

Abstract
The European Commission asked EFSA for a scientific opinion on the risks for animal and human health related to the presence of dioxins (PCDD/Fs) and DL‐PCBs in feed and food. The data from experimental animal and epidemiological studies were reviewed and it was decided to base the human risk assessment on effects observed in humans and to use animal data as supportive evidence. The critical effect was on semen quality, following pre‐ and postnatal exposure. The critical study showed a NOAEL of 7.0 pg WHO2005‐TEQ/g fat in blood sampled at age 9 years based on PCDD/F‐TEQs. No association was observed when including DL‐PCB‐TEQs. Using toxicokinetic modelling and taking into account the exposure from breastfeeding and a twofold higher intake during childhood, it was estimated that daily exposure in adolescents and adults should be below 0.25 pg TEQ/kg bw/day. The CONTAM Panel established a TWI of 2 pg TEQ/kg bw/week. With occurrence and consumption data from European countries, the mean and P95 intake of total TEQ by Adolescents, Adults, Elderly and Very Elderly varied between, respectively, 2.1 to 10.5, and 5.3 to 30.4 pg TEQ/kg bw/week, implying a considerable exceedance of the TWI. Toddlers and Other Children showed a higher exposure than older age groups, but this was accounted for when deriving the TWI. Exposure to PCDD/F‐TEQ only was on average 2.4‐ and 2.7‐fold lower for mean and P95 exposure than for total TEQ. PCDD/Fs and DL‐PCBs are transferred to milk and eggs, and accumulate in fatty tissues and liver. Transfer rates and bioconcentration factors were identified for various species. The CONTAM Panel was not able to identify reference values in most farm and companion animals with the exception of NOAELs for mink, chicken and some fish species. The estimated exposure from feed for these species does not imply a risk.
Keywords: Dioxins, PCDD/Fs, DL‐PCBs, food, feed, risk assessment, transfer
Short abstract
This publication is linked to the following EFSA Supporting Publications articles: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1136/full, http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1137/full, http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1374/full
Summary
The European Commission asked the European Food Safety Authority (EFSA) in accordance with Art. 29 (1) of Regulation (EC) No 178/2002 for a scientific opinion on the risk for animal and human health related to the presence of dioxins (polychlorinated dibenzo‐p‐dioxins and dibenzofurans (PCDD/Fs)) and dioxin‐like polychlorinated biphenyls (DL‐PCBs) in feed and food.
According to the terms of reference provided by European Commission, the scientific opinion should, inter alia, comprise the:
evaluation of the toxicity of dioxins and DL‐PCBs for animals and humans, considering all relevant adverse acute and chronic health effects;
estimation of the dietary exposure (chronic and, if relevant, acute dietary exposure) of the EU population including the consumption patterns of specific (vulnerable) groups of the population (e.g. high consumers, children, people following a specific diet, etc.);
estimation of the exposure of the different animal species to dioxins and DL‐PCBs from feed and the levels of transfer/carry‐over from the feed to the products of animal origin for human consumption;
assessment of the chronic (and if relevant acute) human health risks for the EU population including for specific (vulnerable) groups of the population as the consequence of the estimated dietary exposure;
assessment of the animal health risks for the different animal species as the consequence of the estimated exposure from animal feed.
Although the term ‘dioxin’ is commonly used to refer to both PCDDs and PCDFs, for the sake of clarity in this opinion the term PCDD/Fs is used consistently to refer to this group of compounds. PCDDs and PCDFs are two groups of tricyclic planar compounds. Dependent on the number of chlorine atoms and their positions at the rings, 75 PCDDs and 135 PCDFs, termed ‘congeners’, can occur. Only 17 of these are relatively persistent in animals and humans and therefore considered relevant. They contain at least four chlorines and at positions 2, 3, 7 and 8. In this opinion, they are referred to as the 17 PCDD/Fs. PCDD/Fs have never been produced on an industrial scale and have no technological use. They are formed unintentionally in a number of industrial and thermal processes. In contrast to PCDD/Fs, PCBs had widespread use in open and closed systems, generally in the form of complex technical mixtures. They were produced with an estimated total world production of 1.2–1.5 million tonnes between 1929 and the end of the 1970s, when their production was abandoned in the majority of countries. A subgroup of 12 PCB congeners that are non‐ortho or mono‐ortho chlorine substituted and contain at least four chlorine substituents can easily adopt a coplanar structure and show toxicological properties similar to tetrachlorodibenzo‐p‐dioxin (TCDD) and other PCDD/Fs. This subgroup is termed DL‐PCBs, and in this opinion, they are referred to as the 12 DL‐PCBs. Due to their lipophilic properties and poor degradation, PCDD/Fs and DL‐PCBs accumulate in the food chain.
1.
1.1.
Risk for human health related to the presence of PCDD/Fs and DL‐PCBs in food
The human chronic dietary exposure to PCDD/Fs and DL‐PCBs was estimated using a data set containing:
19,965 food samples with all 29 congeners determined (17 PCDD/Fs and 12 DL‐PCBs)
20,273 food samples with all 17 PCDD/F congeners determined (including samples with the 29 congeners)
22,974 food samples with all 12 DL‐PCB congeners determined (including samples with the 29 congeners)
The mean and P95 lower bound/upper bound (LB/UB) levels of the sum of the 17 PCDD/Fs and 12 DL‐PCBs (29 congeners) in ‘Livestock meat including offal’ were, respectively, 1.43/1.54 and 5.06/5.12 pg WHO2005‐TEQ/g fat weight. In various species within ‘Livestock meat’, the mean levels ranged from 0.12/0.20 to 6.23/6.26 pg WHO2005‐TEQ/g fat weight.
In ‘Milk and milk products’, the mean and P95 LB/UB levels of the sum of the 29 congeners were, respectively, 0.73/0.88 and 1.92/2.04 pg WHO2005‐TEQ/g fat weight, in eggs and egg products 1.17/1.30 and 4.38/4.39 pg WHO2005‐TEQ/g fat weight, in ‘Animal and vegetable fat’ 0.42/0.53 and 1.59/1.65 pg WHO2005‐TEQ/g fat weight, in Vegetables 0.05/0.08 and 0.26/0.28 pg WHO2005‐TEQ/g whole weight, and in ‘Fish and seafood’ 4.35/4.45 and 21.0/21.6 pg WHO2005‐TEQ/g whole weight. For various fish species, the mean LB/UB levels ranged from 0.10/0.10 to 9.17/9.21 pg WHO2005‐TEQ/g whole weight.
For the 17 PCDD/Fs, the mean and P95 LB/UB levels in ‘Livestock meat including offal’ were, respectively, 0.50/0.60 and 1.54/1.61 pg WHO2005‐TEQ/g fat weight. The levels varied between different species of ‘Livestock meat’, showing mean LB/UB levels from 0.08/0.16 to 2.65/2.68 pg WHO2005‐TEQ/g fat weight. In ‘Milk and milk products’, the mean and P95 LB/UB levels were, respectively, 0.28/0.43 and 0.92/1.06 pg WHO2005‐TEQ/g fat weight, in ‘Eggs and egg products’ 0.51/0.62 and 2.02/2.02 pg WHO2005‐TEQ/g fat weight, in ‘Animal and vegetable fat’ 0.20/0.29 and 0.66/0.70 pg WHO2005‐TEQ/g fat weight, in ‘Vegetables’ 0.02/0.05 and 0.12/0.21 pg WHO2005‐TEQ/g whole weight, and in ‘Fish and seafood’ 0.95/1.05 and 4.30/4.66 pg WHO2005‐TEQ/g whole weight. The levels varied between various fish species, showing mean LB/UB levels from 0.01/0.04 to 2.66/2.67 pg WHO2005‐TEQ/g whole weight.
Highest mean LB/UB concentrations for the sum of PCDD/Fs and DL‐PCBs (29 congeners) were found in some rarely consumed foods such as certain game birds (Mallard meat’ and ‘Pheasant meat’ with 39.8/39.8 and 8.29/8.55 pg WHO2005‐TEQ/g fat weight, respectively), ‘Fish liver’ (22.1/22.6 pg WHO2005‐TEQ/g whole weight), and ‘Brown meat of crabs’ (6.10/6.17 pg WHO2005‐TEQ/g whole weight). High mean LB/UB concentrations of the 17 PCDD/F congeners were found in the same categories: game birds (Mallard meat’ and ‘Pheasant meat’ with 2.16/2.19 and 1.76/2.02 pg WHO2005‐TEQ/g fat weight, respectively‘), ‘Fish liver’ (4.41/4.95 pg WHO2005‐TEQ/g whole weight), and ‘Brown meat of crabs’ (3.22/3.29 pg WHO2005‐TEQ/g whole weight).
To estimate the chronic human dietary intake, two exposure assessments were carried out: (i) taking into account the occurrence values of the samples with all the 29 PCDD/F and DL‐PCB congeners, and (ii) taking into account the occurrence values of samples with the 17 PCDD/F congeners (including samples with all 29 congeners analysed).
The difference between the LB and UB estimations across all age classes was small for both exposure assessments. For the sum of PCDD/Fs and DL‐PCBs (29 congeners), the mean UB exposure ranged from 0.4 to 2.6 pg WHO2005‐TEQ/kg body weight (bw) per day. At the 95th percentile exposure, the UB estimates ranged from 0.9 to 6.6 pg WHO2005‐TEQ/kg bw per day. For the sum of PCDD/Fs (17 congeners), the mean UB exposure ranged from 0.2 to 1.3 pg WHO2005‐TEQ/kg bw per day. At the 95th percentile exposure, the UB estimates ranged from 0.4 to 2.4 pg WHO2005‐TEQ/kg bw per day.
The highest exposures to the sum of PCDD/Fs and DL‐PCBs (29 congeners) and to the sum of PCDD/Fs (17 congeners) were estimated for the age classes Toddlers and Other Children, and were about twofold higher than in Adolescents and Adults.
Regarding the average contribution of individual congeners to the overall mean LB WHO2005‐TEQ exposure (29 congeners), PCB‐126 contributes most, followed by 2,3,4,7,8‐PeCDF, 1,2,3,7,8‐PeCDD, 2,3,7,8‐TCDF, PCB‐169 and 2,3,7,8‐TCDD. As a group, the non‐ortho PCBs showed the highest contribution (59%), followed by the PCDFs (23%), PCDDs (14%) and mono‐ortho PCBs (5%). Considering only the sum of PCDDs and PCDFs (17 congeners), the PCDFs contributed 62%.
The main contributors to the mean dietary exposure for the age group Infants were ‘Butter and butter oil’ (contributing from 6.1% to 19.6%) and ‘Fatty fish’ (contributing from 5.8% to 26.3%). For Toddlers, the categories ‘Fatty fish’ (contributing from 5.9% to 13.9%), ‘Cheese’ (contributing from 5.9% to 21.8%) and ‘Livestock meat’ (contributing from 7.7% to 16.2%) were found to be the main sources of exposure. Similarly, for the age groups of Other Children, Adolescents, Adults and Elderly the main contributors were ‘Fatty fish’ (up to 56% contribution), ‘Unspecified fish meat’ (up to 53.4% contribution), ‘Cheese’ (up to 21.8% contribution) and ‘Livestock meat’ (up to 33.8% contribution).
In rodents, PCDD/Fs and DL‐PCBs are well absorbed and distributed to various tissues, and transferred to the fetus. The major accumulation is in adipose tissue and liver, with a liver/adipose tissue ratio that increases with the applied dose. At least in mice, this is shown to be due to binding to CYP1A2 in the liver. In laboratory animals, the biotransformation of TCDD, being slow, mainly consists of hydroxylation at a lateral or peri‐position. It is a detoxification process. There can be some differences in the rate and products of TCDD biotransformation, but these do not seem to account for the strain‐ or species‐specific sensitivities to TCDD toxicity. In rats, 2,3,7,8‐TCDF and 1,2,3,7,8‐PeCDF are effectively metabolised, but higher chlorinated PCDFs are metabolised to a much lower degree. Except for PCB‐77, most of the DL‐PCBs are not readily metabolised. In rats and mice, faecal excretion dominates over excretion via urine. Metabolites are excreted rapidly in bile and urine. At least in rats, the higher chlorinated PCDDs seem to exist predominantly unmetabolised in faeces. Half‐lives are in the order of several weeks and short when compared to humans.
In humans, PCDD/Fs and DL‐PCBs are well absorbed and subsequently distributed to liver and body lipids. The levels of the more relevant congeners in the blood are in equilibrium with those in adipose tissue. At high exposure, PCDD/Fs and DL‐PCBs can show higher lipid‐based levels in the liver than in the adipose tissue. Most PCDD/Fs and DL‐PCBs are poorly metabolised but some hydroxylated metabolites have been identified. Compared to laboratory animals, most PCDD/Fs and DL‐PCBs show long half‐lives (several years) which vary between congeners and depending on the levels, age, BMI and sex.
Concerning adverse effects in experimental animals, the CONTAM Panel selected only those studies that could potentially show effects at lower body burdens than the one used as basis for the tolerable weekly intake (TWI) set by the Scientific Committee on Food (SCF) in their assessment in 2001 (lowest‐observed‐adverse‐effect level (LOAEL) of 40 ng/g bw). The CONTAM Panel decided to focus on studies in which only TCDD had been dosed to the animals. The studies on rodents confirmed that developmental effects were seen at body burdens in a similar range as those that were the basis for the previous risk assessment by the SCF. In rats the adverse effects at such low body burdens were reduced sperm production (LOAEL body burden 25 ng/kg bw), delayed puberty development (LOAEL body burden 42–50 ng/kg bw), altered bone parameters (no‐observed‐adverse‐effect level (NOAEL) body burden 28 ng/kg bw) and hepatopathy (NOAEL body burden 26 ng/kg bw). In mice, the lowest extrapolated body burden at the NOAEL was 9 ng/kg bw, based on embryo loss. Studies in primates treated during gestation and lactation showed dental effects and effects on sperm concentration at high dose.
Concerning effects in humans, the CONTAM Panel selected studies which analysed tissues (e.g. blood, human milk, adipose tissue) of the subjects under study for either (i) TCDD or any other congener dominating the TEQ, e.g. due to a contamination incident, (ii) the 17 PCDD/Fs and 12 DL‐PCBs, (iii) the 17 PCDD/Fs and 4 non‐ortho DL‐PCBs, (iv) the 17 PCDD/Fs and 3 non‐ortho DL‐PCBs (including PCB‐126), or (v) the total TEQs (or BEQs analysed by, e.g. CALUX). Studies assessing dietary exposure with validated methods in relation to outcomes were also included.
The epidemiological studies have been conducted in subjects/cohorts exposed to PCDD/Fs and DL‐PCBs at different life stages under different exposure conditions, e.g. from industrial accidents or contamination incidents, from occupational exposure or from background levels mainly via the diet in the general population.
Chloracne is the most unequivocal toxicity outcome observed in accidental, occupational and unresolved poisoning cases with PCDD/Fs and DL‐PCBs, children appearing to be particularly sensitive. However, chloracne only occurs after high exposures (resulting in serum levels > 20,000 pg/g fat) and is not relevant for deriving a health‐based guidance value (HBGV) for the general population. There is insufficient information with respect to DL‐PCBs, since even in the rice oil incidents with PCB‐oil, 2,3,4,7,8‐PeCDF contributed most to the TEQ level.
Associations between exposure to TCDD during infancy/prepuberty and impaired semen quality were observed in three prospective studies (two after the Seveso incident and one from the Russian Children's Study). Based on weight of evidence, including experimental animal studies, the associations were considered causal. Impaired semen quality was observed in men in Seveso but only in those that were prepubertal at the time of the incident. Even in the lowest quartile the serum levels of TCDD were high compared to present‐day levels in Europe. In another study on adult men born to mothers who were exposed during the Seveso incident, impaired semen quality was observed only in those who had been breastfed. Together, this evidence indicates that there may be a postnatal period of sensitivity that might expand into puberty. In the Russian Children's Study, which included boys exposed to high environmental background levels, associations of serum TCDD with impaired semen quality were observed. Significant associations were observed also for the sum of PCDD‐TEQ and PCDFs‐TEQ, but not for DL‐PCB‐TEQ or total TEQ. The association between TCDD and semen parameters became slightly stronger after adjustment for NDL‐PCBs, but were not changed by adjustment for exposure to organochlorine pesticides.
There is insufficient evidence for an association between PCDD/Fs or DL‐PCBs and cryptorchidism. For changes in time of pubertal onset and sexual maturity, observed in one cohort only (the Russian Children's Study), there was insufficient information to conclude on causal associations.
Regarding female reproductive effects, for endometriosis, the only available prospective study did not observe a dose response, and since the available case–control studies indicating associations had limitations, the available evidence was insufficient to be used as a basis for the risk assessment. The few available studies indicated no association between exposure and pubertal development and the evidence was insufficient for other female reproductive effects (menstrual cycle characteristics, ovarian function, time to pregnancy, uterine leiomyoma and age at menopause).
A relationship between high TCDD exposure in fathers and lower sex ratio in offspring (lower number of boys relative to girls) has been consistently observed across three different cohorts, and is likely to be causal. The studies on other birth outcomes (birth weight, preterm birth, fetal Yusho disease and anogenital distance) were inconclusive and could not be used as a basis for the risk assessment.
Concerning thyroid disease and thyroid hormones, in adults, epidemiological studies provide insufficient support for an association between TCDD, other PCDDs, PCDFs or DL‐PCBs and thyroid disease or thyroid function. A study in children born to mothers highly exposed to TCDD in Seveso indicates a causal association between TCDD and increased neonatal thyroid‐stimulating hormone (TSH). Studies with low‐moderate exposure to TCDD, other PCDDs, PCDFs or DL‐PCBs do not suggest any adverse effects on the thyroid.
The studies on type 2 diabetes and obesity were inconclusive and could not be used as a basis for the risk assessment.
An epidemiological study of very high occupational exposure to TCDD (serum TCDD > 1,000 pg/g fat) indicates increased risk of cardiovascular mortality. At lower exposures to TCDD, other PCDDs, PCDFs or DL‐PCBs, epidemiological studies provide insufficient support for an association with cardiovascular risk.
Following accidental or occupational exposure, evidence for a causal association with hepatic or digestive diseases is insufficient.
Some studies suggest adverse effects on the immune system at background exposure during development, but the available studies do not provide sufficient evidence for an association between PCDD/Fs or DL‐PCBs and the functionality of the immune system.
Various neurodevelopmental outcomes at different ages have been investigated in children, but few outcomes have been assessed in several cohorts and/or at similar age. The available information is not sufficient to form a basis for the risk assessment. There is insufficient information to draw conclusions on effects on the nervous system after exposure in adult life.
In three different population groups, childhood exposure to TCDD and/or other PCDD/Fs was dose‐relatedly associated with tooth enamel hypomineralisation or enamel defects. Hypomineralisation of permanent teeth is likely to be causally related to exposure and is likely to be a postnatal effect. Limited evidence from one cohort indicates associations between PCDD/F and DL‐PCB exposure and some changes in bone parameters.
While several studies (many with multiple co‐exposures) showed a positive association with all cancers combined there was no clear link to any specific cancer site. There was no clear dose–response relationship between exposure and cancer development.
Binding to the aryl hydrocarbon receptor (AHR) is the molecular initiating event of the toxicities of PCDD/Fs and DL‐PCBs. Toxicity is due to inappropriate (in terms of timing, location and/or degree) and sustained activation of this receptor. AHR signalling can proceed via canonical or alternative pathways. The major toxicities of PCDD/Fs and DL‐PCBs appear to be primarily mediated by the canonical pathway, in which the AHR acts as a ligand‐activated transcription factor.
In animal models, structural variations in the ligand‐binding or transactivation domain of the AHR are associated with non‐selective or selective differences, respectively, in sensitivity to the manifestations of TCDD toxicity. The human AHR has a lower binding affinity to TCDD when compared to rats and most mouse strains. This may differ for other PCDD/F and DL‐PCB congeners. PCDD/Fs and DL‐PCBs affect the expression of a large number of genes and these seem to be species‐ and congener‐dependent, indicating additional modes of action.
There is no robust evidence that the development of cancer caused by TCDD and other PCDD/Fs in experimental animals is associated with direct genotoxicity. Rodent studies demonstrate that TCDD is a potent promoter of skin, ovary and liver cancer following initiation with genotoxic agents such as diethylnitrosamine (DEN) and N‐methyl‐N’‐nitrosoguanidine. Hepatic neoplastic changes may be linked to liver regeneration in response to toxicity.
In rats, gestational TCDD exposure abrogates the gender difference in the anteroventral periventricular nucleus expression of glutamic acid decarboxylase 67, a key enzyme in GABA synthesis, and prevents perinatal luteinising hormone (LH) and testosterone surges in male pups. These changes may underlie the alterations in reproductive functions discernible at adult age, including early puberty, constant oestrus and premature reproductive senescence in females, and delayed puberty, feminised sexual behaviour and (possibly) reduced daily sperm production in males. A decreased male‐to‐female ratio has been reported in rat F2 generation after treatment of F0 dams with TCDD, and in the offspring of mouse or human males exposed to TCDD. In mice, suggestive evidence was found of a diminished ability of Y‐bearing sperm to conceive the ova. In adult male rats and marmosets, TCDD impaired testosterone synthesis in Leydig cells and adversely affected spermiogenesis. Similarly, in mice with a constitutively active AHR, epididymal sperm count was reduced by 45%. Exposure of adult female rodents to TCDD has been found to lead to irregular oestrous cycles and reduced ovulatory rate, possibly due to repressed ovarian expression of Cyp17a1, induction of xenobiotic‐metabolising enzymes and inhibition of oestrogen receptor function by the activated AHR.
In rats, TCDD decreases dose‐dependently circulating total and free T4 concentrations, accompanied by an inconsistent impact on serum T3 levels. Functionally, TCDD‐treated rats appear to be euthyroid. The decrease in T4 in rats is primarily due to accelerated hepatic clearance of T4 through biliary excretion as a result of induction of UDP‐glucuronosyltransferase (UGT) (especially UGT1A) activity. In addition to inducing hepatic UGT activity, DL‐PCBs may decrease serum T4 levels via competition of their hydroxylated metabolites with T4 for binding to transthyretin. There is no consistent pattern of thyroid histopathological effects of TCDD. In in vitro studies, TCDD and DL‐PCBs have been shown to reduce the protein or mRNA expression of the sodium‐iodide symporter in animal and human thyroid cells.
In rats, a highly sensitive response to in utero exposure to TCDD is a reduction in size or total missing of third molar teeth in pups. This is associated with an increased susceptibility of their molar teeth to caries. At higher doses in rats, TCDD may also affect the continuously erupting incisor teeth. TCDD especially interferes with mineralisation of the dental matrices in developing teeth, with the most critical window of sensitivity being during the early morphogenesis of teeth. In vitro studies have revealed that at the initiation stage, TCDD blocks mouse molar tooth development by enhancing apoptosis in the dental epithelium and inhibiting the proliferation and differentiation of stem cells of the apical papilla. Epidermal growth factor receptor (EGFR) signalling and the dentin sialophosphoprotein gene appear to be involved in the mineralisation defects caused by TCDD.
The association between serum levels and the decreased sperm concentrations observed in the Russian Children's Study and in the Seveso studies was selected as the critical effect. In the Russian Children's Study, an association between decreased sperm concentrations and increasing serum levels of TCDD, PCDD‐TEQ and PCDD/F‐TEQ was observed. A NOAEL serum level for PCDD/Fs of 7.0 pg WHO2005‐TEQ/g fat at age 9 years was selected, based on the median level in the lowest quartile.
A toxicokinetic model was used to estimate the daily intake leading to a serum level of 7.0 pg WHO2005‐TEQ/g fat at the age of 9 years in boys, taking into account breastfeeding for 12 months by mothers with similar exposure. In the calculations, the twofold higher dietary exposure of Toddlers and Other Children was taken into account. The model includes the concentration‐dependent distribution between the liver and body fat, the degradation in the liver and the direct loss via lipids in the faeces. It was estimated that a level in human milk of 5.9 pg TEQ/g fat, resulting from the constant exposure of mothers to 0.25 pg TEQ/kg bw per day, and subsequent exposure via food to 0.5 pg TEQ/kg bw per day, would result in the NOAEL serum level of 7.0 pg WHO2005‐TEQ/g fat at the age of 9 years.
Taking the uncertainties into account, a TWI of 2 pg WHO2005‐TEQ/kg bw per week was established. The CONTAM Panel decided to base the HBGV on a weekly basis since this is not expected to result in a critical increase in levels in serum. This could not be assumed for extension to a longer, e.g. monthly, intake, in the absence of studies and toxicokinetic models that can exclude that a single high dose with, e.g. half of the tolerable monthly intake could result in a peak in the serum level. The CONTAM Panel noted that in the Russian Children's Study, no association was observed for DL‐PCB‐TEQ or the sum‐TEQ of PCDD/Fs and DL‐PCBs. This might be explained by observations from in vitro studies with human cells, showing that PCB‐126 is much less potent in humans than suggested by the WHO2005‐TEF of 0.1. PCB‐126 is the DL‐PCB contributing most to the current intake of PCDD/Fs and DL‐PCBs, but also in the serum of boys from the Russian Children's Study.
When comparing the mean current exposure to PCDD/Fs and DL‐PCBs of Adolescents, Adults, Elderly and Very Elderly, an up to fivefold exceedance of the TWI was observed (highest UB). At the P95, this ranged from 3 to 15. Toddlers and Other Children showed a factor of 2 higher exceedance than older age groups. When calculating the intake leading to the critical serum level of 7.0 pg WHO2005‐TEQ/g fat at the age of 9 years this factor was taken into account.
Regarding the potentially lower potency of PCB‐126, the CONTAM Panel also evaluated the current exposure to PCDD/Fs only. The mean exposure of Adolescents and adult age groups were up to twofold higher than the TWI (highest UB). At the P95, this was up to sixfold higher.
Breastfed infants are known to have a higher exposure than Toddlers and Other Children. The exposure of breastfed infants should not be compared to the TWI. The reason is that the TWI was set to prevent a level in breast milk that would result in serum levels in children that have been associated with adverse effects.
The CONTAM Panel considered that the impact of the uncertainties on the risk assessment of PCDD/Fs in food is moderate. For the sum of PCDD/Fs and DL‐PCBs, due to the uncertainty in the relative potency of PCB‐126 in humans, the impact of the uncertainties on the risk assessment is high. Overall, the assessment is likely to be conservative.
Transfer of PCDD/Fs and DL‐PCBs in farm animals
The transfer in dairy cows has been studied in a number of controlled experiments and follow‐up studies of incidents. Although to a lesser extent, this also applies for laying hens, growing pigs and sheep. These studies show congener and species specific differences in the excretion and accumulation in meat, body fat and liver.
Long time periods are required to decrease levels after termination of the exposure. Elimination via milk and eggs is a major factor in the decrease of body burdens in lactating ruminants and laying hens, respectively. For meat producing animals, growth of animals contributes to the reduction of the levels. Transfer rates (TRs) and bioconcentration factors (BCFs) were derived for several species, describing the relation between intake and levels in milk and eggs, or accumulation in tissues. At prolonged exposure (steady state), the daily TEQ amount in milk or eggs may be more than one‐third of the daily ingested dose. For dairy cows, laying hens and fattening pigs, toxicokinetic models have been developed that can be used to describe levels in edible products based on levels in feed and duration of exposure and post‐exposure decrease.
PCDD/Fs and DL‐PCBs are accumulated to a greater extent in fillet of farmed oily fish (such as salmon and trout) than in leaner fish such as carp and seabream. BCFs were derived for several fish species, describing the relation between intake and accumulation in fillet. PCDD/Fs and DL‐PCBs accumulate to a greater degree in the liver than in fillet of lean fish, such as cod. Toxicokinetic models have been developed for salmon enabling the prediction of fillet concentrations of PCDD/F and DL‐PCBs from known feed concentrations.
Risk for farm and companion animal health related to the presence of PCDD/Fs and DL‐PCBs in feed
The chronic dietary exposure of farm and companion animals to PCDD/Fs and DL‐PCBs was estimated using a data set containing:
1,830 feed samples with all 29 congeners determined (17 PCDD/Fs and 12 DL‐PCBs);
1,844 feed samples with all 17 PCDD/F congeners determined (including samples with the 29 congeners);
2,131 feed samples with all 12 DL‐PCB congeners determined (including samples with the 29 congeners).
The LB/UB mean levels of the sum of PCDD/Fs and DL‐PCBs (29 congeners) in ‘Fish oil’ were 3.33/3.38 ng WHO2005‐TEQ/kg, in ‘Fish meal’ 0.60/0.62 ng WHO2005‐TEQ/kg, while in complete feed for fish they were 0.54/0.56 ng WHO2005‐TEQ/kg. The LB/UB mean levels in ‘Animal fat (for feed)’ were 0.33/0.37 ng WHO2005‐TEQ/kg and in ‘Vegetable fat and oil’ 0.17/0.22 ng WHO2005‐TEQ/kg (all expressed in 88% dry matter).
The LB/UB mean levels of the PCDD/Fs (17 congeners) in these categories were as follows: ‘Fish oil’ 0.80/0.85 ng WHO2005‐TEQ/kg, ‘Fish meal’ 0.21/0.24 ng WHO2005‐TEQ/kg, and complete feeds for fish 0.13/0.15 ng WHO2005‐TEQ/kg. The levels in ‘Animal fat (for feed)’ and ‘Vegetable fat and oil’ were 0.10/0.13 and 0.10/0.16 ng WHO2005‐TEQ/kg, respectively (all expressed in 88% dry matter).
As for the human dietary exposure, two exposure assessment were carried out, (i) taking into account the occurrence values of the samples with all the 29 PCDD/Fs and DL‐PCBs congeners, and (ii) taking into account the occurrence values of samples with the 17 PCDD/F congeners (including samples with all 29 congeners analysed).
The highest estimated exposure for the sum of PCDD/Fs and DL‐PCBs (29 congeners) was for ‘Salmonids’ (mean LB/UB = 12/13 pg WHO2005‐TEQ/kg bw per day; P95 LB/UB = 27/27 pg WHO2005‐TEQ/kg bw per day). ‘Carp’ had a lower estimated exposure (mean LB/UB = 4.5/5.0 pg WHO2005‐TEQ/kg bw per day; P95 LB/UB = 13/14 pg WHO2005‐TEQ/kg bw per day).
For ruminants, the highest mean and P95 exposures to the sum of PCDD/Fs and DL‐PCBs (in pg WHO2005‐TEQ/kg bw per day) were for ‘Fattening goats’ (mean LB/UB = 3.0/3.6; P95 LB/UB = 9.9/10), and these were approximately three to four times higher than the lowest estimated exposures, that were estimated for ‘Beef cattle on a cereal‐based diet’ (mean UB/LB = 0.75/1.1, P95 LB/UB = 2.2/2.4).
For pigs, the highest exposure for the 29 congeners (in pg WHO2005‐TEQ/kg bw per day) was for ‘Pigs: starters’ (mean LB/UB = 1.3/2.2, P95 LB/UB = 8.6/10), followed by that in ‘Pigs: growing and fattening’ (mean LB/UB = 0.82/1.5, P95 LB/UB = 4.3/5.1) and in ‘Lactating sow’ (mean LB/UB = 0.76/1.3).
For poultry, the highest exposure for the 29 congeners (in pg WHO2005‐TEQ/kg bw per day) was for ‘Fattening chickens’ (mean UB/LB = 1.9/3.1, P95 LB/UB = 11/13), followed by ‘Laying hens’ (mean LB/UB = 1.8/2.8, P95 LB/UB = 10/12) and ‘Starter poultry’ (mean LB/UB = 1.9/3.4, P95 LB/UB = 3.5/7.3). The estimated exposure for ‘Fattening turkeys’ and ‘Fattening ducks’ was lower (mean LB/UB = 0.70/1.2, P95 LB/UB = 4.1/4.9, and mean LB/UB = 1.2/2.0, P95 LB/UB = 8.2/9.7, respectively).
For rabbits, the mean LB/UB exposure was 3.5/4.5 pg WHO2005‐TEQ/kg bw per day, while for mink the values estimated were lower (mean LB/UB = 2.7/3.1; P95 LB/UB = 7.4/7.7). Insufficient data on species‐specific compound feeds for rabbits and mink were available to reliably predict P95 exposures.
For companion animals, the CONTAM Panel noted the marked differences in estimated diet concentrations and exposures between cats and dogs. For dogs, the mean LB/UB exposure was 2.0/2.5 pg WHO2005‐TEQ/kg bw per day, based on data on compound feeds for dogs. In contrast, data on compound feed data were not available for cats but based on individual feed ingredients the mean exposure was estimated to be 0.70/0.88 pg WHO2005‐TEQ/kg bw per day, with P95 LB/UB exposures of 2.4/2.5 pg WHO2005‐TEQ/kg bw per day, respectively.
As for the 29 congeners, the highest exposure to the sum of PCDD/Fs (17 congeners), in pg WHO2005‐TEQ/kg bw per day, was for ‘Salmonids’ (mean LB/UB = 2.9/3.9; P95 LB/UB = 8.2/9.5), and was higher than that of ‘Carp’ (mean LB/UB = 0.78/1.20; P95 LB/UB = 3.0/3.7).
For ruminants, the highest exposures (in pg WHO2005‐TEQ/kg bw per day) were again estimated for ‘Fattening goats’ (mean LB/UB = 1.5/2.1; P95 LB/UB = 6.2/6.5), while the lowest was observed for ‘Beef cattle on a cereal‐based diet’ (mean UB/LB = 0.33/0.62, P95 LB/UB = 1.1/1.5).
For pigs, the highest exposure to the 17 PCDD/Fs (in pg WHO2005‐TEQ/kg bw per day) was for ‘Pigs: starters’ (mean LB/UB = 0.46/1.3; P95 LB/UB = 4.0/6.5) and the lowest for ‘Pigs: growing and fattening’ (mean LB/UB = 0.48/0.94; P95 LB/UB = 1.8/1.9) and ‘Lactating sows’ (mean LB/UB = 0.62/0.93).
For poultry, the highest exposure (in pg WHO2005‐TEQ/kg bw per day) was for ‘Fattening chickens’ (mean UB/LB = 1.0/2.1, P95 LB/UB = 5.7/8.3), while the lowest was estimated for ‘Fattening turkeys’ (mean LB/UB = 0.24/0.69; P95 LB/UB = 1.9/3.1.
For rabbits, the mean LB/UB exposure (based on data for compounds feeds) was 1.9/2.8 pg WHO2005‐TEQ/kg bw per day. In the absence of similar data for mink, exposures were estimated using data for individual feeds, and this resulted in lower estimates of exposure (mean LB/UB = 1.4/1.9; P95 LB/UB = 3.0/3.5).
For companion animals, again the exposure estimated for dogs (mean LB/UB = 1.9/2.0 pg WHO2005‐TEQ/kg bw per day) was higher than that of cats (mean LB/UB = 0.39/0.54; P95 LB/UB = 1.1/1.3).
PCDD/Fs, with the exception of the higher chlorinated congeners, and DL‐PCBs are effectively absorbed. Most are poorly degraded but some metabolites of TCDD and some DL‐PCBs have been identified. The parent compounds are accumulated in body fat and liver in a congener specific manner. They are also transferred to milk and eggs.
For ruminants and pigs, no studies were identified that could be used for the risk assessment, and for rabbits it was not possible to determine a NOAEL from the studies in which they had been exposed to TCDD. In the three studies identified in horses, there was mixed exposure to contaminants and no NOAEL could be identified.
For poultry, chicks treated with PCDD/Fs or DL‐PCBs by gavage had high incidences of mortality during development, which was associated with pericardial, peritoneal and pulmonary oedema as well as atrophy of the thymus and Bursa of Fabricius, depletion of splenic lymphocytes and delayed egg production when mature. After intraperitoneal (i.p.) dosing with TCDD, young chickens showed a decrease in the Bursa of Fabricius after 5 days with a NOAEL of 1 μg/kg bw per day. Studies on eggs following in ovo injection, showed poor hatchability and associated cardiomyopathy and teratogenicity, associated with effects like thymic atrophy and changes in thyroid hormone levels. However, the CONTAM Panel concluded that the in ovo studies could not be used for the risk assessment, since they are confounded by timing and route of administration.
Studies of ducks, turkeys, pheasants and quails and their eggs were not useful for risk assessment but illustrated that these species were less susceptible than chicken to PCDD/Fs and DL‐PCBs for some adverse outcomes.
In fish, fin necrosis, haemorrhages, reduced growth and mortality were the toxicological responses to PCDD/Fs and DL‐PCBs exposure observed. The lowest LOAEL in rainbow trout was 1 μg TCDD/kg bw, with a NOAEL of 0.1 μg TCDD/kg bw. A NOAEL of 1 μg TCDD/kg bw was identified for yellow perch and tilapia, and 0.57 μg TCDD/kg bw for carp.
Several studies in cats and dogs reported non‐adverse effects, e.g. enzyme induction. Lethality was observed in dogs at high dose. Microscopic changes were observed in the liver, kidney and spleen of cats but the route and extent of exposure could not be determined.
