

Abstract
Interleukin-1 receptor–associated kinase-1 (IRAK-1) and IRAK-4, as well as transforming growth factor β–activated kinase 1 (TAK1), are protein kinases essential for transducing inflammatory signals from interleukin receptors. IRAK family proteins and TAK1 have high sequence identity within the ATP-binding pocket, limiting the development of highly selective IRAK-1/4 or TAK1 inhibitors. Beyond kinase activity, IRAKs and TAK1 act as molecular scaffolds along with other signaling proteins, complicating the interpretation of experiments involving knockin or knockout approaches. In contrast, pharmacological manipulation offers the promise of targeting catalysis-mediated signaling without grossly disrupting the cellular architecture. Recently, we reported the discovery of takinib, a potent and highly selective TAK1 inhibitor that has only marginal activity against IRAK-4. On the basis of the TAK1–takinib complex structure and the structure of IRAK-1/4, here we defined critical contact sites of the takinib scaffold within the nucleotide-binding sites of each respective kinase. Kinase activity testing of takinib analogs against IRAK-4 identified a highly potent IRAK-4 inhibitor (HS-243). In a kinome-wide screen of 468 protein kinases, HS-243 had exquisite selectivity toward both IRAK-1 (IC50 = 24 nm) and IRAK-4 (IC50 = 20 nm), with only minimal TAK1-inhibiting activity (IC50 = 0.5 μm). Using HS-243 and takinib, we evaluated the consequences of cytokine/chemokine responses after selective inhibition of IRAK-1/4 or TAK1 in response to lipopolysaccharide challenge in human rheumatoid arthritis fibroblast-like synoviocytes. Our results indicate that HS-243 specifically inhibits intracellular IRAKs without TAK1 inhibition and that these kinases have distinct, nonredundant signaling roles.
Keywords: inhibitor, immunology, autoimmune disease, interleukin, tumor necrosis factor (TNF)
Introduction
The immune response to pathogens and tissue damage is an essential aspect of our biology. However, sometimes as a consequence of direct immune challenge, the immune system remains hyperactivated, leading to autoimmune disorders, which are maladaptive and induce chronic disease. Rheumatoid arthritis (RA),2 inflammatory bowel disease, and ankylosing spondylitis have all been shown to be immune-mediated and associated with local release of proinflammatory cytokines, such as tumor necrosis factor (TNF) and IL-6, at sites of tissue damage (1–3). The development of disease-modifying anti-rheumatic drugs (DMARDs) such as anti-TNF drugs (i.e. Enbrel) and JAK inhibitors (Zeljanz) have transformed the therapeutic options available to autoimmune-affected patients (4, 5). Nevertheless, despite advances in more molecularly targeted DMARDs, a significant portion of patients still fail to respond to current therapeutic options. Often this lack of response is attributed to individual genetic variations, tolerance, and immune sensitization in the case of biologically based antibody therapies, leading to an unmet need for novel DMARD development (6–8).
One target area of great interest is the downstream kinases involved in the inflammatory signaling. In particular, there has been recent interest in the serine/threonine kinase interleukin-1 receptor associated protein kinases IRAK-1 and IRAK-4, as well as transforming growth factor β–activated kinase 1 (TAK1). IRAK-4 is an essential protein kinase in mediating pathogen recognition and local cytokine release (i.e. IL-1, IL-6, TNF) through Toll-like receptor (TLR) signaling in response to prokaryotic lipopolysaccharides (LPSs) (9, 10). Following inflammatory agonist binding (i.e. LPS, IL-1, TNF), MyD88 is recruited to the cytosolic receptor domain, where it triggers downstream phosphorylation of IRAK-1/4, which further stimulates downstream NF-κB signaling and proinflammatory cytokine production (11). It has been shown that aberrant IRAK-4 signaling through hyperactive cytokine signaling is a key regulator of innate and adaptive immune cells in autoimmune diseases (12) (13, 14). Because of its role in inflammatory signaling, IRAK-4 has been the focus of numerous drug development platforms and recently advanced to clinical studies for autoimmune diseases (15–19).
Although previous groups have designed IRAK-4 inhibitors with modest potency, their selectivity within the human kinome has not been examined, begging the question of off-target effects in vitro and in vivo (20). In particular, off-target effects through TAK1 are likely, given the high sequence conservation between the binding pockets of IRAK-1/4 and TAK1; the amino acid sequence identity within the nucleotide-binding pocket of all three protein kinases is ∼93%. Examination of the co-crystal structure of TAK1–takinib identifies critical binding sites of the N-acyl-aminobenzimidazole scaffold within the protein kinase, which when compared in molecular modeling studies with IRAK-1/4 reveals two amino acid variations that may allow for the development of an IRAK discriminatory molecule from the takinib backbone. Herein we identified a previously developed takinib analog, HS-243, as a potent and super-selective IRAK-1/4 inhibitor with minimal TAK1 activity (IC50 = ∼0.5 μm). HS-243's IRAK-1/4 selectivity was further reflected in kinome-wide testing against 468 human protein kinases, which showed 38- and 15-fold change separations from HS-243's IRAK-1/4 activity compared with the next best kinase inhibited at 10 μm. Cellular assays indicate that HS-243 has bioactivity and acts as a strong anti-inflammatory agent with distinct biological responses compared with the parent inhibitor, takinib.
