

Summary
Systemic lupus erythematosus is a chronic inflammatory disease which involves multiple organs. Self‐specific B and T cells play a main role in the pathogenesis of lupus and have been defined as a logical target for selective therapy. The protein annexin A1 (ANX A1) is a modulator of the immune system involving many cell types. An abnormal expression of ANX A1 was found on activated B and T cells during autoimmunity, suggesting its importance as a potential therapeutic target. We hypothesize that it may be possible to down‐regulate the activity of autoreactive T and B cells from lupus patients in a humanized immunodeficient mouse model by treating them with an antibody against ANX A1. When cultured in the presence of anti‐ANX A1, peripheral blood mononuclear cells (PBMC) from lupus patients showed a decreased number of immunoglobulin (Ig)G anti‐dsDNA antibody‐secreting plasma cells, decreased T cell proliferation and expression of activation markers and increased B and T cell apoptosis. We employed a humanized model of SLE by transferring PBMCs from lupus patients to immunodeficient non‐obese diabetic‐severe combined immunodeficient (NOD‐SCID) mice. The humanized animals presented autoantibodies, proteinuria and immunoglobulin deposition in the renal glomeruli. Treatment of these NOD‐SCID mice with an anti‐ANX A1 antibody prevented appearance of anti‐DNA antibodies and proteinuria, while the phosphate‐buffered saline (PBS)‐injected animals had high levels after the transfer. The treatment reduced the levels of autoantibodies to several autoantigens, lupus‐associated cytokines and disease symptoms.
Keywords: anti‐annexin A1 antibody, humanized NOD‐SCID mice, systemic lupus
An abnormal expression of ANX A1 was found on activated B and T cells during autoimmunity suggesting its importance as a potential therapeutic target. We hypothesize that it may be possible to down‐regulate the activity of autoreactive T and B cells from lupus patients in humanized immunodeficient mouse model by treating them with an antibody against ANX A1. The treatment reduced the levels of autoantibodies to several autoantigens, lupus‐associated cytokines and disease symptoms.

Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease that affects multiple organs and systems. The number of genes involved in the onset and the progression of the disease is vast. In addition, various environmental factors contribute to the aetiology of the disorder. The hallmarks of SLE are abnormalities such as generation of autoantibodies against self‐nuclear antigens, glomerulonephritis, vasculitis, inadequate immune complex clearance by phagocytes and shifting towards a T helper type 2 (Th2) response 1.
No cure for SLE currently exists. Nevertheless, it is possible to control the severity of the manifestations through general suppression of the immune response, which significantly increases the survival rate and quality of life of the patients. Glucocorticoid treatment exerts an anti‐inflammatory effect by inhibiting both T and B cells, as well as cells of the innate immune branch 2. Cyclophosphamide is frequently added to the treatment plan, especially in cases of lupus nephritis. However, this approach is non‐specific and is associated with multiple adverse effects; for instance, glucocorticoids are associated with immunodeficiency, myopathy and diabetes, while there are data linking cyclophosphamide with promotion of cancer, leukaemia and lymphoma 3.
Newer, more specific approaches directed to particular immune cells have been developed, and some were approved for clinical trials. Autoreactive B cells are typically considered the principal cause of damage in human and murine SLE. This is primarily a result of their role in the secretion of autoantibodies. B cells are one of the main antigen‐presenting cells (APC) that, in addition, release cytokines 4, where they participate in the regulation of other immune cells. Thus, they are a reasonable target for suppressing disease progression. In particular, most current therapies utilize monoclonal antibodies to CD19, CD20, B lymphocyte stimulator (BLyS), CD22 or other co‐receptors on B cells 5.
As a main B cell regulator, T cells are of crucial importance in SLE pathogenesis 6. Furthermore, T cells govern the whole immune response due to their principle role in cytokine release. Consequently, therapies targeting T cells have been developed 3, 7. Such therapies include targeting of CD28 3 and cytotoxic T lymphocyte antigen‐4 (CTLA‐4) T cell co‐receptors 8, as well as their co‐ligands on B cells (B7‐1 and B7‐2) or inhibition of the CD40–CD40L pathway that is involved in T and B cell interplay 3. So far, however, none of these therapies targeting B and T cells provide a specific treatment for SLE, and therefore an alternative approach for negative signal delivery is needed.
Annexin A1 (ANX A1), also known as lipocortin 1, lipomodulin and macrocortin, is a 37‐kDa protein and a member of the annexin proteins superfamily. ANX A1 is predominantly located on the inner plasma membrane and within the cytoplasm of cells of both innate and adaptive immunity. Similar to other members of this superfamily, ANX A1 binds to acidic phospholipids in the presence of calcium ions 9. The most‐studied functions of this protein are the inhibition of phosholipase A2 (PLA2) activity and its role in the resolution of inflammation. Moreover, ANX A1 has been linked to several different functions 10.
Human and mouse neutrophils, macrophages and monocytes display constitutively high levels of cytoplasmic ANX A1. Upon activation, ANX A1 is secreted in a cell‐specific and calcium‐dependent manner 10, 11, 12. In humans, ANX A1 shares a common G‐protein‐coupled receptor with lipoxin A4, known as formyl peptide receptor 2 (FPR2) or ALXR 13. Glucocorticoids exert some of their effects on monocytes, macrophages and neutrophils by inducing the expression of ANX A1 and its receptor. Furthermore, the ANX A1–FPR2 pathway is a negative regulator of the transmigration of these cells and the generation of proinflammatory mediators such as interleukin (IL)‐1 and IL‐6 11, 14. The immunosuppressive effect of glucocorticoids on T cells is the result of inhibition of ANX A1 expression 15, 16. Interestingly, ANX A1 plays an opposite role compared to innate cells, as the ANX A1–FPR2 pathway aids T cell activation and differentiation. Evidence prompts the idea that ANX A1 has a potential role in fine‐tuning T cell receptor (TCR) signalling and that its action is important for proper T cell activation 12, 17, 18.
Several mouse models spontaneously develop anti‐dsDNA antibodies and SLE‐like disease. However, these animal models do not reproduce the severe complicated clinical manifestations found in humans 19, 20, 21. Severe combined immunodeficient (SCID) mice with T and B immunodeficiency syndrome are a suitable model for transferring human lymphocytes without rejection due to their inability to mount an adaptive immune response 22, 23. SCID mice reconstituted with PBMCs from SLE patients show the presence of autoantibodies against dsDNA and anti‐human immunoglobulin (Ig) deposition in the renal glomeruli. We have utilized such a transferred SLE model in the past and prevented the appearance of anti‐dsDNA antibodies and proteinuria by selective suppression of dsDNA‐specific B cells 24.
Non‐obese diabetic (NOD)‐SCID gamma (NSG) mice are next‐generation immunodeficient animals whose recombinase system mutation leads to the absence of mature T, B and natural killer (NK) cells. These mice cannot develop an adaptive immune response, and are suitable recipients of human lymphoid cells as a model host for therapeutic investigation 22, 25.
Therefore, we hypothesized that in‐vitro and in‐vivo down‐regulation of the activity of disease‐associated T and B cells is possible via blocking the ANX A1–FPR2 pathway by monoclonal antibodies specific to annexin A1 in a humanized NSG mouse model of SLE.
Materials and methods
SLE patients and healthy blood donors
Sixteen patients, who attended the Clinic of Rheumatology, University Hospital St Ivan Rilski, Department of Internal Medicine, Medical University of Sofia, were enrolled into this study. The mean age of the patients (two male, 14 female) was 34·1 years. All the patients met the 1997 update 1982 revised American College of Rheumatology (ACR) criteria for the classification of SLE and were diagnosed with SLE. Major inclusion criteria in the study were positive anti‐nuclear autoantibodies (ANA), positive IgG autoantibodies against dsDNA and proteinuria. Whole venous blood samples for peripheral blood mononuclear cell (PBMC) isolation were collected from all patients before starting systemic glucocorticoid therapy.
The control group consisted of eight age‐ and sex‐matched healthy donors.
