

Abstract
22q11.2 deletion syndrome is characterised by a well defined microdeletion that is associated with a high risk of neuropsychiatric disorders, including intellectual disability, schizophrenia, attention-deficit hyperactivity disorder, autism spectrum disorder, anxiety disorders, seizures and epilepsy, and early-onset Parkinson’s disease. Preclinical and clinical data reveal substantial variability of the neuropsychiatric phenotype despite the shared underlying deletion in this genetic model. Factors that might explain this variability include genetic background effects, additional rare pathogenic variants, and potential regulatory functions of some genes in the 22q11.2 deletion region. These factors might also be relevant to the pathophysiology of these neuropsychiatric disorders in the general population. We review studies that might provide insight into pathophysiological mechanisms underlying the expression of neuropsychiatric disorders in 22q11.2 deletion syndrome, and potential implications for these common disorders in the general (non-deleted) population. The recurrent hemizygous 22q11.2 deletion, associated with 22q11.2 deletion syndrome, has attracted attention as a genetic model for common neuropsychiatric disorders because of its association with substantially increased risk of such disorders.1 Studying such a model has many advantages. First, 22q11.2 deletion has been genetically well characterised.2 Second, most genes present in the region typically deleted at the 22q11.2 locus are expressed in the brain.3–5 Third, genetic diagnosis might be made early in life, long before recognisable neuropsychiatric disorders have emerged. Thus, this genetic condition offers a unique opportunity for early intervention, and monitoring individuals with 22q11.2 deletion syndrome throughout life could provide important information on factors contributing to disease risk and protection. Despite the commonly deleted region being shared by about 90% of individuals with 22q11.2 deletion syndrome, neuropsychiatric outcomes are highly variable between individuals and across the lifespan. A clear link remains to be established between genotype and phenotype.3,5 In this Review, we summarise preclinical and clinical studies investigating biological mechanisms in 22q11.2 deletion syndrome, with a focus on those that might provide insight into mechanisms underlying neuropsychiatric disorders in 22q11.2 deletion syndrome and in the general population.
Neuropsychiatric phenotype in 22q11.2 deletion syndrome
Many psychiatrists will encounter a patient with 22q11.2 deletion syndrome during their career because of the recurrent nature of the associated deletion and high prevalence of psychopathology in this population. However, such an encounter might often occur unknowingly because of ongoing under-recognition of the syndrome. Schizophrenia and related psychotic disorders in 22q11.2 deletion syndrome have been the most widely studied. Lifetime prevalence of schizophrenia in 22q11.2 deletion syndrome is estimated to be about 25%.1 Individuals with 22q11.2 deletion syndrome are also likely to have other neurodevelopmental disorders. Prevalence estimates of autism spectrum disorder (ASD) and attention-deficit hyperactivity disorder (ADHD), typically diagnosed in childhood, are about 35%.1 Estimated prevalence of (mild to severe) intellectual disability is 45%.1 Anxiety disorders are also particularly common among individuals with 22q11.2 deletion syndrome across the lifespan, with reported prevalences of 36% in childhood and around 25% in adulthood.1 However, 22q11.2 deletion syndrome could provide some protection against substance use disorders as they appear to be less common in individuals with this syndrome than in those without.6 ASD, anxiety disorders, and schizophrenia spectrum disorders are seen in all age groups, with a peak prevalence in the age range when these disorders are typically diagnosed, providing some evidence for stability of the psychiatric phenotype across the lifespan.1 Phenotypic presentations of psychiatric disorders are similar to those in the general (non-deleted) population, although subtle differences have been reported with respect to ASD, ADHD, and psychosis.7–9
In addition to the high rate of psychopathology, particular neurological disorders, including movement disorders, seizures (often hypocalcaemic), and epilepsy, are more prevalent in individuals with 22q11.2 deletion syndrome than in the general population.10–12 Individuals with 22q11.2 deletion syndrome are estimated to have at least a 20-times increased risk, compared with estimates for the general population, to develop early-onset Parkinson’s disease, a progressive neurodegenerative disease affecting motor, cognitive, and autonomic functions.13,14 Antipsychotics can stimulate both parkinsonism and seizures, and anticonvulsants might be associated with parkinsonism.15 These side-effects are pertinent as individuals with 22q11.2 deletion syndrome are commonly treated with these medications for underlying psychotic and seizure disorders.10,14,16
Neuropsychiatric disorders associated with 22q11.2 deletion syndrome arise across various stages of life. Some occur during early neurodevelopment (eg, ADHD and ASD), some are best explained as neurodegenerative processes (ie, Parkinson’s disease), and others can emerge at any age across the lifespan (eg, anxiety disorders and psychotic illness). Therefore, 22q11.2 deletion syndrome allows us to study the occurrence of neuropsychiatric disorders in the context of both brain development and ageing.
The molecular neurobiology underlying of the 22q11·.2 deletion syndrome phenotype
Genetics
22q11.2 deletion syndrome is caused by a hemizygous deletion of the long (q) arm of chromosome 22 (figure 1) and is the most common human microdeletion syndrome (OMIM 188400/192430). Estimated prevalence is one in every 4000 live births.17 In about 90% of newly diagnosed cases, the deletion is a de novo occurrence—ie, a spontaneous mutation. In the remaining 10%, the deletion is inherited from one of the parents.2 The 22q11.2 locus has multiple DNA sequences that are highly similar (low copy repeats), rendering this region particularly prone to misalignment during meiosis. This mechanism explains the recurrent nature of copy number variations in this area.18 About 85–90% of cases have a 3 Mb deletion, which is referred to as the commonly deleted region (figure 2). About 90 genes are involved, which include 46 protein-coding genes as well as pseudogenes, non-coding RNAs, and seven microRNAs (figure 2).2,4,19 In about 5% of cases, 22q11.2 deletion involves only the proximal 1·5 Mb. Available data indicate similar phenotypic features as for the typical 3 Mb deletion,2 although evidence also suggests that the nested 1·5 Mb deletion could lead to milder phenotypic expression.20,21
Figure 1: Cytogenetic representation of chromosome 22.
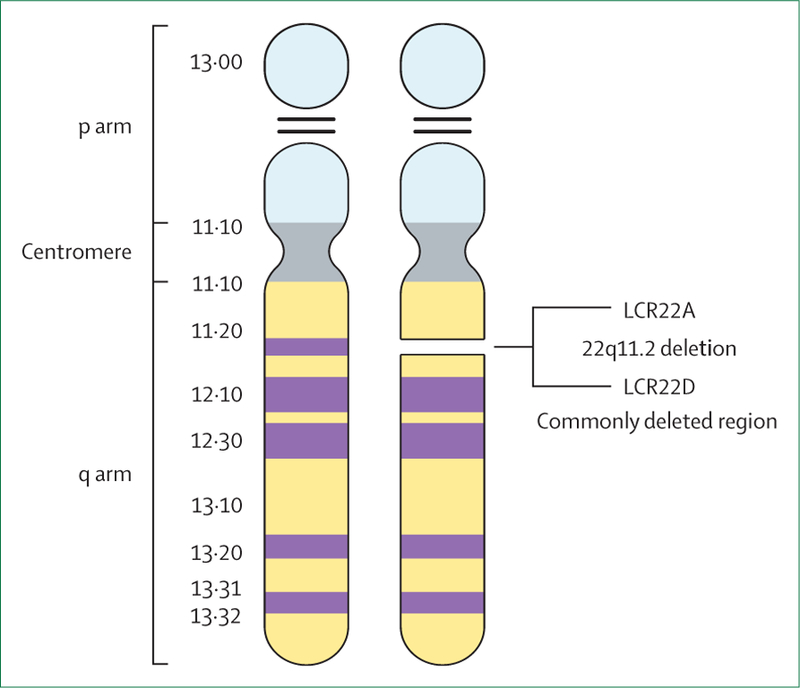
Adapted from McDonald-McGinn et al2 with permission. LCR=low copy repeat.