Results
Takinib analogs display varying potency against IRAK-4
Overlay of crystal structures of IRAK-4 (PDB code 2NRU, shown in blue) and TAK1 (PDB code 5V5N, shown in green) using PyMOL shows high sequence and structural homology in the ATP-binding pocket contributing to the affinity of takinib toward both kinases (Fig. 1a). Takinib fit was assessed with both kinases placed in the DFG-in orientation. A surface representation indicates that takinib lays within the IRAK-4–binding pocket with minimal steric clash (Fig. 1b). However, despite the close 93% overall amino acid identity within the respective ATP-binding pockets of TAK1, IRAK-4 (and IRAK-1) when aligned, significant amino acid variations are present that provide a discriminatory axis for discovery of a takinib analog toward IRAKs over TAK1 (Fig. 1c). Specifically, although the catalytic lysine 63 and aspartate 175 of the DFG motif align, and critical hydrophobic interactions of takinib-TAK1 binding mediated by Tyr-106 and Gly-110 are also congruent between IRAK-1/4 and TAK1, residues preceding the DFG motif are quite distinctive. Furthermore, modeling of IRAK-4 inhibitors suggest π–π stacking interactions with Tyr-262 of IRAK-1/4, which is absent in takinib–TAK1 interactions.
Figure 1.

Structural comparison of TAK1 against IRAK-1 and -4. a, overlay of the ribbon structure of IRAK-4 and TAK1. b, close-up view of the surface representation of takinib in the ATP binding pocket of TAK1 and modeled into IRAK-4. c, amino acid sequence alignment of TAK1, IRAK-4, and IRAK-1 highlighting key amino acid differences.
To identify a novel IRAK-1/4 inhibitor, we tested a series of takinib analogs against IRAK-4. Fig. 2a outlines regions of the parent takinib scaffold subjected to modification (see “Experimental procedures”). Analogs were tested in parallel against purified recombinant IRAK-4 in an in vitro kinase assay and identified HS-238 (643 nm), HS-242 (149 nm), and HS-243 (48 nm) as more potent IRAK-4 inhibitors compared with the parent compound takinib (Fig. 2, b and c). When tested at 1 μm, HS-243 showed 92% inhibition of IRAK-4 compared with takinib at 56% (Fig. 2d). We next sought to compare HS-243 to the previously described N-acyl-aminobenzimidazole (commercially sourced by Sigma; Sigma IRAK-1/4) by Powers and co-workers (18, 21, 22). The potency of HS-243 compared favorably with Sigma IRAK-1/4, which exhibited an IC50 of 134 nm in our assay (Fig. 2e). The improved IRAK-4 potency of HS-243 over the parent compound takinib is related to the R3 position replacement of the carbamide to a nitro group. Other modifications, particularly in the R2 site, reduced potency toward IRAK-4, indicating the addition of functional groups at this position sterically hinders binding (Fig. 2b). It is important to note that IRAK-4 has a relatively high Km for ATP at ∼500 μm (Fig. S1). Therefore, to ensure optimal kinase activity in vitro and meaningful inhibition constants for all our analogs, assays for this protein kinase were conducted well above IRAK-4 Km at 2 mm. Prior studies by others against IRAK-4 compounds, such as Sigma IRAK-1/4 were performed well below the true IRAK-4 Km for ATP, leading to an overestimation of the Ki and potentially a lack of selectivity and potency in vivo at physiological [ATP] (18, 21, 22).
Figure 2.

Structure–activity studies of takinib analogs against IRAK-4. a, core aminobenzimidazole scaffold showing regions targeted for modification. b, outline of modifications made during the SAR campaign and corresponding IC50 determined by mean IRAK-4 radioactive [32P]ATP filter-binding assay inhibition from vehicle. c, titrations of takinib analogs against IRAK-4, presented as percentages of inhibition from vehicle control. The data points represent means ± S.E. (n = 2). d, comparison of percentages of inhibition of DMSO, takinib (1 μm), and HS-243 (1 μm) against IRAK-4. e, dose-dependent response of HS-243 compared with commercially available Sigma IRAK-1/4 compound. The data points represent means ± S.E. (n = 2).
HS-243 displays exquisite selectivity toward IRAK-1 and -4 over all other human kinases
We next sought to test the selectivity of leading IRAK-4 analogs against TAK1 to identify kinase-specific compounds (Fig. 3a). As previously reported, HS-206 (takinib) showed greater affinity toward TAK1 than IRAK-4, whereas HS-238 and HS-242 showed improvements in IRAK-4 potency, exhibiting IC50 values of 643 and 149 nm, respectively, compared with takinib (Fig. 3b). However, they retained significant TAK1 activity and are dual TAK1/IRAK-4 inhibitors (Fig. 3, c and d). In contrast, HS-243 showed exquisite potency toward IRAK-1/4 (20 and 24 nm) with a ∼23-fold difference in IC50 toward TAK1 activity (IC50 > 500 μm), representing a selective IRAK-1/4 inhibitor (Fig. 3e).
Figure 3.

Structure and in vitro profiling of lead compound and other analogs against IRAK-4 and TAK1. a, structures of HS-243 IRAK-1/4 inhibitor, HS-242 dual TAK1/IRAK-4 inhibitor, HS-206 (takinib) TAK1 inhibitor, and HS-238 mixed TAK1/IRAK-4 inhibitor. b–e, data show kinase inhibition generated for each indicated analog against purified IRAK-4 and TAK1. The data points represent means ± S.E. (n = 2).
