

Abstract
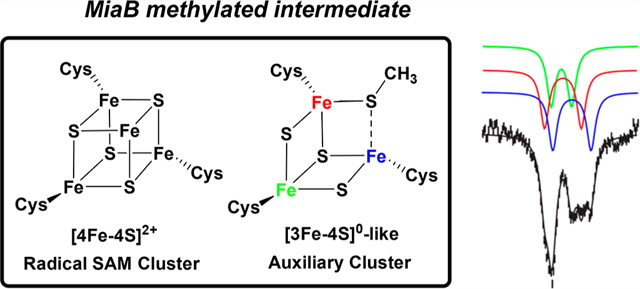
The enzyme MiaB catalyzes the attachment of a methylthio (−SCH3) group at the C2 position of N6-(isopentenyl)adenosine (i6A) in the final step of the biosynthesis of the hypermodified tRNA nucleotide 2-methythio-N6-(isopentenyl)adenosine (ms2i6A). MiaB belongs to the expanding subgroup of enzymes of the radical S-adenosylmethionine (SAM) superfamily that harbor one or more auxiliary [4Fe-4S] clusters in addition to the [4Fe-4S] cluster that all family members require for the reductive cleavage of SAM to afford the common 5′-deoxyadenosyl 5′-radical (5′-dA•) intermediate. While the role of the radical SAM cluster in generating the 5′-dA• is well understood, the detailed role of the auxiliary cluster, which is essential for MiaB catalysis, remains unclear. It has been proposed that the auxiliary cluster may serve as a coordination site for exogenously derived sulfur destined for attachment to the substrate or that the cluster itself provides the sulfur atom and is sacrificed during turnover. In this work, we report spectroscopic and biochemical evidence that the auxiliary [4Fe–4S]2+ cluster in Bacteroides thetaiotaomicron (Bt) MiaB is converted to a [3Fe-4S]0-like cluster during the methylation step of catalysis. Mössbauer characterization of the MiaB [3Fe-4S]0-like cluster revealed unusual spectroscopic properties compared to those of other well-characterized cuboidal [3Fe-4S]0 clusters. Specifically, the Fe sites of the mixed-valent moiety do not have identical Mössbauer parameters. Our results support a mechanism where the auxiliary [4Fe-4S] cluster is the direct sulfur source during catalysis.
Graphical Abstract
INTRODUCTION
The radical S-adenosylmethionine (SAM) (RS) superfamily of enzymes catalyzes a wide variety of reactions using a 5′-deoxyadenosyl 5′-radical (5′-dA•) intermediate, a potent oxidant derived from the reductive cleavage of SAM.1–6 The 5′-dA• typically functions to initiate radical-based catalysis by abstracting a hydrogen atom (H•) from a noncovalently bound substrate. In all instances studied to date, the reductive cleavage of SAM requires a redox-active [4Fe-4S]2+/1+ cluster, termed the RS cluster, which is ligated by three cysteinyl residues found in an almost universally conserved CxxxCxxC motif.7–9 The unique (noncysteinyl ligated) Fe ion of the RS cluster can be coordinated by SAM via SAM’s amino and carboxylate groups.10,11 The reduced RS [4Fe-4S]+ cluster initiates the reductive cleavage of SAM, yielding methionine, 5′-dA•, and the oxidized RS [4Fe-4S]2+ cluster. Recent work has provided evidence for the 5′-dA• intermediate,12,13 which has been shown to be stored as an organometallic species with a C5′–Fe bond.14
Methylthiotransferases (MTTases) are a subfamily of RS enzymes that install a methylthio (−SCH3) group on their substrates. The RS MTTase MiaB, the subject of this study, catalyzes the methylthiolation of N6-isopentenyl adenosine (i6A) nucleotides found at position 37 in tRNAs that decode mRNA codons containing a leading uridine, giving rise to a C2-methylthio-N6-isopentenyl adenosine (ms2i6A) product (Scheme 1).15 The ms2i6A modification imparts enhanced base-pairing interactions between mRNA codons and the cognate tRNA, decreasing slippage and maintaining the ribosomal reading frame during translation.16−20 The reaction catalyzed by MiaB requires the dimethylallyl modification of N6 of the target adenosine, A37, which in Escherichia coli is installed by the enzyme MiaA using dimethylallyl pyrophosphate as a substrate.21−23 Additionally, methylthiolation by CDKAL1, the human ortholog of the MTTase MtaB, which methylthiolates N6-threonylcarbamoyladenosine (t6A) in tRNAs, has been implicated in insulin maturation and the development of type II diabetes.24−26 Furthermore, methylthiolation catalyzed by the human MiaB ortholog CDK5RAP1 has been linked to the development of neurodegenerative diseases via an undetermined mechanism.27−30 Lastly, the MTTase RimO methylthiolates the β carbon of aspartate 89 of the ribosomal S12 protein, a strictly conserved residue near the functional center of the ribosome.31,32 This reaction involves the stereoselective abstraction of the 3 pro-S H• of aspartate 89 with subsequent transfer of a methylthio group with inversion of configuration to give (3R)-methylthioaspartate.33
Scheme 1.
Reaction Catalyzed by MiaB
In addition to the RS MTTases, there are two well-studied RS enzymes that install sulfur onto unactivated carbon centers. These include biotin synthase (BioB), which installs one sulfur atom between C6 and C9 of dethiobiotin, creating the thiophane ring of the molecule,34 and lipoyl synthase (LipA), which installs two sulfhydryl groups, at C6 and C8, on an octanoyllysyl chain of a lipoyl carrier protein to create the lipoyl cofactor.35 BioB, LipA, and the MTTases (MiaB and RimO) have in common that they require two distinct Fe/S clusters in catalysis.6 The first cluster is the RS [4Fe-4S] cluster, which is used for generation of the 5′-dA•, while the second cluster is termed the auxiliary (Aux) cluster. BioB harbors an Aux [2Fe-2S] cluster,36,37 while LipA, RimO, and MiaB contain an Aux [4Fe-4S] cluster.32,38,39 In recent years, it was shown that the Aux [2Fe-2S] cluster in BioB and the Aux [4Fe-4S] cluster in LipA are the immediate sources of sulfur to be incorporated into the product.40−44 However, for the MTTases, the functional role of the Aux cluster remains less well defined.
Catalysis by MiaB appears to take place in two distinct half-reactions (Scheme 1).45,46 In the first stage, which does not require a low-potential reductant, a methyl group is transferred from a SAM molecule to a protein-associated sulfur species via a polar nucleophilic attack to generate a methylthio intermediate.6,45−47 This intermediate is labile in the presence of acid and base and is vulnerable to chaotropic agents, suggesting that the methyl acceptor is an Fe/S cluster rather than a side chain of an amino acid residue.45 Consistent with this assignment, treatment of the methylated protein with acid results in formation of methanethiol. When the intermediate is formed using S-adenosyl-[methyl-2H3]-methionine (d3-SAM) and then isolated and added to a reaction containing a low-potential reductant and unlabeled SAM, a burst of deuterium-containing product is observed before slower formation of unlabeled product.45 Similarly, when methanethiol or meth-aneselenol is included in the MiaB or RimO reactions, a methylthio or methylseleno group derived from these compounds is incorporated intact into ms2i6A (mse2i6A) of tRNA or aspartate 89 of a synthetic peptide substrate.45,48 Electrochemical characterization of Bt MiaB and Tm RimO suggests that the Aux [4Fe-4S] cluster is transformed to a state with an unusually low reduction potential for a [4Fe-4S]2+/1+ redox pair as a direct result of methylation.46
The auxiliary cluster in the MTTases is ligated by three cysteine residues, raising the possibility that the unique noncysteine-ligated Fe could serve as a coordination site for exogenously derived sulfur destined for insertion into the substrate.39,48,49 The crystal structure of holo Tm RimO, obtained in the absence of SAM and its polypeptide substrate, revealed a pentasulfide bridge connecting the unique Fe ions of the RS and Aux clusters.48 While the presence of the pentasulfide bridge is likely an artifact resulting from chemical reconstitution of the Fe/S clusters, given that it occupies the expected binding site for SAM and the polypeptide substrate, its observation suggests the propensity of the unique Fe site of the Aux cluster to bind exogenous (poly)sulfide. Subsequently, a mechanism was proposed involving a (poly)sulfide species bound exogenously to the auxiliary clusters of MiaB and RimO that could be capped by a methyl group transferred from SAM and incorporated into the methylated product.45,48
Recent crystallographic and spectroscopic investigation into the mechanism of the sulfur-inserting enzyme LipA suggests another possible mechanism for the MTTases in which the Aux cluster is sacrificed as the ultimate sulfur source. Mechanistic studies of LipA provided structural and spectroscopic evidence for the disassembly of the Aux cluster during catalysis. In LipA’s first half reaction, a substrate-Fe/S cluster cross-linked intermediate species is generated. Analysis of this species by Mössbauer spectroscopy revealed the presence of a [3Fe-4S]0-like cluster that contains 3 iron ions, 3 sulfide ions, and 1 C6-mercapto-octanoyl ligand, formally affording a [3Fe-3S-1SR]−1 species.42 The cross-linked species and the [3Fe-4S]0-like cluster were subsequently confirmed by X-ray crystallography.50
In this work, we present the Mössbauer-spectroscopic characterization of Bacteroides thetaiotaomicron (Bt) MiaB during the steps leading to the formation of the methylated intermediate. The results reveal conversion of the Aux [4Fe-4S]2+ cluster to a [3Fe-4S]0-like cluster in the absence of a low-potential reductant. The [4Fe-4S]2+ to [3Fe-4S]0-like cluster transformation occurs on the same time scale as that of SAH formation and is only observed under conditions that promote the methylation reaction. Furthermore, the [3Fe-4S]0-like intermediate is chemically and kinetically competent to proceed to product ms2i6A tRNA. The electronic structure of the MiaB [3Fe-4S]0-like cluster is different from that of other spectroscopically characterized cuboidal [3Fe-4S]0 clusters. A potential sacrificial role for the auxiliary [4Fe-4S]2+ cluster as the direct sulfur source will be discussed in light of the observed unusual electronic structure of the [3Fe-4S]0-like cluster. Although the [3Fe-4S]0-like clusters in MiaB and LipA are likely generated by different mechanisms (polar and radical, respectively), its observation in two mechanistically different radical SAM enzymes suggests a potential role for this type of Fe/S cluster as a common intermediate in radical SAM enzymes that catalyze sulfur insertion and that contain auxiliary [4Fe-4S]2+ clusters.
RESULTS
Assessment of the Time Scale of the Methylation Reaction by Bt MiaB.
To facilitate the preparation of Bt MiaB samples for characterization by Mössbauer spectroscopy, the time scale of the MiaB-catalyzed methylation step was determined by liquid chromatography coupled to analysis by mass spectrometry (LC/MS) as previously described.45 Upon incubating Bt MiaB with i6A-containing tRNA and SAM, the formation of SAH was observed to reach a plateau (88 μM, ~0.5 equiv) within 20 min with an initial rate for SAH formation of 86 μM min−1 (Figure 1A). No 5′-dAH or ms2i6A was detected under these reaction conditions, consistent with the requirement for a low-potential reductant and tRNA substrate to initiate the reductive cleavage of SAM.45 This state, herein referred to as the methylated intermediate, is chemically and kinetically competent because addition of sodium dithionite after a 20 min incubation with SAM led to the formation of ms2i6A and 5′-dAH with rates of 28 and 36 μM min−1, respectively, without any lag phase (Figure 1B).
Figure 1.
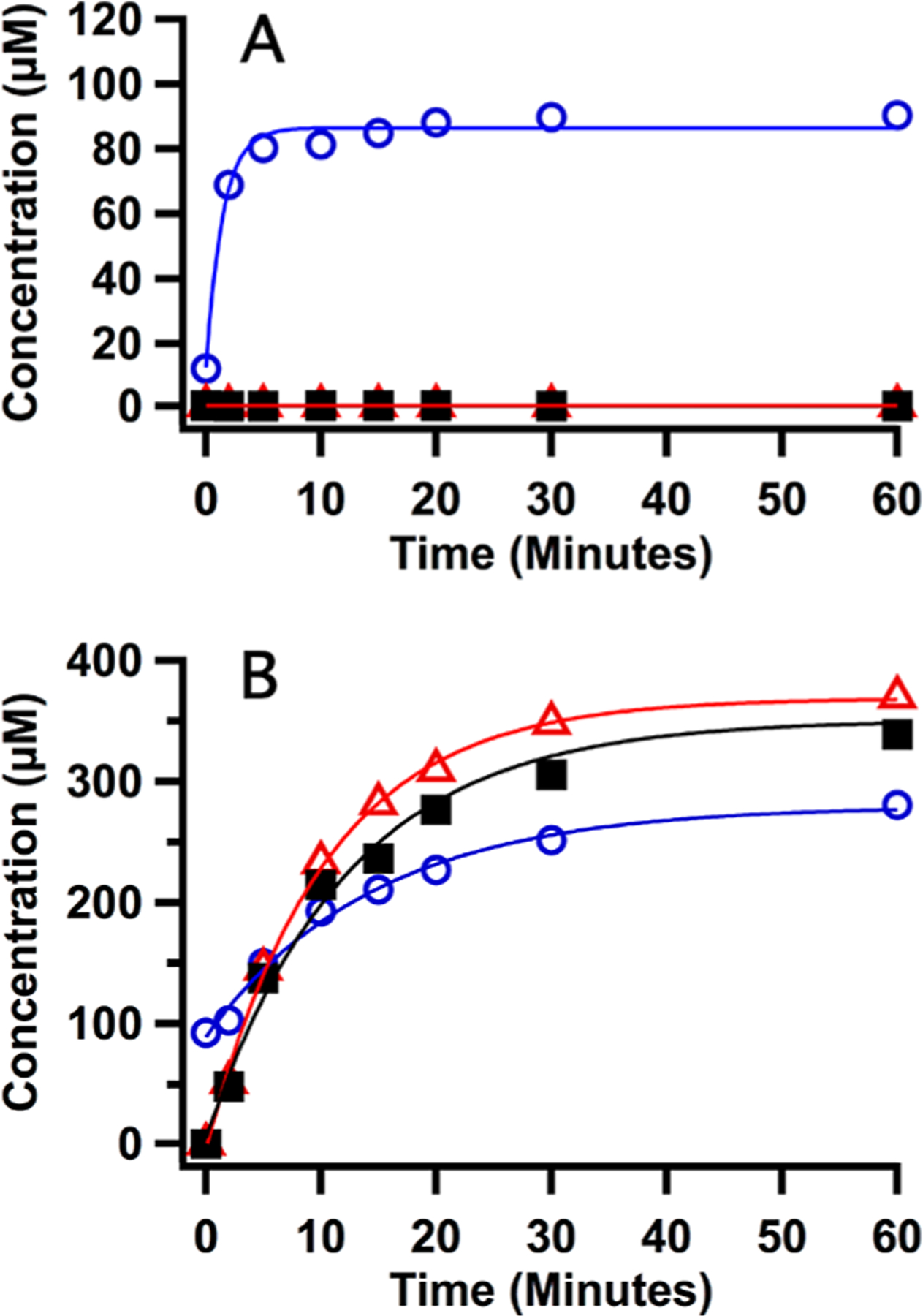
LC/MS analysis of the progress of the Bt MiaB reaction.(A) Reconstituted Bt MiaB (165 μM) was incubated with SAM (600 μM) and tRNA substrate i6A (330 μM) for varying reaction times.(B) Reconstituted MiaB (165 μM) was incubated with SAM (600 μM) and tRNA substrate i6A (330 μM) for 20 min and then reacted with sodium dithionite for varying reaction times. Reaction was monitored by formation of the products SAH (blue circles), 5′-dAH (red triangles), and ms2i6A (black squares). Solid lines are fits to a first-order exponential using parameters mentioned in the text.
