

Abstract
Spatiotemporal localization of protein function is essential for physiological processes from sub-cellular to tissue scales. Genetic and pharmacological approaches have played instrumental roles in isolating molecular components necessary for sub-cellular machinery. However, these approaches have limited capabilities to reveal the nature of spatiotemporal regulation of sub-cellular machineries like those of cytoskeletal organelles. With the recent advancement of optogenetic probes, the field now has a powerful tool to localize cytoskeletal stimuli in both space and time. Here, we detail the use of the tunable light-controlled interacting protein tags (TULIPs) to manipulate RhoA signaling in vivo. This is an optogenetic dimerization system that rapidly, reversibly, and efficiently directs a cytoplasmic RhoGEF to the plasma membrane for activation of RhoA using light. We first compare this probe to other available optogenetic systems and outline the engineering logic for the chosen recruitable RhoGEFs. We also describe how to generate the cell line, spatially control illumination, confirm optogenetic control of RhoA, and mechanically induce cell-cell junction deformation in cultured tissues. Together these basic protocols will detail how to probe mechanochemical circuitry that is downstream of RhoA signaling.
Keywords: Optogenetics, RhoA, Quantitative Imaging, Contractility
Introduction
To gain a better understanding of how signaling achieves spatiotemporally structured sub-cellular protein complexes and cytoskeletal organelles, we must be able to exert experimental control over these signaling pathways. The use of light-sensitive moieties, in combination with structured illumination, provide a promising route. In the past, the availability of photo-responsive elements that allow for switching, binding or uncaging have been limited. Recent advances in optogenetics have allowed for the molecular dissection of spatiotemporal signaling modules. Optogenetics utilizes photosensitive proteins that change conformation upon exposure to specific wavelengths, resulting in altered protein-protein interactions and modulation of downstream signals. This technique is appealing, as it can be used to study the effects of the location, intensity, periodicity, and duration of light pulses and subsequent signaling activity.
Nearly a decade ago, a seminal paper by Strickland et al was published describing how light-sensitive protein domains could be repurposed as optogenetic dimerization tools(1). This initial study introduced the light-oxygen-voltage sensing (LOV) domain of Avena sativa phototropin 1 (AsLOV2). LOV domains’ conformations are light-sensitive, making them ideal for optogenetic use. This system was named TULIPs, as they are tunable, light-controlled interacting protein tags. In their original paper reporting the use of the TULIP system, Strickland et al successfully documented the activation of two cellular signaling modules in yeast. Specifically, they dissected the yeast mating pathway induced by a canonical GPCR pathway. This pathway is responsible for a MAPK cascade associated with both growth arrest and polarized secretion. First, they documented light-dependent recruitment of a truncated Ste5 and full-length Ste11 to activate the MAPK pathway for subsequent cellular growth arrest. They then demonstrated that this system could successfully be used for the control of GTPase signaling. Here, they used light-directed recruitment of Cdc42 to induce mating projections, or shmoos, in a polarized fashion. Together these data showed the effectiveness of the TULIP system in regulating the activity of nucleotide-exchange factors, scaffold proteins, and kinases.
RhoA signaling is an ideal signaling pathway for optogenetic control. Rho-dependent signaling, regulated in space and time, drives a myriad of biological processes (e.g. development, homeostasis, and disease)(2). RhoA is a small, membrane bound, GTPase that largely controls the cellular basis of contractility through activation of its downstream effectors, actin and myosin (3). RhoA activation is achieved by nucleotide exchange mediated by Guanine Nucleotide Exchange factors, or GEFs. The optogenetic strategy here is elegant: drive the localization of a RhoA-specific GEF to the plasma membrane for centralized activation of RhoA and its subsequent downstream effectors (Figure 1). Recent optogenetic tools have subcellularly localized RhoA GEFs for RhoA activation in dividing(4), non-adherent(5, 6), and adherent cells in culture(7), and more recently in tissue both in culture (8–10) and in vivo(11, 12). These studies have successfully probed the complex nature of RhoA-mediated contractility on cell-cell and cell-matrix forces, in addition to deciphering mechanosensitive signaling pathways that regulate cellular morphology and tissue-scale morphogenesis.
Figure 1:
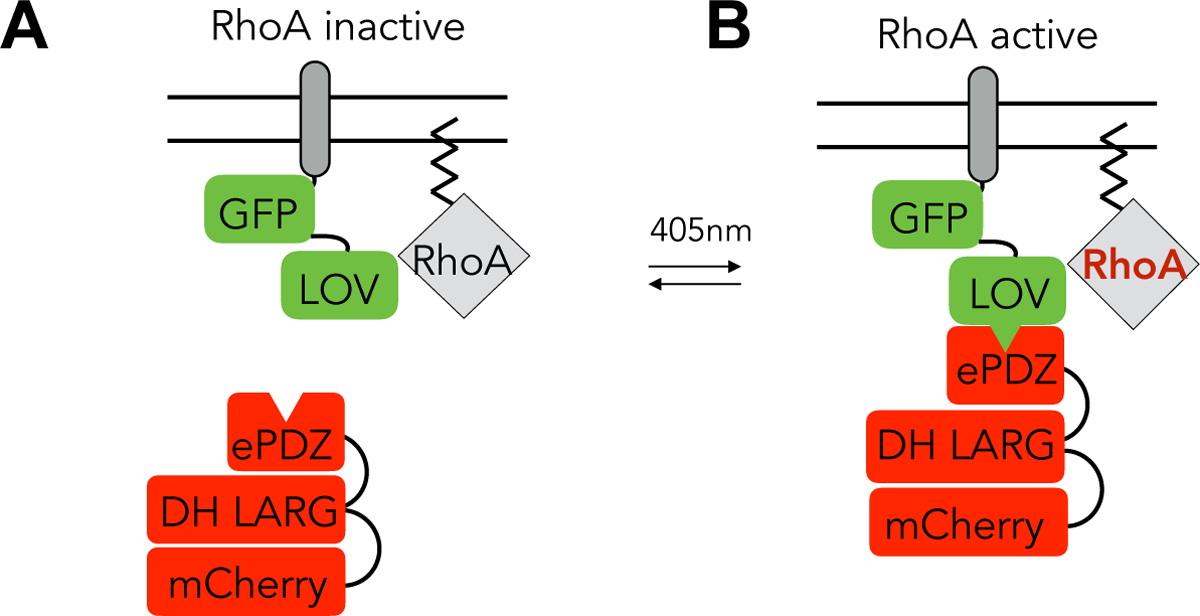
A) Schematic of the TULIP system in the dark state. Stargazin-GFP-LOVpep sits in the closed confirmation at the plasma membrane next to inactive RhoA. The 2x-PDZ-mCherry-LARG sits diffusely in the cytoplasm. B) Schematic of the TULIP system in the activated state. 405nm light causes a conformational change of the Stargazin-GFP-LOVpep that increases the binding affinity to the 2X-PDZmCherry LARG, recruiting it to the membrane where it activates RhoA.
Specifically, the TULIP system has proved to be a versatile tool in this analysis of mechanochemical signaling in driving the formation of sub-cellular cytoskeletal organelles. RhoA activation drives spatiotemporally structured sub-cellular organelles like those of the cytokinetic ring, actin stress fibers, or the contractile actin belt anchored at adherens junctions, to name a few. Using the TULIP system, Wagner and Glotzer exogenously activated RhoA to find that it was sufficient to induce cytokinetic furrow formation in single anaphase cells (4). In Oakes et al 2017, the system was used to probe the molecular basis of actin stress fiber elasticity in single cells, showing a zyxin-dependent mechanism(7). More recently, it was discovered that RhoA was sufficient to induce stable cell-cell junction deformations past a critical strain threshold to trigger mechanosensitive endocytosis in epithelial monolayers (9, 10). Altogether, these studies show the diverse applications of the TULIP system. We believe that with the right engineering, optogenetic RhoA can be used for any application. Further studies using optogenetically activated RhoA will only continue to advance our understanding of cell and tissue mechanics.
