

Abstract
Microbotryum lychnidis-dioicae is an obligate biotrophic parasite of the wildflower species Silene latifolia. This dikaryotic fungus, commonly known as an anther smut, requires that haploid, yeast-like sporidia of opposite mating types fuse and differentiate into dikaryotic hyphae that penetrate host tissue as part of the fungal life cycle. Mating occurs under conditions of cool temperatures and limited nutrients. Further development requires host cues or chemical mimics, including a variety of lipids, e.g. phytols. To identify global changes in transcription associated with developmental shifts, RNA-Seq was conducted at several in vitro stages of fungal propagation, i.e. haploid cells grown independently on rich and nutrient-limited media, mated cells on nutrient-limited media as well as a time course of such mated cells exposed to phytol. Comparison of haploid cells grown under rich and nutrient-limited conditions identified classes of genes probably associated with general nutrient availability, including components of the RNAi machinery. Some gene enrichment patterns comparing the nutrient-limited and mated transcriptomes suggested gene expression changes associated with the mating programme (e.g. homeodomain binding proteins, secreted proteins, proteins unique to M. lychnidis-dioicae¸ multicopper oxidases and RhoGEFs). Analysis for phytol treatment compared with mated cells alone allowed identification of genes likely to be involved in the dikaryotic switch (e.g. oligopeptide transporters). Gene categories of particular note in all three conditions included those in the major facilitator superfamily, proteins containing PFAM domains of the secretory lipase family as well as proteins predicted to be secreted, many of which have the hallmarks of fungal effectors with potential roles in pathogenicity.
Keywords: dimorphism, Microbotryum violaceum, mating, transcriptome
Introduction
Like all organisms, fungi need to perceive environmental cues and respond appropriately for proper growth and development. Those responses are mediated at multiple levels of gene expression regulation, including transcription, post-transcriptional modifications and translation. Changes in steady-state levels of RNA is one level of gene expression that can be assessed in a reliable and sensitive manner to gain insight into responses to changing growth conditions. Microbotryum violaceum sensu lato is a large species complex of plant pathogenic Basidiomycete fungi (anther smuts), which infects members of the Caryophyllaceae (Pinks) family. Each species in the Microbotryum complex is host specific, in most cases, only able to infect one or a few closely related plant species, although a few cryptic generalist species have been reported [1]. Completion of the life cycle and release of diploid spores only occurs in planta and thus sexual reproduction for these species is obligately parasitic. Nevertheless, the haploid basidiospores or sporidial cells are yeast-like and are easily propagated on artificial media in the laboratory. The growth of haploid strains of one species in the complex, Microbotryum lychnidis-dioicae, has been examined for various in vitro conditions [2]. No differences in growth or obvious morphology were observed for individual sporidial strains or in comparisons between different mating-type strains between 2 and 6 days at 15 °C on water agar, considered a limited nutrient stress condition. For mated cells, conjugation was observed from linear tetrad germination, and effects of temperature and nutrient availability on the development and growth of the fungus have also been examined [2].
As a representative of the Microbotryum fungal complex, M. lychnidis-dioicae is a species that infects Silene latifolia. Isolates of this species were the first used to provide published genome sequences at high coverage for members of the Microbotryum complex [3, 4]. Several molecular genetic studies on this species have focused on the mating-type loci and the mating-type chromosome on which they reside; extensive degeneration in the form of transposable elements in the non-recombining regions (NRRs) has been found on that chromosome [5, 6]. Differences between the mating-type chromosomes of haploids of different mating types are found in the mating loci on those chromosomes but additionally throughout the whole NRR which shows losses of genes and differentiation between a1 and a2 alleles [3]. The existence of different mating types is thought to prevent identical haploid clones from mating with each other [7].
Interestingly, some differences have been observed in the behaviours and characteristics of the respective a1 and a2 mating-type strains [3, 8, 9]. Of note, the a2 mating-type partner controls mitochondrial inheritance after mating [10]. To date, there have been no studies that explore extensively whether physiological differences and/or differences in gene expression are associated with haploid cells of different mating type.
The dimorphic switch, usually between a yeast-like form and a filamentous form, is a hallmark of the pathogenic programme of both human and plant pathogenic fungi [11–13]. Since in the life cycles of many pathogenic fungi, including M. lychnidis-dioicae, mating is an important prerequisite for the dimorphic transition and downstream events (e.g. host penetration and subsequent infection success), we were interested in identifying differences in gene expression in haploid strains under distinct conditions, particularly those involved in the dimorphic switch between yeast-like sporidia and filamentous hyphae. Similar studies of haploid response to environmental cues leading to the dimorphic switch in Ustilago maydis, the pathogen of maize, provided important insights into transcriptomic changes induced by differences in pH [14], lipids or low nitrogen availability [15, 16].
In the current study, we examined differences in global gene expression in M. lychnidis-dioicae cells under distinct conditions in vitro, including haploids grown separately on different media, as well as populations of mated cells with and without phytol treatment, a signal known to induce stable dikaryon formation [4, 17]. Comparison of expression profiles helped detect candidate genes with putative roles in mating and/or infection. Further, such analyses suggested differential roles for the two different mating types in these processes. M. lychnidis-dioicae is a particularly good model for such analyses since, unlike other well-studied dimorphic pathogens, unmated haploid cells do not exhibit obvious physiological or morphological changes when starved of nitrogen or exposed to lower temperature. In U. maydis and Saccharomyces cerevisiae, since filamentation as response to low ammonium is associated with up-regulation of the respective ammonium transporters [15, 16], the lack of such a phenotype in M. lychnidis-dioicae might predict that there would be no corresponding up-regulation of ammonium transporters in this species. Moreover, in the current study, comparing haploid cells with mated cells and mated populations exposed to a trigger for stable dikaryon formation allowed further characterization of mating-type-specific gene responses and switches that occur when mated cells produce the dikaryon, which is a prerequisite for successful infection.
Methods
Preparation of fungal cells
Haploid fungal strains (p1A1 and p1A2) (‘Lamole’ strains: Genbank 100-15Lamole [4, 6, 18]) grown under rich conditions (referred to as Rich) were grown for 4–5 days on yeast peptone dextrose media (YPD; 1 % yeast extract, 10 % dextrose, 2 % peptone and 2 % agar) at room temperature before RNA extraction. Two independently extracted RNA samples of each haploid strain were sequenced and analysed as biological replicates.
For the nutrient-limited (referred to as NL) condition, the same isolates were resuspended in sterile distilled water after growing for 4–5 days on YPD at room temperature and were adjusted to 109 cells per millilitre by OD600 determination. Then, 800 µl of the suspension was dispensed to cover the entire agar surface of a ‘Nutrient-Limited (NL)’ agar plate (composition: 2 % Bacto agar) in an even layer and allowed to dry. The plates were incubated at 14 °C for about 48 h before RNA was extracted. Two independently extracted RNA samples of each haploid strain were submitted and analysed as biological replicates. The same was done for mated samples (referred to as Mated) except that the two strains were combined in equal proportions to make the 109 cells per millilitre suspension. Two Mated samples were submitted and analysed as biological replicates.
