

Abstract
Background:
Clonal hematopoiesis of indeterminate potential (CHIP) refers to clonal expansion of hematopoietic stem cells due to acquired leukemic mutations in genes such as DNMT3A or TET2. In humans, CHIP associates with prevalent myocardial infarction. In mice, CHIP accelerates atherosclerosis and increases IL-6/IL-1β expression, raising the hypothesis that IL-6 pathway antagonism in CHIP carriers would decrease cardiovascular disease (CVD) risk.
Methods:
We analyzed exome sequences from 35,416 individuals in the UK Biobank without prevalent CVD, to identify participants with DNMT3A or TET2 CHIP. We used the IL6R p.Asp358Ala coding mutation as a genetic proxy for IL-6 inhibition. We tested the association of CHIP status with incident CVD events (myocardial infarction, coronary revascularization, stroke, or death), and whether it was modified by IL6R p.Asp358Ala.
Results:
We identified 1,079 (3.0%) individuals with CHIP, including 432 (1.2%) with large clones (allele fraction >10%). During 6.9-year median follow-up, CHIP associated with increased incident CVD event risk (HR=1.27, 95% CI: 1.04–1.56, p=0.019), with greater risk from large CHIP clones (HR=1.59, 95% CI: 1.21–2.09, p<0.001). IL6R p.Asp358Ala attenuated CVD event risk among participants with large CHIP clones (HR=0.46, 95% CI: 0.29–0.73, p<0.001) but not in individuals without CHIP (HR=0.95, 95% CI: 0.89–1.06, p=0.08) (pinteraction=0.003). In 9,951 independent participants, the association of CHIP status with myocardial infarction similarly varied by IL6R p.Asp358Ala (pinteraction=0.036).
Conclusions:
CHIP is associated with increased risk of incident CVD. Among carriers of large CHIP clones, genetically reduced IL-6 signaling abrogated this risk.
Keywords: Clonal hematopoiesis, Cardiovascular disease, Interleukin-6, DNMT3A, TET2
Introduction
‘Clonal hematopoiesis of indeterminate potential’ (CHIP) is the presence of clonally expanded acquired leukemogenic mutations primarily in epigenetic regulatory genes, DNMT3A and TET2.1 Large-scale whole exome sequence analysis of DNA derived from blood cells showed that the prevalence of CHIP increases with age and occurs in over 1 in 10 adults > 70 years of age.2–4 While individuals harboring CHIP have a markedly greater relative risk for future hematologic malignancy, the absolute risk remains modest (0.5% to 1% per year).4,5 Heightened risk of mortality among those with CHIP appears to relate to atherosclerotic cardiovascular disease (CVD).6 Indeed, CHIP associates with prevalent coronary artery disease and early-onset myocardial infarction in four case-control studies, with risk principally conferred by large CHIP clones (variant allele fraction >10%).6
Hyperlipidemic mice with experimental CHIP have accelerated atherogenesis and have evidence for heightened IL-1β/IL-6 signaling.6,7 Furthermore, inhibition of the NLRP3 inflammasome that activates IL-1β, a key inducer of IL-6, ameliorates atherosclerosis in such mice.7,8 These observations suggest that inhibiting the IL-6/IL-1β pathway may prove particularly effective in reducing CVD risk in humans with CHIP.
One way to test this hypothesis is through human genetics. A commonly occurring (allele frequency 30–40%) variant in IL-6 receptor gene, IL6R p. Asp358Ala, disrupts IL-6 signaling analogous to tocilizumab 4–8 mg/kg infusions every 4 weeks. Functional studies of the IL6R p.Asp358Ala indicate this mutation reduced expression of membrane-bound IL-6 receptor, impairing responses to classical IL-6 signaling on hepatocytes and leukocytes.9 As such, IL6R p.Asp358Ala provides an attractive genetic proxy to test this therapeutic hypothesis.10 In prior studies IL6R p.Asp358Ala associated with reduced CVD risk in a general population.10,11
Here, we leverage exome sequences in the UK Biobank to first confirm the association of CHIP with incident CVD. We then test whether individuals with IL6R p.Asp358Ala and CHIP would derive greater relative CVD event reduction than those without CHIP.
Methods
Individual level genetic sequence data analyzed in this article are available to qualified researchers for replication of these analyses through the UK Biobank data showcase and the database of Genotypes and Phenotypes (dbGaP) (PROMIS Study Accession: phs000917.v1.p1).
