

Abstract
A 37-year-old woman developed deep venous thrombosis (DVT) of the left lower extremity at 8 weeks of gestation during her second pregnancy. There was no personal or family history of thrombosis. She received intravenous heparin, but heparin resistance was noted. The plasma antithrombin activity decreased to 45% in the acute phase, and it remained low postpartum. Her mother also had low plasma antithrombin activity (46%), and genetic testing revealed a heterozygous SERPINC1 mutation. Even without a family history of thrombosis, we should suspect hereditary antithrombin deficiency in patients with initial DVT and perform thorough investigation.
Keywords: hereditary antithrombin deficiency, deep venous thrombosis, negative family history of thrombosis
Introduction
Antithrombin (AT) is a natural anticoagulant that inactivates thrombin by covalently binding to the active serines of thrombin and activated factor X (1). AT has a binding site for heparin and shows weak inhibition of thrombin in the absence of heparin, while its inhibitory activity is induced by at least 1,000-fold in the presence of heparin (2). Hereditary AT deficiency is a rare disease associated with venous thromboembolism (VTE) that has a reported prevalence ranging between 1 in 500 (3) and 1 in 5,000 (4). Pregnancy is associated with an increased risk of thrombosis, which may be partly due to obstruction of venous return by the enlarged uterus along with the physiological hypercoagulable state that develops in pregnant women (5, 6). Accordingly, a woman with hereditary AT deficiency who becomes pregnant has a markedly increased risk of developing VTE. When hereditary AT deficiency leads to deep venous thrombosis (DVT) during pregnancy, there is usually a family history of thrombosis, and DVT typically occurs with the first pregnancy (7).
We herein report a patient with hereditary AT deficiency who had no personal or family history of thrombosis and developed initial DVT during her second pregnancy, which is a rare presentation of this condition. The present case highlights the importance of suspecting hereditary AT deficiency in patients with VTE, even if there is no family history of thrombosis.
Case Report
A 37-year-old woman developed pain and swelling of the left leg at 8 weeks of gestation in her second pregnancy and was referred to our hospital with suspected DVT. She had no relevant medical history and was not on regular medications. She had undergone laparoscopy for a right ovarian cyst three years earlier, and her first child had been born by normal vaginal delivery without problems two years earlier. There was no personal or family history of thrombosis.
On admission, her blood pressure was 115/74 mmHg, pulse rate was 90/min, temperature was 37.5°C, respiratory rate was 12/min, and oxygen saturation while breathing room air was 100% by pulse oximetry. On an examination, there were no abnormalities of the thorax or abdomen, but her left leg was swollen and tender. Laboratory tests showed a normal renal and liver function, but the C-reactive protein level was elevated to 6.7 mg/dL. Coagulation tests revealed a prothrombin time (PT) of 13.9 seconds and an activated partial thromboplastin time (aPTT) of 38.2 seconds, which were both almost in the normal range. However, D-dimer was elevated to 28.1 μg/mL.
Ultrasonography of the lower limb and intra-abdominal veins detected thrombosis in the left external iliac vein which had spread from a DVT in the completely occluded popliteal vein (Fig. 1). She had no dyspnea. An electrocardiogram showed no abnormalities, including ST-T changes and Q waves, and echocardiography did not demonstrate any evidence of right heart overload, such as right ventricular dilatation or tricuspid regurgitation. Accordingly, we concluded that the patient did not have any critical pulmonary embolism.
Figure 1.

A: Thrombosis of the left external iliac vein. B: Thrombosis of the left superficial femoral vein. C: Thrombosis of the left popliteal vein.
To treat her DVT, we initiated continuous infusion of unfractionated heparin at 10,000 IU daily. However, her aPTT remained at 30 seconds, which was a suboptimal response, and we were unable to achieve a therapeutic aPTT level despite increasing the dose of unfractionated heparin to 20,000 IU daily. Therefore, we investigated coagulation factor abnormalities that could cause thrombophilia. Her plasma AT activity had decreased to 45% (normal range: 80-130%), her protein C activity was 76% (normal range: 70-150%), and her free protein S level was 59% (normal range: 60-150%) during the administration of unfractionated heparin. Antiphospholipid antibody syndrome was excluded because anticardiolipin antibody and lupus anticoagulant were both negative. Because she had a reduced AT activity and heparin resistance, we consulted a hematologist with regard to the administration of AT concentrate, but we did not proceed due to the risk of infection and hypersensitivity reactions. Although a therapeutic aPTT could not be achieved, her leg swelling and symptoms improved over time, and the D-dimer level decreased to 8.1 μg/mL (Fig. 2). She was discharged after 15 days and continued to receive 20,000 IU/day of unfractionated heparin (10,000 IU twice daily subcutaneously) for DVT prophylaxis as an outpatient until childbirth.
