

Summary
In type 1 diabetes (T1D), autoreactive cytotoxic CD8+ T cells are implicated in the destruction of insulin‐producing β cells. The HLA‐B*3906 and HLA‐A*2402 class I genes confer increased risk and promote early disease onset, suggesting that CD8+ T cells that recognize peptides presented by these class I molecules on pancreatic β cells play a pivotal role in the autoimmune response. We examined the frequency and phenotype of circulating preproinsulin (PPI)‐specific and insulin B (InsB)‐specific CD8+ T cells in HLA‐B*3906 + children newly diagnosed with T1D and in high‐risk HLA‐A*2402 + children before the appearance of disease‐specific autoantibodies and before diagnosis of T1D. Antigen‐specific CD8+ T cells were detected using human leucocyte antigen (HLA) class I tetramers and flow cytometry was used to assess memory status. In HLA‐B*3906 + children with T1D, we observed an increase in PPI5–12‐specific transitional memory CD8+ T cells compared to non‐diabetic, age‐ and HLA‐matched subjects. Furthermore, PPI5–12‐specific CD8+ T cells in HLA‐B*3906 + children with T1D showed a significantly more antigen‐experienced phenotype compared to polyclonal CD8+ T cells. In longitudinal samples from high‐risk HLA‐A*2402 + children, the percentage of terminal effector cells within the InsB15–24‐specific CD8+ T cells was increased before diagnosis relative to samples taken before the appearance of autoantibodies. This is the first study, to our knowledge, to report HLA‐B*3906‐restricted autoreactive CD8+ T cells in T1D. Collectively, our results provide evidence that β cell‐reactive CD8+ T cells restricted by disease‐associated HLA class I molecules display an antigen‐experienced phenotype and acquire enhanced effector function during the period leading to clinical diagnosis, implicating these cells in driving disease.
Keywords: CD8+ T cells, HLA‐B*39, HLA‐A*24, type 1 diabetes
In this study, we characterised circulating autoreactive CD8+ T cells in children with type 1 diabetes with HLA-B*3906+ and HLA-A*2402+ genotypes which are associated with increased risk and early onset of disease. In HLA-B*3906+ children with newly diagnosed type 1 diabetes, we observed an increase in PPI5–12‐specific transitional memory CD8+ T cells compared to non‐diabetic, age‐ and HLA‐matched subjects. Furthermore, PPI5–12‐specific CD8+ T cells in HLA‐B*3906+ children with T1D showed a significantly more antigen‐experienced phenotype compared to polyclonal CD8+ T cells. Further, in longitudinal samples from high-risk HLA-A*2402+ children, the percentage of terminal effector cells within the InsB15–24‐specific CD8+ T cell population was increased before diagnosis relative to samples taken before the appearance of autoantibodies.

Introduction
Type 1 diabetes is an autoimmune disease in which insulin‐secreting β cells of the pancreatic islets are selectively destroyed 1, 2. Several lines of evidence point to CD8+ cytotoxic T cells as critical players in β cell destruction. Firstly, CD8+ T cells are the dominant cell type found in insulitic lesions in the pancreas of patients with type 1 diabetes 3, 4, 5, and hyperexpression of human leucocyte antigen (HLA) class I molecules in this lesion has the potential to enhance peptide presentation to infiltrating CD8+ T cells 3, 4, 5, 6, 7, 8. Secondly, CD8+ T cells with specificity for β cell antigens are present in the blood and islets of individuals with type 1 diabetes 9, 10, 11, 12, 13, 14. Thirdly, CD8+ T cell clones recognizing β cell epitopes are able to kill isolated β cells in vitro, exemplifying the potential for CD8+ T cells to constitute major effectors of β cell death 13, 15. Lastly, recent success in delaying β cell loss using immunotherapy targeted at effector CD8+ T cells suggests that CD8+ T cells are a dominant killing pathway 16.
These immunological observations associate the CD8+ T cell pathway with disease, a proposal that is further supported by genetic studies. These highlight specific HLA class I loci with type 1 diabetes predisposition independently of linkage disequilibrium with HLA class II 17. For example, HLA‐B*39 was identified as a major genetic risk locus in a large‐scale study of single nucleotide polymorphisms associated with allelic forms of HLA class I genes 17. HLA‐B*39 is relatively rare, being present in 0·5–1·2% of European, North American and Australian populations and 0·1% of Southeast Asian populations 18, 19. HLA‐B*39 polymorphism is associated with increased susceptibility to type 1 diabetes, providing an odds ratio of 2·41 in a case–control set, and a relative risk of 3·55 in affected sibling‐pair families 17. In addition, HLA‐B*39 polymorphism associates with a lower age of type 1 diabetes diagnosis, and the HLA‐B*3906 subtype is linked to a lower age of diagnosis by an average of 1·7–3·7 years in several independent studies 17, 20, 21, 22, 23, 24. HLA‐B*39 polymorphism is also associated with accelerated disease progression in children from the point of autoantibody development to clinical diagnosis, implying more rapid β cell destruction 25, 26. Furthermore, the HLA‐B*3906 variant significantly enhances the risk of type 1 diabetes in individuals carrying specific HLA‐DR/DQ haplotypes; namely, DRB01*0404‐DQB1*0302 21, 23, 26, 27, 28, 29 and DRB1*08–DQB1*0402 24, 29, 30, 31.
At the HLA‐A locus, the HLA‐A*24 allele has a strong type 1 diabetes disease‐predisposing effect. The HLA‐A*24 supertype is present in 12–20% of Caucasian and 60% of Japanese populations, with HLA‐A*2402 being the most common variant 32, 33. Polymorphisms associated with the HLA‐A*24 allele confer a higher disease risk, with an odds ratio of 1·5 17, and share disease‐influencing features in common with HLA‐B*39. For example, HLA‐A*24 is significantly associated with a younger age at diagnosis 17, 34, 35, 36. HLA‐A*24 polymorphisms are an independent predictor of progression to type 1 diabetes in autoantibody‐positive first‐degree relatives of individuals with type 1 diabetes 37 and are associated with accelerated disease progression from seroconversion to clinical diagnosis 26, 37, 38, 39. Furthermore, the presence of HLA‐A*24 has been associated with early and complete β cell destruction after diagnosis 40, 41, and with poor functional outcome in islet transplant recipients 42.
Collectively, these studies prompt questions in relation to the presentation of β cell autoantigens to CD8+ T cells by HLA‐B*39‐ and HLA‐A*24‐encoded HLA class I molecules and the potential of these events to drive β cell destruction and accelerate type 1 diabetes progression. We recently identified preproinsulin epitopes naturally processed and presented by HLA class I molecules encoded by HLA‐B*3906 and HLA‐A*2402 and associated with disease risk and progression 43, 44. In the present study, we used this insight to examine the repertoires of HLA‐B*3906‐ and HLA‐A*2402‐restricted β cell‐specific CD8+ T cells in children with, or at risk of, type 1 diabetes. We analysed samples from Finnish cohorts in which the independent and additive effects of these high‐risk alleles on type 1 diabetes risk are well documented 21, 23, 26, 27, 29. We provide evidence that autoreactive CD8+ T cells restricted by HLA‐B*3906 and HLA‐A*2402‐encoded HLA class I molecules display an antigen‐experienced phenotype and acquire enhanced effector function during the period leading to clinical diagnosis of type 1 diabetes.
Materials and methods
Subjects
The HLA‐B*3906 + study cohort comprised 10 children with newly diagnosed type 1 diabetes (time after clinical diagnosis 2–10 days; median age 2·7 years) and seven non‐diabetic children recruited as control subjects, with a median age of 2·2 years (Table 1). Additionally, one child had blood drawn at the age of 3·3 years, 5 years before she was diagnosed with type 1 diabetes at the age of 8·6 years. All children with newly diagnosed type 1 diabetes were recruited from the Finnish Pediatric Diabetes Register 45. The Finnish Pediatric Diabetes Register study was approved by the Ethical Committee of the Hospital District of Helsinki and Uusimaa and the register steering committee. The parents of the children gave their written informed consent. Five control subjects were healthy siblings of type 1 diabetes‐affected children recruited from the Finnish Pediatric Diabetes Register, and two control subjects had HLA class II genotypes associated with increased risk for type 1 diabetes and were recruited as participants in the Finnish Type 1 Diabetes Prediction and Prevention (DIPP) study 46, 47. Six of seven control subjects were autoantibody‐negative at the time of blood draw. Although one of the seven control subjects was positive for insulin autoantibodies (IAA) this subject was included, as it known that the number of detectable autoantibodies is related to risk of progression to type 1 diabetes, with seropositivity for two or more islet autoantibodies conferring high risk for developing type 1 diabetes, whereas positivity for a single autoantibody may represent non‐progressive or regressive β cell autoimmunity 48. The DIPP study was approved by local Ethics Committees and all families participating in the study provided written informed consent.
Table 1.
HLA‐B*3906 + subject information
| T1D subjects | Controls | |
|---|---|---|
| n | 10 | 7 |
| Sex n (%) | ||
| Male | 6 (60%) | 3 (43%) |
| Female | 4 (40%) | 4 (57%) |
| Age, median (IQR) (years) | 2·7 (1·8–3·3) | 2·2 (1·8–4·1) |
| Age at diagnosis, median (IQR) (years) | 2·6 (1·8–3·3) | n.a. |
| T1D duration (days) | 5 (3–7) | n.a. |
| Autoantibody‐positive, n (%) | 9 (90%) | 1 (14%) |
T1D = type 1 diabetes; IQR = interquartile range; n.a. = not available.