Study Population
This analysis included individual-level data from 35,416 unrelated European ancestry participants enrolled in the UK Biobank study who had undergone exome sequencing12,13 (Supplemental Figure 1). The overall UK Biobank prospective cohort consists of approximately 500,000 adult participants recruited between 2006 and 2010 from 22 assessment centers across the United Kingdom. Individuals selected for the first tranche of exome sequence analysis were enriched for those with more complete phenotypic information, specifically cardiac magnetic resonance imaging (MRI).12 To exclude pairings within third-degree of relatedness, the KING [Kinship-based Inference for Genome-wide association studies] tool was used to derive kinship coefficients from genome-wide array-derived genotypes.14 European ancestry was defined using principal components of ancestry as before.15 The UK Biobank has institutional review board (IRB) approval from the Northwest Multi-Center Research Ethics Committee and all participants provided written informed consent. Replication analyses utilized individual-level data from 9,951 individuals in the Pakistan Risk of Myocardial Infarction Study (PROMIS), a case-control myocardial infarction cohort designed to understand the determinants of cardiometabolic diseases in individuals from South Asia.16,17 Secondary use of data for the present analysis was approved by the Massachusetts General Hospital IRB 2013P001840. The data that support the findings of this study are available from the UK Biobank (https://www.ukbiobank.ac.uk/) and author upon reasonable request.
Whole Exome Sequencing & CHIP Detection
Exomes were sequenced from blood cell-derived DNA as previously described in the UK Biobank12 and PROMIS17. CHIP detection used previously described methods.2,6 (see Supplemental Methods) Briefly, aligned short-read exome sequences data were re-analyzed using the GATK MuTect2 software18 to detect putative somatic genetic variants. These putative variants were then filtered to exclude common sequencing artifacts and germline polymorphisms frequently observed in the population. Samples were annotated as having CHIP if the MuTect2 output contained one or more of a pre-specified list of putative CHIP variants implicated in the majority of CHIP carriers in DNMT3A or TET2 known to cause myeloid malignancy. (Supplemental Table 1)
Study Outcome
The primary outcome of our analysis, termed CVD events, was a composite of myocardial infarction, coronary artery revascularization, stroke, or death, defined by a combination of inpatient hospital billing ICD codes and UK death registries. Secondary exploratory outcomes included components of the composite outcome, CVD events without death, myeloproliferative neoplasm and myeloid leukemia. (Supplemental Table 2) Myocardial infarction case-control status was the outcome used for the PROMIS replication analysis.16
IL-6 Inhibition Genetic Proxy
We used the common (allele frequency 30–40% in most populations measured reference datasets, but as low as 15% in Africans) IL-6 receptor disruptive coding mutation, IL6R p.Asp358Ala (rs2228145), as a genetic proxy for IL-6 pathway inhibition as before.10 Prior analyses of biomarker changes associated with IL6R p.Asp358Ala indicate a similar magnitude of CRP lowering observed with monthly tocilizumab 4–8 mg/kg infusions in randomized controlled trials.10
Statistical Analysis
We performed a prospective, time-to-event analysis of our CVD outcome stratified by CHIP status in UK Biobank. Cox proportional hazard models were adjusted for age when follow-up began, genetic ancestry (Principal Components 1–5), and clinical covariates at the time of exome sequencing (age, sex, HDL cholesterol, LDL cholesterol (LDL-c), pack years of smoking, current smoking status at the time of enrollment, the interaction of pack years of smoking and smoking status at the time of enrollment, BMI, and diagnoses of type 2 diabetes [T2D] and hypertension). Given the estimated average effect of statins, LDL-c was divided by a correction factor of 0.68 when statins were prescribed, as done previously.19,20 For the subset of individuals selected for exome sequencing due to availability of cardiac MRI, we considered follow-up duration to be from the time of imaging—which was typically several years after exome sequencing—until censorship, disease onset, or death to eliminate immortal time bias.21 We further stratified CHIP status based on the size of the CHIP clone (defining large clones as >10% variant allele fraction). We evaluated the interaction between CHIP and IL6R p.Asp358Ala on the primary outcome. Because of departures from the Cox proportional hazards assumptions beyond 7.5 years of follow-up (beyond which fewer than 5% of participants had data), incident disease was censored at 7.5 years for all analyses. Data is displayed as a cumulative event rate curves based on Kaplan-Meier estimates.
To evaluate whether variants at other IL-6-signaling pathway genes showed similar interactions, we first identified variants previously significantly associated with CRP.22 Variants near known IL-6-signaling pathway genes significantly associated with CRP included IL6R rs4129267 (p.Asp358Ala, used as the primary instrument in the present study), rs1880241 upstream of IL-6, rs6734238 upstream of IL1RN-IL1F10, and rs9284725 within the first intron of IL1R1. We performed similar interaction analyses as described above.
