

Abstract
Premise:
Migraine is a complex neurologic disorder that leads to significant disability, yet remains poorly understood.
Problem:
One potential triggering mechanism in migraine with aura is cortical spreading depression, which can activate the trigeminal nociceptive system both peripherally and centrally in animal models. A primary neuropeptide of the trigeminal system is calcitonin gene-related peptide, which is a potent vasodilatory peptide and is currently a major therapeutic target for migraine treatment. Despite the importance of both cortical spreading depression and calcitonin gene-related peptide in migraine, the relationship between these two players has been relatively unexplored. However, recent data suggest several potential vascular and neural connections between calcitonin gene-related peptide and cortical spreading depression.
Conclusion:
This review will outline calcitonin gene-related peptide-cortical spreading depression connections and propose a model in which cortical spreading depression and calcitonin gene-related peptide act at the intersection of the vasculature and cortical neurons, and thus contribute to migraine pathophysiology.
Keywords: Neurovasculature, cortical spreading depression, trigeminal nerve, vasodilation
Introduction
Migraine is a complex neurologic disorder that leads to significant disability worldwide. A common component of these headaches for many migraineurs is the aura, which precedes migraine symptoms in up to one third of patients (1). Migraine aura may include visual, sensory, language, or brainstem symptoms. The pathologic mechanism by which migraine aura develops has not been fully elucidated. To date, cortical spreading depression (CSD) has been hypothesized to be the phenomenon that causes migraine aura (2,3). Lashley’s description of his own scintillation-scotomas (4) are suggestive of disruption in cortical function with return to normal, akin to CSD. This indeed has been demonstrated in both human and animal models using functional magnetic resonance imaging (fMRI) (5,6). In addition to the role that CSD plays in migraine aura, another key player in migraine pathophysiology that has been identified is calcitonin gene-related peptide (CGRP). CGRP was first noted to be elevated in extracerebral circulation during migraine by Goadsby et al. in 1990 (7). Since that time, CGRP has been recognized as playing a key role in migraine pathophysiology, and thus has become a therapeutic target (8). This review will first describe key aspects of CSD and CGRP, attempt to highlight overlaps between the CGRP actions and CSD events in migraine, and propose a model by which each of these contributes to migraine pathophysiology.
Migraine aura pathophysiology and CSD
The pathogenesis of migraine is complex and involves both the central and peripheral nervous systems (9,10). Centrally, electrophysiology and imaging studies have shown involvement of many areas of the brain, including cerebral cortex, thalamus, hypothalamus, and brainstem (2,11–14). The exact mechanisms of migraine are still undetermined and there is still great debate regarding neural versus vascular based migraine models. Regardless, the trigeminovascular system plays a key role (15–17). In migraine aura, there is evidence to support the hypothesis that CSD is an underlying physiologic process that could contribute to activation of the trigeminal nerve in migraine (18,19).
CSD was first described by Leao in 1944 (20), when he noted “marked, enduring reduction in the spontaneous electrical activity of the cortex” in a rabbit model of experimental epilepsy. CSD is a massive wave of depolarization spreading through grey matter, resulting in a decrease in spontaneous cortical activity. The wave propagates slowly across the cortex at a rate that has generally been reported as 2–5 mm/min (20–22). This results in changes in synaptic activity, extracellular ion concentrations, blood flow, and metabolism (23–25). Spontaneous and evoked synaptic activity is decreased for 5–15 minutes, with a spontaneous return to basal activity under normal circumstances. As detected with electroencephalogram (EEG), there is a marked decrease in activity for 30–60 seconds, followed by a brief period of hyperexcitability (20). This is followed by complete loss of neuronal activity, which can last up to a minute before a return to normal baseline function.
Associated with this wave of depolarization characteristic of CSD are local ionic shifts and release of neurotransmitters. This includes an increase in extracellular potassium, and a decrease in extracellular sodium, chloride, and calcium (23). These ionic shifts in turn cause neuronal swelling, dendritic beading, and a decrease in the extracellular space. Although astrocytes and glia are known to play a role in CSD, their cell volume remains constant during this process (26). Within neurons, swelling and dendritic beading leads to release of amino acids and neurotransmitters, which continue to propagate the spread of depolarization. Glutamate has been identified as one of these excitatory neurotransmitters involved in the initiation and propagation of CSD. Glutamate release and activation of N-methyl-D-aspartic acid (NMDA) receptors possibly leads to sustained depolarization. This increase in glutamate in turn results in an increase in production of nitric oxide (NO) and arachidonic acid metabolites (25).