Mink are sensitive to the toxicity of PCDD/Fs and DL‐PCBs, and the most sensitive response in mink (NOAEL of 2.1 ng TCDD/kg bw per day in a two‐generation feeding study) proved to be proliferation of the squamous gingival epithelium in mouth. This may lead to cyst formation adjacent to teeth and cause osteoporosis in jaw bones. Co‐exposure of mink to a mixture of toxicants (by feeding on contaminated fish) appeared to augment the toxicity of PCDD/Fs and DL‐PCBs, with a LOAEL of 0.4 ng WHO2005‐TEQ/kg bw per day being obtained for mandibular and maxillary squamous cell hyperplastic foci.
Concerning the derivation of reference doses for farm and companion animals, the CONTAM Panel concluded that no studies were identified that could be used to derive a NOAEL or LOAEL for ruminants, pigs, horses, rabbits, ducks, turkeys, quails, pheasants, cats and dogs and that could be compared with the current mean and P95 intake from feed for the risk characterisation.
For laying hens, a NOAEL of 5.6 ng/kg bw per day and corresponding LOAEL of 1.1 μg/kg bw per day was identified, showing that egg production had ceased after 12 days of treatment with a high dose of TCDD. In chicks, a NOAEL of 0.1 μg TCDD/kg bw was observed, a 10‐fold higher dose showing mortality.
In rainbow trout, the lowest LOAEL was 1 μg TCDD/kg bw, with a NOAEL of 0.1 μg TCDD/kg bw, based on growth, fin erosion and survival. In Atlantic salmon, no effects were observed after prolonged exposure to PCDD/Fs and DL‐PCBs at 20 pg WHO2005‐TEQ/kg bw per day via the feed (the highest dose tested). For yellow perch and tilapia, a NOAEL of 1 μg TCDD/kg bw was identified based on growth, fin necrosis and cutaneous haemorrhages. For carp a NOAEL of 0.57 μg TCDD/kg bw was identified based on growth, organ weight and haematological parameters.
For mink, the lowest LOAEL of 4.6 ng TEQ/kg bw per day with corresponding NOAEL of 2.1 ng/kg bw per day was observed in a two‐generation study following oral exposure to TCDD, showing proliferation of the squamous gingival epithelium in the mouth of juveniles.
Comparing the estimated intakes and the reference points identified, for laying hens a large margin is observed between the estimated mean and P95 UB intakes of 2.8 and 12 pg WHO2005‐TEQ/kg bw per day and the NOAEL for reduced egg production of 5.6 ng/kg bw per day. This also applied for young chicks, with similar exposure and a higher NOAEL.
For farmed fish, when the mean and P95 UB exposure of salmonids of, respectively, 13 and 27 pg WHO2005‐TEQ/kg bw per day was compared with the dose of 20 pg WHO2005‐TEQ/kg bw per day reported not to cause any effects in salmon, it appears that the P95 exposure exceeds this level. However, no higher doses were tested and when compared to NOAELs and LOAELs reported for other fish species, including trout (NOAEL of 11 ng TCDD/kg bw), the margin is much larger.
For carp, comparison of the estimated mean and P95 UB intake of, respectively, 5 and 14 pg WHO2005‐TEQ/kg bw per day with the reported NOAEL of 0.57 μg TCDD/kg bw does not imply a risk.
For mink, comparison of the estimated mean and P95 UB exposure of, respectively, 3.1 and 7.7 pg WHO2005‐TEQ/kg bw per day with the NOAEL of 2.1 ng TCDD/kg bw per day does not imply a risk.
The CONTAM Panel concluded that information on levels causing effects in farm and companion animals is limited but that the estimated exposure of various species, based on current levels, does not imply a risk. Exposure from contaminated soil was not included in the calculations.
The CONTAM Panel considered that the impact of the uncertainties on the risk assessment of PCDD/Fs and DL‐PCBs for farm and companion animals is high and that the assessment is incomplete due to lack of data.
Recommendations
In order to improve the risk assessment for both humans and animal and reduce the uncertainties, the CONTAM Panel recommends that:
The current WHO2005‐TEFs should be re‐evaluated in order to take into account new in vivo and in vitro data. In particular, more insight into the relative potency of PCB‐126 in humans is required.
There is a specific need to derive systemic TEFs for PCDD/Fs and DL‐PCBs for use in epidemiological studies, also taking into account the results from human cells.
There should be an evaluation of the relative exposure contribution of other persistent chemicals, acting as agonists on the AHR, taking into account their toxic potencies.
To evaluate the applicability of the TEQ principle, more research and understanding is needed on reported congener‐specific effects of PCDD/Fs and DL‐PCBs, including their relevance at low doses.
Further improvement of toxicokinetic models is needed, including parameters dealing with pregnancy, breastfeeding and occasional exposure to high levels. Inclusion of PCDD/Fs, other than TCDD, and DL‐PCBs is required. The use of in vitro models for further refinement should be considered.
Data from both experimental animal and epidemiological studies should be reported in a way that allows a better dose–response evaluation in order to improve the risk assessment. There is a need to develop a consensus methodology for data sharing between individual researchers and public health authorities.
There is a need for prospective developmental epidemiological studies on PCDD/Fs and DL‐PCBs at low to moderate doses on, in particular, male reproductive outcomes and effects on the thyroid system. Follow‐up studies on existing and previous cohorts with good information on pre‐ and postnatal exposure should be considered.
Validated and cost‐effective methods are needed to assess exposure in small amounts/volumes of biological samples of animals and humans.
Studies on adverse effects at low doses in farm and companion animals are needed.
To better understand the adverse effects of PCDD/Fs and DL‐PCBs, more insight is needed into the mode of action, especially in relation to observed critical effects.
Mechanistic studies on transgenerational (third‐generation) effects are needed.
To improve human exposure estimation, more occurrence data are needed on food of plant origin, especially where individual results of certain foods indicate potential higher contamination.
More data are needed on feed, provided by a greater number of European countries.
There is a need for an updated benefit‐risk assessment of fish consumption that takes exposure to PCDD/Fs and DL‐PCBs into account.
It should be considered whether specific TEFs for farm and companion animal species should be updated or derived.
1. Introduction
1.1. Background and Terms of Reference as provided by the European Commission
1.1.1.
Background
The Scientific Committee for Food (SCF) adopted on 30 May 2001 an opinion on dioxins and dioxin‐like PCBs in food,1 fixing a tolerable weekly intake (TWI) of 14 picogrammes (pg) TEQs/kg body weight (bw) for dioxins and dioxin‐like PCBs.
The Joint Expert Committee on Food Additives (JECFA) of the WHO and from the UN Food and Agriculture Organisation (FAO) established in June 2001 a provisional tolerable monthly intake (PTMI) at 70 pg WHO‐TEQ/kg bw for dioxins and dioxin‐like PCBs.2
Converted to a tolerable daily intake, the SCF health based guidance value of 2 pg WHO‐TEQ/kg bw is in line with the JECFA value of 2.3 pg WHO‐TEQ/kg bw.
The European Food Safety Authority (EFSA) used in 2008 the TWI established by the SCF to estimate the risk for public health due to the presence of dioxins in pork from Ireland.3
In February 2012, the US Environment Protection Agency (EPA) confirmed the oral reference dose (RfD) of 0.7 pg/kg bw per day for 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). In addition, the U.S. Agency for Toxic Substances and Disease Registry/Center for Disease Control and Prevention (ATSDR) have established a chronic‐duration oral Minimal Risk Level (MRL) of 1.0 pg/kg bw per day for TCDD.
It is appropriate that EFSA provides an explanation for the differences in health based guidance values established by different organisations as regards dioxins and dioxin‐like PCBs.
Based on the outcome of the explanation of the differences in these risk assessments, a comprehensive risk assessment on the risk for animal and public health related to the presence of dioxins and dioxin‐like PCBs in feed and food may be needed. If this is the case, the more recent occurrence data of dioxins and dioxin‐like PCBs in feed and food need to be taken into account.
Terms of Reference
In accordance with Art. 31 (1) of Regulation (EC) 178/2002, the Commission asks EFSA for scientific and technical assistance to assess and explain the differences in health based guidance value established by different organisation as regards dioxins and dioxin‐like PCBs.
Based on the outcome of this scientific statement a comprehensive risk assessment might be needed. If this is the case, the Commission asks EFSA, in accordance with Art. 29 (1) of Regulation (EC) No 178/2002, for a scientific opinion on the risks for animal and human health related to the presence of dioxins and dioxin‐like PCBs in feed and food, taking into account the recent occurrence data on the presence of dioxins and dioxin‐like PCBs in feed and food.
The scientific opinion should, inter alia, comprise the:
evaluation of the toxicity of dioxins and dioxin‐like PCBs for animals and humans, considering all relevant adverse acute and chronic health effects;
estimation of the dietary exposure (chronic and, if relevant, acute dietary exposure) of the EU population to dioxins and dioxin‐like PCBs including the consumption patterns of specific (vulnerable) groups of the population (e.g. high consumers, children, people following a specific diet, etc);
estimation of the exposure of the different animal species to dioxins and dioxin‐like PCBs from feed and the levels of transfer/carry‐over of dioxins and dioxin‐like PCBs from the feed to the products of animal origin for human consumption;
assessment of the chronic (and if relevant acute) human health risks for the EU population including for specific (vulnerable) groups of the population as the consequence of the estimated dietary exposure;
assessment of the animal health risks for the different animal species as the consequence of the estimated exposure from animal feed.
1.2. Interpretation of the Terms of Reference
The terms of reference as received by the European Commission asked first for scientific and technical assistance to assess and explain the difference in health‐based guidance values (HBGVs) for polychlorinated dibenzo‐p‐dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs) and DL‐PCBs established by different organisations. This request was tackled in the form of a scientific statement published on the EFSA website on 29 May 2015 (EFSA, 2015). Therefore, the present scientific opinion addresses the request for a scientific opinion on the risks for animal and human health related to the presence of PCDD/Fs and DL‐PCBs in feed and food, taking into account the recent occurrence data on the presence of these contaminants in feed and food.
The toxicity equivalency factors proposed by the World Health Organization in 2005 (WHO2005‐TEFs) (van den Berg et al., 2006) are used in this assessment unless otherwise stated.
Although the term ‘dioxin’ is commonly used to refer to both PCDDs and PCDFs, for the sake of clarity in this opinion the term PCDD/Fs will be used consistently to refer to this group of compounds.
The risk assessment focuses on the seventeen 2,3,7,8‐substituted PCDD/F congeners. In the text, the substitution pattern will not be provided unless necessary for the correct identification of the target compounds (e.g. in the case of the hexaCDD/F congeners).
1.3. Supporting information for the assessment
1.3.1. Sources, characteristics and environmental fate
The sources, characteristics and environmental fate of PCDD/Fs and DL‐PCBs have been extensively reviewed in several earlier scientific EFSA opinions (EFSA CONTAM Panel, 2011, 2012), and EFSA reports (EFSA, 2010a, 2012). The following chapter is an excerpt of these reviews.
Polychlorinated dibenzo‐p‐dioxins (PCDDs) and dibenzofurans (PCDFs)
Polychlorinated dibenzo‐p‐dioxins (PCDDs) and dibenzofurans (PCDFs) are two groups of tricyclic planar compounds (Figure 1) that together are often referred to as ‘dioxins’. Dependent on the number of chlorine atoms and their positions at the rings 75 PCDDs and 135 PCDFs, termed ‘congeners’, can occur.
Figure 1.
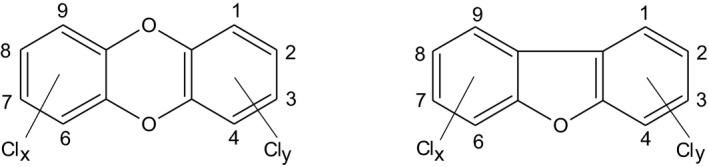
Structure of PCDDs and PCDFs. Clx + Cly = 1–8
PCDD/Fs have never been produced on an industrial scale and have no technological use. They are formed unintentionally in a number of industrial and thermal processes like the burning of certain waste if the process is not controlled appropriately (Olie et al., 1977). Also the production of various chlorinated chemicals, such as the herbicide 2,4,5‐trichlorophenoxy acetic acid (part of Agent Orange), PCBs, and chlorophenols can be sources. Well‐known examples of the latter are trichlorophenols, the source of TCDD in the Seveso incident, and pentachlorophenol (PCP), widely used not only for wood preservation but also as a fungicide (e.g. in the incident with guar gum in 2007; Wahl et al., 2008)). Chlorophenols used to treat cow hides caused one of the first incidents in the food chain because of the use of fat scraped from the hides for production of chicken feed (Higginbotham et al., 1968). Chlorophenols were also involved in the 2010 incident in Germany with industrial grade fatty acids that were used for feed production (Hoogenboom et al., 2015a).
Burning processes include not only large‐scale incineration, such as municipal waste incinerators (Liem et al., 1991), but also local burning of waste at farms and metal recycling. While in the 1980s emissions from solid waste incinerators were one of the major sources of PCDD/Fs in Europe, improved burning techniques in combination with strict regulatory measures resulted in reduction in emissions of more than 90%. As a result, the contribution of PCDD/F emissions from solid waste incinerators is minor (Quaß et al., 2004; BMUB, 2005; Vehlow et al., 2006). Open‐field incineration of waste caused an incident with contaminated mozzarella cheese in the South of Italy between 2001 and 2004 (Diletti et al., 2008).
Other industrial activities caused the contamination of the surrounding area, such as pulp and paper production (UNEP, 2007), treatment of wood with PCP (Harnly et al., 2000), burning of PCB‐waste (Lovett et al., 1998; Turrio‐Baldassarri et al., 2009), metal processing (European Commission, 2001) and chloralkali plants (Svensson et al., 1993; Hansson et al., 1997). Certain accidental fires may also lead to contamination of surrounding farms (Hoogenboom et al., 2012). The drying of bread crumbs used as a feed led to two incidents, one in Germany in 2003 (Hoogenboom et al., 2004a) and one in Ireland in 2008 (Heres et al., 2010; Tlustos et al., 2012). In the first case, painted wood was used and, in the latter case, oil containing PCBs. In 2011, the drying of beet pulp with coal containing plastic parts from a shattered roof caused an incident in Germany. A specific contamination case arose from the use of lime derived from a PVC production plant which was mixed with citrus peels for decreasing the moisture content and increasing the pH. Feeding of these Brazilian citrus pulp pellets to dairy cows led to an incident with contaminated milk in 1998 (Malisch, 2000; Malisch and Kotz, 2014).
PCDD/Fs are also found in certain types of clay materials, due to natural formation under high pressure and temperature (Holmstrand et al., 2006; Horii et al., 2008). Some of these contaminated clays like kaolinite clay (Jobst and Aldag, 2000) and Mississippi ball clay (Hayward et al., 1999) caused incidents because of their use in animal feed. Other clays, called Mabele and Pimba, are consumed by some groups of women during pregnancy to help alleviate the symptoms of morning sickness (Reeuwijk et al., 2013). The use of kaolinite to help remove poor quality potatoes, led to an incident with milk from dairy cows fed potato peel (Hoogenboom et al., 2010). PCDD/Fs have also been detected in other minerals used in animal feed, like zinc oxide (Kim et al., 2011) and so‐called sequestered minerals (Ferrario et al., 2003). In some incidents, the real source of the PCDD/Fs was never established, like an incident with gelatine fat derived from a process where contaminated hydrochloric acid was applied (Hoogenboom et al., 2007). In most cases, PCDD/Fs are not generated as single congeners but as more or less complex mixtures which are often characteristic of the source (Hoogenboom et al., 2015a).
Once released into the environment, PCDD/Fs adhere to soil and sediment particles. Although it was shown that lower chlorinated dioxins can be degraded by aerobic bacteria from the genera of Sphingomonas, Pseudomonas and Burkholderia, and higher chlorinated PCDD/Fs are known to be reductively dechlorinated in anaerobic sediments (Field and Sierra‐Alvarez, 2008), PCDD/Fs are only poorly degradable in the environment. To some extent, both PCDD/Fs in air and soil can be degraded naturally by photodegradation through exposure to ultraviolet radiation. PCDD/Fs in soil are generally bound tightly to particles; hence they are more difficult to destroy by sunlight as compared to PCDD/Fs in the air. As UV radiation cannot penetrate into soil, the photodegradation process of PCDD/Fs in contaminated soil almost exclusively occurs in the top few millimetres of the soil (Binh et al., 2014). PCDD/Fs are highly resistant to acids and bases, possess a low vapour pressure and are thermally stable below 600°C. PCDD/Fs are poorly soluble in water but highly soluble in lipids.
The toxicity of the various congeners depends on the degree of chlorine substitution. Of special importance are those congeners that are substituted in the 2‐, 3‐, 7‐ and 8‐positions and have at least one vicinal hydrogen atom. In general, these are toxic at relatively low doses, and have long biological half‐lives. Due to their lipophilic properties and poor degradation, they accumulate in the food chain. The best known and most intensively studied congener is 2,3,7,8‐TCDD (TCDD).
Because of the numerous sources, PCDD/Fs are ubiquitous. However, due to a number of regulatory measures put in place from the 1980s the emission of PCDD/Fs into the environment in Europe has considerably decreased (Quaß et al., 2004). Investigations into the different pathways of exposure have shown that dietary intake represents the main route of PCDD/F exposure for humans, generally contributing more than 80% of total PCDD/F exposure. Because of the lipophilic properties and the high accumulation potential, products of animal origin are of special importance. These food samples, generally show characteristic PCDD/F profiles in which the toxic 2,3,7,8‐chlorine substituted congeners predominate. In contrast, foodstuffs of plant origin generally contain only low PCDD/F concentrations mostly around the limit of detection (LOD). Because of the reduced emissions and the declining levels in the environment, the PCDD/F concentrations in feed and food have decreased (see Section 3.2.4). Comprehensive monitoring programmes conducted worldwide over the past three decades showed that the human exposure to PCDD/Fs has decreased substantially over time. However, these programmes also detected a number of major contamination incidents (see above) resulting in withdrawal and destruction of thousands of tonnes of food and feed.
Dioxin‐like polychlorinated biphenyls (DL‐PCBs)
The sources, physicochemical characteristics and environmental fate of DL‐PCBs cannot be considered independently from non‐dioxin‐like PCBs (NDL‐PCBs); therefore, the following section gives a general introduction to PCBs.
PCBs are a group of organochlorine compounds that are synthesised by catalysed chlorination of biphenyl. Depending on the number of chlorine atoms (1–10) and their position on the two rings, 209 different compounds, also termed ‘congeners’, are possible. Figure 2 shows the structure of PCBs and the numbering of the carbon atoms in the two rings.
Figure 2.
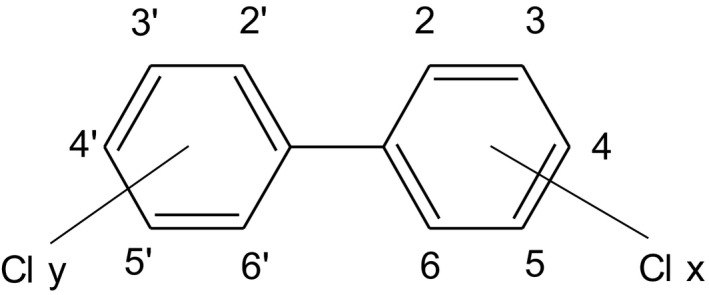
Structure of PCBs. Cly + Clx = 1–10
In contrast to PCDD/Fs, PCBs had widespread use in numerous industrial applications, generally in the form of complex technical mixtures. They were produced with an estimated total world production of 1.2–1.5 million tonnes (Fiedler, 2001; Holoubek, 2001; WHO, 2003) between 1929 and the end of the 1970s, when their production was abandoned in the majority of countries due to their high persistence in the environment and biota. As a result of their physicochemical properties, such as non‐flammability, chemical stability, high boiling point, low heat conductivity and high dielectric constants, PCBs were widely used in a number of industrial and commercial applications (closed and open). The technical PCB mixtures were mobile oils, viscous liquids or sticky resins depending on the degree of chlorination (between 21% and 68% chlorine) (Hutzinger et al., 1974). Various commercial mixtures were produced with different trade names, like Aroclor, Kanechlor, Declor and Clophen. PCBs were not only used in transformers but also as heat exchange fluids in equipment to heat up oils and fats. Due to leakage of PCB oil into the rice oil production, this application caused the Yusho (Kuratsune et al., 1972) and Yucheng (Hsu et al., 1985) rice oil incidents. In addition, PCBs were used in certain paints and sealants, and may as such still be present in buildings. The recycling of building materials and use of granulates in courtyards of chicken farms has been shown to cause incidents with free‐ranging laying hens (Hoogenboom et al., 2014). Major incidents in Europe with PCBs in the food chain were the Belgian incident in 1999 and the Irish incident in 2008 (see above). In the first incident, around 160 kg of PCB oil ended up in 60 tonnes of fat used for production of animal feed (Bernard et al., 1999; Traag et al., 2006). The Irish incident occurred as a result of PCB contaminated fuel being used in an oil‐fired burner (direct flame drying system) that generated the heat to dry dough from industrial bakery facilities and unsold and sell‐by date bread which were intended as feed for pigs (FSAI, 2009).
In fires and other thermal events, PCBs can be converted to PCDFs and other products (Erickson, 1989). As PCBs are often mixed with polychlorobenzenes (for instance, in mixtures for dielectric fluids), their thermal degradation may also be associated with a relevant production of PCDDs (De Felip et al., 1994).
As a result of their widespread use, leakages, improper disposal practices and persistence, PCBs (like PCDD/Fs) also have a global distribution in the environment. Depending on the number and position of the chlorines in the molecule, some of them are poorly degraded and, due to their lipophilic properties, they are bioaccumulated in the food chain. Like PCDD/Fs, PCBs belong to the initial list of 12 persistent organic pollutants (POPs) that are regulated under the Stockholm Convention on POPs. The main pathway of human exposure for the majority of the population is via food consumption with the exception of specific cases of accidental or occupational exposure.
According to Directive 96/59/EC,4 Member States should have taken the necessary measures to ensure that used PCBs are disposed of and equipment containing PCBs are decontaminated or disposed of at the latest by the end of 2010. However, this does not include the more disperse sources like paints and kits used in buildings. It is estimated that approximately 20% of the total amount of PCBs ever produced has been eliminated to date, which means that about 80% remains to be destroyed (UNEP, 2015).
PCBs are divided into two groups based on structural characteristics and toxicological effects. DL‐PCBs consist of 12 congeners that are non‐ortho or mono‐ortho chlorine substituted. Congeners in this group contain at least four chlorine substituents, can easily adopt a coplanar structure and show toxicological properties similar to TCDD. The other 197 PCB congeners are called ‘non‐dioxin‐like PCBs’ (NDL‐PCBs), and include also non‐ or mono‐ortho PCBs that contain less than four chlorines. The chemical structure and toxicity profile of NDL‐PCB congeners differ from that of TCDD (EFSA, 2005; WHO, 2016). As regards the determination of NDL‐PCBs, it was proposed to focus the analysis mainly on the six PCB congeners 28, 52, 101, 138, 153 and 180 (Beck and Mathar, 1985). These six congeners were considered as indicators of the different PCB patterns in various sample types, not based on their toxicology. Hence, they are often termed ‘indicator‐PCBs’ or ‘marker‐PCBs’. In the past, this approach was adopted by a number of countries. Other investigations, especially in connection with the Belgium PCB/dioxin case in 1999 also include PCB‐118 (which is actually a DL‐PCB), as a seventh congener into the group of ‘indicator PCBs’. The sum of the six ‘indicator PCBs’ represents about 50% of total NDL‐PCBs in food (EFSA, 2005). Meanwhile, the six NDL‐PCBs are regulated in the EU by Commission Regulation (EC) 1881/2006 for food and Directive 2002/32/EC for feed.
The toxic equivalency (TEQ) principle
In order to compare the toxicity of a mixture of congeners, the concept of TEQ based on different toxic equivalency factors (TEFs) was introduced. The concept assumes that the relevant PCDD/Fs and DL‐PCBs bind to the intracellular aryl hydrocarbon receptor (AHR) and cause the same type of AHR‐mediated biochemical and adverse effects. Another important requirement of the TEQ concept is the persistence and accumulation of the compounds in the body. Moreover, it is assumed that the effects are purely additive. By definition, TCDD, as the most toxic congener, was assigned a value of 1, and the TEFs for the other 16 toxic PCDD/Fs with 2,3,7,8‐chlorine substitution and 12 DL‐PCBs are between 0.00003 and 1 (see Table 1). Thus, a TEF indicates an order of magnitude estimate of the potency of a dioxin‐like compound relative to TCDD. TEF values have been (re‐)evaluated several times taking into account the multiple endpoints with priority on in vivo responses (e.g. immunosuppression, hepatotoxicity and fetotoxicity) known to be affected by PCDD/Fs and DL‐PCBs.
Table 1.
Toxic equivalency factors (TEFs) established by WHO in 2005 (van den Berg et al., 2006)
| Congener | WHO2005‐TEFs |
|---|---|
| PCDDs | |
| 2,3,7,8‐TCDD | 1 |
| 1,2,3,7,8‐PeCDD | 1 |
| 1,2,3,4,7,8‐HxCDD | 0.1 |
| 1,2,3,6,7,8‐HxCDD | 0.1 |
| 1,2,3,7,8,9‐HxCDD | 0.1 |
| 1,2,3,4,6,7,8‐HpCDD | 0.01 |
| 1,2,3,4,6,7,8,9‐OCDD | 0.0003 |
| PCDFs | |
| 2,3,7,8‐TCDF | 0.1 |
| 1,2,3,7,8‐PeCDF | 0.03 |
| 2,3,4,7,8‐PeCDF | 0.3 |
| 1,2,3,4,7,8‐HxCDF | 0.1 |
| 1,2,3,6,7,8‐HxCDF | 0.1 |
| 2,3,4,6,7,8‐HxCDF | 0.1 |
| 1,2,3,7,8,9‐HxCDF | 0.1 |
| 1,2,3,4,6,7,8‐HpCDF | 0.01 |
| 1,2,3,4,7,8,9‐HpCDF | 0.01 |
| 1,2,3,4,6,7,8,9‐OCDF | 0.0003 |
| Non‐ ortho PCBs | |
| PCB‐77 | 0.0001 |
| PCB‐81 | 0.0003 |
| PCB‐126 | 0.1 |
| PCB‐169 | 0.03 |
| Mono‐ ortho PCBs | |
| PCB‐105 | 0.00003 |
| PCB‐114 | 0.00003 |
| PCB‐118 | 0.00003 |
| PCB‐123 | 0.00003 |
| PCB‐156 | 0.00003 |
| PCB‐157 | 0.00003 |
| PCB‐167 | 0.00003 |
| PCB‐189 | 0.00003 |
To calculate the total TEQ value of a sample, the concentration of each congener is multiplied by its TEF and the products are then added together. The resulting TEQ value expresses the toxicity of PCDD/Fs and DL‐PCBs in a complex sample in terms of TCDD. The current TEF values were proposed by the WHO in 2005 and are termed WHO2005‐TEFs (van den Berg et al., 2006) based on the year of the WHO expert meeting (Table 1). Older analytical data, especially if generated before 2005 are generally reported as WHO1998‐TEQs (van den Berg et al., 1998), I‐TEQs (NATO/CCMS, 1988) or Nordic‐TEQs (Ahlborg, 1988). When interpreting TEQ results and evaluating, e.g. trends in the levels or exposure, it is important to know which TEFs were used. EFSA (2010a) estimated a decrease by 14% following the switch from WHO1998‐TEFs to WHO2005‐TEFs, which is within the interval of 10–25%, estimated by van den Berg et al. (2006). The difference very much depends on the congener pattern, due to large reductions in, e.g. the TEFs for the pentachlorinated PCDFs (40% lower) and mono‐ortho PCBs (threefold lower for the most important congeners PCB‐105 and PCB‐118) during the last change in 2005, and the doubling of the TEF for PeCDD in 1998.
Moreover, in contrast to WHO1998‐TEQ and WHO2005‐TEQ, the I‐TEQs and the initial Nordic‐TEQs did not cover DL‐PCBs, but only PCDD/Fs. Based on a consultation of the European Centre for Environment and Health (WHO‐ECEH) and the International Programme on Chemical Safety (IPCS), TEFs were recommended for DL‐PCBs in 1994 (Ahlborg et al., 1994). The DL‐PCB occurrence data in the exposure assessments of the SCF opinions published in 2000 and 2001 (see Section 1.3.3) were calculated with these TEFs. Compared to the 1994 TEFs, PCB‐170 and ‐180 were withdrawn from the original scheme due to the lack of evidence that they are in vivo AHR agonists, and a TEF was introduced for PCB‐81 which showed similar AHR binding and in vitro CYP1A induction as PCB‐77 and was detected in wildlife and human samples (Van den Berg et al., 1998).
In 1997/1998, WHO did not only re‐evaluate the mammalian TEFs (based on intake through administered dose) but also established separate TEFs for fish and birds. Fish was treated as separate taxa due to the absent or very low response to mono‐ortho PCBs compared to mammals and birds. Moreover, the TEFs for fish were mainly based on tissue concentrations. TEFs for birds could only be derived from egg injection studies, studies with cultured avian hepatocytes, and studies with cultured thymus cells. WHO concluded that the mammalian TEFs are applicable for the human situation as well as for wild mammalian species, and the established TEFs for fish and birds could be used in ecotoxicological risk assessments of these vertebrate classes (Van den Berg et al., 1998). As the WHO1998‐TEFs for fish and birds were not re‐evaluated by international bodies after 1998, and the primary focus in the present risk assessment is on dietary exposure, the CONTAM Panel decided to apply the WHO2005‐TEFs also for fish and birds.
In the European legislation, all regulatory levels for food and feed are presently expressed as TEQs using the WHO2005‐TEFs.
It should be emphasised that the TEF scheme in Table 1 and TEQ methodology are primarily meant for estimating exposure via dietary intake situations, because the TEFs are based largely on oral uptake studies often through the diet. In contrast, ‘application of these ‘intake or ingestion’ ‘TEFs for calculating the TEQ in abiotic environmental matrices has limited toxicological relevance and use for risk assessment, unless the aspect of reduced bioavailability and environmental fate and transport of the various dioxin‐like compounds are taken into account. If human risk assessment is done for abiotic matrices, it is recommended that congener‐specific equations be used throughout the whole model, instead of using a total TEQ basis, because fate and transport properties differ widely between congeners’. (van den Berg et al., 2006).
1.3.2. Sampling and methods of analysis
Sampling and analysis
Detailed requirements for methods of sampling and analysis for the control of levels of PCDD/Fs and DL‐PCBs in certain foodstuffs are laid down in Commission Regulation (EU) No 2017/6445. The provisions apply for official control bodies as well as for food business operators. For feed, respective requirements are laid down in Commission Regulation (EU) No 2017/7716. These Regulations contain, inter alia, a number of provisions concerning methods of sampling depending on the size of the lot, packaging, transport, storage, sealing, labelling, interpretation of analytical results and requirements for assessing the compliance of a lot or sublot with the legislation.
A critical step in PCDD/F and PCB analysis is the determination of lipids7 as the resulting amount is highly dependent on the methodology applied. This is especially true for the analysis of eggs and liver as well as biological samples, such as human milk and blood (see Section 3.2.4). Thus, the analytical reports should include information about the method used for the lipid determination. In order to comply with European legislation, for feed and food of plant origin and of animal origin containing less than 10% fat, the addition of the internal standards is mandatory prior to extraction. For feed and food of animal origin containing more than 10% fat, the internal standards may be added either before or after fat extraction. Furthermore, there is the obligation that an appropriate validation of the extraction efficiency shall be carried out, depending on the stage at which internal standards are introduced and on whether results are reported on product or fat basis.
Regarding analytical methods for the determination of PCDD/Fs and DL‐PCBs in food and feed, the EU generally follows the ‘criteria approach’. This means that no fixed methods are prescribed but detailed and strict performance criteria are established by the European Commission which must be fulfilled. If it can be demonstrated in a traceable manner that these performance criteria are fulfilled and the method is fit for purpose, the analysts can apply their method of choice. The respective performance criteria are laid down in Commission Regulation (EU) No 2017/644 for food and Commission Regulation (EU) No 2017/771 for feed. According to these Regulations, monitoring for the presence of PCDD/Fs and DL‐PCBs in food and feed may be performed by a screening method of analysis (e.g. bioassays and gas chromatography/mass spectrometry (GC/MS) methods) with widely acceptable validation and high throughput to identify the samples exceeding action levels (ALs) and ensuring the selection of samples exceeding maximum levels (MLs). Screening methods compare the analytical result with a cut‐off value, providing a yes/no‐decision over possible exceedance of the maximum or action level. In addition, these methods may give a first indication of the levels of PCDD/Fs and DL‐PCBs present in the sample.
The most commonly used bioassay is the Chemical Activated LUciferase gene eXpression assay (CALUX). It makes use of genetically modified rat and mouse hepatoma cells, in which ‘dioxin responsive elements’ (DREs) (see Section 3.1.4) are coupled to a DNA‐fragment encoding for luciferase (Sanderson et al., 1996). Following exposure of these cells, the increased luciferase levels can be measured by a light reaction. Combining this assay with a sample clean‐up over an acid silica column makes this test specific for stable AHR agonists, like PCDD/Fs and DL‐PCBs. The sensitivity is sufficient to control compliance of samples with the current EU limits with relatively small sample intakes. Another bioassay that is sporadically applied is the ethoxyresorufin‐O‐deethylase (EROD) assay (Kennedy et al., 1993). It measures the increased level of CYP 1 enzymes (caused by exposure of mammalian cell‐lines to PCDD/Fs and DL‐PCBs) by adding specific substrates to the cells, like ethoxyresorufin which is deethylated to resorufin (EROD activity). In practice, there is a good correlation between the response in the bioassays and the levels determined by gas chromatography/high‐resolution mass spectrometry (GC‐HRMS) or gas chromatography/tandem mass spectrometry (GC–MS/MS). However, other AHR agonists that survive the clean‐up may give a response in the test, e.g. brominated dioxins. There are some differences between the relative response of PCDD/Fs and DL‐PCBs in the assays and the WHO‐TEFs. Therefore, for food and feed control, bioassay levels above the cut‐off level must be re‐analysed by a confirmatory method.
In case of application of bioanalytical screening methods, the result is expressed as bioanalytical equivalents (BEQ), whereas in case of application of GC–MS‐based methods it is expressed as TEQ. The concentration of PCDD/Fs and the sum of PCDD/Fs and DL‐PCBs in food and feed samples suspected to be non‐compliant with the legal levels must be determined by a confirmatory method. Confirmatory methods which allow the unequivocal identification and quantification of PCDD/Fs and DL‐PCBs present in a sample and provide full information on the congener pattern are methods applying GC–HRMS and GC–MS/MS. The criteria for confirmatory methods are laid down in Commission Regulation (EU) No 2017/644 for food and Commission Regulation (EU) No 2017/771 for feed. These concern requirements inter alia for trueness and precision, addition of isotope‐labelled standards, gas chromatographic separation of congeners, maximum tolerances for retention times and isotope ratios based on US‐EPA method 1613 B, reporting of results, etc.
In both Commission Regulations for feed and food amended in 2017, the approach applying the decision limit ccα to ensure that an analytical result is above the ML with a certain probability, as provided for in Commission Decision 2002/657/EC, was deleted while only the approach of the expanded measurement uncertainty using the coverage factor of 2, giving a level confidence of approximately 95% was kept.
Analytical quality assurance
In order to contribute to a high quality and uniformity of analytical results, an analytical network of a European Reference Laboratory (EU‐RL), National Reference Laboratories (NRL) and Official Laboratories was designated for PCDD/Fs and PCBs. The activities of reference laboratories cover all areas of feed and food law, in particular those areas where there is a need for reliable analytical results. For example, the EU‐RL for PCDD/Fs and PCBs regularly organises proficiency tests with different matrices for NRLs and Official Laboratories. Respective proficiency tests are also offered by a number of other organisations. Often more than 100 laboratories from all over the world participate in the studies on the determination of PCDD/Fs and PCBs in non‐spiked or spiked food specimens. The results indicate that most of the participating laboratories, although applying different GC–HRMS and GC–MS/MS methods, are capable of reliably analysing PCDD/Fs and PCBs at the level of interest.
1.3.3. Previous risk assessments
Previous human risk assessments and evaluations
The WHO held a consultation in May 1998 on the assessment of the health risk of dioxins and re‐evaluation of the existing tolerable daily intake (TDI) (WHO, 1998). At that stage, the TDI was 10 pg TEQ/kg bw per day, established during a WHO meeting in 1990. WHO considered a number of animal studies with TCDD published since then as the basis for the new TDI, showing a range of effects like neurobehavioural toxicity, immunotoxicity, reproductive toxicity and endometriosis, some in rats, some in monkeys. The accumulated amount of TCDD in the animal rather than the administered dose was taken as the starting point for the extrapolation of toxicity across species. Therefore, the animal body burdens8 corresponding to these effects were estimated, however, without providing any further details on the calculations. These body burdens ranged from 28 to 73 ng/kg bw (Table 2). These levels served as a reference point for the calculation of the chronic daily human intake which would be expected to lead to similar body burdens in humans. The estimated daily intakes (EDIs) were calculated with the aid of a one‐compartmental kinetic model, based on the equation:
Table 2.
