

Abstract
Cytochrome P450 family 102 subfamily A member 1 (CYP102A1) is a self-sufficient flavohemeprotein and a highly active bacterial enzyme capable of fatty acid hydroxylation at a >3,000 min−1 turnover rate. The CYP102A1 architecture has been postulated to be responsible for its extraordinary catalytic prowess. However, the structure of a functional full-length CYP102A1 enzyme remains to be determined. Herein, we used a cryo-EM single-particle approach, revealing that full-length CYP102A1 forms a homodimer in which both the heme and FAD domains contact each other. The FMN domain of one monomer was located close to the heme domain of the other monomer, exhibiting a trans configuration. Moreover, full-length CYP102A1 is highly dynamic, existing in multiple conformational states, including open and closed states. In the closed state, the FMN domain closely contacts the FAD domain, whereas in the open state, one of the FMN domains rotates away from its FAD domain and traverses to the heme domain of the other monomer. This structural arrangement and conformational dynamics may facilitate rapid intraflavin and trans FMN-to-heme electron transfers (ETs). Results with a variant having a 12-amino-acid deletion in the CYP102A1 linker region, connecting the catalytic heme and the diflavin reductase domains, further highlighted the importance of conformational dynamics in the ET process. Cryo-EM revealed that the Δ12 variant homodimer is conformationally more stable and incapable of FMN-to-heme ET. We conclude that closed-to-open alternation is crucial for redox partner recognition and formation of an active ET complex for CYP102A1 catalysis.
Keywords: cytochrome P450, conformational change, electron transfer complex, protein dynamic, cryo-electron microscopy, cytochrome P450 family 102 subfamily A member 1 (CYP102A1), P450 reductase, catalysis, structural biology, redox partner recognition, cytochrome P450 reductase fusion protein
Introduction
WT CYP102A1 enzyme from Bacillus megaterium exhibits high turnover rates for hydroxylating long-chain fatty acids. A turnover rate of ∼17,000 min−1 was reported for hydroxylation of arachidonic acid (1), which is more than 2 orders of magnitude higher than human P450 enzymes known for metabolizing ∼75% of clinically used drugs (2). Catalysis of substrates by P450 enzymes requires transfer of two electrons derived from NADPH to the heme via their redox partners. Homologous to human P450 enzymes and nitric oxide synthase (NOS),3 the redox partner of CYP102A1 is a diflavin reductase containing both FMN and FAD as cofactors. Sequential transfer of two electrons from FMN to the heme leads to scission of dioxygen and subsequent insertion of one oxygen atom into inert C–H bonds (3, 4).
The high turnover rate of CYP102A1 is indicative of rapid redox partner recognition and electron transfer (ET) process and is directly related to its architecture. Similar to NOS, CYP102A1 is a fusion protein where the catalytic heme domain (BMP) is connected to the diflavin reductase domain (BMR) by a linker peptide. Biochemical studies have established that CYP102A1 functions as a homodimer (5–7). Reconstitution with competent individual BMP and BMR domains failed to restore the catalytic activity (8, 9), suggesting an important role of the linker in regulating catalysis. To shed light on redox partner recognition in CYP102A1, Poulos and co-workers (10) determined the crystal structure of a truncated form of CYP102A1 (residue 1–649), referred to as the BMP/FMN construct. The structure revealed both of the heme domains but only one of the FMN domains. This seminal work revealed an ET complex between the FMN and heme domain. Previously, we showed by negative-stain EM and biochemical analysis that the FAD domains are important for dimerization (11). Due to inherent low resolution of negative-stain EM, we were unable to determine the complete architecture of full-length CYP102A1.
To gain further insights, we utilized cryo-EM in this study to investigate the architecture and dynamics of functional full-length CYP102A1 enzyme. For the first time, we have determined the full architecture of a functional full-length enzyme and revealed multiple conformational states important for redox partner recognition. In addition, we have also determined the architecture of a deletion variant lacking peptide 456GGIPSPSTEQSA478 in the linker region, referred to as Δ12, and shown that the linker of CYP102A1 plays an important role in modulating conformational changes required for electron transfer. Our studies demonstrate that cryo-EM is a powerful technique that can provide a glimpse into the conformational dynamics of P450 enzymes.
Results
Cryo-EM sample preparation and data processing
For cryo-EM studies, we used a functional full-length A82F variant enzyme. Like WT CYP102A1, this variant forms a homodimer and exhibits a high rate of NADPH consumption at ∼1,220 min−1 in the presence of omeprazole (11, 12). From this point forward, “full-length CYP102A1” refers to the A82F variant enzyme. To ensure protein quality for cryo-EM sample preparations, we used size-exclusion chromatography–multiangle light scattering (SEC-MALS) to characterize the full-length enzyme. As shown in Fig. 1A, full-length CYP102A1 eluted as a homogeneous peak at 11.2 ml with a molecular weight of 238 ± 8.8, as expected for a homodimer. Well-isolated single particles of full-length CYP102A1 were observed on micrographs (Fig. 1B). 2D classification of ∼900,000 particles revealed distinct classes, indicative of conformational dynamics (Fig. 1C). Three representative classes are shown in Fig. 1D as an expanded view for clarity. A heterogeneous 3D classification process produced multiple EM density maps, three of which exhibit features of homodimer at a global resolution of 7.6 (30%), 8.3 (26%), and 8.5 (29%) Å. More details about cryo-EM data processing are provided in Fig. S1 and Table S1.
Figure 1.