Mössbauer-Spectroscopic Evidence for a [3Fe-4S]0-Like Cluster in the Methylated Intermediate of MiaB.
The 4.2-K/zero-field Mössbauer spectrum of a Bt MiaB sample prior to addition of SAM is shown in Figure 2A (black vertical bars). Analysis of the sample indicates a stoichiometry of 7.6 ±0.1 Fe per protein. Facilitated by the Mössbauer characterization of variants lacking either the RS or the Aux cluster (Figure S2 and associated text in Supporting Information), the spectrum of Bt MiaB prior to addition of SAM can be adequately simulated as a superposition of ~0.85 RS [4Fe-4S]2+ and ~0.80 auxiliary [4Fe-4S]2+ clusters per MiaB monomer. The typical features of a [2Fe-2S]2+ cluster (~0.45 cluster per MiaB monomer, ~12% of total 57Fe) and of a cuboidal [3Fe-4S]+ cluster (less than 0.04 cluster per MiaB monomer, ~1.5% of total 57Fe) are also observed by Mössbauer and EPR spectroscopies, respectively (Figures S1 and S2). However, their presence in the sample likely results from degradation of [4Fe-4S]2+ clusters.51,52 Other common EPR-active Fe/S clusters with a half-integer spin ground state (e.g., [2Fe-2S]+, [4Fe-4S]+, or [4Fe-4S]3+) are not observed. As expected for a sample containing almost exclusively Fe/S clusters with a diamagnetic ground state, the spectrum of Bt MiaB recorded in a weak, externally applied magnetic field of 53 mT is identical to that recorded in the absence of an applied magnetic field (Figure 2A, red line; the small fraction of [3Fe-4S]+ clusters is beyond the detection limit of Mössbauer spectroscopy).
Figure 2.
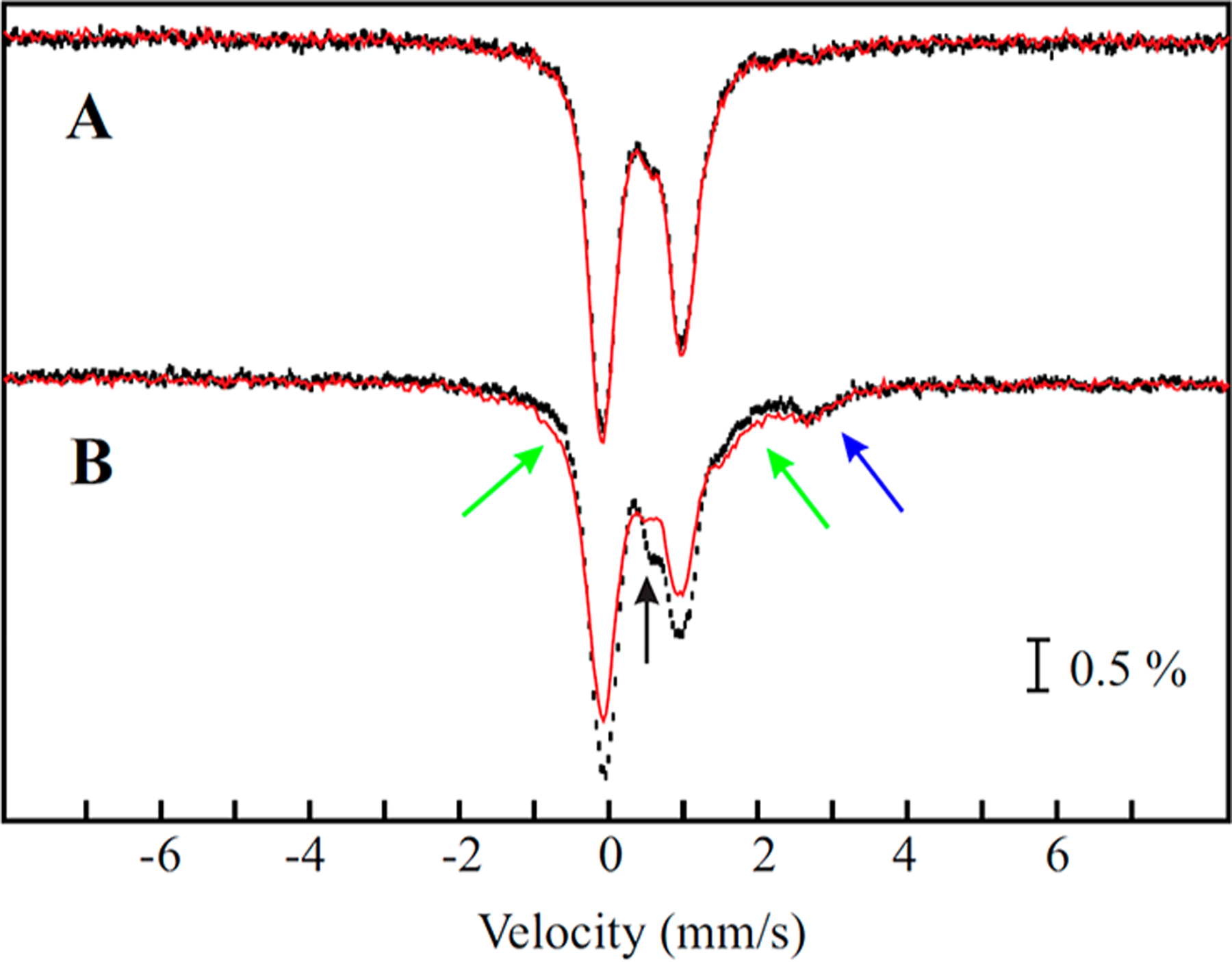
Comparison of Mössbauer spectra of reconstituted Bt MiaB collected at 4.2 K with either no applied magnetic field (black vertical bars) or in a 53 mT magnetic field oriented parallel to the γ beam (red lines) before (A) and after (B) incubation with SAM for 20 min. Black arrow indicates the increased absorption at ~0.6 mm/s. Blue arrow indicates the high-energy line of high-spin FeII. Green arrows indicate the broad unspecific absorption of the component with paramagnetic integer-spin ground state in the 53 mT spectrum.
For the characterization of the methylated intermediate, a sample intended for Mössbauer spectroscopy was prepared by incubating 165 μM Bt MiaB with 330 μM i6A tRNA and 600 μM SAM for 20 min. The conditions were chosen to allow maximum accumulation of SAH based on LC/MS analysis of a sample prepared under identical conditions (Figure 1A). The4.2-K/zero-field Mössbauer spectrum of this sample (Figure 2B, black vertical bars) reveals three distinct differences from that of reconstituted MiaB alone: (1) the prominent high-energy line of the quadrupole doublet associated with the [4Fe-4S]2+ clusters at a position of ~1.0 mm/s is reduced in intensity; (2) the intensity between the two most prominent lines at ~0.6 mm/s increases (Figure 2B, black arrow); and (3) a new absorption feature is observed at ~2.7 mm/s, a position typical for the high-energy line of a quadrupole doublet of high-spin (HS) FeII (blue arrow). The analysis of this spectrum (see below and Figure S3) reveals that this component has an isomer shift (δ) of ~1.3 mm/s and a quadrupole splitting parameter (ΔEQ) of ~2.8 mm/s, which are typical for N/O-coordinated HS FeII ions.53 Interestingly, the spectrum collected in an applied field of 53 mT (Figure 2B, red line) reveals that a fraction of the Fe exhibits unresolved magnetically split features extending from approximately −1.0 to +2 mm/s (Figure 2B, green arrows). This field-strength dependence is typical of a species with a paramagnetic, integer-spin ground state. The fact that treatment of MiaB with SAM results in a reduction of the intensity associated with [4Fe-4S]2+ clusters and yields a HS FeII species along with a new paramagnetic species in an ~1:3 ratio (vide infra) suggests that one [4Fe-4S]2+ cluster is converted to a 3Fe cluster with a paramagnetic integer spin ground state plus an FeII ion.
The spectral features associated with the 3Fe cluster are more easily discernible in the 0–53 mT difference spectrum (Figure 3A, vertical bars), wherein its features in a 53 mT field (broad and featureless, magnetically split absorption extending from −1 to +2 mm/s; see arrows) point upward, while its features in zero applied field (overlapping quadrupole doublets of the three Fe sites of the cluster) point downward. Importantly, the other cluster remains in the [4Fe-4S]2+ configuration, which does not exhibit any field-strength dependence due to its diamagnetic ground state. The [0–53 mT] difference spectrum of the 20 min sample qualitatively resembles that of the cuboidal [3Fe-4S]0-like auxiliary cluster in the E. coli LipA monothiolated intermediate with Stot = 2 (Figure S4),42 although the MiaB difference spectrum does not display the unique 2:1 intensity ratio of the quadrupole doublets associated with the valence-delocalized Fe22.5+ pair and the FeIII site without applied field.54
Figure 3.
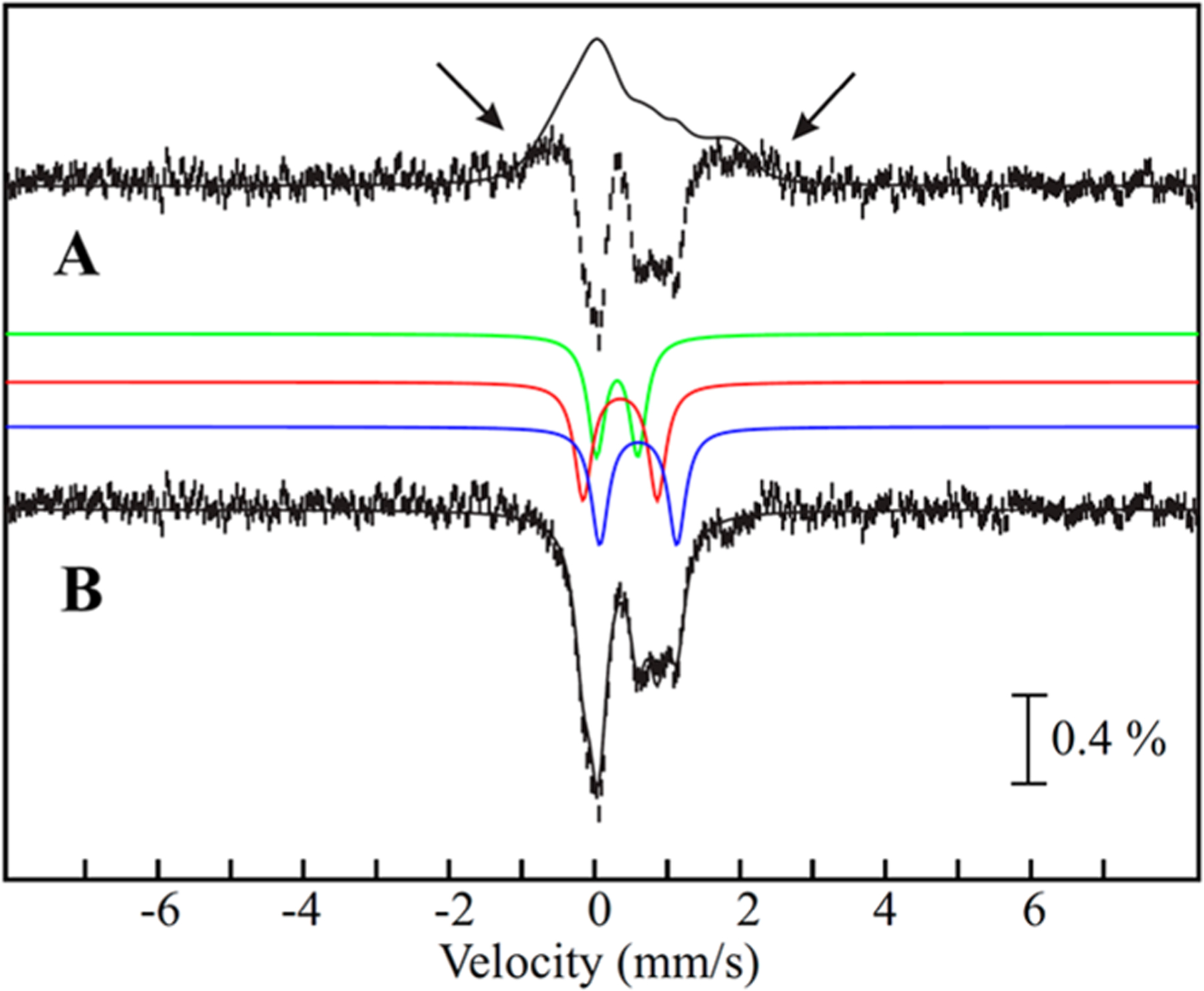
Analysis of the zero-field and 53 mT Mössbauer spectra of reconstituted Bt MiaB after incubation with tRNA and SAM for 20 min. (A) [0–53 mT] difference spectrum (vertical bars), and simulation of the MiaB [3Fe-4S]0-like cluster in 53 mT using the parameters listed in Table 2 and scaled to −27% of total 57Fe absorption (black line). Arrows indicate the onset of the broad absorption features arising from the magnetic hyperfine splitting of the MiaB [3Fe-4S]0-like cluster. (B) Zero-field reference spectrum of the MiaB [3Fe-4S]0-like cluster (vertical bars). Black line is the simulation of the MiaB [3Fe-4S]0-like cluster in zero field with parameters from Table 1, solution 1 (30% of total intensity). Individual contributions from the three sites are shown above the zero-field reference spectrum (site 1 (green), site 2 (red), site 3 (blue); 10% of total intensity each).
