

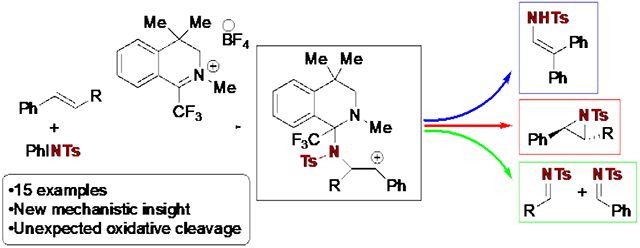
Abstract
Olefin aziridination via organocatalytic nitrene transfer offers potential complementarity to metal-catalyzed methods, however there is a lack of reports of such reactions in the literature. Herein is reported a method that employs an iminium salt to catalyze the aziridination of styrenes by [N-(p-toluenesulfonyl)imino]phenyliodinane (PhINTs). These reactions are hypothesized to proceed via a diaziridinium salt as the active oxidant. In addition to outlining the scope and limitations of the method, evidence for a polar, stepwise mechanism is presented, which provides new insight into the nature of iminium catalysis of nitrene transfer.
Graphical Abstract
INTRODUCTION
Organocatalytic atom-transfer oxidation is well established as a useful approach for enantioselective epoxidation,1 and more recent investigations have extended its capability to site-selective C–H hydroxylation.2 In addition to enabling reactivity and selectivity complementary to organometallic or enzymatic catalysis, the use of organocatalysis can also provide advantages in cost, toxicity, environmental impact, and ease of experimental procedure.4 Consequently, we have explored expanding the scope of this mode of organocatalysis beyond oxygen transfer3 and in 2018 reported a method for organocatalytic nitrene-transfer C(sp3)–H amination (Scheme 1).5 This method employs a trifluoromethyl iminium salt as the catalyst in combination with iminoiodinane nitrene precursors. Preliminary mechanistic evidence suggests that the reaction involves a diaziridinium salt, or related organic nitrenoid, as the active oxidant.
Scheme 1.

Applications of organocatalytic nitrene transfer.
A separate class of reactions enabled by catalytic nitrene transfer is olefin aziridination,6 which provides value for the preparation of bioactive molecules both in isolation and as a means for stepwise olefin functionalization.7 Organocatalysis of aziridination has previously been developed,8 but is limited to α,β-unsaturated carbonyl compounds because of its reliance on a mechanism of conjugate addition of a nucleophilic nitrogen source to an electron-deficient olefin followed by ring closure to provide the aziridine.9 The use of organocatalytic nitrene transfer, which involves the formation of an intermediate bearing an electrophilic nitrogen atom (e.g. an organic nitrenoid), should by virtue of its distinct mechanism provide a complementary organocatalytic aziridination method capable of functionalizing electron-rich substrates (Scheme 1). Given this complementarity to existing organocatalytic methods and further potential complementarity to metal-catalyzed methods, we have therefore endeavored to expand the scope of iminium-catalyzed nitrene transfer to include aziridination. We report herein the development of a first-generation method and exploration of its scope. In addition to establishing the initial capabilities and boundaries of iminium-catalyzed nitrene transfer in the context of substrate-limited aziridination, these studies provide new and important mechanistic insights into the nature of this recently-developed mode of catalysis.
RESULTS AND DISCUSSION
Our preliminary investigations established the ability of the catalytic method developed for C–H amination to be modified to allow for the successful aziridination of trans-stilbene, using iminium salt 110 as the catalyst and PhINTs11 as the nitrene precursor (Scheme 2a).5 Importantly, this un-optimized result served as significant confirmatory evidence for the formation of an organic nitrenoid as the active oxidant. Upon further exploration the reaction conditions, which employed lower temperature as well a reduced amount of PhINTs relative to conditions required to achieve C–H amination, we observed no more than trace amounts of aziridines derived from other olefins, such as styrene (Scheme 2b). Further complicating the extension of this initial observation, attempts to achieve aziridination of styrene at higher temperatures (e.g. 22 °C), led to the complete and nonselective conversion of the olefin to a complex mixture of products. To overcome this challenge required an improved understanding of the factors governing undesired reaction pathways at this comparatively elevated temperature.
Scheme 2.
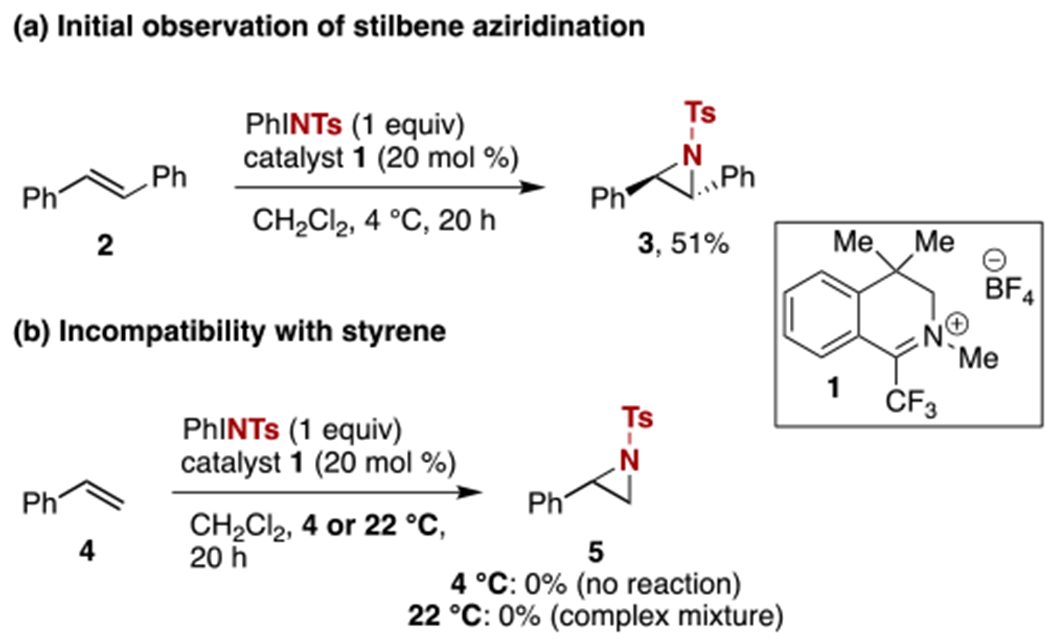
Initial exploration of organocatalytic aziridination.
