

Abstract
Ten-eleven translocation-2 (TET2) is a member of the methylcytosine dioxygenase family of enzymes and has been implicated in cancer and aging because of its role as a global epigenetic modifier. TET2 has a large N-terminal domain and a catalytic C-terminal region. Previous reports have demonstrated that the TET2 catalytic domain remains active independently of the N-terminal domain. As such, the function of the N terminus of this large protein remains poorly characterized. Here, using yeast two-hybrid screening, co-immunoprecipitation, and several biochemical assays, we found that several isoforms of the 14-3-3 family of proteins bind TET2. 14-3-3 proteins bound TET2 when it was phosphorylated at Ser-99. In particular, we observed that AMP-activated protein kinase–mediated phosphorylation at Ser-99 promotes TET2 stability and increases global DNA 5-hydroxymethylcytosine levels. The interaction of 14-3-3 proteins with TET2 protected the Ser-99 phosphorylation, and disruption of this interaction both reduced TET2 phosphorylation and decreased TET2 stability. Furthermore, we noted that protein phosphatase 2A can interact with TET2 and dephosphorylate Ser-99. Collectively, these results provide detailed insights into the role of the TET2 N-terminal domain in TET2 regulation. Moreover, they reveal the dynamic nature of TET2 protein regulation that could have therapeutic implications for disease states resulting from reduced TET2 levels or activity.
Keywords: epigenetics, 5-hydroxymethylcytosine (5hmC), gene regulation, dioxygenase, phosphorylation, 14-3-3, AMP-activated protein kinase (AMPK), Post-translational modification, protein phosphatase 2A (PP2A), ten-eleven-translocation-2 (TET2)
Introduction
Epigenetic modifications of the genome can control gene transcription, thus regulating several molecular and cellular processes. The most well-characterized mark is 5-methylcytosine (5mC),3 which occurs at CpG dinucleotides. Ten-eleven-translocation proteins (TET1 to -3) are α-ketoglutarate–requiring dioxygenases that oxidize 5mC to 5-hydroxymethylcytosine (5hmC) (1). 5hmC can be further oxidized by TETs and eventually converted to demethylated cytosines. TET2 mutations are prevalent in malignancies, including acute myeloid leukemia (AML) (2, 3). Notably, most TET2 mutations in AML are monoallelic (2). Given the presence of a remaining WT allele in these malignancies, characterizing the mechanism that regulates TET2 protein could have therapeutic significance.
Human TET2 is 2002 amino acids in length. The catalytic domain is located in the C-terminal portion of the protein between amino acids 1129 and 1936. The C-terminal catalytic domain comprises a cysteine-rich domain (amino acids 1129–1312) followed by a double-stranded β-helix domain, which make a compact fold to execute the catalytic activity (4). The C-terminal part of TET2 has been reported to be capable of oxidizing 5mC to 5hmC independent of the N terminus. Recent studies have implicated post-translational modifications in the N terminus that can regulate protein stability. Zhang et al. (5) reported that N-terminal acetylation of TET2 promotes both protein stability and in vitro enzymatic activity. More recently, Wu et al. (6) demonstrated that AMP-activated protein kinase (AMPK) phosphorylates TET2 at Ser-99 of the N terminus to promote protein stability.
Here, we report on the functional role of the N terminus in TET2 protein regulation. We demonstrate that several members of the 14-3-3 group of proteins bind to TET2 in a phosphorylation-dependent manner. Chen et al. (7) recently reported this interaction and demonstrated that TET2 mutations that disrupt this association have reduced protein stability. In addition, they demonstrated that 14-3-3 inhibition led to decreased TET2 levels (7). They proposed that this interaction promotes TET2 stability by an undetermined mechanism. Here, we demonstrate that 14-3-3s not only bind TET2 in a phosphorylation-dependent manner, but also protect TET2 Ser-99 phosphorylation, thereby promoting TET2 stability. Correspondingly, we demonstrate that 14-3-3 interaction is required for the maintenance of AMPK-mediated phosphorylation of TET2. Moreover, we report that subunit B (α isoform) of protein phosphatase 2A (PP2A) interacts with TET2 and mediates TET2 dephosphorylation. Collectively, our data not only provide novel insight but also complement recent findings to indicate a role for the N terminus of TET2 in regulating stability. Moreover, our studies demonstrate the dynamic regulation of TET2 via phosphorylation and phosphatase events that may provide new therapeutic avenues to promote TET2 stability.
Results
14-3-3(s) bind phospho-Ser-99 of TET2
Given the lack of information on the N terminus of TET2, we proceeded with a yeast two-hybrid screen to identify partner proteins that directly interact with this portion of TET2 (amino acids 1–1000) in an unbiased manner. Using a spleen cDNA library, we identified multiple distinct clones encoding for 14-3-3 β (data not shown). 14-3-3 proteins are adaptor proteins expressed in all eukaryotes that often bind specifically phosphorylated motifs of interacting proteins. Seven isoforms of 14-3-3 have been described in humans: α/β, ϵ, θ, δ/ζ, τ, γ, and σ. 14-3-3 proteins can bind to two types of phosphorylated consensus motifs, type 1 (RSX(pS/T)XP) and type 2 (RXXX(pS/T)XP), where X is any amino acid and pS/T represents phosphorylated serine or threonine, respectively (8, 9). The TET2 protein sequence was scanned to identify a potential 14-3-3–binding motif using the 14-3-3-Pred web server (10). The site containing the top-scoring motif was Ser-99 of TET2 (data not shown). The interaction between TET2 N terminus and 14-3-3s was assessed via expression of Myc-tagged TET2 (amino acids 1–400) in HEK293T cells followed by co-immunoprecipitation (co-IP) with anti-Myc antibody. IPs were then analyzed by LC-MS. Both WT TET2 and a point mutant in which Ser-99 was mutated to alanine (S99A) were evaluated. We found that multiple 14-3-3 isoforms bound to WT TET2. In contrast, the S99A mutant had no detectable binding to any 14-3-3 isoforms (Fig. 1A). No other proteins demonstrated significant enrichment in WT TET2 pulldown (File S1). Full-length TET2/14-3-3 interaction was examined by co-IP/immunoblotting (IB) using anti-Myc or control IgG antibody for IP and immunoblotting using 14-3-3-θ and 14-3-3-ϵ antibodies (Fig. 1, B and C). 14-3-3-θ– and 14-3-3-ϵ–specific antibodies were validated utilizing siRNA (Fig. S1A). Consistent with our proteomics data, enrichment of these two isoforms of 14-3-3 were readily identified with WT TET2 with minimal to no enrichment in S99A TET2 IP or in control IgG. To eliminate the possibility of nonspecific interaction by anti-Myc antibody, control vector (CV)-transfected HEK293T cell lysate was also subjected to anti-Myc co-IP, but neither Myc-TET2 nor 14-3-3-ϵ was detected by IB (Fig. 1C, last lane). Further, endogenous TET2/14-3-3 interaction was validated by co-IP/IB using anti-TET2 antibody and control IgG for IP and IB using 14-3-3-θ antibody (Fig. 1D). TET2 antibody was validated via immunoblotting (Fig. S1B). The sequence around Ser-99 of TET2 is highly conserved across different species (Fig. 1E) and different isoforms of TET2 as well (Fig. S1C) (2). Moreover, it matches the sequence recognized by a commercially available antibody that detects a phosphoserine-containing motif recognized by 14-3-3: (R/K)XXpSXP. We therefore assessed the ability of this antibody to detect Ser-99 phosphorylation on TET2. Full-length WT and S99A TET2 (Myc tag) were immunoprecipitated by anti-Myc antibody and immunoblotted using the phosphoserine 14-3-3–binding motif antibody. Whereas phosphorylation was readily detectable in WT TET2, no phosphorylation was detected in the S99A mutant (Fig. 1F and Fig. S1D). These data indicate that TET2 is phosphorylated at Ser-99. Moreover, they demonstrate the ability to detect phosphorylation at this residue in a highly specific manner.
