

SUMMARY
The site-specific incorporation of non-canonical amino acids (ncAAs) into proteins via amber suppression provides access to novel protein properties, structures, and functions. Historically, poor protein expression yields resulting from release factor 1 (RF1) competition has limited this technology. To address this limitation, we develop a high-yield, one-pot cell-free platform for synthesizing proteins bearing ncAAs based on genomically recoded Escherichia coli lacking RF1. A key feature of this platform is the independence on the addition of purified T7 DNA-directed RNA polymerase (T7RNAP) to catalyze transcription. Extracts derived from our final strain demonstrate high productivity, synthesizing2.67 ± 0.06 g/L superfolder GFP in batch mode without supplementation of purified T7RNAP. Using an optimized one-pot platform, we demonstrate multi-site incorporation of the ncAA p-acetyl-L-phenylalanine into an elastin-like polypeptide with high accuracy of incorporation and yield. Our work has implications for chemical and synthetic biology.
Graphical Abstract
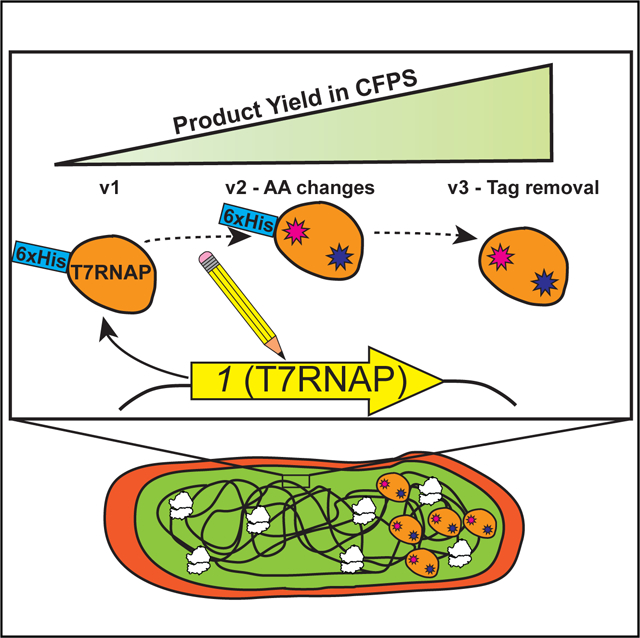
In Brief
Des Soye et al. create and optimize a strain of Escherichia coli that expresses T7 RNA polymerase so that lysates prepared from the strain are enriched with sufficient polymerase to catalyze high-yielding cell-free transcription and translation reactions. Using the resulting platform, the authors synthesize products containing up to 40 non-canonical amino acids.
INTRODUCTION
A burst of recent development has transformed cell-free protein synthesis (CFPS) from a niche tool for molecular biology into a new technology platform with promise for manufacturing proteins at scale and for accelerating biological design (Carlson et al., 2012; Dudley et al., 2015; Garamella et al., 2016; Hodgman and Jewett, 2012; Karim et al., 2015). This rapid advancement has been spurred by the desire to take advantage of the beneficial features unique to CFPS systems, which include easy system access and manipulation, the elimination of competition with cellular growth and adaptation objectives, and a dilute reaction environment that can facilitate folding of complex eukaryotic protein products (Caschera and Noireaux, 2014; Jewett and Swartz, 2004). Batch CFPS reactions now persist for up to a day with yields exceeding 1.5 g/L (Caschera and Noireaux, 2014; Martin et al., 2018), and improvements in scalability culminated recently with the successful completion of a 100-L reaction (Zawada et al., 2011). These impressive advances can be largely attributed to extensive efforts to engineer CFPS systems via chassis organism development, usually by the targeted genetic deletion of genes whose products are known to destabilize key biological substrates (e.g., DNA, mRNA, amino acids, and energy) in cell-free reactions (Hong et al., 2015; Martin et al., 2018; Michel-Reydellet et al., 2004; Yang et al., 2009). As a result of these transformative efforts, CFPS platforms can now be used to complement protein overexpression in vivo, with particular utility in rapid prototyping (Chappell et al., 2013; Dudley et al., 2016; Karim and Jewett, 2016; McManus et al., 2019; Schinn et al., 2017; Takahashi et al., 2015), synthesis of toxic products (Martemyanov et al., 2001; Renesto and Raoult, 2003; Watanabe et al., 2010; Xu et al., 2005; Yim et al., 2018), the production of proteins that are difficult to solubly express in vivo (Heinzelman et al., 2015; Li et al., 2016; Sullivan et al., 2016; Zawada et al., 2011), manufacturing of glycoproteins (Jaroentomeechai et al., 2018; Kightlinger et al., 2018; Schoborg et al., 2018), detection of disease (Chen et al., 2018; Gootenberg et al., 2017; Pardee et al., 2016a; Slomovic et al., 2015; Takahashi et al., 2018), on-demand biomanufacturing (Adiga et al., 2018; Hunt et al., 2017; Karig et al., 2017; Pardee et al., 2016a, 2016b; Smith et al., 2014; Sullivan et al., 2016), and education (Huang et al., 2018; Stark et al., 2018, 2019; Wandera et al., 2019).
One particularly appealing application of CFPS is the production of proteins containing non-canonical amino acids (ncAAs) (Amiram et al., 2015; d’Aquino et al., 2018; Des Soye et al., 2015; Hong et al., 2014a, 2014b, 2015; Liu et al., 2018; Liu et al., 2017; Martin et al., 2018). To date, more than 150 different ncAAs have been incorporated into (poly)peptides (Dumas et al., 2014), enabling the synthesis of proteins featuring novel structures and functions that would otherwise be difficult or even impossible to obtain using only the 20 canonical amino acids. Typically, site-specific ncAA incorporation into proteins is enabled by amber suppression, whereby the amber stop codon (UAG) is recoded as a sense codon designating a ncAA of interest (Liu and Schultz, 2010). This process is mediated by orthogonal translation systems (OTSs), which generally consist of the ncAA, an orthogonal suppressor tRNA (o-tRNA) that has been modified to associate with UAG in the ribosomal A site, and an ncAA-specific aminoacyl-tRNA synthetase (ncAA-RS) that has been evolved to covalently load the ncAA onto the o-tRNA (Des Soye et al., 2015; Santoro et al., 2002), without recognizing natural amino acids. The cytotoxicity of many OTSs (Martin et al., 2018; Nehring et al., 2012), membrane impermeability of some ncAAs (Bundy and Swartz, 2010), and the ability to overcome the relatively poor incorporation efficiencies of OTSs via direct supplementation with OTS components (Des Soye et al., 2015; Hong et al., 2014a, 2015; Martin et al., 2018) makes CFPS an attractive method for the synthesis of peptides featuring ncAAs.
Unfortunately, efforts to apply amber suppression for the incorporation of ncAAs into proteins have long been limited by competition with release factor 1 (RF1), which is responsible for terminating translation in response to the ribosome encountering a UAG codon (Young and Schultz, 2010). In attempting amber suppression, functional RF1 can outcompete ncAA-bearing o-tRNAs at UAG codons, leading to the production of errant truncated products (Hong et al., 2014a, 2014b). Historically, this competition has led to poor protein expression yields, which limits applications in both basic and applied science. Recently, we addressed this limitation. Specifically, we developed a CFPS system derived from a genomically recoded strain of Escherichia coli in which all native instances of the amber codon were changed to the synonymous ochre codon (UAA) followed by elimination of RF1 from the genome (C321.ΔA) (Lajoie et al., 2013; Martin et al., 2018). Extracts derived from the resulting strain (C321.ΔA.759) resulted in CFPS yields of ~1,700 mg/L and 99% suppression efficiency for superfolder green fluorescent protein (sfGFP) with 2 ncAAs, which outperform the best expression of proteins with single or multiple ncAAs in vivo (Martin et al., 2018). While this strain demonstrates high productivity for ncAA incorporation into proteins in vitro, it is limited by its dependence on the addition of purified viral T7 DNA-directed RNA polymerase (T7RNAP) to catalyze transcription. This adds another step to reaction assembly and increases the cost of the system by requiring the addition of purified polymerase to catalyze robust transcription. In principle, one could create a one-pot CFPS system if T7RNAP could be integrated into the genome, and overexpressed in the source strain prior to lysis. Indeed, extracts derived from T7RNAP-expressing strains (most notably BL21(DE3) (Studier and Moffatt, 1986) and its derivatives) are innately enriched in polymerase activity and generally do not require (or even benefit from) supplementation (Kwon and Jewett, 2015). These one-pot CFPS systems, containing all of the biological components necessary to support transcription and translation, are highly attractive due to their convenient plug-and-play nature.
In this study, we developed a high-yielding one-pot CFPS platform for ncAA incorporation into proteins derived from a genomically recoded RF1-deficient strain of E. coli that has been optimized for productivity in CFPS (C321.ΔA.759) (Martin et al., 2018) (Figure 1). Since C321.ΔA.759 does not express T7RNAP, we applied λ-Red-mediated homologous recombination (Datsenko and Wanner, 2000; Mosberg et al., 2010) (λHR) to genomically integrate a series of synthetic constructs featuring the T7RNAP-encoding 1 gene (Studier and Moffatt, 1986) and then assessed the ability of extracts derived from the resulting transformants to catalyze CFPS in the absence of exogenous polymerase supplementation. While native bacterial RNA polymerases and associated sigma factors can be used to catalyze transcription in CFPS reactions (Shin and Noireaux, 2010, 2012), we chose to pursue T7RNAP because of its high productivity, orthogonality, and strong sequence preference (Shin and Noireaux, 2010; Studier and Moffatt, 1986). Two different genomic loci were targeted for integration, with 1 placed under the regulation of three promoters of different strengths. A high-performing strain, C321.ΔA.759.T7, was capable of synthesizing ~1.4 g/L of sfGFP without purified T7RNAP supplementation. We next exploited multiplex automated genome engineering (MAGE) (Wang et al., 2009) to install mutations in the 1 gene of C321.ΔA.759.T7 and make it resistant to proteolytic cleavage during lysate preparation. The resulting strain, C321.ΔA.759.T7.D, yielded ~1.6 g/L sfGFP without T7RNAP supplementation and ~2.2 g/L with supplementation, but remained transcriptionally limited as evidenced by the increase in yields when purified polymerase was supplemented to the system. To address this, we applied a combination of CRISPR/Cas9 and MAGE (CRMAGE) (Ronda et al., 2016) to remove an N-terminal His tag from the polymerase gene in an effort to improve the function of the T7RNAP expressed by the cells. Lysates from the resulting strain, 759.T7.Opt, demonstrate no improvement when supplemented with additional purified polymerase, yielding ~2.7 g/L sfGFP using only the T7RNAP synthesized in-cell. Using an optimized system, we were able to synthesize proteins (elastin-like polypeptides) bearing up to 20, 30, and 40 ncAAs with yields up to ~70 mg/L in the absence of supplemental T7RNAP. When compared with BL21(DE3) and its derivative strains, one-pot CFPS systems derived from 759.T7.Opt are highly productive and superior for applications involving ncAAs.
Figure 1. Simplified Schematic of the Production and Utilization of Crude Lysates from E. coli Cells to Catalyze Cell-Free Protein Synthesis.
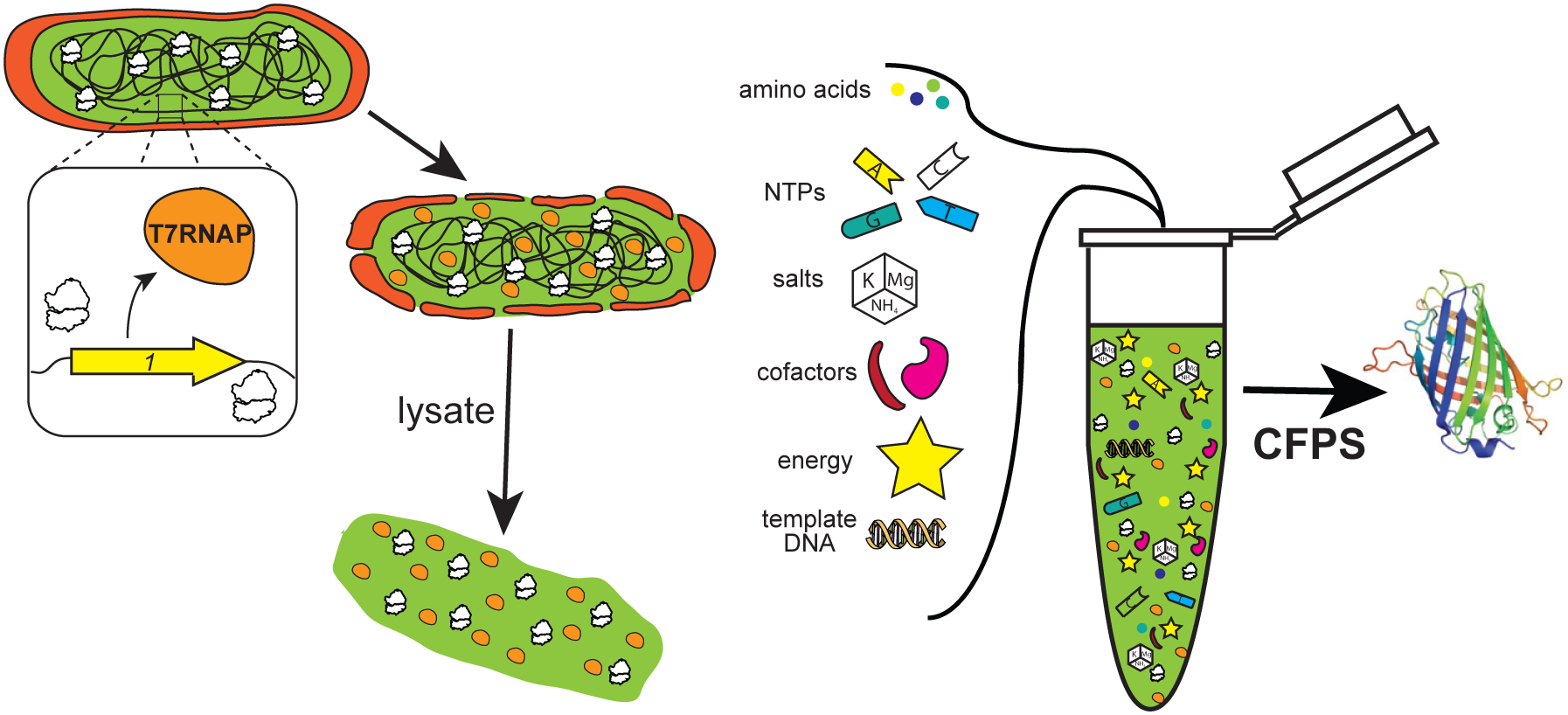
Reactions are supplemented with enzymatic cofactors, energy, and other substrates required for protein synthesis as well as plasmid DNA template directing the system toward the production of a product of interest. The strain illustrated is shown endogenously expressing T7RNAP to enable orthogonal transcription in vitro and generate a one-pot system independent of supplementation with purified protein components. CFPS, cell-free protein synthesis; NTPs, nucleoside triphosphates.
RESULTS
CFPS Activity of C321.ΔA.759 and BL21 Star (DE3) with and without Supplemental T7RNAP
We first set out to establish the extent to which C321.ΔA.759 lysates could perform T7-based transcription. To test this, we prepared batches of crude S12 lysates from C321.ΔA.759 as well as BL21 Star (DE3) which had T7RNAP expression induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Batch CFPS reactions were performed using these lysates, directed to synthesize superfolder green fluorescent protein (sfGFP) both with and without direct supplementation of 16 μg/mL purified T7RNAP (Martin et al., 2018) (Figure 2A). As expected based on our previous work (Martin et al., 2018), the yield from C321.ΔA.759 lysates with T7RNAP added was ~30% higher than either BL21 Star (DE3) condition. There was no observable benefit to supplementing additional T7RNAP into reactions utilizing polymerase-enriched BL21 Star (DE3) lysates. Unsurprisingly, essentially no sfGFP was synthesized by the C321.ΔA.759 lysates when no T7RNAP was supplemented. Thus, we hypothesized that introducing the 1 gene into C321.ΔA.759 would imbue the strain with the ability to synthesize T7RNAP and eliminate its dependence on supplemental polymerase in vitro.
