

Abstract
Background
Stickler syndrome is the most common genetic cause of rhegmatogenous retinal detachment (RRD) in children, and has a high risk of blindness. Type I (STL1) is the most common subtype, caused by COL2A1 mutations. This study aims to analyze the mutation spectrum of COL2A1 and further elucidate the genotype-phenotype relationships in the East Asian populations with STL1, which is poorly studied at present.
Methods
By searching MEDLINE, Web of Science, CNKI, Wanfang Data, HGMD and Clinvar, all publications associated with STL1 were collected. Then, they were carefully screened to obtain all reported STL1-related variants in COL2A1 and clinical features in East Asian patients with STL1.
Results
There were 274 COL2A1 variants identified in 999 patients with STL1 from 466 unrelated families, and more than half of them were truncation mutations. Of the 107 STL1 patients reported in the East Asian population, it was found that patients with truncation mutations had milder systemic phenotypes, whereas patients with splicing mutations had severer phenotypes. In addition, several recurrent variants (c.3106C > T, c.1833 + 1G > A, c.2710C > T and c.1693C > T) were found.
Conclusions
Genotype-phenotype correlations should certainly be studied carefully, contributed to making personalized follow-up plans and predicting prognosis of this disorder. Genome editing holds great potential for treating inherited diseases caused by pathogenic mutations. In this study, several recurrent variants were found, providing potential candidate targets for genetic manipulation in the future.
Keywords: Stickler syndrome, COL2A1, Genotype-phenotype correlation, Retinal detachment, Gene therapy
Background
Stickler syndrome is a clinically variable and genetically heterogeneous disease, first described by Stickler et al [1]. It is featured by ocular, skeletal, auditory and orofacial abnormalities. The incidence among newborns is approximately 1:7500–1:9000 [2]. Ocular findings include early-onset high myopia, retinal detachment (RD), cataract and glaucoma. It was reported that 50–73% of patients developed RD in their lifetime, and up to 75% of patients were bilateral [3–5]. Recurrent RD can cause irreversible vision loss and even blindness. Skeletal changes include joint hypermobility in childhood, mild spondyloepiphyseal dysplasia and premature osteoarthritis. Sensorineural hearing loss (HL) is usually mild, mainly for the high tones [6]. Orofacial features include flat midface, depressed nasal bridge, micrognathia and cleft palate.
At present, six subtypes of Stickler syndrome have been discerned. Type I (STL1, OMIM 108300) is the most common form, accounting for 80–90% [7]. STL1 is a dominantly inherited disorder, caused by mutations in the COL2A1 gene (OMIM 120140). The COL2A1 gene is mapped to human chromosome 12q13.11 and is composed of 54 exons, spanning 31.5 kb of genomic DNA [8]. It encodes α1(II) chain, which mainly expresses in hyaline cartilage, intervertebral discs, adult vitreous and inner ear [9].
To date, numerous patients with STL1 have been reported worldwide [9], but limited data are available in the East Asian population only with a few cases and small cohort studies. In this study, we aim to further elucidate the genotype-phenotype relationships by analyzing the clinical and genetic characteristics of East Asian patients with STL1. Furthermore, all reported variants in COL2A1 associated with STL1 were also analyzed.
Methods
MEDLINE, CNKI, Web of Science and Wanfang Data were searched, applying the following search terms for the period 1965 to October 2019: (mutation OR variant) AND (COL2A1 AND “Stickler syndrome”). In addition, Human Gene Mutation Database (HGMD) and Clinvar were also searched, and the search term was COL2A1. All relevant publications were carefully screened. We included publications that 1) were studies among patients with STL1, 2) provided the variant in COL2A1, and 3) were full-text articles. Reviews and obvious duplicates were excluded, and other types of studies met the inclusion criteria were included. Related information was collected, including variants and ethnicities. For patients in East Asia, detailed clinical features were also collected. All the processes were performed independently by two authors (D.W. and F.G.), including screening, selecting studies, extracting data and assessing the risk of bias. Any discrepancy in the assessment would be resolved by consensus. The study was performed according to the Declaration of Helsinki and approved by the Ethics Committee of the Eye and ENT Hospital of Fudan University.