In this unit, we first describe the strategic planning associated with choosing and designing an optogenetic system. We then describe five basic protocols for optogenetic studies of RhoA in epithelial tissues: (1) Generating stable cell lines (Basic Protocol 1); (2) Preparation of the substrate for imaging (Basic Protocol 2); (3) Visualization of downstream effectors (Basic Protocol 3); (4) Spatial illumination calibration (Basic Protocol 4); and (5) Optogenetic activation of a region of interest (Basic Protocol 5).
Strategic planning
Choosing the optogenetic system
A number of dimerization systems have been developed, each with diverse properties for different biological applications. The choice of optogenetic system will depend on factors such as wavelength compatibility, dynamic range, and requirements for activation speed, reversibility, and depth of tissue to be imaged. We recommend choosing the optogenetic system with the desired reversibility kinetics, as this is important for achieving local, spatially resolved subcellular control of signaling processes. Fast dimerization kinetics can be on the order of seconds, while slower kinetics are on the order of minutes to hours. Systems with slow reversal kinetics may be particularly useful if a more permanent phenotype is desired, or more persistent signaling is needed. However, since physiological signaling occurs within milliseconds to seconds, we recommend using faster dimerization kinetics to mimic in vivo signaling kinetics. The current optogenetic dimerization systems as described in the literature are listed in Table 1.
Table 1:
Optogenetic dimerization probes
| System | Association wavelength | Dissociation wavelength | Tag Sizes (amino acids) | Lifetime | Reference |
|---|---|---|---|---|---|
| PhyB/PIF6 | 660nm | 740nm | 908/100 | Inducible | (13) |
| Cry2-CIBN | 450nm | Dark | 498/170 | 5–10 min | (14) |
| iLID/SspB | 450nm | Dark | 144/110 | <50sec | (15, 16) |
| TULIPs | 450nm | Dark | 153/194 | <50sec | (1) |
| nMag/pMag | 450nm | Dark | 150/150 | Tunable | (17) |
| FKF1/GI | 450nm | Dark | 619/1173 | Hours | (18) |
| LOVTRAP | Dark | 450nm | 143/59 | Tunable | (19) |
| PixD/PixE | Dark | 450nm | 150/380 | Sec-min | (20) |
| BphP1/PpsR2 | 740nm | 650nm | 732/465 | Inducible | (21) |
| UVR8/COP1 | 280nm | N/A | 440/340 | Permanent | (22) |
Many systems have already been published for subcellular control of RhoA. If fast kinetics are desired, we recommend using the iLID/SspB or TULIP system, as these provide high temporal resolution of RhoA activation. The RhoGEF in TULIPs associate within less than 10 seconds and dissociate within 30–60 seconds (1, 4, 7, 9, 10); the iLID/SspB system shows similar association and dissociation kinetics(5, 6). Slower kinetic systems have been seen with the CRY2/CIBN light-gated dimerization system. This CRY2/CIBN system was used to manipulate RhoGEF association within minute timescales but RhoGEF dissociation was on the order of 20 minutes(8). In this case, actin accumulated and dissipated with similar kinetics as the RhoGEF. As such, the CRY2/CIBN system provides for a more permanent phenotype associated with RhoA contractility. This slower system, while not typically physiological, can provide for persistent actomyosin recruitment and RhoA signaling that can be used to study the effects of traction forces on substrates, for example. With fast or slow recruitment kinetics, the major strength of using an optogenetic approach is the ability to image a baseline state prior to activation, the response during activation, and a recovery period following activation. These three periods give key insights into the behavior and response of junctions and/or effector proteins with respect to the activation of RhoA.
Engineering the optogenetic constructs
When engineering the photosensitive protein, it is necessary to consider the desired subcellular location for recruitment. Most studies to date have anchored the photosensitive protein to the plasma membrane where RhoA sits inactive. Here, we describe the use of the TULIP system that utilizes the photosensitive LOVpep domain attached to a transmembrane protein, Stargazin(1) (Figure 1). Recent papers have also probed membrane recruitable RhoGEFs using various photosensitive proteins attached to a CAAX motif, which triggers posttranslational modifications necessary to drive the protein’s plasma membrane association and insertion(5, 6, 8). It is also conceivable to drive RhoGEF localization to other more specific areas within the cell, under the logic of sequestering the RhoGEF away from the plasma membrane. For instance, one study drove RhoGEF activity specifically to the outer mitochondrial membrane by fusion to the mitochondrial matrix targeting sequence from subunit VIII of cytochrome c oxidase (8). Other targetable proteins may be apicojunctional proteins like E-cadherin, ZO-1, or members of the PAR polarity family, although nearly any protein can be conceivably targeted with the right design.
RhoA is activated when the recruitable GEF binds to the photosensitive protein at the plasma membrane upon light activation (Figure 1). GEFs are multidomain proteins capable of catalyzing nucleotide exchange within Rho GTPases(23). The full sequence RhoGEF houses the catalytic DH domain and additional protein and/or lipid interaction motifs, suggesting that these domains act as protein scaffolding complexes and/or localization signals (24). In nearly all isoforms, the catalytic DH domain is found adjacent to a PH domain that commonly binds to phosphoinositide ligands (25) and may even aid in nucleotide exchange (26). Other common functional domains include the SH3 and PDZ protein binding domains and the RGS autoinhibitory domain, to name a few (26). It is therefore vital to consider which type of GEF is used as a dimerizer because different GEFs can result in diverse subcellular behaviors depending on the sequence motifs used (25). As such, designing this optogenetic piece will depend on the nature of the experiment and desired subcellular behaviors.
With TULIPS, the LOVpep’s cognate binding partner is an engineered tandem PDZ domain attached to the catalytic DH domain of the RhoGEF LARG(4) (Figure 1). The DH domain of LARG is a potent RhoA-specific activator and exhibits the highest catalytic activity reported for its GEF family(27). Other groups have used the DHPH domain of the Drosophila-specific RhoGEF2(11), the DHPH domain of LARG(5, 6), or the DHPH domain of ARHGEF11(8). While others have included the PH domain in their recruitable GEF complex, we recommend engineering dimerization constructs that only utilize the GEF’s catalytic DH domain to reduce basal GEF activity. Isolating the DH domain removes functional domain compositions and domain organizations that link GEF activity to specific downstream signaling modules. For example, the PH domain of PDZ-RhoGEF has been shown to bind to activated RhoA to drive a potential feedback loop that either attenuates or enhances RhoA function(28). This effect may not be desirable in an optogenetic system, as activation of a feedback loop may result in unwanted phenotypes resulting from altered RhoA function. Additionally, in some GEF proteins, the PH or the RGS domain may act upon the DH domain in an autoinhibitory fashion, preventing RhoA activation despite any GEF photorecruitment (26, 29).
Visualization and confirmation of these optogenetic proteins depends on their fluorescent tags. Tagging the anchored LOV domain to GFP aids in confirming the uptake of the probe in screening and sorting the cells. Additionally, we recommend tagging the desired RhoGEF with mCherry or another red protein variant to confirm its localization and recruitment. It is also possible to tag the 2XPDZ-LARG with a far-red protein or Halo tag conjugated with the Janelia Fluor far-red protein. There is also a commercially available YFP-2XPDZ-LARG that frees up the red channel. However, confirming the presence of this probe in cells is more difficult and relies on the visualization of either downstream effectors and relocation from the cytosol to the membrane to confirm the presence of the recruitable GEF.
When cloning, be conscious of the linkers between the LOVpep or PDZ domains and their respective proteins of interest. Linkers, or lack thereof, can affect the desired protein’s conformation. This is especially important if tagging a protein that houses specific signaling functions, such apicojunctional proteins or RhoGEFs that localize the cytoskeletal machinery. The design of a suitable linker to join protein domains can often be complicated. Careful attention needs to be paid when designing a linker with the right length, hydrophobicity, amino acid residue, and secondary structure. Flexible linkers have preferable amino acid residues that are composed of small, non-polar (e.g. Gly) or polar (e.g. Ser or Thr) amino acids (31). The most commonly used flexible linkers have stretches of Gly and Ser residues, the length and copy number of which can be optimized to separate the functional domains. For the LOVpep, we have successfully used the flexible linker GGSGGSGGSPR, while for the tandem PDZ we have used QSTVPRARDPPVAT(4, 7, 9). Other linkers for optogenetic tags include GSGGSGSGGT(19) or GSTSGSGKPGSGEGSTKG (30). These published linkers are sufficiently long and flexible so as to not affect the binding of the protein to its downstream effectors. For the anchor protein, when using a CAAX motif or another targeting sequence to a specific subcellular location, linkers are optional.