Figure 2. Engineering a Genomically Recoded E. coli Strain for T7RNAP Overexpression.
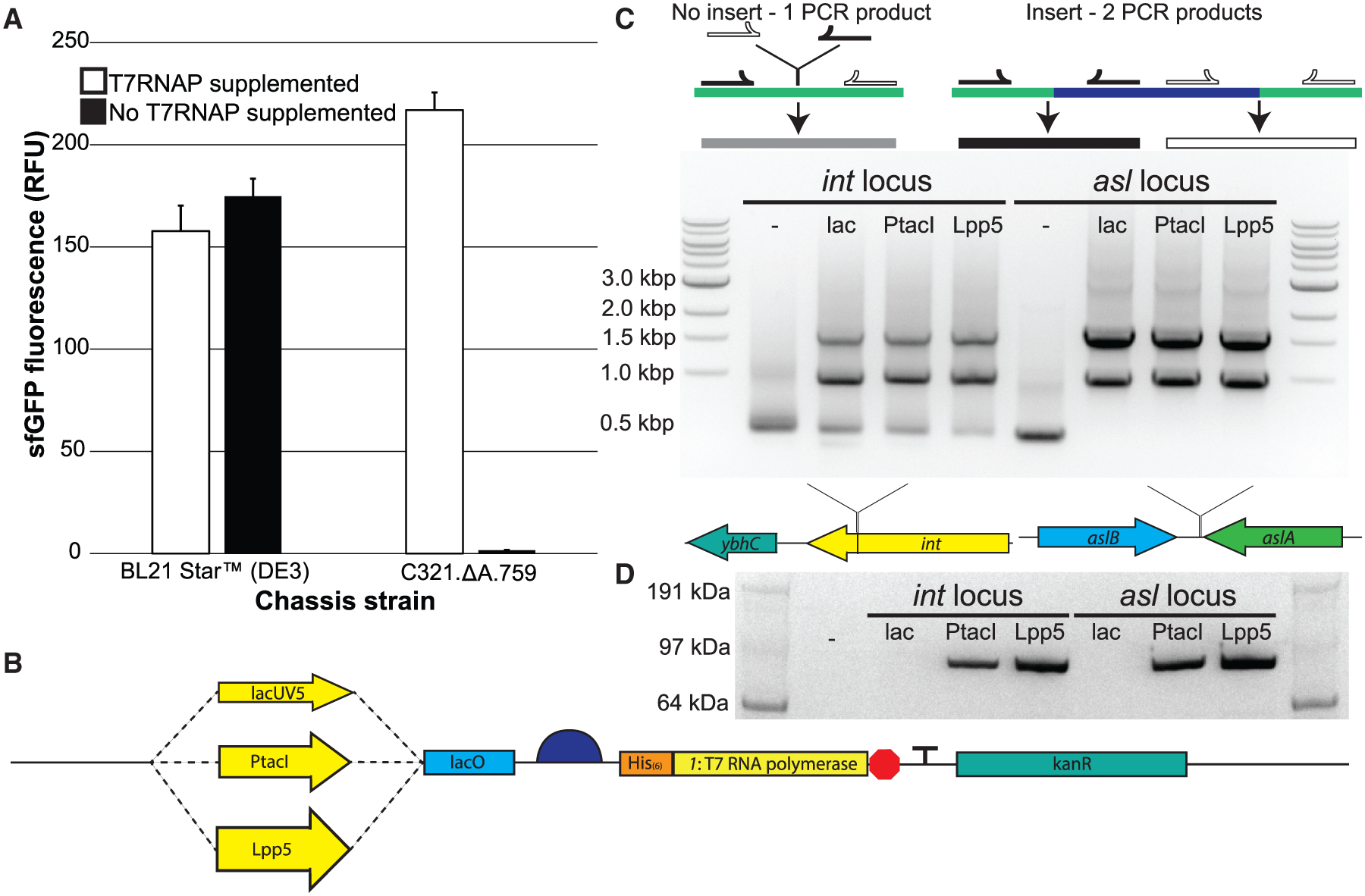
(A) sfGFP fluorescence in vitro from cell extracts derived from induced BL21 Star (DE3) cells as well as C321.ΔA.759 cells, both with and without supplementation with purified T7RNAP.
(B) Schematic of the synthetic genomic insert used in this study to introduce the gene encoding the T7RNAP into the genome of C321.ΔA.759.
(C) Top: diagram illustrating the PCR-based detection scheme for successful genomic integration of the synthetic T7RNAP cassette into strain C321.ΔA.759. Middle: shown are the MASC PCR products generated from C321.ΔA.759 and the six T7RNAP-expressing strains generated in this study, run on an agarose gel. The dash indicates the unaltered strain without any insert incorporated. Bottom: depiction of the two insertion loci used.
(D) α-His western blot analysis of protein samples derived from IPTG-induced populations of C321.ΔA.759 and the six T7RNAP-expressing strains generated in this study. The His-tagged T7RNAP version used has a molecular weight of ~100 kDa. The dash indicates the unaltered strain without any insert incorporated.
T7RNAP Insert Design and Integration
A large body of work has explored various ways of enabling bacteria to produce T7RNAP (Davanloo et al., 1984; McAllister et al., 1981; Studier and Moffatt, 1986). Plasmid-based approaches are simple and effective, but expression levels are high enough to impair plasmid maintenance or otherwise place a significant metabolic burden on host cells, manifesting itself in the form of increased doubling time (Studier and Moffatt, 1986). As increases in doubling time are often indicative of reduced ribosome abundance, this phenomenon is undesirable for CFPS chassis strains (Bremer and Dennis, 1996; Zawada and Swartz, 2006). Another common scheme for 1 gene introduction is via lysogenization with synthetic DE3 bacteriophage (as in BL21(DE3) and its derivatives) (Studier and Moffatt, 1986), but as phage insertion occurs site-specifically at a fixed genomic locus and the viral 1 gene is under the control of a fixed set of cis-regulatory sequences, this approach suffers from a lack of tunability and control. An attractive alternative method for genomic integration is λHR, which site-specifically integrates linear DNA constructs into target genomes using flanking-sequence homology to direct insertion at the desired site (Datsenko and Wanner, 2000; Mosberg et al., 2010). As C321.ΔA.759 natively expresses the requisite λ-Red recombination machinery (Lajoie et al., 2013; Martin et al., 2018), we elected to proceed via λHR.
Several design criteria were considered for genome integration. First, a challenge in protein expression is tuning the expression level: enough protein must be synthesized to adequately perform the desired function, but aggressive overexpression can place too high a metabolic burden on the host organism and/or lead to production of inhibitory levels of the protein. Lacking a priori knowledge as to how to achieve an ideal level of T7RNAP production in C321.ΔA.759, we decided to test a variety of different expression levels. We designed a series of synthetic constructs that placed the 1 gene under the regulation of IPTG-inducible promoters of varying transcriptional strengths, with lacUV5 (Stefano and Gralla, 1979), PtacI (de Boer et al., 1983), and Lpp5 (Inouye and Inouye, 1985) representing relatively low, medium, and high strength, respectively (Figure 2B). Promoter-specific synthetic ribosome binding sites (RBSs) designed for maximal translation using the Ribosome Binding Site Calculator v2.0 were employed for the regulation of translation initiation. In this way, any differences in T7RNAP expression between strains could be predominantly attributed to differences in transcription (Espah Borujeni et al., 2014; Salis et al., 2009). Second, in the interest of easy visualization via western blotting, we added a 6-His tag to the N terminus of the polymerase (a modification that had previously been suggested to have little to no effect on polymerase activity [Ellinger and Ehricht, 1998]). Third, each construct also included the kanamycin kinase (kanR) gene from pKD4 (Datsenko and Wanner, 2000) (which confers resistance to the antibiotic kanamycin) for selection of successful integrants. Finally, to explore influences of genome position on expression, we designed each construct with 50 bp of flanking-sequence homology at each end to facilitate integration at one of two genomic loci: the asl locus, selected because it was previously identified as a highly expressing locus in the E. coli genome (Bryant et al., 2014), and the int locus, selected because it is analogous to the DE3 lysogenization site in BL21(DE3) (Studier and Moffatt, 1986).
A total of six T7RNAP-expressing constructs were assembled (int.lacUV5, int.PtacI, int.Lpp5, asl.lacUV5, asl.PtacI, and asl.Lpp5) and transformed individually into C321.ΔA.759 for site-specific genomic integration. Potential integrants were identified by the ability to survive in the presence of kanamycin and verified via screening by multiplex allele-specific colony (MASC) PCR (Figure 2C). Sanger sequencing of all insert loci confirmed that each construct was integrated at the correct locus, fully intact and free of any unwanted mutations. Finally, western blotting with antibodies against the polymerase’s N-terminal 6-His tag verified that each insert was indeed promoting expression of T7RNAP (Figure 2D). Polymerase expression as determined by western blot band intensity tracked as expected with promoter strength.
Characterization of T7RNAP-Expressing Strains in CFPS
To assess the ability of these strains to independently catalyze T7RNAP-dependent transcription in CFPS, we prepared crude S12 lysates from all six strains for use in cell-free reactions. To promote robust T7RNAP overexpression, we induced all strains with 1 mM IPTG during exponential cell growth. Batch CFPS reactions using each lysate were directed to synthesize sfGFP over 20 h at 30°C both with and without addition of 16 μg/mL of purified T7RNAP to the reactions (Figure 3A). With polymerase supplemented, lysates from all six strains performed within 15% of one another. The strains featuring PtacI- and Lpp5-driven T7RNAP expression demonstrated the ability to perform transcription using only the polymerase expressed by the chassis strain. Not surprisingly, the amount of sfGFP fluorescence appears to be related to the amount of T7RNAP produced in the cells (Figure 3A). At both insertion loci, the amount of fluorescence increases with increasing promoter strength, and for each promoter more fluorescence was observed from the strains featuring inserts at the highly expressing asl locus. The strain capable of generating the most sfGFP fluorescence without T7RNAP supplementation, C321.ΔA.759.asl.Lpp5, achieved ~85% as much sfGFP production without supplementation as with. This strain, hereafter referred to as C321.ΔA.759.T7, was selected for further characterization and development.
Figure 3. Characterization of C321.ΔA.759 T7RNAP-Expressing Variants.
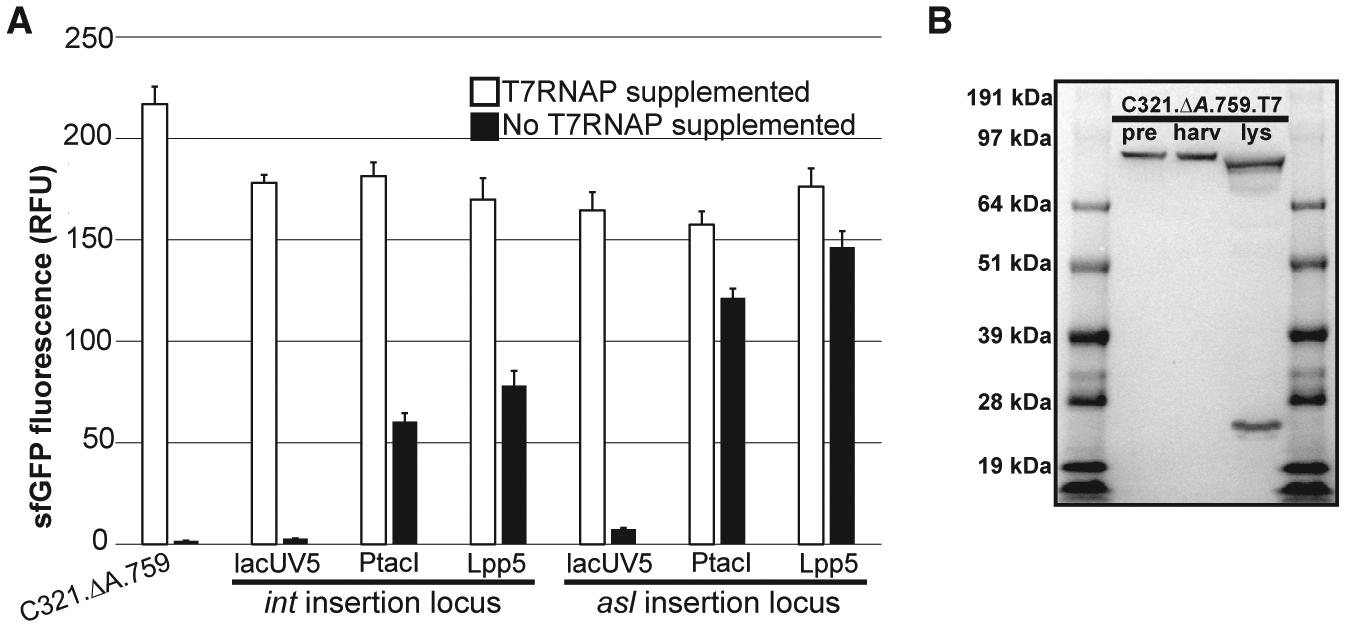
(A) Characterization of the six C321.ΔA.759 T7RNAP-expressing variants generated in this study. Extracts derived from C321.ΔA.759 and its T7RNAP-expressing derivatives were directed to synthesize sfGFP in CFPS both with and without supplementation with purified T7RNAP, and fluorescence was measured after incubation for 20 h at 30°C. Three independent CFPS reactions were performed for each condition, and one standard deviation is shown.
(B) α-His western blot characterization of C321.ΔA.759.T7. pre: samples derived from cells immediately prior to induction. harv: samples derived from mid-exponential phase cells immediately prior to harvest. lys: samples derived from final clarified lysate.
Curiously, western blot analysis of samples derived from C321.ΔA.759.T7 revealed that the T7RNAP produced by the strain is cleaved during lysis near the N terminus to yield a ~21-kDa fragment (Figure 3B). This cleavage is well documented in the literature (Davanloo et al., 1984; Tabor and Richardson, 1985), and previous work has identified the membrane-bound periplasmic protease OmpT as the responsible agent in E. coli (Grodberg and Dunn, 1988). The cleaved polymerase itself has been heavily characterized, and prior work has concluded that the nicked enzyme is impaired by a loss in polymerase activity and efficiency (Ikeda and Richardson, 1987a, 1987b; Muller et al., 1988; Tabor and Richardson, 1985). Thus, we reasoned that OmpT-mediated proteolysis of the T7RNAP expressed by C321.ΔA.759.T7 during cell lysis (when the periplasm and cytoplasm mix) contributed to the reduced capacity of the resulting lysates to support transcription independent of supplemental T7RNAP. To assess this hypothesis, we next sought to inactivate this OmpT activity to protect T7RNAP from proteolysis.
ompT Inactivation to Protect T7RNAP during C321.ΔA.759.T7 Crude Lysate Preparation
BL21(DE3) and its derivative strains all feature a deletion at the ompT locus, which presumably prevents the proteolytic degradation of the T7RNAP produced by those strains (Gottesman, 1996). Based on this, we hypothesized that a deletion at the ompT locus of C321.ΔA.759.T7 would similarly protect strain-synthesized T7RNAP and thus eliminate the strain’s partial dependence on supplemental polymerase in CFPS. To test this, we first “looped” kanR out of the C321.ΔA.759.T7 genome using MAGE (Wang et al., 2009). Next, we applied λHR to replace a ~12-kbp region of the C321.ΔA.759.T7 genome analogous to the spontaneous ompT deletion in BL21(DE3) with a kanR cassette to select for successful integrants. MASC PCR verified the knockout, yielding strain C321.ΔA.759.T7.ΔompT.