The HLA‐A*2402 + study cohort comprised 15 children sampled before diagnosis of type 1 diabetes (median time before clinical diagnosis = 7 months; median age = 5·0 years) and 15 autoantibody‐negative non‐diabetic children recruited as control subjects, with a median age of 4·9 years (Table 2). Of the 15 children who provided samples before diagnosis of type 1 diabetes, 11 had blood drawn before seroconversion to autoantibody positivity (median time before seroconversion = 5 months; median age = 1·5 years) (Table 3). All HLA‐A*2402 + study subjects participated in the DIPP study with regular follow‐up and had HLA class II genotypes associated with increased risk for type 1 diabetes 49. Positivity for islet autoantibodies was analysed in all subjects participating in either the Finnish Pediatric Diabetes Register or the DIPP study at the time of sampling, as previously described 46, 50. Positivity for Epstein–Barr virus (EBV) immunoglobulin (Ig)G antibodies was analysed in 12 of 15 subjects whose samples were stained with EBV tetramers. Serum samples were tested for anti‐viral capsid antigen (VCA) and anti‐early antigen (EA) IgG using an automated immunoanalyzer based on enzyme‐linked fluorescence (ELFA) technology (VIDAS®; bioMérieux, Marcy l’Etoile, France). Results were reported as negative or positive with cut‐offs of 0·1 and 0·23 test value.
Table 2.
HLA‐A*2402 + subject information
| T1D subjects | Controls | |
|---|---|---|
| n | 15 | 15 |
| Sex n (%) | ||
| Male | 10 (66%) | 10 (66%) |
| Female | 5 (33%) | 5 (33%) |
| Age, median (IQR) (years) | 5·0 (1·7–9·0) | 4·9 (1·8–8·5) |
| Age at diagnosis, median (IQR) (years) | 5·9 (3·5–9·3) | n.a. |
| Time before diagnosis, median (IQR) (months) | 7 (4–21) | n.a. |
| Autoantibody‐positive, n (%) | 14 (93%) | 0 (0%) |
T1D = type 1 diabetes; IQR = interquartile range; n.a. = not available.
Table 3.
Pre‐ and post‐seroconversion HLA‐A*2402 + subject information
| T1D subjects pre‐seroconversion | T1D subjects post‐seroconversion | |
|---|---|---|
| n | 11 | 11 |
| Sex n (%) | ||
| Male | 9 (82%) | 9 (82%) |
| Female | 2 (18%) | 2 (18%) |
| Age, median (IQR) (years) | 1·5 (1·0–2·1) | 5·8 (3·4–9·8) |
| Age at seroconversion, median (IQR) (years) | 2·5 (1·3–3·2) | 2·5 (1·3–3·2) |
| Age at diagnosis, median (IQR) (years) | 6·6 (4·0–10·3) | 6·6 (4·0–10·3) |
| Time before seroconversion, median (IQR) (months) | 5 (3–6) | n.a. |
| Time before diagnosis, median (IQR) (months) | 53 (22–79) | 6 (2–19) |
T1D = type 1 diabetes; IQR = interquartile range; n.a. = not available.
Tetramer assembly
Soluble, fluorochrome‐conjugated peptide‐HLA class I tetramers were generated as described previously 51. The peptide–human leucocyte antigen (pHLA)‐B*3906 tetramers were manufactured with PPI5–12 test peptides 43 and EBV BMRF1268–276 control peptides 43, 52. The pHLA‐A*2402 tetramers were manufactured with PPI3–11 and InsB15–24 test peptides 13, 53. Epitope sequences are given in Table 4. Tetramers were assembled over five separate 20‐min steps with the successive addition of streptavidin–allophycocyanin (APC) (Life Technologies, Carlsbad, CA, USA) to monomeric pHLA at a molar streptavidin : pHLA ratio of 1 : 4. Phosphate‐buffered saline (PBS) was added to give a final multimer concentration of 0·1 mg/ml pHLA content. Tetramers were stored in the dark at 4°C and used on the same day as assembly.
Table 4.
HLA‐B*3906 and HLA‐A*2402‐restricted CD8 T cell epitopes
| Epitope | HLA class I restriction | Sequence |
|---|---|---|
| PPI3–11 | A*2402 | LWMRLLPLL |
| InsB15–24 | A*2402 | LYLVCGERGF |
| PPI5–12 | B*3906 | MRLLPLLA |
| EBV BMRF1268–276 | B*3906 | YRSGIIAVV |
PPI = preproinsulin; HLA = human leucocyte antigen; EBV = Epstein–Barr virus.
Cell staining and flow cytometry
Thawed peripheral blood mononuclear cells (PBMC) were washed in RPMI supplemented with 2% human AB serum; 3–4 million PBMCs per stain were transferred to flow cytometry tubes, washed with PBS and stained with a live/dead aqua amine reactive dye (Invitrogen, Carlsbad, CA, USA) for 10 min at room temperature, followed by washing with PBS. PBMCs were incubated with the protein kinase inhibitor dasatinib (Axon Medchem, Reston, VA, USA) at a final concentration of 50 nM for 30 min at 37°C to enhance the detection of low‐avidity T cells 12, 54 and C‐C chemokine receptor type 7 (CCR7) BV421 monoclonal antibody (Biolegend, San Diego, CA, USA). Subsequently, PBMCs were stained for 10 min at 37°C with 1 μg of pHLA tetramer per tube. After washing with fluorescence activated cell sorter (FACS) buffer (PBS supplemented with 2% human AB serum and 3% fetal calf serum), PBMCs were stained with mouse anti‐APC unconjugated monoclonal antibody (Biolegend) at a concentration of 10 mg/ml on ice for 20 min to stabilize binding of APC‐labelled tetramers 55. After washing with FACS buffer, cells were stained with the following surface monoclonal antibodies/fluorophores for 20 min on ice: CD3 BV785, CD8 phycoerythrin cyanin 7 (PECy7), CD27 BV605 (all Biolegend); CD14 V500, CD16 V500, CD19 V500, CD45RA PE‐CF594, CD95 PE, CD57 fluorescein isothiocyanate (FITC) (all BD Biosciences, San Jose, CA, USA) and CD4 PECy5.5 (Invitrogen). Stained PBMCs were kept on ice and in the dark until acquisition on the same day on an LSR Fortessa (BD Biosciences). Gates to define tetramer positivity were set based on PBMC samples which were not stained with tetramer. No CD8 T cells were stained in the absence of tetramers. We elected to use this method to define tetramer‐positive cells as control tetramers incorporating irrelevant self‐peptides are likely to stain some T cells, while control tetramers incorporating foreign peptides from previously unencountered viruses have been found to stain some T cells in seronegative donors 56. Within the antigen‐specific CD8 T cell populations and total CD8 T cell populations, CD8 T cell subsets were assessed using the following cell surface marker combinations: naive (N;CCR7+CD45RA+CD27+CD57–CD95–), stem‐cell memory‐like (SCM; CCR7+CD45RA+CD27+CD57–CD95+), central memory (CM; CCR7+CD45RA–CD27+), transitional memory (TM; CCR7–CD45RA–CD27+), effector memory (EM; CCR7–CD45RA–CD27–), terminal effector (TE; CCR7–CD45RA+). The gating strategy is illustrated in Supporting information, Fig. S1. Flow cytometry data were analysed using FlowJo version 9.4 (Tree Star, Ashland, OR, USA). Due to limitations in cell numbers available of some samples, tetramer staining was limited as follows: two HLA‐B*3906 + control subjects were not stained with EBV tetramer; two HLA‐A*2402 + control subjects, six HLA‐A*2402 + prediagnosis subjects and five HLA‐A*2402 + pre‐seroconversion subjects were not stained with InsB tetramer.
Dual‐colour tetramer staining
Dual‐colour staining was performed with the newly developed pHLA‐B*3906 tetramers to verify the specificity of tetramer‐binding cells (Supporting information, Fig. S2). The pHLA‐B*3906 tetramers manufactured with PPI5–12 and EBV BMRF1268–276 were assembled over five separate 20‐min steps with the successive addition of streptavidin–APC or streptavidin–PE (both Life Technologies) to monomeric pHLA at a molar streptavidin : pHLA ratio of 1 : 4. PBS was added to give a final multimer concentration of 0·1 mg/ml pHLA content. Thawed PBMC were stained as described above.
Statistics
Comparisons between subject groups were performed using non‐parametric Mann–Whitney U‐tests. Paired comparisons between groups were performed using the non‐parametric Wilcoxon paired test. Statistical analysis was performed using GraphPad Prism version 7.0e.
Results
HLA‐B*3906‐restricted autoreactive CD8+ T cells in type 1 diabetes
As HLA‐B*39 carries the strongest type 1 diabetes risk of all HLA class I gene polymorphisms, we sought to determine whether HLA‐B*39‐restricted CD8+ T cells with specificity for β cell antigens could be detected in individuals with type 1 diabetes. In particular, we examined HLA‐B*3906‐restricted CD8+ T cells specific for the preproinsulin epitope PPI5–12 by staining ex‐vivo PBMC samples with pHLA class I tetramers. As a control, we analysed CD8+ T cells specific for the HLA‐B*3906‐restricted EBV lytic cycle protein epitope BMRF1268–276. We assessed the frequency and phenotype of antigen‐specific CD8+ T cells in PBMC samples obtained within 10 days of type 1 diabetes diagnosis from children aged up to 5 years and in non‐diabetic, age‐matched and HLA‐matched control subjects (Table 1). Two subjects with type 1 diabetes and one control subject tested positive for EBV antibodies (data from these subjects are highlighted in Supporting information, Fig. S3). Representative tetramer staining is shown in Fig. 1a. The frequency of PPI5–12‐specific and EBV BMRF1268–276‐specific CD8+ T cells was found to be similar in type 1 diabetes subjects and control subjects (Fig. 1b). Phenotypical analysis revealed that in subjects with type 1 diabetes there was a higher percentage of PPI5–12‐specific transitional memory CD8+ T cells compared to control subjects (Fig. 1c). In contrast, no significant differences were observed in the phenotype of EBV BMRF1268–276‐specific CD8+ T cells in subjects with type 1 diabetes compared to control subjects (Fig. 1c). The median percentages of naïve, memory and terminal effector cells comprising the PPI5–12‐ and EBV BMRF1268–276‐specific CD8+ T cell populations are represented in Fig. 1d. The pie charts illustrate the higher percentage of PPI5–12‐specific memory CD8+ T cells in subjects with type 1 diabetes compared to control subjects and the similarity in the percentages of EBV BMRF1268–276‐specific CD8+ T cell subsets in subjects with type 1 diabetes and control subjects (Fig. 1d).