Monoclonal antibodies
Monoclonal mouse anti‐ANX A1 (human/mouse/rat) IgG1 antibody was provided by R&D Systems (Minneapolis, MN, USA). Unconjugated mouse IgG1 isotype control (eBioscience, Frankfurt, Germany) was used in all experiments. Unconjugated anti‐human CD3 and CD28 antibodies (eBioscience) were used for in‐vitro stimulation. Fluorescein isothiocyanate (FITC)‐conjugated anti‐human CD69 and anti‐human CD3, phycoerythrin‐cyanin 5 (PE‐Cy5)‐conjugated anti‐human CD3, PE‐conjugated anti‐human CD8, anti‐human CD25 and anti‐human CD19, APC‐conjugated anti‐human CD4, biotin‐conjugated anti‐human CD19, streptavidin‐eFluor 710 and streptavidin‐PE (eBioscience) were used for fluorescence‐activated cell sorting (FACS) experiments. Alkaline phosphatase (AP)‐conjugated anti‐human IgG (Sigma‐Aldrich, Taufkirchen, Germany) were used for enzyme‐linked immunosorbent assay (ELISA) and enzyme‐linked immunospot (ELISPOT) assay. FITC‐conjugated goat anti‐human IgG (Sigma‐Aldrich) was used for immunohistology staining.
Biotinylation of anti‐ANX A1 and isotype antibodies
The procedure was performed as previously described 19. Biotin‐conjugated anti‐ANX A1 and control antibodies were used for FACS experiments.
Ethical committee statement
Informed written consent was obtained from all subjects enrolled into the study. The study was conducted according to the Declaration of Helsinki. The protocol was reviewed and approved by the Local Ethics Committee at the University Hospital St Ivan Rilski, Sofia.
PBMC and sera isolation
Venous blood was collected from SLE patients and healthy donors in sterile blood and serum tubes (BD Vacutainer, Franklin Lakes, NJ, USA). Serum samples of 5 ml were collected from each subject using serum separator tubes (BD Vacutainer Systems, Plymouth, UK; 5 ml) and frozen at −80°C. For detection of autoantibodies we used Euroline ANA Profile 3 (IgG) (Euroimmune, Lubeck, Germany). Each test strip contained a panel of: nRNP/Sm, Sm, SS‐A, SS‐B, Scl‐70, PM‐Scl, Jo‐1, CENP‐B, PCNA, dsDNA, nucleosomes, histones, Rib‐P and AMA‐M2. Serum patient samples, positive and negative controls of each series were used according to the manufacturer’s instructions.
We also analysed the sera on automatic ELISA device ‘Alegria’ (Orgentec, Maintz, Germany) for double‐stranded (ds)DNA, anti‐cardiolipin antibody (aCL) and beta‐2‐glycoprotein I (b2GPI) using anti‐dsDNA screen, anti‐cardiolipin screen and anti‐b2GPI screen (Orgentec), respectively.
ANA‐human epithelial type 2 (Hep2) cells (BioSystems S.A., Barcelona, Spain) were used for ANA screening on indirect immunofluorescence. Analyses of the fluorescent images and the ANA titre determination were performed using a fluorescence microscope EUROStar III Plus (Euroimmune, Lubeck, Germany).
PBMCs from patients and healthy controls were isolated using the BD Vacutainer CTPTM (8 ml) by Pancoll human separating medium (Pan‐Biotech GmbH, Aidenbach, Germany). A density gradient centrifugation at 800 g for 20 min at 20°C was performed and isolated PBMCs were cultured in RPMI‐1640 medium (gibco, Gaithersburg, MD, USA) supplemented with 10% heat‐inactivated fetal calf serum (FCS), 100 U/ml penicillin, 100 U/ml streptomycin, 2 mM glutamine and 1 mM sodium pyruvate.
Mice
NOD‐scid IL2rγnull (NSG) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). The animals were kept in a barrier‐type animal house under specific pathogen‐free (SPF) conditions. The national regulations, EU Directive 2010/63/EU for animal experiments and guidance from the Animal Care Commission at the Institute of Microbiology were followed when executing all in‐vivo manipulations.
Flow cytometric analysis
Isolated PBMCs from SLE patients and healthy donors were washed with PBS (containing 2·5% FCS and 0·05% sodium azide) and incubated with biotin‐conjugated anti‐ANX A1 or isotype control antibodies. Next, the cells were incubated with streptavidin‐eFluor 710, washed and incubated either with anti‐CD19‐PE, anti‐CD3‐FITC/anti‐CD4‐APC or anti‐CD3‐PE‐Cy5/anti‐CD8‐PE.
Concomitantly, PBMCs from patients and healthy donors were put under activating conditions using plate‐bound anti‐human CD3 and anti‐human CD28 antibodies at 2·5 μg/ml each. The incubation continued for 20 h with the addition of varying concentrations of anti‐ANX A1 or isotype control antibodies, ranging from 5 to 500 ng/ml. Ten thousand cells from each sample were used for FACS analysis. Stains of choice were anti‐CD3‐PE‐Cy5, anti‐CD25‐APC and anti‐CD69‐PE, all applied at 1 μg/106 cells for 20 min at 4°C. The cells were then analysed on a BD LSR II flow cytometer using Diva version 6.1.1. software (BD Biosciences).
Proliferation assay
PBMCs isolated from lupus patients and healthy donors were cultured (2 × 106 cells/ml) in complete RPMI‐1640 medium only or stimulated simultaneously with plate‐bound anti‐human CD3 and anti‐human CD28 (2·5 μg/well for each) at 37°C/5% CO2. The cells were co‐cultured with rising concentrations of anti‐ANX A1 or control antibodies (5–500 ng/ml). The control cells were stimulated with 10 μg/ml lipopolysaccharide (LPS) (from Escherichia coli; Sigma) or 50 ng/ml phorbol myristate acetate (PMA) plus 0·75 μg/ml ionomycin, or cultured in medium only.
The cells were cultured for 4 days. Proliferation was assessed by formazan absorption at a wavelength of 590 nm, which forms upon 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐ diphenyltetrazolium bromide (MTT) conversion by viable cells. MTT was added for an additional 4 h at a concentration of 0·45 μg/ml. Due to formazan being insoluble in the media, the latter was decanted and 200 μl dimethyl sulphoxide (DMSO) was used to dissolve the crystals. The measurements were corrected by subtracting the absorbance at 620 nm.
ELISPOT assay
ELISPOT was used to determine the influence of anti‐ANX A1 antibody on anti‐dsDNA‐producing plasma cells. PBMCs from the SLE patients and healthy volunteers were isolated (see above) and cultured (2 × 106/ml) in complete RPMI‐1640 medium for 5 days at 37°C/5% CO2. One set of samples was incubated with different concentrations of anti‐ANX A1 and another with control antibodies, both from 5 to 500 ng/ml. A third set was incubated only in medium.
Additionally, PBMCs were activated and treated with anti‐ANX A1 or control antibodies in the same manner as described above for the proliferation assay, except that the incubation lasted 5 days, and then the samples were analysed by ELISpot, as described further below.
In another experiment, the same cells were stimulated with 10 μg/ml LPS and cultured for 5 days with anti‐ANX A1 or control antibodies (ranging from 100 to 500 ng/ml) or with medium only. Cells cultured in LPS or PMA/ionomycin were used as positive controls.
Later, the ELISpot assay was performed as described 19. Briefly, 96‐well ELISpot membrane plates were activated with ethanol, coated with calf thymus DNA and blocked with gelatine. The cells incubated in the culture plates were transferred into respective wells of the ELISpot plates with DNA‐coated membranes and further cultured for an additional 4 h at 37°C/5% CO2. After incubation with an AP‐conjugated anti‐human IgG for 1 h, the membranes were developed by nitro‐blue tetrazolium and 5‐bromo‐4‐chloro‐3'‐indolyphosphate (NBT‐BCIP) substrate and the number of cells producing IgG anti‐dsDNA antibodies was counted by C.T.L. Immunospot S5 Versa Analyzer (Bonn, Germany).
Detection of apoptosis
PBMCs from SLE patients and healthy volunteers (anti‐CD3/CD28 activated or not) were co‐cultured as described above with anti‐ANX A1 or control antibodies (ranging from 5 to 500 ng/ml) for 24 h (2 × 106 cells/ml in complete RPMI‐1640 medium) at 37°C/5% CO2. The lymphocytes were then washed and tagged with streptavidin‐eFluor 710 bound to biotin‐conjugated anti‐human CD19 or PE‐Cy5‐conjugated anti‐human CD3 antibodies. Apoptosis of gated CD19+ B and CD3+ T cells was evaluated by FACS using the annexin V‐FITC apoptosis detection kit (eBioscience).
Cell transfer
Four groups of female NSG mice (12 weeks old, n = two to four per donor) were used for human cell transfer. Isolated PBMCs (1 × 107 cells) from each SLE patient or healthy donor (see above) were transferred to individual NSG mice. The cells from each donor were divided before transfer into equal parts and the first part (0·5 × 107 cells) was transferred intraperitoneally (i.p.), while the second was injected intravenously (i.v.) in the same mice.