To assess the CFPS capabilities of the ompT-deficient strain, we prepared crude S12 extracts from culture induced with 1 mM IPTG for analysis via both western blot and batch CFPS reactions. As expected, a western blot revealed that in the absence of OmpT the T7RNAP is no longer cleaved (i.e., we did not observe the expected 21-kDa band) (Figure 4A, inset). Unfortunately, batch sfGFP CFPS reactions demonstrated that the strain’s ability to perform CFPS suffered significantly overall in response to the ompT knockout (Figure 4A). Compared with C321.Δ3A.759.T7 lysate, C321.ΔA.759.T7.ΔompT lysates show a 2- to 3-fold reduction in CFPS yields both with and without T7RNAP supplementation. This is consistent with earlier work demonstrating that functional OmpT is critical for robust protein synthesis in lysates derived from C321.ΔA and its descendants (Martin et al., 2018). Given our interest in designing a one-pot, high-yield CFPS system, we concluded that this was not a viable strategy for improving C321.ΔA.759.T7 and discontinued our pursuit of this scheme for preventing T7RNAP cleavage during cell lysis.
Figure 4. Engineering an OmpT-Resistant T7 Polymerase.
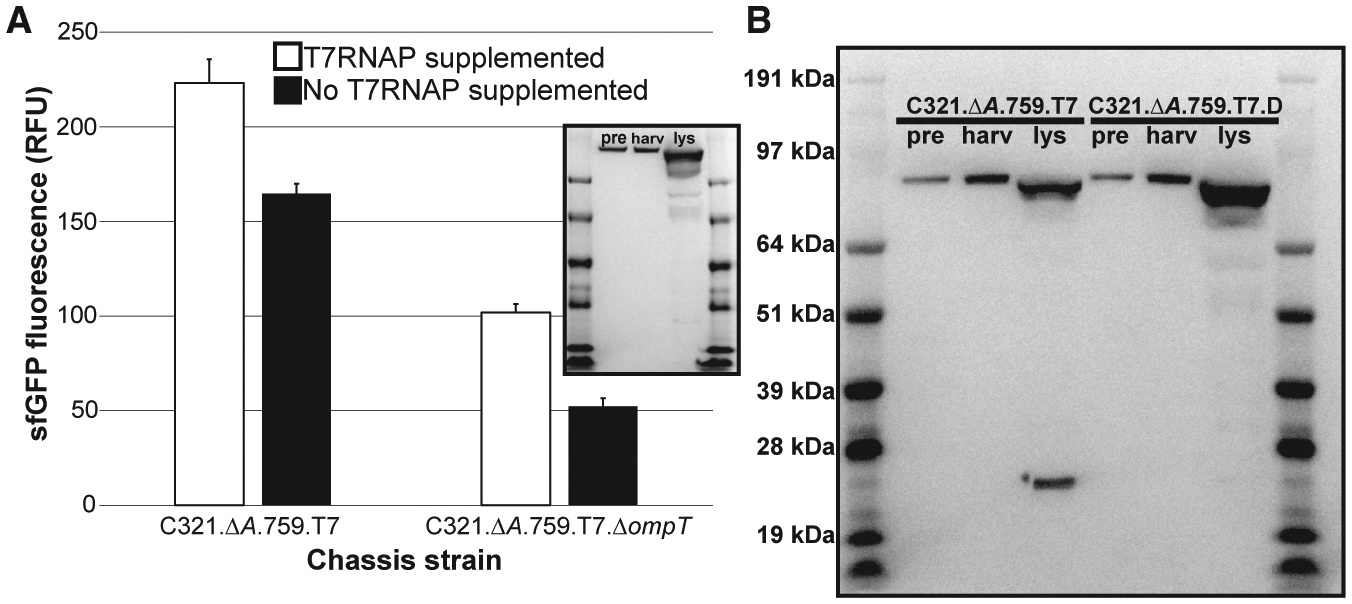
(A) Characterization of C321.ΔA.759.T7 and C321.ΔA.759.T7.DompT. Extracts from each strain were directed to synthesize sfGFP inCFPSboth with and without supplementation with purified T7RNAP, and fluorescence was measured after incubation for 20 h at 30°C. Three independent CFPS reactions were performed for each condition, and one standard deviation is shown. Inset: α-His western blot characterization of C321.ΔA.759.T7.DompT.
(B) α-His western blot comparison of C321. ΔA.759.T7 and C321.ΔA.759.T7.D. pre: samples derived from cells immediately prior to induction. harv: samples derived from mid-exponential phase cells immediately prior to harvest. lys: samples derived from final clarified lysate.
See also Figure S1.
Engineering a Protease-Resistant T7RNAP
We next considered a chemical biology approach to protecting the T7RNAP produced by C321.ΔA.759.T7 from proteolysis during lysate preparation. We reasoned that since the source of the degradation could not be removed without deleterious effects on the strain’s productivity in vitro, perhaps the T7RNAP could be mutated such that it would no longer be an efficient substrate for OmpT. OmpT binds its substrates at pairs of adjacent basic residues and catalyzes hydrolysis of the amide bond linking them (Hwang et al., 2007). The requirement of basic residues for OmpT activity at the cleavage site is fairly rigid—in particular, the 1′ residue residing immediately upstream of the polypeptide cut site must be basic in order for OmpT to facilitate hydrolysis (Hwang et al., 2007). In T7RNAP, two such sites have been identified proximal to the enzyme’s N terminus at K172/R173 (Ikeda and Richardson, 1987a) (K183/R184 in His-tagged mutant polymerase) and K179/K180 (Muller et al., 1988) (K190/K191 in His-tagged mutant polymerase). These sites are relatively close together such that OmpT proteolysis at either would liberate a ~21-kDa N-terminal fragment, consistent with what was observed on our C321.ΔA.759.T7 western blot.
Because K183/R184 was previously identified as the primary site of OmpT activity in T7RNAP (Ikeda and Richardson, 1987a), we hypothesized that mutating K183 to a non-basic residue would abolish the target site and thus protect the polymerase from proteolysis despite the presence of fully functional OmpT in the lysate. To test this, we used MAGE to edit the sequence of the 1 gene on the genome of C321.ΔA.759.T7 to mutate K183 to either glycine or leucine, as these mutants had previously been shown to retain robust polymerase activity (Tunitskaya and Kochetkov, 2002). Mutations were detected using allele-specific primers in MASC PCR and confirmed by Sanger sequencing. When extracts prepared from each mutant strain were directed to synthesize sfGFP in batch CFPS reactions both with and without supplemental T7RNAP, neither performed better than C321.ΔA.759.T7 (Figure S1A). Analysis of the extracts revealed that despite the installed mutations, the polymerase was still being cleaved during cell lysis (Figure S1B).
Next, we reasoned that while the K183/R184 site may be the preferential site for proteolysis when both sites are present, when this site is unavailable OmpT may simply cleave at K190/K191 instead. We hypothesized that the simultaneous elimination of both sites may be necessary to fully prevent the ability of OmpT to bind and cleave the polymerase. To test this, we again exploited MAGE to edit the sequence of 1 on the C321.ΔA.759.T7 genome and install the mutations K183G and K190L. Mutations were detected using allele-specific primers in MASC PCR and confirmed via Sanger sequencing. We prepared crude cell lysates from the resulting strain, C321.ΔA.759.T7.D, for western blot and CFPS analysis. The western blot revealed that the double mutant T7RNAP expressed by C321.ΔA.759.T7.D is not cleaved despite the presence of active OmpT in the cellular lysate (Figure 4B). In batch mode CFPS reactions, C321.ΔA.759.T7.D lysates exhibit a ~15% increase in productivity over C321.ΔA.759.T7, producing ~2.2 g/L and ~1.6 g/L of sfGFP with and without T7RNAP supplementation, respectively (Figure 5).
Figure 5. 759.T7.Opt Is a Highly Productive, One-Pot CFPS System.
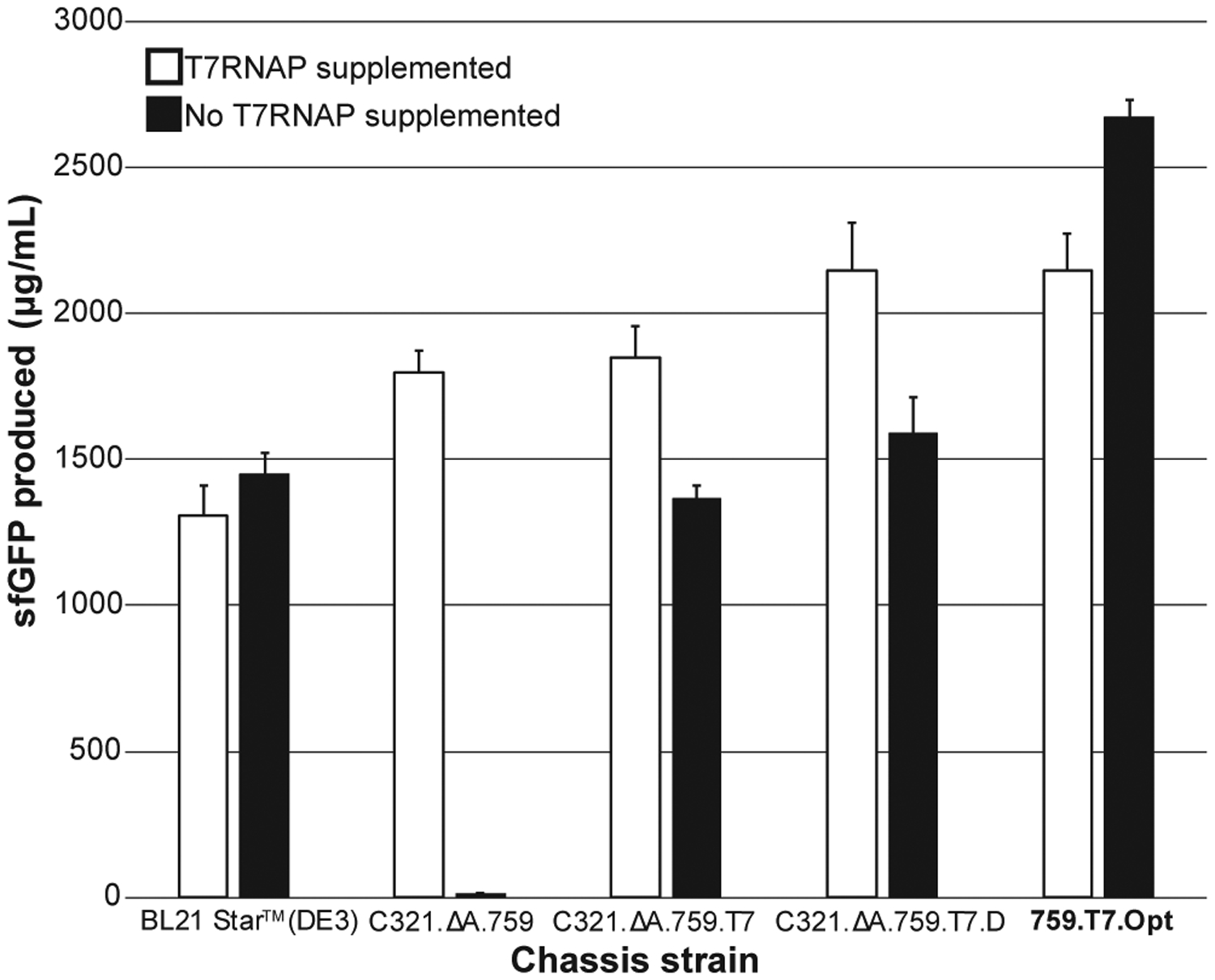
A side-by-side comparison of sfGFP produced in CFPS using crude lysates derived from IPTG-induced BL21 Star (DE3), C321.ΔA.759, C321.ΔA.759.T7, C321.ΔA.759.T7.D, and 759.T7.Opt cells. Shown are results from CFPS reactions performed both with and without supplementation with purified T7RNAP. At least three independent reactions were performed per condition, and one standard deviation is shown.
Optimization of Endogenous T7RNAP Productivity
Noting that CFPS reactions using C321.ΔA.759.T7.D lysates were still partially dependent on supplementation with purified T7RNAP to achieve maximal productivity, we next sought to identify and address the fundamental feature limiting the productivity of the T7RNAP expressed endogenously by the strain. Because the presence of an associated His tag has been shown to impair the function of other recombinantly expressed proteins (Ledent et al., 1997; Sabaty et al., 2013), we hypothesized that the N-terminal tag used throughout this effort as a way of detecting T7RNAP on blots could be reducing the productivity of the T7RNAP expressed by C321.ΔA.759.T7.D, thus causing lysates derived from the strain to be transcription limited. To test this, we applied a combination of CRISPR/Cas9 and MAGE (Ronda et al., 2016) to further edit the sequence of the 1 gene on the genome of C321.ΔA.759.T7.D, removing the His tag from the N terminus of the polymerase to yield strain C321.ΔA.759.T7.D.ΔHis (hereafter referred to simply as 759.T7.Opt). Batch CFPS reactions using 759.T7.Opt demonstrated that, consistent with our hypothesis, lysates derived from this strain no longer benefit from the supplementation of purified polymerase; indeed, in the absence of additional polymerase the system yields ~2.7 g/L of sfGFP, dropping to ~2.1 g/L when the enzyme is supplemented (Figure 5), an observation we do not fully understand. 759.T7.Opt lysates also significantly outperform those derived from BL21 Star (DE3), C321.ΔA.759, and C321.ΔA.759.T7.D regardless of T7RNAP supplementation, establishing 759.T7.Opt as a robust one-pot CFPS system and one of the most productive batch CFPS platforms developed to date.
Demonstration of Capacity for Multiple ncAA Incorporations Using T7RNAP-Expressing Strains
With 759.T7.Opt lysates in hand, we next assessed the capacity of these lysates to produce proteins featuring ncAAs. Because the parent strain had RF1 removed (Lajoie et al., 2013), we expected that ncAA incorporation via amber suppression would be highly efficient—indeed, our previous effort using a recoded strain showed up to 40 ncAA incorporations into a single polypeptide (Martin et al., 2018). To assess ncAA incorporation into proteins, we transformed both 759.T7.Opt and BL21 Star (DE3) with a pEVOL plasmid encoding the OTS components for the ncAA p-acetyl-L-phenylalanine (pAcF) (Young et al., 2010); namely, the pAcF-specific aminoacyl-tRNA synthetase (pAcFRS) and an orthogonal suppressor tRNA engineered to decode the amber codon (o-tRNA) (Wang et al., 2003). We then quantitatively assessed the incorporation of pAcF into sfGFP variants with up to five in-frame amber codons. CFPS reactions were supplemented with additional OTS components based on our previous work (Martin et al., 2018).
As an initial demonstration of ncAA incorporation, we directed the lysates derived from these pEVOL-bearing strains to synthesize amber mutant variants of sfGFP. We first established the optimal concentrations of pAcF OTS components to be supplied to these CFPS reactions via a series of CFPS reactions directed to synthesize an sfGFP variant featuring two amber codons (sfGFP-2UAG) (Figure S2). Using these conditions, the 759.T7.Opt and BL21 Star (DE3) pEVOL-pAcF lysates were used in CFPS to synthesize wild-type sfGFP (sfGFPwt), sfGFP with a single amber codon (sfGFP-T216X), sfGFP with two amber codons (sfGFP-2UAG), or sfGFP with five amber codons (sfGFP-5UAG). Reactions were performed without supplementation with purified T7RNAP (Figure 6A). As expected, the RF1-deficient 759.T7.Opt lysates exhibit a significantly higher capacity for pAcF incorporation than the BL21 Star (DE3) lysates, with the difference becoming more pronounced as the number of pAcF incorporations increases. Indeed, 759.T7.Opt yields 781 ± 33 μg/mL of sfGFP-5UAG, a more than 5-fold improvement over BL21 Star (DE3). 759.T7.Opt lysates remain highly productive for sfGFP variants bearing up to two pAcFs, yielding >2.1 g/L for sfGFPwt, sfGFP-T216X, and sfGFP-2UAG. To assess the degree of pAcF incorporation in these samples, we performed high-resolution top-down mass spectrometry (i.e., mass spectrometric analysis of whole proteins) (Figure 6B). The results clearly indicate the mass shifts associated with the incorporation of one, two, and five pAcF residues. Analysis of these data shows that site-specific incorporation of pAcF was ≥90% in all samples, with ≤2 ppm difference between experimental and theoretical masses for sfGFPwt, sfGFP-T216X, and sfGFP-2UAG, and ~16 ppm difference for sfGFP-5UAG. These results confirm that the dominant species produced in each reaction features a pAcF at every amber codon.