Figure 1.
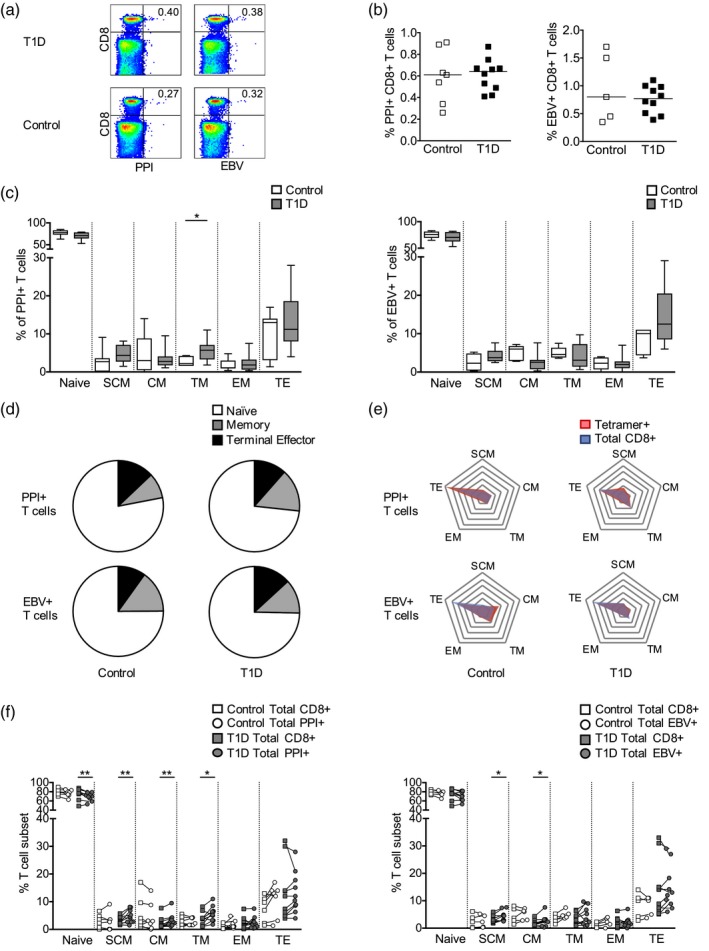
Frequency and phenotype of human leucocyte antigen (HLA)‐B*3906‐restricted preproinsulin (PPI)‐ and Epstein–Barr virus (EBV)‐specific CD8+ T cells in patients with newly diagnosed type 1 diabetes and control subjects. (a) Representative tetramer staining of peripheral blood mononuclear cells (PBMC) from type 1 diabetes (T1D) subjects (n = 10) and controls (n = 7). (b) Frequency of PPI5–12‐ and EBV BMRF1268–276‐specific CD8+ T cells. Mann–Whitney U‐tests P > 0·05. (c) Phenotype of PPI5–12‐ and EBV BMRF1268–276‐specific CD8+ T cells. Mann–Whitney U‐tests; *P < 0·05. (d) Median T cell subset percentages for PPI5–12‐ and EBV BMRF1268–276‐specific CD8+ T cells in T1D and controls. Memory: pooled stem cell‐like memory, central memory, transitional memory and effector memory. (e) Frequency of memory and effector T cell subsets expressed as a percentage of non‐naive T cells within tetramer‐specific (red) and polyclonal (blue) CD8+ T cell populations. Radial lines represent intervals of T cell subset frequencies of 10% from 0 to 60%, with the lowest value at the centre and the highest value at the periphery. Polygons link the frequency of each T cell subset. (f) Phenotype of PPI5–12‐ and EBV BMRF1268–276‐specific CD8+ T cells (circles) compared to polyclonal CD8+ T cells (squares). Wilcoxon paired tests; *P < 0·05, **P < 0·01. Abbreviations: SCM: stem cell‐like memory, CM: central memory; TM: transitional memory; EM: effector memory; TE: terminal effector.
In order to determine whether distinctive phenotypical characteristics were present in the antigen‐specific CD8+ T cells, we compared their phenotypes with those found in the polyclonal CD8+ T cell population within each PBMC sample. We used spider plots to visualize the difference in the memory and effector T cell subsets comprising the PPI5–12‐specific, EBV BMRF1268–276‐specific and polyclonal CD8+ T cell populations in subjects with type 1 diabetes and control subjects (Fig. 1e). For example, in subjects with type 1 diabetes, we observe a larger proportion of memory T cell subsets in the PPI5–12‐specific CD8+ T cell population compared to the polyclonal CD8+ T cell population, illustrated by a red polygon (representing PPI‐specific T cells) that is more expanded along the arcs of the web compared to the blue polygon (representing polyclonal CD8+ T cells) (Fig. 1e). Paired comparisons of tetramer‐specific and polyclonal CD8+ T cells revealed that in subjects with type 1 diabetes, the PPI5–12‐specific CD8+ T cell population was comprised of significantly fewer naive cells and significantly more stem‐cell‐like memory cells, central memory cells and transitional memory cells compared to the polyclonal CD8+ T cell population (Fig. 1f). Therefore, the PPI5–12‐specific CD8+ T cells display a more antigen‐experienced phenotype compared to polyclonal CD8+ T cells in type 1 diabetes subjects. Importantly, these differences were not observed within the PPI5–12‐specific CD8+ T cell population in control subjects, indicating that autoreactive T cells acquire this phenotype as a result of the disease process (Fig. 1f). In EBV‐specific CD8+ T cell populations, there was an increase in stem cell memory‐like and central memory cells compared to the polyclonal CD8+ T cell population in type 1 diabetes subjects, and we did not detect significant phenotypical differences compared to polyclonal CD8+ T cells in control subjects (Fig. 1f). The phenotype of polyclonal CD8+ T cells was not significantly different between type 1 diabetes subjects and control subjects (Supporting information, Fig. S4). A comparison of the data from the autoantibody‐positive and ‐negative control subjects is shown in Supporting information, Fig. S5.
Additionally, we had the opportunity to assess PPI5–12‐specific CD8+ T cells 5 years prior to the diagnosis of type 1 diabetes in an HLA‐B*3906 + child who was aged 3·3 years at the time of blood draw (Supporting information, Fig. S6a). In this individual, we observed a higher frequency of PPI5–12‐specific transitional memory CD8+ T cells compared to control subjects, as we observed in patients with type 1 diabetes (Supporting information, Fig. S6b). Compared to the polyclonal CD8+ T cell population, in the PPI5–12‐specific CD8+ T cell population we observed fewer naive cells and more central memory cells, transitional memory cells, effector memory cells and terminal effector cells, indicative of an antigen‐experienced phenotype (Supporting information, Fig. S6c,d). Although we were not able to compare samples from this case before and after diagnosis, the phenotypical characteristics of PPI5–12‐specific CD8+ T cells in the subject sampled 5 years before diagnosis appear to be similar to the HLA‐B*3906 + subjects sampled at the time of diagnosis of type 1 diabetes.
HLA‐A*2402‐restricted autoreactive CD8+ T cells in type 1 diabetes
Using a similar approach, we studied CD8+ T cells recognizing β cell peptides presented by HLA‐A*2402 in children followed from birth to clinical diagnosis. PPI3–11‐ and InsB15–24‐specific pHLA tetramers were used to stain PBMCs from 15 children ascertained before clinical diagnosis and 15 age‐, sex‐ and HLA‐matched control subjects (Fig. 2a). We found that the frequencies of circulating PPI3–11‐ and InsB15–24‐specific CD8+ T cells were similar in subjects with type 1 diabetes and control subjects (Fig. 2b), and the subject groups did not differ significantly in terms of the phenotype of PPI3–11‐ or InsB15–‐24‐specific CD8+ T cells (Fig. 2c,d). However, when antigen‐specific and polyclonal CD8+ T cell populations were compared, PPI3–11‐ and InsB15–24‐specific CD8+ T cell populations in type 1 diabetes and control subjects contained significantly fewer naive cells and significantly more stem cell‐like memory, central memory and transitional memory cells compared to polyclonal CD8+ T cell populations (Fig. 2e,f). In addition, PPI3–11‐specific T cell populations also contained significantly more effector memory cells than polyclonal CD8+ T cell populations (Fig. 2f). The phenotype of polyclonal CD8+ T cells was not significantly different between type 1 diabetes subjects and control subjects (Supporting information, Fig. S7). Therefore, PPI3–11‐ and InsB15–24‐specific CD8+ T cells in both subject groups showed a more antigen‐experienced phenotype compared to polyclonal CD8+ T cells.
Figure 2.