In a key experiment, we observed that the anticipated aziridination product 5 was unstable to the reaction conditions at ambient temperature (Scheme 3) and would decompose to a complex mixture of products. A series of control experiments revealed that stability was restored when any one of the reagents (styrene, PhINTs, or catalyst) was omitted. Further optimization experiments revealed a dependence of yield of aziridine 5 on reaction time (Table 1). Specifically, at 19 h full consumption of styrene was observed, but no aziridine was produced. At a shorter reaction time of 5 h, up to 59% yield of 5 could be obtained (entry 3). However, we discovered that there was an unusually short window of time in which optimal yield could be achieved – by 6.5 h, all of the aziridine produced had been consumed to a complex mixture of products. Given that this rapid decomposition could complicate reproducibility, we sought to identify alternative conditions that would lead to enhanced product stability. Our earlier observation that the stability of the aziridine to the reaction conditions was dependent on the presence of all components of the reaction mixture led us to explore options to limit the concentration of PhINTs in solution at any given time. Unfortunately, PhINTs has limited solubility in CH2Cl2 to begin with, so portionwise addition of the reagent failed to achieve the desired result. The ultimately successful approach involved the evaluation of solvent mixtures of CH2Cl2 with hexanes, in an attempt to keep the effective concentration of PhINTs at a minimum throughout the reaction by further limiting its solubility. In the event, the use of a 9:1 CH2Cl2:hexanes solvent mixture allowed for better reproducibility, providing a 72% yield when using 2 equivalents of PhINTs and a reaction time of 22 h (entry 8). Both catalyst loading and amount of PhINTs could be reduced by half with only a modest effect on yield (entry 7). Alternatively, acetonitrile could be used as a solvent, although the yield in this case was also modestly lower. Control reactions (not shown) indicate that no styrene is consumed in the absence of catalyst 1, nor when NaBF4 is used in place of 1 to probe for reactivity caused by the counterion rather than the iminium. Finally, evaluation of other iminoiodinanes (e.g. PhINTces, PhINNs, and others) gave no more than trace amounts of aziridine products, in contrast to iminium-catalyzed C–H amination, which is more tolerant of other iminoiodinanes.5,12
Scheme 3.

Instability of aziridine 5 to reaction conditions
Table 1.
Optimization of styrene aziridinationa
 | ||||
|---|---|---|---|---|
| entry | PhINTs (equiv) | solvent | time (h) | yield (%)b |
| 1 | 1 | CH2Cl2 | 19 | 0 |
| 2 | 1 | CH2Cl2 | 3 | 22 |
| 3 | 1 | CH2Cl2 | 5 | 59 |
| 4 | 1 | CH2Cl2 | 6.5 | 0 |
| 5c | 1 | CH2Cl2 | 6 | 25 |
| 6d | 1 | 3:2 CH2Cl2:hexanes | 16 | NR |
| 6d | 1 | 4:1 CH2Cl2:hexanes | 11 | 12 |
| 7d | 1 | 9:1 CH2Cl2:hexanes | 11 | 61 |
| 8 | 2 | 9:1 CH2Cl2:hexanes | 22 | 72 |
| 9 | 2 | CH3CN | 22 | 63 |
| 10 | 2 | benzene | 22 | 5 |
All reactions were conducted on a 0.2 mmol scale of styrene, using 20 mol % of catalyst 1, except where noted.
Isolated yield. NR = no reaction (no consumption of styrene observed).
PhINTs added in four portions at t = 0, 1.5, 3, 4.5 h.
10 mol % loading of catalyst 1 was used.
With optimized conditions in hand, we explored the substrate scope of the method (Table 2). In general, aziridination of monosubstituted styrenes bearing a range of electron-donating and withdrawing groups could be achieved, using the olefin as the limiting reagent. Overall, a positive correlation of reaction yield with increasing substituent resonance contribution was observed, as in the halogen series (products 17-20). The presence of strongly deactivating groups (e.g. –CN, –CF3, –B(pin) as in substrates 10, 11, and 15 respectively) was found to be detrimental to reaction performance, resulting in the lowest isolated yields obtained in the para-substituted series, along with incomplete consumption of the starting material (30%, 48%, and 45% yields based on recovered starting material, respectively). At the other end of the spectrum, ortho- and para-methoxystyrene (not shown) is rapidly and completely consumed under the reaction conditions to a complex mixture of products. In addition to establishing the dependence of reaction outcome on electronic and resonance effects, our initial investigations of substrate scope revealed modest tolerance of acid-sensitive functionality (product 23) and an aryl boronic ester (product 26), the latter of which could in principle be further engaged in cross-coupling reactions to access more complex aziridine products. Confirming the complementarity of nitrene-transfer to the previously reported conjugate addition approaches to organocatalytic aziridination, no aziridination of chalcone (16) was observed.
Table 2.
Scope and limitations of iminium-catalyzed aziridinationa
 |
All reactions were conducted on a 0.5 mmol scale of alkene. Isolated yields are reported.
Discoveries made in this initial study of aziridination proved to be particularly illuminating and have enabled an improved mechanistic understanding of our relatively recent discovery of iminium-catalyzed nitrene transfer. For example, in order to probe whether iminium-catalyzed aziridination is stereospecific, we evaluated aziridination of both trans- and cis-β-methylstyrene (Scheme 4a). In each case, only the trans-aziridine product 29 was observed. The apparent complete stereoinversion in the case of the cis olefin is unusual and might suggest either a long-lived benzylic radical or carbocation intermediate, or a thermodynamically controlled reaction. To rule out the latter, we prepared cis-aziridine 31, using an alternative method, and exposed it to the reaction conditions. No consumption of the aziridine was observed, establishing that selectivity for the trans product is under kinetic control. Additional mechanistic information was gained by reevaluating the performance of trans-stilbene as an alternative 1,2-disubstituted substrate under the modified reaction conditions (Scheme 4b). At the elevated temperature (22 versus 4 °C), rather than the aziridine the observed major product of the reaction was N-tosyl enamine 33, in which an apparent 1,2-phenyl shift has occurred. This provides additional evidence of a long-lived benzylic radical or carbocation intermediate. The change in reaction outcome can be rationalized by the greater migratory aptitude of phenyl (as in 2) as compared to methyl (as in 28 or 30).
