

Abstract
Background:
N1303K, one of the common, severe disease-causing mutations in the CFTR gene, causes both defective biogenesis and gating abnormalities of the CFTR protein. The goals of the present study are to quantitatively assess the gating defects associated with the N1303K mutation and its pharmacological response to CFTR modulators including potentiators VX-770 and GLPG1837 and correctors VX-809, and VX-661.
Methods:
Gating behavior and pharmacological responses to CFTR potentiators were assessed using the patch-clamp technique in the excised, inside-out mode. We also examined the effects of GLPG1837, VX-770, VX-809 and VX-661 on N1303K-CFTR surface expression using Western blot analysis.
Results:
Like wild-type (WT) CFTR, N1303K-CFTR channels were activated by protein kinase A-dependent phosphorylation, but the open probability (Po) of phosphorylated N1303K-CFTR was extremely low (~0.03 vs ~0.45 in WT channels). N1303K mutants showed abnormal responses to ATP analogs or mutations that disrupt ATP hydrolysis and/or dimerization of CFTR’s two nucleotide-binding domains (NBDs). However, the Po of N1303K-CFTR was dramatically increased by GLPG1837 (~17-fold) and VX-770 (~8-fold). VX-809 or VX-661 enhanced N1303K-CFTR maturation by 2 – 3 fold, and co-treatment with GLPG1837 or VX-770 did not show any negative drug-drug interaction.
Conclusion:
N1303K has a severe gating defect, reduced ATP-dependence and aberrant response to ATP analogs. These results suggest a defective function of the NBDs in N1303K-CFTR. An improvement of channel function by GLPG1837 or VX-770 and an increase of Band C protein by VX-809 or VX-661 support a therapeutic strategy of combining CFTR potentiator and corrector for patients carrying the N1303K mutation.
Keywords: Cystic Fibrosis, N1303K, gating, GLPG1837, VX-770, VX-809, VX-661
1. Introduction
Cystic Fibrosis (CF) is an autosomal recessive genetic disease caused by mutations in the gene for the Cystic Fibrosis Transmembrane conductance Regulator (CFTR), a phosphorylation-activated but ATP-gated anion channel predominantly expressed in epithelial cells [1, 2]. CFTR regulates the balance of salt and water across epithelial cells lining the exocrine glands, such as the submucosal glands in the lungs, the pancreatic ducts and the sweat glands. N1303K (asparagine-to-lysine mutation at position 1303) is a common, severe CF disease-causing mutation in the CFTR gene (Cystic Fibrosis Foundation Patient Registry 2016 Annual Data Report). As the newly resolved human CFTR structure has shown [3], N1303 is at the equivalent position, but in the opposite half of the CFTR molecule, as F508, deletion of which (F508del) constitutes the most common pathogenic mutation in CF. Like F508del, N1303K is a Class II folding defect mutation, which results in a reduced number of N1303K channels in the cell membrane [4–6]. Both of these common, Class II folding defect mutations also show gating defects once they reach the cell membrane [7–10]. For mutations with both trafficking and gating defects, one therapeutic strategy is to use a combination of CFTR correctors (e.g., VX-809 or Lumacaftor, [11] that increase the number of channels in the cell membrane and CFTR potentiators (e.g., VX-770 or Ivacaftor, [12]) that enhance the activity or open probability (Po) of the CFTR channel.
Although the FDA approved the use of Orkambi (Lumacaftor plus Ivacaftor) for the treatment of patients homozygous for the F508del mutation [13], the clinical benefits are somehow limited partly due to the undesirable drug-drug interaction: Chronic application of Ivacaftor dampens the effects of Lumacaftor [14, 15] and the mechanisms are complex and may be mutation-specific [16–18]. While clinical trials for use of ivacaftor in combination with other first- and next-generation correctors for F508del heterozygotes bearing a minimal function mutation (e.g. N1303K) on their second allele are ongoing, in vitro results establishing effects of potentiator VX-770 on the N1303K mutation have not been published. In addition, prior to the discovery of efficacious CFTR potentiators, functional studies of CFTR mutants may have underestimated the severity of gating defects associated with these mutations (see ref. [19] for example). Thus, although Berger et. al (2002) showed a PO of ~0.1 for N1303K [7], this should be considered as a maximal value before more thorough studies are possible.
Recent development of new CFTR potentiators provides powerful tools that afford more accurate assessment of the gating abnormalities caused by CFTR mutations. For example, Yeh et al. (2017) showed that the potentiator GLPG1837 is ~3-fold more efficacious than VX-770 (ivacaftor) on G551D-CFTR [20], the third most common pathogenic mutation with distinct gating defects [21]. Interestingly, the same report also provided evidence that GLPG1837 and VX-770 share a similar mechanism of action perhaps by binding to the same binding site. Despite its lower potency than VX-770, the high efficacy of GLPG1837, if also true for the N1303K mutation, should make this compound particularly valuable for a more accurate quantification of the gating defect associated with N1303K-CFTR.
In this present study, we used the CFTR potentiator GLPG1837 as a tool to estimate a maximal PO of ~0.03 for N1303K. This gating defect likely is caused by dysfunction of CFTR’s NBDs as N1303K-CFTR responds to ATP and ATP analogs very differently from WT channels. Furthermore, mutations disrupting ATP hydrolysis or NBD dimerization fail to affect N1303K channel gating. However, CFTR potentiators such as GLPG1837 and VX-770 dramatically improved the gating function of N1303K-CFTR, suggesting a clinical usefulness of these reagents. Furthermore, we showed that the surface expression of N1303K-CFTR can be enhanced by CFTR correctors VX-809 or VX-661. Therapeutic strategy of combining CFTR potentiator and corrector for patients carrying the N1303K mutation, and the potential structural mechanism for the gating defects caused by the mutation will be discussed.
