

Abstract
Dent disease 1 (DD1) is caused by mutations in the CLCN5 gene encoding a voltage-gated electrogenic nCl−/H+ exchanger ClC-5. Using ion-selective microelectrodes and Xenopus oocytes, here we studied Cl−/H+ coupling properties of WT ClC-5 and four DD1-associated variants (S244L, R345W, Q629*, and T657S), along with trafficking and localization of ClC-5. WT ClC-5 had a 2Cl−/H+ exchange ratio at a Vh of +40 mV with a [Cl−]out of 104 mm, but the transport direction did not reverse with a [Cl−]out of 5 mm, indicating that ClC-5-mediated exchange of two Cl− out for one H+ in is not permissible. We hypothesized that ClC-5 and H+-ATPase are functionally coupled during H+-ATPase–mediated endosomal acidification, crucial for ClC-5 activation by depolarizing endosomes. ClC-5 transport that provides three net negative charges appeared self-inhibitory because of ClC-5's voltage-gated properties, but shunt conductance facilitated further H+-ATPase–mediated endosomal acidification. Thus, an on-and-off “burst” of ClC-5 activity was crucial for preventing Cl− exit from endosomes. The subcellular distribution of the ClC-5:S244L variant was comparable with that of WT ClC-5, but the variant had a much slower Cl− and H+ transport and displayed an altered stoichiometry of 1.6:1. The ClC-5:R345W variant exhibited slightly higher Cl−/H+ transport than ClC-5:S244L, but co-localized with early endosomes, suggesting decreased ClC-5:R345W membrane trafficking is perhaps in a fully functional form. The truncated ClC-5:Q629* variant displayed the lowest Cl−/H+ exchange and was retained in the endoplasmic reticulum and cis-Golgi, but not in early endosomes, suggesting the nonsense mutation affects ClC-5 maturation and trafficking.
Keywords: electrophysiology, stoichiometry, epithelial cell, transporter, ion-sensitive electrode, Cl−/H+ exchanger ClC-5, CLCN5, dent disease 1 (DD1), kidney disorder, Xenopus oocyte
Introduction
Dent disease 1 (DD1)2 is an X-linked kidney disorder caused by mutations in the CLCN5 gene, and is characterized by low molecular weight proteinuria (LMWP), hypercalciuria, nephrocalcinosis, nephrolithiasis, rickets, and, most importantly, progressive renal failure in the majority of affected males (1–3). As of 2019 a total of 226 pathogenic CLCN5 mutations have been reported consisting of nonsense, missense, splice site, insertion and deletion mutations (4). Yet, very little is known regarding how these mutations lead to specific disease manifestations.
CLCN5 encodes the ClC-5 protein expressed abundantly in kidney and intestinal epithelial cells (5, 6). In the kidney, immunohistochemistry has localized ClC-5 expression predominantly to epithelial cells lining the proximal tubule (PT), the thick ascending limb of Henle's loop, and to α-intercalated cells of the collecting duct (5, 7, 8). In the PT, expression of ClC-5 is highest below the brush border, where urinary LMW protein is reabsorbed by endocytotic vesicles. ClC-5 colocalizes here with the vacuolar-type (V-type) proton pump (H+-ATPase). The H+-ATPase is ubiquitously expressed in intracellular organelles such as endosomes, lysosomes, secretary granules, and the trans-Golgi network of all eukaryotic cells (9). H+-ATPase pumps H+ across membranes using energy generated by ATP hydrolysis to provide an acidic intraorganellar compartment that is critical for normal membrane trafficking, receptor-mediated endocytosis, and lysosomal degradation of macromolecules (10–12). Interestingly, Clcn5-deficient mice from two independent groups displayed altered endosomal acidification, recapitulating a typical DD1 phenotype including LMWP (13–16). Taken with the fact that ClC-5 colocalizes with H+-ATPase in early endosomes, this evidence strongly suggests that H+-ATPase and ClC-5 are functionally coupled during endosomal acidification and/or endocytosis, likely explaining LMWP in DD1.
Initially, ClC-5 was electrophysiologically characterized as a Cl− channel, a common feature of the CLC gene family (17). More recent studies demonstrated that ClC-5 functions not as a Cl− channel but rather as a voltage-gated, electrogenic nCl−/H+ exchanger (antiporter) similar to the prokaryotic homologue from Escherichia coli (ClC-ec1) and ClC-4 (18–22). The ClC-5 protein sequence is most closely related to the ClC-3 and ClC-4 in the CLC family, however, despite functional differences the overall protein architecture of the whole CLC family is conserved and is made up by two subunits each bearing an ion translocation pathway (23–25).
It is generally assumed that altered endosomal acidification due to dysfunctional ClC-5 impairs proximal tubular endocytosis and degradation of reabsorbed proteins resulting in the characteristic LMWP of DD1. This hypothesis is supported by the altered endosomal acidification observed in Clcn5 KO mice (13–16). Nevertheless, our knowledge of ClC-5 molecular function and biophysical properties remains limited. Further investigation of the exchanger transport stoichiometry, and the effects of DD1 mutations on Cl−/H+ exchanger activity are therefore needed. The bacterial homologue of ClC transporters, ClC-ec1, was the first demonstrated to have strict exchange stoichiometry of 2 Cl− to 1 H+ ratio (26–29). The initial estimates for the Cl−/H+ stoichiometry of ClC-5 were very rough, ranging from 1 to 5 because of extreme outward-rectifying currents (18, 20). Later the relative coupling efficiency of ClC-5 was determined as 2 Cl−/1 H+ by the fluorescence-based measurements using pH-sensitive dye, BCECF, to measure proton flux outside of Xenopus oocytes expressing ClC-5 (30).
Given its initial characterization as a Cl− channel (20, 31), ClC-5 was first hypothesized to transport Cl− to counter and dissipate positive charge (H+) accumulation generated by H+-ATPase, thereby facilitating efficient endosomal acidification. In this model, Cl− shunting by ClC-5 to facilitate H+-ATPase was considered essential for normal endocytosis (32). However, recent studies demonstrate that ClC-5 is a voltage-gated electrogenic Cl−/H+ exchanger (transporter) (18, 20). Valuable cellular energy (ATP) is consumed by H+-ATPase pumping H+ from the cytoplasm into the endosome thereby acidifying it. Why would ClC-5 then move H+ out of the endosome? This would seem to be a very counterproductive, or at least inefficient, process. Therefore, a comprehensive reassessment of the purported ClC-5 physiological roles in endosomal acidification and/or endocytosis and its interaction with H+-ATPase remain important yet unanswered questions (33).
Previously published data on ClC-5 and DD1 mutations have focused on its electrogenic properties (elicited currents) at positive voltages (+60 to +100 mV) (34–38). Consequently, a systematic study of the effects of ClC-5 mutations on Cl−/H+ exchanger activity and coupling properties is lacking. To fill this gap, we used ion selective microelectrodes to monitor intracellular pH (pHi) and Cl− concentration ([Cli]) to investigate Cl−/H+ coupling properties of wildtype (WT) and mutated ClC-5 in Xenopus oocytes under voltage-clamped conditions. We focused on common (S244L) and novel CLCN5 variants (R345W, Q629*) identified by the Rare Kidney Stone Consortium (RKSC). We also examined protein trafficking and cellular localization of WT and these patient-specific mutations in human renal epithelial cells by immunofluorescence microscopy. Together, these findings shed light on functional properties of ClC-5 and the molecular mechanisms leading to Dent disease pathogenicity.