To replicate the CHIP and IL6R p.Asp358Ala interaction, we used cross-sectional data on myocardial infarction outcomes from the PROMIS study. Multivariable logistic regression models of the effect of CHIP, IL6R p.Asp358Ala and the interaction between CHIP and IL6R p.Asp358Ala were adjusted for clinical covariates at the time of exome sequencing (age, sex, LDL-c, T2D, body mass index, hypertension, current and former smoking status, systolic blood pressure) and genetic ancestry (Principal Components 1–10 and estimated proportion of South Asian ancestry). Missing phenotypic covariate data was imputed using the aregImpute algorithm in the Hmisc package23 (see Supplemental Methods).
Statistical significance was assigned at alpha = 0.05. All tests of significance are two-sided. Baseline groups utilized t-test. Analyses were performed in R (R Foundation, Vienna, Austria, version 3.6).
Results
In the UK Biobank cohort, we identified 1,079 (3.0%) individuals with CHIP mutations of whom, 432 (1.2%) had large CHIP clones - detected as variant allele fraction (VAF) > 10%, corresponding to more than 20% of nucleated blood cells harboring the a CHIP mutation. Consistent with prior reports, CHIP presence associated strongly with age (p<0.001, Supplemental Figure 2). Accordingly, individuals with CHIP were on average 3.3 years older than those without CHIP and had a higher rate of cardiovascular comorbidities including hypertension, hyperlipidemia, and T2D. Individuals with CHIP had a modestly elevated hsCRP in unadjusted analyses (Table 1, Supplemental Table 3); however, in a covariate-adjusted model, hsCRP did not associate significantly with CHIP (p=0.08). CHIP was not significantly associated with complete blood cell count with differential indices (Supplemental Table 4). As expected, CHIP associated with incident myeloproliferative neoplasms (HR 3.55, 95% CI 1.38–9.12, p=0.009) and myeloid leukemias (HR 6.25, 95% CI 2.08–18.82, p=0.001).
Table 1:
Characteristics of participants at baseline, stratified by CHIP status
| CHIP noncarriers (N=34,337) | CHIP carriers (N=1,079) | P value | |
|---|---|---|---|
| Women, N (%) | 18823 (54.8) | 599 (55.5) | 0.66 |
| Age at exome sequencing | 57.1±7.87 | 60.7±6.75 | 1.3 x 10−57 |
| Age at the start of follow-up | 58.8±8.08 | 62.1±6.91 | 1.8 x 10−47 |
| BMI (kg/m2) | 27.3±4.75 | 27.4±4.67 | 0.52 |
| Height (cm) | 169±9.21 | 169±9.01 | 0.22 |
| Weight (kg) | 78.3±15.9 | 78.2±15.5 | 0.87 |
| Systolic blood pressure (mmHg) | 137±18.2 | 141±18.6 | 3.5 x 10−9 |
| Diastolic blood pressure (mmHg) | 81.9±9.95 | 82.6±9.47 | 0.01 |
| Type 2 diabetes mellitus, N (%) | 727 (2.12) | 35 (3.24) | 0.02 |
| Hypertension, N (%) | 9743 (28.4) | 354 (32.8) | 1.8 x 10−3 |
| Hypercholesterolemia, N (%) | 5059 (14.7) | 183 (17) | 0.05 |
| Standard drinks/week | 12.1±10.5 | 12.3±10.5 | 0.60 |
| Pack years of tobacco | 6.07±13.1 | 7.24±13.9 | 0.01 |
| Currently using tobacco at enrollment, N (%) | 2976 (8.67) | 113 (10.5) | 0.04 |
| Total cholesterol | 5.75±1.11 | 5.8±1.15 | 0.18 |
| LDL cholesterol, direct | 3.58±0.82 | 3.61±0.86 | 0.21 |
| HDL cholesterol | 1.49±0.36 | 1.49±0.35 | 0.41 |
| Triglycerides | 1.7±0.95 | 1.7±0.9 | 0.89 |
| Apolipoprotein A1 | 1.56±0.27 | 1.58±0.26 | 0.21 |
| Apolipoprotein B | 1.04±0.23 | 1.05±0.24 | 0.22 |
| Lipoprotein (a) | 43.6±49.1 | 45.2±50.3 | 0.37 |
| High-sensitivity C-reactive protein | 2.49±4.26 | 2.85±5.18 | 0.03 |
| Prescribed a statin at enrollment, N (%) | 4846 (14.1) | 193 (17.9) | 6.4 x10−4 |
| Prescribed a blood-pressure lowering medication at enrollment, N (%) | 6184 (18) | 236 (21.9) | 1.5 x 10−3 |
| IL6R p.Asp358Ala minor allele frequency | 0.41 | 0.40 | 0.20 |
CHIP carriers were defined as individuals with CHIP present at any variant allele fraction. Continuous values are presented as mean±standard deviation, and categorical values are presented as count (percentage). BMI, body-mass index; CHIP, clonal hematopoiesis of indeterminate potential; HDL, high-density lipoprotein; LDL, low-density lipoprotein. P-values calculated with two-sample t-test for continuous traits or fisher-exact test for categorical values.
