

Abstract
PACT activates the interferon (IFN)-induced double-stranded (ds) RNA-activated protein kinase (PKR) in response to stress signals. Oxidative stress and endoplasmic reticulum (ER) stress causes PACT mediated PKR activation, which leads to phosphorylation of translation initiation factor eIF2α, inhibition of protein synthesis, and apoptosis. A dominantly inherited form of early-onset dystonia 16 (DYT16) has been identified to arise due to a frameshift (FS) mutation in PACT. In order to examine the effect of the resulting truncated mutant PACT protein on PKR pathway we examined the biochemical properties of the mutant protein and its effect on mammalian cells. Our results indicate that the FS mutant protein loses its ability to bind dsRNA as well as its ability to interact with PKR while surprisingly retaining the ability to interact with PACT and PKR-inhibitory protein TRBP. The truncated FS mutant protein, when expressed as a fusion protein with a N-terminal fluorescent mCherry tag aggregates in mammalian cells to induce apoptosis via activation of caspases both in a PKR- and PACT-dependent as well as independent manner. Our results indicate that interaction of FS mutant protein with PKR inhibitor TRBP can dissociate PACT from the TRBP-PACT complex resulting in PKR activation and consequent apoptosis. These findings are relevant to diseases resulting from protein aggregation especially since PKR activation is a characteristic of several neurodegenerative conditions.
Keywords: PKR, PACT, dystonia, DYT16, caspase, apoptosis
Introduction
PKR (protein kinase, RNA activated) is activated by binding to its Protein Activator PACT in human cells [Patel and Sen, 1998] and its murine homolog is termed PKR associated protein X (RAX) [Ito et al., 1999]. PACT induces autophosphorylation and activation of the interferon (IFN)-inducible, serine/threonine protein kinase PKR in response to cellular stress [Bennett et al., 2006; Ito et al., 1999; Patel et al., 2000; Singh et al., 2009]. Activation of PKR causes phosphorylation of the α subunit of the eukaryotic protein synthesis initiation factor 2 (eIF2α) leading to an inhibition of protein synthesis [Dabo and Meurs, 2012].
PKR is expressed in all cell types at low basal levels in the absence of virus infection, mediates IFN’s antiviral actions in virally infected cells and also regulates cellular survival and apoptosis in response to stress in uninfected cells [Garcia et al., 2006]. Activation of PKR’s kinase activity requires binding to one of its activators to bring about a conformational change causing enzymatic activation [Meurs et al., 1990]. PKR’s two dsRNA-binding motifs (dsRBMs) bind to the dsRNA produced during viral replication [Green and Mathews, 1992; Patel and Sen, 1992] and activate PKR by unmasking the ATP-binding site [Nanduri et al., 2000]. These dsRBMs also mediate dsRNA-independent protein-protein interactions with other proteins that also carry dsRBMs [Chang and Ramos, 2005]. Among such proteins, PACT functions to activate PKR in a dsRNA-independent manner in response to cellular stress [Patel et al., 2000; Patel and Sen, 1998]. There are three copies of dsRBM in PACT (Fig. 1 A), of these the two amino-terminal motifs (M1 and M2) are involved in a direct interaction with the dsRBMs of PKR. The third, carboxy-terminal motif 3 (M3) is dispensable for a high-affinity interaction with PKR but is essential for PKR activation as it contacts a specific region in PKR’s catalytic domain [Huang et al., 2002; Peters et al., 2001]. PACT-dependent PKR activation in cells occurs only in response to oxidative stress signals, growth factor withdrawal, endoplasmic reticulum (ER) stress, to cause phosphorylation of the translation initiation factor eIF2α and cellular apoptosis [Bennett et al., 2006; Ito et al., 1999; Patel et al., 2000; Singh et al., 2009] although the purified, recombinant PACT activates PKR by direct interaction in vitro [Patel and Sen, 1998]. Once the stress conditions have resolved, PACT mediated PKR activation is possibly downregulated by the action of phosphatases that may dephosphorylate PACT. Although the identity of such phosphatases remains unknown, our recent work illustrates that a PKR inhibitory protein TRBP that complexes with PACT in the absence of stress signals is phosphorylated later during the stress response and is involved in the negative regulation of PKR activity at late time points after initial stress signal [Chukwurah and Patel, 2018].
Figure 1: Schematic representation of the frameshift mutation in PACT.
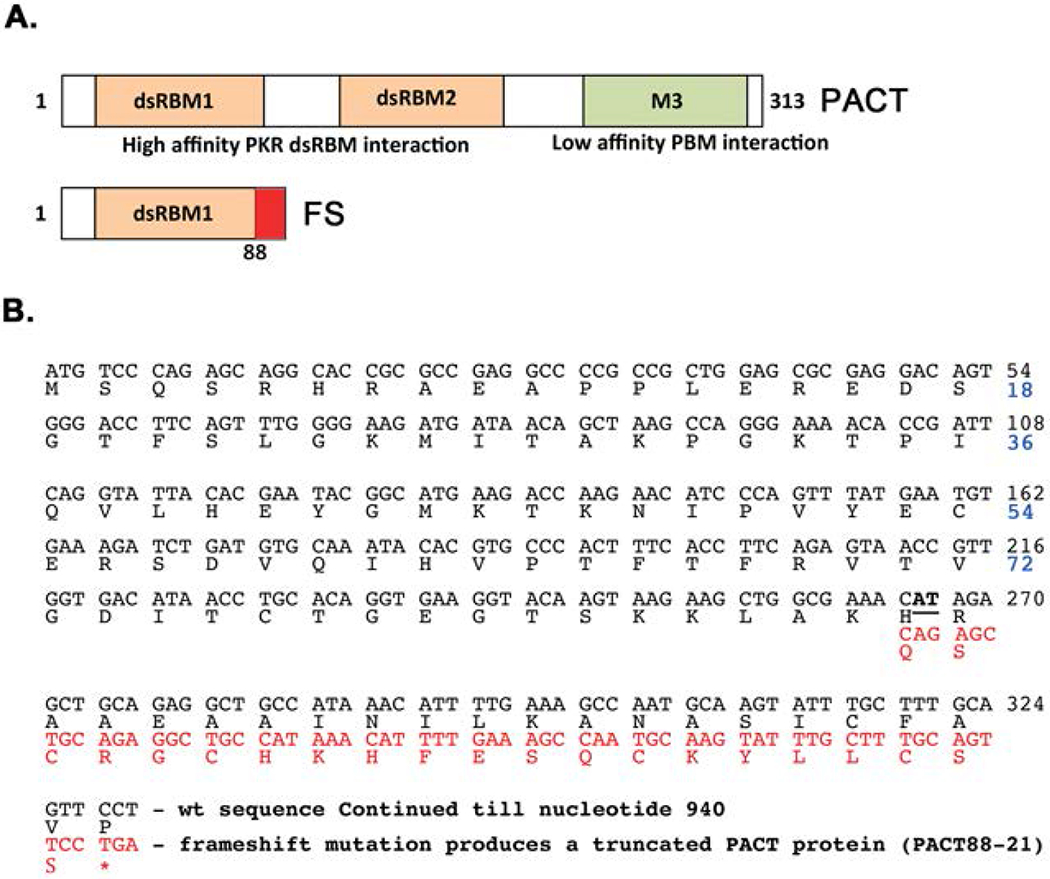
A. Domain structure of PACT and frameshift (FS) mutant. Orange boxes: conserved dsRNA-binding motifs dsRBM1 and dsRBM2 that mediate dsRNA-independent high-affinity interaction with PKR, Green box: M3 motif that does not bind dsRNA and mediates low-affinity interaction with the PACT-binding motif (PBM) within PKR’s catalytic domain. The residues added as a result of frameshift mutation in FS are indicated as a red box. B. FS mutation in PACT ORF. The first 330 nucleotides of the PACT ORF are shown. The two-nucleotide deletion (266-267) is indicated in bold and underlined font. The shifted reading frame is indicated in red font. Thus, the frameshift mutation produces a protein that has amino acids 1-88 and 21 new amino acids (PACT88-21).
PACT is encoded by the Prkra gene and recently many mutations in this gene have been described to cause a movement disorder dystonia 16 (DYT16). Many different dystonia types exist, which result from several diverse genetic and physiological causes, thereby constituting a heterogeneous group of movement disorders in which affected individuals develop sustained and painful involuntary muscle contractions leading to twisted postures [Geyer and Bressman, 2006]. Recently a recessively inherited form of early-onset generalized dystonia (DYT16) has been described to arise from a homozygous missense mutation at amino acid position 222 in PACT [Camargos et al., 2008]. Seven affected members from two unrelated families were originally identified to carry the same P222L mutation [Camargos et al., 2008], which lies between the conserved motifs M2 and M3 within PACT. The same mutation was later reported in many more patients with DYT16 [Quadri et al., 2016]. Subsequently, four more recessive mutations (C77S, C213F, C213R, and S265R) have been identified in DYT16 patients [Brashear, 2013; de Carvalho Aguiar et al., 2015; Lemmon et al., 2013]. Among the dominant mutations, a frameshift mutation which results in truncation of the protein after 88 amino acids [Seibler et al., 2008] and three point mutations reported in Polish and German families (T34S, N102S, and c.-14A>G) indicate that PACT mutations lead to DYT16 in a worldwide distribution [Zech et al., 2014].
