutation could alter B-Raf activity (29).
We used molecular dynamics simulations to test whether engagement of the distal tail segment of B-RafOUT in the active site of B-RafIN is required to maintain the asymmetric interaction between the B-Raf dimer and 14-3-3. We initiated two sets of trajectories starting from the cryo-EM model. In one set, an intact B-Raf:14-3-3 complex, with the distal tail segment of B-RafOUT inserted into the active site of B-RafIN, was used to generate three trajectories, each for 500 ns. In the other set, the distal tail segment was deleted, and three trajectories, each for 500 ns, were generated (see Methods). For simulations with the distal tail segment of B-RafOUT left intact, the interaction between this segment and the active site of B-RafIN is stable in the three independent simulations, supporting our interpretation of the density for the distal tail segment of B-RafOUT (Fig. S12A). The asymmetric organization of the complex was maintained throughout each of these simulations (Fig. S12B). A strikingly different result was obtained in the simulations in which the distal tail segment of B-RafOUT was deleted – here, the quaternary structure was disrupted rapidly, within a few nanoseconds in each simulation, and the N-lobe of the kinase domain of B-RafIN moved away from the close association with 14-3-3 seen in the cryo-EM model (Fig. 4A and S12B). These simulations indicate that the interaction made by the distal tail segment of B-RafOUT and the kinase domain of B-RafIN is the principal determinant of the asymmetric orientation of B-Raf and the 14-3-3 proteins.
Figure 4. Molecular dynamics simulations of the B-Raf:14-3-3 complex.
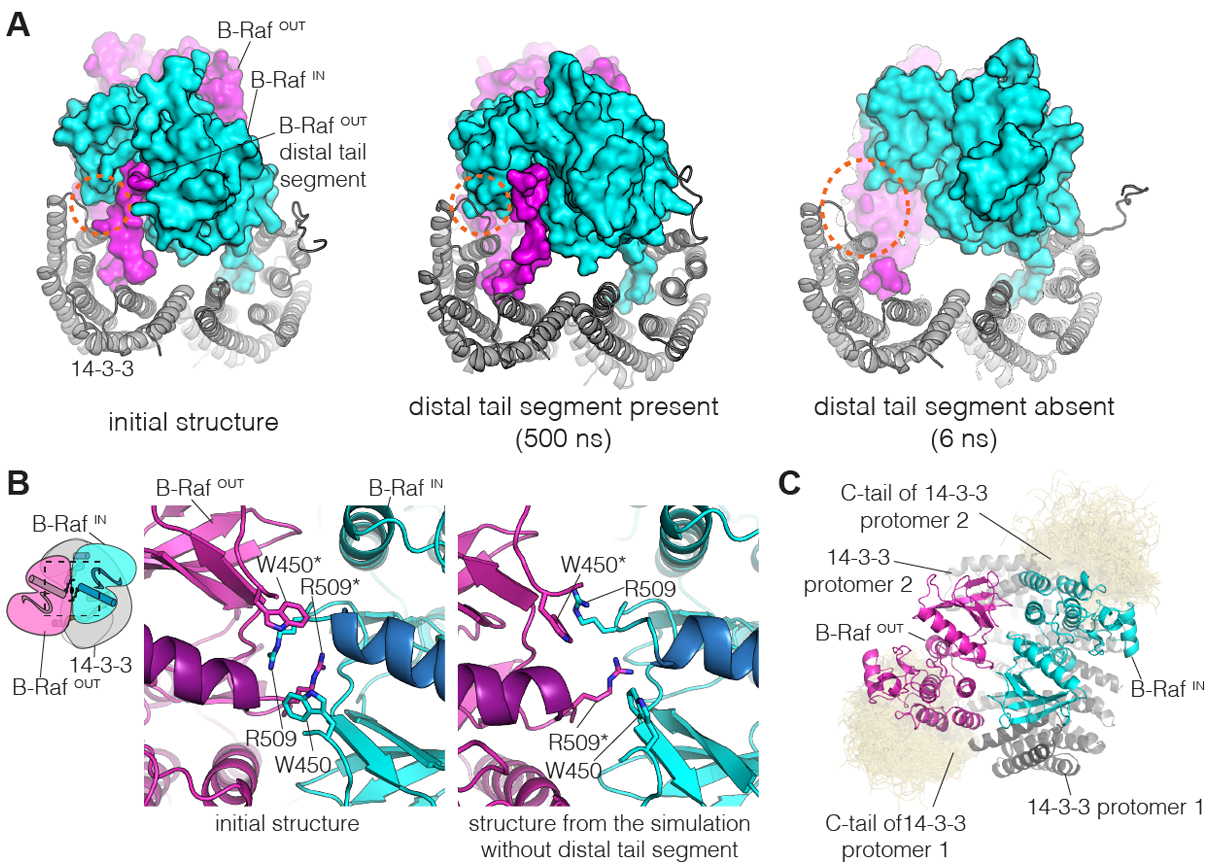
(A) Instantaneous structures from two representative simulations are shown. Left, initial structure. Middle, structure after 500 ns, for one of the simulations with the distal tail segment intact. Right, structure after 6 ns, for one of the simulations with the distal tail segment deleted. Orange dashed circles indicate a region of close contact between B-RafIN and 14-3-3 in the initial structure. (B) Disruption of the B-Raf dimer interface in one of the simulations in which the distal tail segment of B-RafOUT was deleted. The interface between the kinases is shown for the initial structure (left) and the structure after 500 ns of simulation (right). (C) Interactions between the C-terminal tails of 14-3-3 and the B-Raf kinase domains. Shown here is a superposition of the backbone structures of the 14-3-3 tails (yellow) for three simulations with the distal tail segment of B-RafOUT intact, sampled every nanosecond over 500 ns. The 14-3-3 tails cluster around the C-lobes of the two B-Raf kinase domains. This occurs due to electrostatic complementarity, with each instantaneous structure forming two to three ion pairs between each tail segment and the adjacent kinase domain (Fig. S15).