To understand the structural basis for selectivity, we performed unconstrained molecular docking of HS-243 in the ATP-binding pocket of IRAK-4 (PDB code 2NRU) and compared it with the X-ray structure of takinib bound to TAK1. The binding modes were similar overall but highlight interactions that might explain changes in potency and selectivity. In particular, we note hydrogen bonds between the Tyr-262 and a bridging water that interacts with the nitro group of HS-243. Tyr-262, which is a methionine in TAK1, is also predicted to form π–π stacking interactions with the nitrobenzyl component. This is an interaction that cannot occur in TAK1 because of the side-chain difference, which may account for greater specificity of HS-243 toward IRAK-4 (Fig. 4). Furthermore, hydrophobic interaction with residues Val-263, Tyr-264, Met-265, and Pro-266 may contribute to drug stability and potency (Fig. 4a and Fig. S2).
Figure 4.

a, docking pose of HS-243 in the ATP binding pocket of IRAK-4. π–π stacking interactions between HS-243 and Tyr-262 are in pink dots. Other interacting residues are labeled. Hydrogen bonds are shown with green dashes. b, comparison with TAK1 (PDB code 5V5N). c, kinase activity of IRAK-4 (WT), IRAK-4 (Y262A), and IRAK-4 (Y262T) tyrosine gatekeeper mutants. d, titrations of HS-243 against purified IRAK-4 Tyr-262 mutants (n = 2).
We next sought to further validate the π–π stacking interactions between the nitrobenzyl group of HS-243 and Y262 of IRAK-4. Previous work by Wang et al. (22) has shown that Tyr-262 acts as a tyrosine gatekeeper in IRAK-4 and plays a critical role in maintaining the active orientation of the kinase. Consistent with previous literature, we found that IRAK-4 Tyr-262 mutants showed significantly reduced kinase activity compared with WT IRAK-4, further supporting the position that Tyr-262 plays an integral role in maintaining active kinase orientation (Fig. 4c). Additionally, titration of HS-243 against IRAK-4 Y262T or Y262A mutants showed minimal to no percentage of inhibition at 0.3 and 1 μm (Fig. 4d).
Following identification of a lead selective compound with greater potency toward IRAK-4 (HS-243) over TAK1, we further evaluated its selectivity across 468 distinct human protein kinases at 10 μm HS-243. Remarkably, in this study HS-243 showed exquisite selectivity and potency toward IRAK-4 and closely related IRAK-1, inhibiting the kinases 98.6 and 99.35%, respectively (Fig. 5, a–c, and Table S1). To tease out the selectivity of HS-243 further, we carried out titrations against the top five protein kinases identified in the one shot 10 μm screen. The Kd for HS-243 toward IRAK-1 was 24 nm, and that for IRAK-4 was 20 nm, followed by 423 nm for TAK1, 662 nm for CLK4, and 2,278 nm for DYKR1B (Fig. 5d). This study further highlighted the selectivity of HS-243 to the IRAKs over all other protein kinase, with next best inhibited protein kinases being TAK1, and nonrelated CLK4 and DYRK1B at 21-, 33-, and 105-fold less sensitivity, respectively. Importantly, IC50 values for HS-243 against TAK1 determined by our in-house enzymatic assay agreed closely with values determined independently in the commercial kinome-wide study. Finally, to rule out nonprotein kinase off-target binding, we next explored the selectivity of HS-243 against an array of known purine-binding receptors/enzymes including 5-HT receptors; opioid receptors; β receptors; GABA, type A; SERT; and D, H, and M receptors. No significant binding was found within the broader purinome, further highlighting the exquisite selectivity of HS-243 toward IRAK-1 and -4 (Fig. S3).
Figure 5.

Selectivity of HS-243 within the human kinome. a, kinome-wide screening of HS-243 against 468 human kinases. b, percentage of inhibition of top kinase hits from kinase screen (percentage of control from vehicle). c, dendrogram of human nononcogenic kinases show selectivity of HS-243 in the human kinome. The size of the dot is indicative of kinase potency. d, IC50 values of top five kinases as determined by DiscoverX kinase assay. The data points represent means ± S.E. (n = 2). e, IC50 values of HS-243 against IRAK-4, IRAK-1, TAK1, CLK4, and DYKR1B (n = 2).
HS-243 potently reduces the proinflammatory response of RA cells and macrophages
Clinical indications for IRAK-1/4 inhibitors have been in autoimmune diseases in which maladaptive proinflammatory signaling is a key signature of disease. To test and compare the anti-inflammatory effects of HS-243 (IRAK-1/4) against Sigma 1/4, HS-242 (dual IRAK-1/4, TAK1), and HS-206 (takinib, TAK1), we stimulated human isolated RA-FLS cells with LPS followed by treatment with indicated drug at 10 μm. Cytokine array analysis of 36 various cytokines and chemokines indicated that robust changes in drug effects were seen, highlighted by significant effects of HS-243, Sigma 1/4, and HS-242. IRAK-1/4 inhibitors displayed distinct effects on CCL5, CXCL12, MIF, and IL-18 protein expression levels when compared with TAK1 inhibition with takinib (Fig. 6, a–e). Furthermore, overall distinct cytokine profiles were observed with significant differences in overall cytokine/chemokine effects between HS-243–Sigma (p < 0.002), DMSO–HS-243 (p < 0.0002), DMSO–Sigma (p < 0.0001), and DMSO–HS-242 (p < 0.0001) (Fig. 6f). A general schematic of IRAK-1/4 signaling following proinflammatory stimuli is shown (Fig. 6g).
