

Abstract
Pulmonary hypertension and hereditary hemorrhagic telangiectasia (HHT) have an association mediated by activin A receptor type II-like 1 (ACVRL1) gene pathogenic variants. A 30-year-old woman was previously admitted to a hospital due to lung hemorrhage, and was diagnosed with pulmonary hypertension, but stopped follow-up visits. At 48 years of age, she was admitted to our hospital and was diagnosed with HHT. Genetic testing revealed an ACVRL1 pathogenic variant. After the initiation of pulmonary vasodilator treatment, the patient's mean pulmonary artery pressure started to decrease from 43 mmHg, declining to 37 mmHg when she was 58 years of age. This is the first report describing the 28-year follow-up of an HHT and pulmonary hypertension patient with an ACVRL1 mutation.
Keywords: pulmonary hypertension, ACVRL1 mutation, hereditary hemorrhagic telangiectasia
Introduction
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant inherited vascular disease that occurs in approximately 1 in 10,000 people worldwide (1-3); and 85-95% of cases are caused by pathogenic variants of activin A receptor type II-like 1 (ACVRL1) and endoglin (ENG) (1, 4). The ACVRL1 and ENG genes encode members of the transforming growth factor (TGF)-β receptor family, and play important roles in different cellular processes, including proliferation, migration, and apoptosis (5). HHT is characterized by arteriovenous malformation in the lung, liver, and brain, and recurrent epistaxis due to telangiectasia.
Patients with HHT are reported to have lower life expectancy than the general population (6-8). There are no differences regarding the survival rate between HHT patients with ACVRL1 mutations and those with ENG mutations (8). However, one study reported that HHT patients with ACVRL1 mutations had a normal life expectancy (9). Pulmonary hypertension and HHT have an association mediated by pathogenic variants of either ACVRL1 or ENG (10-12). The pathology of HHT is mainly characterized by dilated vessels; however, the presence of HHT and pulmonary hypertension in patients with ACVRL1 mutations is associated with the occlusion of small pulmonary vessels (10). Pulmonary hypertension was detected as a complication in 8-23% of patients with HHT, and was associated with a poor prognosis (6, 13, 14). HHT patients with pulmonary hypertension are also reported to have a lower survival rate in comparison to those without (15, 16). However, the long-term prognosis in patients with pulmonary hypertension and HHT who have ACVRL1 mutations remains unknown. This case report illustrates the 28-year clinical course of a woman with an ACVRL1 mutation who had HHT and pulmonary hypertension.
Case Report
A 30-year-old woman was admitted to a hospital due to lung hemorrhage, and underwent right heart catheterization. Her mean pulmonary artery pressure, mean pulmonary artery wedge pressure, cardiac index, and pulmonary vascular resistance were 47 mmHg, 10 mmHg, 4.22 L/min/m2, and 5.7 Wood units, respectively. She was therefore diagnosed with pulmonary arterial hypertension. After discharge, she was treated in an outpatient clinic; however, she stopped attending the clinic at 31 years of age. At 48 years of age, she was diagnosed with bronchial asthma, and underwent echocardiography, which showed that her transtricuspid pressure gradient was 60 mmHg. She was therefore referred to our hospital for examination of pulmonary hypertension.