Protocol 1 - Generating a stable line
The nature of the experiment necessitates different protein expression systems. To probe the effects of RhoA localization on cell-cell junctions within a tissue in culture, for example, we recommend generating a stable cell line constitutively expressing both dimerization constructs. This is because the likelihood of both optogenetic probes being present in two adjacent cells is very low. Transient transfections of both optogenetic probes may be sufficient for the analysis of RhoA activation in single cells, although generation of a stable cell line will greatly ease experimentation. This protocol will necessitate the cloning of optogenetic constructs into a lentiviral vector (e.g. pWPT), or other vector depending on the cell type (e.g. retroviral, adenoviral), for the generation of stable lines. We recommend using a viral vector with a selectable marker such as Puromycin resistance for cell selection.
This protocol will use the Fugene 6 transfection reagent to produce lentiviral DNA, which will then be used to create a stable cell line constitutively expressing both TULIP constructs. This protocol is designed to generate lentivirus of one optogenetic construct for infection of cells in culture. This experiment will have to be done twice in order to obtain lentivirus of the second optogenetic construct. Following successful expression and sorting of one construct, the cells can be transfected and sorted again with the second construct. Alternatively, you could infect WT cells simultaneously with both viruses and perform dual-channel fluorescence sorting via FACS.
Materials List:
Lentiviral DNA vector containing desired constructs at concentrations 1ug/ul
293T cells (ATCC CRL-3216)
Opti-MEM (Gibco)
pHR1–8.2-delta-R packaging plasmid at concentration 1ug/ul (Addgene #12263)
VSV-G pseudo typing plasmid at concentration 1ug/ul (Addgene #8454)
Polybrene (EMD Millipore)
FuGENE 6 Transfection Reagent (Promega)
0.45um Millex syringe-driven filter unit (Millipore)
30mL Luer Lock tip Disposable syringe (ExellINT)
15mL conical tubes (Corning)
Centrifuge at room temperature
8-well chambers (Ibidi)
Protocol Steps:
- Day 0: Preparation of the cells for infection
- Plate 293T cells at 80% confluence
- Day 1: Transfection of 293T cells.
- Assemble reaction as follows and let sit for 10 minutes
- Note: in general, you want 3:1 ratio of FUGENE to total amount of DNA (in ug)
Item Amount (ul) Lentiviral vector 7.5 dR8.2 5 VSVG 1.25 Optimem 685 FUGENE 33.75 - Add complexes to 10ml of fresh media to the plate of 293T cells
- Place cells into the incubator and let sit for 3 days
- Day 3: Isolating the virus for infection
- Collect supernatant
- At this time, all reagents touching lentivirus should be bleached and put into a biohazard bag
- Wear double gloves and a lab coat to protect the skin
- Spin down virus-infused media to remove any remaining cells
- Carefully collect the supernatant
- Filter sterilize the supernatant with the 0.45um Filter and 30mL syringe to further remove any debris
- Add 2ml of the lentivirus, 4ug/ml Polybrene, to 6ml of fresh media to desired cell line
- The TULIP system has been successfully used in Caco-2, DLD1, HeLa, and NIH 3T3 fibroblast cells, although it is feasible to use this system in other cell lines.
- We recommend snap freezing the rest of the virus for later use in case the infection did not work. Place in the −80 freezer.
- Place cells in the incubator and let sit for a day
- Day 4: Cleaning the optogenetic cells
-
1Remove media from the dish
- Bleach the waste, as the virus is still a hazard
-
2Wash the dish with PBS
- Bleach the waste, as the virus is still a hazard
-
3Replace with fresh media on the optogenetic cells
-
4Place the optogenetic cells in incubator
- From this point on you can culture the cells normally, as the cells are not hazardous.
-
1
- Day 6+: Isolating the optogenetic cell line
-
5Allow the optogenetic cells to grow for a few days
- This will increase the number of cells expressing the optogenetic constructs in the entire cell population
-
6Once the cells have been expanded, sort cells for the desired fluorescence via FACS
- We find that relatively low levels of Stargazin-GFP-LOVpep are tolerated quite well. However, a high expression of mCherry-2xPDZ-LARG is needed to produce a marked cellular response. We recommend sorting the cells for highest 50% expression of the Stargazin-GFP and top 5–10% highest expressing mCherry-2xPDZ-LARG.
- Immediately upon sorting, place cells in the dark. Ambient light can activate the TULIP system. While it is okay to have some exposure to light in the room, prolonged exposure and recruitment of the GEF to the membrane may result in cell blebbing or cell death.
- Expand each clonal population
-
7Screen the cell clones for optimal expression of optogenetic constructs
- Split each of the clonal populations in media, one portion of the cells into a cell culture dish and one portion into one respective well in the 8 well Ibidi chamber. Make sure to keep a record of which clonal population is which. The density of plating can be varied depending on the experiment.
- To confirm expression of both of the constructs, first take an image in the mCherry channel, followed by an image in the GFP channel, and then again in the mCherry channel. If the cytoplasmic RhoGEF shifts localization to the junctions, this clone is primed for optogenetic activation (similar to figure 2).
-
8Expand the selected. clonal population for use in the next Protocols
-
5
Figure 2:
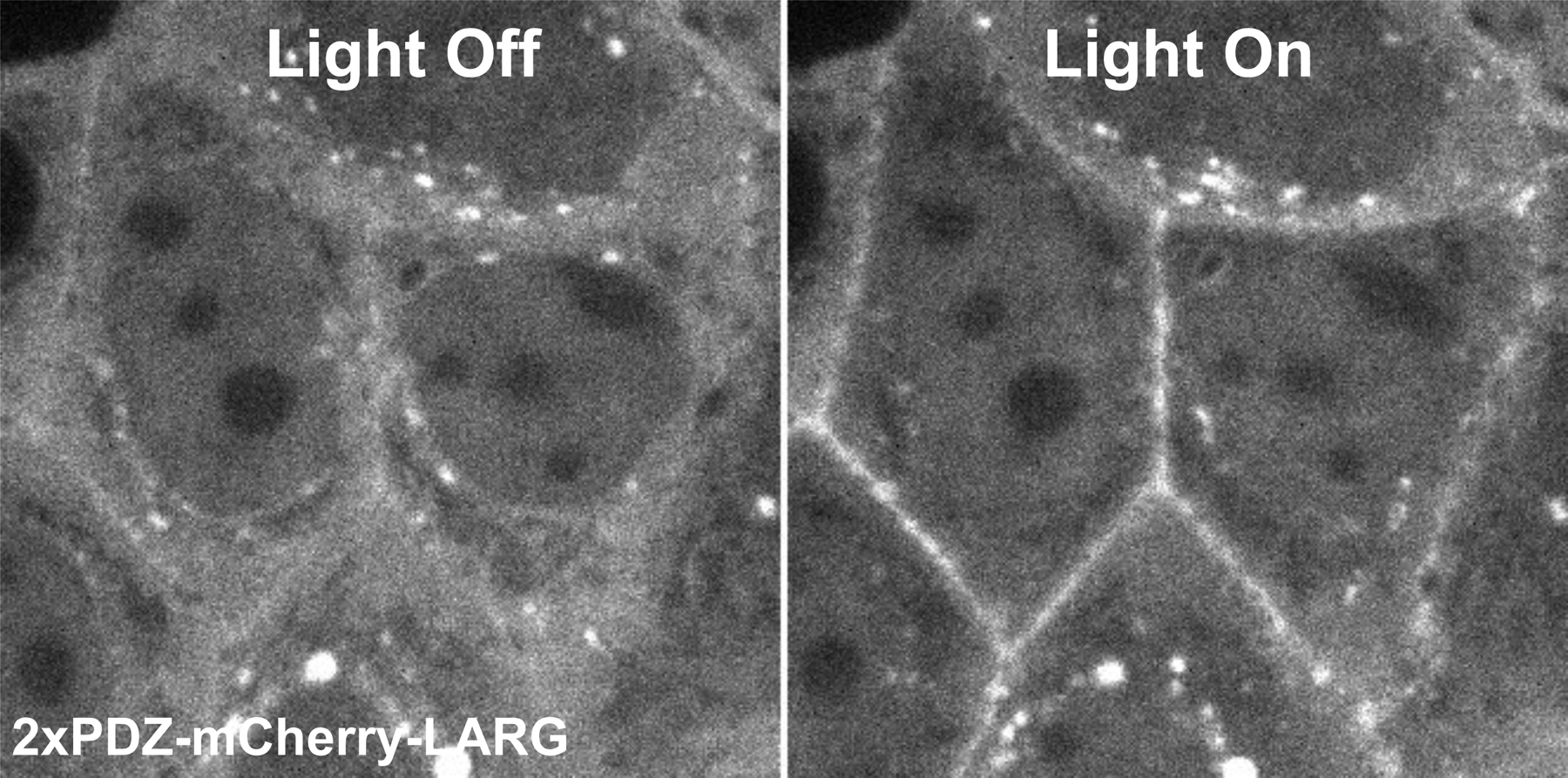
Representative images of a stable optogenetic cell line before and after light recruitment visualized by 2xPDZ-mCherry-LARG expression. Left: Image of the cells with the light off showing diffuse cytoplasmic localization of the 2xPDZ-mCherry-LARG. Right: Image of the cells with the light on showing junctional recruitment of the 2xPDZ-mCherry-LARG.