Figure 2.

Clinical course.
While aPTT was never in the appropriate range, her D-dimer level stabilized at 1 to 2 μg/mL and DVT symptoms did not recur. At 36 weeks of gestation, organized mural thrombosis was detected in the left popliteal vein by ultrasonography, but the left femoral vein and external iliac vein were both patent. She was admitted for delivery at 38 weeks of gestation, and unfractionated heparin was discontinued on the day before admission. An epidural catheter was inserted on the day of admission, and infusion of oxytocin was initiated the next day. She safely delivered a baby girl after 4 hours of oxytocin infusion. The D-dimer level was 1.6 μg/mL before the discontinuation of heparin but rose rapidly to 29.6 μg/mL on the day after delivery. Although the patient was asymptomatic, we immediately restarted 20,000 IU/day of unfractionated heparin and added 2 mg/day of warfarin to prevent recurrence of DVT. The D-dimer level decreased rapidly to 3.6 μg/mL.
The patient was switched from heparin to warfarin after achieving the target PT-INR, and she continued warfarin therapy for six weeks with postpartum outpatient review. After the discontinuation of warfarin for two weeks, we re-measured coagulation factors related to thrombophilia. The free protein S level and protein C activity were both normal, but the AT activity remained low (49%) despite the normalization of the D-dimer level. The AT antigen level was also low at 11.9 mg/dL (normal range: 23.6-33.5 mg/dL), so we suspected hereditary AT deficiency even without a family history of thrombosis. Therefore, we investigated the AT III activity in her parents. While her father showed normal AT III activity (114%), her mother had low AT III activity (46%), and her AT antigen level was also low at 11.6 mg/dL (Fig. 3). Her mother's reproductive history showed two parities, including the present patient, without a history of thrombosis.
Figure 3.
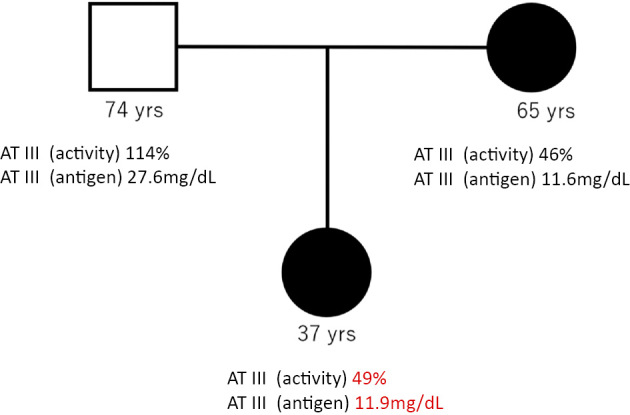
Pedigree of the patient’s family with antithrombin III deficiency (square=male; circles=female; black symbols=AT III deficiency).
To confirm the diagnosis, we performed genetic testing of the patient and her mother. A direct sequence analysis of the serpin family C member 1 (SERPINC1) gene revealed heterozygous duplication of thymine at nucleotide position 767 in exon 5, which caused a frameshift mutation of leucine in codon 256 that resulted in a stop codon after 9 amino acids (Fig. 4). Based on these findings, we made a diagnosis of type I hereditary AT deficiency.
Figure 4.
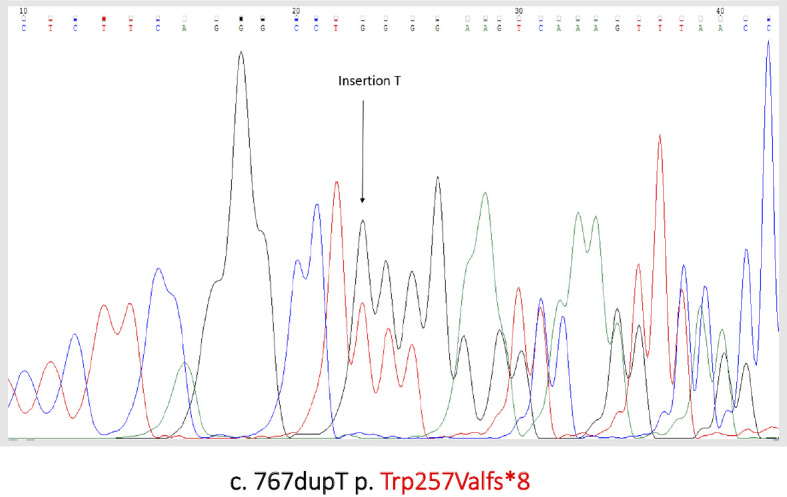
Detection of SERPINC1 gene mutation. DNA mutation numbering is based on cDNA sequence (NM_000488.3). We used the protein numbering system in according to the HSVG guidelines (the +1 aminoacid is the Met coded by ATG initiation codon).