Critical adverse effects observed in animal studies used by WHO (1998) to derive a TDI. Body burdens of TCDD were estimated from the applied oral doses
| Study | Endpoint | Exposure (LOAEL) | Body burdena (ng/kg bw) | Related human EDIe (pg/kg bw per day) |
|---|---|---|---|---|
| Schantz and Bowman (1989) | Rhesus monkey, neurobehavioural toxicity in offspring (decreased learning) | ~ 160 pg/kg bw per day | 42b , d | 21 |
| Gray et al. (1997a), Mably et al. (1992a) | Rat, decreased sperm count in offspring | 64 ng/kg bwc | 28d | 14 |
| Gray et al. (1997b) | Rat increased genital malformations in offspring | 200 ng/kg bwc | 73 | 37 |
| Gehrs et al. (1997), Gehrs and Smialowicz (1999) | Rat immune suppression in offspring | 100 ng/kg bwc | 50 | 25 |
| Rier et al. (1993) | Rhesus monkey, endometriosis | ~ 160 pg/kg bw per day | 69b | 35 |
bw: body weight; EDI: estimated daily intake; LOAEL: lowest‐observed‐adverse‐effect level; TCDD: 2,3,7,8‐ tetrachlorodibenzo‐p‐dioxin; TDI: tolerable daily intake.
Increment to background, reported to be 4 ng/kg (TEQ) for rats and mice.
Body burden at time of delivery.
Single oral dose.
Maternal body burden.
Estimated chronic Daily Intake.
Taking the animal body burdens shown in Table 2, an elimination half‐life (t1/2) in humans of 7.5 years and an assumed fraction absorbed from food (f) to be 0.5, an EDI range of 14–37 pg TCDD/kg bw per day was calculated. Applying a composite uncertainty factor (UF) of 10, the EDI range then resulted in a TDI of 1–4 pg TEQ/kg bw per day (rounded figures).
Regarding the dietary exposure, WHO indicated a daily intake of PCDD/Fs in the order of 1–3 pg I‐TEQ/kg bw per day for a 60‐kg adult based on the available information derived from food surveys from industrialised countries. This value would be greater by a factor of 2–3 if DL‐PCBs were to be considered. WHO recognised that ‘certain subtle effects may be occurring in some sections of the general populations of industrialized countries at current intake levels (2–6 I‐TEQ pg/kg bw per day for PCDD/Fs) and body burdens (4–12 I‐TEQ ng/kg bw), but found it tolerable on a provisional basis as these reported subtle effects were not considered overtly adverse and there were questions as to the contribution of non‐dioxin‐like compounds to the observed effects’.
In 2000, the SCF published its opinion on the risk assessment of dioxins and DL‐PCBs in food (SCF, 2000), which was then updated in 2001 (SCF, 2001). The SCF took as a basis the WHO (1998) assessment but also considered studies published since then. Human studies were also evaluated but, as in the previous WHO evaluations, were not used for deriving an HBGV. Starting with the same critical animal toxicity studies as selected by WHO (Table 2), SCF explicitly recalculated animal lowest‐observed‐adverse‐effect level (LOAEL) body burdens in the rat and the monkey, with corresponding estimated human daily intakes (EHDIs) ranging from 12.5 to 37 pg TCDD/kg bw. Applying an UF of 3 for no‐observed‐adverse‐effect level (NOAEL) to LOAEL extrapolation and a factor of 3.2 for interindividual variation in toxicokinetics within the human population resulted in a TDI ranging from 1 to 3 pg/kg bw per day (TEQ, rounded figures). It was concluded that the lower end of the range, i.e. 1 pg/kg bw per day, was to be considered as a temporary TDI (t‐TDI). Recognising that the compounds like TCDD and related compounds have very long half‐lives in the human body, the SCF found it more appropriate to express the t‐TDI on a weekly rather than a daily basis, thereby arriving at a t‐TWI of 7 pg/kg bw per week (TEQ).
However, new studies revealed a particular sensitivity of the gestation day 15 (GD15) rat embryo to TCDD (Faqi et al., 1998; Ohsako et al., 2001). Furthermore, studies by Hurst et al. (2000a,b) revealed a relatively high exposure of the fetus after a single acute maternal GD15 dose when compared to a chronic maternal exposure. This was corrected by applying a factor of 2.6 on a single GD15 dose when estimating the corresponding maternal animal body burden (Table 3). For example, using 60% absorption of the 64 ng/kg LOAEL of the Mably et al. study resulted in a corresponding maternal body burden of 64 × 0.6 = 38.4 ng/kg bw following treatment with a single high dose. However, a 2.6‐fold higher maternal body burden, i.e. 100 ng/kg bw, is required to obtain the same exposure of the embryo after chronic exposure. In a similar way, body burdens were calculated for the other studies.
Table 3.
Estimated chronic steady‐state body burdens of TCDD and corresponding estimated human daily intakes (EHDI) at NOAEL and LOAELs in the pivotal studies (SCF, 2001)
| Study | Endpoint | NOAEL | LOAEL | Estimated maternal steady‐state body burdena (ng/kg bw) | Associated EHDId (pg/kg bw per day) |
|---|---|---|---|---|---|
| Mably et al. (1992a) | Holzman rats: decreased sperm count in male offspring | – | 64 ng/kg bw single bolus dose by gavage | 100b | 50 |
| Gray et al. (1997a) | Long‐Evans rats: accelerated eye opening and decreased sperm count in male offspring | – | 50 ng/kg bw single bolus dose by gavage | 80b | 40 |
| Faqi et al. (1998) | Wistar rats: decreased sperm production and altered sexual behaviour in male offspring | – | Maintenance of 25 ng/kg bw by s.c. injections | 40b | 20 |
| Ohsako et al. (2001) | Holzman rats: decreased anogenital distance in male offspring | 12.5 ng/kg bw single bolus dose by gavage | – | 20c | 10 |
| – | 50 ng/kg bw single bolus dose by gavage | 80c | 40 |
bw: body weight; GD: gestation day; LOAEL: lowest‐observed‐adverse‐effect level; NOAEL: no‐observed‐adverse‐effect level; TCDD: 2,3,7,8‐ tetrachlorodibenzo‐p‐dioxin; s.c.: subcutaneous.
Increment over background. Estimated background body burden from feed in rats is approximately 4 ng TEQ/kg bw (WHO, 2000). The maternal body burdens were increased by a factor of 2.6 for the difference in the ratio between levels in the fetus and mother after a single vs repeated dose.
Composite value resulting from pseudo steady‐state body burden and acute body burden on GD15.
Maternal body burden at GD16.
Estimated chronic Human Daily Intake.
Based on these body burdens and using the same assumptions as WHO (1998) on absorption from food and human half‐life, the SCF calculated an EHDI of 10 pg/kg bw per day for the body burden (NOAEL) estimated for the Ohsako et al. study. Applying an UF of 3.2 for interindividual differences in toxicokinetics, then resulted in a TDI of 3 pg/kg bw per day. For the Faqi et al. study, applying an UF of 3 × 3.2 on the EHDI derived from the estimated body burden (LOAEL) resulted in a TDI of 2 pg/kg bw per day. As the latter study was performed with the most sensitive rat strain (Wistar), 2 pg/kg bw per day (TEQ) was considered as the TDI. Again, recognising that TCDD and related compounds have very long half‐lives in the human body, the SCF found it more appropriate to express the TDI on a weekly rather than a daily basis, thereby arriving at a tolerable weekly intake (TWI) of 14 pg/kg bw per week (TEQ). The designation ‘temporary’ was removed from the TWI because the new studies provided a firm basis for the evaluation of the pivotal rat toxicity studies.
The SCF based its exposure assessment on the occurrence data compiled by the Scientific Cooperation (EU SCOOP) Task 3.2.5., which included national food contamination data and dietary exposure assessments from ten European countries. The average dietary intake estimate of PCDD/Fs for adults of various European countries ranged from 0.4 to 1.5 pg I‐TEQ/kg bw per day, and of DL‐PCBs ranged from 0.8 to 1.5 pg PCB‐TEQ/kg bw per day. The PCB‐TEQs in this exposure assessment were calculated with the PCB‐TEFs published by Ahlborg et al. (1994). As a result, the total dietary intake of PCDD/Fs and DL‐PCBs was estimated to range from 1.2 to 3.0 pg WHO‐TEQ/kg bw per day. The SCF concluded that a considerable proportion of the European population would exceed the group TWI of 14 pg WHO‐TEQ/kg bw, noting that exceeding it slightly does not necessarily mean that there is an appreciable risk to the health of individuals, but exposure above this tolerable weekly intake leads to an erosion of the protection embedded in the group TWI.
In 2001, JECFA evaluated the risk associated with the presence of dioxins and coplanar PCBs in food (FAO/WHO, 2002). JECFA took into account the previous WHO (1998) evaluation and new studies published afterwards. The Committee chose the same critical studies as the SCF (2000, 2001), i.e. the study by Faqi et al. (1998) and the study by Ohsako et al. (2001) but used both studies to derive an HBGV. However, it decided to use a factor of 1.7 instead of 2.6 to correct for the single dose instead of chronic treatment in these studies resulting in lower reference points. It subsequently applied the same ‘safety’ factors of 3 and 3.2 as the SCF (2000, 2001). JECFA, however, considered that the tolerable intake should be assessed over a period of at least 1 month, and therefore, established provisional tolerable monthly intakes (PTMIs) of 40 and 100 pg WHO1998‐TEQs/kg bw, from which the midpoint of 70 pg WHO1998‐TEQs/kg bw was chosen as the PTMI. JECFA estimated the dietary intake of PCDD/Fs using the GEMS/Food regional diets, and national food consumption data. Estimates using the later consumption data were considered more reliable. For PCDD/Fs, the median estimates ranged from 33 to 42 pg WHO1998‐TEQ/kg bw per month, while for DL‐PCBs it ranged from 9 to 47 pg WHO1998‐TEQ/kg bw per month.
Several other international bodies have performed risk assessments related to the presence of PCDD/Fs and DL‐PCBs in food. The Nordic Council set a TDI of 5 pg TEQ/kg bw (Nordic Ministry Council Report, 1988; Ahlborg et al., 1995; Johansson and Hanberg, 2000). The JEA (1999) established a TDI of 4 pg/kg bw, and the UK Committee on Toxicity (COT, 2001) recommended a TDI of 2 pg/kg bw per day, these values being within the range established by the WHO (1998) and SCF (2001).
In 2012, the US‐EPA published its reanalysis of key issues related to dioxin toxicity, focusing on the non‐cancer endpoints (US‐EPA, 2012). It established an oral reference dose (RfD) of 0.7 pg TCDD/kg bw per day, defining the oral RfD as ‘an estimate (with uncertainty spanning perhaps an order of magnitude) of a daily oral exposure to the human population (including sensitive subgroups) that is likely to be without an appreciable risk of deleterious effects during a lifetime’. The US‐EPA evaluated human and experimental animal studies, and based the risk assessment on data from two studies on the adverse effects in the cohort exposed as a result of the Seveso (Italy) incident in 1976. It was shown that men exposed in childhood showed a reduced sperm count and motility (Mocarelli et al., 2008). TCDD levels were measured in blood taken from the boys within one year after the incident. The study indicated a LOAEL of 68 pg/g fat which was used as one of the points of departure (POD). The other POD was based on elevated levels of thyroid‐stimulating hormone (TSH) observed in 3‐day‐old neonates born to mothers from Seveso exposed during the incident (Baccarelli et al., 2008). In this case, TCDD concentrations measured in maternal blood were used to establish a LOAEL of 235 pg/g fat for the effect on TSH in the neonates. The maternal blood samples were taken 16.5 to 22 years after the incident, and TCDD levels higher than 10 pg/g fat were extrapolated to the time of conception. For TSH levels in blood of neonates, a benchmark of 5 μU/mL established by WHO for medical follow‐up for potential congenital hypothyroidism was used as a cut‐off. Using a physiologically based pharmacokinetic (PBPK) model for humans, the daily TCDD exposures leading to these critical blood concentrations in either boys or mothers were derived. In the case of the boys, it was argued by US‐EPA that it was unclear whether the effects on sperm were due to the peak in the blood just after the incident, or the average blood levels during the years before the boys reached the age of 10 (on average 3.5 years). Therefore, both the initial peak level of TCDD and the level some years after the incident were estimated and the average taken as the POD. For both the effects on sperm and TSH, the PBPK model showed that the levels corresponding to the PODs would be obtained with a continuous daily intake of 20 pg/kg bw per day. An UF of 30 (10 to derive a NOAEL and 3 for intraspecies differences) was used to derive the above‐mentioned oral RfD of 0.7 pg/kg bw per day. TCDD is considered as the index chemical for other PCDD/Fs and DL‐PCBs, using the TEFs to derive the TEQ level. US‐EPA did not estimate the exposure to PCDD/Fs and DL‐PCBs.
In 1994, the ATSDR derived for 2,3,4,7,8‐PeCDF an acute duration oral Minimal Risk Level of 0.001 μg/kg bw per day (based on a LOAEL for thymic effects in guinea pigs (Moore et al., 1979)), and an intermediate duration oral Minimal Risk Level of 0.00003 μg/kg bw per day (Pluess et al., 1988; Poiger et al., 1989a,b)), indicating that applying these minimal risk levels to PCDFs other than 2,3,4,7,8‐PeCDF may lead to overestimating actual risks since 2,3,4,7,8‐PeCDF is more toxic than some other CDF congeners (ATSDR, 1994). In 1998, the ATSDR published a toxicological profile for chlorinated dibenzo‐p‐dioxins (ATSDR, 1998) in which the following HBGVs for TCDD (and extensive to other dioxin‐like compounds expressed as TEQs) were derived: an acute duration oral Minimal Risk Level of 200 pg/kg bw per day (based on its ability to suppress serum total haemolytic complement activity in B6C3F1 mice (White et al., 1986)), an intermediate duration oral Minimal Risk Level of 20 pg/kg bw per day (based on observed decreases in thymus weight in guinea pigs (Decaprio et al., 1986)), and a chronic duration oral Minimal Risk Level of 1 pg/kg bw per day (based on altered social interactions with peers in monkeys exposed to TCDD prenatally and during lactation (Schantz et al., 1992)). The chronic duration oral Minimal Risk Level of 1.0 pg/kg bw per day was confirmed in a later update of the toxicological profile (ATSDR, 2012). In 2000, it published a toxicological profile for PCBs, reporting also on studies on DL‐PCBs, and derived intermediate‐ and chronic duration Minimal Risk Levels for PCBs based on studies in which experimental animals were exposed to PCB congener mixtures.
EFSA has not previously carried out a comprehensive risk assessment on PCDD/Fs and DL‐PCBs in food. In 2008, EFSA used the SCF TWI to estimate the risk for public health related to the presence of dioxins in pork from Ireland (EFSA CONTAM Panel, 2008), and in 2011, in relation to the risk to public health related to the presence of high levels of the target compounds in liver from sheep and deer (EFSA CONTAM Panel, 2011). In 2015, EFSA examined the approaches taken by different organisations for establishing HBGVs in their risk assessment on dioxins and DL‐PCBs (EFSA, 2015). EFSA considered the differing approaches by the SCF (2000, 2001), JECFA (FAO/WHO, 2002) and the US‐EPA (2012), and how these impacted on the final derivation of a numerical value. It was noted that while the SCF and JECFA based the derivation of the HBGV on experimental animal studies, the US‐EPA selected human data. The SCF and JECFA applied a body burden one‐compartment kinetics approach to derive an HBGV from rat data, whereas US‐EPA applied PBPK modelling of blood levels estimated from epidemiology studies. An UF of 3 was applied by the SCF and JECFA as the LOAEL was considered to be close to the NOAEL (observed in another animal study), while US‐EPA applied an UF of 10 for extrapolation from a LOAEL in the absence of a NOAEL. Overall, this resulted in the reference dose set by US‐EPA (0.7 pg/kg bw per day) being threefold lower than the SCF TWI (14 WHO‐TEQ/kg bw, or 2 pg/kg bw per day converted to a tolerable daily basis) or the JECFA PTMI (70 pg/kg bw, or 2.3 pg/kg bw per day converted to a tolerable daily basis).
In 2010 and 2012, EFSA published two scientific reports with the results of the monitoring of PCDD/Fs and PCBs, including DL‐PCBs, in food and feed (EFSA, 2010a, 2012). In these reports, the levels recorded by European Countries were reported and the chronic dietary exposure assessed. Further details on these reports are provided in Section 3.2.2.
Farm and companion animals previous risk assessments
The European Commission's former Scientific Committee on Animal Nutrition (SCAN, 2000) evaluated the contribution of PCDD/Fs and PCBs in feedingstuffs to the contamination of food of animal origin, in order to complement the SCF evaluation of risks related to human dietary exposure to these compounds. The main conclusion of the SCAN (2000) was that fish oil and fishmeal were the most heavily contaminated feed materials. Animal fat (other than fish) had the next highest PCDD/F and DL‐PCB concentrations, depending on the bioaccumulation of PCDD/Fs in fatty tissues along the feed/food chain. The levels vary widely depending on the origin of the foodstuff. Farmed fish, meat, eggs, milk and other dairy products may be contaminated by PCDD/Fs from feedingstuffs. Therefore feedingstuffs, and in some cases soil, are of particular concern as potential sources of PCDD/Fs. The SCAN stressed that direct accidental pollution of feed materials may occur, such as from local discharge of PCDD/Fs from industrial activities, and from the contamination of feed materials, during production, processing and transportation. The remit of the SCAN did not include a risk assessment of PCDD/Fs and/or DL‐PCBs in feed to target animal safety.
EFSA has not previously carried out a comprehensive risk assessment on PCDD/Fs and DL‐PCBs in feed.
1.3.4. Legislation
In this opinion, where reference is made to European legislation (Regulations, Directives, Recommendations, Decisions), the reference should be understood as relating to the most recent amendment, unless otherwise stated.
Food
In order to protect public health, Article 2 of Council Regulation (EEC) No 315/939 of 8 February 1993 laying down Community procedures for contaminants in food stipulates that, where necessary, maximum tolerances for specific contaminants shall be established. Subsequently, a number of MLs for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs for various foodstuffs mainly of animal origin are laid down in the Annex, Section 5 of Commission Regulation (EC) No 1881/200610 of 19 December 2006 setting MLs for certain contaminants in foodstuffs. The Regulation also contains MLs for the sum of six NDL‐PCBs. As these are beyond the terms of reference of this Opinion, they are not considered further in this context. The MLs for PCDD/Fs and PCBs are not toxicologically based but were derived from the frequency distribution of the respective food classes following the principle ‘strict but feasible’. In general, the MLs were set around the 90th percentile of the respective frequency distribution.
The MLs for PCDD/Fs and the sum of PCDD/Fs and DL‐PCBs in food are both expressed as TEQ using the WHO‐toxic equivalency factors (WHO2005‐TEFs) for human risk assessment based on the conclusions of the WHO expert meeting in 2005 (van den Berg et al., 2006) and are given as pg WHO2005‐TEQ/g.
All MLs for PCDD/Fs and PCBs are set as UB concentrations. These are calculated on the assumption that all values of the different congeners below the limit of quantification (LOQ) are equal to the numerical value of the LOQ. Except for certain fish and fish products, liver of fish and terrestrial animals, and foods for infants and young children all other MLs are given on a fat basis.
The currently legally valid MLs are shown in Appendix A (Table A.1). The respective foodstuffs have to comply with the MLs for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs. According to Article 3 of Regulation (EC) No 1881/2006, foodstuffs not complying with the MLs shall not be used as food ingredients and foodstuffs complying with the MLs shall not be mixed with foodstuffs which exceed the MLs.
Table A.1.
Maximum levels for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs in food
| Foodstuff | Maximum levels | ||
|---|---|---|---|
| Sum of PCDD/Fs (WHO‐PCDD/F‐TEQ)a | Sum of PCDD/Fs and DL‐PCBs (WHO‐PCDD/F‐PCB‐TEQ)a | ||
| 5.1 | Meat and meat products (excluding edible offal) of the following animalsb: | ||
|
2.5 pg/g fatc | 4.0 pg/g fatc | |
|
1.75 pg/g fatc | 3.0 pg/g fatc | |
|
1.0 pg/g fatc | 1.25 pg/g fatc | |
| 5.2 | Liver of terrestrial animals referred to in 5.1 with the exception of sheep and derived products thereof | 0.30 pg/g wet weight | 0.50 pg/g wet weight |
| Liver of sheep and derived products thereof | 1.25 pg/g wet weight | 2.00 pg/g wet weight | |
| 5.3 | Muscle meat of fish and fishery products and products thereofd , e, with the exemption ofh: | 3.5 pg/g wet weight | 6.5 pg/g wet weight |
|
|||
|
|||
|
|||
|
|||
| The maximum level for crustaceans applies to muscle meat from appendages and abdomenf. In case of crabs and crab‐like crustaceans (Brachyura and Anomura), it applies to muscle meat from appendages | |||
| 5.4 | Muscle meat of wild caught fresh water fish, with the exception of diadromous fish species caught in fresh water, and products thereofd | 3.5 pg/g wet weight | 6.5 pg/g wet weight |
| 5.4a. | Muscle meat of wild caught spiny dogfish (Squalus acanthias) and products thereof | 3.5 pg/g wet weight | 6.5 pg/g wet weight |
| 5.5 | Muscle meat of wild caught eel (Anguilla anguilla) and products thereof | 3.5 pg/g wet weight | 10.0 pg/g wet weight |
| 5.6 | Fish liver and derived products thereof with the exception of marine oils referred to in point 5.7 | – | 20.0 pg/g wet weight |
| 5.7 | Marine oils (fish body oil, fish liver oil and oils of other marine organisms intended for human consumption) | 1.75 pg/g fatc | 6.0 pg/g fatc |
| 5.8 | Raw milkb and dairy productsb, including butter fat | 2.5 pg/g fatc | 5.5 pg/g fatc |
| 5.9 | Hen eggs and egg productsb | 2.5 pg/g fatc | 5.0 pg/g fatc |
| 5.10 | Fat of the following animals: | ||
|
2.5 pg/g fat | 4.0 pg/g fat | |
|
1.75 pg/g fat | 3.0 pg/g fat | |
|
1.0 pg/g fat | 1.25 pg/g fat | |
| 5.11 | Mixed animal fats | 1.5 pg/g fat | 2.50 pg/g fat |
| 5.12 | Vegetable oils and fats | 0.75 pg/g fat | 1.25 pg/g fat |
| 5.13 | Foods for infants and young childreng | 0.1 pg/g wet weight | 0.2 pg/g wet weight |
PCDD/F: polychlorinated dibenzo‐p‐dioxin and dibenzofuran; DL‐PCB: dioxin‐like polychlorinated biphenyls; WHO: World Health Organization; TEQ: toxic equivalents.
Upper bound concentrations: Upper bound concentrations are calculated on the assumption that all the values of the different congeners below the limit of quantification are equal to the limit of quantification.
Foodstuffs listed in this category as defined in Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 laying down specific hygiene rules for food of animal origin (OJ L 226, 25.6.2004, p. 22).
The maximum level expressed on fat is not applicable for foods containing < 2% fat. For foods containing less than 2% fat, the maximum level applicable is the level on product basis corresponding to the level on product basis for the food containing 2% fat, calculated from the maximum level established on fat basis, making use of following formula: Maximum level expressed on product basis for foods containing less than 2% fat = maximum level expressed on fat for that food × 0.02.
Where fish are intended to be eaten whole, the maximum level shall apply to the whole fish.
Foodstuffs listed in this category as defined in categories (a), (b), (c), (e) and (f) of the list in Article 1 of Regulation (EC) No 104/2000, with the exclusion of fish liver referred to in point 5.11.
This definition excludes the cephalothorax of crustaceans.
The maximum level refers to the products ready to use (marketed as such or after reconstitution as instructed by the manufacturer).
The Maximum Levels for sum of PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs for the categories 5.3, 5.4 and 5.4a are the same. Differences apply to the Maximum Levels of NDL‐PCBs which are not listed in this table as they are beyond the terms of reference.
Article 7 of the Regulation stipulates that by way of derogation, Finland, Sweden and Latvia may authorise the placing on their market of wild caught salmon and products thereof originating in the Baltic region and intended for consumption in their territory with levels of PCDD/Fs and/or DL‐PCB higher than those set out in the Regulation. In addition, by way of derogation, Finland and Sweden may also authorise the placing on their market of wild caught herring larger than 17 cm, wild caught char, wild caught river lamprey and wild caught trout and products thereof originating in the Baltic region and intended for consumption in their territory with levels of PCDD/Fs and/or DL‐PCBs exceeding the respective MLs. These derogations apply only provided that a system is in place to ensure that consumers are fully informed of the dietary recommendations with regard to the restrictions on the consumption of the respective fish species by identified vulnerable sections of the population in order to avoid potential health risks. As it is known that certain fish and fishery products from the Baltic region regularly exceed the respective MLs, Commission Recommendation (EU) 2016/68811 lays down provisions for the monitoring and management of the presence of PCDD/Fs and PCBs in fish and fishery products from the Baltic region. For specific Member States around the Baltic, the Recommendation sets minimum numbers of samples of several fish species to be taken and analysed in the years 2016–2018. It also includes inter alia information on how to measure the size of the fish, provisions on different geographical region (ICES zones) where the fish is caught and risk management measures recommended to be taken by the competent authorities to ensure that fish from the Baltic region placed on the market in the EU complies with the MLs established in Regulation (EC) No 1881/2006. Before 2016, Finland and Sweden had a derogation since 2002 and were obliged to set up specific monitoring programme on the presence of PCDD/Fs and DL‐PCBs in fish and to report yearly to the European Commission the results thereof.
The MLs for PCDD/Fs and the sum of PCDD/Fs and DL‐PCBs expressed on a fat weight basis are not applicable for foods containing < 2% fat. These have to be calculated on a product basis, assuming a fat content of 2%. For details, see footnote (c) in Appendix A (Table A.1).
In addition to MLs, the European Commission has set ALs for both PCDD/Fs and DL‐PCBs in various foods as an early warning tool, the most recent amendment by Commission Recommendation 2014/663/EU12. ALs are meant as a tool for competent authorities and operators to highlight cases where it is appropriate to identify a source of contamination and to take measures for its reduction or elimination. Due to the fact that their sources are generally different, separate ALs for PCDD/Fs and DL‐PCBs were established. The currently effective ALs are given in Appendix A (Table A.2).
Table A.2.
Action levels (ALs) for PCDD/Fs and DL‐PCBs in different foods
| Food | Action level for PCDD/Fs (WHO‐TEQ)a | Action level for DL‐PCBs (WHO‐TEQ)a |
|---|---|---|
| Meat and meat products (excluding edible offal)b of the following animals | ||
|
1.75 pg/g fatc | 1.75 pg/g fatc |
|
1.25 pg/g fatc | 0.75 pg/g fatc |
|
0.75 pg/g fatc | 0.50 pg/g fatc |
| Mixed fats | 1.00 pg/g fatc | 0.75 pg/g fatc |
| Muscle meat of farmed fish and farmed fishery products | 1.50 pg/g wet weight | 2.50 pg/g wet weight |
| Raw milkb and dairy productsb, including butter fat | 1.75 pg/g fatc | 2.00 pg/g fatc |
| Hen eggs and egg productsb | 1.75 pg/g fatc | 1.75 pg/g fatc |
| Clays as food supplement | 0.50 pg/g wet weight | 0.50 pg/g wet weight |
| Cereals and oilseeds | 0.50 pg/g wet weight | 0.35 pg/g wet weight |
| Fruits, vegetables (including fresh herbs)d | 0.30 pg/g wet weight | 0.10 pg/g wet weight |
PCDD/F: polychlorinated dibenzo‐p‐dioxin and dibenzofuran; DL‐PCB: dioxin‐like polychlorinated biphenyls; WHO: World Health Organization; TEQ: toxic equivalents.
Upper bound concentrations: Upper bound concentrations are calculated assuming that all the values of the different congeners less than the limit of quantification are equal to the limit of quantification.
Foodstuffs listed in this category as defined in Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 laying down specific hygiene rules for food of animal origin (OJ L 139, 30.4.2004, p. 55).
The action levels are not applicable for food products containing < 2% fat.
For dried fruits and dried vegetables (including dried herbs), Article 2 of Regulation (EC) No 1881/2006 is applicable. For dried herbs, a concentration factor as the consequence of drying of 7 has been taken into account.
In cases where levels of PCDD/Fs and/or DL‐PCBs in excess of the ALs are determined, the food can still be marketed. However, it is recommended that Member States, in co‐operation with operators, initiate investigations to identify the source of contamination, and take measures to reduce or eliminate the source of contamination.
Feed
The maximum content (also termed MLs) for PCDD/Fs and the sum of PCDD/Fs and DL‐PCBs are laid down in Annex I, Section V of Directive 2002/32/EC on undesirable substances in animal feed.13 These legal limits were also based on existing levels according to the principle ‘strict but feasible’. As for food, all legal limits in feed are UB values expressed as TEQ relative to a feed with a moisture content of 12% using the WHO‐toxic equivalency factors (WHO2005‐TEFs). The currently effective maximum contents for PCDD/Fs and the sum of PCDD/Fs and DL‐PCBs in different animal feeds are illustrated in Appendix A (Table A.3).
Table A.3.
Maximum levels for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs in feed
| Feed | Maximum content in ng WHO‐PCDD/F‐TEQ/kg (ppt)a , b relative to a feed with a moisture content of 12% | Maximum content in ng WHO‐PCDD/F‐DL‐PCBs‐TEQ/kg (ppt)a , b relative to a feed with a moisture content of 12% |
|---|---|---|
| Feed materials of plant origin with the exception of: | 0.75 | 1.25 |
|
0.75 | 1.5 |
| Feed materials of mineral origin | 0.75 | 1.0 |
| Feed materials of animal origin: | ||
|
1.50 | 2.0 |
|
0.75 | 1.25 |
|
5.0 | 20.0 |
|
1.25 | 4.0 |
|
1.75 | 9.0 |
| The feed additives kaolinitic clay, vermiculite, natrolite‐phonolite, synthetic calcium aluminates and clinoptilolite of sedimentary origin belonging to the functional groups of binders and anticaking agents | 0.75 | 1.5 |
| Feed additives belonging to the functional group of compounds of trace elements | 1.0 | 1.5 |
| Premixtures | 1.0 | 1.5 |
| Compound feed with the exception of: | 0.75 | 1.5 |
|
1.75 | 5.5 |
|
− | − |
PCDD/F: polychlorinated dibenzo‐p‐dioxin and dibenzofuran; DL‐PCB: dioxin‐like polychlorinated biphenyls; WHO: World Health Organization; TEQ: toxic equivalents.
Upper bound concentrations: upper bound concentrations are calculated on the assumption that all values of the different congeners below the limit of quantification are equal to the limit of quantification.
Table of TEF (= toxic equivalency factors) for dioxins, furans and DL‐PCBs: WHO‐TEFs for human risk assessment based on the conclusions of the World Health Organisation (WHO) – International Programme on Chemical Safety (IPCS) expert meeting which was held in Geneva in June 2005 (van den Berg et al., 2006).
Fresh fish and other aquatic animals directly delivered and used without intermediate processing for the production of feed for fur animals are not subject to the maximum levels, while maximum levels of 3.5 ng WHO‐PCDD/F‐TEQ/kg product and 6.5 ng WHO‐PCDD/F‐PCB‐TEQ/kg product are applicable to fresh fish and 20.0 ng WHO‐PCDD/F‐PCB‐TEQ/kg product is applicable to fish liver used for the direct feeding of pet animals, zoo and circus animals or used as feed material for the production of pet food. The products or processed animal proteins produced from these animals (fur animals, pet animals, zoo and circus animals) cannot enter the food chain and cannot be fed to farmed animals which are kept, fattened or bred for the production of food.
For crustacean meal only the Maximum content for the sum of PCDD/Fs applies.
Article 5 of Directive 2002/32/EC prescribes that products intended for animal feed containing levels of an undesirable substance, such as PCDD/Fs and DL‐PCBs that exceed the respective maximum content may not be mixed for dilution purposes with the same, or other, products intended for animal feed.
In addition to maximum content, also action thresholds (as early warning tools to trigger investigations by Member States) are set separately for PCDD/Fs and DL‐PCBs, respectively, in Annex II of Directive 2002/32/EC. The action thresholds also refer to products relative to a moisture content of 12%. The current action thresholds for PCDD/Fs and DL‐PCBs are given in Appendix A (Table A.4).
Table A.4.
Action thresholds for PCDD/Fs and DL‐PCBs in different products intended for animal feed
| Products intended for animal feed | Action threshold in ng WHO‐PCDD/F TEQ/kga , b (ppt) relative to a feedingstuff with a moisture content of 12% | Action threshold in ng WHO‐DL‐PCB‐TEQ/kga , b (ppt) relative to a feed with a moisture content of 12% |
|---|---|---|
| Feed materials of plant origin with the exception of: | 0.5c | 0.35c |
|
0.5c | 0.5c |
| Feed materials of mineral origin | 0.5c | 0.35c |
| Feed materials of animal origin: | ||
|
0.75c | 0.75c |
|
0.5c | 0.35c |
|
4.0d | 11.0d |
|
0.75d | 2.0d |
|
1.25d | 5.0d |
| Feed additives belonging to the functional groups of binders and anticaking agents | 0.5c | 0.5c |
| Feed additives belonging to the functional group of compounds of trace elements | 0.5c | 0.35c |
| Premixtures | 0.5c | 0.35c |
| Compound feed with the exception of: | 0.5c | 0.5c |
|
1.25d | 2.5d |
|
− | − |
PCDD/F: polychlorinated dibenzo‐p‐dioxin and dibenzofuran; DL‐PCB: dioxin‐like polychlorinated biphenyls; WHO: World Health Organization; TEQ: toxic equivalents.
Table of TEF (= toxic equivalency factors) for dioxins, furans and dioxin‐like PCBs: WHO‐TEFs for human risk assessment based on the conclusions of the World Health Organisation (WHO) – International Programme on Chemical Safety (IPCS) expert meeting which was held in Geneva in June 2005 (van den Berg et al., 2006).
Upper bound concentrations; upper bound concentrations are calculated on the assumption that all values of the different congeners below the limit of quantification are equal to the limit of quantification.
Identification of source of contamination. Once source is identified, take appropriate measures, where possible, to reduce or eliminate source of contamination.
In many cases it might not be necessary to perform an investigation into the source of contamination as the background level in some areas is close to or above the action level. However, in cases where the action level is exceeded, all information, such as sampling period, geographical origin, fish species etc., shall be recorded with a view to future measures
2. Data and methodologies
The current risk assessment was developed applying a structured methodological approach, which implies developing a priori the strategy of the full risk assessment; performing each step of the risk assessment in line with the strategy, and thoroughly documenting the process.
The strategy in Annex A.1 to this opinion contains the method that was proposed for all steps of the risk assessment process, including any subsequent refinements/changes made.
2.1. Supporting information for the assessment
The steps followed to acquire data for the supporting information is documented in the strategy included in Annex A.1 of this Scientific Opinion.
Information on chemistry, sources, environmental fate, analytical methods, current EU legislation and previous risk assessments by international bodies, was gathered from previous EFSA opinions, previous assessment by international bodies (e.g. SCF, JECFA, ATSDR, US‐EPA) by checking their original websites, from review papers, peer‐reviewed publications and legal text. The information was summarised in a narrative way based on expert knowledge and judgement.
Information on previously reported occurrence data in food and feed, and previous exposure assessments was identified from previous assessments by EFSA and other international bodies (e.g. SCF, JECFA) and by a literature search to gather review studies, and from there, primary research studies (see Annex A.2). The data was summarised in a narrative way based on expert knowledge and judgement.
2.2. Hazard identification and characterisation
The steps followed for the acquisition of data for the hazard identification and characterisation, their selection and appraisal are documented in Annex A.1 of this Scientific Opinion.
2.2.1. Human risk assessment
2.2.1.1. Evidence identification and selection
Studies on the adverse effects of polychlorinated dibenzo‐p‐dioxins and dibenzofurans (PCDD/Fs) and dioxin‐like polychlorinated biphenyls (DL‐PCBs) in humans (subquestions 1 and 8, Structured approach) were identified by performing an Extensive Literature Search (ELS) in two different databases (Web of Science and PubMed14), followed by a selection for relevance according to eligibility criteria and data extraction as outlined in Annex A.1. This task was outsourced to an external contractor (contract: NP/EFSA/BIOCONTAM/2016/06). The search strings and outcome of the selection for relevance steps are shown in the external scientific report of RPA and IEH (2018). The outcome of this exercise (date of search: 5 July 2016) was complemented with further studies found by snowballing (i.e. checking the bibliography of the key full‐text papers for further potential relevant studies) and an update of the literature search performed following the same search strings and criteria on 21 November 2017.
Similarly, studies on the adverse effects of PCDD/Fs and DL‐PCBs in experimental animals (subquestion 2, narrative approach and subquestions 3 and 9, structured approach) were identified by performing an ELS in two different databases (Web of Science and PubMed), followed by a selection for relevance according to the eligibility criteria and data extraction as set in Annex A.1. This task was outsourced to an external contractor (contract: NP/EFSA/BIOCONTAM/2016/07). The search strings and outcome of the selection for relevance steps are shown in the external scientific report of wca environment Ltd (2018). The outcome of this exercise (date of the search: 7 July 2016) was complemented with further studies found by snowballing and an update of the literature search performed following the same search strings and criteria on 31 December 2017.