The depolarization wave causes vasodilation and an increase in regional cerebral blood flow, which has been termed spreading hyperemia. The duration of this increase in blood flow lasts approximately 1–2 minutes, longer than the initial CSD event, and is followed by a prolonged period of hypoperfusion lasting 1–2 hours (23). This has been termed spreading oligemia. These changes in cerebral blood flow and oxygenation, as well as the increased metabolic needs associated with CSD, lead to a mismatch of supply and demand, and normal mechanisms of cerebrovascular homeostasis are overwhelmed. In multiple models of brain injury, these changes in blood flow have been linked to progression of ischemia, worsening secondary injury, and worsening long-term patient outcomes (21,27,28). In migraine, however, it is unclear how the changes in cerebral blood flow associated with CSD contribute to migraine aura and/or headache physiology.
Despite the characteristic changes in cerebral blood flow that are associated with CSD, to date there is not sufficient evidence to draw a direct link between CSD-induced hyperemia/oligemia and migraine aura. Imaging studies during migraine show ischemia via BOLD signal changes in fMRI. Similarly, studies in animals have demonsrated that a single CSD event was sufficient to induce a profound decrease in blood flow to the dura (29). Changes in blood flow to the meninges have subsequently been shown to activate neurons in the trigeminal ganglia (TG) (30). Multiple recent studies have show that CSD can also activate the TG. Thus there are multiple lines of evidence highlighting the role of CSD in migraine pathophysiology.
Role of CGRP in migraine
CGRP is recognized as a key player in migraine. It is a vasoactive neuropeptide that is widely expressed in both the central and peripheral nervous systems, and affects nearly every organ in the body (31). It leads to NO-independent vasodilation through direct action on smooth muscle cells, but can also stimulate endothelial production of NO, thus contributing to vasodilation via two pathways (32). In migraine, CGRP may mediate neurogenic inflammation and modulate nociceptive inputs (8), likely via action in the trigeminovascular system. There is mounting evidence supporting the role of CGRP in migraine physiology, for a comprehensive review of the role of CGRP in migraine, the reader is referred to the review by Russo (8). Consequently, CGRP has become a therapeutic target in the treatment of migraine. Both CGRP receptor antagonists and antibodies against CGRP and its receptor are undergoing clinical trials for migraine treatment (33–36). Examining central and peripheral CGRP, the wide distribution of the molecule and its receptors would allow for it to modulate many aspects of migraine, including pain and the characteristic features of light and sound sensitivity, nausea and central sensitization (8).
How might CGRP and CSD overlap in migraine?
To date, there is little evidence unambiguously connecting CGRP and CSD. Nonetheless, there are several intriguing observations that support connections between the two. The first line of evidence is the role that each plays in changes in cerebral vasculature. Multiple studies in animals and humans have tied CSD events to changes in blood flow, starting with Leao’s pioneering work in CSD (20,37–39). A recent review by Ayata and Lauritzen (40) nicely summarized the characteristic changes in cerebral blood flow during CSD. Another review by Hoffmann et al. described the neurovascular mechanisms underlying migraine and cluster headache (41). CGRP is also known to affect cerebral vasculature (42,43). Release of cortical CGRP has been shown to occur during exposure to elevated concentrations of potassium, which occurs during CSD (44,45). Release of CGRP from trigeminovascular nerves can lead to modulation of pial and meningeal vessels innervated by the trigeminal ganglion (46,47). The initial hyperemia seen in CSD has been shown to be mediated in part by the release of CGRP from ipsilateral trigeminal nerve fibers (39,48), and activation of trigeminal sensory and parasympathetic nerve fibers (39,49). Additionally, by transecting the trigeminal and parasympathetic nerve fibers, CSD-dependent delayed hyperemia was eliminated (50). This reinforces the role that these nerve fibers play in vascular changes, and thus supports the role that CSD plays in activation of the trigeminal nociceptive system in migraine.