Figure 6.

IRAK-1/4 inhibition has distinct cytokine profile from TAK1. Human RA-FLSs were stimulated with LPS and treated with either DMSO, HS-243, Sigma 1/4, takinib, or HS-242 at 10 μm for 24 h before cytokine profiling. a, representative cytokine proteome arrays with indicted cytokine positioning. b–e, quantification of CCL5 (b), CXCL12 (c), MIF (d), and IL-18 (e) expression. *, differences between DMSO and treatment; #, difference from HS-243. The data points represent means ± S.E. (n = 4). f, heat map of 30 cytokines/chemokines expression. g, schematic of IRAK-1/4 and TAK1 signaling. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Because of the selectivity and potency of HS-243 toward IRAK-1/4, we next explored the potential of this compound to reduce inflammatory signaling in THP-1 human macrophages challenged with LPS (100 ng/ml). Following binding to the TLR4 receptor, LPS stimulates the IRAK-1/4 pathway of proinflammatory cytokine signaling that leads to NF-κB activation. Immediately following LPS stimulation, THP-1 macrophages were treated with 10 μm HS-243 or vehicle, and supernatant was collected 24 h later. In a panel of 105 cytokines/chemokines, HS-243 significantly reduced the secretion of 15 cytokines, including IL-8 (p < 0.003), CD14 (p < 0.002), GRO-α (p < 0.001), MIP-1a (p < 0.007), MIP-3a (p < 0.002), uPAR (p < 0.014), Osteopontin (p < 0.018), MMP-9 (p < 0.001), MCP-1 (p < 0.011), I-TAC (p < 0.001), TIM-3 (p < 0.016), IP-10 (p < 0.014), GDF-15 (p < 0.006), and RANTES (p < 0.015) (Fig. 7 and Table S2). This profile contrasts dramatically with that reported previously with takinib, which showed discrete ∼9-fold inhibition of TNF release with little impact on any other cytokines (23, 24). Whereas the discrete actions of takinib in the LPS challenge model are consistent with selective TAK1 inhibition in cells, the broader actions of HS-243 reported herein are consistent with the expected outcomes of IRAK-1/4 inhibition.
Figure 7.

THP-1 human macrophages were differentiated with PMA for 72 h followed by a 24-h rest period in PMA-free medium. The cells were stimulated with LPS (10 ng/ml) immediately followed by 10 μm HS-243. 105 cytokines/chemokines were profiled from the supernatant of the media. a and b, significant changes seen in MIP-3a, Tim-3, uPAR, RANTES, Osteopontin, MMP-9, MIP-1a, MCP-1, I-TAC, IL-8, GDF-15, IP-10, and chitinase-3–like 1 protein expression (n = 3–4). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
IRAK-1/4 inhibition reduces percentage of survival in pancreatic and breast cancer cell lines
Previously TAK1 has been shown to play an integral role in mediating TNF-induced apoptosis representing a novel therapeutic axis in certain cancers (19). Because of high homology between TAK1 and IRAK-1/4, we were interested in the effects of IRAK-1/4 inhibition on cancer cells. To evaluate the potential anti-cancer properties of IRAK-1/4 inhibition, we tested HS-243 against seven cancer cell lines. At 10 μm, HS-243 inhibited cell survival by 21% for AN3-CA, a pancreatic cancer cell line, and 13% for SKOV-3 (Fig. 8). Furthermore, previous studies with TAK1 inhibitors have shown that inhibition of inflammatory kinases in cancers in conjunction with exogenous upstream ligand stimulation can induce apoptosis. In an effort to understand the IRAK-1/4 role in mediating exogenous interleukin signaling and downstream signaling pathways, we stimulated various cancer cell lines with IL-1β (30 ng/ml) and HS-243 (10 μm) for 24 h. The addition of IL-1β in conjunction with HS-243 increased cell death to 46% in SK-OV-3 (ovarian), 33% in AN3-CA (pancreatic), and 31% in H460 (colon) (Fig. 8).
Figure 8.
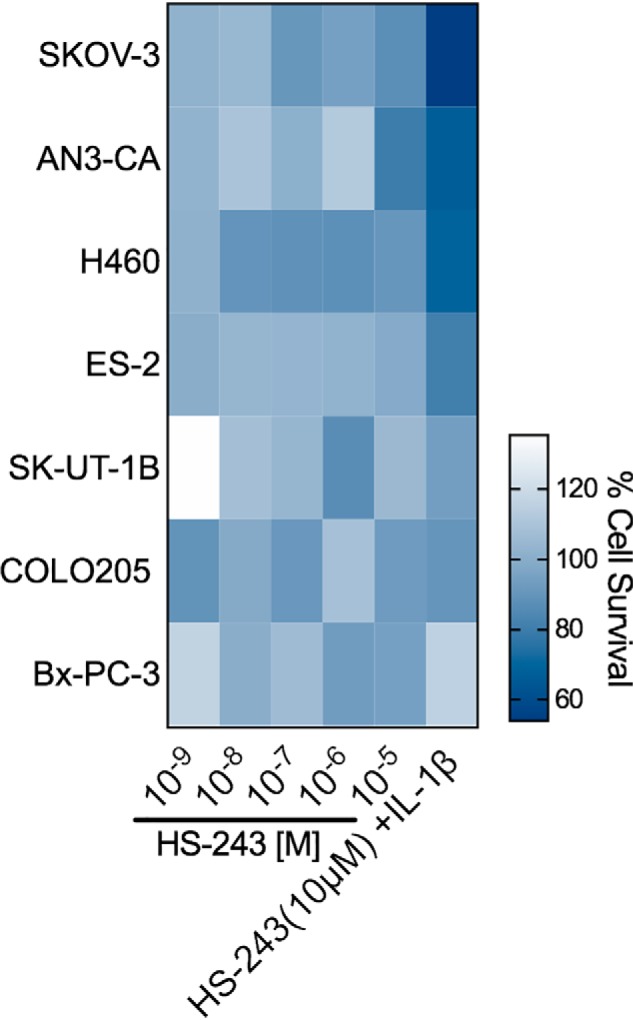
Percentage of survival of IRAK-1/4 inhibited cancer cells. Shown are the percentages of survival of seven cancer cell lines treated for 24 h in the presence of 10 μm HS-243 or HS-243 (10 μm) and IL-1β (30 ng/ml). The data points represent means (n = 2–4).
