

Abstract
PURPOSE
Multiplex genomic profiling is standard of care for patients with advanced lung adenocarcinomas. The Lung Cancer Mutation Consortium (LCMC) is a multi-institutional effort to identify and treat oncogenic driver events in patients with lung adenocarcinomas.
PATIENTS AND METHODS
Sixteen U.S. institutions enrolled 1367 lung cancer patients in LCMC2; 904 were deemed eligible and had at least one of 14 cancer-related genes profiled using validated methods including genotyping, massively parallel sequencing, and immunohistochemistry.
RESULTS
The use of targeted therapies in patients with EGFR, ERBB2, or BRAF p.V600E mutations, ALK, ROS1 or RET rearrangements, or MET amplification was associated with a survival increment of 1.5 years compared to those with such mutations not receiving targeted therapy; and 1.0 year compared to those lacking a targetable driver. Importantly, 60 patients with a history of smoking derived similar survival benefit from targeted therapy for alterations in EGFR/ALK/ROS1, when compared to 75 never smokers with the same alterations. In addition, co-existing TP53 mutations were associated with shorter survival among patients with EGFR, ALK, or ROS1 alterations.
CONCLUSION
Patients with adenocarcinoma of the lung and an oncogenic driver mutation treated with effective targeted therapy have a longer survival, regardless of prior smoking history. Molecular testing should be performed on all individuals with lung adenocarcinomas irrespective of clinical characteristics. Routine use of massively parallel sequencing enables detection of both targetable driver alterations and tumor suppressor gene and other alterations that have potential significance for therapy selection and as predictive markers for the efficacy of treatment.
Keywords: lung adenocarcinoma, TP53, EGFR, targetable, mutation
Statement of translational relevance
Characterization of lung adenocarcinomas by multiplex genomic profiling for multiple mutations is now standard of care. Here we show that the survival benefit of targetable mutation detection and directed therapy is similar for both never smokers and current and former smokers with lung adenocarcinomas. We also demonstrate that concurrent TP53 mutation is associated with poorer survival among lung adenocarcinoma patients with EGFR, ALK, or ROS1 alterations. Hence, routine use of massively parallel sequencing enables rapid detection of all types of clinically significant sequence variants for the care of lung adenocarcinoma patients, accelerating both targeted therapy selection and prognostic assessment.
INTRODUCTION
Lung adenocarcinoma is the most common histologic type of lung cancer and is diagnosed in 130,000 patients in the United States and 1 million persons worldwide each year.1 Lung adenocarcinomas are frequently characterized by different oncogenic driver mutations that affect a variety of kinases and their downstream signaling pathways,2–15 many of which are targetable using both standard-of-care FDA-approved and promising investigational therapies.16 For these reasons, systematic testing for oncogenic driver mutations is standard-of-care at diagnosis of metastatic lung adenocarcinomas and has been formally recommended by multiple molecular pathology guideline panels.16,17
The Lung Cancer Mutation Consortium (LCMC) was established in 2008 as a multi-institutional effort to investigate the frequency of different oncogenic drivers in lung adenocarcinoma, facilitate clinical protocol enrollment especially for rare molecular subsets, enable exchange of information and protocols for reproducibility of molecular testing among institutions, and thereby to accelerate further development of personalized treatment for lung adenocarcinoma across the USA.18–20
Here we report on the tumor genomic patterns and patient outcomes from a second cohort of LCMC subjects (LCMC2). This second cohort of subjects was enrolled because additional oncogenic drivers were identified that could be targeted with novel genotype-specific agents. Patients were prospectively enrolled to perform tumor genotyping of the 10 oncogenic drivers studied in LCMC1, as well as assays for ROS1 (ROS1r) and RET (RETr) rearrangements21–23, and immunohistochemistry (IHC) analysis for PTEN and MET expression. PTEN AND MET IHC analysis were included based on the promise of therapies for these alterations that were in clinical trials, including PI3K inhibitors and antibodies against MET. During the course of LCMC2 enrollment, most institutions switched from focused or serial testing to highly multiplexed genetic testing using massively parallel sequencing (MPS) (also known as next generation sequencing).24–27 This development enabled simultaneous analysis of mutations in several other genes in lung cancer that are biologically important, but not currently targetable (specifically TP53), that may be prognostically relevant when present concomitantly with oncogenic driver mutations.
MATERIALS and METHODS
Patient Recruitment, Enrollment, and IRB approval
These studies were conducted in accordance with the ethical principles present in the Belmont Report. Sixteen clinical sites participated in LCMC2 (Table S1). All sites obtained Institutional Review Board approval for this study. Eligible patients met the following criteria: stage IV or recurrent lung adenocarcinoma; Southwest Oncology Group performance status of 0, 1, or 2; expected survival of more than 6 months; no prior treatment with targeted therapy; diagnosis of metastatic disease after May 1, 2012; and adequate tissue for molecular analyses. All subjects enrolled provided written informed consent. Of 1367 patients enrolled, 1009 were deemed eligible (Figure S1). Epidemiologic and clinicopathologic data were prospectively collected including age, sex, race, cigarette smoking history, stage at diagnosis, metastatic sites, and survival from the time of documented metastatic disease.
Pathology Evaluation
Anatomic pathologists at each institution confirmed a diagnosis of lung adenocarcinoma, assessed tumor content, and determined specimen adequacy for molecular diagnostic testing. Central confirmation of lung adenocarcinoma diagnosis was based on review of an hematoxylin and eosin-stained histology slide or a scanned whole slide image (Leica Biosystems Inc., Buffalo Grove, IL) and the pathology report, when available (I.I.W., or J.F).