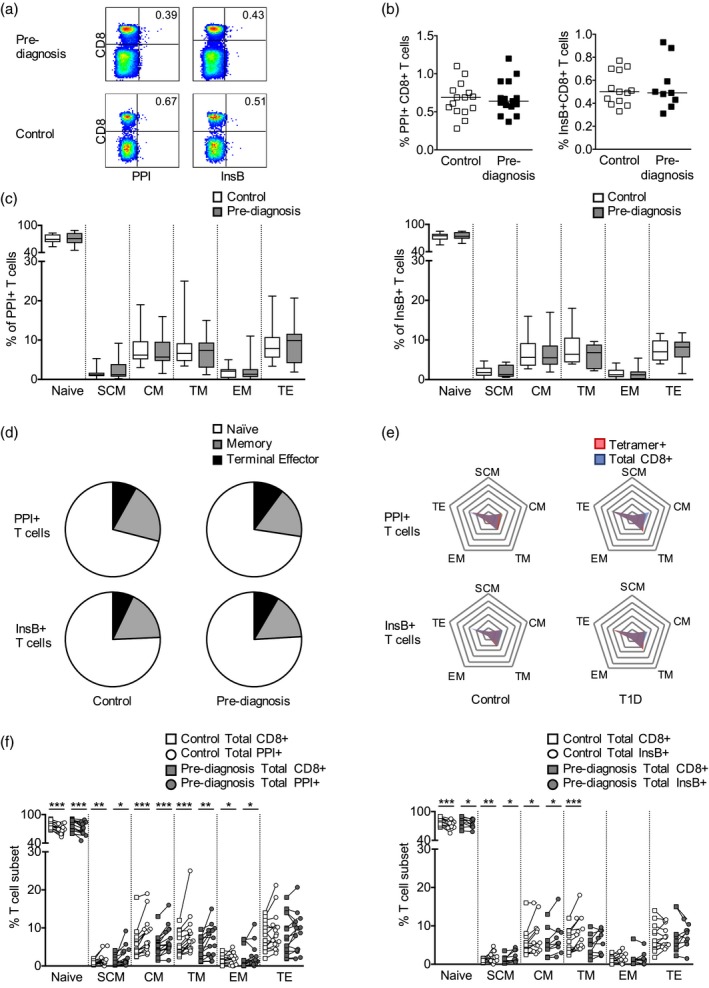
Frequency and phenotype of human leucocyte antigen (HLA)‐A*2402‐restricted preproinsulin (PPI)‐ and insulin B (InsB)‐specific CD8+ T cells before the diagnosis of type 1 diabetes (T1D) in affected subjects and control subjects. (a) Representative tetramer staining of peripheral blood mononuclear cells (PBMC) from subjects before T1D diagnosis (n = 17) and controls (n = 17). (b) Frequency of PPI3–11 and InsB15–24‐specific CD8+ T cells. Mann–Whitney U‐tests P > 0·05. (c) Phenotype of PPI3–11 and InsB15–24‐specific CD8+ T cells. (d) Median T cell subset percentages for PPI3–11 and InsB15–24‐specific CD8+ T cells. Memory: pooled stem cell‐like memory, central memory, transitional memory and effector memory. (e) Frequency of memory and effector T cell subsets expressed as a percentage of non‐naive T cells within tetramer‐specific (red) and polyclonal (blue) CD8+ T cell populations. Radial lines represent intervals of T cell subset frequencies of 5% from 0 to 35%, with the lowest value at the centre and the highest value at the periphery. Polygons link the frequency of each T cell subset. (f) Phenotype of tetramer‐specific CD8+ T cells (squares) compared to polyclonal CD8+ T cells (circle). Wilcoxon paired tests *P < 0·05, **P < 0·01, ***P < 0·001. Abbreviations: SCM: stem cell‐like memory, CM: central memory; TM: transitional memory; EM: effector memory; TE: terminal effector.
In order to evaluate changes in autoreactive HLA‐A*2402‐restricted CD8+ T cell populations before and after the initiation of islet autoimmunity, we analysed the frequency and phenotype of β cell‐specific CD8+ T cells in longitudinal samples from 11 HLA‐A*2402 + children ascertained before seroconversion to autoantibody positivity (median = 5 months) compared to samples taken before clinical diagnosis after the appearance of autoantibodies (median = 2 months before diagnosis) (Table 4, Fig. 3a). The frequencies of PPI3–11‐ and InsB15–24‐specific CD8+ T cells were not found to be significantly different in the samples taken before and after seroconversion (Fig. 3b). When we compared the phenotype of PPI3–11‐specific CD8+ T cells before seroconversion and before diagnosis, we found it was similar (Fig. 3c,d). However, within the InsB15–24‐specific CD8+ T cell population, we observed that the percentage of terminal effector cells was increased and the percentage of central memory cells was decreased in samples taken after seroconversion relative to samples taken before seroconversion (Fig. 3c,e). In contrast, the phenotype of the polyclonal CD8+ T cell population remained similar in pre‐ and post‐seroconversion samples (Fig. 3e). Collectively, these results provide evidence that β cell‐reactive CD8+ T cells restricted by disease‐associated HLA class I molecules display an antigen‐experienced phenotype and acquire enhanced effector function during the period leading to clinical diagnosis.
Figure 3.
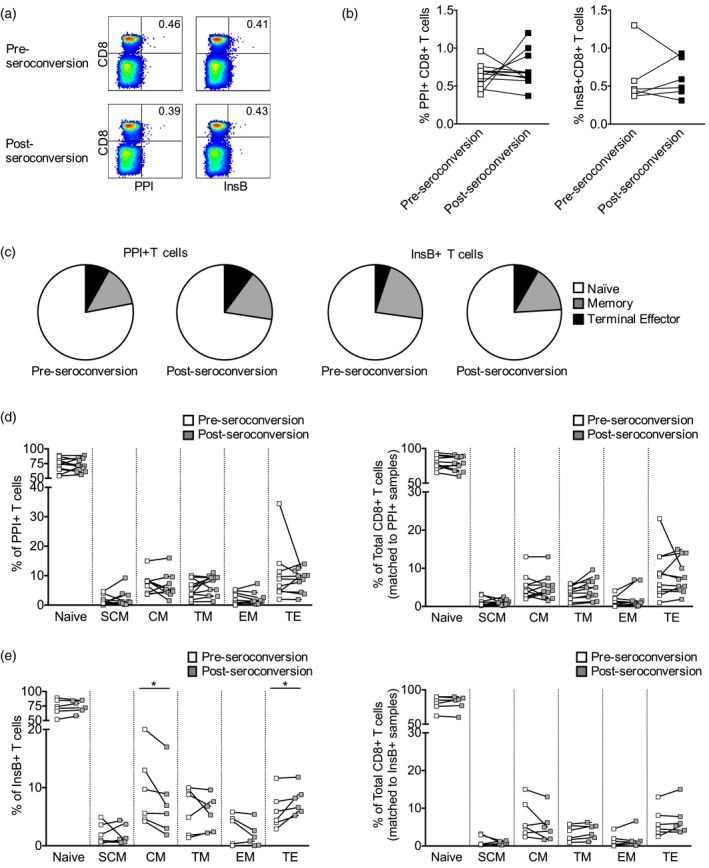
Frequency and phenotype of human leucocyte antigen (HLA)‐A*2402‐restricted preproinsulin (PPI)3–11‐ and InsB15–24‐specific CD8+ T cells before and after seroconversion to autoantibody positivity. (a) Representative tetramer staining of longitudinal peripheral blood mononuclear cells (PBMC) samples (n = 11). (b) Frequency of PPI3–11 and InsB15–24‐specific CD8+ T cells. Wilcoxon paired tests were used to compare groups (P > 0·05). (c) Median T cell subset percentages for PPI3–11 and InsB15–24‐specific CD8+ T cells. Memory: pooled stem cell‐like memory, central memory, transitional memory and effector memory. (d) Phenotype of PPI3–11‐specific and polyclonal CD8+ T cells. Wilcoxon paired tests were used for comparisons (P > 0·05). (e) Phenotype of InsB15–24‐specific and polyclonal CD8+ T cells. Wilcoxon paired tests were used for comparisons; *P < 0·05. Abbreviations: SCM: stem cell‐like memory, CM: central memory; TM: transitional memory; EM: effector memory; TE: terminal effector.
Discussion
The HLA class I HLA‐B*39 allele confers the strongest risk of type 1 diabetes of all HLA class I gene polymorphisms 17. This is the first study to report autoreactive CD8+ T cells restricted by HLA‐B*3906 in blood samples from individuals with type 1 diabetes. Here we show that the transitional memory subset of B*3906‐restricted PPI‐specific transitional memory CD8+ T cells comprises a higher proportion of the total PPI‐specific population in HLA‐B*3906 + children with new‐onset type 1 diabetes compared to HLA‐matched control subjects. Transitional memory cells have a differentiation status that is intermediate between that of central memory cells and effector memory cells in terms of phenotype, transcription factor expression and cytokine‐induced proliferation 57, 58, 59 . According to the recently proposed model of progressive differentiation, a naive T cell will differentiate gradually to different memory subsets through stem cell‐like memory, central memory, transitional memory, effector memory and terminal effector, progressively losing or acquiring specific functions 60. Central memory cells home to secondary lymph nodes and are better equipped to persist and proliferate in response to antigen, whereas effector memory cells are more cytolytic and express integrins and chemokine receptors that facilitate their entry into inflamed tissues 61. So far, a distinct pathophysiological role for the intermediate transitional memory subset has not been described. Here we defined transitional memory cells as having a cell surface expression marker profile of CCR7–CD45RA–CD27+, where the absence of CCR7 expression suggests that these cells have been activated in lymph nodes and are capable of homing to inflamed islets. Studies in non‐human primates have reported that transitional memory CD8+ T cells have higher expression levels of the transcription factors eomesodermin (EOMES), aryl hydrocarbon receptor (AHR) and RAR‐related orphan receptor C (RORC) compared to central memory and effector memory cells 57. The major role of EOMES is to maintain a memory CD8+ T cell repertoire capable of expansion on re‐encountering antigen. EOMES also functions as a master regulator of cell‐mediated immunity that controls genes encoding effector molecules including IFN‐γ, granzyme B and perforin 62, 63, 64. This would indicate that, like effector memory cells, transitional memory cells are able to execute target cell killing. As far as we know, a role for antigen‐specific transitional memory CD8+ T cells in autoimmune pathology has not yet been described. However, the capability of transitional memory cells to home to tissues and mediate cytotoxic effector functions suggests that the higher frequency of PPI5–12‐specific transitional memory cells we have detected in patients affected by type 1 diabetes has the potential to directly mediate β cell killing.
