

Abstract
Permeabilization of the mitochondrial outer membrane is a key step in the intrinsic apoptosis pathway, triggered by the release of mitochondrial intermembrane space proteins into the cytoplasm. The BCL-2–associated X apoptosis regulator (BAX) protein critically contributes to this process by forming pores in the mitochondrial outer membrane. However, the relative roles of the mitochondrial residence of BAX and its oligomerization in promoting membrane permeabilization are unclear. To this end, using both cell-free and cellular experimental systems, including membrane permeabilization, size-exclusion chromatography-based oligomer, and retrotranslocation assays, along with confocal microscopy analysis, here we studied two BAX C-terminal variants, T182I and G179P. Neither variant formed large oligomers when activated in liposomes. Nevertheless, the G179P variant could permeabilize liposome membranes, suggesting that large BAX oligomers are not essential for the permeabilization. However, when G179P was transduced into BAX/BCL2 agonist killer (BAK) double-knockout mouse embryonic fibroblasts, its location was solely cytoplasmic, and it then failed to mediate cell death. In contrast, T182I was inefficient in both liposome insertion and permeabilization. Yet, when transduced into cells, BAXT182I resided predominantly on mitochondria, because of its slow retrotranslocation and mediated apoptosis as efficiently as WT BAX. We conclude that BAX's mitochondrial residence in vivo, regulated by both targeting and retrotranslocation, is more significant for its pro-apoptotic activity than its ability to insert and to form higher-order oligomers in model membranes. We propose that this finding should be taken into account when developing drugs that modulate BAX activity.
Keywords: apoptosis, Bax, mitochondrial apoptosis, molecular cell biology, translocation, anticancer drug, liposome, mitochondrial localization, mitochondrial outer membrane permeabilization (MOMP), protein oligomers
Introduction
Mitochondrial outer membrane permeabilization (MOMP)3 is a critical step in the intrinsic apoptosis pathway. MOMP involves the formation of large protein-conducting pores in the outer membrane. As a result, multiple proteins that are normally restricted to the mitochondrial intermembrane space, such as cytochrome c and SMAC/DIABLO, are released into the cytoplasm, where they trigger the activation of proteases in the caspase family, leading to cell death (1). This process is regulated by Bcl-2 family proteins, which can be either pro- or anti-apoptotic and share up to 4 Bcl-2 homology (BH) domains (2–4). Pro-apoptotic Bcl-2 family proteins are further subdivided into two groups: effector molecules (BAX and BAK), which permeabilize the membrane, and BH3 domain-only proteins (BID, BIM, PUMA, BMF, BAD, BIK, HRK, NOXA etc.), which promote the activation of BAX and BAK.
An intensive body of research has been directed toward unraveling the molecular mechanisms of MOMP, as this step is considered a “point of no return” in most forms of apoptosis and thus may be a good target for therapeutic intervention (5). In particular, BAX/BAK activation is of considerable interest for the development of drugs able to kill cancer cells. One of these drugs, Venetoclax, is specific for heterodimers containing Bcl-2 and has been approved by the FDA to treat a certain form of chronic lymphocytic leukemia (6). In addition, compounds directly activating BAX can also promote MOMP and apoptosis (7). MOMP has therefore been validated as a key process promoting cell death.
Previously, we investigated the molecular mechanisms of MOMP, using systems based on liposomes or vesicles comprised of purified mitochondrial outer membranes (outer membrane vesicles, or OMVs), which can be loaded with fluorescent dextrans (8, 9). These vesicles, when mixed with recombinant BAX and a BH3-only protein such as cleaved BID, can recapitulate the fundamental aspects of the membrane permeabilization process (9, 10) and have been widely used in the community for nearly 20 years (11–16). In further studies using cryo-electron microscopy (cryo-EM), we showed that activated BAX mediates the formation and indefinite enlargement of circular pores in the vesicle membrane, reaching diameters in the hundreds of nanometers (17, 18). We later demonstrated that BAX molecules densely line the pore edges (19). Other groups, using super-resolution microscopy, detected circular arrangements of BAX, proposed to correspond to the pores seen by cryo-EM, in mitochondria in apoptotic cells (20, 21). Together, these findings have supported a model whereby the oligomerization of BAX is an essential part of pore formation.
Although BAX exists in large heterogeneous oligomers in these pores (19), it remains unclear whether this extensive BAX oligomerization is required for pore formation (8, 22). Meanwhile, the retrotranslocation of BAX and BAK from mitochondria to cytosol has been reported as a mechanism by which their residence on the membrane is regulated upstream of oligomerization (23–25). Intensive effort is currently underway on the BAX molecule, employing in vitro systems, to develop drugs to modulate BAX activity for therapeutic purposes (15, 16, 26). Using two BAX mutants, we have investigated the relative roles of BAX mitochondrial residence and oligomerization in promoting membrane permeabilization. In particular, we highlight the importance of BAX targeting and retrotranslocation (i.e. BAX mitochondrial localization) in in vivo regulation of apoptosis.
Results
Membrane insertion of BAX, but not higher-order oligomer formation, is critical for liposome permeabilization
We first asked how critical BAX higher-order oligomer formation is for liposome membrane permeabilization by testing two BAX mutants with substitutions within the C-terminal transmembrane helix 9 of BAX that impact on its ability to oligomerize. The laboratory of Jialing Lin (27) identified BAXT182I as being competent in forming symmetric dimers (28, 29), but defective in higher-order oligomerization (i.e. dimer-dimer interaction). Another study showed that the G179P mutation produced a defect in helix 9 interactions in experiments where the helix 9 sequence was joined to a fusion partner (30).
We generated recombinant T182I or G179P mutant BAX proteins and measured the continuous kinetics of permeabilization in liposomes loaded with fluorescein-conjugated dextrans, using an anti-fluorescein antibody that quenches the fluorescence dextran molecules once the dextrans are released from permeabilized vesicles (8). At any given time, the fluorimeter thus measures only the signal from dextran molecules residing in the vesicles that remain intact. We used the cleaved form of BID (cBID) to activate BAX species. As shown in Fig. 1A, BAXG179P was almost as potent as wildtype (WT) BAX in promoting membrane permeabilization. In contrast, BAXT182I was much less efficient than WT. An earlier study reported that BAXT182I apparently formed “smaller” pores than WT BAX in isolated mitochondria (27), and we attempted to confirm this in our cell-free system. We loaded liposomes with small reporter molecules, ANTS and DPX, ∼500 Da in size. We found that even these small molecules were released only very slowly by BAXT182I (Fig. 1B).
Figure 1.

Two helix 9 mutants of BAX, T182I and G179P, display divergent activities in liposomes. A, BAXT182I is less active than WT in liposomes, whereas BAXG179P is almost as active as WT. Left panel, a representative release kinetics of 70-kDa fluorescein-dextran from liposomes permeabilized with recombinant BAX proteins mixed with the activator, cBID(45 nm). Proteins and liposomes were incubated at 37 °C and dextran release was monitored as quenching by anti-fluorescein antibodies. Right panel, summary of activities of the mutants compared with the WT BAX. ns, not significant; numbers, p values. B, BAXT182I releases small molecules slowly. Left panel, release kinetics for liposomes loaded with quenching concentrations of ANTS and DPX (both ∼0.5 kDa). BAX and cBID were incubated with the liposomes and permeabilization was measured as dequenching of ANTS and DPX resulting from dilution of the dyes once released from the vesicles. Right panel, summary of activities of the mutants compared with the WT BAX. p < 0.0001. C, NBD-labeled BAX species maintain the relative activities of nonlabeled counterparts. Cascade blue (CB)-dextrans (10 kDa) were used to avoid interference with the spectrally overlapping NBD dye. Relative activity of the mutant is shown in the right panel compared with WT BAX. BAXG179P shows no significant difference from WT BAX. BAXT182I did not reach 50% release in time, therefore its activity was considered very close to 0. D, BAXT182I is inefficient in membrane insertion. Membrane insertion was monitored using NBD-labeled BAX (WT versus G179P in the left panel; WT versus T182I in the right panel) with and without cBID (45 nm) on nonloaded liposomes. E, BAXG179P permeabilizes OMVs more slowly. BAX WT, BAXT182, or BAXG179P were incubated with OMVs at 120 nm together with cBID (45 nm). Vesicle permeabilization was monitored as quenching of fluorescein-dextran (70 kDa). The one sample and Wilcoxon test shows borderline significance with BAXG179P (p = 0.054). BAXT182I did not reach 50% release; its activity was considered very close to 0. All the data in Fig. 1 were taken from three to four independent experiments and error bars are the standard deviations.