The second line of evidence that CGRP and CSD overlap in migraine pathophysiology is that CGRP antagonism can modulate CSD. The possibility that CGRP might modulate the extent of CSD events was first suggested with in vitro cortical slice studies by Tozzi et al. (45). These studies showed that blockage of CGRP receptors in vitro resulted in inhibition of CSD. Similar findings have been reported by Wang et al. (51,52). In this most recent preliminary report, intracerebral ventricular perfusion of an anti-CGRP antibody into rat reduced susceptibility to CSD. This preliminary study also showed an antibody against calcitonin receptor-like receptor (CLR) markedly reduced susceptibility to CSD both in vitro and in vivo. In addition, a recent study also found that olcegepant, a CGRP receptor antagonist, administered systemically to mice in vivo, resulted in inhibition of repetitive CSD events and altered the vascular response to CSD (53). Inhibtion of CSD by olcegepant and other small molecule CGRP receptor antagonists was initially believed to demonstrate involvement of the CGRP receptor (CLR and RAMP1), which is expressed in the cerebral cortex (54) and cerebral vasculature (55). However, CGRP can also act via the amylin 1 (AMY1) receptor (56), and although these small molecules have higher affinity at the CGRP receptor than the AMY1 receptor, they may also block CGRP activity at the AMY1 receptor (57). Therefore, the in vivo effects of the CGRP receptor antagonists reported in these studies may be due to blockage of both receptors. Likewise, topical application of a CGRP receptor antagonist has also been shown to decrease CSD-induced vasodilation in pial arteries (58). However, systemic application of MK-8825, a highly selective and potent antagonist at rat CGRP receptors, failed to block CSD and did not alter changes in cerebral blood flow, but did attenuate pain behaviors (59). Further, the anti-CGRP antibody fremanezumab given intravenously in rats was able to prevent activation of central trigeminovascular neurons induced by CSD (60). Together, these data indicate that CGRP or activation of its receptor(s) may contribute to CSD propagation. However, to be clear, there is no evidence to date that CGRP can actually trigger CSD events, only that blockage of CGRP can modulate CSD.
The third line of evidence linking CGRP and CSD is that CSD can increase CGRP synthesis and release in the cerebral cortex. We have recently shown that multiple CSD events were sufficient to induce upregulation of the CGRP gene and increase peptide levels in multiple brain regions (61). In addition, CGRP was released from cortical slices during in vitro generated CSD (45). Similarly, another recent study showed that CSD resulted in an upregulation of CGRP positive cells in the rat trigeminal ganglion (62). Increased CGRP levels following CSD could contribute to a positive feedback loop, as discussed below.
Neurovascular coupling could be the important intersection between these vascular and neural roles. This is the dynamic process by which neuronal interactions with blood vessels result in regulation of regional cerebral blood flow in order to maintain adequate perfusion (63). Evidence suggests local neuronal signaling can act on nearby vessels to regulate vascular tone (64). More recently, this has been shown to be a bi-directional relationship, where changes in vascular tone, at least in vitro, can in turn modulate neuronal activity, termed vascularneuro coupling (65,66). Furthermore, pathologic inverse coupling has been associated with CSD. Hinzman et al. (67) were able to demonstrate that inverse neurovascular coupling was increasingly associated with CSD in patients with traumatic brain injury (TBI), leading to exacerbation of existing ischemic conditions and worse clinical outcomes. Based on the results of this landmark study, they proposed inverse neurovascular coupling as a novel mechanism of secondary injury in TBI.
Proposed model: Bidirectional communication by CGRP at the intersection of cortical neurons and the vasculature
The following model highlights possible links between CSD and CGRP (Figure 1). This model addresses both central and peripheral changes to the vasculature and neurons triggered by CGRP following CSD events. This model does not include the vascular changes triggered by CSD, and does not propose how CGRP might contribute to CSD. However, this model does highlight how CSD could result in elevated CGRP and subsequent alterations in signaling, which in turn could contribute to the pathophysiology of migraine.
Figure 1.
