

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by the loss of motor neurons in the brain and spinal cord. ALS neuropathology is associated with increased oxidative stress, excitotoxicity, and inflammation. We and others reported that the anti-aging and cognition-enhancing protein Klotho is a neuroprotective, antioxidative, anti-inflammatory, and promyelinating protein. In mice, its absence leads to an extremely shortened life span and to multiple phenotypes resembling human aging, including motor and hippocampal neurodegeneration and cognitive impairment. In contrast, its overexpression extends life span, enhances cognition, and confers resistance against oxidative stress; it also reduces premature mortality and cognitive and behavioral abnormalities in an animal model for Alzheimer’s disease (AD). These pleiotropic beneficial properties of Klotho suggest that Klotho could be a potent therapeutic target for preventing neurodegeneration in ALS. Klotho overexpression in the SOD1 mouse model of ALS resulted in delayed onset and progression of the disease and extended survival that was more prominent in females than in males. Klotho reduced the expression of neuroinflammatory markers and prevented neuronal loss with the more profound effect in the spinal cord than in the motor cortex. The effect of Klotho was accompanied by reduced expression of proinflammatory cytokines and enhanced the expression of antioxidative and promyelinating factors in the motor cortex and spinal cord of Klotho × SOD1 compared to SOD1 mice. Our study provides evidence that increased levels of Klotho alleviate ALS-associated pathology in the SOD1 mouse model and may serve as a basis for developing Klotho-based therapeutic strategies for ALS.
Keywords: Amyotrophic lateral sclerosis, Neurodegeneration, Microglia, Neuroinflammation, Motor neurons, Therapeutics
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by the loss of upper and lower motor neurons, leading to progressive muscle atrophy and paralysis, which is fatal within 3–5 years of diagnosis (Paganoni et al. 2014). About 10% of ALS patients exhibit a familial form of ALS (FALS). Mutations in several genes, such as chromosome 9 open reading frame 72 (C9ORF72), Cu, Zn-superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), and fused in sarcoma (FUS), are strongly associated with the classical clinical phenotype of ALS (Sreedharan 2010; Tosolini and Sleigh 2017). The two most prevalent causes of genetic ALS are hexanucleotide repeat expansion in the C9ORF72 gene and mutations in SOD1 (DeJesus-Hernandez et al. 2011; Renton et al. 2011).
Neuroinflammation is one major contributor to motor neuron damage in ALS (Caldeira et al. 2014; Lee et al. 2016). Reactive astrocytes and microglia can trigger neuroinflammation and accelerate disease progression (Henkel et al. 2004), which is further exacerbated by ongoing neuronal injury. Inflammatory cytokines released by astrocytes and microglia may facilitate glutamate excitotoxicity, thereby linking neuroinflammation and excitotoxic death (Caldeira et al. 2014). The metabolism of nitric oxide (NO) provides an additional link between microglial activation and oxidative injury to motor neurons. Mutant SOD1 has the capacity to increase expression of inducible nitric oxide synthase (iNOS) and subsequent production of NO (Almer et al. 1999). iNOS protein expression in SOD1 mice has been reported in microglia cells, especially in the ventral horn (Almer et al. 1999), and the production of NO may play an important role in the switch to a more neurotoxic microglial phenotype and subsequent death to motor neurons (Lewis et al. 2014; Liao et al. 2012). The mutant SOD1 has been demonstrated to interact with the endoplasmic reticulum (ER) and mitochondria, and it has been proposed that mutant SOD1 results in protein misfolding and aggregation and acts primarily via a toxic gain of function in ALS (reviewed in Parakh and Atkin 2016; Pokrishevsky et al. 2017). There is an urgent unmet medical need for effective treatments for this devastating and fatal disease.
A mouse model of ALS based on the overexpression of mutant SOD1G93A (referred by us as SOD1 in this paper) is the most studied animal model of ALS. SOD1 mice exhibit predictably staged, age-dependent degeneration of motor neurons accompanied by extensive damage to nerve fibers and spinal cord tissue, that is detected on the cellular and biochemical levels. These mice exhibit several pathogenic mechanisms that are proposed to be central to ALS pathology, such as protein aggregation of the mutant SOD1, oxidative stress, and neuroinflammation. Biochemical markers of oxidative stress are elevated in tissue specimens from ALS patients, and similarly, SOD1 mice show increased levels of protein and lipid oxidation and nitrosylation (Gurney et al. 1994). Recently, a growing body of evidence has shown that non-neuronal cells such as astrocytes, microglia, and oligodendrocytes directly contribute to damage motor neurons and lead to cell death, suggesting a noncell autonomous effect (reviewed in Lee et al. 2016). Microglial activation is apparent in SOD1 mice (Hall et al. 1998) and is characterized by a series of phenotypic changes associated with morphological transformation. These changes accompany the transition from a stable to progressive phase of ALS in mouse models (Lewis et al. 2014). In the current study, we used this mouse model to perform a validation of the anti-aging and cognition-enhancing protein Klotho as a potential therapeutic target for ALS.
The life-extending protein Klotho, named after the mythical Greek goddess who “spins the thread of life” (Kuro-o et al. 1997), has been shown to counteract oxidative stress and neuroinflammation when it is overexpressed (Kurosu et al. 2005; Zhou et al. 2017). Klotho is predominantly expressed in the kidney, but also in the brain by the choroid plexus and hippocampal neurons (German et al. 2012). Deficiency of the Klotho gene accelerates aging and shortens life span in hypomorphic mice (Kuro-o et al. 1997; Wang 2006), while overexpression of Klotho slows aging and extends life span in mice by up to 30% (Kurosu et al. 2005). Klotho-deficient mice also have impaired cognition and exhibit degeneration of hippocampal pyramidal cells, Purkinje cells, and motor cortex neurons (Anamizu et al. 2005; Shiozaki et al. 2008), as well as lipid and DNA oxidation in the hippocampus (Nagai et al. 2003). For a review on the functions of Klotho in the CNS, see Abraham et al. (2016).
Research from our lab and others provides evidence that Klotho overexpression is neuroprotective against oxidative injury in hippocampal neurons (Cheng et al. 2015; Zeldich et al. 2014), cerebellar granule cells, and cortical (Xin et al. 2015) and dopaminergic neurons (Baluchnejadmojarad et al. 2017; Brobey et al. 2015). We and others reported that Klotho protects hippocampal and dopaminergic neurons by upregulating peroxiredoxin and thioredoxin reductase (Prx/Trx), members of the redox system (Zeldich et al. 2014), and nuclear factor erythroid 2-related factor 2 (Nrf2) (Brobey et al. 2015), respectively, thereby shielding these cells from oxidative stress, thus rendering them resilient against various insults.
In addition, Klotho possesses anti-inflammatory properties which are needed in all neurodegenerative diseases. Klotho was shown to suppress the production of proinflammatory cytokines (TNF-α, interleukin-6 (IL-6), IL-8, IL-12 and IL-1β) and attenuate nuclear factor-κB (NF-κB) activation in in vitro and in vivo models of cardiac inflammation (Guo et al. 2018; Hui et al. 2017) and chronic kidney disease (Jin et al. 2017), as well as in primary cystic fibrosis bronchial epithelium cultures (Krick et al. 2017). Several studies attempted to uncover the molecular features of the mutant SOD1 responsible for initiating ALS (Lee et al. 2015) and the reasons why it specifically accumulates within motor neurons (Shvil et al. 2018); however, these processes have not been fully understood.
Klotho mitigates oxidative stress (Zhou et al. 2018) and inflammation (Guo et al. 2018; Zhu et al. 2018) and decreases neuronal injury (Zeldich et al. 2014). These pleiotropic properties of Klotho prompted us to hypothesize and then test whether Klotho plays a role in lessening ALS symptoms and progression by mitigating the key pathophysiological processes associated with the disease, such as neuroinflammation, oxidative stress, and subsequent neurodegeneration of motor neurons and demyelination.
We examined whether Klotho overexpression can alleviate neurological deficits and extend the life span in the SOD1 mouse model of ALS, and explored the cellular and molecular changes and potential mechanisms underlying Klotho’s beneficial effects. Our data provide strong evidence that enhancing Klotho expression in an ALS mouse model is neuroprotective. These results strengthen and add new support to our previous reports demonstrating the neuroprotective roles Klotho plays generally in the brain and in AD (Dubal et al. 2015) and MS (Zeldich et al. 2015) mouse models and paves the way to explore new therapeutics for neurodegenerative diseases in the form of Klotho enhancing small molecules (Abraham et al. 2012; King et al. 2012) or Klotho gene therapy (Masso et al. 2018).
Results
Klotho Attenuates Neurological Deficits and Prolongs Survival of SOD1 Transgenic Mice
Our aim was to investigate the therapeutic potential of Klotho overexpression on neurological deficits and life span in a mouse model of ALS. Four groups of mice were balanced for age and gender and litters were included in our studies (see Table 1).
Table 1.
Mouse groups and numbers used for the experiments
| Mouse groups | Motor neuron deficits/survival | Biochemical and histochemical assays | ||
|---|---|---|---|---|
| 110 days | 135 days | |||
| Nontransgenic wild-type littermates (WT) | Females | 9 | 5 | 0 |
| Males | 8 | 3 | 0 | |
| Klotho-overexpressing transgenic (KL-OE) | Females | 10 | 3 | 0 |
| Males | 11 | 3 | 0 | |
| SOD1-overexpressing transgenic (SOD) | Females | 9 | 4 | 5 |
| Males | 9 | 4 | 7 | |
| Klotho/SOD1-overexpressing transgenic (KL-OE/SOD) | Females | 10 | 4 | 4 |
| Males | 8 | 4 | 4 | |
Neurologic Scores
We implemented the neurological score (NS) system developed by Hatzipetros et al. (2015) and found that it allowed us an unbiased assessment and clear differentiation between the scores, especially in light of the fact that each hind limb is scored individually. In line with previously published studies (Gill et al. 2009), we defined the onset of neurological disease as when the mice reached NS2. We used “time to event measures” when we analyzed time to reach each of the NSs and analyzed the data by Kaplan–Meier fit for survival and the log-rank test for the assessment of the median (in days) and statistical significance (Kaneb et al. 2011). Female and male SOD1 mice developed an expected disease phenotype characterized by abnormal splay and tremor of the hind limbs during tail suspension, followed by progressive paralysis, gait changes, increased muscle weakness, and weight loss. KL-OE/SOD1 mice from both genders displayed some delay in the development of the first symptoms (NS1, tremor, abnormal splay), with a more noticeable trend observed in females; however, the results did not reach statistical significance (Fig. 1a–c).
Fig. 1.

Graphical representation of the number of days it took for each mouse from the population of SOD1 and KL-OE/SOD1 cohorts to reach NS1 (a–c), NS2 (d–f), and NS3 (g–i). Data from males (a, d, g) and females (b, e, h) were analyzed separately and when combined (c, f, i). The asterisks indicate statistical significance, *p value < 0.05; **p value < 0.01, as analyzed by Kaplan–Meier fit survival and log-rank test for statistical analysis
The differences between bigenic KL-OE/SOD1 and SOD1 mice became more obvious as the mice aged. Disease onset (NS2) was significantly delayed in bigenic KL-OE/SOD1 mice compared to SOD1 mice by about 6 days (median age in days: 135 in SOD1 vs 141 in KL-OE/SOD1 group). The delay in disease onset was observed in the female cohort (median age in days: 135 in SOD1 vs 147 in KL-OE/SOD1 group) but was not detected in the male cohort (median age in days: 135 in SOD1 vs 136 in KL-OE/SOD1 group) (Fig. 1d–f).
The disparity between the KL-OE/SOD1 and SOD1 mice increased further when the time to reach NS3 was measured. In this case, it took the bigenic mice 11 days longer to reach NS3 compared to SOD1 mice (median age in days: from 154 in SOD1 vs 165 in KL-OE/SOD1 mice). Again, the disparity was greater in the female cohort (median age in days: from 157 in SOD1 to 171 in KL-OE/SOD1) than in the male cohort (median age in days: from 152 in SOD1 to 158.5 in KL-OE/SOD1 group; Fig. 1g–i). The inhibition of disease progression by Klotho overexpression was also reflected when reaching the NS4 stage, as shown in the survival graph (Fig. 4).
Fig. 4.