Treatment schedule
The animals from each group (SLE or healthy donors) of PBMC‐transferred NSG mice were separated into two equal subgroups. The animals in the first subgroup were injected i.p. with 200 ng/mouse of anti‐ANX A1 antibody every 6 days. Control animals from the second subgroup were treated with the same quantity of isotype control antibody. Blood samples were collected from the animals every 14 days and the sera were kept frozen at −70°C for additional analyses.
Proteinuria measurement
Urine samples were collected from the urethra and applied to test strips (Combi‐screen strips; Analyticon Biotechnologies, Lichenfels, Germany). The levels of proteinuria were measured weekly and classified semi‐quantitatively as: 0, none; 1, 30–100; 2, 100–300; 3, 300–500 and 4, > 500 mg dL−1.
Detection of anti‐dsDNA antibodies
Serum levels of human anti‐DNA IgG antibodies were evaluated by ELISA, as previously described 24. Briefly, 96‐well immunoplates were coated with methylated bovine serum albumin (BSA), followed by incubation with calf thymus DNA at 4°C. Then, the plates were blocked, incubated with diluted pooled murine sera, followed by AP‐conjugated anti‐human IgG antibody. The reaction, which allows for enumeration of anti‐DNA IgG antibodies, was revealed with p‐nitrophenyl phosphate (pNPP) substrate and the samples were read at 405 nm. Serum from an anti‐dsDNA antibody‐positive SLE patient was analysed by each test and used as a standard to score the rest of the results.
Serological measurements using autoantigen microarrays
Microarrays with panels of SLE antigens and control materials were custom‐printed by Scienion AG (Berlin, Germany) onto nitrocellulose‐covered FAST slides (Maine Manufacturing, Sanford, ME, USA). Different concentrations of the antigens were spotted in triplicates and slides were stored in sealed bags at 4°C until use. Dried arrays were placed in 16‐well slide modules and washed once quickly and then three times for 5 min with 200 μl PBS/chamber, then once for 5 min with 200 μl PBS supplemented with 0·05% Tween 20, 5% BSA and 25 mM ethylenediamine tetraacetic acid (EDTA) on a horizontal shaker (Heidolph Titramax 100) with speed set to 600 RPM.
Microarray slides were then treated with 100 μl of 50‐fold diluted pooled serum samples taken at different time‐points from 12 anti‐ANX A1‐treated humanized mice with PBMC from SLE patients or treated with control antibodies under the scheme described above and incubated at 37°C for 1 h on a shaker. Sera were diluted in 0·05% Tween 20, 5% BSA and 25 mM EDTA‐supplemented PBS buffer. Serum‐treated slides were washed with PBS containing 0·05% Tween 20, then incubated in the mixture of 1 : 2500 diluted DyL488‐conjugated goat anti‐human IgM (μ chain) antibody (Jackson Laboratory) and 1 : 2500 diluted DyL649‐conjugated goat anti‐human IgG (γ chain) antibody (Jackson Laboratory). Dilution was made in PBS containing 5% BSA and 0·05% Tween 20. Slides were removed from the slide modules and placed into one‐well chambers and then labelled at room temperature for 30 min on a shaker in 4 ml antibody cocktail per slide. After washing in PBS containing 0·05% Tween 20, arrays were dried and scanned by an Axon GenePix 4300A scanner (Molecular Devices).
Following visual inspection of images for the exclusion of gross printing or developing failures, data were analysed with GenePixPro 7 software (Molecular Devices). Signal intensities were calculated by subtracting background from medians of three parallel signal intensities using RStudio. To eliminate background noise, two standard deviations (s.d.) of local background signals on a subarray were subtracted from the signal intensities, then values below 100 were rounded‐up to 100. Signals were calculated by taking the base 10 logarithm of relative fluorescence units (RFU) and normalized by expressing as the percentage of initial, first‐sampling values. Taking into account that the coefficient of variation is in the range of 7–14% for the different antigens, we considered more than a 15% difference as methodologically significant.
Cytokine detection
Human IL‐4, IL‐10 and interferon (IFN)‐γ levels were measured in mixed sera of humanized mice using human high‐sensitivity (HS) ELISA kits (Bender MedSystems, Vienna, Austria), according to the manufacturer’s instructions.
Kidney histology and immunofluorescence
Kidneys were isolated from both the test and control animals. They were fixed in 10% buffered formaldehyde solution and paraffin‐embedded. Immune depositions were developed after staining with FITC‐conjugated anti‐human IgG (Sigma). The renal glomeruli were examined in sections under a Nikon Eclipse Ti‐U confocal laser scanning microscopy fluorescent microscope. Sections from the other kidney were also processed by the general staining method with haematoxylin and eosin. We applied a relative score system to judge the pathology, in which a score of 3 represents excellent renal preservation and 0 represents poor renal preservation.
Statistical analysis
All statistical analyses were performed with Prism software from GraphPad (San Diego, CA, USA). The two‐way anova test was used to determine differences between each of the two groups. The continuous variables were presented as means ± s.d. All ELISA and ELISPOT samples were triplicated. A value of P < 0·05 was considered statistically significant.
Results
SLE patients
The clinical characteristics of the patients are shown in Table 1.
Table 1.
SLE patients’ clinical characteristic
| Patient/sex | Diagnosis | Autoantibodies | Proteinuria |
|---|---|---|---|
| Patient 1/f | SLE | ANA, dsDNA, aCL, | − |
| Patient 2/f | SLE | ANA, dsDNA, aCL, b2GPI | + |
| Patient 3/f | SLE | ANA, dsDNA, aCL, b2GPI, nRNP/Sm, Sm, histones, | + |
| Patient 4/f | SLE | ANA, dsDNA, Rib‐P, SS‐B | + |
| Patient 5/f | SLE | ANA, dsDNA, Sm | + |
| Patient 6/f | SLE | ANA, dsDNA, aCL | − |
| Patient 7/f | SLE | ANA, dsDNA, nucleosomes | + |
| Patient 8/f | SLE | ANA, dsDNA, SS‐A, nucleosomes | + |
| Patient 9/f | SLE | ANA, dsDNA, aCL, b2GPI | + |
| Patient 10/m | SLE | ANA, dsDNA | + |
| Patient 11/f | SLE | ANA, dsDNA, Sm, SS‐A, SS‐B | − |
| Patient 12/f | SLE | ANA, dsDNA, Sm, nRNP/Sm | + |
| Patient 13/f | SLE | ANA, dsDNA, Sm | + |
| Patient 14/f | SLE | ANA, dsDNA, nRNP/Sm, SS‐A | − |
| Patient 15/f | SLE | ANA, dsDNA | + |
| Patient 16/m | SLE | ANA, dsDNA, histones, nucleosomes | + |
SLE = systemic lupus erythematosus; ANA = anti‐nuclear autoantibodies; f = female; m = male; dsDNA = double‐stranded DNA; aCL = anti‐cardiolipin antibody; b2GPI = beta‐2‐glycoprotein I.
Human T, and to a lower extent B cells, express ANX A1 on their cell surface
Non‐in‐vitro‐activated PBMCs from healthy donors and lupus patients were analysed by flow cytometry. A high number (21·3% in the representative example) of T cells from patients displayed ANX A1 on their surface, while only 4·1% of the T cells from volunteers expressed the same protein. Both CD4+ and CD8+ T cell subpopulations retained approximately the same considerable difference of ANX A1 expression between SLE patients and healthy donors. Human B cells showed a low percentage of expression of ANX A1, ranging from 1 to 3% for patients, and less than 1% for healthy volunteers (Fig. 1a). No positive cells were found using biotinylated control antibody.
Figure 1.