The aspect of the Raf kinase dimer that is critical for maintenance of the ON-state is a tight interaction between the N-lobes of the two kinases (17). Key interactions are made by the sidechains of Arg509 in each kinase, which form two hydrogen bonds each with the last helical turn of helix αC in the other kinase, a structural element that plays an important regulatory role (Fig. 4B) (17). Notably, in one of the simulations with the distal tail segment deleted, the reciprocal interactions between the N-lobes of the two kinases, centered on Arg509, are disrupted within ~350 ns (Fig. 4B, and S13). Within each kinase, we do not see conformational transitions that correspond to marked departure from the ON-state, such as a reorientation of helix αC or refolding of the activation loop. Nevertheless, the disruption of the interactions pinning down helix αC point to the release of a brake on such transitions, which presumably require much longer simulations to be sampled.
The last 15 residues in each 14-3-3 molecule are not visible in the cryo-EM map, and include several negatively charged residues (Fig. S14). The cryo-EM structure positions positively charged regions in the C-lobes of the kinase domains in the general vicinity of the C-terminal tails of 14-3-3. The molecular dynamics simulations show the formation of transient ion pairs between the 14-3-3 tails and the C-lobes of the kinase domains, resulting in persistent interactions over the course of each of the simulations. This suggests that the 14-3-3 tails may provide additional stabilization to the complex with Raf kinase domains (Fig. 4C, S15).
ATP-bound kinases in the ON-state contain a stack of hydrophobic sidechains termed the catalytic spine (C-spine), which pack on either side of the adenine group of ATP (15, 30). Small-molecule inhibitors of Raf that result in paradoxical activation substitute for the adenine group of ATP, and stabilize the active-like configuration of the C-spine. Our findings are consistent with an asymmetric activation model for B-Raf proposed earlier, in which stabilization of the C-spine by mutation results in activation, even though ATP binding is blocked (31, 32). The distal tail segment of B-RafOUT converts B-RafIN into an “activator” of B-RafOUT, by stabilizing the C-spine during the innate activation process (Fig. 2D). From this we infer that this natural mechanism of activation through stabilization of an asymmetric kinase dimer is mimicked by mutations that introduce bulky sidechains into the active site, or by some small-molecule inhibitors.