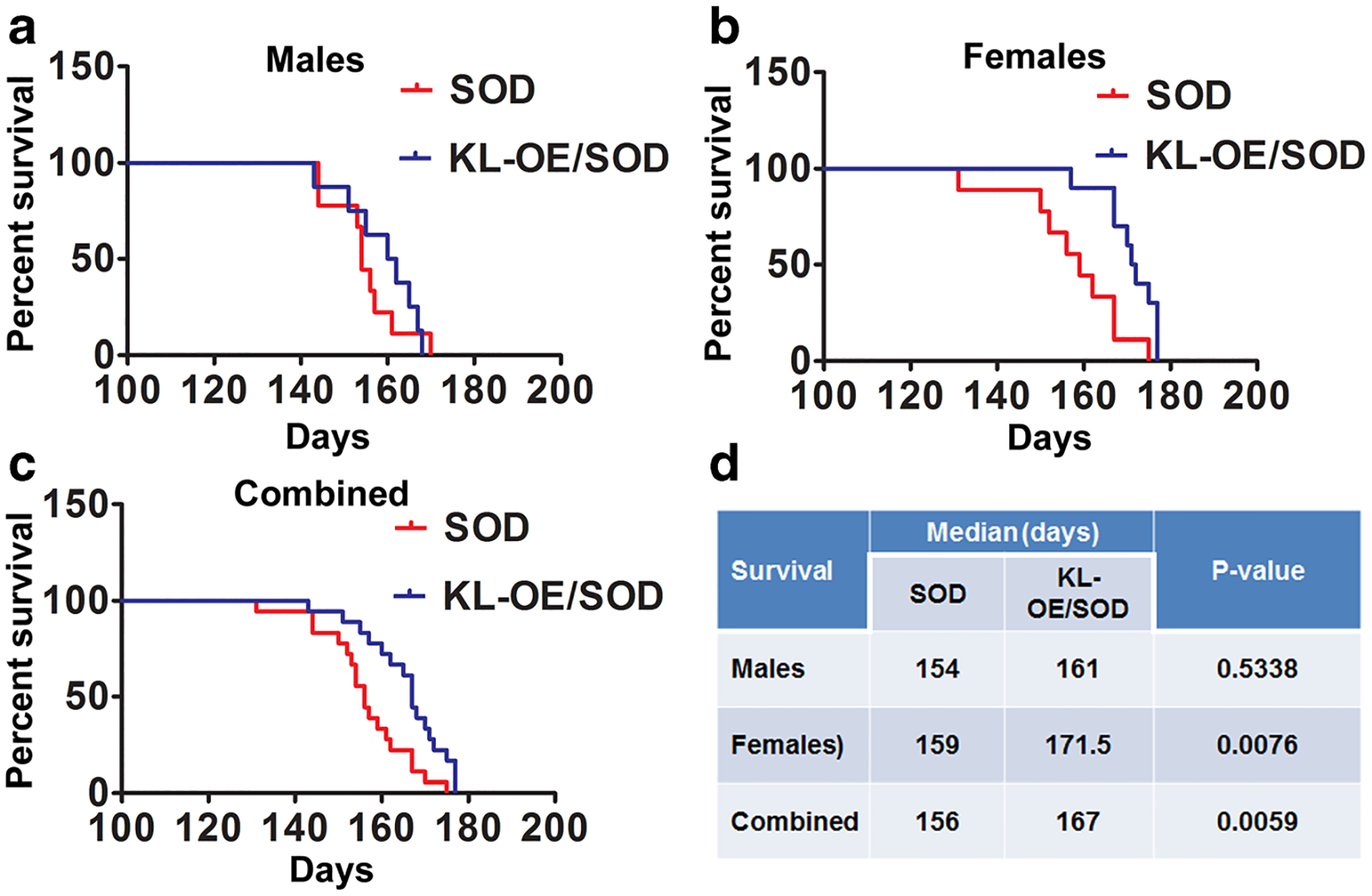
Kaplan–Meier survival plot for time (in days) to reach the humane end point of attaining NS4 representative for survival in males (a), females (b), and when the data from both genders were combined (c). d Results of statistical analysis of log-rank (Mantel–Cox test)
Overall, our results show that Klotho overexpression significantly delayed the disease onset and slowed disease progression, as demonstrated by the lengthening of time needed to reach NS2 and NS3. This effect was more pronounced in the female cohort.
Weight Assessment
Mice carrying the SOD1 mutation are unable to maintain steady body weight from about age day 120 and gradually lose weight from this point until death. Longitudinal weight monitoring provides a good indicator for the onset and progression of the disease and can be used as a measure of the therapeutic effect of various interventions. The changes in mean body weight in all four groups for both genders over time are plotted in Fig. 2a, b from 60 days onwards. At the beginning of the study at age day 60, no significant differences in weight were observed between the four groups in either gender. In females, the average weights at age day 60 were (mean g of the group ± SEM) as follows: WT 19.6 ± 0.35, KL-OE 19.6 ± 0.44, SOD1 19.0 ± 0.25, and KL-OE/SOD1 18.65 ± 0.43, and in the males, WT 24.67 ± 0.83, KL-OE 23.84 ± 0.51, SOD1 24 ± 0.44, and KL-OE/SOD1 23.64 ± 0.49.
Fig. 2.
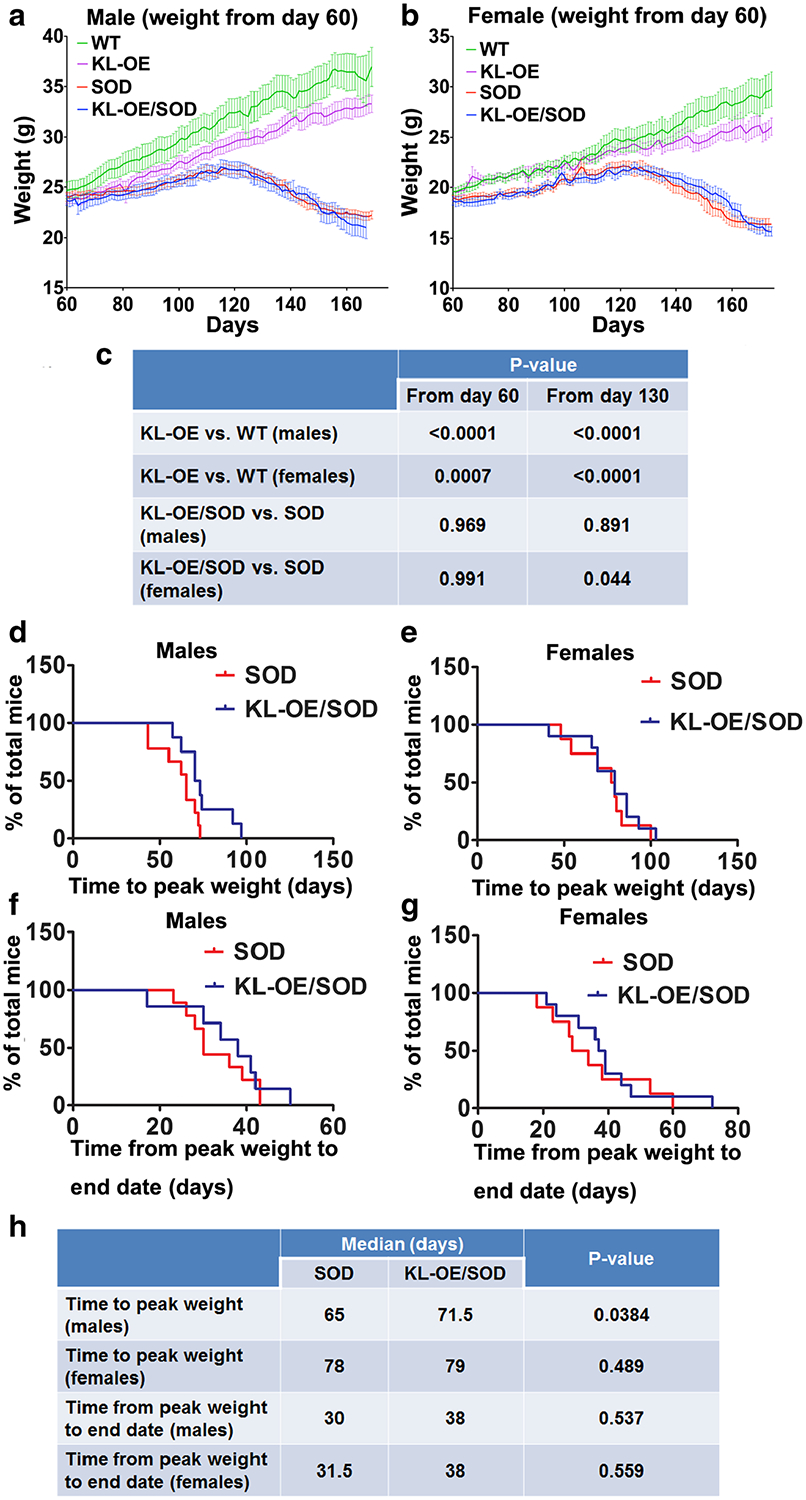
Mean group body weight plotted for male (a) and female (b) mice over time starting from age day 60 (a, b) until the date of last death within each group. Since mice within each individual group died at different ages, the terminal weights for all mice were carried forward for the calculation of the mean values of the group to prevent decomposition of the mean at final stages. c Results of the statistical analysis of one-way ANOVA with post hoc Tukey’s test. d, f Kaplan–Meier survival plot for time (in days) to reach peak weight (as a sign of the onset of the disease) in males (d) and females (e). f, g Kaplan–Meier survival plot for time (in days) from the peak weight to end stage (as a sign of the progression of the disease) in males (f) and females (g). h Results of statistical analysis of log-rank (Mantel–Cox test). The time for the detection of peak body weight for individual mice was assessed after spline smoothing of each mouse’s weight over time using Excel software. In cases where individually smoothed weight curves had numerous equal peaks in weight, the final peak time point was taken for the analysis.
However, as the mice aged, we discovered previously undescribed differences in body weights. Specifically, KL-OE mice of both genders gained weight significantly slower than their WT counterparts. The weight of KL-OE females was increased from 19.6 ± 0.44 at age day 60 to 25.43 ± 0.88 at age day 170, while WT females gained significantly more weight (from 19.6 ± 0.35 at age day 60 to 29.02 ± 1.72 at age day 170 (p value = 0.0007, one-way ANOVA with post hoc Tukey’s test)) (Fig. 2c).
The weight of KL-OE males also increased significantly less than that of WT males: from 23.84 ± 0.51 at age day 60 to 33.28 ± 0.87 at age day 170 in KL-OE and from 24.67 ± 0.83 at age day 60 to 38.09 ± 1.44 at age day 170 in WT males (p value < 0.0001, one-way ANOVA with post hoc Tukey’s test) (Fig. 2c).
Our study demonstrated for the first time that healthy KL-OE mice of either gender weighted significantly less than healthy WT mice starting at age day 60. This difference in weight was not likely caused by a caloric restriction as it was previously determined that food intake in KL-OE is equal to that of WT mice (Kuro-o 2009; Kurosu et al. 2005).
Weight curves for SOD1 and KL-OE/SOD1 male mice showed a very similar pattern with the onset of decline in weight after 120–125 days in each group. This usually occurred just before the mice reached NS2 on both hind limbs. When we compared the weight curves for KL-OE/SOD1 and SOD1 male mice, we did not observe any significant difference at either day 60 or day 130 (Fig. 2a, c). This is not unexpected since initially mice overexpressing Klotho are lighter that the WT mice. Based on these findings, we further investigated the weight parameters and selected to measure time to peak weight as an indicator of the disease onset. KL-OE/SOD1 males displayed significantly extended time to reach the peak weight compared to their SOD1 littermates (Fig. 2d, h) supporting the idea that Klotho delays the disease onset. Increased levels of Klotho also showed a prolonged time from peak weight to end stage (from 30 days in SOD1 mice to 38 days in KL-OE/SOD1 mice); however, the results did not reach significance (Fig. 2f, h).
In contrast, KL-OE/SOD1 females displayed better weight maintenance over time and tended to be heavier as compared to SOD1 females. The weight curve of KL-OE/SOD1 females dropped less drastically and these differences were significant from day 130 (Fig. 2b, c) even though, similar to males, KL-OE females are lighter than WT females. Unlike in males, we did not detect a significant association between Klotho overexpression and time to reach the peak weight in females (Fig. 2e, h). Similar to males, SOD1 females overexpressing Klotho showed a prolonged time from peak weight to end stage (from 31.8 days in SOD1 mice to 38 days in KL-OE/SOD1 mice); however, the results were not significant (Fig. 2g, h).
Hanging Wire Grip Test Assessment
Starting at age day 60, the mice were tested twice weekly for deficits in motor function and strength by the hanging wire grip test (Fig. 3a–c). We have investigated at what time point during the disease progression Klotho overexpression mitigated motor deficits as determined with ANOVA with post hoc Tukey’s test. The grip test revealed statistically significant differences in grip strength, measured by latency to fall, starting at age day 118 between KL-OE/SOD1 and SOD1 males (Fig. 3a, d). In line with our previous observations, a significant effect of Klotho overexpression in SOD1 females was detected already at age day 106 (Fig. 3b, d), earlier than in males, while the effect in both genders was demonstrated to be significant from day 117 (Fig. 3c, d), showing that elevated Klotho levels preserved grip strength (Fig. 3d). KL-OE and WT mice exhibited no decline in the latency to fall.
Fig. 3.