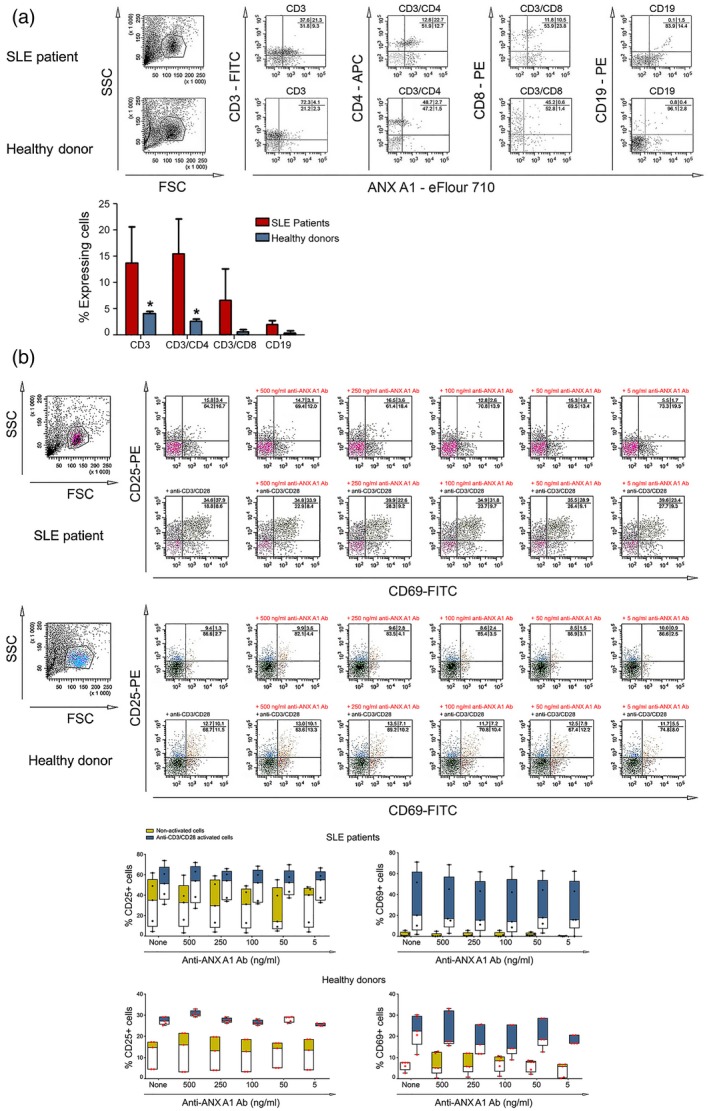
Flow cytometric analyses. (a) Detection of annexin A1 (ANX A1) surface expression. (b) Expression of T cell activation markers is affected by anti‐ANX A1 antibody. Representative data of five experiments are shown (left parts). The extracted results from all experiments are presented graphically (right parts). The data from five systemic lupus erythematosus (SLE) patients and five healthy donors are represented as mean ± standard deviation (n = 5, *P < 0·05).
Anti‐ANX A1 antibody modulates T cell activation marker expression
The effects of the anti‐ANX A1 antibody on T cell activation was investigated using non‐activated or plate‐bound anti‐CD3/CD28 antibody‐activated PBMCs from healthy donors and SLE patients. The cells were incubated in the presence of different concentrations of the anti‐ANX A1 or control antibodies and CD25+/CD69+ expression was registered by flow cytometry. FACS analyses performed for activation markers showed a small reduction in the percentage of anti‐CD3/CD28 antibody‐activated T cells either from healthy donors or SLE patients (up to 5% for CD69+) (Fig. 1b). It is noteworthy that the separate basic levels of CD25+ and CD69+ were at least two times higher in SLE patients compared to healthy donors.
A weak suppression of CD25+ expression of T cells was observed after incubation of PBMCs from SLE patients with anti‐ANX A1 antibody without CD3/CD28 activation, while no change in the activation markers expression was found among healthy donors or after treatment with control antibody (data not shown).
Influence of the anti‐ANX A1 antibody on T cell proliferation in vitro
In order to investigate the inhibitory effect of ANX A1 on cell proliferation, PBMCs from lupus patients and healthy donors were treated in vitro with different concentrations of anti‐ANX A1 or control antibodies with or without anti‐CD3/CD28 antibody activation. The ANX A1 antibody treatment of pre‐activated PBMCs from lupus patients had a weak inhibitory effect achieved with 250 ng/ml, while the same suppressive effect was obtained with 500 ng/ml in samples without anti‐CD3/CD28 stimulation compared to the ANX A1 untreated cells (Fig. 2a, left panel). The anti‐ANX A1 antibody did not significantly influence T cell proliferation of healthy donor cells (Fig. 2a, right panel).
Figure 2.
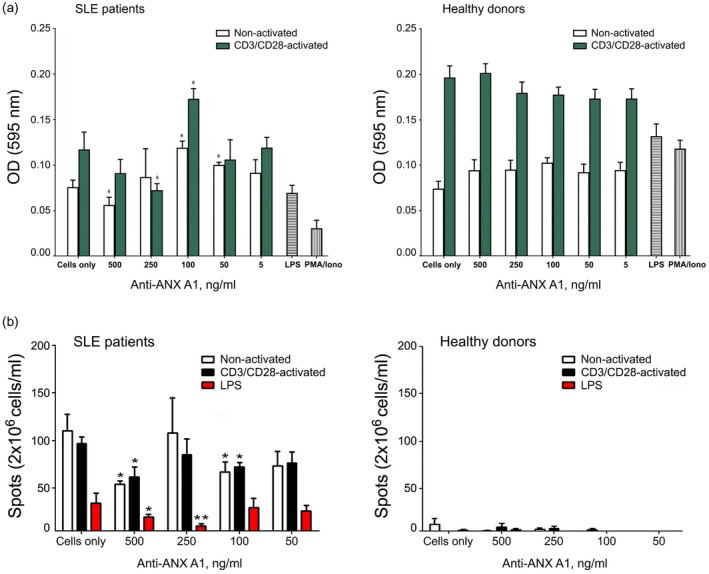
Incubation of peripheral blood mononuclear cells (PBMCs) from systemic lupus erythematosus (SLE) patients and healthy donors with anti‐ annexin A1 (ANX A1) antibody suppresses pathological T and B cells in vitro. Anti‐CD3/CD28 stimulated or non‐activated PBMCs from SLE patients and healthy donors were cultured in the presence of different concentrations of anti‐ANX A1 antibody. All samples were triplicated and average values were used for analysis. Results are expressed as the mean ± standard deviation of triplicate assays [two‐way analysis of variance (anova) test; *P < 0·05; **P < 0·01]. Data are representative of four independent experiments. (a) Anti‐ANX A1 antibody inhibits peripheral blood mononuclear cell (PBMC) proliferation. The samples were compared to untreated proliferating cells. (b) Anti‐ANX A1 antibody administration decreases the number of anti‐dsDNA antibody‐secreting plasmocytes in an enzyme‐linked immunospot (ELISPOT) assay. The number of spots in the test wells was compared with the number of spots in control wells containing medium‐only cultured splenocytes.
Incubation of PBMCs from SLE patients or healthy donors in the presence of different concentrations of control antibody did not affect cell proliferation (data not shown).
Anti‐dsDNA IgG antibody‐secreting plasmocytes are affected by anti‐ANX A1 antibody
The effects of the anti‐ANX A1 antibody treatment on the number of anti‐dsDNA antibody‐producing plasmocytes were assessed using the ELISPOT assay. A non‐concentration‐dependent reduction in the number of anti‐dsDNA‐secreting plasmocytes was observed for all tested groups of cells (non‐activated, anti‐CD3/anti‐CD28 activated and LPS stimulated) treated with anti‐ANX A1 antibody. Weak differences were observed when the cells were stimulated with anti‐CD3/CD28 antibodies. The strongest inhibitory effect was achieved for non‐activated (with 500 ng/ml anti‐ANX A1 antibody) and LPS‐stimulated PBMCs (with 250 ng/ml anti‐ANX A1 antibody) (Fig. 2b, left panel). No reduction in the number of plasmocytes was found using the control antibody (data not shown).
No dsDNA‐specific plasma cells were detected using PBMCs isolated from healthy donors (Fig. 2b, right panel).
Anti‐ANX A1 antibody increased the percentage of apoptotic T and B lymphocytes
The role of anti‐ANX A1 antibody on apoptosis of both T and B lymphocytes was studied using PBMCs from SLE patients and healthy volunteers. We cultured the cells in the presence of increasing concentrations of anti‐ANX A1 or control antibodies and the surface expression of phosphatidylserine and cell permeability was measured.
No modulatory effect on apoptosis was registered in non‐pre‐activated T cells. Although CD3/CD28 pre‐activated T cells from the SLE patients did not show a high level of apoptosis, some concentrations of anti‐ANX A1 antibody induced the process, reaching values that in some patients were twice as high as those in a control sample. Interestingly, although human B cells tend to express low amounts of outer membrane‐bound ANX A1, the pro‐apoptotic effect of the targeting antibody was relatively high (Fig. 3).
Figure 3.