In order to determine the values of δ and ΔEQ of the three Fe sites of the intermediate, the zero-field reference spectrum of the MiaB [3Fe-4S]0-like cluster (Figure 3B, vertical bars) was generated by adding back a spin-Hamiltonian simulation (vide infra) of the [3Fe-4S]0-like cluster in a 53 mT magnetic field (Figure 3A, black line) to the difference spectrum (Figure 3A, vertical bars). We note that due to the broadness of the 53 mT spectrum, the relative amount of the simulation to be added back to generate the zero-field reference spectrum cannot be determined accurately. We therefore generated several reference spectra by adding back varying amounts (21–36% of total 57Fe absorption in 3% increments) of the simulation of the 53 mT spectrum. Analyses of these spectra and spectra collected in large externally applied fields suggest that the subspectra associated with the [3Fe-4S]0-like cluster contribute 27 ± 6% of the total 57Fe absorption. The resulting zero-field reference spectra are qualitatively similar and suggest that the MiaB [3Fe-4S]0-like cluster can be modeled with three symmetrical quadruple doublets having equal intensity. This model yields 15 mathematically equivalent solutions, of which only 6 yield physically reasonable parameters (Table 1). Of those, we prefer solutions 1 (Figure 3B) and 2 (Figure S6) because Fe site 1 has parameters very similar to those of the HS FeIII site of cuboidal [3Fe-4S]0 clusters. The other two Fe sites are associated with the mixed-valent (MV) (Fe2)5+ site. In regular [3Fe-4S]0 clusters, the MV unit is fully valence delocalized and the two Fe sites have indistinguishable Mössbauer parameters, which are the average of typical parameters for FeII and FeIII in a distorted tetrahedral S4 coordination environment, thus establishing formally the oxidation state of +2.5 for both Fe sites.54 In the [3Fe-4S]0-like cluster of MiaB, the two Fe sites of the MV unit have distinct parameters. In solution 1, sites 2 and 3 have virtually identical quadrupole splittings but rather different isomer shifts (0.35 and 0.61 mm/s), which are typical of S4-coordinated FeIII and FeII sites and suggest a valence-localized MV unit. By contrast, in solution 2, sites 2 and 3 have different quadrupole splittings but virtually identical isomer shifts (0.47 and 0.48 mm/s), suggesting a valence-delocalized MV unit, wherein both sites have a formal +2.5 oxidation state but distinct coordination environments leading to different quadrupole splittings. Possible geometric structures of this cluster are presented in the Discussion. Importantly, the parameters obtained from the above analysis of other zero-field reference spectra (obtained by adding back varying amounts of the simulated 53 mT spectrum) do not vary significantly (Figure S5). Taken together, the entire 4.2-K/zero-field spectrum of Bt MiaB after incubation with SAM and i6A tRNA substrate in the absence of reductant can be analyzed as a superposition of a [3Fe-4S]0-like cluster, a [4Fe-4S]2+ cluster, and a small amount of [2Fe-2S]2+ cluster (Figure S3).
Table 1.
Different Sets of Mössbauer Parameters for Simulation of the Zero-Field Reference Spectrum of the [3Fe-4S]0-Like Cluster
| solution 1 | solution 2 | solution 3 | solution 4 | solution 5 | solution 6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| site | δ (mm/s) | ΔEQ (mm/s) | δ (mm/s) | ΔEQ (mm/s) | δ (mm/s) | ΔEQ (mm/s) | δ (mm/s) | ΔEQ (mm/s) | δ (mm/s) | ΔEQ (mm/s) | δ (mm/s) | ΔEQ (mm/s) |
| 1 | 0.31 | 0.58 | 0.31 | 0.58 | 0.34 | 0.51 | 0.34 | 0.51 | 0.22 | 0.75 | 0.22 | 0.75 |
| 2 | 0.35 | 1.02 | 0.47 | 1.28 | 0.35 | 1.02 | 0.44 | 1.28 | 0.44 | 0.84 | 0.47 | 1.11 |
| 3 | 0.61 | 1.04 | 0.48 | 0.78 | 0.57 | 1.11 | 0.48 | 0.84 | 0.61 | 1.04 | 0.57 | 0.78 |
Exploring the Requirements for Generating the [3Fe-4S]0-Like Cluster in MiaB.
Although the [0–53 mT]-difference spectrum is rather broad and difficult to quantify, it is nevertheless a diagnostic feature for the presence of the MiaB [3Fe-4S]0-like cluster. An analysis of the amplitudes of the [0–53 mT]-difference spectra of samples treated for varying amounts of time with SAM in the absence of reductant reveals that the amplitudes, and therefore the amount of the [3Fe-4S]0-like cluster, mirror the trend in the amount of SAH generated in the reaction (Figures 1A and S7). When Bt MiaB is treated with SAM, even in the absence of the tRNA substrate, the [3Fe-4S]0-like cluster is also formed, consistent with the generation of SAH in parallel LC/MS experiments (Figure S8). Incubation of MiaB variants—wherein either the three cysteine residues coordinating the RS cluster (ΔRSMiaB) or the three cysteines coordinating the Aux cluster (ΔAuxMiaB) are changed to alanines—with SAM and the tRNA substrate does not lead to formation of the [3Fe-4S]0-like cluster and does not yield SAH as a product (Figures S9 and S10). Likewise, the characteristic difference spectrum is not observed for a sample of MiaB in the absence of SAM but in the presence of the tRNA substrate (Figure S11). Taken together, these results reveal that the [3Fe-4S]0-like cluster is only observed under conditions that lead to generation of SAH and the methylated intermediate that was previously shown to be chemically and kinetically competent for ms2i6A formation. Because the RS [4Fe-4S] cluster is required in the second step of catalysis for generating the 5′–dA• intermediate, we conclude that it is the Aux [4Fe-4S]2+ cluster that is converted to the [3Fe-4S]0-like cluster.
Finally, we also evaluated whether treatment of Bt MiaB with methanethiol, which was reported to result in a methanethiol-derived ms2i6A product,45,48 would result in generation of the [3Fe-4S]0-like cluster. Evaluation of the Mössbauer spectra suggests that a small amount of the [3Fe-4S]0-like cluster is formed (at most one-half of the amount observed in the sample where MiaB is treated with SAM in the presence of tRNA substrate) along with a small amount (at most ~4% of total 57Fe) of N/O-coordinated FeII (Figure S12). We note that the amounts of both species are at the detection limit of the method. However, the formation of the [3Fe-4S]0-like cluster by exposure of MiaB to methanethiol would invariably involve a mechanism different from that for generating the methylated intermediate upon reacting MiaB with SAM, because the S–Cmethyl bond is already formed in this substrate analog (see Discussion).
Characterization of the [3Fe-4S]0-Like Cluster of the Methylated Bt MiaB Intermediate by Variable-Field Mössbauer Spectroscopy.
The variable-field magnetic Mössbauer spectra of the MiaB [3Fe-4S]0-like cluster were analyzed with the assumption that the cluster has an Stot = 2 ground state (the validity of this assumption is proved below) using the values of δ and ΔEQ obtained from analysis of the zero-field spectrum (solution 1) and published spin Hamiltonian parameters of the cuboidal [3Fe-4S]0 cluster from D. gigas ferredoxin as a starting point.54 Because these simulations depend on up to ~50 parameters with each of the three Fe sites contributing only ~10% of the total intensity to the spectrum, it is impossible to determine the spin Hamiltonian parameters unambiguously. Fortuitously, the initial simulations yielded reasonable agreement, and we then refined the parameters to improve the quality of the simulations (Figures 4 and S13 and Table 2). Simulations using the values of δ and ΔEQ from solution 2 of Table 1 and slightly different spin Hamiltonian parameters fit the experimental data comparably well. In the following section we present salient features of this analysis.
Figure 4.
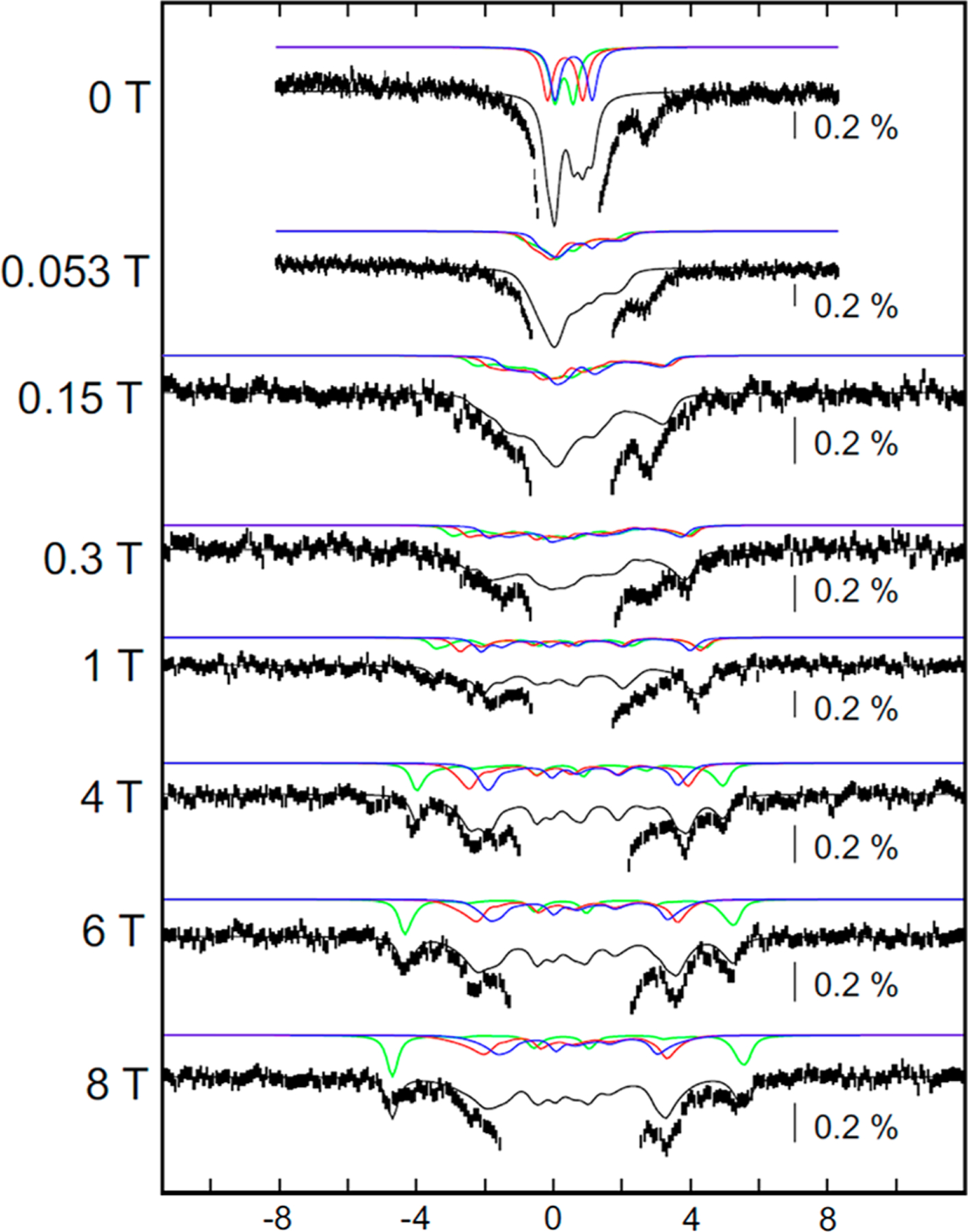
The 4.2-K/variable-field Mössbauer spectra of a sample of reconstituted Bt MiaB incubated with SAM and tRNA substrate for 20 min. Experimental spectra are shown as vertical bars. Strength of the external magnetic field applied parallel to the γ beam is indicated for each spectrum. In all spectra, experimental data are shown as black vertical bars. Central region of the spectra is dominated by the features of the [4Fe-4S]2+ clusters and is cut off in order to amplify the weaker, magnetically split features associated with the [3Fe-4S]0-like cluster. Total simulations of the [3Fe-4S]0-like cluster are shown as black lines using the spin Hamiltonian parameters quoted in Table 2 (27% total intensity). Solid green, red, and blue lines are spin Hamiltonian simulations using the parameters in Table 2 of the unique FeIII site (site 1, 9% of total intensity, green lines), the “FeIII-like” site of the MV unit (site 2, 9% of total intensity, red lines), and the “FeII-like” site of the MV unit (site 3, 9% of total intensity, blue lines). Full spectra are shown in Figure S13A.