Protocol 2 – Preparing the substrate for imaging
The extracellular matrix (ECM) composition can have drastic effects on cellular behavior and morphology. We also find that the substrate greatly affects the cell junctions’ response to exogenous RhoA. Here, we describe how to plate the cells on polymerized collagen gels that allow tissues to be grown on a soft (<2 kPa) fibrillar network, which we have found optimal to construct polarized epithelial monolayers. Alternative substrates, such as those generated using other ECM proteins like Laminin or Fibronectin, Matrigel, or ECM-coated polyacrylamide gels required for applications like traction force microscopy, can also be used. The substrate composition will depend on the specific experiment. Yet for experiments examining cell-cell interactions using monolayers, we recommend making the substrate as soft as possible. Experiments examining interactions between cell-matrix adhesions may require different substrates. Successful completion of this protocol should result in a 2mg/ml collagen gel that is less than ~300 μm thick atop a glass chamber.
Materials list
Ice
DMEM (Sigma-Aldrich) supplemented with 10% FBS (Hyclone; ThermoFisher scientific), 0.2mM L-glutamine (Invitrogen), and 1% pen/strep (Invitrogen)
1.5ml Eppendorf tubes
Rat tail collagen 1 (Corning)
1M Hepes (Mediatech, Inc.)
7.5% NaHCO3 (Thermofisher scientific)
Four-well chambers (Ibidi)
Chilled pipette tips
Cells (see Protocol 1)
Reagents and solutions:
| Reagent | 1500ul total | 1000ul total | 800ul total | 500ul total | 300ul total |
|---|---|---|---|---|---|
| Col1 | 840.3ul | 560.2ul | 448.2ul | 280.1ul | 168.1ul |
| NaHCO3 | 35.8ul | 23.8ul | 19.1ul | 11.9ul | 7.2ul |
| HEPES | 30ul | 20ul | 16ul | 10ul | 6ul |
| DMEM | 593.9ul | 395.9ul | 316.7ul | 198ul | 118.8ul |
Protocol Steps
Place the collagen and an Eppendorf tube on ice to chill.
Add media to the Eppendorf tube and let chill.
Add HEPES to the Eppendorf tube and let chill.
Add NaHCO3 to the Eppendorf tube and let chill.
- Pipette the collagen slowly into the mixture using chilled pipette tips.
- Let the tip equilibrate to adjust to the temperature. Do this while keeping the Eppendorf tube in the ice.
Pipette up and down while stirring the tip in the Eppendorf tube.
- Let the collagen mixture polymerize off the ice.
- Collagen will start polymerizing the instant it is off the ice. For a more meshy collagen, plate immediately onto the chambers. For more bundled collagen, let sit as a liquid for longer than 5 minutes.
- Paint 80–100ul of the collagen mixture onto the glass chamber with the tip of the pipette.
- Make sure to go into all the corners and avoid bubbles.
- The volume used will alter the collagen gel thickness and, if using a different chamber, this will need to be changed. The collagen gel thickness should be confirmed at the time of cell imaging. This can be done using fluorescently labeled collagen and imaging a z-stack of the collagen proper.
Put the chamber slides in the incubator for at least 5 minutes to allow the gels to solidify.
To avoid the collagen drying before plating the cells, put about 50ul of DMEM on top of the gel.
- In the dark cell culture hood, plate the cells in DMEM
- You can plate the cells sparsely, letting them sit for a few days to grow into a confluent monolayer in the dark incubator
- Some ambient light is okay, but the GEF may be recruited to the membrane upon significant light exposure.
Protocol 3- Visualizing downstream effectors
It is critical to confirm the localization of RhoA and any downstream effectors upon RhoA activation. Downstream effector analysis is accomplished by using reporters of RhoA activity typically through transient transfection. These reporters can include RhoA’s direct downstream effectors, Actin and Myosin. RhoA activity can also be confirmed by using the RhoA biosensor, which houses the RhoA binding domain within the C-terminal portion of Anillin. With Myosin and the RhoA biosensor, these constructs can be transiently transfected into the cells for confirmation of RhoA. For visualization of actin structures, there is a commercially available cell permeable far-red SiR-actin, yet we do not detail the use of this probe in this protocol. It is also possible to use non-fluorescent outputs such as traction force microscopy to confirm that RhoA activation is occurring(7).
When performing multi-channel imaging, it is important to take into account the spectral overlap between optogenetic photoexcitation and fluorescence imaging. The broad blue-light sensitivity of the LOV domains prevents imaging in the GFP channel, since the LOVpep can be activated by blue light. Unfortunately, this limits the number of channels for imaging. If possible, the use of far-red fluorescent proteins and probes such as SiR-actin will allow for multispectral imaging. Since the number of far-red proteins is also limited, we commonly use proteins fused with Halo tags and conjugate them with Janelia fluor far-red proteins for visualization.
In our system, we have found that Caco-2 cells are extremely sensitive to electroporation and often result in low transfection efficiencies (<20%), yet this may be dependent on cell type. For monolayers, we do not recommend using electroporation transfection methods, as it also results in marked cell death (>30–40%) that can hinder the growth of a confluent and polarized monolayer. Instead, we recommend forming the monolayer ahead of transfection using Protocol 2 and then using cationic liposome-based reagents like Lipofectamine 3000. For single cells, this is less of an issue and any transfection method can be utilized though efficiency will still vary. Here we describe a protocol that will use Lipofectamine 3000 to produce an epithelial tissue with transient expression of downstream effectors like the RhoA biosensor or myosin. However, this protocol can generally be used to transfect any protein of interest for analysis of the effects of junctional RhoA localization.