Figure 6. One-Pot ncAA Incorporation into sfGFP Using 759.T7.Opt Lysates.
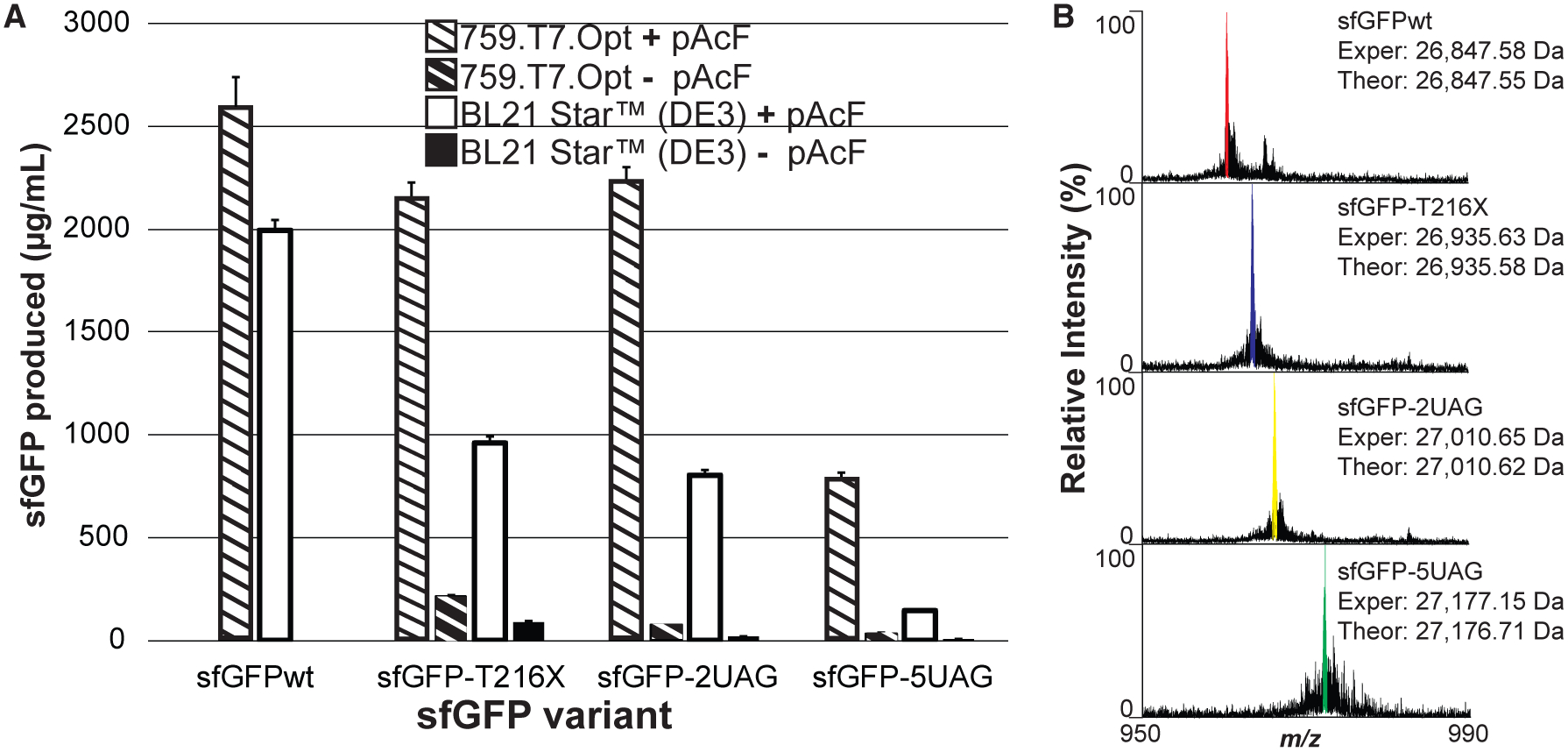
(A) sfGFP produced in vitro from cell extracts derived from induced BL21 Star (DE3) cells as well as 759.T7.Opt cells, without supplementation with purified T7RNAP. The indicated sfGFP amber mutant variants were synthesized in the presence of the complete pAcF OTS. At least three independent reactions were performed per condition, and one standard deviation is shown.
(B) Mass spectra of the +28 charge states of the indicated sfGFP variants obtained by top-down mass spectrometry illustrating site-specific incorporation of one to several pAcF residues. Deconvoluted experimental (Exper) and theoretical (Theor) monoisotopic mass values for each variant are listed. Major peaks are highlighted in each spectrum with color.
See also Figure S2.
We next explored the synthesis of large polypeptides containing multiple identical ncAAs utilizing our one-pot CFPS platform derived from 759.T7.Opt. For our model protein, we sought to produce elastin-like polymers (ELPs) (Martin et al., 2018) containing ncAAs using 759.T7.Opt lysates. ELPs are biocompatible and stimuli-responsive biopolymers that can be applied for drug delivery and tissue engineering (Despanie et al., 2016; Raucher and Ryu, 2015). Previously, we have introduced multiple, identical ncAAs into ELPs by substituting natural amino acids with ncAAs at a guest position in the repeating pentapeptide unit (VPGVG) that can be modified while maintaining ELP structure and function (Amiram et al., 2015; Martin et al., 2018).
Our ELP construct consisted of three pentapeptide repeats per monomer unit with a single valine codon per monomer changed to UAG in amber mutants (Martin et al., 2018) (Figure 7A). Lysates derived from 759.T7.Opt bearing pEVOL-pAcF were directed in the absence of supplemental purified T7RNAP to synthesize wild-type (ELP-WT) and amber mutant (ELP-UAG) ELPs with 20, 30, and 40 monomer units in the presence of pAcF. Products were visualized using an autoradiogram, demonstrating that a high percentage of the protein produced in the presence of the ncAA is full length. Full-length ELP-UAG protein was no longer observed when reactions were performed without the addition of pAcF (Figure 7C). Absolute yields for each of the various ELPs were quantified via [14C]-glycine radioactive scintillation counting (Figure 7B). The lysates generated ~55–70 mg/L of each ELP-WT. For each of the ELP-UAG constructs, the 759.T7.Opt lysates yielded >65 mg/L of product operating in one-pot mode. Of note, we observe some read-through of the UAG codon in the absence of pAcF. This is likely a result of near-cognate suppression via incorporation of natural amino acids, as we and others have observed before (Aerni et al., 2015; Lajoie et al., 2013; Oza et al., 2015). To verify pAcF incorporation in the ELP products, we performed top-down mass spectrometry on intact ELPs to characterize the efficiency of multi-site pAcF incorporation. This analysis confirmed that for 20-, 30-, and 40-mer ELP-UAG constructs, the main products feature pAcF incorporations at all amber codons (Figures 7D–7F, S3, and S4). For example, we observe that site-specific incorporation of pAcF was ≥90% in the 20-mer ELP-UAG construct. Taken together, these results demonstrate that 759.T7.Opt lysates are capable of catalyzing the production of proteins bearing multiple ncAAs independent of supplementation with T7RNAP.
Figure 7. Multi-Site One-Pot ncAA Incorporation Using 759.T7.Opt Lysates.
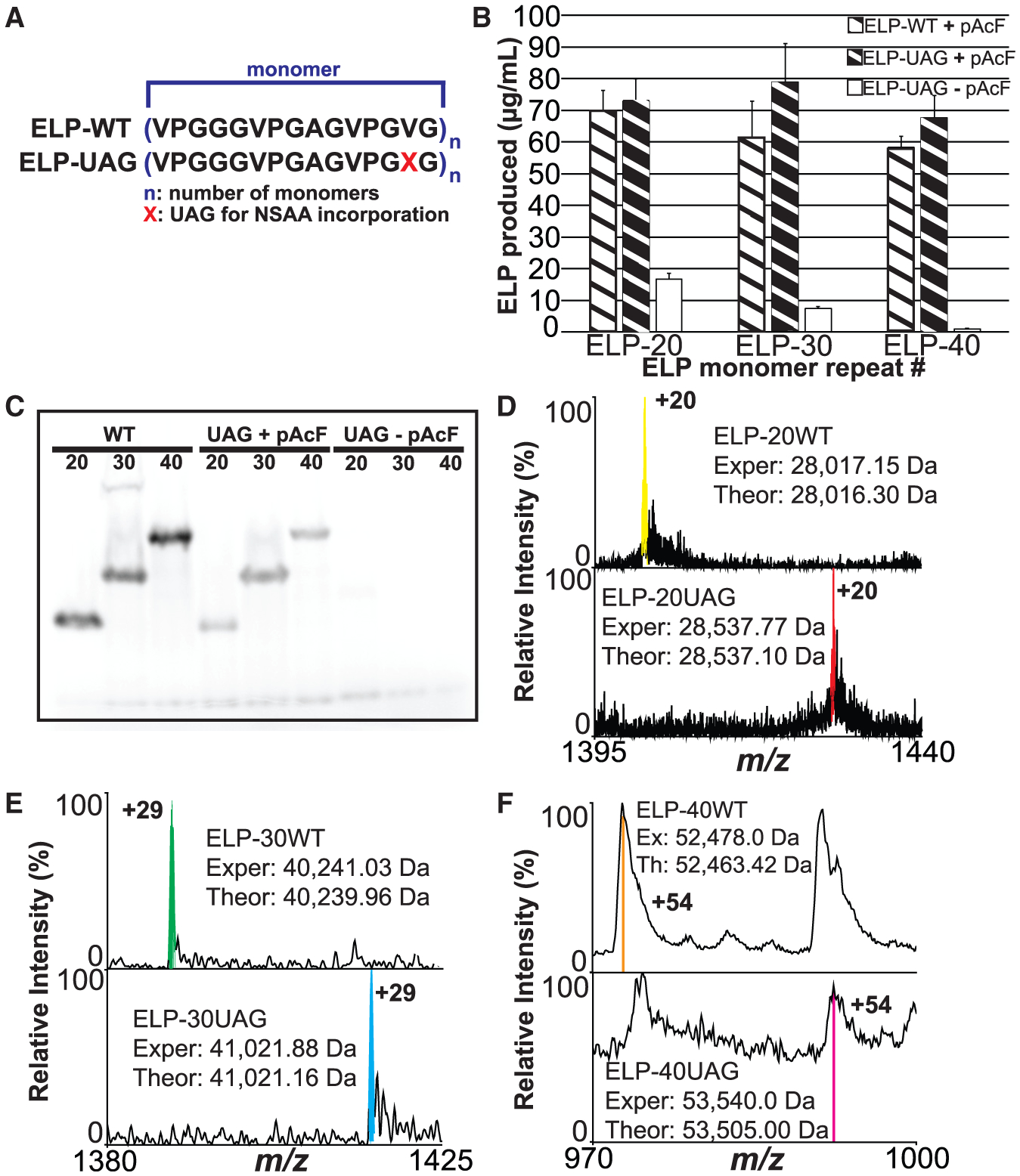
(A) Illustration of both the wild-type (WT) and amber mutant (UAG) ELP monomers used in this study. NSAA, non-standard amino acid.
(B) [14C]Glycine radioactive count quantification of ELPmers produced by 759.T7.Opt lysates under the indicated conditions. At least three independent reactions were performed per condition, and one standard deviation is shown.
(C) Autoradiogram of ELPmers produced by 759.T7.Opt lysates under the indicated reaction conditions.
(D–F) Mass spectra of the indicated charge states for (D) ELP-20, (E) ELP-30, and (F) ELP-40 constructs. Each set of spectra indicates that 759.T7.Opt lysates are catalyzing the site-specific incorporation of the indicated number of pAcF residues. Deconvoluted experimental (Exper, Ex) and theoretical (Theor, Th) average mass values for each variant are listed. Major peaks are highlighted in each spectrum with color. Reactions for all figure panels were performed without supplementation with purified T7RNAP. (D) displays high-resolution data, (E) displays mid-resolution data, and (F) displays low-resolution data. Resolution was constrained by instrumentation limits based on the increasing size of the targets.
See also Figures S3 and S4.
DISCUSSION
One-pot systems, such as those derived from the state-of-theart protein overexpression strain BL21 Star (DE3), are highly desirable for CFPS due to their enrichment with critical enzymes such as T7RNAP. In the specific case of T7RNAP, such systems reduce the cost of CFPS and make the system easier to put together. While robust and versatile, existing one-pot platforms based on BL21 Star (DE3) struggle with the production of proteins containing multiple ncAAs due to the competitive action of RF1 in the reaction environment. In this study, we describe the generation and utilization of a highly productive one-pot CFPS platform beginning with C321.ΔA.759, a genomically recoded RF1-deficient strain that was previously optimized for CFPS. We integrated a series of DNA constructs into the genome of C321.ΔA.759, each of which featured the T7RNAP-encoding gene 1 under the control of one of three different promoter sequences of varying potency. The construct featuring 1 regulated by strong promoter Lpp5 integrated at a previously identified high-expression genomic locus asl (Bryant et al., 2014) yielded C321.ΔA.759.T7, which was capable of supporting in vitro transcription independent of supplementation with purified T7RNAP. When used in CFPS, C321.ΔA.759.T7 lysates yielded 85% as much sfGFP without T7RNAP supplementation as with supplementation. In an effort to address the continuing partial dependence of the system on polymerase supplementation, we explored different strategies to protect the T7RNAP expressed in C321.ΔA.759.T7 from OmpT-mediated proteolysis during lysate preparation. By mutating two lysine residues proximal to the N terminus of 1, we were able to abolish the putative OmpT target sites and establish an OmpT-resistant mutant version of T7RNAP in strain C321.ΔA.759.T7.D. We finally removed the N-terminal His tag from 1 in C321.ΔA.759.T7.D to yield strain 759.T7.Opt. 759.T7.Opt lysates compose a one-pot system, yielding ~2.7 g/L sfGFP in batch mode reactions without supplementation with purified biological components. We also demonstrated the merits of RF1-deficient systems for ncAA incorporation, highlighting the significantly increased capacity for amber suppression in 759.T7.Opt lysates as compared with BL21 Star (DE3). Furthermore, we were able to confirm the synthesis of full-length polypeptides containing up to 40 ncAAs without the addition of purified T7RNAP using 759.T7.Opt lysates. This joins an emerging number of reports pushing the bounds of multiple, identical ncAA incorporations using genomically recoded organisms (Mukai et al., 2015).
Looking forward, one intriguing avenue is the continued development of 759.T7.Opt for improved productivity and enhanced functionality in CFPS. This might be achieved by correcting some of the potentially harmful off-target mutations incurred during the initial recoding of the strain. Additionally, upregulation of other positive effectors of CFPS (e.g., chaperones, elongation factors, energy regeneration enzymes) could be achieved via genomic integration using a strategy similar to that employed in this study. In particular, the development of orthogonal synthetases with improved kinetics and substrate specificity is a critical hurdle that must be overcome before these enzymes can be overexpressed in CFPS chassis strains without deleterious effects on cellular health and lysate performance (Nehring et al., 2012).
Developing efficient CFPS systems specialized for ncAA incorporation is important for synthetic biology for various emerging applications. Powerful one-pot production platforms will support the large-scale synthesis of protein products featuring novel structures and functions, in turn promoting the mass production of potent therapeutics and materials. We anticipate that CFPS platforms such as that derived from 759.T7.Opt are promising for these and other synthetic biology applications.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Plasmids and strains generated in this study are available to interested parties pending a Materials Transfer Agreement (MTA). Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael C. Jewett (m-jewett@northwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe Strains
Escherichia coli Strains
BL21 Star™(DE3): grow in Luria-Bertoni media at 37°C
DH5α: grow in Luria-Bertoni media at 37°C
C321.ΔA.759: E. coli K-strain derivative in which all endogenous amber stop codons (TAG) have been recoded to TAA and release factor 1 has been deleted (ΔA). Strain also features functional deactivations of the following genes: endA, gor, rne, mazF. Grow in Luria-Bertoni media at 34°C, select with 100 μg/mL ampicillin or carbenicillin.
