

Abstract
Following a request from the European Commission, the EFSA Panel on Nutrition, Novel Foods and Allergens (NDA) was asked to deliver an opinion on nicotinamide riboside chloride as a novel food (NF) pursuant to Regulation (EU) 2015/2283, including an evaluation of the safety of its use in food supplements as a source of niacin, and the bioavailability of nicotinamide from this source, in the context of Directive 2002/46/EC. The NF, a synthetic form of nicotinamide riboside, is proposed to be used in food supplements for the healthy adult population at levels up to 300 mg/day. The production process, composition, specifications, batch‐to‐batch variability and stability of the NF do not raise safety concerns. Animal and human data indicate that the NF contributes to the nicotinamide body pool. There are no concerns regarding genotoxicity. Human studies do not raise safety concerns. The proposed maximum use level corresponds to an amount of nicotinamide, which is sixfold lower than the tolerable upper intake level (UL) set for adults, excluding pregnant and lactating women. The margin of exposure (MoE) of 70 derived from repeated dose toxicity studies with rats and dogs is considered sufficient for the adult population, excluding pregnant and lactating women. Regarding these two population groups, the MoE of 76 derived from a developmental toxicity study in rats is considered insufficient in the absence of data which could justify accepting a MoE lower than 100. The Panel concludes that the NF is safe under the proposed conditions of use for the healthy adult population, excluding pregnant and lactating women, and that an intake of the NF up to 230 mg/day is safe for pregnant and lactating women. The Panel also concludes that the NF is a source from which nicotinamide, a form of niacin, is bioavailable.
Keywords: nicotinamide, nicotinamide riboside chloride, niacin, novel food, nutrient source
Summary
Following a request from the European Commission, the EFSA Panel on Nutrition, Novel Foods and Allergies (NDA) was asked to deliver an opinion on nicotinamide riboside chloride as a novel food (NF) pursuant to Regulation (EU) 2015/2283 including an evaluation of the safety of its use in food supplements as a source of niacin, and the bioavailability of nicotinamide, a form of niacin, from this source, in the context of Directive 2002/46/EC. The assessment of the safety of this NF follows the methodology set out in the EFSA Guidance on the preparation and presentation of an application for authorisation of a novel food Regulation (EU) 2015/2283 and in the Commission Implementing Regulation (EU) 2017/2469 and in the EFSA Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources. The assessment is based on the data supplied in the application and the information submitted by the applicant following EFSA's requests for supplementary information.
The NF is obtained through chemical synthesis and contains ≥ 90% nicotinamide riboside chloride, the remaining components being residual solvents, reaction by‐products and degradation products. Information provided on the production process, composition, specifications, batch‐to‐batch variability and stability of the NF is sufficient and does not raise concerns about the safety of the NF.
The applicant proposes to market the NF as a source of niacin in food supplement capsules at levels up to 300 mg/day. The NF is intended for use by the general healthy adult population, including pregnant and lactating women.
Considering the outcomes of the bacterial reverse mutagenicity test, the in vivo mammalian erythrocyte micronucleus test and the in vitro mammalian chromosome aberration test conducted with the NF, as well as its nature, the Panel considers that there are no concerns regarding genotoxicity.
From the data on absorption, distribution, metabolism and elimination available in mice, rats, dogs and humans, the Panel notes that the NF is likely to be absorbed mainly as nicotinamide following hydrolysis in the gut. If a fraction of the NF were absorbed intact, it would be expected to be rapidly metabolised to nicotinamide in the blood. Upon absorption, the NF contributes to the nicotinamide body pool, i.e. acts as a precursor of NAD+ in cells and is primarily metabolised in the liver to 1‐methylnicotinamide through methylation and subsequently to N‐methyl‐2‐pyridone‐carboxamide and N‐methyl‐4‐pyridone‐carboxamide, following oxidation. These metabolites are then excreted in the urine.
The Panel derives a no observed adverse effect level (NOAEL) of 300 mg/kg body weight (bw) per day from the available repeated dose toxicity studies with rats and dogs conducted with the NF. The Panel notes that, in the 90‐day study in rats, the same effects were observed in rats which received a dose of 1,260 mg/kg bw per day nicotinamide (equimolar to 3,000 mg/kg bw per day nicotinamide riboside chloride) as in those which received 3,000 mg/kg bw per day nicotinamide riboside chloride. Reproductive and developmental toxicity studies in rats were also provided, which indicate a NOAEL for fertility and reproductive performance of 675 mg/kg bw per day in males and 1,088 mg/kg bw per day in females and a NOAEL for maternal and embryo/fetotoxicity of 325 mg/kg bw per day. The embryo/fetotoxicity findings are considered secondary to maternal toxicity.
The Panel considers that available human studies on the NF, which were conducted in healthy adult subjects with dosages from 100 mg for 1 day up to 2,000 mg/day for up to 12 weeks, do not raise safety concerns. The proposed maximum use level of 300 mg nicotinamide riboside chloride per day corresponds to an intake of nicotinamide of 126 mg/day. This amount exceeds the population reference intake (PRI) of 1.6 niacin equivalent (NE) mg/MJ for all population groups (in adults ranging from 10.5 to 21.8 mg NE per day) and mean intakes from background diet (27.5–50.1 mg NE/day for adult females and males across EU countries) (EFSA NDA Panel, 2014). However, this intake is sixfold lower than the tolerable upper intake level (UL) for nicotinamide of 900 mg/day in adults (excluding pregnant and lactating women).
The margin of exposure (MoE) between the proposed maximum use level of 300 mg/day (i.e. 4.3 mg/kg bw in a 70‐kg adult) and the NOAEL of 300 mg/kg bw per day derived from animal data is 70. However, in the light of the human data available on nicotinamide riboside chloride and nicotinamide, the Panel considers that this MoE is sufficient for the adult population, excluding pregnant and lactating women.
The MoE between the proposed maximum use level of 300 mg/day (i.e. 4.3 mg/kg bw in a 70‐kg adult) and the NOAEL of 325 mg/kg bw per day for maternal and embryo/fetotoxicity is 76. In the absence of data which could justify accepting a MoE lower than 100 for pregnant and lactating women, the Panel concludes that an intake of 230 mg/day of the NF is safe for these two population groups.
The Panel concludes that the NF, nicotinamide riboside chloride, is safe under the proposed uses and use levels, i.e. up to 300 mg/day, for the healthy adult population, excluding pregnant and lactating women. The Panel concludes that an intake of the NF up to 230 mg/day is safe for pregnant and lactating women.
The Panel also concludes that nicotinamide riboside chloride is a source from which nicotinamide, which is a form of niacin, is bioavailable.
1. Introduction
1.1. Background and Terms of Reference as provided by the European Commission
1.1.1. Background as provided by the European Commission
The European Union legislation lists nutritional substances that may be used for nutritional purposes in certain categories of foods as sources of certain nutrients.
The relevant Union legislative measures are:
-
–
Regulation (EU) 2015/2283 of the European Parliament and the Council on novel foods.1
-
–
Directive 2002/46/EC of the European Parliament and of the Council laying down requirements for food supplements.2
On 14 May 2018, the company ChromaDex Inc., submitted a request to the European Commission in accordance with Article 10 of Regulation (EU) No 2015/2283 to place on the Union market synthetic form of Nicotinamide riboside as a novel food and to be added to the list of niacin forms specified in Annex II of Directive 2002/46/EC as a source of niacin.
1.1.2. Terms of reference as provided by the European Commission
In accordance with Article 29(1)(a) of Regulation (EC) No 178/2002, the European Commission asks the European Food Safety Authority to provide a scientific opinion:
-
–
by carrying out the assessment for synthetic form of Nicotinamide riboside as a novel food in accordance with Article 10(3) of Regulation (EU) 2015/2283, and
-
–
following the outcome of the novel food assessment by evaluating the safety of Nicotinamide riboside, when added for nutritional purposes to food supplements as a source of niacin, and on the bioavailability of nicotinamide from this source, in the context of Directive 2002/46/EC.
1.2. Information on existing evaluations and authorisations
In 2002, the Scientific Committee on Food (SCF) published an opinion on the Tolerable Upper Intake level (UL) for niacin (nicotinic acid (NA) and nicotinamide (NAM)) (SCF, 2002). An UL for NAM of 900 mg/day was established for adults, excluding pregnant and lactating women in view of the lack of data for these population groups.
In 2014, the NDA Panel published an opinion on dietary reference values for niacin (EFSA NDA Panel, 2014).
2. Data and methodologies
2.1. Data
The safety assessment of this novel food (NF) is based on data supplied in the application and information submitted by the applicant following EFSA's requests for supplementary information.
Administrative and scientific requirements for NF applications referred to in Article 10 of Regulation (EU) 2015/2283 are listed in the Commission Implementing Regulation (EU) 2017/2469.3
A common and structured format on the presentation of NF applications is described in the EFSA guidance on the preparation and presentation of a NF application (EFSA NDA Panel, 2016). As indicated in this guidance, it is the duty of the applicant to provide all of the available (proprietary, confidential and published) scientific data (including both data in favour and not in favour) that are pertinent to the safety of the NF.
This NF application includes a request for protection of proprietary data in accordance with Article 26 of Regulation (EU) 2015/2283. The data requested by the applicant to be protected comprise: an in vitro study evaluating the metabolism of nicotinamide riboside in blood (Study No. 160312); an oral 7‐day dose range finding toxicity study in juvenile dogs (Study No. SN17‐921); a hERG screening assay (Study No. 20151223); a 28‐day repeat‐dose oral toxicity study in juvenile dogs (Study No. SN17‐940); a 90‐day repeated‐dose oral toxicity study in Sprague–Dawley rats (Study No. S14022); a reproductive toxicity study (Study No. G10959) and a developmental toxicity study (Study No. G10957) in rats.
2.2. Methodologies
The assessment follows the methodology set out in the EFSA guidance on NF applications (EFSA NDA Panel, 2016) and the principles described in the relevant existing guidance documents from the EFSA Scientific Committee. The legal provisions for the assessment are laid down in Article 11 of Regulation (EU) 2015/2283 and in Article 7 of the Commission Implementing Regulation (EU) 2017/2469.
This assessment concerns only risks that might be associated with the consumption of the NF under the proposed conditions of use and is not an assessment of the efficacy of the NF with regard to any claimed benefit.
The evaluation of the bioavailability of the nutrient (NAM, a form of niacin) from the source (nicotinamide riboside chloride) was conducted in line with the principles contained in the Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources (EFSA ANS Panel, 2018).
3. Assessment
3.1. Introduction
The proposed NF, nicotinamide riboside chloride, is a synthetic form of nicotinamide riboside, proposed to be used in food supplements as a source of niacin. The target population is the general adult population.
The NF falls under the following category, as defined in Art. 3 of Reg. EU 2015/2283: ix) Vitamins, minerals and other substances used in accordance with Directive 2002/46/EC, Regulation (EC) No 1925/2006 or Regulation (EU) No 609/2013.
3.2. Production process
The NF is produced according to Good Manufacturing Practice (GMP) and Hazard Analysis Critical Control Points (HACCP) principles.
The NF is synthesised in a two‐step process. First, the starting material, d‐ribofuranose tetraacetate is reacted at low temperature with hydrochloric acid to generate d‐ribofuranose triacetate chloride. When the reaction is complete, as verified by nuclear magnetic resonance (NMR), NAM is added to the mixture, which is then stirred until the reaction to nicotinamide‐β‐ribofuranoside triacetate chloride is complete.
The filter‐cake is then analysed for nicotinamide‐β‐ribofuranoside triacetate chloride and held for use in Step 2 of the reaction.
In the second step, the nicotinamide‐β‐ribofuranoside triacetate chloride is deacetylated and washed with methanol and acetone to yield nicotinamide‐β‐ribofuranoside chloride (called from now on nicotinamide riboside chloride).
Depending on the quality of the finished product, the NF can be reprocessed by recrystallisation in methanol and/or further slurry in acetone. The product is off‐loaded, vacuum‐dried and transferred directly to food‐grade high‐density polyethylene (HDPE) drums doubly‐lined with an inner food‐grade low‐density polyethylene (LDPE) liner.