2. Materials and Methods
2.1. Cell culture and transfection
Chinese hamster ovary (CHO) cells were grown in 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. The cDNA construct of N1303K-CFTR was co-transfected with pEGFP-C3 (Clontech Laboratories, Inc.) encoding the green fluorescence protein using PolyFect transfection reagent (QIAGEN) according to the manufacturer’s instructions. The transfected CHO cells were plated on sterile glass chips in 35-mm tissue culture dishes and incubated at 27°C for 2 – 6 days before patch-clamp experiments.
2.2. Western Blot Analysis
CHO cells, in 35mm dishes, were transfected with various DNA construct using X-tremeGENE (Roche). Six hours after transfection, drugs were added to the medium to desired concentrations. Cells were lysed 18 hours post drug treatment using 1×SDS loading buffer. Cell lysates were sheared by pushed through 18G needles. Whole cell lysate were separated in 4~20% gradient gels (Bio-Rad Laboratories) and transferred onto nitrocellulose membranes. The membranes were blocked with 5% milk in TBST buffer (20mM Tris, 137mM NaCl, 0.1% Tween 20) at 4°C overnight. The membranes were then probed with anti-CFTR antibody (1:3000 dilution) (AB596 from Cystic Fibrosis Foundation Therapeutics) and anti-Vimentin antibody (1:3000 dilution) (Santa Cruz Biotechnology) at room temperature for two hours. The membranes were washed with TBST five times and then incubated with anti-mouse IgG, HRP linked antibody (Cell Signaling Technology) at room temperature for one hour. The membranes were washed three times with TBST and developed with chemiluminescence reagent (Thermo Scientific). The luminescence was detected by a Molecular Image Chemi Doc (Bio-Rad Laboratories).
2.3. Electrophysiological experiments
Details of patch-clamp experiments were described in our previous publication [22]. The pipette solution contained (in mM): 140 NMDG chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2, and 10 HEPES, pH 7.4 with NMDG. Cells were perfused with a bath solution containing (in mM): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 HEPES, and 20 sucrose, pH 7.4 with NaOH. After the establishment of an inside-out configuration, the patch was perfused with a solution containing (in mM): 150 NMDG-Cl, 2 MgCl2, 10 EGTA, and 8 Tris, pH 7.4 with NMDG.
CFTR channel currents in inside-out patches were recorded at room temperature with an EPC-10 patch clamp amplifier, filtered at 100 Hz with an eight-pole Bessel filter (Warner Instrument Corp.) and captured onto a hard disk at a sampling frequency of 500 Hz. The membrane potential was held at −30 or −50 mV and the inward current was inverted for clear data presentation. CFTR was first activated by cytoplasmic application of 25 U ml−1 PKA catalytic subunit (Sigma) plus 2 mM ATP until the current reached the steady-state (~30 mins.) before changing the cytoplasmic solutions.
2.4. Data analysis and statistics
We used Igor-Pro (WaveMetrics) to measure steady-state mean currents (IM) for macroscopic analyses. For microscopic analyses, recordings with less than 5 visible simultaneously-opening steps in the presence of potentiator were further filtered off-line at 50 Hz with a digital filter and used for single channel kinetic analysis using a program developed by Dr. Csanády [10, 23]. The inverse of the resulting kinetic parameters, mean burst time (τb) and interburst time (τib), correspond to the closing and opening rate of CFTR, respectively. For the sake of convenience, investigators in the field have used τo and τc to represent these kinetic parameters [22, 24, 25]. Of note, because of the uncertainty in assigning the number of active channels in the patch, this method inevitably underestimates the number of functional channels in the patch, hence results in an overestimation of the Po. Therefore, the reported Po value for N1301K-CFTR should be considered as the maximal Po.
Data are presented as means ± SEM. ANOVA and Student’s t test were performed for statistical analysis using Microsoft Excel. p < 0.05 was considered significant.
2.5. Reagents
Mg-ATP, PKA and 2’-deoxy ATP (2’-dATP) were purchased from Sigma (Saint Louis, MO, USA). N6–(2-phenylethyl)-ATP (P-ATP) was purchased from Biolog Life Science Institute (Bremen, Germany). VX-770 and VX-661 were purchased from Selleck Chemicals (Houston, TX, USA). VX-809 was purchased from AOBIOUS INC (Gloucester, MA, USA). GLPG1837 was provided by Galapagos/AbbVie.
3. Results
3.1. Pharmacological responses of N1303K to the CFTR potentiator GLPG1837
Moderate gating defects have been reported for the few N1303K channels that reach the cell membrane [7, 8]. Here, we used the CFTR potentiator GLPG1837 to quantify the gating defects associated with N1303K-CFTR. WT-CFTR data (Fig. 1A, C and D), reported in Yeh et al. (2017) [20], were presented for comparison. Although 3 μM, a saturating concentration of GLPG1837 for WT-CFTR, increased ATP-induced currents of phosphorylated WT-CFTR by ~ 2-fold (Fig. 1A, D), the N1303K channels showed a lower sensitivity to GLPG1837 as seen in Fig. 1B: Increasing [GLPG1837] from 1 to 6 to 20 μM resulted in an incremental increase of the macroscopic N1303K-CFTR currents. A return of the macroscopic currents to the baseline level after application of the selective CFTR inhibitor CFTRInh-172 ensures that the macroscopic currents observed are from CFTR. Fold increase in macroscopic currents was calculated as the ratio of steady-state currents (IM) with potentiator GLPG1837 to IM without potentiator. In contrast to a maximal 2-fold increase of WT-CFTR current by GLPG1837, macroscopic N1303K-CFTR currents increase 16.8 ± 1.8 (n = 6) fold in the presence of 20.0 μM GLPG1837 (summarized in Fig. 1C). This ~17-fold increase of the macroscopic N1303K-CFTR currents sets an upper limit of PO at 0.06 (1/17) in the absence of the potentiator.
Fig. 1.