All variants in COL2A1 were checked to ensure that they were numbered according to the reference cDNA sequence NM_001844.4. If not, nucleotide and codon numbers were converted to ensure that their mutation nomenclature matched with reference transcript NM_001844.4 for COL2A1. For DNA numbering + 1 corresponded to the A of the ATG translation initiation codon. All statistical analyses were performed using SPSS 20.0 (SPSS Inc., Chicago, IL, USA). According to mutation types, East Asian patients were divided into 3 subgroups. Chi-squared test was performed to compare the phenotypes and sex among 3 subgroups. In addition, Kruskal-Wallis test was applied to compare the age among 3 subgroups. P value < 0.05 was considered the threshold of statistical significance.
Results
Spectrum of COL2A1 Mutations
A total of 80 original articles met the inclusion criteria. There were 274 disease-causing variants in COL2A1 identified in diverse ethnicities, which is shown in Additional file 1: Table S1. Of 999 patients with STL1 from 466 unrelated families, most were Europeans (342/466 families, 73.4%), followed by Asians (63/466 families, 13.5%) and North Americans (42/466 families, 9.0%; Fig. 1). All patients carried one heterozygous variant in COL2A1, except for one patient harboring two variants in COL2A1. Most variants (158/274, 57.7%) are nonsense and frameshift mutations (insertions/deletions/indels) that are predicted to cause premature protein truncation, leading to the absence of collagen synthesis (Fig. 2a). The majority of variants were identified in only one family (213/274, 77.7%), suggesting that the mutation spectrum is far from being saturated in spite of numerous COL2A1 mutation reports.
Fig. 1.
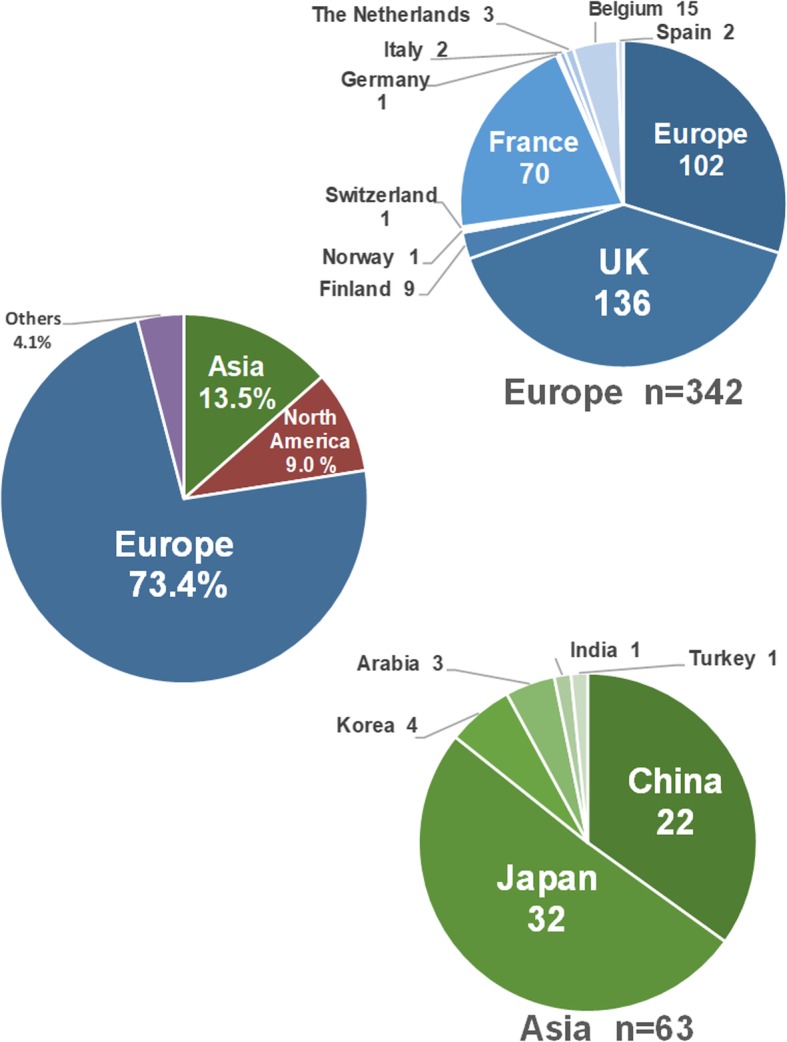
Population distribution of STL1-related COL2A1 mutation reports. The pie-chart shows the number and percentage of STL1 patients harboring COL2A1 variants from different ethnicities
Fig. 2.