The Panel considers that the production process is sufficiently described and does not raise safety concerns.
3.3. Identity of the NF
The proposed NF contains ≥ 90% nicotinamide riboside chloride, predominantly in its β form, the remaining components being residual solvents, reaction by‐products and degradation products.
Nicotinamide riboside chloride is registered with CAS number 23111‐00‐4 and EC number 807‐820‐5. Its IUPAC name is 1‐[(2R,3R,4S,5R)‐3,4‐dihydroxy‐5‐(hydroxymethyl)oxolan‐2‐yl]pyridin‐1‐ium‐3‐carboxamide;chloride. The molecular formula of nicotinamide riboside chloride is C11H15N2O5Cl, and its molecular weight 290.7 g/mol. The molecular structure of nicotinamide riboside chloride is reported in Figure 1.
Figure 1.
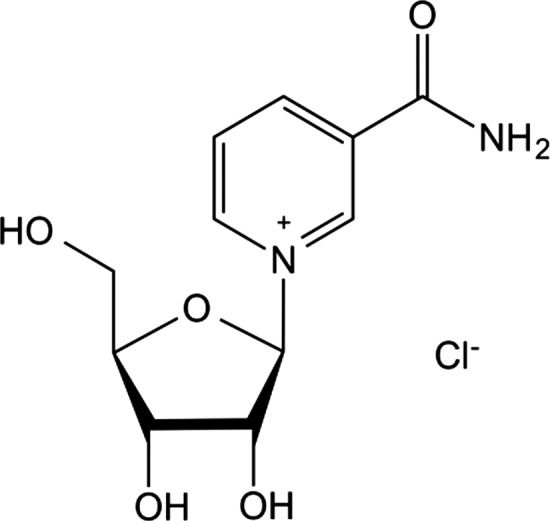
Molecular structure of nicotinamide riboside chloride
To confirm the identity of the NF, the applicant provided analyses by high‐performance liquid chromatography coupled with ultraviolet/visible spectroscopy (HPLC UV‐Vis), electrospray ionisation mass spectrometry (ESI‐MS), and mono‐ and two‐dimensional NMR.
The NMR spectra of five lots of the NF performed in D2O, showed that all expected proton and carbon shifts were present and that the nicotinamide riboside chloride conformed to the β‐anomer.
The particle size distribution of five lots of the NF was determined using a Ro‐Tap sieve shaker. The observed batch‐to‐batch variability appears to be dependent on a variety of factors including the production equipment and whether or not the batch was reprocessed (see Section 3.2).
The crystallinity of five lots of the NF was observed using a polarisation microscope and a digital camera. The crystals were not uniform in size or colour, which is attributed primarily to the production equipment and whether or not the batch was reprocessed.
3.4. Compositional data
The NF is composed primarily of nicotinamide riboside chloride (≥ 90%) together with residual raw materials, residual solvents, reaction by‐products, degradants and heavy metals.
In order to confirm that the manufacturing process is reproducible and adequate to produce on a commercial scale a product with the required characteristics, the applicant provided analytical information for five independent batches of the NF (see Table 1).
Table 1.
Analytical data for five lots of the NF
| Analyte | Lot Number | ||||
|---|---|---|---|---|---|
| 40C910‐16214‐81 | 40C910‐17201‐81 | 40C910‐17202‐81 | 40C910‐17203‐92 | 40C910‐17205‐21 | |
| Nicotinamide riboside chloride (% wt) | 97.9 | 99.9 | 98.1 | 97.1 | 96.0 |
| Nicotinamide (% wt) | 0.24 | 0.25 | 0.25 | 0.35 | 0.33 |
| Acetamide (mg/kg), | < LOQ | < LOQ | < LOQ | < LOQ | < LOQ |
| Water content (% wt) | ND | 0.1 | 0.1 | 0.1 | 0.2 |
| Chloride (%) | 12 | 12 | 12 | 12 | 11 |
| Acetic acid (mg/kg) | ND | 142 | 145 | < LOQ | 137 |
| Acetone (mg/kg) | 953 | 735 | 874 | 955 | 2020 |
| Acetonitrile (mg/kg) | 19 | ND | ND | ND | ND |
| Methanol (mg/kg) | 199 | 170 | 186 | 148 | 379 |
| Methyl acetate (mg/kg) | ND | < LOQ | < LOQ | < LOQ | 19 |
| Methyl t‐butyl ether (mg/kg) | ND | ND | ND | ND | ND |
| Arsenic (mg/kg) | < 0.2 | < 0.2 | < 0.2 | < 0.2 | < 0.2 |
| Mercury (mg/kg) | < 0.025 | < 0.025 | < 0.025 | < 0.025 | < 0.025 |
| Cadmium (mg/kg) | < 0.049 | < 0.05 | < 0.049 | < 0.05 | < 0.05 |
| Lead (mg/kg) | < 0.049 | < 0.05 | < 0.049 | < 0.05 | < 0.05 |
| Total plate count | < 10 CFU/g | < 10 CFU/g | < 10 CFU/g | < 10 CFU/g | < 10 CFU/g |
| Yeast and mould | < 10 CFU/g | < 10 CFU/g | < 10 CFU/g | < 10 CFU/g | < 10 CFU/g |
| Escherichia coli | Absent/10 g | Absent/10 g | Absent/10 g | Absent/10 g | Absent/10 g |
Wt: weight; ND: not detected; LOQ: limit of quantitation; CFU: colony forming units.
Information was provided on the accreditation of the laboratories that conducted the analyses presented in the application.
The solvents used for the manufacturing (acetone, methanol, acetonitrile, and methyl t‐butyl ether), as well as acetic acid and methyl acetate, which are formed during the process, were below their specification limits (see Section 3.5).
The Panel considers that the information provided on the composition of the NF is sufficient and does not raise safety concerns.
3.4.1. Stability
Three studies have been conducted in order to evaluate the stability of the NF under real‐time conditions (25 ± 2°C and 60 ± 5% relative humidity (RH)), three studies under accelerated conditions (40 ± 2°C and 75 ± 5% relative humidity) and two forced degradation studies. The NF was found to be stable for at least 36 months, when stored under ambient conditions. Based on the data available, the applicant recommends that the NF is stored under refrigerated conditions with a shelf‐life of 36 months.
The Panel considers that the data provided are sufficient to establish the stability of the NF.
3.5. Specifications
The specifications of the NF as proposed by the applicant are indicated in Table 2.
Table 2.
Specifications of the NF
| Parameter | Specifications | Method |
|---|---|---|
| Colour | White to Light Brown | Visual |
| Form | Powder | Visual |
| Identification | Conforms by NMR | NMR |
| Nicotinamide riboside chloride | ≥ 90 wt % | HPLC‐UV* |
| Water content | ≤ 2.0 % | Karl Fischer Titration (USP <921>)* |
| Residual solvents | ||
| Acetone | ≤ 5,000 mg/kg | GC Headspace (USP <467>) |
| Methanol | ≤ 1,000 mg/kg | GC Headspace (USP <467>) |
| Acetonitrile | ≤ 50 mg/kg | GC Headspace (USP <467>) |
| Methyl tert‐butyl ether | ≤ 500 mg/kg | GC Headspace (USP <467>) |
| Reaction by‐products | ||
| Methyl acetate | ≤ 1,000 mg/kg | GC Headspace (USP <467>) |
| Acetamide | ≤ 27 mg/kg | GC‐FID* |
| Acetic acid | ≤ 5,000 mg/kg | GC‐FID* |
| Heavy metals | ||
| Arsenic | ≤ 1 mg/kg | ICP‐MS (USP <232>, <233>, <2232>) |
| Mercury | ≤ 0.1 mg/kg | ICP‐MS (USP <232>, <233>, <2232>) |
| Cadmium | ≤ 1 mg/kg | ICP‐MS (USP <232>, <233>, <2232>) |
| Lead | ≤ 0.5 mg/kg | ICP‐MS (USP <232>, <233>, <2232>) |
| Microbiological limits | ||
| Total plate count | ≤ 1,000 CFU/g | AOAC or equivalent |
| Yeast and mould | ≤ 100 CFU/g | AOAC or equivalent |
| Escherichia coli | Absent/10 g | AOAC or equivalent |
AOAC: Association of Analytical Communities; CFU: colony forming units; GC‐FID: gas chromatography coupled with a flame ionisation detector; HPLC‐UV: high‐performance liquid chromatography‐ultraviolet spectroscopy; ICP‐MS: inductively coupled plasma mass spectrometry NMR: nuclear magnetic resonance; USP: United States Pharmacopeia.
In‐house validated analytical methods.
Although the average nicotinamide riboside chloride purity of the NF is approximately 96% at the time of the production, a specification of not less than 90% has been set to account for the degradation of nicotinamide riboside chloride over the course of shelf‐life. Specifications have also been set to control the amount of residual solvents, reaction by‐products, and heavy metals. Forced degradation studies indicate that during shelf‐life NF‐containing products will accumulate small amounts of NAM, ribose and chloride: these have not been included in the specifications because of no safety concern.
The Panel considers that the information provided on the specifications of the NF is sufficient and does not raise safety concerns.
3.6. History of use of the NF and/or of its source
The NF has a generally recognized as safe (GRAS) status in the USA since 2016 for addition to vitamin waters, protein shakes, nutrition bars, gum and chews, as a source of niacin.4 The intended maximum use level is 0.027% by weight. It was also filed to the U.S. Food and Drug Administration as a new dietary ingredient (NDI) for use in dietary supplements in 2015 (daily dose: 180 mg), without objection5; the NDI status was updated in 2017 with new proposed intake level (daily dose 300 mg) and product specifications.6
In 2018, the NF was included in the Licensed Natural Health Products Database (LNHPD) by Health Canada.7
3.7. Proposed uses and use levels and anticipated intake
3.7.1. Proposed uses and use levels
The NF is proposed to be used as a source of niacin in food supplement capsules at levels up to 300 mg/day. The NF is intended for use by the general healthy adult population, including pregnant and lactating women.
3.7.2. Anticipated intake of the NF
The applicant proposes that the NF should be consumed up to a maximum of 300 mg/day.
3.7.3. Combined intake from the NF and other sources
The information on the food sources of nicotinamide riboside is scarce. The applicant provided a publication assessing the presence of NAD+ precursors in cow's milk (Trammell et al., 2016b); average concentrations of nicotinamide riboside ranging from 1.9 to 3.1 were reported, depending on the milk production system. Based on these findings, the applicant estimated that the amount of nicotinamide riboside ingested by humans from the equivalent of 710 mL/day of cow's milk is 545 μg/day. Micromolar amounts of nicotinamide riboside are reported to be present in milk‐derived products, and yeast‐containing food products are presumed to be natural sources of nicotinamide riboside (Chi and Sauve, 2013). From the information available, the Panel considers that the contribution of nicotinamide riboside from food sources other than the NF is too small to be relevant for the safety assessment.
A dose of 300 mg/day of the NF would deliver 126 mg NAM per day, under the assumption that the NF is fully metabolised (see Section 3.8).
Mean intakes of niacin from the background diet were estimated to range from 42.2 to 50.1 mg niacin equivalents (NE) per day in adult men and 27.5 and 35.5 mg NE/day in adult women, across EU countries (EFSA NDA Panel, 2014). Estimates of 95th percentile intakes were up to 78.2 mg NE/day in adult men. These estimates were calculated considering the food contents of preformed niacin (i.e. NAM and NA) as well as of tryptophan (i.e. tryptophan content divided by a factor of 60).
3.7.4. Estimate of exposure to undesirable substances
The Panel identifies no concern from the information provided on the exposure to undesirable substances, as the large majority of the remaining substances from the production process are commonly found in food and the level of exposure to undesirable substances is considered not to pose any safety concern.
3.8. Absorption, distribution, metabolism and excretion (ADME)
The applicant provided study reports which investigated the concentration of nicotinamide riboside or its metabolites in plasma, white blood cells (WBCs) and urine following administration of the NF in rats (Avery, 2017), dogs (Thorsrud, 2017, 2018) and humans (Wilson, 2015; Trammell et al., 2016a; Schacter, 2018; Conze et al., 2019). Table 3 provides an overview of the dose levels of the NF and the main metabolites investigated. Upon EFSA's request about the form(s), mechanism(s), and efficiency of absorption of the NF, the applicant also conducted a systematic search in PubMed to retrieve relevant studies.