Figure 1.

14-3-3 isoforms interact with TET2. A, peptide counts from LC-MS analysis of immunoprecipitates from WT and S99A TET2 (N-terminal 400 amino acids). Analysis of control IgG control is also included. B and C, Myc-TET2 WT and Myc-TET2 S99A were immunoprecipitated from HEK293T cells transiently transfected with the respective plasmids (8 μg of WT TET2, 10 μg of S99A TET2) and analyzed by IB for 14-3-3-θ (B) and 14-3-3-ϵ isoforms (C). Whole-cell extracts were used as inputs. Control IgG and Myc immunoprecipitations were performed from Myc-TET2 and CV-transfected cells, respectively, as negative controls. D, HEK293T cell lysate was immunoprecipitated with anti-TET2 and control IgG antibodies and analyzed by immunoblotting for endogenous 14-3-3 interaction. Data are representative of two independent experiments. E, sequence homology of TET2 around Ser-99 among different species demonstrating a conserved phosphoserine 14-3-3 binding motif ((R/K)XXpSXP). F, WT and S99A TET2 were immunoprecipitated from HEK293T and analyzed by IB with phosphoserine 14-3-3–binding motif antibody.
AMPK phosphorylation of TET2 at Ser-99 promotes protein stability
Of note, we consistently observed that the protein levels of S99A mutant were less than transfected WT TET2. As a result, the plasmid amounts were adjusted during transient transfection of TET2 constructs for the purposes of normalization (e.g. Fig. 1, B and F). Given these data as well as a recent report (6), we assessed the biological significance of TET2 Ser-99 phosphorylation on protein stability. We observed that the steady-state levels of mutant S99A TET2 were consistently lower than transfected WT TET2, even when more S99A mutant construct was transfected (see 0-h time points in Fig. 2A). Consistent with a recent study implicating Ser-99 phosphorylation in promoting protein stability (6), WT TET2 demonstrated significantly increased stability relative to S99A following cycloheximide (CHX) treatment (Fig. 2, A and B) (see Fig. S2A for biological replicate). TET2 degradation has been shown to be protected by calpain inhibitors as well as proteasome inhibitors (5, 11). Consistent with these studies, both calpain and proteasome inhibition increased the level of the S99A mutant (Fig. 2C, right) with less pronounced effects on WT TET2 levels (Fig. 2C, left) (see Fig. 2D and Fig. S2B for statistics and biological replicate, respectively). These same lysates were also analyzed for cyclin-E2, a known target of calpains and the proteasome (12, 13). We next considered potential kinases for TET2. We observed that the TET2 sequence around Ser-99 shared features similar to known substrates of AMPK (Fig. 3A). Both forward and reverse IPs demonstrated that AMPK (FLAG-tagged) and TET2 (Myc-tagged) physically interact (Fig. 3B). We next tested the ability of AMPK to phosphorylate TET2 in vitro. Recombinant AMPK was able to phosphorylate recombinant TET2 (N-terminal 350 amino acids fused with GST) at Ser-99 in an ATP-dependent manner with phosphorylation detected using the phosphoserine 14-3-3–binding motif antibody (Fig. 3C). Moreover, an enzymatically inactive (kinase-dead) form of AMPK-α (AMPK-KD) or catalytically active AMPK-α (AMPK-WT) plasmids were co-transfected with TET2 WT and TET2 S99A plasmids in HEK293T cells, and cell lysates were analyzed by anti-Myc IP followed by immunoblotting with phospho-14-3-3 motif antibody. Consistent with our in vitro kinase assay, AMPK-WT increased TET2 phosphorylation, whereas TET2 phosphorylation was reduced with AMPK-KD (Fig. S3A). In contrast, TET2 phosphorylation was not detectable with the S99A mutant, consistent with Fig. 1F. Furthermore, treatment with the AMPK activator metformin promoted TET2 phosphorylation at Ser-99 in HEK293T cells expressing Myc-TET2 (Fig. 3D) (see Fig. 3E and Fig. S3B for statistics and biological replicate, respectively). Furthermore, AMPK (−/−) (knockout (KO)) mouse embryonic fibroblasts (MEFs) had lower endogenous TET2 than AMPK (+/+) (WT) MEFs (Fig. 3F, compare control lanes). The AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) increased TET2 protein levels in WT but not in AMPK KO MEFs (Fig. 3F). Furthermore, AICAR's effects on TET2 in WT MEFs could be blocked by the AMPK inhibitor compound C. As expected, phosphorylation of the canonical AMPK substrate acetyl-CoA-carboxylase (ACC) (IB: pACC) was promoted and blocked with AICAR and compound C, respectively (Fig. 3F, middle). Similar findings were found upon treatment of WT MEFs with metformin with or without compound C (Fig. S3C). Consistent with TET2's catalytic activity, genomic DNA isolated from HEK293T cells expressing TET2-WT exhibited a higher 5hmC level than that from cells expressing the S99A mutant (Fig. 3G). A biological replicate blot is provided in Fig. S3D. Moreover, AICAR promoted 5hmC accumulation in AMPK (+/+) MEF cells (Fig. 3H). A biological replicate blot is provided in Fig. S3E. Collectively, these data demonstrate that AMPK phosphorylates TET2 at Ser-99, which in turn increases TET2 protein with effects on genomic 5hmC levels.
Figure 2.

WT TET2 is more stable than S99A mutant TET2. A, HEK293T cells (one 10-cm plate) were transfected with Myc-TET2 WT (8 μg) or Myc-TET2 S99A (10 μg). After 36 h, cells were incubated with 50 μg/ml CHX. Whole-cell extracts were collected at the indicated time points and analyzed by IB. The figure is a representative of three biological replicates. Ratios of Myc band intensities normalized to actin band intensities are shown at the top. B, normalized Myc band intensities from biological replicates (n = 3) of A plotted over time. One-way repeated-measures ANOVA was used to compare the difference between the two proteins over time (p = 0.0153; F = 16.49). Post hoc analyses were also performed at individual time points using Student's t test: *, p = 0.0226; #, p = 0.0347; **, p = 0.0121. C, HEK293T cells were transfected with WT and S99A TET2. After 24 h, transfected cells were then treated with either vehicle (untreated), 50 μm calpeptin, or 100 nm bortezomib for 24 h followed by immunoblotting of whole-cell extracts. Ratios of Myc band intensities normalized to GAPDH band intensities are shown at the top. D, quantification of data from C (n = 3 biological replicates). Data were analyzed by one-way ANOVA followed by post hoc Tukey's honestly significant difference test. Left, ANOVA p value is 0.1817 (F = 2.2966). Right, ANOVA p value is 0.0198 (F = 8.0966). Post hoc analysis p values are demonstrated. Error bars, S.E.