Samples where mating occurred in the presence of phytol (referred to as Pmated) were prepared in the same way as Mated except that the solution in which the suspension was prepared contained 5 % ethanol and 1 % phytol (Sigma-Aldrich, cat. no.: P3647). Ethanol and phytol were combined first in order to dissolve the phytol, and then the mixture was added to sterile distilled water containing a total of 109 haploid cells per millilitre final concentration. A similar preparation that included 5 % ethanol but lacked phytol was used as a negative control and did not produce any observable differences compared to untreated Mated cells (data not shown). RNA was extracted from Pmated samples at 12, 24 and 48 h based on observations made in previous pilot experiments. Four Pmated12, three Pmated24 and two Pmated48 samples were submitted to the Broad Institute and analysed as biological replicates.
RNA extraction and sequencing
RNA was extracted and processed as previously published [4]. A minimum of 5 µg of DNA-free RNA was prepared for each sample and strand-specific libraries were constructed for each sample as previously described [19, 20]. Each library was sequenced using Illumina technology to generate 76 base-paired reads. RNA-Seq data from different conditions were processed using Trinity pipeline [21, 22] as follows. RNA-Seq reads from each sample were aligned using Bowtie [23] against annotated transcripts [4]. Fungal gene expression levels were estimated by RSEM [24] and edgeR with TMM normalization [25, 26] was used to identify differentially expressed genes between each pair of conditions with a corrected P-value cutoff of 1E-5. The test used with edgeR was the Exact test, which tests for genewise differences in the means between two groups of negative-binomially distributed counts. Data for correlation between normalized counts for replicates are presented in Fig. S1 (available in the online Supplementary Material) showing the heatmap of Pearson correlation coefficients between libraries, which shows that the counts per gene are very highly correlated between replicates. For all other heatmaps on genes, the clustering on genes was hierarchical clustering (via the programme hclust) based on Euclidean distance; clustering on libraries was also hierarchical clustering based on Spearman correlation with the method ‘complete’.
Analysis of differential gene expression
For differential expression comparisons, the samples were divided into two main groups of contrasts, with Group 1 consisting of NL alone or NL and Mated samples compared to Rich. The Group 2 set of contrasts consisted of the Mated, Pmated12, Pmated24 and Pmated48, where Mated was used as the reference for detection of gene expression changes in Pmated samples at 12, 24 and 48 h. All sequence data for this study are available in NCBI under bioproject PRJNA246470.
Gene set comparisons
Venn diagrams were generated using the online tool Venny [27]. The edgeR results were sorted to extract the list of the predicted genes [4] that were significantly up- or down-regulated (false discovery rate, FDR <1e-5) against a reference condition in the pairwise analysis.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) [28, 29] was performed to discover functional enrichment in differential genes, using functional annotations from PFAM domains, all Gene Ontology (GO) terms [30] after filtering out GO terms with too few or too many genes and KEGG pathway maps [31] using the GenePattern software [32]. In addition, the predicted small secreted proteins (SSPs) (less than 250 amino acids long), secreted proteins (SPs) (more than 250 amino acids long) and proteins found as unique to M. lychnidis-dioicae based on OrthoMCL analysis [4, 33] were also included as gene sets to see if there were any enrichment of these genes among those differentially expressed. The resulting gene set list (.gmt) consisted of 801 gene sets, with a range of 5–1116 members. Preranked gene lists (.rnk) based on significance of differential expression from every pairwise comparison made were processed by the GSEAPreranked module. When running the software, the algorithm was set to allow a minimum gene set of five and maximum of 1200. Gene sets that were significantly enriched at FDR <25 % or nominal P-value <1 % were listed and examined for further hypothesis generation.
Results and Discussion
In this study, our overall goals were to compare gene expression at various stages during the in vitro portion of the life cycle of M. lychnidis-dioicae (Fig. S2). Since the mating process is a prerequisite for plant infection and the complete genome has now been sequenced, understanding the gene expression changes during mating was the next logical step in this kind of investigation. Thus, we compared transcriptomes of haploid cells from each mating-type strain (p1A1 and p1A2), grown on YPD medium (‘Rich’), where they grow vegetatively and reproduce by budding. In addition, the same strains incubated separately on water agar would be exposed to a nutrient-limited condition (NL), allowing a determination of genes differentially regulated by starvation; some of these genes might also be involved in priming the respective mating types in preparation for mating. Under the same conditions, if these strains were inoculated together, mating ensues (‘Mated’) and, thus, transcriptome analysis provided insights into gene expression that is controlled during and after mating. Finally, exposure to lipids, such as phytol or tocopherols, causes mated cells to produce dikarya [17], a prerequisite for penetration of the host plants and successful infection. Thus, mated cells exposed to phytol for a number of hours (e.g. Pmated12 or Pmated24) provided the opportunity to examine altered gene expression in response to this signal and to potentially predict genes whose expression primes the dikaryon for later differentiation.
RNAi pathway differential gene expression during growth in rich media
While differential expression of components of the RNAi pathway has been clearly examined in mammalian cells [34, 35] and plant cells, much less work has described expression of these genes in fungi. In one of the few reports for fungi, dcl-2 of Penicillium marneffei, encoding a Dicer-like protein, was up-regulated in the mycelial form compared to the yeast form [36]. Here, we observed differential expression of predicted components of the RNAi pathway in M. lychnidis-dioicae. None of these genes is located on the mating-type chromosome and they should be nearly identical in both mating-type strains [3, 4]. Nevertheless, both MVLG_06823 (ARGONAUTE) and MVLG_01202 (DICER) were up-regulated in p1A2 Rich compared to all other haploids and compared to Mated cells. Another gene predicted to encode ARGONAUTE, MVLG_06899, did not change expression significantly in any of the in vitro stages but was up-regulated in the early stages of teliosporogenesis [4] (S. S. Toh et al., unpublished). Thus, these RNAi components perhaps play specific roles in the different stages of development of the fungus. Similarly, components of the RNAi machinery of Fusarium graminearum, the causal agent of head blight in cereal crops, appear to be involved in a regulatory pathway for mating-locus-mediated sexual development, in which both mating loci may be activated by several environmental cues, and then control the expression of at least 1245 target genes during sexual development [37].