Table 3.
Overview of animal and human data on the metabolism of the NF
| Reference | Species | Design | Doses | PLASMA | URINE | Notes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NR | NAM | 1‐MN | NA | 2‐PY | 4‐PY | NAD+ | NAM | 1‐MN | 2‐PY | 4‐PY | |||||
| Avery (2017) | Rat |
|
1,000 mg/kg bw per day | nd |

|

|
nd | nd | nd | nd | nd | nd | nd | nd | At the tested dose, 1‐MN reached a higher steady state concentration in plasma over the 90‐day period |
| Thorsrud (2017) | Dog |
|
100 300 1,000 mg/kg bw per day |
nd | nd |
|
nd | nd | nd | nd | nd | nd | nd | nd | – |
| Thorsrud (2018) | Dog |
|
0 100 300 1,000/500b mg/kg bw per day |
nd | nd |
↔
|
nd | nd | nd | nd | nd | nd | nd | nd |
|
| Wilson (2015) and Trammell et al. (2016a) | Human |
|
100 300 1,000 mg, as single doses |
↔ ↔ ↔ |
↔ ↔
|
↔
|
↔ ↔ ↔ |
↔
|
↔
|
↔ ↔ ↔ |
↔ ↔
|
↔ ↔
|
↔ ↔
|
↔ ↔
|
|
| Schacter (2018) and Conze et al. (2019) | Human |
|
0 100 300 1,000 mg/day |
nd |
↔ ↔ ↔
|
↔
|
nd | nd | nd |
↔
|
nd |
↔
|
↔
|
nd | At the mid‐ and high‐dose levels:
|
| Airhart et al. (2017) | Human |
|
Gradual increase from 250 on day 1 to 2,000 mg/day on day 8 | ↔d | nd | nd | nd | nd | nd |
 d
d
|
nd | nd | nd | nd |
|






























↑: increase in metabolite concentration; ↔: no relevant change in metabolite concentration; bw: body weight; 1‐MN: 1‐methylnicotinamide; 2‐PY: N‐methyl‐2‐pyridone‐carboxamide; 4‐PY: N‐methyl‐4‐pyridone‐carboxamide; nd: not determined; NA: nicotinic acid; NAD+: nicotinamide adenine dinucleotide; NAM: nicotinamide; NR: nicotinamide riboside chloride.
Compared to untreated Beagle dogs.
Due to weight loss observed in males in the 1,000 mg/kg bw per day group, dosing was reduced to 500 mg/kg bw per day as of day 9 in females and day 10 in males. Thus, the high‐dose level was 1,000 mg/kg bw on day 1 and 500 mg/kg bw on day 28.
Increase not statistically significant compared to control group.
Day 9 vs day 1.
Animal data
In a rat study investigating the digestion and absorption of labelled NAD, NAD was found to be rapidly hydrolysed to nicotinamide riboside in the intestine (Gross and Henderson, 1983). Further results indicated that nicotinamide riboside was hydrolysed to NAM by an enzyme associated with the mucosal cells before absorption (Baum et al., 1982; Gross and Henderson, 1983). Increasing the NAD dose was associated with a decrease in the percentage of labelled products removed from the intestine and a decrease in the percentage of nicotinamide riboside that was converted to NAM. The uptake of [14C]‐carboxamide‐labelled NAD was compared to the uptake of [14C]NAM and was found to be slower (24% vs 80% in 15 min). This study indicates that, in rats, nicotinamide riboside is converted to NAM before absorption and that this reaction is a rate‐limiting step.
Following the administration of 2H‐ and/or 13C‐labelled nicotinamide riboside to mice by oral gavage (50 mg/kg body weight (bw)), nicotinamide riboside was not detected in blood, while the blood concentration of NAM increased from 2 μM at baseline to 6 μM at the last measurement time point, 135 min after administration (Liu et al., 2018). Examination of tissue NAD+ labelling indicated that a small fraction of doubly‐labelled nicotinamide riboside was assimilated in the NAD+ liver pool. Minimal assimilation of doubly‐labelled nicotinamide riboside was detected in the NAD+ pool of extrahepatic tissues. This study indicates that, in mice, a small fraction of nicotinamide riboside might be absorbed as NAM in the intestine.
Studies in mice were conducted to compare the increase in hepatic NAD+ concentrations following the administration of the NF (185 mg/kg bw) or equimolar amounts of NA or NAM by oral gavage (Trammell et al., 2016a). Both the NF and NAM increased hepatic NAD+ concentrations to levels greater than NA. The NF and NAM administration resulted in increased hepatic concentrations of NAM, 1‐methylnicotinamide (1‐MN) and N‐methyl‐4‐pyridone‐carboxamide (4‐PY), although the maximum concentration achieved (Cmax) and area under the curve (AUC) for these metabolites differed between the NF and NAM groups. In mice receiving the NF, increases in liver concentrations of nicotinamide mononucleotide (NMN), nicotinic acid adenine dinucleotide (NaAD) and nicotinic acid mononucleotide (NAMN) were also observed. The Panel notes that, in mice, the kinetics of the NF and NAM metabolisms differed but results indicate that the NF is metabolised to the same metabolites as NAM.
In the toxicokinetic (TK) arm of the 90‐day subchronic toxicity study in rats (Avery, 2017) (see Section 3.10.2), blood samples were collected before and 0.5, 1, 2, 4, 6, 8, 12 and 24 h after oral administration of 1,000 mg/kg bw per day of the NF (NF group) or an equimolar dose of NAM (420 mg NAM/kg bw per day, NAM group) on days 1 and 90. NAM and 1‐MN were measured in plasma. On day 1, plasma NAM concentration peaked considerably later (time at which the Cmax is observed (Tmax) males: 6.0 ± 0.0 vs 0.67 ± 0.17 h; Tmax females: 6.7 ± 0.7 vs 1.0 ± 0.0 h) and at a lower level (Cmax males: 154.8 ± 13.9 vs 249.8 ± 8.6 μg/mL; Cmax females 118.7 ± 1.6 vs 385.4 ± 15.6 μg/mL) in the NF group than in the NAM group. Systemic exposures to NAM were lower in the NF group than in the NAM group (AUClast male: 1,271.3 ± 121.7 vs 1,713.0 ± 26.1 h*μg/mL; AUClast females: 882.4 ± 109.3 vs 1,872.7 ± 56.8 h*μg/mL). Similar patterns were observed on day 90 although the values reached were higher than on day 1 in both groups (AUClast male: 2,222.5 ± 113.8 vs 4,146 ± 132.6 h*μg/mL; AUClast females: 1,931.5 ± 80.8 vs 3,118.5 ± 69.8 h*μg/mL). On days 1 and 90, plasma NAM concentrations 24 h after dosing were comparable to those before dosing in both groups. On day 1, the plasma concentration of 1‐MN increased within 1 h of dosing and reached similar maximum concentrations in both groups (Cmax males (mean ± SEM): 4.4 ± 1.3 vs 3.5 ± 0.2 μg/mL; Cmax females: 2.6 ± 0.2 vs 2.5 ± 0.1 μg/mL). Systemic exposures to 1‐MN were similar in both groups (AUClast males: 62.5 ± 3.0 vs 70.2 ± 1.2 h*μg/mL; AUClast females: 39.4 ± 0.9 vs 41.2 ± 0.9 h*μg/mL). Similar patterns were observed on day 90; however, the basal (T0) plasma concentration of 1‐MN was higher on day 90 than on day 1 in both groups. The Panel notes that, in rats, ingestion of the NF resulted in slower plasma appearance of NAM and lower systemic exposure, as compared to NAM ingestion. Administration of the NF results in rapid appearance of its metabolite, 1‐MN, in plasma, with kinetic characteristics similar to that following NAM administration. At the tested dose levels, both NF and NAM administration resulted in increase of 1‐MN in plasma at the end of the 90‐day period.
In an oral 7‐day dose range finding toxicity study in juvenile Beagle dogs (Thorsrud, 2017), daily doses of 100, 300 and 1,000 mg/kg bw per day of the NF were administered by gavage (n = 2 per group). On day 7, plasma was collected 2 h after dosing. The plasma 1‐MN concentration increased dose dependently in males (mean ± SD: 416 ± 100, 604 ± 46 and 856 ± 66 ng/mL). Corresponding concentrations in females were 449 ± 71, 818 ± 221 and 575 ± 21 ng/mL. There was no control group in that study. The applicant indicates that concentrations of 1‐MN between 8.3 and 34 ng/mL were reported in plasma samples of untreated Beagle dogs (method validation report by Chen (2017)). The Panel notes that this study indicates that, in dogs, oral administration of the NF results in the appearance of its metabolite, 1‐MN, in plasma.
In a TK study conducted as part of a 28‐day toxicity study in Beagle dogs (Thorsrud, 2018), daily doses of 0, 100, 300 and 1,000 mg/kg bw per day of the NF were administered by gavage (n = 2 per group). Plasma was collected 0, 0.5, 1, 3, 8 and 24 h post‐dose, both on days 1 and 28. On day 1, an increase in plasma concentrations of 1‐MN was detected at the first time point (0.5 h) and following time points, in all dose groups compared to the control (Cmax males (mean ± SD): 141 ± 35, 901 ± 50, 1,004 ± 142, 789 ± 725 ng/mL; Cmax females: 117 ± 1, 860 ± 27, 983 ± 81, 1,072 ± 130 ng/mL). Systemic exposure increased with increasing doses of the NF (AUClast males: 2,827 ± 582, 14,154 ± 449, 21,743 ± 3,467, 17,888 ± 16,728 h*ng/mL; AUClast females: 2,428 ± 55, 17,023 ± 2,353, 24,424 ± 942, 22,783 ± 2,407 h*ng/mL). Mean plasma 1‐MN concentrations at time 0 on day 28 indicated an increase of the metabolite at the end of the 28‐day period in all groups compared to the control (males: 150, 502, 1,650 and 1,541 ng/mL, females: 159, 717, 1,706 and 1,766 ng/mL). On day 28, systemic exposure increased with increasing doses of the NF and reached higher levels than on day 1 (AUClast males: 2,214 ± 440, 25,331 ± 4,780, 41,779 ± 5,827, 38,247 ± 17,201 h*ng/mL; AUClast females: 2,645 ± 35, 28,452 ± 298, 42,256 ± 5,406, 42,218 ± 349 h*ng/mL). The Panel notes that this study indicates that, in dogs, oral administration of the NF results in rapid appearance of its metabolite, 1‐MN, in plasma. An increase of 1‐MN in plasma occurred at the end of the 28‐day period in all dose groups. Similar systemic exposure levels of 1‐MN were reached at 300 and 1,000 mg/kg bw.
Human data
In a single dose, randomised, double‐blind, cross‐over study of the pharmacokinetics of the NF in 12 healthy adults (Wilson, 2015; Trammell et al., 2016a), blood and urine samples were collected over 24 h following the ingestion of 100, 300 or 1,000 mg of the NF in capsules. There was no control group in that study. No significant changes in nicotinamide riboside or NA concentrations were detected in plasma or WBCs at any of the dose tested, at any time point. Compared to baseline, a significant increase in plasma NAM concentration was detected in the high‐dose group, but not in the low‐ and mid‐dose groups. No significant changes in NAM concentration in WBC were detected in any group. Dose‐ and time‐dependent increases in the concentrations of 1‐MN, N‐methyl‐2‐pyridone‐carboxamide (2‐PY) and 4‐PY in plasma were observed. The largest increases were detected for 2‐PY (Cmax (mean ± SD): 3.5 ± 1.3, 6.6 ± 2.0, 17.0 ± 5.0 μM, p < 0.001) and 4‐PY (Cmax (mean ± SD): 0.44 ± 0.35, 0.90 ± 0.37, 2.52 ± 0.64 μM, p < 0.001), while the plasma concentration of 1‐MN increased to a smaller extent (Cmax (mean ± SD): 0.22 ± 0.19, 0.43 ± 0.33, 0.89 ± 0.62 μM, p < 0.001). For the three metabolites, the changes in WBC concentrations followed patterns comparable to those in plasma. No changes in NAD+ or NADP concentrations were detected in either plasma or WBC. Urinary concentrations of NAM, 1‐MN, 2‐PY and 4‐PY significantly increased in the group which received 1,000 mg of the NF, but not in the groups which received 100 and 300 mg of the NF. A relative decrease in urine concentration of nicotinamide riboside was observed in all groups, compared to pre‐dose. No change in urine NA concentration was observed in any group.