In spite of the identification of several PACT mutations, the molecular mechanisms involved in the DYT16 onset or progression have not been studied much [Bragg et al., 2011]. We have previously analyzed the effect of the P222L mutation on PACT’s biochemical properties such as dsRNA binding, PKR interaction, and PKR activation [Vaughn et al., 2015]. The P222L mutation did not affect PACT’s dsRNA-binding, or PKR-interaction properties in vitro. However, in DYT16 patient cells the P222L mutant protein caused a delayed but prolonged activation of PKR in response to ER stress. The altered kinetics of eIF2α phosphorylation brought about by the changes in PKR activation led to defective downstream signaling and a lack of cell recovery and homeostasis. Thus, the DYT16 patient cells underwent enhanced apoptosis in response to the ER stressor tunicamycin in accordance with the altered biochemical properties of P222L protein.
In this report, we analyzed the effect of a dominant acting frameshift mutation in PACT described in a single early onset dystonia case [Seibler et al., 2008]. This frameshift mutation due to deletion of two nucleotides would produce a truncated PACT protein with 1-88 amino acids of the PACT followed by 21 new amino acids as a result of the frameshift. Our results indicate that when expressed transiently in mammalian cells, a fusion protein with N-terminal mCherry tag on the mutant frameshift protein (mCherry-FS) forms aggregates to trigger apoptosis. Furthermore, the PKR+/+ as well as PACT+/+ MEFS show increased caspase activation in response to a transient overexpression of mCherry-FS protein and the caspase activation is reduced, but not absent, in PKR−/− and PACT−/− MEFs indicating that apoptosis caused by the FS mutant protein aggregation may result from both PKR-dependent and PKR-independent mechanisms.
Methods and Materials
Reagent, Cell Lines, and Antibodies:
HeLa (ATCC CCL-2) cells, PKR+/+, PKR−/− [Deb et al., 2001], PACT+/+, and PACT−/− mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco’s modified Eagle’s medium (DMEM), containing 10% [Rowe et al., 2006] fetal bovine serum and penicillin/streptomycin. The anti-mCherry antibodies used were from Abcam (ab183628). Transfections were performed with Effectene Transfection Reagent (Qiagen) according to the manufacturer’s protocol.
Generation of 2 nucleotide deletion frameshift mutation:
The deletion mutation was generated using mutagenic primers for PCR amplification of PACT ORF in two parts to introduce a two-nucleotide deletion as reported in DYT16 patient. The primer sequences were as follows:
PACT-UP1: 5’-GCTCTAGACATATGGAAATGTCCCAGAGCAGGCAC-3’,
DYT16-frameshift-DN1: 5’-GGCAGCCTCTGCAGCTCTGTTTCGCCAGC-3’
DYT16-frameshift-UP1: 5’-GCTGGCGAAACAGAGCTGCAGAGGCTGCC-3’
PACT313M3-AS: 5’-GGGGATCCTTACTTTCTTTCTGCTATTATC-3’
The two PCR products were sub-cloned into pGEMT-easy vector (Promega). Once the sequence of the frameshift mutation was verified, we generated full-length FS ORF in pcDNA3.1− by a three-piece ligation of XbaI–Pst1 restriction piece from the 5’ half of FS point mutant/pGEMT-easy, Pstl-BamHI piece from 3’ half of FS point mutant/pGEMT-easy, and XbaI-BamH1 cut pcDNA3.1−. The full-length FS mutant ORF in pcDNA3.1− has an amino-terminal flag tag that is added from the vector sequence. Full length FS mutant was sub-cloned into pET15b (Novagen) for producing hexahistidine tagged pure recombinant protein and into pmCherryC1 (Clontech) for expression in mammalian cells. The PACT/pmCherryC1 and PACT/pEGFPC1 constructs were generated by sub-cloning the PACT ORF from PACT/pcDNA3.1− and the PKR/pEGFPC1 construct was generated by sub-cloning the PKR ORF with the kinase dead K296R mutation from K296R/pCDNA3.1−. It is well established that the wt PKR overexpression in cells is not feasible as it induces apoptosis very rapidly and thus for all experiments using full length PKR in fluorescence microscopy and co-localization experiments, we used the catalytically dead PKR mutant K296R expression construct.
dsRNA-binding assay:
The dsRNA-binding assay was performed with the in vitro translated, 35S-labeled PACT and FS proteins synthesized using the expression constructs in pcDNA3.1− and TNT-T7 coupled reticulocyte lysate system from Promega as described before [Chukwurah et al., 2018].
Protein–protein interaction assay:
In vitro translated, 35S-labeled PKR, PACT, FS or TRBP proteins were synthesized using the TNT T7 coupled reticulocyte system from Promega as explained before. 5 μl of the in vitro translated 35S-labeled proteins were incubated with either 100 ng of pure recombinant hexahistidine tagged PKR, PACT or FS mutant as indicated in the individual figures and 20 μl of Ni-agarose (Novagen) in 200 μl of binding buffer [5mM imidazole, 20mM Tris–HCl pH 7.9, 200mM NaCl, 0.2mM phenylmethylsulfonyl fluoride (PMSF), 0.5% IGEPAL (Sigma)] at 25°C for 30 min on a rotating wheel. The beads were washed four times in 500 μl of binding buffer each time and the washed beads were then boiled in 20 μl Laemmli buffer (150mM Tris–HCl pH 6.8, 5% SDS, 5% β-mercaptoethanol, 20% glycerol) for 2 min and eluted proteins were analyzed by SDS–PAGE on a 12% gel. Fluorography was performed at 80°C with intensifying screens. To assay if the FS mutant protein can dissociate TRBP-PACT complex, 5 μl of the in vitro translated 35S-labeled TRBP protein was incubated with 100 ng of pure recombinant hexahistidine tagged PACT and anti-PACT rabbit monoclonal antibody bound to 10 μl of protein A sepharose beads in 200 μl of binding buffer [20mM Tris–HCl pH 7.9, 200mM NaCl, 0.2mM phenylmethylsulfonyl fluoride (PMSF), 0.5% IGEPAL (Sigma)] at 25°C for 30 min on a rotating wheel. Increasing amounts of pure recombinant FS protein was then added as indicated in Fig. 6B and the incubation was continued for 30 min longer. The beads were then washed four times in 500 μl of binding buffer each time and the washed beads were then boiled in 20 μl Laemmli buffer (150mM Tris–HCl pH 6.8, 5% SDS, 5% β-mercaptoethanol, 20% glycerol) for 2 min and eluted proteins were analyzed by SDS–PAGE on a 12% gel. The assays were quantified on Typhoon FLA7000 by analyzing the band intensities in the relevant lanes.
Figure 6. Effect of FS mutation on TRBP-PACT interaction.
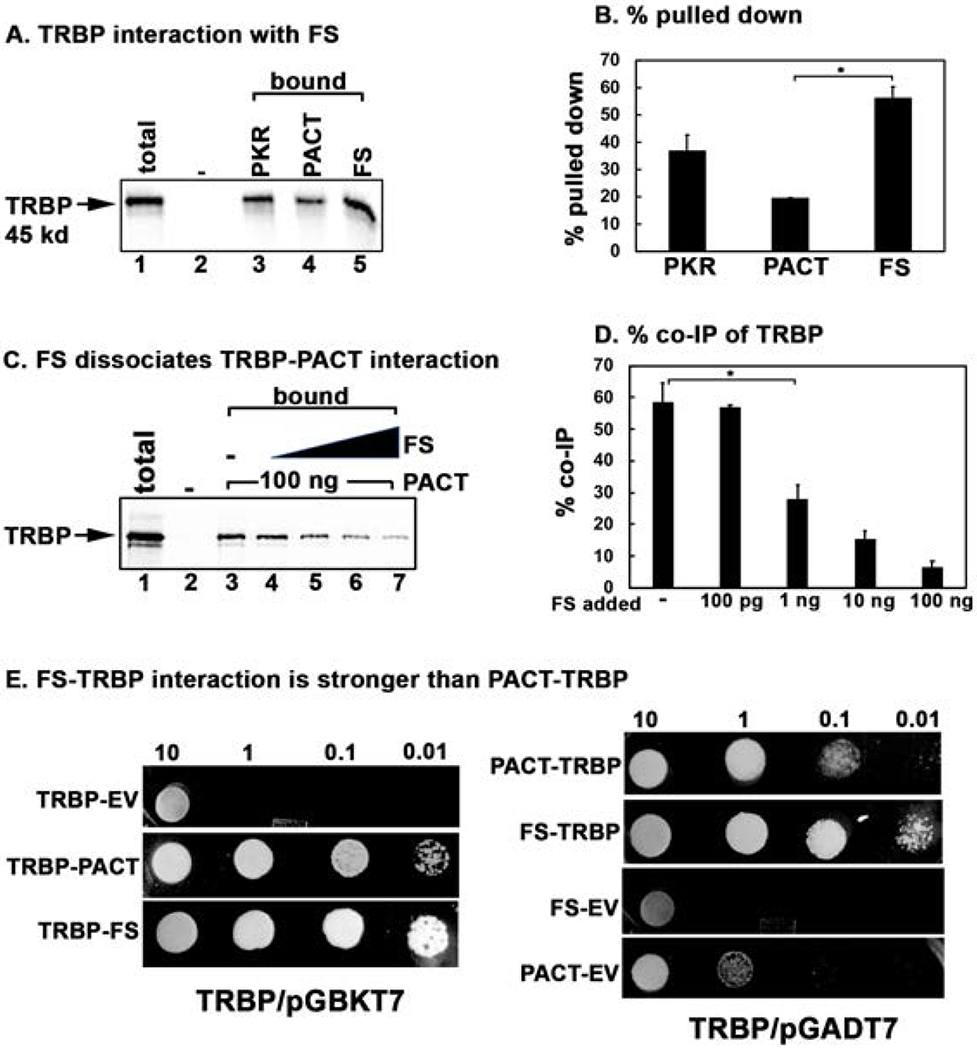
(A) Pull down assay of in vitro translated TRBP protein with pure recombinant PKR, PACT, and FS proteins. 5 μl of in vitro translated, 35S-labeled flag-tagged TRBP protein was mixed with 100 ng of purified, hexahistidine tagged recombinant PKR, PACT, and FS proteins immobilized in Ni-charged Sepharose beads. Pull down of 35S-labelled TRBP was analyzed by SDS-PAGE after washing the beads five times with wash buffer. Total lane: total input TRBP (50% of the bound samples); Bound lanes: pulled down TRBP. The “-” lane shows TRBP pull down with no recombinant protein on Ni-charged Sepharose beads as a negative control. (C) FS mutant protein can dissociate TRBP from PACT. 5 μl of the in vitro translated 35S-labeled TRBP protein was incubated with 100 ng of pure recombinant hexahistidine tagged PACT and anti-PACT rabbit monoclonal antibody bound to 10 μl of protein A sepharose beads. Increasing amounts of pure recombinant FS protein was added as indicated in lanes 4-7. Pull down of 35S-labelled TRBP was analyzed by SDS-PAGE after washing the beads five times with wash buffer. Total lane: total input TRBP (50% of the bound samples); Bound lanes: pulled down TRBP. The “-” lane shows TRBP pull down with no recombinant protein on Ni-charged Sepharose beads as a negative control. Lanes 4-7: pure recombinant FS mutant protein added in increasing amounts (100 pg, 1 ng, 10 ng, 20 ng). (B and D) Quantification of data in 6 A, and 6 C. The radioactivity present in the bands was measured by phosphorimager analysis and the % pull down was calculated as (radioactivity present in the pull down (bound) TRBP bands/the radioactivity present in the TRBP band in the total lane) X 100. Error bars: standard error of mean from 4 independent experiments. Student T-tests were performed, and p values are as follows (B) * = 0.00011, (D) * = 0.000012, n=4. E. TRBP-FS interaction is stronger than TRBP-PACT interaction in yeast two-hybrid assay. PACT/pGADT7, FS/pGADT7 or empty vector (EV) pGADT7 were co-transformed with TRBP/pGBKT7 into AH109 yeast cells and selected on SD double dropout media (-tryptophan, - leucine). Ten microliters of transformed yeast cells (OD600 = 10, 1, 0.1, 0.01) were spotted on SD quadruple dropout media (- tryptophan, - leucine, - histidine, -adenine) containing 10 mM 3-amino-1,2,4-triazole (3-AT). Plates were incubated for 3 days at 30 °C. The assay was also performed with either PACT/pGBKT7 or FS/pGBKT7 and TRBP/pGADT7 in a similar manner (TRBP/pGADT7 panels). Co-transformation of PACT or FS in pGBKT7 and empty vector pGADT7 served as negative controls.