Further observations were made by pairwise comparison of functional phenotypes of HLA‐B*3906‐restricted PPI5–12‐specific CD8+ T cell populations with those of polyclonal CD8+ T cells, the latter representing a synthesis of all CD8+ T cell‐mediated antigen experiences in each subject. The aim of these analyses was to identify functional phenotypes specifically expanded by autoantigens. We noted that multiple memory T cell subsets were enriched within PPI5–12‐specific CD8+ T cell populations in multiple memory T cell subsets in children with type 1 diabetes, but this was not observed in non‐diabetic control subjects. An increase in PPI5–12‐specific stem cell‐like memory cells, central memory cells and transitional memory cells was accompanied by a reduction in naive cells compared to the polyclonal CD8+ T cell population. Memory CD8+ T cells are poised for rapid proliferative responses, and to execute cytotoxic functions and secrete effector cytokines upon re‐encountering their cognate antigen 61, 65, 66, 67. Therefore, the enrichment of β cell‐specific memory CD8+ T cell populations at the time of clinical diagnosis indicates that newly diagnosed type 1 diabetes is characterized by antigen‐driven differentiation of HLA‐B*3906‐restricted CD8+ T cells, probably facilitated by a tissue‐specific inflammatory process. Collectively, our findings suggest that enrichment of PPI5–12‐specific memory CD8+ T cell subsets in subjects with type 1 diabetes compared to non‐diabetic control subjects and polyclonal CD8+ T cells may reflect a driver role in relation to β cell killing, consistent with the accelerated loss of β cells that has been observed in HLA‐B*3906 + individuals, relative to patients with other HLA class I alleles 17, 20, 21, 22, 23, 24. HLA‐B*3906 is also known to associate with a relatively younger age of diagnosis and notably, this allele has been reported to be more frequent in children who progress to clinical type 1 diabetes under the age of 5·5 years 20, an age group which corresponds with the cohort examined in this study.
Clinical diagnosis of type 1 diabetes is an event marking the late stages of the destructive autoimmune process, when the number of functional β cells falls below the threshold required for adequate insulin production. However, the first signs of autoimmunity emerge months or years earlier with the appearance of several islet‐specific autoantibodies. In longitudinal samples from HLA‐A*2402 + children with high‐risk HLA class II genotypes, we found that CD8+ T cells specific for InsB15–24 had reduced frequencies of central memory cells and elevated frequencies of terminal effectors before diagnosis, relative to early‐stage disease before the appearance of autoantibodies. The percentage of terminal effector cells within total CD8+ T cell populations was within the range previously reported for children of a similar age 68, 69, 70, 71. Surprisingly, we observed phenotypical changes in CD8+ T cells with specificity for InsB15–24 but not PPI3–11. We speculate that InsB presentation may be enhanced by autoantibody‐mediated antigen presentation which will increase after seroconversion, whereas PPI, not being a part of mature insulin/proinsulin, may be targeted later by the autoimmune response via epitope spreading. While the concept of epitope spreading of autoantibody responses in preclinical type 1 diabetes has been well established 72, 73, 74, 75, 76, 77, intermolecular spreading of T cell responses has also been described during the preclinical period of type 1 diabetes 78, 79 and in animal models of diabetes 80, 81, 82, 83.
It is important to note that as none of the subjects examined in this study had received exogenous insulin, the changes we observed in InsB‐specific CD8+ T cells reflect the natural history of disease. Our results suggest that the natural evolution of the β cell‐specific CD8+ T cell response during the prediabetic period in young children becomes increasingly dominated by end‐stage cytolytic effector cells which are capable of directly mediating β cell destruction. These data fit with observations that at clinical diagnosis β cell destruction is already well advanced 84. Of interest, previous studies of the DIPP cohort have reported that the HLA‐A*24 allele is associated with accelerated disease progression from seroconversion to clinical disease 26. One interpretation is that HLA‐A*24‐restricted CD8+ T cells are potent mediators of β cell death after autoimmunity has been established. Furthermore, the presence of the HLA‐A*24 allele has also been associated with a reduced frequency of particular autoantibodies at clinical diagnosis 85, 86, 87. To explain this effect, it has been suggested that compared to other allotypes, HLA‐A*24‐restricted CD8+ T cells may mediate a more complete β cell destruction, resulting in a loss of antigenic stimulus, or alternatively, HLA‐A*24‐restricted CD8+ T cells may mediate a more rapid β cell destruction, thus attenuating inter‐ and intramolecular spreading of the autoimmune response 85, 86, 87. Our previous work showed that HLA‐A*2402‐restricted PPI‐specific CD8+ T cell clones derived from patients with type 1 diabetes are able to kill isolated β cells in vitro, exemplifying the potential for such cells to directly mediate β cell death in individuals with type 1 diabetes 88.
In this study, we observed that β cell‐specific CD8+ T cell frequencies in peripheral blood were similar in subjects with type 1 diabetes and control subjects. Similarly, other studies have found no significant differences between type 1 diabetes subjects and healthy donors in the frequencies of circulating CD8+ T cells reactive to multiple HLA‐A2‐restricted β cell epitopes 12, 56, 89. However, some studies have reported higher frequencies of β cell‐reactive CD8+ T cells in HLA‐A*02 + or HLA‐A*24 + type 1 diabetes subjects compared to healthy donors 13, 89, 90, 91, and it is important to take into account differences in methods (such as dasatinib enhancement of tetramer staining) and study populations (i.e. adults versus children).
In summary, we have demonstrated that cytotoxic T cells restricted by disease‐associated HLA class I molecules display an antigen‐experienced phenotype and acquire enhanced effector function during the period leading to clinical diagnosis, suggesting that these cells have a critical role in driving β cell destruction and may provide useful biomarkers of disease activity in monitoring progression to type 1 diabetes.
Disclosures
The authors have declared that no conflicts of interest exist.
Author contributions
L. Y., I. P. A. and R. B. performed the experiments, evaluated data and applied statistical analysis. G. D., K. T., S. H. and A. K. S. contributed to tetramer assembly and staining and flow cytometry experiments. R. V., J. T. and M. K. were responsible for recruitment and follow‐up of children in the DIPP project. R. V. and J. T. also participated in the Finnish Pediatric Diabetes Register as investigators. T. H. and M. K. were responsible for recruitment and follow‐up of children in in the Finnish Pediatric Diabetes Register. M. K. is the principal investigator of the Finnish Pediatric Diabetes Register. J. I., T. H. and M. K. provided clinical samples and M.‐L. M. and J. I. provided HLA genotyping data. L. Y. wrote the draft. M. P. and J. I. designed the study. All authors had the opportunity to discuss the results and comment on the manuscript.
Supporting information
Fig. S1. Flow cytometry gating strategy for determination of antigen‐specific CD8 T cell subsets. (a) Lymphocytes were defined as CD3 positive, ‘Dump’ (dead cell stain, CD14, CD16, CD19) negative gated on FSC and SSC characteristics. Doublets and CD4+CD8+ double‐positive cells were excluded. Cells were gated as CD8+Tetramer+ or CD8+Tetramer‐. (b) Gating of CD8+ T cell subsets: Naïve (N;CCR7+CD45RA+CD27+CD57‐CD95‐), stem cell memory‐like (SCM; CCR7+CD45RA+CD27+CD57‐CD95+), central memory (CM; CCR7+CD45RA‐CD27+), transitional memory (TM; CCR7‐CD45RA‐CD27+), effector memory (EM; CCR7‐CD45RA‐CD27‐) and terminal effector (TE; CCR7‐CD45RA+). (c) Schematic of the CD8+ T cell subsets analysed and the cell surface markers used for their definition.
Fig. S2. Dual‐colour tetramer staining with pHLA‐B*3906 tetramers loaded with PPI5‐12 and EBV BMRF1268‐276. (a) Gating strategy used to identify double‐positive tetramer‐binding CD8+ T cells. Lymphocytes were defined as CD3+, ‘Dump’ (dead cell stain, CD14, CD16, CD19)‐negative, and gated on lymphocyte SSC and FSC characteristics. Doublets were excluded. (b) PBMC from an HLA‐B*3906 + donor stained with PPI5‐12 and EBV BMRF1268‐276 pHLA‐B*3906 tetramers which were dual‐labelled with APC and PE. The majority of tetramer‐binding cells are double‐positive for both tetramers.