Figure 3.

AMPK phosphorylation at Ser-99 promotes TET2 stability. A, sequence analysis of TET2 at Ser-99 relative to known AMPK substrates. Hyd, hydrophobic. The yellow box denotes phosphorylated residues. B, HEK293T cells (one 10-cm plate) were co-transfected with Myc-TET2 (8 μg) and FLAG-AMPK (10 μg) constructs. Protein extracts were immunoprecipitated using either anti-Myc, anti-FLAG, or control IgG and analyzed by IB. C, in vitro kinase assay utilizing N terminus of recombinant TET2 (rTET2; aa 1–350, ∼75 kDa, GST-tagged) as substrate. Phosphorylation was detected with phosphoserine 14-3-3–binding motif antibody. Results are representative of two independent experiments. D and E, HEK293T cells were transfected with Myc-TET2, followed by treatment with or without metformin for 2 h. Myc-TET2 was immunoprecipitated and analyzed by IB with phosphoserine 14-3-3–binding motif antibody. Data from three biological replicates are quantified in E. F, whole-cell lysates from AMPK WT (+/+) and AMPK KO (−/−) MEFs were collected after treatment with the indicated agent (24-h treatment) and analyzed by IB. DMSO-treated cells were set as control. Ratios of Myc band intensities normalized to actin band intensities are shown at the top. G, global 5hmC levels of genomic DNA extracted from HEK293T cells transfected with WT (8 μg) or S99A mutant TET2 (10 μg) plasmids. Methylene blue (MB) blots are included for loading control. 5hmC quantification from three biological replicates is provided below. I, global 5hmC levels of genomic DNA extracted from AMPK (+/+) MEF cells treated with DMSO (control) or AICAR for 24 h. 5hmC quantification from three biological replicates is provided below. Error bars, S.E.
14-3-3 interaction promotes TET2 phosphorylation and stability
We considered the biological significance of 14-3-3 binding to Ser-99–phosphorylated TET2. Prior studies have demonstrated that 14-3-3s can act as adaptor proteins facilitating protein-protein interactions and localization (14, 15). However, our MS data (File S1) did not demonstrate enrichment of other proteins besides 14-3-3 isoforms. We next considered whether TET2's interaction with 14-3-3 might affect cellular localization. However, we did not observe any major change in the localization between WT and S99A (data not shown). We therefore considered whether 14-3-3 interaction had a role in promoting TET2 phosphorylation. To assess this, we utilized a cDNA construct that encodes for a dimeric version of the peptide R18, referred to as difopein. R18 binds 14-3-3 proteins with high affinity and has previously been shown to competitively inhibit 14-3-3 interactions with client proteins (16, 17). HEK293T cells transfected with difopein construct had lower endogenous TET2 than CV-transfected cells (Fig. 4A). A biological replicate blot is provided in Fig. S4A. Moreover, difopein reduced WT TET2 protein without effects on the S99A mutant (Fig. 4B; see Fig. S4 (B and C) for biological replicate and statistics, respectively). As we noted that difopein reduced total TET2 levels, we increased the levels of TET2 construct introduced when co-transfected with difopein to have comparable amounts of protein for analysis of IPs (Fig. 4C). Consistent with difopein's ability to disrupt 14-3-3 interactions, TET2 IP had reduced 14-3-3 levels (IB: 14-3-3-θ). Additionally, difopein treatment significantly reduced TET Ser-99 phosphorylation levels as determined via immunoblotting with the phosphoserine 14-3-3–binding motif antibody (Fig. 4C, top two panels). Given these findings, we next determined the effect of difopein on TET2 protein stability. Difopein treatment reduced TET2 protein stability for both endogenous (Fig. 4D) and transfected Myc-tagged WT TET2 (Fig. 4E). See Fig. S4 (D and E) for biological replicates. However, difopein had virtually no effect on the stability of Myc-tagged TET2 S99A (Fig. S4F). Collectively, these data indicate that disruption of TET2/14-3-3 interaction leads to both reduced TET2 phosphorylation at Ser-99 and diminished protein stability.
Figure 4.

Disruption of 14-3-3 interaction reduces TET2 phosphorylation and stability. A, endogenous TET2 levels in HEK293T cells transfected with control vector (8 μg) or difopein construct (8 μg). Data from three biological replicates are quantified. B, analysis of Myc TET2 WT and Myc-TET2 S99A by immunoblotting from HEK293T cells transfected with different combinations of Myc-TET2 WT (8 μg), Myc-TET2 S99A (8 μg), difopein (8 μg), and CV (8 μg) constructs as displayed in the figure. The figure is a representative of three biological replicates. C, IP analysis of proteins extracted from HEK293T cells transfected with either WT Myc-TET2 and CV (8 μg of TET2 and 8 μg of CV) or with WT Myc-TET2 and difopein (12 μg of TET2 and 8 μg of difopein). S and L, short and long exposures, respectively, following immunoblotting with phosphoserine 14-3-3–binding motif antibody. D, HEK293T cells were transfected with either CV or difopein construct. After 24 h, cells were treated with CHX (50 μg/ml) and harvested at the indicated time points, followed by analysis of endogenous TET2. E, HEK293T cells (one 10-cm plate) were co-transfected with either WT Myc-TET2 (8 μg) and CV (10 μg) (left) or Myc-TET2 WT (10 μg) and difopein (10 μg) constructs (right). After 24 h, cells were treated with CHX (50 μg/ml) and harvested at the indicated time point, followed by IB using anti-Myc antibody. Error bars, S.E.
14-3-3 protects TET2 phosphorylation
Prior studies have demonstrated that phosphospecific binding of 14-3-3 can protect interacting proteins from dephosphorylation (18). We therefore considered whether 14-3-3 binding to TET2 protects Ser-99 phosphorylation. To test this possibility in vitro, we employed a phosphatase protection assay as outlined in Fig. 5A. Immunoprecipitated TET2 (bound to beads) was incubated in vitro in the absence/presence of the 14-3-3 antagonist peptide R18, which can disrupt 14-3-3s' protein interactions. Immunoprecipitates were then washed to remove unbound 14-3-3s. Next, immunoprecipitated TET2 was treated with a broad specificity phosphatase (λ-phosphatase) followed by analysis. Consistent with R18's ability to disrupt 14-3-3 interactions, the addition of R18 to immunoprecipitated TET2 reduced bound 14-3-3 levels (Fig. 5B, middle). In addition, TET2 immunoprecipitates treated with R18 demonstrated significantly reduced Ser-99 phosphorylation following λ-phosphatase treatment as determined using the phosphoserine 14-3-3–binding motif antibody (Fig. 5B, bottom). Densitometric analysis of three independent experiments is shown in Fig. 5C (blot of biological replicate is provided in Fig. S5). Collectively, these data indicate that bound 14-3-3 protects TET2 phosphorylation at Ser-99 in vitro.
Figure 5.