To inform the section on genotoxicity (subquestion 4, narrative approach), a literature search was carried out in Scopus and Web of Science (March 2016) to identify review papers and peer‐reviewed primary research studies published in the open literature relevant to the genotoxicity of PCDD/Fs and DL‐PCBs. The literature search was carried out using search terms such as micronucleus, mutation, mutagenicity, single strand breaks, DNA damage, oxidative DNA damage, DNA repair, chromosomal breaks/deletions/aberrations, unscheduled DNA synthesis, clastogenic, polyploidy and genotoxicity. Additional studies were identified since then and throughout the development of the draft opinion. The outcome of the search was evaluated by relevant experts from the Working Group that described narratively the relevant aspects for the assessment based on expert knowledge and judgement.
Studies on the toxicokinetic in humans, experimental animals (subquestions 5 and 6, structured approach with narrative description) were identified by carrying out a literature search and consulting previous risk assessments. The studies were screened and evaluated by relevant experts from the Working Group that summarised the data in a narrative way based on expert knowledge and judgement.
The toxicokinetic models evaluated in this assessment were the rat and human models previously published by Emond and colleagues (Emond et al., 2006, 2017) and the so‐called concentration‐ and age‐dependant model (CADM), developed by Carrier et al. (1995), further optimised by Aylward et al. (2005) and adapted by Ruiz et al. (2014) to include a growth curve and a breastfeeding period. The Emond model was coded in Berkeley‐Madonna software and this task was outsourced by EFSA (contract NP/EFSA/BIOCONTAM/2016/08) (RIVM and RIKILT Wageningen University & Research, 2018) . Also the CADM was coded in Berkeley‐Madonna software (see Section 3.1.8.2 and Appendix E).
The benchmark dose (BMD) analysis was carried out according to EFSA guidance (EFSA Scientific Committee, 2017a) and using the EFSA BMD modelling application ( https://efsa.openanalytics.eu/app/bmd) (see Section 3.1.7.4 and Appendix C).
To inform the section on mode of action (subquestion 10, narrative approach), a literature search was carried out in Web of Science (August 2015) to identify review papers and peer‐reviewed primary research studies published in the open literature relevant to inform the modes of action of PCDD/Fs and DL‐PCBs. The literature search was carried out using search terms such as differentiation, proliferation, cell cycle, apoptosis, endocrine, hormone, epigenetic, oxidative stress, Ah receptor or aryl hydrocarbon receptor, phylogenesis, signal transduction, (non‐)genomic pathway, alternative pathway, cross‐talk, and other, focusing on publications in English. Dedicated searches were carried out to indentify studies to inform the different subsections in the mode of action chapter. Additional studies were identified since then and throughout the development of the draft opinion. The outcome of the search was evaluated by relevant experts from the Working Group that described narratively the relevant aspects for the assessment based on expert knowledge and judgement.
2.2.1.2. Assessment of the reliability of the studies
Studies on the adverse effects of PCDD/Fs and DL‐PCBs in humans and in experimental animals eligible for the assessment were appraised for reliability. The reliability of the individual studies was appraised by considering the risk of bias, defined as ‘the extent to which the design and conduct of a study are likely to have prevented bias’, i.e. non‐random error (Higgins and Green, 2011).
The elements that were considered for appraising the reliability of each individual study are illustrated in the critical appraisal tools reported in Annex A.1. These tools were developed by tailoring the OHAT Risk of Bias Tool as included in the NTP‐OHAT Approach for Systematic Review (Rooney et al., 2014).
Adjustment for confounding and other modifying variables was taken into account when considering the internal validity of each study.
Expert judgement was translated into a rating scale for each question to be answered as follows:
[++]: definitely low risk of bias;
[+]: probably low risk of bias;
[−]: probably high risk of bias;
[−−]: definitively high risk of bias.
The individual rating for each question was then combined by an algorithm and translated to an overall tier of reliability for each individual study (risk of bias tier 1: low risk of bias; risk of bias tier 2: moderate risk of bias; risk of bias tier 3: high risk of bias) (see Annex A.1 for further details).
It is important to note that the assessment of risk of bias is related to, but distinguished from, the broader concept of assessment of methodological quality. Bias is a systematic error, or deviation from the truth, in results or inferences; an assessment of quality requires evaluation of the extent to which study authors conducted their research to the highest possible standards. As such, assessment of quality includes also other components than bias. Examples of such components, not included in the risk of bias appraisal of the present opinion are poor statistical power due to small sample size, or measures of outcomes that are (although unbiased) not relevant for health risk assessment. These aspects were considered when evaluating the overall confidence in the body of evidence per endpoint.
2.2.2. Farm and companion animals risk assessment
Studies on the adverse effects of the target compounds in farm and companion animals (subquestions 16 and 17, structured approach) were identified by performing an ELS in two different databases (Web of Science and PubMed), followed by a selection of relevance according to agreed eligibility criteria and data extraction as set in Annex A.1 to this scientific opinion. This task was performed by EFSA staff in collaboration with members of the Working Group. The search strings and outcome of the selection for relevance steps are shown in Annex A.2. The outcome of this exercise was complemented with further studies found by snowballing during the development of the opinion.
Studies on the toxicokinetics in farm and companion animals and on the transfer of the target compounds from feed to animal‐derived food products (subquestions 18 and 19, structured approach) were identified by performing an ELS in two different databases (Web of Science and PubMed), and further selection for relevance as set in Annex A.1. This task was performed by EFSA staff in collaboration with members of the Working Group. The search strings are shown in Annex A.2. The outcome of this exercise was complemented with further studies found by snowballing during the development of the opinion.
2.3. Exposure assessment
The steps followed for the acquisition of data for the exposure assessment and subsections therein are documented in Annex A.1 of this Scientific Opinion.
2.3.1. Farm and companion animals exposure assessment
2.3.1.1. Occurrence data in feed submitted to EFSA
Data collection and validation
Following a European Commission mandate to EFSA, a call for annual collection of chemical contaminant occurrence data in food and feed, including PCDD/Fs and DL‐PCBs, was issued by the former EFSA Dietary and Chemical Monitoring Unit (now DATA Unit) in December 2010 with a closing date of 1 October of each year. European national authorities and similar bodies, research institutions, academia, food business operators and other stakeholders were invited to submit analytical data on PCDD/Fs and DL‐PCBs in feed.
The data submission to EFSA followed the requirements of the EFSA Guidance on Standard Sample Description for Food and Feed (EFSA, 2010b) and specific requirements for PCDD/Fs and PCBs15; occurrence data were managed following the EFSA standard operational procedures (SOPs) on ‘Data collection and validation’ and on ‘Data analysis of food consumption and occurrence data’.
Data on PCDD/Fs and DL‐PCBs in feed available in the EFSA database from 2010 until the end of December 2016 were used for the present assessment. Data received after this date were not included.
Data analysis
The data received were carefully evaluated by EFSA in view of cleaning and validating. Special attention was paid to the identification of duplicates and to the accuracy of different parameters, such as ‘Analytical methods’, ‘Reporting unit’ and the coding of the different samples under FoodEx classification. Upon identification of potential inconsistencies, data providers were contacted to provide further clarification. The outcome of the data analysis is shown in Section 3.2.1.
The left‐censored data (analytical data below limit of detection/limit of quantification (LOD/LOQ)) were treated by the substitution method as recommended in the ‘Principles and Methods for the Risk Assessment of Chemicals in Food’ (WHO/IPCS, 2009). The same method is described in the EFSA scientific report ‘Management of left‐censored data in dietary exposure assessment of chemical substances’ (EFSA, 2010c) as an option for the treatment of left‐censored data. The guidance suggests that the lower bound (LB) and upper bound (UB) approach should be used for chemicals likely to be present in the food (e.g. naturally occurring contaminants, nutrients and mycotoxins). At the LB, results below the LOQ or LOD were replaced by zero; at the UB, the results below the LOD were replaced by the numerical values of the LOD and those below the LOQ were replaced by the value reported as LOQ. Additionally, a middle bound (MB) approach was used by assigning a value of LOD/2 or LOQ/2 to the left‐censored data. The use of different cut‐off values on the reported LOQs was also evaluated in order to reduce the uncertainty associated to the exposure estimations
2.3.1.2. Feed consumption data
In the absence of a comprehensive database on the amounts or types of feed consumed by livestock in the EU, estimates of feed consumed for each of the main categories of farmed livestock and companion animals are based on published guidelines on nutrition (Carabano and Piquer et al., 1998; NRC, 2007a,b; Leeson and Summers, 2008; McDonald et al., 2011; EFSA FEEDAP Panel, 2012; OECD, 2013), together with expert knowledge of production systems in Europe.
For many farmed livestock and companion animals, their nutritional requirements are provided in commercially manufactured complete (compound) feeds. Where sufficient (reliable) data on the concentrations of PCDD/Fs and DL‐PCBs in compound feeds have been provided, these have been used to estimate exposure. However, where insufficient compound feed data were available, the CONTAM Panel identified example diets and feed inclusion rates, and used concentrations of PCDD/Fs and DL‐PCBs in individual feed materials to estimate P95 and mean exposure both LB and UB. Details of the intakes and composition of diets used in estimating animal exposure to PCDD/Fs and DL‐PCBs are given in Annex A.3.
2.3.1.3. Feed classification
Feeds were classified based on the catalogue of feed materials specified in Commission Regulation (EU) No 68/2013 as amended by Regulation (EU) 2017/1017 of 15 June 201716 creating the Catalogue of feed materials. Where information was available, compound feedingstuffs were classified in groups based on the species/production categories for which the feed is intended.
2.3.1.4. Farm and companion animals’ exposure assessment
The method used to perform the farm and companion exposure assessment is described in the strategy in Annex A.1 of this Scientific Opinion. The CONTAM Panel considered that only chronic dietary exposure had to be assessed.
2.3.2. Human exposure assessment
2.3.2.1. Occurrence data in food submitted to EFSA
Data collection and validation
Following a European Commission mandate to EFSA, a call for annual collection of chemical contaminant occurrence data in food and feed, including PCDD/Fs and DL‐PCBs, was issued by the former EFSA Dietary and Chemical Monitoring Unit (now DATA Unit) in December 2010 with a closing date of 1 October of each year. European national authorities and similar bodies, research institutions, academia, food business operators and other stakeholders were invited to submit analytical data on PCDD/Fs and DL‐PCBs in food.
The data submission to EFSA followed the requirements of the EFSA Guidance on Standard Sample Description (SSD) for Food and Feed (EFSA, 2010b) and specific requirements for PCDD/Fs and PCBs15; occurrence data were managed following the EFSA SOPs on ‘Data collection and validation’ and on ‘Data analysis of food consumption and occurrence data’.
Data on PCDD/Fs and DL‐PCBs in food available in the EFSA database from 2010 onwards to the end of December 2016 were used for the present assessment. Data received after this date were not included.
Data analysis
The same data analysis regarding cleaning, validating, clarifying of inconsistencies and treatment of left‐censored data was implemented as for food (see Section 2.3.1.1).
2.3.2.2. Food consumption data
The EFSA Comprehensive European Food Consumption Database (Comprehensive Database) provides a compilation of existing national information on food consumption at individual level. It was first built in 2010 (EFSA, 2011a; Huybrechts et al., 2011; Merten et al., 2011). Details on how the Comprehensive Database is used are published in the Guidance of EFSA (EFSA, 2011a). The latest version of the Comprehensive Database17 was used with subjects classified in different age classes as follows:
Infants: < 12 months old
Toddlers: ≥ 12 months to < 36 months old
Other Children: ≥ 36 months to < 10 years old
Adolescents: ≥ 10 years to < 18 years old
Adults: ≥ 18 years to < 65 years old
Elderly: ≥ 65 years to < 75 years old
Very Elderly: ≥ 75 years old.
Two additional surveys provided information on specific population groups: ‘Pregnant women’ (≥ 15 years to ≤ 45 years old; Latvia) and ‘Lactating women’ (≥ 28 years to ≤ 39 years old; Greece).
Overall, the food consumption data gathered by EFSA in the Comprehensive Database are the most complete and detailed data currently available in the EU. Consumption data were collected using single or repeated 24‐ or 48‐h dietary recalls or dietary records covering from 3 to 7 days per subject. As a result of the differences in the methods used for data collection, direct country‐to‐country comparisons can be misleading.
2.3.2.3. Food classification
Food consumption data were classified according to the FoodEx classification system (EFSA, 2011b). FoodEx is a classification system developed by EFSA in 2009 with the objective of simplifying the linkage between occurrence and food consumption data when assessing the exposure to hazardous substances. The system consists of a large number of individual food items aggregated into food groups and broader food categories in a hierarchical parent‐child relationship. It contains 20 main food groups (first level), which are further divided into subgroups having 140 items at the second level, 1,261 items at the third level and reaching about 1,800 items (food names or generic food names) at the fourth level.
2.3.2.4. Human exposure assessment
The method used to perform the human exposure assessment is described in the strategy in Annex A.1 of this Scientific Opinion.
The CONTAM Panel considered that only chronic dietary exposure had to be assessed. As suggested by the EFSA Working Group on Food Consumption and Exposure (EFSA, 2011a), dietary surveys with only one day per subject were excluded from the current assessment because they are not adequate to assess repeated exposure. Similarly, subjects who participated only 1 day in the dietary studies, when the protocol prescribed more reporting days per individual, were also excluded from the chronic exposure assessment. When, for one particular country and age class, two different dietary surveys were available only the most recent one was used.
For calculating the chronic dietary exposure, food consumption and body weight data at the individual level were accessed in the Comprehensive Database. Occurrence data and consumption data were linked at the relevant FoodEx level (see also Section 3.2.1). For each individual of the selected surveys, the mean occurrence values of the different food samples collected (pooled European occurrence data) were combined with the average daily consumption of the corresponding food items, and the resulting exposures per food were summed in order to obtain the total chronic exposure at individual level (divided by the respective individual body weight). The mean and the 95th percentile of the individual exposures were subsequently calculated for each dietary survey and each age class separately. All analyses were performed using the SAS Statistical Software (SAS enterprise guide 5.1).
It is well‐known that fish from certain areas may contain relatively high levels of PCDD/Fs and DL‐PCBs. This applies not only to eel from contaminated rivers and lakes, but also to various fatty fish species from, e.g. the Baltic Sea. When included in the occurrence data this may result in an overestimation of the exposure from such fish in areas where they are not consumed, and vice versa. Fish from, e.g. the Baltic Sea should be monitored to a higher extent and as such there may be a bias towards relatively high levels in the data submitted to EFSA. However, when compliant with the MLs (including measurement certainty), this fish can be put on the EU market and as such it seems not correct to exclude them from the database. It is known that in the EU, the large majority of the salmon and trout on the market is farmed, and it is widely consumed.18 In order to investigate the impact of excluding wild salmon and trout from the data set, exposure was calculated including and excluding wild salmon and trout (see Section 3.5.1).
For matching the occurrence and the consumption data, occurrence data from samples of solid tea and herbs for infusions, and infant formulae were converted to the corresponding beverages by applying specific factors (75 and 8, respectively) which are commonly used in other similar EFSA opinions.
To perform the exposure assessment, the occurrence data that are expressed on a fat weight basis, were combined with the fat per cent of the consumed foods as it is reported in the national consumption surveys in the Comprehensive Database. Fat contents available in the Comprehensive Database are included according to the national composition tables of Member States. Where the fat content was missing in the consumption database, the random hot‐deck imputation method was used to complete this information. This technique consists of replacing the missing value with an observed one, which is randomly drawn from values corresponding to samples sharing ‘similar’ characteristics. In the case of fat content, the ‘similar’ characteristic was defined by the kind of food or food group, according to the different levels of hierarchy of the FoodEx1 catalogue.
2.3.2.5. Effects of food processing
Information on the effects of processing concerning the levels of PCDD/Fs and DL‐PCBs in food (subquestion 12, narrative approach) was addressed narratively, by carrying out a literature search in Web of Science to identify reviews and peer‐reviewed single studies published in the open literature (see Annex A.2). These resulting hits were screened and summarised by relevant domain experts from the Working Group.
2.3.2.6. Levels in humans
For the section on levels of PCDD/Fs and DL‐PCBs in human tissues in the European population (subquestion 15, narrative approach), a literature search was carried out (see Annex A.2). The resulting hits were screened and summarised by relevant domain experts from the Working Group. In addition, data was submitted to EFSA in relation to the PCDD/F‐ and PCB‐related results of the WHO/UNEP‐coordinated exposure studies 2000–2015 performed in European countries (See Documentation submitted to EFSA).
2.4. Risk characterisation
The method used to perform the risk characterisation for humans, farm and companion animals is described in the strategy in Annex A.1 of this Scientific Opinion. The CONTAM Panel applied the general principles of the risk characterisation process for chemicals in food as described by WHO/IPCS (2009) and the relevant EFSA guidance documents (see Section A.1.1 in Annex A.1). For the animal risk characterisation, the same principles were applied.
3. Assessment
3.1. Hazard identification and characterisation
3.1.1. Toxicokinetics
Studies to inform the toxicokinetics and transfer of polychlorinated dibenzo‐p‐dioxins and dibenzofurans (PCDD/Fs) and dioxin‐like polychlorinated biphenyls (DL‐PCBs) in farm and companion animals, laboratory animals and humans were retrieved and selected as described in Section 2.
PCDD/Fs and DL‐PCBs are in general well absorbed, with the exception of the octachlorinated PCDD/F congeners. They are transported to all parts of the body via the blood where they are found in lipid particles and also bound to proteins. Most of the 2,3,7,8‐substituted PCDD/Fs and DL‐PCBs are poorly metabolised but with some species‐ and congener‐specific differences. Metabolism is through hydroxylation followed by sulfation or glucuronidation. Several of the enzymes involved in the hydroxylation by the liver, like cytochrome P450s (CYPs) 1A1, 1A2 and 1B1, are also induced via the aryl hydrocarbon receptor (AHR) pathway and as a result, the elimination is faster in higher exposed animals and humans. Due to their lipophilicity, most of the PCDD/Fs and DL‐PCBs are stored in the adipose tissue, but there is an equilibrium with blood lipids and lipids in other tissues. In the liver, however, these contaminants may show higher levels than in the adipose tissue, when expressed as lipid based. This process resembles a kind of sequestration, since it is thought to be due to specific binding to CYP1A2. The latter, however, has only been proven in mice since it was not observed in knockout mice not expressing this enzyme. Furthermore, in rats, a dose‐related increase in the fraction stored in liver is observed with increasing exposure, in parallel with induction of CYP1A2. Rats not expressing the AHR and hence lacking induction of CYPs of the 1A family, also failed to show sequestration (Harrill et al., 2016). There are clear congener‐specific differences in the accumulation in the liver, which also occurs in other species, including humans. PCDD/Fs and DL‐PCBs can cross the placenta and are transferred to milk and eggs, important routes of exposure of the offspring. For mothers, this results in the loss of these contaminants. Otherwise, elimination is primarily via faeces, partly excreted as metabolites via the bile, but partly via the loss of body fat. More species‐specific details for humans, laboratory, companion and farm animals are described below. For farm animals, most information is presented in a separate section on transfer of PCDD/Fs and DL‐PCBs to edible tissues, milk and eggs, since this is an important feature with respect to human exposure and the relation between feed and food levels (see Section 3.1.1.4).
3.1.1.1. Farm and companion animals
Aspects dealing with absorption and distribution in farm animals are covered in the section on transfer. Concerning metabolism, only one controlled study was retrieved showing the presence of 4‐hydroxy‐PCB‐107 in plasma after treatment of sheep with PCB‐118 (Berg et al., 2010). Sheep were treated during pregnancy, three times per week with two different mixtures of PCB‐118/PCB‐153 (high/low and low/high). Blood samples taken from these sheep at postnatal day 50 (PND50) showed for the high PCB‐118 animals a ratio of 39 between the metabolite and PCB‐118, as compared to 9 in the other group. Other tissues were not analysed for the metabolite. The same metabolite, 4‐hydroxy‐PCB‐107, which can also be derived from PCB‐105, was observed in plasma samples from pigs living on a dump site (Mizukawa et al., 2015).
Molcan et al. (2017) modelled the binding of TCDD, PeCDD, a hexa‐CDD, tri‐ and di‐CDD to porcine CYP1A1 and showed that TCDD has the strongest binding activity. It was also shown that TCDD had the longest distance to the active site of the enzyme, a potential explanation for the very poor degradation. No data were provided on the potential formation of metabolites. Hakk et al. (2001) investigated the metabolism of 1,2,7,8‐TCDD in calves and observed a hydroxylated metabolite in plasma, most likely 2‐hydroxy‐1,3,7,8‐TCDD. However, the relevance for the more persistent 2,3,7,8‐TCDD is unclear.
In rainbow trout, the reported half‐life (t1/2) for whole‐body depuration of tritium after dosing with [3H]‐TCDD was 15 weeks, whereas t1/2 values varied from 8 to 19 weeks for individual organs (Kleeman et al., 1986). TCDD metabolism was examined in rainbow trout injected with [14C]‐TCDD (60 μg/kg, i.p.) but after 1 week only the parent compound was detected in liver, muscle and kidney. In contrast, at least three unidentified TCDD metabolites were present in bile in addition to the parent compound, one being a glucuronide (based on hydrolysation after treatment with glucuronidase). Similarly, Kleeman et al. (1988) reported the presence of at least three TCDD metabolites in addition to the parent compound in the bile of all six species of fish exposed to [14C]‐TCDD (rainbow trout, yellow perch, carp, bluegill, largemouth bass and bullhead).
Regarding companion animals, very few studies on kinetics in cats or dogs were recovered. Kunisue et al. (2005) analysed not only samples of dog and cat feed (see Section 3.2.2), but also tissue samples of two dogs. These showed few detectable PCDD/F congeners but indicated considerable sequestration in the liver. Similar was the case for non‐ortho PCBs but not for mono‐ortho PCBs. Sévère et al. (2015) analysed adipose tissue samples from 115 female pet dogs and detected total TEQ levels of 0.5 pg WHO2005‐TEQ/g fat (range 0.1–4.1). Poiger et al. (1982) treated a dog four times with 1–2 mg [3H]‐TCDD (positions of the label not provided but apparently no exchange to water) on days 0, 2, 7 and 14 by direct introduction into the duodenum. After the third and fourth application, 11% and 8% of the dose was recovered from the bile within 3 days after the application. After the first two applications, this was only 2%, suggesting increased formation and excretion of metabolites. A number of metabolites were observed in bile and tentatively identified, among them 1,3,7,8‐tetrachloro‐2‐hydroxydibenzo‐p‐dioxin and 2,3,7‐trichloro‐8‐hydroxy‐dibenzo‐p‐dioxin.
In a follow‐up study by Poiger and Schlatter (1985), a bile duct cannulated dog was orally treated with lower single doses of [3H]‐TCDD (0.75 μg) at two different days, separated by 3 weeks. Within 110 h after the two dosings, 30% and 41% of the dose was excreted in the faeces, most of it in the first 36 h. This was thought to primarily represent non‐absorbed TCDD. The bile contained 18% and 16% of the dose, urine 2% and 1%. When the animal was pretreated for 15 days with phenobarbital, followed by a similar dose of TCDD, the fraction excreted in faeces increased to 69%, whereas those in bile and urine were 8 and 0.5%, respectively. When pretreated with a single dose of 10 μg TCDD/kg bw (250 μg in total), the fraction excreted in the faeces was 33%, as compared to 34% in the bile and 2% in urine. This clearly indicates an increased metabolism after the pretreatment with TCDD but not phenobarbital. In urine and bile, there was no indication for the presence of TCDD itself. The collection of bile did not allow enterohepatic cycling of metabolites, which could have influenced the fraction excreted via the faeces.
Summary on farm and companion animals
PCDD/Fs, with the exception of the higher chlorinated congeners, and DL‐PCBs are effectively absorbed. Most are poorly degraded but some metabolites of TCDD and some DL‐PCBs have been identified. They are not only accumulated in body fat and liver, but also transferred to milk and eggs.
3.1.1.2. Laboratory animals
Absorption
Subcutaneous administration
After a single subcutaneous (s.c.) dose of 300 ng/kg bw with [14C]‐TCDD, concentrations in rat liver and adipose tissue quickly rose to a maximum around, respectively, 3 and 7 days after administration, to decline afterwards (Abraham et al., 1988). Based on hepatic and adipose tissue levels, the whole body burden was estimated to be 219 ng/kg bw, i.e. 73% of the administered dose.19 When corrected for the expected elimination from the body during a 7‐day period (loss of 21.5% assuming an elimination half‐life of 20 days), this then resulted in a calculated absorbed dose from the injection site of 279 ng/kg bw, i.e. 93% of the administered dose, to be compared with an experimentally observed absorption of 98% (i.e. 2% of the administered dose still present at the injection site 7 days after administration). In a similar way, the fraction absorbed from s.c. doses ranging from 3 to 3,000 ng/kg bw was calculated to be 0.67–0.93.
Oral administration
In mice, the fraction absorbed after a single p.o. dose of 0.1, 1 or 10 μg/kg bw TCDD ranged from 0.70 to 0.88 (Diliberto et al., 1995). In the rat, this fraction ranged from 0.64 to 0.78 (doses of 0.05, 0.20, 0.80 or 1 μg/kg bw: Hurst et al., 2000b). In rats, approximately 90% of a single oral dose of TCDF was absorbed (matrix: 1:1 ethanol:vegetable oil mixture; Birnbaum et al., 1980). Similarly, 70–85% absorption was reported for a single dose of 2,3,4,7,8‐PeCDF (Yoshimura et al., 1986; Brewster and Birnbaum, 1987; Kanimura et al., 1998). In contrast, OCDD is poorly absorbed, 2–15% of a single dose being absorbed after administration by gavage in a 1:1 ortho‐dichlorobenzene:corn oil mixture (Birnbaum and Couture, 1988; Couture et al., 1988).
In mice the fraction absorbed after subchronic p.o. dosing of TCDD (13 weeks/5 days per week; 0.15, 0.45, 1.5, 4.5, 15, 45, 150 and 450 ng/kg bw per day), was found to depend on the administered dose, with highest absorption found at the two lowest doses (0.69 and 0.88, respectively) and lowest absorption at the two highest doses (0.26 at both doses, Diliberto et al., 1995).
Distribution
Since information on accumulation in the body was critical for estimating the body burdens based on either the applied dose or the measured levels in tissues, more details are given in the section on toxicity in experimental animals (see Section 3.1.2). Given lipid partitioning, the distribution of PCDD/Fs and DL‐PCBs in the body is expected to scale according to organ lipid content. For example, since the liver and adipose tissue consist for, respectively, 5% and 80% of lipid, on a wet weight (ww) basis, a ‘steady state’ liver/adipose tissue ratio of 5/80 ≈ 0.06 is expected. Even in case of mild steatosis, characterised by an increase of hepatic lipid content up to 10%, this ratio remains low, i.e. around 0.10. However, in rodents PCDD/Fs and DL‐PCBs clearly do not (solely) distribute according to a lipid partitioning. In the rat, already 1 day after administration of a single subcutaneous dose of 300 ng TCDD/kg bw, a ww liver/adipose tissue ratio of 10 was observed, indicating that a large part of the dose was stored in the liver. This ratio declined to below 1 only 35 days after administration, indicating liver to adipose redistribution at a later stage. This preferential hepatic accumulation, referred to as ‘hepatic sequestration’ was observed at single doses as low as 1 ng/kg bw (Abraham et al., 1988). Hepatic sequestration was confirmed after single TCDD gavage dosing in mice (0.1, 1, 10 μg/kg bw; Diliberto et al., 1995) and rats (0.05, 0.20, 0.80. 1.0 μg/kg bw, Hurst et al., 2000b). Bell et al. (2007a) also observed higher liver to adipose tissue ratios 5 days after a single gavage dose than after daily exposure of rat dams.
After repeated exposure, hepatic sequestration occurs at doses as low as 1 ng/kg bw per day in mice (Diliberto et al., 2001) and rats (Hurst et al., 2000b). Half‐maximal sequestration was observed at repeated dose levels around 30 ng/kg bw per day in mice (Diliberto et al., 2001), as compared to 300 ng/kg bw in rats after a single dose (Abraham et al., 1988). Preferential accumulation of TCDD, i.e. compared to adipose tissue, is restricted to the liver (Santostefano et al., 1996). As a result, the animal body burden is mainly determined by deposition in the liver and adipose tissue (see Appendix B for details). Next to TCDD, hepatic sequestration was also reported for PeCDD, 2,3,4,7,8‐PeCDF, and PCB‐126, but not with the mono‐ortho PCBs ‐118 and ‐156 (Van Ede et al., 2013).
Early experiments by Poland et al. (1989a,b) showed binding of [125I]‐labelled TCDD in rat liver to have two components: binding to the Ah‐receptor with high affinity but low capacity (Kd = 4 × 10−12 M, pool size = 1.2 × 10−12 mol/g liver), and TCDD inducible binding to microsomal proteins with low affinity and high capacity (Kd = 5.6 × 10−8 M, pool size = 2.2 × 10−8 mol/g liver). Subsequent research showed that in CYP 1A2 knockout mice hepatic sequestration was abolished (Diliberto et al., 1999). A study by Hakk et al. (2009) also showed hepatic sequestration in C57BL/6N mice which had been exposed to a single oral dose of 0.1 or 10 μg [1,6‐3H] TCDD/kg bw, whereas their CYP 1A2 (−/−) knockouts did not. Dragin et al. (2006) also showed the lack of sequestration not only in CYP 1A2 knockout mice, but also that in these mice, TCDD caused embryotoxicity at lower doses than in wild‐type mice. The lower storage in the liver of the dams resulted in higher concentrations in the fetus, suggesting a protective role of CYP 1A2 in the liver of the dams. The higher toxicity was not observed in CYP 1A1 and CYP 1B1 knockouts, but mice expressing human CYP 1A2 and no mouse CYP 1A2 showed similar dose response for embryotoxicity as wild‐type mice. This suggests that human CYP 1A2 also sequestrates TCDD in a similar manner as in the mouse. In Ah‐receptor knockout rats, failing to show induction of CYPs of the 1A family, no specific accumulation of TCDD was observed (Harrill et al., 2016).
In pregnant rats, TCDD is also transferred to the fetus and postnatally to offspring via milk. Hurst et al. (2000a,b) showed that a single dose given during pregnancy resulted in a relatively high distribution to the fetus, as compared to a more chronic dose. A similar difference in the ratio mother dams/fetus was observed by Bell et al. (2007a,b,c), based on single and repeated dosing of pregnant rats. It is important to take this into account in the dose–response modelling of effects in offspring of treated laboratory animals. The studies by Diliberto et al. (1999) and Hurst et al. (2000a,b) showed similar levels in the heads of the fetus as in other tissues. Radiolabel was detected in the brain of the mother, but at much lower levels than in the liver and adipose tissue
Metabolism
The bulk of the available literature deals with TCDD, for which a major metabolic conversion appears to be hydroxylation at a lateral or periposition, but the cleavage of one or both of the two ether bonds may also occur, at least in rats (Poiger et al., 1982; Sawahata et al., 1982; Poiger and Buser et al., 1984). As a result, numerous primary polar metabolites can be formed, some of which are excreted in bile as glucuronides (Poiger and Schlatter, 1979; Ramsey et al., 1982; Poiger and Buser et al., 1984). While some interspecies differences in TCDD biotransformation have been described, they do not seem to correlate with sensitivity to TCDD toxicity (Gasiewicz et al., 1983a,b; Poiger and Buser et al., 1984; Wroblewski and Olson, 1985).
Biotransformation of TCDD is regarded as a detoxification process (Beatty et al., 1978; Ramsey et al., 1982; Weber et al., 1982; Mason and Safe, 1986), and TCDD toxicity is ascribed to the unchanged parent compound. As TCDD‐inducible CYPs appear to be involved in the biotransformation step, TCDD may generally be able to induce its own metabolism at high doses (Neal et al., 1982; Poiger and Buser et al., 1984; Poiger and Schlatter, 1985; Wroblewski and Olson, 1985, 1988; Leung et al., 1990a; Shinkyo et al., 2003; Inui et al., 2014). However, although at least guinea pigs seem to be incapable of this autoinduction (Wroblewski and Olson, 1985).
The rate of TCDD biotransformation is sluggish in all species and evidently does not account for the exceptional resistance of hamsters to TCDD, because (1) in the uninduced state, hepatocytes from mice (irrespective of their AHR status) metabolise TCDD at least four times as fast as those from hamsters (Shen and Olson, 1987); and (2) TCDD metabolism occurs at a similar rate in hepatocytes from Sprague–Dawley rats and hamsters in both the uninduced and the induced states (Wroblewski and Olson, 1988). Yet, adult hamsters are some 40–100 times more resistant to TCDD acute lethality than mice possessing a high‐affinity AHR or Sprague–Dawley rats (Pohjanvirta and Tuomisto, 1994). The rate of TCDD metabolism is also highly similar in TCDD‐sensitive Long‐Evans (Turku/AB) and TCDD‐resistant Han Wistar rats (Pohjanvirta et al., 1990a). Nevertheless, guinea pigs have a weaker ability to metabolise TCDD than other laboratory animals, and this may contribute to their high sensitivity to TCDD toxicity (Wroblewski and Olson, 1985; Van den Berg et al., 1994).
In animal tissues, only unmetabolised TCDD can usually be found (Rose et al., 1976; Olson et al., 1980; Koshakji et al., 1984; Curtis et al., 1990; Pohjanvirta et al., 1990a; Weber et al., 1993), implying that its metabolites are excreted rapidly. An exception is the guinea pig, in which a portion (4–28%) of the total radioactivity was reported to stem from TCDD metabolites (Olson, 1986). On the other hand, as in dogs, little if any unmetabolised TCDD occurred in the bile or urine of rats, hamsters, mice and guinea pigs (Olson et al., 1980; Gasiewicz et al., 1983a,b; Neal et al., 1984; Poiger and Schlatter, 1985; Olson, 1986; Pohjanvirta et al., 1990a; Kedderis et al., 1991). In the faeces, TCDD is predominantly excreted as metabolites in hamsters (Neal et al., 1982; Gasiewicz et al., 1983a,b) but as a parent compound in guinea pigs (after i.p. injection; Olson, 1986), suggesting luminal transfer across the intestinal wall in the latter species. For the mouse and rat, the data are inconsistent in this regard (Vinopal and Casida, 1973; Gasiewicz et al., 1983a,b; Koshakji et al., 1984; Pohjanvirta et al., 1990a; Weber et al., 1993).
Much less is known about the biotransformation of other PCDDs. In rat bile, several phenolic metabolites of PeCDD were detected (Wacker et al., 1986), while no metabolites of OCDD were observed (Birnbaum and Couture, 1988). In accordance with these findings, the higher chlorinated PCDDs seem to occur predominantly unmetabolised in rat faeces, which is concomitantly with their low intestinal absorption (Abraham et al., 1989).
In contrast to most other PCDD/F congeners, TCDF is rapidly metabolised in rat liver (at high enough doses to result in induction of CYP1A1, the major metabolising enzyme of TCDF in rats (Tai et al., 1993)), and the metabolites are then mainly excreted in bile. Of the numerous metabolites generated, 4‐hydroxy‐TCDF and 3‐hydroxy‐2,7,8‐triCDF appear to be the principal ones (Burka et al., 1991; Tai et al., 1993). The metabolites are also more abundant than their parent compound in rat faeces (Birnbaum et al., 1980). Similar to rats, in mice (both C57BL/6J and DBA/2J) and in rhesus monkeys, the bulk of TCDF was excreted as metabolites in the faeces (Birnbaum et al., 1981; Decad et al., 1981a), whereas the parent compound prevailed in guinea pig faeces (Decad et al., 1981b).
In rats, also 1,2,3,7,8‐PeCDF is effectively biotransformed. The major metabolites in rat bile proved to be five isomers of dihydroxy‐TCDFs (Pluess et al., 1987; Poiger et al., 1989a,b). However, the presence of a chlorine atom in position 4 of 2,3,4,7,8‐PeCDF strongly inhibited oxidative metabolism compared with 1,2,3,7,8‐PeCDF in rat liver (Brewster and Birnbaum, 1988). Moreover, for 2,3,4,7,8‐PeCDF, ether bond cleavage was an important metabolic transformation (Poiger et al., 1989a,b). Additional chlorine substitution decreases the biotransformation rate substantially. Hence, 1,2,3,6,7,8‐HxCDF was converted in rats to small amounts of only a single biliary metabolite, a dihydroxy‐PeCDF, and no biliary metabolites could be detected of 1,2,3,4,6,7,8‐HpCDF (Poiger et al., 1989a,b). Likewise, no metabolites of OCDF could be found in rat faeces, urine, fat or liver (Veerkamp et al., 1981).