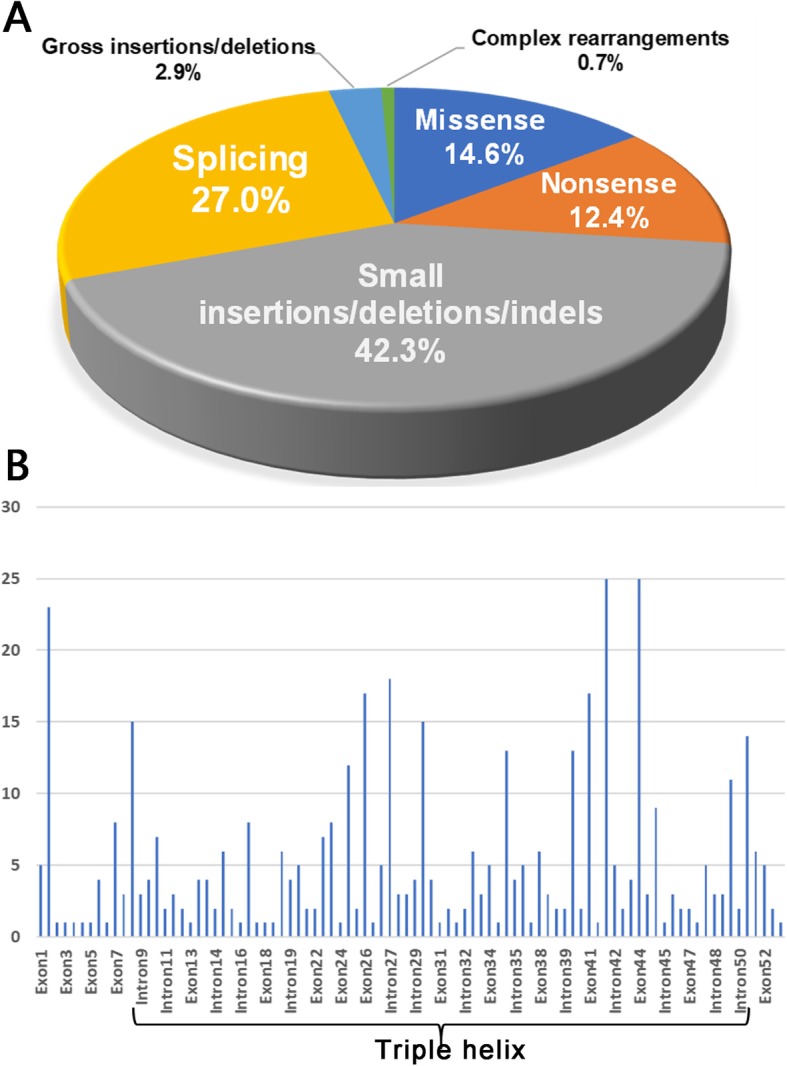
The distribution of variants in the COL2A1 gene. a The distribution of mutation types. b The distribution of variants in different exon/introns
Mutation Locations
Several recurrent variants were identified in the European population, including c.3106C > T (15/342), c.1833 + 1G > A (12/342), c.1693C > T (10/342), c.2710C > T (9/342) and c.2353C > T (9/342). In addition, the variants c.1833 + 1G > A (5/58), c.1693C > T (4/58), c.3106C > T (3/58), c.2353C > T (3/58) and c.1957C > T were common in the East Asian population. Notably, the variant c.1833 + 1G > A was found in multiple ethnicities, but was not found in the North American population in spite of numerous STL1 reports (191 patients, 42 families). In addition, the variant c.258C > A was common in the North American population (4/42), and all 46 individuals carrying this particular allele were from the North America. It suggested that ethnicity contributes significantly to the distribution of mutations. No mutational hot region was found, but most variants in COL2A1 were clustered in exon 42 (25/466, 5.4%), exon 44 (25/466, 5.4%) and exon 2 (23/466, 4.9%; Fig. 2b). To date, no disease-causing variant associated with STL1 has been identified in exons 20, 28, 53 and 54. Variants in these exons can cause severe phenotypes, such as spondyloepiphyseal dysplasia (SED) and spondyloepimetaphyseal dysplasia (SEMD).