Results
Cl−/H+ coupling properties of patient-specific mutations
Oocytes were injected with WT or selected mutant ClC-5 cRNAs to examine effects of mutations on H+ and Cl− transport properties while clamping to +40 mV. WT ClC-5 exhibited a robust influx of Cl− (47.0 ± 7.9 μm s−1) and efflux of H+ (56.7 ± 9.9 × 10−5 pH units s−1) in response to 104 mm [Cl−]out (ND96) in the media (Figs. 1–2 and Table 1). The exchange of Cl− for H+ was effectively inhibited by reduction of extracellular [Cl−] to 5 mm. The final [Cl−]i for WT CLC-5 oocytes increased by 12.4 mm and pHi increased by 0.13 pH units over 5 min. As previously reported (4), the T657S variant exhibited no significant differences from WT ClC-5 in Cl−/H+ exchange activity and in response to extracellular [Cl−] manipulations. In contrast, the S244L variant exhibited defective Cl− for H+ exchange activity. Specifically, the S224L variant blunted H+ transport (16.8 ± 2.7 × 10−5 pH units s− by 70% (p < 0.05) and significantly impaired Cl− influx (8.9 ± 5.0 μm s−1) by 81% (p < 0.001) compared with WT ClC-5 in 104 mm [Cl−]out buffer. S224L-expressing oocytes also exhibited no significant change in [Cl−]i nor pHi in response to experimental solution manipulation from 104 to 5 mm [Cl−]out. Similar trends were observed with the R345W variant demonstrating a decrease in Cl− for H+ exchange activity by 49 (p = 0.07) and 54% (p < 0.05) compared with WT ClC-5, respectively. With these same conditions, the nonsense mutation Q629* dramatically altered both H+ (87% decrease relative to WT; p < 0.01) and Cl− (89% decrease relative to WT; p < 0.01) transport.
Figure 1.

Intracellular chloride ([Cl−]i) measurement of Xenopus oocytes overexpressing ClC-5 while voltage-clamped at +40 mV. The representative intracellular chloride concentration ([Cl−]i) of ClC-5 WT (A) and mutations (B–E) expressing oocytes in response to extracellular chloride concentration, [Cl−]o, substitution from 104 to 5 mm Cl−. F, the rate of [Cl−]i change in response to extracellular chloride substitution from 104 to 5 mm Cl−. Data shown is mean ± S.E. *, p < 0.05 is significantly different from WT ClC-5 in 104 mm Cl−.
Figure 2.

Intracellular pH measurement of Xenopus oocytes overexpressing ClC-5 while voltage-clamped at +40 mV. The representative pHi of ClC-5 WT (A) and mutations (B–E) expressing oocytes in response to extracellular chloride concentration [Cl−]o substitution from 104 to 5 mm Cl−. F, the rate of pHi change in response to extracellular chloride substitution from 104 to 5 mm Cl−. Data shown are mean ± S.E. *, p < 0.05, is significantly different from WT ClC-5 in 104 mm Cl−. *, p < 0.05), is significantly different from WT ClC-5 in 5 mm Cl−.
Table 1.
Voltage clamped intracellular pH (pHi) and intracellular Cl− activity ([Cl−i]), measurements of ClC-5 transport in oocytes
| Condition | Units | ClC-5 WT | S244L | R345W | T657S | Q629* |
|---|---|---|---|---|---|---|
| n = 3 | n = 3 | n = 4 | n = 4 | n = 5 | ||
| Initial pHi (ND96) | 6.97 ± 0.01 | 7.28 ± 0.05 | 7.20 ± 0.04 | 6.98 ± 0.10 | 7.07 ± 0.03 | |
| Final pHi (ND96) | 7.10 ± 0.04 | 7.31 ± 0.05 | 7.26 ± 0.05 | 7.12 ± 0.01 | 7.09 ± 0.03 | |
| ΔpHi (ND96) | 0.13 ± 0.03 | 0.03 ± 0.00 | 0.06 ± 0.01 | 0.14 ± 0.02 | 0.01 ± 0.01 | |
| ΔpHi (5 Cl−-ND96) | 0.00 ± 0.02 | −0.03 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.01 | −0.04 ± 0.04 | |
| ND96 (dpHi/dt) | 10−5pH units s−1 | 56.7 ± 9.9 | 16.8 ± 2.7 | 26.3 ± 2.5 | 61.4 ± 10.3 | 7.6 ± 3.4 |
| 5 Cl−-ND96 (dpHi/dt) | 10−5pH units s−1 | 1.2 ± 9.5 | −32.7 ± 8.1 | 6.8 ± 4.1 | 5.2 ± 2.4 | −31.6 ± 28.6 |
| Im (ND96) | nA | 647 ± 56 | 459 ± 51 | 473 ± 34 | 1196 ± 114 | 395 ± 23 |
| Im (5 Cl−-ND96) | nA | 249 ± 107 | 481 ± 88 | 326 ± 55 | 977 ± 141 | 155 ± 108 |
| n = 3 | n = 5 | n = 3 | n = 3 | n = 3 | ||
| Initial [Cl−]i (ND96) | mm | 41.7 ± 0.4 | 45.6 ± 3.9 | 42.6 ± 3.8 | 43.5 ± 1.7 | 49.6 ± 2.1 |
| Final [Cl−]i (ND96) | mm | 54.1 ± 3.1 | 46.7 ± 3.6 | 46.2 ± 4.5 | 53.9 ± 2.5 | 50.7 ± 1.8 |
| Δ[Cl−]i (ND96) | mm | 12.4 ± 2.7 | 1.1 ± 0.4 | 3.5 ± 0.8 | 10.5 ± 0.8 | 1.1 ± 0.5 |
| Δ[Cl−]i (5 Cl−-ND96) | mm | 0.6 ± 0.6 | −1.3 ± 1.4 | 0.6 ± 1.0 | −1.1 ± 0.1 | −0.2 ± 1.3 |
| ND96 (d[Cl−] i/dt) | μm s−1 | 47.0 ± 7.9 | 8.9 ± 5.0 | 23.9 ± 5.3 | 42.6 ± 1.5 | 5.3 ± 2.0 |
| 5 Cl−-ND96 (d[Cl−]i/dt) | μm s−1 | 2.7 ± 2.6 | −13.6 ± 11.8 | 2.8 ± 4.9 | −6.5 ± 0.9 | 0.6 ± 12.5 |
| Im (ND96) | nA | 914 ± 117 | 385 ± 43 | 532 ± 117 | 1113 ± 162 | 303 ± 11 |
| Im (5 Cl−-ND96) | nA | 529 ± 59 | 222 ± 40 | 186 ± 84 | 755 ± 153 | −15 ± 117 |
Stoichiometry
Because ClC-5 is a voltage-gated transporter that does not have obvious reversal potential to indicate an energetic steady-state, the Nernst equation expression cannot be used to calculate the transport stoichiometry at equilibrium. Instead, we use the more general Gibbs free energy, ΔG, equation to estimate the transport stoichiometry. This ΔG equation for each ion species transport through the ClC-5 is the sum of two energies: the solute concentration gradient and the solute electrical gradient,
| (Eq. 1) |
where Cin and Cout are the respective ion concentration inside and outside the specific compartment, z is the ion valence, T is the absolute temperature (Kelvin), R is the ideal gas constant (8.314 J/mol/K), and F is the Faraday's constant (9.6485 × 104 coulombs/mol), ΔΨ is the holding membrane potential at +40 mV. Of note, when expressed in an oocyte, ClC-5 would not be actively transporting under normal circumstances because the resting membrane potential of an oocyte is around −30 to −60 mV. In other words, using the initial pHi and [Cl−]i (the measurements taken at t = 0) to calculate stoichiometry does not actually represent the state of ClC-5 transport activity. Thus to portray ClC-5 transport activity during active state (Vh = +40 mV) and to account for the biological variations, the ΔpHi and Δ[Cl−]i measurements from each oocyte were used in the equation as Cin to offset the difference of initial pHi and [Cl−]i among oocytes.