Mutational Analyses
All mutational analyses were performed in CLIA-certified diagnostic laboratories, using a variety of methods (Table S2). The mutations studied consisted of four small indels and 93 point mutations occurring in eight genes: AKT1, BRAF, EGFR, ERBB2, KRAS, MAP2K1, NRAS, and PIK3CA (Table S3); hereafter denoted the 8 core genes and 97 core alleles. During the course of this study, many diagnostic laboratories converted from single gene testing to MPS methods (Table S4). MPS technologies at each site were independently validated to CLIA standards for both wet-bench and bioinformatics components. Four hundred sixty subjects’ samples were analyzed by MPS, from which 431 MPS reports or variant call files (VCFs) were centrally reviewed to confirm extent of assay coverage including coverage data for TP53, STK11, and PTEN (Figure S2) and to exclude technical artifacts or germline variants which may have been reported based on automated mutation calling algorithms. Systematic evaluation for MET exon 14 skipping variants was not performed. All results are shown in Table S5.
Rearrangement Detection
Fluorescence in situ hybridization (FISH) was performed using assays for fusions/rearrangements in ALK, ROS1, and RET, as described.12,19,28 Rearrangement results were also accepted from laboratories using hybrid capture-based MPS as the principal detection method. FISH or silver in situ hybridization (Roche/Ventana, Tucson, AZ) for assessment of MET amplification was also performed,29 and amplification was considered to be present when the MET/centromere 7 ratio was at least 2.0.30
Immunohistochemistry for PTEN and MET
Immunohistochemistry (IHC) for PTEN (clone 138G6; Cell Signaling Technology, Danvers, MA) and MET (clone SP44, Roche/Ventana, Tucson, AZ) was performed at 12 study sites. Individual sites were responsible for assay validation on their local staining platforms within a CLIA-certified laboratory. PTEN results were scored as intact (≥90% tumor cells staining), lost (<10% tumor cells staining) or heterogeneous (between 10-90% tumor cells staining). MET immunohistochemistry was defined as positive if the sample had an H score of ≥ 200, following previously published scoring methods.31 Both PTEN and MET IHC scoring involved pathologist training and interlaboratory proficiency testing (Supplemental methods).
Classification of EGFR mutations
We considered EGFR p.L858R, exon 19 in-frame deletions and insertions, p.G719S/C/A, and p.L861Q mutations as sensitizing to therapy with EGFR tyrosine kinase inhibitors (TKIs) (sensitizing EGFR, sEGFR)18. We considered p.E709A, exon 20 in-frame insertion or deletion, and p.T790M mutations as non-sensitizing to TKIs, a category we labeled ‘other’ (oEGFR).32,33,34 With the exception of combinations including de novo p.T790M mutations, all examples of compound sensitizing and non-sensitizing mutations were categorized as sEGFR.
Analysis of TP53 mutations
TP53 mutations were categorized as “disruptive” as described previously: 1) all inactivating mutations (i.e. nonsense, frameshift, splice-site); or 2) non-conservative missense mutations occurring within the DNA-binding domain L2 (codons 163-195) or L3 (codons 236-251) (Table S6).35 All other variants were considered non-disruptive. All combinations of disruptive and non-disruptive mutations were categorized as disruptive.
Targeted therapy
We considered targeted therapy to be any treatment provided as standard of care or within a clinical trial that was a kinase inhibitor or antibody directed at a specific genomic alteration. This included therapies directed at the following alterations: sEGFR, ERBB2 exon 20 insertions or missense mutations, BRAF p.V600E (veBRAF), ALKr, ROS1r, RETr, and METamp, hereafter denoted as ‘the targeted therapy cohort’.
Survival analysis and Statistical Methods
Descriptive statistics, including median for continuous variables, and percentages and frequencies for categorical variables, are presented. Group comparisons were analyzed using the Wilcoxon rank-sum or Kruskal-Wallis tests for continuous variables and chi-square test for categorical variables. Survival curves were calculated from the Kaplan-Meier method and differences in survival were tested by the log-rank test. To evaluate if driver gene mutation effects were similar between smoker and non-smoker groups, Cox Proportional Hazard model analysis was performed including driver gene mutation, smoking status, and their interaction. Statistical analyses were performed using R version 3.3.1.
RESULTS
Subjects and molecular analyses
From January 1, 2013 to December 1, 2015, 1367 subjects were enrolled, of which 1009 (74%) met all eligibility criteria. Reasons for exclusion are indicated in Figure S1.
Of the 907 confirmed adenocarcinomas cases, 904 had at least one mutation analysis, 866 had at least one FISH assay, and 830 had at least one IHC assay completed (Figure S2). Of 904 patients for whom at least one biomarker was assessed (“any genotyping”), 54% were female, 92% had an ECOG performance status of 0 or 1, 63% were former smokers, and 25% of patients were never smokers (Table 1). 423 cases had “full” genotypes reported for all 14 drivers assessed, including MET and PTEN IHC (Table 2).
Table 1.
LCMC2 patient characteristics.