The way in which the distal tail segment of one B-Raf kinase inserts into the active site of the cognate kinase in the dimer has parallels to inhibition mechanisms of other kinases. For example, there is a striking similarity between the binding of the distal tail segment to B-RafIN and the way the cell-cycle inhibitor p27Kip1 blocks the active site of cyclin-dependent kinase 2 (CDK2) (Fig. 2D) (33). A segment of the p27Kip1 protein inhibits CDK2 by entering the ATP-binding site near the hinge region, as does the B-RafOUT distal tail segment. Within the active site, p27Kip1 forms a single helical turn, which is capped by the sidechain of the active site lysine residue. In the B-Raf complex, the corresponding residue (Lys483) is positioned similarly. The helical turn of p27Kip1 presents the sidechain of Tyr88 for hydrogen bonding with the kinase hinge, just as is seen in the B-RafOUT distal tail segment. Although the details are different, Tyr88 of p27Kip1 is flanked by hydrophobic sidechains that fill the space normally occupied by the adenine group of ATP. In twitchin kinase, an autoinhibitory segment follows the last helix in the kinase domain (αI), and the distal portion of this segment enters the ATP-binding site while adopting a helical structure (Fig. 2D) (34). In twitchin kinase, Ile345 replaces the tyrosine residues that form hydrogen bonds with the kinase hinge regions in the B-Raf:14-3-3 and CDK2:Cyclin A:p27Kip1 complexes. These variations may explain the absence of a corresponding tyrosine residue in the distal tail segments of A-Raf and C-Raf, which have other hydrophobic residues in this region but may still be able to form such an interaction (Fig. S10).
These examples of molecular mimicry emphasize the fact that the distal tail segment of B-RafOUT is actually an inhibitor of B-RafIN, and it is the asymmetry of the B-Raf:14-3-3 complex that leaves one of the two active sites in the dimer open and ready for catalysis (Fig. 1 and 2A). The comparison also shows that the interaction of pSer729 with 14-3-3, coupled with dimer formation, forces each distal tail segment of B-Raf to be positioned away from the active site of its own kinase domain, preventing cis-autoinhibition. In B-RafOUT, 20 residues separate the end of helix αI and the part of the distal tail segment that is inserted into the active site of B-RafIN. Thus, it is plausible that in monomeric forms of Raf the C-tail acts as a cis-autoinhibitor, as the corresponding segment does in twitchin kinase (Fig. 2D).
The structure of the B-Raf:14-3-3 complex identifies the importance of the interaction between the proximal tail segment of B-Raf and 14-3-3 for activation. The geometry of this interaction depends on the conformation of the C-terminal helix αI of the kinase domain, suggesting that small molecules that bind to this region of the kinase and alter the disposition of αI could interfere with B-Raf activity. That such an approach might be feasible is suggested by the successful development of drugs that inhibit Abl by altering the conformation of helix αI (35). Abl is regulated differently than B-Raf (36), but in both cases an interaction that is critical for regulation depends on the conformation of helix αI. Our discovery of a natural counterpart to the paradoxical activation of Raf by inhibitors suggests new avenues to explore for inhibition of the Ras-MAP kinase pathway.
Supplementary Material
Acknowledgements:
We thank X. Cao for insect cell culture and J. Paul for helpful discussions and help with experiments. This work used the Vincent J. Proteomics/Mass Spectrometry Laboratory at UC Berkeley, supported in part by NIH S10 Instrumentation Grant S10RR025622. This research was, in part, supported by the National Cancer Institute’s National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH R01-GM129325 and P41-GM103311. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. The authors also acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing grid resources that have contributed to the research results reported within this paper.
Funding: This work was supported in part by a grant from the NIH (P01-AI091580, JPR and JK), a UCSF IRACDA grant (K12GM081266, SB), and by a Canada Excellence Research Chair Award (to SS).
Footnotes
Competing interests: Jeroen Roose is a co-founder and scientific advisor of Seal Biosciences, Inc. and on the scientific advisory committee for the Mark Foundation for Cancer Research. John Kuriyan is a co-founder of Nurix Therapeutics, Inc., and on the Scientific Advisory Boards of Carmot, Inc., and Revolution Medicine, Inc.
Data and materials availability: Accession numbers for the B-Raf:14-3-3 structure is as follows: EMD-20708 (density maps; Electron Microscopy Data Bank) and PDB-6UAN (coordinates of atomic models; Protein Data Bank).