The movement of any molecule or ion up or down a concentration gradient involves a change in free energy, ΔG. When ΔG is positive, the reaction consumes energy, i.e. is not spontaneous. However, if ΔG is negative the reaction releases energy, i.e. is spontaneous. Because both ΔGCl− and ΔGH+ were negative the transporter is predicted to be exchanging spontaneously (active) at the given condition. Thus the ratio between ΔGCl− and ΔGH+ describe how efficiently the transporter is spontaneously transporting the respective ion species (i.e. represents the coupling ratio).
Therefore the apparent stoichiometry was calculated as: (ΔGCl−)/(ΔGH+), or Equation 2.
| (Eq. 2) |
The calculated apparent Cl−:H+ coupling ratio (stoichiometry) for WT CLC-5 and the nonpathogenic T657S variant were 2:1 (Fig. 3) at Vh = +40 mV with [Cl−]out = 104 mm and [H+]out = 3.16 × 10−5 mm. Under the same conditions, the Cl−/H+ coupling ratio of the S244L and R345W variants were reduced to 1.6:1 and 1.7:1, respectively, consistent with decreased current. The Q629* variant has a further decreased apparent stoichiometry of 1.4:1. With 104 mm [Cl−] in the extracellular solution, at +40 mV ClC-5 functions as Cl−/H+ exchanger. However, after lowering extracellular [Cl−] to 5 mm, the discernable ΔpHi and Δ[Cl−]i were too small to make ΔG calculations meaningful. Therefore the Cl−:H+ coupling ratios were not calculated under this condition.
Figure 3.

Stoichiometry (n) Cl−versus H+. The apparent Cl−:H+ coupling ratio was calculated using the Gibbs free energy equation to estimate the free energy for each ion species transport through the ClC-5 transporter.
Of note, this “apparent stoichiometry” is a calculated parameter to describe how efficiently the transporter is spontaneously transporting Cl− relative to transporting H+ within the same ClC-5 variants. The decimal number was the result from mathematical normalization, not the molecular count of the ion species. The ΔpHi recorded with WT ClC-5 was 0.13 pH units versus the ΔpHi for S244L, R345W, and Q629* were 0.03, 0.06, and 0.01 pH units, respectively (Table 1). In other words, the 1 in the apparent stoichiometry of WT ClC-5 is not equivalent to the 1 in the apparent stoichiometry of S244L.
Transport function of EGFP/HA double-tagged ClC-5
To determine whether defective ClC-5 transporter activity is due to failure of ClC-5 trafficking to the plasma membrane, we expressed WT and mutant CLCN5 constructs containing a N-terminal intracellular EGFP tag, as well as an extracellular HA tag for surface labeling, in human immortalized renal cortical tubular epithelial (RCTE) cells. We first sought to validate that transporter activity of these constructs was maintained. We expressed EGFP/HA double-tagged WT ClC-5 (pGEMHE expression vector) in oocytes and detected EGFP signal using an epifluorescence microscope (Fig. 4) indicating that the protein was successfully synthesized by the oocyte. When extracellular [Cl−] is 104 mm, a strong outward-rectifying current was observed that diminished in a 5 mm Cl− solution. These results were not significantly different from the untagged version of ClC-5 counterpart reported previously (4), verifying that the tags (EGFP/HA) do not interfere with innate ClC-5 transport function.
Figure 4.

EGFP/HA double-tagged ClC-5 expressed in Xenopus oocyte. A, strong green fluorescent signal was observed compared with water-injected control oocyte using an epifluorescence microscope (488/509) indicating EGFP/HA-ClC-5 was successfully synthesized. B, transport function of EGFP/HA double-tagged ClC-5 under voltage-clamp (Vh = −60 mv) experimental condition. Current-voltage relationship of EGFP/HA double-tagged ClC-5 in response to extracellular Cl− maneuver is no different from the untagged ClC-5.
Subcellular localization of ClC-5
In human RCTE cells, WT ClC-5 and the S244L variant are both localized to the plasma membrane as illustrated by a robust EGFP signal distributed in a punctate fashion along the cell membrane, as well as positive surface staining of the extracellular HA tag (Fig. 5). Neither cell membrane expression of ClC-5 (EGFP signal) nor surface HA tag labeling were detected in R345W and Q629* expressing cells. Rather, R345W and Q629* proteins appeared to co-localize primarily with the endoplasmic reticulum marker KDEL compared with low co-localization of S244L and WT ClC-5 (Fig. 6). Finally, WT CIC-5, S244L, and R345W variants, but not the Q629* variant, also co-localize with EEA1 (Fig. S1), an early endosome marker, and the signal from the Q629* variant overlapped strongly with a cis-Golgi marker, GM130 (Fig. 7).
Figure 5.
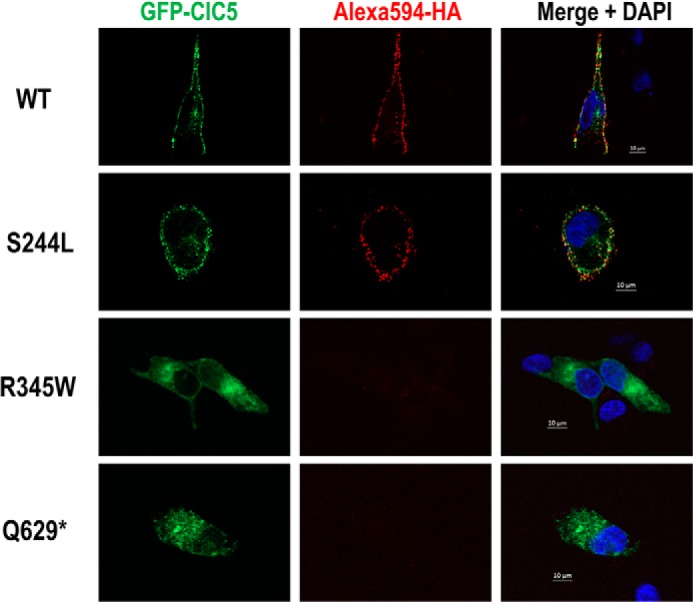
EGFP/HA double-tagged ClC-5 expression in RCTE cells. Human immortalized RCTE cells were transfected with GFP/HA double-tagged ClC-5 WT or patient-specific mutations and incubated overnight at 37 °C. EGFP signal, representing total ClC-5, and Alexa 594-HA tag (red), indicating surface ClC-5 expression, were co-localized along the cell membrane distributed in a punctate fashion in cells expressing WT and S244L. No plasma membrane distribution of ClC-5 protein was detected in R345W and Q629* variant-expressing cells.
Figure 6.
