

ABSTRACT
Antibody discovery using invitro display technologies such as phage and/or yeast display has become acornerstone in many research and development projects, including the creation of new drugs for clinical use. Traditionally, after the selection phase, random clones are isolated for binding validation and Sanger sequencing. More recently, next-generation sequencing (NGS) technology has allowed deeper insight into the antibody population after aselection campaign, enabling the identification of many more specific binders. However, this approach only provides the DNA sequences of potential binders, the properties of which need to be fully elucidated by obtaining corresponding clones and expressing them for further validation. Here we present arapid novel method to harvest potential clones identified by NGS that uses asimple PCR and yeast recombination approach. The protocol was tested in selections against three different targets and was able to recover clones at an abundance level that would be impractical to identify using traditional methods.
KEYWORDS: NGS (Next-generation sequencing), antibody, antibody libraries, phage display, yeast display
Introduction
Monoclonal antibodies are now widely used for the treatment of many different diseases, including cancer, autoimmunity, infection, migraine and, atherosclerosis. By the end of 2018, there were 87 monoclonal antibodies approved in the US and/or Europe, grossing annual revenues of ~$100B (see ref.1 and www.antibodysociety.org), making monoclonal antibodies the fastest growing drug class, and representing half of all worldwide biopharmaceutical sales. Over 150 biotech and pharmaceutical companies are currently discovering or developing antibody drugs, and the high growth rate is in part due to the higher and faster approval rates for antibodies compared to other drugs.2 This reflects their straightforward development pathways, high tolerance and relatively low risks of unexpected safety issues in most human clinical trials, immunotherapy excluded.
In vitro antibody selection platforms such as phage and yeast display assist the development of human recombinant antibodies against virtually any desired antigen or epitope that may have arelevant biological function.3 This, in turn, accelerates the discovery process and helps the continuous expansion of the range of diseases that can be treated with these molecules. Naïve phage antibody libraries generated from the B-cells of nonimmune human donors contain antibodies with the potential to recognize all possible antigen types, including human. The technology has been successfully used in the development of U.S.Food and Drug Administration-approved drugs such as adalimumab (AbbVie/Humira®), necitumumab (Eli Lilly/Portrazza®), ramucirumab (Eli Lilly/Cyramza®). During the lead generation phase of drug discovery, antibodies are commonly displayed on the surface of filamentous phage particles either as antigen-binding fragments (Fab) or as asingle-chain variable fragment (scFv) domains,4 which consist of the antibody heavy and light chain variable regions connected by alinker peptide that preserves the binding properties of the antibody.
When using invitro display, one of the concerns is that the total diversity of antibodies found in these libraries may not be fully tapped during selections. Theoretical5 and experimental6,7 assessments indicate that one would expect to recover thousands of different antibodies from a108 diversity library, and yet, unless heroic efforts are made,8 the number of antibodies identified is usually less than 50. This is mainly due to two factors:1) many antibody libraries are not as diverse as indicated by the number of transformants,9,10 and2) the way potential binders are traditionally screened identifies leads after extensive individual clone screening, an inefficient approach vexed by the redundancy of abundant clones, and sparse, or absent, representation of most clones that occur in lower frequencies within the selected population. Moreover, clones with high binding activity, or with other prominent functional properties, can be found within the lower frequency clone population. Their rarity can be explained by lower expression levels in aprokaryotic system, or by more general biases resulting from the biopanning process.11 An alternative approach is to use next-generation sequencing (NGS), which has massively increased the capacity to sequence millions of clones in avery fast and inexpensive way,12 revolutionizing several aspects of biological research, including invitro and invivo antibody selections.10,13
We routinely use an approach that exploits NGS in invitro display selection analyses. By combining phage display, yeast display, and NGS, we have shown that the number of identified antibodies can exceed 1,000,10,14,15 reflecting the far deeper sampling that NGS provides: millions of reads, rather than hundreds of microtiter plate wells screened. This is particularly true if there is bias in the selected population, which is often the case. However, unless antibodies identified by NGS can be easily produced as clones for further analysis, their specificities and properties cannot be assessed, leaving uncertain the true breadth of available reactivities, and risking the loss of many potentially valuable antibodies. The greater the number of different antibodies available, the greater the number of different epitopes, and consequently biological activities, that can be targeted. The caveat of NGS sequencing is that after identifying the desired sequences, one does not have the isolated clones in hand. scFvs are typically composed of ~250 amino acids, and, for alarge number of potential leads identified after aselection campaign, synthesizing their coding sequences can become very costly. Moreover, this approach requires the full sequence of the scFv domain to be known, and most current NGS platforms do not offer sufficiently long reads to provide more than either the VH or VL domain.
Since generating clones from NGS sequence represents the primary bottleneck in the full exploitation of NGS for invitro antibody selection, we15 and others16 have pioneered the development of efficient methods to rescue even low abundance scFv sequences identified after NGS analysis. Here, we describe arapid, novel, straightforward method to generate antibody clones identified by NGS that can reach deep into the abundance rank. The method consists of isolating clones of interest identified directly from aselected library output by PCR amplification, where the downstream/reverse primer is complementary to the HCDR3 and JH region of the desired clones, while the upstream/forward primer is clone-independent and anchored within the plasmid (Figure 1). The amplification product is subsequently easily cloned directly into yeast by homologous recombination using the specifically designed yeast-display plasmids, allowing the binding properties of the identified antibodies, such as affinity, to be rapidly measured by flow cytometry.14
Figure 1.
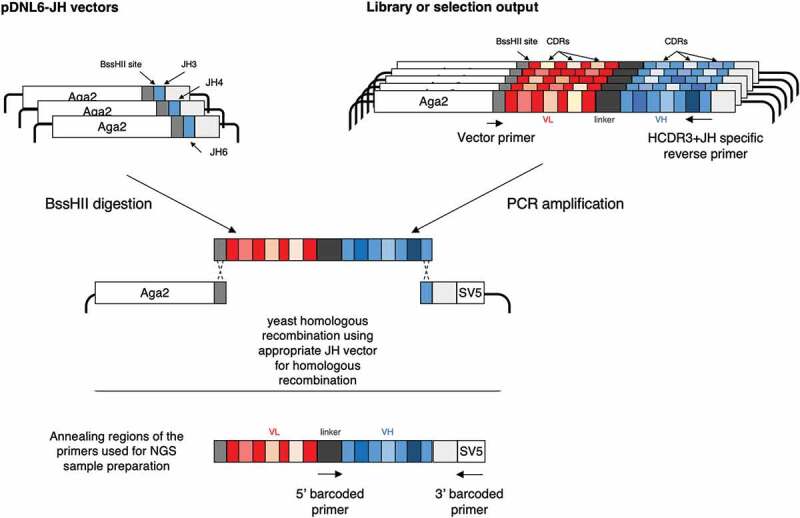
Schematic representation of the strategy. The scFvs containing the HCDR3 of interest is amplified using two oligonucleotides: one specific for the HCDR3 and the JH region and auniversal one annealing on the vector backbone. The amplification product is cloned by homologous recombination in the pDNL6-JH vector matching the JH of the scFv of interest. Barcoded primers are used to prepare samples for NGS analysis of the VH domains of the selected antibodies: one annealing on the linker region, the other annealing on the SV5 tag present on the vector backbone.
Results
PDNL6-JH vectors and ScFv recovery strategy
Three JH regions (corresponding to the ones present in the scFv scaffolds of the phage antibody library used for this study17) were added to the backbone of the pDNL6 yeast display vector.18 The chosen sequences correspond to portions of the JH3, JH4, and JH6 human germlines, which are unique, and in the case of JH4, sufficient to recreate the others (JH1, JH2 and JH5) at the amino acid level (Table 1). Each vector contains aBssHII restriction upstream of the JH sequence to be used for vector linearization. Once linearized, each vector can be used to clone an scFv of interest by yeast homologous recombination (Figure 1), once the sequence, and JH gene, is known.
Table 1.