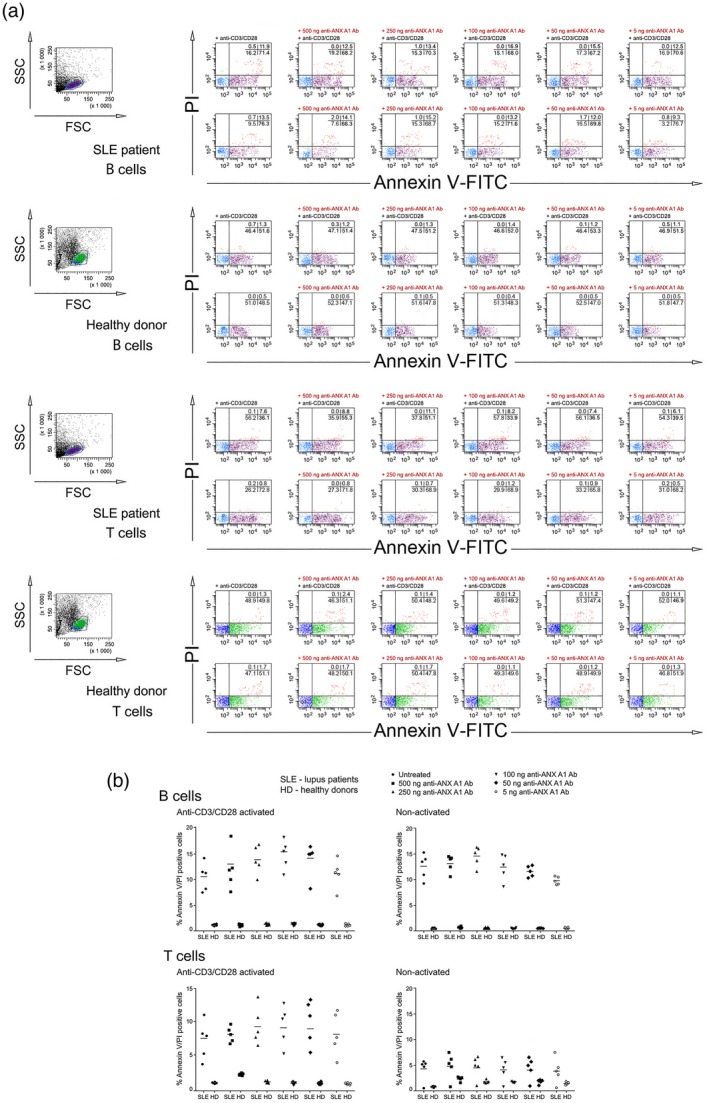
Anti‐annexin A1 (ANX A1) antibody induces T cell apoptosis. (a) Apoptosis was analysed by flow cytometry. The percentage of stained cells is shown in each quadrant. Representative data of five experiments are shown. (b) The extracted results from all experiments are presented graphically and the percentage of apoptotic double‐positive [annexin V‐fluorescein isothiocyanate (FITC)/propidium iodide] is shown. Each symbol represents an individual subject; values are the mean ± standard deviation of five independent experiments using five systemic lupus erythematosus (SLE) patients and five healthy donors; horizontal lines show the median.
The anti‐ANX A1 antibody did not have an effect on the apoptosis of T and B cells from a healthy donor (Fig. 3), while the control antibody did not affect cell apoptosis in any experiment (data not shown).
Influence of anti‐annexin A1 treatment on serum anti‐dsDNA IgG levels and proteinuria
The in‐vivo effects of the anti‐ANX A1 antibody were studied in humanized female NSG mice. Isolated PBMCs from SLE patients or healthy donors were used for the humanization of 40 NSG mice. Half these animals served as a control antibody‐treated group and the other half were treated with anti‐ANX A1 antibody, as described in the Materials and methods section (Fig. 4).
Figure 4.
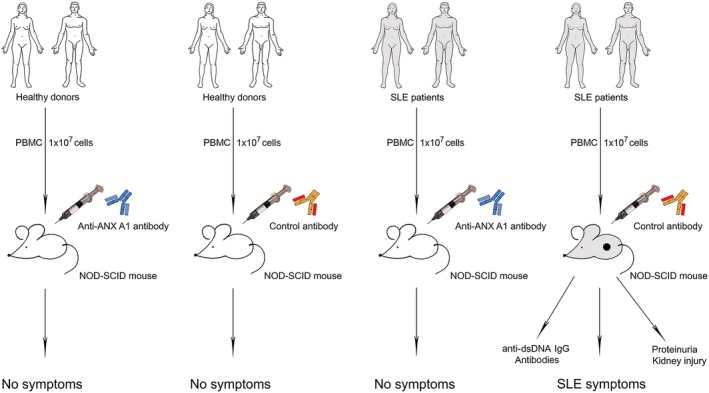
Scheme of peripheral blood mononuclear cell (PBMC) transfer from systemic lupus erythematosus (SLE) patients and healthy donors to non‐obese diabetic‐severe combined immunodeficient (NOD‐SCID) gamma (NSG) mice.
The administration of anti‐ANX A1 antibody to SLE‐PBMC‐transferred mice resulted in a profound decrease of serum anti‐dsDNA antibodies for almost the entire period of treatment (Fig. 5a, upper panel). This effect was strongly manifested between the 41st and 76th days after the cell transfer. Furthermore, individual analysis of the proteinuria showed that some mice were positively affected by the treatment. Collective data for proteinuria levels of all mice are given in Fig. 5a (lower panel). Although the difference between groups was consistent throughout the treatment, the period between days 18 and 42 showed a decrease in albuminuria of anti‐ANX A1‐treated mice.
Figure 5.
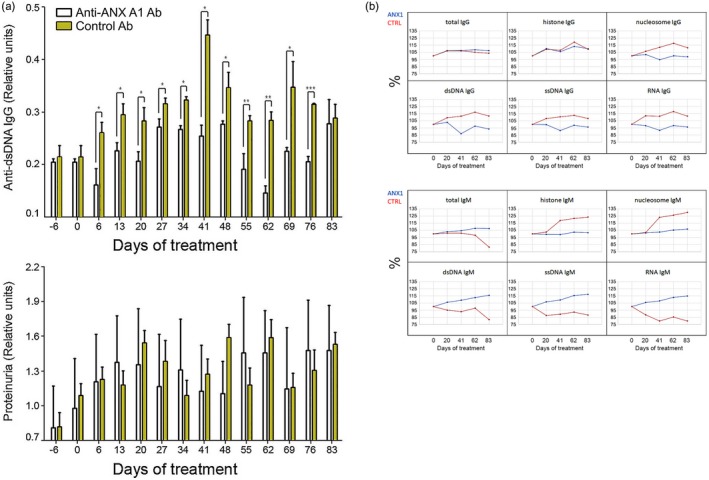
Treatment of humanized non‐obese diabetic‐severe combined immunodeficient (NOD‐SCID) gamma (NSG) mice with anti‐annexin A1 (ANX A1) antibody suppresses the appearance of various lupus immunoglobulin (Ig)G antibodies and of proteinuria. A representative of three independent experiments is shown. All samples were triplicated and average values were used for analysis. Mean ± standard deviation values were calculated for each group; P‐values were calculated using the two‐way analysis of variance (anova) test (*P < 0·05; **P < 0·01; ***P < 0·001) in comparison to control antibody‐treated mice. (a) The anti‐ANX A1 antibody administration prevented the appearance of high titres of IgG anti‐dsDNA antibodies (upper panel) and resulted in a non‐significant change of albuminuria (lower panel). (b) The same treatment of the group influenced various lupus IgG and IgM autoantibody levels measured by microarrays.
The animals transferred with PBMC from healthy donors did not exhibit any pathological symptoms, and no effect was observed after anti‐ANX A1 antibody treatment.
Anti‐ANX A1 administration reduces various serum autoantibody levels
Pooled sera from anti‐ANX A1 antibody‐treated or control antibody‐injected NSG mice humanized with PBMCs from lupus patients were tested for different autoantibodies on autoantigen microarrays comprising lupus‐relevant antigens. Autoreactive IgG antibodies against dsDNA showed significantly reduced levels in sera from treated mice compared to animals treated with control antibody (Fig. 5b). Moreover, the therapy resulted in lower anti‐ssDNA, anti‐RNA, anti‐histone and anti‐nucleosome IgG levels compared to the control ANX A1‐untreated animals. It is noteworthy that anti‐dsDNA, anti‐ssDNA and anti‐RNA IgM levels, reported in several papers to have a protective role in autoimmune conditions, were higher in the sera of treated animals. The anti‐histone and anti‐nucleosome IgM antibody response was stronger in the control group.