Figure 2: 22q11.2 region.
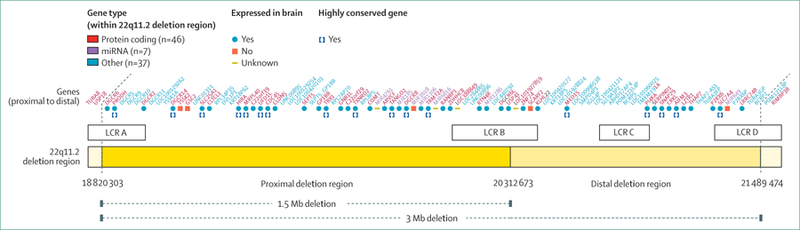
Reproduced from Guna et al.4 The 3 Mb 22q11.2 region (hg19 assembly, coordinates) with genes (red), miRNAs (purple), and others (eg, pseudogenes and non-coding RNA, light blue). The locations of the four 22q11.2 specific breakpoints, mediated by LCRs, are LCR22A, LCR22B, LCR22C, and LCR22D (white boxes). LCR=low copy repeats.
Variable penetrance of brain-related phenotypes
Researchers have long been interested in 22q11.2 deletion syndrome as a molecular subtype of schizophrenia and have investigated genes in the 22q11.2 locus as candidate risk genes.3,5 Hemizygosity of the 22q11.2 region is clearly an important causal mechanism because of decrease of gene dosage. However, this decrease alone is not enough to explain the increased risk of neuropsychiatric disorders in 22q11.2 deletion syndrome, given that none of the associated neuropsychiatric phenotypes show complete penetrance;2,3,5,22 the neuropsychiatric phenotype is highly variable, and correlation between phenotype and deletion size is weak.2 This pleiotropy could be due to activity of proteins coded by the remaining alleles,22 or compensatory mechanisms. In addition, evidence suggests that the 22q11.2 region contains regulatory genes that affect gene expression outside of the 22q11.2 region19 and that genetic background variation might affect phenotypic expression.23 Furthermore, 22q11.2-encoded genes might not equally contribute to all aspects of neuropsychiatric disorders.3,5
A multiple-hit pathway hypothesis postulates that a first hit (22q11.2 deletion) lowers the threshold for expression of genetic variation elsewhere in the genome (additional hits).24 Evidence for such a mechanism in 22q11.2 deletion syndrome comes from several studies.24–27 A proof-of-principle study using whole-genome sequencing in a small but clinically well phenotyped sample of individuals with 22q11.2 deletion syndrome reported a higher burden of additional rare mutations in protein-coding neurofunctional genes in individuals with 22q11.2 deletion syndrome and schizophrenia than in those without schizophrenia,24 and a higher burden of additional rare mutations in genes associated with Parkinson’s disease in individuals with Parkinson’s disease than in individuals without the disease.25 Additionally, individuals with 22q11.2 deletion syndrome and schizophrenia carry more rare deletions that overlap with protein-coding genes than individuals with 22q11.2 deletion syndrome without schizophrenia.26 Rare cases also exist in which a mutation in a gene on the remaining haploid allele of the 22q11.2 locus results in a recessive disorder in 22q11.2 deletion syndrome.27 Together, these findings implicate a role for genome-wide variants in disease risk in 22q11.2 deletion syndrome.
Many studies have investigated the potential effects of 22q11.2 genes using mouse models. Many results from studies that did not control for genetic background have not been replicated, and new genes and mechanistic insights have emerged from later, well controlled studies. This process is being accelerated, as the International Mouse Phenotyping Consortium is screening co-isogenic mouse models (in which genetic background is completely controlled) of many 22q11.2-encoded genes in translational phenotypes, including working memory, vocalisation, and prepulse inhibition, a behavioural phenotype seen in patients with 22q11.2 deletion syndrome and idiopathic cases of schizophrenia, obsessive-compulsive disorder, ADHD, and seizure disorder.28 These studies have revised our understanding of 22q11.2 driver genes that are likely to contribute to aspects of schizophrenia and ASD. Table 1 provides an overview of 22q11.2 genes for which substantial evidence exists for a behavioural phenotype in mice.
Table 1:
Overview of individual 22q11.2 genes for which strong evidence exists from mouse studies for an effect on neuropsychiatric phenotype
| Effect of gene deletions on behavioural phenotypes in mice | Effect of recessive mutation on phenotype in humans | PubMed ID | |
|---|---|---|---|
| Proline dehydrogenase (PRODH) | Altered adult vocalization and prepulse inhibition deficit | Homozygous deletion resulted in hyperprolinaemia type 1: neurological deficits, psychomotor delay, hypotonia, and seizures | 16234811, 24194600 |
| DiGeorge Critical Region 8 (DGCR8) | Deficit in working memory and prepulse inhibition | Unknown | 27892953, 23719809 |
| Cathechol-o-methyltransferase (COMT) | No deficit in prepulse inhibition, working memory, social behaviour, and adult vocalisation | Unknown; studies of Val158Met polymorphism found no clear effect on risk of schizophrenia or other psychiatric disorders, but some association of high-activity Val allele with cognitive dysfunction in schizophrenia | 18753372, 25754081, 24194600, 20631688 |
| T-box 1 (TBX1) | Impaired social interaction and communication, impaired working memory, and heightened anxiety | Point mutations resulted in 22q11.2 deletion syndrome-like physical phenotype | 21908517, 26666205, 11239417, 14585638, 17916582, 24637876, 11748311 |
| Septin 5 (SEPT5) | Impaired social interaction, no prepulse inhibition deficit, and no adult vocalisation deficit | Homozygous deletion of GP1BB and SEPT5 resulted in bleeding disorder, developmental delay, and polymicrogyria | 19240081, 22589251, 21800012 |
All genes are in the commonly deleted 3 Mb (and smaller 1.5 Mb) 22q11.2 region. Genes are listed in order according to map location. Some candidate genes discussed in this Review are not listed because mutant mouse models have not shown evidence for effects of these genes on behavioural phenotypes related to schizophrenia or autism spectrum disorder. In such cases, dissociation exists between behavioural and neuronal phenotypes and further work is needed to identify what behavioural dimensions of neuropsychiatric disorders these neuronal phenotypes might affect. Derived from Hiroi5 and International Mouse Phenotyping Consortium (IMPC), see Koscielny et al, 2014.28
Disruption of miRNA mechanisms
A candidate mechanism with respect to the multiple-hit hypothesis in 22q11.2 deletion syndrome is disruption of miRNA pathways. A miRNA is a small, non-coding RNA molecule that regulates gene expression at the post-transcriptional stage by silencing mRNAs. miRNA-mediated regulation of gene expression plays an important role in fundamental biological processes, such as cell proliferation, differentiation, migration, and apoptosis (cell death).29
The DiGeorge Syndrome Critical Region Gene 8 (DGCR8), located in the commonly deleted region, encodes a key miRNA processing protein.19 Increasing evidence suggests that hemizygosity of DGCR8 might disrupt miRNA functioning.19,30,31 Several miRNAs are encoded in the 22q11.2 region.19 Disruption of miRNA-mediated post-transcriptional regulation of gene expression in 22q11.2 deletion syndrome could directly or indirectly affect expression of neuropsychiatric risk genes elsewhere in the genome through upregulated or downregulated gene expression. DGCR8 expression and the expression of several miRNAs (most of which were located outside the 22q11.2 region) in peripheral blood were shown to be reduced in individuals with 22q11.2 deletion syndrome as compared with controls.32 In a 22q11.2 deletion mouse model (Df(16)A+/−), loss of a copy of micro-RNA, mir-185, which resides in the 22q11.2 locus, resulted in over-expression of a neuronal protein, Mirta22.33 Downregulation of Mirta22 restores the reduced prepulse inhibition in the 22q11.2 deletion mouse model to the levels seen in wild-type mice.34
Mouse studies have connected dysfunctional biogenesis of miRNA in 22q11.2 deletion syndrome to gradual onset of symptoms and to several systems previously implicated in causes of schizophrenia. Dgcr8 haploinsufficiency leads to reduced amounts of miR-338–3p, which downregulates the dopamine D2 receptor in brain regions relevant for schizophrenia.30,31 Failure to downregulate the D2 receptor led to excessive dopaminergic neurotransmission and synaptic defects of thalamo-cortical connections, which were reversible by antipsychotics.30 In a mouse model of 22q11.2 deletion syndrome, sensitivity to antipsychotics was not seen in young animals but only in adolescent and adult mice.31 The underlying mechanism was hypothesised to be gradual decline of miRNA as animals aged.