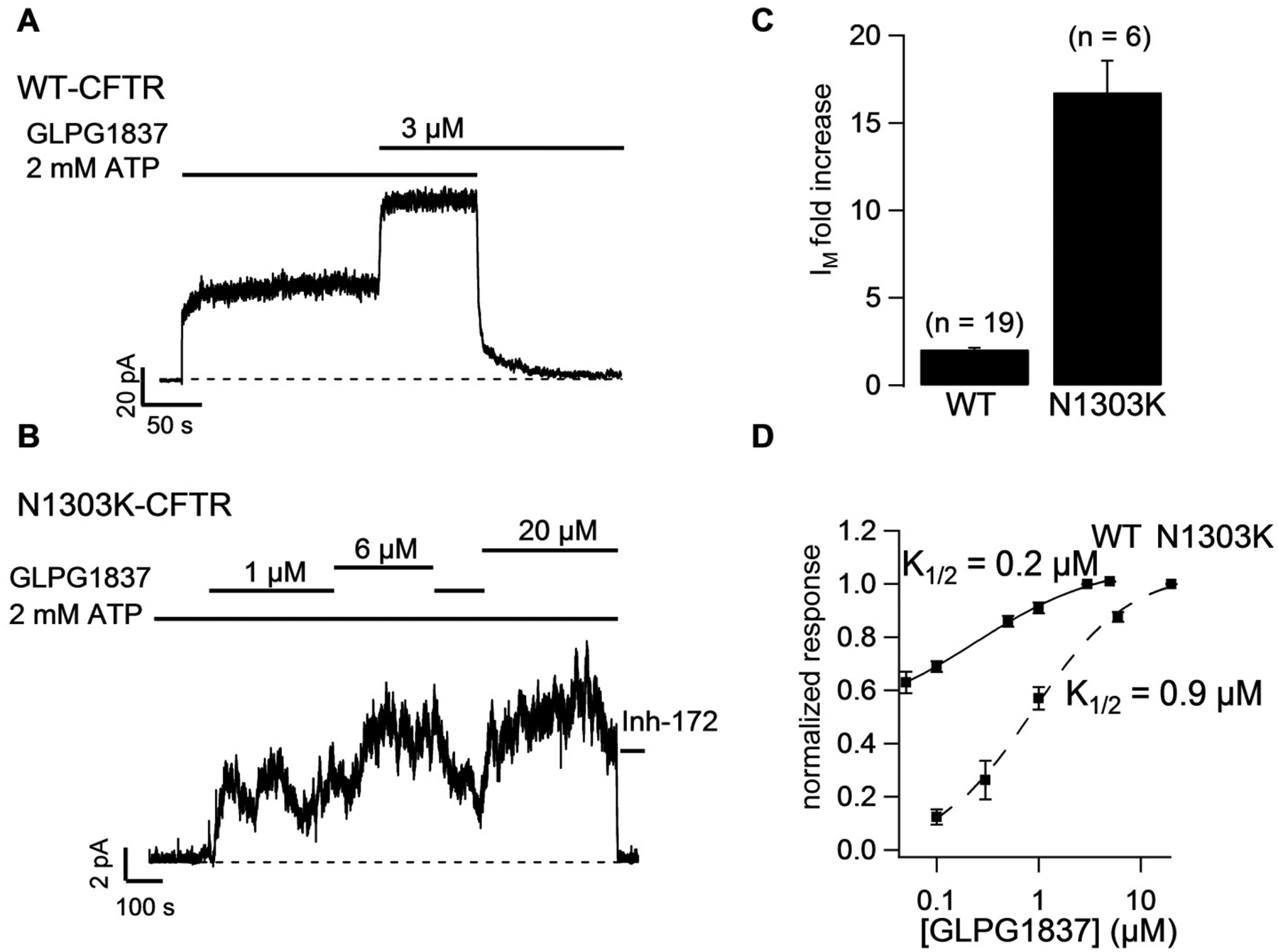
Macroscopic recordings of WT-CFTR (A) or N1303K-CFTR (B) showing response to potentiator GLPG1837. Channels were prephosphorylated with PKA + ATP and then exposed to GLPG1837. The bars indicate the presence of ATP, GLPG1837 and CFTRinh-172 (Inh-172) in the intracellular solution and the dashed lines represent the baseline. N1303K channel currents increase 16.8-fold ± 1.8 (n = 6) in response to a saturating concentration of GLPG1837, indicating a PO for N1303K < 0.06. (C) Quantification of the fold increase in mean current (IM) in response to saturating concentrations of GLPG1837. (D) GLPG1837 concentration response curves (n = 3 – 8 for each data point). The dose response curve is shifted to the right for N1303K-CFTR (K1/2 = 0.9 μM cf. 0.2 μM for WT). Note data on WT-CFTR are extracted from ref. [20].
The fold increase of N1303K-CFTR currents by various concentrations of GLPG1837 was normalized to the effect at the saturating dose (20.0 μM), and subsequently fitted with the Hill equation yielding a K1/2 of 0.9 μM (Fig. 1D; cf. 0.2 μM for WT-CFTR) and a Hill coefficient of 0.92. Of note, our previous studies demonstrated a state-dependent binding of GLPG1837 on CFTR: open states bind GLPG1837 more tightly than closed states [20]. Thus, a rightward shift of the dose response relationship for the N1303K mutation is consistent with the idea that the Po of N1303K-CFTR must be very low.
3.2. Microscopic gating kinetics of N1303K-CFTR
While the macroscopic analyses described above (Fig. 1) provide a reasonable estimation of the maximally possible PO for N1303K channels in the absence of potentiator, kinetic analysis of microscopic current in patches containing fewer than 5 channels provides a much closer assessment of the actual PO for N1303K channels in the presence of potentiator. Figure 2A shows a representative recording of at least three N1303K-CFTR channels. In the absence of potentiator, only one opening event was seen in 30 seconds of the trace, making it virtually impossible to estimate the Po. However, application of 20.0 μM GLPG1837 drastically increased channel activity thus increasing the probability of simultaneous channel opening events. This then allows us to more accurately estimate the number of functional channels in the patch. (Of note, although this method improves accuracy, the number of active channels may still be underestimated.) Kinetic analysis of data of at least 1 – 2 minutes from such patches revealed a PO of 0.44 ± 0.04 (n = 4) in the presence of 20.0 μM GLPG1837 and 2 mM ATP (Fig. 2B). We then back-calculated the PO(app) of N1303K channels in the absence of potentiator by dividing the PO obtained in the presence of potentiator by the fold increase in mean current (IM) by GLPG1837 (17-fold) to yield a PO(app) of ≤ 0.03 for N1303K-CFTR in the absence of potentiator. Interestingly, in addition to a prolonged closed time (Fig. 2B), our analysis also revealed a paradoxical increase of the mean open time (τo) even without GLPG1837 (3.1 ± 0.8 s n = 6 in Fig. 2B; ~400 ms for WT-CFTR), consistent with results in a previous report for N1303K [7]. The mean open time was further prolonged to 9.1 ± 3.1 s (n = 4) by GLPG1837 (vs. ~850 ms for WT-CFTR). Thus, the N1303K mutation causes a more severe gating defect than previously reported [7].