Discussion
Hereditary AT deficiency is strongly associated with VTE. A meta-analysis of case-control studies and cohort studies showed that the odds ratio for VTE was 16.3 (95% confidence interval: 9.9-26.7) in individuals with AT deficiency compared to controls (8). Pregnancy is also associated with an increased risk of thrombosis, and the risk of thromboembolism is four- to five-fold higher in pregnant women than in non-pregnant women (9, 10). Accordingly, a woman with hereditary AT deficiency who becomes pregnant has a correspondingly increased risk of VTE, although there is wide variation in the reported rates (18% to 70%) (11-13).
Our experience with the present case suggests several important points with regard to hereditary AT deficiency. The first point is that we did not strongly suspect hereditary AT deficiency when she first presented with DVT because there was no family history of thrombosis and no problems with delivery in the first pregnancy. James et al. reported that most patients with hereditary AT deficiency who developed DVT during pregnancy had a family history of thrombosis and were affected in their first pregnancy (7). Thus, the initial onset of DVT during the second pregnancy in our patient was different from previous reports and seems to be a rare manifestation in cases of hereditary AT deficiency. Why DVT did not develop in her first pregnancy is unclear. In addition, her mother, who was diagnosed with hereditary AT deficiency at the same time as her daughter, did not have a history of DVT despite having delivered two children, including the present patient. We suspect that the disease activity concerning the development of DVT may differ among patients with hereditary AT deficiency depending on the type of SERPINC1 mutation and the plasma AT activity levels, and there is wide variation in the rate of DVT development, as mentioned above.
In the acute phase of DVT, the AT III activity was extremely low (45%) and heparin resistance was noted in the present patient, which we retrospectively considered to be findings consistent with hereditary AT deficiency. Demonstration of low postpartum AT III activity was a strong clue supporting the diagnosis of hereditary AT deficiency in our patient. Therefore, hereditary AT deficiency should be suspected when very low AT III activity and heparin resistance are noted in the acute phase of DVT, even if there is no personal or family history of thrombosis. We also need to retest the AT III activity in the chronic phase in order to avoid overlooking hereditary AT deficiency.
The second point to note concerning hereditary AT deficiency in the present patient is that abrupt elevation of the D-dimer level occurred postpartum, although heparin was only discontinued for a short period because she was multiparous and the delivery time was only four hours. We restarted anticoagulant therapy promptly, and thromboembolism did not occur, but she might have developed severe thromboembolism if the delivery time and discontinuation of heparin had been longer. Although we considered an inferior vena cava filter for prophylaxis of thromboembolism during discontinuation of heparin, it was not inserted because the findings on pre-delivery ultrasonography were satisfactory. We also considered administering AT concentrate in order to increase her AT III activity, but this was not done either due to the risk of infection and hypersensitivity. James et al. reported that AT concentrate was well tolerated by pregnant patients with hereditary AT deficiency, causing minimal adverse reactions and an extremely low risk of transmission of infectious agents (7). A study on the risk of VTE in 78 women with inherited AT deficiency, protein S deficiency, or protein C deficiency found DVT in 18% of the women during pregnancy and in 33% during the postpartum period (14), showing that patients with inherited thrombophilia have a high risk of thromboembolism around the time of delivery. Considering these reports, it may be better to administer AT concentrate for prophylaxis of fatal thromboembolism during discontinuation of anticoagulant therapy despite the potential side effects. In patients with very low AT III levels and critical thrombosis, we should consider the administration of AT concentrate even if hereditary AT deficiency has not been confirmed.
Finally, although there was no family history of thrombosis, the patient's mother showed a low AT III activity of only 46% when we assessed the levels of coagulation factors in the patient's family members. Subsequent genetic testing of the patient and her mother demonstrated the same SERPINC1 mutation in both, and we diagnosed them with type I hereditary AT deficiency. The SERPINC1 mutation detected in this family has been reported in Japanese patients before (15, 16). Furthermore, sources indicate that there are no reports of this SERPINC1 mutation except for in Japanese patients, suggesting that this SERPINC1 mutation may have a geographical component. Our patient did not remain on anticoagulant therapy after the puerperium, but the D-dimer levels stayed in the normal range, and DVT did not occur.
Regarding future management, we plan to perform temporary anticoagulant therapy when there is an increased risk of thrombophilia, e.g., during acute illness or surgery. Hereditary AT deficiency is an autosomal dominant condition, so the patient's children have an approximately 50% possibility of inheriting it and should be screened for AT III deficiency before puberty, when an increased frequency of thromboembolic events has been reported (17). While hereditary AT deficiency is associated with thromboembolic events and sometimes requires anticoagulant therapy, it is not associated with a higher mortality rate than that of the general population (18, 19).