To determine whether the defect in BAXT182I function occurs at the step of membrane integration, rather than at subsequent events in the pore formation process, we labeled WT and mutant BAX species with nitrobenzoxadiazol (NBD). NBD fluorescence increases if the dye enters a hydrophobic environment, thus signaling that BAX has become inserted into the membrane (8, 13). First, we verified that NBD labeling did not significantly alter either BAX variants activity relative to that of WT BAX (Fig. 1C). Due to the labile nature of NBD-labeled BAX, experiments were performed separately to compare WT and G179P or WT and T182I. Fluorescence of NBD-BAXT182I increased very slowly, compared with WT (Fig. 1D, right panel). On the other hand, fluorescence of NBD-BAXG179P increased as rapidly as WT, although it plateaued at a lower intensity (Fig. 1D, left panel). The labeling efficiency in both mutants were as good as the WT (see “Materials and methods”), therefore the result is unlikely to be due to differential labeling of the BAX variants. Regardless, we can say that the difference in membrane permeabilization activity of these BAX variants correlates well with the rate of membrane insertion. As liposomes are highly artificial membranes, we investigated the activity of the BAX mutants on mitochondrial OMVs (8, 9). These native vesicles contain a full complement of outer membrane proteins and thus provide a more physiological membrane environment. The results (Fig. 1E) showed, surprisingly, that BAXG179P was substantially less potent in permeabilizing OMVs than WT BAX, arguing that this mutant may interact weakly with native mitochondrial outer membranes (further experiments shown below address this in more detail). BAXT182I permeabilized OMVs poorly, similar to what we observed with this mutant in liposomes. In summary, BAXT182I showed very low permeabilization activity, whereas BAXG179P permeabilized model membranes like WT and the activity correlated with the rate of membrane insertion. An alternative explanation for the above findings would be that the BAX C terminus mutants may not form large enough oligomers, as suggested in the original studies (27, 30).
We therefore examined the apparent sizes of mutant and WT BAX oligomers in the liposome membrane using size-exclusion column chromatography (SEC). This technique does not show the exact size of the particle, as the elution is also influenced by the shape of the molecules. However, SEC has been used to reveal major differences between BAX oligomers (9, 31–33). We incubated liposomes with BAX and cBID, then solubilized them with 1.2% CHAPS detergent (Fig. 2A), a treatment thought to better maintain the oligomeric status of BAX (9, 19). WT BAX formed heterogeneous oligomers, the largest of which reached the excluded size of Superdex 200 (>600 kDa). In contrast, BAXT182I and BAXG179P formed much smaller complexes that peaked at an apparent size of 100−200 kDa. BAX in solution was predominantly monomeric (∼25 kDa) with a minor species (possibly dimeric) at around 50 kDa, as seen by others (33). To measure the precise stoichiometry of BAX oligomers, we used a previously described TIRF microscopy approach (34). We fluorescently labeled BAX (BAXWT-488 or BAXG179P-488) and confirmed that they were monomeric in solution (Fig. 2B, a and b). Unfortunately, the labeled T182I mutant aggregated in solution, as demonstrated by the high degree of oligomerization in solution (Fig. 2Bc), precluding an analysis of its stoichiometry in the membrane. BAXWT-488 or BAXG179P-488 were mixed with unilamellar liposomes in the presence of cBID. After 1 h of incubation, these proteoliposomes were fused to obtain a continuous supported lipid bilayer (SLB) containing BAX oligomers (Fig. 2C) on a glass slide. The fluorescence intensities of individual BAX particles (bright spots in the image of Fig. 2C) were then taken to calculate BAX stoichiometry, using monomeric BAX as a calibration reference (34). A direct comparison of the intensity distribution of BAXWT-488 (Fig. 2D) with BAXG179P-488 (Fig. 2F) shows lower values of fluorescence intensity for BAXG179P-488, suggesting a lower oligomeric state of particles for this mutant. Importantly, unlike BAXWT-488 (Fig. 2E), BAXG179–488 was unable to oligomerize into higher-order oligomers at comparable densities in the membrane (Fig. 2G), which is in agreement with the SEC data in Fig. 2A. This conclusion is consistent with the reports that suggest that BAX helix 9 is involved in higher-order oligomer formation (27, 30). Importantly, because BAXG179P could permeabilize liposomes almost as well as WT BAX, we conclude that higher-order oligomers are not required for membrane permeabilization.
Figure 2.

BAX helix 9 mutants form size-restricted oligomers in the membrane. A, size-exclusion chromatography reveals that BAXT182I and BAXG179P form much smaller complexes than WT. BAX species (2 μm each) and cBID (1.3 μm) were incubated with liposomes for 2 h at 37 °C. After the incubation period, the membrane fraction was separated in a sucrose gradient centrifugation step to remove unbound BAX protein. Membranes were dissolved in 1.2% CHAPS and fractionated in a Superdex 200 Increase (10/300) column. Fractions were probed with anti-BAX antibody. The data shown are a representative of three independent experiments. B, an in situ TIRF-based assay for measuring the stoichiometry of BAX complexes in SLBs. Confirmation of monomeric status of BAX species. Schematic representation of the protocol used for confirmation of monomeric status of the BAX species used in the study (upper panel) and the data showing the monomeric status of BAX (lower panels). LUVs (gray) were used to prepare SLBs, which were then incubated with 2.5 nm labeled BAX (light green) for 30 min or 1 h at room temperature, washed carefully with buffer, and immediately imaged by TIRF microcopy. Percentage of occurrence of fluorophore units per particle for BAX (S4C,C62S,C126S)-488 (a), BAX G179P (S4C,C62S,C126S)-488 (b), and BAX T182I (S4C,C62S,C126S)-488 (c) mutants directly added on SLBs calculated as the average value from two different experiments. Due to unspecific interaction of BAX molecules with the SLB glass support, BAX molecules are unable to oligomerize, resulting mainly in monomers and some dimers and trimers. Data provided are the raw values, where no correction for partial labeling was applied. The error bars correspond to the standard deviations from two different experiments. Note that BAX Thr-182 (S4C,C62S,C126S)-488 was already oligomeric in solution. C, schematic representation of the stoichiometry distribution assay on BAX (S4C,C62S,C126S)-488 and BAXG179P (S4C,C62S,C126S)-488. LUVs (gray) were incubated with 2.5 nm BAX (light green) and 5 nm cBID (not shown) at room temperature for 1 h to form proteoliposomes (LUVs containing BAX oligomers). After incubation, these liposomes were used to prepare SLBs with labeled BAX oligomers associated with them. The right panel shows a representative TIRF image of a SLB containing BAX oligomers (bright spots). Scale bar is 10 μm. D and F, representative intensity distribution of BAX-488 (D) and BAX G179P-488 (F) mutant particles (minimum 1600 particles per experiment) bound to SLBs prepared from proteoliposomes after 1 h of incubation with the protein. The obtained brightness distribution was plotted as a probability density function (Pdf, black) or, alternatively, as a histogram and fitted with a linear combination of Gaussians to estimate, from the area under each curve, the percentage of occurrence of particles containing n-mer–labeled molecules (see color code in the graph). The cumulative fit is shown by a dashed black line. E and G, TIRF analysis reveals that BAXG179P fails to form high-order oligomers in SLBs. Percentage of occurrence of different oligomeric species for BAX-488 (E) and BAX G179P-488 (G) mutant particles were calculated as the average value from four (E) and two (G) different experiments. Data provided are the raw values, where no correction for partial labeling (80% for BAX-488 and 50% for BAX G179P-488) was applied (see “Materials and methods”). The error bars correspond to the standard deviations from the different experiments.