Oligonucleotides designed for the study. Oligonucleotides generated to create the three pDNL6-JH(3/4/6) vectors (“vector oligos”) and corresponding sequences present in the oligos to amplify specific scFvs of interest (“constant part of oligos”).
| Sequences and oligos | JH Sequence | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| JH3 (amino acid) | G | T | M | V | T | V | S | S | S | ||
| JH3 vector sequence | cg | cGC | GGG | ACA | ATG | GTC | ACC | GTC | TCT | TCA | G |
| Constant part of HCDR3/JH3 oligo | ct | agC | TGA | AGA | GAC | GGT | GAC | CAT | TGT | CCC | G |
| JH4/1/2/5 (amino acid) | G | T | L | V | T | V | S | S | |||
| JH4/1/2/5 vector sequence | cg | cGC | GGA | ACC | CTG | GTC | ACC | GTC | TCC | TCA | G |
| Constant part of HCDR3/JH4/1/2/5 oligo | ct | agC | TGA | GGA | GAC | GGT | GAC | CAG | GGT | TCC | G |
| JH6 (amino acid) | G | T | T | V | T | V | S | S | |||
| JH6 vector sequence | cg | cGC | GGG | ACC | ACG | GTC | ACC | GTC | TCC | TCA | G |
| Constant part of HCDR3/JH6 oligo | ct | agC | TGA | GGA | GAC | GGT | GAC | CGT | GGT | CCC | G |
| JH1 (amino acid) | G | T | L | V | T | V | S | S | |||
| JH2 (amino acid) | G | T | L | V | T | V | S | S | |||
| JH3 (amino acid) | G | T | L | V | T | V | S | S | |||
To recover an scFv of interest identified by NGS within alibrary or aselection output, we carry out aPCR using acommon forward primer hybridizing to the vector backbone upstream of the scFv gene, and long HCDR3-specific primers that anchor within identified HCDR3-JH sequences. In this way the PCR products will have aregion at the 5ʹ-end matching the vector backbone and the JH region at the 3ʹ-end is homologous to that present in the acceptor vector. This allows the direct cloning of the scFv of interest by yeast homologous recombination (Figure 1).
Antibody selections and deep-sequencing analysis
We tested this novel strategy for rescuing antibodies identified by deep sequencing on the outputs obtained from independent selections on three different targets: B7-H4, CD40 ligand (CD40-L), and OX40. We used anaïve recombinant scFv library17 and aprocedure consisting of two rounds of phage display selection, followed by two rounds of yeast display sorting, which was previously shown to significantly increase the number of identified target-specific antibodies.14,18,19
The selection outputs were analyzed by MiSeq sequencing (2 x250 bp paired-end reads), which provides information on the entire VH domain of enriched scFvs, including the HCDR3 and the JH sequences of each molecule. Barcoded primers were used for the preparation of the samples submitted for the NGS analysis: one primer mapping on the linker region between the VL and the VH, and one mapping downstream the end of the VH, in the SV5 tag region, able to amplify any scFv cloned into the yeast display vector independently of its sequence.
The sequencing depth used covered the maximum possible diversity of the outputs by at least 10-fold. We ranked HCDR3 by abundance for each selection, merging those that were very similar and/or possibly created by PCR mutation or sequencing errors. The HCDR3 sequences identified for each selection, along with their relative abundances, are indicated in Table 2.
Table 2.
HCDR3 sequences and relative abundances identified for each selection. In bold the HCDR3 we tried to rescue using the described strategy.
| Anti B7-H4 HCDR3 |
% of total |
ANTI CD40L HCDR3 |
% of total |
ANTI OX40 HCDR3 |
% of total |
|---|---|---|---|---|---|
| AKVGWQAFDI | 28.429 | ARLAVADPYDY | 26.853 | ARDVGYSYGDNWFDP | 89.603 |
| AKWGLGDLDY | 15.073 | ARLSLDGSGLYGMDV | 16.560 | AKDRSSGWYSDGMDV | 3.045 |
| AKVVVIKGRAFDI | 7.865 | ANWEAVAGTFDY | 9.318 | ARDNSSSWYTSALDV | 0.590 |
| AKGPTVTNVRGFDY | 5.338 | ARGLGGGFGDY | 7.876 | ATGSSSSWFDP | 0.462 |
| ARGHYYDMDV | 5.219 | ATDQISLRYSPDAFDI | 7.221 | ARGEGYYDFWSGGNWFDP | 0.275 |
| ARDRFRNFDY | 3.126 | ARALRGYGDYFDY | 7.085 | TRGGLVVAATHPGAFDI | 0.259 |
| ARKHYYGMDV | 3.056 | ARWEQQLAGGFDP | 2.426 | ARGRYGDY | 0.235 |
| ARATFGAFDI | 2.801 | AADTPSSV | 2.417 | ARAQDYYYYYGMDV | 0.184 |
| ARFPMVRDFDY | 2.400 | ARGYYAADY | 1.611 | ARVRTDWYVDL | 0.135 |
| AKAEWGSFDY | 1.923 | AKEVGYRVYYYGMDV | 1.573 | ARGTAVAGTLVDY | 0.117 |
| AISSWYGFDY | 1.412 | ARLGGDYYDSSLYGMDV | 1.368 | ARGTVTIMRGY | 0.109 |
| VRDSYRFFDH | 1.293 | AKALSSGSYYGSDYYFDY | 1.236 | AHGAYSFDY | 0.108 |
| AKIYYYGMDV | 0.796 | TTVPVVAFDI | 1.204 | ARDVDVGWFDP | 0.078 |
| AKVGWNYFDY | 0.792 | ARHYDHYYYGMDV | 1.173 | ARDWSMTSVTMPDY | 0.076 |
| AKGRWWHFDY | 0.672 | ASTPGRIQLWPLPR | 1.102 | ARAVYGDYASHFDY | 0.074 |
| AKVSVLMVYGLDY | 0.527 | ARRSGDYVSDWFDP | 0.748 | ARASSGWYIDY | 0.074 |
| VRDSYRFLDH | 0.487 | TTVPDYGGNHDY | 0.610 | ATTMVGDDAFDI | 0.061 |
| ARGRFRNFDY | 0.433 | ARLDFWSGYYPEIFDY | 0.497 | ATYYDFSK | 0.060 |
| AKGAWYALDY | 0.393 | ARDLYSYGYREAYDY | 0.417 | AILTMVRGVMVY | 0.060 |
| APNSWYQGWFDP | 0.378 | ARTNYDFWSGPTYYFDY | 0.390 | ARITTRPGGDCYCVDI | 0.042 |
| AKAEWYYFDY | 0.369 | ARLKWGRDGYPDLFDY | 0.358 | ARVRSYYYGMDV | 0.027 |
| AKVPWRFRMDV | 0.299 | ARLDFWSGLYGMDV | 0.088 | ARHKYQPLVDY | 0.014 |
| AKVVVIKGRAFD | 0.248 | ARITTRPGGDCYCVDI | 0.082 | ||
| ARGDIVVVPAAISAY | 0.226 | TTVPNYYYYGMDV | 0.075 | ||
| AKVGWQAFD | 0.196 | VSYLAVAGYYFDY | 0.044 | ||
| ARDGPIDY | 0.154 | ARLGGDYYDY | 0.040 | ||
| ARGRWNYADY | 0.145 | ARLAVADPFDY | 0.033 | ||
| ARGHYY | 0.141 | ATDQISLRYSPDAFD | 0.033 | ||
| VKSAWYAFDI | 0.139 | ARNVERSYYYGMDV | 0.029 | ||
| AKGQWLAFD | 0.120 | ARDVGYSYGDNWFDP | 0.029 | ||
| ARDRFYGMDV | 0.088 | AKALGVVLGTT | 0.019 | ||
| ARATFGAFD | 0.087 | AKALSSGSY | 0.015 | ||
| ARDRFRNFD | 0.073 | ASGGRELDY | 0.015 | ||
| ARKHYYGM | 0.062 | ARGLGGGTFDY | 0.015 | ||
| ARGQAFDI | 0.062 | ANWGWFWGL | 0.015 | ||
| ARGHYYDM | 0.062 | ||||
| ARSLYYSGSYYAYFDY | 0.056 | ||||
| THRWSRYYRDAFDI | 0.055 | ||||
| AREDGPGAFDI | 0.051 | ||||
| ARFPMVRDFD | 0.049 | ||||
| ARHAAIGIGWYY | 0.047 | ||||
| ARGHYYDMD | 0.041 | ||||
| AKYYYYGVDV | 0.034 | ||||
| AKIYYYG | 0.030 | ||||
| AKGGLGDLDY | 0.026 | ||||
| AKWGLGAFDI | 0.026 | ||||
| AKWGLLRYGR | 0.023 | ||||
| ARGEGYYDFWSGGNWFD | 0.015 | ||||
| AKVGGQAFDI | 0.015 | ||||
| AKVGWPLTT | 0.013 | ||||
| AHRVAAADSYYYGM | 0.013 | ||||
| AKVGWNYFD | 0.013 | ||||
| ARGDLGIWTT | 0.011 |
For OX40, the selection was dominated by asingle clone (89.6% of the entire population), which prevented us from reaching too deeply into the selection in terms of percentage abundance, while in the case of the other two proteins amore diverse population was selected (with the top 10 clones consisting of 77.8% of the entire repertoire for B7-H4 and 82.9% for CD40-L) (Table 3). The clonal sequence diversity of the antibodies obtained after each selection campaign was estimated by calculating and plotting the HCDR3 Levenshtein distance (Figure 2).
