

Abstract
Short-acting μ-opioid receptor (MOR) agonists have long been used for the treatment of severe, breakthrough pain. However, selective MOR agonists including fentanyl and morphine derivatives are limited clinically due high risks of dependence, tolerance, and respiratory depression. We recently reported the development of a long-acting, bifunctional MOR agonist/δ-opioid receptor (DOR) antagonist analgesic devoid of tolerance or dependence in mice (AAH8, henceforth referred to as 2B). To address the need for short-acting treatments for breakthrough pain, we present a series of novel, short-acting, high-potency MOR agonist/DOR antagonist ligands with antinociceptive activity in vivo. In this study, we utilized a 2D structure-activity relationship (SAR) matrix to identify pharmacological trends attributable to combinations of two key pharmacophore elements within the chemotype. This work enhances our ability to modulate efficacy at MOR and DOR, accessing a variety of bifunctional profiles while maintaining high affinity and potency at both receptors.
Keywords: Peptidomimetics, structure-activity relationship, MOR agonist, DOR antagonist, bifunctional, short-acting analgesic
Graphical Abstract
Introduction
Opioids such as morphine and its derivatives have been used for pain relief since their isolation in the 19th and early 20th centuries, though preparations of the opium poppy have been used medicinally for millenia.1,2 Unfortunately, pain relief mediated by opioids has been inextricably associated with negative side effects including respiratory depression, analgesic tolerance, physical dependence, and constipation.3 Opioids elicit a pharmacological response through the activation of opioid receptors,4–6 which include the μ-opioid receptor (MOR), δ-opioid receptor (DOR), and κ-opioid receptor (KOR), of which MOR is the primary target of most clinically used opioid analgesics. Recently, ligands with a capacity to preferentially activate one downstream effector over another (i.e. G protein or beta-arrestin) have gained favor as a viable route towards developing analgesics with reduced adverse effects. Specifically, G protein-biased ligands have shown antinociceptive activity in mice and rats with reduced side effects compared to morphine at analgesic doses.7–11 However, recent reports further investigating two G protein-biased MOR agonists of note—Oliceridine (TRV130) and PZM-21— suggest that they may have similar deleterious side effects as morphine.12,13
An alternative strategy—bifunctional or multifunctional ligands that act on more than one subtype of opioid receptors—provides antinociception in animal models with less tolerance and dependence risk compared to morphine.14–16 Much of this work has focused on targeting MOR and DOR, though the nociceptin receptor (NOP) has also been the target of bifunctional opioid approaches toward improved drug profiles.17–19 The targeted profile of this work, MOR agonism/DOR antagonism,20–26 has demonstrated effectiveness at reducing tolerance and dependence associated with selective MOR agonists. This profile has been the focus of several investigations by our laboratory and others, utilizing either a bivalent approach27–29 or a dual-binding single agent.23,30–32
The MOR agonist/DOR antagonist bifunctional profile has been targeted by our lab through a series of small molecule peptidomimetics,33–36 which replaced our previously reported cyclic peptide scaffold with a tetrahydroquinoline (THQ) core to improve drug-like properties, scalability, and ease of synthesis. Compounds in this series feature a dimethyltyrosine moiety, common amongst opioid ligands,37 attached to a THQ core. In Figure 1, the dimethyltyrosine motif is shown in black; the THQ core is in blue. While the dimethyltyrosine moiety enhances ligand affinity for the opioid receptors, modifications to the THQ core can be utilized to further tailor efficacy and relative affinity (selectivity) for each receptor subtype. Of interest to this investigation are those substitutions to the THQ core extending from the N-1 and C-6 positions, corresponding to R1 and R2 respectively (Figure 1).
Figure 1.

General structure of peptidomimetics presented here. The tetrahydroquinoline (THQ) core (blue) features a conserved dimethyltyrosine motif (black) as well as two pharmacophore elements of interest at N-1 (R1) and C-6 (R2).
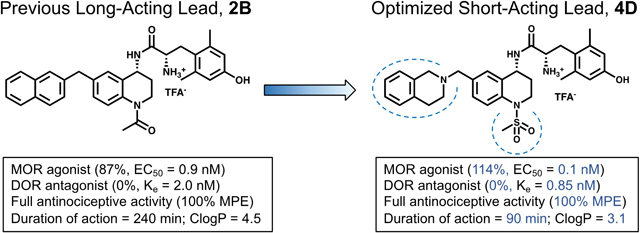
The lead compound of this series 1A (Figure 2), features no N-1 substitution and a benzyl pendant at C-6. Some initial developments in the peptidomimetic series incorporated various bicyclic and bulky aryl substituents at the C-6 position of the THQ core (Figure 2, 1B).34–36 As pharmacophore models predicted,33 these substituents preferentially bind to the MOR active state and DOR inactive state, resulting in a MOR agonist/DOR antagonist profile. However, analogues of this type were generally 10- to 120-fold selective for MOR over DOR. Subsequently, the N-1 position of the scaffold was explored with a series of alkyl and acyl substitutions which generally improved DOR affinity thereby reducing selectivity for MOR over DOR.38 Unfortunately, N-acylated analogues typically displayed partial DOR agonism (Figure 2, 2A). Yet, when a bicyclic C-6 pharmacophore was combined with an acyl N-1 substitution, the MOR agonist/DOR antagonist profile was re-established with high affinity at both receptors. Additionally, compound 2B (previously reported as AAH8, shown in Figure 2), which features both a bicyclic substituent at the C-6 position and an N-1 acetyl moiety, displays full antinociceptive activity in vivo.35 Extending the C-6/N-1 substitution pattern, the work described here further explores the structure-activity relationship (SAR) around the C-6/N-1 chemotype while improving the drug-like properties of the peptidomimetic series.
Figure 2.

Lead peptidomimetic 1A and modifications at N-1 (compound 2A) and C-6 (compound 1B). Favorable changes are shown in bold while unfavorable changes are italicized. Compound 2B combines favorable attributes of both C-6 and N-1 modifications and serves as the 2nd generation lead for this study. 2B has been described previously under the name “AAH8” in reference 39.
Results
Chemistry:
Synthesis of compounds presented in this work began with the THQ intermediate 1 (Scheme 1), which is commercially available or can be synthesized from p-toluidine in 3 steps as described previously.35,36 Compounds in this series are initially acylated at the N-1 position, then C-6 pendants are introduced via benzylic bromination and Suzuki or amide coupling. Reductive amination of the C-4 ketone provides the desired primary amine with (R) stereochemistry. Finally, dimethyltyrosine is coupled to the scaffold and Boc-deprotected to give the final compound 6 depicted in Scheme 1. Final compounds were purified by semi-preparative HPLC as described in the Methods section.
Scheme 1: General Synthetic Scheme for C-6/N-1 Substituted Peptidomimetics.

a Reagents and conditions: (A) Boc anhydride, DMAP, DIPEA, DCM, reflux or (B) Acetic anhydride, cyclopropanecarbonyl chloride, or benzoyl chloride, DIPEA, DCM. (C) NBS, benzoyl peroxide, CCl4, reflux. (D) R2-boronic acid pinacol ester, Pd(dppf)Cl2, K2CO3, 3:1 acetone/water, 80°C, or (E) tetrahydroisoquinoline-HCl, K2CO3, DMF, r.t. (F) (R)-(+)-2-methyl-2-propanesulfinamide, Ti(OEt)4, THF, 0°C to reflux, then NaBH4, THF, −78°C to r.t. (G) HCl, 1,4-dioxane, r.t., then di-Boc 2,6-dimethyl-L-tyrosine, PyBOP, DIPEA, DMF, r.t., then TFA, DCM, r.t.