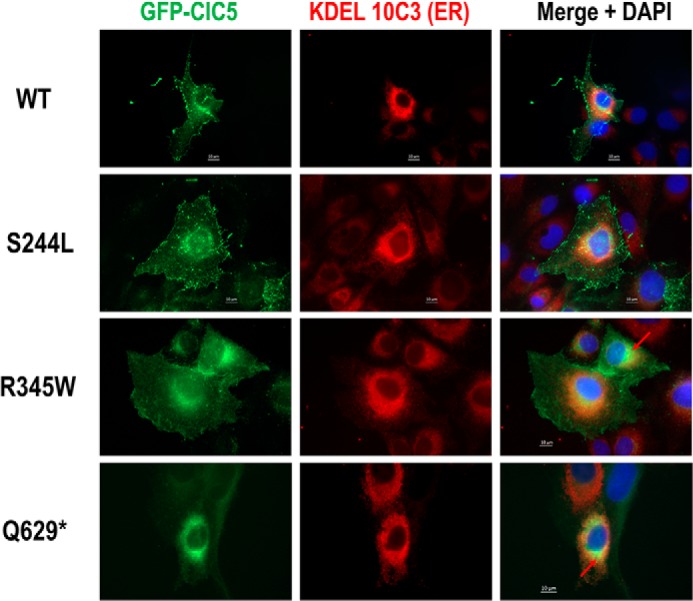
ClC-5 expression and co-localization of ER in RCTE cells. RCTE cells expressing GFP/HA double-tagged ClC-5 WT or patient-specific mutations immunostained with ER marker (KDEL 10C3). Strong ClC-5 GFP signal co-localized with ER (red arrow) were evident in R345W and Q629* mutants.
Figure 7.

ClC-5 (EGFP) double-labeling with (A) cis-Golgi (GM130) or (B) early endosomes marker (EEA1) in renal epithelial cells transfected with GFP/HA double-tagged ClC-5 WT or mutants. Red arrows indicate ClC-5 and marker co-localization. White arrows indicate no apparent co-localization. Q629* appeared to co-localize strongly with the cis-Golgi but not with early endosomes.
Discussion
The current study used ion-selective microelectrodes to directly measure Cl− and H+ transport by ClC-5. The apparent stoichiometry calculated from the Gibbs free energy of Cl− versus H+ transport reveal a 2 Cl− to 1 H+ ratio in agreement with previous findings (30). WT ClC-5 acts as 2Cl−/1H+ exchanger at +40 mV with 104 mm Cl− and pH 7.5 in the extracellular solution. Lowering extracellular [Cl−] to 5 mm, creating a chemical gradient in favor of outward Cl− movement, did not reverse the transport direction because there were no appreciable ΔpHi and Δ[Cl−]i changes observed. This suggests that no meaningful Cl−/H+ exchange occurred as extracellular [Cl−] is reduced to near 0 mm. The nonpathogenic T657S variant also has a 2 Cl− to 1 H+ coupling ratio, consistent with it being a benign single-nucleotide polymorphism prevalent in the African-American population (MIF MAF 0.23%) (4). Compared with WT ClC-5, DD1 patient-specific mutations (S244L, R345W, and Q629*) demonstrate an altered Cl−/H+ exchange stoichiometry ranging from 1.7 to 1.4.
Is the altered Cl−:H+ transport stoichiometry of ClC-5 mutants due to (a) more H+ being now needed to exchange for the same amount of Cl− or (b) due to less Cl− being needed to exchange for the same amount of H+? Our data demonstrated that both H+ transport (ΔpHi and dpHi/dt) and Cl− transport (Δ[Cl−]i and d[Cl−]i/dt) by ClC-5 mutants were much smaller and slower than those recorded for WT ClC-5 or the T657S variant assuming the buffering capacity of the oocytes is consistent (Table 1). Even though mutations have an overall defective ionic exchange, the calculated Cl−/H+ coupling ratio remained >1, suggesting that the mutated ClC-5 transporters still exchange more Cl− for H+ and thus remained electrogenic. We believe the changed amino acids in ClC-5 mutants have altered ClC-5 protein 3D structure. Accordingly, we hypothesize that these changes affect the Cl− and H+ ion binding and releasing by the ClC-5 protein, which ultimately interfere with the ion transporting pathways. A working ClC-5 protein structure model and additional experiments are needed to further test this speculation.
Subcellular localization of ClC-5
Even though the S244L variant displayed a decreased, apparent Cl−/H+ transport ratio, its cell surface expression was not significantly different from WT (previously quantified by chemiluminescence in oocytes (4) or currently by immunocytochemistry in mammalian cells (Fig. 5). Given there is ample expression of the S244L protein at the membrane, the defect of the S244L variant appears solely due to decreased Cl− and H+ transport. The R345W and Q629* variants did not appear at cell surfaces but rather in the ER and/or cis-Golgi suggesting impaired protein trafficking (Figs. 5–7). Although we did not rigorously demonstrate the exact subcellular localization of each variant, our data are consistent with our previous findings using an HEK293 cell expression model (4). It is likely these specific mutated ClC5 proteins never fully matured allowing them to leave the ER and/or Golgi apparatus because Q629* localized to the ER and/or cis-Golgi but not to early endosomes.
Interestingly, despite having lower surface expression, the R345W variant functions slightly better than the S244L variant in terms of current magnitude and Cl−/H+ exchange activity. The R345W variant also localized to early endosomes (Fig. 7B, Fig. S1), suggesting that this variant may be trafficked to the membrane, albeit to a lesser degree, and that R345W molecules that successfully traffic to the plasma membrane are fully functional. This speculation is supported by the slightly higher calculated apparent stoichiometric Cl−/H+ coupling ratio (1.7:1) than that of S244L (1.6:1). These data demonstrate that studying ClC-5 mutational effects on both transport function and cellular localization is essential to better understand divergent pathogenesis of DD1.
Biophysical mechanisms ClC-5 ion transport
The H+-ATPase and ClC-5 are functionally coupled during endosomal-to-lysosomal acidification. However, this critical endosomal acidification does not rely on ClC-5 directly, rather active H+ entry occurs via H+-ATPase with the hydrolysis of ATP. What then is the role of ClC-5 as a 2Cl− (into endosome) for 1 H+ (out to cytoplasm) exchanger? It is this fundamental question that requires elucidating the “control mechanisms” of WT and mutant ClC-5 transport. First, voltage-gating prevents ClC-5 transport at negative voltages (4, 18, 22) as neither inward nor outward rectifying currents were observed in any cellular experimental condition. Second, as illustrated in Figs. 1 and 2 and Table 1, merely creating a chemical gradient (by reducing extracellular [Cl−] to 5 mm) does not result in ClC-5 reversing its transport direction explicitly for membrane potential held at +40 mV, at which the transporter should be active state. However, the physiological role of ClC-5 during endosomal acidification is still more complicated and elusive because impaired endosomal acidification was reported in proximal tubule cells of ClC-5 deficient mice (15).
Immediately after internalization, luminal chloride concentration, [Cl−]lumen, drops from an extracellular value of 120–150 to ∼20 mm inside early endosomes (39). This drop has been attributed to Cl− expulsion by a Donnan potential produced by negative membrane protein charges (40) that face the outside solution of the plasma membrane and the lumen of forming of vesicles (as depicted in Fig. 8) (41). It was also hypothesized that the emerging endosomes were formed as flattened structures with a very high surface/volume ratio to mandate substantial Donnan potential (42).