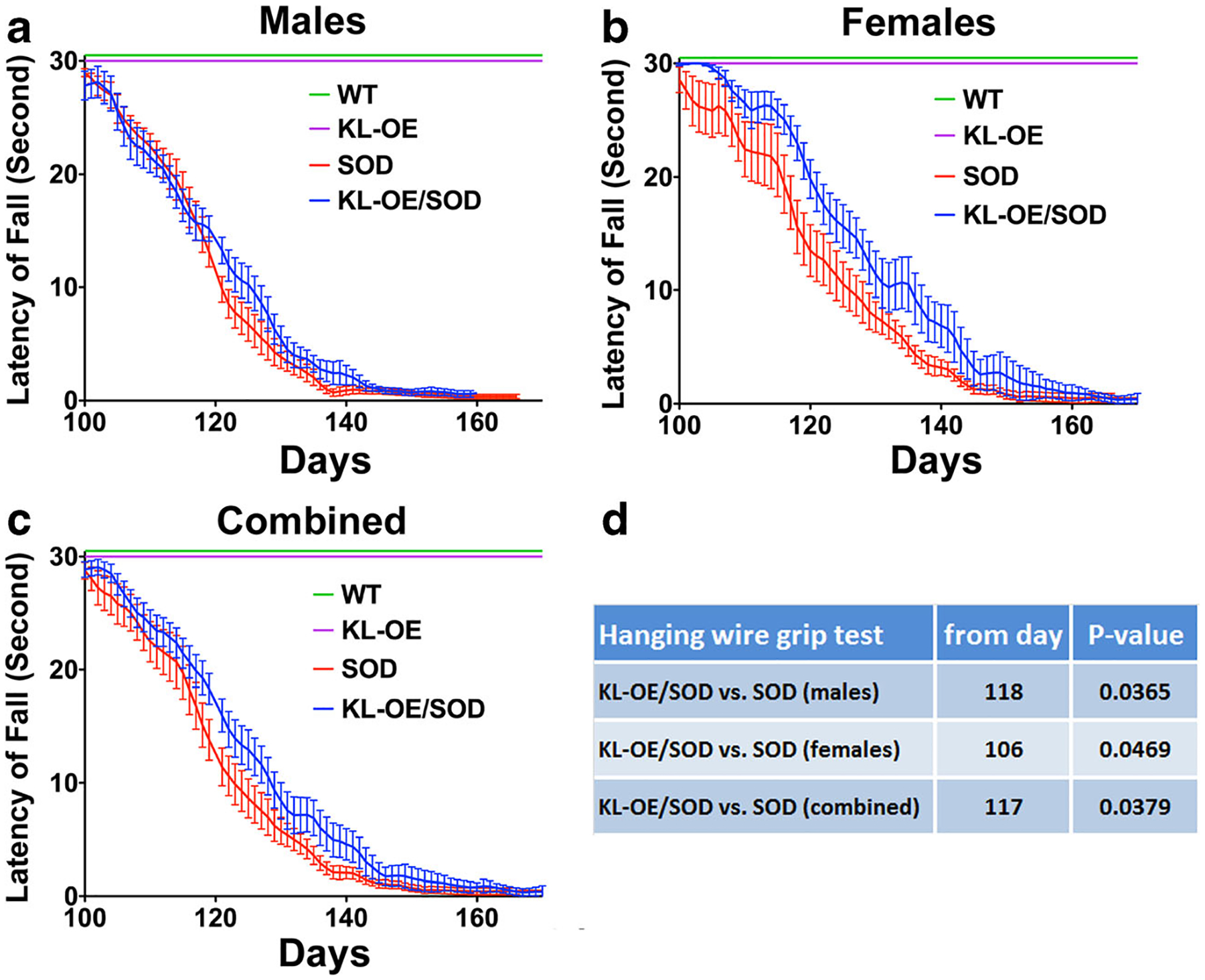
The hanging wire grip test presented as latency to fall (measured in seconds) at indicated ages for data collected from males (a), females (b), and when the data from both genders were combined (c). d Results of the statistical analysis of one-way ANOVA with post hoc Tukey’s test
Survival
Klotho overexpression significantly prolonged survival of more than a week (attaining the NS4, the humane endpoint) in the SOD1 ALS mouse model. The SOD1 mice survived for 156 days, whereas KL-OE/SOD1 mice survived for 167 days (Fig. 4c, d). The positive effect on life expectancy was more profound in the females, where survival significantly increased by almost 2 weeks (from 157 days in SOD1 vs 171.5 days in the KL-OE/SOD1 group; Fig. 4b, d), whereas in the males, the survival increased from 154 days in the SOD1 group to 161 days in the KL-OE/SOD1 group, albeit nonsignificantly (Fig. 4a, d).
Changes in Gene Expression
To elucidate the mechanisms by which Klotho alleviates disease progression and motor deficits, we examined Klotho’s effect on gene regulation from mRNA of the motor cortex and lumbar region of the spinal cord. We examined molecular pathways known to be altered in ALS and those through which Klotho was reported to signal. We collected tissues from SOD1 and bigenic mice at day 110 when the signs of the disease are clearly detected and when the disease was at full progression at age day 135. For the healthy mice (WT and KL-OE), we collected tissues only at 110 days (one time point) since we did not expect to detect changes in the gene expression in these mice between age 110 and 135 days and used the principle of reduction in animal experiments. In the motor cortex of KL-OE and WT mice, we detected a small (1.53-fold) but significant increase in Prx-2 (Fig. 5a), a gene we previously described to be elevated in primary hippocampal neurons by Klotho (Zeldich et al. 2014). We also observed a trend in the downregulation of different members of the Wnt family, such as Wnt2a and Axin2; however, the results did not reach significance (Fig. 5a). When motor cortex areas of bigenic KL-OE/SOD1 and SOD1 mice were compared, more changes were noted (Fig. 5c). Klotho overexpression in bigenic mice showed decreased expression of proinflammatory mediators TNF-α (at both time points), IL-12a at 110 days, and 1L-1β at 135 days. Remarkably, at 110 days, we detected a significant increase in myelin-associated glycoprotein (MAG) accompanied by an increase in myelin basic protein (MBP) that did not reach significance. No increase in PLP1 was detected (Fig. 5b). No changes in these genes were detected at 135 days or when we compared WT and KL-OE motor cortex areas (Fig. 5a, c).
Fig. 5.
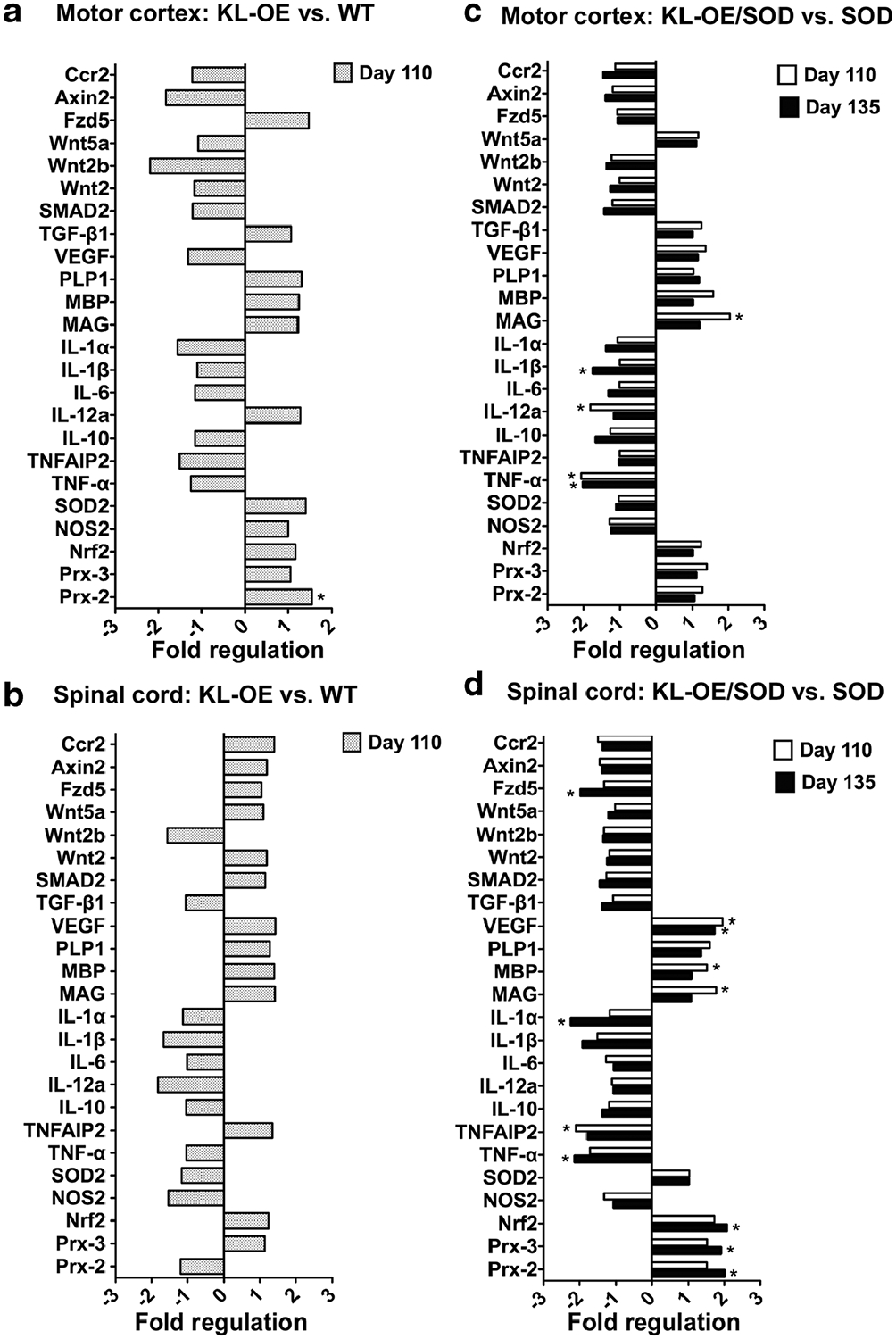
Analysis of the Klotho-induced changes in gene expression in the motor cortex (a, b) and lumbar spinal cord (c, d). The effect of Klotho overexpression in WT (a, c) and SOD1 mice (b, d) on mRNA expression of specific genes was measured using TaqMan gene expression assays (see Table 2) at 110 days (a–d) and 135 days (b, d). Bars represent fold difference (n = 6). The asterisks indicate statistical significance, *p value < 0.05 as analyzed by Bio-Rad CFX Manager 3.1 software
Comparison of gene expression of the lumbar region of the spinal cord between KL-OE and WT mice did not identify any significantly different alterations in the expression of tested genes (Fig. 5b). We reported previously that members of the Prx/Trx system play a key role in Klotho-induced neuroprotection (Zeldich et al. 2014). Prx-2, Prx-3, and their upstream regulator Nrf2 showed an enhanced expression at 110 days that became significant and more prominent at 135 days (Fig. 5d). Similar to the motor cortex area, Klotho overexpression inhibited the expression of proinflammatory cytokine TNF-α and its downstream target TNF alpha induced protein 2 (TNFAIP2), as well as the cytokines 1L-1α and 1L-1β. Remarkably, Klotho increased the expression of vascular endothelial growth factor (VEGF) in the spinal cord of SOD1 mice at 110 and 135 days. In the spinal cord, both MAG and MBP mRNA, but not PLP1, were significantly enhanced by Klotho overexpression in SOD1 mice at 110 days, confirming the supporting effect of Klotho on myelin maintenance. We also observed some increase in the myelin-related genes (MAG, MBP, and PLP1) at 135 days, but the increase did not reach significance (Fig. 5d).
Immunohistochemical Analysis of Microglial, Astrocytic, and Neuronal Markers
To test whether the neuroprotective effect of Klotho in SOD1 mice was accompanied by visible changes at the cellular level, we quantified motor neurons using NeuN, reactive astrocytes with glial fibrillary acidic protein (GFAP), and the number of microglia with Iba1 antibodies in the anterior horns of the spinal cord.
Interestingly, we found that in the motor cortex, the number of Iba1-positive microglia was significantly higher in SOD1 mice, while Klotho overexpression was able to reduce the number of microglia (from 126.2 positive pixels/mm2 in SOD1 mice to 115.1 pixels/mm2 in KL-OE/SOD1 mice, p value 0.037) (Fig. 6a, d).
Fig. 6.
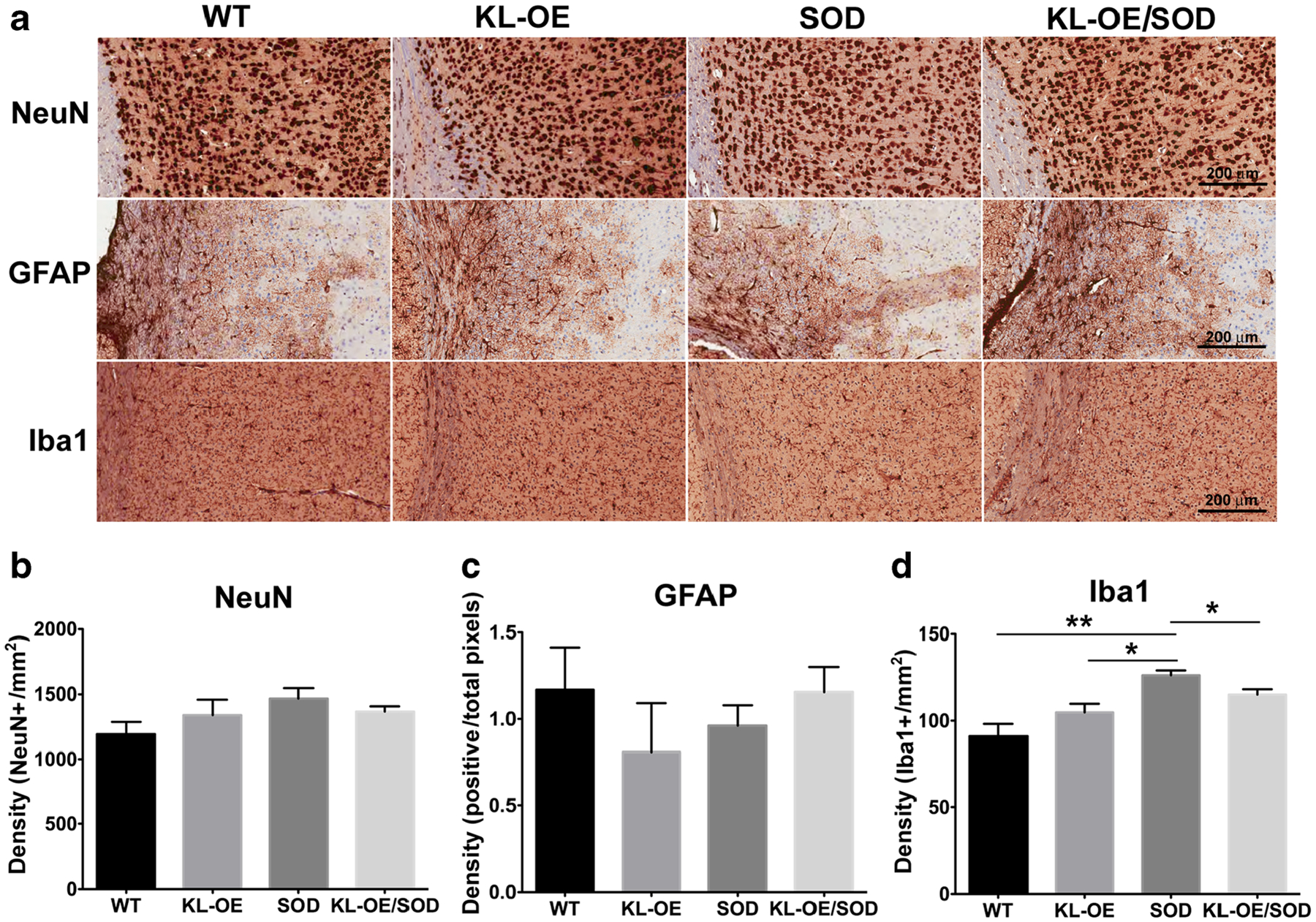
Immunohistochemical analysis of the Klotho-induced changes in the motor cortex at 110 days. a Representative images at ×20 magnification of NeuN, GFAP, and Iba1 staining in the motor cortex. b–d Quantification of the total number of NeuN-positive (b), GFAP- positive (c), and Iba1-positive cells (d) present in the motor cortex. The results are presented as mean ± SE; n = 4–6. The asterisks indicate statistical significance, *p value < 0.05; **p value < 0.01, as analyzed by Student’s t test
When assayed for NeuN positivity, no significant differences were detected between the motor cortex areas of all the different groups (WT, KL-OE, SOD1, KL-OE/SOD1) (Fig. 6a, b) or GFAP (Fig. 6a, c).