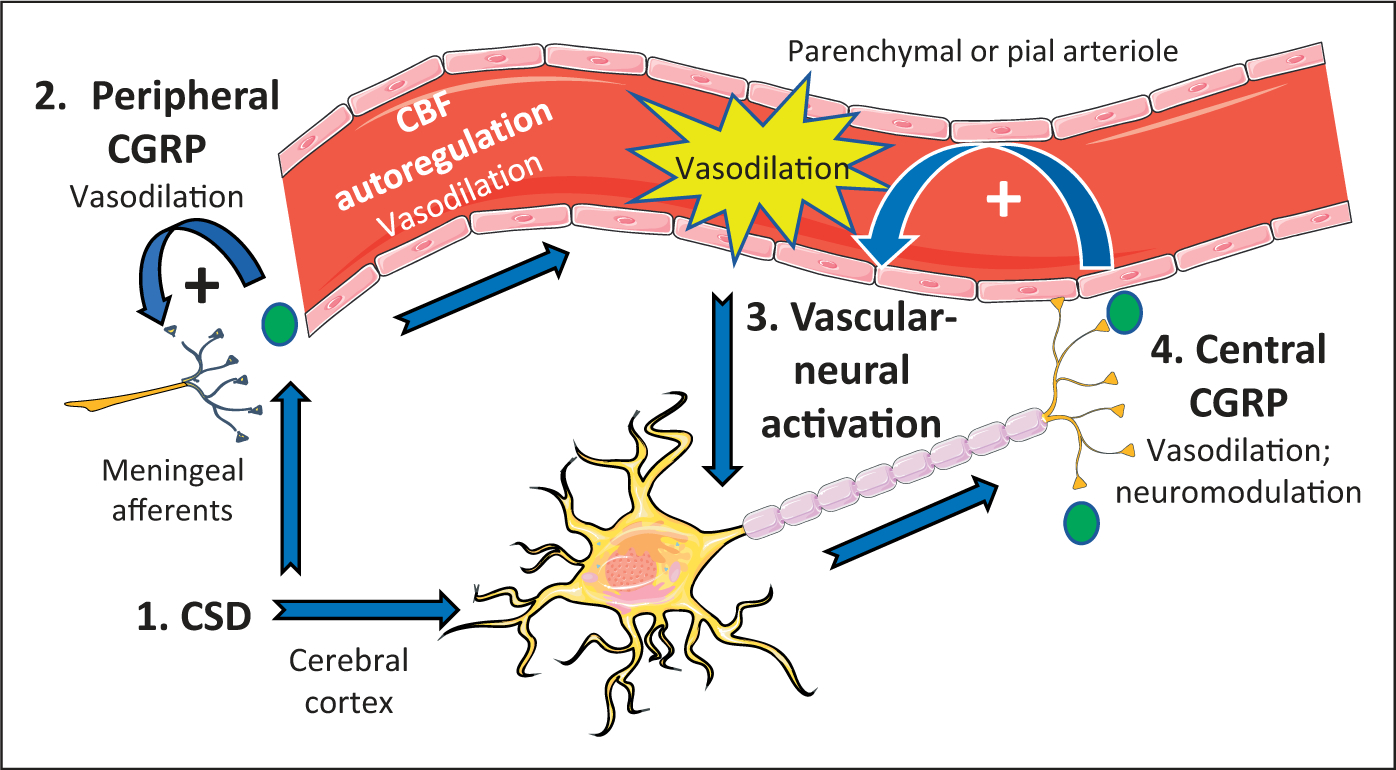
Model of CGRP at the intersection of vascular-neural communication. The steps involved in this bidirectional communication are organized from 1 to 4. Step 1: CSD activates meningeal afferents of the trigeminal nerve and multiple CSD events can increase central CGRP mRNA and peptide release in the cerebral cortex. Step 2: Peripheral CGRP released from trigeminal afferents causes vasodilation and alters the trigeminovascular microenvironment, resulting in more CGRP release. This positive feedback loop would prolong the local vasodilation of meningeal vessels, including pial arterioles, which leads to reflex autoregulation of cerebral blood flow (CBF). This further dilates arterioles in the pia and brain parenchyma. Step 3: Increased flow in parenchymal vessels transmits a signal resulting in increased neuronal firing rates by vascular-neural coupling, as observed in cortical slices. Step 4: Release of central CGRP from cortical neurons acts as a neuromodulator to increase synaptic signaling and causes further vasodilation of parenchymal arterioles. The vasodilation would potentially create a self-sustaining positive feedback loop through vascular-neural activation to maintain increased neural activity and CGRP release. CGRP is represented by the green circles.