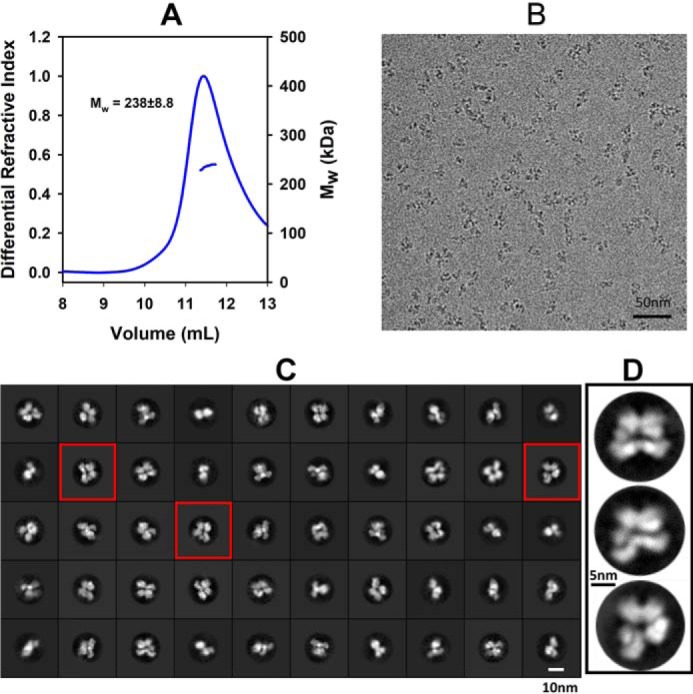
Cryo-EM micrographs and 2D class average images of full-length CYP102A1. A, SEC-MALS data showing full-length CYP102A1 eluted as a homogeneous homodimer with molecular mass of 238 ± 8.8 kDa. The horizontal bars on the right y axis indicate molecular mass in kDa. B, cryo-EM micrograph showing isolated single particles. C, 2D class average images of full-length CYP102A1. D, expanded view of three representative 2D class average images.
Architecture of full-length CYP102A1 homodimer in closed state
We started model building into the density map of the full-length enzyme at 7.6 Å. This density map and constructed model of full-length CYP102A1 are presented in Fig. 2. As shown in Fig. 2A, the EM density map shows four lobes of density that are closely packed with a nearly C2 symmetry. They represent the two BMP and two BMR domains of the homodimer. The density connecting the BMP and BMR domain is visible, which represents the linker, a highly mobile region missing from the crystal structure of CYP102A1 (10). The presence of the linker allowed us to readily assign the two monomers, one in blue and the other in orange.
Figure 2.

Architecture of full-length CYP102A1 homodimer in closed state determined by cryo-EM. A, cryo-EM density map at 7.6 Å for full-length CYP102A1. The density maps of the two monomers are colored in orange and blue, respectively. The diagram above the map shows the sequence and domain arrangement of full-length CYP102A1. BMP and BMR, heme domain and reductase domain, respectively. The red and green lines in the diagram indicate linker and hinge regions, respectively. B, 3D model of full-length CYP102A1 fit into the 7.6 Å EM density map. The map is shown as a transparent surface, and the two monomer chains are colored in orange and blue ribbons, respectively. Cofactors are depicted as space-filled spheres; the heme in red, FMN in green, and FAD in yellow. C, cartoon view of the 3D model to illustrate domain arrangement. D, expanded view of the interface at the BMP domains. Helix D-F and the D/E and E/F loops are labeled. E, expanded view of the interface at the BMR domains. The two areas of contact are labeled as Loop1 (peptide 648–653) and Loop2 (peptide 721–728).
Fig. 2B shows the constructed 3D model of full-length CYP102A1 homodimer. For easy visualization, a cartoon model is presented in Fig. 2C. The 3D model was constructed by fitting individual crystal structure of FAD, BMP, and FMN domains as a rigid body into the density map. The regions missing from the crystal structure, namely peptide 227–229 in the BMP domain, peptide 456–467 in the linker, and peptide 631–659 in the BMR domain, were rebuilt into the density map with a polyalanine chain due to insufficient resolution to model side chains. As shown, the architecture of full-length CYP102A1 clearly exhibits a trans configuration, where BMP is spatially apart from BMR within a monomer but more closely associated with BMR of the other monomer. Based on the rigid-body model, the FMN–heme distance is estimated to be ∼65 Å within a monomer, but shortened to ∼34 Å across the monomer. As observed in the crystal structure of rat cytochrome P450 oxidoreductase (POR), both of the BMR domains are in closed conformation, where the FMN and FAD cofactors are in proximity within a monomer (13).
Fig. 2 (D and E) shows close views of the interfaces between the heme domains and FAD domains. The two BMP domains share extensive contacts that involve helix D and D/E and E/F loops. This interface is nearly identical to that revealed by Poulos and co-workers (see Fig. S2). The interface between the FAD domains, which is important for dimerization as we demonstrated previously (11), is connected by a relatively small area of contacts involving two loop regions (i.e. loop 1 (peptide 648–653) and loop 2 (peptide 721–728)). Caution should be exercised with the exact position of loop 1 because it was rebuilt as a polyalanine chain into the density map at 7.6 Å.
Architecture of full-length CYP102A1 homodimer in open state
In addition to the 7.6 Å density map representing the closed state, we also obtained two additional EM density maps at 8.3 and 8.5 Å. These two density maps are presented in Fig. 3 (A and B). Similar to the 7.6 Å density map, four lobes of EM density are clearly visible. However, the four density lobes are not closely packed, two of which are detached on one side of the homodimer to a different extent. Due to conformational dynamics, the densities for the linkers are missing from both maps.
Figure 3.