Discussion
Evidence supporting the IRAKs as therapeutic targets in autoimmune diseases stems largely from genetic data around IRAK-4. Mutations within the IRAK-4 locus result in premature stop codons and are associated with loss of functional expression of the protein kinase (25). These cause a unique immunodeficiency in early stages of life, rendering adolescents susceptible to certain bacterial infections (25, 26). Interestingly, this impairment resolves during adulthood. IRAK-4–deficient patients show retention of neutrophil function through TLR-9 signaling mechanism, highlighting the role of the protein kinase in regulating inflammatory pathways (27). At the time of writing, ATP competitive inhibitors targeting IRAK-4 are under investigation in clinical trials because of their potential in autoimmune disorders (28, 29). However, because of the close structural similarities within the ATP-binding pocket of the IRAK family and TAK1, the relative contributions and consequences of pharmacological inhibition of both protein kinases in vivo have not been considered. Whereas polypharmacological inhibition of both TAK1 and IRAK-4 may be advantageous, it is equally plausible that such inhibitors could have a limited therapeutic window or result in off-target actions. Development of highly selective pharmacological inhibitors that discriminate between these protein kinase families in vivo is therefore highly desirable. Here, we have shown that the takinib scaffold can serve as an effective starting point to develop inhibitors that are specific for IRAK-1 and -4 while dialing out their closest homolog, TAK1. Significantly, HS-243 retained the exquisite selectivity across the kinome as observed with the original parent scaffold, takinib (19). In the case of HS-243 only, IRAK-1 and -4 are targeted, rather than TAK1, and no other protein kinase is completely inhibited at 10 μm. This study suggests that the core takinib aminobenzimidazole molecule is intrinsically tailored toward TAK1 and closely related IRAK family members and can be effectively utilized to develop super-selective inhibitors for each member and no other protein kinase. This selectivity can be extended to all members of the larger purinome (all proteins that bind purines), as evidenced by the partial purinome screen carried out in this study and the wider purinome specificity study reported previously with takinib by screening against immobilized γ-phosphate ATP charged with whole cellular homogenates (19, 30). To our knowledge, no other protein kinase inhibitor scaffold has exhibited such intrinsic selectivity toward a specific subset of protein kinases. In support of intracellular selectivity, RA-FLS cells immune-challenged with LPS and treated by HS-243 showed reduced proinflammatory signaling that is distinct from takinib (23). These data support the idea that IRAK-1/4 and TAK1, despite participating in similar pathways, have distinct druggable responses, creating discrete patterns of cytokine inhibition. The role of IRAK-1/4 in mediating inflammatory signaling is well-established, and IRAK-4 inhibitors are advancing through clinical trials. However, the kinome-wide selectivity of leading the IRAK-4 inhibitors is not available, opening up the possibility that a significant portion of the observed efficacy with these inhibitors may be due to TAK1 inhibition. Therefore, advancement of HS-243 along the clinical path may provide a second-generation series of inhibitors displaying well-defined kinome selectivity, potentially reducing off-target side effects often conferred upon promiscuous kinase inhibitors.
Experimental procedures
Synthesis of molecules
See the supporting information.
Kinome screens
Kinome-wide screens were carried out independently by DiscoverX Inc. and kinase assay by the University of Dundee's kinome consortium.
Protein purification
TAK1–TAB1 (kinase domain residues 31–303 fused with 36 residues of TAB1 C-terminal domain residues 468–504) was expressed and purified as reported previously (30). In summary, His-TAK1–TAB1 was expressed in insect cells and purified using His-affinity column followed by tobacco etch virus cleavage and finally size-exclusion chromatography. Protein purity was verified using a gradient SDS-PAGE gel stained with Coomassie Blue. Protein concentration was measured using the Bradford method (31).
IRAK-4 N-GST (DU 8853) was purchased from the Medical Research Council 1 PPU, College of Life Sciences, University of Dundee (Dundee, UK). IRAK-4 Y262T (DU4684) and Y262A (DU8865) human plasmid expression vectors were purchased from Medical Research Council 1 PPU at the University of Dundee. HEK293T cells were transfected with 2 μg of HA-IRAK-4 Y262A or Y262T plasmid with X-tremeGENE HP DNA transfection reagent for 24 h and purified using HA-affinity resin.