SAR Studies:
Compounds 1A-F (Table 1), which feature no N-1 substitution, typically display the MOR agonist/DOR antagonist profile, though 1A displays low DOR agonist activity. Compounds in this subset bind to MOR 10- to 100-fold more tightly than DOR, limiting the bifunctional utility of these ligands. In the tables below, binding affinity (Ki), potency (EC50) and efficacy (percent stimulation compared to a standard agonist) are presented at MOR, DOR and KOR. Additionally, the ratio between the MOR and DOR binding affinities (DOR Ki/MOR Ki) is included for each compound as a measure of selectivity.
Table 1.
Opioid Receptor Bindinga and Efficacyb of Compounds 1A-F
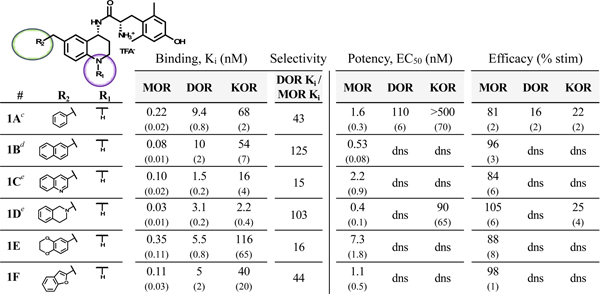 |
Binding affinities (Ki) were obtained by competitive displacement of [3H]-diprenorphine in membrane preparations.
Efficacy and potency data were obtained measuring agonist-induced stimulation of [35S]-GTPγS binding. Efficacy is represented as percent maximal stimulation relative to a standard agonist at 10 μM: DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR). Values are expressed as the mean of three separate assays performed in duplicate with standard error of the mean (SEM) in parentheses. dns = does not stimulate.
First reported in reference 33.
From reference 34.
From reference 36.
In order to improve the DOR binding affinity of these ligands, N-acylated analogues of compounds 1A-F were synthesized, as an N-acyl moiety had improved DOR affinity for the previously reported compounds 2A, 2B and 2D relative to their non-acetylated counterparts.35,36 Compounds 2A-F and 3A-F (Table 2) collectively display subnanomolar affinity for both MOR and DOR. Of note, 2C, 3C and 3E bound to MOR and DOR with less than 2:1 selectivity. Contrary to the DOR antagonist profile established by 2B, the planar aromatic pendants of 2A, 2C, 2F, 3A, 3B, 3C and 3F showed significant efficacy at DOR. Indeed, compounds 3A and 3C were quite efficacious at DOR, while 3C also showed subnanomolar agonist potency at DOR. Compound 2B is a notable exception to the trend of DOR agonism for analogues with planar pendants. The non-planar 1-tetrahydroisoquinolinyl (THIQ) and 6-benzodioxanyl pendants did not stimulate DOR regardless of N-substitution. Regarding MOR activity, most compounds were MOR agonists (greater than 70% stimulation), while 2E, 3C, and 3E were partial agonists (between 30 and 70% stimulation). The 6-benzodioxanyl pendant analogues (2E, 3E, 4E, and 5E) showed the lowest MOR efficacy of any C-6 substitution throughout this SAR study.
Table 2.
Opioid Receptor Bindinga and Efficacyb of Compounds 2A-F and 3A-F
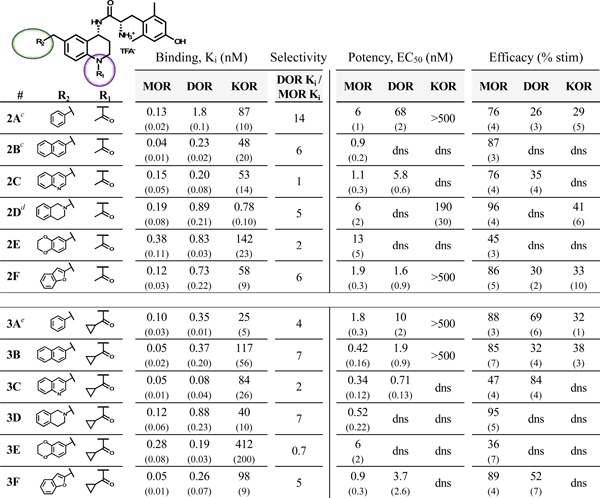 |
Binding affinities (Ki) were obtained by competitive displacement of [3H]-diprenorphine in membrane preparations.
Efficacy and potency data were obtained measuring agonist-induced stimulation of [35S]-GTPγS binding. Efficacy is represented as percent maximal stimulation relative to a standard agonist at 10 μM: DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR). Values are expressed as the mean of three separate assays performed in duplicate with standard error of the mean (SEM) in parentheses. dns = does not stimulate.
First reported in reference 35.
From reference 36.
From reference 38.
Replacement of the N-1 amide with a methyl sulfonamide (mesyl) substituent (compounds 4A-F, Table 3) resulted in an increase in MOR potency and efficacy relative to the unmodified or N-acylated analogues. Analogues in this series generally showed greater than 90% efficacy at MOR with potencies ranging from 0.12 to 0.34 nM; compound 4E was the lone exception, displaying only 47% efficacy and 9 nM potency. Notably, bicyclic analogues 4B-F consistently did not stimulate DOR. MOR affinity varied between 0.04 and 0.23 nM while DOR affinities ranged from 0.41 to 1.5 nM, leading to greater MOR selectivity than observed with the acyl series. Additionally, some selectivity over KOR was lost, as four analogues displayed single-digit nanomolar KOR affinity. The N-benzoyl analogues 5A-F (Table 3) also displayed consistent DOR antagonism, however MOR potency and efficacy were consistently the lowest of any N-1 substitution in this study. Compound 5E, which combined the low-efficacy 6-benzodioxanyl pendant with the low-efficacy N-benzoyl substitution, yielded the only analogue with no significant MOR efficacy within this study.
Table 3.
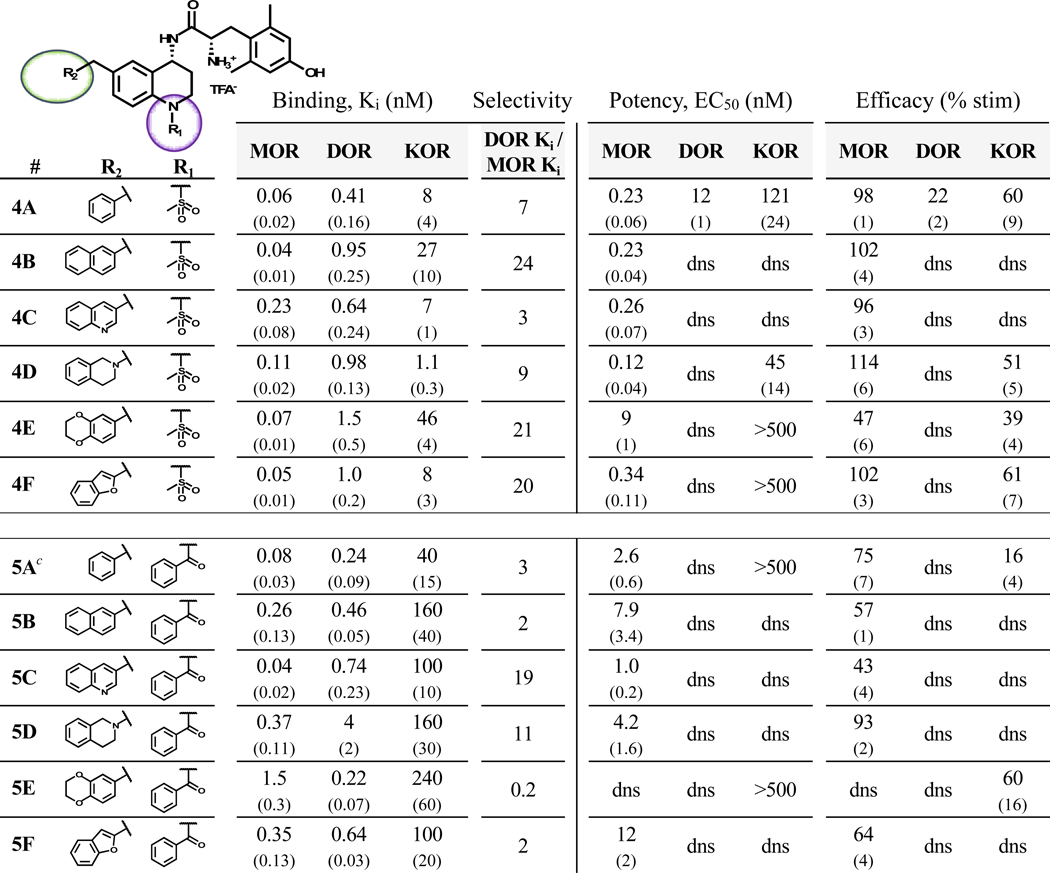 |
Binding affinities (Ki) were obtained by competitive displacement of [3H]-diprenorphine in membrane preparations.