Helix 9 mutations affect BAX mitochondrial localization in vivo, independently from their activities on membrane insertion/oligomerization
To test the effect of the helix 9 mutations in a more physiological setting, we permeabilized BAX/BAK double-knockout (DKO) mouse embryonic fibroblasts (MEFs) with digitonin and incubated them with recombinant BAX and cBID (Fig. 3A). Surprisingly, the results seen were different from what we had observed with liposomes. Although BAXG179P was fairly potent in permeabilizing liposomes, it was less effective than T182I in promoting the release of cytochrome c and SMAC/DIABLO from mitochondria in permeabilized cells. On the other hand, whereas T182I permeabilized liposomes poorly, it was able to release cytochrome c and SMAC/DIABLO from mitochondria in permeabilized cells, although to a lesser extent than observed with the equal amount of WT BAX. A trace amount of SMAC (and cytochrome c to a lesser degree) release with BAX mutants without cBID was seen in the blot, and we suspect that residual BH3 only proteins associated with mitochondria induced some level of activation of these BAX variants. Both cytochrome c and SMAC/DIABLO were released to the same extent with BAXT182I, and thus our data are seemingly inconsistent with those reported by Zhang et al. (27), who observed a differential release of cytochrome c and SMAC, although this could be due to differences in the kinetics of the release. Of note, when we recovered the mitochondria-containing pellet fractions from the permeabilized cells, we saw roughly the same (or higher) levels of BAXT182I as WT BAX (Fig. 3A, bottom panel), whereas cytochrome c/SMAC release by BAXT182I was much less than WT. This suggests that higher levels of BAXT182I may be required to produce the same level of permeabilization as with WT.
Figure 3.

In cells, BAXT182I translocates strongly to mitochondria and, as a result, mediates MOMP and apoptosis as well as WT BAX; BAXG179P remains cytoplasmic and functions poorly. A, permeabilized cells incubated with recombinant BAX species. BAX/BAK DKO MEFs were permeabilized with digitonin and recombinant BAX proteins and cBID were added for 1 h at 30 °C. The supernatant and the pellet fractions were isolated and immunoblotted for cytochrome c, SMAC, or BAX. B, expression of BAX species in transduced cells. Mouse BAX WT, T182I, or G179P were transduced in BAX/BAK DKO MEFs using retroviral transduction with IRES-GFP. We sorted out cell populations with equivalent expression levels of GFP, and therefore of BAX species. Note that each mutant BAX migrated slightly differently from WT. Ponceau S stain was used to verify equivalent sample loading. C, transduced BAXT182I is recruited strongly to mitochondria in permeabilized cells incubated with cBID. BAXWT-, T182I-, or G179P-transduced DKO MEFs were permeabilized with digitonin and incubated with cBID for 1 h at 30 °C. The supernatant and the pellet fractions were immunoblotted for cytochrome c or anti-BAX antibodies. T182I-expressing cells had a higher level of BAX in the pellet than WT and released cytochrome c even more efficiently than WT. Basal BAXT182I levels in the pellet were higher than that of WT BAX and increased dramatically in the presence of cBID, showing that BAXT182I is recruited to mitochondria more efficiently than WT. D, BAXT182I mediates apoptosis effectively, but BAXG179P functions poorly. Staurosporine-induced cell death of transduced cells was measured by propidium iodide (PI) stain. Error bars are S.D. of triplicate samples. Representatives of 2–4 independent experiments are shown.
Mitochondrial residency of BAX determines cellular sensitivity to apoptosis
To explain the difference between our results with permeabilized cells versus liposomes, we considered that in liposomes, which contain none of the other proteins present in or around native mitochondrial membranes, BAX must interact with the membrane directly by itself. We hypothesized that, conversely, cells contain mechanisms to control the mitochondrial localization and accumulation of BAX that are absent in liposomes. Thus, BAX WT and the mutants might interact with membranes differently in these systems.
We reconstituted BAX/BAK DKO MEFs with WT or mutant BAX, using a retroviral IRES-GFP vector. We used fluorescence-activated cell sorting (FACS) to isolate cell populations with equivalent expression levels of GFP, and therefore of the three BAX variants (Fig. 3B). We permeabilized these cells with digitonin, and then incubated them in the presence of cBID protein, to activate the retrovirally expressed WT or mutant BAX. We then analyzed the resulting release of cytochrome c (Fig. 3C). Even in the absence of cBID, the basal level of BAXT182I in the mitochondria-containing pellet fraction was notably higher than that of BAX WT, and T182I levels increased dramatically after cBID addition. Thus, BAXT182I was strongly recruited to mitochondria, both when inactive and active by cBID. In contrast, BAXG179P was undetectable in the pellet, with or without cBID, suggesting that this mutant failed to associate with mitochondria and was therefore washed out during digitonin permeabilization. We conclude that BAX C-terminal mutations can strongly increase or decrease the mitochondrial levels of BAX protein.
Although recombinant BAXT182I protein was inefficient in releasing dextrans and small molecules from liposomes (Fig. 1) and less efficient than WT in releasing cytochrome c from mitochondria in permeabilized cells (Fig. 3A), the mutant protein stably expressed within retrovirally transduced cells released cytochrome c more efficiently than WT BAX. This result suggests that the increased levels of BAXT182I in the mitochondria of the transduced cells could compensate for the strongly decreased efficiency of this mutant BAX in membrane insertion. To test this, we examined the ability of WT and the mutant BAX species to mediate apoptosis in intact DKO MEFs treated with staurosporine. Whether these cells were reconstituted with BAX WT or T182I, they underwent apoptosis to the same extent. On the other hand, cells reconstituted with BAXG179P were significantly resistant to staurosporine-induced apoptosis (Fig. 3D). To summarize, the results seen with DKO MEFs were consistent, whether they were intact or permeabilized. In contrast, the more artificial systems (particularly liposomes and to some extent, OMVs), produced results that diverged from those seen in cells.
To explain the differences between whole cells and vesicle systems, we hypothesized that whole cells contain systems that control the cytoplasm-mitochondrial distribution of BAX, which will not be recapitulated in either liposomes or OMVs. To test this, we fused WT and mutant BAX with GFP and transiently expressed these proteins in DKO MEFs, and compared their localization with that of mitochondrial Hsp70. GFP-BAXT182I was localized preferentially on the mitochondria. GFP-WTBAX showed some co-localization, but this was less than GFP-BAXT182I. In contrast, G179P appeared almost exclusively to be cytoplasmic (Fig. 4A). These observations were consistent with our immunoblot results using digitonin-permeabilized cells (Fig. 3C). Next, we induced apoptosis using staurosporine in DKO cells expressing WT or mutant BAX and stained the cells with a BAX antibody (6A7) that specifically recognizes an N-terminal epitope in BAX that is unmasked after activation. With cells expressing either BAX WT or BAXT182I, we saw 6A7 positive puncta typical of activated BAX in apoptotic cells. In contrast, BAXG179P-expressing cells were not positive for 6A7, showing that BAX G179P failed to be activated (Fig. 4B). Furthermore, immunostaining for SMAC indicated that both WT BAX and BAXT182I were able to promote its release from mitochondria in the 6A7-positive cells. BAXG179P expressing cells retained SMAC within mitochondria following staurosporine treatment.
Figure 4.

