

Abstract
The unwinding of dsRNA intermediates is critical for the replication of flavivirus RNA genomes. This activity is provided by the C-terminal helicase domain of viral nonstructural protein 3 (NS3). As a member of the superfamily 2 (SF2) helicases, NS3 requires the binding and hydrolysis of ATP/NTP to translocate along and unwind double-stranded nucleic acids. However, the mechanism of energy transduction between the ATP- and RNA-binding pockets is not well-understood. Previous molecular dynamics simulations conducted by our group have identified Motif V as a potential “communication hub” for this energy transduction pathway. To investigate the role of Motif V in this process, here we combined molecular dynamics, biochemistry, and virology approaches. We tested Motif V mutations in both the replicon and recombinant protein systems to investigate viral genome replication, RNA-binding affinity, ATP hydrolysis activity, and helicase-mediated unwinding activity. We found that the T407A and S411A substitutions in NS3 reduce viral replication and increase the helicase-unwinding turnover rates by 1.7- and 3.5-fold, respectively, suggesting that flaviviruses may use suboptimal NS3 helicase activity for optimal genome replication. Additionally, we used simulations of each mutant to probe structural changes within NS3 caused by each mutation. These simulations indicate that Motif V controls communication between the ATP-binding pocket and the helical gate. These results help define the linkage between ATP hydrolysis and helicase activities within NS3 and provide insight into the biophysical mechanisms for ATPase-driven NS3 helicase function.
Keywords: flavivirus, RNA helicase, molecular dynamics, computational biology, allosteric regulation, ATPase activity, dengue NS3 helicase, pathway analysis, SF2 helicase, West Nile NS3 helicase, helicase activity, all-atom molecular dynamics, energy transduction, Motif V
Introduction
Arthropod-borne flaviviruses, such as yellow fever virus, Japanese encephalitis virus, Zika virus, West Nile virus (WNV),3 and dengue virus, are a major health concern in the tropical and subtropical regions of the world (1, 2). Infection from these viruses causes disease symptoms ranging from flu-like illness to encephalitis, hemorrhagic fever, coma, and potentially death (3). Over half of the world population is at risk for infection from one or more of these viruses (4, 5). Dengue virus, specifically, infects around 50 million people each year, and of those individuals, 20,000 contract dengue hemorrhagic fever leading to their mortality (6). Additionally, WNV over the past 20 years within the 48 continental United States has around 50,000 clinical infections with a 5% mortality rate (7, 8) (and see https://www.cdc.gov/westnile/statsmaps/index.html (accessed September 12, 2019)). Currently, there are no approved antiviral drugs for treating flaviviral infections, and the vaccines in circulation, like the yellow fever vaccine, are not readily available worldwide for most flaviviruses (10–12). To develop new antiviral treatments (drugs and vaccines) for these viruses, a fundamental understanding of how these flaviviruses replicate is required.
Flaviviruses (Flaviviridae family) have a positive-sense single-stranded RNA (ssRNA) genome that is ∼11 kb in length. The viral RNA genome is translated into a single polyprotein that is subsequently cleaved by host and viral proteases into three structural proteins (C, prM, and E) and eight nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, 2K, NS4B, and NS5) (13, 14). The structural proteins contribute to the formation of mature viral particles, whereas the nonstructural proteins are responsible for replication of the viral genome and protecting the virus from attacks by the host cell innate immune system (13). Once the viral proteins are post-translationally processed, the positive-sense genomic ssRNA is used as a template to create a negative-sense anti-genomic RNA, forming a dsRNA intermediate complex (15, 16). The nascent negative-sense ssRNA serves as a template strand for producing new positive-strand RNAs that can be packaged into viral particles, translated into new proteins, or interfere with the RNA decay pathway (17, 18). Therefore, the unwinding of dsRNA intermediate to produce a free negative ssRNA for positive-strand synthesis is a critical component for the replication of flavivirus RNA genomes (15). This function is achieved by the C-terminal helicase domain of NS3.
Helicases are ubiquitous enzymes that are classified into superfamilies based on their primary structure and highly conserved motifs (19–21). NS3 helicase (NS3h) is a member of superfamily 2 (SF2) helicases (22). The structure of NS3h consists of three subdomains; subdomain 1 and subdomain 2 are RecA-like structures that are highly conserved across SF2 helicases (23). The third subdomain is unique to the viral/DEAH-like subfamily of SF2 helicases and interacts with subdomains 1 and 2 to form the RNA-binding cleft. NS3h is a multifunctional enzyme, possessing three enzymatic activities: RNA helicase, nucleoside triphosphatase (NTPase), and RNA 5′-triphosphatase (RTPase) (24–29). Previous studies have shown that the two latter activities share a catalytic active site where NTP binds between subdomain 1 and subdomain 2 (26, 30–32). The RNA helicase active site is distinct from that of the NTPase/RTPase active site, located at the helical gate between subdomain 2 and subdomain 3 of NS3 (33–35). The helical gate and β-wedge are responsible for splitting the dsRNA intermediate into two ssRNA strands: the positive-sense viral ssRNA and the negative-sense template ssRNA (33). Single-molecule and structural biology studies have suggested that the negative-sense ssRNA template enters the RNA-binding cleft, where the helicase utilizes the energy produced from the hydrolysis of one ATP molecule to power translocation and unwinding of the dsRNA intermediate one base at a time (15, 16, 32, 36–40). A fundamental unanswered question within the SF2 helicase field is how ATP hydrolysis causes structural changes within the helicase to result in translocation and unwinding of dsRNA intermediates.
Between the ATPase active site and the RNA-binding cleft, there are eight structural motifs (Motifs I, Ia, II, III, IV, IVa, V, and VI) that are highly conserved in the viral/DEAH-like subfamily of SF2 helicases (Fig. 1) (22). These structural motifs play a critical role in substrate binding and are responsible for enzymatic activities within the helicase. One of these motifs, Motif V, was reported by Davidson et al. (24) as a potential link between the ATP-binding pocket and the RNA-binding cleft due to strong correlations between residues within Motif V and both binding pockets found within the helicase. Additionally, Mastrangelo et al. (34) reported that Motif V may be one of the main components of the driving force pulling the helicase along the ssRNA due to conformational changes initiated by the binding of ATP, which propagated through Motif V opening the helical gate. At least two residues within Motif V have direct interactions with either the ATP hydrolysis active site or the bound ssRNA molecule. Gly414 coordinates with the lytic water found within the ATPase active site, and Thr408 interacts with the phosphate backbone of the bound ssRNA (24). In addition to these interactions, Motif V has strong coupling between the other highly conserved motifs that interact with the ATP-binding pocket and the RNA-binding cleft, suggesting that Motif V may be central to the communication between these two binding pockets (24, 41). Therefore, we investigated the role of Motif V as a critical link between the two binding pockets. We utilized a combination of all-atom molecular dynamics simulations, biochemical assays, and virological assays to understand the role of Motif V in internal enzymatic communication leading to helicase function. Our results show that residues within Motif V control not only the enzymatic activities of the helicase but also the ability of the flaviviruses to replicate in vitro. This is due to the importance of the secondary structure of Motif V, as indicated through molecular dynamics simulation.
Figure 1.
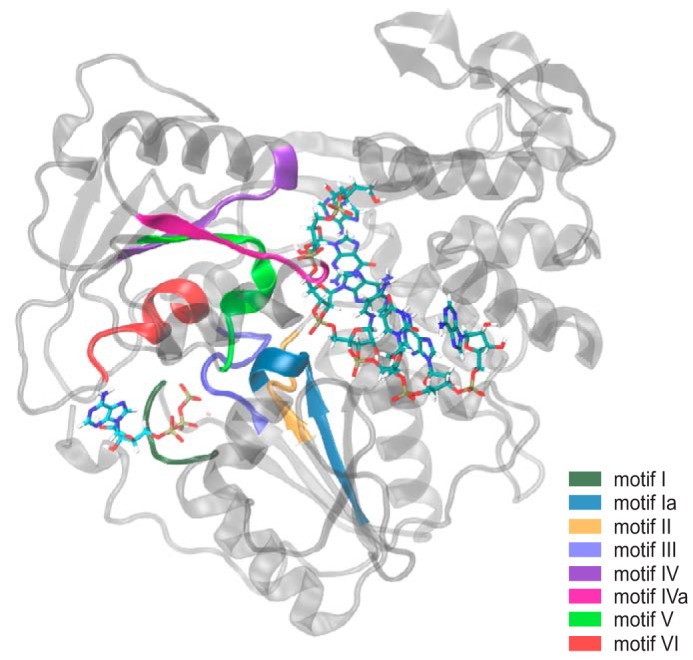
Eight structural motifs of Flavivirus NS3 helicases are highly conserved. The structural motifs (Motifs I, Ia, II, III, IV, IVa, V, and VI) are located between the ATP-binding pocket and RNA-binding cleft. Each motif is highlighted as follows: Motif I in dark green; Motif Ia in blue; Motif II in orange; Motif III in royal blue; Motif IV in purple; Motif IVa in magenta; Motif V in lime green; and Motif VI in red.
Results
Motif V mutants affect viral genome replication
As discussed previously by Davidson et al. (24), Motif V may play a role in the communication between the ATP-binding pocket and the RNA-binding cleft/helical gate. As one of the highly conserved motifs in SF2 helicases, Motif V consists of 13 residues spanning position 404 through position 416 (Fig. 2A). Eight of these residues are 100% conserved across all flaviviruses, both mosquito and tick-borne (Fig. 2B). The other five residues are highly conserved in flaviviruses that infect insects and mammals and are more variable in tick-borne restricted flaviviruses (Table S1). Interestingly, the conserved residues across all of the flaviviruses interact with either the ssRNA or ATP substrates. These highly conserved residues include Thr408, Asp409, Ile410, Glu412, Met413, Gly414, Ala415, Asn416. Of the mostly conserved residues, Thr407 and Ser411 mainly interact with each other through a hydrogen bond that is hypothesized to stabilize the α-helical secondary structure of Motif V (Fig. 2C). The other mostly conserved residues, Phe404, Val405, and Val406, interact primarily with other hydrophobic residues in the surrounding area. Generally, all of Motif V interacts with either a bound substrate or other highly conserved motifs that interact with bound substrate. Therefore, we investigated the importance of each residue in Motif V for viral replication.