Further research is needed to substantiate and explore the biological and clinical implications of disruption of miRNA systems in 22q11.2 deletion syndrome.
Mitochondrial dysfunction
The 22q11.2 region contains at least six genes involved in mitochondrial function (MRPL40, PRODH, SLC25A1, TANGO2, TXNRD2, and ZDDHC8).35 In children with 22q11.2 deletion syndrome, metabolic abnormalities have been found and gene dosage of SLC25A1 was implicated.36 Mitochondrial dysfunction is strongly implicated in the pathogenesis of Parkinson’s disease, particularly in early-onset forms.37 The functional contribution of these genes to neuropsychiatric disorders remains unclear, as prepulse inhibition remains at normal levels in mice deleted for Prodh, Slc25a1, or Tango228,35 and in mice with segmental deletions, including Mrpl40, Prodh, Slc25a1, Tango2, Txnrd2, or Zdhhc8.38
Brain development, structure, and function
Imaging studies
Early imaging studies investigated whole brain, regional, grey matter, and white matter volumes, and generally found whole-brain volume reductions in individuals with 22q11.2 deletion syndrome compared with controls.39 Recent studies have focused on specific cortical measurements, including cortical thickness, surface area, and cortical gyrification, measured by the gyrification index. Gyrification is established early in life as opposed to cortical thinning, which occurs with aging.40 Several studies have reported reduced local gyrification in children and adolescents with 22q11.2 deletion syndrome compared with healthy controls and this finding could be indicative of neurodevelopmental pathology.41–43 Altered trajectories of cortical thickness have been reported in individuals with 22q11.2 deletion syndrome, with slow thinning in children and accelerated thinning in adolescents.44 The largest neuroimaging study to date in children and adults with 22q11.2 deletion syndrome (aged 8–50 years) reported a clear cortical phenotype in individuals with 22q11.2 deletion syndrome in comparison to controls, with thicker cortex bilaterally in major regions, thinner cortex in the superior temporal, cingulate, and parahippocampal cortex, and global reductions of surface area.21 Larger 22q11.2 deletion size (typical 3 Mb vs nested 1·5 Mb) was associated with reduced cortical surface area.
Some evidence exists for differential trajectories of brain development in individuals with 22q11.2 deletion syndrome who develop psychosis compared with those who do not.44,45 Physiological peak and decline of cortical thickness in individuals with 22q11.2 deletion syndrome who developed psychotic symptoms occurred later on average than in children and adolescents with typical development, and the decline in cortical thickness was steeper.45 This decline, which might reflect loss of functional neurons, could be linked to the decrease in verbal IQ observed in children and young people with 22q11.2 deletion syndrome before onset of psychotic symptoms.46 Bakker and colleagues43 found reduced gyrification in young adults with a 22q11.2 deletion (some with a history of psychosis and on antipsychotics) compared with individuals without 22q11.2 deletion at clinical ultra-high risk for psychosis, indicative of early neurodevelopmental pathology. In the latter group, cortical thickness of the insula was consistently lower than in individuals with 22q11.2 deletion syndrome, which could suggest defective pruning processes during adolescence in 22q11.2 deletion syndrome.43 With respect to ASD, a study47 used surface area as a marker for underlying neurobiology of ASD and reported substantial differences between unmedicated individuals with 22q11.2 deletion syndrome with ASD and those without.
As opposed to cortical measures and grey matter which reflect neurons and dendrites, white matter tracts reflect long-range myelinated connecting fibres between parts of the brain. Connectivity in the brain can be measured structurally using diffusion tensor imaging, and functionally using resting-state functional MRI (rs-fMRI). Some diffusion tensor imaging studies that have been done in individuals with 22q11.2 deletion syndrome found evidence for altered myelin and axon integrity in multiple tracts, some of which have been implicated in neuropsychiatric disorders.48–50 Studies using rs-fMRI reported a global decrease in functional connectivity.51,52 Preliminary results from the ENIGMA-22q working group, who collected the largest data set on 22q11.2 deletion syndrome to date, suggest lower diffusivity in individuals with 22q11.2 deletion syndrome than in healthy controls, possibly because of smaller axonal diameter.53 Microstructural white matter differences (increased fractional anisotropy) have been associated with cognitive decline in young people with 22q11.2 deletion syndrome. These microstructural changes were not seen in those without cognitive decline.50 Microstructural white matter changes in individuals with 22q11.2 deletion syndrome were associated with lower axonal integrity, whereas in individuals without deletion at ultra-high risk for psychosis, such changes were suggestive of abnormal myelination.49 Together, these data provide strong evidence that the 22q11.2 deletion has profound effects on brain structure in children and adults.
Neurogenesis and angiogenesis as potential mechanisms underlying the neuropsychiatric phenotype in 22q11.2 deletion syndrome DGCR8 and TBX1 have been consistently implicated in neurogenesis in animal models. Dgcr8 deficiency has been shown to contribute to deficits in working memory and prepulse inhibition in rodents,31,54 potentially by reduced mediation of miRNA biogenesis. Tbx1, a transcription factor important for tissue and organ formation during embryonic development, including brain angiogenesis, might partly be responsible for cortical abnormalities related to 22q11.2 deletion syndrome.55 Point mutations of Tbx1 alone might be enough to cause most of the physical features of the 22q11.2 deletion syndrome phenotype.56,57 However, Tbx1 point mutations are extremely rare and have been scarcely studied in humans. In mice, inactivation of Tbx1 results in abnormalities in brain endothelial cells and different patterns of brain vascularisation.55 Disorganisation of the cerebral vascular network might, therefore, be linked to findings of neuroimaging studies in patients with 22q11.2 deletion syndrome, such as reduced grey matter,58 altered cortical thickness,21 and tortuous vessels.59 Mice heterozygous for Tbx1 have shown abnormal cortical development,60 with altered differentiation of cortical stem cells, also known as neural progenitors, and changes to the distribution of glutamatergic cortical projection neurons and γ-aminobutyric-acid (GABA)ergic interneurons.60 Tbx1-heterozygous mice showed defective social interaction and communication, impaired working memory, and heightened anxiety in a stressful condition.61,62 These ASD-related behavioural phenotypes might suggest a role of TBX1 in the cause of ASD.61 Thus, TBX1 is a strong candidate gene for abnormalities in brain and behaviour seen in 22q11.2 deletion syndrome.