BAXT182I constitutively localizes to mitochondria, whereas G179P is completely cytosolic. A, BAXT182I accumulates on mitochondria to a greater extent than WT, whereas BAXG179P fails to translocate to mitochondria. GFP-BAX was transiently expressed in BAX/BAX DKO MEFs and imaged by epifluorescence microscopy. GFP-BAX was stained with anti-GFP antibody. Mitochondria were stained with mitochondrial HSP70 (mHSP70) and the nuclei, with DAPI. Mitochondria were fragmented as previously reported in these cells (48). WT BAX distributed both at mitochondria and cytoplasm, whereas the localization of BAXT182I was more pronounced in mitochondria. BAXG179P was diffuse cytoplasmic. Scale bar: 10 μm. B, microscopic detection of activated BAX species in apoptotic cells. BAX/BAK DKO MEFs transduced with BAX-IRES-GFP (WT, T182I, or G179P) were treated with staurosporine in the presence of caspase inhibitor Q-VD, then fixed and stained with antibodies 6A7 (for the activated form of BAX) and anti-SMAC. The diffuse green GFP fluorescence served as a marker for BAX co-expression. WT and BAXT182I showed characteristic staining of punctate BAX in apoptotic cells, whereas BAXG179P-expressing cells were not stained with 6A7. Red SMAC staining was only seen in cells with no BAX puncta, i.e. nonapoptotic cells. Note the morphology of the cells regardless of the species of BAX expression was altered due to staurosporin treatment. Scale bar, 10 μm.
As BAXG179P was able to permeabilize liposomes, it is apparently functional with regard to the mechanistic steps of membrane integration and pore formation. However, in cells, this mutant is essentially devoid of apoptotic function, as it fails to translocate to mitochondria. We hypothesize that this mutant is unable to interact with the machinery that targets BAX to mitochondria and that in vivo this is an essential prerequisite for membrane insertion and oligomerization. T182I behaved in the opposite manner, inasmuch as it was inefficient in membrane insertion in vitro, but displayed stronger localization to mitochondria in vivo. Because T182I mediates cell death as potently as WT, its enhanced mitochondrial residency is apparently enough to compensate for any defect in membrane insertion. Our results argue that there are at least two critical steps prior to pore formation: mitochondrial outer membrane residency (defective in G179P and enhanced in T182I) and membrane insertion (inefficient in T182I). We further suggest that the extent of BAX mitochondrial residency is at least as important for cell death as efficiency in the downstream events such as membrane insertion, symmetric dimer formation, and oligomerization.
Slow retrotranslocation increases BAX levels at mitochondria and sensitizes cells to death
Previous reports have shown that BAX retrotranslocates, i.e. is in dynamic equilibrium between the cytoplasm and the mitochondria (23–25, 35). Based on our results, we hypothesized that our helix 9 mutations might alter this equilibrium. As BAXG179P was apparently not targeted to mitochondria, and was therefore entirely cytosolic, we were unable to visualize retrotranslocation for this mutant. However, with WT BAX and BAXT182I, we could assess retrotranslocation of the corresponding GFP fusion proteins from mitochondria, using a modified FLIP (fluorescence loss in photobleaching) protocol, as previously published (24). In this approach, cytosolic regions of GFP-BAX expressing cells are first bleached, and the decline of fluorescence in the mitochondria is measured over time as the fluorescent protein re-equilibrates between the mitochondrial and cytoplasmic fractions. For comparison, we included GFP-BAXS184V, which shows reduced mitochondrial retrotranslocation (24, 36, 37). As previously shown (24), GFP-tagged WT BAX rapidly re-equilibrated between the cytoplasm and mitochondria following photobleaching (Fig. 5, A–C). In contrast, GFP-BAXT182I exhibited significantly reduced retrotranslocation, comparable with GFP-BAXS184V. In this experiment, imaging over time did not result in background photobleaching as measurement of total cell fluorescence in unbleached cells within the same field of view, over the same time course, did not decrease (Fig. 5D).
Figure 5.

BAXT182I retrotranslocates substantially more slowly than WT BAX. A, monitoring of photobleached cells over time. Cytoplasmic portions of cells expressing GFP-BAXWT, BAXS184V, and BAXT182I were photobleached in the yellow-lined area and fluorescence was monitored in the green region of interest. The red line indicates the outline of the imaged cell. BAXS184V is constitutively expressed in mitochondria and was used here as a control for slow retrotranslocation. Scale bars: 10 μm. B and C, BAXT182I retrotranslocates slowly. Fluorescence data from A was normalized to 100% fluorescence pre-bleach and the average decrease in fluorescence over time plotted (B). Data represents values from three independent experiments and error bars represent standard deviation. Data from B was fitted to a one-phase dissociation curve and the average fluorescence recovery calculated. Data were analyzed via ANOVA, *, p < 0.05. Error bars represent standard deviation (C). D, total fluorescence does not decrease in unbleached cells. The total cell fluorescence of unbleached cells within the same region of interest as cells analyzed in A was measured and the average total fluorescence plotted over time.
To confirm the FLIP data, we also developed a permeabilized cell retrotranslocation assay that is more rapid and convenient than a previously reported method using isolated mitochondria (38). We took digitonin-permeabilized BAX-transduced DKO MEFs and monitored the loss of BAX from the cell pellet over time. We observed that WT BAX was quickly released into the soluble fraction, whereas BAXT182I was dissociated from the mitochondrial fraction much more slowly (Fig. 6), consistent with the above FLIP assays using live cells.
Figure 6.
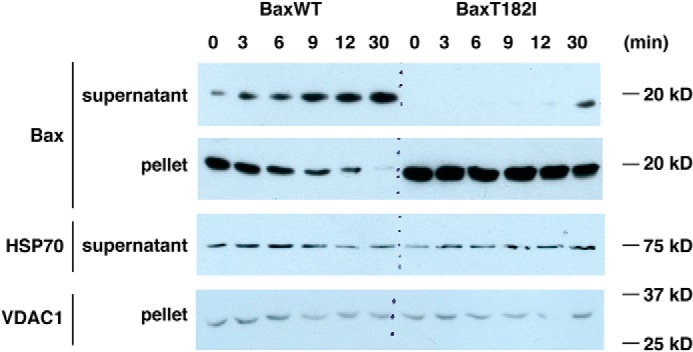
Permeabilized cell system shows BAX retrotranslocation. BAX/BAK DKO MEFs transduced with WT-BAX or BAXT182I were digitonin-permeabilized and incubated at room temperature over time. Supernatant and pellet were separated at each time point and BAX and other proteins were probed by immunoblot. VDAC1 was shown as loading controls for the pellet. A portion of a cytosolic protein, HSP70, still remained in the permeabilized cells, but the level of HSP70 in the soluble fraction was unchanged over time unlike WT BAX. This controls for the genuine dissociation of BAX from the pellet fraction that contains mitochondria. On the other hand, BAXT182I remained in the pellet, and presumably stayed associated with mitochondria, due to slow retrotranslocation.
In summary, our results show that the equilibrium of BAXT182I is shifted toward mitochondria from the cytosolic localization. Furthermore, the increased steady-state content of BAXT182I appears to compensate for its defects in downstream events, including membrane insertion and higher-order oligomer formation.
Discussion
BAX helix 9 mutations show divergent activities in vitro and in vivo
Previous reports showed that helix 9 of BAX can mediate inter-dimer interactions that are involved in higher-order oligomer formation (27, 39). To ask whether such oligomers are required for pore formation, we chose to study BAX helix 9 mutations that disrupt inter-dimer interactions, whereas leaving symmetric dimer formation intact. One previous study identified the T182I mutant as fulfilling this criterion (27). We also took note of another study, showing that the G179P mutation abrogated helix 9-helix 9 interactions, when the C-terminal helix 9 sequence was fused to another polypeptide (30).
These two mutations allowed us to interrogate the BAX oligomer formation measured in the liposome membrane and apoptosis regulation in more complex systems. We found that the disruption of inter-dimer interactions involving helix 9 prevented BAX from forming higher-order oligomers in the liposome membrane, as reported, but surprisingly, did not interfere with liposome membrane permeabilization (as shown by BAXG179P) or cell death (as is the case of BAXT182I). We found instead that helix 9 plays a significant role in BAX mitochondrial association by affecting mitochondrial targeting (G179P) or retrotranslocation (T182I). We conclude that the steady-state amount of BAX associated with mitochondria can be more critical in promoting cell death than membrane-insertion or higher-order oligomer formation characterized in the model membrane system (see below).