Caspase 3/7 assay:
PKR+/+, PKR−/− [Deb et al., 2001], PACT+/+ and PACT−/− [Rowe et al., 2006] mouse embryonic fibroblasts (MEFs) were plated at in 6 well dishes and transfected with flag-FS mutant/pCDNA3.1− or empty vector pCDNA3.1−. Cells were collected at indicated time points and mixed with equal parts of Promega Caspase-Glo 3/7 reagent and incubated for 45 min. Luciferase activity was measured with a negative control of cell culture medium alone used to normalize all readings.
Western Blot analysis:
HeLa cells were transfected in triplicates in 6 well dishes with 500ng DNA per well of pmCherryC1 empty vector, FS mutant/pmCherryC1, and PACT/pmCherryC1. Transfected cells were collected 12 h after transfection and washed twice with ice cold 1× PBS. Harvested cells were lysed in western lysis buffer (2% Triton X-100, 20 mM Tris-HCl pH 7.5, 100 mM KCl, 200 mM NaCl, 4 mM MgCl2, 40% Glycerol, and phosphatase inhibitor cocktail 2 (Sigma) at 1:100 dilution) for 5 min on ice. Lysates were centrifuged at 13,200 rpm for 2 min. Protein concentration in the supernatant was quantified using Bradford reagent. Western blot was performed with the anti-mCherry antibody and western blot images were analyzed using the Typhoon FLA 7000 and ImageQuant LAS 4000 (GE Health).
Expression and purification of recombinant PKR, wt PACT and FS mutant:
The protein coding regions (PKR, wt PACT or FS mutant) were sub-cloned into pET15b (Novagen) to generate PKR/pET15b, PACT/pET15b, and FS mutant/pET15b resulting in the in-frame fusion of the ORFs to the histidine tag. The recombinant proteins were expressed and purified as described [Patel and Sen, 1998; Singh et al., 2011].
Visualization of aggregated FS mutant protein:
HeLa cells were grown on coverslips and transfected with 500 ng of either PACT/pmCherryC1, FS mutant/pmCherryC1 empty vector pmCherryC1 using Effectene (Qiagen). 12 h after transfection, the cells were rinsed with ice-cold phosphate buffered saline (PBS) and fixed in 2% paraformaldehyde for 10 min. The cover slips were mounted in Vectashield mounting medium containing DAPI (Vector Laboratories). Cells were then viewed under the inverted fluorescence microscope (EVOS® FL Imaging System).
Yeast Two-Hybrid Interaction Assay:
To test TRBP-PACT and TRBP-FS interactions, PACT and FS were expressed as GAL4-activation domain fusion proteins from the pGADT7 vector and TRBP was expressed as GAL4 DNA-binding domain fusion proteins from the pGBKT7 vector. We also tested these interactions with TRBP expressed as GAL4-activation domain fusion protein from the pGADT7 vector with PACT and FS proteins expressed as GAL4 DNA-binding domain fusion proteins from the pGBKT7 vector. The appropriate combinations of expression constructs were co-transformed into AH109 yeast cells (Clontech) and the transformed yeast cells were plated on double dropout SD minimal medium lacking tryptophan and leucine. In order to check for the transformants’ ability to grow on quadruple dropout media, transformed yeast cells were grown to an OD600 of 2 in YPD media (yeast extract, peptone, and dextrose). 500 μl of each culture was pelleted and resuspended in an appropriate amount of distilled water to yield an OD600 of 10. Serial dilutions were then made to yield OD600 values of 1, 0.1, and 0.01. 10 μl of each dilution was then spotted onto quadruple dropout SD minimal media lacking histidine, tryptophan, leucine, and adenine in the presence of 10 mM 3-amino-1,2,4-triazole (3-AT). Plates were incubated at 30 °C for 3 days to score the growth.
Quantifications and Statistics:
All binding assays and pull-down assays (Typhoon FLA7000) were quantified using GE Life Sciences ImageQuant TL software. To determine statistical significance of results of the dsRNA-binding, protein interaction and caspase assays a two-tailed Student’s T-test was performed, assuming equal variance. Each figure legend indicates p values as denoted by brackets and special characters. Note that our alpha level was p=0.05.
Results
The FS mutation destroys PACT’s dsRNA binding as well as PKR interaction activity:
The early onset dominant mutation causing DYT16 in one German patient is a deletion of two nucleotides (AT) in codon 89 (Fig. 1 B) of the PACT open reading frame (ORF). This results in a frameshift that would produce a 109 amino acid long truncated protein with 1-88 amino acids of original PACT protein followed by 21 extraneous amino acids due to the frameshift (Fig. 1 A and B). The resulting truncated (mutant FS) protein does not have a complete copy of the evolutionarily conserved dsRNA-binding motif (dsRBM1) and thus is missing the crucial part of the motif that is essential for binding dsRNA as well as protein-protein interactions [Chukwurah et al., 2018; Singh and Patel, 2012]. Therefore, the FS mutant protein is expected to be devoid of dsRNA-binding ability, as well as interaction with PKR and PACT. In order to determine if the frameshift mutation affects dsRNA binding activity, an in vitro dsRNA-binding assay previously well established for PKR and PACT [Patel and Sen, 1998] was performed (Fig. 2 A and B). As seen in Figure 2 A, the wt PACT protein binds to dsRNA (lane 2) but the frameshift mutant protein shows no binding to dsRNA (lane 4). The binding of wt PACT to dsRNA immobilized on the beads could be competed out by exogenously added dsRNA (lane 6) but not ssRNA (lane 5) confirming specific binding to dsRNA. Firefly luciferase, a protein that does not bind dsRNA used as a negative control showed no binding to the beads further demonstrating the binding specificity. The quantification of percentage binding established that FS mutant protein has no dsRNA-binding activity as compared to wt PACT (Fig. 2 B). We next examined if the FS mutant protein can interact with PKR and PACT using protein-protein interaction pull down assay (Fig. 3 A–F). 35S-methionine labeled PKR, and PACT were in vitro translated using rabbit reticulocyte system and mixed with pure recombinant hexahistidine tagged PKR, PACT, or FS mutant proteins bound to Ni-charged sepharose beads to measure the pull down of 35S-labeled proteins. As seen in Fig. 3 A and B, PKR interacts efficiently with PACT (lane 3) and PKR (lane 4) but not with FS mutant protein (lane 5). No 35S-labeled PKR was pulled down by Ni-agarose beads in the absence of any hexahistidine tagged protein (lane 2) indicating absence of any non-specific binding of PKR to the Ni-agarose beads and presence of a specific interaction with PACT and PKR. These results indicate that the mutant FS protein does not interact with PKR in vitro under the conditions of this assay. As seen in Fig. 3 C and D, PACT interacts efficiently with PACT (lane 3), PKR (lane 4) and surprisingly also with mutant FS protein (lane 5). No 35S-labeled PACT was pulled down by Ni-agarose beads in the absence of any hexahistidine tagged protein (lane 2) indicating absence of any non-specific binding of PACT to the Ni-agarose beads and presence of a specific interaction with PACT, PKR, and FS mutant. We further tested the interaction of 35S-labeled, in vitro translated PKR, PACT and FS proteins with pure recombinant FS protein bound to Ni-charged sepharose beads to measure the pull down of 35S-labeled PKR, PACT and FS proteins. As seen in Fig. 3 E and F, in vitro translated PKR showed no interaction with the recombinant FS protein (lane 4), whereas in vitro translated PACT (lane 5) and FS (lane 6) proteins showed a strong interaction with recombinant FS protein bound to the Ni-agarose beads. There was no non-specific binding of PKR, PACT, or FS proteins to the Ni-agarose beads in the absence of FS protein bound to the beads (lanes 7-9) indicating specific interaction between PACT-FS and FS-FS under the conditions of this assay.