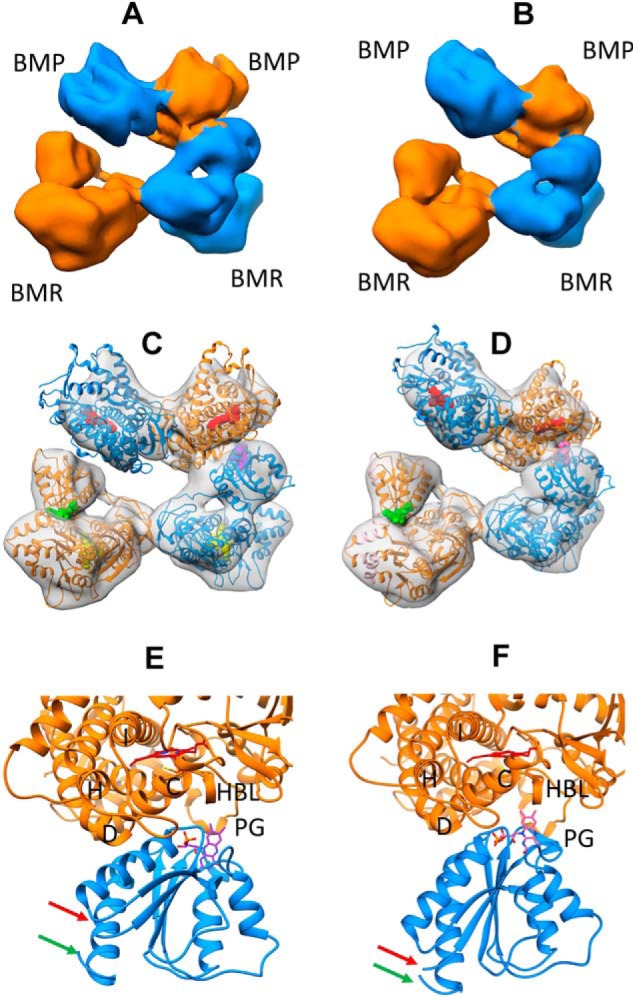
Architecture of full-length CYP102A1 homodimer in open states determined by cryo-EM. A, cryo-EM density map at 8.3 Å for full-length CYP102A1. The two monomers are colored in orange and blue, respectively. B, cryo-EM density map at 8.5 Å for full-length CYP102A1. The two monomers are colored in orange and blue, respectively. C, 3D model of full-length CYP102A1 fit to the 8.3 Å density map (open state I). The map is shown as a transparent surface, and the two monomer chains are colored in orange and blue ribbons, respectively. Cofactor heme and FAD are displayed as space-filled spheres in red and yellow, respectively. Cofactor FMN in closed conformation is displayed as a space-filled sphere in green, whereas FMN in open conformation is shown as a space-filled sphere in purple. D, 3D model of full-length CYP102A1 fit to the 8.5 Å density map (open state II). The color scheme is identical to that in C except that FMN in the open conformation is colored in pink. E, expanded view of ET complex in open state I between the heme domain and open FMN domain. Key structural features are labeled as helix C, D, H, and I, PG (Pro383–Gln387 peptide), and HBL (heme-binding loop). The red and green arrows point to the linker and hinge, respectively. F, expanded view of ET complex in open state II between the heme domain and open FMN domain. The color scheme is identical to that in E.
To construct reliable 3D models, we fit the individual structure of each domain into the two maps sequentially in the order of FAD domain, heme domain, and FMN domain. When the FMN domain was fit to the remaining density last, its initial position was constrained so that Asn479 (linker end) and Leu630 (hinge head) were orientated facing the linker and hinge, as shown in Fig. 2. The constructed 3D models of full-length CYP102A1 are presented in Fig. 3 (C and D). Similar to the closed state, both heme domains and FAD domains are in close contact, holding the homodimer together. One of the BMR domains remains closed where the FMN and FAD cofactors are in proximity, as observed in Fig. 2B. However, the orientation of the other FMN domain is strikingly different where it rotates away from the FAD, bringing the FMN cofactor to the proximal side of the heme. As shown in Fig. 3 (E and F), the isoalloxazine ring of the FMN is located in the vicinity of key structural elements known for affecting electron transfer from FMN to the heme, such as helix C, the heme-binding loop, and peptide Pro383–Gln387. They represent the ET complexes between P450 and diflavin reductase. The ET complex as shown in Fig. 3E is similar to that reported by Poulos and co-workers (10). We refer to this state as open state I. The ET complex as shown in Fig. 3F, referred to as open state II, seems to be moving further into the concave of the proximal side (see Fig. S3). Compared with the closed state, the positions of Asn479 and Leu630 experienced large movements (see Fig. S4).