In conclusion, it is important to make a proper diagnosis of hereditary AT deficiency and provide appropriate management.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia 14: 1229-1239, 2008. [DOI] [PubMed] [Google Scholar]
- 2. Jordan RE, Oosta GM, Gardner WT, Rosenberg RD. The kinetics of hemostatic enzyme-antithrombin interactions in the presence of low molecular weight heparin. J Biol Chem 255: 10081-10090, 1980. [PubMed] [Google Scholar]
- 3. Menache D, O'Malley JP, Schorr JB, et al. Evaluation of the safety, recovery, half-life, and clinical efficacy of antithrombin III (human) in patients with hereditary antithrombin III deficiency. Cooperative Study Group. Blood 75: 33-39, 1990. [PubMed] [Google Scholar]
- 4. Finazzi G, Caccia R, Barbui T. Different prevalence of thromboembolism in the subtypes of congenital antithrombin III deficiency: review of 404 cases. Thromb Haemost 58: 1094, 1987. [PubMed] [Google Scholar]
- 5. Goodrich SM, Wood JE. Peripheral venous distensibility and velocity of venous blood flow during pregnancy or during oral contraceptive therapy. Am J Obstet Gynecol 90: 740-744, 1964. [DOI] [PubMed] [Google Scholar]
- 6. Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 359: 2025-2033, 2008. [DOI] [PubMed] [Google Scholar]
- 7. James AH, Konkle BA, Bauer KA. Prevention and treatment of venous thromboembolism in pregnancy in patients with hereditary antithrombin deficiency. Int J Womens Health 5: 233-241, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Minno MN, Ambrosino P, Ageno W, Rosendaal F, Di Minno G, Dentali F. Natural anticoagulants deficiency and the risk of venous thromboembolism: a meta-analysis of observational studies. Thromb Res 135: 923-932, 2015. [DOI] [PubMed] [Google Scholar]
- 9. Heit JA, Kobbervig CE, James AH, Petterson TM, Bailey KR, Melton LJ 3rd. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 143: 697-706, 2005. [DOI] [PubMed] [Google Scholar]
- 10. Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJ. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 6: 632-637, 2008. [DOI] [PubMed] [Google Scholar]
- 11. Hellgren M, Tengborn L, Abildgaard U. Pregnancy in women with congenital antithrombin III deficiency: experience of treatment with heparin and antithrombin. Gynecol Obstet Invest 14: 127-141, 1982. [DOI] [PubMed] [Google Scholar]
- 12. Conard J, Horellou MH, Van Dreden P, Lecompte T, Samama M. Thrombosis and pregnancy in congenital deficiencies in AT III, protein C or protein S: study of 78 women. Thromb Haemost 63: 319-320, 1990. [PubMed] [Google Scholar]
- 13. Pabinger I, Schneider B. Thrombotic risk in hereditary antithrombin III, protein C, or protein S deficiency. A cooperative, retrospective study. Gesellschaft fur Thrombose- und Hamostaseforschung (GTH) Study Group on Natural Inhibitors. Arterioscler Thromb Vasc Biol 16: 742-748, 1996. [DOI] [PubMed] [Google Scholar]
- 14. Conard J, Horellou MH, Van Dreden P, Lecompte T, Samama M. Thrombosis and pregnancy in congenital deficiencies in AT III, protein C or protein S: study of 78 women. Thromb Haemost 63(2): 319-320, 1990. [PubMed] [Google Scholar]
- 15. Muta T, Okamura T, Kawamoto M, et al. Successful therapy with argatroban for superior mesenteric vein thrombosis in a patient with congenital antithrombin deficiency. Eur J Haematol 75: 167-170, 2005. [DOI] [PubMed] [Google Scholar]
- 16. Miyata T, Sato Y, Ishikawa J, et al. Prevalence of genetic mutations in protein S, protein C and antithrombin genes in Japanese patients with deep vein thrombosis. Thromb Res 124: 14-18, 2009. [DOI] [PubMed] [Google Scholar]
- 17. Thaler E, Lechner K. Antithrombin III deficiency and thromboembolism. Clin Haematol 10: 69-390, 1981. [PubMed] [Google Scholar]
- 18. Rosendaal FR, Heijboer H, Briët E, et al. Mortality in hereditary antithrombin-III deficiency--1830 to 1989. Lancet 337: 260, 1991. [DOI] [PubMed] [Google Scholar]
- 19. van Boven HH, Vandenbroucke JP, Westendorp RG, Rosendaal FR. Mortality and causes of death in inherited antithrombin deficiency. Thromb Haemost 77: 452, 1997. [PubMed] [Google Scholar]