Genes differentially regulated for both NL and Mated conditions suggest media-specific changes in expression, while differential expression among the haploids on NL and the mated samples implicate genes for mating and beyond
During in vitro growth, genes on a1 and a2 strain mating-type chromosomes are differentially expressed [4, 5]. In a previous analysis of Rich, NL and Mated treatments, 177 genes were differentially expressed when comparing the a1 and a2 mating-type strains, and 93 of those were differentially expressed in both NL and Rich. However, some of these changes in gene expression between the two mating types may have been due to use of the a1 genome as reference, without having a complete a2 genome for comparison. Thus, transcripts specific to the a2 mating type may have gone undetected. As we did not map transcripts specifically to the a2 mating-type genome in the current study, we must provide the caveat that a2-specific gene expression for genes on the mating-type chromosome would similarly go undetected in these analyses or perhaps lead to false indications of down-regulation in a2 mating-type cells. Where possible, genes expected to lie on the mating-type chromosomes were thus tested with respect to the a2 mating-type genome [3] when our analysis suggested down-regulation in this background.
Here, we first compared for biological replicates the expression patterns of haploid and mated strains. Among the conditions examined, both media and mating type influenced expression pattern. The number of predicted genes in p1A1 NL, p1A2 NL and Mated that were significantly up- and down-regulated compared to the Rich condition are shown in Fig. 1 and a heatmap of these comparisons is found in Fig. S1. With a cutoff of FDR <1e-5, relative to growth on Rich media, a total of 1508 genes (768 down-regulated and 740 up-regulated) for NL and Mated, or when just comparing NL to Rich, 1551 total (774 down, 777 up) were differentially transcribed (Fig. 1). Among these, a large proportion, 1384, were commonly up- and down-regulated among both NL and Mated conditions, suggesting that these are media-specific changes in expression.
Fig. 1.

Venn diagram of number of predicted genes in each condition that were significantly (a) up- and (b) down-regulated compared to Rich. Venn diagrams were generated using the online tool Venny [27]. The edgeR results were sorted to extract the list of the predicted genes [4] that were significantly up- or down-regulated (FDR <1e-5) against the Rich condition in the pairwise analysis. Note in (a) that there were no p1A1NL genes up-regulated relative to p1A2NL in these analyses.
‘NL’ and Mated treatments share up-regulated gene sets for oligopeptide transporters, secretory lipases, SPs and proteins unique to Microbotryum species
To obtain an objective understanding of the types of genes that were differentially regulated, genes determined to be differentially expressed among the conditions were grouped with respect to functions identified by protein sequence domains, such as PFAM domains. M. lychnidis-dioicae Lamole strain has 56 predicted genes with an RNA recognition motif (PF00076) [4]. Of these, one gene (MVLG_03719) was located on the pseudo-autosomal region of the mating-type chromosome and five genes (MVLG_05905, MVLG_05906, MVLG_05907, MVLG_06413 and MVLG_06570) were found on the NRR. The remainder of these genes are located on the autosomes. When both NL and Mated were compared to Rich (Tables 1 and S2–S5; complete listing, including FDR values, shown in Table S1), the common gene set enrichments among the genes up-regulated (Table 1 and S4) included PFAM domains associated with oligopeptide transporters (OPTs), secretory lipases, alpha/beta hydrolase fold (found in hydrolytic enzymes, including proteases, lipases and esterases), SPs as well as proteins unique to M. lychnidis-dioicae based on comparative genome analysis that included as closest species Sporobolomyces roseus and Rhodotorula graminis [4]. Genes with PFAM or TIGR domains for OPTs (PF03169.8; TIGR00728) were identified as enriched in NL and Mated comparisons with Rich, as were genes with PF03583.7 (secretory lipases). Proteins unique to M. lychnidis-dioicae were up-regulated in p1A2NL and Mated relative to p1A2Rich. The category Secreted Proteins was up-regulated in p1A2NL relative to each haploid strain grown alone in Rich; similarly, Secreted Proteins was an up-regulated gene set in Mated vs. p1A1Rich.
Table 1.
List of enriched gene sets that were up-regulated in NL when compared to Rich, ranked by decreasing magnitude of NES, filtered by nominal P-value of 0.01 and FDR of 0.25
Presented in this table are only enriched gene sets that meet the <0.25 FDR cutoff. All GSEA terms and associated FDR values are found in Table S1.
| Enriched in p1A1 NL vs. p1A1 Rich | Common enrichment both NL vs. both Rich | Enriched in p1A1 NL vs. p1A2 Rich |
|---|---|---|
|
|
|
| Enriched in p1A2 NL vs. p1A1 Rich | Common enrichment p1A1 NL vs. both Rich | Enriched in p1A2 NL vs. p1A2 Rich |
|
|
|
| Common enrichment p1A2 NL vs. both Rich | ||
|
OPTs are extremely versatile peptide transporters with an extended substrate spectrum [38]. M. lychnidis-dioicae is predicted to encode 18 OPT-like proteins [4]. While OPTs were significantly enriched among genes up-regulated in nutrient-limited samples, this gene family displayed a more complex regulation pattern across the conditions tested, and the genes were further differentially regulated during stages in planta (S.S. Toh et al., unpublished) [39]. Five OPTs were significantly up-regulated in Mated and NL compared to Rich, but not different in the phytol-treated groups, compared to Mated; these may be related to nutrient availability.
In NL and Mated treatments, down-regulation of gene sets for cellular components, mitosis and metabolism was found, as was repression of genes bearing multicopper oxidase domains
In the Rich condition, fungal cells are growing and dividing mitotically, whereas on water agar (NL) where mating can take place, lack of nutrients and the low temperature probably slow or arrest mitotic growth. The gene set enrichment is consistent with this prediction where the three main GO categories identified in enrichment analysis (cellular components, biological processes and molecular functions and metabolism) were among those gene sets that were down-regulated in NL and Mated.
Genes bearing three multicopper oxidase domains were significantly enriched among those down-regulated in the NL and Mated conditions. Basidiomycetes tend to have multiple copies of multicopper oxidases, which have been implicated in many different types of processes and regulation [40]. MVLG_00670 and MVLG_01868 appear similar to be ferroxidases; together with MVLG_01284, they were down-regulated in NL and Mated conditions. In contrast, MVLG_03092, probably a laccase, is predicted to be secreted and was up-regulated in NL.