Table 2.
Spin Hamiltonian Parameters for Simulation of Mössbauer Spectra of [3Fe-4S]0 Clusters
| species | ZFS parameters | δ (mm/s) | ΔEQ (mm/s) | η | β (deg)a | A/gnβn (T) | ref | |
|---|---|---|---|---|---|---|---|---|
| Bt MiaB [3Fe-4S]0-like cluster | DS=2 = −2.4 cm−1 | site 1, sol. 1 | 0.31 | 0.58 | 2 | 0 | (+12.3, +12.3, +12.3) | this work |
| sol. 2 | 0.31 | 0.58 | 2 | 0 | (+12.3, +12.5, +12.3) | |||
| site 2, sol. 1 | 0.35 | 1.02 | 1 | 0 | (−11.7, −18.0, −11.6) | |||
| (E/D)S=2 = 0.26 | sol. 2 | 0.47 | 1.28 | 2 | 0 | (−11.2, −18.5, −11.4) | ||
| site 3, sol. 1 | 0.61 | 1.04 | −2 | 0 | (−10.0, −18.0, −10.0) | |||
| sol. 2 | 0.48 | 0.78 | 0 | 0 | (−10.0, −18.4, −10.3) | |||
| Ec LipA [3Fe-4S]0-like cluster | DS=2 = −4.0 cm−1 | FeIII | 0.31 | −0.55 | −2 | 16 | (+10.0, +11.8, +11.8) | 42 |
| (E/D)S=2 = 0.23 | Fe22.5+ | 0.44 | 0.98 | 0.4 | 25 | (−14.1, −14.1, −11.4) | ||
| Dg ferredoxin [3Fe-4S]0 cluster | DS=2 = −2.5 cm−1 | FeIII | 0.32 | −0.52 | −2 | 16 | (+10.0, +11.5, +12.6) | 54 |
| (E/D)S=2 = 0.23 | Fe22.5+ | 0.46 | 1.47 | 0.4 | 20 | (−14.9, −14.9, −11.9) | ||
| Av ferredoxin [3Fe-4S]0 cluster (pH 6) | DS=2 = −2.5 cm−1 | FeIII | 0.29 | −0.47 | 0.8 | 20 | (+10.9, +10.2, +12.6) | 55 |
| (E/D)S=2 = 0.23 | Fe22.5+ | 0.47 | 1.41 | 1.1 | 24 | (−15.2, −18.2, −10.4) | ||
| 0.47 | 1.41 | 0.2 | 24 | (−13.8, −16.0, −12.1) | ||||
| Av ferredoxin [3Fe-4S]0 cluster (pH 8.5) | DS=2 = −2.5 cm−1 | Fem | 0.29 | −0.47 | 5 | 27 | (+11.6, +10.2, +12.5) | 55 |
| Fe22.5+ | 0.47 | 1.41 | 1 | 19 | (−13.8, −19.6, −12.1) | |||
| (E/D)S=2 = 0.23 | 0.47 | 1.41 | 0 | 19 | (−14.5, −16.0, − 12.1) |
Euler angle β as defined in ref 24.
All [3Fe-4S]0 clusters reported to date have an Stot = 2 ground state with an axial zero-field splitting (ZFS) parameter, DS =2, ranging from −2 to −4 cm−1, and a rhombicity, (E/D)S=2, of ~0.2.42,54,55 These ZFS parameters give rise to a large, uniaxial spin expectation value in the z direction, 〈Sz〉, for moderate external fields of up to ~1 T that is particularly sensitive to the values of D and E/D (Figure S14). The sizable magnetic hyperfine splitting induced by an applied field as weak as 53 mT indicates that the Bt MiaB [3Fe-4S]0-like cluster has a Stot = 2 ground state (vide infra for consideration of other possible spin states). With the assumption that the [3Fe-4S]0-like cluster has an Stot = 2 ground state, the analysis of the field-dependent spectra yield DS=2 ≈ −3.0 ± cm−1 and (E/D)S=2 between 0.2 and 0.3. Because the observed splitting depends on the product of the spin expectation value in the z direction and the magnitude of the hyperfine tensor in the z direction, i.e., 〈Sz〉•Az, the Az values can be determined with reasonable accuracy.
In the spectra recorded in strong externally applied magnetic fields of 4, 6, and 8 T, the features of the [3Fe-4S]0-like cluster are dominated by strong internal magnetic fields and are better resolved from the features of the [4Fe-4S]2+ cluster, which has a diamagnetic ground state and no internal field; analysis of these spectra suggests that the [3Fe-4S]0-like cluster contributes ~30% of total intensity, consistent with the analysis from the [0–53 mT] difference spectrum (vide supra). Two subcomponents of an antiferromagnetically (AF) coupled cluster can be readily distinguished in these spectra. One subcomponent, which accounts for ~10% of the total 57Fe absorption, exhibits increased splitting with increasing field strength (green lines in Figure 4 and Figure S13B). This component is associated with an Fe site that has a positive A tensor with respect to the total spin and hence a negative spin projection factor.53 It is assigned to the HS FeIII site with S = 5/2. Conversely, the other component (~20% total 57Fe absorption, thus accounting for two Fe sites) exhibits decreasing splitting with increasing external field (red and blue lines in Figure 4 and Figure S13B). This component is associated with the two Fe sites that have a negative A tensor with respect to the total spin and a positive spin projection factor.53 This component is assigned to the subspectra arising from sites 2 and 3. They exhibit similar splittings and are no longer resolved experimentally because the magnetic hyperfine features are large compared to the small difference in δ and ΔEQ of sites 2 and 3. Importantly, this observation further suggests that Fe sites 2 and 3 have similar spin projection factors (with the assumption that they have the similar intrinsic a tensors). These two sites are assigned to the MV dimeric unit with Smv = 9/2.
Taken together, the variable-field magnetic Mössbauer spectra of the MiaB [3Fe-4S]0-like cluster can be analyzed using the values of isomer shift and quadruple splitting obtained from the zero-field reference spectrum, the assumption that the cluster has an Stot = 2 spin ground state, and the parameters given in Table 2. The fact that the spin Hamiltonian parameters are similar to those observed for other cuboidal [3Fe-4S]0 clusters supports the notion that the electronic structure is adequately described by invoking AF coupling between a HS FeIII center (S = 5/2) and the MV unit (Smv = 9/2), yielding a Stot = 2 ground state.
Evaluation of Possible Electron Spin Ground States of the [3Fe-4S]0-Like Cluster.
Although the variable-field Mössbauer spectra can be satisfactorily simulated with parameters typical of [3Fe-4S]0 clusters, the fact that the δ and ΔEQ values of the two Fe sites of the MV unit are different reveals the presence of either a valence-localized MV unit (solution 1) or a valence-delocalized unit with different coordination environments (solution 2), thus posing the question whether the ground state of the [3Fe-4S]0-like cluster in MiaB is still the |9/2, 2〉 state (spin states are designated as |Smv, Stot〉) as in all other [3Fe-4S]0 clusters. We therefore explored the possibility that other spin states could be the ground state using a spin-coupling model adopted from that introduced by Münck and co-workers.54 The Münck model takes into account the spin-dependent delocalization (viz. double exchange) of the MV (Fe2)5+ unit, parametrized by the double-exchange parameter, Bmv, and further assumes three identical AF superexchange interactions between any two Fe centers (J). Because methylation could potentially occur at one of the μ2-sulfido ligands of a cuboidal [3Fe-4S]0-like cluster, which would be expected to result in a decrease of the exchange coupling constant between the two Fe ions bridged by the methanethiolate ligand, in analogy to the well-established reduction of the exchange coupling of oxo-bridged clusters upon protonation or alkylation,56−58 we expanded the Münck model to explore the effect of one J value being smaller. The case that the exchange coupling within the MV unit, Jmv, is different from the other two exchange coupling constants, J, can be easily solved analytically (see Supporting Information for details on the model).59 Because methanethiolate is a weaker ligand than sulfide it is expected to bind preferentially to the FeII or Fe2.5 sites rather than the FeIII site, i.e., it is expected to bridge the two Fe centers of the MV unit, thus validating the above assumption. The evaluation of possible spin ground states as a function of Jmv/J and Bmv/J, shown in Figure 5, reveals that five states can be the ground state (for Jmv/J > 0 and Bmv/J > 0). The specific case explored by Münck and co-workers (Jmv/J = 1) is indicated by the dashed horizontal line in Figure 5; it can yield |9/2, 2〉, |7/2, 1〉, and |5/2, 0〉 ground states. The |9/2, 2〉 ground state established for the previously characterized [3Fe-4S]0 clusters requires Bmv/J > 2 with the assumption Jmv = J, consistent with the observation of a fully valence-delocalized MV (Fe2)+5 unit.54 For values of Jmv/J > 1, the |1/2, 2〉 and |3/2, 1〉 states can also be the ground state. However, the latter two states are disfavored because the bridging methanethiolate ligand is expected to decrease Jmv compared to J, i.e., Jmv/J < 1.
Figure 5.
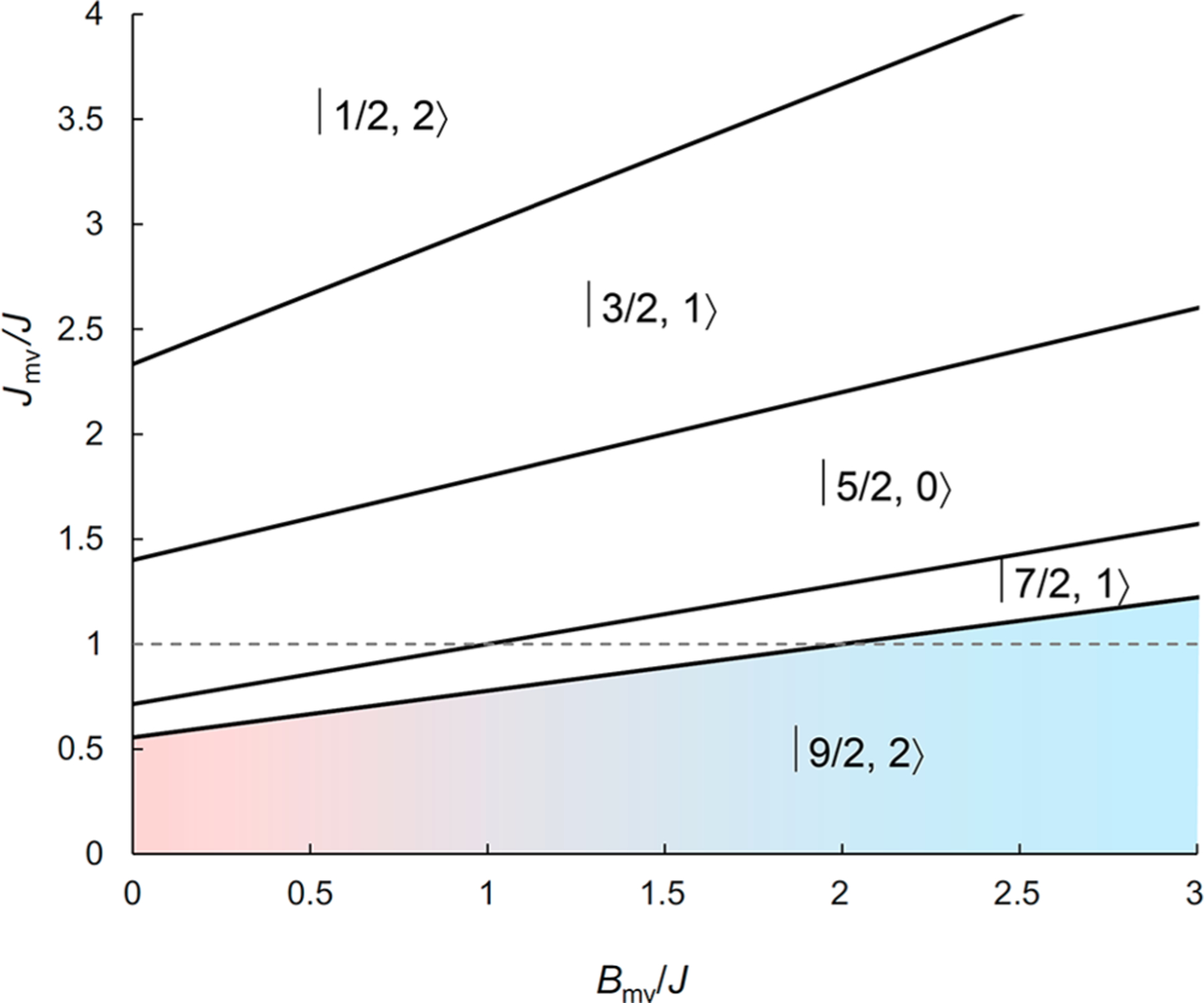
Possible total spin ground states of the [3Fe-4S]0-like cluster as a function of Jmv/J and Bmv/J. Spin states are labeled as |Smv, Stot〉. Parameter space containing values of Jmv/J and Bmv/J yielding the experimentally observed |9/2, 2〉 ground state is shown colored. Dashed line indicates Jmv = J.