14-3-3 protects Ser-99 phosphorylation of TET2 in vitro. A, schematic representation of the workflow for phosphatase protection assay. B, representative Western blot analysis of Myc-TET2 immunoprecipitates following in vitro R18 treatment. C, densitometry ratios of phosphorylated TET2/total Myc-TET2 (left) and total 14-3-3/total Myc-TET2 (right) were calculated from three biological replicates and plotted. Data were analyzed by one-way ANOVA followed by post hoc Tukey's honestly significant difference test. Left, ANOVA p value is 0.0146 (F = 9.2774). Post hoc analysis p values are demonstrated. Right, ANOVA p value is 0.0044 (F = 15.2941). Post hoc analysis p values are demonstrated. Error bars, S.E.
14-3-3 protects TET2 from dephosphorylation by phosphatase PP2A
Given our data demonstrating that 14-3-3 can protect TET2 phosphorylation, we sought to determine potential mechanisms by which TET2 may be dephosphorylated. We examined publicly available MS data sets and identified a potential interaction between TET2 and the α-isoform of the B subunit of protein phosphatase 2A (PP2A-Bα; gene, PPP2R2A; Uniprot, P63151; ∼55 kDa) (19). PP2A is a ubiquitously expressed holoenzyme that consists of three subunits, including catalytic (subunit C), scaffold (subunit A), and regulatory (subunit B) subunits. The B subunits are thought to mediate substrate specificity and consist of four isoforms (α, β, γ, and δ) (20). In HEK293T cells expressing Myc-TET2, co-IP with either anti-Myc or anti-TET2 antibody could pull down PP2A-Bα (Fig. 6A). Control IgG pulldown did not demonstrate the presence of PP2A-Bα. We next assessed interaction between endogenous TET2 and PP2A-Bα by co-IP with anti-TET2 and control IgG antibodies. TET2 IP pulled down PP2A-Bα and 14-3-3-θ, whereas IgG control did not (Fig. 6B). Moreover, PP2A-Bα knockdown using two siRNAs (si-1 and si-2) increased Ser-99 phosphorylation (IB: p-14-3-3 motif) of endogenous TET2 relative to control siRNA (Fig. 6C). As expected, total PP2A-Bα was reduced in si- PP2A-Bα samples (Fig. 6D). Additionally, endogenous total TET2 level was increased following PP2A-Bα knockdown (Fig. 6D, IB: TET2; see Fig. 6E and Fig. S6 for statistics and biological replicate, respectively). For direct evidence of PP2A-mediated TET2 dephosphorylation at Ser-99, an in vitro phosphatase assay was performed with recombinant PP2A (rPP2A) consisting of α-isoforms of A, B, and C subunits. Immunoprecipitated TET2 was found to have significantly reduced Ser-99 phosphorylation when incubated with rPP2A but not in buffer control (Fig. 6F).
Figure 6.

PP2A dephosphorylates TET2 at Ser-99. A, HEK293T cells were transfected with Myc-TET2 followed by IP with anti-Myc, anti-TET2, or IgG control antibodies. IPs were analyzed by IB for the indicated proteins. B, HEK293T cell lysate was immunoprecipitated by either anti-TET2 or control IgG antibodies and analyzed by immunoblot. C and D, control knockdown and si-RNA (si-1, si-2) knockdown of PP2A-Bα cell lysates were immunoprecipitated with anti-TET2 antibody and analyzed by immunoblotting in C. Whole-cell extracts were used as inputs (D). Levels of TET2 Ser-99 phosphorylation were calculated by measuring the ratio of band intensities of phospho-Ser-99-TET2 (IB: p-14-3-3 motif) and total TET2 (IB: TET2). TET2 band intensities in the whole-cell lysates were normalized to loading control tubulin. E, endogenous TET2 levels in HEK293T cells treated with siPP2A-Bα (construct #1). Data from three biological replicates are quantified. F, immunoprecipitated Myc-TET2 was treated in vitro with or without rPP2A. Ser-99 phosphorylation was assessed by immunoblotting with phosphoserine 14-3-3–binding motif antibody. Error bars, S.E.
We next assessed effects of the PP2A inhibitor okadaic acid (OKA) on TET2 phosphorylation in cells. A brief (2-h) OKA treatment of HEK293T cells transfected with Myc-TET2 resulted in significantly increased TET2 phosphorylation at Ser-99, as determined via anti-Myc IP and immunoblot with phosphoserine 14-3-3–binding motif antibody (Fig. 7, A and B) (see Fig. S7A for biological replicate). As PP2A has been reported to inhibit AMPK activity (21), we considered whether the effects of OKA were mediated by AMPK activation. However, we did not see any effect on AMPK activation under these experimental conditions, as demonstrated by the lack of treatment effect on either phospho-AMPKα or phospho-ACC levels (Fig. 7A). As disruption of 14-3-3 interaction with difopein reduced TET2 phosphorylation (Fig. 4C), we assessed whether OKA (via its inhibitory effect on PP2A) could maintain TET2 phosphorylation even under conditions in which 14-3-3 and TET2 interaction is disrupted. Two-hour OKA treatment of cells transfected with both TET2 and difopein resulted in a dose-dependent increase in TET2 phosphorylation (Fig. 7C) (see Fig. S7B for a biological replicate). In contrast, TET2 phosphorylation could not be detected in cells without OKA treatment (lane 1). As a negative control, no phosphorylation of the S99A mutant was found when cultured with difopein with or without OKA (Fig. S7C). These data indicate that TET2 Ser-99 dephosphorylation by PP2A is facilitated by disruption of TET2/14-3-3.
Figure 7.

14-3-3 protects TET2 Ser-99 phosphorylation from PP2A. A and B, HEK293T cells were transfected with Myc-TET2. 24 h following transfection, cells were treated with or without PP2A inhibitor OKA (2-h treatment). Immunoprecipitates and inputs were then analyzed by immunoblotting with the indicated antibodies in A. Levels of Myc-TET2 phosphorylation from three biological replicates are quantified in B. C, HEK293T cells were cotransfected with Myc-TET2 and difopein constructs. Forty-eight hours after transfection, cells were treated with the indicated concentrations of OKA for 2 h. Anti-Myc immunoprecipitates were assessed for Ser-99 phosphorylation with phosphoserine 14-3-3–binding motif antibody. This figure is a representative of two biological replicates. D, HEK293T cells (one 10-cm plate) were cotransfected with either Myc-TET2 (8 μg) and CV (10 μg) (lanes 4–6) or Myc-TET2 (8 μg) and difopein (10 μg) constructs (lanes 1–3). After 48 h, cells were treated with either AICAR (1 mm) or metformin (5 mm) for 2 h before harvesting and analysis of Myc IPs and inputs. E, HEK293T cells were cotransfected with Myc-TET2 and difopein. After 48 h, cells were treated with the indicated treatment for 2 h, followed by analysis of Myc IPs and inputs. F and G, cell lysates from HEK293T cells treated with the indicated agents for 48 h were analyzed by immunoblotting. Total TET2 levels were normalized to loading control actin, and ratios are indicated at the top. Data from three biological replicates are quantified in G. Data were analyzed by one-way ANOVA followed by post-hoc Tukey's honestly significant difference test. ANOVA p value is 0.0101 (F = 10.9000). Post hoc analysis p values are demonstrated. Error bars, S.E.