On admission, the patient had frequent epistaxis. Her blood pressure, heart rate, and percutaneous oxygen saturation (room air) were 110/60 mmHg, 70 ppm, and 99%, respectively, and a physical examination showed no sign of systemic edema or skin disease. A laboratory analysis revealed a B-type natriuretic peptide level of 15 pg/mL, and no sign of collagen disease (Table 1). On room air, her arterial oxygen saturation was 96.3%. Chest X-ray showed dilated pulmonary arteries, and electrocardiography revealed no sign of right ventricular hypertrophy (Fig. 1). Transthoracic echocardiography detected right heart chamber dilatation with distortion of the left ventricle, as well as a transtricuspid pressure gradient of 63 mmHg. Transesophageal echocardiography did not reveal any intra-cardiac shunt, and lung perfusion scintigraphy revealed no sign of pulmonary embolism. Spirometry showed that the percentages of the predicted values of vital capacity and forced vital capacity were 80.5%, and 74.1%, respectively. Right heart catheterization demonstrated that the patient's mean pulmonary artery pressure, mean pulmonary artery wedge pressure, cardiac index, and pulmonary vascular resistance were 43 mmHg, 12 mmHg, 4.05 L/min/m2, and 5.2 Wood units, respectively. An evaluation of oxygen saturation during cardiac catheterization revealed that the pulmonary to systemic blood flow ratio (Qp/Qs) was 1.24 with increased oxygen saturation from the inferior vena cava to the right atrium. Intrahepatic shunts were visualized by dynamic contrast-enhanced computed tomography (Fig. 2). Computed tomography angiography revealed intrahepatic arteriovenous shunts, and upper gastrointestinal endoscopy and colonoscopy showed gastrointestinal telangiectasia (Fig. 3). Magnetic resonance angiography did not suggest arteriovenous malformations in the brain. Contrast transthoracic echocardiography did not reveal any intra-pulmonary shunt. Genetic testing for HHT, including ENG and ACVRL1 genes, identified an ACVRL-1 variant, NM 000020.2: c.1451G>T (p.Arg484Leu), in both the patient and her father. Her sister and nieces did not have the variant (Fig. 4). Her father also had frequent epistaxis. We diagnosed the patient with HHT, and initiated pulmonary vasodilator treatment for her pulmonary hypertension. The clinical course and hemodynamics measured by right heart catheterization are described in Table 2. Beraprost was initially administered as a pulmonary vasodilator. This was later changed to an endothelin-receptor antagonist. After initiating follow up at our hospital, her mean pulmonary artery pressure started to decrease from 43 mmHg, and declined to 37 mmHg over a period of 10 years. During this time she did not require hospitalization due to worsening heart failure. Pulmonary vasodilators might cause vasodilatation of the nasal arteries and increase the risk of epistaxis. The patient is still taking an endothelin-receptor antagonist.
Table 1.
The Laboratory Findings on Admission.
| Parameters | Value | Parameters | Value | ||
|---|---|---|---|---|---|
| Blood count | Troponin I | <0.04 | ng/mL | ||
| White cell count | 6,500 | /µL | BNP | 15 | pg/mL |
| RBC | 418×104 | /µL | Triglyceride | 27 | mg/dL |
| Hemoglobin | 12.5 | g/dL | HDL-C | 105 | mg/dL |
| Hematocrit | 37.6 | % | LDL-C | 108 | mg/dL |
| Platelet count | 28.1×104 | /µL | CRP | 0.03 | mg/dL |
| Biochemistry | FT3 | 2.51 | pg/mL | ||
| AST | 24 | IU/L | FT4 | 1.05 | ng/mL |
| ALT | 16 | IU/L | TSH | 1.186 | U/mL |
| LDH | 209 | IU/L | Glucose | 86 | mg/dL |
| ALP | 228 | IU/L | HbA1c (NGSP) | 5.4 | % |
| Total bilirubin | 1.5 | mg/dL | ANA | Negative | |
| Direct bilirubin | 0.1 | mg/dL | Anti-DNA Ab | Negative | |
| BUN | 16 | mg/dL | Anti-U1 RNP Ab | Negative | |
| Creatinine | 0.78 | mg/dL | Anti-SSA Ab | Negative | |
| eGFR | 91 | mL/min/1.73 m2 | Anti-Scl70 Ab | Negative | |
| Sodium | 139 | mEq/L | C3 | 114 | mg/dL |
| Potassium | 4.0 | mEq/L | C4 | 22 | mg/dL |
| Chlorine | 105 | mEq/L | CH50 | 47 | U/mL |
| Total protein | 7.2 | g/dL | |||
| Albumin | 3.8 | g/dL | |||
| Uric acid | 4.4 | mg/dL | |||
| Creatine kinase | 187 | IU/L | |||
| CK-MB | <0.5 | IU/L | |||
Ab: antibody, ALP: alkaline phosphatase, ALT: alanine aminotransferase, ANA: antinuclear antibody, AST: aspartate transaminase, BNP: B-type natriuretic peptide, BUN: blood urea nitrogen, CK-MB: creatine kinase MB, CH50: 50% hemolytic complement activities, CRP: C-reactive protein, C3: complement component 3, C4: complement component 4, eGFR: estimate glomerular filtration rate, FT3: free triiodothyronine, FT4: free thyroxine, HbA1c: hemoglobin A1c, HDL-C: high-density lipoprotein, LDH: lactate dehydrogenase, LDL-C: low-density lipoprotein, NGSP: national glycohemoglobin standardization program, RBC: red blood cell count, RNP: ribonucleoprotein, Scl70: topoisomerase, SSA: Sjögren’s syndrome-related antigen A, TSH: thyroid stimulating hormone
Figure 1.