Materials list:
Lipofectamine 3000 Reagent (Thermo Fisher Scientific)
P3000 Reagent (2uL/ug DNA) (Thermo Fisher Scientific)
Opti-MEM (Gibco)
Reporter DNA at concentration of 1–5ug/ul
DPBS (Corning)
DMEM (Sigma-Aldrich) supplemented with 10% FBS (ThermoFisher scientific), 0.2mM L-glutamine (Invitrogen), and 1% pen/strep (Invitrogen)
Vortex
Cells in Ibidi chambers (see Protocols 1 and 2)
Protocol steps:
Plate cells in the 4-well Ibidi chambers in DMEM at 100% confluency, or let cells grow into a confluent monolayer
In the dark, gently wash the optogenetic cells with 1ml prewarmed DPBS
Replace media with 250ul opti-MEM 30min-1h before transfection
Place optogenetic cells in the incubator until the transfection reagents are ready
Warm all reagents to room temperature
Make two 125 aliquots of opti-MEM (A and B)
- To tube A: add 6ul lipofectamine 3000
- Vortex briefly and spin down
- To tube B: add 2.5–5ug of DNA then 5ul of P3000 reagent
- Vortex briefly and spin down
- Add all the contents of Tube B to Tube A
- Mix by pipetting gently
Incubate for 10–15 minutes at room temperature
- In the dark cell culture hood, gently add mixture as small drops to one Ibidi well
- The room light can be on, but the hood light should be turned off to prevent any GEF from being recruited to the membrane under ambient light
In the dark, change the opti-MEM for DMEM 12h after transfection
Analyze transfected cells 24–48 hours after transfection
Protocol 4 – Spatial Illumination calibration
Here, we describe our particular microscope set up and calibration protocol for optogenetic activation using MetaMorph software. While each microscope setup will be different, the most important piece of equipment is the digital micromirror device (DMD) coupled to a light source. The DMD is an intricate array of hundreds of thousands of hinge-mounted, adjustable mirrors that are controlled via microscope acquisition software (e.g. MetaMorph). Mirrors that fall within the regions of interest drawn on the computer are rotated into the light path to reflect light from the source onto the sample. This setup has the advantage of illuminating all pixels in the region of interest simultaneously. To ensure accuracy, the system should be calibrated before each experiment and for each objective used. Calibration is typically performed by clicking on a series of markers displayed in sequence by the DMD. This process registers the DMD to the camera pixels. DMD chips are typically smaller than the FOV of the camera, and thus it is often useful to save a region of the fully illuminated chip as a reference for choosing regions during the actual experiment. Successful completion of this protocol will calibrate the DMD system for use in Protocol 5.
Materials List:
Nikon Ti-E inverted microscope (Nikon)
Yokogawa CSU-X confocal scanning head (Yokogawa Electric)
Laser merge model with 491, 561, and 642nm laser lines (Spectral Applied Research)
Zyla 4.2 sCMOS Camera (Andor)
Mosaic digital micromirror device coupled to a 405nm laser (Andor)
60× 1.49 NA ApoTIRF oil immersion objective (Nikon)
MetaMorph Automation and Image Analysis Software (Molecular Devices)
Mirror slide
Protocol steps:
Turn on the microscope, Mosaic DMD, and light source
Turn on the calibration flashlight perpendicular to the Mosaic light path
Insert the 100% mirror in front of the calibration flashlight, and ensure the rest of the light path is clear
Add immersion media to your objective
Place the mirror slide in the slide holder with the mirrored surface closest to the objective
Using transmitted light, focus on scratches in the mirror surface until they are crisp
Rotate the Mosaic filter cube into position below the objective
Under the “Devices” menu, select “Mosaic Targeted Illumination” to open up the control panel (Figure 3A)
Set the Illumination setting (during pulse) to Mosaic (this makes sure to rotate the mosaic filter cube into place below the objective if it’s not already there)
- Set the coordinate system setting to the desired objective (Figure 3A)
- Make sure the objective magnification (Mag setting) in Metamorph matches the coordinate system setting
Click “Activate Test Mask” (Figure 3A)
- In the Acquire Box, click Live to show the Test Mask (Figure 3B)
- At this point you should see the M on the live screen
- Focus so the M becomes crisp and clear
Stop live imaging and click Update setting
- Click on the center of each white dot as it is displayed
- When all 9 dots have been clicked, the system will be calibrated (Figure 3C)
Confirm that that the system is calibrated by drawing an arbitrary region on the image and testing the illumination
Remove the 100% mirror from in front of the calibration flash light
Turn off the calibration flashlight and turn on the illumination light source to the desired intensity by clicking Low and High within the Laser Controller (Figure 4)
Figure 3:
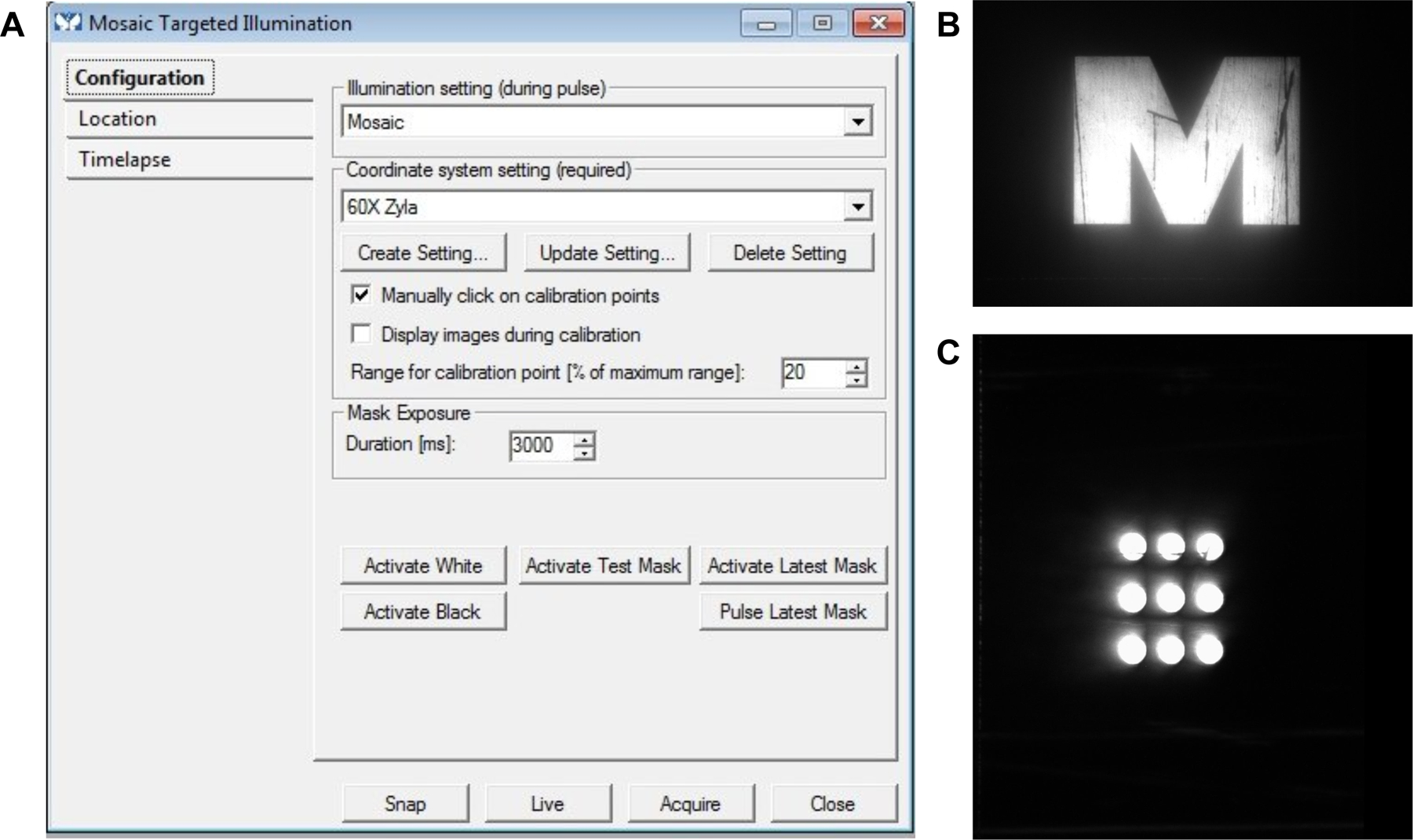
A) Image of the Configuration toolbox within the Mosaic Targeted Illumination device box showing the Illumination setting to the Mosaic. Coordinate setting system is to 60x Zyla. Mask exposure is set to 3 seconds. Activate Test Mask button is shown before the Mask Exposure setting. B) Image showing the activated test mask “M”. C) Image showing the completed, manually clicked calibration points for device calibration.