CHIP associated with increased CVD event risk (HR=1.27, 95% CI: 1.04–1.56, p=0.019, Figure 1A). Since we previously observed that those with large CHIP clones (VAF>10%) have greater incident cancer risk and a greater burden of subclinical atherosclerosis, we tested the hypothesis that carriers of large CHIP clones also have heightened incident CVD event risk. Large CHIP clones conferred most of this risk (HR=1.59, 95% CI 1.21–2.09, p=9.1 x 10−4, Figure 1B, Supplemental Figure 3), which differed significantly from the risk associated with smaller CHIP clones (HR=1.04, 95% CI: 0.78–1.39, p=0.78); heterogeneity test p=0.037. Effects were similar for large CHIP when further adjusting for hsCRP as well (HR=1.59, 95% CI: 1.20–2.10, p=1.6 x 10−3). Over 5-year follow-up, the absolute risk of the primary outcome was 4.7% (95% CI 4.4–4.9%) in the group without CHIP, 6.6% (95% 4.9–8.2%) with any CHIP, and 8.9% (95% CI 6.0–11.8%) with large CHIP. All of the sub-components of the primary outcome showed strong consistency of this association and risk difference with hazard ratios ranging from 1.62–1.82 (Supplemental Figure 4). Excluding all-cause death from the composite outcome, large CHIP clones still associated with 1.50-fold risk for incident CVD events (95% CI 1.06–2.13, p=0.02) Stratifying by DNMT3A and TET2 CHIP driver mutations revealed similar effect sizes (Supplemental Figure 5).
Figure 1. CVD Event Incidence stratified by CHIP carrier status and clone size.
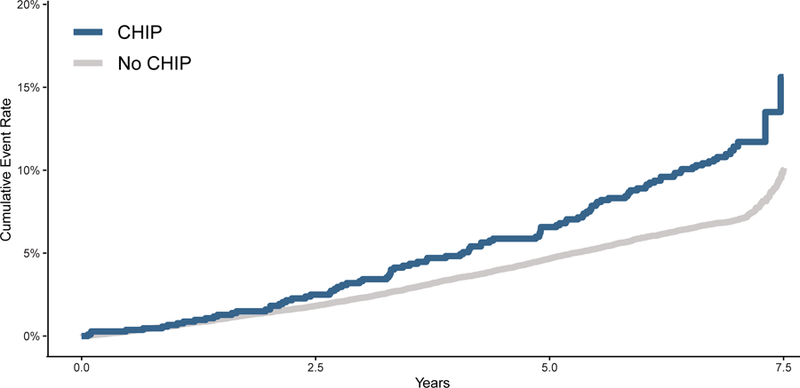
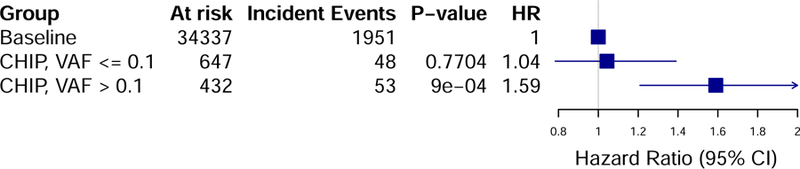
Panel A shows the comparison of time to the primary CVD event outcome of myocardial infarction, coronary artery disease or revascularization, stroke, or death between all CHIP carriers and non-carriers of CHIP. Panel B shows that carriers of large CHIP clones (VAF > 10%) had an increased risk of the primary outcome, while those with small CHIP clones (VAF < 10%) did not have significantly increased risk. VAF = variant allele fraction.