Of these five possible spin ground states, the |5/2, 0〉 state can be easily ruled out because the experimental data unequivocally show that the ground state is paramagnetic. The two states with Stot = 1, |7/2, 1〉 and |3/2, 1〉 can also be ruled out because the smaller limiting value of 〈S〉 = 1 would then require ~2-fold larger average A values (compared to the A values with respect to a Stot = 2 ground state described in the previous section) to reproduce the experimentally observed splitting of the spectra. Calculation of the intrinsic a values using spin projection techniques (see Table 3 for spin projection coefficients)60 yields a values that are significantly larger than typical intrinsic a values of Fe/S clusters.61 Lastly, the |1/2, 2〉 ground state can be ruled out because the spin projection factor of the Fe sites of the MV unit are much smaller than those associated with the |9/2, 2〉 state (see Table 3), resulting in intrinsic a values that are 2-fold smaller than typical intrinsic a values of Fe/S clusters.61 We therefore conclude that the [3Fe-4S]0-like cluster in MiaB has a |9/2, 2〉 ground state.
Table 3.
Spin Projection Factors for Possible Spin Ground States of the [3Fe-4S]0-Like Cluster
| spin state |Smv, Stot〉 | cferric-y,MVa | cferric-y,MVa | cferrica |
|---|---|---|---|
| |9/2, 2〉 | +1.019 | +0.815 | −0.833 |
| [+55/54] | [+22/27] | [−5/6] | |
| |1/2, 2〉 | −0.389 | +0.222 | +1.167 |
| [−7/18] | [+2/9] | [+7/6] | |
| |7/2, 1〉 | +1.321 | +0.929 | −1.25 |
| [+37/28] | [+13/14] | [−5/4] | |
| |3/2, 1〉 | −0.65 | −0.1 | +1.75 |
| [−13/20] | [−1/10] | [+7/4] | |
| |5/2, 0〉b |
iIalicized values are expressed as ratio of smallest integers.
Not defined due to Stot = 0 ground state
DISCUSSION
Previous work has shown that treatment of the RS MTTase MiaB with the cosubstrate, SAM, in the absence of a low-potential reductant leads to an intermediate with an acid-labile methylthio group and the coproduct SAH.45 In this work, we used Mössbauer spectroscopy to monitor the fate of the two [4Fe-4S]2+ clusters of MiaB upon generating the methylated intermediate. Our results show that one [4Fe-4S]2+ cluster remains intact, while the other is converted to a [3Fe-4S]0-like cluster and HS N/O-coordinated FeII. We assign the [4Fe-4S]2+ cluster to the RS cluster, because this cluster is needed for the reductive cleavage of SAM to generate the 5′-dA• oxidant in a subsequent step of the MiaB-catalyzed reaction. Consequently, we assign the [3Fe-4S]0-like cluster to the Aux cluster. The combination of Mössbauer spectroscopy and LC/MS showed that the kinetics of formation of the [3Fe-4S]0-like cluster match those of SAH formation. Furthermore, taking into consideration the previous observation that incubation of the methylated intermediate of MiaB with the tRNA substrate, sodium dithionite (required low-potential reductant), and d3-SAM leads to formation of unlabeled ms2i6A before formation of labeled ms2i6A, the results presented herein indicate that the methylated intermediate is catalytically important.
In contrast to all other characterized [3Fe-4S]0 clusters, which contain a fully valence-delocalized MV unit, of which the two Fe sites have identical Mössbauer parameters,54 the two Fe sites of the MV unit of the MiaB intermediate have different Mössbauer isomer shifts and quadrupole splittings. Of the two solutions we identified, the first has almost identical quadrupole splittings for the two Fe sites of the MV unit (1.02 and 1.04 mm/s) but different isomer shifts (0.35 and 0.61 mm/s). These parameters would suggest valence localization within the MV unit because these isomer shifts are typical of those of FeIII and FeII sites, respectively, in [4Fe-4S] clusters.54,62 The second solution has more different quadrupole splittings (0.78 and 1.28 mm/s) but essentially identical isomer shifts (0.47 and 0.48 mm/s). These parameters would in turn indicate valence delocalization within the MV unit but with different coordination environments of the two Fe sites, resulting in different quadrupole splittings.
The electronic structure of the ground state of MV clusters with two or more paramagnetic sites, such as Fe/S clusters, depends delicately on (1) the strength of the exchange coupling between the paramagnetic centers, (2) the strength of double-exchange interaction between the two paramagnetic centers, and (3) the magnitude of the vibronic coupling of the two sites.63 In particular, the relative magnitude of the double-exchange interaction (which favors valence delocalization) and of the vibronic coupling (which favors valence localization) determines whether the ground state is valence localized or valence delocalized. Only subtle structural perturbations, such as the degree of saturation of a bridging ligand,64 are required to change the ground state from valence localized to valence delocalized. Borshch et al. developed a model for the [3Fe-4S]0 cluster of D. gigas, which takes into account the above three interactions.65 Importantly, this model rationalizes the experimental observation that the excess electron is delocalized over two Fe sites. Subsequently, work by Solomon and co-workers employed ligand K-edge X-ray spectroscopy to measure the average covalency of the Fe–S bonds of various Fe/S clusters with MV units.66–68 These experiments provided further insight into the electronic structures of Fe/S clusters with MV units because the covalency of the Fe–S bonds affects the strength of the exchange coupling and the double-exchange interactions (increased covalency results in stronger AF coupling via the bridging sulfides and decreases the double-exchange interaction via the direct overlap of Fe-based orbitals).
Variable-field Mössbauer spectroscopy revealed that the MiaB [3Fe-4S]0-like cluster has an |9/2, 2〉 electronic ground state resulting from AF coupling between the high-spin FeIII site (S = 5/2) and the MV pair (Smv = 9/2). Münck and co-workers have shown that for the [3Fe-4S]0 cluster the |9/2, 2〉 state is only observed for Bmv/J > 2 (see dashed line and redshaded area in Figure 5). In the absence of the double-exchange interaction (Bmv/J = 0) and for Jmv = J, the ground state would be the |5/2, 0〉 state due to spin frustration caused by three equally strong AF couplings between the three Fe sites. Closer inspection of Figure 5 reveals that the |9/2, 2〉 state can be the ground state for Jmv/J < 1. For Jmv/J ≤ 0.555 (5/9), the |9/2, 2〉 state is the ground state even in the absence of the double-exchange interaction (Bmv/J = 0) (y axis of Figure 5). This behavior can be understood by the loss of spin frustration in the trinuclear cluster. The stronger AF exchange couplings between the HS FeIII site and the two Fe ions of the MV unit therefore result in parallel alignment of the spins of the MV unit to yield Smv = 9/2.
A plausible geometric structure of the MiaB [3Fe-4S]0-like cluster that is in accordance with the electronic structure described by parameters of solution 1 invokes a methanethiolate ligand bridging the two Fe sites of the MV unit asymmetrically with a stronger interaction with FeB than with FeA (Figure 6A). We assign FeB to the FeIII site because the reduced donor strength of the methanethiolate ligand (compared to that of sulfide) for stabilization of the FeIII site would be compensated for by the asymmetric binding mode with a stronger interaction with FeB. The |9/2, 2〉 ground state can be rationalized by the (presumably) less efficient exchange coupling pathway via the μ2-methanethiolate ligand (compared to that involving the μ2-sulfide ligands), thus reducing the AF coupling. For Jmv/J < 0.555, the |9/2, 2〉 will be the ground state (Figure 5), even in the absence of valence delocalization (Bmv = 0).
Figure 6.
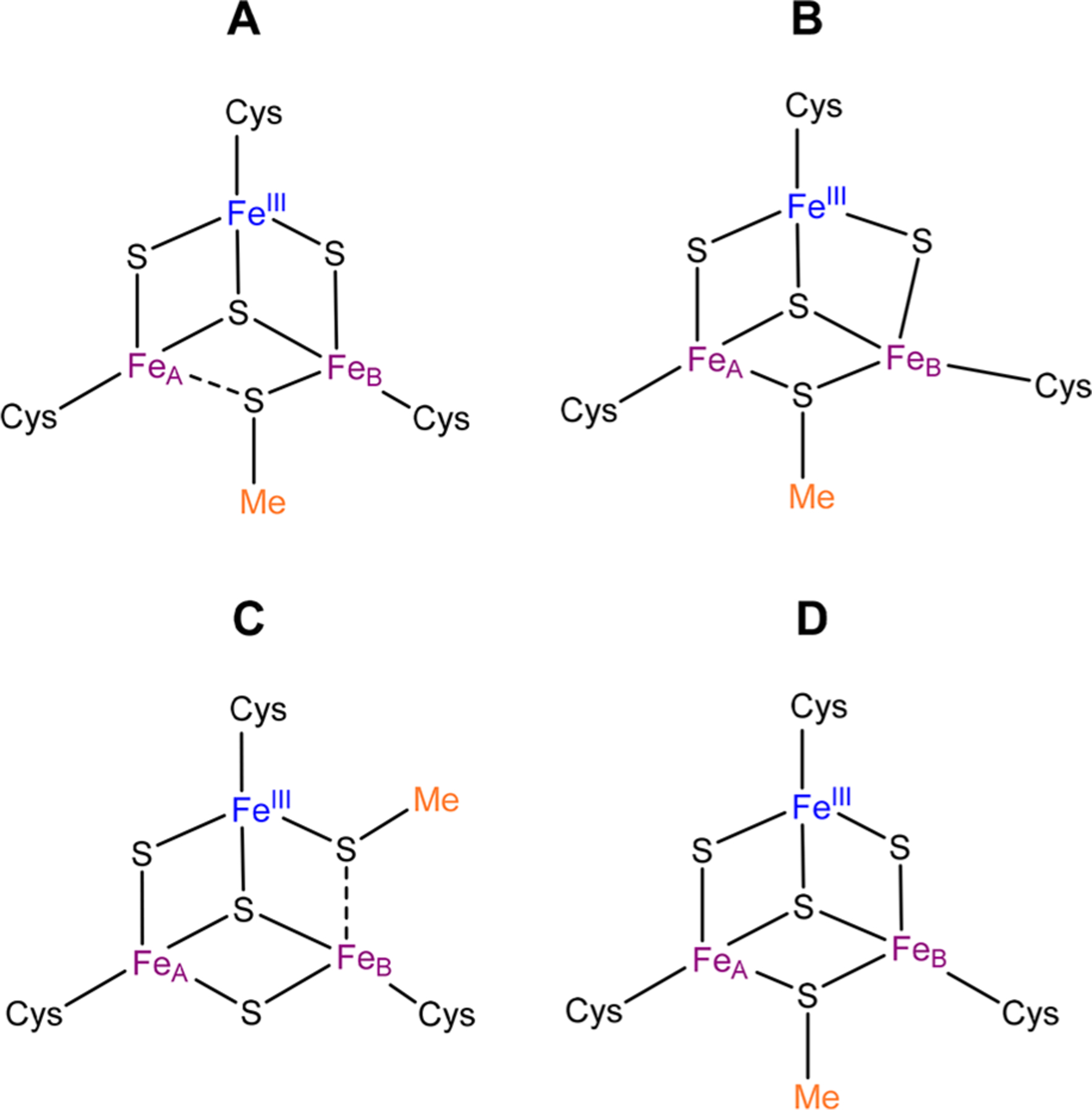
Possible structures of the [3Fe-4S]0-like Aux cluster in MiaB with a valence-localized MV unit (A) or with a valence-delocalized MV unit (B and C) and of the [3Fe-4S]0-like Aux cluster of LipA (D).
Plausible geometric structures of the [3Fe-4S]0-like cluster of MiaB that are in accordance with the electronic structure with the parameters of solution 2 are shown in Figure 6B and 6C. They both feature a symmetric [2Fe-2S] core geometry of the MV unit that would provide an efficient delocalization pathway involving the through-space overlap of the orbitals on FeA and FeB. In structure B, with the methanethiolate ligand bridging the Fe ions of the MV unit, the difference in quadrupole splitting could be attributed to distinct coordination geometries for FeA and FeB (e.g., a distorted FeB-μ2-S-FeIII fragment and/or distorted Cys coordination to FeB). In structure C, the μ2-methanethiolate is depicted as bridging FeB and FeIII asymmetrically with a stronger interaction to FeIII in order to compensate for the weaker donor strength of methanethiolate compared to that of sulfide. This in turn would render the coordination environments of FeA and FeB different, which could rationalize their different quadrupole splittings. For both structures the |9/2, 2〉 ground state can be rationalized by the a strong double-exchange interaction, favoring Smv = 9/2 and AF coupling to FeIII via at least one strong AF coupling pathway. We recognize that the asymmetry in structures A–C could be stabilized by the protein scaffold and that other structures not depicted here can account for the experimentally observed site inequivalence of the MV unit.
Finally, the Aux [3Fe-4S]0-like cluster of the LipA cross-linked intermediate, which has a fully valence-delocalized MV unit, of which both Fe sites have indistinguishable parameters42 and which is known to have a μ2-6-mercapto-octanoyl bridging ligand,50 could be described by the symmetric core structure shown in Figure 6D.
Recent electrochemical studies on Bt MiaB revealed unusual time-dependent redox properties of one of the [4Fe-4S]2+ clusters upon treatment with SAM.46 While both [4Fe-4S]2+ clusters in Bt MiaB exhibit typical reduction potentials (−390 mV for the Aux cluster and −450 mV for the RS cluster), incubation with SAM in the absence of the tRNA substrate significantly shifts the potential of the auxiliary cluster to −600 mV on a time scale that is consistent with that of the generation of the methylated intermediate.46 The authors rationalized the unusual low-potential state as a consequence of methylation of the Aux cluster without change of its chemical nature as a [4Fe-4S] cluster while pointing out that such a low reduction potential is indeed rare for a [4Fe-4S]2+/+ redox couple. In light of our work, we reason that the low reduction potential corresponds to that of the [3Fe-4S]0/−1-like Aux cluster of the methylated intermediate.