Architectures of the Δ12 variant of CYP102A1 in open and closed states
The moderate resolution of the full-length enzyme is likely due to the flexibility of the linker region. Thus, we hypothesized that shortening the linker may restrict the conformational dynamics of CYP102A1 and improve resolution. We expressed and purified a variant enzyme where peptide 456GGIPSPSTEQSA478 is deleted from the linker. We followed the same procedures to acquire and process cryo-EM data for the Δ12 variant as for the full-length enzyme (see Fig. S1 and Table S1). Consistent with our notion that a shorter linker reduces conformational flexibility and enhances structural evaluation, we obtained a 6.7 Å density map in a closed state with fewer single particles (∼59,611) than used for the full-length enzyme (∼155,536). As shown in Fig. 4A, the density map of the Δ12 variant shows a compact homodimer with a trans configuration like the full-length enzyme. At the 6.7 Å resolution, the helical features are clearly visible, such as the F and G helices. The crystal structures of BMP and BMR are fit nicely into the density map with a correlation value of 0.73, indicative of a strong positive correlation (Fig. 4B). The architecture of the Δ12 is nearly identical to that of the full-length enzyme except for the linker and some mobile loop regions (see Fig. S5). In addition to the density map in the closed state, we also determined the density map in the open state at 7.9 Å. As shown in Fig. 4 (C and D), the orientations of the two density maps attributed to the FMN domains are distinctly different. Rigid-body fitting unequivocally reveals that one of the FMN domains rotates away from its FAD domain, approaching the proximal side of the heme domain, as shown in Fig. 4 (E and F). The ET complex shown in Fig. 4F is similar to that observed in open state I of the full-length enzyme and the crystal structure of a truncated BMP/FMN construct (see Fig. S6). This structural agreement validates the 3D models for the full-length enzymes. It is of note that we did not observe the Δ12 variant in open state II, presumably due to reduction in protein flexibility resulting from truncation of the linker region.
Figure 4.

Architecture of the Δ12 variant homodimer in closed and open states determined by Cryo-EM. A, cryo-EM density map at 6.7 Å for the Δ12 variant homodimer in the closed state. The two monomers are colored in orange and blue surface, respectively. B, 3D model of the Δ12 variant fit to the 6.7 Å density map. The color scheme is the same as in Fig. 2B. C, front side of the cryo-EM density map at 7.9 Å for the Δ12 variant homodimer in the open state. D, 180° view of the back side of the cryo-EM density map at 7.9 Å for the Δ12 variant homodimer in the open state (E). Shown is a 3D model of the Δ12 variant fit to the 7.9 Å density map. The color scheme is identical to that used in Fig. 3C. F, zoom view of the ET complex of the Δ12 variant in the open state. HBL and PG, heme-binding loop and Pro383–Gln387 peptide, respectively.
Biochemical analysis of deletion variants of CYP102A1 in the linker region
To better understand the conformational dynamics of full-length CYP102A1 homodimer, we further evaluated the Δ12 variant together with two additional variants, Δ4 (Δ463TEQS466) and Δ6 (Δ462STEQSA467). All three deletion variants remain as homodimers in solution, as revealed by SEC-MALS (see Fig. S7). Compared with the full-length CYP102A1, all variants retain cyt c reductase activity, as shown in Fig. 5A, suggesting that electron transfer from NADPH to the FMN cofactor is intact. However, the rate of NADPH consumption differs dramatically, as shown in Fig. 5B. The Δ4 variant retains full activity, as observed for the full-length CYP102A1, whereas the Δ6 variant retains one-third of the activity. In marked contrast, the Δ12 variant retains only 5% of the activity. This difference is also reflected in the rate of product formation, as shown in Fig. 5C. As reported previously (12), the A82F variant catalyzes omeprazole primarily to form 5-hydroxyomeprazole (5OH-OMP). The rates of 5OH-OMP formation for the Δ6 and Δ12 are 33 and 2% of the full-length enzymes, respectively, consistent with the rate of NADPH consumption. Unlike the full-length enzyme, the rate of product formation for the Δ4 variant was increased by ∼62% compared with the full-length enzyme, indicative of increased coupling. Regardless, these results suggesting that the Δ12 variant is incapable of ET from FMN to the heme. To test this hypothesis, we determined the ET rate directly from FMN to the heme in stopped-flow experiments. As shown in Fig. 5D, the ET rates were determined to be 1.1 ± 0.13 and 11.4 ± 1.5 s−1 for Δ12 and full-length CYP102A1, respectively, confirming the impairment of electron transfer from FMN to the heme in the Δ12 variant.
Figure 5.

Biochemical characterization of full-length and deletion variant enzymes of CYP102A1. A, rate of cyt c reduction. B, rate of NADPH consumption. C, rate of product, 5OH-OMP, formation. D, rate of electron transfer from FMN to the heme determined in stopped-flow experiments for the full-length and Δ12 variant. Experimental details are provided under “Experimental procedures.” Error bars, S.D.
Discussion
For the first time we have determined the architecture of functional full-length CYP102A1 homodimer and revealed its conformational dynamics required for redox partner recognition. We have shown that full-length CYP102A1 exists in closed and open states in solution, suggesting that the structure of CYP102A1 is dynamic.
In the closed state, the architecture of full-length CYP102A1 is a closely packed homodimer. The interfaces between the heme domains and the FAD domains remain in contact. Our BMP dimer model is well-aligned with the crystal structure of the truncated CYP102A1 BMP/FMN construct, as reported previously (10) (see Fig. S2). This agreement validates our 3D model of full-length CYP102A1 homodimer. According to the crystal structure, the heme domain interface is lined up with a number of polar residues, such as Asp121, Asp168, Lys129, Arg132, Arg161, Arg167, and Glu137, and few hydrophobic residues, such as Leu133 and Phe165. The FAD domain interface is located at the connecting domains involving residues in two regions of BMR (i.e. peptide 721–728 and peptide 648–653). We have previously shown that truncation of CYP102A1 beyond residue 650 leads to loss in dimer population by ∼50% (11), indicative of their important roles in dimerization. The 3D BMR model in the closed state resembles the crystal structure of rat POR, where FMN and FAD are in proximity (see Fig. S8). The overall architecture of full-length CYP102A1 exhibits a trans configuration. Based on spatial arrangement of cofactors, the architecture of dimeric CYP102A1 should facilitate intraflavin and intermolecular FMN-to-heme ET, consistent with the electron flow pathway proposed by Dr. Munro's group (7), but argue against interflavin electron transfer, as reported previously (6). However, in the closed state, the FMN is estimated to be ∼34 Å away from the heme, which would prohibit efficient trans ET to the heme. Thus, the closed state of CYP102A1 likely represents a resting state that facilitates intraflavin ET only, but is not ready for electron transfer to the heme.