References
- 1.Lavoie H, Therrien M, Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol 16, 281–298 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Zhang XF et al. , Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 364, 308–313 (1993). [DOI] [PubMed] [Google Scholar]
- 3.Vojtek AB, Hollenberg SM, Cooper JA, Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 74, 205–214 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M, Complex formation between RAS and RAF and other protein kinases. Proc. Natl. Acad. Sci. USA 90, 6213–6217 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moodie SA, Willumsen BM, Weber MJ, Wolfman A, Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 260, 1658–1661 (1993). [DOI] [PubMed] [Google Scholar]
- 6.Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R, Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science. 265, 1713–1716 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Morrison DK, Heidecker G, Rapp UR, Copeland TD, Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem 268, 17309–17316 (1993). [PubMed] [Google Scholar]
- 8.Fu H et al. , Interaction of the protein kinase Raf-1 with 14-3-3 proteins. Science. 266, 126–129 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Davies H et al. , Mutations of the BRAF gene in human cancer. Nature. 417, 949–954 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Flaherty KT et al. , Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med 363, 809–819 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agianian B, Gavathiotis E, Current insights of BRAF inhibitors in cancer. J. Med. Chem 61, 5775–5793 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Heidorn SJ et al. , Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 140, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatzivassiliou G et al. , RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 464, 431–435 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N, RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 464, 427–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS, Kinases and pseudokinases: lessons from RAF. Mol. Cell. Biol 34, 1538–1546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thevakumaran N et al. , Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol 22, 37–43 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M, A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 461, 542–545 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Jura N et al. , Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J, An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 125, 1137–1149 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Wan PTC et al. , Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 116, 855–867 (2004). [DOI] [PubMed] [Google Scholar]
- 21.King AJ et al. , Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 66, 11100–11105 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Haling JR et al. , Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 26, 402–413 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Yaffe MB et al. , The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 91, 961–971 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Zorn JA, Kuriyan J, A structural atlas of kinases inhibited by clinically approved drugs. Meth. Enzymol 548, 23–67 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Palacios R, Steinmetz M, Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. Cell. 41, 727–734 (1985). [DOI] [PubMed] [Google Scholar]
- 26.Whittaker S et al. , Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med 2, 35ra41 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Brummer T et al. , Functional analysis of the regulatory requirements of B-Raf and the B-Raf(V600E) oncoprotein. Oncogene. 25, 6262–6276 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Dhillon AS et al. , The C-terminus of Raf-1 acts as a 14-3-3-dependent activation switch. Cell Signal. 21, 1645–1651 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Ritt DA, Monson DM, Specht SI, Morrison DK, Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol. Cell. Biol 30, 806–819 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor SS, Kornev AP, Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci 36, 65–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J et al. , Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 154, 1036–1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J et al. , Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol 35, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP, Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 382, 325–331 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Kobe B et al. , Giant protein kinases: domain interactions and structural basis of autoregulation. EMBO J. 15, 6810–6821 (1996). [PMC free article] [PubMed] [Google Scholar]
- 35.Wylie AA et al. , The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 543, 733–737 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Nagar B et al. , Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 112, 859–871 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Stevens AJ et al. , Design of a Split Intein with Exceptional Protein Splicing Activity. J. Am. Chem. Soc 138, 2162–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fetics SK et al. , Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure. 23, 505–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer A et al. , Regulation of RAF activity by 14-3-3 proteins: RAF kinases associate functionally with both homo- and heterodimeric forms of 14-3-3 proteins. J. Biol. Chem 284, 3183–3194 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife. 7 (2018), doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng SQ et al. , MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bai X, Rajendra E, Yang G, Shi Y, Scheres SHW, Sampling the conformational space of the catalytic subunit of human γ-secretase. Elife. 4, e11182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanton VP, Nichols DW, Laudano AP, Cooper GM, Definition of the human raf amino-terminal regulatory region by deletion mutagenesis. Mol. Cell. Biol 9, 639–647 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran NH, Wu X, Frost JA, B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem 280, 16244–16253 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Kefford R et al. , Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J. Clin. Oncol 28, 8503–8503 (2010). [Google Scholar]
- 47.Molzan M et al. , Stabilization of physical RAF/14-3-3 interaction by cotylenin A as treatment strategy for RAS mutant cancers. ACS Chem. Biol 8, 1869–1875 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Stevers LM et al. , Characterization and small-molecule stabilization of the multisite tandem binding between 14-3-3 and the R domain of CFTR. Proc. Natl. Acad. Sci. USA 113, E1152–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goddard TD et al. , UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng J et al. , 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr. Sect. D, Biol. Crystallogr 49, 362–365 (1993). [DOI] [PubMed] [Google Scholar]
- 53.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926 (1983). [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K, VMD – Visual Molecular Dynamics. J. of Mol. Graph 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Phillips JC et al. , Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang J et al. , CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darden T, York D, Pedersen L, Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089 (1993). [Google Scholar]
- 58.Martyna GJ, Tobias DJ, Klein ML, Constant pressure molecular dynamics algorithms. J. Chem. Phys 101, 4177 (1994). [Google Scholar]
- 59.Feller SE, Zhang Y, Pastor RW, Brooks BR, Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 103, 4613–4621 (1995). [Google Scholar]
- 60.Case DA et al. , The Amber biomolecular simulation programs. J. Comput. Chem 26, 1668–1688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pettersen EF et al. , UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ, Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.