Fig. 2.
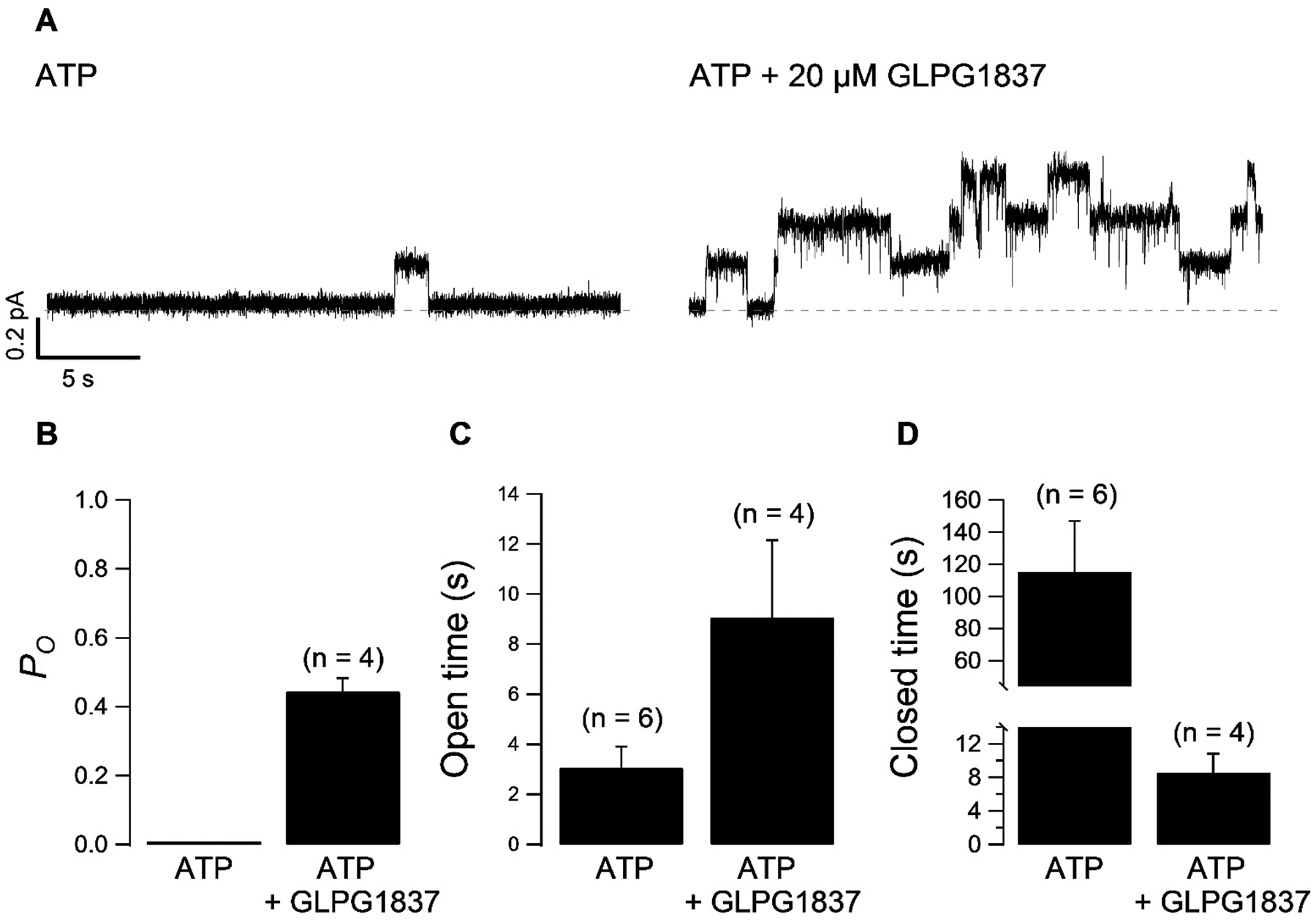
(A) Microscopic recording of N1303K-CFTR in 2 mM ATP or 2 mM ATP + 20 μM GLPG1837. (B) Quantification of gating parameters PO (B) mean open time (τO) (C) mean closed time (τO) (D) in the presence of ATP or ATP + GLPG1837. Mean closed time in ATP was calculated based on PO = τO/(τO + τC).
3.3. Response of N1303K-CFTR to ATP and ATP analogs
N1303 is located in CFTR’s NBD2, which plays a critical role in controlling ATP-dependent opening and closing of the channel [1]. We therefore hypothesized that the gating defect associated with N1303K may be due to problems with how the mutant channel uses ATP as a ligand. We hence tested the N1303K mutant’s response to ATP and to ATP analogs 2’-deoxy ATP (2’-dATP) and N6-(2-phenylethyl)-ATP (P-ATP) in the continuous presence of GLPG1837. (Of note, the constant inclusion of GLPG1837 in the system ensures macroscopic currents that afford more accurate quantification.) In Fig. 3A, washout of ATP in the presence of GLPG1837 resulted in a 35.5 ± 2.8% (n = 4) decrease in potentiated IM, a result in stark contrast to the response of WT-CFTR channels to ATP removal shown in Fig. 1A. This observation indicates that N1303K channels do respond to ATP but the ATP dependence is much less than WT-CFTR. The hypothesis that the N1303K mutation results in an abnormal function of NBDs is further supported by the data shown in Fig. 3B and 3C. While it is known that d-ATP and P-ATP are more efficacious ligands than ATP for WT-CFTR gating [22, 26] exchanging ATP for d-ATP (Fig. 3B) in the presence of GLPG1837 does not increase the currents of N1303K. More strikingly, replacing ATP with the high-affinity ATP analog P-ATP (Fig. 3C) actually decreases the N1303K currents by 39.0 ± 7.0% (n = 4). Interestingly, the effects of the ATP analogues d-ATP and P-ATP on N1303K-CFTR contrast with their actions on other CF mutants (e.g. G551D and G1349D [27] and F508del [10]).