Scheme 4.
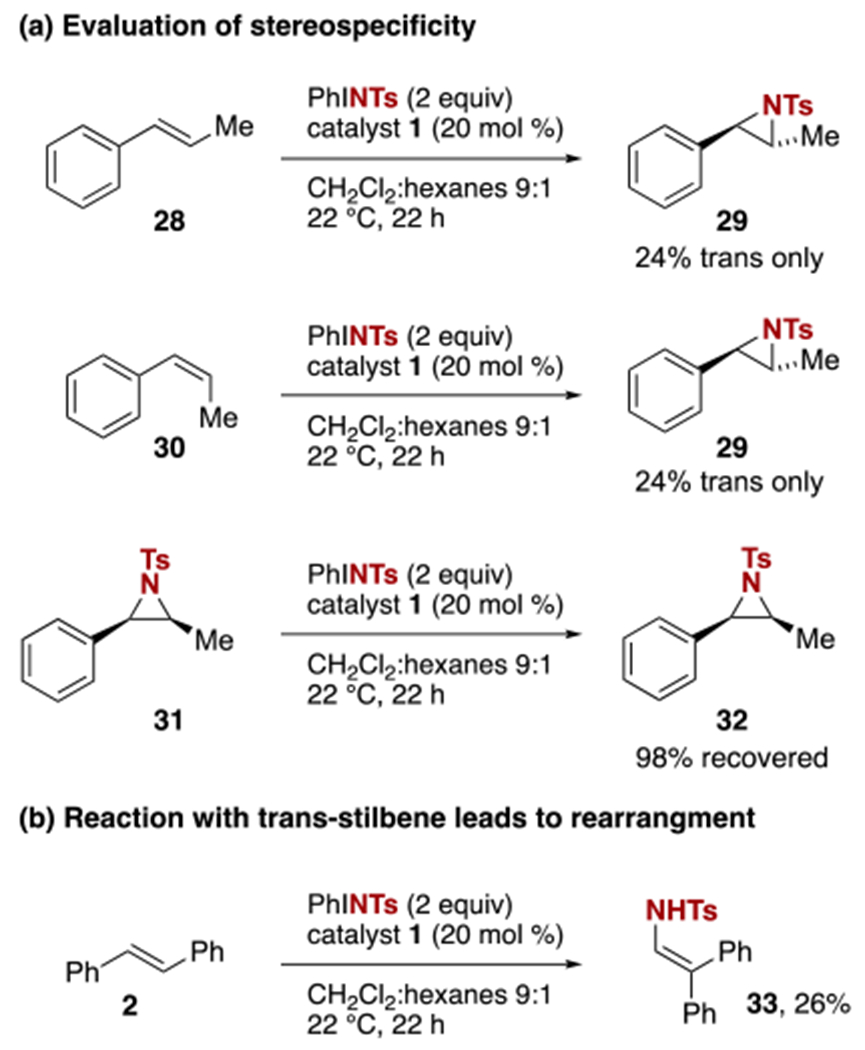
Evidence for a stepwise mechanism
Other key mechanistic insights were gained through the identification of additional products of these reactions. In particular, for all successful reactions disclosed in table 2, a small amount of an imine byproduct (e.g. 34) could be observed and quantified by 1H NMR of the crude reaction mixture (Scheme 5a). This intriguing result led us to consider whether these byproducts were arising from an unusual oxidative cleavage of the olefin directly to an N-tosyl imine.13 If this were the case, one would expect to also observe the formimine as a second product, which we did not. However, if this product were formed in trace amounts, subsequent consumption under the reaction conditions or workup (due to increased electrophilicity compared to 34) might prevent detection. To address this question, we exposed β-cyclohexylstyrene (4.9:1 mixture of trans:cis) to the reaction conditions (Scheme 5b). In addition to the aziridine product (exclusively trans), we observed the two expected imine products of oxidative cleavage (34 and 36) in equal amounts, confirming our suspicions.
Scheme 5.

Oxidative cleavage as a minor reaction pathway
In addition, we chose 6-vinyltetralin (37) as a probe to evaluate the relative rate of aziridination versus benzylic C–H amination (Scheme 6). Despite the fact that in this case, yield of the aziridination product was low, we did not observe any amount of the expected C–H amination product, nor could we identify any other trace products that might arise from subsequent consumption of a benzylic amination product. A small amount of the imine was also formed as was typical for all substrates. The complete chemoselectivity for aziridination over σ-bond insertion observed in this case is consistent with the intermediacy of an organic nitrenoid such as a diaziridinium, as we have previously proposed in the case of C–H amination.
Scheme 6.

Evaluation of C–H amination/aziridination selectivity
A mechanistic proposal consistent with all of these observations is outlined in Scheme 7. We hypothesize that upon nucleophilic attack of catalyst 1 by PhINTs, diaziridinium 40 is formed via intermediate 39.14 To account for the likelihood of a stepwise mechanism, we envision that the diaziridinium, bearing a highly electrophilic nitrogen atom by virtue of its sulfonamide protecting group, its bond to a cationic nitrogen atom, and the adjacent trifluoromethyl group, is likely to react with the olefin in a polar fashion to produce cationic intermediate 41. At least three pathways are available to this intermediate, which lead to the major product and side products observed for different substrates. Via path A, ring closure to the aziridine will occur with selectivity for the trans isomer if 41 is sufficiently long-lived to allow for bond rotation. The cationic intermediate can also account for the product of attempted stilbene aziridination (33) via path B, involving a 1,2-phenyl shift followed by disengagement from the catalyst. In this case only the enamine tautomer of the expected product of this shift is observed. Finally, the unusual oxidative cleavage can be rationalized by invoking the nucleophilic attack of a second equivalent of PhINTs on intermediate 41 (path C), leading to intermediate 42. Driven by the leaving group ability of iodobenzene, C–C bond cleavage can then occur as shown to provide the observed imine side products. In addition to providing a single explanation to account for the variety of products observed, this mechanism is consistent with the current limitation of this method to styrenyl substrates and the observed substituent effects on reaction yield, which correlate with ability to stabilize an intermediate carbocation. The inclusion of radical inhibitors (BHT, TEMPO) had no effect on reaction yield or product distribution, suggesting that a radical pathway is less likely.15 In a larger context, this evidence of a quite electrophilic nature of the diaziridinium (or related organic nitrenoid), which has been shown to effect C–H amination reactions4 as well as the aziridinations demonstrated here, can potentially be exploited in the future for the design of new reactions that are complementary to those that can be achieved using mechanistically distinct metal-catalyzed nitrene transfer methods.