Defects in membrane insertion and oligomerization can be overridden by mitochondrial residency of BAX
We observed that the two helix 9 mutants of BAX (T182I and G179P) exhibited strong phenotypes in cells, corresponding to their mitochondrial localization, rather than their potency in oligomerization or membrane insertion, as measured in model membranes. In particular, we found that BAXG179P is defective in higher-order oligomer formation using two independent approaches: size-exclusion chromatography after CHAPS solubilization (Fig. 2A), and a TIRF microscopic SLB assay (Fig. 2, B–G). G179P, despite its defect in oligomerization, could permeabilize liposomes (and OMVs, albeit to a lesser extent) and formed pores as large as those formed by WT BAX (Fig. 1A). BAXT182I displayed a similar inability to form oligomers, as shown by SEC (Fig. 2A), but unlike G179P, it inserted into the membrane very slowly (Fig. 1D).
In cells, however, these mutants behaved very differently from what we observed with liposomes. BAXG179P was unable to produce MOMP (Fig. 3), apparently because of defective mitochondrial association (Figs. 3A and 4). We can speculate that the proline residue produces a kink in the helix, dislodging helix 9 aberrantly, which might explain the rapid membrane insertion in our liposome assays. However, it appears that this mutation in helix 9 entirely abrogates its association with mitochondria. This suggests that helix 9 in WT BAX interacts with an unknown BAX targeting machinery in mitochondria and that the G179P mutation disrupts this interaction.
On the other hand, BAXT182I accumulated to greater levels on mitochondria than WT, because of its slower retrotranslocation rate, and mediated apoptosis as potently as WT BAX in intact cells. Apparently, the increased concentration of BAXT182I in the mitochondrial membrane compensated for any defect in membrane insertion and oligomerization seen in vitro. Another C terminus mutant, S184V, localizes to mitochondria in nonapoptotic cells, presumably due to the disruption of an intramolecular hydrogen bond between Ser-184 and Asp-98; the mutation to valine at 184 is thought to dislodge helix 9 from the hydrophobic pocket, so that it becomes more available to interact with the mitochondrial outer membrane (37). In the case of T182I, however, the gain in hydrophobicity from threonine to isoleucine could elicit stronger intramolecular hydrophobic interactions with opposing Leu-76 and/or Met-79 residues.4 Alternatively, the increase in helix 9 hydrophobicity itself may elicit stronger affinity with the membrane, resulting in slow retrotranslocation. Therefore, it appears that facilitated exposure of helix 9 cannot explain the enhanced mitochondrial localization of BAXT182I. An additional effect of this potentially tighter association of helix 9 with the rest of the molecule is to impede unfolding of the protein, perhaps explaining why we observed a much slower insertion of BAXT182I into the membrane (Fig. 1D). Why this mutant might interact more strongly with the retrotranslocation machinery (i.e. exhibits a slow off-rate) is unclear.
Regardless of the structural mechanisms at play, our data show that the enhanced mitochondrial association of BAXT182I can overcome this mutant's substantial defect in membrane insertion. We suggest that mitochondrial localization of BAX plays an important role in apoptosis induction more than its ability to insert into the membrane and form higher-order oligomers, although we do not yet know whether BAXT182I shows the same defect in membrane insertion and oligomer formation in vivo. Thus, future studies are needed to clarify how BAX associates with mitochondria in vivo and how this compares with its in vitro interactions with membranes.
Importance of BAX mitochondrial residency regulated by retrotranslocation for apoptosis sensitivity
Multiple studies have shown that, in live cells, BAX and other Bcl-2 family proteins, such as BAK and Bcl-xL, are retrotranslocated continuously from mitochondria outer membranes back to the cytoplasm. The resulting cytoplasm-mitochondria equilibrium regulates the steady-state mitochondrial content of these proteins (23–25). Changes in this rate of retrotranslocation alters the cells sensitivity to pro-apoptotic stimuli (24). We have shown in this report that in vitro model membrane systems appear to lack this BAX translocation process, which is greatly influenced by retrotranslocation. Both of our mutants exhibit discrepancy in their apoptotic activity between model membranes and in cultured cell systems. Based on our data, we postulate that there is a retrotranslocation-related component associated with the mitochondrial outer membrane for which BAXT182I has high affinity (low off-rate) and BAXG179P has low affinity (low on-rate and/or high off-rate).
The molecular mechanisms of retrotranslocation are not completely understood. Both pro- and anti-apoptotic “multidomain” Bcl-2 family proteins (Bcl-xL, Bcl-2, BAX, and BAK) participate in it (23), and its rate can be regulated by the activity of protein kinases with known role in cell survival (24). One mitochondrial protein important for this process is VDAC2, according to a recent report from the Edlich laboratory (38). VDAC2 has also been shown to interact with inactive forms of BAX and BAK in ways that influence MOMP (40–43). The roles of VDAC2 in retrotranslocation and MOMP are therefore legitimate subjects for future study. Because we and others (24, 36, 44) have observed that the BAX equilibrium significantly influences sensitivity to cell death, it is important to unravel the underlying molecular mechanisms of retrotranslocation, and it is conceivable that the retrotranslocation machinery could prove to be a therapeutic target.
Materials and methods
Recombinant proteins and liposomes
Constructs for BAXT182I and G179P were generated with a QuikChange site-directed mutagenesis kit (number 210518; Agilent Technologies, Santa Clara, CA) on the pTYB1 WT-human BAX plasmid (37) and the sequences were validated. Recombinant nontagged BAX, BAXT182I, BAXG179P, and cBID were generated based on published protocols (19, 37). Purity of all the proteins was evaluated to be >90% by Coomassie stained SDS-PAGE. Dextran-loaded or nonloaded large unilamellar vesicles (LUVs) were generated by detergent removal using octylglucoside (OG-LUV) (18). For the BAX oligomerization assay shown in Fig. 2, we made larger liposomes by extrusion through a membrane with a pore size of 400 nm (9). Both liposome preparations responded to BAX and cBID in the same way.
Membrane permeabilization assays
Dextran release assay
Bulk kinetic dextran release assays were performed with OG-LUV loaded with fluorescein-dextrans (70 kDa) or cascade blue-dextrans (10 kDa) and the matched quenching antibodies (8). In this assay, the released dextrans from permeabilized vesicles were rapidly quenched by the quenching antibodies present outside the liposomes and the fluorescence decline in real-time therefore represented vesicle permeabilization. The percentage of release was calculated against 100% release by 0.2% Triton X-100. OMVs incorporated with 70-kDa fluorescein-dextrans were isolated from Xenopus egg extracts as described (9). The BAX activity was determined as the inverted time that took 50% dextran release and is shown as percentage of WT BAX. p values were calculated by one sample t and Wilcoxon tests against 100% (WT) in Prism.
ANTS/DPX release assay
Liposomes containing ANTS/DPX were generated and fluorescence dequenching was measured as previously reported (45, 46). The mutant BAX activity is shown as the slope of the curve relative to WT BAX and statistical analysis was done with one sample t and Wilcoxon tests in Prism.
NBD labeling of BAX and membrane insertion assay
Recombinant BAX was labeled by NBD dye (IANBD amide, Life Technologies) as reported previously (8, 13, 45). 6 μm BAX and 60 μm NBD were incubated in 400 μl of labeling buffer (10 mm HEPES, pH 7.4, 200 mm NaCl, 0.2 mm EDTA) in the presence of 0.25% CHAPS for 2 h at room temperature, in the dark. The sample was eluted from a 10-ml G25 column equilibrated with labeling buffer. The eluted fractions with NBD fluorescence peak (detected by 485 nm excitation and 520 nm emission) were pooled and concentrated. The typical BAX concentration assayed by BCA was 20–30 μm in ∼80 μl. Membrane insertion assays were performed using 720 nm BAX and nonloaded OG-LUVs. Fluorescence at 485 excitation/520 emission was measured immediately after NBD-BAX was added to the mixture consisting of 10 mm potassium buffer, pH 7.4, 50 mm KCl, and 1 mm EDTA together with 45 nm cBID in 100 μl at 37 °C. The fluorescence increase, which indicates the increased hydrophobicity surrounding the NBD dye, was depicted as the fold-increase over the starting value. The labeling efficiency of each BAX mutant was determined by the raw fluorescence value at time 0 in the assay per mg of protein. The values were, then, compared with the labeled WT BAX, which was subjected to the same run. For BAX T182I, it was 92% of the WT (n = 3; 78, 99, and 99%) and for BAXG179P, 86% (n = 2; 97 and 75%).