Figure 8.

Physiological roles of ClC-5 in endosomal acidification of proximal tubule epithelial cells. A, the emerging endosomes form as flattened structures with very high surface/volume ratio, creating substantial Donnan potential that expels Cl− from nascent endosomes. B, active H+ entry via H+-ATPase acidifies the early endosome, and C, accumulates positive charges inside the early endosomes after internalization, whereas ClC-5 remains inactive. D, because ClC-5 is a voltage-gated transporter, it only activates when net positive charges inside the endosomes are sustained. E, ClC-5 activity is self-inhibitory because it hyperpolarizes the endosome by importing three net negative charges (2 Cl− in, 1 H+ out). The hyperpolarization inactivates ClC-5, but facilitates further endosomal acidification by H+-ATPase. F, this alternating activation and inactivation of ClC-5 by voltage gating is consistent with the burst hypothesis proposed by Jentsch and Pusch and colleagues (22). G, H+-ATPase and ClC-5 are functionally coupled; luminal chloride concentration increases and pH decreases in parallel as the endosomes mature.
The difference in [Cl−] from cytoplasm (30–70 mm) (39, 43–45) to nascent endosomes (20 mm) continues to provide a favorable chemical gradient for Cl− entry. However, because ClC-5 is a voltage-gated transporter, it only activates when net positive charges inside of endosomes are sustained, the energy (driving force) from the [Cl−] gradient alone is not sufficient to activate ClC-5 transport (Fig. S2). This critical characteristic was previously reported (4, 18, 20) as strong, outward currents that rectified only when membrane potential inside more positive (i.e. oocytes were depolarized) was controlled by voltage clamping (Fig. 4). Energy consuming active H+ entry driven by the H+-ATPase (making the charge inside early endosomes positive) is crucial for secondary active transport by ClC-5. As portrayed in the model (Fig. 8), ClC-5 removes one H+ from the endosomal lumen to the cytoplasm in exchange for 2 Cl− per transport-cycle providing 3 net negative charges (2Cl−/H+) to dissipate H+ buildup generated by H+-ATPase. Thus the total free energy for ClC-5 transport (ΔGClC-5 = 2×(ΔGCl−) − 1×(ΔGH+)) dissipate the net negative charge accumulation inside the endosome and thereby provides a shunt conductance that facilitates further endosomal acidification by the H+-ATPase without making the endosomal membrane potential (Vendo) too positive. This balance-counterbalance means that ClC-5 transport will resume once Vendo becomes positive due to active H+ entry via H+-ATPase (Fig. S2). ClC-5 activity switched on and off by a gating process is consistent with the “burst” hypothesis proposed by Jentsch and Pusch and colleagues (22) as transport activity of ClC-5 occurs in bursts and transport is very fast within each burst (105 ions/s).
Endosomal [Cl−] increases due to 2Cl−/H+ exchange by ClC-5 as the endosome becomes mature. The Cl− concentration in early endosomes ([Cl−]EE) was reported as 17 or 28 mm in J774 and CHO cells, respectively, quantified using a fluorescent Cl− indicator (39, 41). The Cl− concentration in late endosomes ([Cl−]LE) increased to 58 (J774 cells) and 73 mm (CHO cells). Consistent with these results, an increase in [Cl−]lumen during endosomal maturation, with a mean [Cl−]EE and [Cl−]LE of 37.0 and 60.4 mm, respectively, was also observed in Drosophila S2R+ cells (46). Accompanying the [Cl−]lumen increase, a parallel pH decrease from 6.91–7.1 to 5.2 in J774 cells and from 6.7 to 5.4 in CHO cells occurs (41, 47). Lower intracellular pH (increasing intracellular [H+]) further stimulates ClC-5 transport in an allosteric manner with an apparent pK of ∼7.2 as reported by Zifarelli and Pusch (30). This model is supported by studies that demonstrated impaired endosomal acidification and less Cl− accumulation in proximal tubular cell cultures from ClC-5–deficient mice compared with WT mice (15).
2Cl−/H+ exchangers in the CLC family contain a critical glutamate residue that plays a key role in the coupling of H+ exchange to Cl− transport (48, 49). An artificial mutation of this “gating glutamate” to alanine in ClC-5 (c.632A>C, p.Glu-211–Ala) and in other ClCs (Glu-148 in ClC-ecl and Glu-224 in ClC-4) abolishes H+ coupling and allows the conversion of ClCs to pure Cl− conductance, i.e. Cl− channel (18, 20, 26). However, the removal of gating glutamate not only abolished H+ coupling, but also changes the voltage-gating property, the key characteristic of the ClC-5 transporter. When the “gating” glutamate is removed, Cl− “leaks” out of endosome under a hyperpolarized condition due to a favorable electrochemical gradient (i.e. ΔGClC-5 < 0) through E211A-ClC-5 (20). Intriguingly, mice carrying the E211A mutation displayed the same renal phenotype as Clcn5 knock-out mice (50) including LMWP, despite normal endosomal acidification, suggesting that endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis and proper protein degradation. Another pathogenic mutation identified in DD1 patients that affects the gating glutamate of ClC-5 (c.632A>G, p.Glu-211–Gly, E211G) displays normal endosomal acidification when expressed in HEK293T cells, further indicating that impaired endosomal acidification is not the cause of defective endocytosis in the PT of DD1 patients (21). Nevertheless, it is still possible that the endosomal Cl− concentration in E221A or E211G cells is lower than WT cells because of Cl− leakage, resulting in the LMWP.
Based on the above model, the pathogenic CLCN5 mutations identified in DD1 patients displaying an altered Cl−/H+ transport stoichiometry, have overall lower transport function and apparently do not provide sufficient net negative charges within endosomes to dissipate H+ buildup generated by the H+-ATPase activity. Thus, in these decreased apparent stoichiometry cases, functional coupling of V-ATPase and ClC-5 will be disrupted. As a consequence, impaired endosomal acidification and Cl− accumulation occur, as is seen in PT cell cultures from ClC-5–deficient mice. Finally, and most importantly, how altered endosomal Cl− accumulation impairs renal endocytosis remains to be determined.
Experimental procedures
Molecular biology
Human WT ClC-5 (GenBank NM_000084.4) ORF was subcloned into the pGEMHE expression vector for Xenopus laevis oocyte expression, or into the pEGFP-C2 expression vector for renal epithelial cell expression. The HA epitope (YPYDVPDYA) was introduced into the extracellular loop of ClC-5 between transmembrane domains B and C (48). Four representative mutant CLCN5 constructs (S244L, R345W, Q629*, and T657S) were generated by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) as previously described (4). Capped cRNA were synthesized in vitro from WT and mutant ClC-5 expression vectors linearized with MluI using the T7 mMessage mMachine Kit (Ambion, Austin, TX).
Expression in X. laevis oocytes
Frogs were housed and cared for in accordance and approval of the Institutional Animal Care and Use Committee of the Mayo Clinic College of Medicine. Defolliculated stage V/VI Xenopus oocytes were injected with 10 ng of the specific cRNAs. The oocytes were then kept at 16 °C in OR3 media.