Table 3.
Summary of NGS analysis of each selection campaign.
| Antigen | MiSeq Reads | # HCDR3 | Abundance Top_1 | Abundance Top_10 |
|---|---|---|---|---|
| B7H4 | 47829 | 53 | 28.4% | 77.8% |
| CD40L | 48401 | 35 | 26.5% | 82.9% |
| OX40 | 52766 | 22 | 89.6% | 94.9% |
Figure 2.
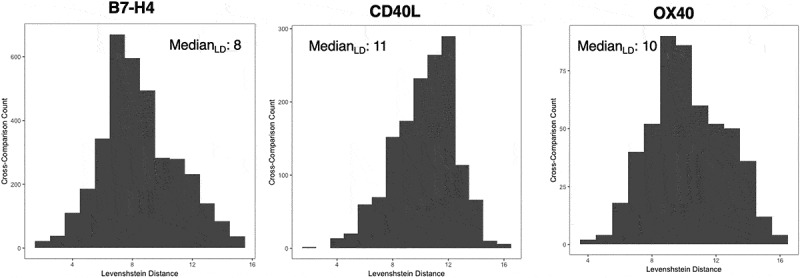
Estimation of the clonal sequence diversity of the antibodies obtained after the selection campaigns against the three targets: B7-H4, CD40-L and OX40. The diversity was represented by calculating and plotting the HCDR3 Levenshtein distance.
As we previously characterized the diversity of the naïve library used in this study by NGS,20 we were able to analyze the initial frequencies of the enriched HCDR3s, obtained after the three selection campaigns. All the identified HCDR3, including the most abundant ones, showed no over-representation in the initial library, indicating no bias advantage during the screening. The abundances of the selected clones in the naïve library are shown in Table 4. All the clones with reported abundances of < 3.16E-09 were so rare that they were not identified in the data set obtained after sequencing the naïve library. The reason we did not find the rare HCDR3s is that with current sequencing platforms the diversity of anaïve library greatly exceeds the number of sequencing reads, but the generated data are large enough to estimate and evaluate the theoretical diversity of the library.20
Table 4.
Frequencies of the antigen specific HCDR3s before and after the selection campaigns.
| B7-H4 |
CD40L |
OX40 |
||||||
|---|---|---|---|---|---|---|---|---|
| HCDR3 | % after selection | % before selection | HCDR3 | % after selection | % before selection | HCDR3 | % after selection | % before selection |
| AKVGWQAFDI | 28.429 | < 3.16E-09 | ARLAVADPYDY | 26.853 | < 3.16E-09 | ARDVGYSYGDNWFDP | 89.603 | < 3.16E-09 |
| AKWGLGDLDY | 15.073 | < 3.16E-09 | ARLSLDGSGLYGMDV | 16.560 | < 3.16E-09 | AKDRSSGWYSDGMDV | 3.045 | < 3.16E-09 |
| AKVVVIKGRAFDI | 7.865 | < 3.16E-09 | ANWEAVAGTFDY | 9.318 | < 3.16E-09 | ARDNSSSWYTSALDV | 0.590 | < 3.16E-09 |
| AKGPTVTNVRGFDY | 5.338 | < 3.16E-09 | ARGLGGGFGDY | 7.876 | < 3.16E-09 | ATGSSSSWFDP | 0.462 | < 3.16E-09 |
| ARGHYYDMDV | 5.219 | 1.90E-08 | ATDQISLRYSPDAFDI | 7.221 | < 3.16E-09 | ARGEGYYDFWSGGNWFDP | 0.275 | < 3.16E-09 |
| ARDRFRNFDY | 3.126 | < 3.16E-09 | ARALRGYGDYFDY | 7.085 | < 3.16E-09 | TRGGLVVAATHPGAFDI | 0.259 | < 3.16E-09 |
| ARKHYYGMDV | 3.056 | < 3.16E-09 | ARWEQQLAGGFDP | 2.426 | < 3.16E-09 | ARGRYGDY | 0.235 | < 3.16E-09 |
| ARATFGAFDI | 2.801 | 1.26E-08 | AADTPSSV | 2.417 | < 3.16E-09 | ARAQDYYYYYGMDV | 0.184 | < 3.16E-09 |
| ARFPMVRDFDY | 2.400 | < 3.16E-09 | ARGYYAADY | 1.611 | < 3.16E-09 | ARVRTDWYVDL | 0.135 | 3.22E-06 |
| AKAEWGSFDY | 1.923 | < 3.16E-09 | AKEVGYRVYYYGMDV | 1.573 | < 3.16E-09 | ARGTAVAGTLVDY | 0.117 | < 3.16E-09 |
| AISSWYGFDY | 1.412 | < 3.16E-09 | ARLGGDYYDSSLYGMDV | 1.368 | < 3.16E-09 | ARGTVTIMRGY | 0.109 | < 3.16E-09 |
| VRDSYRFFDH | 1.293 | < 3.16E-09 | AKALSSGSYYGSDYYFDY | 1.236 | < 3.16E-09 | AHGAYSFDY | 0.108 | < 3.16E-09 |
| AKIYYYGMDV | 0.796 | < 3.16E-09 | TTVPVVAFDI | 1.204 | < 3.16E-09 | ARDVDVGWFDP | 0.078 | < 3.16E-09 |
| AKVGWNYFDY | 0.792 | < 3.16E-09 | ARHYDHYYYGMDV | 1.173 | < 3.16E-09 | ARDWSMTSVTMPDY | 0.076 | < 3.16E-09 |
| AKGRWWHFDY | 0.672 | < 3.16E-09 | ASTPGRIQLWPLPR | 1.102 | 9.48E-09 | ARAVYGDYASHFDY | 0.074 | 6.32E-09 |
| AKVSVLMVYGLDY | 0.527 | < 3.16E-09 | ARRSGDYVSDWFDP | 0.748 | < 3.16E-09 | ARASSGWYIDY | 0.074 | < 3.16E-09 |
| VRDSYRFLDH | 0.487 | < 3.16E-09 | TTVPDYGGNHDY | 0.610 | < 3.16E-09 | ATTMVGDDAFDI | 0.061 | < 3.16E-09 |
| ARGRFRNFDY | 0.433 | < 3.16E-09 | ARLDFWSGYYPEIFDY | 0.497 | < 3.16E-09 | ATYYDFSK | 0.060 | < 3.16E-09 |
| AKGAWYALDY | 0.393 | < 3.16E-09 | ARDLYSYGYREAYDY | 0.417 | < 3.16E-09 | AILTMVRGVMVY | 0.060 | < 3.16E-09 |
| APNSWYQGWFDP | 0.378 | < 3.16E-09 | ARTNYDFWSGPTYYFDY | 0.390 | < 3.16E-09 | ARITTRPGGDCYCVDI | 0.042 | 1.01E-06 |
| AKAEWYYFDY | 0.369 | < 3.16E-09 | ARLKWGRDGYPDLFDY | 0.358 | < 3.16E-09 | ARVRSYYYGMDV | 0.027 | 1.03E-06 |
| AKVPWRFRMDV | 0.299 | < 3.16E-09 | ARLDFWSGLYGMDV | 0.088 | < 3.16E-09 | ARHKYQPLVDY | 0.014 | 3.16E-09 |
| AKVVVIKGRAFD | 0.248 | < 3.16E-09 | ARITTRPGGDCYCVDI | 0.082 | 1.01E-06 | |||
| ARGDIVVVPAAISAY | 0.226 | < 3.16E-09 | TTVPNYYYYGMDV | 0.075 | < 3.16E-09 | |||
| AKVGWQAFD | 0.196 | < 3.16E-09 | VSYLAVAGYYFDY | 0.044 | < 3.16E-09 | |||
| ARDGPIDY | 0.154 | < 3.16E-09 | ARLGGDYYDY | 0.040 | < 3.16E-09 | |||
| ARGRWNYADY | 0.145 | 1.58E-08 | ARLAVADPFDY | 0.033 | < 3.16E-09 | |||
| ARGHYY | 0.141 | < 3.16E-09 | ATDQISLRYSPDAFD | 0.033 | < 3.16E-09 | |||