Disruptions of neurotransmitter systems
Serotonin
In humans with 22q11.2 deletion syndrome, the high prevalence of anxiety disorders, in addition to anecdotal reports of good response to selective serotonin-reuptake inhibitors (SSRIs),63 suggests that serotonergic neurotransmission might be disrupted. However, no clinical trials have been published to date examining treatment response to SSRIs in individuals with 22q11.2 deletion syndrome, and clinical safety and efficacy is based on expert opinion, observational studies, and case series and reports.64,65 The only study investigating serotonergic disruptions in humans with 22q11.2 deletion syndrome reported lower mean urine serotonin concentrations in adults with 22q11.2 deletion syndrome than in healthy controls.66 Serotonin concentrations also showed a weak positive association with full-scale IQ in individuals with 22q11.2 deletion syndrome.66
Catecholamines
Mapping within the commonly deleted region, the catechol-O-methyltransferase (COMT) gene is important in degradation of catecholamines, including dopamine and norepinephrine. COMT expression and enzyme activity levels in peripheral blood cells have been found to be reduced by about 50% in individuals with 22q11.2 deletion syndrome.22 Disturbances of dopaminergic markers in urine, plasma, and cerebrospinal fluid suggest impaired catecholamine metabolism in 22q11.2 deletion syndrome,66,67 which might be more prominent in women than men.68 Systematic in-vivo molecular brain imaging studies of dopamine activity in the striatum provide some evidence for a presynaptic hyperdopaminergic state in 22q11.2 deletion syndrome (table 2).
Table 2:
Studies of in-vivo molecular brain imaging of dopaminergic systems in 22q11.2 deletion syndrome
| Psychotic or non-psychotic illness | Radioligand | Presynaptic or postsynaptic | Main findings | |
|---|---|---|---|---|
| 14 22q11.2 deletion syndrome (4 male, 10 female), 16 healthy controls (5 male, 11 female) | No history of psychotic illness | [18F]-DOPA | Presynaptic | Significantly increased capacity of dopamine synthesis in 22q11.2 deletion syndrome89 |
| 13 22q11.2 deletion syndrome (8 male, 5 female), 12 healthy controls (8 male, 4 female) | Psychotic illness in seven (54%) of 13 patients with 22q11.2 deletion syndrome | [11C]-DTBZ | Presynaptic | Significantly increased binding of [11C]-DTBZ in 22q11.2 deletion syndrome in 12 patients without Parkinson’s disease; severely reduced binding in one patient with Parkinson’s disease75 |
| 12 22q11.2 deletion syndrome (4 male, 8 female), 16 healthy controls (4 male, 12 female) | No history of psychotic illness | [18F]-Fallypride | Postsynaptic | No association between dopamine release and amount of reward in 22q11.2 deletion syndrome in contrast to healthy controls90 |
| 15 22q11.2 deletion syndrome (Met 3 male and 7 female, Val 3 male and 2 female) | No history of psychotic illness | [123I]-IBZM | Postsynaptic | Significantly higher availability of D2/3 receptors in Val carriers91 |
| 12 22q11.2 deletion syndrome (5 male, 7 female),* 12 healthy controls (5 male, 7 female) | No history of psychotic illness | [123I]-IBZM | Postsynaptic | No differences in availability of D2/3 receptors92 |
All studies investigated striatal dopamine. Met=low activity COMT158 allele. Val=high activity COMT158 allele.
Same patients with 22q11.2 deletion syndrome as in Boot and colleagues.91
In mouse models of 22q11.2 deletion syndrome with a hemizygous deletion including Comt (Df(h22q11)/+ and Df1/+), increased concentrations of dopamine metabolite, dihydroxyphenylacetic acid, but normal dopamine concentrations, were found in prefrontal cortex and dorsal striatum.69,70 Comt heterozygosity did not affect behavioural phenotypes, including prepulse inhibition, social behaviours, and anxiety-related behaviours in mice,5 consistent with another mouse model (Df2) in which the deletion includes Comt and also results in normal prepulse inhibition.57 Some evidence exists for sex dichotomous effects of Comt,71 which might be explained by inhibitory regulation of Comt by oestrogens.72
Although COMT activity is reduced in patients with 22q11.2 deletion syndrome and in mouse models for 22q11.2 deletion syndrome, functional relevance to brain function and neuropsychiatric disorders remains unclear.
Parkinson’s disease or hypodopaminergia
22q11.2 deletion was found to be a genetic cause of Parkinson’s disease;13 however, the underlying mechanisms are unknown.13,73 Similarly, little is known about other motor problems that could arise during development, including neurological soft signs, developmental coordination disorder, and parkinsonism not meeting criteria for Parkinson’s disease. Overlapping symptoms and medication-induced side-effects can complicate or delay the correct diagnosis. Large and longitudinal studies on motor functioning in the context of 22q11.2 deletion syndrome are needed to improve delineation of the neurological profile over development and into adulthood. In Parkinson’s disease associated with 22q11.2 deletion syndrome, major clinical characteristics and response to standard treatments, including dopamine replacement therapy, appear to be similar to those in idiopathic Parkinson’s disease. However, onset of Parkinson’s disease in 22q11.2 deletion syndrome is most often early, with a reported average age of about 40 years.14
Preclinical and clinical research on neurobiological aspects of Parkinson’s disease in 22q11.2 deletion syndrome is scarce. One neuropathological study13 found typical loss of dopaminergic neurons in three cases of Parkinson’s disease associated with 22q11.2 deletion syndrome, and Lewy pathology in two of these cases. Raised concentrations of α-synuclein, a primary component of Lewy bodies, and motor coordination deficits have been identified in the Df1/+ mouse model of 22q11.2 deletion syndrome. Reducing α-synuclein gene dosage in Df1/+ mice ameliorated the motor deficits.74 Typical findings of the expected pattern of reduced striatal binding with presynaptic dopaminergic imaging, indicative of severe loss of presynaptic striatal neurons that is a hallmark of Parkinson’s disease, have been shown in Parkinson’s disease associated with 22q11.2 deletion syndrome using molecular neuroimaging.14
Some evidence from simple model organisms shows that the 22q11.2 genes, PRODH and TXNRD2, could be involved in motor functioning.4 A multi-hit mechanism that could explain increased risk of Parkinson’s disease in 22q11.2 deletion syndrome has been postulated and involves a hyperdopaminergic presynaptic state, as suggested by presynaptic dopaminergic neuroimaging findings, which might interact with, or be compounded by, a deficient dopamine clearing mechanism related to the effects of missing one copy of COMT.67,75 Chronic exposure to the neurotoxic properties of dopamine and its metabolites has been proposed to be involved in pathogenesis of Parkinson’s disease.76 Impaired mitochondrial function in 22q11.2 deletion syndrome might contribute to increased oxidative stress and vulnerability to dopaminergic cell death in 22q11.2 deletion syndrome.37,75
Putative effects of PRODH deficiency and hyperprolinaemia
PRODH encodes the enzyme proline dehydrogenase that converts proline to glutamate, the primary excitatory neurotransmitter in the brain. PRODH has been extensively studied because of its location in the commonly deleted region, because hyperprolinaemia is common in 22q11.2 deletion syndrome,77 because hyperprolinaemia has been associated with increased risk of schizophrenia-spectrum disorders, 78 and because hyperprolinaemia has been shown to result in glutamate excess.79 In mice, homozygous loss of Prodh resulted in abnormal vocalisation.28
In humans, reports have occasionally suggested genetic association between PRODH and psychosis.80 Three studies have reported potential interaction between COMT and PRODH. Adolescents with 22q11.2 deletion syndrome and hyperprolinaemia showed abnormal smooth-pursuit eye movements, a proxy for psychosis susceptibility, when they carried the low activity COMT158Met allele.81 In another group of individuals with 22q11.2 deletion syndrome, COMT158Met was associated with increased psychosis risk in those with hyperprolineamia, and proline concentrations were inversely correlated with full-scale IQ.77 Radoeva and colleagues82 reported that within a group of individuals with 22q11.2 deletion syndrome, those with an ASD diagnosis were enriched for the low-activity alleles of COMT and PRODH. Although these findings provide some support for interaction between COMT and PRODH, the effect on the psychiatric phenotype in 22q11.2 deletion syndrome remains unclear.