Fig. S3. Frequency and phenotype of HLA‐B*3906‐restricted EBV‐specific CD8+ T cells in EBV antibody‐positive (n = 3) and antibody‐negative (n = 9) subjects. (a) Frequency of EBV BMRF1268‐276‐specific CD8+ T cells in EBV antibody‐positive (red) and antibody‐negative (white) subjects. (b) Phenotype of EBV BMRF1268‐276‐specific CD8+ T cells in EBV antibody‐positive (red) and antibody‐negative (white) subjects.
Fig. S4. Phenotype of polyclonal CD8+ T cell populations in HLA‐B*3906+ newly diagnosed type 1 diabetes subjects and HLA‐B*3906+ control subjects. The phenotype of polyclonal CD8+ T cells was not found to be significantly different between HLA‐B*3906+ T1D subjects (grey) and healthy controls (white). Mann–Whitney U‐tests P > 0·05.
Fig. S5. Frequency and phenotype of HLA‐B*3906‐restricted PPI‐specific CD8+ T cells in autoantibody positive control (n = 1) and negative controls (n = 6). (a) Frequency and phenotype of PPI5‐12‐specific CD8+ T cells and polyclonal CD8+ T cells from autoantibody‐negative controls (white), autoantibody‐positive control (red) and T1D subjects (black). (b) Frequency and phenotype of EBV BMRF1268‐276‐specific CD8+ T cells and polyclonal CD8+ T cells from autoantibody‐negative controls (white), autoantibody‐positive control (red) and T1D subjects (black).
Fig. S6. Frequency and phenotype of HLA‐B*3906‐restricted PPI‐specific CD8+ T cells before the diagnosis of type 1 diabetes. (a) Frequency of PPI5‐12‐specific CD8+ T cells from an HLA‐B*3906 + type 1 diabetes subject before diagnosis (black) compared to HLA‐B*3906 + newly diagnosed type 1 diabetes subjects (grey) and HLA‐B*3906 + control subjects (white). (b) Phenotype of PPI3‐11‐specific CD8+ T cells from an HLA‐B*3906 + type 1 diabetes subject before diagnosis (black) compared to HLA‐B*3906 + newly diagnosed type 1 diabetes subjects (grey) and HLA‐B*3906 + control subjects (white). (c) Frequency of memory and effector T cell subsets expressed as a percentage of non‐naïve T cells within tetramer‐specific (red) and polyclonal (blue) CD8+ T cell populations. Radial lines represent intervals of T cell subset frequencies of 10% from 0 to 50%, with the lowest value at the centre and the highest value at the periphery. Polygons link the frequency of each T cell subset. (d) Phenotype of PPI5‐12‐specific CD8+ T cells (squares) compared to total polyclonal CD8+ T cells (circles) in an HLA‐B*3906 + type 1 diabetes subject before diagnosis (black), HLA‐B*3906 + newly diagnosed type 1 diabetes subjects (grey) and HLA‐B*3906 + control subjects (white).
Fig. S7. Phenotype of polyclonal CD8+ T cell populations in HLA‐A*2402+ type 1 diabetes subjects before diagnosis and HLA‐A*2402+ control subjects. The phenotype of polyclonal CD8+ T cells was not found to be significantly different between HLA‐A*2402+ pre‐diagnosis subjects (grey) and healthy controls (white). Mann–Whitney U‐tests P > 0.05.
Acknowledgements
This work was supported by the UK Department of Health via the National Institute for Health Research (NIHR) Biomedical Research Centre Award to Guy’s and St Thomas’ National Health Service Foundation Trust in partnership with King’s College London and the Juvenile Diabetes Research Foundation (JDRF) award, ‘Immunological markers of beta cell decline in new onset type 1 diabetes’ (17‐2013‐583). Support from the Innovative Medicines Initiative‐2 Joint Undertaking under grant agreement no. 115797 INNODIA, which receives support from the European Union’s Horizon 2020 research and Innovation Programme and EFPIA, JDRF International and The Leona M. and Harry B. Helmsley Charitable Trust to MP is also acknowledged. IPA is in receipt of a Marie Skłodowska‐Curie Individual Fellowship (Grant Agreement no. 704974). A. K. S. is a Wellcome Senior Investigator (WT100327MA). JDRF, the Academy of Finland and Sigrid Jusélius Foundation have supported the DIPP study and the Finnish Pediatric Diabetes Register. The contents of this article are solely the responsibility of the authors.
References
- 1. Roep BO, Peakman M. Diabetogenic T lymphocytes in human type 1 diabetes. Curr Opin Immunol 2011; 23:746–53. [DOI] [PubMed] [Google Scholar]
- 2. Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med 2012; 2:a007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009; 155:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arif S, Leete P, Nguyen V et al Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 2014; 63:3835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. In’t Veld P, Lievens D, De Grijse J et al Screening for insulitis in adult autoantibody‐positive organ donors. Diabetes 2007; 56:2400–4. [DOI] [PubMed] [Google Scholar]
- 6. Richardson SJ, Rodriguez‐Calvo T, Gerling IC et al Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 2016; 59:2448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Itoh N, Hanafusa T, Miyazaki A et al Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin‐dependent diabetes mellitus patients. J Clin Invest 1993; 92:2313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin‐dependent) diabetes mellitus: a 25‐year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia 1986; 29:267–74. [DOI] [PubMed] [Google Scholar]
- 9. Coppieters KT, Dotta F, Amirian N et al Demonstration of islet‐autoreactive CD8 T cells in insulitic lesions from recent onset and long‐term type 1 diabetes patients. J Exp Med 2012; 209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinkse GG, Tysma OH, Bergen CA et al Autoreactive CD8 T cells associated with beta cell destruction in type 1 diabetes. Proc Natl Acad Sci USA 2005; 102:18425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Babon JA, DeNicola ME, Blodgett DM et al Analysis of self‐antigen specificity of islet‐infiltrating T cells from human donors with type 1 diabetes. Nat Med 2016; 22:1482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Skowera A, Ladell K, McLaren JE et al beta‐cell‐specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure. Diabetes 2015; 64:916–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kronenberg D, Knight RR, Estorninho M et al Circulating preproinsulin signal peptide‐specific CD8 T cells restricted by the susceptibility molecule HLA‐A24 are expanded at onset of type 1 diabetes and kill beta‐cells. Diabetes 2012; 61:1752–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yeo L, Woodwyk A, Sood S et al Autoreactive T effector memory differentiation mirrors beta cell function in type 1 diabetes. J Clin Invest 2018; 128:3460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Skowera A, Ellis RJ, Varela‐Calvino R et al CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose‐regulated preproinsulin epitope. J Clin Invest 2008; 118:3390–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rigby MR, DiMeglio LA, Rendell MS et al Targeting of memory T cells with alefacept in new‐onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Diabetes Endocrinol 2013; 1:284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nejentsev S, Howson JM, Walker NM et al Localization of type 1 diabetes susceptibility to the MHC class I genes HLA‐B and HLA‐A. Nature 2007; 450:887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maiers M, Gragert L, Klitz W. High‐resolution HLA alleles and haplotypes in the United States population. Hum Immunol 2007; 68:779–88. [DOI] [PubMed] [Google Scholar]
- 19. Haimila K, Perasaari J, Linjama T et al HLA antigen, allele and haplotype frequencies and their use in virtual panel reactive antigen calculations in the Finnish population. Tissue Antigens 2013; 81:35–43. [DOI] [PubMed] [Google Scholar]
- 20. Howson JM, Walker NM, Clayton D, Todd JA, Type 1 Diabetes Genetics Consortuim . Confirmation of HLA class II independent type 1 diabetes associations in the major histocompatibility complex including HLA‐B and HLA‐A. Diabetes Obes Metab 2009; 11(Suppl 1):31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nejentsev S, Gombos Z, Laine AP et al Non‐class II HLA gene associated with type 1 diabetes maps to the 240‐kb region near HLA‐B. Diabetes 2000; 49:2217–21. [DOI] [PubMed] [Google Scholar]
- 22. Eike MC, Olsson M, Undlien DE et al Genetic variants of the HLA‐A, HLA‐B and AIF1 loci show independent associations with type 1 diabetes in Norwegian families. Genes Immun 2009; 10:141–50. [DOI] [PubMed] [Google Scholar]
- 23. Gombos Z, Wachowicz J, Veijola R et al Human leukocyte antigen non‐class II determinants for type 1 diabetes in the Finnish population. Hum Immunol 2006; 67:714–21. [DOI] [PubMed] [Google Scholar]
- 24. Valdes AM, Erlich HA, Noble JA. Human leukocyte antigen class I B and C loci contribute to Type 1 Diabetes (T1D) susceptibility and age at T1D onset. Hum Immunol 2005; 66:301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lipponen K, Gombos Z, Kiviniemi M et al Effect of HLA class I and class II alleles on progression from autoantibody positivity to overt type 1 diabetes in children with risk‐associated class II genotypes. Diabetes 2010; 59:3253–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mikk ML, Heikkinen T, El‐Amir MI et al The association of the HLA‐A*24:02, B*39:01 and B*39:06 alleles with type 1 diabetes is restricted to specific HLA‐DR/DQ haplotypes in Finns. HLA 2017; 89:215–24. [DOI] [PubMed] [Google Scholar]