Scheme 7.
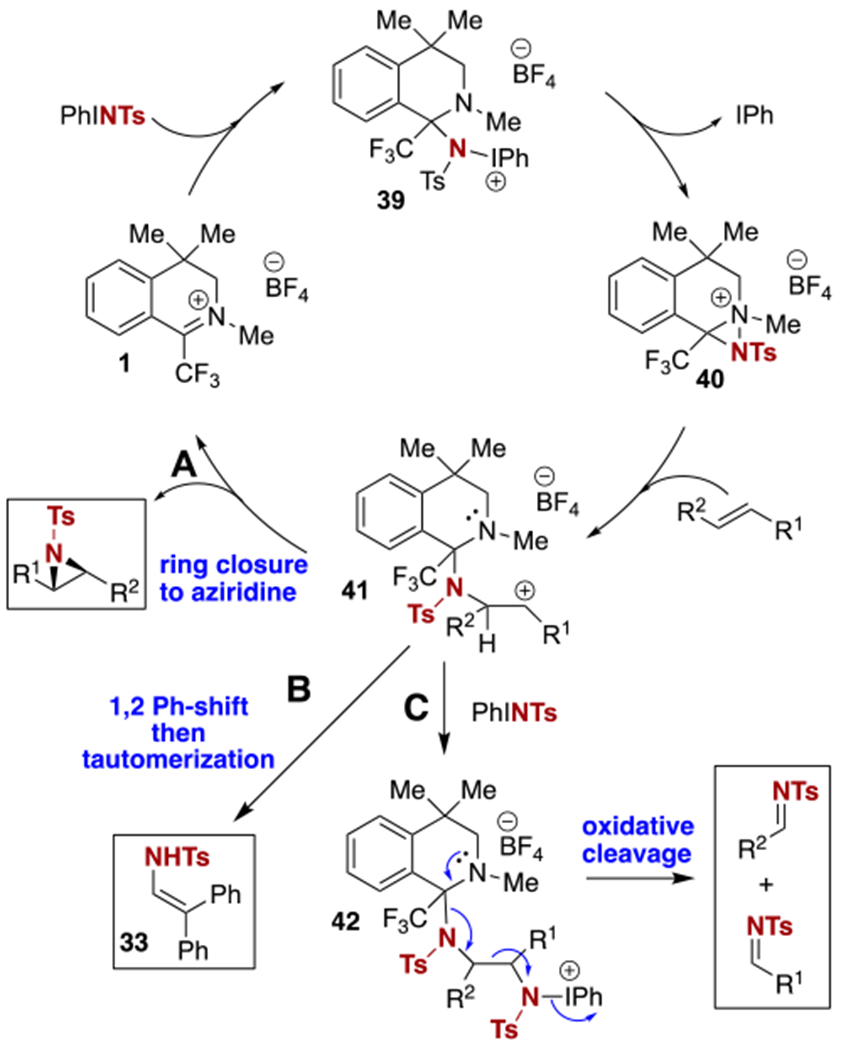
Proposed aziridination mechanism
CONCLUSIONS
In summary, we demonstrated a method for organocatalytic nitrene-transfer aziridination of styrenes using PhINTs as a nitrenoid precursor and an iminium salt as the catalyst. Mechanistic studies shed new light on the nature of iminium-catalyzed nitrene transfer, suggesting a stepwise, polar process driven by the electrophilic nature of the organic nitrenoid. Overall, this offers a complementary approach to metal catalyzed aziridination, which we currently aim to exploit in order to develop new synthetic methods aided by our improved mechanistic understanding.
EXPERIMENTAL DETAILS
General Methods.
All reagents and solvents were obtained commercially in reagent grade or better quality and used without further purification. Anhydrous dichloromethane, tetrahydrofuran and hexanes were obtained by degassing followed by passing through an alumina drying column before use. Unless otherwise noted, reactions were performed under an atmosphere of dry N2. Flash column chromatography was performed using silica gel (230-400 mesh) purchased from Silicycle. 1H and 13C spectra were acquired at 300 K unless otherwise noted on a Broker Avance III (600 MHz) or Varian NMRS (600 MHz) spectrometer. Chemical shifts are reported in parts per million (ppm δ) referenced to the residual 1H resonance of the solvent. The following abbreviations are used singularly or in combination to indicate the multiplicity of signals: s - singlet, d - doublet, t - triplet, q - quartet, m - multiplet and br – broad. NMR yields were determined using Methyl-3-nitrobenzoate as an internal standard. High resolution mass spectrometry was obtained using an Agilent Q-TOF ESI spectrometer. Styrenes 2, 4, 6, 28 and 30 were purchased from Sigma Aldrich Chemicals. Styrenes 7,16 8,17 9,18 10,17 11,16 12,19 14,16 15,20 35,21, 37,22 and aziridine 3123 were prepared using previously reported methods.
Synthesis of N-Tosylphenyimidoliodinane (PhINTs)
p-Toluenesulfonamide (65.4 mmol. 11.2 g) and KOH (165.4 mmol. 9.28g) were dissolved in 240 mL of methanol and cooled to 0 °C. To the stirred solution was slowly added diacetoxyiodobenzene (65.8 mmol. 21.2 g). The now yellow solution was stirred for 30 minutes at 0 °C, then was warmed to room temperature and stirred for 3 hours. The reaction was then cooled to 0 °C and 150 mL ice water was added and the mixture was stirred for an additional 1.5 hours at 0 °C. Following this, the mixture was filtered to collect the precipitate, which was washed with cold methanol followed by ethyl acetate to provide the product (14.5 g. 83%) as an off-white solid. 1H NMR (598 MHz. DMSO-d6) δ 7.69 (dd. J = 8.3, 1.2 Hz, 2H). 7.45 (dd, J = 8.4, 6.7 Hz, 3H). 7.29 (t, J = 7.8 Hz, 2H). 7.06 (d, J = 7.9 Hz, 2H). 2.27 (s, 3H). Matches literature values.24
Synthesis of styrene 13.