Fig. 3.
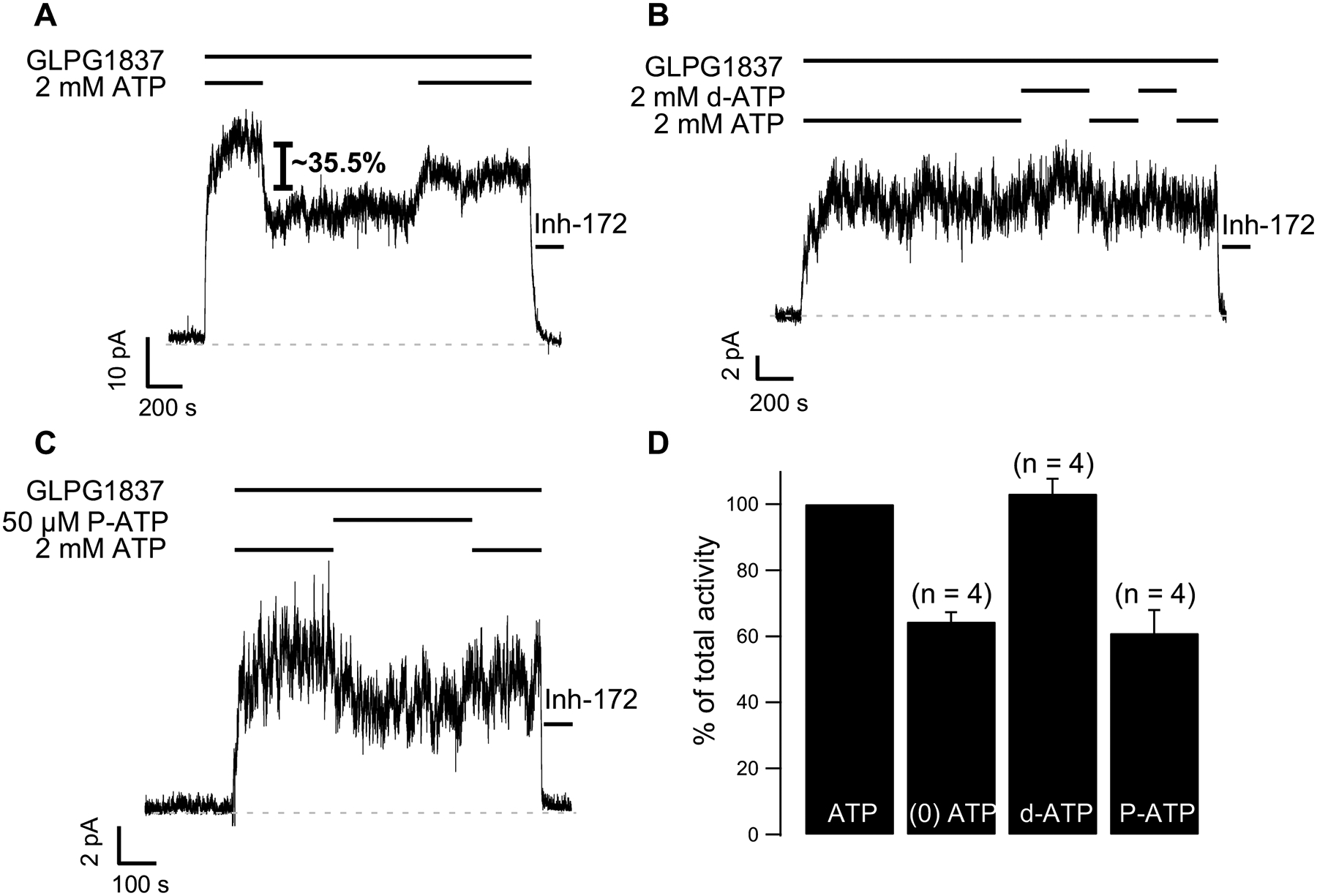
Macroscopic recordings of N1303K-CFTR showing its response to ATP (A) or ATP analogs P-ATP (B) or d-ATP (C) in the continuous presence of GLPG1837. (D) Quantification of steady-state IM in 2 mM ATP, ATP-free solution, d-ATP or P-ATP presented as percent of IM in ATP.
3.4. Second-site mutations G551D, G1349D and E1371S have negligible effect on N1303K mutant’s prolonged open time
CFTR gating involves ATP binding, NBD dimerization, ATP hydrolysis and NBD/TMD coupling. We made several second-site mutations to further investigate the mechanism of gating defects in N1303K. To determine if N1303K’s prolonged open time is mediated by stable NBD dimerization, we introduced second-site mutations G551D and G1349D at the NBD heterodimer interface. Introducing a negatively charged aspartic acid into the signature sequence presumably impedes NBD dimerization via an electrostatic repulsion between the introduced negative charges and the bound ATP molecule. However, these second-site mutations G551D (Fig. 4A) and G1349D (Fig. 4B) did not affect the mean open time. We next mutated the conserved glutamate (E1371) at NBD2, which plays a critical role in ATP hydrolysis for WT-CFTR, under the N1303K background. However, the double mutant E1371S/N1303K shows a similar mean open time as N1303K (Fig. 4C).
Fig. 4.