Other metabolites were also analysed in this study. Increases of NaAD in WBC or nicotinic acid ribose (NaR) in plasma were observed in the 300 mg and 1,000 mg groups at different time points whereas significant time‐ and dose‐dependent increases compared to pre‐dose were observed in urine for all treatment groups. There were significant time‐dependent decreases in NMN in urine in all three groups but no changes in WBC concentrations. No significant dose‐related changes were observed for adenine diphosphate ribose (ADPR) in plasma, cytidine uridine and uridine monophosphate (UMP) in plasma, WBC and urine, inosine in plasma and WBC, and adenosine‐diphosphate (ADP) in urine and plasma.
The Panel notes that ingestion of a single dose of 1,000 mg of the NF resulted in an increase of NAM concentration in plasma, which was not observed at doses of 100 mg or 300 mg. 1‐MN and to a larger extent its oxidation products, 2‐PY and 4‐PY, increased dose‐ and time‐dependently in both plasma and WBC, while urinary concentrations of these metabolites were only increased at a dose of 1,000 mg. The relevance of observed changes at different time points in some of the other metabolites measured such as NaAD in WBC, NaR in plasma and urine or NMN in urine is unclear.
In an 8‐week randomised, double‐blind, placebo‐controlled, parallel group study (Schacter, 2018; Conze et al., 2019), 140 (intention‐to‐threat (ITT)) or 128 (per‐protocol (PP)) healthy subjects ingested capsules containing either silicified microcrystalline cellulose (placebo) or 100, 300 or 1,000 mg/day of the NF (n = 30 per group) for 8 weeks. Fasting blood and urine samples were collected at the end of weeks 1, 2, 4 and 8. In ITT analysis, there was a significantly higher plasma NAM concentration in the high‐dose group compared to the other groups at week 8 (22.3 ± 11.5, 26.6 ± 13.1, 27.9 ± 9.6, 43.7 ± 22.7 ng/mL in the control, low‐, mid‐ and high‐dose groups, respectively). A significantly higher plasma concentration of 1‐MN was found in the mid‐ and high‐dose groups compared to the control (3.1 ± 2.3, 5.6 ± 3.8, 10.1 ± 5.0, 26.6 ± 12.6 ng/mL in the control, low‐, mid‐ and high‐dose groups, respectively). Significant increases in whole blood concentrations of NAD+ were found in the mid‐and high‐dose group compared to the control group, after 1, 2 and 8 weeks of intervention. Maximum blood NAD+ concentrations were reached at week 2 (32.9 ± 7.6 and 50.0 ± 17.2 μg/mL in the mid‐ and high‐dose groups, respectively) and plateaued thereafter. At the end of the study, a significant dose‐related increase was found in urinary 1‐MN concentration (4.1 ± 1.9, 6.6 ± 4.9, 10.6 ± 16.1, 17.8 ± 8.8 ng/μg creatinine for the control, 100 mg/day, 300 mg/day and 1,000 mg/day NIAGEN dose groups, respectively; p < 0.001). Similarly, a significant dose‐related increase was found in urinary 2‐PY concentration (15 ± 7, 30 ± 24, 51 ± 55, 113 ± 50 ng/μg creatinine for the respective groups; p < 0.001). The concentrations of 1‐MN and 2‐PY were significantly higher in the mid‐ and high‐dose groups than in the control group. Results of the PP analyses were comparable. The Panel notes that ingestion of 1,000 mg/day of the NF for 8 weeks resulted in an increase of NAM concentration in plasma, which was not observed at doses of 100 mg/day or 300 mg/day. NAD+ concentration in whole blood significantly increased following administration of 300 and 1,000 mg/day and reached a plateau after two weeks. 1‐MN and its oxidation products increased dose dependently in plasma and urine, indicating saturation of the NAM vitamin body pool and a higher plasma steady state concentration of its metabolites.
Another study of pharmacokinetics of the NF in humans is available in the literature (Airhart et al., 2017). The NF was orally administered to eight healthy volunteers, with gradual increments from a dose of 250 mg/day on days 1 and 2 up to a dose of 2,000 mg/day on days 7 and 8. On day 9, subjects received a dose of 1,000 mg of the NF and whole blood was sampled over 24 h for measurement of nicotinamide riboside and NAD+ concentrations. Mean (± SD) basal concentration of nicotinamide riboside was 0.023 ± 0.007 μM on day 1 and 0.029 ± 0.008 μM on day 9 (p = 0.07). A small transient increase in nicotinamide riboside concentration was observed in four out of eight subjects, reaching a maximum 3 h post‐dose (0.05 ± 0.03 μM, p = 0.04). Basal mean blood concentrations of NAD+ were 27 ± 6 μM on day 1 and 50 ± 20 μM on day 9 (p = 0.001). The Pearson correlation coefficient between the changes in nicotinamide riboside and NAD+ concentrations between day 1 and day 9 was 0.85 (R2 = 0.72, p = 0.008). On day 9, no further change in NAD+ concentration was detected over the 24 h post‐dose. The Panel notes that in this study supplementation of the NF at doses up to 2,000 mg for 9 days significantly increased NAD+ concentrations in blood compared to baseline.
In vitro data
To determine the metabolic fate of the NF in blood, an in vitro study was conducted (Guo, 2018) (Study No. 160312) in which equal amounts of 13C‐ and deuterium‐labelled nicotinamide riboside chloride were added to water, heparinised whole blood, heparinised plasma, and serum at a concentration of 39 μM. The nicotinamide riboside and NAM contents of the samples were analysed immediately (T0) or after incubation at 37°C for 5 and 10 min. Compared to T0, there was no significant change in the amount of nicotinamide riboside or NAM in water, plasma, and serum samples over the course of the 10 min incubation period. In contrast, the amount of nicotinamide riboside in whole blood decreased within 5 min, while the amount of NAM increased. Similar results were obtained when the samples were incubated at 4°C, although the kinetics of the decrease in the amount of nicotinamide riboside and increase in the amount of NAM in whole blood were delayed compared to those observed at 37°C. Similar results have been reported by Liu et al. (2018). The Panel notes that these results indicate that the NF can be metabolised to NAM in a cellular component of whole blood.
In vitro data indicate that nicotinamide riboside can be hydrolysed to nicotinamide and ribose‐1‐phosphate by a purine nucleoside phosphorylase (Rowen and Kornberg, 1951; Kornberg, 1955; Nishizuka, 1971; Wielgus‐Kutrowska et al., 1997), which is an ubiquitous enzyme in mammals (Bzowska et al., 2000).
An alternative metabolic pathway for the contribution of nicotinamide riboside to NAD+ pool, independent from the NAM pathway, has been described in yeast in which nicotinamide riboside was found to act as a precursor for NAD+ through its conversion into NMN via the action of a nicotinamide riboside kinase (Nrk), (Bieganowski and Brenner, 2004). NrK genes were identified in the human genome.
Fate of the ribosyl moiety
Upon EFSA's request about the metabolic fate of the ribosyl moiety, the applicant indicated that, after being released from the NF, ribose is expected to be converted to ribose‐1‐phosphate and enter the pentose phosphate pathway for nucleotide‐base salvage or generation of glyceraldehyde‐3‐phosphate and fructose‐6‐phosphate which can be used as energy source via glycolysis. Assuming a daily intake of 300 mg of the NF, the maximum resulting exposure to free ribose would be 145 mg/day or 2.1 mg/kg bw per day for a 70‐kg adult. In 2018, the EFSA NDA Panel adopted the Scientific Opinion on the safety of D‐ribose as a novel food and concluded that d‐ribose is safe for the general population at intake levels up 36 mg/kg bw per day (EFSA NDA Panel et al., 2018).
Conclusions on ADME
Altogether, the Panel notes that the NF is likely to be absorbed mainly as NAM following hydrolysis in the gut. In case a fraction of the NF would be absorbed intact, it is expected to be rapidly metabolised to NAM in the blood. NAM acts as a precursor of NAD+ in cells and is primarily metabolised in the liver to 1‐MN through methylation and subsequently to 2‐PY, and 4‐PY, following oxidation. In the 8‐week supplementation study in humans, a gradual and significant increase in whole blood concentration of NAD+ was found with doses of 300 mg and 1,000 mg/day of the NF. The single dose pharmacokinetic study and the 8‐week supplementation studies in humans indicated that 1‐MN, 2‐PY and 4‐PY concentrations increase in a time‐ and dose‐dependent fashion in plasma, WBCs and urine following ingestion of 100–1,000 mg/day of the NF. Dose‐dependent increases in the plasma concentration of 1‐MN were also reported in rats and dogs following ingestion of the NF. The Panel notes that at doses of 300 mg and 1,000 mg/day of the NF for 8 weeks, the concentrations of NAD+ and 1‐MN in human plasma were higher than in the control group and increases in urinary excretion of 1‐MN and 2‐PY were observed, indicating a saturation of NAM vitamin body pool.
3.9. Nutritional information
The NF is proposed to be used as a source of niacin. Niacin is a generic term for NA and NAM, which are water‐soluble organic compounds that belong to the group of B vitamins and are present in a wide range of foods (EFSA NDA Panel, 2014). Niacin can be synthesised in the human body from the amino acid tryptophan. The NDA Panel set average requirement (AR) and population reference intake (PRI) for niacin of, respectively, 1.3 mg niacin equivalent (NE8)/MJ and 1.6 NE mg/MJ for all population groups. Dietary reference values (DRV) were expressed on a MJ basis, considering that the niacin requirement depends on energy intake. In adults, the PRIs range from 10.5 mg NE/day for women aged 70–79 years with a sedentary lifestyle (physical activity level (PAL) = 1.4) to 21.8 mg NE/day for men aged 18–29 years with very active lifestyle (PAL = 2.0) (EFSA NDA Panel, 2014). The Panel notes that, at the proposed maximum use level of 300 mg/day, the NF would deliver 126 mg NAM/day under the assumption that nicotinamide riboside chloride is fully metabolised (Section 3.8), which largely exceeds the physiological requirement for niacin.
The SCF set a UL for NAM of 900 mg/day (12.5 mg/kg bw) for the adult population, excluding pregnant and lactating women (SCF, 2002). To derive the UL, the SCF considered that no significant adverse effects had been reported in trials on NAM supplementation in patients with or at risk of diabetes, which had received doses up to the equivalent of 3 g per day for periods up to 3 years. A no observed adverse effect level (NOAEL) of 25 mg/kg bw per day was derived from these studies. The SCF noted that these trials used sensitive markers of hepatic function and glucose homeostasis, and included a range of age groups, with some subjects treated with up to 50 mg/kg bw per day. An uncertainty factor (UF) of 2 was applied to the NOAEL to allow for the fact that adults may eliminate NAM more slowly than the study groups, many of which were children, and that data for children would not reflect the full extent of inter‐subject variability that could occur in an older population. The SCF stated that the UL of 900 mg/day for NAM was not applicable during pregnancy or lactation because of inadequate data relating to this critical life stage.
Based on the NOAEL of 25 mg/kg bw derived from the same studies, the UK Expert Group on Vitamins and Minerals proposed a guidance value of 8.3 mg NAM/kg bw (Expert Group on Vitamins and Minerals, 2003), which corresponds to 580 mg/day for a 70‐kg adult. In deriving this value, the expert group applied an UF of 3 to account for interindividual variability, in view of the nature of the study populations and the small numbers of subjects involved.
High intakes of niacin are not known to inhibit the absorption or modify bioavailability of other nutrients. The Panel notes that available toxicological studies in rats and dogs and human studies on the NF do not specifically address such effects.