Figure 2.

The majority of Motif V residues are highly conserved across all flavivirus NS3 helicases. A, the sequence of Motif V from all flaviviruses, multiple species and insect-restricted. B, the percentage conservation for each residue within Motif V is projected onto the secondary structure. The color bar ranges linearly from red (0.0% conserved) to blue (100.0% conserved). C, Thr407 and Ser411 interact through a hydrogen bond.
We individually mutated every residue within NS3h Motif V in a luciferase-expressing WNV replicon system and determined replication efficiency for each mutant. Each Motif V mutation was designed to interrupt specific WT residue-residue interactions or residue-substrate interactions. As a negative control, Asp664 in the NS5 polymerase catalytic active site was mutated to valine (D664V) to disrupt viral genome replication (42–45). Additionally, the NS3 mutants A286L (Motif II) and R387M (Motif IVa) were used as controls for ablating ATPase activity and RNA-binding affinity, respectively (30, 46–50). The small hydrophobic side chain of Ala286 interacts with other hydrophobic residues stabilizing Motif II interactions with bound ATP. By introducing a bulky hydrophobic residue with A286L, the Motif II interactions with ATP will be perturbed, resulting in a disruption in ATP hydrolysis activity. The guanidium headgroup of Arg387 interacts with the phosphates of the RNA backbone. By removing the guanidium headgroup with R387M, important protein-RNA interactions will be interrupted. NS5 D664V and NS3 A286L and R387M ablated viral genome replication, as expected (Fig. 3A). All of the Motif V mutants, except for V405M, V406M, T407A, T408S, S411A, and A415G, ablated viral genome replication as well. V405M and T407A reduced replication to ∼2.5%, whereas the I406M and A415G mutants reduce viral genome replication to less than 20%. T408S and S411A were ∼48 and 39% active, respectively, compared with WT NS3h. The results from the viral genome replication were projected onto the structure of Motif V (Fig. 3B). We noted that residues with ablated viral replication consisted mostly of the highly conserved residues, suggesting that these highly conserved residues that interact with either the RNA or the ATP play an important role in viral replication. Interestingly, Thr408 and Ala415, both highly conserved residues, still maintain a reduced level of viral replication, suggesting that there may not be a direct correlation between residue conservation and effect on replication.
Figure 3.

Mutations in Motif V negatively affect viral genome replication. A, mutations to remove specific interactions within the central location of Motif V between ATPase active site and the RNA-binding cleft were tested in viral genome replication. B, results from the viral genome replication were projected onto the structure of Motif V with the hydrogen bond highlighted between residues Thr407 and Ser411. The color bar ranges linearly from red (0% replication) to blue (100% replication). These data suggest that the hydrogen bond between Thr407 and Ser411 is important for viral genome replication. Error bars, S.E.
As mentioned before, residues Thr407 and Ser411 interact with each other through a hydrogen bond (Fig. 2C). However, the T407A mutant nearly ablates genome replication, whereas the S411A mutation only reduces genome replication by ∼60%. Both mutations should disrupt the hydrogen bond, but T407A and S411A have significantly different replication levels, suggesting that these two mutations have other interactions that potentially affect one or more of the NS3h functions during viral replication. These NS3h functions include the ability of the helicase to bind RNA, the ability to hydrolyze ATP, and the ability to unwind the dsRNA intermediate. Therefore, to determine whether or not these mutations have an effect on the individual helicase functions, we tested both T407A and S411A biochemically.
NS3h residues Thr407 and Ser411 affect ATPase and helicase functions but not RNA-binding affinity
We designed/adapted three biochemical assays to observe how the Thr407 and Ser411 mutations would affect the individual helicase functions. Recombinant WT, T407A, S411A, A286L (ATPase control), and R387M (RNA-binding control) NS3h were purified and tested in an RNA-binding affinity assay, an ATPase activity assay, and a helicase-unwinding activity assay. We used DENV4 NS3 proteins for this work due to established expression and purification protocols being in place (51) and the high degree of conservation between WNV and DENV in Motif V. To start, the RNA-binding affinity of WT, T407A, S411A, A286L, and R387M were tested via a fluorescence polarization assay (52). Fluorescence polarization for a single-stranded RNA oligonucleotide labeled with an Alexa 488 fluorophore (5 nm) was measured as the NS3 helicase increased in concentration (Fig. 4A). The binding affinity or Kd for WT NS3h was 1.9 ± 0.8 μm, whereas the Kd for R387M was 76.4 ± 31.0 μm (Table 1A). The R387M Kd was significantly different compared with WT, suggesting that R387M does not bind the ssRNA as well as WT. Additionally, a similar trend was observed for WT and A286L. On the other hand, T407A (2.0 ± 1.1 μm) and S411A (1.4 ± 0.2 μm) were not significantly different as compared with WT. These data suggest that neither the T407A nor S411A mutation affected ssRNA binding to NS3.
Figure 4.

Experimental design of biochemical assays. A, RNA-binding affinity of NS3h was measured using fluorescence polarization of a single-stranded RNA (52). WT and NS3h variants are shown. B, ATPase activity was measured using BIOMOL Green absorbance at 650 nm over time. WT NS3h ATPase activity at five concentrations of ATP is shown here. C, RNA helicase-unwinding activity was measured by the increase of fluorescence over time. The WT NS3h data presented here are representative of the ATP-dependent unwinding activity obtained for each NS3h variant. D, WT, A286L, R387M, T407A, and S411A NS3h-unwinding activities were compared at 400 μm ATP. Error bars, S.E.
Table 1.
Enzyme kinetics of WT, A286L, R387M, T407A and S411A NS3 helicase
A, Kd was determined from the RNA-binding affinity assay data. B, kcat, Km, and kcat/Km were determined for the ATPase activity assay through fitting the data to the substrate inhibition equation. C, kcat, Km, and kcat/Km were determined for the helicase-unwinding activity assay for each NS3h variant through fitting the Michaelis–Menten equation to the Kobs versus ATP concentration.
|
A. RNA-binding affinity | ||
|---|---|---|
| NS3h variant | Kd (μm) | -Fold difference |
| WT | 1.9 ± 0.8 | |
| A286L | 6.7 ± 0.4 | 3.5 |
| R387M | 76.4 ± 31.0 | 39.7 |
| T407A | 2.0 ± 1.1 | 1.0 |
| S411A | 1.4 ± 0.2 | 0.7 |
|
B. ATP hydrolysis activity | |||
|---|---|---|---|
| NS3h variant | kcat (s−1) | Km (μm) | kcat/Km (μm−1 s−1) |
| WT | 1.2 ± 0.1 × 10−3 | 17.3 ± 4.6 | 0.0672 ± 0.019 × 10−3 |
| A286L | 0.9 ± 0.4 × 10−3 | 285.1 ± 150.5 | 0.0033 ± 0.0022 × 10−3 |
| R387M | 1.6 ± 0.3 × 10−3 | 49.5 ± 21.2 | 0.0314 ± 0.015 × 10−3 |
| T407A | 1.2 ± 0.2 × 10−3 | 33.7 ± 12.1 | 0.0364 ± 0.0142 × 10−3 |
| S411A | 1.8 ± 0.5 × 10−3 | 71.5 ± 34.8 | 0.0257 ± 0.0143 × 10−3 |
|
C. Helicase-unwinding activity | |||
|---|---|---|---|
| NS3h variant | kcat (s−1) | Km (μm) | kcat/Km (μm−1 s−1) |
| WT | 47.6 ± 4.4 × 10−3 | 24.5 ± 11.3 | 1.9 ± 0.8 × 10−3 |
| A286L | 0.0 | 0.0 | 0.0 |
| R387M | 0.0 | 0.0 | 0.0 |
| T407A | 80.8 ± 6.0 × 10−3 | 14.4 ± 6.2 | 5.6 ± 2.2 × 10−3 |
| S411A | 166.6 ± 15.5 × 10−3 | 58.0 ± 21.6 | 2.9 ± 0.9 × 10−3 |
We next investigated how these mutations affected NS3 ATPase activity. We utilized a colorimetric assay to observe ATP hydrolysis via Pi production and release for WT, T407A, S411A, A286L, and R387M (51). All reactions contained 50 nm NS3h, 5 nm dsRNA, and increasing concentrations of ATP. Progress curves were obtained for each NS3h at various ATP concentrations (Fig. 4B). We noticed at the high concentrations of ATP a substrate inhibition over time. Therefore, we used the substrate inhibition equation to calculate the enzyme kinetics for the ATPase activity assay. The catalytic efficiency (kcat/Km) for A286L (0.0033 ± 0.0022 × 10−3 μm−1 s−1) was significantly reduced as compared with WT (0.0672 ± 0.019 × 10−3 μm−1 s−1). The 20-fold decrease in catalytic efficiency was due to a significant increase in the Km (Km) for A286L (285.1 ± 150.5 μm) as compared with WT (17.3 ± 4.6 μm), whereas the turnover rate (kcat) was consistent between the two systems (Table 1B). These data demonstrate that Ala286 is critical for ATP hydrolysis. Additionally, the kcat/Km for R387M (0.0314 ± 0.015 × 10−3 μm−1 s−1) was reduced by a factor of 2 as compared with WT. Similar to A286L, the slight decrease in the catalytic efficiency of R387M was due to an increase in Km (49.5 ± 21.2 μm), whereas the kcat was not significantly different from WT. Interestingly, the increase in ATPase Km coupled with the decrease in RNA binding Kd for R387M as compared with WT suggests that RNA binding promotes ATP binding, consistent with a previous report that ATP hydrolysis is stimulated by RNA (26). When WT NS3 helicase is compared with T407A and S411A, we observe similar turnover rates yet increased Michaelis–Menten constants. These results suggest that T407A and S411A hydrolyze ATP at a similar rate to WT, but more substrate is needed for the same catalytic efficiency. Overall, however, ATP hydrolysis was not increased or decreased by the T407A or S411A mutations.