First, we propose that CSD triggers CGRP-mediated responses in both the cerebral cortex and peripheral meninges. In the cortex, we have shown that multiple CSD events can increase central CGRP mRNA and peptide release (61). In the meninges, CSD has been shown to activate trigeminal afferents in the meninges (50,60,68,69). Release of CGRP from these afferents could cause local vasodilation and could further alter the trigeminovascular microenvironment by mast cell degranulation, resulting in even more CGRP release. This positive feedback loop would prolong the local vasodilation of meningeal vessels, including pial arterioles. In response to the local drop in blood pressure, cerebral blood flow autoregulation would then lead to further dilation of pial and parenchymal arterioles. This autoregulation is known to involve CGRP, since peripheral administration of a CGRP antibody has been shown to reduce cerebral blood flow autoregulation of pial vessels in rats (43).
As a result of pial vasodilation, we propose that a vascular to neural signal could then potentially activate neurons and create another positive feedback loop. Evidence for such a vascular-neural coupling has recently been demonstrated with in vitro cortical slices in which pyramidal neurons were activated by increased flow through parenchymal arterioles (66). The mechanism underlying activation was not investigated, but signaling in response to decreased flow involved TRPV4 mechanoreceptors on intermediary astrocytes (66). Should vascular-neural coupling occur in vivo, this would provide a mechanism for vascular tone to modulate neural activity. Since the cortex has been shown to express CGRP (54,61,70), vascular activation of cortical neurons would then be predicted to release CGRP in the brain parenchyma. This central CGRP would potentially have two actions. First, synaptic modulation of glutamate transmission via phosphorylation of NMDA and a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, as reported in the dorsal horn and amygdala (71), would potentially increase neural signaling in the cortex. Second, parenchymal release of CGRP could cause dilation of parenchymal arterioles. Evidence that CGRP within the central nervous system can cause vasodilation in vivo has recently been shown by investigators who used CGRP as a physiological contrast tool (72). Parenchymal vasodilation would then create a positive feedback loop leading to further vascular-neural induced cortical neuron activation. The result of this proposed bidirectional communication would be a sustained increase in neural signaling mediated by CGRP, which could contribute to the pathophysiology of migraine.
Conclusion
Many questions still remain unanswered in the quest to fully understand migraine pathophysiology. Multiple lines of evidence exist in vivo and in vitro that support aspects of the hypothesis that CSD induces CGRP activity in migraine. It is likely that the neuronal depolarization and subsequent propagation of CSD leads to stimulation and upregulation of CGRP, which acts to further modulate the vasodilation associated with spreading oligemia via a central mechanism. CSD is a topic of intense research in both clinical and basic science realms, in order to better understand its role in multiple areas of cerebral pathology. Additional work in migraine as well as other models of CSD will continue to further elucidate the relationship between CSD and the associated vascular changes. CGRP, as a therapeutic target in migraine, is similarly a hot topic of research to better understand its role in migraine and migraine aura, as well as its therapeutic potential. A better understanding of this relationship between CSD and CGRP will allow for a better understanding of migraine pathophysiology and the development of more effective treatments for the management of migraine.
Article highlights.
Recent data suggest several potential vascular and neural connections between CGRP and CSD.
A model in which CSD might recruit CGRP to act at the intersection of the vasculature and cortical neurons is proposed, highlighting the overlap between CSD and CGRP in migraine pathophysiology.