Except for PCB‐77, most of the DL‐PCBs are not readily metabolised. They are preferentially metabolised by CYP1A enzymes (Safe, 1992, 1994). Accordingly, only one metabolite was isolated from the faeces of rats treated with PCB‐126 and identified as 4’‐hydroxy‐3,4,5,3’,5’‐PeCB. Its excretion amounted to a mere 1.3% of total dose during 5 days following PCB‐126 administration, but the metabolite exhibited lower toxicity to rats than its parent compound, indicating detoxification (Koga et al., 1990). In vitro, rat CYP1A1 was able not only to generate this but also another metabolite of PCB‐126, 4‐OH‐3,3’,4’,5‐tetrachlorobiphenyl (Yamazaki et al., 2011). In contrast, PCB‐77 was excreted in rat faeces mainly as metabolites of which the major ones were 5‐hydroxy‐3,4,3’,4’‐TCB and 4‐hydroxy‐3,5,3’,4’‐TCB. They were both much less toxic than the parent PCB‐77 and failed to induce the CYP1A1‐associated monooxygenase activity, aryl hydrocarbon (benzo[a]pyrene) hydroxylase (Yoshimura et al., 1987). However, they both exhibited higher transthyretin binding affinity (see Section 3.1.6.10.5) than thyroxine itself (Brouwer et al., 1990). Another toxicologically important metabolite of DL‐PCBs in rats with affinity to transthyretin is 2,3,3′,4′,5‐pentachlorobiphenyl (4‐OH‐CB107), which is generated from both PCB‐105 and PCB‐118 (Sjödin et al., 1998), and has also been found in human blood (Fängström et al., 2005). It is a potent suppressor of circulating free and total thyroxine levels in rats after prenatal exposure (Meerts et al., 2002).
Excretion
Pohjanvirta et al. (1990a) treated Long‐Evans and Han Wistar rats with a single i.p. dose of 5 μg/kg bw [14C]‐TCDD. There was a gradual excretion of the radiolabel, with most being excreted via the faeces but a small amount also via the urine, with no clear strain differences (66.1% vs 58.0% after 32 days in faeces and 5.5% vs 4.1% in urine, respectively). In urine, over 90% of the radiolabel was excreted as unidentified metabolites. It was similar for faeces but there were large interindividual differences.
Diliberto et al. (1996) compared excretion of a single dose of [1,6‐3H]‐TCDD (1 nmol/kg bw) following different routes of administration in 3‐month‐old male Fisher 344 rats (n = 3–4 per treatment). Three days after an oral dose, the fraction of radiolabel excreted in faeces and urine was 32.2% and 1.4%, respectively, as compared to 22.2 and 2.2% after intravenous (i.v.) and 26.3% and 1.3% after intratracheal administration.
In the study by Hakk et al. (2009) in mice, with C57BL/6N mice and CYP 1A2 (−/−) knockouts treated with a single oral dose of 0.1 or 10 μg [1,6‐3H] TCDD/kg bw, faeces was the major route of elimination in both strains. At the high dose 19.8% (wild type), respectively 22.9% (knockouts) was excreted in the faeces during the first 24 h after administration. Corresponding excretion in the urine amounted to, respectively, 1.3% and 1.1%. The combined 96 h faecal and urine excretion amounted to, respectively, 28.2% and 34.3%. A similar excretion was found at the lower dose. In both strains, the bulk of the faecal radioactivity was present as metabolites, the only GC/MS quantifiable metabolite being 1‐hydroxy‐TCDD. At the high dose, 80% of the TCDD‐derived radioactivity in the urine was found to be TCDD in knockout mice, as compared to 40% in wild types, suggesting a carrier mechanism to be operational in urinary TCDD excretion. Indeed, the binding of TCDD to the mouse major urinary protein was demonstrated.
Half‐lives
Emond et al. (2006) reviewed the half‐lives for TCDD in rats, showing a range between 10.5 and 45.2 days (median of 17), depending on strain, sex and applied dose. Concerning other PCDDs, the elimination half‐life increases with the degree of chlorination, with half‐lives ranging from 27.2 to 33.1 days for PeCDD, 83 to 156 days for 1,2,3,4,7,8‐HxCDD, 200 to 314 days for HpCDD, and 173 to 413 days for OCDD (Wacker et al., 1986; Birnbaum and Couture, 1988; Viluksela et al., 1997).
Summary on experimental animals
In rodents, PCDD/Fs and DL‐PCBs are well absorbed and distributed to various tissues, and transferred to the fetus. The major accumulation is in adipose tissue and in the liver, with a ratio between the two that increases with the applied dose, due to binding to CYP1A2 in the liver. Half‐lives are relatively short when compared to humans, in part due to the higher rate of metabolism. Faecal excretion is the more important route of elimination.
3.1.1.3. Humans
Absorption
Poiger and Schlatter (1986) administered an oral dose of 105 ng radiolabelled [1,6‐3H]‐TCDD to one male volunteer and concluded that more than 87% was absorbed. Moser and McLachlan (2001) compared intake and levels in faeces from 5 volunteers with background exposure and estimated absorption to be more than 95% for most PCDD/Fs and DL‐PCBs. Lower absorption was observed for the hepta‐ and especially octachlorinated PCDD/F congeners. Using a toxicokinetic model, Aylward et al. (2005) evaluated data from four of these individuals and concluded that 95–99% of the TCDD was absorbed. These calculations took into account the excretion of TCDD from the body, ‘due to simple lipid partitioning from the circulation across the intestinal lumen into fecal contents’.
McLachlan (1993) determined the 12‐day mass balance, i.e. the difference between the total intake with breast milk and the excretion in the faeces, in a 19 week old boy for 12 PCDD/Fs and 4 DL‐PCBs. TCDD, and penta‐ (2,3,4,7,8‐PeCDF, 1,2,3,7,8‐PeCDD) and hexa‐substituted congeners (1,2,3,4,7,8‐HxCDF, 1,2,3,6,7,8‐HxCDF, 2,3,4,6,7,8‐HxCDF, 1,2,3,4,7,8‐HxCDD, 1,2,3,6,7,8‐HxCDD, 1,2,3,7,8,9‐HxCDD) showed an absorption of 90% or higher. The absorption of the two hepta congeners (HpCDD and 1,2,3,4,6,7,8‐HpCDF) and OCDD was found to be lower, i.e. 61% and 58%, and 23%, respectively.
Dahl et al. (1995) determined the 48‐h mass‐balance for seven PCDDs (TCDD, PeCDD, 1,2,3,4,7,8‐HxCDD, 1,2,3,6,7,8‐HxCDD, 1,2,3,7,8,9‐HxCDD, HpCDD, OCDD), six PCDFs (TCDF, 1,2,3,7,8‐PeCDF, 2,3,4,7,8‐PeCDF, 1,2,3,4,7,8‐HxCDF, 1,2,3,6,7,8‐HxCDF, 1,2,3,4,6,7,8‐HpCDF) and three DL‐PCBs (PCB‐77, ‐126 and ‐169) in four breast fed children at 1, 2, 3 and 6 months post‐partum. For all tetra‐, penta‐ and hexa‐substituted PCDD/Fs and PCB congeners the absorption was found to be over 95%. The absorption of HpCDD, 1,2,3,4,6,7,8‐HpCDF and OCDD was found to be somewhat lower (80%, 93% and 87%, respectively).
Abraham et al. (1996) determined the 5‐day mass balance in two breastfed children at 1 and 5 months of age for TCDD, PeCDD, 2,3,4,7,8‐PeCDF, 1,2,3,6,7,8‐HxCDD, HpCDD, OCDD, and the sum of these PCDD/Fs in I‐TEQ. At the age of 1 month, exposure of the infants was estimated to be 82 and 106 pg I‐TEQ/kg bw per day. For TCDD and the sum in I‐TEQ, the absorption was found to be ≥ 94% and ≥ 91%, respectively. The absorption of HpCDD and OCDD was found to be lower (78% and 51%, respectively). The absorption of dietary fat was found to be ≥ 95%. The results indicate that the absorption of dioxin‐like compounds occurs together with absorption of fat from the food.
Distribution
Once absorbed part of the PCDD/Fs and DL‐PCBs are transported via the serum lipids and part are attached to the protein fraction (Patterson et al., 1989a,b). This depends on the congener, with higher chlorination resulting in higher protein binding. TCDD is primarily associated with lipoproteins. Norén et al. (1999) showed that for a number of PCBs, including some mono‐ortho PCBs (PCBs ‐118, ‐156, ‐157, ‐167, ‐189), also 30–60% in serum is bound to the fraction containing albumins. Compounds can subsequently be taken up by different tissues and in particular adipose tissue, which is the major storage site. Based on a number of studies (see Section 3.2.4), TCDD levels in adipose tissue are in equilibrium with those in the serum, and show a ratio of about 1 when adjusted for lipids20 (Patterson et al., 1988; Schecter et al., 1991, 1992; Iida et al., 1999). For other, lower chlorinated, PCDD/Fs this seems similar but for higher chlorinated ones the ratio serum to adipose tissue was reported to be up to twofold higher (Schecter et al., 1991, 1992). However, Iida et al. (1999) did not observe this. For PCB‐126 and PCB‐169, Iida et al. (1999) observed lower median plasma to adipose tissue rates of 0.4.
In addition to adipose tissue, PCDD/Fs and DL‐PCBs accumulate in the liver. However, lipid adjusted levels of certain PCDD/Fs and DL‐PCBs may be higher than in plasma, especially at high exposure (Kashimoto and Miyata et al., 1987; Kreuzer et al., 1997). Using CYP1A2 knockout mice, this has been shown to be due to binding of PCDD/Fs and DL‐PCBs to CYP1A2 (see Section 3.1.1.2). The level of this enzyme is induced by PCDD/Fs and DL‐PCBs. In humans with background exposure from food (i.e. not due to occupational exposure or specific food incidents), this sequestration has been shown for hepta‐ and octachlorinated PCDDs and especially PCDFs, but much less for the lower chlorinated PCDDs (TCDD, PeCDD and HxCDDs) and non‐ortho DL‐PCBs (reviewed by van Ede et al., 2013). This implies that for TEQ‐based levels, which are driven by tetra‐ and pentachlorinated PCDD/Fs and PCB‐126 (see Section 3.3.1), liver sequestration may be less relevant at current exposure levels. Carrier et al. (1995) used human data from victims of Yusho to develop a toxicokinetic model. Based on data for 2,3,4,7,8‐PeCDF from five persons (Kashimoto and Miyata et al., 1987), a sigmoid curve was obtained for the dose‐related distribution between liver and adipose tissue. The curve started around the lowest observed level in adipose tissue of 100 ng/kg and levelled off at the highest observed level of 5,300 ng/kg. With levels in the liver of 10 to 70,100 ng/kg ww, this corresponds to ratios between 0.1 and 13.2. Based on this study and a rat study with TCDD, a so‐called Kr value of 100 ng/kg bw (or 400 ng/kg fat) was used in the toxicokinetic model for TCDD (Kr being the level at which a half maximal induction occurs). The curve started to rise around 1 and levelled off around 10,000 ng/kg bw (Carrier et al., 1995; Aylward et al., 2005). The model assumes that at low levels, about 1% of the body burden of TCDD is present in the liver (corresponding with a liver to adipose ratio of 0.1, based on ww), and at very high levels 70% (see also Section 3.1.1.5).
PCDD/Fs and DL‐PCBs are transferred via the mother to the fetus in utero and postnatally via breast milk. The prenatal transfer is based on studies showing similar lipid‐based levels in cord blood and that of the mothers. Since cord blood contains less lipids, the wet weight‐based levels are lower (see Section 3.2.4). Kreuzer et al. (1997), analysed adipose tissue and livers of 3 stillborns and 17 infants (up to 44 weeks old, 9 being breastfed). PCDD/Fs could be detected in adipose tissue and livers of the 3 stillborn infants at levels of, respectively, 10 and 7 pg I‐TEQ/g fat, thus confirming the prenatal transfer. In most of the older infants, the lipid‐based I‐TEQ levels were similar in liver and adipose tissue, but in several the ratio was much higher (up to 13), showing increased accumulation in the liver. Based on samples from three babies that had died from SIDS, Beck et al. (1994) concluded that levels of PCDD/Fs in brains were low compared to adipose tissue and that there was no accumulation in the brain. Similar results were obtained for 3 adults by Zober and Päpke (1993).
Metabolism
In general, the 2,3,7,8‐substituted PCDD/Fs are relatively resistant to biotransformation. A clear exception is 1,2,3,7,8‐PeCDF which can hardly be detected in humans. Biotransformation enzymes including CYP1A1 and 1A2 are induced at high exposure, and this is likely to underlie the decreased half‐lives in highly exposed persons. Sorg et al. (2009), investigating the poisoning of Victor Yushchenko, actually reported the presence of two metabolites in faeces, serum and urine, i.e. 2,3,7‐trichloro‐8‐hydroxydibenzo‐p‐dioxin and 1,3,7,8‐tetrachloro‐2‐hydroxydibenzo‐p‐dioxin, at a ratio of about 2 to 1. The excretion of the two metabolites, especially via the faeces, actually accounted for 40% of the reduction of the body burden during a certain period of time, the rest due to loss of TCDD itself via the faeces. The formation of these metabolites is likely to have been accelerated by the high exposure and resulting induction of CYP enzymes. However, Inui et al. (2014) reviewed the literature and reported that in vitro studies with human CYPs 1A1, 1A2 and 1B1 did show metabolism of mono‐, di‐ and tri‐CDDs but no detectable activity towards TCDD. It could be argued that the formation is too low to be detected by in vitro models.
For some DL‐PCBs (PCB‐77, ‐105, ‐118, ‐157), the formation of hydroxy metabolites has been reported (see review by Grimm et al., 2015). These hydroxy metabolites are much more polar and thought not to bind to the AHR. Yamazaki et al. (2011) compared the in vitro metabolism of PCB‐126 and contrary to rats did not observed any formation of metabolites in human liver microsomes.
Excretion
In the above‐mentioned study with [1,6‐3H]‐TCDD by Poiger and Schlatter (1986), no radiolabel was detected in the urine. In the faeces, 11.5% of the radiolabel of the administered single dose of 105 ng was detected in the first 3 days, most of it thought to represent non‐absorbed TCDD. Another 3.5% was excreted during days 3–125. Excretion of metabolites of PCDD/Fs and DL‐PCBs may occur via the bile and subsequently the faeces. However, more important for most congeners, compounds may be excreted unchanged via the faeces, partly following excretion via the liver but possibly more important ‘due to simple lipid partitioning from the circulation across the intestinal lumen into fecal contents’ (Moser and McLachlan, 2001, 2002).
Half‐lives
Several authors have estimated the half‐life of TCDD and other congeners based on measurements of highly exposed people, e.g. from Seveso, Vietnam or chemical plants. These half‐lives were used in several studies for extrapolation of, e.g. blood levels to a relevant point of time, in most cases being a backward extrapolation to higher levels. Estimated half‐lives for TCDD in adults were estimated to be 6.1–11.3 years (Michalek et al., 1992; Wolfe et al., 1994; Flesch‐Janys et al., 1996; Milbrath et al., 2009). Half‐life is decreased at higher levels in the body (Michalek et al., 2002), and in men seems shorter than in females (Aylward et al., 2005). Using the CADM (see Section 3.1.1.5), Aylward and Hays (2002) showed that half‐lives for TCDD can vary from around 4 years at high serum levels to more than 10 years at current background levels.
Some authors have estimated half‐lives for other PCDD/Fs and DL‐PCBs. Aylward et al. (2013) calculated half‐lives for a number of PCDDs based on blood levels of former chlorophenol workers taken in 2004–2005 and 2010. Body fat was taken into account and an estimate of current intake was used to correct the half‐lives. For TCDD, a half‐life of 6.5 years was estimated, for PeCDD 10.7, for 1,2,3,4,7,8‐, 1,2,3,4.6.7.8‐ and 1,2,3,7,8,9‐HxCDD, respectively, 7.0, 9.0 and 6.3, for HpCDD 6.7 and for OCDD 7.3 years. This indicates that half‐lives are very similar for most PCDDs but higher for PeCDD and 1,2,3,4,6,7,8‐HxCDD, two relatively important congeners in terms of contribution to the TEQ (see Section 3.3.1). As a result, the half‐life for PCDD‐TEQ was estimated to be 8.7 years, so longer than for TCDD. These half‐lives were comparable to those obtained for PCDDs by Flesch‐Janys et al. (1996) and Rohde et al. (1999).
Concerning PCDFs, it was reported that the half‐life of 2,3,4,7,8‐PeCDF is rather long in some of the Yusho patients (Matsumoto et al., 2009). Flesch‐Janys et al. (1996) and Rohde et al. (1999) showed for PCDFs similar values as for PCDDs, with the longest half‐life for 2,3,4,7,8‐PeCDF (19.6 and 13.9 years). A review by Milbrath et al. (2009) summarises these half‐lives and confirms that those for TCDF and 1,2,3,7,8‐PeCDF are very short compared to other congeners.
Based on a study by Ogura (2004), the half‐life for PCB‐126 was rather short (2.7 vs. 6.7 years for TCDD). PCB‐77 and PCB‐81 showed even shorter half‐lives, whereas those for PCB‐169 and several other mono‐ortho PCBs were much longer.
Concerning the whole body half‐life of PCDD/Fs in infants, Kreuzer et al. (1997) provided data on the levels of TCDD and sum of PCDD/Fs in adipose tissue and liver from three stillborn children and eight non‐breastfed sudden death infants aged 0.4–26 weeks. Assuming the levels in the stillborns as representative for the levels at birth of the sudden death children, a kinetic analysis of the data resulted in half‐lives of 0.32 year for TCDD and of 0.35 year for I‐TEQ in the body fat mass of neonates.
These half‐lives fit well with half‐lives calculated by Leung et al. (2006) on the basis of the two breastfed infants of the study by Abraham et al. (1996). Taking into account the exposure during the breastfeeding, weaning and post‐weaning periods, as well as body growth and changes in body composition, the following (average) half‐lives for the decrease from the infant's body fat mass were calculated. For TCDD 0.36–0.43 year, for PeCDD 0.28–0.36 year, for 1,2,3,6,7,8‐HxCDD 0.33–0.44 year, for 1,2,3,4,6,7,8‐HpCDD 0.28–0.36 year, for OCDD 0.42–0.50 year and for 2,3,4,7,8‐PeCDF 0.23–0.30 year. It is evident that in growing children, the increase in weight and body fat determines the apparent half‐life in tissues.
Summary on humans
PCDD/Fs and DL‐PCBs are well absorbed in humans and subsequently distributed to liver and in particular adipose tissue. The levels of the more relevant congeners in blood are in equilibrium with those in adipose tissue. In the liver, PCDD/Fs and DL‐PCBs can show lipid‐based levels higher than in the adipose tissue, which can be induced at higher exposure. Compared to laboratory animals, most PCDD/Fs and DL‐PCBs show long half‐lives which vary between different congeners and depending on the levels, age, BMI and sex.
3.1.1.4. Transfer of PCDD/Fs and DL‐PCBs in food‐producing animals
PCDD/Fs and DL‐PCBs accumulate in tissues of food‐producing animals, including fish. In addition, they can be excreted in fat‐containing products such as milk and eggs. Figure 3 shows typical examples for the levels of PCDD/Fs in milk and eggs upon repeated exposure of cows or laying hens to contaminated feed, followed by a period on clean feed. There are clear differences in toxicokinetic behaviour between the various PCDD/F and DL‐PCB congeners, as discussed below. As in other species, certain PCDD/Fs and DL‐PCBs can also specifically accumulate in the liver, in laboratory animals shown to be due to the binding to CYP1A2, rather than accumulation in fat. Since this process, termed sequestration, is congener specific, it will lead to differences between the relative congener composition in liver and fat. High concentrations in the liver are a particular issue in foraging animals like sheep and deer (EFSA CONTAM Panel, 2011). Girolami et al. (2016) investigated the liver expression and protein levels of CYP1A1, CYP1A2 and CYP1B1 in sheep and cows derived from the same area. Although remarkably lower than those reported in other surveys (EFSA CONTAM Panel, 2011) or a controlled study with sheep (Hoogenboom et al., 2015b), much higher levels (up to fivefold) of PCDD/Fs and DL‐PCBs were found in livers from sheep compared to cows (Benedetto et al., 2016). However, gene expression and protein levels of the CYPs were much lower in sheep than in cows, especially those of CYP1A2 (about 10% of the levels in cows), which seems in contrast to the supposed role of this isoform in PCDD/F and DL‐PCB sequestration. However, since levels in adipose tissue were not reported, the possibility that sheep had a higher overall exposure than cows could not be excluded.
Figure 3.
Mean levels of PCDD/Fs (lower bound) in milk and eggs after feeding cows (n = 3) for 33 days and laying hens (n = 5) for 56 days with contaminated feed followed by a similar period on clean feed (modified from Hoogenboom et al., 2006, 2015c). Cows obtained maize silage contaminated by a fire (0.79 ng WHO 2005‐TEQ/kg; 17 kg feed/day), laying hens feed contaminated with a standard mixture (0.40 ng WHO 1998‐TEQ/kg, 0.11 kg feed/day)
The kinetics of contaminants in the animal may be described by factors like the transfer rate (TR, formerly called carry‐over rate or COR), describing the percentage of the daily dose excreted in milk or eggs. An alternative is the bioconcentration factor (BCF), describing the ratio between the level in tissues, milk or eggs, and that in the feed. BCFs are more suitable for tissues, since it is more difficult to obtain the information on the total weight of, e.g. muscle and adipose tissues in the animal required to calculate the TRs. Occasionally also the term biotransfer factor (BTF) is used, describing the ratio between the level in the edible product and the daily intake.
In fish, another parameter used is the accumulation efficiency, which is the net effect of dietary absorption and elimination, calculated as the concentration in fish, divided by the concentration in feed multiplied by the feeding rate (amount per day) and duration of feeding (in days). For a number of animal species, toxicokinetic models have been developed, describing the time‐ and dose‐related levels in meat, milk, eggs or body fat.
Studies on the transfer of these compounds in different food producing animals were retrieved and selected for relevance as described in Section 2.2 and in Annex A.4.
3.1.1.4.1. Milk from cows and buffaloes
A large number of studies have examined the transfer of PCDD/Fs and, to a lesser extent, DL‐PCBs to milk. Only few studies were controlled trials with contaminated feed materials, or included both an exposure and an elimination phase, where in the latter period the cows were put on non‐contaminated feed (Huwe and Smith, 2005; Hoogenboom et al., 2015c). Some older studies focussed on TCDD only, fed via 2,4,5‐T (Jensen and Hummel, 1982) or a purified standard (Jones et al., 1987a, 1989), or on a selected number of congeners, e.g. from PCP or PCP‐treated wood contaminated with PCDD/Fs (Firestone et al., 1979; Fries et al., 1999, 2002). Other studies focussed on the exposure period only, particularly related to grass contaminated by municipal waste incinerators or sewage sludge (McLachlan et al., 1990; Slob et al., 1995; Schuler et al., 1997; McLachlan and Richter, 1998). In a few experimental studies, a single dose was provided and only the elimination phase was examined (Olling et al., 1991). Several authors described field studies dealing with the follow‐up of an incident and hence focussed on the elimination phase only (Tuinstra et al., 1992; Malisch, 2000; Brambilla et al., 2008; Hoogenboom et al., 2010). The latter studies are nevertheless useful to obtain an overall picture on the transfer of PCDD/Fs and DL‐PCBs to milk, especially when both the intake during the incident and excretion via the milk could be estimated. Regarding the terms of reference for this assessment, the primary interest is in the accumulation of PCDD/Fs and DL‐PCBs in tissues, and their excretion into milk during the exposure period, giving insight into the relation between levels in feed and food. Therefore, the CONTAM Panel decided that studies with multiple exposure days should be the basis of the evaluation and that information from other studies can be used to fill specific data gaps.
In the case of dairy cows, the ingested contaminants are absorbed to a certain extent and subsequently distributed in various body compartments (adipose tissue, liver), or excreted into the milk. Jones et al. (1989) treated dairy cows with tritiated TCDD (50 ng/kg bw, single dose) and showed that blood levels increased with a peak level of 6 pg/g blood around day 1, and then slowly decreased in 5 days. Milk levels followed the same pattern. At prolonged exposure, the depot in the tissues will contribute to concentrations of PCDD/Fs and DL‐PCBs in milk. With continued exposure, the milk levels will initially increase rapidly, then gradually level off and eventually reach a plateau level, representing a steady‐state condition (Figure 3). It has been estimated that the steady state is reached after roughly three months, meaning that in practice, either during incidents or in controlled studies, exposure is generally too short to reach this state. When the exposure is terminated, the levels initially decrease rapidly but then the decrease gradually slows down (Figure 3). This decrease can be described with two half‐lives, one for the initial rapid decline and one for the terminal phase with slow decline. Derks et al. (1994) developed a toxicokinetic model for PCDD/Fs for milk and adipose tissue in dairy cows. This model was successfully applied during an incident with contaminated kaolinite clay (Hoogenboom et al., 2010), showing half‐lives for the TEQ levels of, respectively, 1 day and 4 weeks for these two elimination phases.
In dairy cows, TRs describe the fraction of daily ingested PCDD/Fs and DL‐PCBs excreted into the milk on the same day. With unchanged daily intake, the calculated TRs increase during the exposure period and are highest at the steady‐state conditions. Table 4 gives an overview of TRs reported for different PCDD/Fs and to a lesser extent non‐ortho DL‐PCBs in various studies. The last column shows the mean values based on the mean TRs for each study. The lower chlorinated PCDD/Fs show in general higher CORs than the higher chlorinated PCDD/Fs, due to a higher absorption in the gastrointestinal tract (Fries et al., 2002). However, there are some exceptions, like TCDF and 1,2,3,7,8‐PeCDF which appear to be rapidly metabolised. TRs for PCB‐126 and ‐169, the DL‐PCBs with the highest TEFs, show similar or slightly higher TRs than the tetra‐ and pentachlorinated PCDD/Fs. PCB‐81 and especially PCB‐77 show poor transfer to milk, probably due to metabolism or poor absorption. As shown in Table 4, there are also some clear differences between studies in the magnitude of the TRs for a specific congener. This might be related not only to measurement issues (e.g. low levels close to LOQs, measurement uncertainty) but also to factors like the feed matrix or specific characteristics of the animals (milk production, energy status, metabolism, etc.). Since TRs vary for different congeners, the overall transfer rate for the mixture, as expressed in TEQ, is in theory highly dependent on the composition of the mixture. In practice, however, this will only affect some mixtures, in particular those where the TEQ is primarily derived from higher chlorinated PCDD/Fs or from TCDF. For products where the lower chlorinated congeners or PCB‐126 dominate the TEQ level, the TR for the TEQ will be around 30–40%.
Table 4.
Transfer rates (TRs) in % as reported for lactating cows in various studies. TRs are estimated from the intake through the feed and excretion in milk at ‘steady state’.a Missing values indicate that the congener was not determined or detected
| Slob et al. (1995) | Schuler et al. (1997) | McLachlan and Richter (1998) | Malisch (2000) | Huwe and Smith (2005) | Brambilla et al. (2008) | Hoogenboom et al. (2015c) | Meand | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Study design | Field | Field | Stable | Stable | Field | Stable | Field | Stable | Stable | |
| Source | MWI | MWI | Background | Sludge | Citrus pulp | MgO | Minerals | Maize silage | Sugar beet | |
| Dose (ng TEQ/kg bw per day)b | 0.2 | 0.014 | 0.002 | 0.019 | 0.02 | 0.23 | 0.026 | 0.023 | 0.018 | |
| Number of cows | Manyc | 41 | 4 | 4 | Manyc | 2 | 1.604 | 3 | 3 | |
| Duration exposure (days) | 30 | 395 | 84 | 23 | 180 | 40 | 28 | 33 | 33 | |
| Withdrawal period (days) | – | – | – | – | 350 | 40 | 75 | 33 | 33 | |
| Milk fat (kg/day) | 1.0 | 0.9 | 0.9 | 1.0 | 1.2 | 0.7 | 1.0 | 1.5 | 1.4 | |
| 2,3,7,8‐TCDD | 15 | 30 | 38 | 51 | 58 | 30 | 30 | 36 | 35 | |
| 1,2,3,7,8‐PeCDD | 10 | 20 | 39 | 27 | 49 | 27 | 54 | 35 | 38 | 33 |
| 1,2,3,4,7,8‐HxCDD | 5.6 | 8 | 33 | 21 | 51 | 21 | 41 | 27 | 23 | 26 |
| 1,2,3,6,7,8‐HxCDD | 6.4 | 33 | 13 | 77 | 27 | 49 | 30 | 11 | 34 | |
| 1,2,3,7,8,9‐HxCDD | 3.1 | 16 | 10 | 35 | 16 | 29 | 20 | 18 | 19 | |
| 1,2,3,4,6,7,8‐HpCDD | 0.6 | 7 | 3.4 | 2.0 | 18 | 4.8 | 10 | 4.2 | 0.8 | 7 |
| OCDD | 0.1 | 0.8 | 0.7 | 0.3 | 3.7 | 0.7 | 0.4 | 0.2 | 0.0 | 1 |
| 2,3,7,8‐TCDF | 2 | 2.8 | – | 1.8 | 1.1 | 2 | ||||
| 1,2,3,7,8‐PeCDF | 4 | 3.8 | 2.3 | 4.2 | 4.1 | 2.7 | 4 | |||
| 2,3,4,7,8‐PeCDF | 12 | 50 | 40 | 65 | 58 | 24 | 56 | 31 | 25 | 40 |
| 1,2,3,4,7,8‐HxCDF | 4.3 | 7 | 24 | 23 | 33 | 21 | 42 | 24 | 12 | 21 |
| 1,2,3,6,7,8‐HxCDF | 3.6 | 19 | 27 | 30 | 21 | 44 | 26 | 12 | 23 | |
| 2,3,4,6,7,8‐HxCDF | 4.2 | 19 | 20 | 19 | 18 | 39 | 23 | 8.8 | 19 | |
| 1,2,3,7,8,9‐HxCDF | 8.1 | 7.3 | 8 | |||||||
| 1,2,3,4,6,7,8‐HpCDF | 0.4 | 1 | 3.4 | 1.9 | 3.1 | 4.4 | 10 | 4.8 | 1.4 | 4 |
| 1,2,3,4,7,8,9‐HpCDF | 0.5 | 3.6 | 4.2 | 5.6 | 11 | 6.7 | 0.0 | 5 | ||
| OCDF | 1 | 0.3 | 0.4 | 0.5 | 0.4 | 0.0 | 0.0 | 1 | ||
| PCB‐77 | 1.2 | 2.0 | 0.0 | 1 | ||||||
| PCB‐81 | 13 | 9.7 | 11 | |||||||
| PCB‐126 | 35 | 26 | 32 | 36 | 32 | |||||
| PCB‐169 | 31 | 38 | 36 | 36 | 35 | |||||
MWI: Municipal Waste Incinerator. MgO: magnesium oxide. Field: field study. Stable: controlled study.
Steady state is the condition where intake and output are in equilibrium and milk levels no longer rise; study duration is in most cases too short to obtain steady state.
Dose as reported by the authors using the TEFs applied at that time (no effect on TR values).
Milk collected from the whole herd; number of cows not mentioned.
Mean based on the mean values for each study; missing values or zeros were ignored.
De Filippis et al. (2013) compared PCDD/F and DL‐PCB levels in bulk milk from three contaminated farms of buffaloes in the Campania region (Italy) with tissues from individual animals (three animals per farm). Total levels in milk were 48, 42 and 21 pg WHO2005‐TEQ/g fat, most of it coming from the PCDD/Fs. Lipid‐based TEQ levels for both PCDD/Fs and DL‐PCBs in milk and kidney fat were similar, and were twofold higher than in muscle fat. The liver showed 5‐ to 10‐fold higher levels with larger differences for the higher chlorinated PCDD/Fs. For the DL‐PCBs, such sequestration was only clear for PCB‐126. Intake levels were not determined so TRs/BCFs cannot be estimated.
3.1.1.4.2. Meat from cattle
Few studies were performed to investigate the transfer and resulting tissue levels in cattle used for beef production.
Feil et al. (2000) fed four steer calves with 250 g corn/day spiked with nine PCDD/Fs and three DL‐PCBs at levels varying between 83.3 and 750 ng for the different congeners. The mixture was given daily for 120 days, after which the animals were slaughtered. In addition, animals received on average 7.7 kg of corn and some hay. During this period, animals grew from 250 to 350 kg, body fat increased from 4% to 10.5%. The levels in control animals showed that there was a second source of PCDD/Fs, i.e. PCP‐treated wood in the stable. This affected in particular the overall intake of higher chlorinated PCDD/Fs. Blood levels of tetra‐ and penta‐congeners were at or close to maximum levels at the first sampling point (after 4 weeks). Both in this paper and a follow‐up report (Huwe et al., 2001), it was concluded that back fat and perirenal fat reflected the lipid‐based levels in the meat. In the liver, there was a clear accumulation, especially of the higher chlorinated PCDD/Fs. Ratios for the 2,3,7,8‐substituted congeners of lipid‐based levels for liver to adipose tissue were 2 for TCDD/Fs, 10 for PeCDD/Fs, 20 for HxCDD, 50–100 for HpCDD/Fs and > 300 for OCDD/Fs. Based on overall intake during the study, the levels in fat at day 120 and the estimated total body fat, it was calculated that 53% of the TCDD was retained in the body, 37% of the PeCDD, 40% of the 2,3,4,7,8‐PeCDF, but only 1% of the TCDF. Levels in faeces collected on the last day were similar for these four congeners, despite the almost twofold higher TCDF level in the feed. This suggests that in cattle, TCDF is quickly metabolised. For the lower chlorinated PCDD/Fs, BCFs could be calculated assuming that the applied dose could be distributed over the whole meal of 7.7 kg/day. When expressed on lipid base, BCFs in back fat for TCDD, PeCDD, 2,3,4,7,8‐PeCDF and TCDF were 12.9, 8.4, 9.9 and 0.3. These BCFs present the situation in growing animals and after 120 days of exposure. Based on the blood levels, at this stage a kind of steady‐state level might have been reached, but this cannot be seen from the tissue data with only one time‐point. The levels of the DL‐PCBs were not presented in this paper.
Thorpe et al. (2001) treated 10 growing steers with a cocktail of 5 PCDD/Fs (TCDD, PeCDD, 1,2,3,6,7,8‐HxCDD, 2,3,4,7,8‐PeCDF and 1,2,3,4,7,8‐HxCDF; 150 ng/day per congener) for 4 weeks, followed by 1, 14 and 27 weeks without treatment. At 5, 18 and 31 weeks after the start of the dosing, 3, 3 and 4 animals were slaughtered, respectively, with average body weights at slaughter of 342, 430 and 489 kg. In those slaughtered 1 week after the last treatment, levels in perirenal and subcutaneous fat were very similar for the five PCDD/Fs, with average levels of 49, 41, 26, 38 and 31 pg/g fat for TCDD, PeCDD, 1,2,3,6,7,8‐HxCDD, 2,3,4,7,8‐PeCDF and 1,2,3,4,7,8‐HxCDF, respectively. Lipid‐based levels in the liver were, respectively, 9‐, 12‐, 17‐, 14‐ and 15‐fold higher, again demonstrating the sequestration in this tissue. Contrary to Feil et al. (2000), lipid‐based levels in muscle tissue were five‐ to sevenfold higher than those in adipose fat. The levels in both types of fat tissue after the depuration period of 14 weeks were lower by a factor of 2 to 5 for all congeners, which could not be explained by only the increase in body weight. However, as shown by Feil et al. (2000), also the percentage of body fat may have increased, but this was not investigated. In muscle tissue, the decrease was much higher, suggesting some redistribution between muscle and body fat. However, depending on the congener, the muscle levels were still up to threefold higher than in adipose tissue, both after 14 and 27 weeks after the last treatment. Levels in animals slaughtered after 14 or 27 weeks were very similar, despite the further weight gain. To calculate BCFs, feed consumption is required, which was not reported. When assuming a similar daily amount as in the study by Feil et al. (2000), i.e. 8 kg, BCFs of 2.7, 2.5, 1.6, 1.8, and 1.6 were obtained for TCDD, PeCDD, 1,2,3,6,7,8‐HxCDD, 2,3,4,7,8‐PeCDF and 1,2,3,4,7,8‐HxCDF, as compared to, respectively, 13, 13, 7, 14 and 8 for muscle. The BCFs for fat tissue are clearly lower than those by Feil et al. (2000), but the exposure period was 3.4‐fold shorter and 5 weeks may be too short to reach steady‐state conditions.
3.1.1.4.3. Sheep
Sheep meat can be derived both from young animals (less than one year‐old) and older ones (e.g. ewes). In 2011, EFSA evaluated the risks of consumption of sheep livers (EFSA CONTAM Panel, 2011) (see Section 1.3.3), following reports that these can contain high TEQ levels (Bruns‐Weller et al., 2010). Comparison with lipid‐based levels in meat and fat showed that the high levels point to sequestration in the liver.