Because both RNA binding and ATP hydrolysis were minimally affected by T407A and S411A, we then examined how these mutations affected overall helicase-unwinding activity. We utilized a molecular beacon–based helicase activity assay to identify the effects of translocation and unwinding of dsRNA for WT, A286L, R387M, T407A, and S411A (51, 53). Progress curves were obtained for each reaction containing NS3h variants (50 nm), dsRNA (5 nm), and increasing concentrations of ATP from the absence of ATP to 1 mm ATP (Fig. 4C). Each reaction was preincubated before the addition of ATP. For initial comparison, progress curves of WT, A286L, R387M, T407A, and S411A NS3h at 400 μm ATP were normalized to the maximum relative fluorescence unit of WT, resulting in a relative unwinding activity compared with WT (Fig. 4D). The A286L (ATPase-defective) and R387M (RNA binding–defective) mutations ablated helicase-unwinding activity, as expected. The progress curves for T407A and S411A indicate that the mutations deviate from WT activity. To quantify the enzyme kinetics for each NS3h variant, we utilized a nonlinear regression fit on the progress curves that were not normalized. From the nonlinear regression, we determined an initial rate of reaction (kobs) for each NS3h variant at all ATP concentrations. A plot consisting of kobs versus ATP concentration was fit to the Michaelis–Menten equation to determine the kcat, Km, and kcat/Km (Table 1C). WT NS3 helicase unwound dsRNA with a kcat of 47.6 ± 4.4 × 10−3 s−1 and Km of 24.5 ± 11.3 μm (Table 1C). The dsRNA was unwound at a kcat of 80.8 ± 6.0 × 10−3 s−1 and a Km of 14.4 ± 6.2 μm for the T407A mutant, and at a kcat of 166.6 ± 15.5 × 10−3 s−1 and a Km of 58.0 ± 21.6 μm for S411A. These kinetics for T407A and S411A were significantly different compared with WT, which suggests that both mutations are catalytically more efficient than WT NS3h.
The interaction between Thr407 and Ser411 controls NS3 helicase function
T407A and S411A exhibit increased helicase activity, so we wanted to investigate if this was due to the absence or presence of a hydrogen bond between the two residues. Therefore, we constructed three additional mutations (T407C, S411C, and a double mutant T407C/S411C) in both the replicon and recombinant protein systems to test viral genome replication, RNA-binding affinity, ATPase activity, and helicase-unwinding activity. Replacing residues Thr407 and Ser411 with cysteine residues would largely retain the hydrogen-bonding potential for each residue but would remove the methyl group from Thr407. T407C/S411C can be used to create a disulfide bond (∼2 Å) under oxidized conditions, locking 407 and 411 into a closer and more rigid interaction than a hydrogen bond (2.5–3 Å). First, we investigated the effects of T407C, S411C, and T407C/S411C on viral genome replication in our replicon system. The viral genome replication of T407C was significantly reduced (13.0 ± 8.0%), whereas the double mutant indicated a reduction to genome replication at 52.7 ± 30.0% as compared with WT NS3 helicase (Fig. 5). On the other hand, S411C exhibited genome replication activity similar to that of WT. These results suggest that the hydroxyl group of Thr407 is responsible for part of the effect on genome replication because the cysteine mutation recovers replication by ∼2–3-fold compared with T407A. Additionally, these results indicate that the Thr407 methyl group may be the important component of the threonine side chain due to the absence of viral replication in both T407A and T407C, which both lack the methyl group. The S411C mutant shows a complete recovery of replication efficiency, so the effect seen in S411A is due to the removal of the hydroxyl group. These data suggest overall that the hydrogen bond between Thr407 and Ser411 plays some role in replication, but the methyl in Thr407 plays a different and potentially more important role in replication. Therefore, we investigated the individual NS3 helicase functions for these mutations in the recombinant protein system to further understand the role of the methyl and hydroxyl groups found in Thr407 and Ser411.
Figure 5.
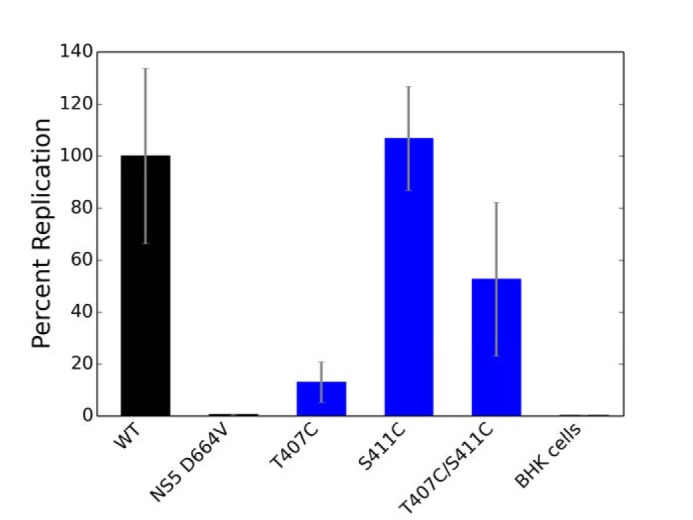
Examining the role of Thr407 and Ser411 interaction in NS3h function. Additional mutations to force covalent bonds between the Thr407 and Ser411 residues were tested in viral genome replication. Error bars, S.E.
The T407C, S411C, and T407C/S411C mutants were expressed in BL21 DE3 pLysS competent cells to promote a reducing environment. WT and T407C/S411C mutant were also expressed in T7 Shuffle competent cells to promote an oxidizing environment for disulfide bond formation. All of the purified NS3 helicase variants were then tested in the three biochemical assays: RNA-binding affinity, ATPase activity, and helicase-unwinding activity. These cysteine mutations were tested under reducing conditions (TCEP) with the exception of the oxidized WT and T407C/S411C expressed in T7 Shuffle, which were tested in both reducing (TCEP) and nonreducing conditions. In all three assays, the BL21-expressed WT compared with T7 Shuffle–expressed WT have differing Kd, kcat, and Km values, suggesting that the expression cell lines provide a different environment and therefore the T7 Shuffle–expressed NS3 helicases cannot be directly compared with the BL21-expressed helicases (Table 2). In the RNA-binding affinity assay, the BL21-expressed S411C and T407C/S411C were not significantly different in their binding affinities compared with WT, whereas T407C indicated a weaker RNA-binding affinity of 8.0 ± 2.5 μm, which was a 4.2-fold difference compared with WT (Table 2A). Additionally, the reduced T7-expressed T407C/S411C showed a 1.3-fold increase in Kd as compared with the reduced T7 Shuffle–expressed WT. Similarly, the WT and T407C/S411C expressed in T7 Shuffle cells under nonreducing conditions were also not significantly different, with a 0.6-fold difference. All of these data suggest that T407C slightly decreases RNA-binding affinity potentially due to the lack of the methyl group, which may play a role in RNA binding to the helicase.
Table 2.
Enzyme kinetics of T407C, S411C, and T407C/S411C NS3 helicase
A, Kd was determined from the RNA-binding affinity assay data. B, kcat, Km, and kcat/Km were determined for the ATPase activity assay through fitting the data to the substrate inhibition equation. Km is related to the ATP concentration. C, kcat, Km, and kcat/Km were determined for the helicase-unwinding activity assay for each NS3h variant through fitting the Michaelis–Menten equation to the Kobs versus ATP concentration.
|
A. RNA-binding affinity | ||
|---|---|---|
| NS3h variant | Kd (μm) | -Fold difference |
| WT | 1.9 ± 0.8 | |
| T407C | 8.0 ± 2.5 | 4.2 |
| S411C | 0.6 ± 0.3 | 0.3 |
| T407C/S411C | 1.4 ± 0.8 | 0.7 |
| WT reduceda | 3.4 ± 1.4 | |
| WT oxidizedb | 4.5 ± 1.5 | |
| T407C/S411C reduceda | 4.4 ± 1.1 | 1.3 |
| T407C/S411C oxidizedb | 2.6 ± 1.4 | 0.6 |
|
B. ATP hydrolysis activity | |||
|---|---|---|---|
| NS3h variant | kcat (s−1) | Km (μm) | kcat/Km (μm−1 s−1) |
| WT | 1.2 ± 0.1 × 10−3 | 17.3 ± 4.6 | 0.0671 ± 0.0189 × 10−3 |
| T407C | 1.4 ± 0.2 × 10−3 | 24.4 ± 7.4 | 0.0565 ± 0.0184 × 10−3 |
| S411C | 1.2 ± 0.1 × 10−3 | 23.4 ± 4.8 | 0.0498 ± 0.0109 × 10−3 |
| T407C/S411C | 0.9 ± 0.1 × 10−3 | 19.3 ± 6.9 | 0.0443 ± 0.0168 × 10−3 |
| WT reduceda | 1.8 ± 0.2 × 10−3 | 43.0 ± 8.5 | 0.0428 ± 0.0094 × 10−3 |
| WT oxidizedb | 2.9 ± 0.8 × 10−3 | 116.5 ± 49.7 | 0.0249 ± 0.0128 × 10−3 |
| T407C/S411C reduceda | 1.8 ± 0.1 × 10−3 | 35.1 ± 6.5 | 0.0512 ± 0.0104 × 10−3 |
| T407C/S411C oxidizedb | 1.7 ± 0.2 × 10−3 | 49.9 ± 12.1 | 0.0341 ± 0.0093 × 10−3 |
|
C. Helicase-unwinding activity | |||
|---|---|---|---|
| NS3h variant | kcat (s−1) | Km (μm) | kcat/Km (μm−1 s−1) |
| WT | 47.6 ± 4.4 × 10−3 | 24.5 ± 11.3 | 1.9 ± 0.8 × 10−3 |
| T407C | 64.2 ± 5.2 × 10−3 | 4.1 ± 3.0 | 15.7 ± 10.8 × 10−3 |
| S411C | 219.4 ± 16.2 × 10−3 | 166.3 ± 33.7 | 2.8 ± 1.2 × 10−3 |
| T407C/S411C | 81.8 ± 46.7 × 10−3 | 81.7 ± 113.1 | 1.0 ± 0.9 × 10−3 |
| WT reduceda | 39.4 ± 2.7 × 10−3 | 21.9 ± 7.7 | 1.8 ± 0.6 × 10−3 |
| WT oxidizedb | 32.5 ± 1.7 × 10−3 | 10.8 ± 3.4 | 3.0 ± 0.9 × 10−3 |
| T407C/S411C reduceda | 21.1 ± 1.7 × 10−3 | 14.7 ± 6.6 | 1.4 ± 0.6 × 10−3 |
| T407C/S411C oxidizedb | 21.2 ± 2.0 × 10−3 | 4.2 ± 4.4 | 5.1 ± 5.2 × 10−3 |
a WT reduced and T407C/S411C reduced are compared.
b WT oxidized and T407C/S411C oxidized are compared.