Acknowledgements
The authors thank members of their labs for interesting discussions and grant support from NIH (NS075599), Veterans Affairs Medical Center (1I01RX002101), and Department of Defense USAMRAA (W81XWH-16-1-0071 and W81XWH-16-1-0211).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Cutrer FM and Huerter K. Migraine aura. Neurologist 2007; 13: 118–125. [DOI] [PubMed] [Google Scholar]
- 2.Lauritzen M Pathophysiology of the migraine aura. The spreading depression theory. Brain 1994; 117: 199–210. [DOI] [PubMed] [Google Scholar]
- 3.Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K, Liu C, et al. Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 2010; 67: 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lashley K Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiatry 1941; 46: 331–339. [Google Scholar]
- 5.Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA 2001; 98: 4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arngrim N, Hougaard A, Ahmadi K, et al. Heterogenous migraine aura symptoms correlate with visual cortex functional magnetic resonance imaging responses. Ann Neurol 2017; 82: 925–939. [DOI] [PubMed] [Google Scholar]
- 7.Goadsby PJ, Edvinsson L and Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 1990; 28: 183–187. [DOI] [PubMed] [Google Scholar]
- 8.Russo AF. Calcitonin gene-related peptide (CGRP): A new target for migraine. Annu Rev Pharmacol Toxicol 2015; 55: 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olesen J, Burstein R, Ashina M, et al. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol 2009; 8: 679–690. [DOI] [PubMed] [Google Scholar]
- 10.de Tommaso M, Ambrosini A, Brighina F, et al. Altered processing of sensory stimuli in patients with migraine. Nat Rev Neurol 2014; 10: 144–155. [DOI] [PubMed] [Google Scholar]
- 11.Schoenen J Cortical electrophysiology in migraine and possible pathogenetic implications. Clin Neurosci 1998; 5: 10–17. [PubMed] [Google Scholar]
- 12.Moulton EA, Becerra L, Johnson A, et al. Altered hypothalamic functional connectivity with autonomic circuits and the locus coeruleus in migraine. PLoS One 2014; 9: e95508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coppola G, Tinelli E, Lepre C, et al. Dynamic changes in thalamic microstructure of migraine without aura patients: A diffusion tensor magnetic resonance imaging study. Eur J Neurol 2014; 21: 287–292. [DOI] [PubMed] [Google Scholar]
- 14.Chong CD, Plasencia JD, Frakes DH, et al. Structural alterations of the brainstem in migraine. Neuroimage Clin 2017; 13: 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goadsby PJ, Lipton RB and Ferrari MD. Migraine – current understanding and treatment. N Engl J Med 2002; 346: 257–270. [DOI] [PubMed] [Google Scholar]
- 16.Messlinger K Migraine: Where and how does the pain originate? Exp Brain Res 2009; 196: 179–193. [DOI] [PubMed] [Google Scholar]
- 17.Pietrobon D and Striessnig J. Neurobiology of migraine. Nat Rev Neurosci 2003; 4: 386–398. [DOI] [PubMed] [Google Scholar]
- 18.Charles AC and Baca SM. Cortical spreading depression and migraine. Nat Rev Neurol 2013; 9: 637–644. [DOI] [PubMed] [Google Scholar]
- 19.Ayata C Spreading depression and neurovascular coupling. Stroke 2013; 44: S87–S89. [DOI] [PubMed] [Google Scholar]
- 20.Leao AA. Spreading depression of activity in the cerebral cortex. J Neurophysiology 1944; 7: 359–390. [DOI] [PubMed] [Google Scholar]
- 21.Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 2002; 33: 2738–2743. [DOI] [PubMed] [Google Scholar]
- 22.Dohmen C, Sakowitz OW, Fabricius M, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol 2008; 63: 720–728. [DOI] [PubMed] [Google Scholar]
- 23.Lauritzen M, Dreier JP, Fabricius M, et al. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 2011; 31: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charles A and Brennan K. Cortical spreading depression – new insights and persistent questions. Cephalalgia 2009; 29: 1115–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–447. [DOI] [PubMed] [Google Scholar]
- 26.Zhou N, Gordon GR, Feighan D, et al. Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex 2010; 20: 2614–2624. [DOI] [PubMed] [Google Scholar]