Roles of NBD dimerization and ATP hydrolysis in N1303K-CFTR gating. Representative single channel recordings of G551D/N1303K (A), G1349D/N1303K (B) and E1371S/N1303K at 2 mM ATP. Microscopic kinetic analysis revealed mean open times (τO) for G551D/N1303K, G1349D/N1303K and E1371S/N1303K are 3.0 ± 0.5 s (n = 3), 3.0 ± 1.1 s (n = 3) and 3.0 ± 0.3 s (n = 6), respectively. Note: τO = 3.1 ± 0.8 s for N1303K-CFTR (Fig. 2).
3.5. Pharmacological response of N1303K-CFTR to CFTR potentiators and correctors
We next tested if N1303K responds to VX-770, the only FDA approved CFTR potentiator for the treatment of patients with CF. A representative recording in Fig. 4A shows 200 nM VX-770 increased the channel activity of N1303K-CFTR. In Fig. 4B, the steady-state N1303K-CFTR IM did not increase when the concentration of VX-770 was raised from 200 to 500 nM, suggesting 200 nM is the maximally effective concentration of VX-770 for N1303K-CFTR. Using the maximally effective concentrations of GLPG1837 and VX-770, we compared the potentiators in the experimental protocol shown in Fig. 4C. Once the GLPG1837-potentiated N1303K currents reached a steady-state, the perfusion solution was switched to VX-770 + ATP. The steady-state IM in GLPG1837 decreased about 50% upon switching to VX-770, indicating that GLPG1837 is ~2-fold more efficacious than VX-770.
N1303K is classified as a Class II mutation due to a decrease in Band C yield in Western blot analysis [4–6]. Consistent with these previous reports, we found N1303K has decreased surface expression compared to WT-CFTR. However, this trafficking defect caused by the N1303K mutation can be partly rectified by treatment (18 h) of the cells with VX-809 (lane 2 in Fig. S1) or VX-661 (lane 5 in Fig. S1). We also tested the effects of CFTR correctors in the presence of CFTR potentiators. Two CFTR potentiators, VX-770 and GLPG1837, were examined together with VX-809 or VX-661. Co-treatment with VX-770 or GLPG1837 did not appear to counteract the effect of the correctors on the N1303K mutation (Fig. S1), in striking contrast to the reported effect of co-treatment with potentiator on pharmacologically corrected F508del [14, 15].
4. Discussion
In this study, we have assessed the N1303K mutant’s pharmacological response to CFTR modulators GLPG1837, VX-770, VX-809 and VX-661. Specifically, we quantified the N1303K mutant’s gating defects and showed a more severe defect than reported previously. The N1303K mutant’s mostly ATP-independent channel activity and its abnormal response to ATP analogs P-ATP and d-ATP indicate the origin of its gating defects likely resides in CFTR’s NBDs. We also provided evidence that GLPG1837 is a more efficacious but less potent potentiator of N1303K-CFTR than VX-770, as was previously reported for G551D-CFTR [17]. In this section, we will discuss the mechanism for the gating defects in N1303K-CFTR and clinical implications of our results.
N1303 and F508 are located at equivalent but opposite positions within the CFTR protein structure (Fig. S2A). F508 sits in the interface between NBD1 and the fourth intracellular loop (ICL4), also known as the coupling helix of the CFTR’s second transmembrane domains (TMD2) [3, 28, 29]. N1303, on the other hand, resides in the interface between NBD2 and ICL2 [3], and is thought to stabilize the Q-loop of NBD2 [27]. These two domain interfaces play important roles in CFTR gating because they serve to relay signals from the cytosolic NBDs to trigger conformational changes in the TMDs to open and close the gate [30, 31]. Not surprisingly, both F508del and N1303K mutations cause gating defects [7–9]. What is surprising however is that the two interface mutations show distinct gating patterns.
Despite >15-fold reduction in Po mostly due to a lower opening rate, F508del retains ATP-dependence [10, 32, 33]. The function of F508del can be improved by maneuvers that promote NBD dimerization [10, 32] or abolish ATP hydrolysis [33]. It was proposed that F508del’s low opening rate is due to a high enthalpy of the opening transition state reflecting strain at the NBD1-TMD interface [30]. Alternatively, ATP-binding induced dimerization of the NBDs in F508del is no longer tightly coupled to the opening of the gate in the TMDs. Although N1303K has about the same Po as F508del, the gating function of N1303K is mostly unresponsive to the two ATP analogs tested. In addition, the prolonged open time of N1303K-CFTR is not altered by mutations that disrupt ATP hydrolysis or NBD dimerization. This nearly complete uncoupling between NBDs and TMDs in N1303K-CFTR may result from a disruption of the Q-loop in NBD2, which plays a critical role in aligning the bound ATP at site 2 (composed of the head of NBD2 and the tail of NBD1) for catalyzing NBD dimerization [34]. Thus, despite the apparently opposite locations for F508 and N1303, mutations at these two positions result in similar but not identical dysfunction of the critical site 2 (see Fig. S2B & C for more detailed discussion).
Regardless of the mechanism underlying the gating defect caused by the N1303K mutation, our results bear important clinical implications. Here, we report that VX-770 increases the Po of N1303K channels by ~ 8-fold. This, plus a ~3-fold improvement of N1303K biogenesis by either VX-809 or VX-661, and the absence of negative drug-drug interaction with potentiator co-treatment suggest that a combination of corrector and potentiator may be a viable therapeutic option for patients bearing the N1303K allele. Indeed, a Phase II clinical trial with a combination of VX-661 (tezacaftor), a second-generation corrector VX-440 and VX-770 (ivacaftor) reported favorable clinical outcomes for F508del heterozygotes carrying a minimal function mutation (e.g. N1303K) on their second allele [35]. Future work on other mutations with minimal function may provide more in vitro evidence supporting the strategies of using a combination of CFTR correctors and potentiators for CF treatment.
Supplementary Material
Fig. 5.