To uncover the mechanism by which FMN transfers electron to the heme, we have identified full-length CYP102A1 in open states (see Fig. 3). In the open state, one FMN domain rotates away from its FAD and interacts with the proximal side of the heme, whereas the other FMN domain remains closed in the vicinity of the FAD. We did not observe both of the FMN domains rotating out simultaneously, suggesting that the FMN domain likely opens up one at a time. The sequential closed-to-open transition should be energetically favored over a concerted transition that may disrupt the integrity of the homodimer because large conformational changes are required to reach the open state, and dissociation of dimeric CYP102A1 would lead to enzyme inactivation. We postulate that the two FMN domains alternate closed-to-open transition for electron transfer between FMN and the heme. In the closed state, FMN is located in proximity to FAD and capable of accepting an electron from FAD. After accepting the electron, the FMN domain opens up and traverses to the proximal side of the heme. This should facilitate formation of an active ET complex for electron delivery to the heme. Upon electron transfer to the heme, the FMN domain returns to the closed conformation, and the process repeats itself by the other FMN domain. The alternating transition of the two monomers in synchronized motion underpins the importance of protein dynamics in highly efficient catalysis by CYP102A1. The two open states may represent conformational progression en route to deliver electron to the heme.
Structural information on the dynamic process of redox partner recognition and electron transfer in P450 and NOS systems has been difficult to obtain. Our cryo-EM studies of full-length CYP102A1 offer important clues. We have identified three conformational states in full-length enzyme, one closed and two open states. The open states are likely involved in redox partner recognition and the electron transfer process. As reported previously (10), the recognition site likely includes key structural elements on the proximal side of the heme, such as helix C, the heme-binding loop, and peptide Pro383–Gln387. Studies have shown that mutations in helix C and peptide Pro383–Gln387 significantly decrease electron transfer to the heme. For instance, introduction of a bulky dansylcystine at position 104 in helix C and position 387 in peptide Pro382–Gln387 led to >10- and 2-fold decrease in the FMN-to-heme electron transfer rate (14). The importance of the heme-binding loop in modulating electron transfer has been demonstrated in both CYP102A1 and CYP2B4 (15–17). Residues in these regions modulate redox potential of the heme and thus electron transfer. Electron transfer from FMN to the heme may occur via space jump, as proposed for the complex of mitochondrial CYP11A1 with adrenodoxin (18) and/or via peptides depending on the distance between electron donor and acceptor. The ET complex in open state I (see Fig. 3E) is similar to that reported for truncated CYP102A1 BMP/FMN construct. The FMN-to-heme distance is approximately ∼17 Å (Fig. 3E). Interestingly, the FMN in the ET complex in open state II, as shown in Fig. 3F, appears to insert further into the concave of the proximal side with an FMN-to-heme distance of ∼11 Å. We estimated that the ET rates from FMN to the heme would be ∼72 and 2.8 × 105 s−1 in open state I and II, respectively, based solely on Dutton's ruler. These results suggest an alternative pathway more directly to the heme via the Gly396–Arg398 in the heme-binding loop (see Fig. S9).
The relevance of open state II in electron transfer to the heme seems consistent with results from the structural analysis of the Δ12 variant (see Fig. 4). By truncating 12 amino acids from the linker, we achieved better map resolution with fewer than half of the single particles used to determine the density map of full-length enzyme, suggesting that shorter linker limits the conformational dynamics of CYP102A1. It also suggests that the dynamic nature of full-length enzyme is responsible, at least in part, for the modest map resolution of full-length enzyme. Due to restricted conformational dynamics, we did not observe open state II in the Δ12 variant. As discussed below, the Δ12 variant is incapable of electron transfer from FMN to the heme, indicative of the critical role of conformational changes in the FMN-to-heme ET.
The relatively large conformational change in closed-to-open transition requires extension and contraction of the linker. To investigate the role of the linker, we constructed three deletion variants. All three deletion variants remain as homodimers in solution and retain full cyt c reductase activity, indicative of intact ET from NADPH to FMN. Deletion of 12 residues results in almost complete loss of NADPH consumption. This loss of activity is due to impaired ET from FMN to the heme (see Fig. 5D). Our results suggest that the linker length likely affects the optimal geometry to form a functional ET complex. Truncated linker likely restricts motions of the FMN domain, leading to unfavorable geometry for ET. This is consistent with the report that the length, not the specific secondary structure, of the linker is critical for controlling FMN-to-heme electron transfer (19).
The conformational changes revealed by cryo-EM in this work may also occur in mammalian P450 and NOS, both of which utilize diflavin reductase as redox partners (20, 21). It was previously reported that rat POR locked in closed conformation by de novo disulfide bond is incapable of ET from FMN to the heme (22). However, a ΔTGEE variant in the hinge region exhibited an open conformation where the FMN domain swings away from the FAD domain as observed in full-length CYP102A1 (22, 23). The ΔTGEE variant is capable of ET from FMN to the heme under single-turnover conditions, suggesting that a closed-to-open conformational transition likely occurs in mammalian P450 or NOS. Our cryo-EM work may facilitate further interrogation of the mechanism for redox partner recognition and electron transfer in mammalian P450 and NOS systems.