| Patient Characteristics | Genotyping | Driver* and Treatment Status | |||
|---|---|---|---|---|---|
| Any | Full | No Driver | Driver + Tx | Driver No Tx | |
| Sex | n=904 | n=423 | n=337 | n=162 | n=74 |
| Male | 416 (46) | 213 (50.4) | 176 (52.2) | 67 (41.4) | 34 (45.9) |
| Female | 488 (54) | 210 (49.6) | 161 (47.8) | 95 (58.6) | 40 (54.1) |
| Age at enrollment,mean (range) | 64 (22-90) | 64 (22-90) | 64 (34-90) | 61 (35-86) | 63 (22-90) |
| Performance Status | |||||
| 0 | 247 (27.6) | 109 (25.8) | 77 (22.9) | 47 (29.2) | 21 (28.4) |
| 1 | 574 (64.1) | 273 (64.7) | 226 (67.3) | 105 (65.2) | 44 (59.5) |
| 2 | 74 (8.3) | 40 (9.5) | 33 (9.8) | 9 (5.6) | 9 (12.2) |
| missing | 9 (1.0) | 1 (0.2) | 1 (0.3) | 1 (0.7) | 0 |
| Cigarette smoking history | |||||
| Never | 219(24.6) | 101 (24.1) | 45 (13.4) | 86 (53.4) | 22 (29.7) |
| Former | 556 (62.5) | 274 (65.4) | 242 (72.2) | 69 (42.9) | 41 (55.4) |
| Current | 115 (12.9) | 44 (10.5) | 48 (14.3) | 6 (3.7) | 11 (14.9) |
| Missing | 14 (1.5) | 4 (1) | 2 (0.6) | 1 (0.7) | 0 |
| Prior therapy | |||||
| Surgery | 389 (43.9) | 175 (41.6) | 149 (44.3) | 49 (30.4) | 33 (45.2) |
| Chest radiotherapy | 137 (15.5) | 55 (13.1) | 54 (16.2) | 14 (8.7) | 14 (18.9) |
| Chemotherapy | 487 (61.6) | 223 (57.5) | 205 (65.7) | 81 (53.6) | 40 (59.7) |
| Time from metastatic disease dx to enrollment, mean (years) | 0.31 | 0.3 | 0.34 | 0.28 | 0.31 |
Driver in this table refers to sensitizing EGFR, BRAF V600E, and ERBB2 mutation, ALK, ROS1, and RET rearrangement, and MET amplification.
Table 2.
Summary of mutation and expression findings in LCMC II
| Any Genotyping (n*) | % (based on n for each assay) | CI | Full Genotyping (n=423) | % | CI | Targeted Therapy | |
|---|---|---|---|---|---|---|---|
| Major Targetable Alterations | |||||||
| EGFR | |||||||
| sEGFR | 116 (862) | 13.5% | 11-16 | 65 | 16.7% | 12-19 | 100 |
| L858R | 50 | 31 | |||||
| Exon 19 in/del | 56 | 30 | |||||
| G719X or L861Q | 10 | 4 | |||||
| MET amplification | 33 (689) | 4.8% | 3-7 | 19 | 4.9% | 3-7 | 6 |
| ALK rearrangement | 36 (843) | 4.3% | 3-6 | 17 | 4.4% | 2-6 | 30 |
| BRAF V600E | 26 (860) | 3.0% | 2-4 | 17 | 4.4% | 2-6 | 10 |
| ROS1 rearrangement | 18 (832) | 2.2% | 1.3-3 | 11 | 2.8% | 1.3-5 | 8 |
| RET rearrangement | 18 (817) | 2.2% | 1.3-4 | 11 | 2.8% | 1.3-5 | 9 |
| ERBB2 | 16 (647) | 2.5% | 1.4-4 | 12 | 3.1% | 1.5-5 | 6 |
| Other Alterations | |||||||
| KRAS | 269 (862) | 31.2% | 28-34 | 113 | 29.0% | 23-31 | 2 |
| oEGFR | 20 (861) | 2.3% | 1.4-4 | 11 | 2.8% | 1.3-5 | 0 |
| NRAS | 6 (860) | 0.7% | 0.3-2 | 5 | 1.3% | 0.4-3 | 0 |
| BRAF (non-V600E) | 8 (860) | 0.9% | 0.4-2 | 2 | 0.5% | 0.1-2 | 0 |
| AKT1 | 0 (708) | 0.0% | 0 | ||||
| Known Co-Occuring Alterations | |||||||
| MET expression (IHC) | 482 (827) | 58.3% | 55-62 | 235 | 60.3% | 51-60 | |
| TP53 mutation | 218 (431)** | 50.5% | 46-56 | 136 (274)** | 49.6% | 44-56 | |
| PTEN loss (IHC) | 54 (646) | 8.3% | 6-11 | 40 | 10.3% | 10-18 | |
| PIK3CA | 23 (860) | 2.7% | 2-4 | 15 | 3.8% | 2-6 | |
| MAP2K1 | 2 (765) | 0.3% | 0-1 | 0 |
n denotes the number of subjects whose cancers were tested for each alteration;
for TP53 mutation detection rate, only cases in which NGS testing was performed are considered
Mutation findings
Rates of genomic alterations among patients with “any” genotyping (n=904) and “full” genotyping (n=423) and numbers of patients enrolled on targeted therapies are shown in Table 2. A driver oncogenic alteration, when including KRAS mutations, was observed in 544 (60%) patients overall and in 273 (65%) patients with full genotyping (Figure 1A). RETr and ROS1r each were seen in 11 cases (2.8%; 95% CI, 1.3-3%) of the full genotyping cohort. Tumors containing two putative oncogenic drivers were detected in 22/904 (2.4%) in the overall cohort, and in 10/423 (2.4%) in the full genotyping cohort (Table S7). MET amplification (METamp) was observed as a concurrent oncogenic driver event in 8% of veBRAF, 3.0% of KRAS, and 2.5% of EGFR-mutated cases and was present at a low level (MET to CEP7 ratio of 2-3.3) in all KRAS and veBRAF cases. Three tumor specimens with sEGFR mutation also had de novo METamp, of which two were high level (ratios of 15 and 4.7). Combined sEGFR and KRAS activating mutations were observed in three cases. Dual EGFR and fusion alterations were observed in three cases (sEGFR/ALKr = 2, sEGFR/RETr =1) and KRAS mutations/ROS1 fusions were observed in two cases; corroborating evidence for a rearrangement was limited in all cases.