| VKSAWYAFDI | 0.139 | < 3.16E-09 | ARNVERSYYYGMDV | 0.029 | < 3.16E-09 | |||
| AKGQWLAFD | 0.120 | < 3.16E-09 | ARDVGYSYGDNWFDP | 0.029 | < 3.16E-09 | |||
| ARDRFYGMDV | 0.088 | < 3.16E-09 | AKALGVVLGTT | 0.019 | < 3.16E-09 | |||
| ARATFGAFD | 0.087 | 7.58E-08 | AKALSSGSY | 0.015 | < 3.16E-09 | |||
| ARDRFRNFD | 0.073 | < 3.16E-09 | ASGGRELDY | 0.015 | 1.58E-08 | |||
| ARKHYYGM | 0.062 | < 3.16E-09 | ARGLGGGTFDY | 0.015 | < 3.16E-09 | |||
| ARGQAFDI | 0.062 | < 3.16E-09 | ANWGWFWGL | 0.015 | < 3.16E-09 | |||
| ARGHYYDM | 0.062 | 3.92E-07 | ||||||
| ARSLYYSGSYYAYFDY | 0.056 | < 3.16E-09 | ||||||
| THRWSRYYRDAFDI | 0.055 | < 3.16E-09 | ||||||
| AREDGPGAFDI | 0.051 | 1.80E-05 | ||||||
| ARFPMVRDFD | 0.049 | 2.69E-07 | ||||||
| ARHAAIGIGWYY | 0.047 | < 3.16E-09 | ||||||
| ARGHYYDMD | 0.041 | < 3.16E-09 | ||||||
| AKYYYYGVDV | 0.034 | < 3.16E-09 | ||||||
| AKIYYYG | 0.030 | < 3.16E-09 | ||||||
| AKGGLGDLDY | 0.026 | < 3.16E-09 | ||||||
| AKWGLGAFDI | 0.026 | < 3.16E-09 | ||||||
| AKWGLLRYGR | 0.023 | < 3.16E-09 | ||||||
| ARGEGYYDFWSGGNWFD | 0.015 | < 3.16E-09 | ||||||
| AKVGGQAFDI | 0.015 | 6.32E-09 | ||||||
| AKVGWPLTT | 0.013 | 1.66E-06 | ||||||
| AHRVAAADSYYYGM | 0.013 | < 3.16E-09 | ||||||
| AKVGWNYFD | 0.013 | < 3.16E-09 | ||||||
| ARGDLGIWTT | 0.011 | < 3.16E-09 | ||||||
| AKVGWQAFDI | 28.429 | < 3.16E-09 | ||||||
| AKWGLGDLDY | 15.073 | < 3.16E-09 | ||||||
| AKVVVIKGRAFDI | 7.865 | < 3.16E-09 | ||||||
| AKGPTVTNVRGFDY | 5.338 | < 3.16E-09 | ||||||
| ARGHYYDMDV | 5.219 | < 3.16E-09 | ||||||
| ARDRFRNFDY | 3.126 | < 3.16E-09 | ||||||
| ARKHYYGMDV | 3.056 | < 3.16E-09 | ||||||
ScFv rescue approach
The isolation strategy of specific clones from selection outputs is depicted in Figure 1. Our approach consisted of rescuing the clones containing:1) the most abundant HCDR3 sequence, (28.4% abundance for the anti-B7H4 clone, 26.8% for CD40-L and 89.6% for OX40);2) clones for which HCDR3s are represented at ~5% of the population (5.33% abundance for the anti-B7H4 clone, 6.9% for CD40-L and 3.0% for OX40– for OX40 the clone with 3% abundance was also thesecond most abundant clone, the selection being heavily dominated by the most abundant one);3) ~1% of the population (1.29% abundance for the anti-B7H4 clone, 1.16% for CD40-L and 0.6% for OX40);4) ~0.5% of the population (0.52% abundance for the anti-B7H4 clone, 0.50% for CD40-L, while no attempt was performed on OX40 because of the clonal dominance);5) ~0.1% of the population (0.14% abundance for the anti-B7H4 clone, 0.09% for CD40-L);6) ~0.05% of the population (0.056% abundance for the antiB7H4 clone, 0.044% for CD40-L); and7) ~0.01% of the population (0.015% abundance for the anti-B7H4 clone, 0.015% for CD40-L). These are all shown in Table 2. The goal of testing this broad clone abundance range was to assess, first, the lowest HCDR3 clone abundance that could be effectively isolated using this approach, andsecond, the lowest abundance at which antibodies reliably recognizing the selection target could be isolated.
Having identified clones from the NGS, we designed oligonucleotides that matched the HCDR3 DNA sequence of each of the clones of interest plus its corresponding JH segment. Each primer was 60 nucleotides in length, with 30 nucleotides matching the HCDR3, and 30 nucleotides matching the adjacent JH region. The PCR reactions were performed using the total plasmids prepared from the yeast display selection outputs as templates. To reduce the introduction of unwanted mutations, ahigh-fidelity polymerase was used in the PCR reactions.
PCR using the designed primers produced fragments of the appropriate size for all abundance ranges. The amplification products were retransformed into the yeast cells along with the appropriate linearized vector containing the corresponding JH sequence. Each of the yeast populations obtained, comprising yeast clones all sharing the same HCDR3, was induced to display scFv on the yeast surface, and the binding of biotinylated antigen was tested by flow cytometry with fluorescently labeled streptavidin. For all cloned outputs except those at the lowest abundance (< 0.02%), abinding population was obtained. For the B7-H4 output obtained by amplifying the 0.015% HCDR3 clonotype, there was some scFv display on the yeast surface, but no target binding, while for CD40-L, the 0.015% population showed no binding and poor antibody display (Figure 3).
Figure 3.
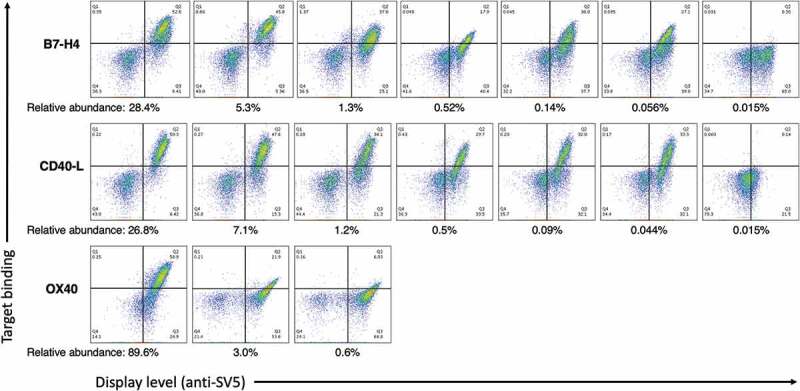
The binding profile of the antibody populations rescued using the present strategy was analyzed by yeast display and flow cytometry analysis. All clones but the ones with 0.015% abundance show expression and binding activity when cloned into the yeast display system.