Only one study with sheep was identified that had been performed under controlled conditions (Hoogenboom et al., 2015b). Young blackhead sheep were fed up to 112 days with grass obtained from a flood plain, containing an elevated level of PCDD/Fs (1.7 ng WHO2005‐TEQ/kg; 2.5 times the ML) and to some extent also DL‐PCBs (0.3 ng WHO2005‐TEQ/kg) due to the presence of contaminated soil. PCDD/F levels in adipose tissue showed an initial rapid increase which then levelled off around day 30, with a maximum PCDD/F level of 2.1 pg WHO2005‐TEQ/g fat. The levels for DL‐PCBs appeared to peak at 30 days (2.3 pg WHO2005‐TEQ/g fat) and then slightly decreased to about 1.7 pg WHO2005‐TEQ/g fat (at the end of the study). Table 5 shows the BCF values for PCDD/Fs in adipose tissue on day 112 based on the ratio of the levels with those in the grass. It is evident that higher chlorinated PCDD/Fs show lower BCFs than lower chlorinated ones, with the exception of TCDF and 1,2,3,7,8‐PeCDF, similar as observed for cows. Although DL‐PCBs were detected in fat, the straw contributed to the exposure, making an estimation of BCFs difficult.
Table 5.
Bioconcentration factors (BCFs) for adipose tissue of sheep and pigsa
| Hoogenboom et al. (2015b)c | Hoogenboom et al. (2004b) | Spitaler et al. (2005)b | |
|---|---|---|---|
| Sheep | Pigs | Pigs | |
| Duration (days) | 112 | 7 | 84 |
| 2,3,7,8‐TCDD | 2.35 | 0.86 | 1.41 |
| 1,2,3,7,8‐PeCDD | 2.73 | 0.60 | 1.39 |
| 1,2,3,4,7,8‐HxCDD | 2.04 | 0.78 | 3.15 |
| 1,2,3,6,7,8‐HxCDD | 2.04 | 0.15 | 1.71 |
| 1,2,3,7,8,9‐HxCDD | 0.47 | 0.29 | 0.30 |
| 1,2,3,4,6,7,8‐HpCDD | 0.26 | 0.11 | 2.96 |
| OCDD | 0.04 | 0.06 | 2.60 |
| 2,3,7,8‐TCDF | 0.11 | 0.11 | 0.17 |
| 1,2,3,7,8‐PeCDF | 0.09 | < 0.01 | 0.06 |
| 2,3,4,7,8‐PeCDF | 1.66 | 0.62 | 1.87 |
| 1,2,3,4,7,8‐HxCDF | 0.72 | 0.58 | 2.42 |
| 1,2,3,6,7,8‐HxCDF | 0.58 | 0.54 | 1.63 |
| 2,3,4,6,7,8‐HxCDF | 0.42 | 0.29 | 0.90 |
| 1,2,3,7,8,9‐HxCDF | nd | < 0.04 | |
| 1,2,3,4,6,7,8‐HpCDF | 0.12 | 0.33 | 1.56 |
| 1,2,3,4,7,8,9‐HpCDF | 0.15 | 0.21 | 0.56 |
| OCDF | 0.01 | < 0.04 | 0.08 |
| PCB‐77 | 0.12 | ||
| PCB‐81 | nd | ||
| PCB‐126 | 0.78 | ||
| PCB‐169 | 1.15 | ||
| PCB‐105 | 0.69 | ||
| PCB‐114 | 0.71 | ||
| PCB‐118 | 0.80 | ||
| PCB‐123 | nd | ||
| PCB‐156 | 0.93 | ||
| PCB‐157 | 1.13 | ||
| PCB‐167 | 0.76 | ||
| PCB‐189 | 0.89 |
nd: not detected in the feed.
Bioconcentration factors based on the ratio between the level in adipose tissue at the end of the exposure and that in feed.
Based on the highest dose group of 4 ng WHO1998‐TEQ/kg feed.
BCFs for kidney fat and grass pellets. PCBs also derived from straw and therefore not presented.
The corresponding levels in the liver were 59 and 25 pg WHO2005‐TEQ/g fat, respectively. The high TEQ levels in the liver clearly demonstrated sequestration. Ratios of lipid‐based levels in liver and fat were relatively low for most PCDDs, TCDF and 1,2,3,7,8‐PeCDF, but higher than 50 for HpCDD, OCDD and most of the PCDFs. Among DL‐PCBs, PCB‐126 showed the highest ratio (around 19), followed by PCB‐81 (around 8) and PCB‐169 and ‐77 (both around 2), whereas the mono‐ortho PCBs showed ratios around 1. There was also some background contamination from the straw, in the control animals resulting at day 112 in levels in meat of 0.35 and 1.09 pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs, respectively, and in liver of 13 and 20 pg WHO2005‐TEQ/g fat. The levels of DL‐PCBs were similar to those in exposed animals. As such the DL‐PCB levels are difficult to evaluate. In meat, lipid‐based levels of PCDD/Fs were similar to those in adipose tissue, and about two‐thirds of those for DL‐PCBs. Most animals were given clean feed from day 56 to 112. Levels in both livers and fat initially decreased rapidly and then the decrease slowed down, reaching the levels observed in the control animals (112 days on clean feed) within the additional period of 56 days. Part of the decrease could be explained by the gain in body and fat mass.
Similar differences in sequestration of PCDD/Fs and DL‐PCBs in the liver were reported by Fernandes et al. (2011) for sheep exposed at background levels under conventional farming practices.
3.1.1.4.4. Goats
Grova et al. (2002) treated two female goats with a high single oral dose of 0.48 mg 14C‐labelled TCDD and measured the levels of radiolabel in blood, milk, urine and faeces at different time points up to 103 h. Goats weighed around 50 kg and produced 3.3 L milk/ day. Blood levels showed a peak of around 4 ng/mL at the first time point (7 h) and then concentrations gradually decreased over a period of about 70 h. Milk showed the highest level of 10 ng/mL at 22 h, then showed a rapid decrease by about 50% over 24 h, followed by a much slower decrease. This pattern is similar to those observed in dairy cows (see Figure 3). Around 20% of the label was recovered in the faeces up to 103 h, 0.7% in the urine, and 7.8% in milk. The rest was assumed to still be in the tissues.
Costera et al. (2006) investigated the transfer to milk in three goats fed for 10 weeks with contaminated hay (0.8 kg/day) from an area around a hazardous municipal waste incinerator. The level in the hay was 2 and 0.38 ng WHO1998‐TEQ/kg for PCDD/Fs and DL‐PCBs, respectively. Milk levels for PCDD/Fs were 0.86, 2.84, 4.26 and 6.93 pg WHO1998‐TEQ/g fat at day 0, 8, 15 and 71. Levels for the sum of PCDD/Fs and DL‐PCBs at these sampling days were, respectively, 2.46, 4.85, 6.84 and 9.24 pg WHO1998‐TEQ/g fat. The relatively high DL‐PCB level at the start was caused by some contamination of the control hay fed during the adaptation period and the concentrate fed during the whole study period. For most PCDD/Fs, steady‐state levels in milk were estimated to be reached after 37 to 74 days, but earlier for TCDF (20 days) and 1,2,3,7,8‐PeCDF (26 days). The latter congeners also showed a low transfer to milk, as observed in cows and is probably due to metabolism. For DL‐PCBs, steady state was reached within 8–15 days. TRs, estimated from a graph presented in the study, were 60–100% for PCB‐105, ‐114, ‐118, ‐156, ‐157 and ‐189. For PCB‐126, the TR was around 50%, as compared to around 40%, 10% and 25% for the non‐ortho PCB‐77, ‐81 and ‐169. The three most potent PCDD/F congeners, TCDD, PeCDD and 2,3,4,7,8‐PeCDF, showed TRs around 48%, 34% and 28%. TRs decreased with degree of chlorination, with the exception of the two PCDFs mentioned before.
Ounnas et al. (2010) studied the transfer of PCBs from contaminated soil in three lactating goats and observed that steady‐state conditions in milk were obtained after two weeks (total duration 76 days). TRs were estimated at day 45 for part of the PCBs, being 6% and 47% for PCB‐77 and ‐126, and 57%, 59%, 21%, 62%, 52% and 48% for the mono‐ortho PCB‐105, ‐118, ‐123, ‐156, ‐157 and ‐167. TRs were similar to those observed by Costera et al. (2006) with contaminated hay. Goats were slaughtered at the end of the study and body fat and livers were analysed to determine BCFs. For PCB‐126, these were around 4% and 23% for fat and liver, respectively. For other detectable DL‐PCBs, BCFs in liver and adipose tissue were very similar. This suggests that PCB‐126 is the only DL‐PCB that shows clear sequestration in goat liver. Milk levels at the time of slaughter were 0.8‐ to 1.3‐fold of those in adipose tissue for most congeners, that for PCB‐77 showing the highest ratio, being 1.7.
Fournier et al. (2013) fed four lactating goats (66 kg, 3.5 L milk/day) with contaminated corn silage for 39 days followed by a period of 20 days on clean feed. The corn contained PCB contaminated soil due to a fire. The corn level of PCDD/Fs and DL‐PCBs was 4.49 ng WHO2005‐TEQ/kg, primarily due to DL‐PCBs (83%). The corn silage was mixed with commercial feed, hay and soybean meal to give a level of 1.9 ng WHO2005‐TEQ/kg. The levels in milk increased rapidly, then levelled off to reach a highest level of 17.9 pg WHO2005‐TEQ/g fat at day 39. The estimated TRs at the end of the exposure period were 37% for the sum‐TEQ, and 1‐42% for individual PCDD/Fs and 5–73% for DL‐PCBs (no further details provided). After the switch to clean corn silage, the levels decreased rapidly and then entered a slow phase, with estimated half‐lives of 4–7 and 33–59 days (the short depuration period precluded a more precise estimate).
3.1.1.4.5. Pigs
Meat‐producing pigs are normally reared for a period of about 6 months, a period during which they grow to just over 100 kg. In this period, they also increase their body fat content. This has important consequences for the kinetics of PCDD/Fs and DL‐PCBs, since this will slow down the increase in contaminant levels and cause a decrease when the animals are transferred to uncontaminated clean feed. In addition, there are strong indications that some PCDD/Fs are metabolised, in particular TCDF and 1,2,3,7,8‐PeCDF (see below). As a consequence, an increased exposure in the later phase of life will determine the levels in meat and adipose tissue, unless the initial exposure is very high.
A limited number of studies were performed with pigs, both field and controlled. Watanabe et al. (2010) analysed livers, adipose tissue and muscle from adult pigs living near a dumpsite as compared to a reference area. Total TEQ levels in adipose tissue from boars and sows from the first group were both around 5 pg WHO2005‐TEQ/g fat, as compared to 110 (sows) and 170 (boars) pg WHO2005‐TEQ/g fat in the liver, showing sequestration even with relatively low levels in fat tissue. This was also observed in the reference group. The liver to fat ratios increased with higher chlorination, at least for PCDDs (less clear for PCDFs) and was relatively high for PCB‐126 when compared to other DL‐PCBs. Brambilla et al. (2011), studying various groups of farmed and wild pigs on Sicily, also observed liver sequestration of PCDD/Fs and PCB‐126 at low exposure levels, and also reported sequestration for other DL‐PCBs.
Laurent et al. (2002) treated two 40‐kg pigs each with 1 L milk containing 0.35 mg 14C‐labelled TCDD. Levels of radiolabel determined in the portal vein and brachiocephalic artery showed similar level and pattern with a peak at 6 h around 0.34 ng/mL. The levels gradually decreased, at 24 h being 0.2 ng/mL (with an assumed fat level in blood of 0.5% corresponding to 40 ng/g fat). The absorption rate was estimated to be 9%. The CONTAM Panel noted that when assuming 10% body fat and total distribution of the TCDD in the fat, a level around 8 ng/g fat would be expected. This indicates that at 24 h blood fat and adipose tissue are not in equilibrium and that only part of the TCDD is distributed from the blood to the adipose tissue.
Hoogenboom et al. (2004b) exposed five 3‐month‐old piglets for 1 week with diluted feed from the Belgian incident, followed by another 12 weeks on clean feed. At 1, 2, 4 and 8 weeks, biopsy samples of back fat were taken. Feed levels of PCDD/Fs and DL‐PCBs were 48 and 116 ng WHO1998‐TEQ/kg, respectively, again with a high contribution of the mono‐ortho PCBs. Levels of PCDD/Fs just after the exposure were 26 pg WHO1998‐TEQ/g fat, decreasing to 22, 15, 7.4, 3.3 and 1.3 pg TEQ/g fat after 1, 2, 4, 8 and 12 weeks. The corresponding DL‐PCB levels were 97, 73, 55, 32, 19 and 11 pg WHO1998‐TEQ/g fat, respectively, with a contribution of the non‐ortho PCBs of initially 16% and decreasing to 6%. The study shows that after 12 weeks, even with the high PCDD/F level in feed (60 times the ML), the level in fat decreased to around the ML. BCFs were calculated based on the ratio between the fat level measured on day 7 and the feed level (Table 5). The BCFs decrease with higher chlorination, with the exception of TCDF and 1,2,3,7,8‐PeCDF. DL‐PCBs show similar BCFs as the lower chlorinated PCDD/Fs, except for PCB‐77. For longer exposure times, application of these BCFs will result in an overestimation of the levels, as also predicted by the kinetic model. Data from this study were used to develop a toxicokinetic model (Hoogenboom et al., 2007). This model includes growth curves for pigs and should also apply to older pigs but this has not been validated. According to this model, the levels in the fat following exposure for an 8‐week period would be about 4 times that obtained after 1 week.
Spitaler et al. (2005) treated female pigs with feeds spiked with a mixture of PCDD/Fs at total TEQ levels around 0, 0.75, 2 and 4 ng WHO1998‐TEQ/kg feed for the last 12 weeks in the fattening period. This resulted in levels in the belly fat of 0.1, 1.4, 4.0 and 5.4 pg WHO1998‐TEQ/g fat. Lipid‐based concentrations in fore‐end and loin were not consistent with those in belly fat. Livers of these animals fed with 0.75 and 4 ng WHO1998‐TEQ/kg feed contained levels of 82 and 525 pg WHO1998‐TEQ/g fat clearly indicating sequestration at feed levels one‐ and fivefold the ML. Table 5 includes BCFs calculated for this group based on the levels in belly fat after 12 weeks. In general, BCFs are only two‐ to threefold higher than those observed after 1 week in the above‐mentioned study, which can be explained by the longer exposure period and the plateau of the levels. When comparing BCFs for congeners, there is a reasonable agreement between the two studies. Clear exceptions are HpCDD, OCDD, 1,2,3,6,7,8‐HxCDD and TCDD. Such differences may be caused by the relatively low levels measured in the meat and the inherent uncertainty in measuring the levels.
In another study, weaned pigs were treated orally for 11 weeks with capsules containing a mixture of eight PCDD/Fs and PCB‐126 (3 dose levels plus control; dosing equivalent to 0, 1, 10 and 100 ng WHO1998 WHO1998‐TEQ/kg feed) (Shen et al., 2012a,b).21 Biopsy samples of fat taken at the end of the exposure period contained 0.02, 3.23, 37.1 and 193 pg WHO1998‐TEQ/g fat. Animals were killed after an additional 10 weeks on clean feed, showing levels in adipose tissue of, respectively, 0.0, 0.7, 6.6 and 27 pg WHO1998‐TEQ/g fat (Schramm et al., 2009). Shen et al. (2012a) reported these levels in adipose tissue to be 0.0, 0.7, 6.4 and 22 pg WHO2005‐TEQ/g fat, as compared to 1.3, 5.9, 224 and 351 pg WHO2005‐TEQ/g fat in livers (fat levels possibly lower due to use of WHO2005‐TEF). These levels are lower than observed by Spitaler et al. (2005) but may be related to differences in the study (age, congener mixture). Nevertheless, the higher lipid‐based levels in liver are in line with the data of Spitaler et al. (2005) and Watanabe et al. (2010) showing liver sequestration even at low doses, which seems to persist after a long period on clean feed. The high liver sequestration at background levels was also shown by Fernandes et al. (2011), who studied pigs kept indoors and outdoors under routine conditions.
Wittsiepe et al. (2007a) investigated the bioavailability of PCDD/Fs from highly contaminated soil in minipigs treated for 28 days. The absorption, based on levels in adipose tissue and liver, was compared with levels observed in pigs receiving an extract from the same soil. The exposure levels were, respectively, 2.63 and 1.58 ng WHO1998‐TEQ/kg bw per day. Animals were killed on day 29 and various tissues were collected. The obtained results showed that the absorption from soil was relatively poor, on average 28% compared to the extract. TCDF, 1,2,3,7,8‐PeCDF, 1,2,3,7,8,9‐HxCDD and ‐HxCDF showed low levels in tissues, as did TCDD in the case of the soil. The study also showed a high sequestration in the liver for all PCDD/Fs and a liver to adipose tissue ratio which was increasing with higher chlorination. Since the exposure was via a small amount of soil or extract, no BCFs can be calculated from this study.
In addition to the toxicokinetic model described above, the studies by Hoogenboom et al. (2004b) were used by Adolphs et al. (2013) for building a probabilistic model to describe the fate of PCDD/Fs in pigs, although the study focusses on one congener, 1,2,3,4,7,8‐HxCDD. The model was successfully applied using data from the German incident that was discovered at the end of 2010 and concerned the use of industrial fatty acids for producing feeds (see Section 1.3.1). In this incident, two HxCDDs contributed most to the PCDD/F‐TEQ level. The model was also tested on the data from Spitaler et al. (2005) but could not describe them equally well.
3.1.1.4.6. Eggs from chicken and ducks
A number of studies have been published on the transfer of PCDD/Fs and DL‐PCBs in eggs of laying hens. The production of an egg, and in particular the fat containing egg yolk, requires about ten days. Still, there is a rapid increase in the levels in eggs in time, which gradually levels off when reaching a steady state (Figure 3). After termination of the exposure, concentrations initially decline rapidly and then level off again. Table 6 shows TRs and BCFs described in a number of studies for laying hens under (semi‐)steady‐state conditions.
Table 6.
Transfer rates (TRs) and bioconcentration factors (BCFs) for the transfer of PCDD/Fs and DL‐PCBs from feed to eggs or meat in laying hens, broilers and ducks
| Hoogenboom et al. (2006) | Pirard and De Pauw (2006) | Shih et al. (2009)b , c | Iben et al. (2003) | Hoogenboom et al. (2004b) | |
|---|---|---|---|---|---|
| Study design | Stable | Stable | Stable | Stable | Stable |
| Animals | Laying hens | Laying ducks | Laying ducks | Broilers | Broilers |
| Number/group | 12–26 | 3 | 6 | 7 | |
| Source | Standards | Fly ash | Fly ash | Standards | PCB feed |
| Level feed (ng TEQ/kg) | 1.85/3.95 | 30 | 0.70/1.3 | 2.1/3.9 | 48 |
| Duration exposure (days) | 56 | 98 | 41 | 14–42 | 7 |
| Withdrawal period (days) | 56 | – | 19 | 28 | 21 |
| TR or BCF | BCF (TR)e eggs | BCFa eggs | BCF meat | BCF eggs | BCF meat | BCF meat | BCF meat |
|---|---|---|---|---|---|---|---|
| Time point (days) | 56 | 70 | 98 | 33+41d | 41 | 42 | 7 |
| 2,3,7,8‐TCDD | 10.0 (44) | 4.3/2.3 | 5.0/19.2 | 4.0/5.9 | 2.6 | ||
| 1,2,3,7,8‐PeCDD | 9.7 (43) | 9.0 | 9.4 | 3.1/2.7 | 4.4/11.5 | 6.8/4.2 | 1.8 |
| 1,2,3,4,7,8‐HxCDD | 9.7 (43) | 10.6 | 7.0 | 2.9/2.4 | 3.5/5.2 | 3.0/3.5 | 2.3 |
| 1,2,3,6,7,8‐HxCDD | 10.0 (44) | 9.5 | 6.1 | 3.6/2.9 | 3.8/5.6 | 3.0/4.1 | 1.4 |
| 1,2,3,7,8,9‐HxCDD | 7.5 (33) | 5.8 | 4.1 | 1.5/1.2 | 1.6/1.3 | 2.2/2.7 | 1.5 |
| 1,2,3,4,6,7,8‐HpCDD | 4.9 (22) | 2.6 | 2.7 | 1.5/0.9 | 0.7/0.4 | 1.1/1.1 | 0.9 |
| OCDD | 2.4 (11) | 0.6 | 2.7 | 1.1/0.8 | 2.7/0.4 | 0.2/0.7 | < 0.1 |
| 2,3,7,8‐TCDF | 9.1 (40) | 10.2 | 11.3 | 7.6/5.0 | 11.1/29.8 | 5.2/4.8 | 0.3 |
| 1,2,3,7,8‐PeCDF | 9.8 (44) | 8.8 | 8.2 | 3.8/3.7 | 4.6/6.8 | 6.1/4.3 | 0.4 |
| 2,3,4,7,8‐PeCDF | 9.3 (41) | 8.1 | 7.0 | 4.1/3.9 | 5.0/11.0 | 4.7/3.8 | < 0.5 |
| 1,2,3,4,7,8‐HxCDF | 9.9 (44) | 9.2 | 3.6 | 2.1/2.1 | 2.1/1.9 | 3.2/3.3 | 3.0 |
| 1,2,3,6,7,8‐HxCDF | 9.3 (41) | 7.1 | 3.3 | 2.2/2.1 | 2.0/1.8 | 3.2/2.5 | 2.2 |
| 2,3,4,6,7,8‐HxCDF | 7.6 (33) | 5.0 | 2.1 | 0.5/0.4 | 0.4/0.6 | 2.4/2.0 | 1.7 |
| 1,2,3,7,8,9‐HxCDF | 9.1 (40) | 2.5 | 1.2 | 2.0/1.7 | 1.7/1.2 | 3.4/2.6 | 0.6 |
| 1,2,3,4,6,7,8‐HpCDF | 4.0 (18) | 1.6 | 1.1 | 0.9/0.6 | 0.3/0.2 | 0.6/0.6 | 1.0 |
| 1,2,3,4,7,8,9‐HpCDF | 4.6 (21) | 1.9 | 0.5 | 1.1/0.9 | 0.4/0.5 | 0.6/1.2 | < 0.3 |
| OCDF | 1.3 (6) | 0.4 | 1.2 | 0.3/0.2 | 0.8/0.2 | 0.3/0.5 | 0.04 |
| PCB‐77 | 10.9 (48) | 2.9 | |||||
| PCB‐81 | 9.3 (41) | nd | |||||
| PCB‐126 | 11.1 (49) | 4.3 | |||||
| PCB‐169 | 11.5 (51) | 3.7 | |||||
| PCB‐105 | 11.2 (49) | nd | |||||
| PCB‐114 | 11.8 (52) | 2.1 | |||||
| PCB‐118 | 11.3 (50) | 2.0 | |||||
| PCB‐123 | 16.9 (74) | 2.5 | |||||
| PCB‐156 | 12.9 (57) | 2.7 | |||||
| PCB‐157 | 13.2 (58) | 2.5 | |||||
| PCB‐167 | 18.2 (80) | 2.3 | |||||
| PCB‐189 | 13.9 (61) | 1.9 |
TR: Transfer rate; BCF: bioconcentration factor.
BCF based on lipid levels in eggs in week 10 and feed levels in dry weight.
Dose as reported by the authors using the TEFs applied at that time (no effect on COR values).
BCFs eggs based on mean day 33 and 44, BCFs meat based on day 41.
TRs for eggs could not be derived due to a lack of information on feed consumption, egg production and egg weights.
Levels in eggs for the two dose levels, averaged for days 33 and 41.
Average TR and BCF for the two highest dose groups, showing similar values.
Hoogenboom et al. (2006) fed six groups of hens (n = 12–26/group) for 56 days with feeds containing six different levels of a mixture of PCDD/Fs and DL‐PCBs in a range of 0.04 to 3.95 ng WHO1998‐TEQ/kg (contribution PCDD/Fs about 50%). This was followed by a 56‐day withdrawal period on clean feed. Half of the hens were slaughtered after the exposure period, and one group also at intermediate time‐points. The study showed linearity between the levels in feed and eggs. Levels in abdominal fat were about 50–70% of those in egg fat during the exposure period, but slightly higher after the 56‐day withdrawal period. Data from this study were used to develop a toxicokinetic model (Van Eijkeren et al., 2006), which showed that a steady state would require about 200 days, when levels in eggs and abdominal fat would be similar. The steady‐state concentration in eggs would be about 1.5 times higher than the one observed at 56 days in the study. For eggs, half‐lives were calculated being 2.5 and 50 days, respectively, for the initial phase and terminal phase during the elimination period. The levels in body fat did not show biphasic behaviour. Table 6 shows both the TRs and BCFs for the eggs based on the levels at day 56 in the two highest dose groups. In particular the transfer of the hepta‐ and octachlorinated PCDD/Fs was lower than that of the other congeners, whereas DL‐PCBs showed a comparable to slightly higher transfer.
Brambilla et al. (2009) observed an average half‐life of 3.8 weeks (27 days) for the PCDD/F‐TEQ level in eggs during an incident with wood shavings contaminated with PCP. The initial level before the switch to clean feed was 47 pg TEQ/g fat (TEF scheme not reported). There were clear differences in the half‐lives of the PCDD/Fs, being 8.7 and 9.7 weeks for PeCDD and 2,3,4,7,8‐PeCDF, as compared to 1.3 and 1.4 weeks for OCDD and OCDF.
Pirard and De Pauw (2006) performed a study in which fly ash from a steel company was fed to 3 hens for 14 weeks. The study showed that 41 to 93% of the dose was present in excreta, with a higher percentage of the higher chlorinated congeners excreted. This suggests a lower absorption of the higher chlorinated PCDD/Fs, although uptake and subsequent excretion via bile cannot be excluded. As in previous studies, the opposite was observed for the eggs, as shown by the BCFs presented in Table 6. For the lower chlorinated PCDD/Fs, these BCFs are comparable to those described for the study by Hoogenboom et al. (2006), but a bit lower for the higher chlorinated PCDD/Fs. BCFs in breast meat (Table 6) and adipose tissue were in general comparable to those in eggs, although those for the HxCDFs were clearly lower. Eggs reached a highest PCDD/F‐TEQ level of 71, compared to 53 in breast muscle, 64 in abdominal fat and 63 in liver, all expressed in pg WHO1998‐TEQ/g fat. The latter indicates no sequestration, even at this relatively high dose level.
This is contrary to Traag et al. (2006), who fed laying hens with feed from the Belgian incident in 1999 (see Section 1.3.1) (10‐fold diluted; 200 ng WHO1998‐TEQ per kg) for 7 days followed by 42 days on clean feed. Animals were slaughtered at the end of the treatment and after 1, 3 and 6 weeks on clean feed. In particular, the hexa‐ to octachlorinated congeners showed sequestration in the liver but overall, the majority of the TEQs was stored in the body fat. Total TEQ levels in adipose tissue were slightly (1.2‐fold) higher in animals killed after the first week on clean feed than those killed just after the last treatment, whereas TEQ levels in the liver showed a fourfold decrease. This decrease was stronger for PCDD/Fs than for DL‐PCBs, but these showed less sequestration. These data suggest a clear redistribution of PCDD/Fs and DL‐PCBs in the body, in addition to the excretion into the eggs.
Shih et al. (2009) fed three groups of six ducks each with control feed or feed containing 0.3 or 0.6% fly ash for 41 days followed by 19 days on clean feed. The levels of PCDD/Fs were 0.09, 0.70 and 1.3 ng WHO1998‐TEQ/kg with highest contributions of 2,3,7,8,9‐PeCDF and PeCDD, followed by the HxCDFs. Egg levels for the groups fed 0.3% or 0.6% fly ash reached a plateau at 15–21 days with levels around 2 and 3.5 pg WHO1998‐TEQ/g fat, as compared to 1.4 in the controls. After 19 days on clean feed, levels were 2.0, 2.6 and 1.3 pg WHO1998‐TEQ/g fat, respectively. In meat from ducks killed after the last treatment, levels in control, low and high fly ash groups were, respectively, 0.9, 2.4 and 9.5 pg WHO1998‐TEQ/g fat, as compared to 1.3, 3.1 and 2.4 pg WHO1998‐TEQ/g fat after 19 days on clean feed. Variation between ducks may explain some of the inconsistent observations. However, compared to chickens, it appears that steady state is reached sooner in ducks, and that the decrease after the switch to clean feed is low. Table 6 shows the BCFs for both eggs and meat at the end of the exposure period. The BCF for TCDF seems quite high. BCFs decrease with higher chlorination as observed for other species.
3.1.1.4.7. Broilers
Broilers are normally reared for a period of about six weeks with a final body weight around 2 kg. This implies that after an exposure early in life levels will decrease due to the rapid growth.
Iben et al. (2003) fed 10 groups of 7 broilers for 2, 4 or 6 weeks with feed contaminated with standards of PCDD/Fs at levels around 1, 2 or 4 ng WHO1998‐TEQ/kg (confirmed by analysis). Animals fed for 2 or 4 weeks were transferred to clean feed for the remaining period. All animals were killed after the 6 weeks of treatment and all edible meat was collected, mixed and analysed. Lipid‐based levels in the meat of animals fed for 6 weeks with the three contaminated feeds were, respectively, 4.2, 9.1 and 17.1 pg WHO1998‐TEQ/g fat, as compared to 0.5, 1.9 and 2.6 pg WHO1998‐TEQ/g fat for the ones that received contaminated feed only during the first 2 weeks, followed by clean feed (level in controls was 0.0). Table 6 shows the BCFs calculated for the lipid‐based levels divided by the feed levels for animals in the two highest groups fed with contaminated feed for the whole 6 weeks. BCFs decrease with higher chlorination of the congener, from the hexa‐congeners onwards. It was estimated that 21% 20% and 21% of the administered dose (TEQ) was retained in the edible parts after the 6 weeks of treatment. In animals treated for 2 weeks followed by 4 weeks on clean feed 14%, 15% and 15% of the dose was still present at slaughter, suggesting that only a limited part of the dose is lost during the period on clean feed.
Hoogenboom et al. (2004b) treated fifteen 3‐week‐old broilers for 1 week with a diluted feed from the Belgium incident (see Section 1.3.1), followed by a period of up to 3 weeks on clean feed to study the depletion. The level of PCDD/Fs in feed was 48 ng WHO1998‐TEQ/kg. In broilers killed just after the exposure or after another 1 or 3 weeks on clean feed, the levels were 105, 55 and 26 pg WHO1998‐TEQ/g fat, respectively. Corresponding DL‐PCB levels were 116 ng WHO1998‐TEQ/kg feed and 300, 162 and 83 pg WHO1998‐TEQ/g fat in adipose tissue. Overall, the study demonstrates the rapid decrease after termination of the exposure. BCFs for this study are shown in Table 6. As in laying hens, the higher chlorinated PCDD/Fs show a relatively poor accumulation in the fat. DL‐PCBs in general show BCFs which are comparable to those for the lower chlorinated PCDD/Fs. A BCF for total TEQ might be calculated but such a BCF will vary for different congener patterns, and also for different TEF schemes. Application of the WHO2005‐TEFs in the above‐mentioned study would, e.g. cause a considerable change in the TEQ levels, due to the 59% contribution of the mono‐ortho PCBs to the total TEQ level. These factors (congener pattern, TEF scheme) should be considered when establishing transfer rates for total TEQs.
Parera et al. (2008) studied the potential increase of PCDD/Fs in the livers of broilers following treatment with feed containing naturally contaminated sepiolite, a clay widely used in feed. Four groups of animals (n = 6 per group) were fed from day 8 to 39 with either control feed, a feed supplemented with 3% sepiolite, a feed with added PCDD/Fs (via soy oil), or a diet with 2% contaminated kaolinite clay. The levels in feed were, respectively, 0.05, 0.1, 2.9 and 9.2 ng WHO1998‐TEQ/kg (levels in second and fourth feed not determined but estimated from those in clay). The resulting levels in liver were 1.7, 1.6, 42.2 and 20.1 pg WHO1998‐TEQ/g fat. Based on fat levels of 2.7%, 2.4%, 2.7% and 2.5%, this corresponds to 0.04, 0.04, 1.12 and 0.49 pg WHO1998‐TEQ/g liver, respectively. The results show that the level in sepiolite was too low to show an increase of levels in liver, contrary to the kaolinite. However, for kaolinite, the increase in liver was not as expected when compared with the spiked feed, which contained a threefold higher level but resulted in half the liver level. These relative low levels could not be explained by a difference in the congener pattern with the congeners contributing most to the TEQ level in the kaolinite, TCDD and PeCDD, showing levels of 2.81 and 3.51 ng/kg as compared to 0.23 and 1.15 ng/kg, respectively, in the spiked feed. There was a strong increase in levels of HpCDD and OCDD in the liver, the congeners showing the highest levels in clay on an absolute base, but again less than could be expected from the spiked feed. This indicates that the clay reduces the absorption of the PCDD/Fs. No BCFs were provided or could be calculated from this study.
3.1.1.4.8. Summary for ruminants, pigs and poultry
For most farm animal species, there are data, either from controlled studies or incidents, which give insight into the transfer of PCDD/Fs and DL‐PCBs. Studies show that PCDD/Fs and DL‐PCBs are not only accumulated in body fat and liver, but also excreted into eggs and milk. This excretion contributes to lower accumulation in the body and a stronger decrease in the levels after termination of the exposure. In growing animals, the increase in body fat mass is also an important factor in the tissue levels obtained during exposure, and a decrease in levels after termination of the exposure. However, in addition, metabolism and excretion is expected to be involved, although no specific data on these processes in farm animals were identified.
Some of the studies allow the calculation of transfer parameters like TRs or BCFs for dairy cows, laying hens, ducks and pigs. In addition, data have been used to develop toxicokinetic models for dairy cows, laying hens and pigs. These models are more suitable to describe the relation between feed and food, since they are more flexible in terms of exposure duration. In addition, they are useful during incidents in order to estimate the time required to obtain food levels below the regulatory limits. The developed models, and in particular, the model parameters for absorption and metabolism are normally estimated based on the data from one study and require validation against other data sets.
With the exception of one study in beef cows, showing much higher levels in meat, lipid‐based levels of adipose tissue and meat are reported to be quite similar. This is an important observation when using adipose tissue for testing whether non‐compliant levels may be found in other edible tissues. Another specific issue is the sequestration in the liver, since this may result in relatively high lipid‐based levels when compared to meat or adipose tissue. Such sequestration has been observed in almost all animal species. In practice, this is particularly relevant for foraging animals with a relatively high exposure due to e.g. soil consumption, such as sheep.
The data presented above show either TRs or BCFs. The values can be used to estimate the levels in milk, eggs or meat based on levels measured in feed or a feed ingredient. TRs present the fraction of the ingested contaminant that is excreted into eggs or milk. BCFs are the ratio between the level in the food product and that in feed, in most cases based on the level in compound feed or total ration consumed. Both TRs and BCFs will increase with prolonged exposure until the phase where a steady state is obtained. At that stage, the TRs/BCFs are at their maximum value. Levels in edible products will be overestimated when applying TRs/BCFs determined at steady state for only a short‐term exposure. However, the major increase in the levels occurs during the first week of the exposure.
When using TRs to estimate the level in, e.g. milk fat or egg fat, it is important to first estimate the intake level by multiplying the level in the feed (or ingredient) with the daily amount ingested. Subsequently, this intake level can be multiplied with the TR to estimate the total amount excreted into eggs or milk. Based on milk or egg production per day and their fat levels, the total amount of egg or milk fat excreted can be estimated (typical values are 5 g/day for egg fat or 1 kg/day for milk fat). Combining these with absolute amounts excreted will result in an estimate of the level in milk or egg fat.
In the case of BCFs, the level in the feed can be multiplied with the BCF to obtain the level in the edible product of interest. When detected in an ingredient, the level should be extrapolated to the level in the daily ration. As in the case of TRs it is important to realise if the BCFs were determined under steady‐state conditions or after a short‐term exposure.
TRs and BCFs differ for each congener but in practice those for the lower chlorinated and more persistent congeners are more relevant because they contribute most to the TEQ, like PeCDD, 2,3,4,7,8‐PeCDF, TCDD, TCDF (in the case of chickens) and to a lesser extent the hexachlorinated PCDD/Fs. Only in some cases, like where PCP is the contamination source, will higher chlorinated congeners like HpCDD make a significant contribution to the TEQ level. In the case of DL‐PCBs, PCB‐126 and to some extent PCB‐169 are the most relevant congeners in terms of contribution to the TEQ levels.
3.1.1.4.9. Fish
Since muscle (or fillet) is the tissue relevant as food, this section will focus on transfer to this tissue, in particular in the case of salmonids. For other types of fish, liver or liver products may also be consumed (e.g. cod liver or cod liver oil), and accumulation of PCDD/Fs and DL‐PCBs in this tissue will be included where data are available.
Several studies have examined uptake of PCDD/Fs and DL‐PCBs in the field, which reflects dietary and aquatic exposure to mixtures of contaminants (Kuehl et al., 1985, 1987; Servos et al., 1989; Frakes et al., 1993; Hellou et al., 1999; Wu et al., 2001; Bhavsar et al., 2010; Berge et al., 2011; Ruus et al., 2012; Zhu et al., 2015). Other studies have been performed in controlled conditions, following aquatic exposure to PCDD/Fs and/or DL‐PCBs (Sijm et al., 1993; Loonen et al., 1994; Koponen et al., 1998, 2000). However, transfer of PCDD/Fs and DL‐PCBs to fish in field studies and from waterborne exposure are not considered by the CONTAM Panel to be within the scope of the current Opinion.