Microelectrodes
The voltage electrodes fabricated from a P-97 Flaming Brown Micropipette Puller (Sutter Instrument, Novato, CA) with a borosilcate fiber capillary had a resistance of 0.5–1 MΩ and were back filled with 3 m KCl (51). For ion-selective experiments, the electrodes were silanized with bis-(dimethylamino)-dimethylsilane, and filled with the Fluka H+ ionophore I, mixture B (pH), or Fluka Cl− ionophore I, mixture A (aCl−i). The finished electrodes were backfilled with buffer solution (pH backfill is phosphate buffer, pH 7.0; Cl− backfill is 500 mm KCl).
Two-electrode voltage-clamp electrophysiology
Two-electrode voltage-clamp experiments were performed 2 or 3 days after cRNAs injection at room temperature using an OC-725C voltage clamp (Warner instruments, Hamden, CT) and HEKA software (Wiesenstrasse, Germany). A ClC-5 expressing oocyte, visualized with a dissecting microscope, was held on a nylon mesh in a chamber, through which saline flows continuously. Currents were recorded in either 104 mm Cl− (ND96) solution (high Cl−: 96 mm NaCl, 2.0 mm KCl, 1.8 mm CaCl2, 1.0 mm MgCl2, 5.0 mm HEPES, pH 7.5) or 5 mm Cl− (5 Cl−-ND96) solution (low Cl−: iso-osmotic Cl− replacements with gluconate). Currents were recorded in response to a voltage protocol consisting of 20-mV steps from −120 to +80 mV during 75 ms/step from a holding potential of −60 mV; the resulting I-V traces were filtered at 2 kHz (8 pole Bessel filter) and sampled at 10 kHz. Data were acquired and analyzed using Pulse and PulseFit (HEKA Instruments, Germany).
Measurement of ion transport in oocytes
pH electrodes were calibrated to standard solutions (pH 6.0 and 8.0) and Cl− electrodes were calibrated to 10 and 100 mm NaCl standard solutions. All ion-selective microelectrodes had slopes of −54 to −57 mV/decade ion concentration (or activity). Ion-selective electrodes were connected to a high-impedance electrometer (WPI FD-223). The Vm signal (from the voltage-clamp apparatus) was subtracted from the ion selective electrode voltage yielding a true voltage change due solely to pHi or aCl−i. With all three electrodes impaled into the oocyte, Vm and pHi or Cl−i were allowed to stabilize. The oocyte was then clamped to a holding potential (Vh) at +40 mV. At the steady state, clamping current and pHi or Cl−i were filtered (20 Hz) and continuously monitored. Solutions were switched by computer, which records intracellular pH (pHi), intracellular Cl− activity (aCli), membrane potential (Vm), and membrane current (Im) at 0.5–1 Hz; and controls the voltage-clamp (52–54).
Cell culture and transfection
Human immortalized RCTE cell lines were cultured in Dulbecco's modified Eagle's medium (supplemented with GlutaMAX, 5% fetal bovine serum and 5% penicillin-streptomycin; GIBCO, Invitrogen) as previously described (55) and maintained in a humidified atmosphere containing 5% CO2 at 37 °C. For transient expression, RCTC cells were grown to 80–90% confluence and transfected with 1.25 μg of DNA/million cells in 100 μl of transfection buffer (135 mm KCl, 2 mm MgCl2, 20 mm HEPES, 0.5% Ficoll 400, pH 7.6) using Gene Pulser XcellTM Electroporation Systems (Bio-Rad).
Immunocytochemistry staining
Transiently transfected RCTE cells expressing EGFP/HA double-tagged ClC-5 were grown on glass coverslips for immunostaining. Primary and secondary antibody incubations were performed in a humidified chamber at room temperature unless specified otherwise. For surface HA tag staining, cells were cooled to 4 °C prior to staining to prevent endocytosis of antibodies. Monoclonal anti-HA antibody produced in mouse (HA-7, IgG1; Sigma) was diluted to 1:500 in serum-free media and applied to the cells and incubated at 4 °C for 1 h. After fixing with 4% paraformaldehyde in PBS (10 min in room temperature), cells were blocked (1% BSA, 10% normal growth serum in PBS) and stained with a secondary antibody. For organelle labeling, cells were first fixed in 4% paraformaldehyde and permeabilized using 0.5% Triton X-100, 1% BSA, 10% normal growth serum in PBS. All primary antibodies were diluted to 1:200 as a working concentration. Mouse monoclonal KDEL antibody (10C3, IgG2a; Novus) was used for endoplasmic reticulum (ER) labeling, rabbit monoclonal GM130 antibody (EP892Y, IgG; Abcam) was used as a cis-Golgi marker. For early endosome staining, rabbit polyclonal EEA1 antibody (IgG; Abcam) was used. Secondary antibodies (goat anti-mouse Alexa Fluor® 594 or goat anti-rabbit Alexa Fluor® 647) were used at a dilution of 1:1000. Controls for specificity and autofluorescence staining were performed using secondary antibodies alone. Labeled cells were imaged using an inverted epifluorescence microscope system (Zeiss, Germany) and analyzed using ImageJ software.
Author contributions
M.-H. C. and P. C. H. conceptualization; M.-H. C., P. C. H., and M. F. R. resources; M.-H. C., M. R. B., Y. L., and P. C. H. data curation; M.-H. C. and M. R. B. software; M.-H. C., M. R. B., and Y. L. formal analysis; M.-H. C., P. C. H., and J. C. L. supervision; M.-H. C., P. C. H., and M. F. R. funding acquisition; M.-H. C. validation; M.-H. C. investigation; M.-H. C. visualization; M.-H. C., M. R. B., Y. L., and V. G. G. methodology; M.-H. C. writing-original draft; M.-H. C. and J. C. L. project administration; M.-H. C., P. C. H., M. F. R., and J. C. L. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Heather L. Holmes for excellent technical support and Dr. Chris Gillen for proofreading the manuscript.
This work was supported by Rare Kidney Stone Consortium Grant U54 DK083908, a member of the National Institutes of Health Rare Diseases Clinical Research Network (RDCRN), funded by the NIDDK and the National Center For Advancing Translational Sciences (NCATS), Mayo Clinic O'Brien Urology Research Center Grant U54 DK100227, Mayo Clinic nuSURF program Grant R25 DK101405, and the Mayo Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- DD1
- Dent disease 1
- LMWP
- low molecular weight proteinuria
- PT
- proximal tubule
- RCTE
- renal cortical tubular epithelial
- EGFP
- enhanced green fluorescent protein
- HA
- Human influenza hemagglutinin
- CHO
- Chinese hamster ovary
- ER
- endoplasmic reticulum
- Vh
- holding voltage.