Kinase assay
Activity of purified IRAK-4 protein was measured as previously described (32). In brief, IRAK-4 (20 ng/well) was incubated with 2 mm ATP containing radiolabeled [32P]ATP in the presence of 300 μm substrate peptide in a final volume of 40 μl in the presence of buffer (containing 50 mm Tris, pH 7.5, 0.1 mm EGTA, 0.1% β-Mercaptoethanol, 10 mm magnesium acetate, 0.5 mm MnCl) and indicated compounds. The reaction was terminated at 20 min with 10 μl of 1 m H3PO4. The remaining activity was measured using a scintillation counter. Dose–response curves were repeated two times and averaged.
Binding and functional assays of purinome-binding proteins
Binding of 5-HT1A; 5-HT1B; 5-HT1D; 5-HT1E; 5-HT2A; 5-HT2B; 5-HT2C; 5-HT3; 5-HT5A; 5-HT6; 5-HT7A; Alpha 1A; Alpha 1B; Alpha 1D; Alpha 2A; Alpha 2B; Alpha 2C; Beta 1; Beta 2; Beta 3; BZP Rat Brian Site; D1; D2; D3; D4; D5; DAR; DOR; GABA, type A; H1; H2; H3; H4; KOR; M1; M2; M3; M4; M5; MOR; NET; PBR; SERT; Sigma 1; Sigma 2; and NOP were tested by the University of North Carolina Psychoactive Drug Screening Program following previously established radioligand-binding assays (33).
Cell culture
THP-1 human macrophages were obtained from American Type Culture Collection. The cells were incubated at 37 °C in 5% CO2. THP-1 were cultured in RPMI 1640X, 10% fetal bovine serum, 1% penicillin–streptomycin, HEPES, pyruvate, glucose, and beta mercaptoethanol. RA-FLS cells were isolated as previously reported and cultured for no more than nine passages in Connaught Medical Research Laboratories (CRML) medium, 10% fetal bovine serum, and 1% penicillin–streptomycin. Cancer cell lines AN3 CA, BxPC-3, SK-UT-1B, ES-2, SKOV-3, H460, and COLO 205 were cultured according to American Type Culture Collection media guidelines.
Macrophage differentiation
THP-1 cells were treated with 100 nm phorbol 12-myristate 13-acetate (PMA) for 72 h in RPMI 1640X medium. The cells were rested in PMA-free medium 24 h prior to treatments. LPS (10 ng/ml) was used for proinflammatory stimulation.
Cytokine/chemokine proteome profile
RA-FLS cells or THP-1 cells were treated with 10 μm HS-243 or vehicle (DMSO), and 24 h after treatment, supernatant was added to human cytokine XL proteome array (R&D Systems) in accordance with the manufacturer's protocol. Chemiluminescence was used to visualize protein quantities.
Cell viability assay
The cells were cultured and treated as previously described. Briefly, the cells were plated at 80% confluency ∼104 in 96-well plates and treated with either takinib at various concentrations or takinib + IL-1β (30 ng/ml) for 24 h and compared with vehicle treated samples. Cell death was quantified using Cell Titer Glo 2.0 (Promega) according to the manufacturer's protocol.
Quantification and statistical analysis
GraphPad Prism 8 was used for statistical analysis. For each analysis, total n and S.E. are presented in the figure legends. An α of 0.05 was used for all statistical analysis.
Author contributions
S. A. S. and T. A. J. H. conceptualization; S. A. S., K. W. Y., D. A. C., D. G., and T. A. J. H. data curation; S. A. S. supervision; S. A. S. investigation; S. A. S., D. A. C., D. G., and K. D. W. visualization; S. A. S., P. F. H., and T. A. J. H. methodology; S. A. S., K. W. Y., D. A. C., D. G., K. D. W., and T. A. J. H. writing-original draft; S. A. S., P. F. H., K. W. Y., D. A. C., D. G., K. D. W., and T. A. J. H. writing-review and editing; P. F. H. and K. D. W. resources; K. W. Y. formal analysis; T. A. J. H. funding acquisition.
Supplementary Material
Acknowledgments
We thank the Medical Research Council PPU Reagents and Service Facility, College of Life Sciences, University of Dundee for the reagents and services indicated in this publication. Ki determinations, receptor binding profiles, agonist, and/or antagonist functional data were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program under Contract HHSN-271-2018-00023-C. The National Institute of Mental Health's Psychoactive Drug Screening Program is directed by Bryan L. Roth at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at the National Institute of Mental Health (Bethesda MD). T. A. J. H., P. F. H., and S. A. S. have filed patent claims on all molecules related to the takinib scaffold described in this body of work.
This work was supported by NIGMS, National Institutes of Health T32GM007105-45 (to S. A. S.), by a grant from the North Carolina Biotechnology Center (to T. A. J. H.), and by Welch Foundation Grant I-1829 (to D. G. and K. D. W.). T. A. J. H., S. A. S., and P. F. H. are part owners of EydisBio (Durham, NC), which options the takinib patent from Duke University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting schemes, Tables S1 and S2, and Figs. S1–S3.
- RA
- rheumatoid arthritis
- TNF
- tumor necrosis factor
- IL
- interleukin
- DMARD
- disease-modifying anti-rheumatic drug
- TLR
- Toll-like receptor
- LPS
- lipopolysaccharide
- FLS
- fibroblast-like synoviocyte
- PDB
- Protein Data Bank
- PMA
- phorbol 12-myristate 13-acetate
- PPU
- protein phosphorylation and ubiquitylation unit
- 5-HT
- 5-hydroxytryptamine.