Experimental procedures
Construction, overexpression, and purification of full-length and deletion variant CYP102A1
The gene encoding the full-length A82F variant of CYP102A1, in addition to a His6 tag at the N terminus, was optimized and synthesized by GeneScript (Piscataway, NJ). The optimized gene was then cloned into pCWori vector to construct pCW-A82F-6xHis plasmid, as described previously (11). Deletion of residues in the linker region from full-length A82F CYP102A1 was achieved by PCR using a Q5 mutagenesis kit (New England Biolabs, Ipswich). PCR primers used for PCRs are shown in Table S2. Deletion was confirmed by DNA sequencing at the University of Michigan Biomedical Research Core Facility. Both full-length and deletion variants were overexpressed in C41(DE3) cells and purified to homogeneity using an HP Histrap column on an ÄKTA FPLC protein purification system, as reported previously (11). The concentrations of CYP102A1 were determined according to the method of Omura and Sato (24).
Determination of oligomeric state of full-length and deletion variant CYP102A1 by SEC-MALS
To ensure that deletion in the linker does not alter the oligomeric state, we determined the molecular weight of both full-length and deletion variant CYP102A1 by SEC-MALS on a Bio-Rad NGC Quest chromatography system equipped with a miniDAWN TREOS MALS detector and an Optilab TrEX differential refractive index detector, as described previously (11). The system was calibrated with bovine serum album. An aliquot of full-length or deletion variant CYP102A1 (∼50 μm) was loaded onto a WTC-050S5 column (7.8 × 300 mm, 5 μm; Wyatt Technology) and eluted at 0.5 ml/min in PBS buffer. The molecular weight values were determined from the Raleigh ratio calculated by measuring the static light scattering and corresponding protein concentration of a selected peak using ASTRA VI software (Wyatt Technology).
Cryo-EM data acquisition and image processing
Prior to preparation of vitrified samples, full-length or Δ12 CYP102A1 was prepurified by SEC (Superdex Increase 10/300GL, GE Health Sciences). Central fractions were pooled and concentrated to ∼3 mg/ml, to which omeprazole was added to 0.1 mm. An aliquot (∼2.5 μl) of the sample was applied to glow-discharged Quantifoil® Holey carbon film (Q250-CR2, EMS). The sample was plunge-frozen in liquid ethane with Vitrobot (Thermo Fisher Scientific). The samples were visualized on a Titan Krios electron microscope operating at 300 kV (Thermo Fisher Scientific). Cryo-EM images were recorded at a magnification of ×29,000 using a K2 Summit direct electron detector in counted mode (Gatan, Inc.), corresponding to a pixel size of 1.01 Å/pixel with a dose rate of ∼6.0 electrons/Å2/s. The total exposure time was 8.0 s, and intermediate frames were recorded in 0.2-s intervals, resulting in an accumulated dose of ∼48 electrons/Å2 and a total of 40 frames/micrograph.
In the case of full-length enzyme, a total of 7,048 cryo-EM images were recorded. Dose-fractionated image stacks were subjected to motion correction using Motioncor2 (25) with CTF parameters determined by CTFFIND4 (26). A total of 2,158,774 particles were selected using crYOLO (27). The subsequent data processing, including 2D classification, ab initio reconstruction, 3D classification, and refinement, was carried out using cryoSPARC (28). Multiple rounds of 2D classifications were performed to remove false positive particles. In the second step, ab initio reconstruction was conducted. The classes representing the dimeric form of the protein were selected and subjected to heterogeneous refinement, which resulted in three conformational states (i.e. closed state (30%), open state I (26%), and open state II (29%)). In the next step, the classes representing the closed form of the proteins were selected and subjected to another round of ab initio/heterogeneous refinement. After visual inspection, one class showing solid linker connectivity was selected and subjected to homogeneous refinement, which results in a final reconstruction with a global resolution of 7.6 Å. Reported resolutions are based on the gold standard Fourier shell correlation using the 0.143 criterion. In the case of the Δ12 variant, we used the same procedures to determine two EM maps, one in the closed state at 6.7 Å (31%) and the other in the open state at 7.9 Å (24%). More details are provided in the cryo-EM workflow, as shown in Fig. S1 and Table S1. The EM density maps for full-length enzyme and the Δ12 variant have been deposited to the EM Data Bank with EMDB ID of 20785, 20786, 20787, 21099, and 21100.
Construction of 3D model of dimeric CYP102A1
The 3D model of dimeric CYP102A1 was constructed by rigid-body docking the crystal structure of individual domain into the EM density map using the fitmap function of Chimera (29). The crystal structures of individual heme domains of the A82F variant (PDB ID 4KEW:A), FMN domain (PDB ID, 1BVY:F), and FAD domain (PDB ID, 4DQK:A) were obtained from the PDB. It is of note that the crystal structure of BMR is not available, so that the structures of the FMN and FAD domains were docked separately. Rigid-body fitting was performed sequentially in the order of FAD domains, BMP domains, and FMN domains. The densities of BMR and BMP are readily differentiated due to their geometric differences. Similar to rat POR, BMR is shaped like a “bowl” in contrast to a more globular shape of BMP. For the purpose of illustration, polyalanine chains were built into the 7.6 Å EM density map in the regions where polypeptide chains were missing from the crystal structure using Coot (30). These missing chains include peptide 227–229 in the G/H loop, peptide 456–478 in the linker region, and peptide 631–659 in the hinge region.