Figure 2: Effect of FS mutation on dsRNA-binding.
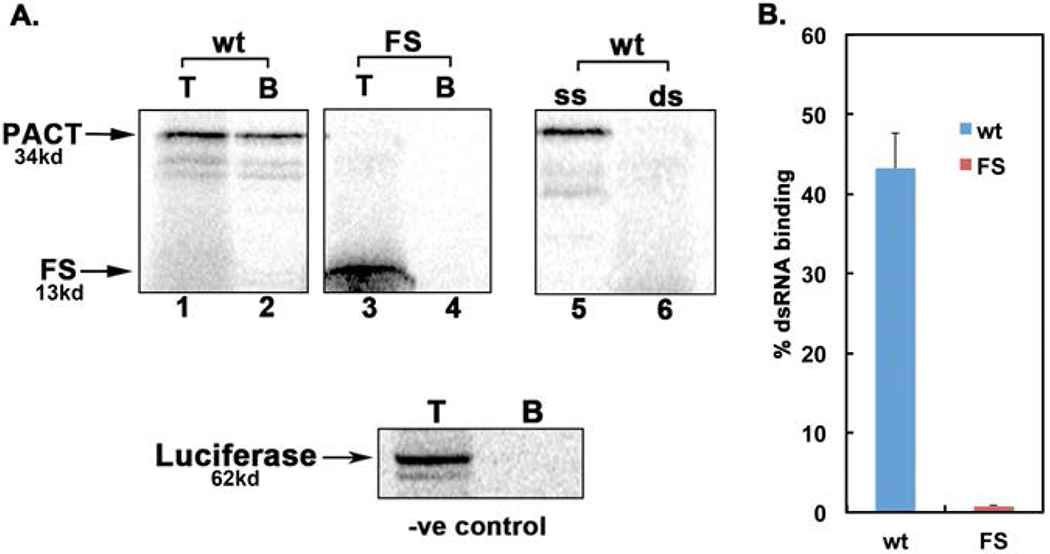
dsRNA-binding activity of wt PACT and FS mutant was measured by poly(I)-poly(C)-agarose binding assay with in vitro translated 35S-labeled proteins. The positions of PACT and FS bands are indicated by arrows and the molecular weights are as indicated. T, total input; B, proteins bound to poly(I)-poly(C)-agarose. Competition lanes 5 and 6: competition with 100-fold molar excess of ssRNA (ss) or dsRNA (ds). The fainter bands below the parent PACT bands represent products of in vitro translation from internal methionine codons. The firefly luciferase, which does not bind dsRNA, was used as a negative control. B. Quantification of dsRNA-binding assay. Bands were quantified by phosphorimager analysis and % bound was calculated. Error bars: standard error of mean from 4 independent experiments. wt PACT: blue bar and FS mutant: red bar.
Figure 3: Effect of FS mutation on interaction with PKR and PACT.
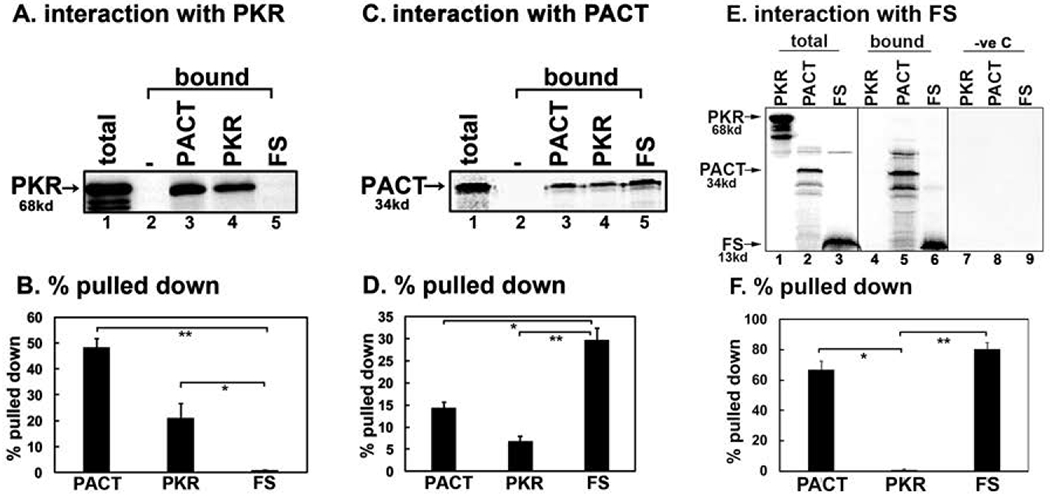
(A, C, and E) Pull down assay of in vitro translated proteins with pure recombinant PACT, PKR, and FS proteins. 5 μl of in vitro translated, 35S-labeled flag-tagged PKR (A) or PACT (C) or FS (E) protein was mixed with 100 ng of purified, hexahistidine tagged recombinant PKR, PACT, and FS proteins immobilized in Ni-charged Sepharose beads. Pull down of 35S-labelled PKR, PACT and FS was analyzed by SDS-PAGE after washing the beads five times with wash buffer. Total lane: total input PKR, PACT, and FS as indicated (50% of the bound samples); Bound lanes: the pulled down PKR (A) or PACT (C) or PKR, PACT and FS (E). B, D, and F. Quantification of data in 3 A, 3 C, and 3E. The radioactivity present in the bands was measured by phosphorimager analysis and the % pull down was calculated as (radioactivity present in the pull down (bound) PKR, PACT, FS bands/the radioactivity present in the PKR, PACT, FS bands in the total lane) X 100. Error bars: standard error of mean from 4 independent experiments. Student T-tests were performed, and p values are as follows (B) * = 0.00018, ** = 0.00013, (D) * = 0.00019, and ** = 0.00009, (F) * = 0.00002 and ** = 0.00001, n=4.
FS mutant protein forms aggregates in mammalian cells:
In order to test the effect of FS mutant protein expression on mammalian cells, we transfected HeLa cells with mCherry-PACT and mCherry-FS expression constructs. Observing the transfected cells under a fluorescence microscope 12 h after transfection indicated that mCherry-PACT protein shows primarily cytoplasmic localization without aggregates (Fig. 4 A, panel a), whereas the mCherry-FS mutant protein forms cytoplasmic aggregates (Fig. 4 A, panel b). The mCherry protein itself shows localization both to cytoplasm and nucleus (Fig. 4 A, panel c). The mCherry tag is known to be monomeric and any aggregation of mCherry-FS fusion protein observed in mammalian cells would thus be driven by the FS portion of the fusion protein and not by the mCherry tag. We also analyzed the expression of mCherry fusion proteins by a western blot analysis with anti-mCherry antibody 12 h after transfection. As seen in Fig. 4B, the expression of mCherry fusion proteins can be detected on the western blot. The presence of higher molecular weight bands in mCherry-FS lane indicated that the aggregates formed by the mCherry-FS protein persist even in the presence of SDS and during gel electrophoresis (Fig 4 B, lane 2). Such aggregates could be formed by the mCherry-FS protein molecules alone or with endogenous PACT protein and at present we do not know the exact composition of these protein aggregates. At longer time points after transfection, the cells overexpressing mCherry-FS mutant protein showed cell death around 24-36h. Despite multiple efforts, it was not feasible to establish a stable expression of mCherry-FS to achieve 100% cells in transfected population expressing the mutant mCherry-FS. We also attempted to express the FS mutant protein using a smaller tag such as the 8-amino acid long flag tag. Expression of the flag tagged FS mutant protein was not detectable by either immunofluorescence or western blot in the transient transfection system, which may be due to more efficient and quicker induction of cell death or due to nonsense-mediated decay (NMD) of the mRNA [Karousis and Muhlemann, 2018]. Addition of the mCherry tag on the amino terminus makes the fusion protein size larger and this may allow the mRNA to escape NMD, making detection of the mCherry-FS mutant protein possible. In order to detect any change in PKR and eIF2α phosphorylation in response to FS mutant protein expression, we performed western blot analysis at shorter time points after transfection. As seen in Fig. 5 A, we detected a time-dependent small increase in eIF2α phosphorylation (lanes 2-4) in cells expressing mCherry-FS protein starting at 1-8 h (lanes 2-5) and the cells transfected with empty vector mCherry did not show a similar increase (lanes 7-10). PKR phosphorylation also was detected in mCherry-FS transfected cells (lanes 2-5) but not in cells transfected with empty vector mCherry (lanes 7-10). These results suggest that expression of mCherry-FS protein causes PKR activation leading to eIF2α phosphorylation indicating that PKR activation could be the reason for cell death observed at later time points in response to mCherry-FS expression.
Figure 4.