759.T7.int.lacUV5: C321.ΔA.759 with a synthetic construct encoding T7 RNA polymerase under control of the lacUV5 promoter inserted into the genome at the int locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.int.PtacI: C321.ΔA.759 with a synthetic construct encoding T7 RNA polymerase under control of the PtacI promoter inserted into the genome at the int locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.int.Lpp5: C321.ΔA.759 with a synthetic construct encoding T7 RNA polymerase under control of the Lpp5 promoter inserted into the genome at the int locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.asl.lacUV5: C321.ΔA.759 with a synthetic construct encoding T7 RNA polymerase under control of the lacUV5 promoter inserted into the genome at the asl locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.asl.PtacI: C321.ΔA.759 with a synthetic construct encoding T7 RNA polymerase under control of the PtacI promoter inserted into the genome at the asl locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.asl.Lpp5: C321.ΔA.759 with a synthetic construct encoding T7 RNA polymerase under control of the Lpp5 promoter inserted into the genome at the asl locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.ΔkanR: 759.T7.asl.Lpp5 with kanamycin resistance gene removed. Grow in Luria-Bertoni media at 34°C, select with 100 μg/mL ampicillin or carbenicillin.
759.T7.ΔompT: 759.T7.asl.Lpp5 with deletion at genomic ompT locus. Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.K172L: 759.T7.asl.Lpp5 with a K172L mutation installed in the strain’s genomic 1 gene (encoding T7 RNA polymerase). Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.K172G: 759.T7.asl.Lpp5 with a K172G mutation installed in the strain’s genomic 1 gene (encoding T7 RNA polymerase). Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.D: 759.T7.asl.Lpp5 with mutations K172G and K179A installed in the strain’s genomic 1 gene (encoding T7 RNA polymerase). Grow in Luria-Bertoni media at 34°C, select with 50 μg/mL kanamycin.
759.T7.D.ΔAbR: 759.T7.D with resistances to ampicillin/carbenicillin and kanamycin removed. Grow in Luria-Bertoni media at 34°C.
759.T7.Opt: 759.T7.D.ΔAbR with the N-terminal 6xHistag removed from the strain’s genomic 1 gene (encoding T7 RNA polymerase). Grow in Luria-Bertoni media at 34°C. Strain is not resistant to ampicillin/carbenicillin or kanamycin.
Cell lines produced in this study have not been authenticated.
METHOD DETAILS
Strains and Plasmids
The bacterial strains and plasmids used in this study are listed in Table S2. Carbenicillin (50 μg/mL) was used for culturing C321.ΔA.759 and to maintain plasmid pMA7CR_2.0, kanamycin (50 μg/mL) was used for culturing C321.ΔA.759 T7RNAP linear insert transformants and maintaining pY71/pJL1/pMAZ-based plasmids, and chloramphenicol (34 μg/mL) was used to maintain the pEVOL-pAcF plasmid.
PCR Reactions
Reactions using purified DNA species as template were performed using Phusion™ polymerase along with dNTP solution mix and Phusion™ HF Buffer (all from New England Biolabs, Ipswich, MA). Reactions from cellular genomic DNA (colony PCR, multiplex allele-specific colony (MASC) PCR) were performed using 2x colony PCR master mix (Thermo Fisher Scientific, Inc., Waltham, MA).
Plasmid DNA Purification
For applications requiring small amounts of plasmid DNA (PCR, cloning, moving between strains, etc.) plasmid DNA was purified from cells using E.Z.N.A.® Plasmid Mini Kit (Omega Bio-tek, Norcross, GA). For applications requiring large amounts of plasmid DNA (templating CFPS reactions) plasmid DNA was purified from cells using Hi-Speed® Plasmid Maxi Kit (Qiagen, Venlo, The Netherlands).
DNA Gel Electrophoresis
Unless otherwise stated, all DNA electrophoresis was done in 1% agarose gels stained with SYBR® Safe (Thermo Fisher Scientific, Inc., Waltham, MA). Samples were run at 100V for 30–60 minutes on a Mini Gel II Complete Electrophoresis System (VWR, Radnor, PA). 100 bp and 1 kb Quick-load® DNA Ladders (New England Biolabs, Ipswich, MA) were used for fragment size reference.
Plasmid Assembly Using Gibson Assembly
To assemble plasmids from linear DNA pieces, throughout this work we use the method of Gibson et al.(Gibson et al., 2009). Individual pieces were synthesized by PCR such that adjacent fragments had ~20 bp of flanking homology with each neighboring fragment. Fragments were assembled by incubating 50 ng of each fragment with assembly mix (6.7 mM PEG-8000, 107 mM Tris-HCL pH 7.5, 10.7 mM magnesium chloride, 213 μM dATP, 213 μM dGTP, 213 μM dCTP, 213 μM dTTP, 10.7 mM dithiothreitol, 1 mM nicotinamide adenine dinucleotide, 0.0043U/μL T5 exonuclease (New England Biolabs, Ipswich, MA), 4.3 U/μL Taq ligase (New England Biolabs, Ipswich, MA), and 0.023 U/μL Phusion polymerase (New England Biolabs, Ipswich, MA)) for 1 hr at 50°C. Following this incubation, 2 μL of reaction volume were transformed into electrocompetent DH5α cells using a Micropulse electroporator (Bio-Rad, Hercules, CA). Transformed cells were recovered for 1 hr in LB media at 37°C and 250 rpm. 100 μL of recovered culture was spread onto selective plates and put at 37°C overnight. Resulting colonies contained the assembled plasmid of interest.
T7RNAP Linear Insert Construction
The six T7RNAP-encoding inserts used in this study were assembled from PCR products obtained using primers listed in Table S1. The insert loci were at coordinates 3,986,255 (asl) and 805,473 (int) in the genome of C321.ΔA.759. In brief, each insert was assembled from four segments of linear DNA – a promoter segment featuring 50 base pairs of sequence homology to the genome of C321.ΔA.759 upstream of the targeted insert site, a T7RNAP segment containing the 1 gene, a terminator segment encoding the synthetic terminator sequence L3S2P21(Chen et al., 2013), and a kanR segment featuring the kanamycin resistance cassette from pKD4(Datsenko and Wanner, 2000) as well as 50 bp of sequence homology to the genome of C321.ΔA.759 downstream of the targeted insert site. All constructs were designed such that the coding strand would be integrated into the leading strand during genome replication. Adjacent segments featured at least 20 bp of sequence homology to one another to facilitate their assembly into a single unit of DNA, and novel sequence elements (e.g. 6His-tag and synthetic RBS sequences) were built into the 5’ tails of primers. Inserts featuring the promoter Lpp5 were assembled via overlap assembly PCR (SOEing)(Horton, 1995) and amplified with end primers. End primer PCR reactions for these species generated a large number of off-target sequences, so full-length insert DNA was separated from other products via electrophoresis and extracted from 1% agarose gel using a DNA gel extraction kit (Product No. D2500; Omega Bio-tek, Norcross, GA). Inserts featuring promoters PtacI and lacUV5 were assembled together with plasmid origins of replication (p15a and pUC, respectively) via Gibson assembly (described above) to yield plasmid DNA, and these plasmids were used as template with end primers to yield the linear insert DNA via PCR. We found that the PCR products generated using this approach had a significant reduction in the prevalence of off products observed for the other inserts, accelerating our workflow.
Strain Transformation and Insert Verification
The T7RNAP cassettes were inserted into the genome of C321.ΔA.759 via λHR following the protocol of Datsenko and Wanner (Datsenko and Wanner, 2000). In brief, for each cassette a 5 mL culture of C321.ΔA.759 was grown in LB media (10 g/L tryptone, 5 g/L yeast extract and 10 g/L NaCl)(Wang and Church, 2011) to an OD600 of 0.6, after which it was incubated at 42°C for 15 min. 1.5 mL of this culture was washed twice in ice cold, sterile nuclease-free water and resuspended in 30 μL of insert DNA at a concentration of 70 μg/mL. The cell suspension was transferred to a 2 mL electroporation cuvette and DNA was introduced into cells using a Micropulse electroporator (Bio-Rad, Hercules, CA). Immediately following electroporation, cells were resuspended in 1 mL sterile LB media and recovered for 3 hrs at 34°C at 250 rpm. The recovered cell culture was plated on kanamycin selective plates and permitted to grow overnight at 34°C. The following day, colonies to be screened were picked and inoculated into 100 μL of kanamycin media on a 96-well plate (Costar 3370; Corning, Corning, NY) and cultured for 3 hrs at 34°C at 250 rpm. 1 μL of each miniature culture was used as template to detect successful genomic integration of each insert by colony PCR using Colony PCR Master Mix (Thermo Fisher Scientific, Inc., Waltham, MA) with primers listed in Table S1. Two primer pairs were used for detection such that if no insert was present, the outermost pair of primers would anneal to the flanking genomic sequence and generate a single ~500 bp product; however, if the insert was present intact at the locus both pairs of detection primers would be able to anneal and generate two products of ~1250 and ~1750 bp (Figure 2C).
Cell Extract Preparation
For rapid prototyping of engineered strains, cells were grown in 1 L of 2xYTPG media (16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl, 7 g/L K2HPO4, 3 g/L KH2PO4, pH 7.2) in a 2.5 L Tunair® shake flask and incubated at 34°C at 220 rpm. Unless otherwise stated, cultures were inoculated with 1 mM IPTG at an OD600 of 0.6 and permitted to continue to grow to an OD600 of 3.0. Cells were pelleted by centrifuging for 15 min at 5000 × g at 4°C, washed three times with cold S30 buffer (10 mM tris-acetate pH 8.2, 14 mM magnesium acetate, 60 mM potassium acetate, 2 mM dithiothreitol(DTT))(Swartz et al., 2004), and stored at −80°C. To make cell extract, cell pellets were thawed and suspended in 0.8 mL of S30 buffer per gram of wet cell mass and 1.4 mL of cell slurry was transferred into1.5 mL microtubes. The cells were lysed using a Q125 Sonicator (Qsonica, Newtown, CT) with 3.175 mm diameter probe at a 20 kHz frequency and 50 % amplitude for three cycles of 45s ON/59s OFF. To minimize heat damage during sonication, samples were placed in an ice-water bath. For each 1.4 mL sample, the input energy was ~844 Joules and was monitored during sonication. Immediately following sonication, DTT was added to each tube to a final concentration of 3 mM. Extract was then centrifuged at 12,000 × g at 4°C for 10 min. For strain derivatives of C321.ΔA.759, a run-off reaction (37°C at 250 rpm for 1 h) and second centrifugation (10,000 × g at 4°C for 10 min) were performed(Kwon and Jewett, 2015). The supernatant was flash-frozen using liquid nitrogen and stored at −80°C until use.
CFPS Reaction
A modified PANOx-SP system was utilized for CFPS reactions (Jewett and Swartz, 2004),(Jewett et al., 2008). Briefly, each 15 μL CFPS reaction was assembled in a 2.0 mL microtube by mixing the following components: 1.2 mM ATP; 0.85 mM each of GTP, UTP, and CTP; 34 μg/mL folinic acid; 170 μg/mL of E. coli tRNA mixture; 13.3 μg/mL plasmid; 16 μg/mL T7 RNA polymerase; 2 mM for each of the 20 standard amino acids; 0.33 mM nicotinamide adenine dinucleotide (NAD); 0.27 mM coenzyme-A (CoA);1.5 mM spermidine; 1 mM putrescine; 4 mM sodium oxalate; 130 mM potassium glutamate; 10 mM ammonium glutamate; 12 mM magnesium glutamate; 57 mM HEPES, pH 7.2; 33 mM phosphoenolpyruvate (PEP), and 27% v/v of cell extract. For ncAA incorporation, 2 mM pAcF, 0.5 μg/mL pAcFRS, and 30 μg/mL of o-tz-tRNA linear DNA were supplemented to cell-free reactions. o-tRNA linear DNA was amplified from pY71-T7-tz-o-tRNA plasmid via PCR and transcribed during the cell-free reaction. Furthermore, the o-tRNA was expressed in the source strain prior to extract preparation. Each CFPS reaction was incubated for 20 h at 30°C unless noted otherwise. E. coli total tRNA mixture (from strain MRE600) and phosphoenolpyruvate was purchased from Roche Applied Science (Indianapolis, IN). ATP, GTP, CTP, UTP, 20 amino acids and other materials were purchased from Sigma (St. Louis, MO) without further purification. T7RNAP was purified in house as described below.
Quantification of Active sfGFP
CFPS reactions were diluted 1:25 in nanopure water and active full-length sfGFP protein yields were quantified by measuring fluorescence using a Synergy 2 plate reader (BioTek, Winooski, VT) with excitation at 485 nm, emission at 528 nm, and cut-off at 510 nm in 96-well half area black plates (Costar 3694; Corning, Corning, NY). sfGFP fluorescence units were converted to concentration using a standard curve established with 14C-Leucine quantification.
Detection of His-tagged T7RNAP by Western Blot
To visualize T7 RNA polymerase overexpression in vivo, cell samples were collected during harvest. Pre-induction cell samples were derived from 1 mL of culture at OD600 of 0.6, harvest samples were derived from 200 μL of culture at OD600 of 3.0. To prepare samples for gel electrophoresis, cells were pelleted and resuspended in 200 μL of nuclease-free water. 100 μL of this suspension was mixed with 34 μL of 4x NuPAGE® LDS Sample Buffer (Thermo Fisher Scientific, Inc., Waltham, MA) and boiled for 10 minutes. Following the boil, samples were spun at >13,500 × g. Samples derived from lysates were prepared by diluting 1 μL of extract in 8 μL of nuclease-free water and boiling for 10 minutes with 3 μL of 4x NuPAGE® LDS Sample Buffer. 12 μL of each sample was loaded into 12% Bis-Tris NuPAGE® gel (Thermo Fisher Scientific, Inc., Waltham, MA) and run at 130 V for 90 min using 1X MOPS running buffer (diluted from 20X MOPS SDS Running Buffer, Thermo Fisher Scientific, Inc., Waltham, MA). For reference, SeeBlue® Plus2 Pre-Stained Protein Standard (Thermo Fisher Scientific, Inc., Waltham, MA) was loaded into wells flanking the samples. Following electrophoresis, gels were washed in nanopure water. Proteins were transferred to Immun-Blot® PVDF membrane (Bio-rad, Hercules, CA) using a semi-dry protocol in 20% methanol/80% 1x MOPS. Transfer proceeded at 80 mA per gel for 55 min using a Trans-Blot® SD Semi-Dry Transfer Cell (Bio-rad, Hercules, CA). Blots were blocked overnight in 5% (m/v) fat-free dry milk at 4°C. Primary antibody (Sigma, Cat. #H1029, St. Louis, MO) was diluted 10,000x in PBS and applied to blots for 2 hrs. Secondary antibody conjugated to horseradish peroxidase (Bio-rad, Cat. #1701011, Hercules, CA) was diluted 3,000x in PBS-T and applied to blots for 1 hr. Finally, His-tagged proteins were visualized using the Immun-Blot® Opti-4CN™ Colorimetric kit (Bio-rad, Hercules, CA).
Multiplex Advanced Genome Engineering (MAGE) Cycling
To perform a single cycle of MAGE, a 5 mL liquid culture of the strain of interest was grown to an OD600 of 0.6–0.8 in LB media at 32°C and 250 rpm, after which the culture was transferred to a 42°C water bath for 15 minutes. Next, the culture was immediately put on ice for at least 5 minutes. 1.5 mL of culture was transferred to a microcentrifuge tube and washed 3x in ice-cold nuclease-free water by pelleting cells for 1 min at 13,500 × g followed by resuspension in water. After the third wash, cells were resuspended directly in a solution of MAGE oligo (5–20 μM). Oligo was introduced into cells using a Micropulse electroporator (Bio-Rad, Hercules, CA). Immediately following electroporation, cells were resuspended in 5 mL sterile LB media – the resulting 5 mL culture was used to begin the next cycle.