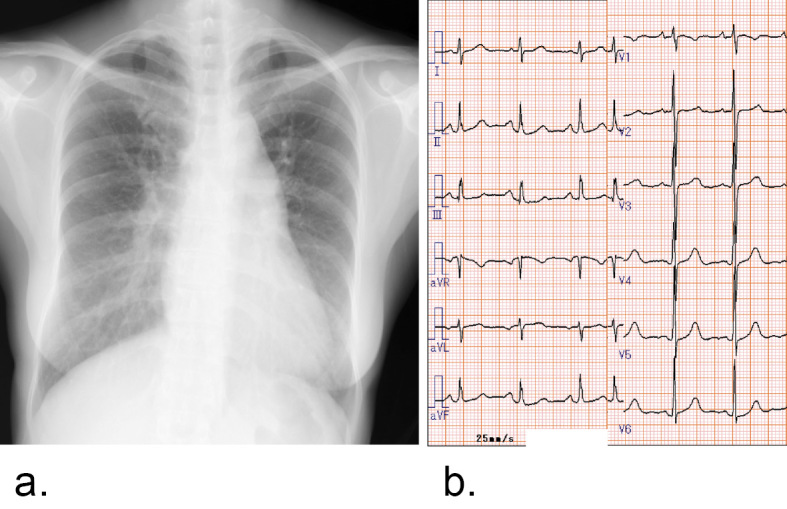
a: Chest X-ray. The cardiothoracic ratio was 52%. A prominent left central pulmonary artery and dilated right descending pulmonary artery were detected. b: Electrocardiogram. The heart rate was 60 bpm with sinus rhythm. There was no sign of right ventricular hypertrophy.
Figure 2.

Abdominal dynamic computed tomography. a: Arterial phase. b: Venous phase. White arrows show the hepatic vein at an early stage of the arterial phase. Intrahepatic shunts were visualized.
Figure 3.

a: Upper gastrointestinal endoscopy. Telangiectasia was detected in the duodenum, as shown in the white circle. b: Colonoscopy. Telangiectasia was detected in the descending colon, as shown in the white circle.
Figure 4.
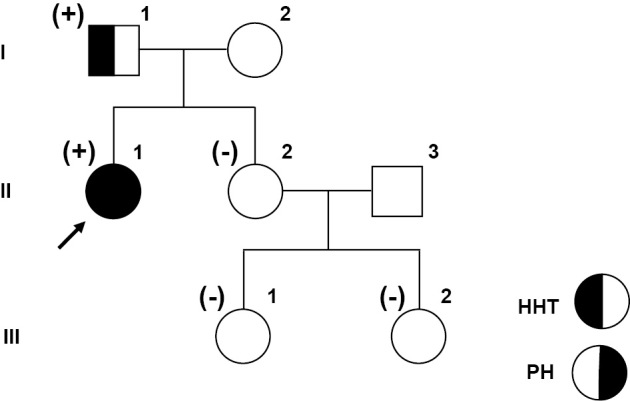
Pedigree and the results of genetic testing. Genetic testing was performed in I-1, II-1, II-2, III-1, and III-2. An ACVRL1 variant, NM 000020.2: c.1451G>t (p.Arg484Leu), was identified in I-1 and II-1. The black arrow shows the proband. Cases with the ACVRL1 variant are shown as (+), and those without the mutation are shown as (-). Squares represent males, and circles represent females. HHT: hereditary hemorrhagic telangiectasia, PH: pulmonary hypertension
Table 2.
Hemodynamic Parameters.
| 30 years | 48 years | 51 years | 53 years | 55 years | 56 years | 58 years | |
|---|---|---|---|---|---|---|---|
| CI, L/min/m2 | 4.22 | 4.05 | 3.67 | 3.76 | 3.46 | 3.31 | 2.97 |
| Mean PAP, mmHg | 47 | 43 | 35 | 35 | 32 | 32 | 37 |
| Mean PAWP, mmHg | 10 | 12 | 10 | 12 | 8 | 10 | 14 |
| Mean RAP, mmHg | 9 | 9 | 8 | 10 | 5 | 6 | 11 |
| PVR, Wood unit | 5.7 | 5.2 | 4.6 | 4.1 | 4.7 | 3.31 | 5.09 |
| Medication | None | None | ERA | ERA | ERA | ERA | ERA |
CI: cardiac index, ERA: endothelin-receptor antagonist, PAP: pulmonary artery pressure, PAWP: pulmonary artery wedge pressure, PVR: pulmonary vascular resistance, RAP: right atrial pressure
Discussion
This is the first report to demonstrate the long-term (28 years) follow-up of an HHT and pulmonary hypertension patient with an ACVRL1 mutation.