Given that prior studies have demonstrated that AMPK phosphorylates TET2 at Ser-99, we assessed the importance of 14-3-3s in maintaining TET2 phosphorylation upon AMPK activation. AMPK activators AICAR and metformin promoted TET2 phosphorylation (Fig. 7D, compare lanes 5 and 6 with lane 4). In contrast, we were unable to detect TET2 phosphorylation by either AICAR or metformin when cells were co-transfected with difopein (Fig. 7D, lanes 2 and 3). On the other hand, PP2A inhibition with OKA (2-h treatment) was readily able to restore TET2 phosphorylation, even in the presence of difopein (Fig. 7E). Moreover, 48-h treatment with OKA resulted in a dose-dependent increase in endogenous TET2 cells in HEK293T (Fig. 7F). Additionally, OKA treatment significantly increased endogenous TET2 protein, whereas metformin had little effect, which did not reach statistical significance (Fig. 7 (F and G) and Fig. S7D). Collectively, these data demonstrate the role of 14-3-3 interaction in maintaining AMPK's effects on TET2 phosphorylation.
Discussion
In this report, we demonstrate a critical role for 14-3-3s in protecting TET2 phosphorylation and thereby promoting its stability. Our data indicate the dynamic regulation of TET2 at the post-translational level with regard to its phosphorylation. Our data are in line with recent studies demonstrating a role for AMPK-mediated phosphorylation of TET2 at Ser-99 (6). Additionally, the data herein demonstrate a key role for 14-3-3s in protecting this phosphorylation residue from the phosphatase activity of PP2A.
We are aware of a recent communication that reported on 14-3-3 binding to TET2 (7). In this study, exogenous treatment of cells with 14-3-3 inhibitors led to reduced protein levels of WT TET2; however, no effect on TET2 phosphorylation was observed. The effects of 14-3-3 inhibition on TET2 protein stability were not reported, yet the authors proposed that 14-3-3 proteins are TET2 phosphoreaders that promote TET2 stability by an undetermined mechanism. Our studies add new insights into the biological significance of 14-3-3/TET2 interactions (see Fig. 8). First, our studies demonstrate that multiple 14-3-3 isoforms can bind TET2 and that this interaction depends on Ser-99 phosphorylation of TET2. These data are significant as they demonstrate significant redundancy between 14-3-3 family members with respect to TET2 binding. Additionally, our studies demonstrate a key role for 14-3-3 binding in maintaining TET2 phosphorylation and thereby promoting protein stability. Moreover, our studies demonstrate the importance of 14-3-3 binding to TET2 in maintaining AMPK's phosphorylation of TET2. We also identify that PP2A is a TET2 phosphatase. We demonstrate an interaction between TET2 and PP2A-Bα subunit, which is known to regulate substrate specificity of the PP2A complex. Furthermore, PP2A-Bα subunit knockdown and the use of PP2A inhibitor were able to increase TET2 Ser-99 phosphorylation. Collectively, our studies demonstrate the dynamic regulation of TET2 via phosphorylation by AMPK and dephosphorylation by PP2A. 14-3-3s serve as the phosphoreader adaptor proteins that protect TET2 phosphorylation and therefore promote its stability.
Figure 8.
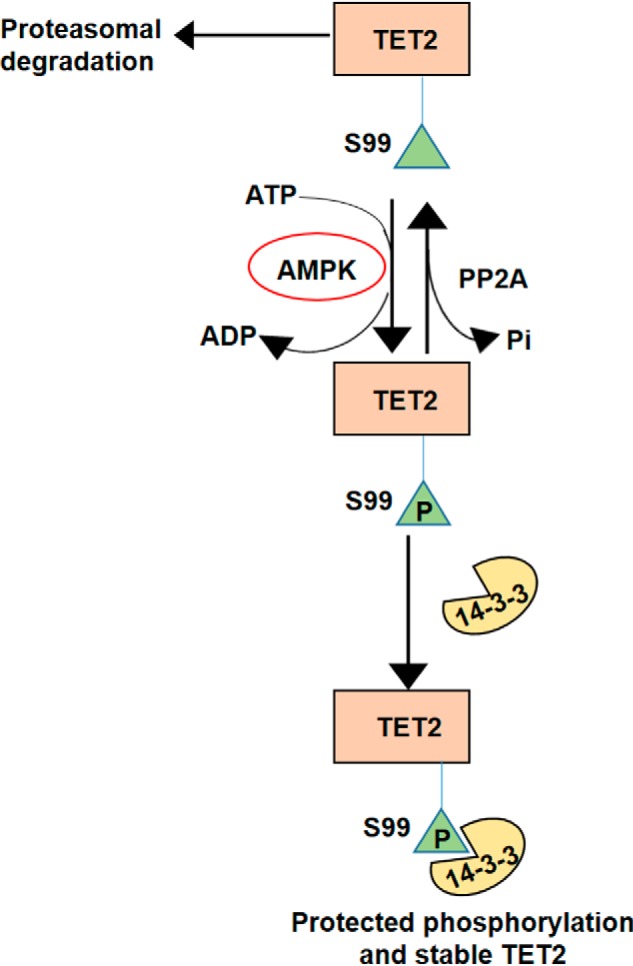
Proposed model for dynamic regulation of TET2 by 14-3-3.
TET2 is a tumor suppressor gene commonly mutated in myeloid malignancies, including AML as well as other hematologic malignancies. Notably, most TET2 mutations impact only one allele. This has led to interest in therapeutic approaches that could activate the function of the remaining WT allele. The TET proteins (TET1 to -3) are members of the α-ketoglutarate–requiring dioxygenase family of enzymes. These enzymes utilize vitamin C as a required cofactor. Recent studies have now reported that vitamin C treatment can reactivate TET2 with demonstrable antitumor activity in preclinical models of leukemia and renal cell carcinoma (22–24). Hence, our studies could point to new strategies by which TET2 function could be promoted via enhanced stability. This is particularly relevant as stabilizers (e.g. fusicoccin) of the interaction between 14-3-3 and client proteins have been reported (25, 26). Such stabilizers may have particular relevance to TET2 mutant AML as a cluster of mutations around the Ser-99 site has been recently reported, which have diminished 14-3-3 binding and reduced protein stability (7). This group of TET2 mutant tumors may be more amenable to approaches that foster interaction with 14-3-3s. A much broader strategy would be to promote stability of the remaining WT TET2 protein via PP2A inhibition. PP2A has both tumor-promoting and -suppressive activities, indicating that its role in malignancy is context-dependent. Notably, a PP2A small-molecule inhibitor (LB-100) is currently being evaluated in patients with myelodysplastic syndrome, in which TET2 mutations are common (NCT03886662).
We acknowledge limitations of our study. Our MS data indicate that mutation of Ser-99 only impacts 14-3-3 binding. However, we would like to point out that the MS-based studies utilized the N-terminal 400 amino acids of TET2. Hence, we cannot exclude effects on the interactions of proteins outside of this region. Alternatively, 14-3-3 binding could impact TET2's interaction with its substrate DNA and/or catalytic activity. Recent studies by Zhang et al. (5) demonstrate that TET2 acetylation at nearby lysine residues, including Lys-110, promotes TET2 protein stability and activity. Hence, it may be possible that the TET2/14-3-3 interaction impacts acetylation as well as other post-translational modifications.