Replica Plating of Bacterial Colonies
To identify colonies that had regained sensitivity to a given antibiotic, plates were replicated. To begin, cells from a mixed population (some of which would be sensitive to the antibiotic, and some of which would not be) were spread onto LB agar plates lacking the antibiotic and grown overnight at 34°C to produce a series of source plates. Then, for each source plate a sterile velvet was fastened to a replica plating tool (VWR, Radnor, PA). A source plate was gently pressed down onto the surface of the velvet such that the velvet picked up some cells from each colony present. Next, a new plate lacking the antibiotic was pressed onto the velvet, after which a new plate containing the antibiotic was pressed onto the velvet. All plates were returned to 34°C overnight. Ultimately, colonies that were present on the source plate as well as the replica plate lacking antibiotic but absent from the replica plate containing antibiotic were identified as having regained sensitivity to the antibiotic.
Knockout of ompT Locus
In order to use kanamycin resistance to select for successful knockout of the ompT locus in C321.ΔA.759.T7, the kanR cassette first employed to select for integration of the T7RNAP insert needed to be removed from the genome. This DNA was physically looped out of the genome using the oligo listed in Table S1 for MAGE. Cultures were grown in LB media at 32°C and 250 rpm throughout 8 MAGE cycling steps as described above. To identify colonies that regained sensitivity to kanamycin, cells were replica plated as described above. Colony PCR using the protocol described above confirmed that the kanamycin resistance cassette DNA was no longer present in the genome. The kanamycin resistant cassette from pKD4 was then amplified with primers containing up- and downstream homology to the genomic region targeted for deletion in their 5’ tails. The knockout construct was given flanking homology such that coordinates 580,650–592,260 in the genome of C321.ΔA.759.T7 would be replaced by the resistance cassette. LHR followed by colony PCR detection of the knockout were performed as described above to yield C321.ΔA.759.T7.DompT.
Generation and Verification of OmpT-resistant T7RNAP-expressing Strains
Nucleotide changes designed to introduce mutations of K183 to glycine/leucine and K190 to alanine were installed into C321.ΔA.759.T7’s genomic copy of the N-terminally 6His-tagged 1 gene via MAGE (described above) using the oligos listed in Table S1. Cultures were grown in selective LB media at 32°C and 250 rpm throughout 6 MAGE cycling steps . Putative mutant colonies were picked and cultured as described above prior to screening for the desired mutation. Multiplex allele-specific colony (MASC) PCR was performed with Colony PCR Master Mix (Thermo Fisher Scientific, Inc., Waltham, MA) to verify mutations(Wang and Church, 2011) using wild-type forward or mutant forward primers and reverse primers (Table S1). Wild-type and mutant forward primers were identical except at the 3’-ends of the oligonucleotides which featured allele-specific sequence such that stable annealing of the end of the primer should only be possible when paired with the corresponding genomic allele. In this way the mutant allele could be amplified using the mutant forward and reverse primer set but not amplified by the wild-type forward and reverse primer set, and vice versa. The reverse primers were used for detection of both wild-type and mutant alleles.
pMAZ Plasmid Assembly
All primers and oligos described in this section are presented in Table S1. pMAZ-DHis was assembled from two parts: linear plasmid backbone derived from PCR amplification of pMAZ-SK (primers pMAZbb_F/pMAZbb_R), and annealed oligos encoding the target gRNA sequence (T7delHis_oligo1 and T7delHis_oligo2). To anneal, 10 μL of a 100 μM stock for each oligo were combined with 10 μL CutSmart® buffer (New England Biolabs, Ipswich, MA) and 70 μL nuclease-free water, and the mix was heated to 95°C for 5 mins before being returned to room temperature. Insertion of the annealed oligos into the plasmid backbone was performed with USER® Enzyme (New England Biolabs, Ipswich, MA). To assemble pMAZ-Cure, pMAZ backbone DNA was amplified using primers pMAZCurebb_F/pMAZCurebb_R. DNA insert encoding gRNAs against the origin of replication and ampicillin resistance gene of pMA7CR_2.0 was purchased from Life Technologies (Carlsbad, CA). This pMAZ backbone and gRNA cassette insert were assembled via Gibson assembly to yield pMAZ-Cure. Sanger sequencing confirmed correct sequences of both pMAZ plasmids.
CRMAGE Removal of N-terminal His-tag from C321.ΔA.759.T7.D
All primers and oligos described in this section are listed in Table S1. In preparation for CRMAGE, genomically-encoded resistances to ampicillin and kanamycin were removed from C321.ΔA.759.T7.D to allow for the CRMAGE plasmids to be selected for. This was achieved via MAGE to loop the resistance genes out of the genome followed by replica plating to identify sensitized colonies, yielding strain C321.ΔA.759.T7.D.DAbR. To begin CRMAGE, pMA7CR_2.0 was transformed into C321.ΔA.759.T7.D.DAbR where it was stably maintained throughout the process. For each cycle of CRMAGE, the strain was grown to an OD600 of ~0.6–0.8, incubated for 15 minutes at 42°C, and made electrocompetent via washing with cold nuclease-free water. Next, pMAZ-ΔHis and MAGE oligo T7delHis_MAGE were transformed into the strain simultaneously. After electroporation, cells were allowed to recover for 1 hour in 5 mL SOC media at 34°C at 250 RPM. After 1 hr, 50 μg/mL kanamycin was added to the recovery media to begin selecting for the pMAZ plasmid and the culture was left to continue shaking for 2 hours. Next, a total of 3 hours post-transformation, 1 mL of recovery culture was transferred to 4 mL of fresh SOC media with 50 μg/mL carbenicillin, 50 μg/mL kanamycin, and 400 ng/mL anhydrotetracycline (aTc), and put back at 34°C, 250 RPM for another 3 hours. Finally, the culture was diluted 1:100 in nanopure water and plated on LB plates with 50 μg/mL carbenicillin, 50 μg/mL kanamycin, and 400 ng/mL anhydrotetracycline. Plates recovered at 34°C overnight. Individual colonies were sequenced to verify incorporation of T7delHis_MAGE and, by extension, removal of the N-terminal His-tag from the genomic 1 gene in the strain. Once removal of the His-tag was detected, cells were grown to an OD600 of ~0.6–0.8 and induced with 400 ng/mL aTc and 2% (m/v) rhamnose to induce destruction of pMAZ-ΔHis. Replica plating identified cells that had lost pMAZ-ΔHis via their regained sensitivity to kanamycin. To remove all CRMAGE-associated plasmids from the strain, plasmid pMAZ-Cure was introduced into the cells. pMAZ-Cure encodes a gRNA cassette targeting the selectable marker and origin of replication of pMA7CR_2.0. such that induction of the system with both aTc and rhamnose triggers elimination of both plasmids. pMAZ-Cure-bearing cells were grown to an OD600 of ~0.6–0.8 and induced with 400 ng/mL aTc and 2% (m/v) rhamnose to trigger the simultaneous destruction of both pMAZ-Cure and pMA7CR_2.0 in the cells. Replica plating identified colonies that had lost both plasmids to yield strain 759.T7.Opt.
Expression and Purification of His-tagged Orthogonal pAcF tRNA Synthetase
BL21 (DE3) harboring a pY71 plasmid encoding pAcFRS were grown in 1L of 2xYTPG to an OD600 of 0.6 at 250 rpm and 37°C. At that point, synthetase expression was induced by adding 1mM IPTG and the culture was moved to 30°C and allowed to continue to grow for 4 hrs. Cells were pelleted by centrifuging for 15 min at 5000 × g at 4°C, washed three times with cold S30 buffer, and stored at −80°C overnight. To purify pAcFRS, cell pellets were thawed on ice and resuspended in 5 mL of 50 mM Tris/300 mM NaCl per gram of wet cell pellet. Cells were lysed using an EmulsiFlex-B15 homogenizer (Avestin, Ottawa, Canada) with three passes at a pressure of 12,000 psig and centrifuged at 10,000 × g for 10 mins. To purify pAcF synthetase from the supernatant, Ni-NTA agarose (Qiagen, Venlo, The Netherlands) was used following manufacturer protocol. Following elution, purified synthetase was dialyzed overnight at 4°C in a Slide-a-Lyzer™ cassette (10 kDA MWCO; Thermo Fisher Scientific, Inc., Waltham, MA) against S30 buffer with 25% glycerol. After dialysis, synthetase was concentrated with Amicon® Ultracel centrifugal spin filters (3 kDa MWCO; MilliporeSigma, Burlington, MA). Final synthetase concentration was determined with a NanoDrop™ spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA) using an extinction coefficient of 20985, determined using ExPASy ProtParam tool(Gasteiger et al., 2005).
ELP Radioactive Quantitation
Radioactive 14C-Glycine was added into 15 μL CFPS reactions. After incubation, yields were quantified by determining radioactive 14C-Gly incorporation into trichloroacetic acid (TCA) -precipitated protein(Swartz et al., 2004). Briefly, following CFPS reactions were quenched with 100 μL 0.1N sodium hydroxide and put at 37°C for 30 minutes. Two small tabs of Whatman 3MM chromatography paper (GE Healthcare Life Sciences, Little Chalfont, United Kingdom) were prepared for each individual radioactive CFPS reaction and suspended on pins above a block of Styrofoam wrapped with aluminum foil. Following the sodium hydroxide incubation, reactions were mixed by pipetting. Next, 50 mL of material from each reaction vessel was deposited onto both paper tabs prepared for that vessel. The population of loaded paper tabs was split in half, into a “washed” subpopulation and an “unwashed” subpopulation, such that a single tab derived from each CFPS reaction was present in each subpopulation. Tabs were allowed to dry under a heat lamp for ~2 hr. After drying, all “washed” subpopulation tabs were combined in a beaker and washed 3x with ice-cold 5% TCA at 4°C. Following the third wash, these tabs were washed with 100% ethanol for 10 mins at room temperature. Washed tabs were remounted on Styrofoam and dried under a heat lamp for ~2 hr. Finally, all tabs (“washed” and “unwashed”) were individually deposited into 1.5 mL microcentrifuge tubes and submerged into 1 mL of scintillation fluid. Radioactivity of TCA-precipitated samples in terms of counts per minute (cpm) was measured using liquid scintillation counting (MicroBeta2, PerkinElmer, Waltham, MA). With radioactive counts in hand, protein yield for each sample could be calculated according to the following equation:
Autoradiogram Analysis
For autoradiogram analysis of ELP constructs, CFPS reactions were performed supplemented with 10 μM of radioactive 14C-glycine. Samples were prepared for polyacrylamide gel electrophoresis as described above from 4 mL of each reaction and run on a 12% Bis-Tris NuPAGE® gel (Thermo Fisher Scientific, Inc., Waltham, MA). After electrophoresis, the gel was soaked in Gel Drying solution (Bio-Rad, Hercules, CA) for 30 min, fixed with cellophane films, dried overnight in GelAir Dryer (Bio-Rad, Hercules, CA), and exposed for 3 days on Storage Phosphor Screen (GE Healthcare Biosciences, Pittsburgh, PA). Autoradiograms were scanned using Typhoon FLA 7000 Imager (GE Healthcare Biosciences, Pittsburgh, PA).
Purification and Top Down Mass Spectrometry of sfGFP and ELP Constructs
To prepare sfGFP and ELP-20mer/30mer samples for top-down mass spectrometry, products were purified out of CFPS reactions using Strep-Tactin®XT magnetic beads (IBA Lifesciences, Göttingen, Germany) precipitated in methanol/chloroform and water(Toby et al., 2019), dried, and resuspended in Buffer A (95% water, 5% acetonitrile, 0.2% formic acid). ELP-40mers were purified using a modified inverse transition cycling (ITC) method as previously described(Martin et al., 2018), which was sufficient to purify ELP-40WT. ELP-40UAG products required further enrichment, and for this construct ITC was followed up with affinity purification using Strep-Tactin®XT magnetic beads following manufacturer’s instructions to generate a highly pure sample of the protein. Purified proteins were injected onto a trap column (150 μm ID × 3 cm) coupled with a nanobore analytical column (75 mm ID × 15 cm). Trap and column were packed with polymeric reverse phase (PLRP-S, Phenomenex, Torrance, CA) media (5 μm, 1,000Å pore size). Samples were separated using a linear gradient of Buffer A and Buffer B (5% water, 95% acetonitrile, 0.2% formic acid). Samples were loaded for 10 min onto the trap column and subsequently separated using a linear gradient from 5% to 95% of solvent B (80 min for sfGFP samples and ELP-20mer/30mer, 60 min for ELP-40mer). Mass spectrometric data for all sfGFP and ELP-20mer/30mer samples were obtained on a Orbitrap Fusion™ Lumos™ (Thermo Fisher Scientific, Inc., Waltham, MA) instrument fitted with a custom nanospray ionization source. The acquisition method was essentially a full scan FTMS experiment, with data obtained from 500–2000 m/z at a resolving power of 120,000 at m/z 400. sfGFP data were deconvoluted using Xtract (Thermo Fisher) and monoisotopic masses were reported. ELP-20mer/30mer data were deconvoluted using Esiprot(Winkler, 2010) and average masses were reported. Mass spectrometric data for ELP-40mer samples were obtained on a Velos Pro ion trap (Thermo Fisher Scientific, Inc., Waltham, MA) fitted with a custom nanospray ionization source. The acquisition method was a full scan using the ion trap. ELP-40mer data were deconvoluted using UniDec (Marty et al., 2015).
DNA Sequencing
To sequence the genomic T7RNAP inserts, the entire region was PCR amplified using end primers listed in Table S1. Amplified linear insert DNA was submitted to the NUSeq Core facility along with forward primers spaced ~700 bp apart, and the sequence for each region was determined using traditional Sanger sequencing. pMAZ plasmids were sequenced by submitting purified plasmid samples as well as primer pMAZ_seq to the NUSeq Core facility.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantified data in this work consist exclusively of protein yields from various CFPS platforms. As outlined in figure legends, these data present average yields from at least three independent CFPS reactions along with one standard deviation. All calculations were performed using Microsoft Excel.