To date, the long-term prognosis of HHT and pulmonary hypertension in patients with ACVRL1 mutations has not yet been fully described. Table 3 illustrates the reported cases of HHT and pulmonary hypertension in patients with ACVRL1 mutations. Only three cases have been followed for more than 15 years: a 36-year-old woman, whose mean pulmonary artery pressure was 38 mmHg, and who survived for 18.8 years (17); a 41-year-old man, whose mean pulmonary artery pressure was 40 mmHg, and who survived for 18.4 years (17); and a woman in her thirties, whose mean pulmonary artery pressure was 52 mmHg, and who died due to right heart failure after 21 years of treatment with vasodilator and thalidomide (18). The current case was followed for 28 years, which-to the best of our knowledge-is the longest reported follow-up period among all reported cases of HHT and pulmonary hypertension in patients with ACVRL1 mutations.
Table 3.
Prognosis of Hereditary Hemorrhagic Telangiectasia and Pulmonary Hypertension Patients with ACVRL1 mutations in the Literature.
| References | Number of cases | Age at diagnosis, sex | Duration of survival (years) | Results | Cause of death | Types of mutation |
|---|---|---|---|---|---|---|
| (19) | 1 | 36 years, a male | Unknown | Dead | PH | Unknown |
| (25) | 3 | 4 to 16 years, females | 1 and 3 (Two patient data was available) | Alive | None | L273P (c. T818C) A352D (c. C1055A) |
| (20) | 1 | 29 years, a female | 1.8 | Dead | Rupture of AVM | p.Asp427Val (c. 1280A>T) |
| (16) | 9 | One year to 40 years, males and females | 0.1 to 6.7 | Alive (5 patients) Dead (4 patients) |
RHF, Rupture of AVM | Unknown |
| (24) | 1 | 37 years, a female | 1 | Alive | None | p.Gly309Val (c.926G>T) |
| (22) | 8 | 15 to 63 years, a male and females | 2 to 11 | Alive (4 patents) Dead (4 patients) |
RHF, bleeding | p.Val198Glu (c.593T>A) p.Arg67Trp (c.199C>T) p.Arg218Pro (c.653_654delinsCC) p.Arg374Gln (c.1121G>A) p.Tyr375Gln (c.1124A>G) p.Typ399Arg (c.1195T>C) p.Arg479X (c.1435C>T) p.Arg484Gln (c.1451G>A) |
| (23) | 1 | 47 years, a female | 0.8 | Alive | None | His 328 Thr (928C>T) |
| (17) | 22 | 4 to 70 years, males and females | 0.2 to 18.8 | Alive (15 patients) Dead (7 patients) |
Sepsis, PH, car accident | Unknown |
| (18) | 1 | 17 years, a female | 21 | Dead | RHF | Unknown |
| (21) | 3 | 42 to 55 years, females | 2 to 14 | Alive (2 patients) Dead (One patient) |
PH | p.A199P (c.595G>C) |
| (28) | 1 | One year, a male | 0.3 | Alive | None | p.Arg484Trp (c.1450C>T) |
| (29) | 1 | 51 years, a male | 7 | Alive | None | c.1451G>A |
AVM: arteriovenous malformation, PH: pulmonary hypertension, RHF: right heart failure
A previous report described that HHT patients with ACVRL1 mutations had a normal life expectancy; however, that report did not examine pulmonary hypertension (9). More cases of HHT and pulmonary hypertension were reported among patients with ACVRL1 mutations than among patients with ENG mutations (5). Previous studies, that did not include genetic information about ACVRL1 mutations, found that HHT and pulmonary hypertension patients had a higher mortality, and died due to cardiac diseases associated with pulmonary hypertension, sepsis, and major bleeding related to HHT (6, 14, 15). The prognostic information in Table 3, which describes the reported cases of HHT and pulmonary hypertension in patients with ACVRL1 mutations, indicates that patients died due to right heart failure, sepsis, bleeding, and rupture of arteriovenous malformation after 1.8 to 21 years' follow-up (16-22). At the time of writing, the patient of the current case report is still alive at 28 years after the initial diagnosis of pulmonary hypertension, although she has a history of frequent epistaxis and lung hemorrhage. Taking this case and previous reports into account, we should remain aware of pulmonary hypertension, sepsis, and major bleeding in the management of HHT and pulmonary hypertension in patients with ACVRL1 mutations.