In summary, our data provide novel insights into the regulation of TET2. Our data demonstrate a key role for TET2's interaction with 14-3-3s in maintaining TET2 stability. In particular, our studies indicate that 14-3-3 protects TET2 from dephosphorylation by the PP2A phosphatase. These data may inform new strategies for malignancies driven by loss of TET2 expression and/or activity.
Materials and methods
Reagents
AMPKα1 WT, KD expression constructs (via Addgene), AMPK WT (+/+), and AMPK KO (−/−) MEF cell lines were obtained from Reuben Shaw (Salk Institute). Cell culture media and fetal bovine serum were from Cellgro (Corning) and Atlanta Biologicals, respectively. TET2 and 14-3-3 antagonist difopein expression plasmid constructs were kind gifts from Dr. Xiaochun Yu (University of Michigan) and Dr. Talene Yacoubian (University of Alabama, Birmingham, AL), respectively. Genomic DNA isolation kit was from Promega (#A1120). λ-protein phosphatase was purchased from New England Biolabs (#P0753S). Recombinant proteins used in this study include N-terminal (aa 1–350) TET2 (Proteintech), AMPK (SignalChem), and PP2A (Abcam).
Antibodies
Primary antibodies used in this study were purchased from commercial vendors and are indicated below. These antibodies include 1) c-Myc (#M4439; origin, mouse; source, monoclonal; 1:3000), FLAG (#F1804; origin, mouse; source, monoclonal; 1:3000), PP2A-Bα (#SAB4200241; origin, mouse; source, monoclonal; 1:1000), and tubulin (#T9026; origin, mouse; source, monoclonal; 1:5000) from Sigma-Aldrich; 2) 14-3-3-θ from Proteintech (#14503–1-AP; origin, rabbit; source, polyclonal; 1:1000); 3) β-actin conjugated with HRP (#ab49900; origin, mouse; source, monoclonal; 1:10,000) and GAPDH (#ab181602; origin, rabbit; source, polyclonal; 1:5000) from Abcam; 4) anti-5hmC from Active Motif (#39069; origin, rabbit; source, polyclonal; 1:2500); 5) anti-TET2 from Diagenode (#C15200179; origin, mouse; source, monoclonal; 1 μg/1 mg lysate for immunoprecipitation); and 6) 14-3-3 isoform sampler kit (#9769; origin, rabbit; source, monoclonal and polyclonal; 1:1000), phosphoserine 14-3-3–binding motif (#9601; origin, rabbit; source, monoclonal; 1:1000), AMPKα (#5831; origin, rabbit; source, monoclonal; 1:1000), phospho-AMPKα (#2535; origin, rabbit; source, monoclonal; 1:1000), phospho-ACC (#3661; origin, rabbit; source, monoclonal; 1:1000), anti-protein phosphatase 2A (#9780; origin, rabbit; source, monoclonal; 1:1000) sampler kit, and TET2 (#18950; origin, rabbit; source, monoclonal; 1:1000) from Cell Signaling Technology. Secondary antibodies used in this work were HRP-conjugated anti-mouse (GE Healthcare, #NA931V; 1:5000) and HRP-conjugated anti-rabbit (GE Healthcare, #NA9340V; 1:5000) antibodies. TET2 (Cell Signaling) antibody was validated by TET2 overexpression (2 μg of pCMV-Myc-TET2 plasmid was transfected to HEK293T cells grown in a 10-cm plate) (Fig. S1B). 14-3-3-θ, 14-3-3-ϵ, and PP2A-Bα antibodies were validated by siRNA knockdown and Western blot analysis (Fig. S1B and Fig. 6D). Phospho-14-3-3 motif antibody was validated by point mutation (S99A) in the antibody-binding motif of TET2 followed by immunoprecipitation and Western blotting (Fig. 1F and Fig. S1D).
Cell culture, cloning, and transfection
Human embryonic kidney cells (HEK293T) and MEF were maintained at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 4.5 g/liter glucose, 2 mm l-glutamine, 10% fetal bovine serum and without antibiotics. TET2 S99A mutant was made following the standard procedures. TET2 WT and TET2 mutant cDNAs were cloned into pCMV-Myc (N terminus) plasmid vector (Clontech) and verified by Sanger sequencing. Fugene 6 (Promega) and Opti-MEM (Thermo Fisher Scientific) reduced serum media were used for transient transfection of plasmid DNAs in the above cell lines following the manufacturer's protocol. Any treatments to transfected cells including cycloheximide (Sigma-Aldrich), AICAR (#ab120358, Abcam), Metformin (#ab120847, Abcam), Compound C (#ab120843, Abcam), and okadaic acid (#5934, Cell Signaling Technology) were added 24–48 h post-transfection, as indicated in the respective figure legends.
Yeast two-hybrid screen
The ProQuest yeast two-hybrid system with Gateway technology (Invitrogen) was used to identify proteins interacting with the TET2 N-terminal part (amino acids 1–1000), according to the manufacturer's instructions. The TET2 region was PCR-amplified using primers containing 5′ attB1 and 3′ attB2 recombination sites (forward, GGGGACAAGTTTGTACAAAAAAGCAGGCTCCATGGAACAGGATAGAACCAACCATGTTG; reverse, GGGGACCACTTTGTACAAGAAAGCTGGGTTTATGAGCTTTGCTTGAAGTAAGCACCATTC) and cloned into the pDONR222.1 vector (Invitrogen) to make the entry clones, followed by recombinatorial cloning in frame with the GAL4 DNA-binding domain in the pDEST32 yeast expression vector (Invitrogen). The TET2 bait clones were transformed into the MaV203 strain of yeast, and colonies that grew on selective SC-Leu medium were screened by PCR for the TET2 gene. Yeasts containing the TET2-pDEST32 construct were transformed with a Proquest human spleen c-DNA library, which contains cDNAs fused to the GAL4 activation domain of pEXP-AD502 (Invitrogen). Interactions between fusion proteins in this system activate three reporter genes: β-gal, HIS3, and URA3. Screening of the library of clones resulted in the isolation of colonies that fulfilled the selection criteria of His prototropy on yeast dropout medium lacking Trp, Leu, and His in the presence of 3-aminotriazole. Positive colonies were confirmed by expression of β-gal by X-gal filter assays and for growth on yeast dropout medium lacking Leu, Trp, and Ura. Selected yeast colonies were grown, plasmids were isolated, and the insert cDNA was PCR-amplified with primers specific for pEXP-AD502 (Invitrogen). The PCR products were sequenced using pEXP-AD502 sequencing primers and analyzed using the NCBI BLAST software to identify the interacting proteins.
Co-immunoprecipitation
All immunoprecipitation steps were carried out at 4 °C. Cells expressing Myc-tagged TET2 WT or S99A TET2 were lysed in buffer X (50 mm HEPES, pH 7.5, 150 mm NaCl, 0.1% Nonidet P-40, and 1 mm EDTA) containing 1× protease/phosphatase inhibitor (Thermo Fisher Scientific). After clearing, a 50-μl slurry of protein A/G-agarose (Santa Cruz Biotechnology, Inc.) and 2 μg of the anti-Myc or anti-TET2 or control IgG antibody were added to 1–2 mg of the extracted proteins and rotated for 16 h. Next, the beads were spun down at 600 × g, and the immunoprecipitates were washed three times with buffer Y (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm EDTA) for 10 min each with rotation at 4 °C. Finally, the beads were boiled in 2× Laemmli buffer to elute the immunoprecipitated proteins.