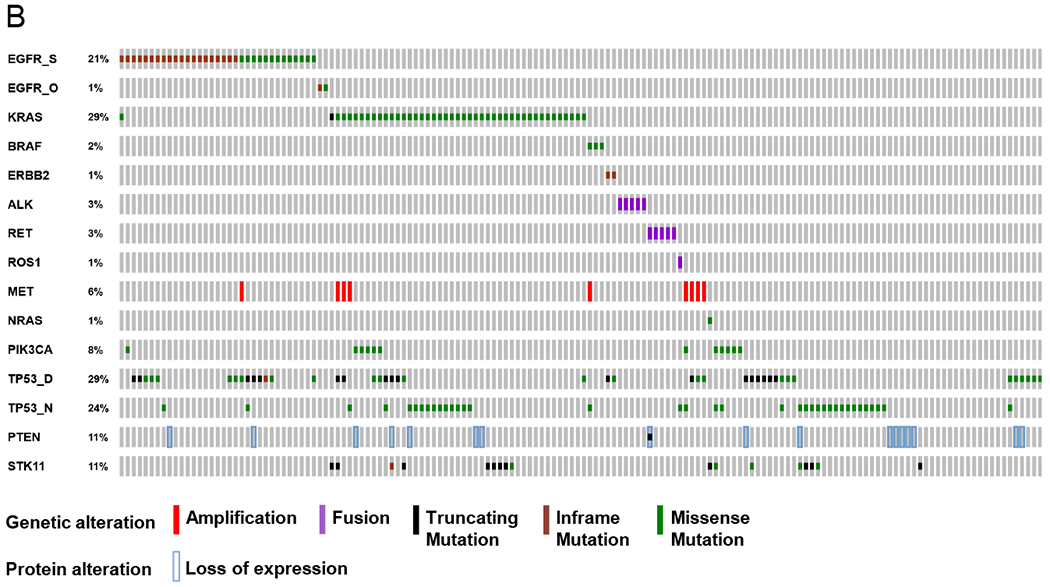
Figure 1. Mutations and co-mutation plot in LCMC II.
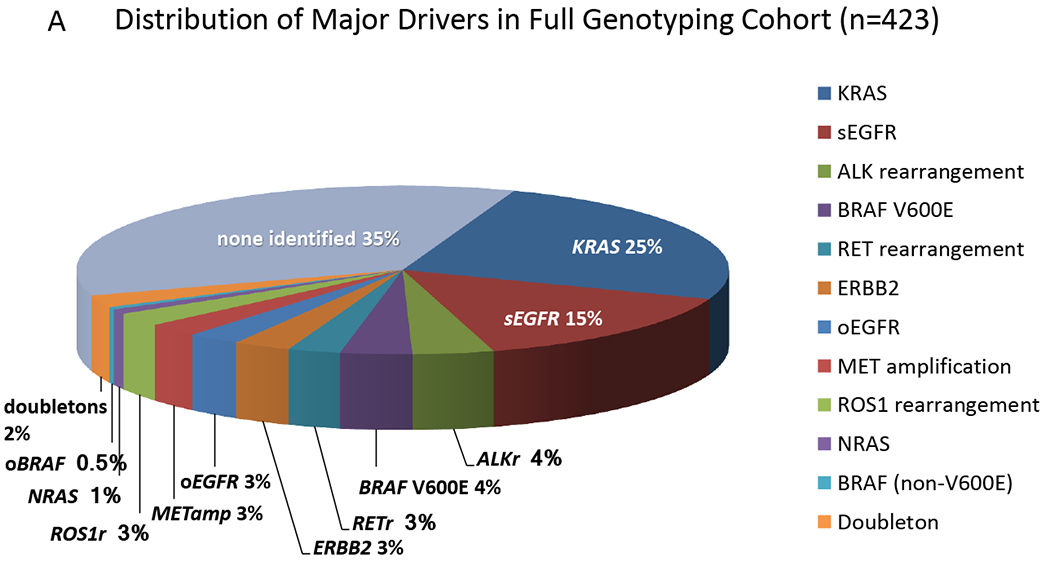
A. Distribution of Oncogenic Drivers in Full Genotyping Cohort. The relative proportion of the various driver mutations are shown for the 423 subjects with complete testing for 12 genes. No AKT1 orMAP2K1 mutations were detected in this set. sEGFR = sensitizing EGFR mutation; oEGFR = other EGFR mutations; ALKr, RETr, and ROS1r denote rearrangements in the respective genes; METamp denotes amplification of MET; oBRAF denotes a mutation other than V600E; and doubletons denotes samples with two or more of the oncogenic drivers shown here.
B. Co-mutation plot.Genetic and expression alterations in the 14 core genes plus key tumor suppressor genes in 154 lung adenocarcinomas with complete analysis. No AKT1 or MAP2K1 mutations were detected in this set. EGFR_S = sensitizing EGFR mutations; EGFR_O = other EGFR mutations; TP53_D = disruptive alterations; TP53_N = nondisruptive alterations. Prepared using http://www.cbioportal.org/oncoprinter.jsp#.
Co-mutation plot and analysis
Use of MPS allowed us to perform analyses of mutations in other genes in 460 samples, including tumor suppressor genes TP53, STK11, and PTEN. We examined concurrence of mutations in detail in 154 subjects with complete genotyping of core alleles, as well as TP53, STK11 and PTEN. In this set, mutations in the core alleles were mutually exclusive, except for one sample with an sEGFR and KRAS p.Q61R mutation, and 5 samples that had both METamp and another driver mutation (Figure 1B; Table S7). TP53 mutations were identified in 14 of 35 (40%) EGFR and 22 of 44 (50%) KRAS mutant tumors, and were rare in ALKr, ROS1r or RETr tumors (1/11, 9%, ROS1r). STK11 mutations were observed in 11% of cases, exclusively in KRAS-mutated and driver oncogene-negative cases. Only one of 17 cases with PTEN loss of expression by IHC (see below) had an identifiable PTEN mutation. In 41 (27%) cases, PIK3CA, TP53, or STK11 mutation and/or PTEN loss was identified in the absence of a co-existing oncogenic driver alteration. No variants were detected in the examined genes in 14 (9%) of cases.
PTEN and MET immunohistochemistry
Central pathology review was performed for 646 PTEN-stained tumors: PTEN was lost in 54 (8%, 95% CI, 6-11%), intact in 526 (81%) and heterogeneous in 66 (11%) (Figure S3). Heterogeneous cases rarely demonstrated abrupt loss of expression, as has been reported in prostate adenocarcinoma. 36 Instead, these cases typically showed a gradient of staining, which was interpreted as intact expression. MET IHC results were reported in 827 cases and were considered positive (H score ≥ 200) in 482 (58%, 95% CI 55-62%) (to be reported in detail elsewhere).