Cytokine measures
The pooled sera from humanized NSG mice from the above experiment were also used to determine whether anti‐ANX A1 administration affected IFN‐γ, IL‐4 and IL‐10 serum levels. Serum IFN‐γ of SLE‐PBMC‐transferred mice was significantly influenced at the beginning and end of treatment only (Fig. 6). IL‐4 levels were not considerably affected except on day 62, when a significantly lower value was registered compared to control animals, similar to IFN‐γ results. A sharp increase in IL‐10 levels was observed after anti‐ANX A1 introduction, while the level of control antibody‐treated animals was constantly high after cell transfer. Cytokines of the control group of animals transferred with PBMCs from healthy volunteers were not affected by anti‐ANXA A1. The control antibody treatment had no effect on either group and cytokine levels were under the detectable limits (data not shown).
Figure 6.
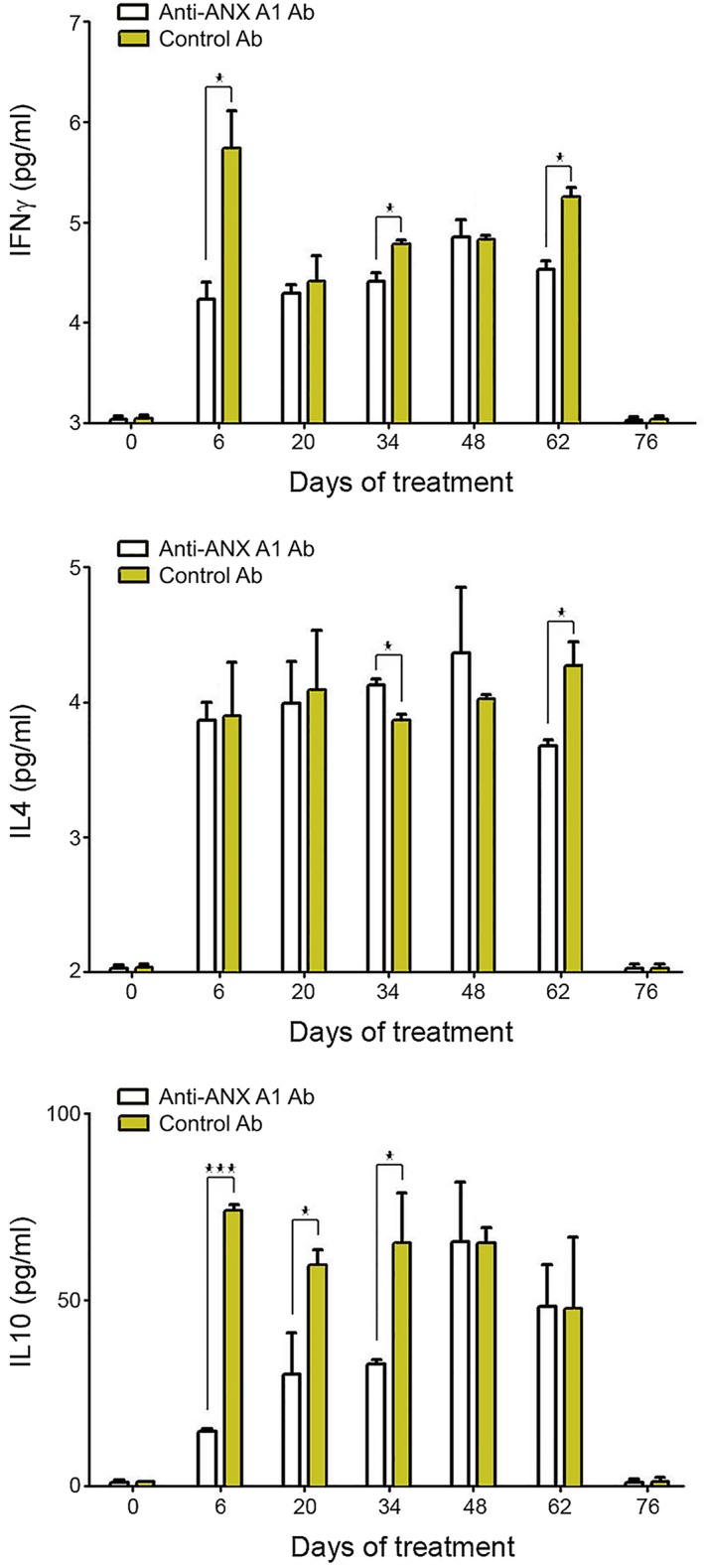
Anti‐annexin A1 (ANX A1) antibody treatment of humanized non‐obese diabetic‐severe combined immunodeficient (NOD‐SCID) gamma (NSG) mice affects cytokine production. Serum levels of interleukin (IL)‐4, IL‐10 and interferon (IFN)‐γ in all groups were measured by sandwich enzyme‐linked immunosorbent assay (ELISA). A representative of three independent experiments is shown. All samples were triplicated and average values were used for analysis. Mean ± standard deviation values were calculated for each group; P‐values were calculated using the two‐way analysis of variance (anova) test. (*P < 0·05) in comparison to phosphate‐buffered saline (PBS)‐treated controls.
The anti‐ANX A1 antibody treatment decreases disease symptoms in humanized NSG mice
At the end of the in‐vivo experiments, kidneys from all animals were removed for histopathological and immunofluorescent analysis (Fig. 7). All control antibody‐injected SLE‐PBMC‐transferred NSG mice had massive mesangial glomerular IgG‐containing immune complex depositions, demonstrated by immunofluorescence of the paraffin kidney sections. The kidneys from the same mice presented segmental mesangial cell hyperplasia within the glomeruli, mononuclear infiltration and histological injuries (score 1). At the same time, the renal histology of anti‐ANX A1 antibody‐treated mice was preserved (score 3) and significantly fewer cell infiltrates were observed. The anti‐ANX A1 antibody‐injected SLE‐PBMC‐transferred NSG animals also had no immune complex depositions.
Figure 7.
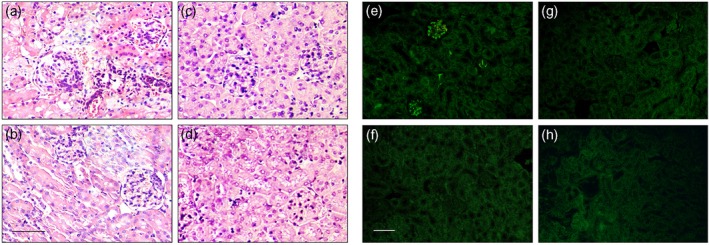
Histological analyses. Paraffin‐embedded kidney sections of humanized non‐obese diabetic‐severe combined immunodeficient (NOD‐SCID) gamma (NSG) mice stained with haematoxylin and eosin. Animals transferred with peripheral blood mononuclear cells (PBMCs) from systemic lupus erythematosus (SLE) patients and treated with control antibody (a) or anti‐annexin A1 (ANX A1) antibody (b). Mice transferred with PBMCs from healthy donors and treated with control antibody (c) or anti‐ANX A1 antibody (d). Scale bar, 200 μm. Representative images are shown. Original magnification×400. Immunofluorescence analysis of immunoglobulin (Ig)G deposition in the glomeruli of mice transferred with PBMCs from SLE patients and treated with control antibody (e) or anti‐ANX A1 antibody (f). Mice transferred with PBMCs from healthy donors and treated with control antibody (g) or anti‐ANX A1 antibody (h). Paraffin kidney sections from five mice per group were stained with fluorescein isothiocyanate (FITC)‐labelled goat anti‐mouse IgG. Representative images are shown. Scale bar, 200 μm. Original magnification × 250.
Control antibody‐injected and anti‐ANX A1 antibody‐treated NSG mice transferred with PBMCs from healthy donors did not exhibit kidney damage and IgG immune complex depositions.
Discussion
The autoimmune process is an unregulated immune response of recognizing self‐structures as harmful substances, as a result injuring normal cells and tissues. SLE is a prototype autoimmune syndrome characterized by the development of autoantibodies to a wide range of nuclear antigens which form pathological immune complexes. Many different cell types have an independent contribution during lupus development, but autoreactive B and T lymphocytes form the most effective tandem for breaking T/B cell tolerance to autoantigens and switching on the autoimmune cascade of inflammation. Both cell types exhibit regulatory and effector functions in accelerating the disease progression and SLE pathogenesis 1. The successful interruption of T/B cell autoantigen presentation, inflammatory cytokines and autoantibody release by disturbing T/B communication could promote a suppression of lupus symptoms.
During disease progression, SLE represents a number of abnormalities concerning lymphocyte activation and proliferation, cytokine level alteration and surface marker expression, e.g. B and T cell surface expression and secretion of ANX A1. Normally, ANX A1 is localized within the cytoplasm of T and B lymphocytes, which exhibit low surface expression.