Methyl triphenylphosphonium iodide (11 mmol) was suspended in anhydrous THF (50 mL) under inert atmosphere. After cooling to 0°C. n-butyl lithium (11 mmol) was added dropwise and the resulting solution was stirred for 5 minutes. The 2-bromo-6-methoxybenzaldehyde (10 mmol) was added in a single portion and the reaction mixture was allowed to warm to ambient temperature. The reaction was monitored by TLC until the benzaldehyde was consumed, at which time the reaction was diluted with 50 mL Et2O and filtered through a pad of silica. The filtrate was concentrated and purified by silica gel chromatography (2% acetone in hexanes) to afford the product 13 as a colorless oil (1.83g, 86%).
2-bromo-6-methoxystyrene (13)
1H NMR (600 MHz, CDCl3) δ 7.55 (d. J = 2.5 Hz, 1H), 7.32 (dd, J = 8.7, 2.5 Hz, 1H), 6.96 (dd, J = 17.7, 11.1 Hz, 1H), 6.74 (d, J = 8.7 Hz, 1H), 5.72 (d, J = 17.7 Hz, 1H), 5.30 (d, J = 11.1 Hz, 1H), 3.83 (s, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 155.9, 131.4, 130.6, 129.2, 128.9, 115.8, 113.2, 112.7, 55.8, HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H10BrO 212.9915; Found 212.9922.
General procedure for optimization of aziridination.
Under inert atmosphere, iminium catalyst 1 and nitrene-precursor were suspended in anhydrous solvent. Styrene (0.1 mmol. 11.5 μL) was added and the reaction was stirred at room temperature. Upon completion, the reaction was diluted with 2 mL of ethyl acetate and filtered through a short silica plug. The crude reaction mixture was concentrated on a rotary evaporator and analyzed by NMR using methyl-3-nitrobenzoate as an internal standard.
General procedure for aziridine synthesis.
Under inert atmosphere. PhINTs (1 mmol. 373 mg) and iminium catalyst 1 (0.1 mmol. 33 mg) were suspended in a 9:1 DCM:Hexanes mixture (2.5mL total). The corresponding styrene (0.5 mmol) was added, and the reaction was stirred at room temperature for 22 hours; upon which time the mixture was diluted with 5 mL EtOAc and filtered through a short pad of silica gel. The filtrate was carefully concentrated in vacuo, and was purified by silica gel chromatography (10-15% acetone in hexanes) to afford the corresponding aziridine.
2-phenyl-N-tosylaziridine (5).
Prepared from styrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. White solid (97.6 mg, 0.357 mmol, 72%). 1H NMR (600 MHz, CDCl3) δ 7.87 (d, J = 8.3 Hz, 2H), 7.35 – 7.31 (m, 2H), 7.29 (d, J = 7.5 Hz, 3H), 7.22 (d, J = 2.1 Hz, 2H), 3.78 (dd, J = 7.2, 4.4 Hz, 1H), 2.99 (d, J = 7.2 Hz, 1H), 2.43 (s, 3H), 2.39 (d, J = 4.4 Hz, 1H) ppm. Matches literature values.25
2-(4-fluorophenyl)-N-tosylaziridine (17).
Prepared from 4-fluorostyrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Off-white solid (59.8 mg,, 0.205 mmol, 41%). 1H-NMR (598 MHz, CDCl3) δ 7.86 (d, J = 8.4 Hz, 2H), 7.35 – 7.32 (m, 2H), 7.19 (dd, J = 8.8, 5.3 Hz, 2H), 6.98 (t, J = 8.6 Hz, 2H), 3.75 (dd, J = 7.2, 4.4 Hz, 1H), 2.97 (d, J = 7.2 Hz, 1H), 2.44 (s, 3H), 2.35 (d, J = 4.4 Hz, 1H). Matches literature values.24
2-(4-chlorophenyl)-N-tosylaziridine (18).
Prepared from 4-chlorostyrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Yellow solid (72.5 mg 0.236 mmol, 47%). 1H NMR (598 MHz, CDCl3) δ 7.86 (d, J = 8.3 Hz, 2H), 7.35 – 7.32 (m, 2H), 7.28 – 7.25 (m, 3H), 7.15 (d, J = 8.5 Hz, 2H), 3.73 (dd, J = 7.1, 4.4 Hz, 1H), 2.98 (d, J = 7.1 Hz, 1H), 2.44 (s, 3H), 2.34 (d, J = 4.4 Hz, 1H). Matches literature values.25
2-(4-bromophenyl)-N-tosylaziridine (19)
Prepared from 4-bromostyrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Yellow solid (57.9 mg, 0.165 mmol, 33%). 1H NMR (598 MHz, CDCl3) δ 7.85 (d, J = 7.9 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 7.09 (d, J = Hz, 2H), 3.72 (dd, J = 7.0, 4.5 Hz, 1H), 2.98 (d, J = 8.1 Hz, 1H), 2.44 (s, 3H), 2.34 (d, J = 4.4 Hz, 1H). Matches literature values.26
2-(4-iodophenyl)-N-tosylaziridine (20).
Prepared from 4-iodostyrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Yellow solid (64.4 mg, 0.161 mmol, 32%). 1H NMR (600 MHz, CDCl3) δ 7.85 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.2 Hz, 2H). (dd, J = 7.2, 4.4 Hz, 1H), 2.98 (d, J = 7.2 Hz, 1H), 2.43 (s, 3H), 2.33 (d, J = 4.4 Hz, 1H). Matches literature values.26
2-(4-cyanophenyl)-N-tosylaziridine (21).
Prepared from 4-cyanostyrene using the general procedure for aziridination. Purified on silica using 20% acetone in hexanes. Pale yellow semi-solid (36.2 mg, 0.121 mmol, 24%). 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.35 (dd, J = 8.3, 4.2 Hz, 4H), 3.80 (dd, J = 6.6, 3.7 Hz, 1H), 3.02 (d, J = 7.2 Hz, 1H), 2.45 (s, 3H), 2.35 (d, J = 4.3 Hz, 1H). Matches literature values.23
2-(4-trifluoromethylphenyl)-N-Tosylaziridine (22).