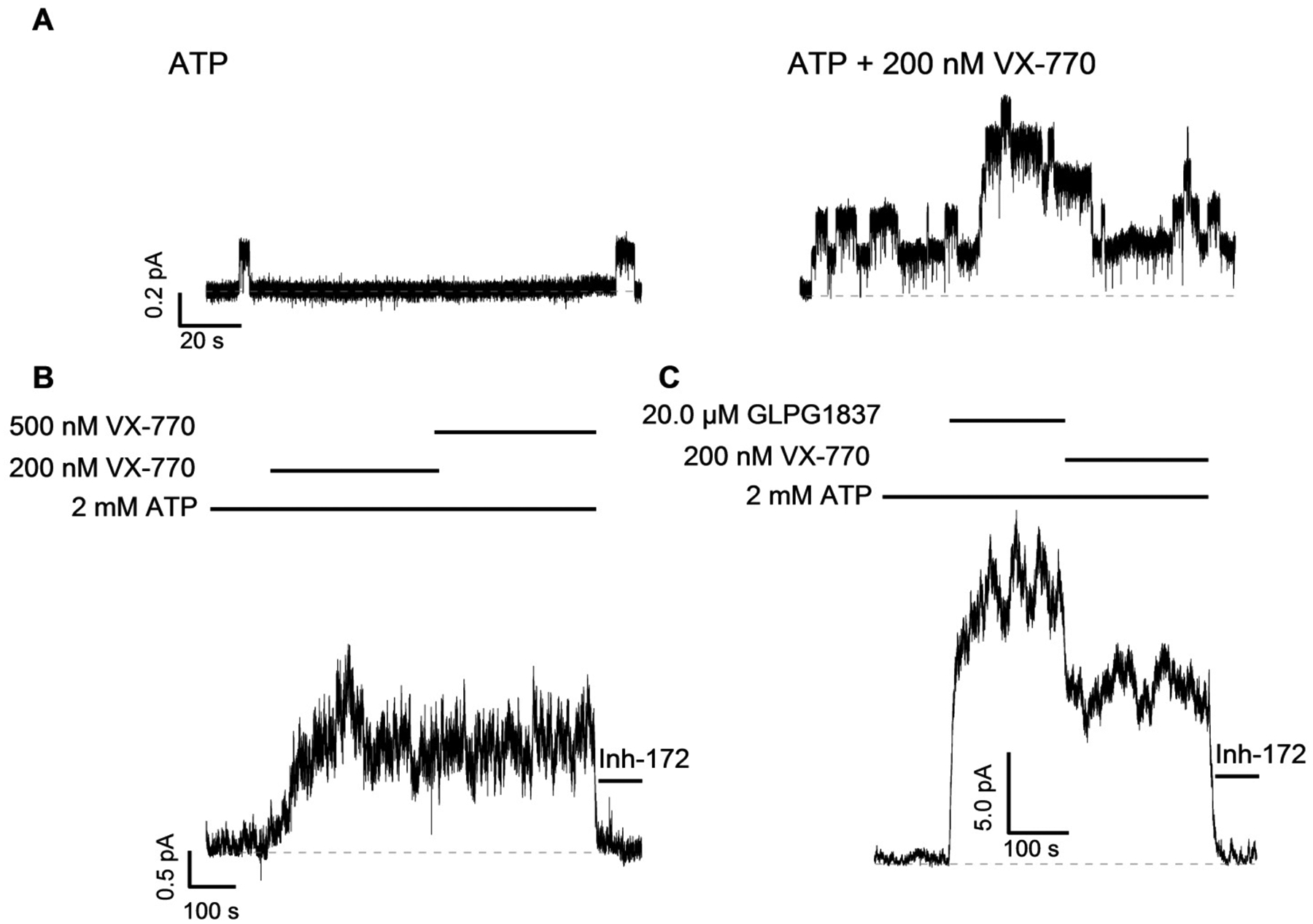
(A) Microscopic recording of N1303K-CFTR in ATP or ATP + VX-770. VX-770 can increase the channel activity of N1303K-CFTR. In (B), steady-state IM potentiated with 200 nM VX-770 does not increase when the concentration of VX-770 is increased to 500 nM. (C) Macroscopic recording of N1303K-CFTR exposed first to GLPG1837 and then to VX-770. GLPG1837-potentiated IM is reduced by 48.8 ± 4.8 % (n = 4), when the potentiator is switched to VX-770.
Acknowledgements
We thank Cindy Chu and Shenghui Hu for their technical assistance. This work is supported by the National Institutes of Health (grant R01DK55835) and the Cystic Fibrosis Foundation (grant Hwang11P0) to T.-C. Hwang. Part of the work is supported by a service agreement with AbbVie Inc. The authors declare no additional competing financial interests. S. Destefano is supported by a gift fund from Nanova Inc, and a traineeship from the Cystic Fibrosis Foundation (DESTEF17H0).