Immunohistochemical analysis of the same markers was performed on the anterior horns from cervical and lumbar spinal sections. The cervical region showed a trend toward reduced NeuN-positive cells in SOD1 mice and increased NeuN-positive cells in the KL-OE/SOD1 mice, but the differences were not statistically significant (Fig. 7a). No changes in GFAP staining was detected from either KL-OE/SOD1 or SOD1 mice, suggesting that the astrocyte-mediated neuroinflammation is not prominent in the cervical spinal cord at this stage of the disease (Fig. 7b). In contrast, an increased number of Iba1-positive cells was observed in the cervical region of the spinal cord of SOD1 mice, while Klotho overexpression greatly normalized this alteration (from 137.5 Iba1-positive cells/mm2 in SOD1 mice to 73.4 Iba1-positive cells/mm2 in KL-OE/SOD1 mice, p value 0.059) (Fig. 7c).
Fig. 7.
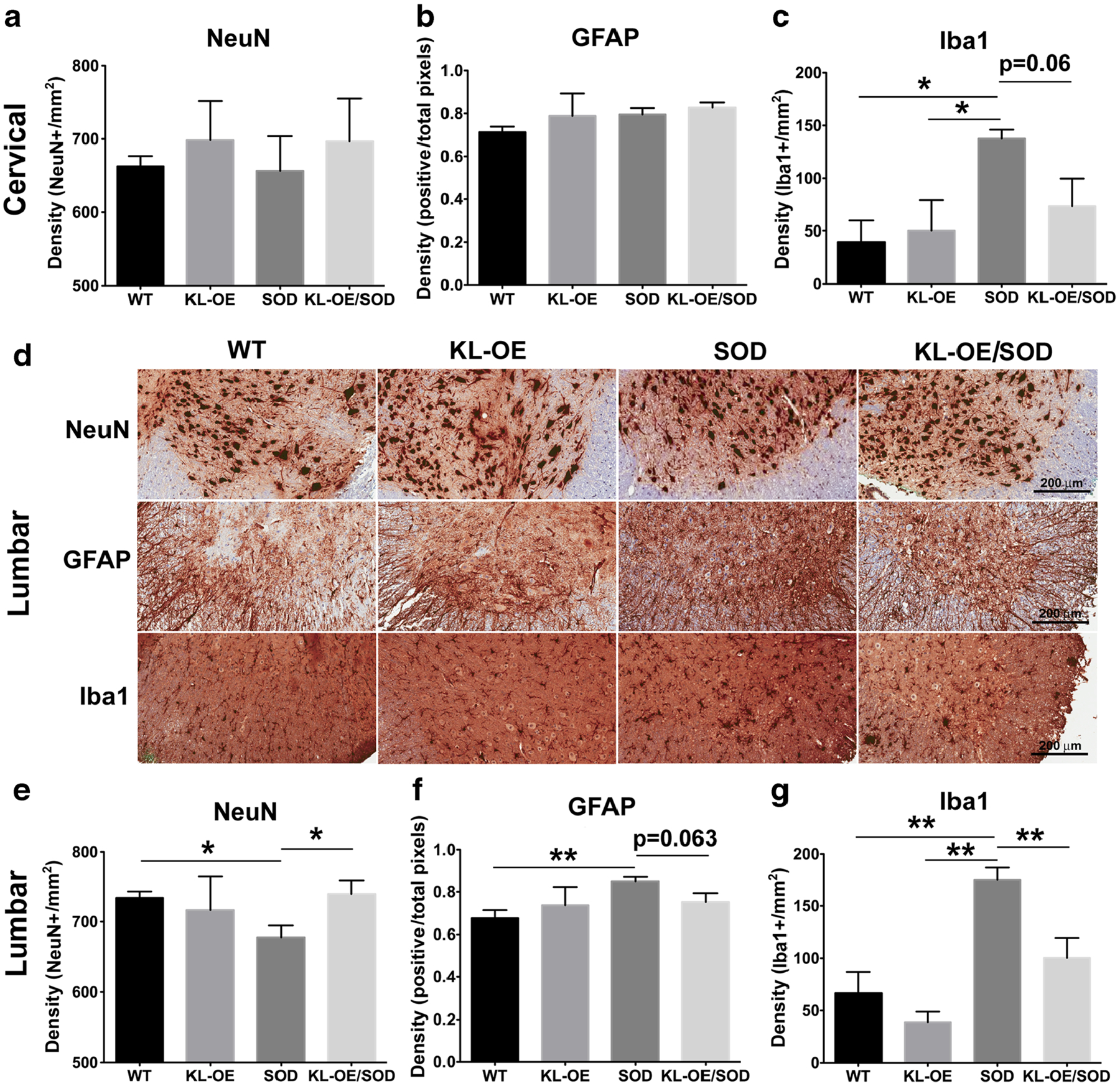
Immunohistochemical analysis of the Klotho-induced changes in the spinal cord at 110 days. a–c Quantification of the total number of NeuN-positive (a), GFAP-positive (b), and Iba1-positive cells (c) present in the anterior horn of the cervical spinal cord. d Representative images at ×20 magnification of NeuN, GFAP, and Iba1 staining of the lumbar spinal cord. e–g Quantification of the total number of NeuN-positive (e), GFAP-positive (f), and Iba1-positive cells (g) present in the anterior horn of the lumbar spinal cord. The results are presented as mean ± SE; n = 4–6. The asterisks indicate statistical significance, *p value < 0.05; **p value < 0.01, as analyzed by Student’s t test
The differences were even more dramatic in the anterior horns of the lumbar areas of the spinal cord. The number of NeuN-positive cells were significantly reduced in the SOD1 mice, while Klotho overexpression fostered a remarkable neuronal rescue (Fig. 7d, e). Noticeably, in the lumbar area of SOD1 mice, the number of GFAP-positive cells was significantly increased, while Klotho overexpression showed a trend toward bringing the extent of GFAP-immunolabeled cells back to normal (p value 0.063) (Fig. 7d, f). The effect of the SOD1 and Klotho transgenes on the numbers of microglial cells was more dramatic than that of astrocytes and neurons. The number of Iba1-immunopositive microglia was significantly higher in the lumbar spinal cord of SOD1 mice, while Klotho overexpression restored the alterations in the number of microglial cells in SOD1 mice by reducing it from 175.2 positive pixels/mm2 in SOD1 mice to 100.4 pixels/mm2 in KL-OE/SOD1 mice (p value 0.007) (Fig. 7d, g). Our findings suggest that Klotho promotes motor neuronal survival at least partially through the reduction of neuroinflammation in the lumbar part of the spinal cord of SOD1 mice.
Klotho Attenuates Proinflammatory Mediators in an In Vitro Model of Neuroinflammation
Our in vivo results described above showed the ability of Klotho overexpression to reduce the mRNA expression of proinflammatory cytokines and the number of Iba1-positive cells in the motor cortex and spinal cord of KL-OE/SOD1 mice. In order to understand whether the anti-inflammatory effect of Klotho in vivo can be attributed to the regulation of the microglial response, we utilized primary microglia cultures to determine the production of inflammatory modulators associated with the SOD1 phenotype. Crossing of heterozygous Klotho-overexpressing females with heterozygous SOD1 males resulted in litters carrying four genotypes: WT, KL-OE, SOD1, and KL-OE/SOD1. The levels of Klotho mRNA from microglia obtained from KL-OE pups were 10.2-fold higher (p value 1.07 × 10−11) than those from microglia isolated from WT littermates, as assessed by RNA sequencing (RNAseq), indicating that these cells maintained their genotype in culture.
Proinflammatory mediators TNF-α, IL-6, and nitric oxide (NO) were quantitated from the conditioned media of microglial cultures from each group of mice. Basal levels of NO were similar in microglia obtained from all four genotypes and were significantly increased following LPS/IFN-γ treatment. Increased production of NO in response to LPS/IFN-γ treatment was significantly less in Klotho-overexpressing cultures. Furthermore, while LPS stimulation of microglia obtained from SOD1 mice resulted in even greater production of NO than from WT microglia, Klotho overexpression in microglia from bigenic KL-OE/SOD1 pups led to significantly reduced NO levels (Fig. 8a). Concentrations of TNF-α and IL-6 followed a similar pattern where we observed diminished levels of these cytokines in microglia from KL-OE/SOD1 compared to microglia from both WT and SOD1 mice (Fig. 8b, c). The addition of recombinant mouse Klotho (rmKL) had only a marginal effect on the expression of TNF-α and IL-6 in microglia obtained from either SOD1 or WT mice, and the results reached significance for TNF-α, but not for IL-6 (p = 0.08).
Fig. 8.

Primary microglia from four different genotypes were exposed to LPS (100 ng/ml) and IFN-γ (30 ng/ml) for 24 (a–c) or 18 h (d–f). a NO production was measured by the Griess reagent and normalized to the cell number in individual wells, as assessed by crystal violet staining. b, c TNF-α and IL-6 levels were evaluated by ELISA and normalized to the cell number in individual wells, as assessed by crystal violet staining. d–f Western blot analysis of the expression of Cox-2 and iNOS (d) and diagram of the normalized results for Cox-2 (e) and iNOS (f). The results are presented as mean ± SE; n = 4–6. The asterisks indicate statistical significance, *p value < 0.05; **p value < 0.01 for WT vs KL-OE and WT vs WT+rmKL, #p value < 0.05; ##p value < 0.01; ###p value < 0.001 for SOD1 vs KL-OE/SOD1 and SOD1 vs SOD1+ rmKL, as analyzed by one-way ANOVA with post hoc Tukey’s test
Two inducible enzymatic pathways iNOS and Cox-2 are induced by a large number of the same stimuli and produce proinflammatory mediators NO and prostaglandins, respectively. iNOS and Cox-2 are often expressed concomitantly in inflamed tissues (Needleman and Manning 1999). iNOS generates toxic levels of NO using the amino acid arginine, while Cox-2 regulates the enzymatic production of prostaglandin E(2) (PGE(2)). To further confirm Klotho’s anti-inflammatory effects, we evaluated the expression of iNOS and Cox-2 after LPS/IFN-γ stim- ulation. In line with the previous results, LPS treatment of microglia resulted in increased levels of these two enzymes, and Klotho overexpression significantly reduced the expression of both enzymes compared to WT and SOD1 cultures (Fig. 8d–f).
Discussion
In the current study, we present the neuroprotective effects of Klotho in the SOD1 mouse model of ALS. Klotho overexpression delayed the onset and slowed down the progression of the disease, alleviated motor deficits, and extended survival in this mouse model. Recent studies on Klotho, performed by us and by others in other mouse models of neurodegenerative diseases, indicate that Klotho may be able to reduce pathological progression of the disease in the SOD1 model as well by fostering neuronal survival (Zeldich et al. 2014), counteracting oxidative stress (Kurosu et al. 2005), diminishing the proinflammatory immune response (Guo et al. 2018), and upregulating oligodendrocyte differentiation and maturation (Chen et al. 2013).
Klotho Delays Disease Onset in SOD Mice and Improves Performance in the Grip Test
The degeneration of cranial and spinal motor neurons, subsequently leading to muscular atrophy, is the ultimate cause of death in mice with the SOD1 mutation (Gould et al. 2006; Lee et al. 2016). Therefore, preservation of motor function is a crucial aspect of mitigating disease progression and pathology in these mice. The phenotypic screening protocol used in this study provides a reliable assessment of neuromuscular functions as indicated by the NS protocol developed by the ALS Therapy Development Institute (TDI). Since hind limb deficit is the earliest sign of disease in SOD1 mice, the NS assessment of the hind limbs provided a reliable measurement of disease onset. We reasoned that the analysis should start on day 60 when the mice are asymptomatic. We discovered that Klotho overexpression in KL-OE/SOD1 bigenic mice resulted in delayed onset and progression of the disease. Even though the benefits of increased levels of Klotho were more obvious in females, there were significant positive outcomes in males, such as time to reach the peak weight and the hanging wire grip test. Remarkably, when we combined the data from males and females, KL-OE/SOD1 mice reached the NS2 stage significantly later than the SOD1 mice, representing a delay in disease onset. Additionally, the statistically significant delayed time to reach the NS3 stage provided evidence that Klotho overexpression slowed the progression of disease. Our assessment that Klotho preserves motor functions was further supported by the hanging wire grip test in KL-OE/SOD1 and SOD1 mice, which was significantly improved for males and females and when the data from both genders were combined.