Prepared from 4-trifluomethylstyrene using the general procedure for aziridination. Purified on silica using 10-15% acetone in hexanes. Off white solid. (50.8 mg, 0.149 mmol, 30%). 1H NMR (598 MHz, CDCl3) δ 7.87 (d, J = 8.3 Hz, 2H), 7.55 (d, J = 8.1 Hz, 2H), 7.36 – 7.323 (m, 4H), 3.81 (dd, J = 7.2, 4.3 Hz, 1H), 3.02 (dd, J = 7.2, 0.7 Hz, 1H), 2.45 (s, 3H), 2.37 (d, J = 4.4 Hz, 1H). Matches literature values.25
2-(3-tertbutyldimethylsiloxyphenyl)-N-tosylaziridine (23).
Prepared 3-tertbutyldimethylsiloxystyrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Light brown solid (92.8 mg, 0.230 mmol, 46%) 1H NMR (600 MHz, CDCl3) δ 7.86 (d, J = 8.2 Hz, 2H), 7.35 – 7.31 (m, 2H), 7.13 (t, J = 7.9 Hz, 1H), 6.84 – 6.78 (m, 1H), 6.73 (ddd, J = 8.1, 2.5, 1.0 Hz, 1H), 6.66 – 6.62 (m, 1H), 3.69 (dd, J = 7.2, 4.4 Hz, 1H), 2.98 (d, J = 7.2 Hz, 1H), 2.43 (s, 4H), 2.37 (d, J = 4.5 Hz, 1H), 0.95 (s, 9H), 0.14 (d, J = 1.4 Hz, 6H). 13C{1H} NMR (150 MHz, CDCl3) δ 155.9, 144.7, 136.6, 135.12, 129.9, 129.7, 128.0, 120.1, 119.6, 118.3, 41.1, 35.8, 25.7, 21.74 18.3, −4.36. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H30NO3SSi 404.1710; Found 404.1706.
2-(2-bromo-6-methoxyphenyl)-N-tosylaziridine (24).
Prepared 2-bromo-6-methoxystyrene using the general procedure for aziridination. Purified on silica using 15-20% acetone in hexanes. White solid (97.4 mg, 0.255 mmol, 51%). 1H NMR (598 MHz, CDCl3) δ 7.88 (s, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.32 (dd, J = 8.7, 2.5 Hz, 1H), 7.17 (d, J = 2.5 Hz, 1H), 6.70 (d, J = 8.7 Hz, 1H), 4.04 (dd, J = 7.2, 4.4 Hz, 1H), 3.79 (s, 3H), 2.96 (d, J = 7.3 Hz, 1H), 2.45 (s, 3H), 2.27 (d, J = 4.5 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 157.3, 135.0, 132.0, 130.0, 129.5, 128.2, 126.0, 113.0, 112.1, 55.8, 36.4, 35.8, 21.8. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H17BrNO3S 382.0107; Found 382.0125.
2-(3,5-dimethylphenyl)-N-tosylaziridine (25).
Prepared from 3,5-dimethylstyrene using the general procedure for aziridination. Purified on silica using 10-15% acetone in hexanes. White semi-solid (58.8 mg,0.195 mmol, 39%). 1H NMR (600 MHz, CDCl3) δ 7.87 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 6.91 (s, 1H), 6.83 (s, 2H), (dd, J = 7.2, 4.5 Hz, 1H), 2.94 (d, J = 7.2 Hz, 1H), 2.44 (s, 3H), 2.37 (d, J = 4.5 Hz, 1H), 2.26 (s, 6H). 13C{H} NMR (150 MHz, CDCl3) δ 144.7, 138.3, 135.2, 135.0, 130.1, 129.9, 128.1, 124.5, 41.2, 36.0, 21.8, 21.3. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H20NO2S: 302.1209; Found 302.1211.
2-(4-phenylboronic acid pinacol ester)-N-tosylaziridine (26).
Prepared from 4-(boronic acid pinacol ester)-styrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Yellow solid (60.9 mg, 0.152 mmol, 31%). 1H NMR (598 MHz, CDCl3) δ 7.86 (d, J = 7.1 Hz, 2H), 7.72 (d, J = 6.8 Hz, 2H), (d, J = 7.6 Hz, 2H), 7.20 (d, J = 7.0 Hz, 2H), 3.75 (dd, J = 7.0, 4.5 Hz, 1H), 3.01 (d, J = 8.6 Hz, 1H), 2.42 (s, 3H), 2.39 (d, J = 4.4 Hz, 1H), 1.32 (s, 12H). 13C{1H} NMR (150 MHz, CDCl3) δ 144.8, 138.2, 135.1, 129.9, 128.1, 126.0 , 84.0, 41.3, 36.1, 25.0, 25.0, 21.8. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H27BNO4S 400.1748; Found 400.1750.
2-methyl-3-phenyl-N-tosylaziridine (29).
Prepared from trans- β-methylstyrene or cis-β-methylstyrene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Off-white solid (34.6 mg, 0.121 mmol, 24%). 1H NMR (598 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 2H), 7.29 – 7.25 (m, 5H), 7.18 – 7.14 (m, 2H), 3.81 (d, J = 4.2 Hz, 1H), 2.93 (p, J = 6.0 Hz, 1H), 2.41 (s, 3H), 1.86 (d, J = 6.0 Hz, 3H). Matches literature values.23
2-(1,2,3,4-tetrahydronaphthalen-2-yl)-1-tosylaziridine (38).