Catalytic activities of full-length and deletion variant CYP102A1
The rate of NADPH consumption was determined at 25 °C as we described previously (11). Under the same conditions, we also determined the rate of product formation. After 1 min of incubation, aliquots of the reaction mixture (50 μl) were quenched with an equal volume of acetonitrile containing 0.5 μm carbamazepine as internal standard. After a brief centrifugation, an aliquot of 5 μl of the supernatant was analyzed by an LC tandem mass spectrometer. Product 5OH-OMP was chromatographed on an XBridge C18 column (4.6 × 50 mm, 3.5 μm; Waters, Milford, MA) using a binary mobile phase consisting of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile. Ion transitions from m/z 362.0 → m/z 213.9 for 5OH-OMP and m/z 237.1 → m/z 194.1 for internal standard were used for quantification. Standard solutions containing a known amount of 5OH-OMP were prepared in the reaction mixture without NADPH and analyzed under the same conditions. The rate of cyt c reduction was determined in 0.5 ml of PBS buffer containing 20 μm cyt c, 0.1 mm NADPH, and 0.1 mm omeprazole. A small aliquot of CYP102A1 enzyme was added to 10 nm to initiate electron transfer. The absorbance change at 550 nm was monitored over time, and the initial linear region was utilized to determine the rate using an extinction coefficient of 21.1 mm−1 cm−1 at 550 nm (31).
The rate of electron transfer from FMN to the heme was determined under pre-steady-state conditions in a stopped-flow spectrophotometer (model SF61DX2, Hi-Tech Co) by rapidly mixing an equal volume of 5 μm CYP102A1 enzyme with 0.4 mm NADPH at 25 °C in the presence of omeprazole. Prior to the mixing, both the CYP102A1 and NADPH solutions were saturated with CO gas (Cryogenic Gas, Detroit, MI). After the mixing, time-evolving spectra from 700 to 300 nm were recorded with a photodiode array detector, and the absorbance increase at 449 nm over time was fit to a single exponential function to obtain the rate constant as described previously (32).
Author contributions
M. S., Y. O., and H. Z. resources; M. S., S. C., and H. Z. data curation; M. S., S. C., and H. Z. formal analysis; M. S. and H. Z. investigation; M. S., S. C., and H. Z. methodology; M. S. and H. Z. writing-original draft; M. S., Y. O., and H. Z. writing-review and editing; Y. O. and H. Z. funding acquisition; H. Z. conceptualization; H. Z. supervision; H. Z. validation; H. Z. visualization; H. Z. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Sangchoul Im and Lucy Waskell for help with stopped-flow experiments.
This work was supported in part by National Institutes of Health Grant GM077430, the University of Michigan Protein Folding Diseases Initiative, and the UMHS-PUHSC Joint Research Grant. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S9.
The EM density maps for full-length enzyme and the Δ12 variant have been deposited to the EM Data Bank with EMDB ID of 20785, 20786, 20787, 21099, and 21100.
- NOS
- nitric oxide synthase
- POR
- cytochrome P450 oxidoreductase
- ET
- electron transfer
- SEC-MALS
- size-exclusion chromatography–multiangle light scattering
- 2D and 3D
- two- and three-dimensional, respectively
- cyt c
- cytochrome c
- 5OH-OMP
- 5-hydroxyomeprazole
- PDB
- Protein Data Bank.
References
- 1. Noble M. A., Miles C. S., Chapman S. K., Lysek D. A., MacKay A. C., Reid G. A., Hanzlik R. P., and Munro A. W. (1999) Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J. 339, 371–379 10.1042/bj3390371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Evans W. E., and Relling M. V. (1999) Pharmacogenomics: translating functional genomics into rational therapeutics. Science 286, 487–491 10.1126/science.286.5439.487 [DOI] [PubMed] [Google Scholar]
- 3. Rittle J., and Green M. T. (2010) Cytochrome P450 compound I: capture, characterization, and C–H bond activation kinetics. Science 330, 933–937 10.1126/science.1193478 [DOI] [PubMed] [Google Scholar]
- 4. Guengerich F. P. (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650 10.1021/tx0002583 [DOI] [PubMed] [Google Scholar]
- 5. Black S. D., and Martin S. T. (1994) Evidence for conformational dynamics and molecular aggregation in cytochrome P450 102 (BM-3). Biochemistry 33, 12056–12062 10.1021/bi00206a007 [DOI] [PubMed] [Google Scholar]
- 6. Kitazume T., Haines D. C., Estabrook R. W., Chen B., and Peterson J. A. (2007) Obligatory intermolecular electron-transfer from FAD to FMN in dimeric P450BM-3. Biochemistry 46, 11892–11901 10.1021/bi701031r [DOI] [PubMed] [Google Scholar]
- 7. Neeli R., Girvan H. M., Lawrence A., Warren M. J., Leys D., Scrutton N. S., and Munro A. W. (2005) The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett. 579, 5582–5588 10.1016/j.febslet.2005.09.023 [DOI] [PubMed] [Google Scholar]
- 8. Sevrioukova I., Truan G., and Peterson J. A. (1997) Reconstitution of the fatty acid hydroxylase activity of cytochrome P450BM-3 utilizing its functional domains. Arch. Biochem. Biophys. 340, 231–238 10.1006/abbi.1997.9895 [DOI] [PubMed] [Google Scholar]
- 9. Munro A. W., Lindsay J. G., Coggins J. R., Kelly S. M., and Price N. C. (1994) Structural and enzymological analysis of the interaction of isolated domains of cytochrome P-450 BM3. FEBS Lett. 343, 70–74 10.1016/0014-5793(94)80609-8 [DOI] [PubMed] [Google Scholar]