Genes differentially expressed in NL compared to Mated conditions: specialized roles respectively for pre- and post-mating functions
In contrast to genes expressed similarly in NL and Mated conditions, genes differentially expressed uniquely in NL but not in Mated could reflect a response as preparation for mating, while those unique to Mating reflect genes needed for early responses of mating cells. Relative to the Mated condition, the haploid p1A1NL and p1A2NL conditions shared more common differential expression (324 up- and 247 down-regulated) than mating-type-specific changes (100 up- and 2 down-regulated uniquely in p1A1 vs. 162 up- and 97 down-regulated uniquely in p1A2). Among the genes differentially expressed in the haploids in NL compared to Mated were two whose predicted proteins contained PFAM domains associated with lipases [MVLG_00914, PF03583.7, Secretory lipase; MVLG_01816, PF01764.18, Lipase (class 3)]; these genes were up-regulated in NL (but not Mated) compared to Rich (FDR approximately 2e-5). Additionally, genes encoding proteins bearing PFAM domains for CAAX amino-terminal protease (MVLG_01370), isoprenylcysteine carboxyl methyltransferase and prenylcysteine lyase (MVLG_05930) were up-regulated in one mating type or in both mating types in NL relative to Mated. It is possible that these genes are involved in post-translational modifications of pheromone precursors in order to facilitate mating [41]. There were also classes of proteins differentially represented by unique genes in the NL or Mated conditions, including those encoding PFAM domains for mitochondrial carrier proteins, RhoGEFs [e.g. the Mated category was enriched for the RhoGEF domain (PF00621), a motif that includes components involved in G-protein signalling], fungus-specific Zn(2)-Cys(6) cluster zinc finger transcription factors, MFS/sugar transporters, glycosyl hydrolases and response regulator receiver domains. Interestingly, 192 genes were uniquely down-regulated in p1A1NL relative to p1A2NL and no genes were up-regulated in p1A1NL vs. p1A2NL. Twelve of the genes up-regulated in p1A2NL were located on the mating-type chromosome (i.e. MVLG_07258, MVLG_7161, MVLG_6563, MVLG_6231, MVLG_6236, MVLG_6416, MVLG_6572, MVLG_6794, MVLG_7142, MVLG_7146, MVLG_7267 and MVLG_7321). Additional differentially regulated clusters of genes were found on the ‘autosomes’ (i.e.not on the mating-type chromosome): e.g. MVLG_06710-17, MVLG_7020–23 and MVLG_7051–7055 (except MVLG_07054, located on the mating-type chromosome). This is consistent with the hypothesis that the a2 mating type contributes more in the initiation of the mating relationship since the NL treatment is conducive to mating but the mating partner is absent. This is also consistent with previous reports that the a2 mating type produces the conjugation tube earlier and to a greater length [8, 9]. Further support is provided by the presence of two homologues of STE12 on the a2 mating-type chromosome [3] and its absence in the a1 mating-type chromosome.
In these analyses, 126 differentially regulated genes specific to the Mated condition were identified. Nearly 50 of these were annotated as hypothetical proteins, whose function has yet to be determined. Perhaps somewhat surprisingly, a gene encoding a high-affinity ammonium transporter (MVLG_00253) was up-regulated in the Mated condition, but not significantly in the NL condition relative to Rich. In a number of other fungi, such genes are often highly up-regulated in haploid cells grown under conditions of low ammonium [15, 42]. Based on previous analysis [5], genes located on NRRs were not more frequently up-regulated than those in the non-mating-type chromosomes in the Mated condition, nor were the differentially regulated genes between a1 and a2 mating types up-regulated in the Mated condition. While these genes could have mating-type-specific roles, genes with weak or no expression in one of the mating-type strains may have been inactivated during the evolution of the respective mating-type chromosomes that included extensive rearrangements and degeneration [43]. In fact, 10 genes lacked any expression under the p1A2NL condition, whereas they were expressed in Mated cells. Of these, five (MVLG_06188, MVLG_6229, MVLG_ 6269, MVLG_6270 and MVLG_6750) showed no match to the p1A2 genome [3] (European Molecular Biology Laboratory European Nucleotide Archive, accession no. PRJEB7910).
In the typical process of mating for Basidiomycetes, only the pheromones/receptors and homeodomain genes are involved in mating [44]. However, genes involved in initiation of mating could be expressed specifically in haploid NL cells rather than Mated. In NL, the fungal cells were in a conducive environment for mating (i.e. stressful both in terms of temperature and nutrient availability) but lacked the mating partner; in contrast, in Mated samples, mating had already taken place and conjugation tube formation had already occurred; thus, transcripts related to initiation of mating may already have been lost. We investigated this possibility with further GSEA.
Importantly, five of the OPT group (MVLG_04056, MVLG_04057, MVLG_04545, MVLG_05201 and MVLG_07217), none of which were located on the mating-type chromosomes, were significantly up-regulated in p1A2 NL even when compared to Mated, suggesting possible involvement with pheromone sensing due to the ‘initiative’ taken by a2 in mating; thus, they exhibit higher transcription levels in the p1A2NL condition, whereas in Mated, their activity would no longer be required, since the mating process would have been completed.
In basidiomycete fungi, where post-cell fusion events of mating are controlled by genes other than those encoding the pheromone and receptor, b mating-type genes encode two subunits of a homeodomain (HD) transcription factor, consisting of an HD1 class and an HD2 class protein [45]. In general, the HD1 and HD2 proteins share a common domain organization, though they are not related to each other in primary sequence. Also, compatible mating partners must have both different HD1 and HD2 alleles [45]. The identified HD proteins, MVLG_07149 and MVLG_07150, which determine post-mating compatibility, had nearly undetectable expression in Rich. Mated samples showed additional increases beyond those of p1A1 NL, of 2.21 and 5.91 log2 fold, respectively, for these two genes. This suggests that there is transcription of the HD genes in haploids under NL conditions, even in the absence of a mating partner, and that the gene expression level increases post-mating, which is consistent with its function. Given the importance of HD function in downstream events in both basidiomycetes [46] and in ascomycetes [47], expression of HD genes as a response to environmental cues appears more generally to be crucial to completion of the mating process. Becker et al. [48] found evidence for mating-type transcription factor functions that extend beyond their previously understood role in sexual development. These new roles include regulation of hyphal morphology, asexual development as well as amino acid, iron and secondary metabolism. Furthermore, in vitro DNA protein binding studies and downstream analysis in yeast and Penicillium chrysogenum enabled the identification of a MAT1-1-1 DNA binding motif, which is highly conserved among euascomycetes. An additional gene identified in our comparisons between NL and Mated conditions (MVLG_01557, located on an autosome) was predicted to encode a protein with an HMG box domain, associated with transcriptional regulation, especially for sexual development [49].
Phytol-mated samples showed enrichment for up-regulation of MFS, secretory lipase, SP, SSPs and proteins unique to Microbotryum species
Dikaryon formation is required for entry into the plant host. M. lychnidis-dioicae reacts to chemicals like phytol and α-tocopherol [17] in vitro to produce a dikaryon/hyphae on artificial medium. A filamentous appearance following formation of conjugation tube (Fig. 2d) is observed compared to no phytol control (Fig. 2c).
Fig. 2.