Several new studies have reported increased ANX A1 expression and high serum levels of the same protein in SLE patients, combined with high levels of anti‐ANX A1 autoantibodies, which is itself a paradox by suggesting a role of ANX A1 in autoimmunity. Some authors suggest that the idea involving high serum ANX A1 levels is a compensation to anti‐ANX A1 antibody over‐production, which strongly correlates with the appearance of specific antibodies – biomarkers for SLE activity, such as anti‐dsDNA 26. Conversely, without post‐translational modifications ANX A1 is not immunogenic, and the high levels of circulating protein cannot explain the development of autoantibodies against it.
The precise mechanism of ANX A1 action on innate and adaptive immunity is still under discussion. Within the frame of innate immunity, ANX A1 has multi‐functional roles for control and resolution of inflammation by negative regulation of neutrophil activity, and its absence leads to prolonged inflammation 10, 12, 27. It is involved in the resolution of inflammation by induction of neutrophil apoptosis 11, reduced proinflammatory Toll‐like receptor activation on dendritic cells, promoting tolerogenic cell characteristics [reduced expression of CD80, CD86, major histocompatibility complex (MHC) class II and low secretion of tumour necrosis factor (TNF) and IL‐12], and acts as a chemoattractant for monocytes, aiding necrotic debris removal and reduction of the exposure time for molecules that potentially stimulate the autoimmune reaction 28.
More disputable are the ANX A1 effects on T and B lymphocytes and adaptive immunity in general. The main reason for the contradictory interpretations is the low level of surface ANX A1 expression by T cells 16, 17. Nevertheless, we found that more than 20% of SLE patients’ T cells and only 4% of healthy donors’ T cells expressed ANX A1 on the outer membrane leaflet. We studied the effect of anti‐ANX A1 in the Murphy Roths large/lymphoproliferation (MRL/lpr) and pristane‐induced lupus models, and discovered that ANX A1 is also expressed on the surface of murine B cells, reaching significant levels for the BALB/c mice (up to 20% of splenocytes of animals with induced SLE and approximately 10% of healthy animals) 19. Contrary to our murine experiments, human B cells exhibited low expression of ANX A1 (1–3%) in SLE patients and less than 1% in healthy donors. Despite the low percentage of ANX A1 expression from human B cells, remarkable differences were observed in both groups.
The proinflammatory effect of ANX A1 is promoted by stimulation of T cell differentiation to Th1 subtype and increased IFN‐γ synthesis, which is a result of the cooperation between ANX A1 receptor (ALXR) signalling and T cell receptor activation 26. In‐vitro results linked enhanced T cell receptor signalling and transcription activation after T cell stimulation with exogenous ANX A1 17. In a mouse model of collagen‐induced arthritis, treatment with ANX A1 promoted the Th1 cell profile and suppressed the Th2 cell subtype, which mediated intensive joint inflammation 29.
An important limitation for strong adaptive immunity control by ANX A1 could be the weak ALXR expression by T cells and even lower expression by B cells 9, 12. ANX A1 participates in several signalling pathways, among them up‐regulation of sphingosine‐1‐phosphate phosphatase (SGPP2), an enzyme involved in the degradation of sphingosine‐1‐phosphate (S1P), and the up‐regulation of jagged 1 protein (JAG1) genes after ANX A1–ALXR engagement on human cells 13, 15. Both pathways are involved in T cell maturation, differentiation and migration from lymphoid organs, but not in B cell development, suggesting an indirect ANX A1 impact to B lymphocytes via T cell activation, although a direct B cell effect is also possible 16, 30, 31.
A correlation in T cell activation, inhibition of cell proliferation and autoantibody production has been found in human and mouse lupus after in‐vitro treatment with anti‐ANX A1 antibody. During lupus progression, over‐activated and proliferating self‐reactive T cells stimulate respective B cell clones, leading to generation of plasmocytes producing autoantibodies with a wide range of self‐specificity. Incubation of PBMCs isolated from SLE patients with anti‐ANX A1 antibody reduced the expression of CD25+/CD69+ activation markers on activated T cells, suppressed the cell proliferation of dsDNA‐specific IgG antibody‐secreting plasma cells in a dose‐dependent manner, and induced B and T cells apoptosis of activated lymphocytes. Similar results were observed in pristane‐induced murine lupus using the same antibody, suggesting shared mechanisms of T cell activity regulation through the ANX A1–FPR2 signalling pathway 19. The accumulated data suggest a more complex relation between ANX A1, the monoclonal antibody and apoptosis, respectively, rather than a simple pro‐ or anti‐apoptotic effect 32.
Suppression of disease‐associated human B and T cells in vivo is a more ambitious task. Different levels of mouse immunodeficiency have been used in recent years to generate humanized mouse SLE models 33, 34. SCID mice lacking both B and T cells and engrafted with PBMCs from SLE patients have been used for lupus reconstitution and exhibition of disease symptoms 24.
In the present study, we explored a next level of immunodeficiency for humanized model host generation, namely NSG mice. These animals lack mature T, B and NK cells, which provides more advantages for human cell reconstitution 22, 25. The potential drawback of graft‐versus‐host disease (GVHD), mediated by the transferred human over‐activated T cells, exists without native resistance. Thus, the increased deficiency of this model makes it the most appropriate recipient for foreign cell transfer.
The balance between regulatory Th1/Th2 cytokines in the healthy immune system is upset in autoimmune diseases and the abnormal levels of some cytokines rebuild new configurations. The specific cytokine combination for lupus development still remains unclear, and a great variety of levels and combinations are found among SLE patients during different stages of the disease 35.
In our study, NSG mice engrafted with PBMCs from SLE patients exhibited IgG autoantibodies against a wide range of self‐antigens, unstable proteinuria and high serum levels of human IFN‐γ and IL‐10. The lupus symptoms were not affected after treatment with the control antibody, and severe glomerulonephritis and mononuclear cell infiltration were found in the kidneys of the animals.
The clonal activation of autoreactive plasmocytes is probably regulated by different mechanisms, so we cannot expect to down‐regulate the specific self‐response to all antigens 36. We investigated the autoantibody production specific to lupus‐relevant antigens by microarrays as a very sensitive analytical tool. Both ELISA and the microarray measurement confirmed the prevention of development of pathogenic IgG antibodies against nucleic acids and suppression of proinflammatory cytokine production after the anti‐ANX A1 antibody treatment of SLE‐PBMC humanized animals. In addition, the same treatment induced production of IgM self‐reactive antibodies against most of the autoantigens, suggesting introduction of tolerogenic effect after T cell activation disfunction 37. It remains to be clarified why anti‐histone antibodies show a different behaviour and are not affected by anti‐ANX A1 treatment.
Abnormal IgG‐containing immune complexes and kidney damage were found in NSG mice engrafted with PBMCs from SLE patients. The control animals exhibited massive glomerular depositions of IgG‐containing immune complexes, kidney injury and mononuclear infiltrate, while the anti‐ANX A1 antibody‐treated group had a significant improvement in kidney histology without any notable immune complex depositions. We did not observe any different pathological symptoms than those resulting from the lupus, suggesting the absence of GVHD. No disease symptoms were found in the mouse groups transferred with PBMC from healthy donors and treated with anti‐ANX A1 or control antibodies.
Witnessing long‐term plasma persistence of anti‐ANX A1 antibody after low‐dose administration in the mice (0·2 μg/mouse) is unexpected. Theoretically, a high‐avidity antibody has multiple targets in vivo, but we expected to obtain a biological effect of this low dose due to interaction with the limited number of ANX A1‐expressing T cells, rather than the huge number of ANX A1‐positive innate cells.
We have already used the same approach (0·2 μg antibody/mouse) and a similar design of treatment (every 6 days) to explore the monoclonal anti‐ANX A1 antibody in a pristane‐induced mouse model of SLE in BALB/c mice, as well as in a lupus‐prone MRL/lpr model. This treatment suppressed disease symptoms and autoantibody production, protected kidney histology and prolonged survival compared to the control groups 19. Other authors have used even lower concentrations of antibody with the same specificity for the same period 38. The low‐dose treatment must also exclude any strong effect due to the Fc portion of the antibody.
Annexin A1 has multiple functions with contradictory effects dependent on tissue and cell diversity, but its roles as a Th1 cell promotor and Th2 cell profile suppressor is of great interest. The ANX A1 quantity‐dependent fine‐tuning of T cell activation is an additional mechanism for modulation of the adaptive immunity, being both a bridge and a regulator of the innate immunity functions.