Affinity determination of the rescued clones
For each of the scFv-clonotype populations transformed into yeast containing the rescued HCDR3 of interest, and demonstrating binding activity, we sequenced anumber of clones to determine whether the rescued population was homogenous or comprised a“mini-library” in which acommon HCDR3 was combined with additional mutations elsewhere. We found that, with one exception, clones shared the same HCDR3, but had different point mutations in either one of the other CDRs or in the framework. The one exception had amutation at the N-terminus of the HCDR3 in one of the clones obtained from the most abundant binder in the CD40-L campaign. Interestingly, but perhaps not surprisingly, such mutations resulted in different affinities. In the case of the clones selected against B7-H4, the antibody affinities ranged from 75 to 310 nM (Table 5), with the most abundant antibodies correlated to the better affinity values, while for CD40-L the range was 92 to 125 nM with no apparent correlation with antibody abundance (Table 5). For the OX40 selection, significant binding activity was observed only for the clones sharing the most abundant HCDR3, with ameasured affinity value of 125 nM. Two other rescued HCDR3s had affinities > 1 µM (Table 5). None of the tested antibodies that bound their target showed activity toward an irrelevant negative control, indicating the specificity of all selected antibodies.
Table 5.
Affinities and correlations of the individual clones obtained from each clonotype. Clonotype indicated with * represent the ones rescued using longer primers.
| Clones | Affinity Kd (nM) | R2 (correlation) | Clones | Affinity Kd (nM) | R2 (correlation) | Clones | Affinity Kd (nM) | R2 (correlation) |
|---|---|---|---|---|---|---|---|---|
| Clonotype A | Clonotype A | Clonotype A | ||||||
| #1 | 95 | 0.9926 | #1 | 95 | 0.9847 | #1 | 125 | 0.9811 |
| #2 | 110 | 0.9837 | #2 | 103 | 0.9883 | #2 | 125 | 0.9711 |
| #3 | 75 | 0.9882 | #3 | 102 | 0.9906 | Clonotype B | ||
| #4 | 78 | 0.9834 | #4 | 101 | 0.9794 | #1 | >1 µM | Not calculated |
| #5 | 109 | 0.9790 | Clonotype B | Clonotype C | ||||
| #6 | 102 | 0.9872 | #1 | 105 | 0.9873 | #1 | >1 µM | Not calculated |
| #7 | 115 | 0.9882 | #2 | 103 | 0.9810 | |||
| Clonotype B | #3 | 105 | 0.9864 | |||||
| #1 | 105 | 0.9911 | Clonotype C | |||||
| #2 | 155 | 0.9817 | #1 | 125 | 0.9904 | |||
| #3 | 154 | 0.9802 | #2 | 125 | 0.9721 | |||
| Clonotype C | Clonotype D | |||||||
| #1 | 175 | 0.9915 | #1 | 110 | 0.9826 | |||
| #2 | 180 | 0.9716 | #2 | 110 | 0.9870 | |||
| #3 | 195 | 0.9793 | #3 | 110 | 0.9869 | |||
| #4 | 178 | 0.9856 | Clonotype E | |||||
| Clonotype D | #1 | 92 | 0.9902 | |||||
| #1 | 310 | 0.9827 | #2 | 92 | ||||
| #2 | 310 | 0.9811 | #3 | 92 | ||||
| Clonotype E | Clonotype E* | |||||||
| #1 | 245 | 0.9821 | #1 | 360 | 0.9701 | |||
| #2 | 200 | 0.9802 | Clonotype F* | |||||
| #3 | 195 | 0.9910 | #1 | 125 | 0.9802 | |||
| #4 | 285 | 0.9806 | ||||||
For B7-H4, we were able to correctly rescue clones with an HCDR3 abundance as low as 0.1% abundance in the NGS analysis. When we tried to rescue clones at 0.05%, we inadvertently amplified amore abundant clonotype (0.792%) that shared four amino acids at the 3ʹ of the HCDR3 of interest (Figure 4a).
Figure 4.

Analysis of the rescued clones. Between 2 and 8 clones from each rescued antibody population were Sanger sequenced and their affinities measured. Underlined are the parts of the HCDR3 used to design our clone specific primers. (a) During the B7H4 campaign the clone at 0.056% abundance was not amplified, and instead aclone at higher abundance (0.792%), sharing the same JH domain and part of the HCDR3 region, was obtained. (b) During the CD40-L campaign the primer designed to amplify the clone with 0.088% abundance perfectly matched amore abundant clones (~16%) that was hence amplified. The primer used to amplify aclone at 0.044% abundance was not specific enough and amplified amore abundant clone (~7%).
For the CD40-L campaign, the limit of valid clone isolation was 0.497%, since when we tried to rescue clones at an abundance of 0.088% and 0.044% we again amplified more abundant clonotypes (15.89 and 7.085%, respectively) sharing seven and four amino acids at the 3ʹ of the HCDR3 of interest, respectively (Figure 4b).
Selection of OX40 provided avery dominant clone with affinity in the nM range. Clonotypes with abundances of 3.04% and 0.59% showed aweak binding signal with affinity above 1 µM (Figures 3 and 4c). For the OX40 selection we decided to also analyze the population of yeast obtained after the first yeast sort, to see if less clonal dominance was present and if it was possible to recover more clones (see Table 6). The results show the most abundant clone in the final yeast sorting was also the most dominant in the previous selection step (from 72.87% to 89.6%), and also thesecond-highest ranked clone was the same after both sorts with an increase in abundance at the end of the enrichment process (from 1.72% to 3.05%). Two other clones were also shared between the two data sets: one clone showing aslight enrichment (from 0.24% to 0.59%) representing the third-high ranked clone after two sorts and showing minimal binding activity (Figure 3). The fourth one dropped in abundance after thesecond sort, when rescued using the described approach, and no binding activity was detected (data not shown). The remaining clones were present in either of the two data sets (but not both) at very low abundance, probably indicating their presence as “background noise”.
Table 6.
Comparison of the relative abundances of clones present after the first andsecond yeast sort during the selection campaign against OX40.
| After first sort |
Aftersecond sort |
||
|---|---|---|---|
| HCDR3 | Abundance (%) | HCDR3 | Abundance (%) |
| ARDVGYSYGDNWFDP | 72.87 | ARDVGYSYGDNWFDP | 89.60 |
| AKDRSSGWYSDGMDV | 1.72 | AKDRSSGWYSDGMDV | 3.05 |
| ATDRGCSSTSCYTGVYYFDY | 1.42 | ARDNSSSWYTSALDV | 0.59 |
| ARYRSAYGLDV | 1.11 | ATGSSSSWFDP | 0.46 |
| ARDVDVGWFDP | 0.79 | ARGRYGDY | 0.28 |
| ATHSSRWGDYGMDV | 0.66 | ARGEGYYDFWSGGNWFDP | 0.26 |
| ARDLSYGDYVPDY | 0.56 | TRGGLVVAATHPGAFDI | 0.24 |
| ARSGDYYDSPFDY | 0.42 | ARGTVTIMRGY | 0.18 |
| ARDSGLYGMDV | 0.37 | ARAQDYYYYYGMDV | 0.14 |
| ARRNPGGWFDH | 0.36 | AHGAYSFDY | 0.12 |
| ARAGTSMADDAFDI | 0.34 | ARVRTDWYVDL | 0.11 |
| AREDGVLMVEDY | 0.31 | ARGTAVAGTLVDY | 0.11 |
| ARDNSSSWYTSALDV | 0.24 | ARDWSMTSVTMPDY | 0.08 |
| ARPVLGAMNV | 0.21 | ATYYDFSK | 0.08 |
| ARPTSSESYLDDAFDI | 0.19 | ARDVDVGWFDP | 0.07 |
| ARHSSYYYDSSFDY | 0.18 | ATTMVGDDAFDI | 0.07 |
| ARMRVGGFDY | 0.15 | ARAVYGDYASHFDY | 0.06 |
| AREGYSGYYYYGMDV | 0.10 | ARASSGWYIDY | 0.06 |
| VAEGVSGSYATFDP | 0.08 | AILTMVRGVMVY | 0.06 |
| TTEGFHSRGGYYYYGMDV | 0.04 | ARITTRPGGDCYCVDI | 0.04 |
| AKDFPLDYSNYYYYYGMDV | 0.04 | ARVRSYYYGMDV | 0.03 |
| ARDHVVVPAAFEY | 0.02 | ARHKYQPLVDY | 0.01 |
| AREGDYPYGMDV | 0.02 | ||
| ARGGYSSSPDAFDI | 0.02 | ||
| ATDPGGFGYYFDY | 0.01 | ||
| AFGSIAARPLGY | 0.01 | ||
Using longer primers for rescuing low abundance clones
The use of 60 base primers was unable to rescue clones with abundances < 0.1%. In order to determine whether primers annealing to the entire HCDR3 sequences of rare clones (plus the clone-specific JH region) could overcome this issue, we carried out an additional set of experiments in which primers were much longer. We focused this analysis on clones we failed to rescue using the 60mer oligonucleotides: the clone with 0.056% abundance obtained during the selection on B7-H4, and the clones obtained from the CD40-L campaign with an abundance of 0.088% and 0.044%. The designed primers ranged from 78 to 87 base pairs. Such long oligonucleotides are more expensive, partly because of their length, but also due to the extra purification required to reduce errors accumulating during the synthesis process. PCR reactions were performed using plasmids prepared from the total yeast display selection outputs obtained after thesecond sort, and the correctly-sized fragments were transformed into yeast, together with the corresponding JH-specific linearized vector. The obtained yeast populations were analyzed for binding by flow cytometry. The population obtained from the B7-H4 clone did not show any binding activity, reflecting the previously observed correlation between abundance and affinity we saw for this selection campaign, while both populations derived from the CD40-L clonotypes showed specific binding (Figure 5a). For each of the populations transformed into yeast, including the one not showing binding activity, we sequenced four clones. We found that clones with the specific HCDR3s of interest were rescued. For each clonotype, four identical clones were rescued (Figure 5b). The affinities were measured only for the CD40-L clonotypes (Table 5).