The Panel considers that consumption of the NF is not nutritionally disadvantageous.
3.10. Toxicological information
The applicant provided several toxicological studies on the NF. These studies are listed in Table 4.
Table 4.
Summary of toxicological studies with the NF
| Test item | Reference | Type of study | Test system | Dose |
|---|---|---|---|---|
| NF |
Study No. S15004 (Unpublished) |
Bacterial reverse mutation test (GLP, OECD TG 471) | Salmonella Typhimurium and Escherichia coli | Up to 5 mg/plate (absence and presence of S9 mix) |
| NF |
Study No. S15005 (Unpublished) |
In vitro mammalian chromosome aberration test (GLP, OECD TG 473) | Human peripheral blood lymphocyte | Up to 5 mg/mL |
| NF |
Study No. S15006 (Unpublished) |
In vivo mammalian erythrocyte micronucleus test (GLP, OECD TG 474) | Sprague–Dawley rats | Up to 2,000 mg/kg bw |
| NF | Study No. 20151223 (Unpublished) | hERG screening assay (non‐GLP) | Human embryonic kidney 293 cells | Up to 1.03 mM |
| NF |
Study No. S13101 (Unpublished) |
Single dose oral toxicity study (GLP) | Sprague–Dawley rats | 5,000 mg/kg bw |
| NF |
Study No. SN17‐921 (Unpublished) (Thorsrud, 2017) |
7‐day dose range finding oral toxicity study (GLP) | Juvenile dogs | Up to 1,000 mg/kg bw per day |
| NF |
Study No. S13120 (Unpublished) |
14‐day dose range finding oral toxicity study (non‐GLP) | Sprague–Dawley rats | Up to 5,000 mg/kg bw per day |
| NF |
Study No. SN17‐940 (Unpublished) (Thorsrud, 2018) |
28‐day oral toxicity study (GLP) | Juvenile dogs | Up to 1,000 mg/kg bw per day |
| NF and nicotinamide |
Study No. S14022 (Unpublished) |
90‐day repeated dose oral toxicity study (GLP, OECD TG 408) | Sprague–Dawley rats | Up to 3,000 mg/kg bw per day |
| NF |
Study No. G10959 (Unpublished) (Ganiger, 2016) |
One generation reproduction toxicity study (GLP, OECD TG 415) | Sprague–Dawley rats | Up to 12,000 mg/kg in the diet (ad libitum) |
| NF |
Study No. G10957 (Unpublished) (Geetha Rao, 2016) |
Embryo‐fetal developmental toxicity study (GLP, OECD TG 414) | Sprague–Dawley rats | Up to 1,500 mg/kg bw per day |
bw: body weight; GLP: Good Laboratory Practice; OECD: Organisation for Economic Co‐operation and Development; hERG: human ether‐ago‐go‐related gene.
3.10.1. Genotoxicity
The applicant provided the results of a bacterial reverse mutagenicity test, an in vivo mammalian erythrocyte micronucleus test and an in vitro mammalian chromosome aberration test performed with the NF.
In a bacterial reverse mutagenicity test (Study No. S15004, unpublished, GLP, OECD TG 471; (Kamath, 2015; Conze et al., 2016)) with Salmonella Typhimurium TA98, TA100, TA1535 and TA1537 and Escherichia coli strain WP2 uvrA (pKM101) the NF did not increase the number of revertant colonies in any of the tester strains at concentration up to 5,000 μg/plate, either when incubated in the presence or absence of a metabolic activation system or using the plate incorporation or preincubation methods. The results of the study indicated that the NF is non‐mutagenic.
In an in vitro mammalian chromosome aberration test with human peripheral blood lymphocytes (HPBLs) (Study No. S15005, unpublished, GLP, OECD TG 473; (Conze et al., 2016; Kamath, 2016)), no increase in the number or type of aberrant metaphases was detected up to 5 mg/mL, either in the presence or absence of a metabolic activation system. No cytotoxicity was observed at any concentration. The results of the study indicated that the NF is not clastogenic at doses up to 5 mg/mL.
In an in vivo mammalian erythrocyte micronucleus test with Sprague–Dawley rats (Study No. S15006, unpublished, GLP, OECD TG 474; (Conze et al., 2016; Pandey, 2016)), the NF had no effect on the percentage of polychromatic erythrocytes with micronuclei or on the mean polychromatic erythrocyte/total erythrocyte (P/E) ratio compared to the vehicle control 24 or 48 h after a single dose of 500, 1,000 and 2,000 mg/kg bw. In view of the ADME data in rats (see Section 3.10.2), it can be assumed that the target, the bone marrow has been reached. The results of the study indicated that the NF is non‐cytotoxic and does not induce micronuclei.
Even though the genotoxicity testing strategy is not fully in line with current EFSA recommendations (EFSA Scientific Committee, 2011), as no in vitro mammalian micronucleus test was provided, in view of the nature of the test substance, the Panel considers that there are no concerns regarding genotoxicity of the NF.
3.10.2. Acute, subacute and subchronic toxicity studies
The applicant provided several toxicity studies performed with the NF: a single dose oral toxicity study in rats, a 7‐day dose range finding toxicity study in juvenile dogs, a 14‐day dose range finding toxicity study in rats, a 28‐day toxicity study in juvenile dogs and a 90‐day repeated dose toxicity study in rats.
In an acute toxicity study in rats (Study No. S13101, unpublished, GLP (Bhoite and Jayachandra, 2014b; Conze et al., 2016), five male and female Sprague–Dawley rats were administered 0 or 5,000 mg/kg bw of the NF as a single dose by oral gavage. The only finding was a significantly lower cumulative body weight gain in female rats (3%) compared to control group.
In a 7‐day dose range finding oral toxicity study (Study No. SN17‐921, unpublished, GLP (Thorsrud, 2017)), the NF was administered to groups of male and female juvenile dogs (2/sex per group) for 7 days via oral gavage at dose levels of 100, 300, and 1,000 mg/kg bw per day, respectively. No toxicologically relevant findings were observed in any of the groups tested.
In a 14‐day dose range finding oral toxicology study (Study No. S13120, unpublished, non‐GLP (Bhoite and Jayachandra, 2014a; Conze et al., 2016)), the NF was administered to five groups of male and female Sprague–Dawley rats (5/sex per group) for 14 days by oral gavage at dose levels of 0, 750, 1,500, 2,500 or 5,000 mg/kg bw per day, respectively. The only relevant finding was a reduction in mean body weight in relation to the control group observed in male rats administered 1,500, 2,500 and 5,000 mg/kg bw per day at different time points (all < 10%) and a decrease in overall food consumption in male rats administered 5,000 mg/kg bw per day (8%).
In a 28‐day repeated dose toxicity study (Study No. SN17‐940, unpublished, GLP (Thorsrud, 2018)), the NF was administered to six groups of male and female juvenile dogs (test animals: 4/sex per group; TK satellite animals: 2/sex per group) for 28 days by oral gavage at dose levels of 0, 100, 300 and 1,000 mg/kg bw per day, respectively. Because of decreased body weights and adverse clinical findings in the animals dosed at the highest level, the high dose was decreased from 1,000 to 500 mg/kg bw per day (on day 9 for females and on day 10 for males). The evaluation included clinical observations, body weight, body weight gain and qualitative feed consumption. Clinical pathology parameters reported for core animals included haematology, clinical chemistry, coagulation and urinalysis. The core animals were sacrificed, a full gross necropsy was performed, and tissues were collected for organ weights and histopathology. In addition, blood samples were collected on the first and last day of dosing for evaluation of the metabolite, 1‐MN, in satellite TK animals. The results of this part of the study are described in Section 3.8. Effects that are considered as substance related are summarised in Table 5. Clinical findings observed at 1,000 mg/kg bw included salivation after dosing, abdominal contractions, diarrhoea, vomiting and reduced body weight. After lowering the dose to 500 mg/kg bw, still salivation and vomiting occurred, and several other parameters were affected. In the male dogs, glucose, sodium, potassium, prothrombin time, eosinophils, absolute and relative testes and thyroid weights were decreased. In female dogs, AST was increased, phosphate decreased, albumin and fibrin decreased. Increases in glucose concentrations in the mid‐dose group and decreases in prothrombin time in the mid‐ and low‐dose groups were not very pronounced and not clearly dose related. Therefore, overall, the NOAEL in juvenile dogs was 300 mg/kg bw per day.
Table 5.
Results of repeated dose toxicity studies on NR that are relevant for deriving the NOAEL
| Reference | Species, strain, n/sex Study duration Guideline GLP | Test substance Dose levels (mg/kg bw) | Dose mg/kg bw | Lowest dose with statistically significant effect, considered as substance related, affected sex % change if relevant for the decision on a NOAEL |
|---|---|---|---|---|
|
Study No. SN17‐940 (Unpublished) (Thorsrud, 2018) |
Dog, Beagle, 4m/4f 28 days similar to OECD 408 GLP |
NF 0, 100, 300, 500/1,000 |
100 | m: prothrombin time ↓ (9%, not clearly dose related) |
| 300 |
m: glucose ↑ (22%, not clearly dose related) NOAEL |
|||
| 500/1,000 |
Salivation after dosing, abdominal contractions, diarrhoea, vomiting, bw ↓ m: sodium ↓, potassium ↓, eosinophils ↓, absolute and relative testes weights ↓ (not significant), abs and relative thyroid weights ↓ (not significant) f: AST ↑, phosphate ↓, fibrin ↓, rel. ovary weight ↑ |
|||
|
Study No. S14022 (Unpublished) (Bhoite et al., 2015) |
Rat, Sprague–Dawley, 10m/10f 90 days OECD 408 GLP |
NF 0,300, 1,000, 3,000 |
300 |
m: bw ↓ (8%), feed consumption ↓ (sometimes) NOAEL |
| 1,000 |
m: bw ↓ (9%), feed consumption ↓ (sometimes), leucocytes ↑ f: AST ↑, ALP ↑ m + f: neutrophils ↑, ALT ↑, triglycerides ↑, rel. liver weight ↑, rel. kidney weight ↑ |
|||
| 3,000 |
m: bw ↓ (17%), feed consumption ↓ (9‐14%), ALP ↑, bile acids ↑, abs liver weight ↑, abs and rel. testes weight ↓, abs and rel. epididymis weight ↓, chronic progressive nephropathy, tubular degeneration/atrophy of testes, reduced luminal sperm in epididymis and cellular debris f: feed consumption ↓ (sometimes), leucocytes ↑, monocytes ↑, GGT ↑, rel. ovary weight ↑, hypertrophy of corpora lutea m + f: hepatocellular hypertrophy and necrosis, thyroid follicular cell hypertrophy, hypertrophy of cortical zona glomerulosa in adrenals |
Abs: absolute; ALP: alkaline phosphatase; AST: aspartate aminotransferase; bw: body weight; f: females; GLP: Good Laboratory Practice; GGT: gamma‐glutamyltransferase; m: males; NOAEL: no observed adverse effect level; OECD: Organisation for Economic Co‐operation and Development; rel: relative.
3.10.2.1. 90‐day study
In a 90‐day repeated dose toxicity study in male and female Sprague–Dawley rats (Study No. 14022, unpublished, GLP, OECD TG 408; (Bhoite et al., 2015; Conze et al., 2016)), the NF was administered by gavage at doses of 0, 300, 1,000 or 3,000 mg/kg bw per day to 10 male and female Sprague–Dawley rats per group. One additional group was administered NAM at 1,260 mg/kg bw which was equimolar to 3,000 mg/kg bw of the NF. The results of the TK part of this study are described in Section 3.8.
Effects that are considered as substance‐related are summarised in Table 5. Compared to controls, lower body weights were noted in all male rats that received the test items. The effect was dose related (−8%, −9%, −17%) after 90 days, and statistically significant at several time points throughout the study for all dose levels. No effect was found for female rats (0, −5%, −2%). Feed consumption was decreased in male rats at 3,000 mg/kg bw per day of the NF (9‐14%). Isolated decreases in feed consumption were observed at 300 and 1,000 mg/kg bw per day of the NF in male rats. The Panel notes that the effects on body weight are related to reduced food consumption and not very pronounced in the low and mid dose and are therefore not considered as adverse.