BAX oligomer assay with SEC
The size of BAX oligomers in the membrane was analyzed as previously reported (19). BAX (2 μm) and cBID (1.3 μm) were incubated with extruded liposomes for 2 h at 37 °C. Liposomes were floated up by sucrose step gradient centrifugation to separate them from soluble BAX and then collected in a Microcon filtration device with 0.1-μm pore size. The retained membrane fraction was solubilized in a running buffer consisting of 20 mm HEPES, pH 7.4, 150 mm NaCl, and 1.2% CHAPS and size fractionated on a Superdex 200 Increase 10/300 GL column (GE Healthcare, Pittsburgh, PA). Fractions were immunoblotted with anti-BAX antibody (Santa Cruz; N20).
BAX oligomer assay in supported lipid bilayers
BAX purification and labeling
Full-length human BID and single-cysteine, full-length human BAX mutant (S4C, C62S, and C126S), as well as BAX mutants (S4C, C62S, C126S, and G179P) were expressed in Escherichia coli, purified, and labeled with Atto 488 (Attotec, Siegen, Germany) as previously described (34). Excess label was removed by size-exclusion chromatography, and only monomeric fractions were collected for experiments. The activity of the labeled proteins was measured using a giant unilamellar vesicle membrane permeabilization assay. Labeling efficiency was calculated to be 80% for BAX-488 and 50% for BAX G179P-488 by comparing protein and label concentrations with Bradford, spectrometer, and fluorescence correlation spectroscopy measurements.
Supported lipid bilayers
All lipids were purchased from Avanti Polar Lipids. Lipid mixtures containing egg phosphatidylcholine and cardiolipin in a 80:20 ratio were used. To obtain proteoliposomes, LUVs (100 nm) were prepared as described elsewhere (34, 47). LUVs were incubated with 5 nm cBID and 2.5 nm BAX (S4C,C62S,C126S)-488 or BAX G179P (S4C,C62S,C126S)-488 mutants for 1 h at room temperature. After the indicated incubation time, labeled BAX-containing proteoliposomes were diluted to a 1:3 ratio with LUVs to be in the single-molecule regime. The resulting solution was immediately used to create SLBs on piranha-cleaned glass slides (0.13–0.16-mm thickness; Menzel) (34). Unbound proteins and nonfused vesicles were removed by careful washing with buffer and the SLBs were immediately imaged.
Microscopy and stoichiometry analysis
All experiments were performed using a modified Zeiss Axiovert 200M epifluorescence microscope using a 488 laser (Ichrome MLE-LFA multi laser, Toptica) equipped with a α Plan-Fluor ×100/1.46 oil objective (Zeiss), a Laser-TIRF 3 Imaging System (Zeiss), and a EM-CCD camera (iXon 897, Andor). Samples were illuminated for 35 ms with a delay time between frames of 25 ms (number of frames 1200) with an intensity of ∼0.1 kW/cm2.
Stoichiometry analysis was performed as described elsewhere (34, 47). Data in Fig. 2 are provided as raw values and no correction for partial labeling was applied, because this maneuver would introduce uncertainty, particularly with BAXG179P, which had a relatively low labeling efficiency. (We also provide the data corrected for partial labeling in Fig. S1, which are in agreement with the noncorrected data.)
Digitonin-permeabilized cells for cytochrome c and SMAC release and retrotranslocation
BAX/BAK DKO MEFs (∼10 million cells) were harvested and permeabilized on ice for 5 min with 200 μg/ml of digitonin in digitonin-lysis buffer (DLB; 20 mm HEPES, pH 7.4, 100 mm KCl, 250 mm sucrose, 5 mm MgCl2, 1 mm EDTA, 1 mm EGTA). DLB containing 1% BSA was added to stop the reaction, and the cells were pelleted, washed with DLB, and resuspended in DLB. 20 μl of digitonin-permeabilized cell suspension was incubated with recombinant BAX (80–120 nm) with and without cBID (45 nm) at 30 °C for 1 h. After the reaction, the cells were pelleted and the supernatant was immunoblotted with anti-cytochrome c antibody (BD; 556433). The membrane was then stripped and probed with anti-SMAC antibody (MBL; JM3298). The supernatant was removed completely from the pellet, which was resuspended in DLB, sonicated briefly, and loaded onto SDS-PAGE. BAX in the membrane fraction was detected by anti-BAX antibody (Santa Cruz; N20). Anti-mouse (Santa Cruz; sc-2005) or rabbit (Invitrogen; 31460) secondary antibodies conjugated to horseradish peroxidase were used to detect primary antibodies.
BAX-transduced MEFs were treated the same way as above and incubated with cBID alone (45 or 90 nm) at 30 °C for 1 h. The incubation mix was processed as above for cytochrome c or BAX immunoblotting. For the biochemical retrotranslocation assay, the permeabilized cell suspension was incubated at room temperature and 50 μl was collected at each time point on ice. The supernatant and the pellet of the suspension were processed the same way as above and immunoblotted with anti-BAX, HSP70 (BD Biosciences; 610607), or VDAC1 (Calbiochem; 31HL) antibodies. All primary antibodies were used at 1:1000 and the secondary antibodies, 1:2000.
Transduction of WT-BAX, T182I, or G179P in BAX/BAK DKO MEFs
Mouse WT-BAX, T182I, and G179P were cloned into pMIG vector (a gift from Dr. Emily Cheng) and retrovirus was produced by transfecting Plat-E cells with the plasmid. BAX/BAK DKO MEFs were infected with the virus and the cells were sorted for high level GFP expression. The cell lysates were prepared in PBS containing 1% Nonidet P-40 with protease inhibitors and BAX levels were examined by immunoblotting with anti-BAX antibody (N20). Equal loading was confirmed by Ponceau stain on the transferred nitrocellulose membrane.
Cell death assay
BAX-transduced MEFs (20,000/well) were seeded into a 96-well-plate and treated with staurosporine at 0.05 and 0.1 μm in duplicate for 20 h. The cells were detached with Accutase (Innovative Cell Technologies, San Diego, CA), washed with PBS in the plate, and stained with propidium iodide (Invitrogen)(1 μg/ml) in 100 μl of PBS. Propidium iodide fluorescence was detected by FACSCanto II (BD Biosciences, San Jose, CA) in a 96-well-plate format.
Transient expression of GFP-BAX and retrotranslocation assay
Mouse WT BAX, BAXT182, and BAX G179P were cloned in pAcGFP1-C1 (Clontech) and human BAXS184V was cloned in pEGFP-C1 (Clontech). BAX/BAK DKO MEFs were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were seeded onto glass-bottom dishes (IBL) 24 h before transfection to ensure 60–70% confluence on the day of transfection. Transfections were carried out using Lipofectamine LTX Plus (Life Technologies) following the manufacturer's instructions, using 0.5 μg of DNA. Cells were imaged 24 h post-transfection.
For immunofluorescence, cells were fixed in 4% formaldehyde and permeabilized in PBS, 0.1% Triton X-100. Anti-GFP antibodies (Life Technologies; A-11122) were diluted to 2 μg/ml in PBS, 10% horse serum (37 °C for 1 h). After washing in PBS, secondary antibodies were diluted in the above buffer (37 °C for 1 h). After washing in PBS, DAPI was diluted to 1 μg/ml in PBS and incubated for 5 min. Coverslips were mounted using Dako Fluorescence Mounting Medium (Agilent), imaged using an Axioplan 2 imaging microscope with a ×63/1.4NA Plan Apochromat objective (Zeiss MicroImaging), and quantified with ImageJ software.