Consistent with prior reports10,11, among all individuals, each additional IL6R p.Asp358Ala allele was associated with reduced risk of CVD events (HR=0.94, 95% CI: 0.88–1.00, p=0.05). In participants with large CHIP clones, each additional IL6R p.Asp358Ala allele attenuated the risk of CVD events (HR=0.46, 95% CI: 0.29–0.73, p=9.6 x 10−4); in contrast, the presence of IL6R p.Asp358Ala alleles had no significant effect on CVD event risk in individuals without CHIP (HR=0.95, 95% CI: 0.89–1.01, p=0.08), revealing a statistical interaction between CHIP carrier status and IL6R p.Asp358Ala alleles (pinteraction=0.003; Figure 2). When considering all CHIP (i.e., large and small) carriers, there was no significant interaction with IL6R p.Asp358Ala for incident CVD event risk (pinteraction=0.42). The MI, revascularization and death subcomponents of the primary outcome showed strongly consistent results, while stroke did not (Supplemental Table 5). TET2 and DNMT3A status showed strongly consistent results as well (Supplemental Table 6). When including CHIP carriers (n=30) of the next two commonly implicated genes (i.e., JAK2 and ASXL1), results were unchanged (Supplemental Table 7).
Figure 2: CVD Event Incidence stratified by CHIP carrier status and genetically determined impairment in IL-6 signaling.
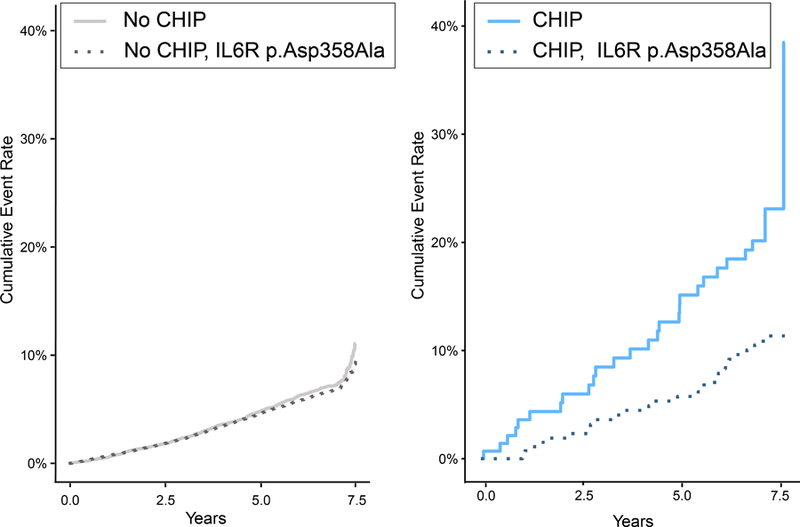
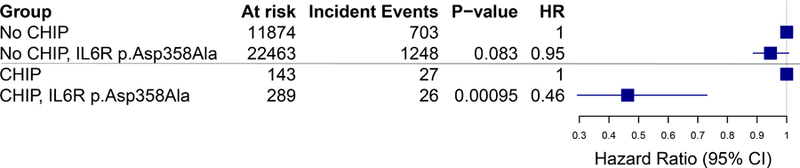
Panel A. Comparison of time to the primary outcome of myocardial infarction, coronary artery disease or revascularization, stroke, or death stratified by IL6R p.Asp358Ala carrier status in (left panel) individuals without CHIP or (right panel) carriers of large CHIP clones (VAF > 10%). Panel B. Among those with large CHIP clones, but not those without CHIP, the presence of IL6R p.Asp358Ala variants conferred a reduced risk of incidence of the primary outcome (pinteraction=0.003).
We evaluated three non-coding variants near IL-6 signaling pathway genes that were previously significantly associated with CRP. None of these variants were significantly associated with incident CVD events among all individuals. However, one variant (rs1880241) which is upstream of IL6, showed reduced CVD events only among large CHIP carriers and not all others (pinteraction = 0.025) (Supplemental Table 8).
The UK Biobank cohort consists primarily of individuals of British descent. We sought to replicate the interaction between CHIP and IL6R p.Asp358Ala and its association with myocardial infarction in the sequenced subset of the PROMIS cohort over 50 years old (n=9,951; 2,275 MI cases), a cohort of adults living in Pakistan (Supplemental Table 9). 52 individuals had large (VAF>10%) DNMT3A or TET2 CHIP clones. In participants with these large CHIP clones, each additional IL6R p.Asp358Ala allele attenuated the risk of CVD events (OR=0.27, 95% CI: 0.07–0.92, p=0.047); in contrast, the presence of IL6R p.Asp358Ala alleles had no significant effect on MI risk in individuals without CHIP (OR=0.96, 95% CI: 0.89–1.03, p=0.32). The apparent difference in odds ratios was statistically different (pinteraction=0.036).