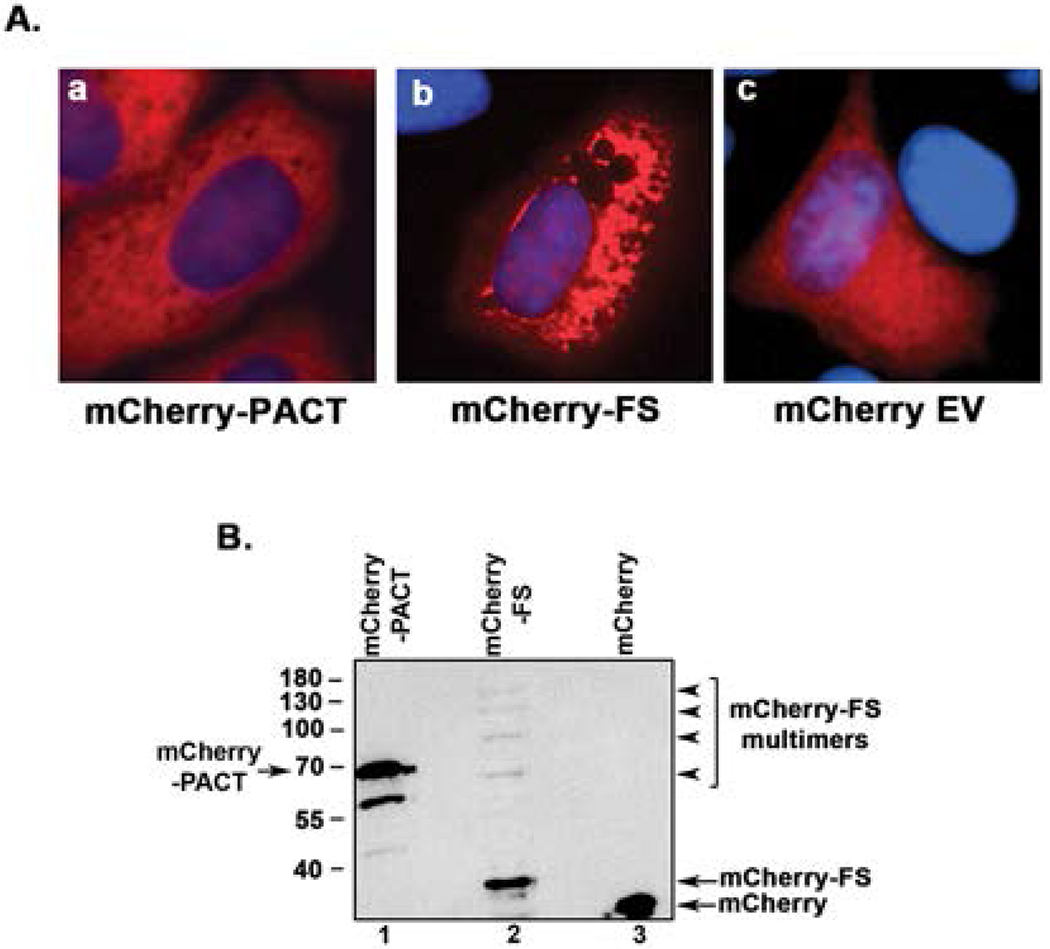
A. The mCherry-FS mutant protein aggregates in mammalian cells. HeLa cells were transfected with mCherry-PACT/pmCherryC1, Cherry-FS/pmCherryC1, and pmCherryC1 empty vector (EV). Expression of proteins was examined 12 h after transfection using a fluorescence microscope. a: overlay of mCherry-PACT (red) and DAPI nuclear stain (blue), b: overlay of mCherry-FS fusion protein (red) DAPI nuclear stain (blue), c: overlay of mCherry (red) and DAPI nuclear stain (blue). B. Western blot analysis of mCherry tagged proteins. HeLa cells were transfected with mCherry-PACT/pmCherryC1, Cherry-FS/pmCherryC1, and pmCherryC1 empty vector (EV). 12 h post transfection, the cells were harvested, cell extracts were made and analyzed by western blot analysis with anti-mCherry antibody. Lane 1: mCherry-PACT, lane2: mCherry-FS, lane 3: mCherry EV. The positions of mCherry-PACT, mCherry-FS and mCherry are as indicated by arrows. The multimers of mCherry-FS seen in lane 2 are indicated by arrowheads.
Figure 5.
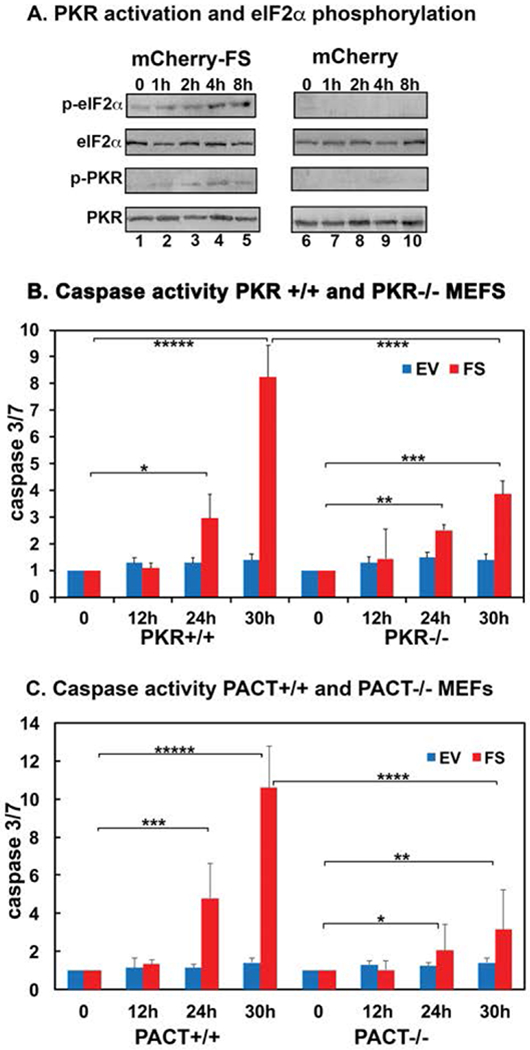
A. PKR activation and eIF2α phosphorylation in response to transient overexpression of FS mutant protein. HeLa cells were transfected with Cherry-FS/pmCherryC1, and pmCherryC1 empty vector. At 1h-8h post transfection the cells were harvested, cell extracts were made and analyzed by western blot analysis with the indicated antibodies. Lanes 1-5: mCherry-PACT/pmCherryC1 transfected cell extracts, lanes 6-10: pmCherryC1 empty vector transfected cell extracts. B. mCherry-FS mutant induces caspase3/7 activation in a PKR-dependent as well as PKR-independent manner. The PKR+/+ and PKR−/− mouse embryonic fibroblasts were transfected with either the mCherry-FS/pmCherryC1 expression construct or empty vector pmCherryC1. Caspase 3 and 7 activities were measured, at indicated time points. blue bars: EV (pmCherryC1) transfected cells and the red bars: mCherry-FS/pmCherryC1 transfected cells. Student T-tests were performed, and p values are as follows * = 0.0073 (significant), ** = 0.0065 (significant), *** = 0.0027 (significant), **** = 0.00051, and ***** = 0.00023 (significant), n=4. C. FS mutant induces caspase 3/7 activation in a PACT-dependent as well as PACT-independent manner. The PACT+/+ and PACT−/− mouse embryonic fibroblasts were transfected with either the mCherry-FS/pmCherryC1 expression construct or empty vector pmCherryC1. Caspase 3 and 7 activities were measured, at indicated time points. blue bars: EV (pmCherryC1) transfected cells and the red bars: mCherry-FS/pmCherryC1 transfected cells. Student T-tests were performed, and p values are as follows * = 0.0067 (significant), ** = 0.0051 (significant), *** = 0.0043 (significant), **** = 0.00065, and ***** = 0.00027 (significant), n=4.
Transient overexpression of FS mutant protein activates caspase 3/7:
Activation of PKR in mammalian cells causes apoptosis via activation of caspases [Gil and Esteban, 2000]. Thus, we tested if transient overexpression of mCherry-FS mutant protein results in activation of caspases. Overexpression of mCherry-FS mutant in mouse embryonic fibroblasts from PKR+/+ and PKR−/− mice resulted in significant induction of caspase activity and apoptosis of transfected cells. As seen in Figure 5 B, there is a significant increase in caspase 3/7 activity in PKR+/+ MEFs transfected with mCherry-FS mutant expression construct at 24 and 30 h (red bars) as compared to the cells transfected with empty vector (blue bars). Compared to the PKR +/+ MEFs, the PKR −/− MEFs showed significantly less increase in caspase 3/7 activity at equivalent time points after the transfection. However, the PKR−/− cells overexpressing mCherry-FS mutant also underwent apoptosis similar to PKR +/+ cells, but with a slower kinetics. We also tested if PACT was essential for caspase activation in response to FS mutant protein expression. Overexpression of mCherry-FS mutant in mouse embryonic fibroblasts from PACT+/+ and PACT−/− mice resulted in significant induction of caspase activity. As seen in Figure 5 C, similar to PKR+/+ MEFS, there is a significant increase in caspase 3/7 activity in PACT+/+ MEFs transfected with mCherry-FS mutant expression construct at 24 and 30h (red bars) as compared to the cells transfected with empty vector (blue bars). Compared to the PACT +/+ MEFs, the PACT −/− MEFs show significantly less increase in caspase 3/7 activity at equivalent time points after the transfection. These results suggest that the FS mutant protein aggregates depend on PACT and PKR only in part to induce caspase activation and apoptosis.