The present case had intrahepatic arteriovenous shunts, which increased oxygen saturation from the inferior vena cava to the right atrium and Qp/Qs during cardiac catheterization. However, the effect of the intrahepatic shunts on the patient's pulmonary hypertension was considered to be limited because of the slightly high cardiac index and high pulmonary vascular resistance. We believe that the pulmonary hypertension in our patient was mainly caused by pulmonary artery lesions, such as idiopathic pulmonary hypertension.
Evidence on treatment for pulmonary hypertension associated with HHT is limited. Pulmonary vasodilators, which are a well-known treatment for pulmonary arterial hypertension, are challenging to administer when treating patients with HHT and pulmonary hypertension. Pulmonary atrioventricular fistula is common in HHT patients, and vasodilators are associated with the risk of increased shunt flow. Ambrisentan, tadalafil, and sildenafil have previously been used to treat HHT and pulmonary hypertension in patients with ACVRL1 mutations (18, 23). Bosentan treatment has also been reported for pulmonary hypertension in several HHT patients with ACVRL1 mutations (22, 24, 25). Among the patients treated with bosentan, only one patient is reported to have died due to right heart failure (22). In addition, only two HHT and pulmonary hypertension patients with ACVRL1 mutations are reported to have undergone intravenous epoprostenol treatment (16). In the current case, the mean pulmonary artery pressure continued to decrease for approximately 10 years under treatment with an endothelin-receptor antagonist, as shown in Table 2.
The ACVRL1 variant in the current study, c.1451G>T (p.Arg484Leu), was not archived in the ClinVar database. It was previously described in two reports related to either HHT or pulmonary hypertension (26, 27). Other ACVRL1 variants that affect the same amino acid as the ACVRL1 variant of c.1451G>T (p.Arg484Leu) [c.1450C>G (p.Arg484Gly), c.1450C>T (p.Arg484Trp), and c.1451G>A (p.Arg484Gln)] have been reported in the ClinVar database, and are considered to be pathogenic variants related to pulmonary hypertension and/or HHT. All in silico computational prediction models (Polyphen2, SIFT, CADD, MutationTaster, FATHMM) predict the pathogenicity of the ACVRL1 variant. Moreover, the ACVRL1 variant, c.1451G>T (p.Arg484Leu) is not included in the general population databases (Exome Aggregation Consortium, 1000 Genomes Project, and Genome Aggregation database). Thus, our ACVRL1 variant, c.1451G>T (p.Arg484Leu) is considered to be pathogenic.
In conclusion, we describe the first case in which an HHT and pulmonary hypertension patient with an ACVRL1 mutation was followed for 28 years. Some HHT and pulmonary hypertension patients with ACVRL1 mutations may require long-term careful follow-up for pulmonary hypertension under pulmonary vasodilator treatment.
Author's disclosure of potential Conflicts of Interest (COI).
Tetsuro Yokokawa: Employment, Actelion Pharmaceuticals Japan. Koichi Sugimoto: Employment, Actelion Pharmaceuticals Japan. Tomofumi Misaka: Employment, Fukuda Denshi. Akiomi Yoshihisa: Employment, Fukuda Denshi.