An exploratory analysis showed no association of IL6R p.Asp358Ala with incident hematologic malignancy risk (p=0.94 for acute myeloid leukemia; p=0.86 for myeloproliferative neoplasm). Similarly, specifically among individuals with large CHIP clones, IL6R p.Asp358Ala alleles did not associate with incident hematologic malignancy risk (p=0.19 for acute myeloid leukemia; p=0.82 for myeloproliferative neoplasm). Nor did we observe a significantly altered risk of sepsis among those with large CHIP clones carrying IL6R p.Asp358Ala versus those with large CHIP clones without IL6R p.Asp358Ala (p=0.11). Lastly, IL6R p.Asp358Ala was not associated with the presence of CHIP (p=0.16), with large CHIP (p=0.98), or with clonal expansion as measured by variant allele fraction among individuals with CHIP (p=0.41).
Discussion
Central goals of precision medicine include refinement in both risk assessment and targeting of therapies.24 This prospective study of CHIP showed that the presence of clonally expanded somatic mutations in DNMT3A and TET2 among asymptomatic carriers independently and significantly associated with CVD event risk. Using genetic proxies for putative CVD preventive therapies, we show that individuals with large CHIP clones (1 in 80 middle-aged adults in the UK Biobank)—but not those without CHIP—have reduced CVD events by a genetically-mediated reduction in IL-6 signaling. These results provide several insights into atherosclerosis as well as preventive cardiovascular medicine.
First, building upon prior work6, these new analyses affirm DNMT3A and TET2 CHIP as a previously unrecognized risk factor for future CVD events independent of conventional CVD risk factors. While those with CHIP have modestly higher hsCRP concentrations, risk for CVD events persists even after adjusting for hsCRP concentrations. Other clonal hematopoietic phenomena have recently been described, including clonal hematopoiesis without identified driver mutations4,25 and somatic chromosomal mosaicism26, however these other forms of clonal hematopoiesis have not been linked to CVD risk.
Second, this study showed that degree of clonal expansion refines CVD risk. Larger CHIP clones (VAF>10%) confer most of the CVD risk (Supplemental Figure 6). While ultra-deep sequencing methods can identify very small clones in many more individuals, limited longitudinal analyses indicate that few will substantial clonal expansion at least over a decade.27 Recent work suggests that analyses of mutation fitness may better predict clonal progression, and future longitudinal analyses are required to confirm this hypothesis.28
Third, IL-6 signaling blockade may prove particularly beneficial in individuals with large CHIP clones. Experimental inactivation of Tet2 or Dnmt3a in macrophages augments IL-6 expression.29 Furthermore, our observation agrees with recent experiments showing that an NLRP3 inflammasome (activator of IL-1β, situated upstream of IL-6 signaling) inhibitor mitigates the development of atherosclerosis in atherogenic mice with transplanted Tet2−/− bone marrow to a greater degree than in control mice.7 CANTOS showed that individuals with prevalent CVD and elevated hsCRP have fewer incident CVD events when given canakinumab, an IL-1β inhibitory antibody, versus placebo.30 IL-1β inhibition strikingly induces IL-6.31,32 Indeed, in CANTOS, the degree of benefit correlated strongly with on-treatment IL-6 levels.33 The relative benefit conferred from canakinumab was HR 0.85 in the overall trial, but a preliminary targeted sequencing analysis in CANTOS suggested a markedly greater relative benefit among trial participants with TET2 CHIP (HR=0.36, p=0.034) consistent with our findings.33
Fourth, IL-6 blockade among those with large CHIP clones may yield distinct effects compared to IL-1β inhibition. Tocilizumab, an IL6-receptor inhibitor, is associated with increased LDL cholesterol concentrations. The ENTRACTE trial demonstrated that tocilizumab was not associated with greater CVD risk than etanercept in rheumatoid arthritis patients.34 Our data indicate that similar IL-6 inhibition, without adversely affecting LDL cholesterol may be associated with reduced CVD risk among those with large CHIP. Intriguing exploratory findings from CANTOS that canakinumab may reduce lung cancer risk.30 The current study did not observe that genetically reduced IL-6 signaling alters future CHIP-associated risk for myeloid malignancies. IL-1β neutralization in CANTOS associated with a small but significant risk of infection was not observed in our study. Thus, targeting IL-6, distal to IL-1β might interrupt a causal pathway in atherothombosis while leaving some of IL-1s host defense functions unopposed.