While the slight effects on body weight and food consumption were the only effects at 300 mg/kg bw, numerous effects were found at 1,000 and 3,000 mg/kg bw, which are summarised in Table 5. They refer to haematology, liver, kidneys and the hormonal system/genital organs. The number and magnitude of effects increase considerably from the mid‐ to the high‐dose group, indicating a steep dose–response relationship. At 3,000 mg/kg bw in several organs also findings in histopathological examinations are reported. The Panel notes that with 1,260 mg/kg bw NAM, equimolar to 3,000 mg/kg bw nicotinamide riboside chloride, the same effects occurred as for the NF. Only the effect sizes differed slightly and were sometimes higher or lower with NAM compared to the NF.
Based on the results of this study, the NOAEL is 300 mg/kg bw.
3.10.3. Reproductive and developmental toxicity
3.10.3.1. One generation reproduction toxicity study in rats
In a one‐generation reproduction toxicity study (Study No. G10959, unpublished, GLP, OECD TG 415 (Ganiger, 2016)), the reproductive performance of male and female Sprague–Dawley rats and potential developmental toxic effects of the NF were investigated. Four groups of animals (25/sex per group) where administered diets (ad libitum) containing different concentrations of the NF (0, 3,000, 6,000 and 12,000 mg/kg feed, respectively, corresponding to 169, 334, and 675 mg/kg bw per day in male rats and 273, 543, and 1,088 mg/kg bw per day in female rats) before mating, during pregnancy and through weaning. The evaluation included clinical signs, body weight and food consumption. The number, weight, survival and mortality of pups were recorded during the lactation period. Animals were subjected to necropsy and histopathological examination of the male and female reproductive organs was carried out.
At 12,000 mg/kg feed, lower body weight was observed only in males from treatment day 43 to 92, with reductions of 4.6–6.5%, when compared to the control group. Food consumption in the male group receiving 12,000 mg/kg feed was significantly reduced at two time points. Due to the weak effect, the finding on body weight was not considered as adverse by the authors of the study. The Panel agrees with this view.
Other endpoints including precoital time, gestation length, fertility parameters, pathological and histopathological examinations of reproductive organs of adult rats, survival and abnormalities in life and death were not affected.
Based on the above results, it is concluded that the NOAEL for fertility and reproductive performance is 12,000 mg/kg feed (675 mg/kg bw per day in males and 1,088 mg/kg bw per day in females) under the testing conditions and doses employed.
The Panel notes that slight changes in body weight in male rats were consistent with the findings in the repeated dose toxicity studies and support the NOAEL of 300 mg/kg bw per day.
3.10.3.2. Developmental toxicity study in rats
In an embryo‐fetal developmental toxicity study (Study No. G10957, unpublished, GLP, OECD TG 414; (Geetha Rao, 2016)), the NF was administered to four groups of pregnant Sprague–Dawley rats (24/group) at dose levels of 0, 325, 750 and 1,500 mg/kg bw per day by gavage, from gestation day 5 to 19. The evaluation included clinical signs, mortality, morbidity, body weight and food consumption. After caesarean section dams were examined for gross pathological changes. Fetuses were examined for sex, weight and external, visceral and skeletal malformations.
At doses of, respectively, 750 and 1,500 mg/kg bw of the NF, lower maternal feed consumption (respectively, −6%, −12%), maternal body weight (−4%, −8%), maternal body weight gain (−11%, −28%), and maternal corrected body weight gain9 (−55%, −107%) during gestation, were observed as compared to the control group. Gravid uterine weights were reduced by 13% at 1,500 mg/kg bw per day. Late resorptions were dose‐relatedly increased (0.05, 0.29, 0.48, 0.52%), statistically significant at 1,500 mg/kg bw. The mean fetal weight was significantly reduced by 6% and 17% at 750 and 1,500 mg/kg per day, respectively. The Panel considers these findings secondary to maternal toxicity.
At the 1,500 mg/kg per day dose, there was a significant increase in the incidence of fetal anasarca. In addition, there were incidences of two small fetuses, an incidence of a fetus with moderate flexed right forelimb and one fetus with a thread‐like tail.
Fetal skeletal examination revealed dose‐dependent increases in delayed, incomplete or poor ossification that were statistically significant in the high dose for some sternebrae, some cervical and thoracic vertebrae, pubis and in some phalanges of fore and hindlimbs. In addition, as minor anomaly, there was an increase of extra, accessory, and rudimentary rib No.14 (10.32; 10.26; 16.47 and 30.72% of the fetuses, which was statistically significant at 1,500 mg/kg bw), and a hypoplastic sternum No. 2 (0, 1.28, 4.12, 24.84% of the fetuses, statistically significant at 750 and 1,500 mg/kg bw). The incidence of the finding at 750 mg/kg bw was within the historical control range and therefore not considered as adverse by the investigating laboratory. The Panel notes that these skeletal findings are often associated with general toxicity/reduced body weight gain of the dams.
Based on the above findings, it is concluded that the NOAEL for maternal toxicity is 325 mg/kg bw per day. The NOAEL for embryo/fetotoxicity is also 325 mg/kg bw per day; effects observed at 750 mg/kg bw per day are considered secondary to maternal toxicity.
3.10.3.3. Other studies
In a test performed on human embryonic kidney (HEK) 293 cells transfected with a human ether‐ago‐go‐related gene (hERG) (Study No. 20151223, unpublished), the NF did not affect the functioning of the human potassium channel as no inhibition of the hERG tail current density was observed.
3.10.4. Human data
The applicant provided one single dose pharmacokinetic study (Wilson, 2015; Trammell et al., 2016a) and four clinical trials (Airhart et al., 2017; Martens, 2017; Dollerup et al., 2018; Martens et al., 2018; Schacter, 2018; Conze et al., 2019) in which safety‐related parameters following the consumption of the NF were addressed (Table 6).
Table 6.
Overview table of human studies
| Reference | Study design | Study population | Duration of Study | Doses | Safety‐related parameters investigated | Summary of Results |
|---|---|---|---|---|---|---|
| Schacter (2018) and Conze et al. (2019) | Randomised, double‐blind, placebo‐controlled parallel study |
140 healthy men and women (30/dose group) Age: 40–60 years BMI: 25.0–30.1 kg/m2 |
2‐week run‐in and 8‐week supplementation period |
100 mg NF/day 300 mg NF/day 1,000 mg NF/day Placebo |
Anthropometric measures and vital signs, haematology and clinical chemistry (blood lipids, CBC, Na, K, Cl, HbA1c, creatinine, BUN, AST, ALT, GGT, bilirubin), and incidence of adverse events (AE) |
AEs classified as possibly related to the treatment 100 mg NR (2 AEs): leg pain, high blood pressure; 300 mg NR (2 AEs): nausea, leg pain; 1,000 mg NR (3 AEs): sore back, muscle soreness, nausea; placebo (4 AEs): rash, raised liver function tests, nausea, upset stomach. Haematology and clinical chemistry Significant between‐group difference in participants’ WBC count between the 300 mg NR and placebo at week 8 (p = 0.048) within clinical reference ranges Significant between‐group difference in the change in RBC distribution width from screening to week 8 between the 1,000 mg NR group and placebo (p = 0.038) within clinical reference ranges Significant between‐group difference eosinophil count between the 1,000 mg NR group and 100 mg NR group, 300 mg NR group, and placebo group at week 4 (p = 0.037, p = 0.009 and p = 0.047, respectively) within clinical reference ranges Liver function Significant between‐group difference in the change in ALT from baseline to week 1 between the 1,000 mg NR group and the 100 mg NR group (p = 0.013) and placebo group (p = 0.033) and in the change in ALT from baseline to week 2 between the 1,000 mg NR group and 100 mg NR group (p = 0.025) and placebo group (p < 0.001) within clinical reference ranges Significant between‐group difference in AST between the 1,000 mg NR group and placebo (p = 0.019) at week 2 within clinical reference ranges Significant between‐group difference in the change in AST from baseline to week 2 between the 1,000 mg NR group and 300 mg NR group (p = 0.040) within clinical reference ranges Vital signs and anthropometric measures No significant between‐group differences |
| Wilson (2015) | Single centre, three arm, randomised, double‐blind cross‐over study |
16 healthy men or women Age: 30–55 years BMI: 18.5–29.9 kg/m2 |
Each individual receives each dose on separate study days with a 7‐day wash‐out period between study days |
100 mg NF 300 mg NF 1,000 mg NF |
Vital signs, haematology and clinical chemistry (fasting glucose, eGFR, CBC, Na, K, Cl, creatinine, urate, AST, ALT, GGT, bilirubin), and monitoring of AEs |
AEs classified as possibly related to the treatment 100 mg NR (1 AE): decrease in haemoglobin (participant withdrawn); 300 mg NR (1 AE): decrease in WBC count (participant withdrawn) Haematology and clinical chemistry Significant between‐group difference in participants’ neutrophils count between the 300 mg NR group and 1,000 mg NR group (p = 0.026) within clinical reference ranges Vital signs No significant between‐group differences |
| Martens (2017) and Martens et al. (2018) | Randomised, placebo‐controlled, double blind, crossover study |
30 healthy men and women Age: 55–79 years BMI: < 40 kg/m2 |
Each group received placebo or NR for 6 weeks (2×/day) followed by opposite treatment for 6 weeks. No washout period was included; carryover not expected |
500 mg NF per day Placebo |
AEs and self‐reported side effects, anthropometric measures, haematology and clinical chemistry (CBC, blood lipids, total protein, albumin, AST, ALT, creatinine, BUN, eGFR, Ca, glucose, Na, K, Cl, CO2) |
AEs classified as possibly related to the treatment NR group (3 AEs): nausea, muscle cramps, and increased bruising Haematology and clinical chemistry Significant between‐group differences in mean platelet volume and relative (%) eosinophil and basophil composition after 6 weeks, within the normal reference range |
| Dollerup et al. (2018) |
Randomised, double‐blinded, placebo controlled, parallel‐group study |
40 healthy obese (BMI > 30 kg/m2) Caucasian males Age: 40–70 years Sedentary (< 30 min exercise per day), non‐smokers, and no prescribed medicine |
Each group received treatment or placebo twice daily for 12 weeks |
1,000 mg NF, 2 times per day Placebo |
Anthropometric measures, haematology and clinical chemistry (creatinine, Na, K, urea nitrogen, albumin, ALT, AST, bilirubin, haemoglobin, WBC count and platelets, fasting glucose, fasting insulin, HbA1c, blood lipids) and monitoring of AEs |
AEs classified as possibly related to the treatment NR group (4 AEs): pruritus, excessive sweating, bloating, and transient changes in stool Placebo group (2 AEs) Haematology and clinical chemistry Three participants from the placebo group displayed mild thrombocytosis (< 700 × 109/L) Significant increase in plasma triglycerides from a pretreatment mean of 1.5 mmol/L to a post‐treatment mean of 1.8 mmol/L in the NR group (within‐group p = 0.01), within the normal reference No significant between‐group differences reported |
| Airhart et al. (2017) | Open‐label, non‐randomised study | 8 healthy men and women, ages 21–50 | 10 days | 250 mg NF was orally administered on days 1 and 2, then up‐titrated to peak dose of 1,000 mg twice daily on days 7 and 8. On the morning of day 9, subjects completed a 24‐h pharmacokinetic study after receiving 1,000 mg of the NF at t = 0 | Anthropometric measures, haematology and clinical chemistry (CBC, Na, Cl, glucose, BUN, creatinine, creatine kinase, AST, ALT, uric acid, lactate dehydrogenase) and self‐reported AEs |
No AEs or side effects were reported. Haematology and clinical chemistry (between baseline and day 9) Serum K decreased by an average of 0.4 mEq/L (p = 0.015), within normal range Decrease in haematocrit (−2%, p = 0.005), haemoglobin (−0.4 g/dL, p = 0.04), and platelet count (−20,000/μL, p = 0.03) |
AE: adverse event; ALT: alanine transaminase; AST: aspartate transaminase; BMI: body mass index; BUN: blood urea nitrogen; Ca: calcium; CBC: complete blood count; Cl: chloride; eGFR: estimated glomerular filtration rate; GGT: gamma‐glutamyltransferase; HbA1c: haemoglobin A1c; K: potassium; Na: sodium; NR: nicotinamide riboside chloride; RBC: red blood cell; WBC: white blood cell.