Efficacy and potency data were obtained measuring agonist-induced stimulation of [35S]-GTPγS binding. Efficacy is represented as percent maximal stimulation relative to a standard agonist at 10 μM: DAMGO (MOR), DPDPE (DOR), or U69,593 (KOR). Values are expressed as the mean of three separate assays performed in duplicate with standard error of the mean (SEM) in parentheses. dns = does not stimulate.
First reported in reference 38.
In Vivo Antinociceptive Activity:
All compounds in this series with MOR agonist activity in vitro were evaluated for their antinociceptive activity in mice via the warm water tail withdrawal (WWTW) assay (Table 4).
Table 4.
Mouse Warm Water Tail Withdrawal Assay for Antinociception (% MPE)a
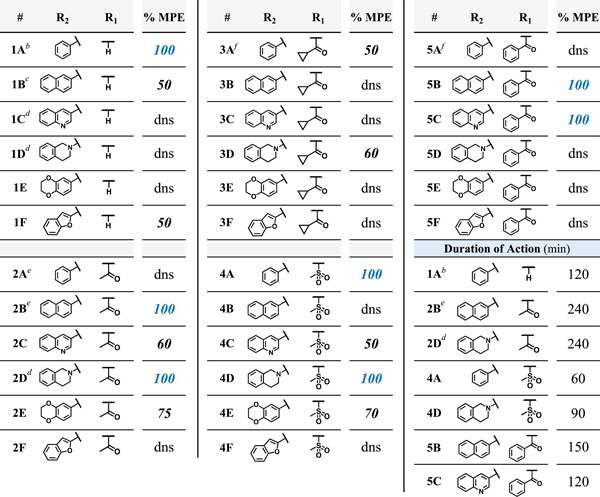 |
Results from the mouse WWTW assay after cumulative dosing of test compound up to 10 mg/kg ip. Antinociceptive activity represented as percent maximum possible effect (% MPE), with MPE being a 20 s latency to tail withdrawal. Baseline tail withdrawal latency is ~5 s, or 25% MPE. Duration of action for compounds with full antinociceptive actviity (100% MPE) is calculated as the amount of time between administration and return to baseline following a bolus 10 mg/kg dose of compound ip.
First reported in reference 33.
From reference 34.
From reference 36. Compound 2D was previously reported under the name AMB-47 in reference 39.
From reference 35.
From reference 38.
Test compounds (10 mg/kg cumulative dosing) were administered via intraperitoneal injection at 30-minute intervals, as described in the Methods section. Of the 21 novel analogues presented here, four reached the maximal possible effect (100% MPE) while six others showed partial activity (50–75% MPE); the remaining eleven compounds showed no significant difference from baseline at the doses tested. Duration of action, or the amount of time between administration of test compound and the test subject’s return to baseline latency to tail flick, was measured for all of the fully active analogues. The mesyl analogues 4A and 4D were slightly shorter-acting than the lead 1A (120 min), featuring a duration of less than 90 min. Meanwhile, the benzoyl analogues 5B and 5C displayed durations of 120 to 150 min (see Figure 3). The previously reported acetyl analogues 2B and 2D remained the longest-acting ligands in this study with a duration of 240 min.
Figure 3.

Time course data for compounds 4A (n=6), 4D (n=6), 5B (n=4), and 5C (n=3) in the 50C WWTW assay in C57BL6 male mice. Animals were injected with saline and baseline latencies were established 30 minutes later. Animals were then injected with test compound at 10 mg/kg ip and latency to tail flick was measured at the times indicated.
Antagonist Potency of In Vivo Candidates:
Compounds displaying a full antinociceptive effect in vivo and DOR antagonism in vitro were further evaluated in order to determine the potency of their DOR antagonist effects. Analogues 4D, 5B, and 5C affected a rightward shift in the EC50 of the standard DOR agonist SNC80 which equated to Ke values of 0.85 nM, 15 nM, and 8 nM respectively (calculated as described in Methods).
Discussion and Conclusions
Previous work in our lab has investigated the effects of various substituents at the C-6 position of the THQ core in conjunction with an unmodified N-1 amine.33,34 Through these earlier studies, we have developed pharmacophore models that establish a preference for an aryl moiety at C-6 in order to achieve nanomolar affinity at MOR and DOR. While our lead peptidomimetic 1A featured a benzyl pendant at C-6 and acted as a potent MOR agonist with low efficacy at DOR, it was discovered that a 2-naphthyl C-6 pendant abolished all DOR efficacy.34 Further work showed that other bulky groups at C-6, typically in the form of two fused rings with varying degrees of aromaticity and saturation, preferentially bound and stabilized the DOR inactive state.34–36 The work described here expands the SAR on four previously reported C-6 pendants—the benzyl pendant from our lead compound 1A, as well as the 2-naphthyl, 3-quinolinyl, and 1-tetrahydroisoquinolinyl (THIQ) pendants. Additionally, two new bicyclics that incorporate oxygen heteroatoms—6-benzodioxanyl and 2-benzofuranyl—were added to this series for further study.
Prior to this work, it was noted that various N-1 substitutions incorporated in conjunction with the C-6 benzyl pendant displayed improved DOR affinity at the expense of increasing DOR efficacy.38 However, combining an N-1 substitution with a bicyclic pendant at C-6 improved the efficacy profile by reducing DOR agonism while retaining high MOR efficacy. Table 5 summarizes the trends in efficacy at MOR and DOR with respect to both C-6 and N-1 substitutions.
Table 5.
SAR Trends Illustrated by 2D Matrices: Efficacy at MOR and DOR
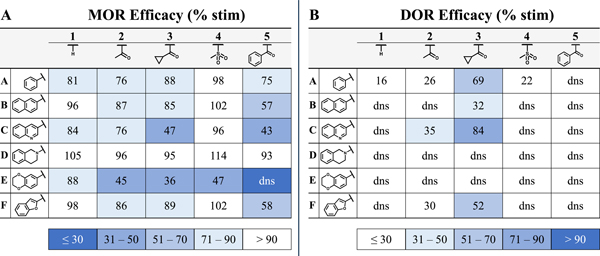 |
(A) SAR matrix highlights trends in MOR efficacy. Darker shades of blue correspond to lower efficacy (least desirable) whereas those with white background are most efficacious. (B) DOR efficacy matrix displays ligands with the highest efficacy (least desirable) in blue whereas antagonists or low-efficacy agonists are shown in white. dns = does not stimulate.