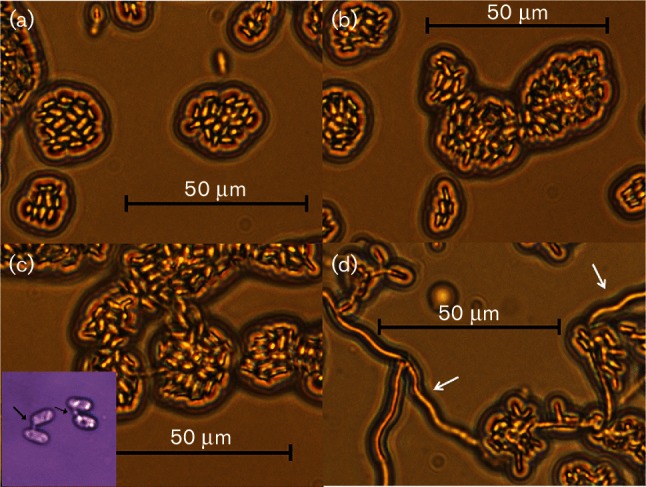
Morphology of haploid and mated cells in the absence or presence of phytol. (a and b) Phytol-treated haploid cell control (p1A1 and p1A2, respectively); (c) Nutrient-Limited (NL)-treated mated cell control (Inset: higher magnification showing cells with conjugation bridge, black arrows); (d) mated cells treated with phytol showing filamentation (white arrows). Size bars, 50 µm. This figure was adapted, with permission, from our previously published work (Fig. 4b–e; [4]).
Assessing the number of genes that were differentially regulated in mated cells in the presence of phytol over 12 h (Pmated12), 24 h (Pmated24) and 48 h (Pmated48) compared to mated cells in the absence of phytol at 48 h (Mated) identified a total of 964 (13.1 %) predicted genes that were up-regulated compared to Mated and 445 (6.0 %) were down-regulated compared to Mated. Of these, a smaller proportion were up-regulated (22.1 %) and down-regulated (2.7 %) throughout the entire course of treatment compared to Mated (Fig. 3; heatmap Fig. S3). These comparisons show that the majority of the gene expression changes happened within the first 24 h of phytol treatment.
Fig. 3.
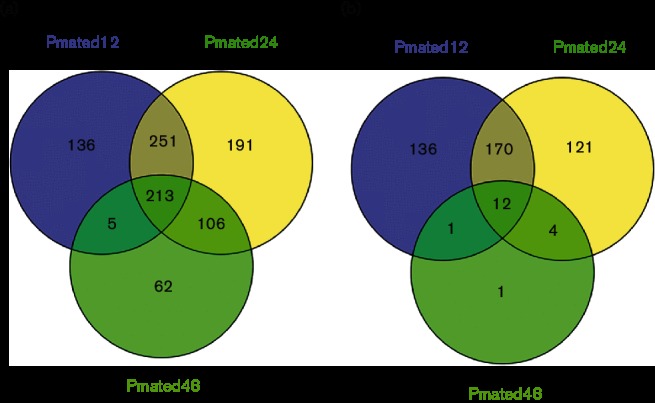
Venn diagram of number of predicted genes that were (a) up-regulated and (b) down-regulated in Phytol-mated (Pmated) compared to Mated condition. Venn diagrams were generated using the online tool Venny [27]. The edgeR results were sorted to extract the list of the predicted genes [4] that were significantly up- or down-regulated (FDR <1e-5) against the Mated condition in the pairwise analysis.
Genes that presented a detectable trend over the time course were identified by examining all the pairwise comparisons and taking all significant gene expression changes into account (Table 2; gene set enrichments of note in Tables 3 and 4). However, it should be noted that these trends may or may not be exclusive to mating cells in the presence of phytol since the time-course data for corresponding mating cells in the absence of phytol were not obtained.
Table 2.
Summary of trends observed in genes of M. lychnidis-dioicae over time when treated with phytol
| Trend* | Description | Number of genes | Percentage† (%) |
|---|---|---|---|
| 12<24<48 h | Increasing from 12 to 48 h | 147 | 2.0 |
| 12>24>48 h | Decreasing from 12 to 48 h | 123 | 1.7 |
| 12<24=48 h | Increasing from 12 to 24 h and plateaued | 215 | 2.9 |
| 12>24=48 h | Decreasing from 12 to 24 h and plateaued | 127 | 1.7 |
| 12<24>48 h | Highest at 24 h | 223 | 3.0 |
| 12>24<48 h | Lowest at 24 h | 158 | 2.1 |
| 12=24<48 h | Constant from 12 to 24 h then increase | 24 | 0.3 |
| 12=24>48 h | Constant from 12 to 24 h then decrease | 150 | 2.0 |
*The numbers represent the hours at which the phytol-treated sample were extracted. < represents an increase; > represents a decrease; = represents no detectable changes.
†Percentages were calculated based on total gene set of 7364 genes.
Table 3.
List of enriched gene sets that were up-regulated in Pmated12, Pmated24 and Pmated48 when compared to Mated, ranked by decreasing magnitude of NES, filtered by nominal P-value of 0.01 and FDR of 0.25
| Enriched in all Pmated vs. Mated | |
|---|---|
| • SECRETED PROTEIN • UNIQUE TO M. lychnidis-dioicae | |
| Common enrichment in Pmated12 and Pmated24 vs. Mated | Common enrichment in Pmated24 and Pmated48 vs. Mated |
|
|
| Enriched in Pmated12 only vs. Mated | Enriched in Pmated48 only vs. Mated |
|
|
*Presented in this table are only enriched gene sets that meet the <0.25 FDR cutoff or were of particular note (indicated by * if >0.25 FDR). All GSEA terms and associated FDR values are found in Table S1.
Table 4.
List of enriched gene sets that were up-regulated in Pmated12, Pmated24 and Pmated48 when compared to each other, ranked by decreasing magnitude of NES, filtered by nominal P-value of 0.01 and FDR of 0.25
Presented in this table are only enriched gene sets that meet the <0.25 FDR cutoff. All GSEA terms and associated FDR values are found in Table S1. CC, cellular component; BP, biological process; MF, molecular function.
| Enriched in Pmated12 vs. Pmated24 | Enriched in Pmated24 vs. Pmated12 |
|---|---|
|
|
| Enriched in Pmated12 vs. Pmated48 | Enriched in Pmated48 vs. Pmated12 |
|
|
Major facilitator superfamily
Several OPTs were differentially expressed during treatment of mated cells with phytol. Three predicted OPTs (MVLG_00150, MVLG_03106 and MVLG_05200), though up-regulated in NL and Mated, were reduced in expression in Pmated, while three others (MVLG_02728, MVLG_04056 and MVLG_04057) were further up-regulated in Pmated relative to their levels in Mated. Moreover, we note that M. lychnidis-dioicae has a large inventory (i.e. 118) of MFS domain-containing genes [4]. A cluster of genes was identified (MVLG_06790–MVLG_06794) containing either only a major facilitator domain (MFS, PF07690) or an additional sugar transporter domain (PF00083; for MVLG_06794). They were most closely related to MFS domain-containing proteins of Microbotryomycetes and belong together as one paralogous family, suggesting similar functions, yet they had varying expression patterns. Three of these transporters (MVLG_06790–MVLG_06792) are potentially involved in the filamentation process, as they were increasingly expressed from 12 to 48 h phytol treatment, while the Mated samples expressed them at nearly the same levels as at 12 h. Interestingly, MVLG_06792 is located on the mating-type chromosome.