The glutamate/GABA system in patients with a 22q11.2 deletion
In-vivo studies on the glutamate system in 22q11.2 deletion syndrome are scarce.83,84 One study83 using 1H-MR spectroscopy reported no differences in glutamate concentrations in the prefrontal cortex between adults with 22q11.2 deletion syndrome with and without a history of psychosis, or with healthy controls. The same group reported that glutamate concentrations in the hippocampus were higher in individuals with 22q11.2 deletion syndrome with psychosis than in individuals without psychosis.83 Another cross-sectional study84 investigating adults with 22q11.2 deletion syndrome reported a negative association between serum glutamate concentrations and full-scale IQ, a positive association between glutamate concentrations and dose of antipsychotic medication, and no association between serum glutamate concentrations and history of psychosis.
SEPT5, located in the commonly deleted region, encodes septin-5 protein, which is thought to inhibit exocytosis of dopamine and glutamate85 and to be involved in serotonin release from platelets.86 Therefore, SEPT5 deficiency might contribute to some of the altered dopamine, glutamate, and serotonin availability observed in 22q11.2 deletion syndrome.66 In mice, Sept5 deficiency and over-expression selectively impair and enhance reciprocal social interaction, respectively; Sept5 deficiency does not impair prepulse inhibition, working memory, vocalisation, or anxiety.23,28,87
Outlook
For neuroscientists, 22q11.2 deletion syndrome offers a unique view into the neurobiology of common developmental and neurodegenerative disorders. For clinicians, recognising 22q11.2 deletion syndrome is relevant because of the high prevalence of such disorders and the specific knowledge required for optimal treatment.63
The common theme arising from research of the past 20 years is that the phenotype of 22q11.2 deletion syndrome varies substantially. Investigating effects of genetic variants, as well as environmental factors (eg, trauma and stress) known to affect psychiatric functioning in the general population, and how they contribute to increased risk of psychopathology in 22q11.2 deletion syndrome, will provide valuable information for those with and without the syndrome (figure 3).88 Multiple complex processes are probably involved, including development of the cortex and white matter, dopaminergic neurotransmission, and the balance between glutamate and GABA signalling. Hemizygous deletion at the 22q11.2 locus probably disrupts essential developmental aspects of these processes. Many studies have investigated brain structure, function, and development in 22q11.2 deletion syndrome, with sometimes contradictory results.39 This discrepancy is likely to be due to methodological differences and underpowered studies. Results from (for example) the ENIGMA 22q11.2 Working Group88 for human brain imaging studies, and the efforts of the International Mouse Phenotyping Consortium28 to optimise mouse assays, should address these problems and increase our understanding of human brain structure and function in 22q11.2 deletion syndrome.
Figure 3: Clinical and translational phenotypes relevant to neuropsychiatric disorders in 22q11.2 deletion syndrome.
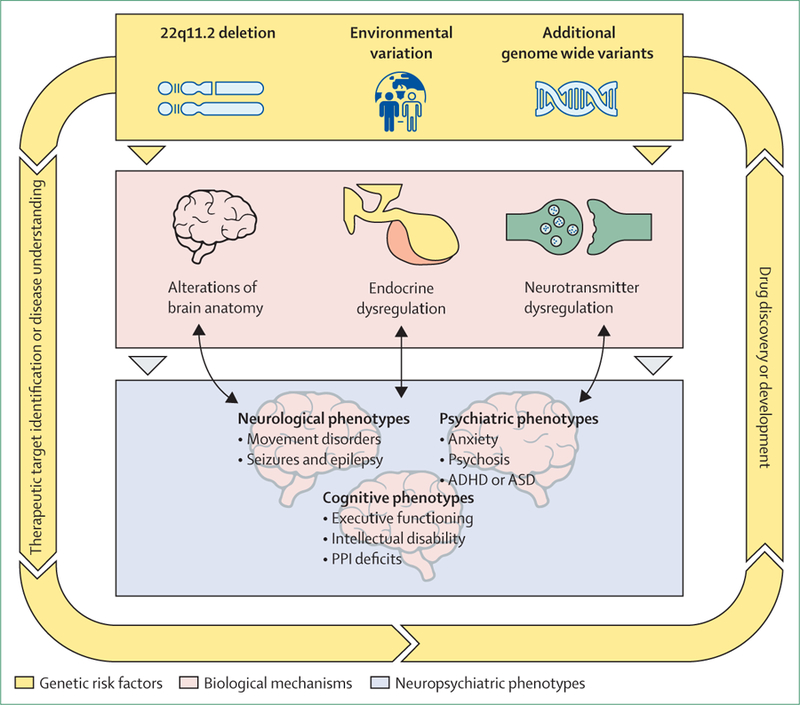
The coloured boxes represent different levels that illustrate how known genetic variability might improve knowledge on pathophysiological mechanisms underlying neuropsychiatric disorders. The top level (yellow) indicates genetic variation (22q11.2 deletion and additional genetic variation). The middle level (pink) indicates biological systems that are known to be altered in 22q11.2 deletion syndrome. Alterations in biological mechanisms can lead to the clinical and translational phenotypes pictured in the bottom level (lilac). The reciprocal arrows between the biological level and clinical phenotypes illustrate that clinical phenotypes might, in turn, affect biological processes. Arrows around the figure indicate that 22q11.2 deletion or known additional genetic variation, or both, might contribute to disease understanding and could lead to therapeutic target identification, and, ultimately, to drug discovery and development. ADHD=attention-deficit hyperactivity disorder. ASD=autism spectrum disorder. PPI=prepulse inhibition.