- 10. Sevrioukova I. F., Li H., Zhang H., Peterson J. A., and Poulos T. L. (1999) Structure of a cytochrome P450-redox partner electron-transfer complex. Proc. Natl. Acad. Sci. U.S.A. 96, 1863–1868 10.1073/pnas.96.5.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang H., Yokom A. L., Cheng S., Su M., Hollenberg P. F., Southworth D. R., and Osawa Y. (2018) The full-length cytochrome P450 enzyme CYP102A1 dimerizes at its reductase domains and has flexible heme domains for efficient catalysis. J. Biol. Chem. 293, 7727–7736 10.1074/jbc.RA117.000600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Butler C. F., Peet C., Mason A. E., Voice M. W., Leys D., and Munro A. W. (2013) Key mutations alter the cytochrome P450 BM3 conformational landscape and remove inherent substrate bias. J. Biol. Chem. 288, 25387–25399 10.1074/jbc.M113.479717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang M., Roberts D. L., Paschke R., Shea T. M., Masters B. S., and Kim J. J. (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. U.S.A. 94, 8411–8416 10.1073/pnas.94.16.8411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sevrioukova I. F., Hazzard J. T., Tollin G., and Poulos T. L. (1999) The FMN to heme electron transfer in cytochrome P450BM-3. Effect of chemical modification of cysteines engineered at the FMN-heme domain interaction site. J. Biol. Chem. 274, 36097–36106 10.1074/jbc.274.51.36097 [DOI] [PubMed] [Google Scholar]
- 15. Yang Y., Zhang H., Usharani D., Bu W., Im S., Tarasev M., Rwere F., Pearl N. M., Meagher J., Sun C., Stuckey J., Shaik S., and Waskell L. (2014) Structural and functional characterization of a cytochrome P450 2B4 F429H mutant with an axial thiolate-histidine hydrogen bond. Biochemistry 53, 5080–5091 10.1021/bi5003794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ost T. W., Miles C. S., Munro A. W., Murdoch J., Reid G. A., and Chapman S. K. (2001) Phenylalanine 393 exerts thermodynamic control over the heme of flavocytochrome P450 BM3. Biochemistry 40, 13421–13429 10.1021/bi010716m [DOI] [PubMed] [Google Scholar]
- 17. Ost T. W., Munro A. W., Mowat C. G., Taylor P. R., Pesseguiero A., Fulco A. J., Cho A. K., Cheesman M. A., Walkinshaw M. D., and Chapman S. K. (2001) Structural and spectroscopic analysis of the F393H mutant of flavocytochrome P450 BM3. Biochemistry 40, 13430–13438 10.1021/bi010717e [DOI] [PubMed] [Google Scholar]
- 18. Strushkevich N., MacKenzie F., Cherkesova T., Grabovec I., Usanov S., and Park H. W. (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc. Natl. Acad. Sci. U.S.A. 108, 10139–10143 10.1073/pnas.1019441108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Govindaraj S., and Poulos T. L. (1996) Probing the structure of the linker connecting the reductase and heme domains of cytochrome P450BM-3 using site-directed mutagenesis. Protein Sci. 5, 1389–1393 10.1002/pro.5560050717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haque M. M., Bayachou M., Tejero J., Kenney C. T., Pearl N. M., Im S. C., Waskell L., and Stuehr D. J. (2014) Distinct conformational behaviors of four mammalian dual-flavin reductases (cytochrome P450 reductase, methionine synthase reductase, neuronal nitric oxide synthase, endothelial nitric oxide synthase) determine their unique catalytic profiles. FEBS J. 281, 5325–5340 10.1111/febs.13073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hedison T. M., Hay S., and Scrutton N. S. (2017) A perspective on conformational control of electron transfer in nitric oxide synthases. Nitric Oxide 63, 61–67 10.1016/j.niox.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamdane D., Xia C., Im S. C., Zhang H., Kim J. J., and Waskell L. (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 284, 11374–11384 10.1074/jbc.M807868200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xia C., Hamdane D., Shen A. L., Choi V., Kasper C. B., Pearl N. M., Zhang H., Im S. C., Waskell L., and Kim J. J. (2011) Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem. 286, 16246–16260 10.1074/jbc.M111.230532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Omura T., and Sato R. (1963) Fractional solubilization of haemoproteins and partial purification of carbon monoxide-binding cytochrome from liver microsomes. Biochim. Biophys. Acta 71, 224–226 10.1016/0006-3002(63)91015-1 [DOI] [PubMed] [Google Scholar]
- 25. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., and Agard D. A. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rohou A., and Grigorieff N. (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 10.1016/j.jsb.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wagner T., Merino F., Stabrin M., Morlya T., Gatsogiannis C., and Raunser S. (2019) SPHIRE-crYOLO: a fast and well-centering automated particle picker for cryo-EM. Commun. Biol. 2 10.1038/s42003-019-437-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Punjani A., Rubinstein J. L., Fleet D. J., and Brubaker M. A. (2017) cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 10.1038/nmeth.4169 [DOI] [PubMed] [Google Scholar]
- 29. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Gelder B., and Slater E. C. (1962) The extinction coefficient of cytochrome c. Biochim. Biophys. Acta 58, 593–595 10.1016/0006-3002(62)90073-2 [DOI] [PubMed] [Google Scholar]
- 32. Zhang H., Gruenke L., Arscott D., Shen A., Kasper C., Harris D. L., Glavanovich M., Johnson R., and Waskell L. (2003) Determination of the rate of reduction of oxyferrous cytochrome P450 2B4 by 5-deazariboflavin adenine dinucleotide T491V cytochrome P450 reductase. Biochemistry 42, 11594–11603 10.1021/bi034968u [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.