Clinicopathologic associations with specific mutations
Figure S5 displays associations between oncogenic driver mutations and clinical characteristics. Multiple nominally significant associations were identified that should be considered exploratory in this analysis but are consistent with previously published observations.
Survival in the Presence of Targetable Alteration
Survival was longer in 162 subjects with mutations in any targetable driver gene (sEGFR (n=95), ERBB2 (n=6) , veBRAF(n=9) , ALKr (n=28), ROS1r (n=8), RETr (n=8), METamp (n=2), or multiple drivers (n=6) who received targeted therapy in comparison to patients with such mutations who did not received targeted therapy, and in comparison to those without a driver identified (p<0.001, Figure 2). As expected, patients with sEGFR alterations received benefit from EGFR targeted therapy, compared to those who did not receive therapy (p<0.001), with 1.7 years improvement in median survival from 1.3 to 3 years (Figure S6).
Figure 2. Survival comparisons according to targeted therapy.
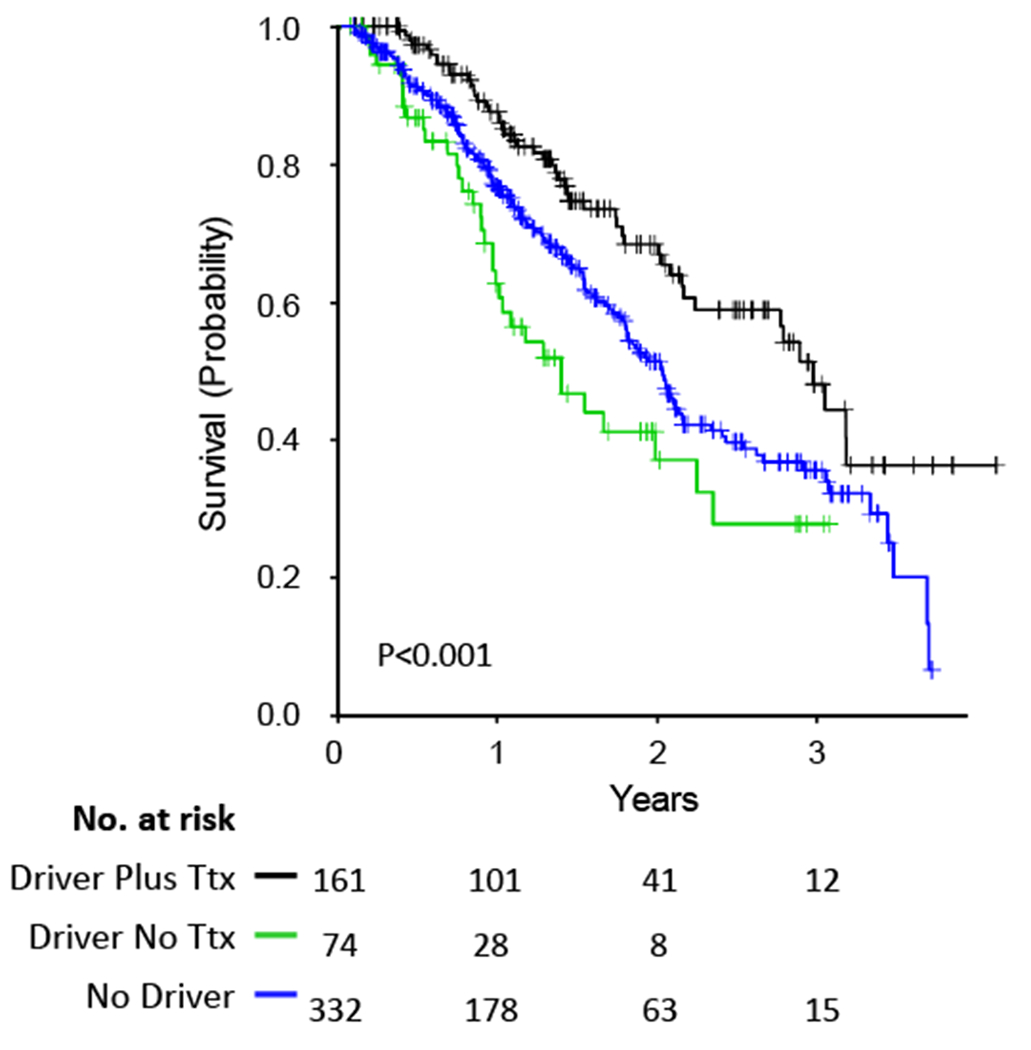
Survival curves for subjects with any of sEGFR, ERBB2, BRAF p.V600E (veBRAF), ALKr, ROS1r, RETr, or METamp alterations who received targeted therapy (Ttx), versus those with similar alterations who did not receive targeted therapy (No Ttx), versus those with no mutations in any of these genes.
Molecular Modulators of Survival in Targeted Therapies
Neither PTEN loss nor MET positivity by IHC were associated with a difference in overall survival for the targeted therapy cohort (p=0.944, Figure S7A; p=0.729, Figure S7B); however analysis of PTEN is limited by the small number of cases with loss of expression. In addition, when considering the following events as a class: PTEN loss by IHC, PTEN mutation, PIK3CA mutation or TP53 mutation; no significant effect on survival was noted in the overall targeted therapy cohort (Figure S7C).