Multidisciplinary strategies should include studies of post-mortem brain tissue of individuals with 22q11.2 deletion syndrome, animal models, and studies in humans, connecting in-vivo measurements such as imaging, electrophysiology, and behavioural assessments, with in-vitro models such as induced pluripotent stem cells and brain organoids. Results from large-scale prospective studies following well phenotyped individuals with 22q11.2 deletion syndrome from early in life (before symptoms emerge) can provide information on pathways and mechanisms that underlie the transition to brain-related phenotypes and deliver an unprecedented opportunity to study the effects of early interventions. Studying individuals with 22q11.2 deletion syndrome who do not develop neuropsychiatric symptoms is equally important to identify biological factors associated with potential protective effects. Some mechanisms we have discussed have promising therapeutic implications, which we hope researchers will capitalise on. The next few decades might deliver novel strategies and identification of early predictors relevant for treatment of neuropsychiatric disorders in individuals with and without 22q11.2 deletion syndrome. 22q11.2 deletion syndrome might provide an excellent opportunity for studying disease-modifying treatments for common neuropsychiatric disorders.
Search strategy and selection criteria.
We searched PubMed for articles published between Jan 1, 1971, and Jan 7, 2019, with combinations of the following search terms: “22q11”, “22q11.2DS”, “22q11.2 deletion”, “22q11 deletion syndrome”, “22q11.2 deletion syndrome”, “velocardiofacial syndrome”, “DiGeorge syndrome”, “dopamine”, “catecholamine”, “monoamine”, “Parkinson’s disease”, “psychiatric”, “mental disorder”, “serotonin”, “proline”, “glutamate”, “GABA”, “epilepsy”, “seizures”, “EEG”, “imaging”, “MRI”, “MR spectroscopy”, “genetics”, “genomic”, “miRNA”, “iPSCs”, “post-mortem”, “anatomy”, “cortical development”, “cortical layer”, and “electrophysiology”. We selected and reviewed articles from this search, and reviewed relevant references cited in the selected papers. We included peer-reviewed publications, and some published abstracts, and excluded articles not written in English.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (MOP #97800, MOP #111238), Dalglish Chair, a McLaughlin Centre Accelerator Grant, and National Institute of Mental Health grants U01 MH101723–01(3/5), R01 MH099660, R01 DC015776, and U54 HD090260. These funding sources had no role in interpretation of data, preparation, review, or approval of the manuscript, or decision to submit the manuscript for publication. We would like to thank Geoffrey Cramm for his assistance with design of the figures.
Footnotes
Declaration of interests
NH received funding from Astellas on a topic unrelated to this Review, outside the submitted work. All other authors declare no competing interests.
Contributor Information
Janneke R Zinkstok, Department of Psychiatry and Brain Center, University Medical Center, Utrecht, Netherlands.
Erik Boot, ‘s Heeren Loo Zorggroep, Amersfoort, Netherlands; The Dalglish Family 22q Clinic for Adults with 22q11.2 Deletion Syndrome, University Health Network, Toronto, ON, Canada; Department of Psychiatry & Neuropsychology, Maastricht University, Maastricht, Netherlands; Department of Radiology and Nuclear Medicine, Amsterdam University Medical Center, Amsterdam, Netherlands.
Anne S Bassett, The Dalglish Family 22q Clinic for Adults with 22q11.2 Deletion Syndrome, University Health Network, Toronto, ON, Canadal; Clinical Genetics Research Program, Centre for Addiction and Mental Health, Toronto, ON, Canada, Department of Psychiatry, University of Toronto, Toronto, ON, Canada; Campbell Family Mental Health Research Institute, Toronto, ON, Canada; Division of Cardiology & Toronto General Hospital Research Institute, University Health Network, Toronto, ON, Canada.
Noboru Hiroi, Department of Pharmacology, Department of Cellular and Integrative Physiology, Department of Cell Systems and Anatomy, Department of Psychiatry, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA.
Nancy J Butcher, Child Health Evaluative Sciences, The Hospital for Sick Children Research Institute, Toronto, ON, Canada.
Claudia Vingerhoets, Department of Psychiatry & Neuropsychology, Maastricht University, Maastricht, Netherlands, Department of Radiology and Nuclear Medicine, Amsterdam University Medical Center, Amsterdam, Netherlands.
Jacob A S Vorstman, Sick Children Research Institute, Genetics & Genome Biology Program, Toronto, ON, Canada.
Therese A M J van Amelsvoort, Department of Psychiatry & Neuropsychology, Maastricht University, Maastricht, Netherlands.
References
- 1.Schneider M, Debbane M, Bassett AS, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry 2014; 171: 627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDonald-McGinn DM, Sullivan KE, Marino B, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers 2015; 1: 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiroi N, Takahashi T, Hishimoto A, Izumi T, Boku S, Hiramoto T. Copy number variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol Psychiatry 2013; 18: 1153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guna A, Butcher NJ, Bassett AS. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J Neurodev Disord 2015; 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiroi N Critical reappraisal of mechanistic links of copy number variants to dimensional constructs of neuropsychiatric disorders in mouse models. Psychiatry Clin Neurosci 2018; 72: 301–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vingerhoets C, van Oudenaren MJF, Bloemen OJN, et al. Low prevalence of substance use in people with 22q11.2 deletion syndrome. Br J Psychiatry 2019; published online Jan 3. DOI: 10.1192/bjp.2018.258. [DOI] [PubMed] [Google Scholar]
- 7.Tang KL, Antshel KM, Fremont WP, Kates WR. Behavioral and psychiatric phenotypes in 22q11.2 deletion syndrome. J Dev Behav Pediatr 2015; 36: 639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiksinski AM, Schneider M, Murphy CM, et al. Understanding the pediatric psychiatric phenotype of 22q11.2 deletion syndrome. Am J Med Genet A 2018; published online Sept 8. DOI: 10.1002/ajmg.a.40387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armando M, Girardi P, Vicari S, et al. Adolescents at ultra-high risk for psychosis with and without 22q11 deletion syndrome: a comparison of prodromal psychotic symptoms and general functioning. Schizophr Res 2012; 139: 151–56. [DOI] [PubMed] [Google Scholar]
- 10.Boot E, Butcher NJ, van Amelsvoort TA, et al. Movement disorders and other motor abnormalities in adults with 22q11.2 deletion syndrome. Am J Med Genet A 2015; 167: 639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim EH, Yum MS, Lee BH, et al. Epilepsy and other neuropsychiatric manifestations in children and adolescents with 22q11.2 deletion syndrome. J Clin Neurol 2016; 12: 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung EN, George SR, Costain GA, et al. Prevalence of hypocalcaemia and its associated features in 22q11.2 deletion syndrome. Clin Endocrinol (Oxf) 2014; 81: 190–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butcher NJ, Kiehl TR, Hazrati LN, et al. Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: identification of a novel genetic form of Parkinson disease and its clinical implications. JAMA Neurol 2013; 70: 1359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boot E, Butcher NJ, Udow S, et al. Typical features of Parkinson disease and diagnostic challenges with microdeletion 22q11.2. Neurology 2018; 90: e2059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zadikoff C, Munhoz RP, Asante AN, et al. Movement disorders in patients taking anticonvulsants. J Neurol Neurosurg Psychiatry 2007; 78: 147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wither RG, Borlot F, MacDonald A, et al. 22q11.2 deletion syndrome lowers seizure threshold in adult patients without epilepsy. Epilepsia 2017; 58: 1095–101. [DOI] [PubMed] [Google Scholar]