Figure 4:
Left) Image of the laser controller with the laser in the OFF state. Right) Image of the laser controller in the ON state set to 1000AU.
Critical notes: Any change in the optical setup (e.g. objectives, filters, etc…) will require recalibration of the DMD. It is a good habit to simply perform the calibration before each experiment to ensure proper alignment and thus targeting of the activation.
Protocol 5 – Optogenetic activation of a region of interest
In this protocol, we describe how to use the mosaic micromirror device to induce RhoA activation by light localization within a region of interest (ROI). We generally recommend imaging three periods: a baseline state prior to activation, the response during activation, and the recovery period following activation. For example, we have imaged the junction’s steady state for 10 minutes before a 5-minute activation and then documented junction recovery for 15 minutes post-activation(9, 10) (Figure 5). This scheme allowed us to evaluate any cellular or protein response to exogenous RhoA activation. However, this protocol necessitates optimization. Within the activation period, there may be variations in pulse duration, pulse frequency, and interval spacing of RhoA activation that is dependent on the nature of the experiment. We recommend taking some time to establish optimal activation schemes as necessary for each experiment. Successful completion of this protocol will produce timelapse images of cell junctions undergoing deformations resulting from exogenous RhoA.
Figure 5:
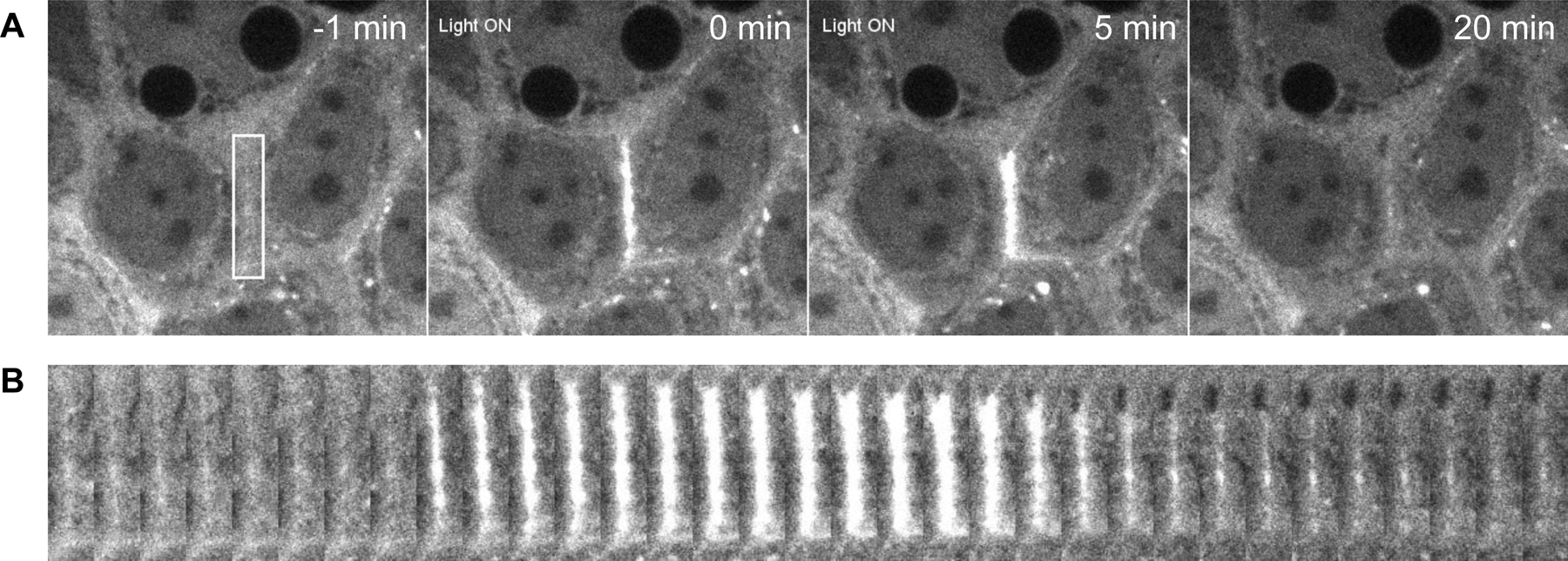
A) Representative images of a stable optogenetic cell line expressing 2xPDZ-mCherry-LARG before, during, and after targeted junctional optogenetic activation. Image shows ROI of activation (White box) inducing junctional localization of the GEF and shortening of the targeted junction over a 5 minute activation, with a 15 minute relaxation period showing reversal of any junction contraction. B) Representative kymograph showing expression of the 2xPDZ-mCherry-LARG before, during, and after targeted junctional activation.
Additional Materials List (See Basic Protocol 4):
Stage incubator (Chamlide TC and FC-5N; Quorum Technologies).
Protocol steps:
Make sure the light path from the mosaic light source to the sample is free of any obstruction or mirrors.
- Turn on the mosaic laser using the Laser Diode Control (Figure 4)
- The laser power here needs to be determined empirically so that the optogenetic cells can be activated. This will be determined in part by the optics of the individual microscope. Due to its high sensitivity, only a very small amount of light is actually needed to activate the protein. We, and the lab of Tobin Sosnick, also found that too much light hinders the activation of the LOV protein. We have had success generating contractile responses at junctions using light intensities between 6.7uW and 10.5uW (750–1000AU), with a minimal junctional response at 4.3uW (500AU) (see junctional responses as a function of the light intensity in (9, 10)).
- Place sample in the stage incubator, keeping sample in the dark and allow to equilibrate
- The stage incubator will maintain the cells at 37°C, whereas humidified 5% CO2 will be maintained at 50°C at its source to prevent condensation within its tubing.
- Scan the cells for optimal expression of 2xPDZ-mCherry-LARG
- To confirm expression of both of the constructs, first take an image in the mCherry channel, followed by an image in the GFP channel, and then again in the mCherry channel. If the cytoplasmic RhoGEF shifts to the junctions, this cell is primed for optogenetic activation (similar to Figure 2).
- Wait > 5 min until all the 2xPDZ-mCherry-LARG has dissociated from the junctions and has returned to the cytoplasm. You can confirm this by taking another image in the mCherry channel to visualize cytoplasmic RhoGEF.
- Draw an ROI to be targeted on the previously acquired image
- The ROI should be similar to the region drawn in Figure 5
- In the Mosaic Targeted Illumination box, click Location (Figure 6A)
- Make sure the coordinate system setting matches with the Mag setting
- Select the region in the image and click the Active Region button in the Target Location box
- This will specify the drawn ROI as the mask
- It is also possible to click All regions if multiple ROI’s are drawn.
In the Mosaic Targeted Illumination box, click Configuration (Figure 3A)
- Specify Mask Exposure Duration in milliseconds
- We use 1000ms but this can be subject to change depending on the nature of the experiment
In the mosaic targeted illumination box, click Timelapse (Figure 6B)
- Specify the acquisition cycles
- This step should be determined by the nature of the experiment, since different activation schemes require pulses at different timepoints. Typically during a period of activation we illuminate the ROI of interest with blue light prior to each timepoint. In Figure 6B, we have 100 timepoints at intervals of 30 seconds, with blue light pulsing before time point 10–13.
- Here, you can also use a journal for acquisition like using different color channels for acquisition. In our example in Figure 6B we have a journal specifying images in both 561 and 647 to image the recruitable GEF and any far-red effector protein or far-red membrane stain. See Table 2 for an example protocol.
- Click Acquire in the mosaic targeted illumination box
- Cell-cell junctions should undergo contraction, similar to Figure 5.