Prepared from 2,3,4,5-tetrahydro-2-vinylnapthalene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes (21% 1H-NMR yield). Imine isolated as yellow oil (22.7mg, 0.073 mmol, 15%). 1H NMR (600 MHz, CDCl3) δ 8.95 (s, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.63 (s, 1H), 7.61 (d, J = 7.9 Hz, 1H), (d, J = 7.8 Hz, 2H), 7.16 (d, J = 7.9 Hz, 1H), 2.80 (d, J = 21.6 Hz, 4H), 2.43 (s, 3H), 1.80 (p, J = 3.3 Hz, 4H). 13C{1H} NMR (150 MHz, CDCl3) δ 170.5, 146.0, 144.5, 138.5, 135.7, 132.3, 130.1, 129.9, 128.9, 128.1, 77.2, 30.1, 29.3, 22.9, 22.8, 21.8. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H20NO2S 314.1209; found 314.1201.N-(2,2-diphenylvinyl)-4-methylbenzenesufonamide (33). Prepared from trans-stilbene using the general procedure for aziridination. Purified on silica using 10-20% acetone in hexanes. Off-white solid (26.1 mg, 0.075 mmol, 15%). 1H NMR (598 MHz, CDCl3) δ (d, J = 8.3 Hz, 2H), 7.36 – 7.33 (m, 4H). 7.25 – 7.21 (m, 2H). 7.12 – 7.08 (m, 2H), 6.92 (dd, J = 7.6, 1.8 Hz, 2H), 6.79. (d, J = 11.6 Hz, 1H), 6.25 (d, J = 11.6 Hz, 1H), 2.45 (s, 3H). Matches literature values.27
Aziridination procedure (1 mmol scale).
Under inert atmosphere. PhINTs (2 mmol, 746 mg) and iminium catalyst 1 (0.2 mmol, 66 mg) were suspended in a 9:1 DCM:Hexanes mixture (5 mL total). 4-chlorostyrene (1 mmol, 139 mg) was added, and the reaction was stirred at room temperature for 22 hours; upon which time the mixture was diluted with 10 111F EtOAc and filtered through a short pad of silica gel. The filtrate was concentrated and purified by silica gel chromatography (10-15% acetone in hexanes) to afford aziridine 18 (154.5 mg, 0.503 mmol, 50%) as a yellow solid.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (R01 GM124092) for funding.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
NMR spectra for all isolated products (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).For some recent reviews on organocatalytic epoxidation see:; (a) Zhu Y; Wang Q; Cornwall RC; Shi Y Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev 2014, 114, 8199–8256. [DOI] [PubMed] [Google Scholar]; (b) Davis RF; Stiller J; Naicker T; Jiang H; Jorgensen KA Asymmetric organocatalytic epoxidations: Reactions, scope, mechanisms, and applications. Angew. Chem. Int. Ed 2014, 53, 7406–7426. [DOI] [PubMed] [Google Scholar]
- (2).(a) Shuler WG; Johnson SL; Hilinski MK Organocatalytic, dioxirane-mediated C-H hydroxylation under mild conditions using oxone. Org. Lett 2017, 19, 4790–4793. [DOI] [PubMed] [Google Scholar]; (b) Wang D; Shuler WG; Pierce CJ; Hilinski MK An iminium salt organocatalyst for selective aliphatic C-H hydroxylation. Org. Lett 2016, 18, 3826–3829. [DOI] [PubMed] [Google Scholar]; (c) Pierce CJ; Hilinski MK Chemoselective hydroxylation of aliphatic sp3 C–H bonds using a ketone catalyst and aqueous H2O2. Org. Lett 2014. 16, 6504–6507. [DOI] [PubMed] [Google Scholar]; (d) Adams AM; Du Bois J Organocatalytic C–H hydroxylation with oxone enabled by an aqueous fluoroalcohol solvent system. Chem. Sci 2014, 5, 656–659. [Google Scholar]; (e) Litvinas ND; Brodsky BH; Du Bois J C–H hydroxylation using a heterocyclic catalyst and aqueous H2O2. Angew. Chem. Lnt. Ed 2009, 48, 4513–4516. [DOI] [PubMed] [Google Scholar]; (f) Brodsky BH; Du Bois J Oxaziridine-mediated catalytic hydroxylation of unactivated 3° C–H bonds using hydrogen peroxide. J. Am. Chem. Soc 2005, 127, 15391–15393. [DOI] [PubMed] [Google Scholar]
- (3).Johnson S; Combee L; Hilinski M Organocatalytic atom-transfer C(sp3)–H oxidation. Synlett 2018, 29, 2331–2336. [Google Scholar]
- (4).MacMillan DWC The Advent and Development of Organocatalysis. Nature 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- (5).Combee LA; Raya B; Wang D; Hilinski MK Organocatalytic nitrenoid transfer: Metal-free selective intermolecular C(sp3)–H amination catalyzed by an iminium salt. Chem. Sci 2018, 9, 935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For a recent review on aziridination see:; Degennaro L; Trinchera P; Luisi R Recent advances in the stereoselective synthesis of aziridines. Chem. Rev 2014, 114, 7881–7929. [DOI] [PubMed] [Google Scholar]
- (7).For helpful reviews on functionalization of aziridines, see:; (a) Stankovic S; D’hooge M; Catak S; Eum H; Waroquier M; Van Speybroeck V; De Kimpe N; Ha H-J Regioselectivity in the ring opening of non-activated aziridines. Chem. Soc. Rev 2012, 41, 643–665. [DOI] [PubMed] [Google Scholar]; (b) Lu P Recent developments in regioselective ring opening of aziridines. Tetrahedron 2010, 66, 2549–2560. [Google Scholar]; (c) Hu XE Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. [Google Scholar]
- (8).For a recent review on the subject of organocatalytic aziridination, see:; Roma E; Tosi E; Micelli M; Gasperi T Asymmetric Organocatalytic Aziridination: Recent Advances. Asian J. Org. Chem 2018. 7 2357–2367. [Google Scholar]