The triple helix is the common structural feature of collagens, composed of a core repeat of Gly-X-Y [10]. The majority of variants in COL2A1 were located in the triple helix region (encoded from codon 201–1214), accounting for 81.0% (222/274), followed by N-terminal propeptide (encoded from codon 26–181, 10.2%, 28/274). Corresponding to the enrichment of Gly-X-Y repeat, glycine replacement accounted for 22.6% (58/274), showing a predominant position. Variants located in other regions of type II collagen were rare.
Clinical Features
A total of 107 STL1 patients (55 males, 52 females) harboring one heterozygous variant in COL2A1 have been reported in the East Asian population. Thirty-two families from Japan, 22 from China, and four from Korea were included (Additional file 2: Table S2). The average age was 21.2 ± 16.2 years (range, 0.3–63.0; median, 15.0). All patients presented with ocular manifestations, and 82.8% (24/29) of eyes were high myopia before 6 years old. RD occurred in 41.7% (43/103) of the patients, and 46.5% (20/43) were bilateral. Membrane vitreous abnormalities (MVA) is a characteristic finding in patients with STL1 [10], observed in 45.8% (44/96) of the patients. In addition, 22.1% (23/104) of the patients had cataract, and only one patient had glaucoma. Skeletal changes occurred in 35.4% (34/96) of the patients, including precocious osteoarthrosis and joint hypermobility. Midfacial dysplasia, such as flat midface and depressed nasal bridge, is also a common manifestation, observed in 60.6% (60/99) of the patients, and 31.1% (19/61) of the patients presented with micrognathia. In addition, 30.7% (31/101) of the patients had cleft palate, 2 of which were submucous cleft palate. Sensorineural hearing loss was found in 18.1% (17/94) of the patients (Fig. 3a).
Fig. 3.
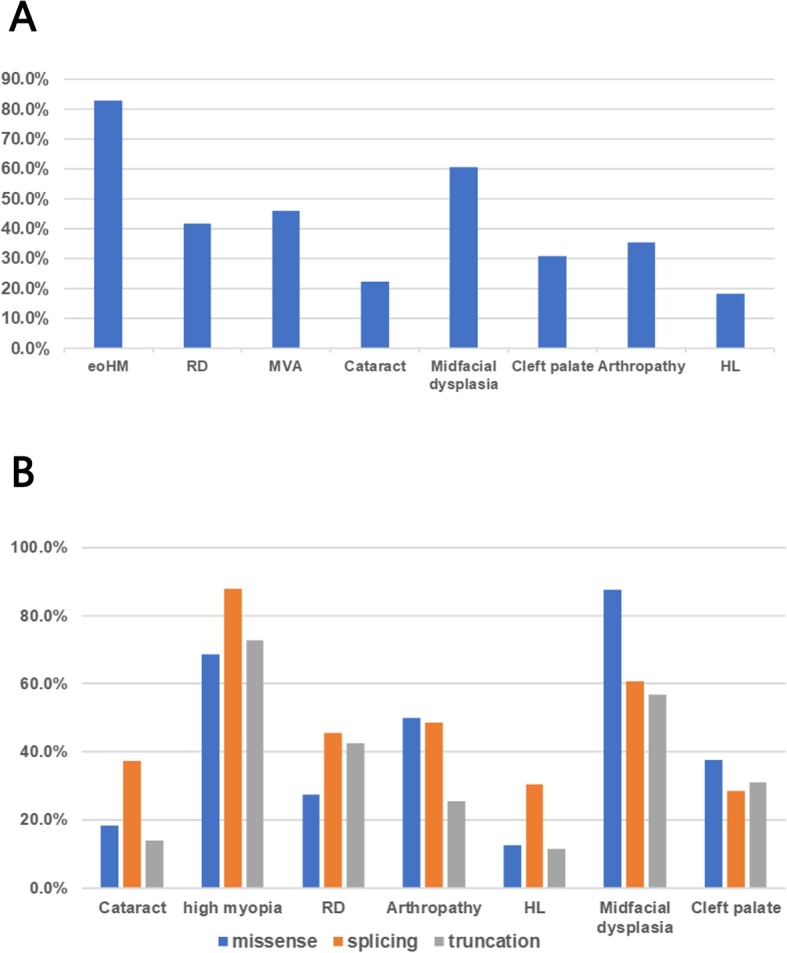
The clinical characteristics of 107 patients with STL1 in East Asia. a The prevalence of different phenotypes in STL1. b The prevalence of different phenotypes among 3 subgroups. eoHM: early-onset high myopia; RD: retinal detachment; MVA: membrane vitreous abnormalities; HL: hearing loss
Genotype-Phenotype Correlations
All East Asian patients were divided into 3 subgroups according to mutation types (Table 1, Fig. 3b). No apparent difference in age or sex was observed among 3 subgroups (P = 0.301 and 0.692, respectively). Compared with the other two groups, patients harboring splicing mutations were more likely to develop cataract (P = 0.004) and had severer systemic phenotypes (P = 0.010 in arthropathy, P = 0.022 in HL), whereas patients with truncation mutations had milder systemic phenotypes. In addition, there was no significant difference in the occurrence of high myopia, RD, midfacial dysplasia and cleft palate among 3 subgroups (P = 0.510, 0.575, 0.246 and 0.743, respectively).