- 27.Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 2006; 129: 778–790. [DOI] [PubMed] [Google Scholar]
- 28.Hartings JA, Strong AJ, Fabricius M, et al. Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma 2009; 26: 1857–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert GA and Michalicek J. Cortical spreading depression reduces dural blood flow – a possible mechanism for migraine pain? Cephalalgia 1994; 14: 430–436, discussion 393–394. [DOI] [PubMed] [Google Scholar]
- 30.Goadsby PJ, Knight YE, Hoskin KL, et al. Stimulation of an intracranial trigeminally-innervated structure selectively increases cerebral blood flow. Brain Res 1997; 751: 247–252. [DOI] [PubMed] [Google Scholar]
- 31.Raddant AC and Russo AF. Calcitonin gene-related peptide in migraine: Intersection of peripheral inflammation and central modulation. Expert Rev Mol Med 2011; 13: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brain SD and Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev 2004; 84: 903–934. [DOI] [PubMed] [Google Scholar]
- 33.Goadsby PJ, Reuter U, Hallstrom Y, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med 2017; 377: 2123–2132. [DOI] [PubMed] [Google Scholar]
- 34.Silberstein SD, Dodick DW, Bigal ME, et al. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med 2017; 377: 2113–2122. [DOI] [PubMed] [Google Scholar]
- 35.Skljarevski V, Oakes TM, Zhang Q, et al. Effect of different doses of galcanezumab vs placebo for episodic migraine prevention: A randomized clinical trial. JAMA Neurol 2018; 75: 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong JJY, Wei DY and Goadsby PJ. Recent advances in pharmacotherapy for migraine prevention: From pathophysiology to new drugs. Drugs 2018; 78: 411–437. [DOI] [PubMed] [Google Scholar]
- 37.Leao AA. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 1947; 10: 409–414. [DOI] [PubMed] [Google Scholar]
- 38.Takano T, Tian GF, Peng W, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 2007; 10: 754–762. [DOI] [PubMed] [Google Scholar]
- 39.Reuter U, Weber JR, Gold L, et al. Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol 1998; 274: H1979–H1987. [DOI] [PubMed] [Google Scholar]
- 40.Ayata C and Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann J, Baca SM and Akerman S. Neurovascular mechanisms of migraine and cluster headache. J Cereb Blood Flow Metab. Epub ahead of print 26 September 2017. DOI: 10.1177/0271678X17733655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansen-Olesen I, Jorgensen L, Engel U, et al. In-depth characterization of CGRP receptors in human intracranial arteries. Eur J Pharmacol 2003; 481: 207–216. [DOI] [PubMed] [Google Scholar]
- 43.Hong KW, Pyo KM, Lee WS, et al. Pharmacological evidence that calcitonin gene-related peptide is implicated in cerebral autoregulation. Am J Physiol 1994; 266: H11–H16. [DOI] [PubMed] [Google Scholar]
- 44.Ebersberger A, Schaible HG, Averbeck B, et al. Is there a correlation between spreading depression, neurogenic inflammation, and nociception that might cause migraine headache? Ann Neurol 2001; 49: 7–13. [PubMed] [Google Scholar]
- 45.Tozzi A, de Iure A, Di Filippo M, et al. Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc Natl Acad Sci USA 2012; 109: 18985–18990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goadsby PJ, Edvinsson L and Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol 1988; 23: 193–196. [DOI] [PubMed] [Google Scholar]
- 47.Moreno MJ, Cohen Z, Stanimirovic DB, et al. Functional calcitonin gene-related peptide type 1 and adrenomedullin receptors in human trigeminal ganglia, brain vessels, and cerebromicrovascular or astroglial cells in culture. J Cereb Blood Flow Metab 1999; 19: 1270–1278. [DOI] [PubMed] [Google Scholar]
- 48.Wahl M, Schilling L, Parsons AA, et al. Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilatation elicited by cortical spreading depression. Brain Res 1994; 637: 204–210. [DOI] [PubMed] [Google Scholar]
- 49.Bergerot A, Holland PR, Akerman S, et al. Animal models of migraine: Looking at the component parts of a complex disorder. Eur J Neurosci 2006; 24: 1517–1534. [DOI] [PubMed] [Google Scholar]