Live cell imaging was carried out using a Zeiss Axio-Observer Z1 microscope with CSU-X1 spinning disc confocal (Yokagowa), using a ×63/1.40 Plan Apochromat objective, Evolve EMCCD camera (Photometrics), and a motorized XYZ stage (ASI). The 488-nm laser was controlled via AOTF through the laserstack (Intelligent Imaging Innovations). Images were captured using Slidebook 6.0 software (Intelligent Imaging Innovations). FLIP was carried out by photobleaching two regions within the cell, one cytosolic and one overlapping the nucleus, for two 50-ms iterations, 100% laser power. Images were captured every 5 s. Captured images were analyzed using ImageJ. In brief, the background was subtracted and the cytosolic content exclusive of the bleached ROIs was selected. Signal decay was quantified and normalized to 100% fluorescence post-bleaching. Statistical analysis was carried out by ANOVA, using GraphPad Prism. The imaging did not result in photobleaching when we analyzed other cells in each field of view in the vicinity of the bleached cells over the same time course (Fig. S1).
Confocal microscopy
WT BAX, BAXT182, or BAXG179P-transduced BAX/BAK DKO MEFs were plated out in 8-chamber coverglasses (Lab-Tek II Chambered coverglass number 1.5; 155409) and treated with 1 μm staurosporine and 20 μm Q-VD (Selleckchem) for 5 h. Cells were fixed with 0.5% glutaraldehyde and quenched with 0.5% sodium borohydride in PBS. After permeabilization with 0.5% Triton X-100, cells were stained with anti-BAX antibody (6A7; Biolegend) and anti-SMAC antibody (Sigma; PRS2411) at 1:100, with 10% BSA in PBS. Primary antibodies were detected by anti-rabbit antibody conjugated to Alexa Fluor 568 and anti-mouse antibody conjugated to AF488 (Life Technologies). The cells were imaged in a Zeiss LSM780 microscope.
Author contributions
T. K. conceptualization; T. K. resources; T. K., L. E. K., K. C., and J. S. data curation; T. K., L. E. K., K. C., A. J. G.-S., A. P. G., and D. D. N. formal analysis; T. K. supervision; T. K. validation; T. K. investigation; T. K. visualization; T. K. and A. P. G. methodology; T. K. and D. D. N. writing-original draft; T. K. project administration; T. K., L. E. K., K. C., A. J. G.-S., A.P.G., and D. D. N. writing-review and editing; A. J. G.-S., A. P. G., and D. D. N. funding acquisition.
Supplementary Material
Acknowledgment
We thank Dr. Emily Cheng (Memorial Slone-Kettering Institute, NY) for providing the pMIG vector and the protocols for retroviral transduction.
This work was supported by grants from the Eliteprogramme for Postdocs of the Baden-Wurttemberg Stiftung (to K. C.), ERC-St12 309966 (to A. J. G.-S.), Deutsche Forschungsgemeinschaft Grant GA1641/2–2 (to A. J. G.-S.), a Manchester Cancer Research Centre CRUK training award (to L. E. K.), a Wellcome Center for Cell-Matrix Research Core Grant 203128/Z/16/Z from the Wellcome Trust, and National Institutes of Health Grant R01CA179087 (to D. D. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
D. Zajonc, La Jolla Institute for Immunology, personal communication.
- MOMP
- mitochondrial outer membrane permeabilization
- BH
- Bcl-2 homology
- OMV
- outer membrane vesicles
- NBD
- nitrobenzoxadiazol
- SEC
- size-exclusion column
- SLB
- supported lipid bilayer
- DKO
- double-knockout
- MEF
- mouse embryonic fibroblast
- cBID
- cleaved BID
- FLIP
- fluorescence loss in photobleaching
- LUV
- large unilamellar vesicle
- OG
- octylglucoside
- DLB
- digitonin-lysis buffer
- DAPI
- 4′,6-diamidino-2-phenylindole
- ANOVA
- analysis of variance
- TIRF
- total internal reflection fluorescence.
References
- 1. Danial N. N., and Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 10.1016/S0092-8674(04)00046-7 [DOI] [PubMed] [Google Scholar]
- 2. Chipuk J. E., Moldoveanu T., Llambi F., Parsons M. J., and Green D. R. (2010) The BCL-2 family reunion. Mol. Cell 37, 299–310 10.1016/j.molcel.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gillies L. A., and Kuwana T. (2014) Apoptosis regulation at the mitochondrial outer membrane. J. Cell Biochem. 115, 632–640 10.1002/jcb.24709 [DOI] [PubMed] [Google Scholar]
- 4. Youle R. J., and Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 5. Adams J. M., and Cory S. (2018) The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36 10.1038/cdd.2017.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cang S., Iragavarapu C., Savooji J., Song Y., and Liu D. (2015) ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 8, 129 10.1186/s13045-015-0224-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reyna D. E., Garner T. P., Lopez A., Kopp F., Choudhary G. S., Sridharan A., Narayanagari S. R., Mitchell K., Dong B., Bartholdy B. A., Walensky L. D., Verma A., Steidl U., and Gavathiotis E. (2017) Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell 32, 490–505.e10 10.1016/j.ccell.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kushnareva Y., Andreyev A. Y., Kuwana T., and Newmeyer D. D. (2012) BAX activation initiates the assembly of a multimeric catalyst that facilitates BAX pore formation in mitochondrial outer membranes. PLos Biol. 10, e1001394 10.1371/journal.pbio.1001394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuwana T., Mackey M. R., Perkins G., Ellisman M. H., Latterich M., Schneiter R., Green D. R., and Newmeyer D. D. (2002) BID, BAX, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342 10.1016/S0092-8674(02)01036-X [DOI] [PubMed] [Google Scholar]
- 10. Kuwana T., Bouchier-Hayes L., Chipuk J. E., Bonzon C., Sullivan B. A., Green D. R., and Newmeyer D. D. (2005) BH3 domains of BH3-only proteins differentially regulate BAX-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535 10.1016/j.molcel.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 11. Chipuk J. E., Kuwana T., Bouchier-Hayes L., Droin N. M., Newmeyer D. D., Schuler M., and Green D. R. (2004) Direct activation of BAX by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014 10.1126/science.1092734 [DOI] [PubMed] [Google Scholar]
- 12. Billen L. P., Kokoski C. L., Lovell J. F., Leber B., and Andrews D. W. (2008) Bcl-XL inhibits membrane permeabilization by competing with BAX. PLos Biol. 6, e147 10.1371/journal.pbio.0060147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., and Andrews D. W. (2008) Membrane binding by tBID initiates an ordered series of events culminating in membrane permeabilization by BAX. Cell 135, 1074–1084 10.1016/j.cell.2008.11.010 [DOI] [PubMed] [Google Scholar]
- 14. Gavathiotis E., Suzuki M., Davis M. L., Pitter K., Bird G. H., Katz S. G., Tu H. C., Kim H., Cheng E. H., Tjandra N., and Walensky L. D. (2008) BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081 10.1038/nature07396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gavathiotis E., Reyna D. E., Bellairs J. A., Leshchiner E. S., and Walensky L. D. (2012) Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol. 8, 639–645 10.1038/nchembio.995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niu X., Brahmbhatt H., Mergenthaler P., Zhang Z., Sang J., Daude M., Ehlert F. G. R., Diederich W. E., Wong E., Zhu W., Pogmore J., Nandy J. P., Satyanarayana M., Jimmidi R. K., Arya P., Leber B., Lin J., Culmsee C., Yi J., and Andrews D. W. (2017) A small-molecule inhibitor of BAX and BAK oligomerization prevents genotoxic cell death and promotes neuroprotection. Cell Chem. Biol. 24, 493–506.e495 10.1016/j.chembiol.