The NS3 helicase mutants expressed in BL21 and T7 Shuffle competent cells were tested for their ability to hydrolyze ATP. T7 Shuffle–expressed NS3 helicases were tested in both oxidizing and reducing conditions. T407C, S411C, and T407C/S411C expressed in BL21 cells all showed insignificant differences in both kcat and Km as compared with WT. The kcat for oxidized WT and T407C/S411C were not significantly different, with a kcat of 2.9 ± 0.8 × 10−3 s−1 and a kcat of 1.7 ± 0.2 × 10−3 s−1, respectively, whereas the Km for WT (116.5 ± 49.7 μm) and T407C/S411C (49.9 ± 12.1 μm) under oxidized conditions were significantly different (Table 2B), suggesting that the disulfide bond present in the oxidized form of T407C/S411C increases ATP hydrolysis activity. On the other hand, WT and T407C/S411C expressed in T7 Shuffle cells under reduced conditions exhibit an insignificant difference for either kcat or Km. Overall, these data suggest that hydrolysis of ATP is similar between WT and the cysteine mutations, meaning that the cysteine residues do not influence ATP hydrolysis directly.
Finally, the various cysteine NS3 helicase mutants as well as WT expressed in both BL21 and T7 Shuffle cells were tested in the helicase-unwinding activity assay. When comparing T407C, S411C, and T407C/S411C expressed in BL21 cells with WT, we observed a higher kcat with a lower Km for each mutant except for T407C, which exhibited a higher Km value (Table 2C). These data suggest that both S411C and T407C/S411C unwind dsRNA faster than WT although they do not bind ssRNA as well. T407C also unwinds dsRNA faster than WT, but the affinity for ssRNA is stronger, suggesting that T407C is a more catalytically efficient helicase overall compared with WT. The catalytic efficiency of WT and T407C/S411C expressed in T7 cells indicates no significant difference in the ability to unwind dsRNA substrates, suggesting that the double mutant exhibits similar overall unwinding activity compared with WT.
In silico mutations of Thr407 and Ser411 validate experimental assays
We further investigated how each mutant, alanine and cysteine, affected either the binding energy of the substrates or the overall secondary structure of NS3 helicase in the ssRNA + ATP substrate state using all-atom molecular dynamics simulations. Each mutant (A286L, R387M, T407A, T407C, S411A, S411C, and T407C/S411C) was simulated in triplicate for 1 μs using the ff14SB force field within the AMBER18 software package. Once the simulations were completed, the root mean square deviation was calculated in reference to the initial starting structure to determine the equilibration time that would be excluded for all other analyses. The three analyses performed on the simulations were a nonbonding interaction energy analysis, a probability of finding a lytic water in the ATP-binding pocket, and a projected covariance magnitude analysis. These analyses were specifically designed to compare results between simulation and the three biochemical assays.
First, we utilized the nonbonding interaction energy analysis to provide insight into how the mutations affect RNA-binding affinity from simulation. The nonbonding interaction energy was calculated between the bound ssRNA and the entire protein for each NS3 helicase variant. The more negative the linear interaction energy, the more strongly bound the ssRNA substrate is in the helicase RNA-binding cleft. All of the mutations exhibit strong ssRNA binding as compared with WT with the exception of A286L, R387M, and T407C mutants (Table 3A). The A286L and R387M mutations increase in their nonbonding interaction energies as compared with WT by 5.3 and 13.8%, respectively. These data suggest that A286L and R387M bind ssRNA weakly compared with WT. Additionally, T407C exhibits a nonbonding interaction energy of −708.6 ± 36.7 kcal/mol. The more positive nonbonding interaction energy suggests that T407C binds ssRNA more weakly than WT. The lower nonbonding interaction energy correlates well with the reduced RNA binding affinity for T407C (Table 2A), validating our computational approach. Additionally, we observed an increase in side-chain mobility in the T407C mutant simulation compared with WT (Fig. S1), suggesting that the threonine at position 407 may control fluctuations within Motif V, allowing for the helicase to bind ssRNA strongly.
Table 3.
Thr407 and Ser411 mutations do not affect nonbonding interaction energies of bound ssRNA to NS3 helicase
A, the nonbonding interaction energies were calculated between all residues in the protein and all nucleotides of the single-stranded RNA substrate bound into the RNA-binding cleft of NS3 helicase. The post-analysis AMBER18 Tools package, cpptraj, was utilized to calculate the linear interaction energy (kcal/mol) for each mutation run in triplicate with an interaction cutoff of 12 Å, and short-range, electrostatic energies were calculated with a dielectric of 1. The resulting linear interaction energies were the average over the three simulations. B, the projected covariance magnitude represents the fluctuations of Gly414 and residues Ser364 and Lys366 projected onto the helical gate access site axis. Each mutant is reported here as well as the -fold difference compared with WT NS3h.
|
A. RNA-binding energy | |
|---|---|
| NS3h variant | Linear interaction energy (kcal/mol) |
| WT | −747.4 ± 3.9 |
| A286L | −707.7 ± 0.0 |
| R387M | −644.5 ± 0.0 |
| T407A | −744.8 ± 23.0 |
| T407C | −708.6 ± 36.7 |
| S411A | −743.3 ± 11.9 |
| S411C | −736.7 ± 11.7 |
| T407C/S411C reduced | −730.6 ± 27.2 |
| T407C/S411C oxidized | −744.6 ± 18.4 |
|
B. Projected covariance between Gly414 and the helical gate | ||
|---|---|---|
| NS3h variant | Response magnitude (Å2) | -Fold difference |
| WT | 0.032 ± 0.008 | 1.0 |
| A286L | 0.023 ± 0.0 | 0.7 |
| R387M | 0.060 ± 0.0 | 1.9 |
| T407A | 0.224 ± 0.088 | 7.0 |
| T407C | 0.045 ± 0.010 | 1.4 |
| S411A | 0.049 ± 0.007 | 1.5 |
| S411C | 0.057 ± 0.023 | 1.8 |
| T407C/S411C reduced | 0.019 ± 0.001 | 0.6 |
| T407C/S411C oxidized | 0.062 ± 0.021 | 1.9 |
Next, we investigated how the mutations affect the ATP-binding pocket through determining the probability of finding a lytic water in the ATP hydrolysis active site. In order to do this, we utilized three collective variables to select for lytic waters: 1) the nucleophilic attack distance between the water oxygen atom and the ATP-γ-phosphate atom, 2) the nucleophilic attack angle between the water oxygen atom and the terminal phosphoanhydride bond of ATP, and 3) the angle between the water dipole moment and the terminal phosphoanhydride bond of ATP. Once the lytic waters were determined, the simulations were analyzed to determine the probability of finding a lytic water throughout the entire equilibrated portion of the simulations. The WT simulations indicate that 63 ± 5% of the time, a lytic water was found within the ATP-binding pocket. All of the mutations are not significantly different compared with the WT, except for A286L (44.9 ± 0.11%), R387M (52.5 ± 0.12%), and T407A (34.7 ± 16.8%). These probabilities suggest that A286L, R387M, and T407A mutations have negatively affected the ATP hydrolysis active site due to the low probability of finding a lytic water in the ATP-binding pocket during the simulations. However, when we compare the probability of finding a lytic water with the turnover rate determined from the ATPase assay, we observe no significant difference between the mutations and the WT NS3 helicase (Fig. 6A). On the other hand, comparing the probability of finding the lytic water with the turnover rate determined from the helicase assay, we observe that S411A and S411C are set apart from the rest of the mutations, suggesting that these two mutations are catalytically more efficient than WT and the other mutations because they are more likely to have a lytic water in the ATPase active site (Fig. 6B).
Figure 6.

Simulations of mutants do not indicate altered probability of finding a lytic water in the ATPase active site. A, the average kcat (s−1) from the ATP hydrolysis activity assay is plotted against finding a lytic water in the ATP binding pocket. B, the average kcat (s−1) from the helicase-unwinding activity assay is plotted against the probability of finding a lytic water in the ATP-binding pocket. Error bars, S.E.
Last, we implemented a projected covariance analysis to examine how the mutants affect the dynamics of the helical gate through changes in fluctuations between the ATPase active site and the helical gate. As mentioned previously, during replication, the helical gate and β-wedge are responsible for unwinding the dsRNA intermediate into two ssRNA molecules (33). One of the two strands enters into the RNA-binding cleft through the helical gate access site. The ssRNA strand in the access site interacts with surrounding residues Ser364, Ile365, Lys366, Asp603, Pro604, and Met605 found within two α-helices (α-helix 2 in subdomain 2 (α2′) and α-helix 6 in subdomain 3 (α6″)) that flank either side of the incoming ssRNA (34). Previous studies have suggested that the dynamics of the helical gate are controlled by the presence of bound ATP (34). Therefore, we wanted to determine how the mutations within Motif V affect the connection between the ATP-binding pocket and the helical gate. We quantified this connection as the positional covariance of ATP pocket residues (sources) and helical gate residues (sinks). Specifically, we investigated the covariance of Gly414 (Motif V, source) and Ser364 and Lys366 (α2′, sink residues) along the helical gate opening/closing coordinate. This quantified as the average of the projections of the covariance tensors between source and sinks along helical gate opening/closing direction (Table 3B).