Figure 5.
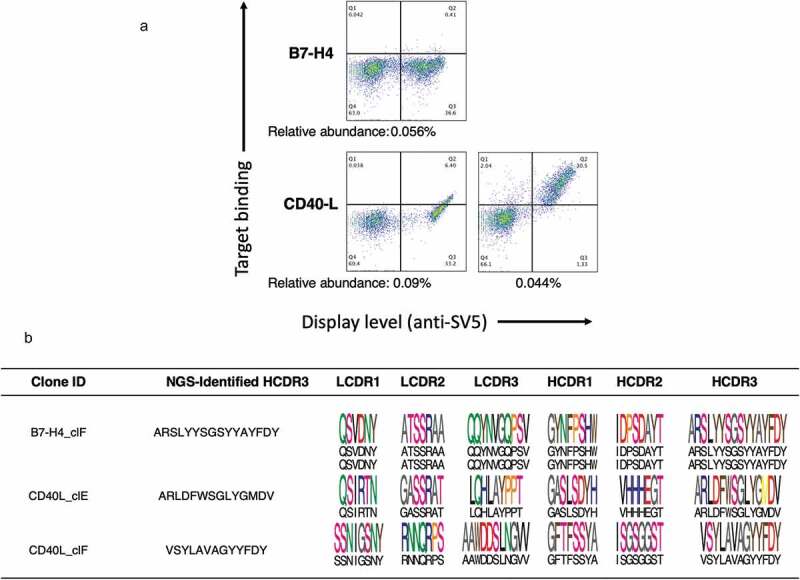
Analysis of the rescued clones using primers covering the entire HCDR3 sequences of rare clones. (a) Binding profile of the antibody populations rescued using the longer primer strategy analyzed by yeast display and flow cytometry analysis. All clones show expression but binding activity was present only in the clones rescued from the CD40-L campaign. (b) Four clones from each rescued antibody population, including the non-binding one, were Sanger Sequenced. Represented are the HCDR3 used to design our clone specific primers. The scFvs containing the HCDR3s of interest were specifically amplified, including the one with 0.044% of abundance.
Discussion
Display-based technologies, in particular phage display, have been successfully applied to the discovery of novel therapeutic antibodies. The importance of this platform was recognized by the award of the 2018 Noble Prize to scientists that developed the technology.21 One of the main limitations of the standard screening method is that, during the biopanning process, some binding clones become dominant (as was observed for the OX40 selection), resulting in identification of alimited number of different candidates after screening. For example, in our study only 40 of 384 clones would not have been derived from the dominant clone in the OX40 selection. The advent of NGS has dramatically increased the depth and breadth of the analysis of display library selection outputs, making it possible to obtain information not only on the clones identified using aconventional colony screening method, but also additional, less abundant, candidates.14,15,20,22 Among the most commonly used NGS platforms, there has been asteady increase in the number and length of reads that can be obtained, but only PacBio has invested in increasing read length to alevel commensurate with obtaining full-length sequences of displayed antibody-like molecules, i.e., scFvs and Fabs. For this reason, isolating clones identified by NGS from aselection campaign, to test their functional properties, continues to be achallenge. With PacBio now able to provide full-length clone sequences, gene synthesis is avaluable option, but remains prohibitive on alarge scale, even if the cost of gene synthesis is becoming significantly cheaper. Furthermore, gene synthesis usually takes weeks. We previously described an approach allowing the direct isolation of NGS identified clones of interest from aselected library using an inverse PCR23 and ligation-based strategy that exploits the unique barcode nature of the HCDR3 sequence to synthesize pairs of outward-facing primers that are used to amplify plasmids containing the HCDR3.15 However, that approach was not very effective for short HCDR3s or clones with abundances less than 1%.
Here, we explored afaster, cheaper method, requiring the design of asingle specific primer for each HCDR3 of interest, to quickly translate an NGS analysis into actual clones. We focused our approach on the HCDR3 sequence, since this is considered to be the most important element of an antibody molecule, particularly for binding activity and specificity.24–27 This explains why HCDR3s are frequently used as unique molecular identifiers when dissecting the adaptive immune response invivo, as well as when characterizing invitro selection outputs of display systems. However, we have observed22 that, while all antibodies in aselected binding population with aspecific HCDR3 bind the target, even if at low abundance, 99.85% of antibodies containing the same HCDR3 in anaïve library do not. This reflects our analysis of both invivo28,29 and invitro22 NGS datasets, which indicates that identical HCDR3s can be generated by different VDJ recombination events and are expected to result in very different antibody binding properties.
We demonstrated the approach on three different selection outputs. The selections were performed using arelatively high (100 nM) target concentration in order to preserve diversity and test our working hypothesis with clones at different abundance levels. We were able to isolate and characterize clones present down to 0.1% of the total population obtained by NGS using 60 base primers. Such athreshold is reasonable, considering that in our selection strategy, which combines phage and yeast display, we perform the final steps of the enrichment by sorting 10,000 antigen-binding yeast events. As we were unable to generate binding clones with abundances less than 0.1%, we believe many of these less abundant clones may represent NGS artifacts, although it is likely that rare binders do exist, but are more difficult to isolate. Initially, we limited our primer lengths to 60 bases, in order to optimize the costs of the strategy (longer primers required higher purification standards due to the greater possibility of errors during synthesis). Using 60 base primers we obtained one clone with amutation at the N-terminus of HCDR3 for the most abundant CD40-L binder subpopulation, and when we tried to target clones with an abundance <0.1% using 60mer primers, we obtained more abundant clones that partially shared the targeted HCDR3 sequence. We hypothesize that the use of the Q5 proof-reading polymerase, which has apowerful 3ʹ to 5ʹ exonuclease activity, to rescue clones of interest from the enriched populations probably allowed primers to be degraded from the 3ʹ end until they were able to amplify more abundant HCDR3s, which in the case of the least abundant B7H4 clone, resulted in the removal of 18 bases from the 3ʹ end. The use of anon-proof-reading polymerase (e.g., Taq polymerase) would probably overcome this problem, but at the cost of introducing additional mutations. As an alternate solution, we tested whether longer primers, more stringent for aparticular HCDR3, would help in the isolation of lower abundance clones. This strategy was successful in the rescue of aclone with 0.044% abundance, that was still able to bind its target (CD40-L) with an affinity similar to the most abundant antibodies in the selection.