Numerous studies have been performed on the transfer of PCDD/Fs and DL‐PCBs from feed to fish (Isosaari et al., 2002, 2004; Lundebye et al., 2004; Bell et al., 2005, 2012; Berntssen et al., 2007, 2010a, 2011; Drew et al., 2007; Sprague et al., 2010, 2015; Friesen et al., 2015). Only a few have included both an exposure and an elimination phase (Brambilla et al., 2007; Ábalos et al., 2011; Berntssen et al., 2016). Several studies focused only on TCDD (Branson et al., 1985; Kleeman et al., 1986; Tietge et al., 1998; Jones et al., 2001) or on a selected number of individual PCDD/F or DL‐PCB congeners (Sijm et al., 1990; Coristine et al., 1996; Huuskonen et al., 1996; Brown et al., 2002).
Salmonids
Few of the studies on the transfer of PCDD/Fs and DL‐PCBs from feed to Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss) fillets have reported congener‐specific concentrations in both feed and fish, those which have are described below.
Isosaari et al. (2004) fed graded levels of PCDD/Fs and DL‐PCBs (by varying inclusion levels of different fish oils: (A) 100% Pacific fish oil, (B) 75% Pacific oil + 25% Baltic fish oil, (C) 75% Baltic fish oil + 25% Pacific fish oil or (D) 100% Baltic fish oil) to Atlantic salmon (Salmo salar) for 30 weeks. Levels for the sum of PCDD/Fs and DL‐PCBs were 2.9, 5.0, 8.7 and 10 pg WHO1998‐TEQ/g feed, respectively, with DL‐PCBs contributing 93%, 69%, 55% and 53% of the TEQ. The PCDD/F and DL‐PCB concentrations in both fillet and whole body of salmon increased with increasing dietary exposure, in fillets ranging from 2.1 to 5.2 pg WHO1998‐TEQ/g ww after 30 weeks. A higher proportion of the sum of DL‐PCBs (83%) were retained in the fillet, than for the sum of PCDD/Fs (43%). Tetra‐ and pentachlorinated PCDD/F congeners were preferentially accumulated in salmon compared to the higher chlorinated congeners. At the end of the exposure period, whole fish BCFs for PCDD/Fs ranged from 0.52 (HpCDD) to 1.38 (TCDD). For DL‐PCBs, the BCFs ranged from 1.35 (PCB‐123) to 1.89 (PCB‐81). Lipid‐normalised PCDD/F and DL‐PCB concentrations indicated that congeners were equally partitioned in whole fish and fillets. Approximately 30% of the total PCDD/F and PCB content in salmon was present in skinned fillet. Absorption and lipid‐normalised BCFs were not affected by the PCDD/F and PCB levels in the feeds.
In a study by Sprague et al. (2010), Atlantic salmon were fed one of three diets for 11 weeks to compare the fatty acid composition and levels of POPs in the fillets. The feeds contained either northern fish oil (NFO), the same oil after decontamination (deNFO) or a mixture of southern fish oil, rapeseed and soya bean oils (SFO/RO/SO): levels for the sum of PCDD/Fs and DL‐PCBs were 17.4, 0.45 and 0.54 pg WHO2005‐TEQ/g feed, respectively. At the end of the study, the fillet concentration of the sum of PCDD/Fs and DL‐PCBs in salmon fed the NFO was 6.42 pg WHO2005‐TEQ/g, whereas the fillet levels in salmon fed the deNFO and SFO/RO/SO diets were 0.34 and 0.41 pg WHO2005‐TEQ/g, respectively. BCFs calculated from this study are given in Table 7.
Table 7.
Bioconcentration factors (BCFs) in Atlantic salmon fillets from controlled feeding trials with varying PCDD/F and DL‐PCB concentrations depending on the type of oil (vegetable vs fish oil) incorporated in the feed
| Sprague et al. (2010) | Bell et al. (2012) | Sprague et al. (2015) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Main source PCDD/F + DL‐PCB | NFO | deNFO | VO | Lean FO | Fat FO | Com FO | Lean VO | Fat VO | Com VO | NFO | SFO | 11% AM | 5.5% AM |
| Number of fish | 9 | 9 | 9 | 20 | 20 | 20 | 20 | 20 | 20 | 18 | 18 | 18 | 18 |
| Exposure duration (weeks) | 11 | 11 | 11 | 55 | 55 | 55 | 55 | 55 | 55 | 19 | 19 | 19 | 19 |
| Feed sum PCDD/F (pg TEQ/g) | 8.64 | 0.27 | 0.25 | 1.10 | 1.10 | 1.10 | 0.37 | 0.37 | 0.37 | 0.52 | 0.24 | 0.04 | 0.05 |
| Feed sum DL‐PCB (pg TEQ/g) | 8.76 | 0.18 | 0.29 | 4.25 | 4.25 | 4.25 | 1.06 | 1.06 | 1.06 | 1.30 | 0.73 | 0.03 | 0.04 |
| Fillet sum PCDD/F + DL‐PCB | 6.42 | 0.34 | 0.41 | 1.93 | 1.94 | 1.95 | 0.44 | 0.48 | 0.45 | ||||
| 2,3,7,8‐TCDD | 0.31 | 0.33 | 0.18 | 0.23 | 0.17 | 0.25 | 0.38 | 0.36 | 0.21 | 0.33 | 0.94 | 1.00 | 1.00 |
| 1,2,3,7,8‐PeCDD | 0.26 | 0.16 | 0.79 | 0.42 | 0.37 | 0.44 | 0.28 | 0.28 | 0.18 | 0.28 | 0.57 | 1.10 | 1.20 |
| 1,2,3,4,7,8‐HxCDD | 0.14 | 0.22 | 0.29 | 0.44 | 0.44 | 0.22 | 0.40 | 0.40 | 0.40 | 0.53 | 1.00 | 1.00 | 1.00 |
| 1,2,3,6,7,8‐HxCDD | 0.07 | 0.15 | 0.17 | 0.09 | 0.09 | 0.12 | 0.22 | 0.22 | 0.22 | 0.10 | 0.50 | 0.37 | 0.43 |
| 1,2,3,7,8,9‐HxCDD | 0.14 | 0.43 | 0.40 | 0.11 | 0.11 | 0.11 | 0.67 | 0.67 | 1.33 | 0.91 | 1.00 | 1.00 | 1.00 |
| 1,2,3,4,6,7,8‐HpCDD | 0.07 | 0.25 | 0.29 | 0.14 | |||||||||
| OCDD | |||||||||||||
| 2,3,7,8‐TCDF | 0.34 | 1.00 | 0.87 | 0.87 | 0.21 | 0.20 | 0.20 | 0.04 | 0.26 | 0.33 | 0.53 | 3.57 | 2.54 |
| 1,2,3,7,8‐PeCDF | 0.17 | 0.33 | 1.00 | 1.00 | 0.11 | 0.14 | 0.09 | 0.00 | 0.16 | 0.33 | 0.23 | 1.33 | 2.00 |
| 2,3,4,7,8‐PeCDF | 0.21 | 0.31 | 0.52 | 0.52 | 0.56 | 0.54 | 0.55 | 0.02 | 0.55 | 0.31 | 0.31 | 2.00 | 1.00 |
| 1,2,3,4,7,8‐HxCDF | 0.07 | 0.08 | 0.40 | 0.40 | 0.10 | 0.10 | 0.10 | 0.00 | 0.12 | 1.00 | 0.50 | 1.00 | 1.00 |
| 1,2,3,6,7,8‐HxCDF | 0.06 | 0.17 | 0.40 | 0.40 | 0.08 | 0.08 | 0.08 | 0.00 | 0.11 | 0.22 | 0.31 | 1.00 | 1.10 |
| 2,3,4,6,7,8‐HxCDF | 0.08 | 0.13 | 0.13 | 0.13 | 0.15 | 0.15 | 0.15 | 0.11 | 0.22 | 0.24 | 0.85 | 1.00 | 1.00 |
| 1,2,3,7,8,9‐HxCDF | 0.17 | 0.08 | 0.22 | 0.22 | 0.15 | 0.07 | 0.15 | 0.20 | 0.20 | 1.00 | 1.00 | 1.00 | 1.20 |
| 1,2,3,4,6,7,8‐HpCDF | 0.10 | 0.33 | 1.75 | 0.17 | |||||||||
| 1,2,3,4,7,8,9‐HpCDF | 0.33 | 1.00 | 0.25 | 0.50 | |||||||||
| OCDF | |||||||||||||
| PCB‐77 | 14.00 | 2.00 | 0.46 | 0.46 | 0.46 | 0.40 | 0.40 | 0.40 | 0.42 | 0.43 | 5.50 | 1.60 | |
| PCB‐81 | 0.05 | 0.33 | 0.43 | 3.00 | |||||||||
| PCB‐126 | 0.49 | 1.63 | 0.96 | 0.34 | 1.11 | 0.34 | 0.28 | 0.32 | 0.31 | 0.50 | 0.59 | 7.04 | 4.76 |
| PCB‐169 | 0.80 | 0.29 | 1.00 | 0.30 | 0.29 | 0.29 | 0.43 | 0.43 | 0.35 | 0.40 | 0.34 | 5.00 | 3.00 |
| PCB‐105 | 0.60 | 3.75 | 1.86 | 0.51 | 0.53 | 0.52 | 0.43 | 0.46 | 0.46 | 0.49 | 0.59 | 7.00 | 3.00 |
| PCB‐114 | 0.57 | 1.50 | 2.00 | 0.53 | 0.53 | 0.53 | 0.54 | 0.54 | 0.54 | 0.56 | 0.50 | ||
| PCB‐118 | 0.45 | 3.30 | 2.00 | 0.51 | 0.52 | 0.52 | 0.04 | 0.39 | 0.41 | 0.49 | 0.57 | 9.00 | 3.50 |
| PCB‐123 | 0.80 | 0.81 | 0.89 | 0.89 | 1.00 | 1.00 | 1.00 | 0.30 | 0.29 | ||||
| PCB‐156 | 0.58 | 1.07 | 1.42 | 0.58 | 0.61 | 0.61 | 0.48 | 0.48 | 0.51 | 0.54 | 0.75 | 7.00 | |
| PCB‐157 | 0.59 | 1.38 | 2.00 | 0.54 | 0.58 | 0.54 | 0.43 | 0.43 | 0.43 | 0.53 | 0.63 | ||
| PCB‐167 | 0.80 | 0.67 | 0.67 | 0.67 | 0.57 | 0.73 | |||||||
| PCB‐189 | 0.60 | 0.67 | 0.67 | 0.67 | 0.67 | 0.57 | 0.67 | ||||||
FO: fish oil; VO: vegetable oil; NFO: northern fish oil; deNFO: decontaminated northern fish oil; Lean: Salmon strain with low flesh adiposity; Fat: Salmon strain with high flesh adiposity; Com: Non‐pedigreed commercial families of Atlantic salmon; Number of fish: 9: triplicate samples analysed of 3 fish; 20: duplicate samples of 10 fish; 30: triplicate samples of 10 fish.
In another study, Sprague et al. (2015) fed Atlantic salmon a diet containing either fish oil of a northern (NFO), southern (SFO) hemisphere origin or DHA‐rich (Schizochytrium sp.) algal meal at two inclusion levels (11% and 5.5% of diet) for 19 weeks. Concentrations of the sum of PCDD/Fs and DL‐PCBs in feed differed substantially among treatments in the order of NFO > SFO > 11% algal meal/5.5% algal meal and this was reflected in the fillets. BCFs for the various PCDD/F and DL‐PCB congeners from feed to fillet are given in Table 7.
Bell et al. (2012) fed genetically different families of salmon, diets with low fishmeal contents and either 100% fish oil (FO) or a blend of vegetable oils (VO), over a full production cycle. Three families of salmon were used; ‘Lean’ or ‘Fat’, based on fillet lipid deposition (from a breeding programme) or a representative of ‘commercial’, farmed salmon. Levels for the sum of PCDD/Fs and DL‐PCBs were 4.25 and 1.06 pg WHO2005‐TEQ/g for the FO and VO feeds, respectively. Fish were fed these diets for 55 weeks until they reached the size of about 3 kg. Average fillet PCDD/F levels across the three salmon strains were 1.94 pg WHO2005‐TEQ/g in the fish‐oil fed salmon and 0.46 pg WHO2005‐TEQ/g in the fish fed vegetable oil. Salmon reared on vegetable oil had on average 52% lower PCDD/Fs concentration and 79% lower DL‐PCB level than fish reared on feed which contained fish oil. BCFs (from feed to fillet) for the specific PCDD/F and DL‐PCB congeners in the different groups of salmon are given in Table 7. The main impact on fillet levels of PCDD/Fs and DL‐PCBs was feed composition; the effects of genetic differences among the salmon families were not statistically significant.
Berntssen et al. (2010a) assessed levels of chemical contaminants in Atlantic salmon reared for an entire seawater production period (12 months; from 0.3 to approximately 4 kg) on traditional feed (containing fishmeal and FO) or feed based on a combination of alternative ingredients (including wheat‐ and corn gluten, extracted soybean meal, krill meal, linseed‐, palm‐ and rapeseed oil). The use of alternative feed ingredients reduced the fillet levels of both sum PCDD/Fs and DL‐PCBs by 71% compared to traditionally raised salmon.
Berntssen et al. (2007) performed a cross‐over design feeding trial with growing Atlantic salmon (for 5 months), which had previously been reared on either VO‐ or FO‐based feeds. The results of these experiments were used to develop a one‐compartmental transfer model for PCDD/Fs and DL‐PCBs from feed to whole fish. Tetra‐ and pentachlorinated PCDD/Fs had a higher absorption than higher chlorinated congeners, the fraction absorbed ranging from 89% for TCDD, to 9% for 1,2,3,4,6,7,8‐HpCDF, whereas in the case of OCDD and OCDF no absorption could be detected. All investigated DL‐PCBs showed an absorption fraction comparable to tetra‐ and pentachlorinated PCDD/Fs (range: 73–83%). Elimination rate constants were twofold higher for PCDD/Fs than for DL‐PCBs. Lower chlorinated PCDDs had a twofold lower elimination rate than the higher chlorinated PCDDs, whereas no differences were apparent for either PCDF or DL‐PCB congeners. For each of the analysed PCDD/Fs and DL‐PCBs, the analysis allowed for the calculation of congener specific BCFs. For PCDDs, BCFs were low and ranged from 1.6 for PeCDD to 0.2 for HpCDD. Similarly, BCFs for PCDFs ranged from 0.98 (TCDF) to 0.13 (1,2,3,4,6,7,8‐HpCDF), whereas BCFs for DL‐PCBs were higher (range: 1.1–14.8). The calibrated model (Berntssen et al., 2007) was used to predict PCDD/F and DL‐PCB levels in Atlantic salmon reared under commercial conditions. Predictions were based on pre‐analysed concentrations of PCDD/Fs and DL‐PCBs in feeds with three different inclusion levels of VO (0, 30% or 60%). The difference between the predicted and the measured concentrations varied between 0 and 11% in the commercially reared salmon.
The previously calibrated one‐compartmental transfer model for PCDD/Fs and DL‐PCBs in Atlantic salmon (Berntssen et al., 2007, see above) was used by Berntssen et al. (2016) to simulate levels of PCDD/Fs and DL‐PCB in farmed salmon fillets based on different feed composition. The transfer model was validated by two independent feeding trials, with a significant linear correlation (r2 = 0.96; n = 116; p < 0.0001) between observed and predicted values. Model fillet predictions were conducted for four different scenarios:
Commercial feed composition in 1999 (high fish oil, approx. 30% FO, typical feed sum of PCDD/Fs and DL‐PCBs was 3 ng WHO2005‐TEQ/kg),
Commercial feed composition in 2013 (low fish oil, 11%, typical feed sum of PCDD/Fs and DL‐PCBs was 1 ng WHO2005‐TEQ/kg),
Expected feed future composition with low fish oil and fishmeal,
Expected future feed composition with decontaminated fish oil and low fishmeal.
Model predictions of the sum of PCDD/F and DL‐PCB concentrations in 1999 and 2013 of 1.05 and 0.57 ng WHO2005‐TEQ/kg, respectively, corresponded well with data from the national monitoring programmes for these years which gave estimates of 1.1 and 0.52 ng WHO2005‐TEQ/kg, respectively. Concentrations of PCDD/Fs and DL‐PCBs in commercial salmonid feed were reduced by approximately 60% from 2003–2013 (Sissener et al., 2013; VKM, 2014). More recent data showed a further reduction, from 1.09 in 2013 to 0.76 ng WHO2005‐TEQ/kg (sum of PCDD/Fs and DL‐PCBs) (Sanden et al., 2016).
The scenarios with low fishmeal and oil, and low fishmeal and decontaminated fish oil gave predicted concentrations of 0.41 and 0.27 ng WHO2005‐TEQ/kg salmon fillet (Berntssen et al., 2016).
Other fish
Accumulation (30 days) and depuration (180 days) of three dietary concentrations of 2,3,7,8‐[3H]‐TCDD were compared in rainbow trout (Oncorhynchus mykiss) and lake whitefish (Coregonus clupeaformis) by Fisk et al. (1997). Despite similar body size and muscle lipid content. Absorption of TCDD was higher in lake whitefish (66–76%) than in rainbow trout (43–58%), whereas BCFs ranged from 1.6 to 1.8 in rainbow trout and from 0.8 to 0.9 in lake whitefish. TCDD half‐lives were not concentration dependent, but were shorter in lake whitefish (32–39 days) than in rainbow trout (73–83 days) which indicates either a greater capacity for biotransformation of the TCDD or other physiological differences between the two species such as growth rate.
Few studies in fish other than salmonids have simultaneously reported concentrations of all PCDD/F and DL‐PCB congeners assigned WHO‐TEFs in both feed and fish. Blanco et al. (2007) examined the transfer of PCDD/Fs and DL‐PCBs in turbot (Psetta maxima) raised on commercial fish feed for 1–2 years. Levels of PCDD/Fs and DL‐PCBs in turbot were 0.13–0.27 pg WHO1998‐TEQ/g and 0.35–1.2 pg WHO1998‐TEQ/g, respectively. The absorption of DL‐PCBs ranged between 30% and 46%, and between 11% and 42% for PCDD/Fs (see Table 8). DL‐PCBs had higher accumulation than PCDDs in turbot as reported for salmonids (see section above). Lipid‐based BCFs were approximately 1.5 for all DL‐PCBs and tetra‐ and pentachlorinated PCDD/Fs, but were lower for higher chlorinated PCDD/Fs (Table 8).
Table 8.
Bioconcentration factors (BCFs) and fraction absorbed (FA) of PCDD/Fs and DL‐PCBs in fish from a controlled feeding trial on turbot
| Blanco et al. (2007) | ||
|---|---|---|
| FA (%)c | BCF | |
| Feed | Commercial | Commercial |
| Number of fisha | 21 | 21 |
| Exposure duration (weeks) | 1–2 years | 1–2 years |
| Feed sum PCDD/Fb (pg TEQ/g) | 0.5 | 0.5 |
| Feed sum DL‐PCBb (pg TEQ/g) | 1.6 | 1.6 |
| 2,3,7,8‐TCDD | 28 | 1.58 |
| 1,2,3,7,8‐PeCDD | 34 | 1.48 |
| 1,2,3,4,7,8‐HxCDD | ||
| 1,2,3,6,7,8‐HxCDD | 21 | 0.98 |
| 1,2,3,7,8,9‐HxCDD | 42 | 1.14 |
| 1,2,3,4,6,7,8‐HpCDD | 25 | 1.40 |
| OCDD | 11 | 0.81 |
| 2,3,7,8‐TCDF | 33 | 1.55 |
| 1,2,3,7,8‐PeCDF | 29 | 1.41 |
| 2,3,4,7,8‐PeCDF | 31 | 1.26 |
| 1,2,3,4,7,8‐HxCDF | 15 | 0.75 |
| 1,2,3,6,7,8‐HxCDF | 28 | 1.29 |
| 2,3,4,6,7,8‐HxCDF | 13 | 0.77 |
| 1,2,3,7,8,9‐HxCDF | ||
| 1,2,3,4,6,7,8‐HpCDF | 19 | 1.69 |
| 1,2,3,4,7,8,9‐HpCDF | ||
| PCB‐77 | 42 | 1.42 |
| PCB‐81 | 38 | |
| PCB‐126 | 35 | 1.60 |
| PCB‐169 | 30 | 0.74 |
| PCB‐105 | 39 | 1.34 |
| PCB‐114 | 40 | 1.44 |
| PCB‐118 | 39 | 1.51 |
| PCB‐123 | 47 | 1.46 |
| PCB‐156 | 32 | 1.48 |
| PCB‐157 | 33 | 1.51 |
| PCB‐167 | 31 | 1.53 |
| PCB‐189 | 26 | 1.62 |
BCF: bioconcentration factor; NFO: northern fish oil; deNFO: decontaminated northern fish oil; 11% AM: 11% algal meal; 55% AM: 5.5% algal meal.
Number of fish: 19, triplicate samples analysed of 6 fish.
Sum of PCDD/F and DL‐PCB in pg WHO2005‐TEQ/g in Sprague et al. (2015), and in pg WHO1998‐TEQ/g in Blanco et al. (2007).
AE, assimilation efficiency: Cfish/(Ft Cfeed),where Cfish and, Cfeed is concentration in fish and feed, F is feeding rate, and t is time.
The transfer of PCDD/Fs and DL‐PCBs from feed to farmed orange spotted grouper (Ephinephelus coioides) over time (in juveniles and after 2, 15, 24 and 36 months) was investigated by Wang and Lee (2010). Bioaccumulation in fish of low chlorinated PCDD/F congeners increased, whereas bioaccumulation of highly chlorinated PCDD/Fs decreased with age. There were no obvious changes in bioaccumulation of DL‐PCBs with time. The contribution of DL‐PCBs to total WHO1998‐TEQ increased from 76% to 91% in fillets from 2‐ month‐old and 3‐year‐old grouper, respectively.
Since cod liver is consumed, it is of interest to assess transfer of PCDD/Fs and DL‐PCB to this tissue. However, since controlled studies are not available for this species, data from wild‐caught fish is considered. Karl et al. (2016) reported PCDD/F concentrations in cod muscle between 0.001 and 0.103 ng WHO2005‐TEQ/kg ww and DL‐PCB concentrations between 0.004 and 0.445 ng WHO2005‐TEQ/kg ww. Liver concentrations of sum PCDD/Fs and DL‐PCB vary considerably depending on the fishing location; between 141–290 ng WHO2005‐PCDD/F‐PCB‐TEQ/kg ww in the Baltic Sea and 19.9–263 ng WHO2005‐PCDD/F‐PCB‐TEQ/kg ww in the North Sea (Karl and Lahrssen‐Wiederholt, 2009). Similarly, Julshamn et al. (2013) reported large variations in levels of sum PCDD/Fs and DL‐PCB in cod liver sampled from different fishing grounds in the Barent Sea (1–151 ng WHO2005‐PCDD/F‐PCB‐TEQ/kg ww).
3.1.1.4.10. Summary for fish
As for terrestrial farm animal species, there are data from experimental‐ and field studies in fish which give insight into the transfer of PCDD/Fs and DL‐PCBs to fish. Studies show that PCDD/Fs and DL‐PCBs are accumulated to a greater extent in fillet of oily fish (such as salmon and trout) than leaner fish, the latter having higher concentrations of these compounds in the liver (as is the case for cod). Limited data indicate that metabolism and excretion of PCDD/Fs and DL‐PCBs occur, depending on the congener in question. The main source of PCDD/Fs and DL‐PCBs in farmed fish is marine feed ingredients in fish feeds including fish oil and fishmeal. In addition to the feed composition, the transfer of PCDD/Fs and DL‐PCBs to fillet depends on other factors such as species, and animal growth.
Some of the studies allow the calculation of the transfer parameter BCF, mainly in salmonids. Toxicokinetic models have been developed for salmon enabling the prediction of fillet concentrations of PCDD/F and DL‐PCBs from known feed concentrations. One of the models has been validated and may be useful for harmonising the regulatory limits for these compounds in feed and fish fillet.
3.1.1.5. Toxicokinetic modelling
Toxicokinetic modelling quantitatively describes absorption, distribution, metabolism and elimination (ADME) of chemicals. In the case of food, the latter comprises the entrance into, and absorption from, the gastrointestinal tract, the uptake in the blood, the distribution by the blood, the interaction of the blood with the organs, i.e. uptake from the blood and release from the organ (determined by the chemical's and organ's biological and physicochemical properties), enzymatic metabolism and excretion via the urine, bile and faeces.
Since 1980 several toxicokinetic models for TCDD and related compounds have been developed. Early toxicokinetic models on PCBs and PCDD/Fs took into account lipid solubility as the central disposition mechanism of these compounds (Tuey and Matthews, 1980; King et al., 1983). These models however could not describe the observed preferential accumulation of TCDD in the rodent liver over the adipose tissue at increasing dose and exposure duration, referred to as ‘hepatic sequestration’. This has led to toxicokinetic models which describe hepatic TCDD disposition in terms of binding to basal and AHR dependent inducible hepatic microsomal protein, in particular CYP1A2 (Leung et al., 1988, 1990a,b). Andersen et al. (1993) were the first to describe TCDD kinetics on the basis of induction of hepatic binding protein in response to interactions of TCDD with the AHR and of the TCDD‐AHR complex with DNA, leading to de novo CYP1A1 and CYP1A2 synthesis and subsequent binding of TCDD to CYP 1A2, based on rats. In addition, blood tissue diffusion was incorporated as part of the model's distribution mechanism. Next to rodent models, Van der Molen et al. (1996) developed a generic lipid‐based toxicokinetic model for background levels of TCDD in the blood of humans.
Wang et al. (1997, 2000) improved previous rodent toxicokinetic models with respect to consistency of model structure, model parametrisation and interspecies scaling. However, next to the liver the Wang model included the lungs, the kidneys, the spleen and the skin as compartments with AHR‐dependent sequestration capacity. As these compartments are no target tissue during gestation and in the absence of human data for these compartments, Emond et al. removed these organs from the toxicokinetic model structure in developing a gestational rat model, i.e. a model including a fetal/placental compartment. Furthermore, TCDD elimination depending on CYP1A2 induction was introduced (Emond et al., 2004, 2006). The rat model was extrapolated to humans and used to analyse the fate of TCDD in the blood of high exposed humans (Ranch Hand cohort, Geusau study) (Emond et al., 2005). The model for humans, as developed by Emond, was used by US‐EPA to derive its chronic RfD for TCDD (US‐EPA, 2012). Emond et al. (2016) extended the human gestational model for breastfeeding by including the mammary gland and the formation/excretion of human milk.
Carrier et al. (1995) developed a relatively simple model for TCDD that includes absorption and subsequent distribution between body fat and liver. The model, called CADM, includes dose‐dependent liver sequestration and increased metabolism due to induced CYP1A enzyme levels. As a result, half‐life in the model increases with decreased exposure, similar as the model by Emond et al. (2006). Aylward et al. (2005) included excretion via the daily loss of small amounts of fat in the faeces ‘due to simple lipid partitioning from the circulation across the intestinal lumen into fecal contents’. The model was validated using data from Seveso victims and highly exposed Austrian women. Ruiz et al. (2014) included body growth and breastfeeding into this model.
The CONTAM Panel evaluated the model developed by Emond et al. (2005) and the one by Carrier et al. (1995) further optimised by Aylward et al. (2005) and adapted by Ruiz et al. (2014). Details about this can be found in Section 3.1.8.2.
3.1.2. Toxicity in experimental animals
A large number of studies in experimental animals have been performed. The CONTAM Panel decided to select only studies that could potentially lead to body burdens lower than the one in the animal studies used by the SCF in 2001 to derive the current TWI of 14 pg TEQ/kg bw. Studies providing mechanistic information in support of effects observed in human studies were selected using a different approach and are included in the mode of action (see Section 3.1.5).
Several criteria were applied in the selection process, some during the literature search and others at a later stage (see Section 2.2 and Annex A.1):
Only studies not evaluated by the SCF (2000, 2001) in their previous assessment, i.e. published after 1998, were considered,
Only studies with one or more exposure levels that could result in body burdens lower than the ones on which the SCF based its risk assessment (NOAEL of 20 or LOAEL of 40 ng/kg bw, respectively) were considered by applying a two‐step selection process (see below),
Only studies with exposure validated, i.e. levels measured in tissues or concentrations of the compounds in the administered dosing solutions or feed analysed, were considered,
Only studies showing endpoints regarded as adverse were selected. Effects such as differential gene expression or enzyme induction could not be linked directly to adverse endpoints and in accordance with the EFSA Guidance on Biological Relevance (EFSA Scientific Committee, 2017b) were therefore excluded in the literature search. On the basis of the retrieved papers, the CONTAM Panel decided to also exclude other identified endpoints considered as ‘intermediate’ and not clearly acknowledged as a risk factor for specific diseases. Although these ‘intermediary endpoints’ (i.e. fat pad weight, reactive oxygen species (ROS), hepatic vitamin A (retinoid) concentrations may provide important support to the health risk identification and characterisation steps of the risk assessment process, they cannot per se be used to derive the HBGV. Nevertheless, information on some of the ‘intermediary endpoints’ in the studies selected for this Scientific Opinion is provided in the corresponding tables,
In the case of studies performed in ovariectomised animals, it was decided that the hormonal status of these animals was not considered suitable for the risk assessment,
At a later stage, after preliminary assessment, the CONTAM Panel decided to exclude animal studies with exposure to PCDD/Fs and DL‐PCBs other than TCDD, or with exposure to mixtures. Body burdens estimated from some of these studies (see Annex A.5) would be associated with higher uncertainty, as TEFs are weighted factors based on a range of relative potencies from various studies and endpoints, and determined to discrete points on a log scale.
Studies were carried out mainly in rodents (rats, mice, hamsters and guinea pigs) and primates.
3.1.2.1. Studies in rodents
A total of 58 rat studies, 19 mice studies, two hamster studies and two guinea pig studies had exposure validated and were further evaluated (see Annex A.6).
As described above, the CONTAM Panel decided to select from the retrieved literature only studies with effects considered relevant and occurring at body burdens at or below those estimated for the NOAEL in the study by Ohsako et al. (2001) of 20 ng/kg bw, or for the LOAEL in the study by Faqi et al. (1998) of 40 ng/kg bw, both dealing with developmental reproductive effects in male rat offspring of dams treated with TCDD on GD15 (see Section 1.3.3).
To select such studies, it was decided to apply a (screening) ‘body burden cut‐off’ of 100 ng/kg bw. So all retrieved studies in which either a single dose, or repeated doses could lead to a body burden at or below 100 ng/kg bw were selected. As discussed below, in rodents a body burden of 100 ng/kg bw is expected after chronic gavage dosing of 10 ng/kg bw per day, therefore selected as the chronic intake cut‐off value. In a similar way, intake cut‐off values for less than subacute, subacute and subchronic exposure duration were developed and used for screening such studies (see below Section 3.1.2.1.1).
In the second step, within the retrieved studies, the body burden corresponding to the reported NOAEL/LOAEL was calculated (see below). Studies with body burdens at or below the ones used by the SCF for deriving the TWI were appraised and described in more detail. Studies using doses below the cut‐off but without showing any effect were not considered suitable for the risk assessment but were nevertheless retained in the tables presented in Annex A.6.
3.1.2.1.1. Selection criteria for low‐dose rodent toxicity studies
For the first screening step, it was required to derive doses that could lead to a body burden of 100 ng/kg bw for various dosing regimens, i.e. single dose, subacute, subchronic or chronic studies (for details see Appendix B). The methods used to derive such cut‐off doses are presented below.
In Figure 4, the mice body burden of TCDD after subchronic exposure is shown (Abraham et al., 1988; Diliberto et al., 1995, 2001; Santostefano et al., 1996). Given the elimination half‐life of TCDD in mice (see Figure 5), the body burden represents a ‘steady‐state’ situation. As shown, a subchronic exposure of 10 ng/kg bw per day results in a body burden of 100 ng/kg bw. Consequently, 10 ng/kg bw per day was selected as the intake cut‐off for any exposure duration beyond subchronic in mice. The dose–response relationship of the body burden in the rat equals that of mouse (Figure 4). From this, it can be concluded that 10 ng/kg bw per day is also an intake cut‐off for exposure duration beyond subchronic in rats as well.
Figure 4.
Dose response of the experimentally observed body burden (BB) of [3H]‐TCDD in female B6C3F1 mice and Long‐Evans rats after subchronic p.o. exposure (13 weeks, 5 days per week). Mice were exposed with a dose range of 0.15–450 ng/kg bw per day (vehicle corn; oil, Diliberto et al., 2001). Rats were exposed (including GD0–9) to doses of 1, 10, 30 ng/kg bw per day (Hurst et al., 2000a). Modelling: solid line
Figure 5.
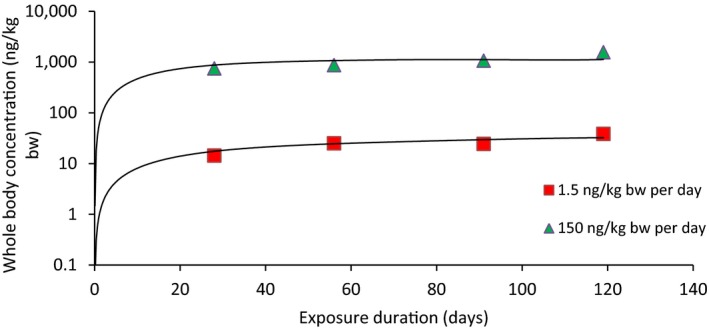
Time course of the body burden of [3H]‐TCDD in female B6C3F1 mice after p.o. exposure for 4, 8, 13 and 17 weeks, 5 days per week. Mice were dosed 1.5 and 150 ng/kg bw per day (Diliberto et al., 2001). Modelling: solid lines (high order polynomial function)
Diliberto et al. (2001) reported that in mice near ‘steady‐state’ kinetics for TCDD are already achieved after a subacute exposure period of 4 weeks (see Figure 5). From this, it can be concluded that the dose–response relationship shown in Figure 4 approximates to any exposure duration longer than 4 weeks. As the time period at which a ‘steady‐state’ is expected does not differ between mice (elimination half‐life: 15–31 days, Figure 5) and rats (elimination half‐life: 20 days), this conclusion holds for the latter species too. Consequently, 10 ng/kg bw per day was selected as the intake cut‐off for any exposure duration beyond subacute in rodents.
As shown in Figure 5, the body burden depends both on dose and exposure duration in case of exposures shorter than 4 weeks. Figure 6 illustrates this principle for the various dose/exposure duration combinations which, at the end of the exposure period, are expected to lead to a body burden of 100 ng/kg bw. For example, 24 ng/kg bw per day is needed for a 10‐day exposure period, and 100 ng/kg bw in case of a single dose.
Figure 6.
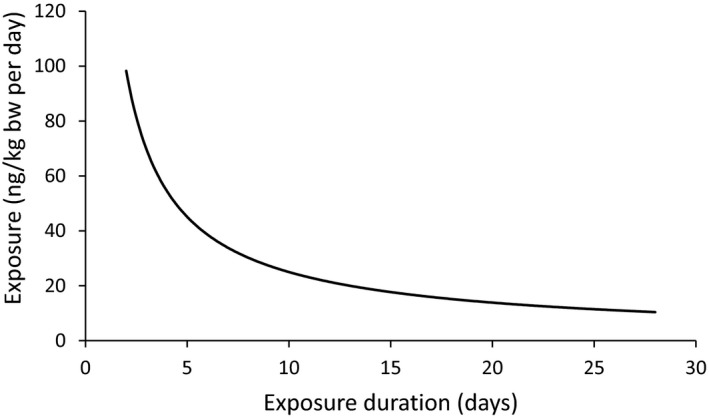
The exposure vs. exposure duration relationship to obtain a body burden of 100 ng/kg bw. Modelling: solid line, one‐compartment model with an absorption fraction of 0.5 and a half‐life of 20 days.
3.1.2.1.2. Estimation of the body burden of TCDD in rodent toxicity studies
The intake cut‐offs were used to select rodent toxicity studies with NOAELs/LOAELs which are expected to lead to a body burden at or below 100 ng TCDD/kg bw. As a second step, the actual body burden corresponding to the reported NOAEL/LOAEL/BMD was estimated. The principles underlying such body burden estimates are presented below.
Ideally, the animal body burden is obtained from experimental studies which provide information on the levels of TCDD in the body. Examples are the studies of Diliberto et al. (1995, 2001) and Hurst et al. (2000a,b). Although the latter studies are crucial in defining the (generic) relationship between exposure duration and TCDD's body burden (see Figure 4), they are not exemplary for the kinetic information provided in regular animal toxicity studies. At least, toxicity studies should provide concentration data in the liver and/or the adipose tissue. Nevertheless, some studies without such information were evaluated based on applied dose and assumed absorption fraction and half‐life.
In rodents, the body burden of TCDD predominantly rests in the liver and the adipose tissue (Diliberto et al., 1995, 2001; Hurst et al., 2000a,b). At low dose, the adipose tissue dominates, whereas at increasing dose and exposure duration liver deposition increases and ultimately this may dominate the body burden (hepatic sequestration, see Appendix B, Figure B.1‐B/C and B.2‐B/C). The summing of deposition in liver and adipose tissue provides a fair estimation of the body burden (see Appendix B, Figure B.2‐A and B.2‐A).