The WT simulations indicated a projected covariance of 0.032 ± 0.008 Å2, suggesting that there was a correlation between the fluctuations seen between the ATP-binding pocket and the helical gate. Both T407C (0.045 ± 0.010 Å2) and S411C (0.057 ± 0.023 Å2) were not significantly different compared with WT, although their -fold differences indicated an increase in projected covariance along the helical access. All of the rest of the mutations were significantly different compared with WT (Table 3B). Both A286L (0.023 ± 0.0 Å2) and reduced T407C/S411C (0.019 ± 0.001 Å2) showed a decrease in -fold difference (0.7 and 0.6, respectively) compared with WT, suggesting that these mutations negatively affect the dynamics of the helical gate (Table 3B). R387M, T407A, S411A, and oxidized T407C/S411C indicate an increase in -fold difference (1.9, 7.0, 1.5, and 1.9, respectively) compared with WT (Table 3B), suggesting that the fluctuations through Motif V positively affect the motion of the helical gate promoting translocation and unwinding of the dsRNA intermediate. Interestingly, when we compared the projected covariance magnitude -fold differences with the turnover rate -fold differences of the helicase-unwinding activity assay, all of the data supported each other except for A286L, R387M, and oxidized T407C/S411C. Both A286L and R387M were unable to unwind dsRNA in the helicase-unwinding assay, but in simulation, these mutants indicated a decrease and an increase in the projected covariance along the helical gate axis, respectively. The difference observed between simulation and experiment may be due to the prebound state of the simulation, whereas in experiment, the helicase still needs to bind the RNA. Additionally, the turnover rate of the oxidized double mutant indicated a decreased rate compared with WT, whereas the projected covariance magnitude indicated an increased fluctuation projected onto the helical axis compared with WT. This discrepancy might be due to the inability to control the state of the disulfide bond in experiment.
Discussion
In this study, a combination of computational, biochemical, and virological experiments were utilized to investigate the role of Motif V in NS3 helicase functions. WT simulations pointed to Motif V as a key player in the communication between the ATP-binding pocket and the RNA-binding cleft. To determine how Motif V may play a role in the communication, mutations were introduced in both replicon and recombinant protein systems. The mutations were tested in a viral genome replication assay, and results suggested that the residues within Motif V affect how the virus replicates. From the viral replication assay, residues Thr407 and Ser411 sparked our interest due to the differing replication activity with prior knowledge of their interaction through a hydrogen bond that may stabilize Motif V structure. We tested both alanine and cysteine mutations in an RNA-binding affinity assay, an ATP hydrolysis activity assay, and a helicase-unwinding activity assay as well as simulated theses mutations in all-atom molecular dynamics. Results in this paper suggest that the methyl group in the side chain of Thr407 may play a role in decreasing helicase-unwinding activity through stabilizing interactions with residues Thr408, Lys366, and Arg387. These residues all interact with the bound RNA, allowing for viral replication. We can directly compare between the helicase-unwinding assay and viral replication assay because the helicase rates we observed biochemically appear to be in line with how fast dsRNA unwinding would likely need to occur in vivo during genome replication. Overall, our data suggest that Motif V is critical for the communication between the ATP binding pocket and the RNA-binding cleft in NS3 helicase.
Initial analyses of WT simulations supported findings reported by Davidson et al. (24) that Motif V may play a critical role in the communication between the ATP-binding pocket and the RNA-binding cleft. This led us to mutating all of the Motif V residues (residues 404–416) in the replicon system. Interestingly, only two Motif V mutations have been studied previously within the Flaviviridae virus family: T411A in HCV and G414A in dengue 2 virus (30, 54). Both of these mutations were tested biochemically but were not tested for their effect on viral genome replication. Therefore, our replicon data represent the first replication-based examination of the effect of Motif V mutations on viral genome replication. We observed that the majority of the mutations ablated viral replication activity with the exception of residues Val405, Val406, Thr407, Thr408, Ser411, and Ala415. Interestingly, most of the highly conserved residues ablated viral replication, but not all did, suggesting that the highly conserved residues can have some variability with the type of amino acid at those positions. Of the residues that did not ablate activity, Thr407 and Ser411 were of most interest due to the variable nature of these residues across all flaviviruses and the hydrogen bond that may stabilize the secondary structure of Motif V during replication. Therefore, we focus on these two positions for the remainder of the discussion.
Thr407 and Ser411 were mutated to alanine residues in the recombinant NS3 helicase and were expressed, purified, and tested in RNA-binding affinity, ATPase activity, and helicase-unwinding activity assays along with two controls, A286L and R387M. These three assays provide specific insight into how the helicase binds ssRNA and the rates of 1) ATP hydrolysis and 2) dsRNA unwinding. The rates determined from the latter two assays cannot be directly compared because the time scales are different because the ATPase assay is an end-point assay, whereas the helicase-unwinding activity assay is a continuous assay. The results from the RNA-binding affinity assay indicated that T407A and S411A bind ssRNA as strongly as WT while in the absence of ATP. We noted that ATP inhibited RNA binding in the fluorescence polarization assay, and thus ATP was left out of the reactions. The results from the ATP hydrolysis activity assay indicated that T407A and S411A hydrolyze ATP as well as WT. On the other hand, T407A and S411A seem to unwind dsRNA more quickly than WT in the molecular beacon helicase assay, suggesting that Thr407 and Ser411 are involved in the linkage between the ATP-binding pocket and the RNA-binding cleft. If the individual activities of RNA binding and ATP hydrolysis are unaffected by these mutations but the overall ability for the helicase to unwind dsRNA is affected, then these two mutations must play a role in the communication between the substrate-binding pockets. The observation that Thr407 and Ser411 increased the helicase turnover rate was surprising and suggested that one or both of these residues may naturally act to weaken the effect of ATP hydrolysis on helicase activity, giving rise to the intriguing possibility that the helicase has evolved to slow down its ATPase-dependent helicase activity. Position 407 is found predominately as a threonine, and 411 is found as serine (Fig. 2A and Table S1), but in a small group of flaviviruses, position 407 can be alanine or serine and 411 can be alanine. This may indicate that the helicase function in other flaviviruses (yellow fever virus, Sepik virus, Entebbe bat virus, cell-fusing agent, Kamiti River virus, and Culex flavivirus) could be faster than most other flaviviruses.
We further investigated which aspects of the threonine and serine residues may play a critical role in the regulation of helicase-unwinding activity by introducing cysteines at positions 407 and 411 individually or as a double-mutant, T407C/S411C. T407C, S411C, and T407C/S411C were mutated in both the replicon and recombinant protein systems. We observed an increase in replication activity for the T407C mutation, a full recovery for the S411C mutation, and an increase in replication activity for the double mutant. T407C was the only mutation that weakened the ability of the helicase to bind ssRNA. The ATP hydrolysis activity is unchanged for all of the cysteine mutations, and the helicase-unwinding activity indicates that T407C and S411C increase unwinding activity, whereas the double mutant under oxidizing and reducing conditions remains as active as WT. The combination of these data suggests that the methyl group in the side chain of Thr407 may be important for slowing down helicase function. We observed a catalytically more efficient T407A in unwinding dsRNA, but a significant reduction in viral replication activity. When we mutate Thr407 to a cysteine (similar to a threonine but lacking in the methyl group), we recover some replication activity. When we mutated Ser411 to cysteine, we completely recover viral replication, suggesting that whereas the hydrogen bond between Thr407 and Ser411 is important, Thr407 has a separate function related to its methyl group. The methyl group in the Thr407 side chain normally interacts with the phenylalanine ring of Phe361 via hydrophobic interactions, potentially stabilizing the residues Thr408, Lys366, and Arg387, which all interact with the RNA phosphate backbone (Fig. S1A). By removing the methyl from Thr407 with the T407C mutation, the side chain of position 407 is able to rotate and interact not only with Phe361 but also Leu385, potentially causing RNA-interacting residues to become more variable with their interactions with bound ssRNA. As a result, these three residues that interact with the RNA may become more catalytically efficient in unwinding the dsRNA substrate. Results reported by Appleby et al. (37) examining residues equivalent to Arg387 and Thr408 (Arg393 and Thr411) suggest that both residues play a direct role in the translocation along the ssRNA through conformational changes that are observed between nucleotide-bound and nucleotide-free states of HCV NS3. When comparing our results with HCV NS3, mutations at position 407 may disrupt the interactions that Arg387 and Thr408 have with the phosphodiester backbone of ssRNA such that Motif V does not undergo the same structural rearrangements to allow for Thr408 to shift to the next nucleotide in the bound ssRNA molecule as described previously (37). In the T407C mutation, the viral replication recovers some activity, suggesting that the hydroxyl group in Thr407 is also potentially important for proper viral replication due to the ability to form a hydrogen bond-like interaction with Ser411. Overall, when the helicase becomes catalytically more efficient in the absence of the Thr407 methyl, viral genome replication decreases. It is unclear why this is the case, but one hypothesis is that reduced replication may be due to a rate mismatch between T407A, T407C, and S411A NS3 and the RNA-dependent RNA polymerase of NS5. If this is the case, it may suggest that the NS3 helicase and NS5 polymerase have co-evolved to match their catalytic rates so that RNA is fed at an optimal rate between the two enzymes.
All of the NS3 helicase mutations were simulated using the ff14SB force field within the AMBER18 software package to better define how the mutations affected NS3 helicase function. Three analyses were performed on the simulations to determine how the structure of the helicase has changed due to the presence of the mutations. The first analysis we utilized was the nonbonding interaction energy analysis. The interaction energies determined from this analysis were compared with the RNA-binding affinity (Kd) obtained from experiment. We observed that in both computation and experiment, the mutations (T407A, T407C, S411A, S411C, and T407C/S411C) did not significantly change the ability of the helicase to bind ssRNA. The only mutation that weakened the binding of ssRNA in the helicase was R387M, which was expected because this mutation was a control for RNA binding. As we moved forward with our analysis of the simulations, we investigated how the mutations affected the probability of finding a lytic water in the ATPase active site (Fig. 6). When we compared the probability of finding a lytic water in the ATP-binding pocket with the turnover rate determined from the ATPase assay, we observed that none of the mutations significantly changed the probability of finding a lytic water. When comparing the probability of a lytic water with the turnover rate of the helicase activity, we observed that S411A and S411C were set apart from the rest of the mutations, suggesting that these two mutations play a role in controlling the rate of unwinding activity through the hydrogen bond with Thr407. Interestingly, when Thr407 is mutated to a cysteine, we observe an increase in mobility at position 407 (Fig. S1). The cysteine residue lacks the methyl group and contains a thiol group instead of a hydroxyl group found in the threonine side chain. The lack of the methyl group seems to allow for the thiol group to fluctuate frequently throughout the simulation (Fig. S1, B and C), suggesting a potential hydrophobic interaction between Thr407 and nearby hydrophobic residues (such as Leu385) that may influence the position of Thr407 within Motif V. This supports our previous results for Thr407 in that the orientation of the methyl groups is critical for helicase function. Additionally, the projected covariance magnitude analysis indicated that T407A increased by 7.0 compared with WT, suggesting that the methyl and hydroxyl group of threonine are important for stabilizing the interactions of position 407 with its surrounding residues. Without either of those functional groups at position 407, Motif V may allow more structural changes to occur that positively affect the helical gate. This is evident when we take into account the projected covariance of T407C. T407C indicated a slight increase compared with WT, which is similar to what is observed in the kcat of T407C from the helicase-unwinding assay. The thiol group of the cysteine potentially interacts with Ser411, stabilizing Motif V enough to control the structural changes within the helicase to allow for optimal translocation and unwinding of dsRNA.