There are four known oxidation states of the cubane-type [3Fe-4S] clusters. The [3Fe-4S]+/0 couple is the most physiologically relevant redox pair in biological systems, with reported potentials spanning approximately 550 mV (from −460 to +90 mV).69 To the best of our knowledge, [3Fe-4S] clusters in the −1 core oxidation state have not been observed in a discrete form in any biological system. However, a [3Fe-4S]− moiety has been deduced from studies on heterometallic cuboidal [M-3Fe-4S] clusters where M is a redox-inert, divalent metal, such as Zn2+ or Cd2+.70 The potentials reported for the [M-3Fe-4S]2+/+ redox couple, where the Fe/S fragment formally corresponds to a [3Fe-4S]0/− unit, are −492 mV for [Zn-3Fe-4S]2+/+ and −569 mV for [Cd-3Fe-4S]2+/+.70 It has been demonstrated that [3Fe-4S]0 clusters can be reduced electrochemically in the region of ca. −700 mV to a [3Fe-4S]2− state in a process that involves a cooperative proton-coupled two-electron reduction and thus bypasses the [3Fe-4S]− state.71 It has been postulated that a discrete [3Fe-4S]− state is not accessible under physiological conditions without binding to a positively charged moiety, such as divalent metal ions or protons.70,71 Similarly, replacement of a dianionic sulfido ligand (S2−) with a monoanionic methanethiolato ligand is expected to increase the reduction potential of the [3Fe-4S]0/− couple in the MiaB [3Fe-4S]0-like cluster, perhaps to an extent that the [3Fe-4S]− state becomes accessible under physiologically relevant conditions. In this instance, the one-electron reduction event at −600 mV detected electrochemically in the study by Maiocco et al. might correspond to the [3Fe-4S]0/− redox couple.46
Previously, the proposed structures for the methylated intermediate of the MTTases MiaB and RimO were depicted as having a terminal methanethiolato ligand to the unique, noncysteinyl-coordinated Fe ion of the Aux [4Fe-4S] cluster based primarily on two experimental observations by Atta and co-workers.45,48,72 The first was the X-ray crystal structure of RimO, which showed that the unique Fe sites of the Aux and RS clusters are connected by a pentasulfide bridge.48 While the pentasulfide ligand detected in the X-ray structure is almost certainly an artifact of the crystallization conditions, given that the unique Fe site of the RS cluster binds SAM, this result nevertheless underscores the ability of the unique Fe site of the Aux cluster to be coordinated by a terminal (hydro)sulfido ligand. Second, hyperfine sublevel correlation (HYSCORE) spectroscopy was used to demonstrate that exogenously supplied methanethiolate or methaneselenolate can bind as a terminal ligand to the unique Fe site of the Aux [4Fe-4S]+ cluster.48 Thus, it was proposed that MiaB has a terminal sulfide ligand on the unique Fe site of its [4Fe-4S]2+ Aux cluster, which can attack the methyl carbon of SAM to generate the coproduct, SAH, and yield a terminal methanethiolato ligand (Scheme 2A). It is expected that the Aux cluster of the methylated intermediate would be a stable [4Fe-4S]2+ cluster given that it would be coordinated by four thiolate ligands (three cysteines from the protein and the methanethiolate). However, this structure of the methylated intermediate is in contrast with our observation of a [3Fe-4S]0-like Aux cluster, the formation of which would require an unstable Aux cluster capable of losing the unique Fe site together with its terminal methanethiolate ligand. We disfavor this scenario because (1) the Mössbauer parameters of the FeII observed in the sample containing the methylated intermediate are typical of HS N/O-coordinated FeII 53 and do not support coordination by sulfur (in this case the isomer shift would be expected to decrease to ~1.1 mm/s,73 which is not supported by our data), (2) the remaining cluster would be expected to be a regular cuboidal [3Fe-4S]0 cluster with a fully valence-delocalized MV unit, which is not observed, and (3) there is thus far no evidence for a binding site for a FeII–SMe fragment on MiaB.
Scheme 2.
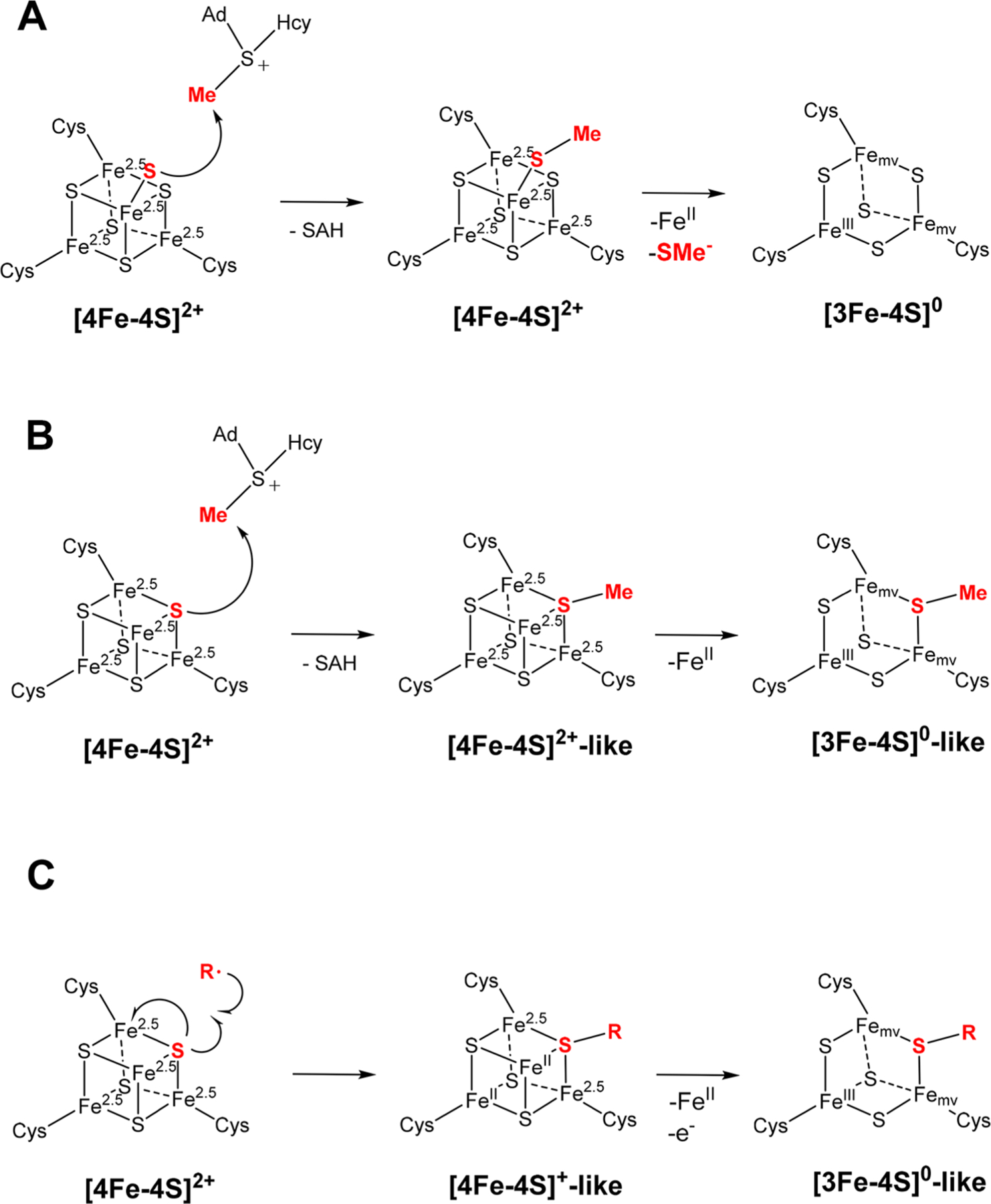
Two Possible Mechanisms for Formation of the [3Fe-4S]0-Like Auxiliary Cluster in MiaB by Nucleophilic Attack of Either a Terminal Sulfide on the Unique Fe Site (A) or a μ3-Bridging Sulfide (B) from the [4Fe-4S]2+ Cluster on the Methyl Group of SAM and the Mechanism for Generation of the [3Fe-4S]0-Like Cluster by a Radical Mechanism in LipA (C)a
aThe sulfur atom and the methyl group forming the methylthio moiety of the intermediate are emphasized in bold red.
An alternative sulfur source to be methylated would be one of the μ3-sulfido ligands of the Aux [4Fe-4S]2+ cluster (Scheme 2B). Nucleophilic attack of the μ3-sulfido ligand on Cmethyl of SAM would yield SAH and a [4Fe-4S]2+-like cluster, wherein one μ3-sulfido ligand is converted to a μ3-methanethiolato ligand. Loss of the unique iron ion as FeII would then yield the [3Fe-4S]0-like cluster, which we assume is effected by a weakening of the Feunique–SMe bond. We note that the order of these two steps could also be reversed, i.e., loss of FeII to make a [3Fe-4S]0 cluster followed by attack of one of the μ2-sulfides on the methyl group of SAM. This mechanism is consistent with the results obtained in this work by Mössbauer spectroscopy, namely, observation of FeII in solution and a [3Fe-4S]0-like cluster with an unusual electronic structure, which is attributed to the fact that one of the sulfides is converted to a methanethiolate.
At face value the results from previous work using HYSCORE spectroscopy48 contradict our results from Mössbauer spectroscopy. We speculate that the discrepancy arises from the way the samples were prepared. In our study, wild-type Bt MiaB was used under conditions that lead to generation of the methylated intermediate by a nucleophilic substitution on the cosubstrate, SAM, which is known to be chemically competent to make the methylthiolated product. By contrast, the HYSCORE study was carried out on a sample of a MiaB variant that lacks the RS cluster. More importantly, the sample was generated by treatment with methanethiol, a reagent wherein the S–Cmethyl bond is already formed. We speculate that the formation of the experimentally observed Aux [4Fe-4S]+ cluster featuring a terminal methanethiolato/selenolato ligand involves a ligand substitution wherein the methanethiolato/selenolato replaces the ligand (perhaps a water or buffer molecule) to the unique Fe of the Aux [4Fe-4S] cluster. Thus, the unequivocal experimental observation that a methanethiolato/selenolato ligand can serve as exogenous ligand to one Fe site (likely the unique Fe site) of the [4Fe-4S] Aux cluster is nevertheless no proof that this state is an intermediate on the pathway and hence not in contrast to our observation of a [3Fe-4S]0-like Aux cluster.
The formation of a related [3Fe-4S]0-like cluster has recently been demonstrated in the catalytically competent intermediate on the Aux cluster of the RS enzyme LipA,42 which features a μ2-6-mercapto-octanoyl ligand.50 Although the reactions of LipA and MiaB feature the conversion of a [4Fe-4S]2+ Aux cluster to a [3Fe-4S]0-like Aux cluster, the mechanisms of this conversion in the two enzymes are fundamentally different. In the LipA-catalyzed reaction, the formation of the new C–S bond is initiated by attack of the C6 octanoyl radical—generated by abstraction of the C6 proR hydrogen by the 5′-dA•—on one of the μ3-sulfido ligands of the Aux [4Fe-4S]2+ cluster. Formally, in this reaction the new C–S bond is formed by one electron from the substrate radical and the other from the lone pair on the sulfide ligand. The remaining electron of the sulfido lone pair then formally reduces the cluster by one electron, yielding a [4Fe-4S]+-like intermediate, which is not observed in the native LipA reaction. Loss of one FeII and an additional electron then yields the experimentally observed [3Fe-4S]0-like cluster (Scheme 2C).
Previous studies indicated that MiaB and RimO may share a similar mechanism for the first step of their reactions: generation of the methylated intermediate.45 Considering the biochemical and electrochemical similarities between the proposed methylated intermediates in MiaB and RimO, it will be interesting to explore if a [3Fe-4S]0-like intermediate is also observed in RimO. A recent study by Molle et al. on Tm RimO reported observation of HS FeII species and unspecified paramagnetic species by Mössbauer spectroscopy following incubation of samples of Tm RimO containing predominantly [4Fe-4S]2+ clusters with SAM.72 In the study by Molle et al., the Mössbauer spectra were recorded in an applied field of 60 mT and the remaining spectral features were modeled as a superposition of valence-delocalized [4Fe-4S]2+, site-differentiated [4Fe-4S]2+, and [2Fe-2S]2+ clusters. We acknowledge the complexity associated with spectral simulation of multiple Fe-containing species with heavily overlapping features. However, it will be interesting to see if the observed spectral changes in Tm RimO could also be rationalized by invoking conversion of a [4Fe-4S]2+ cluster to a potential [3Fe-4S]0-like cluster.
The reaction mechanisms of the second step of the reactions of RimO and MiaB, the conversion of the methylated intermediate to the respective products, wherein SAM functions as the source of the 5′-dA• intermediate, are likely different. In RimO, the second phase of the reaction is initiated by abstraction of the aliphatic 3-pro-S hydrogen of the substrate by the 5′-dA• intermediate followed by transfer of the methylthio group to the C3 substrate radical.33 While the mechanism of the latter radical coupling reaction resulting in generation of the C3–S bond remains to be elucidated, there is precedent for such a reaction in the related RS enzyme BioB.41,43 In the second step of the BioB reaction the substrate is 9-mercapto-dethiobiotin, which is generated during the first step of the reaction and involves attack of the C9 dethiobiotinyl radical (generated by abstraction of hydrogen from C9 by 5′-dA•) on one of the μ2-sulfido ligands of the Aux [2Fe-2S]2+ cluster, which gets reduced to the [2Fe-2S]+ form in the process. A second equivalent of 5′-dA• abstracts the 6-pro-S hydrogen from the substrate, followed by attack of the ensuing radical on the sulfur of the coordinated thiolate of the substrate, yielding the thiophane ring of biotin.5,41,43 Again, this radical coupling step results in further reduction of the Aux [2Fe-2S] cluster by one electron. The second part of the RimO reaction has been proposed to proceed according to that mechanism, namely, attack of the C3 radical from the substrate on the coordinated methanethiolate, yielding a one-electron-reduced Aux cluster.33,72
The second step of the MiaB reaction, installment of the methylthio moiety on C2 of the substrate, is likely different because the homolytic bond dissociation energy (BDE) of the C2–H bond of the substrate (estimated to be ~105 kcal/mol)74 is expected to be too high for the hydrogen to be abstracted by the 5′-dA• oxidant. Recent work by Arcinas et al. demonstrated that, nevertheless, the hydrogen atom from C2 is removed by the 5′-dA•.75 The proposed mechanism for the second part of the MiaB reaction (Scheme 3) is initiated by nucleophilic attack by the coordinated methanethiolate on C2 of the substrate, which would decrease the BDE of the C2–H bond due to rehybridization of C2 from sp2 to sp3. In the next step the 5′-dA• is proposed to abstract the hydrogen from C2 followed by loss of an electron from the substrate to generate the 2-methylthiolated product. Importantly, in this mechanism, formation of the new C–S bond occurs before hydrogen abstraction, while in RimO (and all other RS enzymes that catalyze C–S bond formation via radical-based mechanisms involving the 5′-dA• radical, like BioB, LipA, and RimO) formation of the new C–S bond is known or proposed to occur after hydrogen-atom abstraction.