In Table 5B, the effect that a C-6 pendant has on DOR efficacy can be observed on the horizontal axis. The benzyl pendant analogues in line A typically display partial DOR agonism while the 2-naphthyl pendant analogues in line B are typically antagonists at DOR, denoted as dns (does not stimulate) in Table 5B. All analogues in lines A and B in Table 5A were MOR agonists. To further map the efficacy landscape of this chemotype, additional bicyclic analogues were designed and synthesized for this study (see lines C-F, Table 5). In line C, a 3-quinolinyl pendant was selected as a less lipophilic mimic of the 2-naphthyl pendant (line B). However, the 3-quinolinyl pendant was associated with some DOR agonism in analogues 2C and 3C (Table 5B), making this pendant a less suitable lead in pursuit of the MOR agonist/DOR antagonist profile. The THIQ pendant of line D probed nonplanar chemical space with a semi-saturated bicyclic ring system. The THIQ pendant consistently displayed the highest MOR efficacy of any pendant investigated here (Table 5A, line D) and maintained DOR antagonism throughout the series (Table 5B, line D), though analogues 2D and 4D (Tables 2 and 3 respectively) also display relatively high KOR affinity and efficacy. Incorporation of oxygen heteroatoms with the 6-benzodioxanyl and 2-benzofuranyl pendants proved deleterious towards both in vitro and in vivo profiles. The 6-benzodioxanyl pendant, consistent with previously reported analogues featuring heteroatoms distal to the THQ core,36 decreased MOR efficacy considerably (Table 5, line E) but showed favorable MOR and DOR affinity with significantly lower KOR affinity in analogues 2E and 3E (Table 2). The 2-benzofuranyl pendant produced a wide variety of multifunctional profiles, with 1F and 4F acting as MOR agonists/DOR antagonists, 2F and 3F acting as MOR agonists/DOR partial agonists, and 5F displaying MOR partial agonist/DOR antagonist activity (Table 5, line F). This unpredictability, paired with high lipophilicity and limited activity in vivo at the doses tested, minimized the utility of the 2-benzofuranyl pendant. The lipophilicity (measured by ClogP) and in vivo activity (%MPE) for each analogue described above are listed in Table 6. Notably, analogues 2C-E and 4C-E, which have the lowest ClogP throughout the series (Table 6A), also display substantial in vivo activity (Table 6B).
Table 6.
2D Matrix Indicates Low ClogP is Favorable for Achieving Antinociceptive Activity
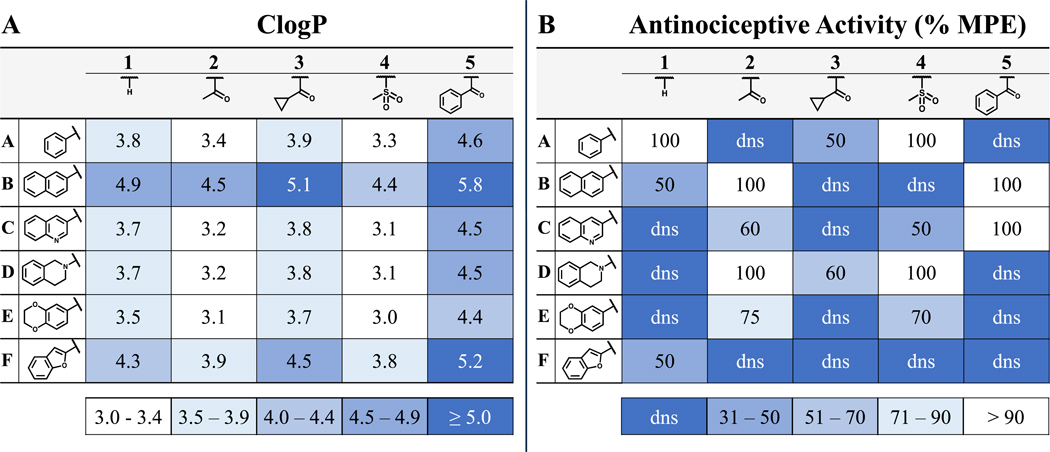 |
(A) Ligand matrix indicates trends in lipophilicity, as measured by ClogP. Compounds with highest ClogP (least desirable) are displayed in darker shades of blue whereas more polar analogues are shown in white. (B) Antinociceptive activity, measured by the WWTW assay, is displayed as the percent maximal possible effect (%MPE). Compounds with the least antinocicetive activity in this assay (least desirable) are depicted in darker shades of blue whereas those with the highest activity are shown in white.
A separate arm of the early peptidomimetic SAR campaign focused on N-1 substitutions.38 An initial N-acetyl modification was incorporated with the goal of improving metabolic stability by blocking oxidation of the THQ core. However, this N-acetylation had the beneficial effect of increasing DOR affinity. When the N-acetyl group was incorporated in tandem with the 2-naphthyl or THIQ pendants, the resultant compounds, 2B and 2D respectively (Table 2), showed significantly improved DOR affinities relative to the unsubstituted analogues 1B and 1D (Table 1). The 100-fold selectivity of 1B and 1D for MOR over DOR was reduced to 5- to 6-fold MOR selectivity in analogues 2B and 2D. Furthermore, these C-6/N-1 substituted analogues provided full antinociceptive efficacy in mice after intraperitoneal (ip) administration with a duration of action greater than four hours.31,35,36 As such, further N-1 substitutions were evaluated early in the peptidomimetic campaign—primarily in the context of the C-6 benzyl THQ scaffold.38 In the work presented here, four previously studied N-1 motifs (unsubstituted, acetyl, cyclopropanecarbonyl and benzoyl) and one novel motif (the mesyl group) were further explored, combining each N-1 motif with the five bicyclic pendants discussed previously.
The acetyl and cyclopropanecarbonyl motifs of compounds 2A-F and 3A-F (Table 2) were most effective at improving DOR affinity and reducing the selectivity for MOR, though these motifs were also associated with DOR agonism (Table 5B, columns 2 and 3). The mesyl group was selected as a bioisostere to the acetyl group and showed promising in vivo activity in the initial mesyl-substituted analogue 4A (Table 6B, column 4). The mesyl subset 4A-F displayed the highest MOR efficacy (Table 5A, column 4) and potency of any N-1 substitution. Additionally, the mesyl group was generally effective at maintaining DOR antagonism (Table 5B, column 4), though 4A did elicit partial DOR agonism.
The benzoyl derivatives 5A-F also did not stimulate DOR (Table 5B, column 5) and afforded two fully efficacious analogues in vivo—5B and 5C (Table 6B, column 5). Although these two compounds exhibited only partial MOR efficacy in vitro (Table 5A, column 5), they produced robust antinociceptive activity in vivo. Evidently, full MOR agonism in vitro is not required for a full antinociceptive response in this in vivo assay. Further pharmacokinetics studies are required to better elucidate the basis for the unexpected in vivo activity of 5B and 5C.
By combining advantageous C-6 and N-1 moieties from past SAR campaigns and expanding those with novel substituents at both positions, we have developed an SAR matrix of 30 analogues that further expand the available toolkit of multifunctional opioid ligands. Of the 21 novel ligands presented here, eight displayed the desired MOR agonist/DOR antagonist profile (>70% MOR efficacy, <10% DOR efficacy). Though not our initial goal, compounds such as 3A and 3C showed surprisingly high potency and efficacy for DOR. Such compounds may be useful in the further pharmacological evaluation of compounds bearing a MOR agonist/DOR agonist profile—a profile that is also purported to reduce opioid-related tolerance while improving analgesic potency and efficacy.40,41 Four novel compounds from this series, 4A, 4D, 5B, and 5C, showed full antinociceptive activity in mice, and will be carried forward for evaluation in tolerance and dependence models. Comparing these with the bicyclic lead 2B (ClogP = 4.5), we observed a significant reduction in lipophilicity for analogues 4A (ClogP = 3.2) and 4D (ClogP = 3.1), while 5B (ClogP = 5.8) was significantly more lipophilic and 5C (ClogP = 4.5) showed no change. In addition to reducing lipophilicity, compound 4D also displays the highest in vitro potency and efficacy at MOR and maintains potent DOR antagonist activity (Ke = 0.85 nM).