References
- 1. Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev 24: 203-219, 2010. [DOI] [PubMed] [Google Scholar]
- 2. Marchuk DA, Guttmacher AE, Penner JA, Ganguly P. Report on the workshop on Hereditary Hemorrhagic Telangiectasia, July 10-11, 1997. Am J Med Genet 76: 269-273, 1998. [PubMed] [Google Scholar]
- 3. Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med 245: 31-39, 1999. [DOI] [PubMed] [Google Scholar]
- 4. McDonald J, Wooderchak-Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet 6: 1, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vorselaars VMM, Hosman AE, Westermann CJJ, et al. Pulmonary arterial hypertension and hereditary haemorrhagic telangiectasia. Int J Mol Sci 19: 3203, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Droege F, Thangavelu K, Stuck BA, Stang A, Lang S, Geisthoff U. Life expectancy and comorbidities in patients with hereditary hemorrhagic telangiectasia. Vasc Med 23: 377-383, 2018. [DOI] [PubMed] [Google Scholar]
- 7. Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. Complications and mortality in hereditary hemorrhagic telangiectasia: a population-based study. Neurology 84: 1886-1893, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sabba C, Pasculli G, Suppressa P, et al. Life expectancy in patients with hereditary haemorrhagic telangiectasia. QJM 99: 327-334, 2006. [DOI] [PubMed] [Google Scholar]
- 9. de Gussem EM, Edwards CP, Hosman AE, et al. Life expextancy of parents with hereditary haemorrhagic telangiectasia. Orphanet J Rare Dis 11: 46, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 345: 325-334, 2001. [DOI] [PubMed] [Google Scholar]
- 11. Harrison RE, Flanagan JA, Sankelo M, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 40: 865-871, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pousada G, Baloira A, Fontan D, Nunez M, Valverde D. Mutational and clinical analysis of the ENG gene in patients with pulmonary arterial hypertension. BMC Genet 17: 72, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vorselaars V, Velthuis S, van Gent M, et al. Pulmonary hypertension in a large cohort with hereditary hemorrhagic telangiectasia. Respiration 94: 242-250, 2017. [DOI] [PubMed] [Google Scholar]
- 14. Harder EM, Fares WH. Hospitalizations with hereditary hemorrhagic telangiectasia and pulmonary hypertension in the United States from 2000 to 2014. Respir Med 147: 26-30, 2019. [DOI] [PubMed] [Google Scholar]
- 15. Li W, Xiong CM, Gu Q, et al. The clinical characteristics and long-term prognosis of pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Pulm Circ 8: 2045894018759918, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 181: 851-861, 2010. [DOI] [PubMed] [Google Scholar]
- 17. Revuz S, Decullier E, Ginon I, et al. Pulmonary hypertension subtypes associated with hereditary haemorrhagic telangiectasia: haemodynamic profiles and survival probability. PLoS One 12: e0184227, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakamura T, Ogo T, Tahara N, et al. Thalidomide for hereditary hemorrhagic telangiectasia with pulmonary arterial hypertension. Circ J 82: 1205-1207, 2018. [DOI] [PubMed] [Google Scholar]
- 19. Abdalla SA, Gallione CJ, Barst RJ, et al. Primary pulmonary hypertension in families with hereditary haemorrhagic telangiectasia. Eur Respir J 23: 373-377, 2004. [DOI] [PubMed] [Google Scholar]
- 20. Montani D, Price LC, Girerd B, et al. Fatal rupture of pulmonary arteriovenous malformation in hereditary haemorrhagic telangiectasis and severe PAH. Eur Respir Rev 18: 42-46, 2009. [DOI] [PubMed] [Google Scholar]
- 21. Greco A, Plumitallo S, Scelsi L, et al. Different forms of pulmonary hypertension in a family with clinical and genetic evidence for hereditary hemorrhagic teleangectasia type 2. Pulm Circ. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen YJ, Yang QH, Liu D, et al. Clinical and genetic characteristics of Chinese patients with hereditary haemorrhagic telangiectasia-associated pulmonary hypertension. Eur J Clin Invest 43: 1016-1024, 2013. [DOI] [PubMed] [Google Scholar]
- 23. Miyake R, Fujino T, Abe K, et al. Pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia successfully treated with sildenafil. Int J Cardiol 214: 275-276, 2016. [DOI] [PubMed] [Google Scholar]
- 24. Chang SA, Jang SY, Ki CS, Kang IS, Kim DK. Successful bosentan therapy for pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Heart Vessels 26: 231-234, 2011. [DOI] [PubMed] [Google Scholar]
- 25. Smoot LB, Obler D, McElhinney DB, et al. Clinical features of pulmonary arterial hypertension in young people with an ALK1 mutation and hereditary haemorrhagic telangiectasia. Arch Dis Child 94: 506-511, 2009. [DOI] [PubMed] [Google Scholar]
- 26. Brakensiek K, Frye-Boukhriss H, Malzer M, et al. Detection of a significant association between mutations in the ACVRL1 gene and hepatic involvement in German patients with hereditary haemorrhagic telangiectasia. Clin Genet 74: 171-177, 2008. [DOI] [PubMed] [Google Scholar]
- 27. Graf S, Haimel M, Bleda M, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 9: 1416, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alsheikh B, Aljohani O, Coufal NG. Paediatric pulmonary hypertension caused by an ACVRL1 mutation presenting as Ortner syndrome. Cardiol Young 28: 1475-1476, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sommer N, Droege F, Gamen KE, et al. Treatment with low-dose tacrolimus inhibits bleeding complications in a patient with hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension. Pulm Circ. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]