DATA AND CODE AVAILABILITY
Data for this study will be provided upon reasonable request and agreement. Such requests will be fulfilled by the Lead Contact, Michael C. Jewett (m-jewett@northwestern.edu).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| Monoclonal Anti-polyhistidine antibody produced in mouse | Sigma-Aldrich | Cat#H1029; RRID: AB_260015 | ||
| Goat Anti-Mouse IgG (H+L)-HRP Conjugate | Bio-Rad | Cat#1721011; RRID: AB_11125936 | ||
| Bacterial and Virus Strains | ||||
| E. coli BL21 Star™ (DE3) | ThermoFisher | Cat#C601003 | ||
| E. coli BL21 (DE3) | New England Biolabs | Cat#C2527I | ||
| E. coli DH5α | ThermoFisher | Cat#18265017 | ||
| E. coli C321.ΔA.759 | Martin et al., 2018 | N/A | ||
| E. coli 759.T7.int.lacUV5 | This paper | N/A | ||
| E. coli 759.T7.int.PtacI | This paper | N/A | ||
| E. coli 759.T7.int.Lpp5 | This paper | N/A | ||
| E. coli 759.T7.asl.lacUV5 | This paper | N/A | ||
| E. coli 759.T7.asl.PtacI | This paper | N/A | ||
| E. coli 759.T7.asl.Lpp5 | This paper | N/A | ||
| E. coli 759.T7.ΔkanR | This paper | N/A | ||
| E. coli 759.T7.ΔompT | This paper | N/A | ||
| E. coli 759.T7.K172L | This paper | N/A | ||
| E. coli 759.T7.K172G | This paper | N/A | ||
| E. coli 759.T7.D | This paper | N/A | ||
| E. coli 759.T7.D.ΔAbR | This paper | N/A | ||
| E. coli 759.T7.Opt | This paper | N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||||
| T7 RNA polymerase | Prepared in-house | N/A | ||
| pAcF aminoacyl-tRNA synthetase | Prepared in-house | N/A | ||
| Potassium glutamate | Sigma-Aldrich | Cat#G1501 | ||
| Magnesium glutamate | Sigma-Aldrich | Cat#49605 | ||
| Ammonium glutamate | MP Biomedicals | Cat#02180595.1 | ||
| HEPES Buffer | Sigma-Aldrich | Cat#H3375 | ||
| PEG-8000 | Sigma-Aldrich | Cat#202452-500G | ||
| Dithiothreitol | Sigma-Aldrich | Cat#43816 | ||
| Putrescine | Sigma-Aldrich | Cat#P5780 | ||
| Spermidine | Sigma-Aldrich | Cat#S0266 | ||
| Glycerol | Sigma-Aldrich | Cat#G5516 | ||
| Folinic acid | Sigma-Aldrich | Cat#47612 | ||
| tRNA from E. coli MRE600 | Roche | Cat#10109541001 | ||
| Phosphoenol pyruvate | Roche | Cat#10108294 | ||
| NAD | Sigma-Aldrich | Cat#N8535 | ||
| CoA | Sigma-Aldrich | Cat#C3144 | ||
| Oxalic acid | Sigma-Aldrich | Cat#P0963 | ||
| Nuclease-free water | Ambion | Cat#AM9937 | ||
| ATP | Sigma-Aldrich | Cat#A2383 | ||
| GTP | Sigma-Aldrich | Cat#G8877 | ||
| UTP | Sigma-Aldrich | Cat#U6625 | ||
| CTP | Sigma-Aldrich | Cat#C1506 | ||
| L-Valine | Sigma-Aldrich | Cat#V0500 | ||
| L-Tryptophan | Sigma-Aldrich | Cat#T0254 | ||
| L-Phenylalanine | Sigma-Aldrich | Cat#P2126 | ||
| L-Isoleucine | Sigma-Aldrich | Cat#I2752 | ||
| L-Leucine | Sigma-Aldrich | Cat#L8000 | ||
| L-Cysteine | Sigma-Aldrich | Cat#C7352 | ||
| L-Methionine | Sigma-Aldrich | Cat#M9625 | ||
| DL-Alanine | Sigma-Aldrich | Cat#A7627 | ||
| L-Arginine | Sigma-Aldrich | Cat#A8094 | ||
| L-Asparagine | Sigma-Aldrich | Cat#A0884 | ||
| L-Aspartic Acid | Sigma-Aldrich | Cat#A9256 | ||
| L-Glutamic acid | Sigma-Aldrich | Cat#G1501 | ||
| L-Glycine | Sigma-Aldrich | Cat#G7126 | ||
| L-Glutamine | Sigma-Aldrich | Cat#G3126 | ||
| L-Histidine | Sigma-Aldrich | Cat#H8000 | ||
| L-Lysine | Sigma-Aldrich | Cat#L5501 | ||
| L-Proline | Sigma-Aldrich | Cat#P0380 | ||
| L-Serine | Sigma-Aldrich | Cat#S4500 | ||
| L-Threonine | Sigma-Aldrich | Cat#T8625 | ||
| L-Tyrosine | Sigma-Aldrich | Cat#T3754 | ||
| Phusion™ polymerase | New England Biolabs | Cat#M0530L | ||
| SYBR® Safe | ThermoFisher | Cat#S33102 | ||
| Quick-Load® 1 kb DNA ladder | New England Biolabs | Cat#N0468S | ||
| Quick-Load® 100 bp DNA ladder | New England Biolabs | Cat#N0467S | ||
| T5 Exonuclease | New England Biolabs | Cat#M0363S | ||
| Taq Ligase | New England Biolabs | Cat#M0208L | ||
| dNTP Solution Mix | New England Biolabs | Cat#N0447L | ||
| Phusion™ HF Buffer | New England Biolabs | Cat#B0518S | ||
| Kanamycin | Sigma-Aldrich | Cat#K4000-25G | ||
| Carbenicillin | IBI Scientific | Cat#IB02025 | ||
| Chloramphenicol | Sigma-Aldrich | Cat#C0378-25G | ||
| IPTG | Sigma-Aldrich | Cat#I6758-10G | ||
| Tris-acetate | Sigma-Aldrich | Cat#T1258-250G | ||
| Tris | Sigma-Aldrich | Cat#T1503-500G | ||
| 14C Glycine | Perkin Elmer | Cat#NEC276E050UC | ||
| Scintillation fluid | MP Biomedicals | Cat#0188245304 | ||
| Bacto™ Tryptone | ThermoFisher | Cat#211705 | ||
| Bacto™ Yeast Extract | ThermoFisher | Cat#211929 | ||
| Sodium Chloride | Sigma-Aldrich | Cat#S9888 | ||
| Bacto™ Agar | VWR | Cat#90000-762 | ||
| Potassium phosphate monobasic | Sigma-Aldrich | Cat#P0662-500G | ||
| Potassium phosphate dibasic | Sigma-Aldrich | Cat#P3786-500G | ||
| Magnesium acetate | Sigma-Aldrich | Cat#M5661-250G | ||
| Potassium acetate | Sigma-Aldrich | Cat#P1190-1KG | ||
| L-p-acetylphenylalanine | Chem-Impex International Inc. | Cat#24756 | ||
| 4x NuPAGE LDS Sample Buffer | ThermoFisher | Cat#NP0007 | ||
| SeeBlue™ Plus2 Pre-stained Protein Standard | ThermoFisher | Cat#LC5925 | ||
| Methanol | Sigma-Aldrich | Cat#34860-1L-R | ||
| 2x Colony PCR Master mix | ThermoFisher | Cat#K0172 | ||
| Cutsmart® buffer | New England Biolabs | Cat#B7204S | ||
| USER™ Enzyme | New England Biolabs | Cat#M5505S | ||
| anhydrotetracycline | Sigma-Aldrich | Cat#37919-100MG-R | ||
| rhamnose | Sigma-Aldrich | Cat#83650-10G | ||
| Trichloroacetic acid | Sigma-Aldrich | Cat#T6399-500G | ||
| Sodium Hydroxide | Sigma-Aldrich | Cat#221465-500G | ||
| 200-proof ethanol | Sigma-Aldrich | Cat#E7023-6X500ML | ||
| Gel drying solution | Bio-Rad | Cat#1610752 | ||
| LC-MS Grade methanol | Fisher Scientific | Cat#A456-4 | ||
| LC-MS Grade water | Fisher Scientific | Cat#W6-4 | ||
| LC-MS Grade acetonitrile | Fisher Scientific | Cat#A955-4 | ||
| LC-MS Grade formic acid | Fisher Scientific | Cat#A1171-AMP | ||
| NuPAGE MOPS SDS running buffer | ThermoFisher | Cat#NP0001 | ||
| Critical Commercial Assays | ||||
| HiSpeed® Plasmid Maxi Kit | Qiagen | Cat#12643 | ||
| Ni-NTA Agarose | Qiagen | Cat#30210 | ||
| MagStrep “type3” XT beads 5% suspension | IBA Life Sciences | Cat#2-4090-002; Cat#2-1003-100; Cat#2-1042-025 | ||
| E.Z.N.A.® Plasmid Mini Kit | Omega Biotek | Cat#D6943-01 | ||
| E.Z.N.A.® Gel Extraction Kit | Omega Biotek | Cat#D2500-01 | ||
| Immun-Blot® Opti-4CN™ Colorimetric Kit | Bio-Rad | Cat#1708235 | ||
| Oligonucleotides | ||||
| See Table S1 | ||||
| Recombinant DNA | ||||
| pKD4 | Datsenko and Wanner (2000). | N/A | ||
| pAR1219 | Davanloo et al., 1984. | N/A | ||
| pDPtacIAcRSTT1 | de Boer et al., 1983. | N/A | ||
| pDTT1-Lpp5-EF-Tu | Inouye and Inouye (1985). Gan et al., 2017. | N/A | ||
| pY71-sfGFP | Bundy and Swartz (2010). | N/A | ||
| pY71-sfGFP-T216amb | Bundy and Swartz (2010). | N/A | ||
| pY71-sfGFP-2amb | Hong et al., 2014b. | N/A | ||
| pY71-sfGFP-5amb | Hong et al., 2014b. | N/A | ||
| pY71-pAcFRS | Hong et al., 2014b. | N/A | ||
| pEVOL-pAcF | Young et al., 2010. | N/A | ||
| pY71-T7-tz-o-tRNA | Hong et al., 2014b. | N/A | ||
| pUC-T7-int.lacUV5 | This paper | N/A | ||
| pUC-T7-asl.lacUV5 | This paper | N/A | ||
| pUC-T7-asl.PtacI | This paper | N/A | ||
| pUC-T7-asl.Lpp5 | This paper | N/A | ||
| p15a-T7-asl.Lpp5 | This paper | N/A | ||
| p15a-T7-asl.PtacI | This paper | N/A | ||
| p15a-T7-int.Lpp5 | This paper | N/A | ||
| pMA7CR_2.0 | Ronda et al., 2016 | N/A | ||
| pMAZ-SK | Ronda et al., 2016 | N/A | ||
| pMAZ-His | This paper | N/A | ||
| pMAZ-Cure | This paper | N/A | ||
| pY71-FI-ELP20 | Martin et al., 2018 | N/A | ||
| pY71-FI-ELP30 | Martin et al., 2018 | N/A | ||
| pY71-FI-ELP40 | Martin et al., 2018 | N/A | ||
| pY71-FI-ELP20X | Martin et al., 2018 | N/A | ||
| pY71-FI-ELP30X | Martin et al., 2018 | N/A | ||
| pY71-FI-ELP40X | Martin et al., 2018 | N/A | ||
Highlights.
A one-pot, high-yield CFPS system was developed from a recoded strain of E. coli
Genomic edits improved the function of a T7 RNA polymerase expressed in the strain
Lysates prepared from the strain function without exogenous biological components
The platform is capable of incorporating up to 40 non-canonical amino acids per protein
SIGNIFICANCE.
The addition of non-canonical amino acids (ncAAs) to the genetic code has been transformative for protein design and engineering, unlocking protein properties, structures, and functions that would be otherwise be difficult or even impossible to achieve. To better leverage the power of these ncAAs, a cell-free protein synthesis (CFPS) platform comprising crude lysates derived from a genomically recoded strain of Escherichia coli that is capable of high-level expression of proteins bearing ncAAs was recently developed. While an extremely potent synthesis tool, this platform is dependent upon supplementation with purified polymerase enzyme to catalyze transcription, limiting its use in both academic laboratories and industrial-scale syntheses. To address this limitation, our work sought to develop this existing platform into a “one-pot” system containing all of the biological components required for transcription and translation, with a specific focus on the viral T7 RNA polymerase (T7RNAP) that is commonly used to perform transcription in CFPS systems. By genomically incorporating the gene 1 (encoding T7RNAP) into the genome of the source strain and editing its coding sequence, we developed a novel source strain, which expresses a mutant T7RNAP variant that is highly active and resistant to proteolysis. Crude lysates derived from this strain are enriched with this polymerase and are capable of high levels of protein synthesis in vitro independent of supplementation with purified enzymes. This platform facilitates the use of CFPS systems and presents a strategy for the introduction of new capabilities to E. coli strains for use both in vivo and in vitro.
ACKNOWLEDGMENTS
This work was supported by the Army Research Office W911NF-18-1-0200 and W911NF-18-1-0181, National Science Foundation (NSF) grant MCB-1716766, the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust, the David and Lucille Packard Foundation, and the Camille Dreyfus Teacher-Scholar Program (to M.C.J.). This research was carried out in collaboration with the National Resource for Translational and Developmental Proteomics under grant P41 GM108569 from the National Institute of General Medical Sciences, National Institutes of Health. B.J.D. is a recipient of the NSF Graduate Research Fellowship and was supported in part by NIH Predoctoral Biotechnology training grant T32GM008449. We thank Prof. Brad Bundy for providing pY71 plasmids and Prof. Peter G. Schultz for providing the pEVOL-pAcF plasmid. Research reported in this publication was made possible in part by the services of the NUSeq Core Facility, which is supported by the Northwestern University Center for Genetic Medicine, Feinberg School of Medicine, and Shared and Core Facilities of the University’s Office for Research. The US Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of the US Government.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chembiol.2019.10.008.
DECLARATION OF INTERESTS
The authors have filed a patent application related to this work with US Serial No. 15/651484.