Secretory lipase (PF03583)
In previous work [4], expression of the secretory lipase gene family (named for the presence of this PFAM domain, PF03583.7, in the predicted proteins) presented interesting patterns. With these new data, we have evidence of enrichment of up-regulated secretory lipases both in Mated and NL when compared to Rich and, perhaps more importantly, members of this group were up-regulated even more so in Pmated24 and Pmated48 when compared to Mated.
Three cytoplasmic secretory lipase proteins (MVLG_07229, MVLG_07284 and MVLG_07291) were found to be highly induced in the NL condition, as found previously [4].
Expression of two proteins lacking a secretion signal was significantly up-regulated in Mated stages. MVLG_00914 showed 9 and 11 log2 fold increase in Pmated24 and Pmated48, respectively, when compared to Mated. MVLG_05549 showed about a 3 log2 fold increase throughout the 48 h period when compared to Mated and the three cytoplasmic proteins were not differently expressed than in Mated. Thus, expression of the three cytoplasmic proteins may be related to nutrient availability.
In addition, two other secretory lipases, MVLG_00933 and MVLG_00934, were also up-regulated in Pmated. Previous quantitative real-time PCR experiments (M.H. Perlin et al., unpublished) were not able to establish the expression level of these two secretory lipases. From these new data, we found that significant up-regulation was only detectable at 24 h in RNA-Seq and transcript abundance was only about 20 transcripts per million, compared to more than 100 with the other secretory lipases that were detected in the quantitative real-time PCR experiment. Moreover, these two secretory lipases showed more than 9 and 11 log2 fold increases in Pmated24 and further increases to 12 and 15 log2 fold increases, respectively, in Pmated48. Hence, we propose that MVLG_00914, MVLG_00933, MVLG_00934 and MVLG_05 549 are directly involved in the response to phytol and filamentation of the fungus. It is also interesting to note that the MVLG_00914, MVLG_00933 and MVLG_00 934 are located close together in the genome; of these, only MVLG_00934 was not predicted to be secreted. Also of interest, MVLG_04698, which was predicted to encode a fatty acid synthase, was up-regulated in NL and Mated when compared to Rich, but the expression was the same for this gene in Pmated compared to Mated.
Secretory or extracellular lipases from plant pathogenic fungi likely function in acquiring nutrients or in penetrating or otherwise negotiating interaction with the host target tissue. As such, these enzymes can be involved in breaking down the physical defence barrier presented by the plant cuticular layer. Differential expression of some of the M. lychnidis-dioicae genes with this PFAM domain (i.e. MVLG_00914, MVLG_00933, MVLG_00934 and MVLG_05549) is consistent with that prediction, as their expression appeared to increase with phytol treatment that would lead to dikaryon formation. However, their role may have additional or alternative roles in the interaction of M. lychnidis-dioicae with its host. Two of the genes predicted to encode secreted lipases, MVLG_00914 and MVLG_05549, were also significantly up-regulated in late-stage infected flowers (S.S. Toh et al., unpublished) [39]. We speculate that some of these lipases may have a role in the induced morphological changes in Silene flowers. In particular, it is interesting that lipases can produce localized pools of α-linolenic acid which through a series of plant-based chemical reactions is converted to jasmonic acid (JA). Since JA plays a role in development of inflorescence, including sex determination and fertility [50, 51], it is plausible that M. lychnidis-dioicae lipases could create altered metabolic pools of α-linolenic acid in the developing meristematic region, thereby altering JA levels and effecting sex determination/development of both male and female inflorescence. This would provide a novel functional consequence of secretory lipase for an endophytic/biotrophic fungus to affect growth and development of plant reproductive structures.
SPs, SSPs and genes unique to Microbotryum species
In conducting the gene enrichment analysis, by designating genes encoding secreted (SP), small and secreted (SSP) and products unique to Microbotryum species in genomic comparisons [4] as gene sets, we were able to discover their enrichment, which would not have otherwise been possible due to the lack of known domains. A subset of this group, 71, were predicted SSPs, small proteins which bear the hallmarks of fungal effectors that would be postulated to play roles in manipulating the host during infection [4, 52]. Similarly, 1534 predicted proteins were unique to M. lychnidis-dioicae, based on the comparisons made to other fungi [4]; of these, 46 were predicted to be SSPs.
To understand whether the genes were up-regulated in Mated and NL and further up-regulated in Pmated, we compared the SPs that contributed to core enrichment of each of the three gene sets in each treatment. Out of the 65 SPs that were up-regulated relative to Mated during the Pmated time course, 21 (32.3 %) of them were found at all time points (Fig. S4a); of the SPs, 26 were SSPs found up-regulated in Pmated compared to Mated, and 38.5 % of these were induced at all time points (Fig. S5a). It is interesting that ‘Pmated12’ and ‘Pmated48’ did not share any up-regulated SPs other than those that were up-regulated through ‘Pmated24’, suggesting that there were specific roles for individual SPs at different time points. For instance, the SSPs produced during early exposure to phytol appeared to be different from those used in the last stage of Pmated treatment. As examples, MVLG_01138, MVLG_05717, MVLG_05398, MVLG_00443 and MVLG_05835 were all significantly up-regulated in ‘Pmated24’ and ‘Pmated48’ compared with ‘Pmated12’. Additionally, some genes of note that were significantly expressed more highly at 48 h of exposure to phytol compared with 12 or 24 h included MVLG_00566, MVLG_00784, MVLG_1310, MVLG_06949, MVLG_03368 and MVLG_01690. Of course, due the limitation of sampling times in this study, we must point out that additional subsets of genes could be differentially regulated at different timepoints (not specifically measured here) throughout the course of phytol exposure. It is also important to note that, although few of the SSPs contain PFAM domains, MVLG_01690 contains a PFAM domain for a GPI-anchored CFEM; proteins containing this domain are often associated with fungal pathogenesis. A total of 109 SPs are potentially involved in the mating and filamentation process (Fig. S4b), of which 29 were SSPs (Fig. S5b). NL and Mated uniquely shared 30 (29.1 %) of these genes (3 were SSPs) and only 17 (16.5 %; of which 9 were SSPs) overlapped with what was also up-regulated in Pmated. Out of the 65 proteins (26 SSPs) that were up-regulated in Pmated, 48 (73.8 %) were used exclusively in Pmated. A similar pattern of differential expression was observed for the ‘unique’ gene set as well. Out of the 141 unique proteins that were up-regulated during the Pmated time course compared to Mated, 79.4 % were used exclusively in Pmated, while 42 (29.8 %) of these were used across all three time points (Fig. S6a); only two were common between Pmated12 and Pmated48. There appeared to be substantial discrete and overlapping gene expression throughout the course of the treatment (Table 4). A total of 393 unique proteins were involved in the mating and filamentation process (Fig. S6b). NL and Mated shared 148 (37.7 %) of these and only 29 (7.4 %) overlapped with those also up-regulated in Pmated (Tables 3 and 4).