In order to understand how the mutant FS mutant protein could bring about PKR activation in mammalian cells when it shows no interaction with PKR, we explored the possibility that FS mutant protein may promote PACT-PKR interaction by interacting with TRBP. Our previous work has suggested that in unstressed cells, PACT is complexed with TRBP, a PKR inhibitory protein [Daher et al., 2009; Vaughn et al., 2015]. TRBP inhibits PKR by directly interacting with PKR as well as by sequestering dsRNA and PACT [Daniels and Gatignol, 2012]. In unstressed cells PACT-TRBP heterodimers prevail and PKR remains inactive. In response to stress signals, once PACT is phosphorylated, it dissociates from TRBP to form PACT-PACT homodimers which bind to PKR at higher affinity and activate PKR catalytically [Singh et al., 2011]. Thus, we investigated the possibility that mutant FS protein could interact with TRBP to prevent its association with PACT and thereby releasing PACT to activate PKR. In order to investigate this, we first tested if FS mutant protein interacts with TRBP using protein-protein interaction pull down assay (Fig. 6 A). 35S-methionine labeled TRBP was in vitro translated using rabbit reticulocyte system and mixed with pure recombinant hexahistidine tagged PKR, PACT, or FS mutant proteins bound to Ni-charged sepharose beads to measure the pull down of 35S-labeled TRBP. As seen in Fig. 6 A and B, TRBP interacts efficiently with PKR (lane 3), PACT (lane 4) as well as with FS mutant protein (lane 5). No 35S-labeled TRBP was pulled down by Ni-agarose beads in the absence of any hexahistidine tagged protein (lane 2) indicating absence of any non-specific binding of TRBP to the Ni-agarose beads and presence of a specific interaction with PKR, PACT and FS. These results indicate that TRBP interacts with FS mutant protein similar to its interaction with PKR and PACT in vitro under the conditions of this assay. We reasoned that if FS mutant protein can displace PACT from PACT-TRBP complex, it may be able to activate PKR in cells by releasing PACT from TRBP. In order to test this, we explored if FS mutant protein can dissociate PACT-TRBP interaction. 35S-methionine labeled TRBP was in vitro translated using rabbit reticulocyte system and mixed with pure recombinant hexahistidine tagged PACT protein bound to anti-PACT monoclonal antibody attached to protein A-sepharose beads in the absence of any FS mutant protein. Increasing amounts of pure recombinant FS mutant protein was then added (lanes 4-7) to measure the pull down of 35S-labeled TRBP with PACT bound to beads. As seen in Fig. 6 C and D, the amount of TRBP pulled down with PACT was inversely correlated to the amount of FS mutant protein added (lanes 4-7 compared to lane 3). In order to compare the relative strengths of TRBP-PACT and TRBP-FS interactions, we used the yeast two hybrid protein-protein interaction assay. As seen in Fig. 6 E, TRBP-FS interaction was stronger than the TRBP-PACT interaction (compare TRBP-PACT panel with TRBP-FS panel in the TRBP/pGBKT7 set). When TRBP was expressed as the GAL4-activation domain fusion protein, similar results were seen (compare PACT-TRBP panel with FS-TRBP panel in the TRBP/pGADT7 set). These results indicate that FS mutant protein can bind to TRBP to release PACT, which than could bring about PKR activation leading to caspase activation and apoptosis. The FS protein also may bind to PACT to prevent PACT-TRBP association and the FS-PACT heteromeric interactions could bring about PKR activation. Figure 7 depicts these possibilities in a schematic model.
Figure 7. A schematic model of PKR activation in cells expressing FS mutant.
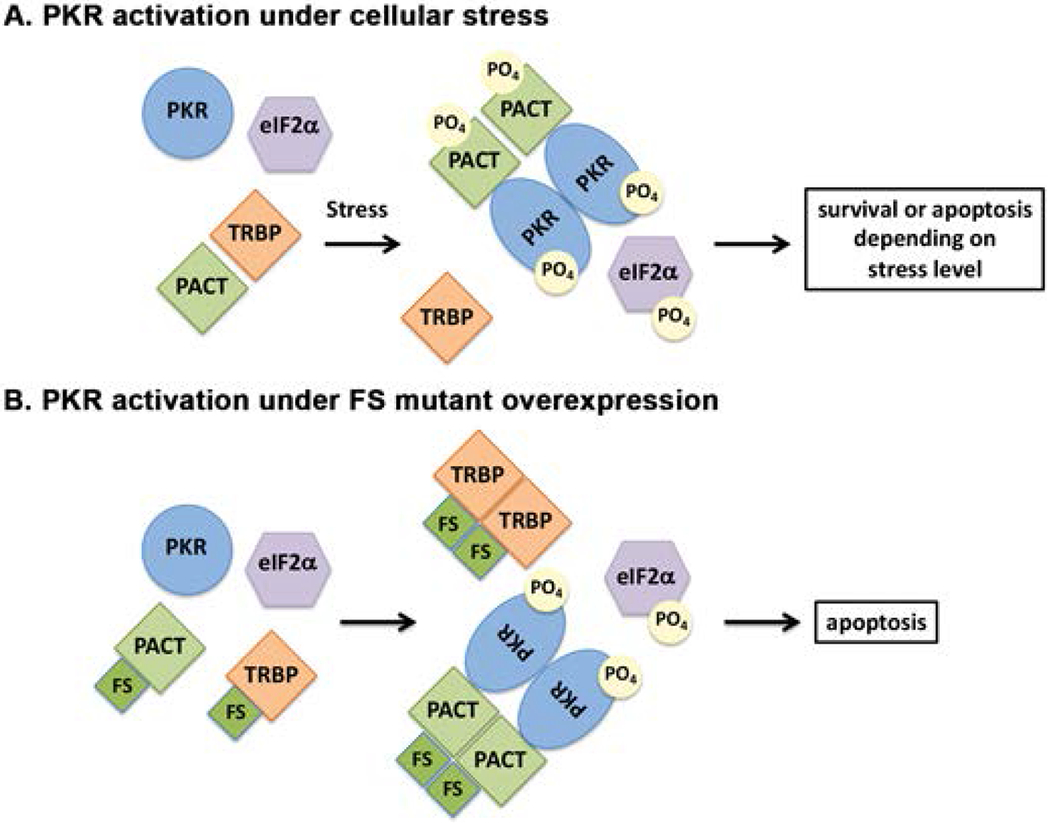
(A) wt cells. As previously established, in the absence of stress, PACT heterodimerizes with TRBP, PKR is catalytically inactive and eIF2α is not phosphorylated. In response to a stress signal, PACT dissociates from TRBP due to its phosphorylation, forms homodimers that bind to PKR with high affinity, activate its kinase activity leading to eIF2α phosphorylation. (B) Cells expressing FS mutant protein. In the absence of stress, FS mutant protein heterodimerizes with TRBP with high affinity (and possibly also with PACT) to displace PACT from TRBP-PACT heterodimers. Consequently, PACT homodimers form due to high affinity interactions between FS mutant protein molecules. Such homodimers bind to PKR with high affinity and activate its kinase activity leading to eIF2α phosphorylation leading to apoptosis via caspase activation.
Discussion
PKR activation in response to cellular stress is regulated by PACT and is involved in modulating cellular survival [Bennett et al., 2006; Ito et al., 1999; Patel et al., 2000; Singh et al., 2009; Vaughn et al., 2015]. Although many mutations in PACT have now been identified in a worldwide occurrence of DYT16 cases, the pathomechanisms involved in DYT16 remain poorly understood. Our previous work on the most prevalent P222L mutation in DYT16 has revealed that this mutation leads to a dysregulation of eIF2α phosphorylation in response to cellular stress [Vaughn et al., 2015]. Cells homozygous for P222L mutation exhibited enhanced sensitivity to ER stressor tunicamycin due to a delayed but more robust eIF2α phosphorylation. In this study, we aimed to examine the effect of a dominant PACT mutation reported in a single DYT16 patient. We started by characterizing the biochemical properties of the FS mutant protein in vitro. The FS mutant protein exhibited no dsRNA-binding ability as it is truncated at residue 88 and lacks the carboxy-terminal part of M1, the first conserved motif, and also the amino acid residues critical for interaction with dsRNA [Chukwurah et al., 2018; Patel and Sen, 1992; Patel and Sen, 1998]. We have previously characterized the contribution of individual amino acids in this motif to dsRNA-binding and protein-protein interaction and the results obtained with FS mutant are in accordance with our previous work [Chukwurah et al., 2018]. The mutant FS protein also showed no interaction with PKR, although it showed interaction with PACT. Based on our previous work on PACT-PKR and PACT-PACT interactions, it is expected that FS mutant protein may not interact with PKR or PACT as it lacks the alanine residues at positions 91 and 92 [Chukwurah et al., 2018; Singh and Patel, 2012]. Thus, it was surprising that the FS mutant protein interacted with PACT in vitro and the FS protein aggregates showed co-localization of PKR and PACT in HeLa cells. It is worth noting here is that the pure recombinant hexahistidine tagged FS protein showed aggregate forms on an SDS-PAGE gel indicating that it forms aggregates and such aggregated forms are present in the FS mutant we used to bind to Ni-agarose beads in our pull-down assays. This aggregation may possibly contribute to interaction with PACT in this assay and similarly also in mammalian cells.