Western blotting
Protein samples prepared in Laemmli buffer were resolved by 4–15% SDS-PAGE (Bio-Rad). Proteins were transferred onto polyvinylidene difluoride (Immobilon P, #IPVH00010, Millipore) using standard procedures. The membranes were blocked either with 3% BSA or with 5% skim milk and probed with specific antibodies. Mild stripping buffer (Abcam) was used to reprobe Western blotting membranes. Western blots were incubated with primary antibodies at room temperature for 1 h or at 4 °C for overnight, washed three times (10 min each) with 1× TBST buffer containing 0.1% Tween 20, and then incubated with secondary antibodies for 1 h at room temperature. Western blots were developed using Immobilon Classico Western HRP substrate (#WBLUC0500, Millipore) and exposing the blots to X-ray films (Hawkins, #HXR0507) or to an Amersham Biosciences Imager 600 (GE Healthcare). Images were quantified by ImageJ software (National Institutes of Health, RRID: SCR_003070). To validate the linear range of detection by TET2 antibody compared with actin/tubulin and total protein loading, TET2 band intensities were plotted along with total protein, actin, and tubulin band intensities against different loading volumes. TET2 band and Ponceau stain intensities were found to be linear from 5 to 20 μl of the loading volumes. However, the linear range of band intensities for both actin and tubulin was within 5–15 μl, which corresponded to 8–25 μg of total cell lysates (data not shown). In all of our experiments, TET2 band intensities were calculated from less than 25 μg of cell lysates, which were further normalized to actin or tubulin band intensities. Further, we observed no change in actin, tubulin, and GAPDH band intensities by treatments in our experiments, and thus we preferred to use them as normalizing signals during quantifications. Statistics were done from three biological replicates and presented as mean ± S.E. For statistical analysis of two experimental groups, Student's t test was performed, and differences were considered significant if p was <0.05. For multiple-group comparisons, ANOVA was performed with the indicated post hoc tests.
LC-MS analysis
cDNA for the first 400 amino acids from WT TET2 N terminus was cloned into N-Myc-pCMV vector (Clontech). cDNA for the S99A mutation in the same region of TET2 was also generated and cloned into the same vector as that of WT TET2. 10 μg of these plasmids were used to transfect HEK293T cells, and cell lysates were co-immunoprecipitated by anti-Myc and control IgG antibody as described earlier. Eluted protein samples were run in 10% BisTris SDS-PAGE and stained with colloidal Coomassie Blue. Following destaining, the whole lane of each sample was cut into multiple molecular weight fractions, equilibrated in 100 mm ammonium bicarbonate, and then digested overnight with PierceTM trypsin protease, MS Grade (#90058, Thermo Scientific), following the manufacturer's instructions. Peptide extracts were reconstituted in 0.1% formic acid/double-distilled H2O at 0.1 μg/μl and then analyzed by LC-MS at the UAB Cancer Center Mass Spectrometry/Proteomics Shared Facility using a 1260 Infinity nHPLC stack (Agilent Technologies) and Thermo Orbitrap Velos Pro hybrid mass spectrometer, equipped with a Nanospray FlexTM ion source (Thermo Fisher Scientific). The raw files were analyzed by SEQUEST using a specific subset of the UniProtKB database. Detailed methods of LC-MS analysis are documented in File 2. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD016160.
In vitro kinase assay
0.5 μg of recombinant AMPK was added to 5 μg of recombinant TET2 in the presence or absence of ATP (500 μm) in a reaction buffer (#K01-09, SignalChem) containing 25 mm MOPS, pH 7.2, 12.5 mm β-glycerol phosphate, 25 mm MgCl2, 5 mm EGTA, 2 mm EDTA, 250 μm DTT and incubated for 30 min at 30 °C. The samples were boiled in Laemmli buffer and resolved in an 8% SDS-polyacrylamide gel, followed by blotting onto a polyvinylidene difluoride membrane, and probed with phosphoserine 14-3-3–binding motif antibody.
Phosphatase protection assay
Immunoprecipitated Myc-TET2 (WT) bound on protein A/G-agarose beads was washed three times in buffer Y as above and incubated with 0, 100, and 500 nm R18 peptide (in buffer Y) for 3 h with rotation at 4 °C. The beads were washed once with buffer Y and incubated with λ-protein phosphatase (200 units each reaction) at 15 °C for 30 min. The reactions were stopped by adding 2× Laemmli buffer and boiling the samples at 95 °C for 5 min. Samples were resolved in 4–15% SDS-PAGE followed by Western blotting using the indicated antibodies.
siRNA knockdown
Two siRNA constructs were used to knock down the PP2A-Bα isoform in HEK293T cells. si-1 (#hs.Ri.PPP2R2A.13.1), si-2 (#hs.Ri.PPP2R2A.13.2), and control siRNA (#51-01-14-04) from IDT were transfected into HEK293T cells using Lipofectamine RNAiMAX (Thermo Scientific) following the manufacturer's protocol, and cells were harvested 24 h post-transfection for analysis. Similarly, 14-3-3-ϵ (hs.Ri.YWHAE.13.1) and 14-3-3-θ (hs.ri.YWHAQ.13.1) were knocked down by siRNAs from IDT.
In vitro PP2A assay
Protein A/G-bound Myc-TET2 (WT) was washed three times in buffer Y and incubated in a buffer containing 25 mm Tris-HCl, pH 8.0, 150 mm NaCl, 3 mm DTT, 50 μm MnCl2, and 1 mm CaCl2 with or without 1.0 μg of recombinant PP2A (which consists of α-isoforms of A, B, and C subunits) at room temperature for 30 min. Reactions were stopped by adding Laemmli buffer and analyzed by immunoblotting.
5hMC dot blot
Global 5hmC levels were analyzed from extracted genomic DNA following the protocol described by Zhang et al. (5).
Author contributions
A. K. and S. Sudarshan conceptualization; A. K., S. Shelar, A. G., M. B., R. K., H.-Y. N., G. B., and S. K. data curation; A. K. validation; A. K., J. A. M., and S. B. methodology; A. K. writing-original draft; A. K. and S. Sudarshan writing-review and editing; S. V. formal analysis; S. Sudarshan funding acquisition.
Supplementary Material
This work was supported by National Institutes of Health Grant R01CA200653, Department of Veterans Affairs Grant I01BX002930, and a Mike Slive Foundation Grant (to S. S.) and in part by UAB Comprehensive Cancer Center Grant P30CA013148. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Files S1 and S2 and Figs. S1–S7.
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD016160.
- 5mC
- 5-methylcytosine
- 5hmC
- 5-hydroxymethylcytosine
- TET
- ten-eleven translocation
- AML
- acute myeloid leukemia
- PP2A
- protein phosphatase 2A
- IP
- immunoprecipitation
- IB
- immunoblotting
- CV
- control vector
- CHX
- cycloheximide
- KD
- kinase-dead
- KO
- knockout
- MEF
- mouse embryonic fibroblast
- AICAR
- 5-aminoimidazole-4-carboxamide ribonucleotide
- rPP2A
- recombinant PP2A
- OKA
- okadaic acid
- HRP
- horseradish peroxidase
- ACC
- acetyl-CoA-carboxylase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ANOVA
- analysis of variance
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- AMPK
- AMP-activated protein kinase.