The durations of action in vivo for ligands 4A, 4D, 5B, and 5C are considerably shorter than the bicyclic lead 2B. Whereas the previously reported ligands 2B and 2D both displayed a duration of action of over 200 min, compounds 5B and 5C persisted for 120 min or less while 4A and 4D were particularly short-acting with a duration of 60 to 90 min. Such short-acting agents could be useful for the treatment of breakthrough pain, especially if used in conjunction with a lower dose of a longer-acting opioid agent such as 2B. Further investigation of the tolerance and dependence liabilities associated with the long-term use of these novel, short-acting agents is to follow. Future studies will aim to establish whether these ligands show fewer side-effects than classic short-acting pain treatments such as fentanyl.
In summary, we have further investigated the bicyclic C-6/N-1 chemotype established by 2B and 2D,35,36 expanding the published C-6 and N-1 chemical space. The various C-6 and N-1 modifications reported here have been combined in an SAR matrix to further elucidate the chemical motifs that govern ligand binding and receptor activation in the context of the THQ peptidomimetic core. Furthermore, the number and duration of ligands with in vivo antinociceptive activity is expanded with the addition of novel ligands 4A, 4D, 5B, and 5C. This SAR study reinforces previous findings and refines our ability to develop potent bifunctional opioid ligands with a range of mixed-efficacy profiles in order to further probe the unique pharmacology of the opioid receptor family.
Experimental Section
Chemistry.
Final compounds were characterized by 1H NMR, electrospray ionizing mass spectrometry (ESI-MS), and HPLC retention time. 1H NMR data were obtained on a 500 MHz Varian spectrometer using CD3OD as the solvent. ESI-MS was obtained using an Agilent 6130 LC–MS mass spectrometer in positive ion mode. The retention time and purity of final compounds were assessed using a Waters Alliance 2690 analytical HPLC instrument with a Vydac protein and peptide C18 reverse phase column. Retention times were obtained by running a linear gradient starting at 0% solvent B (99.9% acetonitrile, 0.1% TFA) and 100% solvent A (99.9% water, 0.1% TFA) to 70% solvent B and 30% solvent A in 70 min, measuring UV absorbance at 230 nm. All final compounds used for testing were ≥95% pure, as determined by analytical HPLC. Purification of final compounds was performed using a Waters semipreparative HPLC with a Vydac protein and peptide C18 reverse phase column, using a linear gradient of 0% solvent B (0.1% TFA in acetonitrile) in solvent A (0.1% TFA in water) to 100% solvent B in solvent A at a rate 1% per minute, monitoring UV absorbance at 230 nm. Full synthetic procedures and characterization data can be found in the Supplemental Information section.
General Procedure (A): Boc protection of the tetrahydroquinoline (THQ) core.
To a flame-dried round bottom flask under Ar was added tetrahydroquinolin-4-one intermediate (1.0 eq), Boc2O (1.5 eq), and DMAP (0.1 eq). The reaction vessel was placed under vacuum for 5 min, then anhydrous DCM was added via syringe and the solution stirred for 5 min under vacuum. The round bottom flask was flooded with Ar, and DIPEA (1.5 eq) was added via syringe. The reaction vessel was equipped with a condenser and placed in oil bath at 60°C. The reaction stirred at reflux for 12–16 h under Ar and was monitored by TLC. Once significant conversion to product was seen, the reaction was quenched using deionized (DI) H2O (20 mL) and the layers were separated. The organic layer was washed with sat. NaHCO3 and sat. NaCl solutions then dried over MgSO4. Organic layer was filtered and concentrated under reduced pressure, then purified using silica gel chromatography.
General Procedure (B): N-Acylation or mesylation of the THQ core.
To a round-bottom flask containing THQ intermediate (1.0 eq) under Ar atmosphere was added DCM. Reaction flask was then cooled to 0°C before adding Et3N (1.2 eq), followed by acyl or sulfonyl chloride (1.2 eq). When starting material showed complete conversion to product by TLC, solvent was removed under reduced pressure and reaction mixture was purified by silica chromatography.
General Procedure (C): Benzylic bromination of the C-6 methyl group.
To a round-bottom flask containing N-protected 6-methyl THQ intermediate (1.00 eq) under Ar atmosphere was added degassed, Ar-sparged CCl4, followed by N-bromosuccinimide (1.05 eq) and benzoyl peroxide (0.1 eq). Reaction was then heated to reflux, monitored by TLC. Quantitative conversion of starting material was generally not observed, so reaction was halted when side-product began to form. Reaction was halted by cooling to −20°C, and precipitate was filtered from solution (washing with additional cold CCl4). Filtrate was then concentrated onto silica and purified by silica chromatography.
General Procedure (D): Suzuki coupling of benzylic bromide to R2-boronic acid.
To a round-bottom flask under Ar atmosphere was added 3:1 acetone/water and stirred under vacuum for 10 minutes. Next, Ar was bubbled through solvent for an additional 10 minutes before adding benzylic bromide intermediate (1.0 eq), boronic acid (1.2–2.0 eq), K2CO3 (3 eq) and Pd(dppf)Cl2 (0.1 eq). Reaction was heated to 80°C for 6–12 hours, after which the reaction mixture was cooled and diluted with ethyl acetate and aqueous NaHCO3. The organic layer was separated and dried over MgSO4, then filtered and concentrated onto silica. Product was purified by silica chromatography.
General Procedure (E): Substitution of benzylic bromide with tetrahydroisoquinline (THIQ).
To a round-bottom flask under inert atmosphere was added DMF, followed by K2CO3 (1.2 eq) and THIQ (1.2 eq), then benzylic bromide (1.0 eq) stirring at room temperature (r.t.). After 6–12 hours, solvent was removed under reduced pressure and residual oil was resuspended in ethyl acetate and sat. NaHCO3. The organic layer was separated and dried over MgSO4, then filtered and concentrated onto silica. Product was purified by silica chromatography.
General Procedure (F): Reductive amination of THQ ketone to sulfinamide using Ellman’s sulfinamide.
To a round bottom flask already containing desiccated THQ intermediate (1.0 eq) under Ar atmosphere was added (R)-2-methylpropane-2-sulfinamide (3.0 eq). Meanwhile, a reflux condenser was flame-dried under vacuum, and then flooded with Ar. Next, anhydrous THF (5–10 mL) was added to the reaction vessel containing starting reagents via syringe. The round bottom flask was placed in an ice bath and allowed to equilibrate to 0°C. Next, Ti(OEt)4 (6.0 eq) was added slowly via syringe. Once addition was complete, the reaction vessel was taken out of ice bath and placed in oil bath at 70°C-75°C, affixed condenser, and stirred for 16–48 h under Ar. The reaction was monitored by TLC for loss of ketone. Once sufficient conversion to the tert-butanesulfinyl imine was observed, reaction vessel was taken out of oil bath and cooled to r.t. Meanwhile, an additional round bottom flask was flame-dried under vacuum, then flooded with Ar. NaBH4 (6.0 eq) was added quickly, and anhydrous THF was added (5–10 mL). The round bottom flask was placed in dry ice/acetone bath and allowed to equilibrate to −78°C. Contents from the round bottom flask containing the imine intermediate were transferred to flask containing NaBH4 via cannula. Once contents were completely added, the reaction was taken out of dry ice/acetone bath and was allowed to warm to room temperature. The reaction stirred at r.t. for 2–3 h. To quench, 5–10 mL of saturated NaCl in H2O was added. Reaction mixture was diluted with ethyl acetate and DI H2O. The organic layer was then isolated and dried over MgSO4 and filtered. Organic extract was then concentrated onto silica and purified by silica chromatography.