Klotho Prolongs Survival in SOD Mice
Klotho overexpression also prolonged survival. Similar to the NS results, the effect on survival was evident and significant in females, and when the male and female data were combined, they reached significance (p value 0.0076 and 0.0059, respectively, employing the Kaplan–Meier survival fit followed by the log-rank test). However, the survival benefit was not significant in males. These findings are in line with other published results demonstrating gender-specific differences. In humans, the prevalence and incidence of ALS are known be higher in men than in women (McCombe and Henderson 2010). This gender-related difference is observed in extensive studies of both sporadic and familial ALS cases. Men tend to develop the disease at a younger age with onset predominantly in the spinal regions, while in women the onset is presented in the bulbar region (McCombe and Henderson 2010). In ALS mice, the gender-related discrepancies in spinal cord mitochondrial function can result in less aggressive disease phenotypes in female (Cacabelos et al. 2016), and the gender-related therapeutic response to a variety of potential therapeutics, with a greater effect achieved in females, was demonstrated previously in SOD1 mice (Bame et al. 2012; Butovsky et al. 2015). Furthermore, treatment of SOD1 mice with methionine sulfoximine resulted in increased survival of both male and female mice, with a significantly greater effect on females (Bame et al. 2012). Ovariectomy or castration completely eliminated the differences in the therapeutic effect of the methionine sulfoximine on survival and neuromuscular deterioration, suggesting that the estrogen receptor plays a role in SOD1 mice disease progression and response to potential therapeutics (Bame et al. 2012). These findings are of interest in relation to Klotho since Klotho-overexpressing males exhibit a more prolonged life span than females, 30 vs 19%, respectively (Kurosu et al. 2005). In humans, however, the levels of serum Klotho are lower in boys and men than in girls and women (Pedersen et al. 2013; Semba et al. 2011) and are lower in male bonobos than in females (Behringer et al. 2018).
Klotho Delays Weight Loss in the SOD Mice
Weight assessment further supported Klotho’s ability to extend life span in SOD1 mice. The curves of mean group body weight from bigenic KL-OE/SOD1 females show that they maintained their weight significantly better than SOD1 females starting from day 130. No such difference was observed in males. However, another finding was striking: the very clear and significant separation in weight gain starting from day 60 (when we started to measure the weight) between KL-OE and WT mice from both sexes. Kurosu et al. (2005) found no difference either in growth and weight maintenance or in food intake and oxygen consumption in KL-OE mice. The possible explanation could be the C3H mouse background used by Kurosu et al. vs the C57BL background of our mice. A literature search did not reveal any previously known reports of such differences (Razzaque 2012; Samms et al. 2016). Since Klotho was reported to inhibit the insulin and IGF-1 signaling pathways, possibly mimicking a calorie-restriction model, it is reasonable to assume that KL-OE mice would be lighter. However, to our knowledge, this is the first time that these differences are being reported. In light of these findings, the effect of Klotho overexpression on weight maintenance in bigenic mice is even greater. Since Klotho overexpression causes the KL-OE/SOD1 mice of both genders to be lighter, therefore, even maintaining the same weight as SOD1 mice (as in the case with males) could be considered as meaningful. In other words, since KL-OE mice exhibit consistently lower weight than WT of both genders, the positive effect on the weight maintenance in KL-OE/SOD1 mice should be considered even more dramatic. This is further supported by the fact that KL-OE/SOD1 males reached peak body weight later than SOD1 males, indicating that KL-OE/SOD1 remained healthy for a longer period of time and that the development of disease was significantly slowed by Klotho. The same trend was observed in females; however, the results did not reach significance.
Klotho Reduces Microglia-Derived Inflammation in the SOD Mice
Primary microglia and astrocytes isolated from SOD1 mice exhibit a variety of upregulated proinflammatory genes, including biologically active enzymes, such as Cox-2 and iNOS and cytokines (particularly TNF-α) (Caldeira et al. 2014). In support of the role glia play in the mouse model of ALS, survival in SOD1 transgenic ALS mice is significantly prolonged by knocking down the expression of the SOD1 gene from astrocytes (Yamanaka et al. 2008), microglia (Boillée et al. 2006; Peters et al. 2015), or oligodendrocytes (Kang et al. 2013), suggesting that mutant SOD1 is also detrimental to glia and not only to motor neurons.
Our findings that Klotho overexpression in the KL-OE/SOD1 mice reduced proinflammatory cytokines in the motor cortex and spinal cord suggest that Klotho modulates microglia responses. It has been previously reported that the changes in the proinflammatory genes in microglia are detected in the spinal cord of SOD1 mice as early as 70 days of age, and this activation precedes neuronal damage and the appearance of motor neuron dysfunction (Butovsky et al. 2015; Lewis et al. 2014; Solomonov et al. 2016).
To detect the changes that may point to the main effectors in Klotho’s neuroprotective mechanisms, we performed immunohistochemical (IHC) analyses at 110 days. At this time point, the signs of the disease, such as tremor, are clearly detected, but other motor deficits are relatively mild. In line with the previous findings, IHC analysis of SOD1 mice revealed an increased number of Iba1-positive cells in the motor cortex and cervical region and, to a greater extent, in the lumbar anterior horn compared to WT mice. Klotho overexpression reversed the increased number of microglial cells in the motor cortex and spinal cord. Our mRNA analysis of the motor cortex and spinal cord included mixed populations of cells, while the IHC results implied that the anti-inflammatory effect of Klotho is mediated mainly through the effect on the microglia. We were able to further confirm this hypothesis by showing in vitro that Klotho overexpression in WT and SOD1 primary microglia led to the diminished release of proinflammatory mediators in response to LPS. The expressions of TNF-α, IL-6, Cox-2, and iNOS were significantly reduced in microglia from KL-OE pups. These findings indicate that Klotho can alleviate the microglia responses and suggest a new role for Klotho as a modulator of inflammation in the CNS. Interestingly, the application of rmKL in vitro to SOD1 microglia resulted in only marginal decrease in the production of the proinflammatory cytokines. This observation suggests the possible involvement of the transmembrane form of Klotho or, alternatively, that the Klotho levels and the duration to its exposure did not match the conditions of the endogenously overexpressed Klotho.
Our findings are further supported by a recently published study where Klotho depletion in the choroid plexus achieved through viral vector-induced knockout resulted in increased number of Iba1-positive cells and enhanced Iba1 staining intensity in the mouse hippocampus after injection of low-dose LPS and triggered the infiltration of macrophages. This study suggested that Klotho produced by the choroid plexus controls the junction between the brain and immune system (Zhu et al. 2018).
IHC with the astrocytic marker GFAP did not show any significant differences in the motor cortex and the cervical region of the spinal cord of ALS mice. However, an increased number of GFAP-positive cells was detected in the lumbar region of the spinal cord, and even though these changes were not dramatic, Klotho overexpression showed a trend toward normalization of this alteration in the number of astrocytes (p value 0.06).
Multiple lines of evidence indicate that microglial activation plays a pivotal role in the disease progression (Beers et al. 2011; Lewis et al. 2014) since an increased number of microglial cells precedes the onset and advancement of the disease toward more profound functional deficits (Lewis et al. 2014). Thus, we can speculate that Klotho’s ability to alleviate motor neuronal deficits and delay the disease onset can be attributed to its effect on microglia.
To our knowledge, this is also the first IHC study specifically comparing the motor cortex and spinal cord areas between WT and KL-OE mice. No changes were identified in the microglia, astrocytes, or motor neurons when these two groups of mice were compared.
Klotho Rescues Motor Neurons in the SOD Mice
The ultimate goal of the therapeutic approaches aimed to combat ALS is the rescue of motor neurons. In our study, we found that the number of motor neurons in the spinal cord was reduced in SOD1 mice. Klotho overexpression had a remarkable effect on neuronal rescue in the lumbar spinal cord by preserving the number of NeuN-positive cells to the numbers observed in WT mice. A similar pattern was observed in the cervical spinal cord; however, the results did not reach significance. This profound effect of Klotho on fostering motor neuronal survival was detected through IHC studies at 110 days when the process of neuron cell death is less aggressive and the effects on cell survival are more detectable. This Klotho-induced motor neuronal survival can account for the alterations in neuronal deficits and extended survival and can be attributed to the ability of Klotho to modulate the inflammatory response of microglia and counteract the oxidative stress. Since not all neurons in the motor cortex and spinal cord are motor neurons, staining with a motor neuron-specific marker (CTIP-2 and AchT) might show an even larger rescue by Klotho in the bigenic mice.
Klotho Effects on Gene Expression
In our examination of the neuroprotective effect of Klotho on gene expression in the motor cortex and spinal cord, we have chosen a panel of genes implicated in ALS pathology and known Klotho downstream targets. We did not observe any significant differences in gene expression when we compared mRNA from the spinal cord and motor cortex of healthy WT and KL-OE mice, besides a small, but significant increase in Prx-2 in the motor cortex of KL-OE mice, confirming our previous results in primary neurons (Zeldich et al. 2014). To our knowledge, this is the first comparison of a limited gene profiling of isolated areas of CNS tissues in these mice. When comparing tissues from KL-OE/SOD1 and SOD1 mice from the same regions, we discovered a significant decrease in the proinflammatory transcripts of TNF-α, IL-1β, and IL-12a, further confirming the immunoregulatory role of Klotho. We also tested other inflammatory modulators such as IL-6, IL-10, IL-1α, and iNOS that are altered in the pathogenesis of ALS (Almer et al. 1999; Henkel et al. 2004; Lewis et al. 2014), but no significant changes were detected in our mouse model.
Klotho inhibits the activation of TGFβ-R (Doi et al. 2011; Yamamoto et al. 2005) and suppresses Wnt pathway activation (Liu et al. 2007). Both of these pathways are associated with proinflammatory microglia (Halleskog et al. 2011) and are implicated in glial activation and pathogenesis in the spinal cord of SOD1 mice (Chen et al. 2012; Li et al. 2013; Yu et al. 2013). We did not observe any significant changes induced by Klotho overexpression in this pathways in either the motor cortex or spinal cord of healthy or SOD1 mice.
Early explorations of Klotho’s effects on the brain reported that impaired cognition in Klotho-deficient mice was accompanied by apoptotic cells and increased levels of oxidized proteins, lipids, and DNA in the hippocampus (Kuro-o 2009). In addition, KL-OE mice express increased levels of SOD2 in muscles and exhibit lower levels of DNA oxidative damage than WT mice (Yamamoto et al. 2005). Moreover, when cultured cells were treated with the shed form of the Klotho protein, it resulted in induction of SOD2 expression and reduction of oxidative injury and apoptosis in HeLa cells (Yamamoto et al. 2005) and in vascular endothelial cells (Ikushima et al. 2006). The results from our studies deviated from the expectations since we did not detect any significant effect of Klotho overexpression on the levels of SOD2 when RNA expression was compared from WT and KL-OE mice and from KL-OE/SOD1 to SOD1 mice. However, this is in line with our previous studies of Klotho-induced neuroprotection in primary hippocampal neurons from glutamate and oligomeric amyloid β cytotoxicity that pointed to the Prx/Trx family members and not to SOD2 as key regulators in Klotho-mediated neuronal survival (Zeldich et al. 2014). In the current study, we found that Prx-2, Prx-3, and their upstream regulator Nrf2 were upregulated about 2-fold by Klotho in the spinal cord of KL-OE/SOD1 mice as compared to SOD1 mice. In the motor cortex, Prx-2 was increased by Klotho under naive conditions in healthy mice. These findings make Klotho an especially relevant therapeutic target for ALS, where oxidative stress was shown to be one of the leading contributors to disease progression (Gurney et al. 1994). In order to confirm that key role of Prx/Trx family members in the effect of Klotho in SOD1 model of ALS, future specific knockdown studies are required.
Klotho overexpression upregulated myelin-related genes such as MBP and MAG in the motor cortex and spinal cord of SOD1 mice at 110 days. This effect was less profound and not significant at 135 days. These findings suggest that Klotho promotes oligodendrocyte differentiation and maturation and protects myelin integrity at the earlier stages of the disease when the neurodegeneration is less profound. This is further supported by a recent study revealing extensive mobilization of oligodendrocyte progenitor cells (OPCs) in the anterior gray matter area, where motor neurons are located. This mobilization precedes the motor neuronal deficits associated with the disease; however, these newly formed oligodendrocytes demonstrated insufficient differentiation and abnormal morphologies, thus leaving motor neuron axons demyelinated (Kang et al. 2013).
We have recently shown that Klotho plays an important role in oligodendrocyte biology by enhancing OPCs’ differentiation and myelin protein production (Chen et al. 2013). Klotho is downregulated in the white matter of the aging brain (Duce et al. 2008), which parallels the age-related myelin loss and damage we observed in rhesus monkeys (Duce et al. 2006; Hinman et al. 2004, 2006; Hinman and Abraham 2007; Sloane et al. 2003). Also, in Klotho knockout (KL-KO) mice, we reported a striking reduction of myelinated axons as well as oligodendrocytes in the corpus callosum and optic nerve, compared to WT mice (Chen et al. 2013). In vivo, Klotho overexpression in KL-OE mice leads to a significant 2-fold increase in the level of spontaneous remyelination in the corpus callosum compared to WT littermates in the cuprizone model of MS (Zeldich et al. 2015).
Common inflammatory pathways were identified between ALS and MS, and the types of degenerative changes that MS causes in oligodendrocytes are widely present in ALS patients and in the mutant SOD1 mouse model (Philips et al. 2013). Research by others has demonstrated that demyelination in ALS is induced by proinflammatory mediators and NO, which are released by microglia and astrocytes. These mediators were recently shown to inhibit the expression of myelin genes, such as MBP, preceding oligodendrocyte death (Jana and Pahan 2013). The positive effect of Klotho on the promyelinating properties of oligodendrocytes supports its potential therapeutic role in ALS.