- (9).For selected recent examples, see:; (a) Frankowski S; Bojanowski J; Saktura M; Romaniszyn M; Drelich P; Albrecht L Organocatalytic synthesis of cis-2,3-aziridine aldehydes by a postreaction isomerization. Org. Lett 2017, 19, 5000–5003. [DOI] [PubMed] [Google Scholar]; (b) Molnár IG; Tanzer E-M; Daniliuc C; Gilmour R Enantioselective Aziridination of Cyclic Enals Facilitated by the Fluorine-Iminium Ion Gauche Effect. Chem. Eur. J 2014. 20, 794–800. [DOI] [PubMed] [Google Scholar]; (c) Halskov KS; Naicker T; Jensen ME; Jorgensen KA Organocatalytic asymmetric remote aziridination of 2,4-dienals. Chem. Commun 2013, 49, 6382–6383. [DOI] [PubMed] [Google Scholar]; (d) Page PCB; Bordogna C; Strutt I; Chan Y; Buckley B Asymmetric aziridination of chalcone promoted by binaphthalene-based chiral amines. Synlett 2013, 24, 2067–2072. [Google Scholar]; (e) De Fusco C; Fuoco T; Croce G; Lattanzi A Noncovalent organocatalytic synthesis of enantioenriched terminal aziridines with a quaternary stereogenic center. Org. Lett 2012, 14, 4078–4081. [DOI] [PubMed] [Google Scholar]; (f) Desmarchelier A; Pereira de Sant’Ana D; Terrasson V; Campagne JM; Moreau X; Greck C; Marcia de Figueiredo R Organocatalyzed aziridination of α-branched enals: Enantioselective synthesis of aziridines with a quaternary stereocenter. Eur. J. Org. Chem 2011, 2011, 4046–4052. [Google Scholar]; (g) Deiana L; Dziedzic P; Zhao GL; Vesely J; Ibrahem I; Rios R; Sun J; Córdova A Catalytic asymmetric aziridination of α,β-unsaturated aldehydes. Chem. Eur. J 2011, 17, 7904–7917. [DOI] [PubMed] [Google Scholar]; (h) Murakami Y; Takeda Y; Minakata S Diastereoselective aziridination of chiral electron-deficient olefins with N-chloro-N-sodiocarbamates catalyzed by chiral quaternary ammonium salts. J. Org. Chem 2011, 76, 6277–6285. [DOI] [PubMed] [Google Scholar]; (i) De Vincentiis F; Bencivenni G; Pesciaioli F; Mazzanti A; Bartoli G; Galzerano P; Melchiorre P Asymmetric catalytic aziridination of cyclic enones. Chem. Asian J 2010. 5, 1652–1656. [DOI] [PubMed] [Google Scholar]
- (10).Wang D; Shuler WG; Pierce CJ; Hilinski MK An Iminium Salt Organocatalyst for Selective Aliphatic C-H Hydroxylation. Org. Lett 201618, 3826–3829. [DOI] [PubMed] [Google Scholar]
- (11).Yamada Y; Yamamoto T; Okawara M Synthesis and reaction of new type I-N ylide, N-tosyliminoiodinane, Chem. Lett 1975, 4, 361–362. [Google Scholar]
- (12).Of the iminoiodinanes used for this study, only PhINTces had been previously shown to be compatible with iminium-catalyzed aziridination. Attempted aziridination of styrene using PhINTces resulted in nearly complete consumption of the olefin (~5% remaining), with no production of either aziridine or imine byproduct. No products arising from styrene consumption could be conclusively identified; oligomerization is suspected.
- (13).For a related oxidative cleavage reaction, see:; Ding Y; Li H; Meng Y; Zhang T; Li J; Chen Q-Y; Zhu C Direct synthesis of hydrazones by visible light mediated aerobic oxidative cleavage of the C=C bond. Org. Chem. Front 2017, 4, 1611–1614. [Google Scholar]
- (14).For the first report of the isolation and unambiguous characterization, of a diaziridinium salt, see:; Allen JM; Lambert TH Synthesis and characterization of a diaziridinium ion. Conversion of 3,4-dihydroisoquinolines to 4,5-dihydro-3H-benzo[2,3]diazepines via a formal N-insertion process. Tetrahedron 2014, 70, 4111–4117. [Google Scholar]
- (15).Three concurrent reactions using the standard conditions outlined in the general procedure, those conditions plus 1 equiv of BHT, and those conditions plus 1 equiv of TEMPO, gave identical yields for the aziridination of styrene. Imine 34 was also formed in each case, in roughly equivalent amounts by crude 1H NMR.
- (16).Haubenreisser S; Woste TH; Martinez C; Ishihara K; Muniz K Sturcturally Defined Molecular hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed 201555413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Movahhed S; Westphal J; Dindaroglu M; Falk A; Schmalz H-G Low-Pressure Cobalt-Catalyzed Enatioselective Hydrovinylation of Vinylarenes. Chem. - Eur. J 2016, 22, 7381–7384. [DOI] [PubMed] [Google Scholar]
- (18).Cao H; Jiang H; Feng H; Kwan JJC; Liu X; Wu J Photo-induced Decarboxylative Heck-Type Coupling of Unactivated Aliphatic Acids and Terminal Alkenes in the Absence of Sacrificial Hydrogen Accceptors. J. Am. Chem. Soc 2018, 140, 16360–16367. [DOI] [PubMed] [Google Scholar]
- (19).Kumar GDK; Natarajan A total synthesis of ovalifoliolatin B, acerogenins A and C. Tet. Lett 2008, 49, 2103–2105. [Google Scholar]
- (20).Wang G-Z; Shang R; Fu Y Irradiation-Induced Palladium-Catalyzed Decarboxylative Heck Reaction of Aliphatic N-(Acyloxy)phthalimides at Room Temperature. Org. Lett 2018, 20, 888–891’. [DOI] [PubMed] [Google Scholar]
- (21).Antonioletti R; Bonadies F; Ciammaichella A; Viglianti A Tet. Lett 2008. 64, 4644–4648. [Google Scholar]
- (22).Jian W; Ge L; Jiao Y; Qian B; Bao H Iron-Catalyzed Decarboxylatice Alkyl Etherification of Vinylarenes with Aliphatic Acids as the Alkyl Source. Angew. Chem. Int. Ed 2017, 56, 3650–3654. [DOI] [PubMed] [Google Scholar]
- (23).Sharpless KB; Jeong J Patent US 1998-97845 1999.
- (24).Ito M; Tanaka A; Higuchi K; Sugiyama S; Eur. J. Org. Chem. 2017. 9, 1272–1276. [Google Scholar]
- (25).Shukla P; Mahata S; Sahu A; Singh M; Rai VK; Rai A First graphene oxide promoted metal-free nitrene insertion into olefins in water: towards facile synthesis of activated aziridines. RSC Adr. 2017. 748723–48729. [Google Scholar]
- (26).Yin J; Hyland CJT. Palladium(II)-Catalyzed C3-Selective Friedel-Crafts Reaction of Indoles with Aziridines. Asian. J. Org. Chem 2016. 5 1368–1377. [Google Scholar]
- (27).Selander N; Worrell WT; Chuprakov S; Velaparthi S; Fokin VV J. Am. Chem. Soc 2012134, 14670–14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.