This work provides detailed computational, biochemical, and virological insight into how the helicase utilizes the energy produced from ATP hydrolysis to power translocation and unwinding of the viral dsRNA intermediates. We have learned that the methyl group in Thr407 is potentially critical for regulating the helicase to maintain an optimal rate of translocation and unwinding during replication. If the Thr407 is mutated to an alanine or a cysteine, the viruses we examined are not able to replicate the viral genome efficiently in vitro. This Thr407 residue within Motif V interacts with Ser411, which seems to properly orient Thr407, potentially stabilizing the structure of Motif V. When we remove that interaction between Thr407 and Ser411 with a mutation to Ser411, the helicase is able to unwind dsRNA intermediates with more catalytic efficiency, suggesting that Thr407 is responsible for regulating translocation and unwinding dsRNA intermediates. As previously mentioned, the role of Motif V in SF2 helicases is not well-understood. This work has provided computationally driven experimental insight into how Motif V plays a role in the communication between the ATP-binding pocket and the RNA-binding cleft in the viral DEAH subfamily of SF2 helicases (22). Additional work will be required to determine whether the results we observe here for flaviviruses hold in other subfamilies of SF2 helicases. Ultimately, the data from this paper suggest that flaviviruses may utilize suboptimal NS3 helicase activity for optimal genome replication and that Motif V may play an inhibitory role for helicase activity during viral infection in vitro and in vivo. The role of Motif V in controlling helicase function will need to be further defined, which may eventually lead to the development of novel vaccine candidates or antiviral drug targets.
Experimental procedures
Plasmids and mutagenesis
The DNA expression plasmid used in the protein purification protocol for dengue 4 NS3 (accession number AQV12689) was adapted from Luo et al. (6). The QuikChange mutagenesis (Agilent Technologies) method was used to mutate specific residues (A286L, R387M, T407A, T407C, S411A, S411C, and T407C/S411C) within the WT dengue NS3h plasmid optimized for expression in Escherichia coli. Plasmids for each mutation were submitted to GENEWIZ for sequencing to verify each sequence.
Protein expression and purification
WT dengue NS3h gene with an N-terminal thioredoxin, His6 tag, and thrombin cleavage site was synthesized and cloned into a T7 expression plasmid. The WT NS3h construct was transformed into BL21 DE3 pLysS or T7 Shuffle E. coli competent cells. 5 ml of Luria broth cultures containing ampicillin (50 μg/ml) and chloramphenicol (34 μg/ml) were grown overnight at 37 °C, and the next day, the entire 5-ml culture was added to a 750-ml Luria broth culture containing 50 μg/ml ampicillin. 750-ml cultures were incubated until an A600 of 0.6 was reached, the cultures were induced with a final concentration of 400 μm isopropyl 1-thio-β-d-galactopyranoside for 18 h, and bacteria were collected by centrifugation. Bacteria were resuspended with 20 ml of low-imidazole buffer (LIB) (50 mm Tris-base, pH 8.00, 400 mm NaCl, 10 mm imidazole, 5% glycerol, and 12 mm CaCl2) and stored at −80 °C until use. Pre- and postinduction samples were collected and used to verify protein expression by SDS-PAGE.
For the protein purification, LIB (20 ml) was added to the frozen bacterial pellets. Once thawed, bacterial cells were disrupted three times on a 110S Microfluidizer (Microfluidics), the lysate was clarified by centrifugation using a JA-25.50 rotor for 20 min at 17,000 rpm at 4 °C, and supernatant was filtered using a 0.45-μm syringe filter. Protein purification was achieved using a multistep approach. The thioredoxin-dengue NS3h complex was first purified from the filtered lysate with nickel affinity chromatography using a HisTrap HP (GE Healthcare) column on an AKTA Pure FPLC system (GE Healthcare). The protein complex was eluted off the nickel column with a linear gradient of high-imidazole buffer (HIB) (50 mm Tris-base, pH 8.00, 400 mm NaCl, 10 mm imidazole, and 5% glycerol). Fractions containing the NS3h complex were collected and dialyzed against 350 ml of LIB containing CaCl2 for 2 h using a Slide-A-Lyzer dialysis cassette (3–12 ml (Life Technologies, Inc.)). 75 μl of soluble thrombin (GE Life Sciences) was added to the dialyzed protein for an overnight incubation at 4 °C. A second nickel column was subsequently used to remove thioredoxin from NS3h, which was collected in the flow-through fraction. The flow-through fraction was concentrated using a Vivaspin Turbo 15 ultrafiltration spin column (Satorius) and then applied to a HiLoad 16/600 Superdex 200 pg (GE Life Sciences) gel filtration column to further purify and buffer-exchange the isolated NS3h in gel filtration buffer (50 mm Hepes at pH 8.00, 400 mm NaCl, and 20% glycerol). Purified NS3h was concentrated in Vivaspin spin columns before storage at −80 °C in single-use aliquots. All buffers were made with RNase-free H2O. The concentrations of NS3h variants were confirmed on by SDS-PAGE (Fig. S2A). The recombinant WT NS3h was also confirmed by MS (data not shown).
RNA helicase–unwinding activity assay
For the ATP-dependent helicase assay (51, 53), all reactions were performed in 96-well black microplates (Thermo Scientific) in a final volume of 120 μl, and they contained 50 nm NS3h, 0.05 mm TCEP, 0.01% Tween 20, 5 μg/μl BSA, 1.25 mm MgCl2, 25 mm MOPS (pH 6.5), 5 nm RNA complex, and varying concentrations of ATP (Jena Bioscience) ranging from 1 μm to 1 mm. The RNA complex consists of an RNA-tagged Alexa 488 oligonucleotide (5′-AGUGCGCU-GUAUCGUCAAGG-CACU-3′-AlexF488N) and a DNA-tagged Iowa Black fluorescent quencher (5′-IABkFQ-CCTACGCCACCAGCTCCGTAGG-3′) bound to an RNA template (5′-GGAGCUGGUGGCGUAGGCAAGAGUGCCUUGACGAUACAGCUUUUUUUUUUUUUUUUUUUU-3′). The RNA complex was annealed through a slow cooling process by boiling H2O to 95 °C and allowing it to cool to room temperature over 1 h. The control with no MgCl2 contained 20 mm EDTA. Reactions were scanned individually at 37 °C using a Victor X5 multilabel plate reader (PerkinElmer Life Sciences) instrument for 300 s. Rates were determined from where the progress curves were exponential and linear with time. Data were then fit to the Michaelis–Menten equation (Equation 1) using Python2.7,
| (Eq. 1) |
where Vmax = kcat[E]t, v is the velocity, kcat is the apparent first-order rate constant in s−1, [E]t is the concentration of the enzyme, [S] is the concentration of the ATP substrate, and Km is the concentration of the substrate at one-half kcat. The Km, turnover rate (kcat), and specificity constant (kcat/Km) were determined from fitting to Equation 1.
RNA ATPase activity assay
A colorimetric ATPase activity assay was derived from the RNA helicase assay (51). The contents of the reactions remained the same. Using a 96-well clear plate, reactions were injected with BIOMOL Green (Enzo) and incubated at 37 °C for 15 s before measuring the absorbance at 650 nm on a Victor X5 multilabel plate reader. To obtain an entire curve for an individual ATP concentration, reactions were measured every 90 s for 12.5 min. Rates were determined from progress curves. Data were then fit to the substrate inhibition equation (Equation 2) using the R software environment (55),
| (Eq. 2) |
where Ki is the inhibition constant. From the fit, the kcat, Km, kcat/Km, and Ki were determined for the ATPase assay. Ki data are reported in Table S2.
RNA-binding affinity assay
A fluorescence polarization assay was also adapted from the RNA helicase assay to determine RNA-binding Kd as described previously (52). The fluorescence polarization assay reactions contained increasing concentrations of NS3h, 0.05 mm TCEP, 0.01% Tween 20, 5 μg/μl BSA, 1.25 mm MgCl2, 25 mm MOPS (pH 6.5), and 5 nm ppAGUAA-tagged Alexa 488 RNA oligonucleotide. All reactions were incubated at 37 °C for 10 min in a 384-well black plate within the plate reader before measuring the fluorescence polarization of the Alexa 488 fluorophore. Data were analyzed using nonlinear regression analysis in Prism8 using a maximum value of 400.0 based on protein binding reported previously (52).
Cell culture and virus replicon plasmids
Baby hamster kidney (BHK) cells were maintained in Hyclone Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 50 mm HEPES (pH 7.5), 5% penicillin/streptomycin, and 5% l-glutamine. Cells were grown in humidified incubators at 37 °C with 5% CO2. West Nile virus replicon utilized in the viral genome replication assay was designed as a plasmid-launched virus replicon expressing the firefly luciferase as described previously (56, 57). Additional site-directed mutagenesis was performed on the WT WNV replicon using the QuikChange mutagenesis (Agilent Technologies) method to produce the following mutations: NS5 D664V and NS3h A286L, R387M, F404K, V405M, V406M, T407A, T407C, T408S, D409A, I410G, S411A, S411C, E412D, M413A, G414A, A415G, N416D, and T407C/S411C double mutant. The presence of the mutations was confirmed through GENEWIZ sequencing.