References
- 1. Wu X., and Zhang Y. (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 10.1038/nrg.2017.33 [DOI] [PubMed] [Google Scholar]
- 2. Langemeijer S. M., Kuiper R. P., Berends M., Knops R., Aslanyan M. G., Massop M., Stevens-Linders E., van Hoogen P., van Kessel A. G., Raymakers R. A., Kamping E. J., Verhoef G. E., Verburgh E., Hagemeijer A., Vandenberghe P., et al. (2009) Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 41, 838–842 10.1038/ng.391 [DOI] [PubMed] [Google Scholar]
- 3. Delhommeau F., Dupont S., Della Valle V., James C., Trannoy S., Massé A., Kosmider O., Le Couedic J. P., Robert F., Alberdi A., Lécluse Y., Plo I., Dreyfus F. J., Marzac C., Casadevall N., et al. (2009) Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 360, 2289–2301 10.1056/NEJMoa0810069 [DOI] [PubMed] [Google Scholar]
- 4. Hu L., Li Z., Cheng J., Rao Q., Gong W., Liu M., Shi Y. G., Zhu J., Wang P., and Xu Y. (2013) Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155, 1545–1555 10.1016/j.cell.2013.11.020 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y. W., Wang Z., Xie W., Cai Y., Xia L., Easwaran H., Luo J., Yen R. C., Li Y., and Baylin S. B. (2017) Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol. Cell 65, 323–335 10.1016/j.molcel.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu D., Hu D., Chen H., Shi G., Fetahu I. S., Wu F., Rabidou K., Fang R., Tan L., Xu S., Liu H., Argueta C., Zhang L., Mao F., Yan G., et al. (2018) Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641 10.1038/s41586-018-0350-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen H., Yu D., Fang R., Rabidou K., Wu D., Hu D., Jia P., Zhao Z., Wu Z., Peng J., Shi Y., and Shi Y. G. (2019) TET2 stabilization by 14-3-3 binding to the phosphorylated serine 99 is deregulated by mutations in cancer. Cell Res. 29, 248–250 10.1038/s41422-018-0132-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mackintosh C. (2004) Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 381, 329–342 10.1042/BJ20031332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muslin A. J., Tanner J. W., Allen P. M., and Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 10.1016/S0092-8674(00)81067-3 [DOI] [PubMed] [Google Scholar]
- 10. Madeira F., Tinti M., Murugesan G., Berrett E., Stafford M., Toth R., Cole C., MacKintosh C., and Barton G. J. (2015) 14-3-3-Pred: improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 31, 2276–2283 10.1093/bioinformatics/btv133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y., and Zhang Y. (2014) Regulation of TET protein stability by calpains. Cell Rep. 6, 278–284 10.1016/j.celrep.2013.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Libertini S. J., Robinson B. S., Dhillon N. K., Glick D., George M., Dandekar S., Gregg J. P., Sawai E., and Mudryj M. (2005) Cyclin E both regulates and is regulated by calpain 2, a protease associated with metastatic breast cancer phenotype. Cancer Res. 65, 10700–10708 10.1158/0008-5472.CAN-05-1666 [DOI] [PubMed] [Google Scholar]
- 13. Clurman B. E., Sheaff R. J., Thress K., Groudine M., and Roberts J. M. (1996) Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 10, 1979–1990 10.1101/gad.10.16.1979 [DOI] [PubMed] [Google Scholar]
- 14. Brunet A., Kanai F., Stehn J., Xu J., Sarbassova D., Frangioni J. V., Dalal S. N., DeCaprio J. A., Greenberg M. E., and Yaffe M. B. (2002) 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 156, 817–828 10.1083/jcb.200112059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun S., Wong E. W., Li M. W., Lee W. M., and Cheng C. Y. (2009) 14-3-3 and its binding partners are regulators of protein-protein interactions during spermatogenesis. J. Endocrinol. 202, 327–336 10.1677/JOE-09-0041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Masters S. C., and Fu H. (2001) 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276, 45193–45200 10.1074/jbc.M105971200 [DOI] [PubMed] [Google Scholar]
- 17. Wang B., Yang H., Liu Y. C., Jelinek T., Zhang L., Ruoslahti E., and Fu H. (1999) Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry 38, 12499–12504 10.1021/bi991353h [DOI] [PubMed] [Google Scholar]
- 18. Hausser A., Link G., Hoene M., Russo C., Selchow O., and Pfizenmaier K. (2006) Phospho-specific binding of 14-3-3 proteins to phosphatidylinositol 4-kinase III β protects from dephosphorylation and stabilizes lipid kinase activity. J. Cell Sci. 119, 3613–3621 10.1242/jcs.03104 [DOI] [PubMed] [Google Scholar]
- 19. Nickerson M. L., Das S., Im K. M., Turan S., Berndt S. I., Li H., Lou H., Brodie S. A., Billaud J. N., Zhang T., Bouk A. J., Butcher D., Wang Z., Sun L., Misner K., et al. (2017) TET2 binds the androgen receptor and loss is associated with prostate cancer. Oncogene 36, 2172–2183 10.1038/onc.2016.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seshacharyulu P., Pandey P., Datta K., and Batra S. K. (2013) Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 335, 9–18 10.1016/j.canlet.2013.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joseph B. K., Liu H. Y., Francisco J., Pandya D., Donigan M., Gallo-Ebert C., Giordano C., Bata A., and Nickels J. T. Jr. (2015) Inhibition of AMP kinase by the protein phosphatase 2A heterotrimer, PP2APpp2r2d. J. Biol. Chem. 290, 10588–10598 10.1074/jbc.M114.626259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ge G., Peng D., Xu Z., Guan B., Xin Z., He Q., Zhou Y., Li X., Zhou L., and Ci W. (2018) Restoration of 5-hydroxymethylcytosine by ascorbate blocks kidney tumour growth. EMBO Rep. 19, e45401 10.15252/embr.201745401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shenoy N., Bhagat T. D., Cheville J., Lohse C., Bhattacharyya S., Tischer A., Machha V., Gordon-Mitchell S., Choudhary G., Wong L. F., Gross L., Ressigue E., Leibovich B., Boorjian S. A., Steidl U., et al. (2019) Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Invest. 130, 1612–1625 10.1172/JCI98747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cimmino L., Dolgalev I., Wang Y., Yoshimi A., Martin G. H., Wang J., Ng V., Xia B., Witkowski M. T., Mitchell-Flack M., Grillo I., Bakogianni S., Ndiaye-Lobry D., Martín M. T., Guillamot M., et al. (2017) Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20 10.1016/j.cell.2017.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bunney T. D., De Boer A. H., and Levin M. (2003) Fusicoccin signaling reveals 14-3-3 protein function as a novel step in left-right patterning during amphibian embryogenesis. Development 130, 4847–4858 10.1242/dev.00698 [DOI] [PubMed] [Google Scholar]
- 26. Skwarczynska M., Molzan M., and Ottmann C. (2013) Activation of NF-κB signalling by fusicoccin-induced dimerization. Proc. Natl. Acad. Sci. U.S.A. 110, E377–E386 10.1073/pnas.1212990110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.