Save Images under desired name
Save ROI for use in image analysis
Figure 6:
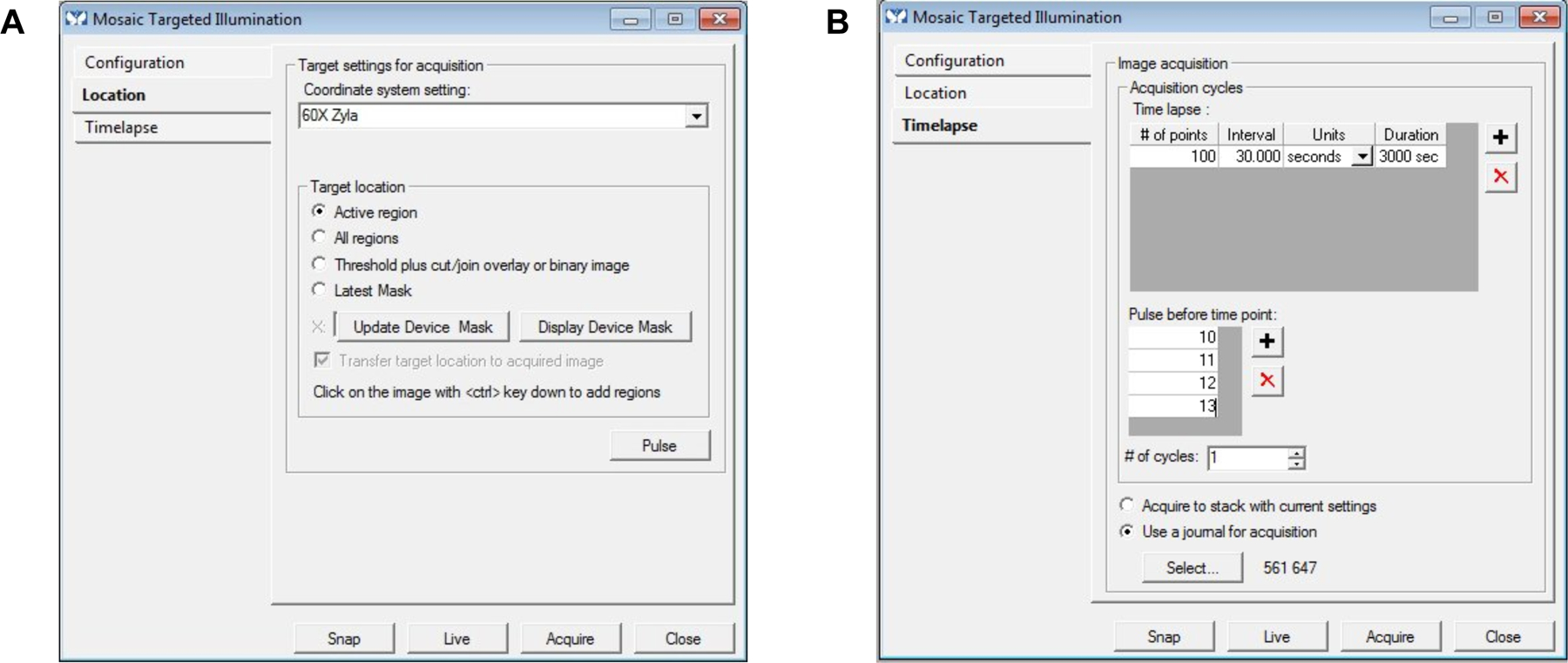
A) Image of the Location toolbox within the Mosaic Targeted Illumination device setting. Coordinate system setting is to 60x Zyla, with target location set to the Active Region. B) Time-lapse toolbox within the Mosaic Targeted Illumination device setting showing the number of timepoints set to 100 at 30 second intervals with pulses before timepoints 10–13. A journal is used to image the 561 and 647 channels during each of these timepoints.
Table 2:
Example protocols for optogenetic experiments
| Goal: | To confirm presence and functionality of optogenetic probes: | Monolayer (imaging mCherry Larg and Cell Mask Deep Red) : | Single Cell (imaging mApple-Myosin and Alexa 647 Beads for Traction Force Microscopy): |
|---|---|---|---|
| Journal: | Acquire mCherry (GEF) | Timelapse Loop (30 second intervals for 10 min)
|
Timelapse Loop (20 second intervals for 15 min)
|
Critical notes: While we detail here the use of the mosaic micromirror with the Targeted Illuminated system using MetaMorph, other acquisition software and hardware setups exist. Acquisition can be greatly streamlined through use of journals which are beyond the scope of this protocol.
Commentary
Background information
LOV domains of the phototropin blue light receptors contain a flavin-based blue-light sensing chromophore and regulate light-mediated biological processes in microbes and plants(32). Specifically, the AsLOV2 protein is made up of a core per-arnt-sim (PAS) fold with flanking alpha helicies on both the N- and C-termini (33). Upon blue light absorption, a conserved cysteine residue in the AsLOV2 core covalently binds to the flavin cofactor. This binding causes conformational changes that propagate along the PAS fold, leading to the uncaging of a ~20 amino acid amphipathic C-terminal alpha helix (34). This C-terminal α-helix is known as the Jα helix. The beauty of this system lies in its reversibility; in the dark-state the Jα is left intact but light stimulation can expose linear motifs in its amino acid sequence. Therefore, reversible caging of the Jα can lead to masking or unmasking of signaling protein activity with light.
The nature of Jα is such that it can block certain peptide epitopes in the AsLOV2 core. Upon photoexcitation, the Jα can undock and expose this specific epitope. This feature of the AsLOV2 protein is useful because it can be used to design a binding partner to create a dimerizable protein complex. For the AsLOV2’s binding partner, the original Strickland paper used the high-affinity, high-specificity engineered variant of the Erbin PDZ domain, ePDZ-b1(35). This domain is small at about 194 amino acids. The ePDZ domain’s affinity and lifetime in the photoexcited state are tunable by various mutations, effectively tuning downstream signaling events(1). Recently, this system has been improved upon by substituting the ePDZ-b1 domain with a tandem PDZ tag that is functional in more diverse protein fusions(4).
Troubleshooting:
The RhoGEF is not recruiting:
The RhoGEF should be visibly recruited to the membrane upon photoactivation (Similar to Figure 2). If this is not the case, there may be a problem with the expression levels of the optogenetic probes. It is possible that the ratio of Stargazin-GFP-LOVpep and 2xPDZ-mCherry-LARG is not optimal. We then recommend choosing cells that have higher expression of either protein.
The cells are constitutively activating:
It is a possibility that the light from bright computer monitors or ambient room light can activate the cells. This is because of the presence of blue light. This can be easily solved by keeping a far distance between the monitors and the sample or turning the computer away from the microscope. Keep the cells in the dark whenever possible.
Activation is occurring outside the targeted ROI:
Make sure that the “active region” button is toggled in the Targeted Illumination control panel, and that the correct region is chosen.
The cells are blebbing or dying:
We find it a common occurrence that the cells may bleb in other regions of the cell upon photorecruitment of the GEF. If this is the case, or if the cells are dying, it is wise to ease off the 405nm laser intensity. Too much laser power may result in an excess amount of RhoA activation that weakens the opposite membranes to cause cell blebbing or bursting.
The RhoGEF is aggregating:
We see that the RhoGEF often aggregates, showing up as clumps in fluorescence. This is natural and a feature of the 2xPDZ-mCherry-LARG.
The junctions are not contracting:
While we expect robust junction contraction upon RhoA activation, it is possible that the junctions may simply not contract. This can arise for a few reasons. First, it is possible that the light intensity levels are not high enough to produce a marked cellular response. Light intensities would then need to be increased. Second, it is possible that the RhoGEF expression levels are not optimal for RhoA activation. Here, we suggest choosing cells with higher RhoGEF expression. Third, we have also found clones of cells may express a variant of the recruitable RhoGEF that renders it unrecruitable, although this is rare. It is possible this is due to spontaneously occurring mutations in the DNA encoding the photosensitive dimerization domains, which are present in the DNA stock used to generate the stable cell line. We recommend sequencing the DNA used to make sure there are no mutations, otherwise use another isolated cell clone.