Table 1.
The Difference of Phenotypes in Patients with Different Mutation Types
| Mutation types | Missense | Splicing | Truncation | P value |
|---|---|---|---|---|
| Cases | 12 | 35 | 60 | |
| Male/Female | 5/7 | 20/15 | 30/30 | 0.692 |
| Age, years | 16.0 ± 13.8 (1.0–45.0) | 23.2 ± 15.2 (2.3–62.0) | 21.0 ± 17.1 (0.3–63.0) | 0.301 |
| Cataract | 18.2% | 37.1% | 13.8% | 0.004* |
| High myopia | 68.8% | 87.8% | 72.8% | 0.510 |
| RD | 27.3% | 45.5% | 42.4% | 0.575 |
| Arthropathy | 50.0% | 48.5% | 25.5% | 0.010* |
| HL | 12.5% | 30.3% | 11.3% | 0.022* |
| Midfacial dysplasia | 87.5% | 60.6% | 56.9% | 0.246 |
| Cleft palate | 37.5% | 28.6% | 31.0% | 0.743 |
RD retinal detachment; HL: hearing loss; *: P < 0.05
Type II collagen can be alternatively spliced, and its long form including exon 2 mainly expresses in vitreous [11]. Therefore, patients with exon 2 mutations have few extraocular manifestations [12]. Nevertheless, seven patients harboring other exon mutations also had no systemic manifestation. In addition, phenotypic variation, such as differences in spherical equivalent refraction and sensorineural hearing loss or presence versus absence of cleft palate, and varying skeletal manifestations, was observed in siblings or in unrelated families with the same variants. This suggested the high clinical heterogeneity in this disorder, and there were other factors affecting the severity of the phenotypic spectrum, such as modifier genes and environmental exposures.
Diagnostic Relevance
Of 107 patients with STL1, 10 patients were initially diagnosed with early-onset high myopia (eoHM), and diagnosis was not made correctly until they presented with systemic symptoms of STL1 after years of follow-up [13]. There were seven patients still diagnosed with eoHM, even if their COL2A1 variants have been recorded in HGMD as STL1-related variants (Additional file 3: Table S3) [14, 15], because the diagnosis of STL1 is clinically based at present [16]. In addition, nine other patients with eoHM, harboring COL2A1 variants, have been reported, and these variants have been recorded in HGMD as eoHM-related variants (Additional file 3: Table S3) [14, 15].
Discussion
In this study, it was found that patients with splicing mutations had severer systemic phenotypes when compared with patients harboring truncation mutations and missense mutations. In addition, most STL1 patients carried with truncation mutations, and they were less likely to develop arthropathy. In light of the previous study, the STL1 spectrum was mostly attributed to truncation mutations, SED to missense mutations and Kniest dysplasia (KND) to splicing mutations [9, 17], which is consistent with our finding. This also explains why patients with STL1-related variants have milder phenotypes, especially arthropathy, than patients harboring other variants in COL2A1. Furthermore, this genotype-phenotype correlation provides a reference for personalized follow-up plans in spite of high clinical heterogeneity in STL1.
Genome editing shows great potential for treating inherited diseases caused by pathogenic mutations. Currently, multiple Cas9-based clinical trials are in progress or beginning soon, which is likely to guide future use for somatic cell editing both ex vivo and in patients [18]. Several recurrent variants were found in this study, including the variants c.3106C > T (18/466), c.1833 + 1G > A (18/466), c.2710C > T (15/466) and c.1693C > T (14/466), providing potential candidate targets for genetic manipulation in the future.