- 27. Reijonen H, Nejentsev S, Tuokko J et al HLA‐DR4 subtype and ‐B alleles in DQB1*0302‐positive haplotypes associated with IDDM. The Childhood Diabetes in Finland Study Group. Eur J Immunogenet 1997; 24:357–63. [DOI] [PubMed] [Google Scholar]
- 28. Nejentsev S, Reijonen H, Adojaan B et al The effect of HLA‐B allele on the IDDM risk defined by DRB1*04 subtypes and DQB1*0302. Diabetes 1997; 46:1888–92. [DOI] [PubMed] [Google Scholar]
- 29. Mikk ML, Kiviniemi M, Laine AP et al The HLA‐B*39 allele increases type 1 diabetes risk conferred by HLA‐DRB1*04:04‐DQB1*03:02 and HLA‐DRB1*08‐DQB1*04 class II haplotypes. Hum Immunol 2014; 75:65–70. [DOI] [PubMed] [Google Scholar]
- 30. Baschal EE, Baker PR, Eyring KR, Siebert JC, Jasinski JM, Eisenbarth GS. The HLA‐B 3906 allele imparts a high risk of diabetes only on specific HLA‐DR/DQ haplotypes. Diabetologia 2011; 54:1702–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noble JA, Valdes AM, Varney MD et al HLA class I and genetic susceptibility to type 1 diabetes: results from the Type 1 Diabetes Genetics Consortium. Diabetes 2010; 59:2972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams F, Meenagh A, Maxwell AP, Middleton D. Allele resolution of HLA‐A using oligonucleotide probes in a two‐stage typing strategy. Tissue Antigens 1999; 54:59–68. [DOI] [PubMed] [Google Scholar]
- 33. Middleton D, Williams F, Meenagh A et al Analysis of the distribution of HLA‐A alleles in populations from five continents. Hum Immunol 2000; 61:1048–52. [DOI] [PubMed] [Google Scholar]
- 34. Fujisawa T, Ikegami H, Kawaguchi Y et al Class I HLA is associated with age‐at‐onset of IDDM, while class II HLA confers susceptibility to IDDM. Diabetologia 1995; 38:1493–5. [DOI] [PubMed] [Google Scholar]
- 35. Noble JA, Valdes AM, Bugawan TL, Apple RJ, Thomson G, Erlich HA. The HLA class I A locus affects susceptibility to type 1 diabetes. Hum Immunol 2002; 63:657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valdes AM, Erlich HA, Carlson J, Varney M, Moonsamy PV, Noble JA. Use of class I and class II HLA loci for predicting age at onset of type 1 diabetes in multiple populations. Diabetologia 2012; 55:2394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mbunwe E, Van der Auwera BJ, Vermeulen I et al HLA‐A*24 is an independent predictor of 5‐year progression to diabetes in autoantibody‐positive first‐degree relatives of type 1 diabetic patients. Diabetes 2013; 62:1345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tait BD, Colman PG, Morahan G et al HLA genes associated with autoimmunity and progression to disease in type 1 diabetes. Tissue Antigens 2003; 61:146–53. [DOI] [PubMed] [Google Scholar]
- 39. Balke EM, Balti EV, Van der Auwera B et al Accelerated progression to type 1 diabetes in the presence of HLA‐A*24 and ‐B*18 is restricted to multiple islet autoantibody‐positive individuals with distinct HLA‐DQ and autoantibody risk profiles. Diabetes Care 2018; 41:1076–83. [DOI] [PubMed] [Google Scholar]
- 40. Nakanishi K, Kobayashi T, Murase T, Naruse T, Nose Y, Inoko H. Human leukocyte antigen‐A24 and ‐DQA1*0301 in Japanese insulin‐dependent diabetes mellitus: independent contributions to susceptibility to the disease and additive contributions to acceleration of beta‐cell destruction. J Clin Endocrinol Metab 1999; 84:3721–5. [DOI] [PubMed] [Google Scholar]
- 41. Nakanishi K, Kobayashi T, Murase T et al Association of HLA‐A24 with complete beta‐cell destruction in IDDM. Diabetes 1993; 42:1086–93. [DOI] [PubMed] [Google Scholar]
- 42. Demeester S, Balke EM, Van der Auwera BJ et al HLA‐A*24 carrier status and autoantibody surges posttransplantation associate with poor functional outcome in recipients of an islet allograft. Diabetes Care 2016; 39:1060–4. [DOI] [PubMed] [Google Scholar]
- 43. Kronenberg‐Versteeg D, Eichmann M, Russell MA et al Molecular pathways for immune recognition of preproinsulin signal peptide in type 1 diabetes. Diabetes 2018; 67:687–96. [DOI] [PubMed] [Google Scholar]
- 44. Eichmann M, de Ru A, van Veelen PA, Peakman M, Kronenberg‐Versteeg D. Identification and characterisation of peptide binding motifs of six autoimmune disease‐associated human leukocyte antigen‐class I molecules including HLA‐B*39:06. Tissue Antigens 2014; 84:378–88. [DOI] [PubMed] [Google Scholar]
- 45. Makinen A, Harkonen T, Ilonen J, Knip M, Finnish Pediatric Diabetes Register . Characterization of the humoral immune response to islet antigen 2 in children with newly diagnosed type 1 diabetes. Eur J Endocrinol 2008; 159:19–26. [DOI] [PubMed] [Google Scholar]
- 46. Parikka V, Nanto‐Salonen K, Saarinen M et al Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia 2012; 55:1926–36. [DOI] [PubMed] [Google Scholar]
- 47. Nanto‐Salonen K, Kupila A, Simell S et al Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double‐blind, randomised controlled trial. Lancet 2008; 372:1746–55. [DOI] [PubMed] [Google Scholar]
- 48. Kukko M, Kimpimaki T, Korhonen S et al Dynamics of diabetes‐associated autoantibodies in young children with human leukocyte antigen‐conferred risk of type 1 diabetes recruited from the general population. J Clin Endocrinol Metab 2005; 90:2712–7. [DOI] [PubMed] [Google Scholar]
- 49. Helminen O, Aspholm S, Pokka T et al HbA1c predicts time to diagnosis of type 1 diabetes in children at risk. Diabetes 2015; 64:1719–27. [DOI] [PubMed] [Google Scholar]
- 50. Salonen KM, Ryhanen S, Harkonen T, Ilonen J, Knip M, Finnish Pediatric Diabetes Register . Autoantibodies against zinc transporter 8 are related to age, metabolic state and HLA DR genotype in children with newly diagnosed type 1 diabetes. Diabetes Metab Res Rev 2013; 29:646–54. [DOI] [PubMed] [Google Scholar]
- 51. Wooldridge L, van den Berg HA, Glick M et al Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor‐antigen complexes at the cell surface. J Biol Chem 2005; 280:27491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pudney VA, Leese AM, Rickinson AB, Hislop AD. CD8+ immunodominance among Epstein–Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J Exp Med 2005; 201:349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Motozono C, Pearson JA, De Leenheer E et al Distortion of the major histocompatibility complex class I binding groove to accommodate an insulin‐derived 10‐mer peptide. J Biol Chem 2015; 290:18924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lissina A, Ladell K, Skowera A et al Protein kinase inhibitors substantially improve the physical detection of T‐cells with peptide‐MHC tetramers. J Immunol Methods 2009; 340:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tungatt K, Bianchi V, Crowther MD et al Antibody stabilization of peptide‐MHC multimers reveals functional T cells bearing extremely low‐affinity TCRs. J Immunol 2015; 194:463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Culina S, Lalanne AI, Afonso G et al Islet‐reactive CD8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol 2018; 3:eaao4013. doi: 10.1126/sciimmunol.aao4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Billingsley JM, Rajakumar PA, Connole MA et al Characterization of CD8+ T cell differentiation following SIVDeltanef vaccination by transcription factor expression profiling. PLOS Pathog 2015; 11:e1004740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Picker LJ, Reed‐Inderbitzin EF, Hagen SI et al IL‐15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. J Clin Invest 2006; 116:1514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jeng MY, Hull PA, Fei M et al Metabolic reprogramming of human CD8(+) memory T cells through loss of SIRT1. J Exp Med 2018; 215:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T‐cell differentiation: human memory T‐cell subsets. Eur J Immunol 2013; 43:2797–809. [DOI] [PubMed] [Google Scholar]
- 61. Martin MD, Badovinac VP. Defining memory CD8 T cell. Front Immunol 2018; 9:2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Banerjee A, Gordon SM, Intlekofer AM et al Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol 2010; 185:4988–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pipkin ME, Sacks JA, Cruz‐Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin‐2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 2010; 32:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Intlekofer AM, Takemoto N, Wherry EJ et al Effector and memory CD8+ T cell fate coupled by T‐bet and eomesodermin. Nat Immunol 2005; 6:1236–44. [DOI] [PubMed] [Google Scholar]
- 65. Abdelsamed HA, Moustaki A, Fan Y et al Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med 2017; 214:1593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Akondy RS, Fitch M, Edupuganti S et al Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017; 552:362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol 2003; 171:27–31. [DOI] [PubMed] [Google Scholar]
- 68. Ding Y, Zhou L, Xia Y et al Reference values for peripheral blood lymphocyte subsets of healthy children in China. J Allergy Clin Immunol 2018; 142:970–3.e8. [DOI] [PubMed] [Google Scholar]