The Panel notes that in these studies on healthy, adult human subjects, with doses of the NF from 100 mg for one day up to 2,000 mg/day for 12 weeks, changes in haematology and clinical chemistry remained within reference ranges and that no dose‐dependent adverse effects in the safety parameter examined were observed.
Studies suggest that high NAM intake, that undergoes methylation‐mediated degradation, could affect the methyl group pool balance (Sun et al., 2012, 2017; Li et al., 2013; Tian et al., 2013, 2014). However, the Panel considers that these studies on NAM do not allow firm conclusions on potential adverse effects.
The applicant conducted a post hoc analysis of the blood samples collected in the 8‐week supplementation study by (Schacter, 2018; Conze et al., 2019) using sodium citrate‐treated plasma collected before and after 8 weeks of NF ingestion up to 1,000 mg/day. Plasma homocysteine concentrations were quantified by liquid chromatography coupled with tandem mass spectrometry. The ingestion of up to 1,000 mg NF/day had no effect on plasma homocysteine concentrations.
3.11. Allergenicity
The NF is a synthetic product containing > 90 % nicotinamide riboside chloride. Potential process impurities have been well characterised. Since the NF does not contain any protein, the risk of allergenicity is low.
4. Discussion
The NF, nicotinamide riboside chloride, is a synthetic form of nicotinamide riboside, proposed to be used in food supplements at levels up to 300 mg/day. The NF is intended for use by the healthy adult population, including pregnant and lactating women.
Information provided on the production process, composition, specifications, batch‐to‐batch variability and stability of the NF is sufficient and does not raise concerns about the safety of the NF.
Considering the outcomes of the bacterial reverse mutagenicity test, the in vivo mammalian erythrocyte micronucleus test and the in vitro mammalian chromosome aberration test conducted with the NF, as well as its nature, the Panel considers that there are no concerns regarding genotoxicity.
From the ADME data available in mice, rats, dogs, and humans, the Panel notes that the NF is likely to be absorbed mainly as NAM following hydrolysis in the gut. If a fraction of the NF were absorbed intact, it would be expected to be rapidly metabolised to NAM in the blood. Animal and human data indicate that, upon absorption, the NF contributes to the NAM body pool, i.e. acts as a precursor of NAD+ in cells and is primarily metabolised in the liver to 1‐MN through methylation and subsequently to 2‐PY, and 4‐PY, following oxidation. These metabolites are then excreted in the urine.
The Panel derives a NOAEL of 300 mg/kg bw per day from the repeated dose toxicity studies with rats and dogs conducted with the NF. The Panel notes that in the 90‐day study in rats, equimolar doses of NAM and the NF (1,260 mg/kg bw per day of NAM vs 3,000 mg/kg bw per day nicotinamide riboside chloride) caused the same effects. Reproductive and developmental toxicity studies in rats with the NF were also provided, which indicate a NOAEL for fertility and reproductive performance of 675 mg/kg bw per day in males and 1,088 mg/kg bw per day in females and a NOAEL for maternal and embryo/fetotoxicity of 325 mg/kg bw per day. The embryo/fetotoxicity findings observed at a dose of 750 mg/kg bw per day are considered secondary to maternal toxicity.
The Panel considers that available human studies on the NF, which were conducted in healthy adult subjects with dosages from 100 mg for one day up to 2,000 mg/day for up to 12 weeks, do not raise safety concerns. The safety parameters addressed in these studies were vital signs, haematology and clinical chemistry.
The proposed maximum use level of 300 mg nicotinamide riboside chloride per day corresponds to an intake of NAM of up to 126 mg/day. This intake is sixfold lower than the current UL for NAM of 900 mg/day for adults (excluding pregnant and lactating women) in the EU (SCF, 2002).
The margin of exposure (MoE) between the proposed maximum use level of 300 mg/day (i.e. 4.3 mg/kg bw in a 70‐kg adult) and the NOAEL of 300 mg/kg bw per day derived from animal data is 70. In the light of the human data available on nicotinamide riboside chloride and NAM, the Panel considers that this MoE is sufficient for the adult population, excluding pregnant and lactating women.
The MoE between the proposed maximum use level of 300 mg/day (i.e. 4.3 mg/kg bw in a 70‐kg adult) and the NOAEL of 325 mg/kg bw per day for maternal and embryo/fetotoxicity derived from the rat study, is 76. In the absence of data which could justify a MoE lower than 100 for pregnant and lactating women, the Panel concludes that an intake of 230 mg/day of the NF (rounded to the nearest ten) is safe for these population groups.
5. Conclusions
The Panel concludes that the NF, nicotinamide riboside chloride, is safe under the proposed uses and use levels, i.e. up to 300 mg/day, for the healthy adult population, excluding pregnant and lactating women. The Panel concludes that an intake of the NF up to 230 mg/day is safe for pregnant and lactating women.
The Panel also concludes that nicotinamide riboside chloride is a source from which NAM, which is a form of niacin, is bioavailable.
The Panel could not have reached the conclusion on the safety of the NF under the proposed conditions of use without the following data claimed as proprietary by the applicant: an in vitro study evaluating the metabolism of nicotinamide riboside in blood (Study No. 160312); an oral 7‐day dose range finding toxicity study in juvenile dogs (study No. SN17‐921); a 28‐day repeated‐dose oral toxicity study in juvenile dogs (Study No. SN17‐940); a 90‐day repeated‐dose oral toxicity study in Sprague–Dawley rats (Study No. S14022); a reproductive toxicity study (Study No. G10959) and a developmental toxicity study (Study No. G10957) in rats.
Steps taken by EFSA
Letter from the European Commission to the European Food Safety Authority with the request for a scientific opinion on synthetic form of nicotinamide riboside. Ref. Ares(2018)5155905, dated 8 October 2018.
On 08 October 2018, a valid application on the Use of NIAGEN® for General Use as a Novel Food Ingredient in Supplements per Commission Implementing Regulation (EU) 2017/2469, which was submitted by ChromaDex Inc., was made available to EFSA by the European Commission through the Commission e‐submission portal (NF 2018/0233) and the scientific evaluation procedure was initiated.
On 27 February 2019, EFSA requested the applicant to provide additional information to accompany the application and the scientific evaluation was suspended.
On 20 March 2019, additional information was provided by the applicant through the Commission e‐submission portal and the scientific evaluation was restarted.
During its meeting on 04 July 2019, the NDA Panel, having evaluated the data, adopted a scientific opinion on the safety of nicotinamide riboside chloride as a NF pursuant to Regulation (EU) 2015/2283.
Abbreviations
- 1‐MN
1‐methylnicotinamide
- 2‐PY
N‐methyl‐2‐pyridone‐carboxamide
- 4‐PY
N‐methyl‐4‐pyridone‐carboxamide
- ADME
asorption, distribution, metabolism and excretion
- ADPR
adenine diphosphate ribose
- AE
adverse events
- ALP
alkaline phosphatase
- ALT
alanine transaminase
- AOAC
Association of Analytical Communities
- AR
average requirement
- AST
aspartate transaminase
- AUC
area under the curve
- BMI
body mass index
- BUN
blood urea nitrogen
- bw
body weight
- CAS
Chemical Abstracts Service
- CBC
complete blood count
- CFU
colony forming units
- Cmax
maximum concentration
- DRV
dietary reference values
- EC
Enzyme Commission
- eGFR
estimated glomerular filtration rate
- ESI‐MS
electrospray ionisation mass spectrometry
- GC‐FID
gas chromatography coupled with a flame ionisation detector
- GGT
gamma‐glutamyltransferase
- GLP
Good Laboratory Practice
- GMP
Good Manufacturing Practice
- GRAS
generally recognized as safe
- HACCP
Hazard Analysis Critical Control Points
- HbA1c
haemoglobin A1c
- HDPE
high‐density polyethylene
- hERG
human ether‐ago‐go‐related gene
- HPBL
human peripheral blood lymphocyte
- HPLC‐UV
high‐performance liquid chromatography‐ultraviolet spectroscopy
- HPLC UV‐Vis
high‐performance liquid chromatography coupled with ultraviolet/visible spectroscopy
- ICP‐MS
inductively coupled plasma mass spectrometry
- ITT
intention‐to‐threat
- IUPAC
International Union of Pure and Applied Chemistry
- LDPE
low‐density polyethylene
- LNHPD
Licensed Natural Health Products Database
- LOQ
limit of quantitation
- MoE
margin of exposure
- NA
nicotinic acid
- NaAD
nicotinic acid adenine dinucleotide
- NAD+
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NaR
nicotinic acid ribose
- NDA
EFSA Panel on Nutrition, Novel foods and Food Allergens
- NDI
new dietary ingredient
- NE
niacin equivalent
- NF
novel food
- NMN
nicotinamide mononucleotide
- NMR
nuclear magnetic resonance
- NOAEL
no observed adverse effect level
- NR
nicotinamide riboside chloride
- Nrk
nicotinamide riboside kinase
- OECD
Organisation for Economic Co‐operation and Development
- P/E
polychromatic erythrocyte/total erythrocyte
- PAL
physical activity level
- PP
per‐protocol
- PRI
population reference intake
- RBC
red blood cell
- RH
relative humidity
- TK
toxicokinetic
- Tmax
time at which the Cmax is observed
- UF
uncertainty factor
- UL
tolerable upper intake level
- UMP
uridine monophosphate
- USP
United States Pharmacopeia
- WBC
white blood cell
Suggested citation: EFSA NDA Panel (EFSA Panel on Nutrition, Novel foods and Food allergens) , Turck D, Castenmiller J, de Henauw S, Hirsch‐Ernst KI, Kearney J, Maciuk A, Mangelsdorf I, McArdle HJ, Naska A, Pelaez C, Pentieva K, Siani A, Thies F, Tsabouri S, Vinceti M, Cubadda F, Engel K‐H, Frenzel T, Heinonen M, Marchelli R, Neuhäuser‐Berthold M, Pöting A, Poulsen M, Sanz Y, Schlatter JR, van Loveren H, de Sesmaisons‐Lecarré A, Germini A and Knutsen HK, 2019. Scientific Opinion on the safety of nicotinamide riboside chloride as a novel food pursuant to Regulation (EU) 2015/2283 and bioavailability of nicotinamide from this source, in the context of Directive 2002/46/EC. EFSA Journal 2019;17(8):5775, 30 pp. 10.2903/j.efsa.2019.5775
Requestor: European Commission
Question number: EFSA‐Q‐2018‐00480
Panel members: Dominique Turck, Jacqueline Castenmiller, Stefaan De Henauw, Karen Ildico Hirsch‐Ernst, John Kearney, Helle Katrine Knutsen, Alexandre Maciuk, Inge Mangelsdorf, Harry J McArdle, Androniki Naska, Carmen Pelaez, Kristina Pentieva, Alfonso Siani, Frank Thies, Sophia Tsabouri, Marco Vinceti.
Adopted: 4 July 2019
Amended: 8 December 2019
Notes
Regulation (EU) 2015,2283 of the European Parliament and of the Council of 25 November 2015 on novel foods, amending Regulation (EU) No 1169.’2011 of the European Parliament and of the Council and repealing Regulation (EC) No 258:97 of the European Parliament and of the Council and Commission Regulation (EC) No 1852/2001 (0J L 327, 11.12.2015, p. 1).
Directive 2001,16/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements (0.11, 183, 12.7.2002, p. 51).
Commission Implementing Regulation (EU) 2017/2469 of 20 December 2017 laying down administrative and scientific requirements for applications referred to in Article 10 of Regulation (EU) 2015/2283 of the European Parliament and of the Council on novel foods. OJ L 351, 30.12.2017, pp. 64–71.