References
- 1. Wang X., Anglani F., Beara-Lasic L., Mehta A. J., Vaughan L. E., Herrera Hernandez L., Cogal A., Scheinman S. J., Ariceta G., Isom R., Copelovitch L., Enders F. T., Del Prete D., Vezzoli G., Paglialonga F., Harris P. C., Lieske J. C., and Investigators of the Rare Kidney Stone Consortium (2016) Glomerular pathology in Dent disease and its association with kidney function. Clin. J. Am. Soc. Nephrol. 11, 2168–2176 10.2215/CJN.03710416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Devuyst O., and Thakker R. V. (2010) Dent's disease. Orphanet. J. Rare Dis. 5, 28 10.1186/1750-1172-5-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Claverie-Martín F., Ramos-Trujillo E., and García-Nieto V. (2011) Dent's disease: clinical features and molecular basis. Pediatr. Nephrol. 26, 693–704 10.1007/s00467-010-1657-0 [DOI] [PubMed] [Google Scholar]
- 4. Tang X., Brown M. R., Cogal A. G., Gauvin D., Harris P. C., Lieske J. C., Romero M. F., and Chang M. H. (2016) Functional and transport analyses of CLCN5 genetic changes identified in Dent disease patients. Physiol. Rep. 4, e12776 10.14814/phy2.12776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Günther W., Lüchow A., Cluzeaud F., Vandewalle A., and Jentsch T. J. (1998) ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. U.S.A. 95, 8075–8080 10.1073/pnas.95.14.8075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vandewalle A., Cluzeaud F., Peng K. C., Bens M., Lüchow A., Günther W., and Jentsch T. J. (2001) Tissue distribution and subcellular localization of the ClC-5 chloride channel in rat intestinal cells. Am. J. Physiol. Cell Physiol. 280, C373–C381 10.1152/ajpcell.2001.280.2.C373 [DOI] [PubMed] [Google Scholar]
- 7. Sakamoto H., Sado Y., Naito I., Kwon T. H., Inoue S., Endo K., Kawasaki M., Uchida S., Nielsen S., Sasaki S., and Marumo F. (1999) Cellular and subcellular immunolocalization of ClC-5 channel in mouse kidney: colocalization with H+-ATPase. Am. J. Physiol. 277, F957–F965 [DOI] [PubMed] [Google Scholar]
- 8. Devuyst O., Christie P. T., Courtoy P. J., Beauwens R., and Thakker R. V. (1999) Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease. Hum. Mol. Genet. 8, 247–257 10.1093/hmg/8.2.247 [DOI] [PubMed] [Google Scholar]
- 9. Forgac M. (1999) The vacuolar H+-ATPase of clathrin-coated vesicles is reversibly inhibited by S-nitrosoglutathione. J. Biol. Chem. 274, 1301–1305 10.1074/jbc.274.3.1301 [DOI] [PubMed] [Google Scholar]
- 10. Brown D., Paunescu T. G., Breton S., and Marshansky V. (2009) Regulation of the V-ATPase in kidney epithelial cells: dual role in acid-base homeostasis and vesicle trafficking. J. Exp. Biol. 212, 1762–1772 10.1242/jeb.028803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gluck S., and Nelson R. (1992) The role of the V-ATPase in renal epithelial H+ transport. J. Exp. Biol. 172, 205–218 [DOI] [PubMed] [Google Scholar]
- 12. Sun-Wada G. H., and Wada Y. (2013) Vacuolar-type proton pump ATPases: acidification and pathological relationships. Histol. Histopathol. 28, 805–815 10.14670/HH-28.805 [DOI] [PubMed] [Google Scholar]
- 13. Piwon N., Günther W., Schwake M., Bösl M. R., and Jentsch T. J. (2000) ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 408, 369–373 10.1038/35042597 [DOI] [PubMed] [Google Scholar]
- 14. Wang S. S., Devuyst O., Courtoy P. J., Wang X. T., Wang H., Wang Y., Thakker R. V., Guggino S., and Guggino W. B. (2000) Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum. Mol. Genet. 9, 2937–2945 10.1093/hmg/9.20.2937 [DOI] [PubMed] [Google Scholar]
- 15. Hara-Chikuma M., Wang Y., Guggino S. E., Guggino W. B., and Verkman A. S. (2005) Impaired acidification in early endosomes of ClC-5 deficient proximal tubule. Biochem. Biophys. Res. Commun. 329, 941–946 10.1016/j.bbrc.2005.02.060 [DOI] [PubMed] [Google Scholar]
- 16. Günther W., Piwon N., and Jentsch T. J. (2003) The ClC-5 chloride channel knock-out mouse: an animal model for Dent's disease. Pflugers Arch. 445, 456–462 10.1007/s00424-002-0950-6 [DOI] [PubMed] [Google Scholar]
- 17. Steinmeyer K., Schwappach B., Bens M., Vandewalle A., and Jentsch T. J. (1995) Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J. Biol. Chem. 270, 31172–31177 10.1074/jbc.270.52.31172 [DOI] [PubMed] [Google Scholar]
- 18. Picollo A., and Pusch M. (2005) Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436, 420–423 10.1038/nature03720 [DOI] [PubMed] [Google Scholar]
- 19. Zifarelli G., and Pusch M. (2009) Intracellular regulation of human ClC-5 by adenine nucleotides. EMBO Rep. 10, 1111–1116 10.1038/embor.2009.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scheel O., Zdebik A. A., Lourdel S., and Jentsch T. J. (2005) Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436, 424–427 10.1038/nature03860 [DOI] [PubMed] [Google Scholar]
- 21. Bignon Y., Alekov A., Frachon N., Lahuna O., Jean-Baptiste Doh-Egueli C., Deschênes G., Vargas-Poussou R., and Lourdel S. (2018) A novel CLCN5 pathogenic mutation supports Dent disease with normal endosomal acidification. Hum. Mutat. 39, 1139–1149 10.1002/humu.23556 [DOI] [PubMed] [Google Scholar]
- 22. Zdebik A. A., Zifarelli G., Bergsdorf E. Y., Soliani P., Scheel O., Jentsch T. J., and Pusch M. (2008) Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J. Biol. Chem. 283, 4219–4227 10.1074/jbc.M708368200 [DOI] [PubMed] [Google Scholar]
- 23. Middleton R. E., Pheasant D. J., and Miller C. (1996) Homodimeric architecture of a ClC-type chloride ion channel. Nature 383, 337–340 10.1038/383337a0 [DOI] [PubMed] [Google Scholar]
- 24. Ludewig U., Pusch M., and Jentsch T. J. (1996) Two physically distinct pores in the dimeric ClC-0 chloride channel (see comments). Nature 383, 340–343 10.1038/383340a0 [DOI] [PubMed] [Google Scholar]
- 25. Weinreich F., and Jentsch T. J. (2001) Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem. 276, 2347–2353 10.1074/jbc.M005733200 [DOI] [PubMed] [Google Scholar]
- 26. Accardi A., and Miller C. (2004) Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 427, 803–807 10.1038/nature02314 [DOI] [PubMed] [Google Scholar]
- 27. Walden M., Accardi A., Wu F., Xu C., Williams C., and Miller C. (2007) Uncoupling and turnover in a Cl−/H+ exchange transporter. J. Gen. Physiol. 129, 317–329 10.1085/jgp.200709756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Accardi A., Lobet S., Williams C., Miller C., and Dutzler R. (2006) Synergism between halide binding and proton transport in a CLC-type exchanger. J. Mol. Biol. 362, 691–699 10.1016/j.jmb.2006.07.081 [DOI] [PubMed] [Google Scholar]
- 29. Nguitragool W., and Miller C. (2006) Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J. Mol. Biol. 362, 682–690 10.1016/j.jmb.2006.07.006 [DOI] [PubMed] [Google Scholar]