SSP is the only gene set that had more overlap among NL, Mated and Pmated than when comparing NL and Mated only. Genes up-regulated in common by NL and Mated and exclusively used by Pmated were the major categories in the three gene sets. There is also a suggestion of tight control over which genes were used at each stage of the filamentation. From these observations, we deduce that discrete sets of genes were responsible for initiation of mating and the hyphae formation that follows, likely in a very systematic manner.
Conclusion
Availability of nutrients is often a trigger for a developmental switch. Fungal mating, for example, may be associated with low nitrogen [44] and/or low temperature [4, 8, 17]. For pathogenic fungi, additional downstream events in the pathogenic programme require compounds that serve as host cues [17, 53]. In this study, RNA-Seq analysis was used to examine a range of in vitro conditions that are associated with a pathogenic lifestyle. This examination allowed us to parse out differential transcription of genes that were expressed preferentially in one mating-type background compared with the other, including differences associated with nutrient availability. Moreover, some of these differences persisted in downstream stages, including mating and the initiation of the switch to filamentous dikaryon. In comparing the two different mating-type strains, proteins that were differentially expressed between the mating types in NL may be related to preparation for mating and may suggest division of labour for each of the two mating types in this endeavour. Genes that were responsive to phytol may be important for dikaryon formation, and those that are differentially expressed at different time points of phytol treatment may have specific functions at each stage of the switching process.
This investigation uncovered several categories of differentially regulated genes (i.e. OPTs, secretory lipases, MFS, SPs/SSP and unique to M. lychnidis-dioicae) worthy of further investigation, as well as evidence that the RNAi pathway is also affected differentially under the conditions examined. These findings provide the basis to generate further hypotheses aimed at elucidating the mating pathway of M. lychnidis-dioicae and its sequelae leading to the successful infection of its host plant. It will be necessary to move beyond computational analysis and experimentally target the classes of genes identified in this study.
Supplementary Data
Supplementary File 1
Funding information
This work was supported by National Science Foundation Grant No. 0947963 to M.H.P., D.J.S. and C.A.C. and by a Dissertation Completion Grant to S.S.T. from the School of Interdisciplinary and Graduate Studies at the University of Louisville.
Acknowledgements
The authors would like to thank T. Giraud, Universite du Paris-Sud, Orsay, France, for helpful comments and suggestions on the manuscript prior to submission. We thank the Broad Institute Genomics Platform for constructing libraries and sequencing the RNA samples. The technical assistance of D. Razeeq, University of Louisville, in preliminary experiments using phytol-treated cells is also appreciated.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
The authors declare that this manuscript follows the Ethical Responsibilities of Authors, as indicated in the Instructions to Authors (including not being previously published or simultaneously submitted elsewhere, and consent of all authors).
Footnotes
Abbreviations: FDR, false discovery rate; GSEA, gene set enrichment analysis; JA, jasmonic acid; NRR, non-recombining region; OPT, oligopeptide transporter; SP, secreted protein; SSP, small secreted protein.
All sequence data for this study are available in NCBI under bioproject PRJNA246470.
Six supplementary figures and five supplementary tables are available with the online Supplementary Material.
Edited by: S. Pöggeler and V. J. Cid
References
- 1.Refrégier G, Le Gac M, Jabbour F, Widmer A, Shykoff JA, et al. Cophylogeny of the anther smut fungi and their caryophyllaceous hosts: prevalence of host shifts and importance of delimiting parasite species for inferring cospeciation. BMC Evol Biol. 2008;8:100. doi: 10.1186/1471-2148-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hood ME, Antonovics J. Two-celled promycelia and mating-type segregation in Ustilago violacea (Microbotryum violaceum) Int J Plant Sci. 1998;159:199–205. doi: 10.1086/297539. [DOI] [Google Scholar]
- 3.Badouin H, Hood ME, Gouzy J, Aguileta G, Siguenza S, et al. Chaos of rearrangements in the mating-type chromosomes of the anther-smut fungus Microbotryum lychnidis-dioicae . Genetics. 2015;200:1275–1284. doi: 10.1534/genetics.115.177709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perlin MH, Amselem J, Fontanillas E, Toh SS, Chen Z, et al. Sex and parasites: genomic and transcriptomic analysis of Microbotryum lychnidis-dioicae, the biotrophic and plant-castrating anther smut fungus. BMC Genomics. 2015;16:461. doi: 10.1186/s12864-015-1660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fontanillas E, Hood ME, Badouin H, Petit E, Barbe V, et al. Degeneration of the nonrecombining regions in the mating-type chromosomes of the anther-smut fungi. Mol Biol Evol. 2015;32:928–943. doi: 10.1093/molbev/msu396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hood ME, Antonovics J, Koskella B. Shared forces of sex chromosome evolution in haploid-mating and diploid-mating organisms: Microbotryum violaceum and other model organisms. Genetics. 2004;168:141–146. doi: 10.1534/genetics.104.029900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Billiard S, López-Villavicencio M, Devier B, Hood ME, Fairhead C, et al. Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biol Rev Camb Philos Soc. 2011;86:421–442. doi: 10.1111/j.1469-185X.2010.00153.x. [DOI] [PubMed] [Google Scholar]
- 8.Day AW. Communication through fimbriae during conjugation in a fungus. Nature. 1976;262:583–584. doi: 10.1038/262583a0. [DOI] [PubMed] [Google Scholar]
- 9.Xu L, Petit E, Hood ME. Variation in mate-recognition pheromones of the fungal genus Microbotryum . Heredity. 2016;116:44–51. doi: 10.1038/hdy.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilch G, Ward S, Castle A. Transmission of mitochondrial DNA in Ustilago violacea . Curr Genet. 1992;22:135–140. doi: 10.1007/BF00351473. [DOI] [PubMed] [Google Scholar]
- 11.Gauthier GM. Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog. 2015;11:e1004608. doi: 10.1371/journal.ppat.1004608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lengeler KB, Davidson RC, D'Souza C, Harashima T, Shen W-C, et al. Signal transduction cascades regulating fungal development and virulence. Microbiol Biol Rev. 2000;64:746–785. doi: 10.1128/MMBR.64.4.746-785.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sánchez-Martínez C, Pérez-Martín J. Dimorphism in fungal pathogens: Candida albicans and U stilago maydis – similar inputs, different outputs. Curr Opin Microbiol. 2001;4:214–221. doi: 10.1016/S1369-5274(00)00191-0. [DOI] [PubMed] [Google Scholar]