Perfect focus is not working:
Painting 80–100ul of collagen should be sufficient to produce an even layer of polymerized gel. This is advantageous because this gel thickness is conducive to using Perfect Focus on many microscopes. If Perfect Focus is not working, it is possible that the gel is too thick. If so, consider using less collagen for a thinner gel. It is also possible to coat the chamber with 80–100ul and then aspirate the excess collagen to produce a very thin layer of a collagen gel.
The stable cell line is losing expression:
A common problem that we have is dilution of the optogenetic constructs over time. This can be for a few reasons, although we believe that the cells expressing the optogenetic proteins are more prone to cellular extrusion because they are hypercontractile. To alleviate this, we recommend using cells at a low passage number. If the cells are still losing expression, we recommend fluorescently sorting the cells again to achieve better protein expression.
Anticipated results
Cells with good expression of both optogenetic constructs can be photoactivated such that there is a marked increase in junctional RhoGEF compared to cytoplasmic RhoGEF, the latter which should reduce in fluorescence (Figure 2). The photoactivated junction will be visually distinguishable when compared to other cells that have not been activated with 405nm light. The unactivated cells should have diffuse cytoplasmic localization of the RhoGEF. Upon RhoGEF recruitment, the activated junction will undergo contraction and noticeably shorten (Figure 5), although the extent of this shortening may be dependent on the cell type used and light intensity. With this approach, junction lengths can be analyzed under WT and various inhibitor conditions. Additionally, the use of far-red protein labeling can allow for the visualization of the membrane, junctional components, or effector proteins as a result of RhoA localization and subsequent junction contraction.
Time considerations
The largest time investment comes from obtaining the stable cell line. This may take a few weeks from infection to isolation and expansion of a clonal cell line. Careful attention needs to be paid when sorting and screening the cells for optimal fluorescence intensities, and multiple clones may need to be isolated to obtain the recruitable cell line with optimal expression levels of the optogenetic constructs. Time is also spent on optimizing the microscopy, including determining the light intensity and pulse duration. This may take a few optogenetic experiments to determine optimal microscopy parameters. Imaging can take anywhere from minutes to hours, depending on the desired activation scheme driving the wanted cellular behavior. A typical experiment, from plating the cells to the onset of imaging, should only take a few days. This is limited by how fast the cultured cells can grow into a confluent monolayer if testing cell mechanics at the tissue scale. At the cellular scale, cells can be plated the night before to ensure proper attachment to the desired substrate.
Acknowledgements
KEC acknowledges an HHMI Gilliam Fellowship, National Academies of Sciences Ford Foundation Fellowship, and NIH training grant GM007183. PWO acknowledges funding from NSF CAREER Award #1749302. MLG acknowledges funding from NIH RO1 GM104032.
REFERENCES
- 1.Strickland D, et al. , TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nature Methods 9, 379–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lecuit T, Lenne P-F, Munro E, Force Generation, Transmission, and Integration during Cell and Tissue Morphogenesis. Annual Review of Cell and Developmental Biology 27, 157–184 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Lessey EC, Guilluy C, Burridge K, From Mechanical Force to RhoA Activation. Biochemistry 51, 7420–7432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner E, Glotzer M, Local RhoA activation induces cytokinetic furrows independent of spindle position and cell cycle stage. The Journal of Cell Biology 213, 641–649 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Neill PR, et al. , Membrane Flow Drives an Adhesion-Independent Amoeboid Cell Migration Mode. Developmental Cell 46, 9–22.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meshik X, O’Neill PR, Gautam N, Physical Plasma Membrane Perturbation Using Subcellular Optogenetics Drives Integrin-Activated Cell Migration. ACS Synthetic Biology 8, 498–510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oakes PW, et al. , Optogenetic control of RhoA reveals zyxin-mediated elasticity of stress fibres. Nature Communications 8, 15817 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valon L, Marín-Llauradó A, Wyatt T, Charras G, Trepat X, Optogenetic control of cellular forces and mechanotransduction. Nature Communications 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavanaugh KE, Staddon MF, Munro E, Banerjee S, Gardel ML, RhoA mediates epithelial cell shape changes via mechanosensitive endocytosis. bioRxiv (2019) 10.1101/605485 (September 18, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Staddon MF, Cavanaugh KE, Munro EM, Gardel ML, Banerjee S, Mechanosensitive junction remodelling promotes robust epithelial morphogenesis. bioRxiv (2019) 10.1101/648980 (September 9, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izquierdo E, Quinkler T, Renzis SD, Guided morphogenesis through optogenetic activation of Rho signalling during early Drosophila embryogenesis. Nat Commun 9, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krueger D, Quinkler T, Mortensen SA, Sachse C, Renzis SD, Cross-linker–mediated regulation of actin network organization controls tissue morphogenesis. J Cell Biol 218, 2743–2761 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levskaya A, Weiner OD, Lim WA, Voigt CA, Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy MJ, et al. , Rapid blue-light–mediated induction of protein interactions in living cells. Nat Methods 7, 973–975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guntas G, et al. , Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proceedings of the National Academy of Sciences 112, 112–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmerman SP, et al. , Tuning the Binding Affinities and Reversion Kinetics of a Light Inducible Dimer Allows Control of Transmembrane Protein Localization. Biochemistry 55, 5264–5271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawano F, Suzuki H, Furuya A, Sato M, Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nature Communications 6 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE, Induction of protein-protein interactions in live cells using light. Nature Biotechnology 27, 941–945 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Hahn KM, LOVTRAP: A Versatile Method to Control Protein Function with Light. Current Protocols in Cell Biology 73, 21.10.1–21.10.14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dine E, Gil AA, Uribe G, Brangwynne CP, Toettcher JE, Protein Phase Separation Provides Long-Term Memory of Transient Spatial Stimuli. cels 6, 655–663.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaberniuk AA, Shemetov AA, Verkhusha VV, A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nature Methods 13, 591–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crefcoeur RP, Yin R, Ulm R, Halazonetis TD, Ultraviolet-B-mediated induction of protein–protein interactions in mammalian cells. Nature Communications 4 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Schmidt A, Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes & Development 16, 1587–1609 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Bos JL, Rehmann H, Wittinghofer A, GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 129, 865–877 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Cook DR, Rossman KL, Der CJ, Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33, 4021–4035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cherfils J, Zeghouf M, Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiological Reviews 93, 269–309 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Jaiswal M, et al. , Mechanistic Insights into Specificity, Activity, and Regulatory Elements of the Regulator of G-protein Signaling (RGS)-containing Rho-specific Guanine Nucleotide Exchange Factors (GEFs) p115, PDZ-RhoGEF (PRG), and Leukemia-associated RhoGEF (LARG). Journal of Biological Chemistry 286, 18202–18212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, et al. , Activated RhoA Binds to the Pleckstrin Homology (PH) Domain of PDZ-RhoGEF, a Potential Site for Autoregulation. J. Biol. Chem 285, 21070–21081 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Guo L, Sprang SR, Sternweis PC, Modulation of a GEF switch: Autoinhibition of the intrinsic guanine nucleotide exchange activity of p115-RhoGEF. Protein Science 20, 107–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitlow M, et al. , An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng Des Sel 6, 989–995 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Zaro J, Shen W-C, Fusion Protein Linkers: Property, Design and Functionality. Adv Drug Deliv Rev 65, 1357–1369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Möglich A, Moffat K, Engineered photoreceptors as novel optogenetic tools. Photochem. Photobiol. Sci 9, 1286–1300 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Halavaty AS, Moffat K, N- and C-Terminal Flanking Regions Modulate Light-Induced Signal Transduction in the LOV2 Domain of the Blue Light Sensor Phototropin 1 from Avena sativa,. Biochemistry 46, 14001–14009 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Harper SM, Neil LC, Gardner KH, Structural Basis of a Phototropin Light Switch. Science 301, 1541–1544 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Huang J, Koide A, Makabe K, Koide S, Design of protein function leaps by directed domain interface evolution. PNAS 105, 6578–6583 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]