- 17.Botto LD, May K, Fernhoff PM, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 2003; 112: 101–07. [DOI] [PubMed] [Google Scholar]
- 18.Shaikh TH, Kurahashi H, Saitta SC, et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 2000; 9: 489–501. [DOI] [PubMed] [Google Scholar]
- 19.Merico D, Costain G, Butcher NJ, et al. MicroRNA dysregulation, gene networks, and risk for schizophrenia in 22q11.2 deletion syndrome. Front Neurol 2014; 5: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Guo T, Fiksinski A, et al. Variance of IQ is partially dependent on deletion type among 1,427 22q11.2 deletion syndrome subjects. Am J Med Genet A 2018; published online Oct 5. DOI: 10.1002/ajmg.a.40359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun D, Ching CRK, Lin A, et al. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: convergence with idiopathic psychosis and effects of deletion size. Mol Psychiatry 2018; published online June 13. DOI: 10.1038/s41380-018-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gothelf D, Law AJ, Frisch A, et al. Biological effects of COMT haplotypes and psychosis risk in 22q11.2 deletion syndrome. Biol Psychiatry 2014; 75: 406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki G, Harper KM, Hiramoto T, et al. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum Mol Genet 2009; 18: 1652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merico D, Zarrei M, Costain G, et al. Whole-genome sequencing suggests schizophrenia risk mechanisms in humans with 22q11.2 deletion syndrome. G3 (Bethesda) 2015; 5: 2453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butcher NJ, Merico D, Zarrei M, et al. Whole-genome sequencing suggests mechanisms for 22q11.2 deletion-associated Parkinson’s disease. PLoS One 2017; 12: e0173944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bassett AS, Lowther C, Merico D, et al. Rare genome-wide copy number variation and expression of schizophrenia in 22q11.2 deletion syndrome. Am J Psychiatry 2017; 174: 1054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ. Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A 2018; 176: 2070–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koscielny G, Yaikhom G, Iyer V, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res 2014; 42: D802–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahmoudi E, Cairns MJ. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry 2017; 22: 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chun S, Westmoreland JJ, Bayazitov IT, et al. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 2014; 344: 1178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chun S, Du F, Westmoreland JJ, et al. Thalamic miR-338–3p mediates auditory thalamocortical disruption and its late onset in models of 22q11.2 microdeletion. Nat Med 2017; 23: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sellier C, Hwang VJ, Dandekar R, et al. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLoS One 2014; 9: e103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu B, Hsu PK, Stark KL, Karayiorgou M, Gogos JA. Derepression of a neuronal inhibitor due to miRNA dysregulation in a schizophrenia-related microdeletion. Cell 2013; 152: 262–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diamantopoulou A, Sun Z, Mukai J, et al. Loss-of-function mutation in Mirta22/Emc10 rescues specific schizophrenia-related phenotypes in a mouse model of the 22q11.2 deletion. Proc Natl Acad Sci USA 2017; 114: e6127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devaraju P, Zakharenko SS. Mitochondria in complex psychiatric disorders: lessons from mouse models of 22q11.2 deletion syndrome. Bioessays 2017; 39: 1600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Napoli E, Tassone F, Wong S, et al. Mitochondrial citrate transporter-dependent metabolic signature in the 22q11.2 deletion syndrome. J Biol Chem 2015; 290: 23240–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J 2012; 31: 3038–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paylor R, Lindsay E. Mouse models of 22q11 deletion syndrome. Biol Psychiatry 2006; 59: 1172–79. [DOI] [PubMed] [Google Scholar]
- 39.Boot E, van Amelsvoort TA. Neuroimaging correlates of 22q11.2 deletion syndrome: implications for schizophrenia research. Curr Top Med Chem 2012; 12: 2303–13. [DOI] [PubMed] [Google Scholar]
- 40.Dennis EL, Thompson PM. Typical and atypical brain development: a review of neuroimaging studies. Dialogues Clin Neurosci 2013; 15: 359–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kunwar A, Ramanathan S, Nelson J, et al. Cortical gyrification in velo-cardio-facial (22q11.2 deletion) syndrome: a longitudinal study. Schizophr Res 2012; 137: 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitt JE, Vandekar S, Yi J, et al. Aberrant cortical morphometry in the 22q11.2 deletion syndrome. Biol Psychiatry 2015; 78: 135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakker G, Caan MW, Vingerhoets WA, et al. Cortical morphology differences in subjects at increased vulnerability for developing a psychotic disorder: a comparison between subjects with ultra-high risk and 22q11.2 deletion syndrome. PLoS One 2016; 11: e0159928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaer M, Debbane M, Bach Cuadra M, et al. Deviant trajectories of cortical maturation in 22q11.2 deletion syndrome (22q11DS): a cross-sectional and longitudinal study. Schizophr Res 2009; 115: 182–90. [DOI] [PubMed] [Google Scholar]
- 45.Ramanathan S, Mattiaccio LM, Coman IL, et al. Longitudinal trajectories of cortical thickness as a biomarker for psychosis in individuals with 22q11.2 deletion syndrome. Schizophr Res 2017; 188: 35–41. [DOI] [PubMed] [Google Scholar]
- 46.Vorstman JA, Breetvelt EJ, Duijff SN, et al. Cognitive decline preceding the onset of psychosis in patients with 22q11.2 deletion syndrome. JAMA Psychiatry 2015; 72: 377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gudbrandsen M, Daly E, Murphy CM, et al. The neuroanatomy of autism spectrum disorder symptomatology in 22q11.2 deletion syndrome. Cereb Cortex 2018; published online Oct 1. DOI: 10.1093/cercor/bhy239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Padula MC, Schaer M, Scariati E, et al. Structural and functional connectivity in the default mode network in 22q11.2 deletion syndrome. J Neurodev Disord 2015; 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakker G, Caan MW, Schluter RS, et al. Distinct white-matter aberrations in 22q11.2 deletion syndrome and patients at ultra-high risk for psychosis. Psychol Med 2016; 46: 2299–311. [DOI] [PubMed] [Google Scholar]
- 50.Nuninga JO, Bohlken MM, Koops S, et al. White matter abnormalities in 22q11.2 deletion syndrome patients showing cognitive decline. Psychol Med 2018; 48: 1655–63. [DOI] [PubMed] [Google Scholar]
- 51.Schreiner MJ, Karlsgodt KH, Uddin LQ, et al. Default mode network connectivity and reciprocal social behavior in 22q11.2 deletion syndrome. Soc Cogn Affect Neurosci 2014; 9: 1261–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scariati E, Schaer M, Richiardi J, et al. Identifying 22q11.2 deletion syndrome and psychosis using resting-state connectivity patterns. Brain Topogr 2014; 27: 808–21. [DOI] [PubMed] [Google Scholar]
- 53.Villalon-Reina J, Martínez K, Qui X, et al. Highly atypical white matter in 22q11.2 deletion syndrome: an ENIGMA-DTI consortium study. Hum Brain Mapp (in press). [Google Scholar]
- 54.Ouchi Y, Banno Y, Shimizu Y, et al. Reduced adult hippocampal neurogenesis and working memory deficits in the DGCR8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J Neurosci 2013; 33: 9408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cioffi S, Martucciello S, Fulcoli FG, et al. Tbx1 regulates brain vascularization. Hum Mol Genet 2014; 23: 78–89. [DOI] [PubMed] [Google Scholar]