References
- 1. Feldmann M., Brennan F. M., Elliott M., Katsikis P., and Maini R. N. (1994) TNFα as a therapeutic target in rheumatoid arthritis. Circ. Shock 43, 179–184 [PubMed] [Google Scholar]
- 2. Elliott M. J., Maini R. N., Feldmann M., Long-Fox A., Charles P., Katsikis P., Brennan F. M., Walker J., Bijl H., and Ghrayeb J. (1993) Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor α. Arthritis Rheum. 36, 1681–1690 10.1002/art.1780361206 [DOI] [PubMed] [Google Scholar]
- 3. Clark I. A. (2007) How TNF was recognized as a key mechanism of disease. Cytokine Growth Factor Rev. 18, 335–343 10.1016/j.cytogfr.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 4. Boyle D. L., Soma K., Hodge J., Kavanaugh A., Mandel D., Mease P., Shurmur R., Singhal A. K., Wei N., Rosengren S., Kaplan I., Krishnaswami S., Luo Z., Bradley J., and Firestein G. S. (2015) The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 74, 1311–1316 10.1136/annrheumdis-2014-206028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Danese S., Grisham M., Hodge J., and Telliez J. B. (2016) JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G155–G162 10.1152/ajpgi.00311.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feldmann M. (2002) Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2, 364–371 10.1038/nri802 [DOI] [PubMed] [Google Scholar]
- 7. Rubbert-Roth A., and Finckh A. (2009) Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Res. Ther. 11, (Suppl. 1) S1 10.1186/ar2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lam S. (2016) JAK inhibitors: A broadening approach in rheumatoid arthritis. Drugs Today (Barc.) 52, 467–469 10.1358/dot.2016.52.8.2543995 [DOI] [PubMed] [Google Scholar]
- 9. Henderson C., and Goldbach-Mansky R. (2010) Monogenic IL-1 mediated autoinflammatory and immunodeficiency syndromes: finding the right balance in response to danger signals. Clin. Immunol. 135, 210–222 10.1016/j.clim.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koziczak-Holbro M., Littlewood-Evans A., Pöllinger B., Kovarik J., Dawson J., Zenke G., Burkhart C., Müller M., and Gram H. (2009) The critical role of kinase activity of interleukin-1 receptor–associated kinase 4 in animal models of joint inflammation. Arthritis Rheum. 60, 1661–1671 10.1002/art.24552 [DOI] [PubMed] [Google Scholar]
- 11. Warner N., and Núñez G. (2013) MyD88: a critical adaptor protein in innate immunity signal transduction. J. Immunol. 190, 3–4 10.4049/jimmunol.1203103 [DOI] [PubMed] [Google Scholar]
- 12. Staschke K. A., Dong S., Saha J., Zhao J., Brooks N. A., Hepburn D. L., Xia J., Gulen M. F., Kang Z., Altuntas C. Z., Tuohy V. K., Gilmour R., Li X., and Na S. (2009) IRAK4 kinase activity is required for Th17 differentiation and Th17-mediated disease. J. Immunol. 183, 568–577 10.4049/jimmunol.0802361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Netea M. G., van der Graaf C., Van der Meer J. W., and Kullberg B. J. (2004) Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J. Leukoc. Biol. 75, 749–755 10.1189/jlb.1103543 [DOI] [PubMed] [Google Scholar]
- 14. Silver M. R., Margulis A., Wood N., Goldman S. J., Kasaian M., and Chaudhary D. (2010) IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm. Res. 59, 207–218 10.1007/s00011-009-0088-5 [DOI] [PubMed] [Google Scholar]
- 15. Buckley G. M., Ceska T. A., Fraser J. L., Gowers L., Groom C. R., Higueruelo A. P., Jenkins K., Mack S. R., Morgan T., Parry D. M., Pitt W. R., Rausch O., Richard M. D., and Sabin V. (2008) IRAK-4 inhibitors: II. A structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorg Med. Chem. Lett. 18, 3291–3295 10.1016/j.bmcl.2008.04.039 [DOI] [PubMed] [Google Scholar]
- 16. Buckley G. M., Gowers L., Higueruelo A. P., Jenkins K., Mack S. R., Morgan T., Parry D. M., Pitt W. R., Rausch O., Richard M. D., Sabin V., and Fraser J. L. (2008) IRAK-4 inhibitors: Part 1. A series of amides. Bioorg Med. Chem. Lett. 18, 3211–3214 10.1016/j.bmcl.2008.04.058 [DOI] [PubMed] [Google Scholar]
- 17. Buckley G. M., Fosbeary R., Fraser J. L., Gowers L., Higueruelo A. P., James L. A., Jenkins K., Mack S. R., Morgan T., Parry D. M., Pitt W. R., Rausch O., Richard M. D., and Sabin V. (2008) IRAK-4 inhibitors: Part III. A series of imidazo[1,2-a]pyridines. Bioorg. Med. Chem. Lett. 18, 3656–3660 10.1016/j.bmcl.2008.04.042 [DOI] [PubMed] [Google Scholar]
- 18. Wang Z., Sun D., Johnstone S., Cao Z., Gao X., Jaen J. C., Liu J., Lively S., Miao S., Sudom A., Tomooka C., Walker N. P., Wright M., Yan X., Ye Q., et al. (2015) Discovery of potent, selective, and orally bioavailable inhibitors of interleukin-1 receptor-associate kinase-4. Bioorg Med. Chem. Lett. 25, 5546–5550 10.1016/j.bmcl.2015.10.060 [DOI] [PubMed] [Google Scholar]