We also tested two additional overlapping pathways between genes that Klotho targets and genes that are altered in ALS pathology, that seemed relevant to our studies: C-C chemokine receptor type 2 (CCR2) (Molina et al. 1989) and VEGF (Li et al. 2003). In our study, Klotho overexpression did not induce any significant changes in the CCR2 mRNA but did increase VEGF expression in the spinal cord. In vitro studies demonstrated that VEGF fosters motor neuron survival in the SOD1 mouse model of ALS (Li et al. 2003), while the deficiency in VEGF correlates with progressive loss of motor neurons (Oosthuyse et al. 2001). VEGF levels are significantly lowered in the CSF of ALS patients during the initial stages of the disease (Devos et al. 2004). The expression of VEGF in the spinal cord, but not in the motor cortex, of KL-OE/SOD1 mice was significantly increased by Klotho. This finding opens another route of exploration to the mechanisms of Klotho-mediated effects in SOD1 mice.
About 5–10% of ALS patients exhibit FALS. FALS is associated with mutations found in the genes coding for SOD1 (Gurney et al. 1994), the C9orf72 gene (Sreedharan 2010), and to a lesser extent, in the FUS/TLS and TDP-43 genes (reviewed in Tosolini and Sleigh 2017). With further insight into the gain- and loss-of-function mechanisms of FALS, oligonucleotide-mediated therapeutics such as antisense oligonucleotide (ASO) and RNAi have emerged. These oligonucleotide-mediated therapeutics are specifically targeting the underlying genetic mutations in genes such as C9ORF72, SOD1, TDP-43, and FUS and have been tested in SOD1 and C9ORF72 rodent models (reviewed in Tosolini and Sleigh 2017). FALS and sporadic ALS are known to share pathogenic mechanisms (as in the case with WT-SOD1 and mutant SOD1; Bosco et al. 2010), which may broaden the scope of ASO and RNAi therapies. However, this approach targets only known mutations, while Klotho could be a more applicable target for idiopathic–sporadic ALS cases that do not display any known genetic mutations and account for the majority of ALS. Due to its multifunctional role in the nervous system, Klotho can potentially target a number of ALS-associated pathological processes at once, such as motor neuron death, oxidative stress, neuroinflammation, and oligodendrocyte dysfunction. Our study reveals for the first time the beneficial effect of Klotho in an ALS mouse model. Although the results were statistically significant, the effect was moderate, as expected in this aggressive mouse model. Further studies targeting specific signaling pathways are required to reveal the precise beneficial mechanisms of Klotho-induced changes in the SOD1 mouse model. These studies can be expanded to other animal models of ALS presently available. We believe that our findings will prompt in the near future the development of Klotho-based therapeutics for ALS in the form of either small molecule Klotho-enhancing compounds or Klotho gene therapy.
Experimental Procedures
Mice
All animal procedures were performed in accordance with a protocol approved by the Boston University Institutional Animal Care and Use Committee. SOD1G93A male mice with the toxic gain-of-function SOD1 Gly 93 to Ala substitution (G93A) were obtained from The Jackson Laboratory (stock number 004435) (Bar Harbor, ME, USA). The colony was bred and maintained in the animal facility at Boston University. Hemizygous SOD1 males were crossed with hemizygous Klotho-overexpressing females. Both transgenic mice are bred on a C57BL/6J background and the F1 generation was used for all the experiments. Isolated genomic tail DNAwas used to genotype mice using a protocol provided by the Jackson Lab, and the expression of Klotho was detected by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) as described (Zeldich et al. 2014). Littermates of both sexes (approximately equal numbers of males and females) were included in each group. The number of mice used for the assessment of motor neuron deficits and survival as well as for immunohistochemistry and biochemical assays is shown in Table 1.
Tissue Acquisition
For the biochemical and histochemical studies, mice from the four groups were perfused transcardially with 0.1 M phosphate-buffered saline, pH 7.4, at 4 °C and sacrificed at age 110 (onset of paralysis) and 135 days (end stage) (Butovsky et al. 2015), in order to detect Klotho’s effects during the different stages of the disease. These time points were chosen in accordance with disease progression and previously published results describing the gene alterations expected at these time points in SOD1 mice (Butovsky et al. 2015). After being removed, a portion of the lumbar region and the isolated motor cortex from one hemisphere were immediately frozen in liquid nitrogen and stored at − 80 °C for biochemical evaluation by qRT-PCR. The remaining spinal cord and the second hemisphere were fixed in 4% paraformaldehyde and used for IHC as previously described (Crosio et al. 2011).
Genotyping of Mouse Tails and SOD1 Copy Number Assessment
All mice were genotyped twice: first at the time of weaning and secondly, following euthanasia, in order to confirm that all genotyping is in agreement with the ear punching. The tail from each animal was clipped individually into a microcentrifuge tube and subjected for genomic DNA isolation according to the manufacturer’s protocol employing the DirectAmp™ tissue genomic DNA amplification kit (Denville Scientific, Metuchen, NJ) as we described before (Zeldich et al. 2014). Briefly, 100 μl of extraction solution was added to the tail, followed by the addition of 25 μl of preparation solution. The samples were allowed to incubate at RT for 10 min and then boiled at 95 °C for 3 min. Then, 100 μl of neutralization solution was added to each sample. After mixing all the samples by vortex, 4 μl of the tissue DNA extract was used for qRT-PCR amplification using Applied Biosystems FastTaq Polymerase master mix (Denville Scientific, Metuchen, NJ) in a 20-μl final volume. The Klotho primers used were as follows: forward, 5′-GGTT G C C C A C A A C C TA C T T-3 ′ r e v e r s e, 5 ′ - T G G G AGCTTAAGGCGATAGA-3′ with the probe: 5′-TCTC TACAA/ZEN/CACCTCTTTCCGCCC/3IABKFQ-3′. The SOD1 transgene primers were designed according to the recommendations provided by Jackson Lab (https://www2.jax.org/protocolsdb/f?p=116:5:0::NO:5:P5_MASTER_PROTOCOL_ID,P5_JRS_CODE:9877,004435) as follows: forward, 5′-GGGAAGCTGTTGTCCCAAG-3′; reverse, 5′-CAAGGGGAGGTAAAAGAGAGC-3′ with the probe: 5′−56-FAM/CTCCATCTG/ZEN/GTTCTTGCAAAACACCA/3IABKFQ-3′. For the endogenous control reference gene (mIL2), the following primers were used: forward, 5′-CTAG GCCACAGAATTGAAAGATCT-3′; reverse, 5′-GTAG GTGGAAATTCTAGCATCATCC-3′ with the probe: 5′−5Cys5/CCAATGGTC/TAO/GGGCACTGCTCAA/3IAbRQSp/−3′. PCR was carried out using Bio-Rad CFX96 Real-Time PCR system utilizing Fast Advanced Master Mix (Life Technologies), in accordance with the manufacturer’s protocol.
Copy number assessment was done for all mice in the study, as previously described (Alexander et al. 2004; Kaneb et al. 2011). Average CT values for human SOD1 and endogenous reference (internal control (IC)) were determined for each sample, and ΔCT values were calculated by subtracting the average CT SOD1 value of a sample from its corresponding average of CT IC value to calculate the transgene copy number. This assessment was performed for all the mice in the study, and any mice showing ΔCT value that differed by more than half a cycle from the average value of the cohort were supposed to be excluded from the study; however, no mouse met this criterion. One female mouse carrying the SOD1 transgene was excluded from the study after it reached 190 days and developed a complete paralysis of the left side exclusively.
qRT-PCR Experiments and Analysis
RNA was extracted using the combination of the TRIzol protocol with subsequent utilization of the reagents from RNAeasy plus mini kit (Qiagen, USA). Motor cortex and spinal cord tissues were homogenized in 1 ml of TRIzol and incubated for 5 min at RT, with frequent vortexing. After the addition of 0.2 ml of chloroform to the TRIzol, and vortexing for 15 s, the samples were incubated for 1 min at RT, vortexed again for 15 s, and centrifuged at 15,000×g for 10 min to separate phases. Two hundred microliters from the top layer was removed and mixed with 700 μl of Qiagen RLT buffer (from RNAeasy plus mini kit (Qiagen, USA)) in a new tube. The rest of the top layer was removed and frozen at − 20 °C to serve as a backup in case the initial yield is low. Then, 200 μl of the sample was combined with 700 μl RLT buffer and with 500 μl of 96–100% ethanol and applied to a Qiagen MinElute spin column placed in a 2-ml microfuge tube. This step was followed by a repeated wash with RPE buffer (from RNAeasy plus mini kit (Qiagen, USA)) and subsequent elution. One microgram of total RNA was reversed transcribed using the SuperScript™ VILO™ cDNA Synthesis Kit as previously described (Chen et al. 2018) and according to the manufacturer’s instructions (cat. 11754050, ThermoFisher Scientific). qRT-PCR was carried out for all genes of interest in each sample using human TaqMan Gene Expression Assays (Life Technologies) (see Table 2), and the transcripts of genes of interest were normalized to the endogenous controls, represented by actin B and PPIA. Samples were run in triplicates and the data analyses were performed by the ΔΔCt method as previously described (Chen et al. 2018). Gene expression fold changes were determined by Bio-Rad Gene Expression Module of CFX Manager 3.1 software, and a p value ≤ 0.05 was considered significant.
Table 2.
TaqMan gene expression assays
| Catalog number | Gene expression assay |
|---|---|
| Mm00446190_m1 | IL-6 |
| Mm01288386_m1 | IL-10 |
| Mm00434169_m1 | IL-12a |
| Mm00439620_m1 | IL-1α |
| Mm00434228_m1 | IL-1β |
| Mm00443258_m1 | TNF-α |
| Mm00447578_m1 | Tnfaip2 |
| Mm04208213_g1 | Prdx-2 |
| Mm00545848_m1 | Prdx-3 |
| Mm00477784_m1 | NRF2 |
| Mm01313000_m1 | SOD2 |
| Mm00440502_m1 | NOS2 |
| Mm00470018_m1 | Wnt2 |
| Mm00437330_m1 | Wnt2b |
| Mm00437347_m1 | Wnt5a |
| Mm00443610_m1 | Axin2 |
| Mm00445623_s1 | Fzd5 |
| Mm01178820_m | TGF-β1 |
| Mm00487530_m1 | SMAD 2 |
| Mm99999051_gH | CCR-2 |
| Mm00437306_m1 | VEGF |
| Mm01266402_m1 | MBP |
| Mm00487538_m1 | MAG |
| Mn01297210_m1 | PLP1 |
| Mm02619580_m1 | Actin B |
| Mm02342430_m1 | PPIA |
Immunohistochemical Analysis
The genders of all the mice used for IHC are listed in Table 1. The genders were mixed and analyzed together. We performed the immunohistochemical analysis at 110 days. At this time point, the signs of the disease, such as tremor, are clearly detected, but other motor deficits are relatively mild. The intention was to detect early change that may point to the main effectors in Klotho’s neuroprotective mechanisms.
Mouse brains and cervical and lumbar regions of the spinal cords were fixed in 4% paraformaldehyde, embedded in paraffin, and serially sectioned at 10 μm in the coronal plane. The sections were mounted and stained with the following antibodies: mouse monoclonal NeuN for neurons (antibody raised against N-terminal 100 amino acids of human Fox3 (Biolegend, USA)), rabbit polyclonal antibody Iba1 (ionized calcium-binding adaptor molecule 1 (human Iba1, aa135–147)) for microglial staining, and mouse monoclonal antibody GA5 (Millipore) for GFAP for reactive astrocytes (Solomonov et al. 2016). Incubation with an avidin-biotin complex (Vector Laboratories) was used as secondary antibodies and the signal was developed using 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich). For each mouse, two to three sections were analyzed and three to four fields in the motor cortex and spinal cord were examined for each section. There were four mouse groups: WT, KL-OE, SOD1, and KL-OE/SOD1, and six mice per group.
Analysis of GFAP, Iba1, and NeuN immunostaining was as previously described (Cherry et al. 2016). Briefly, whole-stained brain and spinal cord sections were scanned and digitized using an Aperio ScanScope AT Turbo. Digital images were viewed and analyzed using Aperio ImageScope (Leica). Analyses of digital images were limited to the motor cortex and left and right anterior horns of the cervical and lumbar regions of the spinal cord. The areas of analysis were outlined using the Allen mouse atlas (http://mouse.brain-map.org/and http://mousespinal.brain-map.org/). A customized version of the Aperio positive pixel count algorithm (Version 9) was used to determine total GFAP-positive staining and to count the total number of Iba1- and NeuN-positive cells. Densities in units of count per square millimeter or pixels were obtained by standardizing results to the total area measured.
Monitoring Disease Progression
Males were singly housed and females were housed two per cage for all the experiments in accordance with previously published methods (Hatzipetros et al. 2015). The researchers were blind to genotype throughout the experiment.
Phenotypic Neurological Scoring System
Mice were assessed twice a week starting from age day 60 and daily from age day 80 for NS and weight. NS was determined based on the protocol described in Hatzipetros et al. (2015) on the scale of 0–4. A NS of 0 was assigned when mice had a normal gait during the walk as well as normal splay after they were suspended by the tail. We assigned a NS of 1 (the appearance of the first symptoms) if the gait was still normal but if we observed an abnormal splay, such as collapsed or partially collapsed hind limbs toward lateral midline or a hind limbs tremor during the suspension by the tail. The NS2 (onset of the disease) was assigned when during the tail suspension, the hind limbs are partially or completely collapsed, and the toes curl downwards or when the mouse drags any part of the foot during the walk. We defined NS3 (paralysis) when the mouse displayed a rigid paralysis of the hind limbs when suspended by the tail and the hind limbs were not being used to achieve forward motion. An NS3 mouse is still able to right itself up within 10 s when placed on either side. A humane end point (NS4) was assigned when no forward motion was observed due to extensive hind limbs paralysis and progressive weakness in the upper extremities that subsequently prevented the mouse from righting itself from either side within 10 s. This happens because of the extensive paralysis of the hind limbs and weakness in the upper extremities as a result of advancement of the disease. The mice were euthanized when they reached NS4.