However, since previous reports have suggested that TP53 mutation might adversely impact the survival of patients treated with targeted therapy for oncogenic driver mutations in lung cancers,37,38 we explored this possibility, specifically focusing on patients in whom MPS testing had been performed, TP53 status manually curated, and survival data was available. Patients with sEGFR treated with targeted therapy harboring a TP53 mutation displayed a trend toward shorter survival compared to those without a TP53 mutation (2.9 years vs. not reached (p=0.06)) (Figure 3A). To examine this further, we divided TP53 mutations into disruptive and non-disruptive types (see Methods). Disruptive TP53 mutations were associated with a reduction in survival (median survival = 2.6 years) in comparison to no TP53 mutation (median survival not reached) in those with sEGFR mutations (p=0.055) (Figure 3B, Figure S8A). Given these results, we extended this analysis to the set of patients with any of sEGFR, ALKr, and ROS1r alterations. Any TP53 mutation was associated with reduced survival (median 2.6 years) in the EGFR-ALKr-ROS1r subset, compared to no TP53 mutation (median not reached, p=0.014, Figure 3C), and this difference was enhanced by consideration of TP53 disruptive mutations only (median survival 2.6 years versus not reached, p=0.009, Figure 3D). In addition, for the EGFR-ALKr-ROS1r subset, survival differed according to the presence of a TP53 disruptive mutation versus a TP53 nondisruptive mutation versus no TP53 mutations (p=0.033, Figure S8B).
Figure 3. Survival comparisons according to presence or absence of TP53 mutation.
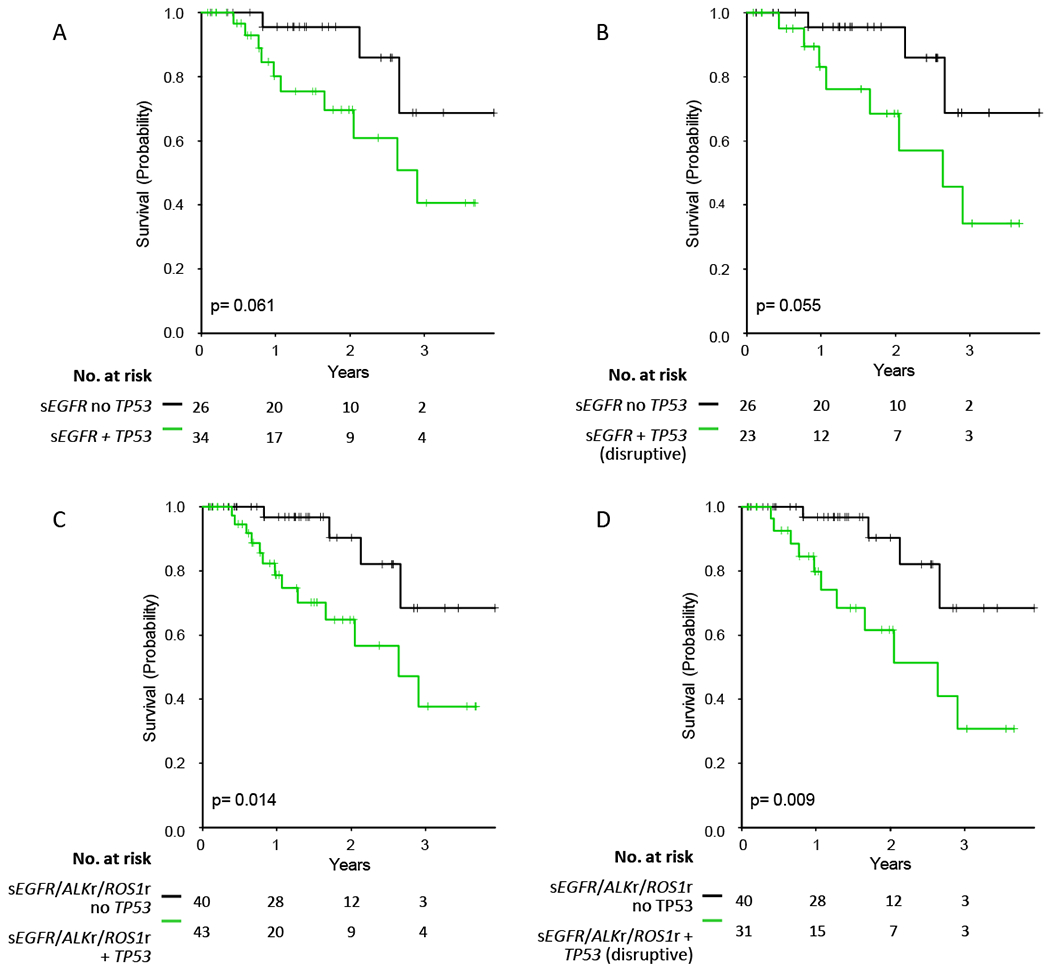
A. Comparison of survival among 60 subjects with sEGFR mutations with and without TP53 mutation.
B. Comparison of survival among 49 subjects with sEGFR mutations with a disruptive TP53 versus those without any TP53 mutation.
C. Comparison of survival among 83 subjects with sEGFR, ALKr, or ROS1r mutations with and without TP53 mutation.
D. Comparison of survival among 71 subjects with sEGFR, ALKr, or ROS1r mutations with a disruptive TP53 versus those without any TP53 mutation.
Effect of Smoking History on mutation frequency and targeted therapy benefit
Despite a correlation between targetable driver mutations and non-smoking status, all mutation types were also seen in current and/or former smokers (Figure S4). We examined the benefit of targeted therapy for sEGFR-ALKr-ROS1r alterations in patients with and without a cigarette smoking history. As expected, targeted therapy conferred a major survival benefit to never smoker patients with an sEGFR-ALKr-ROS1r alteration (Figure 4A, p=0.011). Notably, a similar improvement in survival was seen in former and current smokers with an sEGFR-ALKr-ROS1r alteration who received targeted therapy in comparison to those who did not (Figure 4B, p=0.003). Furthermore, the survival benefit from targeted therapy was similar in the never smoking and current/former smoking subgroups (p=0.975, Cox Proportional Hazards Model).
Figure 4. Survival comparisons among subjects with sEGFR, ALKr, or ROS1r mutations according to smoking status.