General Procedure (G): Conversion of sulfinamide to final compound – 1) Boc removal 2) sulfinamide cleavage 3) amide coupling 4) Boc deprotection.
Step 1: If N-1 substitution is Boc, sulfinamide is first treated with 1:1 TFA/DCM (10 mL), which is then removed under reduced pressure. Otherwise, proceed to step 2. Step 2: To a round bottom flask containing sulfinamide (1.0 eq) was added 1,4-dioxane, followed by conc. HCl (6.0 eq), cleaving the sulfinamide to the primary amine. The reaction stirred at r.t. for up to 3 h. Solvent was removed under reduced pressure and residue was re-suspended in Et2O. The resultant white solid precipitate (the HCl salt of the amine) was isolated by decanting and washing with Et2O up to three times. After desiccation, the solid residue was used without further purification. Step 3: To a pear-shaped flask under inert atmosphere containing amine salt (1.0 eq) was added di-Boc-Dmt (1.1 eq), PyBOP (1.1 eq), and, when specified, 6-Cl HOBt (1.1 eq), followed by DMF (10 mL) and DIPEA (10 eq) at r.t. After stirring for 6 hours, solvent was removed under reduced pressure and residual oil was dry-loaded onto silica. Boc-protected intermediate was purified by silica chromatography. Step 4: Boc-protected intermediate was suspended in DCM (10 mL), then TFA (3–5 mL) was added. After 1 hour, solvent was removed under vacuum. Product was resuspended in a solution of 99.9% acetonitrile, 0.1% TFA, then diluted with DI H2O. Final products were purified by reverse-phase semi-preparative HPLC. Final yield not calculated.
Cell Lines and Membrane Preparations.
All tissue culture reagents were purchased from Gibco Life Sciences (Grand Island, NY, U.S.). C6-rat glioma cells stably transfected with a rat MOR (C6-MOR) or rat DOR (C6-DOR) and Chinese hamster ovary (CHO) cells stably expressing a human KOR (CHO-KOR) were used for all in vitro assays. Cells were grown to confluence at 37 °C in 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum and 5% penicillin/streptomycin. Membranes were prepared by washing confluent cells three times with ice cold phosphate buffered saline (0.9% NaCl, 0.61 mM Na2HPO4, 0.38 mM KH2PO4, pH 7.4). Cells were detached from the plates by incubation in warm harvesting buffer (20 mM HEPES, 150 mM NaCl, 0.68 mM EDTA, pH 7.4) and pelleted by centrifugation at 1600 rpm for 3 min. The cell pellet was suspended in ice-cold 50 mM Tris- HCl buffer, pH 7.4, and homogenized with a Tissue Tearor (Biospec Products, Inc., Bartlesville, OK, U.S.) for 20 s. The homogenate was centrifuged at 15,000 rpm for 20 min at 4°C. The pellet was rehomogenized in 50 mM Tris-HCl with a Tissue Tearor for 10 s, followed by recentrifugation. The final pellet was resuspended in 50 mM Tris-HCl and frozen in aliquots at 80°C. Protein concentration was determined via a bicinchonic assay (BCA) protein assay42 (Thermo Scientific Pierce, Waltham, MA, U.S.) using bovine serum albumin as the standard. Briefly, 5 mL of reagent A was combined with 100 uL of Reagent B and mixed thoroughly. In a clear 96 well plate, 200 uL of the activated reagent was added to each well, along with 25 uL of test sample. Both membrane preparations and samples for the standard curve were run in triplicate. The plate was then incubated for 30 mins at 37C. The amount of protein present in solution was quantified by measuring the absorption spectra at 562nm and an equation for the standard curve was generated using a linear regression fit. The concentration of protein in each membrane preparation was calculated using this equation.
Radioligand Competition Binding Assays.
Radiolabeled compounds were purchased from Perkin-Elmer (Waltham, MA, U.S.). Opioid ligand binding assays were performed by competitive displacement of 0.2 nM [3H]-diprenorphine (250 μCi, 1.85 TBq/mmol) by the peptidomimetic from membrane preparations containing opioid receptors as described above. The assay mixture, containing membranes (20 μg protein/tube) in 50 mM Tris-HCl buffer (pH 7.4), [3H]-diprenorphine, and various concentrations of test peptidomimetic, was incubated at room temperature on a shaker for 1 h to allow binding to reach equilibrium. Samples were rapidly filtered through Whatman GF/C filters using a Brandel harvester (Brandel, Gaithersburg, MD, U.S.) and washed three times with 50 mM Tris-HCl buffer. Bound radioactivity on dried filters was determined by liquid scintillation counting, after saturation with EcoLume liquid scintillation cocktail, in a Wallac 1450 MicroBeta (Perkin-Elmer, Waltham, MA, U.S.). Nonspecific binding was determined using 10 μM naloxone. The results presented are the mean ± standard error (S.E.M.) from at least three separate assays performed in duplicate. Ki (nM) values were calculated using nonlinear regression analysis to fit a logistic equation to the competition data using GraphPad Prism, version 6.0c (GraphPad Software Inc., La Jolla, CA).
[35S]-GTPγS Binding Assays.
Agonist stimulation of [35S]guanosine 5′-O-[γ- thio]triphosphate [35S]-GTPγS, 1250 Ci, 46.2 TBq/mmol) binding to G protein was measured as described previously.43 Briefly, membranes (10–20 μg of protein/tube) were incubated for 1 h at 25°C in GTPγS buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, pH 7.4) containing 0.1 nM [35S]-GTPγS, 30 μM guanosine diphosphate (GDP), and varying concentrations of test peptidomimetic. G protein activation following receptor activation with peptidomimetic was compared with 10 μM of the standard compounds [D-Ala2,N-MePhe4,Gly-ol]enkephalin (DAMGO) at MOR, D-Pen2,5- enkephalin (DPDPE) at DOR, or U69,593 at KOR. The reaction was terminated by vacuum filtration of GF/C filters that were washed 10 times with GTPγS buffer. Bound radioactivity was measured as previously described. The results are presented as the mean ± standard error (S.E.M.) from at least three separate assays performed in duplicate; potency (EC50 (nM)) and percent stimulation were determined using nonlinear regression analysis with GraphPad Prism, as above.
Ke Determination.
Agonist stimulation of [35S]-GTPγS binding by the known standard agonist SNC80 at DOR was measured as described above. This was then compared to [35S]-GTPγS binding stimulated by SNC80 in the presence of test compound (1 μM). Both conditions produced 100% stimulation relative to SNC80. The difference between the EC50 of SNC80 alone and in the presence of test antagonist is the shift in concentration response. The Ke was then calculated as Ke = (concentration of compound)/ (concentration response shift – 1). The results presented are the mean from at least three separate assays performed in duplicate.
In Vivo Drug Preparation.
All compounds were administered by intraperitoneal (ip) injection in a volume of 10 mL/kg of body weight. Test compounds were dissolved in 5% DMSO (v/v) in sterile saline (0.9% NaCl w/v).
Animals.
Male C57BL/6 wild type mice (Stock number 000664, Jackson Laboratory, Sacramento CA, USA) bred in-house from breeding pairs and weighing between 20–30 g at 8–16 weeks old, were used for behavioral experiments. Mice were group-housed with free access to food and water at all times. Experiments were conducted in the housing room, maintained on a 12 h light/dark cycle with lights on at 7:00 am; all experiments were conducted during the light cycle. Studies were performed in accordance with the University of Michigan Committee on the Use and Care of Animals and the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011 publication).
Antinociception.