REFERENCES
- Adiga R, Al-adhami M, Andar A, Borhani S, Brown S, Burgenson D, Cooper MA, Deldari S, Frey DD, Ge X, et al. (2018). Point-of-care production of therapeutic proteins of good-manufacturing-practice quality. Nat. Biomed. Eng 2, 675–686. [DOI] [PubMed] [Google Scholar]
- Aerni HR, Shifman MA, Rogulina S, O’Donoghue P, and Rinehart J (2015). Revealing the amino acid composition of proteins within an expanded genetic code. Nucleic Acids Res. 43, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiram M, Haimovich AD, Fan C, Wang Y-S, Aerni H-R, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, et al. (2015). Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol 33, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer H, and Dennis P (1996). Escherichia coli and Salmonella: Cellular and Molecular Biology, Vol 1, Second Edition (ASM Press; ). [Google Scholar]
- Bryant JA, Sellars LE, Busby SJ, and Lee DJ (2014). Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res. 42, 11383–11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundy BC, and Swartz JR (2010). Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein-protein click conjugation. Bioconjug. Chem 21, 255–263. [DOI] [PubMed] [Google Scholar]
- Carlson ED, Gan R, Hodgman CE, and Jewett MC (2012). Cell-free protein synthesis: applications come of age. Biotechnol. Adv 30, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caschera F, and Noireaux V (2014). Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 99, 162–168. [DOI] [PubMed] [Google Scholar]
- Chappell J, Jensen K, and Freemont PS (2013). Validation of an entirely in vitro approach for rapid prototyping of DNA regulatory elements for synthetic biology. Nucleic Acids Res. 41, 3471–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, and Doudna JA (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Liu P, Nielsen AA, Brophy JA, Clancy K, Peterson T, and Voigt CA (2013). Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 10, 659–664. [DOI] [PubMed] [Google Scholar]
- d’Aquino AE, Kim DS, and Jewett MC (2018). Engineered ribosomes for basic science and synthetic biology. Annu. Rev. Chem. Biomol. Eng 9, 311–340. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U S A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davanloo P, Rosenberg AH, Dunn JJ, and Studier FW (1984). Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U S A 81, 2035–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer HA, Comstock LJ, and Vasser M (1983). The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc. Natl. Acad. Sci. U S A 80, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Soye BJ, Patel JR, Isaacs FJ, and Jewett MC (2015). Repurposing the translation apparatus for synthetic biology. Curr. Opin. Chem. Biol 28, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despanie J, Dhandhukia JP, Hamm-Alvarez SF, and MacKay JA (2016). Elastin-like polypeptides: therapeutic applications for an emerging class of nanomedicines. J. Control. Release 240, 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley QM, Anderson KC, and Jewett MC (2016). Cell-free mixing of Escherichia coli crude extracts to prototype and rationally engineer high-titer mevalonate synthesis. ACS Synth. Biol 5, 1578–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley QM, Karim AS, and Jewett MC (2015). Cell-free metabolic engineering: biomanufacturing beyond the cell. Biotechnol. J 10, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas A.e., Lercher L, Spicer CD, and Davis BG (2014). Designing logical codon reassignment—expanding the chemistry in biology. Chem. Sci 6, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger T, and Ehricht R (1998). Single-step purification of T7 RNA polymerase with a 6-histidine tag. BioTechniques 24, 718–720. [DOI] [PubMed] [Google Scholar]
- Espah Borujeni A, Channarasappa AS, and Salis HM (2014). Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 42, 2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan R, Perez JG, Carlson ED, Ntai I, Isaacs FJ, Kelleher NL, and Jewett MC (2017). Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol. Bioeng 114, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garamella J, Marshall R, Rustad M, and Noireaux V (2016). The all E. coli TX-TL toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth. Biol 5, 344–355. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S.e., Wilkins MR, Appel RD, and Bairoch A (2005). Protein identification and analysis tools on the ExPASy server In The Proteomics Protocols Handbook, Walker JM, ed. (Humana Press; ), pp. 571–607. [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S (1996). Proteases and their targets in Escherichia coli. Annu. Rev. Genet 30, 465–506. [DOI] [PubMed] [Google Scholar]
- Grodberg J, and Dunn JJ (1988). ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification.J. Bacteriol 170, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzelman P, Schoborg JA, and Jewett MC (2015). pH responsive granulocyte colony-stimulating factor variants with implications for treating Alzheimer’s disease and other central nervous system disorders. Protein Eng. Des. Sel 28, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgman CE, and Jewett MC (2012). Cell-free synthetic biology: thinking outside the cell. Metab. Eng 14, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Kwon YC, and Jewett MC (2014a). Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front. Chem 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Kwon YC, Martin RW, Soye BJ, de Paz AM, Swonger KN, Ntai I, Kelleher NL, and Jewett MC (2015). Improving cell-free protein synthesis through genome engineering of Escherichia coli lacking release factor 1. Chembiochem 16, 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Ntai I, Haimovich AD, Kelleher NL, Isaacs FJ, and Jewett MC (2014b). Cell-free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site-specific nonstandard amino acid incorporation. ACS Synth. Biol 3, 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM (1995). PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol. Biotechnol 3, 93–99. [DOI] [PubMed] [Google Scholar]
- Huang A, Nguyen PQ, Stark JC, Takahashi MK, Donghia N, Ferrante T, Dy AJ, Hsu KJ, Dubner RS, Pardee K, et al. (2018). BioBits explorer: a modular synthetic biology education kit. Sci. Adv 4, eaat5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt JP, Yang SO, Wilding KM, and Bundy BC (2017). The growing impact of lyophilized cell-free protein expression systems. Bioengineered 8, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang BY, Varadarajan N, Li H, Rodriguez S, Iverson BL, and Georgiou G (2007). Substrate specificity of the Escherichia coli outer membrane protease OmpP. J. Bacteriol 189, 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda RA, and Richardson CC (1987a). Enzymatic properties of a proteolytically nicked RNA polymerase of bacteriophage T7. J. Biol. Chem 262, 3790–3799. [PubMed] [Google Scholar]
- Ikeda RA, and Richardson CC (1987b). Interactions of a proteolytically nicked RNA polymerase of bacteriophage T7 with its promoter. J. Biol. Chem 262, 3800–3808. [PubMed] [Google Scholar]
- Inouye S, and Inouye M (1985). Up-promoter mutations in the lpp gene of Escherichia coli. Nucleic Acids Res. 13, 3101–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroentomeechai T, Stark JC, Natarajan A, Glasscock CJ, Yates LE, Hsu KJ, Mrksich M, Jewett MC, and DeLisa MP (2018). Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat. Commun 9, 2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, and Swartz JR (2008). An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol 4, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett MC, and Swartz JR (2004). Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng 86, 19–26. [DOI] [PubMed] [Google Scholar]
- Karig DK, Bessling S, Thielen P, Zhang S, and Wolfe J (2017). Preservation of protein expression systems at elevated temperatures for portable therapeutic production. J. R. Soc. Interfaces 14, 10.1098/rsif.2016.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim AS, Dudley QM, and Jewett MC (2015). Cell-free synthetic systems for metabolic engineering and biosynthetic pathway prototyping In Industrial Biotechnology: Microorganisms, Liao JC and Wittmann C, eds. (Wiley-VCH; ), pp. 125–148. [Google Scholar]
- Karim AS, and Jewett MC (2016). A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab. Eng 36, 116–126. [DOI] [PubMed] [Google Scholar]
- Kightlinger W, Lin L, Rosztoczy M, Li W, DeLisa MP, Mrksich M, and Jewett MC (2018). Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat. Chem. Biol 14, 627–635. [DOI] [PubMed] [Google Scholar]
- Kwon YC, and Jewett MC (2015). High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep 5, 8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, et al. (2013). Genomically recoded organisms expand biological functions. Science 342, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent P, Duez C, Vanhove M, Lejeune A, Fonzé E, Charlier P, Rhazi-Filali F, Thamm I, Guillaume G, Samyn B, et al. (1997). Unexpected influence of a C-terminal-fused His-tag on the processing of an enzyme and on the kinetic and folding parameters. FEBS Lett. 413, 194–196. [DOI] [PubMed] [Google Scholar]
- Li J, Lawton TJ, Kostecki JS, Nisthal A, Fang J, Mayo SL, Rosenzweig AC, and Jewett MC (2016). Cell-free protein synthesis enables high yielding synthesis of an active multicopper oxidase. Biotechnol.J 11, 212–218. [DOI] [PubMed] [Google Scholar]
- Liu CC, Jewett MC, Chin JW, and Voigt CA (2018). Toward an orthogonal central dogma. Nat. Chem. Biol 14, 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, and Schultz PG (2010). Adding new chemistries to the genetic code. Annu. Rev. Biochem 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kim DS, and Jewett MC (2017). Repurposing ribosomes for synthetic biology. Curr. Opin. Chem. Biol 40, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martemyanov KA, Shirokov VA, Kurnasov OV, Gudkov AT, and Spirin AS (2001). Cell-free production of biologically active polypeptides: application to the synthesis of antibacterial peptide cecropin. Protein Expr. Purif 21, 456–461. [DOI] [PubMed] [Google Scholar]
- Martin RW, Des Soye BJ, Kwon YC, Kay J, Davis RG, Thomas PM, Majewska NI, Chen CX, Marcum RD, Weiss MG, et al. (2018). Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nat. Commun 9, 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, and Robinson CV (2015). Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister WT, Morris C, Rosenberg AH, and Studier FW (1981). Utilization of bacteriophage T7 late promoters in recombinant plasmids during infection. J. Mol. Biol 153, 527–544. [DOI] [PubMed] [Google Scholar]
- McManus JB, Murray RM, Emanuel PA, and Lux MW (2019). A method for cost-effective and rapid characterization of engineered T7-based transcription factors by cell-free protein synthesis reveals insights into the regulation of T7 RNA polymerase-driven expression. Arch. Biochem. Biophys 674, 108045. [DOI] [PubMed] [Google Scholar]
- Michel-Reydellet N, Calhoun K, and Swartz J (2004). Amino acid stabilization for cell-free protein synthesis by modification of the Escherichia coli genome. Metab. Eng 6, 197–203. [DOI] [PubMed] [Google Scholar]
- Mosberg JA, Lajoie MJ, and Church GM (2010). Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics 186, 791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T, Hoshi H, Ohtake K, Takahashi M, Yamaguchi A, Hayashi A, Yokoyama S, and Sakamoto K (2015). Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep 5, 9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller DK, Martin CT, and Coleman JE (1988). Processivity of proteolytically modified forms of T7 RNA polymerase. Biochemistry 27, 5763–5771. [DOI] [PubMed] [Google Scholar]
- Nehring S, Budisa N, and Wiltschi B (2012). Performance analysis of orthogonal pairs designed for an expanded eukaryotic genetic code. PLoS One 7, e31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza JP, Aerni HR, Pirman NL, Barber KW, Ter Haar CM, Rogulina S, Amrofell MB, Isaacs FJ, Rinehart J, and Jewett MC (2015). Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat. Commun 6, 8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee K, Green AA, Takahashi MK, Braff D, Lambert G, Lee JW, Ferrante T, Ma D, Donghia N, Fan M, et al. (2016a). Rapid, low-cost detection of zika virus using programmable biomolecular components. Cell 165, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Pardee K, Slomovic S, Nguyen PQ, Lee JW, Donghia N, Burrill D, Ferrante T, McSorley FR, Furuta Y, Vernet A, et al. (2016b). Portable, on-demand biomolecular manufacturing. Cell 167, 248–259.e12. [DOI] [PubMed] [Google Scholar]
- Raucher D, and Ryu JS (2015). Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol. Med 21, 560–570. [DOI] [PubMed] [Google Scholar]
- Renesto P, and Raoult D (2003). From genes to proteins: in vitro expression of rickettsial proteins. Ann. N. Y. Acad. Sci 990, 642–652. [DOI] [PubMed] [Google Scholar]
- Ronda C, Pedersen LE, Sommer MOA, and Nielsen AT (2016). CRMAGE: CRISPR optimized MAGE recombineering. Sci. Rep 6, 19452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabaty M, Grosse S, Adryanczyk G, Boiry S, Biaso F, Arnoux P, and Pignol D (2013). Detrimental effect of the 6 His C-terminal tag on YedY enzymatic activity and influence of the TAT signal sequence on YedY synthesis. BMC Biochem. 14, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis HM, Mirsky EA, and Voigt CA (2009). Automated design of synthetic ribosome bindingsites to control protein expression.Nat. Biotechnol 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro SW, Wang L, Herberich B, King DS, and Schultz PG (2002). An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat. Biotechnol 20, 1044–1048. [DOI] [PubMed] [Google Scholar]
- Schinn S-M, Bradley W, Groesbeck A, Wu JC, Broadbent A, and Bundy BC (2017). Rapid in vitro screening for the location-dependent effects of unnatural amino acids on protein expression and activity. Biotechnol. Bioeng 114, 2412–2417. [DOI] [PubMed] [Google Scholar]
- Schoborg JA, Hershewe JM, Stark JC, Kightlinger W, Kath JE, Jaroentomeechai T, Natarajan A, DeLisa MP, and Jewett MC (2018). A cell-free platform for rapid synthesis and testing of active oligosaccharyltransferases. Biotechnol. Bioeng 115, 739–750. [DOI] [PubMed] [Google Scholar]
- Shin J, and Noireaux V (2010). Efficient cell-free expression with the endogenous E. coli RNA polymerase and sigma factor 70. J. Biol. Eng 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, and Noireaux V (2012). An E. coli cell-free expression toolbox: application to synthetic gene circuits and artificial cells. ACS Synth. Biol 1, 29–41. [DOI] [PubMed] [Google Scholar]
- Slomovic S, Pardee K, and Collins JJ (2015). Synthetic biology devices for in vitro and in vivo diagnostics. Proc. Natl. Acad. Sci. U S A 112, 14429–14435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Berkheimer SD, Werner CJ, and Bundy BC (2014). Lyophilized Escherichia coli-based cell-free systems for robust, high-density, long-term storage. BioTechniques 56, 186–193. [DOI] [PubMed] [Google Scholar]
- Stark JC, Huang A, Hsu KJ, Dubner RS, Forbrook J, Marshalla S, Rodriguez F, Washington M, Rybnicky GA, Nguyen PQ, et al. (2019). BioBits health: classroom activities exploring engineering, biology, and human health with fluorescent readouts. ACS Synth. Biol 8, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Stark JC, Huang A, Nguyen PQ, Dubner RS, Hsu KJ, Ferrante TC, Anderson M, Kanapskyte A, Mucha Q, Packett JS, et al. (2018). BioBits Bright: a fluorescent synthetic biology education kit. Sci. Adv 4, eaat5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefano JE, and Gralla J (1979). Lac UV5 transcription in vitro. Rate limitation subsequent to formation of an RNA polymerase-DNA complex. Biochemistry 18, 1063–1067. [DOI] [PubMed] [Google Scholar]
- Studier FW, and Moffatt BA (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol 189, 113–130. [DOI] [PubMed] [Google Scholar]
- Sullivan CJ, Pendleton ED, Sasmor HH, Hicks WL, Farnum JB, Muto M, Amendt EM, Schoborg JA, Martin RW, Clark LG, et al. (2016). A cell-free expression and purification process for rapid production of protein biologics. Biotechnol. J 11, 238–248. [DOI] [PubMed] [Google Scholar]
- Swartz JR, Jewett MC, and Woodrow KA (2004). Cell-free protein synthesis with prokaryotic combined transcription-translation. Methods Mol. Biol 267, 169–182. [DOI] [PubMed] [Google Scholar]
- Tabor S, and Richardson CC (1985). A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U S A 82, 1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi MK, Hayes CA, Chappell J, Sun ZZ, Murray RM, Noireaux V, and Lucks JB (2015). Characterizing and prototyping genetic networks with cell-free transcription-translation reactions. Methods 86, 60–72. [DOI] [PubMed] [Google Scholar]
- Takahashi MK, Tan X, Dy AJ, Braff D, Akana RT, Furuta Y, Donghia N, Ananthakrishnan A, and Collins JJ (2018). A low-cost paper-based synthetic biology platform for analyzing gut microbiota and host biomarkers. Nat. Commun 9, 3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toby TK, Fornelli L, Srzentić K, DeHart CJ, Levitsky J, Friedewald J, and Kelleher NL (2019). A comprehensive pipeline for translational top-down proteomics from a single blood draw. Nat. Protoc 14, 119–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunitskaya VL, and Kochetkov SN (2002). Structural-functional analysis of bacteriophage T7 RNA polymerase. Biochemistry (Mosc.) 67, 1124–1135. [DOI] [PubMed] [Google Scholar]
- Wandera KG, Collins SP, Wimmer F, Marshall R, Noireaux V, and Beisel CL (2019). An enhanced assay to characterize anti-CRISPR proteins using a cell-free transcription-translation system. Methods. 10.1016/j.ymeth.2019.05.014. [DOI] [PubMed]
- Wang HH, and Church GM (2011). Multiplexed genome engineering and genotyping methods applications for synthetic biology and metabolic engineering. Methods Enzymol. 498, 409–426. [DOI] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, and Church GM (2009). Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang Z, Brock A, and Schultz PG (2003). Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U S A 100, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Miyazono K, Tanokura M, Sawasaki T, Endo Y, and Kobayashi I (2010). Cell-free protein synthesis for structure determination by X-ray crystallography. Methods Mol. Biol 607, 149–160. [DOI] [PubMed] [Google Scholar]
- Winkler R (2010). ESIprot: a universal tool for charge state determination and molecular weight calculation of proteins from electrospray ionization mass spectrometry data. Rapid Commun. Mass Spectrom 24, 285–294. [DOI] [PubMed] [Google Scholar]
- Xu Z, Chen H, Yin X, Xu N, and Cen P (2005). High-level expression of soluble human beta-defensin-2 fused with green fluorescent protein in Escherichia coli cell-free system. Appl. Biochem. Biotechnol 127, 53–62. [DOI] [PubMed] [Google Scholar]
- Yang WC, Patel KG, Lee J, Ghebremariam YT, Wong HE, Cooke JP, and Swartz JR (2009). Cell-free production of transducible transcription factors for nuclear reprogramming. Biotechnol. Bioeng 104, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim SS, Johns NI, Park J, Gomes ALC, McBee RM, Richardson M, Ronda C, Chen SP, Garenne D, Noireaux V, et al. (2018). Multiplex transcriptional characterizations across diverse and hybrid bacterial cell-free expression systems. Mol. Syst. Biol 15, e8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TS, Ahmad I, Yin JA, and Schultz PG (2010). An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol 395, 361–374. [DOI] [PubMed] [Google Scholar]
- Young TS, and Schultz PG (2010). Beyond the canonical 20 amino acids: expanding the genetic lexicon. J. Biol. Chem 285, 11039–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawada J, and Swartz J (2006). Effects of growth rate on cell extract performance in cell-free protein synthesis. Biotechnol. Bioeng 94, 618–624. [DOI] [PubMed] [Google Scholar]
- Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, Gold DS, Heinsohn HG, and Murray CJ (2011). Microscale to manufacturing scale-up of cell-free cytokine production–a new approach for shortening protein production development timelines. Biotechnol. Bioeng 108, 1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for this study will be provided upon reasonable request and agreement. Such requests will be fulfilled by the Lead Contact, Michael C. Jewett (m-jewett@northwestern.edu).