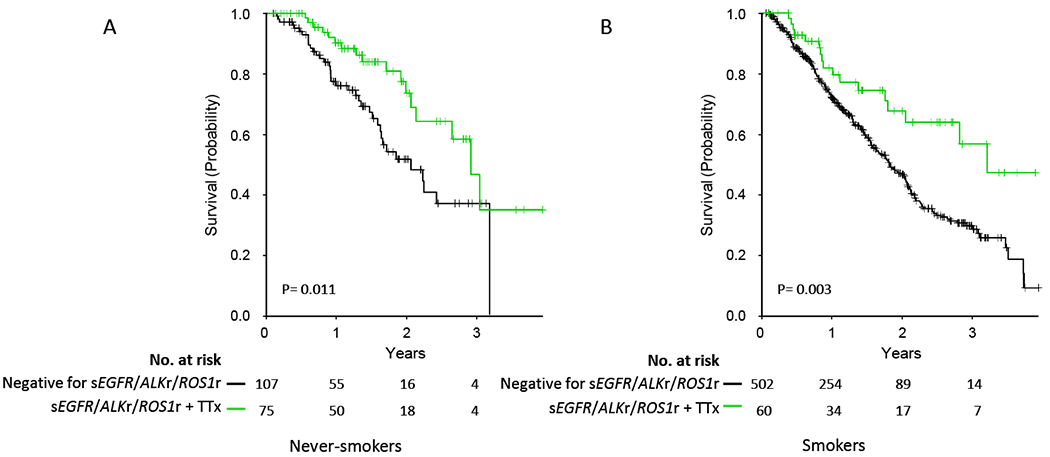
A. Survival of never smokers without sEGFR-ALKr-ROS1r mutation, or with sEGFR-ALKr-ROS1r mutation who received targeted therapy.
B. Survival of former and current smokers without sEGFR-ALKr-ROS1r mutation, or with sEGFR-ALKr-ROS1r mutation who received targeted therapy.
DISCUSSION
The LCMC was formed to expand and formalize molecular genetic testing of lung adenocarcinoma specimens for targetable driver mutations to enable broader dissemination of personalized therapy for this malignancy. In the current study we have expanded the panel of genetic alterations examined to include ROS1r and RETr, and added additional assays to explore PTEN expression and MET expression in this new cohort of 904 patients. The PTEN and MET analyses were added since at the time the study was planned, both PI3 Kinase inhibitors and onartuzumab were promising therapies for alterations in these two genes, respectively. Subsequent studies failed to show benefit from these agents in patients with lung carcinoma. However newer compounds and treatment strategies targeting these alterations are under continuing investigation. 39,40 Currently, MET-directed TKIs are thought to have potential benefit for patients with MET amplification, with a suggestion that high-level MET amplification may be the most predictive marker. 41 MPS-based panel testing was incorporated into routine diagnostic practice at most sites during LCMC2, enabling us to examine the effect of co-mutations on outcomes following targeted therapy in that subset.
Similar to our analyses of the LCMC1 population, we found that persons with oncogenic drivers in their tumors who were treated with targeted therapy experienced a longer survival than those who did not receive such therapy.18 Although patients with an identified driver mutation typically receive targeted therapy, a variety of factors may prevent the therapeutic intervention including rapid clinical decline after enrollment and loss to follow-up at the institution where the testing was performed. However, the reduced survival of untreated patients was not clearly attributable to early death after enrollment (Figure 2). 40 We acknowledge that since this population did not derive from a randomized trial, there is potential for bias, and all observations made here should be considered in that context. 25,26,42,43
Although prior studies have suggested a correlation between TP53 mutation and worse outcomes among EGFR-mutated lung adenocarcinomas,38,44,45 this is the first study to demonstrate the adverse prognostic impact of TP53 mutations on patients treated with targeted therapy directed against sEGFR, ALKr, or ROS1r alterations. Similar findings have been recently observed in a cohort of EGFR mutation positive patients. 45 In our study, this association was enhanced when disruptive TP53 mutations only were considered in comparison to subjects with no TP53 mutation (p=0.009). However, the total number of evaluable subjects for this analysis was small, and therefore this correlation should be considered preliminary. Additional studies are needed to confirm the prognostic impact of TP53 mutation in this setting. TP53 mutation testing is included in many MPS assays used currently in the USA, so this information is often available to clinicians. The molecular basis which underlies the potential prognostic value of TP53 mutations in this setting is not certain. Mechanistically, we suggest that TP53 mutation leads to genome instability in lung adenocarcinoma, and thus may accelerate the development of multiple mechanisms of resistance to targeted therapy in these patients, leading to shorter survival. 46
Although MPS is a powerful and informative technology now in wide use for lung cancer care, caution is appropriate in the interpretation of reported findings. In this data set, we performed manual curation to review findings in TP53, STK11, and PTEN, due to the inclusion of both germline variants and artifacts in the initial molecular reports (and/or variant call files). Although automated approaches to this review process may be helpful, careful review by a knowledgeable human expert is still required at this time. Nonetheless, we strongly advocate MPS analysis of lung adenocarcinoma specimens for all patients with advanced disease, since it is the most efficient means to rapidly identify diverse driver mutations, enabling access to a broader portfolio of targeted therapies. We note that the panel of genes with proven targetability continues to expand, with the most recent additions being veBRAF and MET exon 14 skipping mutations.7,8,47–51
We observed that the presence of a sEGFR, ALKr or ROS1 alteration that was treated with targeted therapy led to benefit in both smoking and never smoking populations of equivalent magnitude. Although these targetable alterations are much more prevalent in never smokers, to our knowledge this is the first study to directly compare outcomes between smokers and never smokers. These findings underscore the importance of testing patients regardless of smoking history, as all patients with a targetable alteration such as sEGFR, ALKr or ROS1r stand to benefit from targeted therapy.