In order to study how FS protein affects the PACT-PKR pathway during cellular stress, we wanted to investigate the effect of FS mutant protein expression in mammalian cells. As the mutation is reported to be dominant, we wanted to establish a cell line with a stable expression of the mutant protein in order to analyze its effects on the PKR pathway. Despite multiple attempts with several different smaller epitope tags (Flag, Myc, HA) on the FS mutant protein, we were unable to detect its expression at early or late time points post-transfection using western blot analyses. Thus, we reasoned that an overexpression of the FS mutant protein may be toxic to cells or the mRNA may be undergoing NMD [Karousis and Muhlemann, 2018]. We next studied its expression by tagging the FS protein with a mCherry tag as the expression could be detected in live cells using fluorescence microscopy. We could detect mCherry-FS protein expression as early as 6 h after transfection and the cells expressing mCherry-FS protein showed increasing cell death at later time points. Most strikingly, the mCherry-FS protein showed aggregate formation in cells. The high molecular weight forms of aggregated FS protein observed by us on SDS-PAGE are similar to aggregate forms of other proteins such as mutant Cu/Zn superoxide dismutase that are aggregate prone and cause ALS [Wang et al., 2014]. The misfolded α-synuclein involved in the pathogenesis of Lewy body diseases isolated from the brains of dementia patients also shows similar aggregates that are not disaggregated in presence of SDS [Sano et al., 2018]. Since PKR shows no interaction with FS mutant protein but FS mutant protein can interact strongly with TRBP to dissociate PACT from the PACT-TRBP heterodimers, we propose that PKR is activated by PACT released from TRBP when FS mutant protein is present. A schematic model depicting this is presented in Fig. 7, which outlines PKR- and PACT-dependent processes leading to apoptosis. In the absence of FS mutant protein when PACT mediated PKR activation takes place in response to stress (Fig. 7 A), PACT is phosphorylated and forms homodimers efficiently after its dissociation from TRBP. When overexpressed, the FS mutant protein can cause dissociation of PACT and the “free” PACT released from TRBP-PACT heterodimers could bring about PKR activation (Fig. 7 B). Any possible phosphorylation of PACT due to overexpression of FS mutant protein remains to be explored in future. Under such conditions, PKR activation leads to caspase activation and apoptosis as we observed in cells overexpressing mCherry-FS protein. Our results demonstrated that FS mutant protein expression led to caspase activation in murine fibroblasts which was partly dependent on the presence of PACT and PKR. However the FS protein aggregates also induced caspase activation in a PKR and PACT independent manner as both PKR as well as PACT null cells showed a slower kinetics of caspase induction. These results are indicative that FS mutant protein aggregates induce apoptosis in both a PACT-PKR-dependent and PACT-PKR-independent manner.
Activation of PKR by protein aggregates observed with the FS mutant could be indicative of a more general process rather than a specific process driven by the FS mutation. In this regard, it could be worth investigating if known aggregation prone proteins involved in neurodegenerative diseases trigger PACT dependent PKR activation. A significant amount of evidence exists to indicate a possible connection between PACT and PKR recruitment to such aggregates. A number of studies have implicated PKR in the pathogenesis of neurodegenerative diseases, such as Alzheimer’s disease (AD) [Hugon et al., 2017], Parkinson’s disease (PD) [Bando et al., 2005; Reimer et al., 2018], Huntington’s disease (HD) [Bando et al., 2005], and amyotrophic lateral sclerosis (ALS) [Hu et al., 2003]. Of these, PKR activation has been most investigated in Alzheimer’s disease. The earliest reports of PKR activation in AD demonstrated an accumulation of activated (phosphorylated) PKR in degenerating neurons [Chang et al., 2002] mainly using histological methods. A direct implication of PKR activation in the pathology of the disease is indicated based on the recent evidence and inhibition of PKR prevents the neuronal apoptosis in AD mouse models as well as in neuronal cultures [Carret-Rebillat et al., 2015; Couturier et al., 2012]. Activated PKR aggregates are reported in hippocampal neurons in PD and HD patient brains [Bando et al., 2005]. More relevantly, there is evidence for PACT being present in potential aggregates in addition to presence of activated PKR in the degenerating neurons. Immunohistochemical studies with the hippocampal neurons showed a co-localization of PACT with phosphorylated PKR in the post-mortem brains of AD patients [Paquet et al., 2012]. A mass spectrometry analysis showed an increased association of PACT with the aggregation prone mutant HTT protein in the affinity-purified complexes from huntingtin mutant (HTT) juvenile mouse brain [Culver et al., 2012]. Elevated PKR expression or activation is also observed in ALS although co-localization of PACT and PKR has not been investigated so far for ALS [Hu et al., 2003]. There is no clear evidence for neurodegeneration in monogenic inherited forms dystonia. Although neurodegeneration has been investigated and found to be absent in DYT6 [Paudel et al., 2016], evidence of neurodegeneration has been reported in X-linked dystonia-parkinsonism [Bruggemann et al., 2017] and in DYT16 [Lemmon et al., 2013].
It also remains an open question if dysregulation of eIF2α pathway observed in a few primary dystonias is the primary cause of the pathology or a secondary effect related to activation of stress signaling pathways. Our research identified the dysregulation of PKR-eIF2α signaling pathway as a consequence of P222L mutation in DYT16 [Vaughn et al., 2015]. Subsequently DYT1, DYT6 as well as the sporadic cervical dystonia also were reported to show an impairment of the eIF2α signaling pathway [Beauvais et al., 2016; Beauvais et al., 2018; Rittiner et al., 2016; Zakirova et al., 2018]. Although we could not analyze eIF2α dysregulation with expression of FS mutant protein in the transient expression system, it is worth a mention that a spontaneously arisen, recessive insertion mutation in Prkra identified at the Jackson Laboratory (JAX) results in a progressive dystonia [Palmer et al., 2015], kinked tails, and mortality in mice. Some neurons in the dorsal root ganglia and the trigeminal ganglion were apoptotic in the homozygous mutant mice, consistent with the observed neurodegenerative phenotype. The described mutation could result in the production of a truncated PACT/RAX protein with seven extraneous amino acids after first 178 AA before a premature stop codon. It remains to be determined if such a truncated protein is present in these mice and may cause the observed dystonia phenotype. The Prkra null mice show no dystonia symptoms [Rowe and Sen, 2001], thereby raising the possibility that the dystonia phenotype may be directly correlated to presence of mutant PACT proteins. On the contrary, it is also possible that the mutant truncated PACT protein is not produced in patient cells or in mice due to NMD of the mRNA due to the presence of an early termination codon. As no patient cells are available for the reported FS mutation, this spontaneously created mouse model can be utilized to detect if the truncated protein is present in brain or any other cell types and may help understand the pathomechanisms involved in DYT16.
In summary, our results presented here report the biochemical properties of a truncated PACT protein resulting from a frameshift mutation in a DYT16 patient. Interestingly, these results indicate that although the FS mutant protein shows no direct interaction with PKR, the aggregates of the FS mutant protein formed in mammalian cells result in PKR activation to cause caspase activation leading to apoptosis. In the context of neurodegenerative diseases caused by protein aggregates in neurons, these findings beg for more in depth investigations for the possible role of PKR activation in triggering neuronal loss.
Acknowledgements
We would like to thank Victoria Willingham for technical help with caspase assays. This work was supported in part by a Dystonia Medical Research Foundation grant to R. P. and a Magellan Undergraduate Research Grant to V. W. The authors declare that they have no conflicts of interest with the contents of this article.
References:
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, Tohyama M. 2005. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochem Int 46:11–18 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Beauvais G, Bode NM, Watson JL, Wen H, Glenn KA, Kawano H, Harata NC, Ehrlich ME, Gonzalez-Alegre P. 2016. Disruption of Protein Processing in the Endoplasmic Reticulum of DYT1 Knock-in Mice Implicates Novel Pathways in Dystonia Pathogenesis. J Neurosci 36:10245–10256 10.1523/jneurosci.0669-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais G, Rodriguez-Losada N, Ying L, Zakirova Z, Watson JL, Readhead B, Gadue P, French DL, Ehrlich ME, Gonzalez-Alegre P. 2018. Exploring the Interaction Between eIF2alpha Dysregulation, Acute Endoplasmic Reticulum Stress and DYT1 Dystonia in the Mammalian Brain. Neuroscience 371:455–468 10.1016/j.neuroscience.2017.12.033. [DOI] [PubMed] [Google Scholar]
- Bennett RL, Blalock WL, Abtahi DM, Pan Y, Moyer SA, May WS. 2006. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood 108:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg DC, Armata IA, Nery FC, Breakefield XO, Sharma N. 2011. Molecular pathways in dystonia. Neurobiol Dis 42:136–147 10.1016/j.nbd.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brashear A 2013. Commentary. Mov Disord 28:1939 10.1002/mds.25774. [DOI] [PubMed] [Google Scholar]
- Bruggemann N, Rosales RL, Waugh JL, Blood AJ, Domingo A, Heldmann M, Jamora RD, Munchau A, Munte TF, Lee LV, Buchmann I, Klein C. 2017. Striatal dysfunction in X-linked dystonia-parkinsonism is associated with disease progression. Eur J Neurol 24:680–686 10.1111/ene.13256. [DOI] [PubMed] [Google Scholar]