The humanized NSG model of SLE presented here explores a novel approach for the suppression of autoreactive B and T cell cooperation during disease progression. Using a low‐dose monoclonal antibody against ANX A1 we restricted disease symptom progression and pathological kidney damage, thus providing insight into a new potential therapy for human SLE.
Disclosures
None of the authors has any potential financial conflict of interest related to this paper.
Author contributions
A. T., N. M., J. P. and D. K. designed the experiments. N. M., S. B., P. C., S. C., E. I.‐T., M. H., T. V. and D. K. performed the experiments. A. T., J. P. and N. M. analysed the data. N. M., P. C. and A. T. wrote and edited the paper.
Acknowledgements
This work was supported by the Bulgarian National Science Fund (grant DDVU 02/34) and a bilateral grant between Bulgarian Academy of Sciences and Hungarian Academy of Sciences (grant contract number SNK‐73/2013)]. We thank Professor Fulvio D’Acquisto (William Harvey Research Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK) and Associate Professor Alexander Shinkov (Department of Endocrinology, Medical Faculty, Medical University, Sofia, Bulgaria) for helpful discussion and ideas.
References
- 1. Tiffin N, Adeyemo A, Okpechi I. A diverse array of genetic factors contribute to the pathogenesis of systemic lupus erythematosus. Orphanet J Rare Dis 2013; 8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ugarte A, Danza A, Ruiz‐Irastorza G. Glucocorticoids and antimalarials in systemic lupus erythematosus an update and future directions. Curr Opin Rheumatol 2018; 30:482–9. [DOI] [PubMed] [Google Scholar]
- 3. Yildirim‐Toruner C, Diamond B. Current and novel therapeutics in treatment of SLE. J Allergy Clin Immunol 2012; 127:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lund FE. Cytokine ‐ producing B lymphocytes – key regulators of immunity. Curr Opin Immunol 2008; 20:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan VSF, Tsang HHL, Tam RCY, Lu L, Lau CS. B‐cell‐targeted therapies in systemic lupus erythematosus. Cell Mol Immunol 2013; 10:133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moulten VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res Ther 2011; 13:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T cells as therapeutic targets in SLE. Nat Rev Immunol 2010; 6:317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dahal LN, Basu N, Youssef H et al Immunoregulatory soluble CTLA‐4 modifies effector T‐cell responses in systemic lupus erythematosus. Arthritis Res Ther 2016; 18:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gavins FN, Hickey MJ. Annexin A1 and the regulation of innate and adaptive immunity. Front Immunol 2012; 3:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res 2016; 2016:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vago JP, Nogueira CR, Tavares LP et al Annexin A1 modulates natural and glucocorticoid‐induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol 2012; 92:249–58. [DOI] [PubMed] [Google Scholar]
- 12. Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol 2009; 9:62–70. [DOI] [PubMed] [Google Scholar]
- 13. Renshaw D, Montero‐Melendez T, Dalli J et al Downstream gene activation of the receptor ALX by the agonist annexin A1. PLOS ONE 2010; 5:e12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang YH, Toh ML, Clyne CD et al Annexin A1 negatively regulates IL‐6 expression via effects on p38 MAPK and MAPK phosphatase 1. J Immunol 2006; 177:8148–53. [DOI] [PubMed] [Google Scholar]
- 15. Pham TH, Okada M, Matloubian M et al S1P1 receptor signaling overrides retention mediated by Gαi‐coupled receptors to promote T cell egress. Immunity 2008; 28:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Young L‐H, Jeong Y‐S, Lee M et al Intracellular formyl peptide receptor regulates naïve CD4 T cell migration. Biochem Biophys Res Commun 2018; 497:226–32. [DOI] [PubMed] [Google Scholar]
- 17. D’Acquisto F, Paschalidis N, Raza K, Buckley CD, Flower RJ, Perretti M. Glucocorticoid treatment inhibits annexin‐1 expression in rheumatoid arthritis CD4+ T cells. Rheumatology 2008; 47:636–9. [DOI] [PubMed] [Google Scholar]
- 18. D’Acquisto F. On the adaptive nature of annexin‐A1. Curr Opin Pharmacol 2009; 9:521–8. [DOI] [PubMed] [Google Scholar]
- 19. Mihaylova N, Bradyanova S, Chipinski P et al Annexin A1 as a target for managing murine pristane‐induced systemic lupus erythematosus. Autoimmunity 2017; 50:257–68. [DOI] [PubMed] [Google Scholar]
- 20. Dimitrova I, Gesheva V, Nikolova K et al Target silencing of disease‐associated B‐lymphocytes by chimeric molecules in SCID model of pristane‐induced autoimmunity. Lupus 2010; 19:1261–71. [DOI] [PubMed] [Google Scholar]
- 21. Papp K, Végh P, Tchorbanov A et al Progression of lupus‐like disease drives the appearance of complement‐activating IgG antibodies in MRL/lpr mice. Rheumatology 2010; 49:2273–80. [DOI] [PubMed] [Google Scholar]
- 22. Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol 2012; 9:208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koboziev I, Jones‐Hall Y, Valentine JF, Webb CR, Furr KL, Grisham MB. Use of humanized mice to study the pathogenesis of autoimmune and inflammatory diseases. Inflamm Bowel Dis 2015; 21:1652–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kerekov N, Mihaylova N, Grozdev I et al Elimination of autoreactive B cells in humanized SCID mouse model of SLE. Eur J Immunol 2011; 41:3301–11. [DOI] [PubMed] [Google Scholar]
- 25. Brehm M, Racki W, Leif J et al Engraftment of human HSCs in nonirradiated newborn NOD‐scid IL2rγnull mice is enhanced by transgenic expression of membrane‐bound human SCF. Blood 2012; 119:2778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bruschi M, Petretto A, Vaglio A, Santucci L, Candiano G, Ghiggeri GM. Annexin A1 and autoimmunity: from basic science to clinical applications. Int J Mol Sci 2018; 19:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Solito E, Christian HC, Festa M et al Post‐translational modification plays an essential role in the translocation of annexin A1 from the cytoplasm to the cell surface. FASEB J 2006; 20:1498–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blume KE, Soeroes S, Keppeler H et al Cleavage of annexin A1 by ADAM10 during secondary necrosis generates a monocytic ‘find‐me’ signal. J Immunol 2012; 188:135–45. [DOI] [PubMed] [Google Scholar]
- 29. D’Acquisto F, Paschalidis N, Sampaio AL, Merghani A, Flower RJ, Perretti M. Impaired T cell activation and increased Th2 lineage commitment in annexin‐1‐deficient T cells. Eur J Immunol 2007; 37:3131–42. [DOI] [PubMed] [Google Scholar]
- 30. Odobasic D, Jia Y, Kao W et al Formyl peptide receptor activation inhibits the expansion of effector T cells and synovial fibroblasts and attenuates joint injury in models of rheumatoid arthritis. Int Immunopharmacol 2018; 61:140–9. [DOI] [PubMed] [Google Scholar]
- 31. Schepetkin I, Kirpotina L, Khlebnikov A, Cheng N, Ye R, Quinn M. Antagonism of human formyl peptide receptor 1 (FPR1) by chromones and related isoflavones. Biochem Pharmacol 2014; 92:627–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Su Y‐J, Cheng T‐T, Chen C‐J et al The association among leukocyte apoptosis, autoantibodies and disease severity in systemic lupus erythematosus. J Transl Med 2013; 11:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gunawan M, Her Z, Liu M et al A novel human systemic lupus erythematosus model in humanised mice. Scient Rep 2017; 7:16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrade D, Redecha P, Vukelic M et al Engraftment of peripheral blood mononuclear cells from systemic lupus erythematosus and antiphospholipid syndrome patient donors into BALB‐RAG‐2–/–IL‐2Rγ–/– mice. Arthritis Rheum 2011; 63:2764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lourenço EV, La Cava A. Cytokines in systemic lupus erythematosus. Curr Mol Med 2009; 9:242–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shlomchik M. Sites and stages of autoreactive B cell activation and regulation. Immunity 2008; 28:18–28. [DOI] [PubMed] [Google Scholar]
- 37. Lobo PI. Role of natural IgM autoantibodies (IgM‐NAA) and IgM anti‐leukocyte antibodies (IgM‐ALA) in regulating inflammation. Curr Top Microbiol Immunol 2017; 408:89–117. [DOI] [PubMed] [Google Scholar]
- 38. Piras G. From the bench to the pipeline: testing the immunosuppressive potential of novel therapy targeting Annexin A1. Queen Mary University of London, 2014. Available at: https://books.google.bg/books?xml:id=APsxvwEACAAJ (accessed 13 July 2016). [Google Scholar]