- 19. Totzke J., Gurbani D., Raphemot R., Hughes P. F., Bodoor K., Carlson D. A., Loiselle D. R., Bera A. K., Eibschutz L. S., Perkins M. M., Eubanks A. L., Campbell P. L., Fox D. A., Westover K. D., Haystead T. A. J., et al. (2017) Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNF-α inhibition for cancer and autoimmune disease. Cell Chem. Biol. 24, 1029–1039.e7 10.1016/j.chembiol.2017.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaudhary D., Robinson S., and Romero D. L. (2015) Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor– associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J. Med. Chem. 58, 96–110 10.1021/jm5016044 [DOI] [PubMed] [Google Scholar]
- 21. Powers J. P., Li S., Jaen J. C., Liu J., Walker N. P., Wang Z., and Wesche H. (2006) Discovery and initial SAR of inhibitors of interleukin-1 receptor–associated kinase-4. Bioorg Med. Chem. Lett. 16, 2842–2845 10.1016/j.bmcl.2006.03.020 [DOI] [PubMed] [Google Scholar]
- 22. Wang Z., Liu J., Sudom A., Ayres M., Li S., Wesche H., Powers J. P., and Walker N. P. (2006) Crystal structures of IRAK-4 kinase in complex with inhibitors: a serine/threonine kinase with tyrosine as a gatekeeper. Structure 14, 1835–1844 10.1016/j.str.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 23. Scarneo S. A., Mansourati A., Eibschutz L. S., Totzke J., Roques J. R., Loiselle D., Carlson D., Hughes P., and Haystead T. A. J. (2018) Genetic and pharmacological validation of TAK1 inhibition in macrophages as a therapeutic strategy to effectively inhibit TNF secretion. Sci. Rep. 8, 17058 10.1038/s41598-018-35189-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scarneo S. A., Eibschutz L. S., Bendele P. J., Yang K. W., Totzke J., Hughes P., Fox D. A., and Haystead T. A. J. (2019) Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res. Ther. 21, 292 10.1186/s13075-019-2073-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Picard C., Puel A., Bonnet M., Ku C. L., Bustamante J., Yang K., Soudais C., Dupuis S., Feinberg J., Fieschi C., Elbim C., Hitchcock R., Lammas D., Davies G., Al-Ghonaium A., et al. (2003) Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 299, 2076–2079 10.1126/science.1081902 [DOI] [PubMed] [Google Scholar]
- 26. Isnardi I., Ng Y. S., Srdanovic I., Motaghedi R., Rudchenko S., von Bernuth H., Zhang S. Y., Puel A., Jouanguy E., Picard C., Garty B. Z., Camcioglu Y., Doffinger R., Kumararatne D., Davies G., et al. (2008) IRAK-4– and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 29, 746–757 10.1016/j.immuni.2008.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoarau C., Gérard B., Lescanne E., Henry D., François S., Lacapère J. J., El Benna J., Dang P. M., Grandchamp B., Lebranchu Y., Gougerot-Pocidalo M. A., and Elbim C. (2007) TLR9 activation induces normal neutrophil responses in a child with IRAK-4 deficiency: involvement of the direct PI3K pathway. J. Immunol. 179, 4754–4765 10.4049/jimmunol.179.7.4754 [DOI] [PubMed] [Google Scholar]
- 28. Lee K. L., Ambler C. M., Anderson D. R., Boscoe B. P., Bree A. G., Brodfuehrer J. I., Chang J. S., Choi C., Chung S., Curran K. J., Day J. E., Dehnhardt C. M., Dower K., Drozda S. E., Frisbie R. K., et al. (2017) Discovery of clinical candidate 1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoli ne-6-carboxamide (PF-06650833), a potent, selective inhibitor of interleukin-1 receptor associated kinase 4 (IRAK4), by fragment-based drug design. J. Med. Chem. 60, 5521–5542 10.1021/acs.jmedchem.7b00231 [DOI] [PubMed] [Google Scholar]
- 29. Lee M. H., Balupuri A., Jung Y. R., Choi S., Lee A., Cho Y. S., and Kang N. S. (2018) Design of a novel and selective IRAK4 inhibitor using topological water network analysis and molecular modeling approaches. Molecules 23, E3136 10.3390/molecules23123136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan L., Nomanbhoy T., Gurbani D., Patricelli M., Hunter J., Geng J., Herhaus L., Zhang J., Pauls E., Ham Y., Choi H. G., Xie T., Deng X., Buhrlage S. J., Sim T., et al. (2015) Discovery of type II inhibitors of TGFβ-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2). J. Med. Chem. 58, 183–196 10.1021/jm500480k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 32. Hastie C. J., McLauchlan H. J., and Cohen P. (2006) Assay of protein kinases using radiolabeled ATP: a protocol. Nat. Protoc. 1, 968–971 10.1038/nprot.2006.149 [DOI] [PubMed] [Google Scholar]
- 33. Besnard J., Ruda G. F., Setola V., Abecassis K., Rodriguiz R. M., Huang X. P., Norval S., Sassano M. F., Shin A. I., Webster L. A., Simeons F. R., Stojanovski L., Prat A., Seidah N. G., Constam D. B., et al. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 10.1038/nature11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.