- Camargos S, Scholz S, Simon-Sanchez J, Paisan-Ruiz C, Lewis P, Hernandez D, Ding J, Gibbs JR, Cookson MR, Bras J, Guerreiro R, Oliveira CR, Lees A, Hardy J, Cardoso F, Singleton AB. 2008. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol 7:207–215 S1474-4422(08)70022-X [pii] 10.1016/S1474-4422(08)70022-X [doi]. [DOI] [PubMed] [Google Scholar]
- Carret-Rebillat AS, Pace C, Gourmaud S, Ravasi L, Montagne-Stora S, Longueville S, Tible M, Sudol E, Chang RC, Paquet C, Mouton-Liger F, Hugon J. 2015. Neuroinflammation and Abeta accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci Rep 5:8489 10.1038/srep08489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KY, Ramos A. 2005. The double-stranded RNA-binding motif, a versatile macromolecular docking platform. Febs J 272:2109–2117 [DOI] [PubMed] [Google Scholar]
- Chang RC, Wong AK, Ng HK, Hugon J. 2002. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13:2429–2432 [DOI] [PubMed] [Google Scholar]
- Chukwurah E, Patel RC. 2018. Stress-induced TRBP phosphorylation enhances its interaction with PKR to regulate cellular survival. Sci Rep 8:1020 10.1038/s41598-018-19360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukwurah E, Willingham V, Singh M, Castillo-Azofeifa D, Patel RC. 2018. Contribution of the two dsRBM motifs to the double-stranded RNA binding and protein interactions of PACT. J Cell Biochem 119:3598–3607 10.1002/jcb.26561. [DOI] [PubMed] [Google Scholar]
- Couturier J, Paccalin M, Lafay-Chebassier C, Chalon S, Ingrand I, Pinguet J, Pontcharraud R, Guillard O, Fauconneau B, Page G. 2012. Pharmacological inhibition of PKR in APPswePS1dE9 mice transiently prevents inflammation at 12 months of age but increases Abeta42 levels in the late stages of the Alzheimer’s disease. Curr Alzheimer Res 9:344–360 [DOI] [PubMed] [Google Scholar]
- Culver BP, Savas JN, Park SK, Choi JH, Zheng S, Zeitlin SO, Yates JR 3rd, Tanese N. 2012. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem 287:21599–21614 10.1074/jbc.M112.359307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabo S, Meurs EF. 2012. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4:2598–2635 10.3390/v4112598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher A, Laraki G, Singh M, Melendez-Pena CE, Bannwarth S, Peters AH, Meurs EF, Braun RE, Patel RC, Gatignol A. 2009. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Molecular and Cellular Biology 29:254–265 10.1128/MCB.01030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels SM, Gatignol A. 2012. The multiple functions of TRBP, at the hub of cell responses to viruses, stress, and cancer. Microbiol Mol Biol Rev 76:652–666 10.1128/mmbr.00012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Aguiar P, Borges V, Ferraz HB, Ozelius LJ. 2015. Novel compound heterozygous mutations in PRKRA cause pure dystonia. Mov Disord 30:877–878 10.1002/mds.26175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb A, Haque SJ, Mogensen T, Silverman RH, Williams BR. 2001. RNA-dependent protein kinase PKR is required for activation of NF-kappa B by IFN-gamma in a STAT1-independent pathway. J Immunol 166:6170–6180. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer HL, Bressman SB. 2006. The diagnosis of dystonia. Lancet Neurol 5:780–790 10.1016/s1474-4422(06)70547-6. [DOI] [PubMed] [Google Scholar]
- Gil J, Esteban M. 2000. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis 5:107–114. [DOI] [PubMed] [Google Scholar]
- Green SR, Mathews MB. 1992. Two RNA-binding motifs in the double-stranded RNA-activated protein kinase, DAI. Genes Dev 6:2478–2490 [DOI] [PubMed] [Google Scholar]
- Hu JH, Zhang H, Wagey R, Krieger C, Pelech SL. 2003. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem 85:432–442 [DOI] [PubMed] [Google Scholar]
- Huang X, Hutchins B, Patel RC. 2002. The C-terminal, third conserved motif of the protein activator PACT plays an essential role in the activation of double-stranded-RNA-dependent protein kinase (PKR). Biochem J 366:175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugon J, Mouton-Liger F, Dumurgier J, Paquet C. 2017. PKR involvement in Alzheimer’s disease. Alzheimers Res Ther 9:83 10.1186/s13195-017-0308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Yang M, May WS. 1999. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem 274:15427–15432. [DOI] [PubMed] [Google Scholar]
- Karousis ED, Muhlemann O. 2018. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a032862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon ME, Lavenstein B, Applegate CD, Hamosh A, Tekes A, Singer HS. 2013. A novel presentation of DYT 16: acute onset in infancy and association with MRI abnormalities. Mov Disord 28:1937–1938 10.1002/mds.25703. [DOI] [PubMed] [Google Scholar]
- Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. 1990. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62:379–390 [DOI] [PubMed] [Google Scholar]
- Nanduri S, Rahman F, Williams BR, Qin J. 2000. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. Embo J 19:5567–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer K, Fairfield H, Borgeia S, Curtain M, Hassan MG, Dionne L, Yong Karst S, Coombs H, Bronson RT, Reinholdt LG, Bergstrom DE, Donahue LR, Cox TC, Murray SA. 2015. Discovery and characterization of spontaneous mouse models of craniofacial dysmorphology. Dev Biol 10.1016/j.ydbio.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet C, Mouton-Liger F, Meurs EF, Mazot P, Bouras C, Pradier L, Gray F, Hugon J. 2012. The PKR activator PACT is induced by Abeta: involvement in Alzheimer’s disease. Brain Pathol 22:219–229 10.1111/j.1750-3639.2011.00520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CV, Handy I, Goldsmith T, Patel RC. 2000. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem 275:37993–37998. [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC. 1992. Identification of the double-stranded RNA-binding domain of the human interferon-inducible protein kinase. J Biol Chem 267:7671–7676 [PubMed] [Google Scholar]
- Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. Embo J 17:4379–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel R, Li A, Hardy J, Bhatia KP, Houlden H, Holton J. 2016. DYT6 Dystonia: A Neuropathological Study. Neurodegener Dis 16:273–278 10.1159/000440863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Hartmann R, Qin J, Sen GC. 2001. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol 21:1908–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri M, Olgiati S, Sensi M, Gualandi F, Groppo E, Rispoli V, Graafland J, Breedveld GJ, Fabbrini G, Berardelli A, Bonifati V. 2016. PRKRA Mutation Causing Early-Onset Generalized Dystonia-Parkinsonism (DYT16) in an Italian Family. Mov Disord 31:765–767 10.1002/mds.26583. [DOI] [PubMed] [Google Scholar]
- Reimer L, Vesterager LB, Betzer C, Zheng J, Nielsen LD, Kofoed RH, Lassen LB, Bolcho U, Paludan SR, Fog K, Jensen PH. 2018. Inflammation kinase PKR phosphorylates alpha-synuclein and causes alpha-synuclein-dependent cell death. Neurobiol Dis 115:17–28 10.1016/j.nbd.2018.03.001. [DOI] [PubMed] [Google Scholar]
- Rittiner JE, Caffall ZF, Hernandez-Martinez R, Sanderson SM, Pearson JL, Tsukayama KK, Liu AY, Xiao C, Tracy S, Shipman MK, Hickey P, Johnson J, Scott B, Stacy M, Saunders-Pullman R, Bressman S, Simonyan K, Sharma N, Ozelius LJ, Cirulli ET, Calakos N. 2016. Functional Genomic Analyses of Mendelian and Sporadic Disease Identify Impaired eIF2alpha Signaling as a Generalizable Mechanism for Dystonia. Neuron 92:1238–1251 10.1016/j.neuron.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe TM, Rizzi M, Hirose K, Peters GA, Sen GC. 2006. A role of the double-stranded RNA-binding protein PACT in mouse ear development and hearing. Proc Natl Acad Sci U S A 103:5823–5828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe TM, Sen GC. 2001. Organizations and promoter analyses of the human and the mouse genes for PACT, the protein-activator of the interferon-induced protein kinase, PKR. Gene 273:215–225. [DOI] [PubMed] [Google Scholar]
- Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y, Yoshida M, Murayama S, Mishima K, Nishida N. 2018. Prion-Like Seeding of Misfolded alpha-Synuclein in the Brains of Dementia with Lewy Body Patients in RT-QUIC. Mol Neurobiol 55:3916–3930 10.1007/s12035-017-0624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibler P, Djarmati A, Langpap B, Hagenah J, Schmidt A, Bruggemann N, Siebner H, Jabusch HC, Altenmuller E, Munchau A, Lohmann K, Klein C. 2008. A heterozygous frameshift mutation in PRKRA (DYT16) associated with generalised dystonia in a German patient. Lancet Neurol 7:380–381 S1474-4422(08)70075-9 [pii] 10.1016/S1474-4422(08)70075-9 [doi]. [DOI] [PubMed] [Google Scholar]
- Singh M, Castillo D, Patel CV, Patel RC. 2011. Stress-induced phosphorylation of PACT reduces its interaction with TRBP and leads to PKR activation. Biochemistry 50:4550–4560 10.1021/bi200104h. [DOI] [PubMed] [Google Scholar]
- Singh M, Fowlkes V, Handy I, Patel CV, Patel RC. 2009. Essential role of PACT-mediated PKR activation in tunicamycin-induced apoptosis. Journal of Molecular Biology 385:457–468 10.1016/j.jmb.2008.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Patel RC. 2012. Increased interaction between PACT molecules in response to stress signals is required for PKR activation. J Cell Biochem 113:2754–2764 10.1002/jcb.24152. [DOI] [PubMed] [Google Scholar]
- Vaughn LS, Bragg DC, Sharma N, Camargos S, Cardoso F, Patel RC. 2015. Altered Activation of Protein Kinase PKR and Enhanced Apoptosis in Dystonia Cells Carrying a Mutation in PKR Activator Protein PACT. J Biol Chem 290:22543–22557 10.1074/jbc.M115.669408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Popko B, Roos RP. 2014. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum Mol Genet 23:2629–2638 10.1093/hmg/ddt658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakirova Z, Fanutza T, Bonet J, Readhead B, Zhang W, Yi Z, Beauvais G, Zwaka TP, Ozelius LJ, Blitzer RD, Gonzalez-Alegre P, Ehrlich ME. 2018. Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet 14:e1007169 10.1371/journal.pgen.1007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech M, Castrop F, Schormair B, Jochim A, Wieland T, Gross N, Lichtner P, Peters A, Gieger C, Meitinger T, Strom TM, Oexle K, Haslinger B, Winkelmann J. 2014. DYT16 revisited: exome sequencing identifies PRKRA mutations in a European dystonia family. Mov Disord 29:1504–1510 10.1002/mds.25981. [DOI] [PubMed] [Google Scholar]