- 56.Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003; 362: 1366–73. [DOI] [PubMed] [Google Scholar]
- 57.Paylor R, Glaser B, Mupo A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA 2006; 103: 7729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bearden CE, van Erp TG, Dutton RA, et al. Mapping cortical thickness in children with 22q11.2 deletions. Cereb Cortex 2007; 17: 1889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu P, Teot L, Murdoch G, Monaghan-Nichols AP, McFadden K. Neuropathology of 22q11 deletion syndrome in an infant. Pediatr Dev Pathol 2014; 17: 386–92. [DOI] [PubMed] [Google Scholar]
- 60.Flore G, Cioffi S, Bilio M, Illingworth E. Cortical development requires mesodermal expression of TBX1, a gene haploinsufficient in 22q11.2 deletion syndrome. Cereb Cortex 2017; 27: 2210–25. [DOI] [PubMed] [Google Scholar]
- 61.Hiramoto T, Kang G, Suzuki G, et al. Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum Mol Genet 2011; 20: 4775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi T, Okabe S, Broin PO, et al. Structure and function of neonatal social communication in a genetic mouse model of autism. Mol Psychiatry 2016; 21: 1208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fung WL, Butcher NJ, Costain G, et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med 2015; 17: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stachon AC, De Souza C. Anxiety disorders and perceptual disturbances in adolescents with 22q11.2 deletion syndrome treated with SSRI: a case series. J Can Acad Child Adolesc Psychiatry 2011; 20: 305–10. [PMC free article] [PubMed] [Google Scholar]
- 65.Dori N, Green T, Weizman A, Gothelf D. The effectiveness and safety of antipsychotic and antidepressant medications in individuals with 22q11.2 deletion syndrome. J Child Adolesc Psychopharmacol 2017; 27: 83–90. [DOI] [PubMed] [Google Scholar]
- 66.Evers LJ, Curfs LM, Bakker JA, et al. Serotonergic, noradrenergic and dopaminergic markers are related to cognitive function in adults with 22q11 deletion syndrome. Int J Neuropsychopharmacol 2014; 17: 1159–65. [DOI] [PubMed] [Google Scholar]
- 67.Boot E, Booij J, Zinkstok J, et al. Disrupted dopaminergic neurotransmission in 22q11 deletion syndrome. Neuropsychopharmacology 2008; 33: 1252–58. [DOI] [PubMed] [Google Scholar]
- 68.Boot E, Booij J, Abeling N, et al. Dopamine metabolism in adults with 22q11 deletion syndrome, with and without schizophrenia—relationship with COMT Val(1)(0)(8)/(1)(5)(8)Met polymorphism, gender and symptomatology. J Psychopharmacol 2011; 25: 888–95. [DOI] [PubMed] [Google Scholar]
- 69.Didriksen M, Fejgin K, Nilsson SR, et al. Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: a study in male mice. J Psychiatry Neurosci 2016; 41: 150381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kimoto S, Muraki K, Toritsuka M, et al. Selective overexpression of Comt in prefrontal cortex rescues schizophrenia-like phenotypes in a mouse model of 22q11 deletion syndrome. Transl Psychiatry 2012; 2: e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sannino S, Gozzi A, Cerasa A, et al. COMT genetic reduction produces sexually divergent effects on cortical anatomy and working memory in mice and humans. Cereb Cortex 2015; 25: 2529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrison PJ, Tunbridge EM. Catechol-O-methyltransferase (COMT): a gene contributing to sex differences in brain function, and to sexual dimorphism in the predisposition to psychiatric disorders. Neuropsychopharmacology 2008; 33: 3037–45. [DOI] [PubMed] [Google Scholar]
- 73.Mok KY, Sheerin U, Simon-Sanchez J, et al. Deletions at 22q11.2 in idiopathic Parkinson’s disease: a combined analysis of genome-wide association data. Lancet Neurol 2016; 15: 585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sumitomo A, Horike K, Hirai K, et al. A mouse model of 22q11.2 deletions: molecular and behavioral signatures of Parkinson’s disease and schizophrenia. Sci Adv 2018; published online Aug 15. DOI: 10.1126/sciadv.aar6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Butcher NJ, Marras C, Pondal M, et al. Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain 2017; 140: 1371–83. [DOI] [PubMed] [Google Scholar]
- 76.Goldstein DS, Sullivan P, Holmes C, et al. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem 2013; 126: 591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raux G, Bumsel E, Hecketsweiler B, et al. Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum Mol Genet 2007; 16: 83–91. [DOI] [PubMed] [Google Scholar]
- 78.Jacquet H, Raux G, Thibaut F, et al. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum Mol Genet 2002; 11: 2243–49. [DOI] [PubMed] [Google Scholar]
- 79.Loureiro SO, Sidegum DS, Biasibetti H, et al. Crosstalk among disrupted glutamatergic and cholinergic homeostasis and inflammatory response in mechanisms elicited by proline in astrocytes. Mol Neurobiol 2016; 53: 1065–79. [DOI] [PubMed] [Google Scholar]
- 80.Roussos P, Giakoumaki SG, Bitsios P. A risk PRODH haplotype affects sensorimotor gating, memory, schizotypy, and anxiety in healthy male subjects. Biol Psychiatry 2009; 65: 1063–70. [DOI] [PubMed] [Google Scholar]
- 81.Vorstman JA, Turetsky BI, Sijmens-Morcus ME, et al. Proline affects brain function in 22q11DS children with the low activity COMT 158 allele. Neuropsychopharmacology 2009; 34: 739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Radoeva PD, Coman IL, Salazar CA, et al. Association between autism spectrum disorder in individuals with velocardiofacial (22q11.2 deletion) syndrome and PRODH and COMT genotypes. Psychiatr Genet 2014; 24: 269–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.da Silva Alves F, Boot E, Schmitz N, et al. Proton magnetic resonance spectroscopy in 22q11 deletion syndrome. PLoS One 2011; 6: e21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Evers LJ, van Amelsvoort TA, Bakker JA, de Koning M, Drukker M, Curfs LM. Glutamatergic markers, age, intellectual functioning and psychosis in 22q11 deletion syndrome. Psychopharmacology (Berl) 2015; 232: 3319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang YM, Fedchyshyn MJ, Grande G, et al. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca(2+) influx to neurotransmitter release at a central synapse. Neuron 2010; 67: 100–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dent J, Kato K, Peng XR, et al. A prototypic platelet septin and its participation in secretion. Proc Natl Acad Sci USA 2002; 99: 3064–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harper KM, Hiramoto T, Tanigaki K, et al. Alterations of social interaction through genetic and environmental manipulation of the 22q11.2 gene SEPT5 in the mouse brain. Hum Mol Genet 2012; 21: 3489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gur RE, Bassett AS, McDonald-McGinn DM, et al. A neurogenetic model for the study of schizophrenia spectrum disorders: the International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol Psychiatry 2017; 22: 1664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rogdaki M, Gudbrandsen M, Daly E, et al. State or trait? Investigation of dopamine function in individuals with 22q11deletion. Schizophr Bull 2017; 43 (suppl 1): S75. [Google Scholar]
- 90.van Duin ED, Kasanova Z, Hernaus D, et al. Striatal dopamine release and impaired reinforcement learning in adults with 22q11.2 deletion syndrome. Eur Neuropsychopharmacol 2018; 28: 732–42. [DOI] [PubMed] [Google Scholar]
- 91.Boot E, Booij J, Zinkstok JR, et al. COMT Val158Met genotype and striatal D2/3 receptor binding in adults with 22q11 deletion syndrome. Synapse 2011; 65: 967–70. [DOI] [PubMed] [Google Scholar]
- 92.Boot E, Booij J, Zinkstok JR, et al. Striatal D₂ receptor binding in 22q11 deletion syndrome: an [123I]IBZM SPECT study. J Psychopharmacol 2010; 24: 1525–31. [DOI] [PubMed] [Google Scholar]