References
- 1.Hwang TC, Yeh JT, Zhang J, Yu YC, Yeh HI, Destefano S. Structural mechanisms of CFTR function and dysfunction. The Journal of general physiology. 2018. Epub 2018/03/28. doi: 10.1085/jgp.201711946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science (New York, NY). 1989;245(4922):1066–73. [DOI] [PubMed] [Google Scholar]
- 3.Liu F, Zhang Z, Csanady L, Gadsby DC, Chen J. Molecular Structure of the Human CFTR Ion Channel. Cell. 2017;169(1):85–95.e8. Epub 2017/03/25. doi: 10.1016/j.cell.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 4.Gregory RJ, Rich DP, Cheng SH, Souza DW, Paul S, Manavalan P, et al. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Molecular and cellular biology. 1991;11(8):3886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rapino D, Sabirzhanova I, Lopes-Pacheco M, Grover R, Guggino WB, Cebotaru L. Rescue of NBD2 mutants N1303K and S1235R of CFTR by small-molecule correctors and transcomplementation. PloS one. 2015;10(3):e0119796 Epub 2015/03/24. doi: 10.1371/journal.pone.0119796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dekkers JF, Gogorza Gondra RA, Kruisselbrink E, Vonk AM, Janssens HM, de Winter-de Groot KM, et al. Optimal correction of distinct CFTR folding mutants in rectal cystic fibrosis organoids. The European respiratory journal. 2016;48(2):451–8. Epub 2016/04/23. doi: 10.1183/13993003.01192-2015. [DOI] [PubMed] [Google Scholar]
- 7.Berger AL, Ikuma M, Hunt JF, Thomas PJ, Welsh MJ. Mutations that change the position of the putative gamma-phosphate linker in the nucleotide binding domains of CFTR alter channel gating. The Journal of biological chemistry. 2002;277(3):2125–31. [DOI] [PubMed] [Google Scholar]
- 8.Randak C, Welsh MJ. An intrinsic adenylate kinase activity regulates gating of the ABC transporter CFTR. Cell. 2003;115(7):837–50. [DOI] [PubMed] [Google Scholar]
- 9.Randak CO, Welsh MJ. ADP inhibits function of the ABC transporter cystic fibrosis transmembrane conductance regulator via its adenylate kinase activity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(6):2216–20. Epub 2005/02/03. doi: 10.1073/pnas.0409787102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miki H, Zhou Z, Li M, Hwang TC, Bompadre SG. Potentiation of disease-associated cystic fibrosis transmembrane conductance regulator mutants by hydrolyzable ATP analogs. The Journal of biological chemistry. 2010;285(26):19967–75. doi: 10.1074/jbc.M109.092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(46):18843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18825–30. Epub 2009/10/23. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. The New England journal of medicine. 2015;373(3):220–31. Epub 2015/05/20. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cholon DM, Quinney NL, Fulcher ML, Esther CR Jr., Das J, Dokholyan NV, et al. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Science translational medicine. 2014;6(246):246ra96 Epub 2014/08/08. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Science translational medicine. 2014;6(246):246ra97 Epub 2014/08/08. doi: 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthes E, Goepp J, Carlile GW, Luo Y, Dejgaard K, Billet A, et al. Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). British journal of pharmacology. 2016;173(3):459–70. Epub 2015/10/24. doi: 10.1111/bph.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng X, Wang Y, Wang X, Wrennall JA, Rimington TL, Li H, et al. Two Small Molecules Restore Stability to a Subpopulation of the Cystic Fibrosis Transmembrane Conductance Regulator with the Predominant Disease-causing Mutation. The Journal of biological chemistry. 2017;292(9):3706–19. Epub 2017/01/15. doi: 10.1074/jbc.M116.751537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Avramescu RG, Kai Y, Xu H, Bidaud-Meynard A, Schnur A, Frenkiel S, et al. Mutation-specific downregulation of CFTR2 variants by gating potentiators. Human molecular genetics. 2017;26(24):4873–85. Epub 2017/10/19. doi: 10.1093/hmg/ddx367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bompadre SG, Li M, Hwang TC. Mechanism of G551D-CFTR (cystic fibrosis transmembrane conductance regulator) potentiation by a high affinity ATP analog. The Journal of biological chemistry. 2008;283(9):5364–9. Epub 2008/01/03. doi: 10.1074/jbc.M709417200. [DOI] [PubMed] [Google Scholar]
- 20.Yeh HI, Sohma Y, Conrath K, Hwang TC. A common mechanism for CFTR potentiators. The Journal of general physiology. 2017;149(12):1105–18. Epub 2017/10/29. doi: 10.1085/jgp.201711886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bompadre SG, Sohma Y, Li M, Hwang TC. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. The Journal of general physiology. 2007;129(4):285–98. Epub 2007/03/14. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai MF, Li M, Hwang TC. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. The Journal of general physiology. 2010;135(5):399–414. Epub 2010/04/28. doi: 10.1085/jgp.201010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csanady L Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophysical Journal. 2000;78(2):785–99. doi: 10.1016/S0006-3495(00)76636-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(11):4404–9. Epub 2013/02/27. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433(7028):876–80. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aleksandrov AA, Aleksandrov L, Riordan JR. Nucleoside triphosphate pentose ring impact on CFTR gating and hydrolysis. FEBS letters. 2002;518(1–3):183–8. [DOI] [PubMed] [Google Scholar]
- 27.Cai Z, Taddei A, Sheppard DN. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl-channel. The Journal of biological chemistry. 2006;281(4):1970–7. Epub 2005/11/29. doi: 10.1074/jbc.M510576200. [DOI] [PubMed] [Google Scholar]
- 28.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(9):3256–61. Epub 2008/02/29. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mornon JP, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cellular and molecular life sciences : CMLS. 2008;65(16):2594–612. Epub 2008/07/04. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorum B, Czege D, Csanady L. Timing of CFTR pore opening and structure of its transition state. Cell. 2015;163(3):724–33. Epub 2015/10/27. doi: 10.1016/j.cell.2015.09.052. [DOI] [PubMed] [Google Scholar]
- 31.Sorum B, Torocsik B, Csanady L. Asymmetry of movements in CFTR’s two ATP sites during pore opening serves their distinct functions. eLife. 2017;6 Epub 2017/09/26. doi: 10.7554/eLife.29013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopeikin Z, Yuksek Z, Yang HY, Bompadre SG. Combined effects of VX-770 and VX-809 on several functional abnormalities of F508del-CFTR channels. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2014;13(5):508–14. Epub 2014/05/07. doi: 10.1016/j.jcf.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Jih KY, Li M, Hwang TC, Bompadre SG. The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. The Journal of physiology. 2011;589(Pt 11):2719–31. Epub 2011/04/14. doi: 10.1113/jphysiol.2010.202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nature structural & molecular biology. 2004;11(10):918–26. Epub 2004/09/29. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 35.https://investors.vrtx.com/news-releases/news-release-details/vertex-announces-positive-phase-1-phase-2-data-three-different?ReleaseID=1033559. [cited 2018 May 10].
- 36.Zhang Z, Chen J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell. 2016;167(6):1586–97.e9. Epub 2016/12/03. doi: 10.1016/j.cell.2016.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.