Supplementary Material
Acknowledgements
We gratefully acknowledge Free to Breathe, Madison, WI for funding support for this research. We thank Lisa Litzenberger (University of Colorado) for assistance with preparation of figures.
Financial Support: We gratefully acknowledge Free to Breathe, Madison, WI for funding support for this research.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A: Cancer Statistics, 2017. CA Cancer J Clin 67:7–30, 2017 [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, et al. : EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–500, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, et al. : Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine 350:2129–39, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Soda M, Choi YL, Enomoto M, et al. : Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–6, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Campbell JD, Alexandrov A, Kim J, et al. : Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607–16, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imielinski M, Berger AH, Hammerman PS, et al. : Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150:1107–20, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Planchard D, Besse B, Groen HJ, et al. : Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 17:984–93, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Planchard D, Kim TM, Mazieres J, et al. : Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 17:642–50, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kris MG, Camidge DR, Giaccone G, et al. : Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol 26:1421–7, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mok TS, Wu YL, Thongprasert S, et al. : Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361:947–57, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Kwak EL, Bang YJ, Camidge DR, et al. : Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–703, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw AT, Ou SH, Bang YJ, et al. : Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 371:1963–71, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gautschi O, Milia J, Filleron T, et al. : Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J Clin Oncol 35:1403–1410, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pao W, Miller V, Zakowski M, et al. : EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 101:13306–11, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, Hu H, Pan Y, et al. : RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol 30:4352–9, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Ettinger DS, Wood DE, Aisner DL, et al. : Non-Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 15:504–535, 2017 [DOI] [PubMed] [Google Scholar]
- 17.Lindeman NI, Cagle PT, Beasley MB, et al. : Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn 15:415–53, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Kris MG, Johnson BE, Berry LD, et al. : Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311:1998–2006, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sholl LM, Aisner DL, Varella-Garcia M, et al. : Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol 10:768–77, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villaruz LC, Socinski MA, Abberbock S, et al. : Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 121:448–56, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergethon K, Shaw AT, Ou SH, et al. : ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 30:863–70, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li C, Fang R, Sun Y, et al. : Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS One 6:e28204, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi K, Soda M, Togashi Y, et al. : RET, ROS1 and ALK fusions in lung cancer. Nat Med 18:378–81, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Cheng DT, Mitchell TN, Zehir A, et al. : Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17:251–64, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sholl LM, Do K, Shivdasani P, et al. : Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1:e87062, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordan EJ, Kim HR, Arcila ME, et al. : Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov 7:596–609, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zehir A, Benayed R, Shah RH, et al. : Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23:703–713, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner J, Couts K, Sheren J, et al. : Kinase gene fusions in defined subsets of melanoma. Pigment Cell Melanoma Res 30:53–62, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dziadziuszko R, Wynes MW, Singh S, et al. : Correlation between MET gene copy number by silver in situ hybridization and protein expression by immunohistochemistry in non-small cell lung cancer. J Thorac Oncol 7:340–7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Go H, Jeon YK, Park HJ, et al. : High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J Thorac Oncol 5:305–13, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Koeppen H, Yu W, Zha J, et al. : Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib+/−onartuzumab in advanced non-small cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res 20:4488–98, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He M, Capelletti M, Nafa K, et al. : EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res 18:1790–7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi Y, Togashi Y, Yatabe Y, et al. : EGFR Exon 18 Mutations in Lung Cancer: Molecular Predictors of Augmented Sensitivity to Afatinib or Neratinib as Compared with First- or Third-Generation TKIs. Clin Cancer Res 21:5305–13, 2015 [DOI] [PubMed] [Google Scholar]
- 34.Wu JY, Yu CJ, Chang YC, et al. : Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 17:3812–21, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Poeta ML, Manola J, Goldwasser MA, et al. : TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 357:2552–61, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lotan TL, Wei W, Morais CL, et al. : PTEN Loss as Determined by Clinical-grade Immunohistochemistry Assay Is Associated with Worse Recurrence-free Survival in Prostate Cancer. Eur Urol Focus 2:180–188, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molina-Vila MA, Bertran-Alamillo J, Gasco A, et al. : Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res 20:4647–59, 2014 [DOI] [PubMed] [Google Scholar]
- 38.Clinical Lung Cancer Genome P, Network Genomic M: A genomics-based classification of human lung tumors. Sci Transl Med 5:209ra153, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spigel DR, Edelman MJ, O’Byrne K, et al. : Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J Clin Oncol 35:412–420, 2017 [DOI] [PubMed] [Google Scholar]
- 40.Patnaik A, Appleman LJ, Tolcher AW, et al. : First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol 27:1928–40, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Camidge DR, Ou SH, Shapiro GI, et al. : Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol 32:(suppl; abstr 8001), 2014 [Google Scholar]
- 42.Barlesi F, Mazieres J, Merlio JP, et al. : Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 387:1415–1426, 2016 [DOI] [PubMed] [Google Scholar]
- 43.Meric-Bernstam F, Brusco L, Shaw K, et al. : Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol 33:2753–62, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.VanderLaan PA, Rangachari D, Mockus SM, et al. : Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer 106:17–21, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Labbe C, Cabanero M, Korpanty GJ, et al. : Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 111:23–29, 2017 [DOI] [PubMed] [Google Scholar]
- 46.Muller PA, Vousden KH: p53 mutations in cancer. Nat Cell Biol 15:2–8, 2013 [DOI] [PubMed] [Google Scholar]
- 47.Awad MM, Oxnard GR, Jackman DM, et al. : MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 34:721–30, 2016 [DOI] [PubMed] [Google Scholar]
- 48.Frampton GM, Ali SM, Rosenzweig M, et al. : Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 5:850–9, 2015 [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Jia Y, Stoopler MB, et al. : Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J Clin Oncol 34:794–802, 2016 [DOI] [PubMed] [Google Scholar]
- 50.Paik PK, Drilon A, Fan PD, et al. : Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 5:842–9, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm564331.htm?platform=hootsuite:
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.