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gillies L. A., Du H., Peters B., Knudson C. M., Newmeyer D. D., and Kuwana T. (2015) Visual and functional demonstration of growing BAX-induced pores in mitochondrial outer membranes. Mol. Biol. Cell 26, 339–349 10.1091/mbc.E13-11-0638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schafer B., Quispe J., Choudhary V., Chipuk J. E., Ajero T. G., Du H., Schneiter R., and Kuwana T. (2009) Mitochondrial outer membrane proteins assist BID in BAX-mediated lipidic pore formation. Mol. Biol. Cell 20, 2276–2285 10.1091/mbc.e08-10-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuwana T., Olson N. H., Kiosses W. B., Peters B., and Newmeyer D. D. (2016) Pro-apoptotic BAX molecules densely populate the edges of membrane pores. Sci. Rep. 6, 27299 10.1038/srep27299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Große L., Wurm C. A., Brüser C., Neumann D., Jans D. C., and Jakobs S. (2016) BAX assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 35, 402–413 10.15252/embj.201592789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salvador-Gallego R., Mund M., Cosentino K., Schneider J., Unsay J., Schraermeyer U., Engelhardt J., Ries J., and Garcíia-Sáez A. J. (2016) BAX assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 35, 389–401 10.15252/embj.201593384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu X. P., Zhai D., Kim E., Swift M., Reed J. C., Volkmann N., and Hanein D. (2013) Three-dimensional structure of BAX-mediated pores in membrane bilayers. Cell Death Dis. 4, e683 10.1038/cddis.2013.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edlich F., Banerjee S., Suzuki M., Cleland M. M., Arnoult D., Wang C., Neutzner A., Tjandra N., and Youle R. J. (2011) Bcl-x(L) retrotranslocates BAX from the mitochondria into the cytosol. Cell 145, 104–116 10.1016/j.cell.2011.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schellenberg B., Wang P., Keeble J. A., Rodriguez-Enriquez R., Walker S., Owens T. W., Foster F., Tanianis-Hughes J., Brennan K., Streuli C. H., and Gilmore A. P. (2013) BAX exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell 49, 959–971 10.1016/j.molcel.2012.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Todt F., Cakir Z., Reichenbach F., Emschermann F., Lauterwasser J., Kaiser A., Ichim G., Tait S. W., Frank S., Langer H. F., and Edlich F. (2015) Differential retrotranslocation of mitochondrial BAX and BAK. EMBO J. 34, 67–80 10.15252/embj.201488806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dengler M. A., Robin A. Y., Gibson L., Li M. X., Sandow J. J., Iyer S., Webb A. I., Westphal D., Dewson G., and Adams J. M. (2019) BAX activation: mutations near its proposed non-canonical BH3 binding site reveal allosteric changes controlling mitochondrial association. Cell Rep. 27, 359–373.e6 10.1016/j.celrep.2019.03.040 [DOI] [PubMed] [Google Scholar]
- 27. Zhang Z., Subramaniam S., Kale J., Liao C., Huang B., Brahmbhatt H., Condon S. G., Lapolla S. M., Hays F. A., Ding J., He F., Zhang X. C., Li J., Senes A., Andrews D. W., and Lin J. (2016) BH3-in-groove dimerization initiates and helix 9 dimerization expands BAX pore assembly in membranes. EMBO J. 35, 208–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dewson G., Ma S., Frederick P., Hockings C., Tan I., Kratina T., and Kluck R. M. (2012) BAX dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 19, 661–670 10.1038/cdd.2011.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dewson G., Kratina T., Sim H. W., Puthalakath H., Adams J. M., Colman P. M., and Kluck R. M. (2008) To trigger apoptosis, BAK exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 30, 369–380 10.1016/j.molcel.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 30. Andreu-Fernández V., Sancho M., Genovés A., Lucendo E., Todt F., Lauterwasser J., Funk K., Jahreis G., Pérez-Payá E., Mingarro I., Edlich F., and Orzáez M. (2017) BAX transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc. Natl. Acad. Sci. U.S.A. 114, 310–315 10.1073/pnas.1612322114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Antonsson B., Montessuit S., Sanchez B., and Martinou J. C. (2001) BAX is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 276, 11615–11623 10.1074/jbc.M010810200 [DOI] [PubMed] [Google Scholar]
- 32. Kim H., Tu H. C., Ren D., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J., and Cheng E. H. (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499 10.1016/j.molcel.2009.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garner T. P., Reyna D. E., Priyadarshi A., Chen H. C., Li S., Wu Y., Ganesan Y. T., Malashkevich V. N., Cheng E. H., and Gavathiotis E. (2016) An autoinhibited dimeric form of BAX regulates the BAX activation pathway. Mol. Cell 64, 431 10.1016/j.molcel.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 34. Subburaj Y., Cosentino K., Axmann M., Pedrueza-Villalmanzo E., Hermann E., Bleicken S., Spatz J., and García-Sáez A. J. (2015) BAX monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 6, 8042 10.1038/ncomms9042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Todt F., Cakir Z., Reichenbach F., Youle R. J., and Edlich F. (2013) The C-terminal helix of Bcl-x(L) mediates BAX retrotranslocation from the mitochondria. Cell Death Differ. 20, 333–342 10.1038/cdd.2012.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nechushtan A., Smith C. L., Hsu Y. T., and Youle R. J. (1999) Conformation of the BAX C-terminus regulates subcellular location and cell death. EMBO J. 18, 2330–2341 10.1093/emboj/18.9.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suzuki M., Youle R. J., and Tjandra N. (2000) Structure of BAX: coregulation Of dimer formation and intracellular localization. Cell 103, 645–654 10.1016/S0092-8674(00)00167-7 [DOI] [PubMed] [Google Scholar]
- 38. Lauterwasser J., Todt F., Zerbes R. M., Nguyen T. N., Craigen W., Lazarou M., van der Laan M., and Edlich F. (2016) The porin VDAC2 is the mitochondrial platform for BAX retrotranslocation. Sci. Rep. 6, 32994 10.1038/srep32994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bleicken S., Jeschke G., Stegmueller C., Salvador-Gallego R., García-Sáez A. J., and Bordignon E. (2014) Structural model of active BAX at the membrane. Mol. Cell 56, 496–505 10.1016/j.molcel.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lazarou M., Stojanovski D., Frazier A. E., Kotevski A., Dewson G., Craigen W. J., Kluck R. M., Vaux D. L., and Ryan M. T. (2010) Inhibition of BAK activation by VDAC2 is dependent on the BAK transmembrane anchor. J. Biol. Chem. 285, 36876–36883 10.1074/jbc.M110.159301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma S. B., Nguyen T. N., Tan I., Ninnis R., Iyer S., Stroud D. A., Menard M., Kluck R. M., Ryan M. T., and Dewson G. (2014) BAX targets mitochondria by distinct mechanisms before or during apoptotic cell death: a requirement for VDAC2 or BAK for efficient BAX apoptotic function. Cell Death Differ. 21, 1925–1935 10.1038/cdd.2014.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng E. H., Sheiko T. V., Fisher J. K., Craigen W. J., and Korsmeyer S. J. (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517 10.1126/science.1083995 [DOI] [PubMed] [Google Scholar]
- 43. Naghdi S., and Hajnóczky G. (2016) VDAC2-specific cellular functions and the underlying structure. Biochim. Biophys. Acta 1863, 2503–2514 10.1016/j.bbamcr.2016.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valentijn A. J., Metcalfe A. D., Kott J., Streuli C. H., and Gilmore A. P. (2003) Spatial and temporal changes in BAX subcellular localization during anoikis. J. Cell Biol. 162, 599–612 10.1083/jcb.200302154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kale J., Chi X., Leber B., and Andrews D. (2014) Examining the molecular mechanism of bcl-2 family proteins at membranes by fluorescence spectroscopy. Methods Enzymol. 544, 1–23 10.1016/B978-0-12-417158-9.00001-7 [DOI] [PubMed] [Google Scholar]
- 46. Asciolla J. J., Renault T. T., and Chipuk J. E. (2012) Examining BCL-2 family function with large unilamellar vesicles. J. Vis. Exp. 4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Flores-Romero H., Landeta O., Ugarte-Uribe B., Cosentino K., García-Porras M., García-Sáez A. J., and Basañez G. (2019) BFL1 modulates apoptosis at the membrane level through a bifunctional and multimodal mechanism showing key differences with BCLXL. Cell Death Differ. 26, 1880–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karbowski M., Norris K. L., Cleland M. M., Jeong S. Y., and Youle R. J. (2006) Role of BAX and BAK in mitochondrial morphogenesis. Nature 443, 658–662 10.1038/nature05111 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.