- 14.Martínez-Soto D, Ruiz-Herrera J. Transcriptomic analysis of the dimorphic transition of Ustilago maydis induced in vitro by a change in pH. Fungal Genet Biol. 2013;58–59:116–125. doi: 10.1016/j.fgb.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Paul JA, Barati MT, Cooper M, Perlin MH. Physical and genetic interaction between ammonium transporters and the signaling protein Rho1 in the plant pathogen Ustilago maydis . Eukaryot Cell. 2014;13:1328–1336. doi: 10.1128/EC.00150-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith DG, Garcia-Pedrajas MD, Gold SE, Perlin MH. Isolation and characterization from pathogenic fungi of genes encoding ammonium permeases and their roles in dimorphism. Mol Microbiol. 2003;50:259–275. doi: 10.1046/j.1365-2958.2003.03680.x. [DOI] [PubMed] [Google Scholar]
- 17.Castle AJ. Isolation and identification of α-tocopherol as an inducer of the parasitic phase of Ustilago violacea . Phytopathology. 1984;74:1194–1200. doi: 10.1094/Phyto-74-1194. [DOI] [Google Scholar]
- 18.Antonovics J, Hood M, Partain J. The ecology and genetics of a host shift: microbotryum as a model system. Am Nat. 2002;160:S40–S53. doi: 10.1086/342143. [DOI] [PubMed] [Google Scholar]
- 19.Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010;7:709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadota K, Nishiyama T, Shimizu K. A normalization strategy for comparing tag count data. Algorithms Mol Biol. 2012;7:5. doi: 10.1186/1748-7188-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliveros JC. VENNY. An Interactive Tool for Comparing Lists with Venn's Diagrams. 2.0 ed. 2007–2015. [Google Scholar]
- 28.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 31.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, et al. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurzynska-Kokorniak A, Koralewska N, Pokornowska M, Urbanowicz A, Tworak A, et al. The many faces of Dicer: the complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 2015;43:4365–4380. doi: 10.1093/nar/gkv328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Potenza N, Papa U, Russo A. Differential expression of Dicer and Argonaute genes during the differentiation of human neuroblastoma cells. Cell Biol Int. 2009;33:734–738. doi: 10.1016/j.cellbi.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Lau SK, Chow WN, Wong AY, Yeung JM, Bao J, et al. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei . PLoS Negl Trop Dis. 2013;7:e2398. doi: 10.1371/journal.pntd.0002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HK, Jo SM, Kim GY, Kim DW, Kim YK, et al. A large-scale functional analysis of putative target genes of mating-type loci provides insight into the regulation of sexual development of the cereal pathogen Fusarium graminearum . PLoS Genet. 2015;11:e1005486. doi: 10.1371/journal.pgen.1005486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunkel N, Hertlein T, Franz R, Reuß O, Sasse C, et al. Roles of different peptide transporters in nutrient acquisition in Candida albicans . Eukaryot Cell. 2013;12:520–528. doi: 10.1128/EC.00008-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toh SS, Perlin MH. Size does matter: staging of Silene latifolia floral buds for transcriptome studies. Int J Mol Sci. 2015;16:22027–22045. doi: 10.3390/ijms160922027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kües U, Rühl M. Multiple multi-copper oxidase gene families in basidiomycetes—what for? Curr Genomics. 2011;12:72–94. doi: 10.2174/138920211795564377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones SK, Jr, Bennett RJ. Fungal mating pheromones: choreographing the dating game. Fungal Genet Biol. 2011;48:668–676. doi: 10.1016/j.fgb.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rutherford JC, Lin X, Nielsen K, Heitman J. Amt2 permease is required to induce ammonium-responsive invasive growth and mating in Cryptococcus neoformans . Eukaryot Cell. 2008;7:237–246. doi: 10.1128/EC.00079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petit E, Giraud T, De Vienne DM, Coelho MA, Aguileta G, et al. Linkage to the mating-type locus across the genus Microbotryum: insights into nonrecombining chromosomes. Evolution. 2012;66:3519–3533. doi: 10.1111/j.1558-5646.2012.01703.x. [DOI] [PubMed] [Google Scholar]
- 44.Feldbrügge M, Kämper J, Steinberg G, Kahmann R. Regulation of mating and pathogenic development in Ustilago maydis . Curr Opin Microbiol. 2004;7:666–672. doi: 10.1016/j.mib.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Bakkeren G, Kämper J, Schirawski J. Sex in smut fungi: structure, function and evolution of mating-type complexes. Fungal Genet Biol. 2008;45:S15–S21. doi: 10.1016/j.fgb.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Heimel K, Scherer M, Schuler D, Kämper J. The Ustilago maydis Clp1 protein orchestrates pheromone and b-dependent signaling pathways to coordinate the cell cycle and pathogenic development. Plant Cell. 2010;22:2908–2922. doi: 10.1105/tpc.110.076265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pöggeler S, Nowrousian M, Ringelberg C, Loros JJ, Dunlap JC, et al. Microarray and real-time PCR analyses reveal mating type-dependent gene expression in a homothallic fungus. Mol Genet Genomics. 2006;275:492–503. doi: 10.1007/s00438-006-0107-y. [DOI] [PubMed] [Google Scholar]
- 48.Becker K, Beer C, Freitag M, Kück U. Genome-wide identification of target genes of a mating-type α-domain transcription factor reveals functions beyond sexual development. Mol Microbiol. 2015;96:1002–1022. doi: 10.1111/mmi.12987. [DOI] [PubMed] [Google Scholar]
- 49.Ait Benkhali J, Coppin E, Brun S, Peraza-Reyes L, Martin T, et al. A network of HMG-box transcription factors regulates sexual cycle in the fungus Podospora anserina . PLoS Genet. 2013;9:e1003642. doi: 10.1371/journal.pgen.1003642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jewell JB, Browse J. Epidermal jasmonate perception is sufficient for all aspects of jasmonate-mediated male fertility in Arabidopsis . Plant J. 2016;85:634–647. doi: 10.1111/tpj.13131. [DOI] [PubMed] [Google Scholar]
- 51.Yuan Z, Zhang D. Roles of jasmonate signalling in plant inflorescence and flower development. Curr Opin Plant Biol. 2015;27:44–51. doi: 10.1016/j.pbi.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 52.Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, et al. Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol Ecol. 2014;23:753–773. doi: 10.1111/mec.12631. [DOI] [PubMed] [Google Scholar]
- 53.Klose J, De Sá MM, Kronstad JW. Lipid-induced filamentous growth in Ustilago maydis . Mol Microbiol. 2004;52:823–835. doi: 10.1111/j.1365-2958.2004.04019.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1