This method allows the relatively rapid isolation of clones corresponding to HCDR3s identified by the most common NGS platforms. This requires asingle PCR reaction, no purification, and direct transformation into ayeast display vector thanks to the homologous recombination system. The use of yeast display, in combination with fluorescence-activated cell sorting, also allows further refinement of any particular HCDR3 clone set, by, for instance, generating apanel of antibodies with similar structure but different affinities. If yeast display and flow cytometry are not available, we believe asimilar approach can also be applied to phage antibody libraries. In this case, after identified antibody genes are amplified, they would be cloned into acorresponding phage display vector using aGibson assembly30 (or similar) approach.
The method described here is amenable only for antibodies displayed as scFvs in the VL-VH orientation, with the HCDR3 present at the 3ʹ end of the molecule. In asituation where the molecules are displayed with aVH-VL orientation, the strategy can be optimized around the LCDR3, but, unlike the HCDR3, this is not expected to provide sufficient diversity to distinguish different clones.
Beyond its use for the isolation of NGS identified clones selected from naïve libraries, we believe this approach will also be suitable for other library selections in which NGS is used, such as:1) in error-prone PCR affinity maturation campaigns where preservation of the antibody HCDR3 is important;2) selection from semi-synthetic libraries in which diversity is found in the CDRs embedded within constant scaffolds; or3) in synthetic combinatorial libraries where the CDRs are also synthetically generated. In all these examples the same primer design strategy can be applied to rescue antibodies with HCDR3s of interest, with the advantage that all clones in these libraries will have the same JH region (i.e., of the chosen scaffold for asemi-synthetic library). The power of NGS lies in the ability to analyze the entire depth of an antibody output after aselection campaign. However, the challenge of going rapidly from “sequence to clone” has remained aconsistent bottleneck. Our latest approach to overcome this problem, described here, has clear advantages compared to previous methods. For instance, compared to the previous methodology described by our group and others, based on inverse PCR,15,16,31 the present strategy requires the design and synthesis of asingle primer, no purification of the inverse PCR products and subsequent enzymatic ligation to reconstitute aworking plasmid, and no bacterial transformation and subsequent cloning into yeast cells for validation of the binding activity. The isolation limit when using 60 base primers is ~0.14%, decreasing to 0.044% when longer primers are used. In both cases binding antibodies could be isolated at these levels, and the lower limit remains to be discovered. The transformation and cloning of PCR products directly into yeast cells by homologous recombination, allows the rapid validation of the system by flow cytometry with further enrichment of higher affinity clones in populations with the same HCDR3. At apractical level, we recommend that 60 base primers are suitable for the isolation of clones >0.2%, but, below this, longer primers that span the whole HCDR3 should be used. When using the 60 base primers, it is important that proposed primer sequences are screened against the whole NGS output to ensure that more abundant similar clones are not amplified, bearing in mind that up to 18 bases may be removed by the polymerase exonuclease activity.
The direct transformation of yeast cells with adisplay vector and PCR products obtained with auniversal primer specific for the backbone of the vector and aprimer homologous to the heavy chain CDR3 and framework 4 regions of the scFv sequence is here shown to be an effective methodology for the recovery of scFvs, even for clones at very low abundance clones within aselection output.
Materials and methods
Bacterial and yeast strains
Omnimax™ (ThermoFisher Scientific, C854003): F’[proAB lacIq lacZΔM15 Tn10(TetR) Δ(ccdAB)] mcrAΔ(mrr hsdRMS-mcrBC) Φ 80(lacZ)ΔM15 Δ(lacZYA-argF) U169 endA1 recA1 supE44 thi-1 gyrA96 relA1 tonApanD
EBY100 (ATCC®, MYA-4941): MATa AGA1::GAL1-AGA1::URA3 ura3-52 trp1 leu2delta200 his3- delta200 pep4::HIS3 prb11.6R can1 GAL
Generation of pDNL6-JH vectors
We generated three different vectors to match the terminal part of the JH sequences present in our library: JH3 (GTMVTVSS), JH4 (GTLVTVSS) and JH6 (GTTVTVSS). The JH1, JH2 and JH5 germlines share the same amino acid sequence with JH4, making them compatible with the vectors. Synthetic oligo nucleotides (Genewiz, NJ) encoding the two strands of each JH regions
(Table 1) were annealed and ligated to the pDNL6 vector previously digested with the enzyme.
We transformed the ligation products into E.coli Omnimax™ (ThermoFisher Scientific, C854003) by heat shock and single colonies were sequenced to confirm the presence of the desired sequence.
ScFv antibody selections
The antigens used for the scFv selections were B7-H4 (ACRObiosystem, B74-H5222), CD40-L (Shenandoah Biotechnology, 100-25AF), and OX40 (ACRObiosystem, OXO-H5224). The targets were biotinylated using the EZ-Link NHS-LC-LC-Biotin system (Thermo Fisher Scientific), following the manufacturer’s instructions. The phage naïve scFv library17 was used for two rounds of phage display against the antigens followed by two rounds of yeast display sorting. The detailed protocols for antibody selections against biotinylated proteins are described in Ferrara etal.14,18
Deep sequencing
Plasmid DNA was extracted from the final yeast populations, specifically enriched for their binding to the antigens of interest, using the QIAprep spin miniprep kit (Qiagen, 27104), adding 50–100 µl of acid-washed glass beads (Sigma, G-8772) during the resuspension step and vortexing for 5 min before proceeding with the miniprep protocol.
The plasmid DNA was used directly as template for NGS sample preparation, which consisted of PCR amplification of the VH domains of the selected scFv. The designed primers contain different barcodes that allowed us to pool different amplicons together in order to ligate Illumina adaptors to asingle pool. The PCR amplification was performed using Q5 high fidelity polymerase (NEB, M0491) following the manufacturer’s protocol. PCR products were gel purified, quantified and sent to Genewiz® for sequencing using their Amplicon-EZ service. This is asimpler, accelerated version of MiSeq NGS, providing afast and convenient way to sequence up to 50,000 reads per sample.
Briefly, for the bioinformatic analysis, all the paired-end sequence reads derived from MiSeq runs were assembled using PEAR32 and the Vregion germlines and CDRs were annotated using the IgBlast command-line tool. After appropriate alignment, all HCDR3 amino acid sequences were aligned in pairs using the Needleman–Wunsch global alignment algorithm and the PAM30 substitution matrix,33 which assigned ascore to each pair, the higher the score the higher the similarity between the HCDR3 sequences. Subsequently, the alignment scores were stored in asquare matrix and clustered using the DBSCAN algorithm (e = 135),34 which created groups
(clusters) based on the similarity between the different HCDR3 sequences.
Primer design and PCR amplification
From the plasmid preps obtained from the final yeast-sorted populations, specific scFv clones containing the HCDR3 of interest were amplified using ahigh-fidelity polymerase (Q5 Polymerase, NEB, M0491) and 0.1 ng of template DNA, which represents 100–1000 times the diversity of the selection output. The 5ʹ primer consist of a“universal” primer that anneals to the yeast display vector with enough homology for the subsequent homologous recombination of the PCR product into the pDNL6-JH vector of choice, determined by the JH sequence of the scFv of interest. The 3ʹ primers were designed on the DNA consensus sequence for the HCDR3s of interest: each HCDR3 represent acluster consisting of merged, highly homologous, HCDR3s; the most frequent sequence was used as the consensus sequence within acluster to design the primers employed in our strategy. The first set of 3ʹ primer length was limited to 60bp, with the primer matching the identified JH region of the scFv of interest and the rest of the oligonucleotide consisting of the consensus sequence of the targeted HCDR3. Longer 3ʹ primers were designed matching the identified JH region of the scFv of interest and entire consensus sequence of the HCDR3 of interest.
Yeast transformation and sequencing
The partially amplified scFvs were sub-cloned into the pDNL6-JH yeast display vector as previously described,18 this was possible thanks to the overlapping regions introduced by PCR: within the vector upstream of the scFv cloning site, and in the JH region of the antibody, which is also present in each specific pDNL6-JH vector. The vector and the fragments were cotransformed into yeast cells to allow cloning by gap repair.35
To test the binding of the recovered scFvs, the transformed yeast cells were induced and 1 × 106 cells were stained with 100 nM of biotinylated antigen. Subsequently, the cells were labeled with anti-SV5-PE to detect the scFv display level on the cell surface and streptavidin-AlexaFluor633 to detect the antigen bound by the expressing scFvs.