Antinociceptive effects were evaluated in the mouse WWTW assay. Withdrawal latencies were determined by briefly placing a mouse into a cylindrical plastic restrainer and immersing 2–3 cm of the tail tip into a water bath maintained at 50°C. The latency to tail withdrawal or rapidly flicking the tail back and forth was recorded with a maximum cutoff time of 20 s to prevent tissue damage. Antinociceptive effects were determined using a cumulative dosing procedure. Each mouse received an injection of saline ip and then 30 min later baseline withdrawal latencies were recorded. Following baseline determinations, cumulative doses of each test compound (1, 3.2, and 10 mg/kg) were given ip at 30 min intervals. Thirty min after each injection, the tail withdrawal latency was measured as described above. To determine the duration of antinociceptive action, baseline latencies were determined as described above. Thirty minutes after baseline determination, animals were given a 10 mg/kg bolus injection of test compound ip. Latency to tail withdrawal was then determined at 5, 15, and 30 min after injections, and every 30 min thereafter until latencies returned to baseline values.
Supplementary Material
Acknowledgments
Funding Sources
This study was supported by NIH grant DA003910 (H.I.M, E.M.J and J.R.T). J.P.A. was supported by DA003910 and the Biology of Drug Abuse Training Program DA007268. D.J.M. was funded through ACS MEDI Predoctoral Fellowship. N.W.G. was supported by the NIDA Training Program in Neuroscience (NTPiN, DA007281–22) and the Endowment for the Development of Graduate Education (EDGE) award. T.J.F. was funded by a PREP R25 grant.
Abbreviations
- MOR
μ-opioid receptor
- DOR
δ-opioid receptor
- KOR
κ-opioid receptor
- ClogP
calculated logarithm of the partition coefficient between octanol and water
- THQ
tetrahydroquinoline
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
- DPDPE,
[D-Pen2,D-Pen5]-enkephalin
- WWTW
warm water tail withdrawal
- MPE
maximum possible effect
- THIQ
tetrahydroisoquinoline
- SAR
structure-activity relationship
Footnotes
Supporting information
Synthesis and characterization of test compounds
Molecular formula strings (CSV)
Conflict of Interest
The authors declare no competing financial interest.
References
- (1).Brownstein MJ A Brief History of Opiates, Opioid Peptides, and Opioid Receptors. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (June), 5391–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Devereaux AL; Mercer SL; Cunningham CW DARK Classics in Chemical Neuroscience : Morphine. ACS Chem. Neurosci. 2018, 9 (10), 2395–2407. [DOI] [PubMed] [Google Scholar]
- (3).Bailey CP; Connor M Opioids : Cellular Mechanisms of Tolerance and Physical Dependence. Curr. Opin. Pharmacol. 2005, 5, 60–68. [DOI] [PubMed] [Google Scholar]
- (4).Beckett AH; Casy AF Synthetic Analgesics: Stereochemical Considerations. J. Pharm. Pharmacol. 1954, 6, 986–999. [DOI] [PubMed] [Google Scholar]
- (5).Portoghese PS A New Concept on the Mode of Interaction of Narcotic Analgesics with Receptors. J. Med. Chem. 1965, 8, 609–616. [DOI] [PubMed] [Google Scholar]
- (6).Martin WR; Eades CG; Thompson JA; Huppler RE; Gilbert PE The Effects of Morphine- and Nalorphine-like Drugs in the Nondependent and Morphine-Dependent Chronic Spinal Dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [PubMed] [Google Scholar]
- (7).Bohn LM; Lefkowitz RJ; Gainetdinov RR; Peppel K; Caron MG; Lin FT Enhanced Morphine Analgesia in Mice Lacking β-Arrestin 2. Science (80-. ). 1999, 286 (5449), 2495–2498. [DOI] [PubMed] [Google Scholar]
- (8).Raehal KM; Schmid CL; Groer CE; Bohn LM Functional Selectivity at the -Opioid Receptor: Implications for Understanding Opioid Analgesia and Tolerance. Pharmacol. Rev. 2011, 63 (4), 1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171 (5), 1165.e13–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Koblish M; Carr R; Siuda ER; Rominger DH; Gowen-MacDonald W; Cowan CL; Crombie AL; Violin JD; Lark MW TRV0109101, a G Protein-Biased Agonist of the μ -Opioid Receptor, Does Not Promote Opioid-Induced Mechanical Allodynia Following Chronic Administration. J. Pharmacol. Exp. Ther. 2017, 362 (2), 254–262. [DOI] [PubMed] [Google Scholar]
- (11).Manglik A; Lin H; Aryal DK; McCorvy JD; Dengler D; Corder G; Levit A; Kling RC; Bernat V; Hübner H; Huang X-P; Sassano MF; Giguère PM; Löber S; Da Duan; Scherrer G; Kobilka BK; Gmeiner P; Roth BL; Shoichet BK Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537 (7619), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hertz S AADPAC Briefing Document NDA 210730 Oliceridine Injection; 2018. [Google Scholar]
- (13).Hill R; Disney A; Conibear A; Sutcliffe K; Dewey W; Husbands S; Bailey C; Kelly E; Henderson G The Novel μ-Opioid Receptor Agonist PZM21 Depresses Respiration and Induces Tolerance to Antinociception. Br. J. Pharmacol. 2018, 175 (13), 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Turnaturi R; Aricò G; Ronsisvalle G; Parenti C; Pasquinucci L Multitarget Opioid Ligands in Pain Relief: New Players in an Old Game. Eur. J. Med. Chem. 2016, 108, 211–228. [DOI] [PubMed] [Google Scholar]
- (15).Schiller PW Bi- or Multifunctional Opioid Peptide Drugs. Life Sci. 2010, 86 (15–16), 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dietis N; Guerrini R; Calo G; Salvadori S; Rowbotham DJ; Lambert DG Simultaneous Targeting of Multiple Opioid Receptors: A Strategy to Improve Side-Effect Profile. Br. J. Anaesth. 2009, 103 (1), 38–49. [DOI] [PubMed] [Google Scholar]
- (17).Lagard C; Chevillard L; Guillemyn K; Risède P; Laplanche JL; Spetea M; Ballet S; Mégarbane B Bifunctional Peptide-Based Opioid Agonist/Nociceptin Antagonist Ligand for Dual Treatment of Nociceptive and Neuropathic Pain. Pain 2017, 158 (3), 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ding H; Czoty PW; Kiguchi N; Cami-Kobeci G; Sukhtankar DD; Nader MA; Husbands SM; Ko M-C A Novel Orvinol Analog, BU08028, as a Safe Opioid Analgesic without Abuse Liability in Primates. Proc. Natl. Acad. Sci. 2016, 113 (37), E5511–E5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ding H; Kiguchi N; Yasuda D; Daga PR; Polgar WE; Lu JJ; Czoty PW; Kishioka S; Zaveri NT; Ko MC A Bifunctional Nociceptin and Mu Opioid Receptor Agonist Is Analgesic without Opioid Side Effects in Nonhuman Primates. Sci. Transl. Med. 2018, 10 (456), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Abdelhamid EE; Sultana M; Portoghese PS; Takemori AE Selective Blockage of Delta Opioid Receptors Prevents the Development of Morphine Tolerance and Dependence in Mice. J. Pharmacol. Exp. Ther. 1991, 258 (1), 299–303. [PubMed] [Google Scholar]