We reviewed clinical features of 107 STL1 patients in East Asia. There were 98.9% (88/89) of the patients with myopia, higher than that in the European population (89%) [19]. In addition, 82.8% (24/29) of eyes were high myopia before 6 years old. Zhou L et al. proposed that eoHM was the most easily recognizable sign for children with potential STL, usually presenting earliest [13]. Midfacial dysplasia was observed in 60.6% (60/99) of the patients, second only to myopia. However, it is also a common facial feature in Asians, which is easily ignored by clinicians. Therefore, some patients with STL1 are initially diagnosed with eoHM. Most phenotypes of STL1 become more prevalent with advancing age, such as RD, sensorineural hearing loss and arthropathy [20]. So, it is not wise to draw a conclusion too early, and a regular follow-up for multisystem is essential for patients harboring COL2A1 variants.
STL is the most common genetic cause of rhegmatogenous retinal detachment (RRD) in children [21]. In this study, RRD was observed in 41.7% (43/103) of the patients and 46.5% (20/43) were bilateral, which is lower than previous studies (50%~ 73 and 75%, respectively) [3–5]. This may be related to the younger age of the patients included, because the prevalence of RRD is a function of age [20]. In addition, the prevalence of MVA was 45.8% (44/96), similar to the previous study in the European population (42%) [19]. There are some limitations in this study. The phenotypic data in some articles is incomplete, which limits the number of evaluable patients. Furthermore, the reporting bias should be inherent. The information based on the number of reports cannot reflect the prevalence of STL1 in East Asia. Lastly, a long-term, controlled prospective study is required to substantiate the genotype-phenotype correlation found in this study.
Conclusions
In this study, our findings revealed that patients with splicing mutations had severer systemic phenotypes when compared with patients harboring other types of mutations, whereas patients with truncation mutations had milder phenotypes. This helps clinicians develop personalized follow-up plans for patients with STL1. In addition, recurrent variants c.3106C > T, c.1833 + 1G > A, c.2710C > T and c.1693C > T were found, which provides potential candidate targets for future gene therapy. Finally, high myopia before 6 years old is a key sign. For patients with high myopia, a regular follow-up for multisystem is essential if a heterozygous variant in COL2A1 is identified.
Supplementary information
Additional file 1 : Table S1. Variants of COL2A1 in STL1 patients.
Additional file 2 : Table S2. Clinical characteristics in East Asian patients.
Additional file 3 : Table S3. The patients suspected with STL1.
Acknowledgements
We would like to thank all the participants for their invaluable devotion to this study.
Abbreviations
- eoHM
early-onset high myopia
- HGMD
Human gene mutation database
- HL
Hearing loss
- KND
Kniest dysplasia
- MVA
Membrane vitreous abnormalities
- RRD
Rhegmatogenous retinal detachment
- SED
Spondyloepiphyseal dysplasia
- SEMD
Spondyloepimetaphyseal dysplasia
- STL1
Stickler syndrome type I
Authors’ contributions
JW designed this study. PX, FH, SZ and DW collected the clinical and genetic information. FG and DW analyzed the data. JW and DW wrote and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81770925, No. 81790641), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018PT32019).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The study was in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Eye and ENT Hospital of Fudan University.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12881-020-0963-z.
References
- 1.Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, Ward LE. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc. 1965;40:433–455. [PubMed] [Google Scholar]
- 2.Printzlau A, Andersen M. Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J. 2004;41(1):47–52. doi: 10.1597/02-055. [DOI] [PubMed] [Google Scholar]
- 3.Ang A, Poulson AV, Goodburn SF, Richards AJ, Scott JD, Snead MP. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology. 2008;115(1):164–168. doi: 10.1016/j.ophtha.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 4.Donoso LA, Edwards AO, Frost AT, Ritter RR, Ahmad N, Vrabec T, Rogers J, Meyer D, Parma S. Clinical variability of Stickler syndrome: role of exon 2 of the collagen COL2A1 gene. Surv Ophthalmol. 2003;48(2):191–203. doi: 10.1016/S0039-6257(02)00460-5. [DOI] [PubMed] [Google Scholar]