GRAS No 635, available at https://www.accessdata.fda.gov/scripts/fdcc/index.cfm?set=GRASNotices%26id=635
NDIN 882, available at https://www.regulations.gov/document?D=FDA-2015-S-0023-0087
NDIN 1062, available at https://www.regulations.gov/document?D=FDA-2018-S-0023-0032
NPN 80088977, available at https://health-products.canada.ca/lnhpd-bdpsnh/info.do?licence=80088977
1 mg NE = 1 mg nicotinamide = 1 mg nicotinic acid = 60 mg tryptophan.
Corrected body weight gain = (maternal weight change between day 5 and day 20) – gravid uterus weight.
References
- Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, Tian R, Shen DD and O'Brien KD, 2017. An open‐label, non‐randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE, 12, e0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery B (Department of Pharmaceutics and Drug Delivery, The University of Mississippi), 2017. Pharmacokinetic Analysis of Plasma Nicotinamide and 1‐Methyl Nicotinamide Concentrations in Toxicokinetic Study of NIAGEN (Nicotinamide Riboside). Study Report No AV151003. Unpublished Study Report, 9 pp.
- Baum CL, Selhub J and Rosenberg IH, 1982. The hydrolysis of nicotinamide adenine nucleotide by brush border membranes of rat intestine. The Biochemical Journal, 204, 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoite P and Jayachandra K (Syngene International Limited), 2014a. Niagen: 14‐Day Dose Range Finding Oral Toxicity Study in Sprague Dawley Rats (Non‐GLP Study). Study No S13120. Unpublished Study Report, 74 pp.
- Bhoite P and Jayachandra K (Syngene International Limited), 2014b. Single Dose Oral Toxicity Study of Niagen in Sprague Dawley Rats. Study No. S13101. Unpublished Study Report, 60 pp.
- Bhoite P, Jayachandra K and Suman M (Syngene International Limited), 2015. Comparative 90‐Day Oral Toxicity Study of Niagen and Nicotinamide in Sprague Dawley Rats. Study No S14022. Unpublished Study Report, 291 pp.
- Bieganowski P and Brenner C, 2004. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss‐Handler independent route to NAD+ in fungi and humans. Cell, 117, 495–502. [DOI] [PubMed] [Google Scholar]
- Bzowska A, Kulikowska E and Shugar D, 2000. Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacology & Therapeutics, 88, 349–425. [DOI] [PubMed] [Google Scholar]
- Chen G (Keystone Bioanalytical, Inc.), 2017. Validation Report on an Analytical Procedure for the Quantification of 1‐Methyl Nicotinamide in Dog Citrate Plasma by LC/MS/MS (Calibration Range: 20–2000 ng/mL). Study Report No V170821.00. Unpublished Study Report, 41 pp.
- Chi Y and Sauve AA, 2013. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Current Opinion in Clinical Nutrition and Metabolic Care, 16, 657–661. [DOI] [PubMed] [Google Scholar]
- Conze DB, Crespo‐Barreto J and Kruger CL, 2016. Safety assessment of nicotinamide riboside, a form of vitamin B3. Human and Experimental Toxicology, 35, 1149–1160. [DOI] [PubMed] [Google Scholar]
- Conze D, Brenner C and Kruger CL, 2019. Safety and metabolism of long‐term administration of NIAGEN (nicotinamide riboside chloride) in a randomized, double‐blind, placebo‐controlled clinical trial of healthy overweight adults. Scientific Reports, 9, 9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollerup OL, Christensen B, Svart M, Schmidt MS, Sulek K, Ringgaard S, Stodkilde‐Jorgensen H, Moller N, Brenner C, Treebak JT and Jessen N, 2018. A randomized placebo‐controlled clinical trial of nicotinamide riboside in obese men: safety, insulin‐sensitivity, and lipid‐mobilizing effects. American Journal of Clinical Nutrition, 108, 343–353. [DOI] [PubMed] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), Younes M, Aggett P, Aguilar F, Crebelli R, Dusemund B, Filipicč M, Frutos MJ, Galtier P, Gundert‐Remy U, Kuhnle GG, Lambré C, Leblanc J‐C, Lillegaard IT, Moldeus P, Mortensen A, Oskarsson A, Stankovic I, Waalkens‐Berendsen I, Woutersen RA, Wright M, Di Domenico A, Fairweather‐Tait S, McArdle H, Smeraldi C and Gott D, 2018. Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources. EFSA Journal 2018;16(6):5294, 35 pp. 10.2903/j.efsa.2018.5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2014. Scientific Opinion on Dietary Reference Values for niacin. EFSA Journal 2014;12(7):3759, 42 pp. 10.2903/j.efsa.2014.3759 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), Turck D, Bresson J‐L, Burlingame B, Dean T, Fairweather‐Tait S, Heinonen M, Hirsch‐Ernst K, Mangelsdorf I, McArdle H, Naska A, Neuhäuser‐Berthold M, Nowicka G, Pentieva K, Sanz Y, Siani A, Sjödin A, Stern M, Tomé D, Vinceti M, Willatts P, Engel K‐H, Marchelli R, Pöting A, Poulsen M, Salminen S, Schlatter J, Arcella D, Gelbmann W, de Sesmaisons‐Lecarré A, Verhagen H and van Loveren H, 2016. Guidance on the preparation and presentation of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283. EFSA Journal 2016;14(11):4594, 24 pp. 10.2903/j.efsa.2016.4594 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), Turck D, Bresson J‐L, Burlingame B, Dean T, Fairweather‐Tait S, Heinonen M, Hirsch‐Ernst K, Mangelsdorf I, McArdle H, Naska A, Neuhäuser‐Berthold M, Nowicka G, Pentieva K, Sanz Y, Siani A, Sjödin A, Stern M, Tomé D, Vinceti M, Willatts P, Engel K‐H, Marchelli R, Pöting A, Poulsen M, Schlatter J, Germini A and Van Loveren H, 2018. Scientific opinion on safety of d‐ribose as a novel food pursuant to Regulation (EU) 2015/2283. EFSA Journal 2018;16(5):5265, 28 pp. 10.2903/j.efsa.2018.5265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- Expert Group on Vitamins and Minerals , 2003. Safe Upper Levels for Vitamins and Minerals. Food Standards Agency, London, UK. 360 pp. [Google Scholar]
- Ganiger S (Advinus Therapeutics Limited), 2016. Nicotinamide Riboside Chloride: One Generation Reproduction Toxicity Study Through Diet in Sprague‐Dawley Rats. Study No G10959, 313 pp.
- Geetha Rao G (Advinus Therapeutics Limited), 2016. Nicotinamide Riboside Chloride: Embryo‐Fetal Developmental Toxicity Study in Sprague Dawley Rats by Oral Route. Study No G10957. Unpublished Study Report, 319 pp.
- Gross CJ and Henderson LM, 1983. Digestion and absorption of NAD by the small intestine of the rat. Journal of Nutrition, 113, 412–420. [DOI] [PubMed] [Google Scholar]
- Guo T (Keystone Bioanalytical, Inc.), 2018. The Metabolism of Nicotinamide Riboside in Human Blood. Study No.: 160312. Unpublished Study Report, 14 pp.
- Kamath G (Syngene International Limited), 2015. Bacterial Reverse Mutation Test with Niagen. Study Number S15004. Unpublished Study Report, 71 pp.
- Kamath G (Syngene International Limited), 2016. In Vitro Mammalian Chromosome Aberration Test Using Human Peripheral Blood Lymphocyte with Niagen. Study No S15005, Unpublished Study Report, 69 pp.
- Kornberg A, 1955. Nicotinamide riboside phosphorylase Methods in Enzymology, 2, pp. 454–456. [Google Scholar]
- Li D, Tian YJ, Guo J, Sun WP, Lun YZ, Guo M, Luo N, Cao Y, Cao JM, Gong XJ and Zhou SS, 2013. Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. British Journal of Nutrition, 110, 2156–2164. [DOI] [PubMed] [Google Scholar]
- Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, Migaud M, Mitchison TJ, Baur JA and Rabinowitz JD, 2018. Quantitative Analysis of NAD Synthesis‐Breakdown Fluxes. Cell Metabolism, 27, 1067–1080.e1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens C (The Integrative Physiology of Aging Laboratory, University of Colorado Boulder), 2017. NIAGEN® Supplementation for Improving Physical and Metabolic Function in Midlife and Older Adult Humans. Unpublished Study Report. 28 pp.
- Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M and Seals DR, 2018. Chronic nicotinamide riboside supplementation is well‐tolerated and elevates NAD(+) in healthy middle‐aged and older adults. Nature Communications, 9, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y, 1971. Enzymatic phosphorolysis of pyridine ribonucleosides by rat liver nucleoside phosphorylase. Nicotinic Acid: Analogs and Coenzymes, 204–210. [Google Scholar]
- Pandey A (Syngene International Limited), 2016. In vivo Mammalian Erythrocyte Micronucleus Test in Rat with Niagen. Study No S15006. Unpublished Study Report. 78 pp.
- Rowen JW and Kornberg A, 1951. The phosphorolysis of nicotinamide riboside. Journal of Biological Chemistry, 193, 497–507. [PubMed] [Google Scholar]
- SCF (Scientific Committee on Food), 2002. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Nicotinic Acid and Nicotinamide (Niacin). https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80j_en.pdf [Google Scholar]
- Schacter G (KGK Science, Inc.), 2018. A Randomized, Double‐Blind, Placebo‐Controlled Parallel Study Investigating the Effects of NIAGEN® (Nicotinamide Riboside Chloride) on NIAGEN® Metabolites in Healthy Adults. Study Report No 15NRHC. Unpublished Study Report. 313 pp.
- Sun WP, Li D, Lun YZ, Gong XJ, Sun SX, Guo M, Jing LX, Zhang LB, Xiao FC and Zhou SS, 2012. Excess nicotinamide inhibits methylation‐mediated degradation of catecholamines in normotensives and hypertensives. Hypertension Research, 35, 180–185. [DOI] [PubMed] [Google Scholar]
- Sun WP, Zhai MZ, Li D, Zhou Y, Chen NN, Guo M and Zhou SS, 2017. Comparison of the effects of nicotinic acid and nicotinamide degradation on plasma betaine and choline levels. Clinical Nutrition, 36, 1136–1142. [DOI] [PubMed] [Google Scholar]
- Thorsrud B (Experimur), 2017. Oral 7‐Day Dose Range Finding Toxicity Study in Juvenile Dogs with NIAGEN®. Experimur Study No.: 17‐921. Unpublished Study Report. 172 pp.
- Thorsrud B (Experimur), 2018. Oral 28‐Day Toxicity Study in Juvenile Dogs with NIAGEN®. Experimur Study No.: 17‐940. Unpublished Study Report. 388 pp.
- Tian YJ, Li D, Ma Q, Gu XY, Guo M, Lun YZ, Sun WP, Wang XY, Cao Y and Zhou SS, 2013. Excess nicotinamide increases plasma serotonin and histamine levels. Sheng li xue Bao: [Acta physiologica Sinica], 65, 33–38. [PubMed] [Google Scholar]
- Tian YJ, Luo N, Chen NN, Lun YZ, Gu XY, Li Z, Ma Q and Zhou SS, 2014. Maternal nicotinamide supplementation causes global DNA hypomethylation, uracil hypo‐incorporation and gene expression changes in fetal rats. British Journal of Nutrition, 111, 1594–1601. [DOI] [PubMed] [Google Scholar]
- Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME and Brenner C, 2016a. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nature Communications, 7, 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Yu L, Redpath P, Migaud ME and Brenner C, 2016b. Nicotinamide riboside is a major NAD+ precursor vitamin in cow milk. Journal of Nutrition, 146, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgus‐Kutrowska B, Kulikowska E, Wierzchowski J, Bzowska A and Shugar D, 1997. Nicotinamide riboside, an unusual, non‐typical, substrate of purified purine‐nucleoside phosphorylases. European Journal of Biochemistry, 243, 408–414. [DOI] [PubMed] [Google Scholar]
- Wilson D (KGK Synergize, Inc.), 2015. Randomized, Double‐blind, Cross‐over Study of the Pharmacokinetics of Three Dosages of Niagen™ in Healthy Subjects. Study Report No 14NBHC. Unpublished Study Report. 212 pp.