Viral genome replication assay
BHK cells were plated into 24-well plates at 50,000 cells/well and allowed to adhere to the plates overnight. The next day, WT and mutant WNV replicon plasmids were transfected into the BHK cells at 250 ng/well using Lipofectamine2000 (Life Technologies). 48 h post-transfection, the cells were lysed with 1× lysis buffer from the Luciferase Assay Kit (Promega), and 20 μl of the lysates were transferred into a white 96-well plate. Using the plate reader, 100 μl of the luciferase assay reagent from the Luciferase Assay Kit was injected into each sample before measuring the luminescence of the firefly luciferase signal. Data were obtained with an n ≥ 3. The mean and S.E. were plotted using Matplotlib (58).
All-atom molecular dynamics simulations
An exemplar structure of the simulated DENV4 NS3h ssRNA + ATP substrate state (Protein Data Bank code 2JLV) was used for all simulations (24, 33). The ssRNA + ATP state was run for nine systems of NS3h (WT, A286L, R387M, T407A, T407C, S411A, S411C, and the double mutant T407C/S411C in oxidized and reduced conditions) in triplicate for 1 μs each. All of the mutations were generated from the WT NS3 helicase.
All of the NS3h systems were simulated in explicit solvent MD using the AMBER18 software package: ff14SB (protein) and RNA.OL3 (RNA) (59). Parameterization files for the ATP molecule (60) were obtained from the AMBER parameter database. Each system was solvated with a cubic box of TIP3P water with an average dimension of 85 Å. Sodium and chloride ions were added at a concentration of 100 mm to neutralize the system. The simulations were performed with periodic boundary conditions in an isothermal-isobaric (NPT) ensemble with a stochastic barostat of 1 bar and a Langevin thermostat of 310.0 K. The nonbonding interactions cutoff was 12 Å; particle mesh Ewald was used for treating long-range electrostatics; and hydrogens were constrained with the SHAKE algorithm. An integration time step of 2 fs was used. Positions and energies were written every 2 ps.
All systems were initially minimized with a 10,000-step minimization with harmonic restraints (force constant of 75 kcal mol−1 Å−2) placed on all solute atoms and a 10,000-step minimization with no restraints. Following the minimization, each system was heated from 50.0 to 310.0 K in 10-K increments with a harmonic restraint of 75 kcal mol−1 Å−2 on all solute atoms. Last, a series of five equilibration steps were implemented to slowly remove the harmonic restraint.
Data analysis of MD simulations
All analyses of MD simulations were performed using Python 2.7 and the MDAnalysis package (61, 62). These analyses include the linear interaction energy analysis, the lytic water analysis, and the projected covariance magnitude analysis. Further details of each analysis, except for the linear interaction energy, are described below. Plots were created using Matplotlib (58), and VMD (9) was utilized to visualize simulations and generate structural images. All scripts for the analyses are available on GitHub (https://github.com/mccullaghlab/T407_S411_Mutants_of_NS3h).4
Waters within the NTPase active site were defined as “lytic” using three collective variables: 1) the nucleophilic attack distance between the water oxygen atom and the ATP γ-phosphorous atom, 2) the nucleophilic attack angle between the water oxygen atom and the terminal phosphoanhydride bond of ATP, and 3) the dipole moment angle between the water molecule's dipole moment vector and the terminal phosphoanhydride bond of ATP. The first two metrics describe the geometric positioning of waters within the hydrolysis active site. Waters that have a nucleophilic attack distance less than 5 Å and a nucleophilic attack angle greater than 160° have the potential to be the lytic water due to their positioning relative to the terminal phosphate group. The third metric describes the chemically relevant orientation of a water in regard to its nucleophilic attack on this phosphate group. An ideally positioned water with a dipole moment angle of greater than 90° is defined as “lytic.” A water that meets all three of these collective variable conditions is positioned in a small volume of the NTPase active site, is hydrogen-bonding with the active site's proton acceptor (Glu285) (data not shown), and the water's dipole moment vector is approximately facing the terminal phosphoanhydride bond vector, as proposed in the SN2 mechanism for the hydrolysis reaction.
The projected covariance magnitude represents the fluctuation between Gly414 (source residue) and Ser364 and Lys366 (sink residues) projected onto the vector spanning the helical gate between subdomain 2 and subdomain 3 of NS3h. Gly414 was chosen as the source residue due to its coordination with the lytic water within the ATP binding pocket (24), and Ser364 and Lys366 were chosen as the sink residues due to their location in the helical gate and large magnitude covariance with Gly414. The covariance tensors between Gly414–Ser364 and Gly414–Lys366 were then dotted into the helical gate open/closing vector to obtain how the fluctuation perturbs the opening/closing of the helical gate.
Author contributions
K. E. D., M. M., and B. J. G. conceptualization; K. E. D. data curation; K. E. D. formal analysis; K. E. D. and B. J. G. investigation; K. E. D. methodology; K. E. D. writing-original draft; R. B. D. software; R. B. D., M. M., and B. J. G. writing-review and editing; M. M. and B. J. G. supervision; M. M. and B. J. G. funding acquisition; M. M. and B. J. G. project administration.
Supplementary Material
Acknowledgments
We acknowledge the support of NVIDIA Corporation with the donation of one of the Titan X Maxwell GPUs used for this research. We also acknowledge the help of Dr. Christopher Berndsen in analyzing the kinetic data, as well as the helpful discussions with Dr. Olve Peersen for analyzing the kinetic data, and other helpful discussions with members of the Geiss and McCullagh laboratories.
This work was supported by National Institutes of Health Grant R01 AI132668 (to B. J. G.) and XSEDE resources under the allocation CHE160008 as well as Colorado State University funding (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- WNV
- West Nile virus
- ssRNA
- single-stranded RNA
- NS3
- nonstructural protein 3
- NS3h
- NS3 helicase
- SF2
- superfamily 2
- NTPase
- nucleoside triphosphatase
- RTPase
- RNA 5′-triphosphatase
- TCEP
- tris(2-carboxyethyl)phosphine
- LIB
- low-imidazole buffer
- HIB
- high-imidazole buffer
- BHK
- baby hamster kidney.
References
- 1. Bhatt S., Gething P. W., Brady O. J., Messina J. P., Farlow A. W., Moyes C. L., Drake J. M., Brownstein J. S., Hoen A. G., Sankoh O., Myers M. F., George D. B., Jaenisch T., Wint G. R. W., Simmons C. P., et al. (2013) The global distribution and burden of dengue. Nature 496, 504–507 10.1038/nature12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Messina J. P., Brady O. J., Scott T. W., Zou C., Pigott D. M., Duda K. A., Bhatt S., Katzelnick L., Howes R. E., Battle K. E., Simmons C. P., and Hay S. I. (2014) Global spread of dengue virus types: mapping the 70 year history. Trends Microbiol. 22, 138–146 10.1016/j.tim.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Henchal E. A., and Putnak J. R. (1990) The dengue viruses. Clin. Microbiol. Rev. 3, 376–396 10.1128/CMR.3.4.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization. (2017) Vector-borne diseases Key facts, Geneva, Switzerland [Google Scholar]
- 5. World Health Organization (2018) Dengue and Severe Dengue Key Facts, World Health Organization, Geneva [Google Scholar]
- 6. Luo D., Xu T., Hunke C., Grüber G., Vasudevan S. G., and Lescar J. (2008) Crystal structure of the NS3 protease-helicase from dengue virus. J. Virol. 82, 173–183 10.1128/JVI.01788-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. World Health Organization (2017) West Nile Virus Key Facts, World Health Organization, Geneva [Google Scholar]
- 8. Noueiry A. O., Olivo P. D., Slomczynska U., Zhou Y., Buscher B., Geiss B., Engle M., Roth R. M., Chung K. M., Samuel M., and Diamond M. S. (2007) Identification of novel small-molecule inhibitors of West Nile virus infection. J. Virol. 81, 11992–12004 10.1128/JVI.01358-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 10. Low J. G. H., Ooi E. E., and Vasudevan S. G. (2017) Current status of dengue therapeutics research and development. J. Infect. Dis. 215, S96–S102 10.1093/infdis/jiw423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Noble C. G., Chen Y. L., Dong H., Gu F., Lim S. P., Schul W., Wang Q. Y., and Shi P. Y. (2010) Strategies for development of dengue virus inhibitors. Antiviral Res. 85, 450–462 10.1016/j.antiviral.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 12. Barrett A. D. T. (2017) Yellow fever live attenuated vaccine: a very successful live attenuated vaccine but still we have problems controlling the disease. Vaccine 35, 5951–5955 10.1016/j.vaccine.2017.03.032 [DOI] [PubMed] [Google Scholar]
- 13. Lindenbach B. D., Heinz-Jurgen T., and Rice C. M. (2007) Flaviviridae: the viruses and their replication. in Fields Virology, 5th Ed (Knipe D. M., and Howley P. M., eds) pp. 1101–1152, Lippincott-Raven Publishers, Philadelphia [Google Scholar]
- 14. Chambers T. J., Hahn C. S., Galler R., and Rice C. M. (1990) Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 44, 649–688 10.1146/annurev.mi.44.100190.003245 [DOI] [PubMed] [Google Scholar]
- 15. Brand C., Bisaillon M., and Geiss B. J. (2017) Organization of the flavivirus RNA replicase complex. Wiley Interdiscip. Rev. RNA 8, e1437 10.1002/wrna.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saeedi B. J., and Geiss B. J. (2013) Regulation of flavivirus RNA synthesis and capping. Wiley Interdiscip. Rev. RNA 4, 723–735 10.1002/wrna.1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heck A. M., and Wilusz J. (2018) The interplay between the RNA decay and translation machinery in eukaryotes. Cold Spring Harb. Perspect. Biol. 10, a032839 10.1101/cshperspect.a032839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cross S. T., Michalski D., Miller M. R., and Wilusz J. (2019) RNA regulatory processes in RNA virus biology. Wiley Interdiscip. Rev. RNA 10, e1536 10.1002/wrna.1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Byrd A. K., and Raney K. D. (2012) Superfamily 2 helicases. Front. Biosci. (Landmark Ed.) 17, 2070–2088 10.2741/4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singleton M. R., Dillingham M. S., and Wigley D. B. (2007) Structure and Mechanism of Helicases and Nucleic Acid Translocases. Annu. Rev. Biochem. 76, 23–50 10.1146/annurev.biochem.76.052305.115300 [DOI] [PubMed] [Google Scholar]