- 5.Stickler GB, Hughes W, Houchin P. Clinical features of hereditary progressive arthro-ophthalmopathy (Stickler syndrome): a survey. Genet Med. 2001;3(3):192–196. doi: 10.1097/00125817-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Majava M, Hoornaert KP, Bartholdi D, Bouma MC, Bouman K, Carrera M, Devriendt K, Hurst J, Kitsos G, Niedrist D, et al. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am J Med Genet A. 2007;143A(3):258–264. doi: 10.1002/ajmg.a.31586. [DOI] [PubMed] [Google Scholar]
- 7.Robin NH, Moran RT, Ala-Kokko L. Stickler Syndrome. 1993. [Google Scholar]
- 8.Deng H, Huang X, Yuan L. Molecular genetics of the COL2A1-related disorders. Mutat Res Rev Mutat Res. 2016;768:1–13. doi: 10.1016/j.mrrev.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, Touitou I. Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies. Hum Mutat. 2016;37(1):7–15. doi: 10.1002/humu.22915. [DOI] [PubMed] [Google Scholar]
- 10.Richards AJ, Baguley DM, Yates JR, Lane C, Nicol M, Harper PS, Scott JD, Snead MP. Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen Gly-X-Y triple helix. Am J Hum Genet. 2000;67(5):1083–1094. doi: 10.1016/S0002-9297(07)62938-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reardon A, Sandell L, Jones CJ, McLeod D, Bishop PN. Localization of pN-type IIA procollagen on adult bovine vitreous collagen fibrils. Matrix Biol. 2000;19(2):169–173. doi: 10.1016/S0945-053X(00)00058-5. [DOI] [PubMed] [Google Scholar]
- 12.McAlinden A, Majava M, Bishop PN, Perveen R, Black GC, Pierpont ME, Ala-Kokko L, Mannikko M. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome. Hum Mutat. 2008;29(1):83–90. doi: 10.1002/humu.20603. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L, Xiao X, Li S, Jia X, Wang P, Sun W, Zhang F, Li J, Li T, Zhang Q. Phenotypic characterization of patients with early-onset high myopia due to mutations in COL2A1 or COL11A1: Why not Stickler syndrome? Mol Vis. 2018;24:560–573. [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou L, Xiao X, Li S, Jia X, Zhang Q. Frequent mutations of RetNet genes in eoHM: Further confirmation in 325 probands and comparison with late-onset high myopia based on exome sequencing. Exp Eye Res. 2018;171:76–91. doi: 10.1016/j.exer.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Sun W, Huang L, Xu Y, Xiao X, Li S, Jia X, Gao B, Wang P, Guo X, Zhang Q. Exome Sequencing on 298 Probands With Early-Onset High Myopia: Approximately One-Fourth Show Potential Pathogenic Mutations in RetNet Genes. Invest Ophthalmol Vis Sci. 2015;56(13):8365–8372. doi: 10.1167/iovs.15-17555. [DOI] [PubMed] [Google Scholar]
- 16.Rose PS, Levy HP, Liberfarb RM, Davis J, Szymko-Bennett Y, Rubin BI, Tsilou E, Griffith AJ, Francomano CA. Stickler syndrome: clinical characteristics and diagnostic criteria. Am J Med Genet A. 2005;138A(3):199–207. doi: 10.1002/ajmg.a.30955. [DOI] [PubMed] [Google Scholar]
- 17.Nishimura G, Haga N, Kitoh H, Tanaka Y, Sonoda T, Kitamura M, Shirahama S, Itoh T, Nakashima E, Ohashi H, et al. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26(1):36–43. doi: 10.1002/humu.20179. [DOI] [PubMed] [Google Scholar]
- 18.Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361(6405):866–869. doi: 10.1126/science.aat5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoornaert KP, Vereecke I, Dewinter C, Rosenberg T, Beemer FA, Leroy JG, Bendix L, Bjorck E, Bonduelle M, Boute O, et al. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010;18(8):872–880. doi: 10.1038/ejhg.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liberfarb RM, Levy HP, Rose PS, Wilkin DJ, Davis J, Balog JZ, Griffith AJ, Szymko-Bennett YM, Johnston JJ, Francomano CA, et al. The Stickler syndrome: genotype/phenotype correlation in 10 families with Stickler syndrome resulting from seven mutations in the type II collagen gene locus COL2A1. Genet Med. 2003;5(1):21–27. doi: 10.1097/00125817-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Richards AJ, Scott JD, Snead MP. Molecular genetics of rhegmatogenous retinal detachment. Eye (Lond) 2002;16(4):388–392. doi: 10.1038/sj.eye.6700195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 : Table S1. Variants of COL2A1 in STL1 patients.
Additional file 2 : Table S2. Clinical characteristics in East Asian patients.
Additional file 3 : Table S3. The patients suspected with STL1.
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.