Scheme 3.
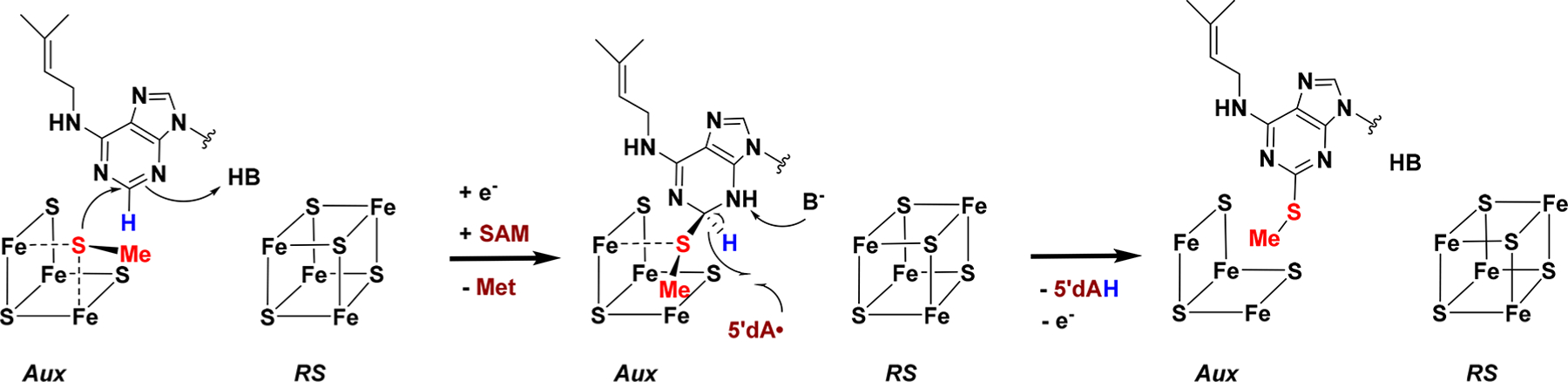
Proposed Mechanism for the Second Part of the MiaB Reaction, viz. the Transfer of the Methylthio Group from the Aux Fe/S Cluster to C2 of A37 of the tRNA Substrate
Our proposal that a methylated [3Fe-4S]0-like cluster is the immediate source of sulfide to be incorporated into the final product is also consistent with two previous observations. It has been previously demonstrated that Tm MiaB containing [4Fe-4Se]2+ instead of [4Fe-4S]2+ clusters is able to catalyze formation of mse2i6A.76 Furthermore, in E. coli, a decrease in MiaB activity in vivo has been associated with the inactivation of the ygf Z gene.77,78 E. coli YgfZ and its yeast homologue Iba57 have recently been recognized as playing prominent roles in the biogenesis of Fe/S clusters.78,79 In particular, although yeast Iba57 is not required for the de novo assembly of Fe/S clusters, it is required for the in vivo functions of both BioB and LipA.79 Taken together, these observations are consistent with the MiaB auxiliary [4Fe-4S]2+ cluster as the most likely ultimate source of the sulfide to be incorporated into the final ms2i6A product. Due to the importance of tRNA methylthiolation, a sacrificial role of the MiaB auxiliary cluster was considered unfavorable due to concerns that inclusion of cluster generation during catalysis may not be able to sustain the cellular demand of the MiaB reaction product.48 Recently, it has been demonstrated that the E. coli Fe/S cluster carrier protein NfuA is able to efficiently restore the Aux [4Fe-4S] cluster in LipA during catalysis in a process that is not rate limiting.44 Intriguingly, studies on Fe/S cluster trafficking in E. coli have demonstrated that NfuA is able to physically interact with MiaB and to deliver a [4Fe-4S]2+ cluster to both RS and auxiliary cluster domains in MiaB.80 Therefore, it is tempting to speculate that NfuA might play a role in restoration of the Aux cluster in MiaB, like it does in LipA.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (awards GM-122595 to S.J.B. and GM-127079 to C.K.), the National Science Foundation (MCB-1716686 to S.J.B.), and the Eberly Family Distinguished Chair in Science (S.J.B). S.J.B is an Investigator of the Howard Hughes Medical Institute. We thank Reviewer 1 for helpful discussion concerning plausible geometric structures of the [3Fe-4S]0-like cluster in MiaB.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b11093.
Materials and Methods, Mössbauer-spectroscopic characterization of wt MiaB, ΔAuxMiaB, and ΔRSMiaB, evaluation of alternative spin states as ground state of the [3Fe-4S]0-like cluster, Figures S1–S15, and references (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.9b11093
The authors declare no competing financial interest.
REFERENCES
- (1).Frey PA; Hegeman AD; Ruzicka FJ The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol 2008, 43, 63. [DOI] [PubMed] [Google Scholar]
- (2).Atta M; Mulliez E; Arragain S; Forouhar F; Hunt JF; Fontecave M S-Adenosylmethionine-dependent radical-based modification of biological macromolecules. Curr. Opin. Struct. Biol 2010, 20, 684. [DOI] [PubMed] [Google Scholar]
- (3).Vey JL; Drennan CL Structural Insights into Radical Generation by the Radical SAM Superfamily. Chem. Rev 2011, 111, 2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-Adenosylmethionine Enzymes. Chem. Rev 2014, 114, 4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Jarrett JT The Biosynthesis of Thiol- and Thioether-containing Cofactors and Secondary Metabolites Catalyzed by Radical S-Adenosylmethionine Enzymes. J. Biol. Chem 2015, 290, 3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lanz ND; Booker SJ Auxiliary iron-sulfur cofactors in radical SAM enzymes. Biochim. Biophys. Acta, Mol. Cell Res 2015, 1853, 1316. [DOI] [PubMed] [Google Scholar]
- (7).Sofia HJ; Chen G; Hetzler BG; Reyes-Spindola JF; Miller NE Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Holliday GL; Akiva E; Meng EC; Brown SD; Calhoun S; Pieper U; Sali A; Booker SJ; Babbitt PC In Radical SAM Enzymes; Bandarian V, Ed.; Elsevier Academic Press Inc: San Diego, CA, 2018; Vol. 606, p 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Akiva E; Brown S; Almonacid DE; Barber AE; Custer AF; Hicks MA; Huang CC; Lauck F; Mashiyama ST; Meng EC; Mischel D; Morris JH; Ojha S; Schnoes AM; Stryke D; Yunes JM; Ferrin TE; Holliday GL; Babbitt PC The Structure-Function Linkage Database. Nucleic Acids Res. 2014, 42, D521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Krebs C; Broderick WE; Henshaw TF; Broderick JB; Huynh BH Coordination of Adenosylmethionine to a Unique Iron Site of the [4Fe-4S] of Pyruvate Formate-Lyase Activating Enzyme: A Mössbauer Spectroscopic Study. J. Am. Chem. Soc 2002, 124, 912. [DOI] [PubMed] [Google Scholar]
- (11).Walsby CJ; Hong W; Broderick WE; Cheek J; Ortillo D; Broderick JB; Hoffman BM Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active 4Fe-4S+ cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc 2002, 124, 3143. [DOI] [PubMed] [Google Scholar]
- (12).Yang H; McDaniel EC; Impano S; Byer AS; Jodts R; Yokoyama K; Broderick WE; Broderick JB; Hoffman BM The Elusive 5′-Deoxyadenosyl Radical: Captured and Characterized by EPR and ENDOR Spectroscopies. J. Am. Chem. Soc 2019, 141, 12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sayler RI; Stich TA; Joshi S; Cooper N; Shaw JT; Begley TP; Tantillo DJ; Britt RD Trapping and Electron Paramagnetic Resonance Characterization of the 5′dAdo• Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate. ACS Cent. Sci 2019, 5, 1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Horitani M; Shisler K; Broderick WE; Hutcheson RU; Duschene KS; Marts AR; Hoffman BM; Broderick JB Radical SAM catalysis via an organometallic intermediate with an Fe-5 ′-C-deoxyadenosyl bond. Science 2016, 352, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Atta M; Arragain S; Fontecave M; Mulliez E; Hunt JF; Luff JD; Forouhar F The methylthiolation reaction mediated by the Radical-SAM enzymes. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Urbonavičius J; Qian O; Durand JMB; Hagervall TG; Björk GR Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001, 20, 4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gustilo EM; Vendeix FA; Agris PF tRNA’s modifications bring order to gene expression. Curr. Opin. Microbiol 2008, 11, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hagervall TG; Ericson JU; Esberg KB; Ji-nong L; Björk GR Role of transfer-RNA modification in translational fidelity. Biochim. Biophys. Acta, Gene Struct. Expression 1990, 1050, 263. [DOI] [PubMed] [Google Scholar]
- (19).Torres AG; Batlle E; Ribas de Pouplana L Role of tRNA modifications in human diseases. Trends Mol. Med 2014, 20, 306. [DOI] [PubMed] [Google Scholar]
- (20).Durand JMB; Björk GR; Kuwae A; Yoshikawa M; Sasakawa C The modified nucleoside 2-methylthio-N-6-isopentenyl-adenosine in tRNA of Shigella flexneri is required for expression of virulence genes. J. Bacteriol 1997, 179, 5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bartz JK; Kline LK; Söll D N6-(Δ2-Isopentenyl)adenosine - Biosynthesis in vitro in transfer RNA by an enzyme purified from Escherichia coli. Biochem. Biophys. Res. Commun 1970, 40, 1481. [DOI] [PubMed] [Google Scholar]
- (22).Caillet J; Droogmans L Molecular cloning of the Escherichia coli miaA gene involved in the formation of the Δ2-isopentenyl adenosine in transfer RNA. J. Bacteriol 1988, 170, 4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rosenbaum N; Gefter ML Δ2-Isopentenylpyrophosphate - transfer ribonucleic acid Δ2-isopentenyltransferase from Escherichia coli-Purification and properties of enzyme. J. Biol. Chem 1972, 247, 5675. [PubMed] [Google Scholar]
- (24).Steinthorsdottir V; Thorleifsson G; Reynisdottir I; Benediktsson R; Jonsdottir T; Walters GB; Styrkarsdottir U; Gretarsdottir S; Emilsson V; Ghosh S; Baker A; Snorradottir S; Bjarnason H; Ng MCY; Hansen T; Bagger Y; Wilensky RL; Reilly MP; Adeyemo A; Chen YX; Zhou J; Gudnason V; Chen GJ; Huang HX; Lashley K; Doumatey A; So W; Ma RCY; Andersen G; Borch-Johnsen K; Jorgensen T; van Vliet-Ostaptchouk JV; Hofker MH; Wijmenga C; Christiansen C; Rader DJ; Rotimi C; Gurney M; Chan JCN; Pedersen O; Sigurdsson G; Gulcher JR; Thorsteinsdottir U; Kong A; Stefansson K A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet 2007, 39, 770. [DOI] [PubMed] [Google Scholar]
- (25).Ohara-Imaizumi M; Yoshida M; Aoyagi K; Saito T; Okamura T; Takenaka H; Akimoto Y; Nakamichi Y; Takanashi-Yanobu R; Nishiwaki C; Kawakami H; Kato N; Hisanaga S; Kakei M; Nagamatsu S Deletion of CDKAL1 Affects Mitochondrial ATP Generation and First-Phase Insulin Exocytosis. PLoS One 2010, 5, e15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Landgraf BJ; McCarthy EL; Booker SJ Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem 2016, 85, 485. [DOI] [PubMed] [Google Scholar]
- (27).Jessberger S; Gage FH; Eisch AJ; Lagace DC Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci. 2009, 32, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liebl J; Fürst R; Vollmar AM; Zahler S Twice switched at birth: Cell cycle-independent roles of the ″neuron-specific″ cyclin-dependent kinase 5 (Cdk5) in non-neuronal cells. Cell. Signalling 2011, 23, 1698. [DOI] [PubMed] [Google Scholar]
- (29).Reiter V; Matschkal DMS; Wagner M; Globisch D; Kneuttinger AC; Müller M; Carell T The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res. 2012, 40, 6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wei F-Y; Zhou B; Suzuki T; Miyata K; Ujihara Y; Horiguchi H; Takahashi N; Xie P; Michiue H; Fujimura A; Kaitsuka T; Matsui H; Koga Y; Mohri S; Suzuki T; Oike Y; Tomizawa K Cdk5rap1-Mediated 2-Methylthio Modification of Mitochondrial tRNAs Governs Protein Translation and Contributes to Myopathy in Mice and Humans. Cell Metab. 2015, 21, 428. [DOI] [PubMed] [Google Scholar]
- (31).Anton BP; Saleh L; Benner JS; Raleigh EA; Kasif S; Roberts RJ RimO, a MiaB-like enzyme, methylthiolates the universally conserved Asp88 residue of ribosomal protein S12 in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee KH; Saleh L; Anton BP; Madinger CL; Benner JS; Iwig DF; Roberts RJ; Krebs C; Booker SJ Characterization of RimO, a New Member of the Methylthiotransferase Subclass of the Radical SAM Superfamily. Biochemistry 2009, 48, 10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Landgraf BJ; Booker SJ Stereochemical Course of the Reaction Catalyzed by RimO, a Radical SAM Methylthiotransferase. J. Am. Chem. Soc 2016, 138, 2889. [DOI] [PubMed] [Google Scholar]
- (34).Sanyal I; Cohen G; Flint DH Biotin synthase - purification, characterization as a [2Fe-2S] cluster protein, and in vitro activity of the Escherichia coli bioB gene product. Biochemistry 1994, 33, 3625. [DOI] [PubMed] [Google Scholar]