- 50.Bolay H, Reuter U, Dunn AK, et al. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 2002; 8: 136–142. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Li Y and Wang M. Involvement of CGRP receptors in retinal spreading depression. Pharmacol Rep 2016; 68: 935–938. [DOI] [PubMed] [Google Scholar]
- 52.Wang M, Jiang L, Wang Y, et al. Both anti-CGRP and anti-CALCRL antibodies suppress cortical spreading depression. Cephalalgia 2017; 37: 209–303.28880580 [Google Scholar]
- 53.Eftekhari S, Kechechyan G, Faas G, et al. The CGRP receptor antagonist Olcegepant modulates cortical spreading depression in vivo. Cephalalgia 2017; 37: 295–296. [Google Scholar]
- 54.Warfvinge K and Edvinsson L. Distribution of CGRP and CGRP receptor components in the rat brain. Cephalalgia 2019; 39: 342–353. [DOI] [PubMed] [Google Scholar]
- 55.Oliver KR, Wainwright A, Edvinsson L, et al. Immunohistochemical localization of calcitonin receptor-like receptor and receptor activity-modifying proteins in the human cerebral vasculature. J Cereb Blood Flow Metab 2002; 22: 620–629. [DOI] [PubMed] [Google Scholar]
- 56.Walker CS, Eftekhari S, Bower RL, et al. A second trigeminal CGRP receptor: Function and expression of the AMY1 receptor. Ann Clin Transl Neurol 2015; 2: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hay DL and Walker CS. CGRP and its receptors. Headache 2017; 57: 625–636. [DOI] [PubMed] [Google Scholar]
- 58.Colonna DM, Meng W, Deal DD, et al. Calcitonin gene-related peptide promotes cerebrovascular dilation during cortical spreading depression in rabbits. Am J Physiol 1994; 266: H1095–H1102. [DOI] [PubMed] [Google Scholar]
- 59.Filiz A, Tepe N, Eftekhari S, et al. CGRP receptor antagonist MK-8825 attenuates cortical spreading depression induced pain behavior. Cephalalgia 2017; 333102417735845. [DOI] [PubMed] [Google Scholar]
- 60.Melo-Carrillo A, Noseda R, Nir R, et al. Selective inhibition of trigeminovascular neurons by fremanezumab: A humanized monoclonal anti-CGRP antibody. J Neurosci 2017; 37: 7149–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Tye AE, Zhao J, et al. Induction of calcitonin gene-related peptide expression in rats by cortical spreading depression. Cephalalgia 2019; 39: 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yisarakun W, Chantong C, Supornsilpchai W, et al. Up-regulation of calcitonin gene-related peptide in trigeminal ganglion following chronic exposure to paracetamol in a CSD migraine animal model. Neuropeptides 2015; 51: 9–16. [DOI] [PubMed] [Google Scholar]
- 63.Girouard H and Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer diseaseJ Appl Physiol 2006; 100: 328–335. [DOI] [PubMed] [Google Scholar]
- 64.Wahl M Local chemical, neural, and humoral regulation of cerebrovascular resistance vessels. J Cardiovasc Pharmacol 1985; 7: S36–S46. [DOI] [PubMed] [Google Scholar]
- 65.Moore CI and Cao R. The hemo-neural hypothesis: On the role of blood flow in information processing. J Neurophysiol 2008; 99: 2035–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim KJ, Ramiro Diaz J, Iddings JA, et al. Vasculoneuronal coupling: Retrograde vascular communication to brain neurons. J Neurosci 2016; 36: 12624–12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hinzman JM, Andaluz N, Shutter LA, et al. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 2014; 137: 2960–2972. [DOI] [PubMed] [Google Scholar]
- 68.Zhang X, Levy D, Noseda R, et al. Activation of meningeal nociceptors by cortical spreading depression: Implications for migraine with aura. J Neurosci 2010; 30: 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karatas H, Erdener SE, Gursoy-Ozdemir Y, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013; 339: 1092–1095. [DOI] [PubMed] [Google Scholar]
- 70.Ma W, Chabot JG, Powell KJ, et al. Localization and modulation of calcitonin gene-related peptide-receptor component protein-immunoreactive cells in the rat central and peripheral nervous systems. Neuroscience 2003; 120: 677–694. [DOI] [PubMed] [Google Scholar]
- 71.Han JS, Li W and Neugebauer V. Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci 2005; 25: 10717–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Desai M, Slusarczyk AL, Chapin A, et al. Molecular imaging with engineered physiology. Nat Commun 2016; 7: 13607. [DOI] [PMC free article] [PubMed] [Google Scholar]