- 69. Jansen MA, van den Heuvel D, van Zelm MC et al Decreased memory B cells and increased CD8 memory T cells in blood of breastfed children: the generation R study. PLOS ONE 2015; 10:e0126019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Caccamo N, Meraviglia S, La Mendola C, Guggino G, Dieli F, Salerno A. Phenotypical and functional analysis of memory and effector human CD8 T cells specific for mycobacterial antigens. J Immunol 2006; 177:1780–5. [DOI] [PubMed] [Google Scholar]
- 71. Longwe H, Phiri KS, Mbeye NM, Gondwe T, Jambo KC, Mandala WL. Proportions of CD4+, CD8+ and B cell subsets are not affected by exposure to HIV or to Cotrimoxazole prophylaxis in Malawian HIV‐uninfected but exposed children. BMC Immunol 2015; 16:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kimpimaki T, Kulmala P, Savola K et al Natural history of beta‐cell autoimmunity in young children with increased genetic susceptibility to type 1 diabetes recruited from the general population. J Clin Endocrinol Metab 2002; 87:4572–9. [DOI] [PubMed] [Google Scholar]
- 73. Ziegler AG, Hummel M, Schenker M, Bonifacio E. Autoantibody appearance and risk for development of childhood diabetes in offspring of parents with type 1 diabetes: the 2‐year analysis of the German BABYDIAB Study. Diabetes 1999; 48:460–8. [DOI] [PubMed] [Google Scholar]
- 74. Bonifacio E, Lampasona V, Bernasconi L, Ziegler AG. Maturation of the humoral autoimmune response to epitopes of GAD in preclinical childhood type 1 diabetes. Diabetes 2000; 49:202–8. [DOI] [PubMed] [Google Scholar]
- 75. Naserke HE, Ziegler AG, Lampasona V, Bonifacio E. Early development and spreading of autoantibodies to epitopes of IA‐2 and their association with progression to type 1 diabetes. J Immunol 1998; 161:6963–9. [PubMed] [Google Scholar]
- 76. Kawasaki E, Yu L, Rewers MJ, Hutton JC, Eisenbarth GS. Definition of multiple ICA512/phogrin autoantibody epitopes and detection of intramolecular epitope spreading in relatives of patients with type 1 diabetes. Diabetes 1998; 47:733–42. [DOI] [PubMed] [Google Scholar]
- 77. Yu L, Rewers M, Gianani R et al Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab 1996; 81:4264–7. [DOI] [PubMed] [Google Scholar]
- 78. Brooks‐Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol 2001; 166:5265–70. [DOI] [PubMed] [Google Scholar]
- 79. Ott PA, Dittrich MT, Herzog BA et al T cells recognize multiple GAD65 and proinsulin epitopes in human type 1 diabetes, suggesting determinant spreading. J Clin Immunol 2004; 24:327–39. [DOI] [PubMed] [Google Scholar]
- 80. Kaufman DL, Clare‐Salzler M, Tian J et al Spontaneous loss of T‐cell tolerance to glutamic acid decarboxylase in murine insulin‐dependent diabetes. Nature 1993; 366:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gelber C, Paborsky L, Singer S et al Isolation of nonobese diabetic mouse T‐cells that recognize novel autoantigens involved in the early events of diabetes. Diabetes 1994; 43:33–9. [DOI] [PubMed] [Google Scholar]
- 82. Zechel MA, Krawetz MD, Singh B. Epitope dominance: evidence for reciprocal determinant spreading to glutamic acid decarboxylase in non‐obese diabetic mice. Immunol Rev 1998; 164:111–8. [DOI] [PubMed] [Google Scholar]
- 83. Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non‐obese diabetic mice. Nature 1993; 366:72–5. [DOI] [PubMed] [Google Scholar]
- 84. Oram RA, Sims EK, Evans‐Molina C. Beta cells in type 1 diabetes: mass and function; sleeping or dead? Diabetologia 2019; 62:567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Howson JM, Stevens H, Smyth DJ et al Evidence that HLA class I and II associations with type 1 diabetes, autoantibodies to GAD and autoantibodies to IA‐2, are distinct. Diabetes 2011; 60:2635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Qu HQ, Polychronakos C. The effect of the MHC locus on autoantibodies in type 1 diabetes. J Med Genet 2009; 46:469–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Long AE, Gillespie KM, Aitken RJ, Goode JC, Bingley PJ, Williams AJ. Humoral responses to islet antigen‐2 and zinc transporter 8 are attenuated in patients carrying HLA‐A*24 alleles at the onset of type 1 diabetes. Diabetes 2013; 62:2067–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Knight RR, Kronenberg D, Zhao M et al Human beta‐cell killing by autoreactive preproinsulin‐specific CD8 T cells is predominantly granule‐mediated with the potency dependent upon T‐cell receptor avidity. Diabetes 2013; 62:205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Luce S, Lemonnier F, Briand JP et al Single insulin‐specific CD8+ T cells show characteristic gene expression profiles in human type 1 diabetes. Diabetes 2011; 60:3289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Velthuis JH, Unger WW, Abreu JR et al Simultaneous detection of circulating autoreactive CD8+ T‐cells specific for different islet cell‐associated epitopes using combinatorial MHC multimers. Diabetes 2010; 59:1721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yu W, Jiang N, Ebert PJ et al Clonal deletion prunes but does not eliminate self‐specific alphabeta CD8(+) T lymphocytes. Immunity 2015; 42:929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flow cytometry gating strategy for determination of antigen‐specific CD8 T cell subsets. (a) Lymphocytes were defined as CD3 positive, ‘Dump’ (dead cell stain, CD14, CD16, CD19) negative gated on FSC and SSC characteristics. Doublets and CD4+CD8+ double‐positive cells were excluded. Cells were gated as CD8+Tetramer+ or CD8+Tetramer‐. (b) Gating of CD8+ T cell subsets: Naïve (N;CCR7+CD45RA+CD27+CD57‐CD95‐), stem cell memory‐like (SCM; CCR7+CD45RA+CD27+CD57‐CD95+), central memory (CM; CCR7+CD45RA‐CD27+), transitional memory (TM; CCR7‐CD45RA‐CD27+), effector memory (EM; CCR7‐CD45RA‐CD27‐) and terminal effector (TE; CCR7‐CD45RA+). (c) Schematic of the CD8+ T cell subsets analysed and the cell surface markers used for their definition.
Fig. S2. Dual‐colour tetramer staining with pHLA‐B*3906 tetramers loaded with PPI5‐12 and EBV BMRF1268‐276. (a) Gating strategy used to identify double‐positive tetramer‐binding CD8+ T cells. Lymphocytes were defined as CD3+, ‘Dump’ (dead cell stain, CD14, CD16, CD19)‐negative, and gated on lymphocyte SSC and FSC characteristics. Doublets were excluded. (b) PBMC from an HLA‐B*3906 + donor stained with PPI5‐12 and EBV BMRF1268‐276 pHLA‐B*3906 tetramers which were dual‐labelled with APC and PE. The majority of tetramer‐binding cells are double‐positive for both tetramers.
Fig. S3. Frequency and phenotype of HLA‐B*3906‐restricted EBV‐specific CD8+ T cells in EBV antibody‐positive (n = 3) and antibody‐negative (n = 9) subjects. (a) Frequency of EBV BMRF1268‐276‐specific CD8+ T cells in EBV antibody‐positive (red) and antibody‐negative (white) subjects. (b) Phenotype of EBV BMRF1268‐276‐specific CD8+ T cells in EBV antibody‐positive (red) and antibody‐negative (white) subjects.
Fig. S4. Phenotype of polyclonal CD8+ T cell populations in HLA‐B*3906+ newly diagnosed type 1 diabetes subjects and HLA‐B*3906+ control subjects. The phenotype of polyclonal CD8+ T cells was not found to be significantly different between HLA‐B*3906+ T1D subjects (grey) and healthy controls (white). Mann–Whitney U‐tests P > 0·05.
Fig. S5. Frequency and phenotype of HLA‐B*3906‐restricted PPI‐specific CD8+ T cells in autoantibody positive control (n = 1) and negative controls (n = 6). (a) Frequency and phenotype of PPI5‐12‐specific CD8+ T cells and polyclonal CD8+ T cells from autoantibody‐negative controls (white), autoantibody‐positive control (red) and T1D subjects (black). (b) Frequency and phenotype of EBV BMRF1268‐276‐specific CD8+ T cells and polyclonal CD8+ T cells from autoantibody‐negative controls (white), autoantibody‐positive control (red) and T1D subjects (black).
Fig. S6. Frequency and phenotype of HLA‐B*3906‐restricted PPI‐specific CD8+ T cells before the diagnosis of type 1 diabetes. (a) Frequency of PPI5‐12‐specific CD8+ T cells from an HLA‐B*3906 + type 1 diabetes subject before diagnosis (black) compared to HLA‐B*3906 + newly diagnosed type 1 diabetes subjects (grey) and HLA‐B*3906 + control subjects (white). (b) Phenotype of PPI3‐11‐specific CD8+ T cells from an HLA‐B*3906 + type 1 diabetes subject before diagnosis (black) compared to HLA‐B*3906 + newly diagnosed type 1 diabetes subjects (grey) and HLA‐B*3906 + control subjects (white). (c) Frequency of memory and effector T cell subsets expressed as a percentage of non‐naïve T cells within tetramer‐specific (red) and polyclonal (blue) CD8+ T cell populations. Radial lines represent intervals of T cell subset frequencies of 10% from 0 to 50%, with the lowest value at the centre and the highest value at the periphery. Polygons link the frequency of each T cell subset. (d) Phenotype of PPI5‐12‐specific CD8+ T cells (squares) compared to total polyclonal CD8+ T cells (circles) in an HLA‐B*3906 + type 1 diabetes subject before diagnosis (black), HLA‐B*3906 + newly diagnosed type 1 diabetes subjects (grey) and HLA‐B*3906 + control subjects (white).
Fig. S7. Phenotype of polyclonal CD8+ T cell populations in HLA‐A*2402+ type 1 diabetes subjects before diagnosis and HLA‐A*2402+ control subjects. The phenotype of polyclonal CD8+ T cells was not found to be significantly different between HLA‐A*2402+ pre‐diagnosis subjects (grey) and healthy controls (white). Mann–Whitney U‐tests P > 0.05.