- 30. Zifarelli G., and Pusch M. (2009) Conversion of the 2 Cl−/1 H+ antiporter ClC-5 in a NO3− /H+ antiporter by a single point mutation. EMBO J. 28, 175–182 10.1038/emboj.2008.284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jentsch T. J., Neagoe I., and Scheel O. (2005) CLC chloride channels and transporters. Curr. Opin. Neurobiol. 15, 319–325 10.1016/j.conb.2005.05.002 [DOI] [PubMed] [Google Scholar]
- 32. Pusch M., and Zifarelli G. (2015) ClC-5: physiological role and biophysical mechanisms. Cell Calcium 58, 57–66 10.1016/j.ceca.2014.09.007 [DOI] [PubMed] [Google Scholar]
- 33. Satoh N., Suzuki M., Nakamura M., Suzuki A., Horita S., Seki G., and Moriya K. (2017) Functional coupling of V-ATPase and CLC-5. World J. Nephrol. 6, 14–20 10.5527/wjn.v6.i1.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamamoto K., Cox J. P., Friedrich T., Christie P. T., Bald M., Houtman P. N., Lapsley M. J., Patzer L., Tsimaratos M., Van'T Hoff W. G., Yamaoka K., Jentsch T. J., and Thakker R. V. (2000) Characterization of renal chloride channel (CLCN5) mutations in Dent's disease. J. Am. Soc. Nephrol. 11, 1460–1468 [DOI] [PubMed] [Google Scholar]
- 35. Igarashi T., Günther W., Sekine T., Inatomi J., Shiraga H., Takahashi S., Suzuki J., Tsuru N., Yanagihara T., Shimazu M., Jentsch T. J., and Thakker R. V. (1998) Functional characterization of renal chloride channel, CLCN5, mutations associated with Dent's Japan disease. Kidney Int. 54, 1850–1856 10.1046/j.1523-1755.1998.00203.x [DOI] [PubMed] [Google Scholar]
- 36. Ludwig M., Doroszewicz J., Seyberth H. W., Bökenkamp A., Balluch B., Nuutinen M., Utsch B., and Waldegger S. (2005) Functional evaluation of Dent's disease-causing mutations: implications for ClC-5 channel trafficking and internalization. Hum. Genet. 117, 228–237 10.1007/s00439-005-1303-2 [DOI] [PubMed] [Google Scholar]
- 37. Tanuma A., Sato H., Takeda T., Hosojima M., Obayashi H., Hama H., Iino N., Hosaka K., Kaseda R., Imai N., Ueno M., Yamazaki M., Sakimura K., Gejyo F., and Saito A. (2007) Functional characterization of a novel missense CLCN5 mutation causing alterations in proximal tubular endocytic machinery in Dent's disease. Nephron Physiol. 107, p87–p97 10.1159/000111253 [DOI] [PubMed] [Google Scholar]
- 38. Grand T., Mordasini D., L'Hoste S., Pennaforte T., Genete M., Biyeyeme M. J., Vargas-Poussou R., Blanchard A., Teulon J., and Lourdel S. (2009) Novel CLCN5 mutations in patients with Dent's disease result in altered ion currents or impaired exchanger processing. Kidney Int. 76, 999–1005 10.1038/ki.2009.305 [DOI] [PubMed] [Google Scholar]
- 39. Sonawane N. D., Thiagarajah J. R., and Verkman A. S. (2002) Chloride concentration in endosomes measured using a ratioable fluorescent Cl− indicator: evidence for chloride accumulation during acidification. J. Biol. Chem. 277, 5506–5513 10.1074/jbc.M110818200 [DOI] [PubMed] [Google Scholar]
- 40. Hryciw D. H., Jenkin K. A., Simcocks A. C., Grinfeld E., McAinch A. J., and Poronnik P. (2012) The interaction between megalin and ClC-5 is scaffolded by the Na+-H+ exchanger regulatory factor 2 (NHERF2) in proximal tubule cells. Int. J. Biochem. Cell Biol. 44, 815–823 10.1016/j.biocel.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 41. Sonawane N. D., and Verkman A. S. (2003) Determinants of [Cl-] in recycling and late endosomes and Golgi complex measured using fluorescent ligands. J. Cell Biol. 160, 1129–1138 10.1083/jcb.200211098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohshima H., and Ohki S. (1985) Donnan potential and surface potential of a charged membrane. Biophys. J. 47, 673–678 10.1016/S0006-3495(85)83963-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka S., Miyazaki H., Shiozaki A., Ichikawa D., Otsuji E., and Marunaka Y. (2017) Cytosolic Cl− affects the anticancer activity of paclitaxel in the gastric cancer cell line, MKN28 cell. Cell Physiol. Biochem. 42, 68–80 10.1159/000477116 [DOI] [PubMed] [Google Scholar]
- 44. Bregestovski P., Waseem T., and Mukhtarov M. (2009) Genetically encoded optical sensors for monitoring of intracellular chloride and chloride-selective channel activity. Front. Mol. Neurosci. 2, 15 10.3389/neuro.02.015.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salomonsson M., Gonzalez E., Kornfeld M., and Persson A. E. (1993) The cytosolic chloride concentration in macula densa and cortical thick ascending limb cells. Acta Physiol. Scand. 147, 305–313 10.1111/j.1748-1716.1993.tb09503.x [DOI] [PubMed] [Google Scholar]
- 46. Saha S., Prakash V., Halder S., Chakraborty K., and Krishnan Y. (2015) A pH-independent DNA nanodevice for quantifying chloride transport in organelles of living cells. Nat. Nanotechnol. 10, 645–651 10.1038/nnano.2015.130 [DOI] [PubMed] [Google Scholar]
- 47. Modi S., Nizak C., Surana S., Halder S., and Krishnan Y. (2013) Two DNA nanomachines map pH changes along intersecting endocytic pathways inside the same cell. Nat. Nanotechnol. 8, 459–467 10.1038/nnano.2013.92 [DOI] [PubMed] [Google Scholar]
- 48. Dutzler R., Campbell E. B., Cadene M., Chait B. T., and MacKinnon R. (2002) X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 415, 287–294 10.1038/415287a [DOI] [PubMed] [Google Scholar]
- 49. Feng L., Campbell E. B., Hsiung Y., and MacKinnon R. (2010) Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 330, 635–641 10.1126/science.1195230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Novarino G., Weinert S., Rickheit G., and Jentsch T. J. (2010) Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 328, 1398–1401 10.1126/science.1188070 [DOI] [PubMed] [Google Scholar]
- 51. Chang M. H., DiPiero J., Sönnichsen F. D., and Romero M. F. (2008) Entry to “formula tunnel” revealed by SLC4A4 human mutation and structural model. J. Biol. Chem. 283, 18402–18410 10.1074/jbc.M709819200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sciortino C. M. (2001) Characterization and localization of the sodium mediated bicarbonate transporters NBC and NDAE1. Ph.D. thesis, Case Western Reserve University [Google Scholar]
- 53. Romero M. F., Henry D., Nelson S., Harte P. J., Dillon A. K., and Sciortino C. M. (2000) Cloning and characterization of a Na+-driven anion exchanger (NDAE1): a new bicarbonate transporter. J. Biol. Chem. 275, 24552–24559 10.1074/jbc.M003476200 [DOI] [PubMed] [Google Scholar]
- 54. Sciortino C. M., and Romero M. F. (1999) Cation and voltage dependence of rat kidney, electrogenic Na+/HCO3− cotransporter, rkNBC, expressed in oocytes. Am. J. Physiol. 277, F611–F623 10.1152/ajprenal.1999.277.4.F611 [DOI] [PubMed] [Google Scholar]
- 55. Nauli S. M., Rossetti S., Kolb R. J., Alenghat F. J., Consugar M. B., Harris P. C., Ingber D. E., Loghman-Adham M., and Zhou J. (2006) Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol 17, 1015–1025 10.1681/ASN.2005080830 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.