- (21).Hepburn MJ; Little PJ; Gingras J; Kuhn CM Differential Effects of Naltrindole on Morphine-Induced Tolerance and Physical Dependence in Rats. J. Pharmacol. Exp. Ther. 1997, 281 (3), 1350–1356. [PubMed] [Google Scholar]
- (22).Fundytus ME; Schiller PW; Shapiro M; Weltrowska G; Coderre TJ Attenuation of Morphine Tolerance and Dependence with the Highly Selective δ-Opioid Receptor Antagonist TIPP[ψ]. Eur. J. Pharmacol. 1995, 286 (1), 105–108. [DOI] [PubMed] [Google Scholar]
- (23).Schiller PW; Fundytus ME; Merovitz L; Weltrowska G; Nguyen TM; Lemieux C; Chung NN; Coderre TJ The Opioid Mu Agonist/Delta Antagonist DIPP-NH(2)[Psi] Produces a Potent Analgesic Effect, No Physical Dependence, and Less Tolerance than Morphine in Rats. J. Med. Chem. 1999, 42 (18), 3520–3526. [DOI] [PubMed] [Google Scholar]
- (24).Wells JL; Bartlett JL; Ananthan S; Bilsky EJ In Vivo Pharmacological Characterization of SoRI 9409, a Nonpeptidic Opioid Mu-Agonist/Delta-Antagonist That Produces Limited Antinociceptive Tolerance and Attenuates Morphine Physical Dependence. J. Pharmacol. Exp. Ther. 2001, 297 (2), 597–605. [PubMed] [Google Scholar]
- (25).Zhu Y; King M. a.; Schuller AGP; Nitsche JF; Reidl M; Elde RP; Unterwald E; Pasternak GW; Pintar JE Retention of Supraspinal Delta-like Analgesia and Loss of Morphine Tolerance in δ Opioid Receptor Knockout Mice. Neuron 1999, 24 (1), 243–252. [DOI] [PubMed] [Google Scholar]
- (26).Ananthan S Opioid Ligands With Mixed Mu / Delta Opioid Receptor Interactions : An Emerging Approach to Novel Analgesics. AAPS J. 2006, 8 (1), 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Deekonda S; Wugalter L; Rankin D; Largent-Milnes TM; Davis P; Wang Y; Bassirirad NM; Lai J; Kulkarni V; Vanderah TW; Porreca F; Hruby VJ Design and Synthesis of Novel Bivalent Ligands (MOR and DOR) by Conjugation of Enkephalin Analogues with 4-Anilidopiperidine Derivatives. Bioorganic Med. Chem. Lett. 2015, 25 (20), 4683–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Daniels DJ; Lenard NR; Etienne CL; Law P-Y; Roerig SC; Portoghese PS Opioid-Induced Tolerance and Dependence in Mice Is Modulated by the Distance between Pharmacophores in a Bivalent Ligand Series. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (52), 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lenard NR; Daniels DJ; Portoghese PS; Roerig SC Absence of Conditioned Place Preference or Reinstatement with Bivalent Ligands Containing Mu-Opioid Receptor Agonist and Delta-Opioid Receptor Antagonist Pharmacophores. Eur. J. Pharmacol. 2007, 566 (1–3), 75–82. [DOI] [PubMed] [Google Scholar]
- (30).Mosberg HI; Yeomans L; Anand JP; Porter V; Sobczyk-Kojiro K; Traynor JR; Jutkiewicz EM Development of a Bioavailable μ Opioid Receptor (MOPr) Agonist, δ Opioid Receptor (DOPr) Antagonist Peptide That Evokes Antinociception without Development of Acute Tolerance. J. Med. Chem. 2014, 57 (7), 3148–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Anand JP; Kochan KE; Nastase AF; Montgomery D; Griggs NW; Traynor JR; Mosberg HI; Jutkiewicz EM In Vivo Effects of μ-Opioid Receptor Agonist/δ-Opioid Receptor Antagonist Peptidomimetics Following Acute and Repeated Administration. Br. J. Pharmacol. 2018, 175 (11), 2013–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Healy JR; Bezawada P; Shim J; Jones JW; Kane MA; MacKerell AD; Coop A; Matsumoto RR Synthesis, Modeling, and Pharmacological Evaluation of UMB 425, a Mixed μ Agonist/δ Antagonist Opioid Analgesic with Reduced Tolerance Liabilities. ACS Chem. Neurosci. 2013, 4 (9), 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wang C; McFayden I; Traynor JR; Mosberg HI Design of a High Affinity Peptidomimetic Opioid Agonist from Peptide Pharmacophore Models. Bioorganic Med. Chem. Lett. 1998, 8, 2685–2688. [DOI] [PubMed] [Google Scholar]
- (34).Mosberg HI; Yeomans L; Harland A. a.; Bender AM; Sobczyk-Kojiro K; Anand JP; Clark MJ; Jutkiewicz EM; Traynor JR Opioid Peptidomimetics: Leads for the Design of Bioavailable Mixed Efficacy μ Opioid Receptor (MOR) Agonist/δ Opioid Receptor (DOR) Antagonist Ligands. J. Med. Chem. 2013, 56 (5), 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Harland AA; Yeomans L; Griggs NW; Anand JP; Pogozheva ID; Jutkiewicz EM; Traynor JR; Mosberg HI Further Optimization and Evaluation of Bioavailable, Mixed-Efficacy Mu-Opioid Receptor (MOR) Agonists/Delta-Opioid Receptor (DOR) Antagonists: Balancing MOR and DOR Affinities. J. Med. Chem. 2015, 58 (22), 8952–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bender AM; Griggs NW; Anand JP; Traynor JR; Jutkiewicz EM; Mosberg HI Asymmetric Synthesis and in Vitro and in Vivo Activity of Tetrahydroquinolines Featuring a Diverse Set of Polar Substitutions at the 6 Position as Mixed-E Fficacy μ Opioid Receptor/ δ Opioid Receptor Ligands. ACS Chem. Neurosci. 2015, 6, 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Balboni G; Marzola E; Sasaki Y; Ambo A; Marczak ED; Lazarus LH; Salvadori S Role of 2′,6′-Dimethyl-l-Tyrosine (Dmt) in Some Opioid Lead Compounds. Bioorganic Med. Chem. 2010, 18 (16), 6024–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Harland AA; Bender AM; Griggs NW; Gao C; Anand JP; Pogozheva ID; Traynor JR; Jutkiewicz EM; Mosberg HI Effects of N-Substitutions on the Tetrahydroquinoline (THQ) Core of Mixed-Efficacy μ-Opioid Receptor (MOR)/δ-Opioid Receptor (DOR) Ligands. J. Med. Chem. 2016, 59 (10), 4985–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Anand JP; Kochan KE; Nastase AF; Montgomery D; Griggs NW; Traynor JR; Mosberg HI; Jutkiewicz EM In Vivo Effects of μ Opioid Receptor Agonist/δ Opioid Receptor Antagonist Peptidomimetics Following Acute and Repeated Administration. Br. J. Pharmacol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Stevenson GW; Folk JE; Linsenmayer DC; Rice KC; Negus SS Opioid Interactions in Rhesus Monkeys : Effects of Delta + Mu and Delta + Kappa Agonists on Schedule-Controlled Responding and Thermal Nociception. J. Pharmacol. Exp. Ther. 2003, 307 (3), 1054–1064. [DOI] [PubMed] [Google Scholar]
- (41).Lowery JJ; Raymond TJ; Giuvelis D; Bidlack JM; Polt R; Bilsky EJ In Vivo Characterization of MMP-2200, a Mixed Delta/Mu Opioid Agonist, in Mice. J. Pharmacol. Exp. Ther. 2011, 336 (3), 767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Smith PK; Krohn RI; Hermanson GT; Mallia AK; Gartner FH; Provenzano MD; Fujimoto EK; Goeke NM; Olson BJ; Klenk DC Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, No. 150, 76–85. [DOI] [PubMed] [Google Scholar]
- (43).Traynor JR; Nahorski SR Modulation by Mu-Opioid Agonists of Guanosine-5’-O-(3-[35S]Thio)Triphosphate Binding to Membranes from Human Neuroblastoma SH-SY5Y Cells. Mol. Pharmacol. 1995, 47 (4), 848–854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.