- (35).Miller JR; Busby RW; Jordan SW; Cheek J; Henshaw TF; Ashley GW; Broderick JB; Cronan JE; Marletta MA Escherichia coli LipA is a lipoyl synthase: In vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry 2000, 39, 15166. [DOI] [PubMed] [Google Scholar]
- (36).Ugulava NB; Surerus KK; Jarrett JT Evidence from Mössbauer spectroscopy for distinct 2Fe-2S (2+) and 4Fe-4S (2+) cluster binding sites in biotin synthase from Escherichia coli. J. Am. Chem. Soc 2002, 124, 9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cosper MM; Jameson GNL; Hernandez HL; Krebs C; Huynh BH; Johnson MK Characterization of the Cofactor Composition of Escherichia coli Biotin Synthase. Biochemistry 2004, 43, 2007. [DOI] [PubMed] [Google Scholar]
- (38).Cicchillo RM; Lee KH; Baleanu-Gogonea C; Nesbitt NM; Krebs C; Booker SJ Escherichia coli lipoyl synthase binds two distinct [4Fe-4S] clusters per polypeptide. Biochemistry 2004, 43, 11770. [DOI] [PubMed] [Google Scholar]
- (39).Hernández HL; Pierrel F; Elleingand E; García-Serres R; Huynh BH; Johnson MK; Fontecave M; Atta M MiaB, a bifunctional radical-S-adenosylmethionine enzyme involved in the thiolation and methylation of tRNA, contains two essential [4Fe-4S] clusters. Biochemistry 2007, 46, 5140. [DOI] [PubMed] [Google Scholar]
- (40).Ugulava NB; Sacanell CJ; Jarrett JT Spectroscopic changes during a single turnover of biotin synthase: Destruction of a 2Fe-2S cluster accompanies sulfur insertion. Biochemistry 2001, 40, 8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jameson GNL; Cosper MM; Hernández HL; Johnson MK; Huynh BH Role of the [2Fe-2S] Cluster in Recombinant Escherichia coli Biotin Synthase. Biochemistry 2004, 43, 2022. [DOI] [PubMed] [Google Scholar]
- (42).Lanz ND; Pandelia M-E; Kakar ES; Lee KH; Krebs C; Booker SJ Evidence for a Catalytically and Kinetically Competent Enzyme-Substrate Cross-Linked Intermediate in Catalysis by Lipoyl Synthase. Biochemistry 2014, 53, 4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Tao LZ; Stich TA; Fugate CJ; Jarrett JT; Britt RD EPR-Derived Structure of a Paramagnetic Intermediate Generated by Biotin Synthase BioB. J. Am. Chem. Soc 2018, 140, 12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).McCarthy EL; Booker SJ Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science 2017, 358, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Landgraf BJ; Arcinas AJ; Lee KH; Booker SJ Identification of an Intermediate Methyl Carrier in the Radical S-Adenosylmethionine Methylthiotransferases RimO and MiaB. J. Am. Chem. Soc 2013, 135, 15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Maiocco SJ; Arcinas AJ; Landgraf BJ; Lee KH; Booker SJ; Elliott SJ Transformations of the FeS Clusters of the Methylthiotransferases MiaB and RimO, Detected by Direct Electro-chemistry. Biochemistry 2016, 55, 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mulliez E; Duarte V; Arragain S; Fontecave M; Atta M On the Role of Additional 4Fe-4S Clusters with a Free Coordination Site in Radical-SAM Enzymes, Front. Chem 2017, 5 DOI: 10.3389/fchem.2017.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Forouhar F; Arragain S; Atta M; Gambarelli S; Mouesca JM; Hussain M; Xiao R; Kieffer-Jaquinod S; Seetharaman J; Acton TB; Montelione GT; Mulliez E; Hunt JF; Fontecave M Two Fe-S clusters catalyze sulfur insertion by radical-SAM methylthiotransferases. Nat. Chem. Biol 2013, 9, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Molle T; Clémancey M; Latour JM; Kathirvelu V; Sicoli G; Forouhar F; Mulliez E; Gambarelli S; Atta M Unanticipated coordination of tris buffer to the Radical SAM cluster of the RimO methylthiotransferase. J. Biol. Inorg. Chem 2016, 21, 549. [DOI] [PubMed] [Google Scholar]
- (50).McLaughlin MI; Lanz ND; Goldman PJ; Lee KH; Booker SJ; Drennan CL Crystallographic snapshots of sulfur insertion by lipoyl synthase. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Krebs C; Henshaw TF; Cheek J; Huynh BH; Broderick JB Conversion of 3Fe-4S to 4Fe-4S clusters in native pyruvate formate-lyase activating enzyme: Mössbauer characterization and implications for mechanism. J. Am. Chem. Soc 2000, 122, 12497. [Google Scholar]
- (52).Lanz ND; Grove TL; Gogonea CB; Lee K-H; Krebs C; Booker SJ RImN and AtsB as Models for the Overproduction and Characterization Radical SAM Proteins Natural Product Biosynthesis By Microorganisms and Plants, Part B; Methods in Enzymology; Elsevier, 2012; Vol. 516, p 125. [DOI] [PubMed] [Google Scholar]
- (53).Münck E In Physical Methods in Bioinorganic Chemistry; Que L Jr., Ed.; University Science Books: Sausalito, CA, 2000; p 287. [Google Scholar]
- (54).Papaefthymiou V; Girerd JJ; Moura I; Moura JJG; Münck E Mössbauer study of D. gigas ferredoxin-II and spin-coupling model for the Fe3S4 cluster with valence delocalization. J. Am. Chem. Soc 1987, 109, 4703. [Google Scholar]
- (55).Hu ZG; Jollie D; Burgess BK; Stephens PJ; Münck E Mössbauer and EPR studies of Azotobacter vinelandii ferredoxin-I. Biochemistry 1994, 33, 14475. [DOI] [PubMed] [Google Scholar]
- (56).Gorun SM; Lippard SJ Magnetostructural correlations in magnetically coupled (μ-oxo)diiron(III) complexes. Inorg. Chem 1991, 30, 1625. [Google Scholar]
- (57).Baldwin MJ; Stemmler TL; Riggs Gelasco PJ; Kirk ML; Penner Hahn JE; Pecoraro VL Structural and magnetic effects of successive protonations of oxo bridges in high-valent manganese dimers. J. Am. Chem. Soc 1994, 116, 11349. [Google Scholar]
- (58).Pantazis DA; Krewald V; Orio M; Neese F Theoretical magnetochemistry of dinuclear manganese complexes: broken symmetry density functional theory investigation on the influence of bridging motifs on structure and magnetism. Dalton Trans. 2010, 39, 4959. [DOI] [PubMed] [Google Scholar]
- (59).Kambe K On the paramagnetic susceptibilities of some polynuclear complex salts. J. Phys. Soc. Jpn 1950, 5, 48. [Google Scholar]
- (60).Bencini A; Gatteschi D EPR of Exchange Coupled Systems; Springer-Verlag: Berlin, 1990. [Google Scholar]
- (61).Mouesca JM; Noodleman L; Case DA; Lamotte B Spin-densities and spin coupling in iron-sulfur clusters - a new analysis of hyperfine coupling constants. Inorg. Chem 1995, 34, 4347. [Google Scholar]
- (62).Pandelia M-E; Lanz ND; Booker SJ; Krebs C Mössbauer spectroscopy of Fe/S proteins. Biochim. Biophys. Acta, Mol. Cell Res 2015, 1853, 1395. [DOI] [PubMed] [Google Scholar]
- (63).Blondin G; Girerd JJ Interplay of electron exchange and electron transfer in metal polynuclear complexes in proteins or chemical models. Chem. Rev 1990, 90, 1359. [Google Scholar]
- (64).Dutta SK; Ensling J; Werner R; Flörke U; Haase W; Gütlich P; Nag K Valence-delocalized and valence-trapped Fe(II)Fe(III) complexes: Drastic influence of the ligands. Angew. Chem., Int. Ed. Engl 1997, 36, 152. [Google Scholar]
- (65).Borshch SA; Bominaar EL; Blondin G; Girerd JJ Double exchange and vibronic coupling in mixed-valent systems - origin of the broken-symmetry ground state of Fe3S40 cores in proteins and models. J. Am. Chem. Soc 1993, 115, 5155. [Google Scholar]
- (66).Gamelin DR; Bominaar EL; Kirk ML; Wieghardt K; Solomon EI Excited-state contributions to ground-state properties of mixed-valence dimers: Spectral and electronic-structural studies of [Fe2(OH)3(tmtacn)2]2+ related to the Fe2S2+ active sites of plant-type ferredoxins. J. Am. Chem. Soc 1996, 118, 8085. [Google Scholar]
- (67).Glaser T; Rose K; Shadle SE; Hedman B; Hodgson KO; Solomon EI S K-edge X-ray absorption studies of tetranuclear iron-sulfur clusters: μ-sulfide bonding and its contribution to electron delocalization. J. Am. Chem. Soc 2001, 123, 442. [DOI] [PubMed] [Google Scholar]
- (68).Dey A; Glaser T; Moura JJG; Holm RH; Hedman B; Hodgson KO; Solomon EI Ligand K-edge X-ray absorption spectroscopy and DFT calculations on [Fe3S4]0,+ clusters: Delocalization, redox, and effect of the protein environment. J. Am. Chem. Soc 2004, 126, 16868. [DOI] [PubMed] [Google Scholar]
- (69).Johnson MK; Smith AD Iron-sulfur proteins. Encyclopedia of Inorganic Chemistry, 2nd ed.; John Wiley & Sons: Chichester, 2005; p 2589. [Google Scholar]
- (70).Butt JN; Armstrong FA; Breton J; George SJ; Thomson AJ; Hatchikian EC Investigation of metalion uptake reactivities of [3Fe-4S] clusters in proteins - voltammerty of coadsorbed ferredoxin aminocyclitol films at graphite electrodes and spectroscopic identification of transformed clusters. J. Am. Chem. Soc 1991, 113, 6663. [Google Scholar]
- (71).Duff JLC; Breton JLJ; Butt JN; Armstrong FA; Thomson AJ Novel redox chemistry of [3Fe-4S] clusters: Electrochemical characterization of the all-Fe(II) form of the [3Fe-4S] cluster generated reversibly in various proteins and its spectroscopic investigation in Sulfolobus acidocaldarius ferredoxin. J. Am. Chem. Soc 1996, 118, 8593. [Google Scholar]
- (72).Molle T; Moreau Y; Clémancey M; Forouhar F; Ravanat JL; Duraffourg N; Fourmond V; Latour JM; Gambarelli S; Mulliez E; Atta M Redox Behavior of the S-Adenosylmethionine (SAM)-Binding Fe-S Cluster in Methylthiotransferase RimO, toward Understanding Dual SAM Activity. Biochemistry 2016, 55, 5798. [DOI] [PubMed] [Google Scholar]
- (73).Tamanaha E; Zhang B; Guo Y; Chang W.-c.; Barr EW; Xing G; St. Clair J; Ye S; Neese F; Bollinger JM Jr.; Krebs C Spectroscopic Evidence for the Two C-H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. J. Am. Chem. Soc 2016, 138, 8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).McMillen DF; Golden DM Hydrocarbon bond-dissociation energies. Annu. Rev. Phys. Chem 1982, 33, 493. [Google Scholar]
- (75).Arcinas AJ Mechanistic Studies of the Radical S-Adenosylmethinine tRNA Methylthiotransferase MiaB, Ph.D. Thesis, The Pennsylvania State University, 2016. [Google Scholar]
- (76).Pierrel F; Douki T; Fontecave M; Atta M MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. J. Biol. Chem 2004, 279, 47555. [DOI] [PubMed] [Google Scholar]
- (77).Ote T; Hashimoto M; Ikeuchi Y; Su’etsugu M; Suzuki T; Katayama T; Kato JI Involvement of the Escherichia coli folate-binding protein YgfZ in RNA modification and regulation of chromosomal replication initiation. Mol. Microbiol 2006, 60, 252. [DOI] [PubMed] [Google Scholar]
- (78).Waller J; Alvarez S; Naponelli V; Lara-Nuñez A; Blaby I; Da Silva V; Ziemak M; Vickers T; Beverley S; Edison A; Rocca J; Gregory J; de Crécy-Lagard V; Hanson A A role for tetrahydrofolates in the metabolism of iron-sulfur clusters in all domains of life. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Gelling C; Dawes IW; Richhardt N; Lill R; Mühlenhoff U Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes. Mol. Cell. Biol 2008, 28, 1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Boutigny S; Saini A; Baidoo EEK; Yeung N; Keasling JD; Butland G Physical and Functional Interactions of a Monothiol Glutaredoxin and an Iron Sulfur Cluster Carrier Protein with the Sulfur-donating Radical S-Adenosyl-L-methionine Enzyme MiaB. J. Biol. Chem 2013, 288, 14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.