Hanging Wire Grip Test
Grip testing scores were determined by latency of the hind limbs to detach from a wire grid panel after inversion. Briefly, mice were positioned on a panel and allowed to accommodate for 3 to 5 s before the panel was inverted over a cage containing 5 to 7 cm of soft bedding. Holding time was determined and recorded as the time it takes for the hind limbs to detach. The test was stopped after 30 s if the mouse did not detach. Mice were given three consecutive attempts and the final scores were calculated as the average of all three attempts with a highest possible score of 30 s. The test was performed twice a week (Ross et al. 2014; Weydt et al. 2003). Scores are presented as the mean of the mice cohort final scores ± standard error of the mean (SEM) for each time point.
Microglial Cultures
P2 mixed astroglia cells were isolated from KL-OE/SOD1 and SOD1 pups, as described previously (Chen et al. 2013). At the same time, the tail from individual pups was clipped into a microcentrifuge tube, and genomic DNA was isolated as described below in order to determine the genotype and correlate it with the astroglia which were collected into separate tubes until the genotype was assayed. The cells from each pup were plated in individual flasks and cultures were maintained in high-glucose DMEM culture medium (1% L-glutamine, 1% penicillin/streptavidin, and 20% fetal bovine serum). The medium for bathing of the cells was replaced with fresh medium every other day. At day 18 or 19, the microglia and astrocytes were separated using mild trypsinization as described (Saura et al. 2003). Briefly, after washing the cells with PBS, the mixed glial cultures were incubated with a trypsin solution (0.25% trypsin, 1 mM EDTA in HBSS; named henceforth trypsin 0.25%) and diluted 1:3 in DMEM-F12. The incubation for 30 min at 37 °C in the incubator resulted in the complete detachment of an upper cell layer in one piece which consisted of the astrocytes and was removed. At this point, the microglia remained attached to the bottom of the well and these adherent microglia were trypsinized and reseeded in 12-well plates (one well corresponding to the amount of cells obtained from one individual pup (collected and grown previously in individual flasks). Five to six pups for each condition were used in each experiment. This method generates a high yield and purity of microglia. Twenty-four hours after seeding, the microglia were incubated with LPS (200 ng/ml) and interferon gamma (30 ng/ml) for an additional 18–24 h (as indicated for each experiment), and media and cell lysates were collected for further analysis. For the exogenously added Klotho, we utilized rmKL that contains the shed ectodomain of Klotho (Ala35-Lys982, recombinant mouse Klotho, catalog no. 1819-KL-050 from R&D Systems, Minneapolis, MN) that was applied at a concentration of 0.4 μg/ml, 4 h before the exposure to LPS (Zeldich et al. 2014).
Nitrite Assay
Nitric oxide production by microglia was assessed by the Griess reagent kit (cat. number G-7921, Promega, USA) according to the manufacturer’s instructions. With this kit, a colorimetric detection of nitrite (NO2−) is achieved. Briefly, 150 μl of conditioned medium was incubated with 20 μl of Griess reagent and 130 μl of deionized water at RT for 30 min, and optical density was measured at 540 nm. The nitrite concentration was established based on a standard curve and normalized to the number of cells in the well as assessed by crystal violet staining.
ELISA for TNF-α, IL-6, and IL-1β
The amount of TNF-α, IL-6, and IL-1β released in 100 μl of conditioned medium was determined using ELISA duo set kits specific for the following mouse cytokines: DY410–05 for TNF-α, DY406–05 for IL-6, and DY401–05 for IL-1β (R&D Systems, USA). ELISA measurements were performed using the standard curves generated using standard proteins and instructions provided by the manufacturer. The results were normalized to the cell number in the well as assessed by crystal violet staining.
Crystal Violet Staining
Crystal violet staining of cells was performed as described before (Zeldich et al. 2007) and was used to assess the cell number used for normalization of the inflammatory mediators. Briefly, the cells were washed with PBS, fixed in 70% ethanol, and stained with 1% crystal violet. Unincorporated stain was removed by washing, cells were air-dried, and the dye was extracted with 70% ethanol and its absorbance at 550 nm was measured in a Microplate Reader (GloMax-Multi Detection System, Promega, CA, USA).
Western Blotting
Protein concentrations of microglial lysates were measured using the Micro BCA Protein Assay Reagent Kit (Pierce, Rockford, IL) according to the manufacturer’s protocol. For SDS-PAGE and western blots, 10 μg of cell lysates were processed as previously described. Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse, anti-rat, or anti-rabbit (1:5000, Kirkegaard & Perry Laboratories, Gaithersburg, MD). The primary antibodies used were anti-cyclooxygenase-2 (Cox-2) mouse monoclonal antibody (1:1000, Abcam), anti-iNOS mouse monoclonal antibody (1:1000, Abcam), and anti-β-actin monoclonal antibody (1:6000, Cell Signaling).
RNA Sequencing
Total RNA from three biologically independent samples each of WT and KL-OE microglia was submitted to GENEWIZ for ribosomal RNA depletion and library construction for Illumina sequencing. RNAseq libraries were all determined to be greater than 40 million reads deep and passing quality control by FastQC (Brown et al. 2017). After TMM normalization (trimmed mean of M values), differential gene expression analysis was performed with the EdgeR package (Anders et al. 2013) to derive expression fold changes and p value statistics.
Statistical Analyses and Graphical Representations of Data
All statistical analyses and visual and graphical representations were carried out separately for male and female cohorts. For visual representation of the comprehensive changes in body weight and hanging wire grip test, mean body weight and hanging wire grip test data were plotted for each cohort over the period of time until the point where the last animal in the group was euthanized. To avoid decomposition of average weight values as mice reached the humane end point, final weight measurements for each individual mouse were carried forward until the point in time when the last mouse in their cohort was euthanized. This practice is applied when analyzing survival through the longitudinal monitoring of weight and NS (Choi et al. 2008; Kaneb et al. 2011). We focused on the relevant summary measurements for weight, hanging wire grip test, and NS data as published before (Kaneb et al. 2011) and as recommended by Matthews et al. (1990) and performed statistical analyses on these measurements. We choose the time to attain a NS of 1, 2, and 3 in both hind limbs. The time to reach the humane end point (NS4) was taken to represent survival.
For the statistical analysis, one-way ANOVA with post hoc Tukey’s test was used for hanging wire grip test and weight analysis. The differences in days to reach NS was performed using Kaplan–Meier survival fit analyses followed by log-rank test that was utilized for statistical significance of survival. For the statistical analysis of the differences in the expression of inflammation and neuronal markers, we applied Student’s t test. The threshold for statistical significance was defined as p value < 0.05. The graphical representation was performed using GraphPad Prism 5 software.
Acknowledgments
The authors wish to thank Dr. Douglas Rosene for his help with statistical analysis.
Funding Information This work was supported by NIH grants R01-AG052465 to NCL and R56-AG051638 to CRA.
Abbreviations
- ADAM
A disintegrin and metalloprotease domain
- ALS
Amyotrophic lateral sclerosis
- ALS TDI
ALS Therapy Development Institute
- AD
Alzheimer’s disease
- Axin 2
Axin-related protein 2
- AHCs
Anterior horn cells
- C9ORF72
Chromosome 9 open reading frame 72
- CSF
Cerebrospinal fluid
- CCR2
C-C chemokine receptor type 2
- Cox-2
Cyclooxygenase-2
- FALS
Familial amyotrophic lateral sclerosis
- FGFR
Fibroblast growth factor receptors
- FOXO
Forkhead box O
- Fzd5
Frizzled gene family encoding 7-transmembrane domain proteins
- GFAP
Glial fibrillary acidic protein
- HBSS
Hank’s balanced salt solution
- Iba1
Ionized calcium-binding adaptor molecule 1
- KL-OE
Klotho-overexpressing
- IGF-1
Insulin-like growth factor 1
- IHC
Immunohistochemistry
- IL-1α
Interleukin-1α
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- IL-10
Interleukin-10
- IL-12a
Interleukin-12a
- iNOS
Inducible nitric oxide synthase
- MAG
Myelin-associated glycoprotein
- MBP
Myelin basic protein
- MN
Motor neurons
- MS
Multiple sclerosis
- Prx-2
Peroxiredoxin-2
- Prx-3
Peroxiredoxin-3
- PLP1
Proteolipid protein 1
- rmKL
Recombinant mouse Klotho
- NeuN
Neuronal nucle
- NF-κB
Nuclear factor-κB
- NOS
Nitric oxide synthase
- Nrf2
Nuclear factor erythroid 2-related factor 2
- NS
Neurological score
- SMAD
The activated type I receptors interact with and phosphorylate SMAD (an acronym for the fusion of Caenorhabditis elegans Sma genes and the Drosophila Mad) proteins to transduce signals
- SOD
Superoxide dismutase
- TDP-43
TAR DNA-binding protein 43
- TGF-β
Transforming growth factor-β
- TNF-α
Tumor necrosis factor-α
- TNFAIP2
TNF alpha-induced protein 2
- qRT-PCR
Quantitative reverse transcription polymerase chain reaction
- VEGF
Vascular endothelial growth factor
- Wnt
Wnt is an acronym that stands for “Wingless/Integrated”
- WT
Wild type
Footnotes
All animal procedures were performed in accordance with a protocol approved by the Boston University Institutional Animal Care and Use Committee.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abraham CR, Chen C, Cuny GD, Glicksman MA, Zeldich E (2012) Small-molecule Klotho enhancers as novel treatment of neurodegeneration. Future Med Chem 4:1671–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham CR, Mullen PC, Tucker-Zhou T, Chen CD, Zeldich E (2016) Klotho is a neuroprotective and cognition-enhancing protein. Vitam Horm 101:215–238 [DOI] [PubMed] [Google Scholar]
- Alexander GM, Erwin KL, Byers N, Deitch JS, Augelli BJ, Blankenhorn EP, Heiman-Patterson TD (2004) Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res 130:7–15 [DOI] [PubMed] [Google Scholar]
- Almer G, Vukosavic S, Romero N, Przedborski S (1999) Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 72:2415–2425 [DOI] [PubMed] [Google Scholar]
- Anamizu Y, Kawaguchi H, Seichi A, Yamaguchi S, Kawakami E, Kanda N, Matsubara S, Kuro-o M, Nabeshima Y, Nakamura K, Oyanagi K (2005) Klotho insufficiency causes decrease of ribosomal RNA gene transcription activity, cytoplasmic RNA and rough ER in the spinal anterior horn cells. Acta Neuropathol 109:457–466 [DOI] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD (2013) Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786 [DOI] [PubMed] [Google Scholar]
- Baluchnejadmojarad T, Eftekhari SM, Jamali-Raeufy N, Haghani S, Zeinali H, Roghani M (2017) The anti-aging protein klotho alleviates injury of nigrostriatal dopaminergic pathway in 6-hydroxydopamine rat model of Parkinson’s disease: involvement of PKA/CaMKII/CREB signaling. Exp Gerontol 100:70–76 [DOI] [PubMed] [Google Scholar]
- Bame M, Pentiak PA, Needleman R, Brusilow WS (2012) Effect of sex on lifespan, disease progression, and the response to methionine sulfoximine in the SOD1 G93A mouse model for ALS. Gend Med. 9:524–535 [DOI] [PubMed] [Google Scholar]
- Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, Appel SH, Henkel JS (2011) Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun 25:1025–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer V, Stevens JMG, Deschner T, Sonnweber R, Hohmann G (2018) Aging and sex affect soluble alpha klotho levels in bonobos and chimpanzees. Front Zool 15:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 312:1389–1392 [DOI] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH Jr (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13:1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brobey RK, German D, Sonsalla PK, Gurnani P, Pastor J, Hsieh CC, Papaconstantinou J, Foster PP, Kuro-o M, Rosenblatt KP (2015) Klotho protects dopaminergic neuron oxidant-induced degeneration by modulating ASK1 and p38 MAPK signaling pathways. PLoS One 10:e0139914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Pirrung M, McCue LA (2017) FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33:3137–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O, Lawson RJ, Frosch MP, Pochet N, Fatimy RE, Krichevsky AM, Gygi SP, Lassmann H, Berry J, Cudkowicz ME, Weiner HL (2015) Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol 77:75–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacabelos D, Ramirez-Nunez O, Granado-Serrano AB, Torres P, Ayala V, Moiseeva V, Povedano M, Ferrer I, Pamplona R, Portero-Otin M, Boada J (2016) Early and gender-specific differences in spinal cord mitochondrial function and oxidative stress markers in a mouse model of ALS. Acta Neuropathol Commun 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira C, Oliveira AF, Cunha C, Vaz AR, Falcao AS, Fernandes A, Brites D (2014) Microglia change from a reactive to an age-like phenotype with the time in culture. Front Cell Neurosci 8:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Guan Y, Zhang Z, Liu H, Wang S, Yu L, Wu X, Wang X (2012) Wnt signaling pathway is involved in the pathogenesis of amyotrophic lateral sclerosis in adult transgenic mice. Neurol Res 34:390–399 [DOI] [PubMed] [Google Scholar]
- Chen CD, Sloane JA, Li H, Aytan N, Giannaris EL, Zeldich E, Hinman JD, Dedeoglu A, Rosene DL, Bansal R, Luebke JI, Kuro-o M, Abraham CR (2013) The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci 33: 1927–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Zeldich E, Li Y, Yuste A, Abraham CR (2018) Activation of the anti-aging and cognition-enhancing gene klotho by CRISPR-dCas9 transcriptional effector complex. J Mol Neurosci 64:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MF, Chen LJ, Niu HS, Yang TT, Lin KC, Cheng JT (2015) Signals mediating Klotho-induced neuroprotection in hippocampal neuronal cells. Acta Neurobiol Exp (Wars) 75:60–71 [PubMed] [Google Scholar]
- Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 4:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CI, Lee YD, Gwag BJ, Cho SI, Kim SS, Suh-Kim H (2008) Effects of estrogen on lifespan and motor functions in female hSOD1 G93A transgenic mice. J Neurol Sci 268:40–47 [DOI] [PubMed] [Google Scholar]
- Crosio C, Valle C, Casciati A, Iaccarino C, Carri MT (2011) Astroglial inhibition of NF-kappaB does not ameliorate disease onset and progression in a mouse model for amyotrophic lateral sclerosis (ALS). PLoS One 6:e17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 72:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Moreau C, Lassalle P, Perez T, De Seze J, Brunaud-Danel V, Destee A, Tonnel AB, Just N (2004) Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 62:2127–2129 [DOI] [PubMed] [Google Scholar]
- Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, Shiizaki K, Gotschall R, Schiavi S, Yorioka N, Takahashi M, Boothman DA, Kuro-o M (2011) Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem 286:8655–8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Zhu L, Sanchez PE, Worden K, Broestl L, Johnson E, Ho K, Yu GQ, Kim D, Betourne A, Kuro OM, Masliah E, Abraham CR, Mucke L (2015) Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J Neurosci 35:2358–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duce JA, Hollander W, Jaffe R, Abraham CR (2006) Activation of early components of complement targets myelin and oligodendrocytes in the aged rhesus monkey brain. Neurobiol Aging 27:633–644 [DOI] [PubMed] [Google Scholar]
- Duce JA, Podvin S, Hollander W, Kipling D, Rosene DL, Abraham CR (2008) Gene profile analysis implicates Klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia. 56:106–117 [DOI] [PubMed] [Google Scholar]
- German DC, Khobahy I, Pastor J, Kuro OM, Liu X (2012) Nuclear localization of Klotho in brain: an anti-aging protein. Neurobiol Aging 33(1483):e25–e30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill A, Kidd J, Vieira F, Thompson K, Perrin S (2009) No benefit from chronic lithium dosing in a sibling-matched, gender balanced, investigator-blinded trial using a standard mouse model of familial ALS. PLoS One 4:e6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26:8774–8786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhuang X, Huang Z, Zou J, Yang D, Hu X, Du Z, Wang L, Liao X (2018) Klotho protects the heart from hyperglycemia-induced injury by inactivating ROS and NF-kappaB-mediated inflammation both in vitro and in vivo. Biochim Biophys Acta 1864:238–251 [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 264:1772–1775 [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME (1998) Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 23:249–256 [DOI] [PubMed] [Google Scholar]
- Halleskog C, Mulder J, Dahlstrom J, Mackie K, Hortobagyi T, Tanila H, Kumar Puli L, Farber K, Harkany T, Schulte G (2011) WNT signaling in activated microglia is proinflammatory. Glia. 59:119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzipetros T, Kidd JD, Moreno AJ, Thompson K, Gill A, Vieira FG (2015) A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH (2004) Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 55:221–235 [DOI] [PubMed] [Google Scholar]
- Hinman JD, Abraham CR (2007) What’s behind the decline? The role of white matter in brain aging. Neurochem Res 32:2023–2031 [DOI] [PubMed] [Google Scholar]
- Hinman JD, Duce JA, Siman RA, Hollander W, Abraham CR (2004) Activation of calpain-1 in myelin and microglia in the white matter of the aged rhesus monkey. J Neurochem 89:430–441 [DOI] [PubMed] [Google Scholar]
- Hinman JD, Peters A, Cabral H, Rosene DL, Hollander W, Rasband MN, Abraham CR (2006) Age-related molecular reorganization at the node of Ranvier. J Comp Neurol 495:351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui H, Zhai Y, Ao L, Cleveland JC Jr, Liu H, Fullerton DA, Meng X (2017) Klotho suppresses the inflammatory responses and ameliorates cardiac dysfunction in aging endotoxemic mice. Oncotarget. 8:15663–15676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima M, Rakugi H, Ishikawa K, Maekawa Y, Yamamoto K, Ohta J, Chihara Y, Kida I, Ogihara T (2006) Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun 339:827–832 [DOI] [PubMed] [Google Scholar]
- Jana M, Pahan K (2013) Down-regulation of myelin gene expression in human oligodendrocytes by nitric oxide: implications for demyelination in multiple sclerosis. J Clin Cell Immunol 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Lv P, Chen G, Wang P, Zuo Z, Ren L, Bi J, Yang CW, Mei X, Han D (2017) Klotho ameliorates cyclosporine A-induced nephropathy via PDLIM2/NF-kB p65 signaling pathway. Biochem Biophys Res Commun 486:451–457 [DOI] [PubMed] [Google Scholar]
- Kaneb HM, Sharp PS, Rahmani-Kondori N, Wells DJ (2011) Metformin treatment has no beneficial effect in a dose-response survival study in the SOD1(G93A) mouse model of ALS and is harmful in female mice. PLoS One 6:e24189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE (2013) Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16:571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, Chen C, Huang MM, Zeldich E, Brazee PL, Schuman ER, Robin M, Cuny GD, Glicksman MA, Abraham CR (2012) Identification of novel small molecules that elevate Klotho expression. Biochem J 441:453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krick S, Baumlin N, Aller SP, Aguiar C, Grabner A, Sailland J, Mendes E, Schmid A, Qi L, David NV, Geraghty P, King G, Birket SE, Rowe SM, Faul C, Salathe M (2017) Klotho inhibits interleukin-8 secretion from cystic fibrosis airway epithelia. Sci Rep 7:14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M (2009) Klotho and aging. Biochim Biophys Acta 1790:1049–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 390:45–51 [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M (2005) Suppression of aging in mice by the hormone Klotho. Science. 309:1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Jeon GS, Shim YM, Seong SY, Lee KW, Sung JJ (2015) Modulation of SOD1 subcellular localization by transfection with wild- or mutant-type SOD1 in primary neuron and astrocyte cultures from ALS mice. Exp Neurobiol. 24:226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hyeon SJ, Im H, Ryu H, Kim Y, Ryu H (2016) Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp Neurobiol 25:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KE, Rasmussen AL, Bennett W, King A, West AK, Chung RS, Chuah MI (2014) Microglia and motor neurons during disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis: changes in arginase1 and inducible nitric oxide synthase. J Neuroinflammation 11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Xu W, Luo C, Gozal D, Liu R (2003) VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Brain Res Mol Brain Res 111:155–164 [DOI] [PubMed] [Google Scholar]
- Li X, Guan Y, Chen Y, Zhang C, Shi C, Zhou F, Yu L, Juan J, Wang X (2013) Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int J Clin Exp Pathol 6:1245–1260 [PMC free article] [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH (2012) Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol 237:147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T (2007) Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 317:803–806 [DOI] [PubMed] [Google Scholar]
- Masso A, Sanchez A, Bosch A, Gimenez-Llort L, Chillon M (2018) Secreted alphaKlotho isoform protects against age-dependent memory deficits. Mol Psychiatry 23(9):1937–1947 [DOI] [PubMed] [Google Scholar]
- Matthews JN, Altman DG, Campbell MJ, Royston P (1990) Analysis of serial measurements in medical research. BMJ. 300:230–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCombe PA, Henderson RD (2010) Effects of gender in amyotrophic lateral sclerosis. Gend Med 7:557–570 [DOI] [PubMed] [Google Scholar]
- Molina M, Ortega G, Perez Gracia A, Saez JA (1989) Spontaneous bacterial empyema and hepatic cirrhosis. Enferm Infecc Microbiol Clin 7:516. [PubMed] [Google Scholar]
- Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima Y, Nabeshima T (2003) Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J 17:50–52 [DOI] [PubMed] [Google Scholar]
- Needleman P, Manning PT (1999) Interactions between the inducible cyclooxygenase (COX-2) and nitric oxide synthase (iNOS) pathways: implications for therapeutic intervention in osteoarthritis. Osteoarthr Cartil 7:367–370 [DOI] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D, Carmeliet P (2001) Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 28:131–138 [DOI] [PubMed] [Google Scholar]
- Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A, Cudkowicz M, Atassi N (2014) Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener 15:453–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parakh S, Atkin JD (2016) Protein folding alterations in amyotrophic lateral sclerosis. Brain Res 1648:633–649 [DOI] [PubMed] [Google Scholar]
- Pedersen L, Pedersen SM, Brasen CL, Rasmussen LM (2013) Soluble serum Klotho levels in healthy subjects. Comparison of two different immunoassays. Clin Biochem 46:1079–1083 [DOI] [PubMed] [Google Scholar]
- Peters OM, Ghasemi M, Brown RH Jr (2015) Emerging mechanisms of molecular pathology in ALS. J Clin Invest 125:1767–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Bento-Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, Hersmus N, Kusters B, Van Den Bosch L, Van Damme P, Richardson WD, Robberecht W (2013) Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain. 136: 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrishevsky E, Hong RH, Mackenzie IR, Cashman NR (2017) Spinal cord homogenates from SOD1 familial amyotrophic lateral sclerosis induce SOD1 aggregation in living cells. PLoS One 12:e0184384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MS (2012) The role of Klotho in energy metabolism. Nat Rev Endocrinol 8:579–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EK, Winter AN, Wilkins HM, Sumner WA, Duval N, Patterson D, Linseman DA (2014) A cystine-rich whey supplement (Immunocal((R))) delays disease onset and prevents spinal cord glutathione depletion in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Antioxidants (Basel) 3:843–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samms RJ, Cheng CC, Kharitonenkov A, Gimeno RE, Adams AC (2016) Overexpression of beta-klotho in adipose tissue sensitizes male mice to endogenous FGF21 and provides protection from diet-induced obesity. Endocrinology. 157:1467–1480 [DOI] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J (2003) High-yield isolation of murine microglia by mild trypsinization. Glia. 44:183–189 [DOI] [PubMed] [Google Scholar]
- Semba RD, Cappola AR, Sun K, Bandinelli S, Dalal M, Crasto C, Guralnik JM, Ferrucci L (2011) Plasma klotho and cardiovascular disease in adults. J Am Geriatr Soc 59:1596–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki M, Yoshimura K, Shibata M, Koike M, Matsuura N, Uchiyama Y, Gotow T (2008) Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience. 152:924–941 [DOI] [PubMed] [Google Scholar]
- Shvil N, Banerjee V, Zoltsman G, Shani T, Kahn J, Abu-Hamad S, Papo N, Engel S, Bernhagen J, Israelson A (2018) MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: implications for familial ALS. Cell Death Dis 9:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane JA, Hinman JD, Lubonia M, Hollander W, Abraham CR (2003) Age-dependent myelin degeneration and proteolysis of oligodendrocyte proteins is associated with the activation of calpain-1 in the rhesus monkey. J Neurochem 84:157–168 [DOI] [PubMed] [Google Scholar]
- Solomonov Y, Hadad N, Levy R (2016) Reduction of cytosolic phospholipase A2alpha upregulation delays the onset of symptoms in SOD1G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation 13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J (2010) Neuronal death in amyotrophic lateral sclerosis (ALS): what can we learn from genetics? CNS Neurol Disord Drug Targets 9:259–267 [DOI] [PubMed] [Google Scholar]
- Tosolini AP, Sleigh JN (2017) Motor neuron gene therapy: lessons from spinal muscular atrophy for amyotrophic lateral sclerosis. Front Mol Neurosci 10:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YA (2006) Klotho, the long sought-after elixir and a novel tumor suppressor? Cancer Biol Ther 5:20–21 [DOI] [PubMed] [Google Scholar]
- Weydt P, Hong SY, Kliot M, Moller T (2003) Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 14: 1051–1054 [DOI] [PubMed] [Google Scholar]
- Xin YJ, Yuan B, Yu B, Wang YQ, Wu JJ, Zhou WH, Qiu Z (2015) Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress. Sci Rep 5:7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, Miyoshi M, Ogawa Y, Castrillon DH, Rosenblatt KP, Kuro-o M (2005) Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem 280:38029–38034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Guan Y, Wu X, Chen Y, Liu Z, Du H, Wang X (2013) Wnt signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem Res 38:1904–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldich E, Koren R, Nemcovsky C, Weinreb M (2007) Enamel matrix derivative stimulates human gingival fibroblast proliferation via ERK. J Dent Res 86:41–46 [DOI] [PubMed] [Google Scholar]
- Zeldich E, Chen CD, Colvin TA, Bove-Fenderson EA, Liang J, Tucker Zhou TB, Harris DA, Abraham CR (2014) The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem 289:24700–24715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldich E, Chen CD, Avila R, Medicetty S, Abraham CR (2015) The anti-aging protein Klotho enhances remyelination following cuprizone-induced demyelination. J Mol Neurosci 57:185–196 [DOI] [PubMed] [Google Scholar]
- Zhou HJ, Li H, Shi MQ, Mao XN, Liu DL, Chang YR, Gan YM, Kuang X, Du JR (2017) Protective effect of Klotho against ischemic brain injury is associated with inhibition of RIG-I/NF-kappaB signaling. Front Pharmacol 8:950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HJ, Zeng CY, Yang TT, Long FY, Kuang X, Du JR (2018) Lentivirus-mediated klotho up-regulation improves aging-related memory deficits and oxidative stress in senescence-accelerated mouse prone-8 mice. Life Sci 200:56–62 [DOI] [PubMed] [Google Scholar]
- Zhu L, Stein LR, Kim D, Ho K, Yu GQ, Zhan L, Larsson TE, Mucke L (2018) Klotho controls the brain-immune system interface in the choroid plexus. Proc Natl Acad Sci U S A 115:E11388–E11396 [DOI] [PMC free article] [PubMed] [Google Scholar]