Our study has important limitations. First, to minimize potential heterogeneity that exists by CHIP driver mutation we focused here only on CHIP caused by the two most common driver mutations, DNMT3A and TET2. These two genes together comprise >75% of CHIP in prior analyses2,6,35, have a similar pro-inflammatory profile35 and have prior data supporting the importance of IL-6/IL-1β in atherosclerotic disease pathogenesis in mice.6,7,29 It remains to be determined whether our observations generalize to other CHIP genes (eg JAK2, ASXL1, etc) or other clonal hematopoietic conditions in the absence of driver mutations25,26. A second limitation is the use of a prespecified composite endpoint, chosen for consistency with the CANTOS analysis. Reassuringly, the secondary analyses of the sub-components of the CVD event endpoint showed strong consistency across all subcomponents of the composite endpoint. Third, our germline genetic proxy for therapeutically reduced IL-6 signaling inherently models reduced signaling from birth, well before the development of CHIP, a condition that arises from acquired somatic mutations that accumulate with age. The analyses presented here do not inform regarding the optimal timing of putative IL-6 signaling blockade to reduce CVD risk. Yet, prior mouse studies and post hoc analyses in CANTOS suggest that the efficacy of initiation of therapy after CHIP development.
In conclusion, DNMT3A and TET2 CHIP, particularly with larger clone size, was independently associated with incident CVD events. This risk was specifically mitigated in the setting of a disruptive genetic variant in IL6R.
Supplementary Material
Clinical Perspective.
What is new?
In a prospective cohort, clonal hematopoiesis of indeterminate potential (CHIP) [defined as clonally expanded mutations in DNMT3A and TET2 in blood cells], is independently associated future risk of cardiovascular disease.
The heightened cardiovascular disease risk conferred by CHIP is restricted to those with greater clonal expansion (‘large CHIP’), defined as a variant allele fraction greater than 10%.
Individuals who develop CHIP with simultaneous genetic deficiency of IL-6 signaling (by carrying IL6R p.Asp358Ala versus wild-type) had greater cardiovascular disease risk reduction compared to those without CHIP with genetic IL-6 signaling deficiency.
What are the clinical implications?
Patients with large CHIP, asymptomatic leukemogenic blood cell mutations in DNMT3A or TET2 and variant allele fraction is greater than 10%, have an increased future risk of cardiovascular disease.
Therapies inhibiting IL-6 signaling may more efficiently reduce cardiovascular disease risk among individuals with large CHIP. Prospective clinical trials are required to confirm this hypothesis.
Acknowledgements
This research was conducted using the UK Biobank resource, application 7089.
Funding Sources:
This work was supported by grant (R01HL148565) from the National Heart, Lung, and Blood Institute (NHLBI) to Dr. Natarajan; NHLBI grant (R01HL148050) to Drs. Natarajan and Kathiresan; Fondation Leducq Transatlantic Networks of Excellence grant (TNE-18CVD04) to Drs. Natarajan, Kathiresan and Libby; the Massachusetts General Hospital Department of Medicine Stanbury Physician-Scientist Pathway Program (to Dr. Bick); the John S. LaDue Memorial Fellowship in Cardiology at Harvard Medical School (to Dr. Pirruccello); funding from the NHLBI (R01HL080472 and R01HL134892), the American Heart Association (18CSA34080399), and the RRM Charitable Fund to Dr. Libby; the Ofer and Shelly Nemirovsky Research Scholar Award from Massachusetts General Hospital to Dr. Kathiresan.
P.L. has received has served as an unpaid consultant or has been involved in clinical trials for Amgen, AstraZeneca, Esperion Therapeutics, Ionis Pharmaceuticals, Kowa Pharmaceuticals, Novartis, Pfizer, Sanofi-Regeneron, and XBiotech, Inc.; has served on the Scientific Advisory Board for Amgen, Athera Biotechnologies, Corvidia Therapeutics, DalCor Pharmaceuticals, IFM Therapeutics, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, and Novartis; and has received laboratory funding in the last 2 years from Novartis. S.K. has received a research grant from Bayer Healthcare; and consulting fees from Merck, Novartis, Sanofi, AstraZeneca, Alnylam Pharmaceuticals, Leerink Partners, Noble Insights, MedGenome, Aegerion Pharmaceuticals, Regeneron Pharmaceuticals, Quest Diagnostics, Color Genomics, Genomics PLC, and Eli Lilly and Company; and holds equity in San Therapeutics, Catabasis Pharmaceuticals, and Endcadia. SK. is an employee of Verve Therapeutics. P.N. has received research grants from Amgen, Apple, and Boston Scientific, and is a scientific advisor to Apple and Blackstone Life Sciences.