Amplicon sequencing and sanger sequencing
After testing the binding activity of the transformed cells, plasmid DNA was extracted from the yeast cells and used as template to prepare PCR products to be analyzed by NGS, where we obtained approximately 50,000 sequences per sample, enough to prove the efficacy of our scFv rescuing strategy. Data was analyzed as described before. In parallel, the same plasmid preps were used to transform competent bacterial cells to obtain single clone sequences and to obtain single clones plasmid DNA to be transformed back into yeast if necessary.
Binding assay
Taking advantage of the flexibility of flow cytometry, we performed antigen dose response experiments on single yeast clones to determine the affinity of the displayed scFv, without subcloning, expression and purification of the recombinant antibody, following published methods.36
Funding Statement
This work was supported by the National Institutes of Health under Grant R43CA224843 awarded to ARMB; National Institutes of Health [1R43CA224843-01].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Grilo AL, Mantalaris A.. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9–13. doi: 10.1016/j.tibtech.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11:219–38. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of invitro display technologies. Nat Biotechnol. 2011;29:245–54. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, etal. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci USA. 1988;85:5879–83. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perelson AS, Oster GF. Theoretical studies of clonal selection: minimal antibody repertoire size and reliability of self-non-self discrimination. JTheor Biol. 1979;81:645–70. doi: 10.1016/0022-5193(79)90275-3. [DOI] [PubMed] [Google Scholar]
- 6.Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, Kontermann RE, Jones PT, Low NM, Allison TJ, etal. Isolation of high affinity human antibodies directly from large synthetic repertoires. Embo J. 1994;13:3245–60. doi: 10.1002/embj.1994.13.issue-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. Bypassing immunization. Human antibodies from V-gene libraries displayed on phage. JMol Biol. 1991;222:581–97. doi: 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- 8.Edwards BM, Barash SC, Main SH, Choi GH, Minter R, Ullrich S, Williams E, Du Fou L, Wilton J, Albert VR, etal. The remarkable flexibility of the human antibody repertoire; isolation of over one thousand different antibodies to asingle protein, BLyS. JMol Biol. 2003;334:103–18. doi: 10.1016/j.jmb.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 9.Glanville J, D’Angelo S, Khan TA, Reddy ST, Naranjo L, Ferrara F, etal. Deep sequencing in library selection projects: what insight does it bring? Curr Opin Struct Biol. 2015;33:146–60. doi: 10.1016/j.sbi.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GMR, etal. Precise determination of the diversity of acombinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci USA. 2009;106:20216–21. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang W, Yoon A, Lee S, Kim S, Han J, Chung J. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp Mol Med. 2017;49:e308. doi: 10.1038/emm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niedringhaus TP, Milanova D, Kerby MB, Snyder MP, Barron AE. Landscape of nextgeneration sequencing technologies. Anal Chem. 2011;83:4327–41. doi: 10.1021/ac2010857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32:158–68. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara F, D’Angelo S, Gaiotto T, Naranjo L, Tian H, Graslund S, Dobrovetsky E, Hraber P, Lund-Johansen F, Saragozza S, etal. Recombinant renewable polyclonal antibodies. MAbs. 2015;7:32–41. doi: 10.4161/19420862.2015.989047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Angelo S, Kumar S, Naranjo L, Ferrara F, Kiss C, Bradbury AR. From deep sequencing to actual clones. Protein Eng Des Sel. 2014;27:301–07. doi: 10.1093/protein/gzu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spiliotopoulos A, Owen JP, Maddison BC, Dreveny I, Rees HC, Gough KC. Sensitive recovery of recombinant antibody clones after their in silico identification within NGS datasets. JImmunol Methods. 2015;420:50–55. doi: 10.1016/j.jim.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Sblattero D, Bradbury A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 2000;18:75–80. doi: 10.1038/71958. [DOI] [PubMed] [Google Scholar]
- 18.Ferrara F, Naranjo LA, Kumar S, Gaiotto T, Mukundan H, Swanson B, etal. Using phage and yeast display to select hundreds of monoclonal antibodies: application to antigen 85, atuberculosis biomarker. PLoS One. 2012;7:e49535. doi: 10.1371/journal.pone.0049535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Angelo S, Staquicini FI, Ferrara F, Staquicini DI, Sharma G, Tarleton CA, Nguyen H, Naranjo LA, Sidman RL, Arap W, etal. Selection of phage-displayed accessible recombinant targeted antibodies (SPARTA): methodology and applications. JCI Insight. 2018;3. doi: 10.1172/jci.insight.98305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Angelo S, Glanville J, Ferrara F, Naranjo L, Gleasner CD, Shen X, etal. The antibody mining toolbox: an open source tool for the rapid analysis of antibody repertoires. MAbs. 2014;6:160–72. doi: 10.4161/mabs.27105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan CE, Lim AP, MacAry PA, Hanson BJ. The role of phage display in therapeutic antibody discovery. IntImmunol. 2014;26:649–57. doi: 10.1093/intimm/dxu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Angelo S, Ferrara F, Naranjo L, Erasmus MF, Hraber P, Bradbury ARM. Many routes to an antibody heavy-chain CDR3: necessary, yet insufficient, for specific binding. Front Immunol. 2018;9:395. doi: 10.3389/fimmu.2018.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoskins RA, Stapleton M, George RA, Yu C, Wan KH, Carlson JW. Rapid and efficient cDNA library screening by self-ligation of inverse PCR products (SLIP). Nucleic Acids Res. 2005;33:e185. doi: 10.1093/nar/gni184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbas CF 3rd, Bain JD, Hoekstra DM, Lerner RA. Semisynthetic combinatorial antibody libraries: achemical solution to the diversity problem. Proc Natl Acad Sci USA. 1992;89:4457–61. doi: 10.1073/pnas.89.10.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braunagel M, Little M. Construction of asemisynthetic antibody library using trinucleotide oligos. Nucleic Acids Res. 1997;25:4690–91. doi: 10.1093/nar/25.22.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahon CM, Lambert MA, Glanville J, Wade JM, Fennell BJ, Krebs MR, Armellino D, Yang S, Liu X, O’Sullivan CM, etal. Comprehensive interrogation of aminimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. JMol Biol. 2013;425:1712–30. doi: 10.1016/j.jmb.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi: 10.1016/S1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 28.DeKosky BJ, Lungu OI, Park D, Johnson EL, Charab W, Chrysostomou C, Kuroda D, Ellington AD, Ippolito GC, Gray JJ, etal. Largescale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc Natl Acad Sci USA. 2016;113:E2636–45. doi: 10.1073/pnas.1525510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeWitt WS, Lindau P, Snyder TM, Sherwood AM, Vignali M, Carlson CS, Greenberg PD, Duerkopp N, Emerson RO, Robins HS, etal. Apublic database of memory and naive B-cell receptor sequences. PLoS One. 2016;11:e0160853. doi: 10.1371/journal.pone.0160853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–45. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 31.Ferrara F, Bradbury ARM, D’Angelo S. Primer design and inverse PCR on yeast display antibody selection outputs. Methods Mol Biol. 2018;1721:35–45. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: afast and accurate Illumina PairedEnd reAd mergeR. Bioinformatics. 2014;30:614–20. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Needleman SB, Wunsch CD. Ageneral method applicable to the search for similarities in the amino acid sequence of two proteins. JMol Biol. 1970;48:443–53. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 34.Ester M, Kriegel H-P, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. KDD’96 Proceedings of theSecond International Conference on Knowledge Discovery and Data Mining; Portland, OR; 1996:226–31. [Google Scholar]
- 35.Boder ET, Wittrup KD. Optimal screening of surface-displayed polypeptide libraries. Biotechnol Prog. 1998;14:55–62. doi: 10.1021/bp970144q. [DOI] [PubMed] [Google Scholar]
- 36.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, etal. Flow-cytometric isolation of human antibodies from anonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21:163–70. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]