- 21. Gorbalenya A. E., and Koonin E. V. (1993) Helicases: amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 3, 419–429 10.1016/S0959-440X(05)80116-2 [DOI] [Google Scholar]
- 22. Fairman-Williams M. E., Guenther U. P., and Jankowsky E. (2010) SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 20, 313–324 10.1016/j.sbi.2010.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Story R. M., and Steitz T. A. (1992) Structure of the recA protein-ADP complex. Nature. 355, 374–376 10.1038/355374a0 [DOI] [PubMed] [Google Scholar]
- 24. Davidson R. B., Hendrix J., Geiss B. J., and McCullagh M. (2018) Allostery in the dengue virus NS3 helicase: insights into the NTPase cycle from molecular simulations. PLoS Comput. Biol. 14, e1006103 10.1371/journal.pcbi.1006103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matusan A. E., Pryor M. J., Davidson A. D., and Wright P. J. (2001) Mutagenesis of the dengue virus type 2 NS3 protein within and outside helicase motifs: effects on enzyme activity and virus replication. J. Virol. 75, 9633–9643 10.1128/JVI.75.20.9633-9643.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang C.-C., Huang Z.-S., Chiang P.-L., Chen C.-T., and Wu H.-N. (2009) Analysis of the nucleoside triphosphatase, RNA triphosphatase, and unwinding activities of the helicase domain of dengue virus NS3 protein. FEBS Lett. 583, 691–696 10.1016/j.febslet.2009.01.008 [DOI] [PubMed] [Google Scholar]
- 27. Warrener P., Tamura J. K., and Collett M. S. (1993) RNA-stimulated NTPase activity associated with yellow fever virus NS3 protein expressed in bacteria. J. Virol. 67, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wengler G., and Wengler G. (1991) The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 184, 707–715 10.1016/0042-6822(91)90440-M [DOI] [PubMed] [Google Scholar]
- 29. Wengler G., and Wengler G. (1993) The NS 3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology 197, 265–273 10.1006/viro.1993.1587 [DOI] [PubMed] [Google Scholar]
- 30. Sampath A., Xu T., Chao A., Luo D., Lescar J., and Vasudevan S. G. (2006) Structure-based mutational analysis of the NS3 helicase from dengue virus. J. Virol. 80, 6686–6690 10.1128/JVI.02215-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benarroch D., Selisko B., Locatelli G. A., Maga G., Romette J.-L., and Canard B. (2004) The RNA helicase, nucleotide 5′-triphosphatase, and RNA 5′-triphosphatase activities of Dengue virus protein NS3 are Mg2+-dependent and require a functional Walker B motif in the helicase catalytic core. Virology 328, 208–218 10.1016/j.virol.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 32. Bartelma G., and Padmanabhan R. (2002) Expression, purification, and characterization of the RNA 5′-triphosphatase activity of dengue virus type 2 nonstructural protein 3. Virology 299, 122–132 10.1006/viro.2002.1504 [DOI] [PubMed] [Google Scholar]
- 33. Luo D., Xu T., Watson R. P., Scherer-Becker D., Sampath A., Jahnke W., Yeong S. S., Wang C. H., Lim S. P., Strongin A., Vasudevan S. G., and Lescar J. (2008) Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 27, 3209–3219 10.1038/emboj.2008.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mastrangelo E., Bolognesi M., and Milani M. (2012) Flaviviral helicase: insights into the mechanism of action of a motor protein. Biochem. Biophys. Res. Commun. 417, 84–87 10.1016/j.bbrc.2011.11.060 [DOI] [PubMed] [Google Scholar]
- 35. Pérez-Villa A., Darvas M., and Bussi G. (2015) ATP dependent NS3 helicase interaction with RNA: insights from molecular simulations. Nucleic Acids Res. 43, 8725–8734 10.1093/nar/gkv872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo D., Vasudevan S. G., and Lescar J. (2015) The flavivirus NS2B-NS3 protease-helicase as a target for antiviral drug development. Antiviral Res. 118, 148–158 10.1016/j.antiviral.2015.03.014 [DOI] [PubMed] [Google Scholar]
- 37. Appleby T. C., Anderson R., Fedorova O., Pyle A. M., Wang R., Liu X., Brendza K. M., and Somoza J. R. (2011) Visualizing ATP-dependent RNA translocation by the NS3 helicase from HCV. J. Mol. Biol. 405, 1139–1153 10.1016/j.jmb.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dumont S., Cheng W., Serebrov V., Beran R. K., Tinoco I. Jr., Pyle A. M., and Bustamante C. (2006) RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature 439, 105–108 10.1038/nature04331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gu M., and Rice C. M. (2010) Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 521–528 10.1073/pnas.0913380107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Myong S., Bruno M. M., Pyle A. M., and Ha T. (2007) Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science 317, 513–516 10.1126/science.1144130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Banroques J., Cordin O., Doère M., Linder P., and Tanner N. K. (2008) A conserved phenylalanine of motif IV in superfamily 2 helicases is required for cooperative, ATP-dependent binding of RNA substrates in DEAD-box proteins. Mol. Cell. Biol. 28, 3359–3371 10.1128/MCB.01555-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Teramoto T., Boonyasuppayakorn S., Handley M., Choi K. H., and Padmanabhan R. (2014) Substitution of NS5 N-terminal domain of dengue virus type 2 RNA with type 4 domain caused impaired replication and emergence of adaptive mutants with enhanced fitness. J. Biol. Chem. 289, 22385–22400 10.1074/jbc.M114.584466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guyatt K. J., Westaway E. G., and Khromykh A. A. (2001) Expression and purification of enzymatically active recombinant RNA-dependent RNA polymerase (NS5) of the flavivirus Kunjin. J. Virol. Methods 92, 37–44 10.1016/S0166-0934(00)00270-6 [DOI] [PubMed] [Google Scholar]
- 44. Yap T. L., Xu T., Chen Y.-L., Malet H., Egloff M.-P., Canard B., Vasudevan S. G., and Lescar J. (2007) Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 81, 4753–4765 10.1128/JVI.02283-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Selisko B., Papageorgiou N., Ferron F., and Canard B. (2018) Structural and functional basis of the fidelity of nucleotide selection by flavivirus RNA-dependent RNA polymerases. Viruses 10, E59 10.3390/v10020059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tai C. L., Pan W. C., Liaw S. H., Yang U. C., Hwang L. H., and Chen D. S. (2001) Structure-based mutational analysis of the hepatitis C virus NS3 helicase. J. Virol. 75, 8289–8297 10.1128/JVI.75.17.8289-8297.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wardell A. D., Errington W., Ciaramella G., Merson J., and McGarvey M. J. (1999) Characterization and mutational analysis of the helicase and NTPase activities of hepatitis C virus full-length NS3 protein. J. Gen. Virol. 80, 701–709 10.1099/0022-1317-80-3-701 [DOI] [PubMed] [Google Scholar]
- 48. Kim D. W., Kim J., Gwack Y., Han J. H., and Choe J. (1997) Mutational analysis of the hepatitis C virus RNA helicase. J. Virol. 71, 9400–9409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lam A. M. I., Keeney D., and Frick D. N. (2003) Two novel conserved motifs in the hepatitis C virus NS3 protein critical for helicase action. J. Biol. Chem. 278, 44514–44524 10.1074/jbc.M306444200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ashour J., Laurent-Rolle M., Shi P.-Y., and García-Sastre A. (2009) NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 83, 5408–5418 10.1128/JVI.02188-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sweeney N. L., Hanson A. M., Mukherjee S., Ndjomou J., Geiss B. J., Steel J. J., Frankowski K. J., Li K., Schoenen F. J., and Frick D. N. (2015) Benzothiazole and pyrrolone flavivirus inhibitors targeting the viral helicase. ACS Infect. Dis. 1, 140–148 10.1021/id5000458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Henderson B. R., Saeedi B. J., Campagnola G., and Geiss B. J. (2011) Analysis of RNA binding by the dengue virus NS5 RNA capping enzyme. PLoS One 6, e25795 10.1371/journal.pone.0025795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sweeney N. L., Shadrick W. R., Mukherjee S., Li K., Frankowski K. J., Schoenen F. J., and Frick D. N. (2013) Primuline derivatives that mimic RNA to stimulate hepatitis C virus NS3 helicase-catalyzed ATP hydrolysis. J. Biol. Chem. 288, 19949–19957 10.1074/jbc.M113.463166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin C., and Kim J. L. (1999) Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 73, 8798–8807 10.1128/JVI.73.10.8798-8807.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. R Core Team (2014) R: A Language and Environment for Statistical Computing, R Project for Statistical Computing, Vienna, Austria [Google Scholar]
- 56. Pierson T. C., Sánchez M. D., Puffer B. A., Ahmed A. A., Geiss B. J., Valentine L. E., Altamura L. A., Diamond M. S., and Doms R. W. (2006) A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346, 53–65 10.1016/j.virol.2005.10.030 [DOI] [PubMed] [Google Scholar]
- 57. Gullberg R. C., Jordan Steel J., Moon S. L., Soltani E., and Geiss B. J. (2015) Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology 475, 219–229 10.1016/j.virol.2014.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hunter J. D. (2007) Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 10.1109/MCSE.2007.55 [DOI] [Google Scholar]
- 59. Case D. A., Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., Cheatham T. E. I., Cruzeiro V. W. D., Darden T. A., Duke R. E., Ghoreishi D., Gilson M. K., Gohlke H., Goetz A. W., Greene D., Harris R., Homeyer N., et al. (2018) AMBER 2018, University of California, San Francisco [Google Scholar]
- 60. Meagher K. L., Redman L. T., and Carlson H. A. (2003) Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 24, 1016–1025 10.1002/jcc.10262 [DOI] [PubMed] [Google Scholar]
- 61. Gowers R., Linke M., Barnoud J., Reddy T., Melo M., Seyler S., Domański J., Dotson D., Buchoux S., Kenney I., and Beckstein O. (2016) MDAnalysis: a Python package for the rapid analysis of molecular dynamics simulations. in Proceedings of the 15th Python in Science Conference, pp. 98–105 10.25080/Majora-629e541a-00e [DOI] [Google Scholar]
- 62. Michaud-Agrawal N., Denning E. J., Woolf T. B., and Beckstein O. (2011) MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 10.1002/jcc.21787 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.