Non-standard Abbreviations and Acronyms
- CHIP
Clonal hematopoiesis of indeterminate potential
- CVD
cardiovascular disease
- PROMIS
Pakistan Risk of Myocardial Infarction Study
- VAF
variant allele fraction
Footnotes
Conflict of Interest Disclosures: All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
References
- 1.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014; 371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014; 20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. New England Journal of Medicine 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C4, Vuong J, Jacob S, Muralidhar V, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017; 355:842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J Am Coll Cardiol. 2018; 71:875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, Kaptoge S, Clarke P, Boreham C, Coulson RM, et al. Functional IL6R 358Ala Allele Impairs Classical IL-6 Receptor Signaling and Influences Risk of Diverse Inflammatory Diseases. PLoS Genetics. 2013;9: e1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, Guo Y, Chung C, Peasey A, Pfister R, Mooijaart SP, Ireland HA, Leusink M, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet. 2012;379:1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai T, Zhang Y, Ho YL, Link N1, Sun J, Huang J, Cai TA, Damrauer S, Ahuja Y, Honerlaw J, Huang J, Costa L, Schubert P, et al. Association of Interleukin 6 Receptor Variant with Cardiovascular Disease Effects of Interleukin 6 Receptor Blocking Therapy: A Phenome-Wide Association Study. JAMA Cardiology 2018;3:849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hout CV van, Tachmazidou I, Backman JD, Hoffman JX, Ye B, Pandey AK, Gonzaga-Jauregui C, Khalid S, Liu D, Banerjee N, et al. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. bioRxiv 2019. 10.1101/572347v1. [DOI] [PMC free article] [PubMed]
- 13.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas ME, Aragam KG, Emdin CA, Bick AG; International Consortium for Blood Pressure, Hemani G, Davey Smith G, Kathiresan S. Genetic Association of Albuminuria with Cardiometabolic Disease and Blood Pressure. Am J Hum Genet. 2018;103:461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleheen D, Zaidi M, Rasheed A, Ahmad U, Hakeem A, Murtaza M, Kayani W, Faruqui A, Kundi A, Zaman KS, Yaqoob Z, Cheema LA, Samad A, et al. The Pakistan Risk of Myocardial Infarction Study: a resource for the study of genetic, lifestyle and other determinants of myocardial infarction in South Asia. Eur J Epidemiol. 2009; 24:329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won HH, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, Weisburd B, Gupta N, Zaidi M, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017; 544:235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 2013; 31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars NJ, Aguirre M, Venkataraman GR, Wainberg M, Ollila HM, Pirruccello JP, Qian J, et al. Genetics of 38 blood and urine biomarkers in the UK Biobank. bioRxiv 2019. 10.1101/660506. [DOI]
- 20.Li T, Ma H, Chiang JYL. TGFb1, TNFa, and insulin signaling crosstalk in regulation of the rat cholesterol 7a-hydroxylase gene expression. J Lipid Res. 2008;49:1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suissa S. Immortal time bias in pharmacoepidemiology. Am J Epidemiol. 2008;167:492–9. [DOI] [PubMed] [Google Scholar]
- 22.Ligthart S, Vaez A, Võsa U, Stathopoulou MG, de Vries PS, Prins BP, Van der Most PJ, Tanaka T, Naderi E, Rose LM, et al. Genome Analyses of >200,000 Individuals Identify 58 Loci for Chronic Inflammation and Highlight Pathways that Link Inflammation and Complex Disorders. Am J Hum Genet. 2018;103:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breiman L, Friedman J. Estimating Optimal Transformations for Multiple Regression and Correlation. J Am. Statistical Assn 1985;391:580–598. [Google Scholar]
- 24.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017; 130:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loh PR, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018; 559:350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016; 7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson CJ, Papula A, Poon YPG, Wong WH, Young AL, Druley TE, Fisher DS, Blundell JR. The evolutionary dynamics and fitness landscape of clonal haematopoiesis. bioRxiv 2019. 10.1101/569566 [DOI] [PubMed]
- 29.Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res 2018; 123:335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. [DOI] [PubMed] [Google Scholar]
- 31.Loppnow H, Libby P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cellular immunology. 1989; 122:493–503. [DOI] [PubMed] [Google Scholar]
- 32.Loppnow H, Libby P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J Clin Invest. 1990; 85:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svensson EC, Madar A, Campbell CD, et al. Abstract 15111: TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation 2018;138(Supplement 1):A15111. [Google Scholar]
- 34.Giles JT, Sattar N, Gabriel S, Ridker PM, Gay S, Warne C, Musselman D, Brockwell L, Shittu E, Klearman M, et al. Cardiovascular Safety of Tocilizumab Versus Etanercept in Rheumatoid Arthritis: A Randomized Controlled Trial. Arthritis & Rheumatology 2019. 10.1002/art.41095 [DOI] [PubMed]
- 35.Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Leventhal MJ, Bao EL, Nasser J, Zekavat SM, Szeto MD, Laurie C, et al. Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes. bioRxiv 2019. 10.1101/782748 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.