In rats, the fraction of the body burden residing in the liver and adipose tissue after single dose exposure, ranges from 0.84 (at a dose as low as 1 ng/kg bw) to 0.91 (at a dose of 10 μg/kg bw). The corresponding range in mice is 0.65–0.90 (see Appendix B, Figure B.2‐A). After chronic exposure, these ranges are 0.78–0.87 (rat) and 0.69–0.82 (mouse) (see Appendix B, Figure B.2‐A). Clearly, for any rodent toxicity study which provides hepatic and/or adipose tissue levels, the relationships depicted in Figures D2‐1 and D2‐2 can be used to estimate the corresponding body burden. This principle is illustrated on the studies of Bell et al. (2007a,b,c).
In Bell et al. (2007a,b), pregnant rats were given a single oral gavage dose of 0, 50, 200 or 1,000 ng TCDD/kg bw at GD15 (Bell et al., 2007a). The 50 ng/kg bw dose is below the intake cut‐off of 100 ng/kg bw, as set for single dose exposure (see above). At this lowest dose of 50 ng/kg bw, the GD16 and GD21 body burdens were experimentally determined at 18.8 and 19.6 ng/kg bw, respectively, based on the summation of liver, adipose tissue and (negligible) fetal deposition (Table 5; Bell et al., 2007a). Correcting these body burdens for the fraction of the body burden residing in the liver and adipose tissue ((0.87 as calculated with Figure A.2‐1‐A, y = 0.7548ln0.05) + 88.77) then gives a single dose GD16 body burden of 18.8/0.87 = 21.6, and a GD21 body burden of 19.6/0.87 = 22.5 ng TCDD/kg bw. As mentioned in Section 1.3.3, a 2.6‐fold higher maternal body burden is required to obtain the same exposure in the embryo after chronic exposure. Hence, applying this factor on these body burdens resulted in an estimated chronic GD16 body burden of 21.6 × 2.6 = 56.2 ng/kg bw, and a GD21 body burden of 22.5 × 2.6 = 58.5 ng/kg bw.
In Bell et al. (2007c), female rats were exposed via the diet to 0, 2.4, 8 or 46 ng TCDD/kg bw per day during 12 weeks, including pregnancy. The 2.4 and 8 ng/kg bw doses are below the intake cut‐off of 10 ng/kg bw as set for subchronic exposure (see above). At the lowest dose of 2.4 ng/kg bw, GD16 and GD21 body burdens were experimentally determined to be 35.4 and 42.4 ng TCDD/kg bw, respectively, based on the summation of liver, adipose tissue and (negligible) fetal deposition (Table 5; Bell et al., 2007a). Correcting these body burdens for the fraction of the body burden residing in the liver and adipose tissue (0.84 as calculated from Figure B.2A, y = 1.934ln(2.4) + 82.39), then gives subchronic body burden at GD16 of 35.4/0.84 = 42.1 ng/kg bw, and at GD21 of 42.4/0.84 = 50.4 ng/kg bw.
3.1.2.1.3. Studies in rats
Based on the first step in the selection process (see Section 3.1.2.1.1), the following TCDD studies had exposure levels above the intake cut‐off and were not further considered for the current assessment: Kransler et al. (2007a,b, 2008, 2009), Tuomisto et al. (2000), Nishimura et al. (2001, 2002, 2003), Ishimura et al. (2002), Shirota et al. (2007), Kakeyama et al. (2003) and Fletcher et al. (2001). The remaining rat studies that were at or below the intake cut‐off were further considered and are described in this section and in Annex A.6.1 (Tables 55 and 56 therein). The reliability of each individual study was appraised by considering the internal validity, especially the risk of bias, and each study was allocated to one of the three risk of bias tiers as described in Annex A.1 and Section 2.2.1.2. Details about the risk of bias appraisal can be found in Annex A.7.1. Details on the calculation of the body burden for each study are shown in Appendix B).
In the search and selection also, the studies by Faqi et al. (1998) and Ohsako et al. (2001) were retrieved. These studies formed the basis for the derivation of the TWI by the SCF in 2001 (see Section 1.3.3). Since the study by Faqi et al. (1998) resulted in the lowest TWI, the CONTAM Panel decided to include this study in the current assessment in order to evaluate the critical body burdens obtained from other studies.
Subacute and subchronic studies
In a study by Harrill et al. (2016) on AHR knockout Sprague–Dawley rats to explore pathological changes associated with AHR‐mediated tumorigenesis, 8‐week‐old wild‐type and null female animals (10 females per group) were administered by oral gavage 0, 3, 22, 100, 300 or 1,000 ng TCDD/kg bw every 4 or 5 days a week for 4 weeks to a total of 19 doses. AHR knockout rats were unaffected. Mild effects on liver to body weight ratios, hepatocellular hypertrophy and increased serum cholesterol were noted in wild‐type females at a dose of 22 ng/kg bw per day or higher. This occurred in the absence of serum clinical chemistry or enzymes measurements consistent with hepatocellular injury, including serum alanine transaminase (ALT) and aspartate aminotransferase (AST) (not observed until 1,000 ng TCDD/kg bw). Based on bile duct hyperplasia and thymus atrophy, a NOAEL of 100 ng TCDD kg/bw per day was identified. At the end of the exposure period, a NOAEL body burden of 789 ng/kg bw was calculated (see Table 9).
Table 9.
Body burden (BB) of TCDD corresponding with the NOAEL/LOAEL/BMD identified in the rat and mice toxicity studies. For details see Appendix B
| Study exposure duration | NOAEL, LOAEL (applied dose) | Extrapolated BB for chronic exposure (ng/kg bw) |
|---|---|---|
| Studies in rats | ||
| Viluksela et al. ( 2000 ) Subchronic (20 weeks) |
Long‐Evans (Turku/AB) rats: NOAEL = 1 ng/kg bw per day (based on increase in liver weight and AST and increased incidence of histopathological findings in the liver) Han Wistar rats: NOAEL = 100 ng/kg bw per day (based on decreased body weight gain, increased liver weight and a decrease in thymus weight and histopathological findings in the liver) |
Long‐Evans rats: 26a |
| Jämsä et al. ( 2001 ) Subchronic |
Long‐Evans (Turku/AB) rats: NOAEL= 1 ng TCDD/kg bw per day (based on decreased tibia length, tibia geometry parameters, tibia ash weight, and increased plasma ALP activity) |
28a |
| Harrill et al. ( 2016 ) Subacute |
Sprague–Dawley rats: NOAEL = 100 ng TCDD/kg bw per day (based on bile duct hyperplasia and thymus atrophy) |
789b |
| Phadnis‐Moghe et al. ( 2016 ) Subacute |
Sprague–Dawley rats: NOAEL = 22 ng/kg bw per day (based on distribution of lymphocytes and NK cells) |
250b |
| NTP (2006a) Chronic |
Sprague–Dawley rats: NOAEL = 3 ng/kg bw per day (5 days/week: extrapolated to 2.1 ng/kg bw per day) (based on hepatopathy) |
85c |
| Bell et al. ( 2007b ) Single dose at GD15 Dams/male offspring |
Wistar Han rats: NOAEL = 50 ng/kg bw (based on the decreased pup weight from PND1 to PND7) |
GD16: 56d GD21: 59d |
| Bell et al. ( 2007c ) Repeated dosing Dams/male offspring |
Wistar Han rats: LOAEL = 2.4 ng/kg bw per day (based on the delay in balanopreputial separation) |
GD16: 42e GD21: 50e |
| Faqi et al. ( 1998 ) Repeated dosing Dams/male offspring |
Wistar rats: LOAEL = 25/5 ng/kg bw (single loading dose/weekly maintenance dose, s.c.) (based on decreased sperm production) |
GD21: 25c |
| Studies in mice | ||
| Li et al. ( 2006 ) Embryotoxicity study Repeated dosing during GD1‐8, GD1‐3, GD4‐8 |
NIH mice: NOAEL = 2 ng/kg bw per day (based on embryonic loss) |
9f |
NOAEL: no‐observed‐adverse‐effect level; LOAEL: lowest‐observed‐adverse‐effect level; BMD: benchmark dose; bw: body weight; AST: aspartate aminotransferase; TCDD: 2,3,7,8‐ tetrachlorodibenzo‐p‐dioxin; ALP: alkaline phosphatase; PND: postnatal day; s.c.: subcutaneous.
Based on levels in liver and extrapolated to the whole body.
Based on levels in liver and adipose tissue, extrapolated to whole body.
Based on levels in liver and adipose tissue, extrapolated to whole body.
Based on levels in liver and adipose tissue, extrapolated to whole body and correction for acute to chronic exposure.
Based on measurements in liver, adipose tissue and fetus and extrapolated to whole body.
Based on applied oral dose, fraction absorbed: single dose 0.6, and correction for acute to chronic exposure.
Phadnis‐Moghe et al. (2016) compared effects on the immune system of TCDD in the same study by Harrill et al. (2016) of Sprague–Dawley AHR knockout rats with wild types (3, 22, 100, 300 or 1,000 ng/kg per day for 4 weeks, 5 days per week by gavage). Wild‐type rats had more pronounced effects than the knockout rats. In wild‐type rats, lymphocyte subpopulations were influenced, with a marked suppression of the percentage of lipopolysaccharide (LPS)‐induced IgM+ cells, yet an increment of (ex vivo LPS‐induced) proliferation, indicating a dysregulation of the humoral immune response with an effect level of 100 ng/kg. Also, the NK percentage was decreased at this dose. The NOAEL was therefore 22 ng/kg bw per day. Attributing a daily dose rate of 15.7 ng/kg bw per day this resulted in a body burden of 250 ng/kg bw at the end of the 28‐day exposure period.
The effects of TCDD were studied by Viluksela et al. (2000) in TCDD‐sensitive (inbred Long‐Evans, Turku/AB) and in TCDD‐resistant rats (outbred Han Wistar, Kuopio), strains known to differ in sensitivity to TCDD‐induced toxicity due to a difference in AHR structure (see Section 3.1.6.4). Starting at an age of 10 weeks, TCDD was dosed subcutaneously, first a single loading dose and thereafter a weekly dose for 19 weeks. The female Han Wistar rats were dosed with loading doses of 0, 0.035, 0.35, 3.5 and 35 μg TCDD/kg bw followed by weekly maintenance doses of, respectively, 0, 0.007, 0.07, 0.7 and 7 μg TCDD/kg bw. This resulted in total doses of 0, 0.17, 1.7, 17 or 170 μg TCDD/kg bw. In a similar way, the female Long‐Evans rats were dosed with a total dose of 0, 0.17, 1.7 or 17 μg TCDD/kg bw. These doses correspond to calculated daily doses of 0, 1, 10, 100 or 1,000 ng TCDD/kg bw (the highest dose applied on Han Wistar rats only). These dose levels will be used in the further description of this study and those by Jämsä et al. (2001) based on this same study.
At the highest doses, body weight gain was decreased, i.e. in Long‐Evans rats at the dose of 100 ng/kg bw per day, and Han Wistar at the dose of 1,000 ng/kg bw per day. Increases in plasma enzyme activities (ALT, AST) were observed in Long‐Evans rats: ALT from 100 ng/kg bw per day, AST from 10 ng/kg bw per day. In Han Wistar rats: ALT not significantly increased and AST from 1,000 ng/kg bw per day. GGT was not increased in either strain. Maximum liver EROD activity was reached in Long‐Evans rats at 10 ng/kg bw per day and in Han Wistar rats at 100 ng/kg bw per day, showing the strain difference in response. An increase in liver weight was observed in Long‐Evans rats from 10 ng/kg bw per day and in Han Wistar from 100 ng/kg bw per day. In both strains relative thymus weight was decreased from 100 ng/kg bw per day. Histopathology findings (foci in the liver, multinuclear hepatocytes, mitotic figures, bile duct hyperplasia, extramedullary haematopoiesis) were observed in the Long‐Evans rats from 10 ng/kg bw per day and in the Han Wistar rats from 100 ng/kg bw per day. The CONTAM Panel considered 1 ng TCDD/kg bw per day as the NOAEL in Long‐Evans rats based on increase in liver weight and AST and increased incidence of histopathological findings in the liver of the 10 ng TCDD/kg bw per day group. In the Han Wistar rats the NOAEL was 10 ng TCDD/kg bw per day based on decreased body weight gain, increased liver weight and a decrease in thymus weight and histopathological findings in the livers of the 100 ng TCDD/kg bw per day group. At the end of the exposure period, the NOAEL body burden for the Long‐Evans rats was calculated to be 26 ng/kg bw (Table 9).
Jämsä et al. (2001) reported on bone effects found in the study of Viluksela et al. (2000). A significant reduction of bone growth was seen at 10 ng/kg bw per day (p < 0.01) in the Long‐Evans rats, while in Han Wistar rats the effect of TCDD was seen only at the high dose of 1,000 ng/kg bw per day (p < 0.05). The biomechanical measurements stiffness and three‐point bending breaking force of the tibia were reduced by TCDD exposure at the dose of 100 ng/kg bw per day in the Long‐Evans rats, while three‐point bending breaking force of the tibia was significantly reduced in Han Wistar rats at 1,000 ng/kg bw per day. Plasma alkaline phosphatase (ALP) activities were dose dependently increased in Long‐Evans rats at 10 and 100 ng/kg bw per day, and in Han Wistar rats at 100 and 1,000 ng/kg bw per day. According to the authors, this parameter was related to leakage of the enzyme from the liver due to hepatotoxicity in parallel with an increase in ALT and AST as reported previously by Viluksela et al. (2000). In the Long‐Evans rats the NOAEL was 1 ng/kg bw per day based on several bone parameters. The obtained results are in agreement with the sensitivity difference between the two rat strains, which is ascribed to a difference in the transactivation domain of AHR. At the end of the exposure period, a NOAEL body burden of 28 ng/kg bw was calculated for the Long‐Evans rats (see Table 9).
Carcinogenicity studies
NTP carried out a series of toxicological and carcinogenesis studies with various individual PCDD/F and DL‐PCB congeners to determine the suitability of the TEF methodology for predicting and comparing carcinogenicity and toxicity of dioxin‐like compounds (NTP, 2006a, 2006b, 2006c, 2006d–e, 2010).
In the study on TCDD (NTP, 2006a; Walker et al., 2006), female Sprague–Dawley rats were administered 0, 3, 10, 22, 46 or 100 ng/kg bw per day, 5 days a week, by gavage for 14, 31, 53 or 105 weeks. Survival of dosed animals was similar to the control group. At the end of the study, most tumours were observed at the highest dose of 100 ng/kg bw per day and were mainly hepatocellular adenomas and cholangiocarcinomas, but also lung cystic keratinising epithelium and uterine squamous cell carcinoma. The principal non‐neoplastic finding in this study was a significant increase in incidence and severity of hepatotoxicity. By 105 weeks there was a dose‐related increase in toxic hepatopathy encompassing a variety of non‐neoplastic toxic changes (including focal cellular alteration, multinucleated hepatocytes, fatty change, necrosis, bile duct hyperplasia, hepatocyte degeneration, oval cell hyperplasia, and portal fibrosis) at doses of 10 ng/kg bw per day and above. Increased incidences above control of gingival hyperplasia, thymus atrophy, cardiomyopathy, cholangiofibrosis were observed, as well as microscopic lesions in lung, oral mucosa, pancreas, thymus, adrenal cortex, heart, clitoral gland, kidney, forestomach, mesentery and thyroid, which were not always dose‐related.
Interim studies at 14 and 31 weeks showed significant decreases in serum total and free T4 levels from 22 ng/kg bw per day or greater and increases in serum total T3 at 46 ng/kg bw per day but these were not dose related. TSH was elevated after 14 weeks at 46 and 100 ng/kg bw per day but not after 31 weeks. Liver weights were increased in all dose groups at 14 and 31 weeks. At 31 weeks, multinucleated liver cells were observed at 46 ng/kg bw per day and clear toxic hepatopathy at 100 ng/kg bw per day.
The CONTAM Panel noted that the NOAEL decreased from 100 ng/kg bw at 14 weeks to 3 ng/kg bw at 105 weeks. Based on the findings after 105 weeks exposure, a NOAEL was agreed by the Panel of 3 ng/kg bw per day, based on hepatopathy. Since the dosing was only for 5 days per week this corresponds to a NOAEL of 2.1 ng/kg bw per day. The body burden at this NOAEL was estimated to be 85 ng/kg bw.
Reproductive and developmental studies
In the study by Faqi et al. (1998), female Wistar rats (25 per group) were treated s.c. with a single dose of 0, 25, 60 or 300 ng 14C‐labelled TCDD/kg bw two weeks prior to mating (loading dose), followed by weekly injections of 0, 5, 12 and 60 ng/kg bw during mating, pregnancy and lactation as a maintenance dose, aiming at stable body burdens. Three animals from each group were killed on GD21 and levels of the radiolabel in liver and adipose tissue of dams, and livers of fetuses were determined. Male offspring was killed on either PND70 or 170 (n = 20 per group and time‐point). It was shown that based on the number of spermatids in the testes, daily sperm production was reduced at all three dose levels, showing a larger decrease (50% vs 30%) at PND170. At this day, also sperm number per cauda epididymis was significantly decreased in all treatment groups. No further reduction in semen parameters was observed at higher exposure. There was no effect on weight of testes, epididymes, prostate or seminal vesicles on PND70 or 170, and neither on anogenital distance as measured on PND 2, 12 and 22. There was a slight delay in preputial separation in the low‐ and high dose. Treatment had no effect on reproductive performance. No NOAEL could be derived from this study but the LOAEL was 25/5 ng TCDD/kg bw (maternal dose). The body burden at this LOAEL was estimated to be 25 ng/kg bw. The CONTAM Panel noted that spermatids were counted manually with a haemocytometer and that it was not reported whether this evaluation was performed blinded.
In a study by Nohara et al. (2000), a single oral dose of 0, 12.5, 50, 200 or 800 ng TCDD/kg bw given to pregnant Holztman rats on GD15, had no effect on the weights of thymus or spleen in male offspring at weaning (PND21), puberty (PND49) or in adulthood (PND120). At any time, TCDD had no impact on thymus cell number or cellular population defined by CD4 and CD8 molecules. In spleen, the number of splenocytes decreased in a dose‐dependent manner on PND49. The decreased spleen cellularity was not detected on PNDs 21 or 120 after TCDD‐exposure. Based on the dose‐related decrease in the number of splenocytes on PND49, the NOAEL of the study was 200 ng TCDD/kg bw (maternal dose). The CONTAM Panel noted that the study had some limitations. For example, the number of pregnant females was not stated and the randomisation and numbers of selected F1‐offspring used for observations was not described. Due to these limitations this study was not further considered.
Bell and colleagues performed two studies in female Wistar (Han) rats (n = 55–70 per group) administered TCDD either on GD15 (Bell et al., 2007b) or during premating, mating and gestation (Bell et al., 2007c). Both studies were performed according to OECD/GLP guidelines. Reproductive effects and abnormalities were studied in F1 male animals.
In Bell et al. (2007b), mated female rats were dosed by gavage on GD15 with 0, 50, 200 or 1,000 ng TCDD/kg bw. No effect on body weight of the dams was observed. Four dams of the high‐dose group showed total litter loss. The litter size in the high‐dose group was significantly decreased compared to controls during lactation. Pup weights were reduced during the lactation period in the high‐dose group. No effects were seen in the functional observational battery and learning tests. Balanopreputial separation was delayed in the high‐dose group (average delay of 2.8 days) compared to the control group. Fertility of the F1‐males was not affected. On PND70 and PND120 the sperm parameters were comparable between the TCDD‐treated F1‐males except for the increased number of abnormal sperm in the high‐dose group on PND70 and an increased number of mean epididymal sperm on PND120 in the mid‐ and high‐dose groups. The abnormal sperm effect on PND70 was related to the delayed puberty of this group. Testis weights in the high‐dose group were decreased at PND70 and 120, and at PND120 brain weight was lower in the high‐dose group (2.2%). On PND120, the liver weight of the mid‐dose group was increased (10.6%). Liver to body weight ratios were increased for all three dose groups by approximately 3–3.5%. This increase was not dose‐related and considered not treatment related. Furthermore, an increase in inflammatory cell foci in the epididymis in the high‐dose group was observed. The LOAEL for this study was 200 ng TCDD/kg bw (based on the decreased pup weight from PND1 to PND7 in this group) and the NOAEL 50 ng/kg bw. The maternal NOAEL body burden at GD16 and GD21 was calculated to be 56 and 59 ng/kg bw, respectively, taking into account the single dosing (see Table 9).
In Bell et al. (2007c), female rats were fed diets containing TCDD resulting in a dose of 0, 2.4, 8 or 46 ng TCDD/kg bw per day during 12 weeks premating, mating and gestation. No effect on body weight of the dams was observed. Food intake of the high‐dose group was increased during the premating period. Dams of the high‐dose group showed an increased total litter loss. The number of pups alive on PND1 per litter and the number of pups surviving on PND4 was reduced in the high‐dose group. Pup weights were reduced during the lactation period in the high‐dose group and gained less weight than control F1‐males during the study. The low‐ and mid‐dose group showed also a decrease in pup weight which was most marked at PND4. However, on PND21 the pup weights of these groups were similar to the controls. On PND120, the F1‐males of these groups showed lower body weights (dose‐related trend). Apart from a decrease in motor activity observed in the F1‐males of the high‐dose group, no effects were seen in the functional observational battery and learning tests. Balanopreputial separation was delayed by 1.8, 1.9 and 4.4 days for the low‐, mid‐ and high‐dose groups, respectively, compared to the control group. Fertility of the F1‐males was not affected. On PND70 and PND120, the sperm parameters were comparable between the TCDD‐treated F1‐males and controls except for the increased number of abnormal sperm in the high‐dose group on PND70. Terminal body weight of the high‐dose group was decreased on PND120. The testis weight of the F1‐males of the high‐dose group was reduced at PND70 (but not PND120). Ventral prostate weight was not reduced. The small (3.3–4.6%) increase in liver weight‐to‐body weight ratio in the exposed groups was not dose‐related and considered not treatment related. Histopathological examination revealed no treatment‐related effects. The LOAEL for this study was 2.4 ng TCDD/kg bw (the lowest dose tested), based on the balanopreputial separation delay (delayed puberty). The maternal LOAEL at GD16 and GD21 body burden was calculated to be 42 and 50 ng/kg bw, respectively (see Table 9).
Mated female Sprague–Dawley rats (n = 3–9 per group) were dosed by gavage on GD15 with 0, 10, 100 or 200 ng TCDD/kg bw (Rebourcet et al., 2010). No differences in number or weight of pups or sex ratio were observed between TCDD‐treated and control groups on litter basis. However, when individual data were used, the pup weights of the high‐dose group were decreased from PND5 onwards. At necropsy on PND28, 40, 67 and 145, intra‐testicular levels of testosterone and 4‐androstenedione of the 200 ng TCDD/kg bw group were in the normal range. Testicular and epididymal weights of the TCDD‐treated F1‐males were comparable to the control males; in the testes, no treatment‐related histopathological changes were observed. Effects in the high‐dose group on epididymal sperm reserves and daily sperm production were only observed on PND67 and not on PND145. No effects on these parameters were observed in the other TCDD‐treated groups. Reproductive performance of the F1‐males of the high‐dose group was considered within the normal range (no control group used). Sperm reserves and daily sperm production were measured in F2‐males on PND67 and 145 and were in the same range as the F1‐controls. The LOAEL of the study was 200 ng TCDD/kg bw and the NOAEL was 100 ng TCDD/kg bw. The LOAEL was based on decreased pup weights and decreased sperm reserves. The CONTAM Panel noted that the study had some limitations. For example, the number of pregnant females which was only 3 in the low‐ and mid‐dose groups. Furthermore, no control group was used for the F2‐generation. Due to these limitations this study was not further considered
3.1.2.1.4. Studies in mice
Studies retrieved from the literature search were evaluated to identify those in which the exposure levels were below or in the range of the intake cut‐off for the specific exposure duration (see Section 3.1.2.1.1). The following TCDD studies were above the intake cut‐off, and were not further considered: Patterson et al. (2003), Aragon et al. (2008), Morris et al. (1998), DeKrey et al. (2013), Kimura et al. (2015), and Fletcher et al. (2001).
The remaining studies were at or below the intake cut‐off and are discussed in this section and in Annex A.6.1 (Table 57 and 58 therein) (Slezak et al., 2000; Li et al., 2006; Fader et al., 2015; Van Esterik et al., 2015; Nault et al., 2016). The studies by Fader et al. (2015) and Nault et al. (2016) showed NOAELs/LOAELs above the intake cut‐off. Slezak et al. (2000) and Van Esterik et al. (2015) were not further considered as oxidative stress and fat pad weight were considered an adverse endpoint but rather an intermediate effect.
Li et al. (2006) exposed pregnant NIH mice (data were reported from 10 pregnant females per group) to 0, 2, 50 or 100 ng TCDD/kg bw per day from GD1–8 (early gestation), GD1–3 (pre‐implantation) or GD4–8 (peri‐implantation to early post‐implantation). The embryonic loss, oestradiol and progesterone levels were measured on GD9. The embryonic loss was observed in the mid‐ and high‐dose groups of all exposure regimens but was mainly seen in the pre‐implantation period. Oestradiol levels were not affected and progesterone levels were decreased at all dose levels. The NOAEL of the study was 2 ng/kg bw per day based on embryonic loss. The corresponding chronic body burden was 9 ng/kg bw (see Table 9). The CONTAM Panel noted the large gap between the low‐ and mid dose.
3.1.2.1.5. Studies in hamsters
Three studies on the effects of TCDD in hamsters were identified by the literature search carried out as described in Section 2.2 and Annex A.6.1 (Table 59 therein). Two had exposure validated (Fletcher et al., 2001; Kransler et al., 2007a,b) and one did not (Yellon et al., 2000).
The CONTAM Panel decided not to consider these studies in hamsters for the human risk assessment because they were conducted at high doses of TCDD.
3.1.2.1.6. Studies in guinea pigs
Two studies on the effects of TCDD in guinea pigs were identified by the literature search carried out as described in Section 2.2 and Annex A.6.1 (Table 60 therein). Dose levels in Kransler et al. (2007a,b) were above the intake cut‐off and the study was therefore not further considered, while in Fletcher et al. (2001) the increase in relative liver weight was not considered as an adverse endpoint, and as such the study was not suitable for risk assessment.
3.1.2.1.7. Conclusion for rodents studies
The studies on rodents selected by the CONTAM Panel confirmed that various effects were seen at body burdens in a similar range to the ones which the SCF based its previous risk assessment on. These effects included male reproductive effects, effects on embryonic loss, on bones and hepatopathy.
The lowest estimated body burdens related to adverse effects (i.e. LOAEL) were observed for the study by Faqi et al. (1998), being 25 ng/kg bw for reduced sperm production, followed by the study by Bell et al. (2007c), showing a LOAEL body burden of 42 ng/kg bw at GD16 for a delay in balanopreputial separation. At somewhat higher body burdens effects on bone development, pup weight and embryonic loss were observed, with NOAEL body burdens of, respectively, 26, 56 and 42 ng/kg bw. The lowest NOAEL was estimated for the study by Li et al. (2006) but with a LOAEL of 50 ng/kg bw per day, corresponding to a chronic body burden of 234 ng/kg bw, it cannot be excluded that the actual NOAEL is much higher.
3.1.2.2. Studies in primates
In 2000, the SCF considered that the studies by Schantz and Bowman (1989) and Rier et al. (1993) in female rhesus monkeys could not be used to base the HBGV on, due to problems in reporting (SCF, 2000). In its re‐evaluation in 2001, the SCF considered two additional studies of these monkeys by Rier et al. (2001a,b) and supplemental unpublished data. These new data addressed some of the concerns the SCF had identified in the study on endometriosis by Rier et al. (1993). Due to the uncertainties raised by the new studies, the SCF had less confidence in the quantitative relationship between exposure to TCDD and the incidence of endometriosis in monkeys. Therefore, it was decided not to include this study as a pivotal study in the assessment. For a more detailed description see Annex A.6.2.
The literature search and the selection for relevance as described in Section 2.2 resulted in a total of 17 studies on the effects of TCDD in primates. Nine of these had no validation of the exposure (either levels measured in tissues or concentration of the dosing solutions analysed) and were not further considered (Enan et al., 1998a,b; Guo et al., 1999a, 2000; Yang et al., 2000; Moran et al., 2001; Scott et al., 2001; Shridhar et al., 2001; Riecke et al., 2002; Nottebrock et al., 2006). The remaining eight studies had exposure validated. The list included the studies by Rier et al. (2001a,b) considered by the SCF (2001), in addition to another six studies published after 1998. These additional six studies were considered in this assessment (Yasuda et al., 2005; Negishi et al., 2006; Korenaga et al., 2007; Hermsen et al., 2008; Arima et al., 2009, 2010). Details of these studies are shown in Annex A.6.2 (Table 61 therein), including considerations of the internal validity, especially the risk of bias and each study was allocated to one of the three risk of bias tiers as described in Annex A.1 and Section 2.2.1.2.
The studies by Yasuda et al. (2005), Negishi et al. (2006), Hermsen et al. (2008), Arima et al. (2009, 2010) and Korenaga et al. (2007) reported results on the same pregnant female rhesus monkeys and/or their offspring. Pregnant females (20–23/group) were dosed on GD20 with an initial s.c. dose of 0, 30 or 300 ng TCDD/kg bw (purity > 98%; dose solution analysed to confirm concentrations, but outcome not described). Seven or eight additional doses of TCDD of 0, 1.5 or 15 ng/kg bw were administered every 30 days during gestation and lactation periods until PND90 after delivery. The number of doses depended on the length of gestation. An additional high‐dose group (9 pregnant females) was added to increase the number of offspring. The total TCDD dose was 0, 40.5‐42 or 405–420 ng/kg bw for the control, low‐ and high‐dose group, respectively. No effects on maternal general condition, body weight, food consumption and length of gestation were observed after TCDD exposure. Abortions, stillbirth and early postnatal deaths occurred with higher frequency in the high‐dose group, but was not significant, as in all groups (including control groups), these effects were found at a relatively high frequency. The survival rate at approximately 7 years was comparable between the groups.
In the offspring of these, rhesus monkeys which were treated with TCDD during the gestation and lactation period, dental effects (Yasuda et al., 2005) and effects on sperm concentration (Arima et al., 2009) were observed in the high‐dose group. Furthermore, histopathology of the epididymides (ductus epididymis) and decrease of the epithelial height of the gland cells of the prostate was observed in the low‐ and high‐dose group (dose relation was not evident) (Arima et al., 2010). No effects were observed on anogenital distance, bone development (Hermsen et al., 2008), reproductive organ weights, serum testosterone level, intra‐testicular testosterone level, 5‐alpha‐dihydrotestosterone level in the testis, and other sperm parameters apart from sperm concentration (Arima et al., 2009). Effects were observed in one of the 4 tests for social behaviour after 12–15 months but not after 24–27 months (Negishi et al., 2006). In adult females, histopathological changes in the liver were observed in the low‐ and high‐dose group (not dose‐related) (Korenaga et al., 2007). Electron microscopy revealed sinusoidal endothelial cell injuries in the liver of TCDD‐treated animals.
In general, it should be noted that in several parts of these studies with pregnant females, only a few adult animals or offspring were used for each determination and that the selection of these animals was not described. In addition the survival rate in the control and treated groups was low. Therefore, the CONTAM Panel concluded that these studies are not suitable for deriving an HBGV.
3.1.3. Genotoxicity
The genotoxicity of TCDD has been studied intensively over the last five decades. The evidence for the direct genotoxicity of TCDD is negative or equivocal for a large array of in vitro and in vivo endpoints (Giri, 1986; IARC, 1997; ATSDR, 1998; NTP, 2006a; Budinsky et al., 2014). These include aneuploidy, chromosomal aberrations, DNA damage, dominant lethal mutation, gene mutation, micronuclei, mitotic recombination and gene conversion, sister chromatid exchange (SCE) and cell transformation. Studies have shown induction of oxidative stress‐related DNA damage by high‐dose acute exposure to TCDD. It is hypothesised that TCDD‐mediated persistent activation of AHR may be responsible for inducing oxidative stress and associated indirect genotoxicity (NTP, 2006a).
Few studies have recently addressed the potential genotoxicity of PCDD/Fs. In an interlaboratory comparison of TCDD among five laboratories, no significant increase in the induction of micronuclei formation was detected in human peripheral blood cells exposed in vitro (Katic et al., 2010). In vivo, no increase in mutation frequency or change in mutational spectra was observed after 6 weeks of exposure to 2 μg TCDD/kg bw twice a week for 6 weeks, in both male and female Big Blue® lacI transgenic rats (Thornton et al., 2001).
Although there is considerably less information for DL‐PCBs, again there is little evidence for direct genotoxicity. PCB‐77 caused DNA damage to human peripheral lymphocytes at the highest dose tested as assessed by the Comet assay, but was significantly less potent than the non‐dioxin‐like congener PCB‐52 (Sandal et al., 2008). No increase in mutation frequency was observed in an in vivo transgenic transgenerational mutagenicity assay using Muta(M) mice administered single doses of PCB‐126 (125, 250 or 500 μg/kg) during pregnancy (Inomata et al., 2009).
Studies of peripheral lymphocytes from sheep, buffalo and cattle livestock exposed to PCDD/Fs and DL‐PCBs via contaminated pasterage or feed have yielded results that are regarded as inconclusive with respect to chromosome instability (see Section 3.1.5.1).
Overall, the evidence is robust that PCDD/Fs and DL‐PCBs are not directly genotoxic in standard assays (Schwartz and Appel, 2005; Knerr and Schrenk, 2006; Budinsky et al., 2014).
3.1.4. Observations in humans
Studies in humans in which the exposure to PCDD/Fs and DL‐PCBs has been evaluated in relation to adverse effects were retrieved and selected for relevance as described in Section 2.2.
From those, the CONTAM Panel considered that studies which analysed the following set of compounds in tissues (e.g. blood, human milk, adipose tissue) of the subjects under study were those relevant for the hazard identification and characterisation (see Annex A.1):
TCDD or any other congener dominating the TEQ, e.g. due to a contamination incident,
17 PCDD/Fs and 12 DL‐PCBs,
17 PCDD/Fs and 4 non‐ortho DL‐PCBs,
17 PCDD/Fs and 3 non‐ortho DL‐PCBs (including PCB‐126),
Total TEQs (or BEQs by, e.g. CALUX).
The individual studies resulting from this selection were grouped according to endpoint health categories (i.e. chloracne and other dermal effects, reproductive effects, thyroid disease and thyroid hormones, type 2 diabetes and obesity, cardiovascular effects, hepatic disorders and digestive effects, effects on the immune system, effects on the nervous system, effects on teeth and bones, cancer, and other effects). The reliability of each individual study was appraised by considering the internal validity, especially the risk of bias, and each study was allocated to one of the three risk of bias tiers as described in Annex A.1 and Section 2.2.1.2. Adjustment for confounding and other modifying variables was taken into account when considering the internal validity of each study. The CONTAM Panel focussed the discussion on studies appraised as having a definitely low or probably low risk of bias (tier 1 and 2). Studies having a definitely high or probably high risk of bias (tier 3) are described in the tables but not further considered in the discussion.
The epidemiological studies have been conducted in subjects/cohorts exposed to PCDD/Fs and DL‐PCBs at different life stages under different exposure conditions. Such factors may influence the adverse outcomes reported. The main sources of exposure have been from:
industrial accidents, such as the Seveso Cohort, or contamination incidents, such as the Yusho or Yucheng Cohorts,
occupational exposure, such as chemical workers or military personnel serving in the Vietnam War,
background levels mainly via the diet in the general population.
A brief description of the main characteristics of some of these cohorts is given in Section 3.1.4.1. In Sections 3.1.4.2 to 3.1.4.12, the evidence available per endpoint is presented and discussed, with detailed information about each of the studies provided in Annex A.8. Since some papers cover several different endpoints, some of these studies appear in more than one table. Details about the risk of bias appraisal can be found in Annex A.9.
Because details are provided in the tables, and in order to avoid repetitions, the descriptions in Sections 3.1.4.3 to 3.1.4.12 are provided at a low level of detail. More details are given in the text for those studies that were considered potentially critical for the hazard characterisation. The overall discussion and identification of critical effects is provided in Section 3.1.7.1.
3.1.4.1. Description of cohorts
3.1.4.1.1. Cohorts based on industrial accidents or contamination incidents
Seveso Cohort
In 1976, there was an explosion at a TCP manufacturing plant in Seveso (Italy) that released up to 30 kg22 of TCDD over the surrounding area. The contaminated area was divided into three major zones: A (100 ha, n = 736 residents23), B (200 ha, n = 4,737 residents23) and R (1,400 ha, n = 31,800 residents23) based on decreasing concentrations of TCDD in surface soils. The area surrounding the A, B and R zones had more than 180,000 residents and was denoted zone non‐ABR.
A number of studies have been carried out in the Seveso Cohort to investigate the relation between exposure to TCDD and adverse effects at different life stages. Analysed effects include chloracne, reproductive effects, dental effects, effects on thyroid function, diabetes, immunological effects, mortality and incidence of cancer (Bertazzi et al., 1998; Alaluusua et al., 2004; Mocarelli et al., 2011).
Serum samples from the exposed subjects from the different zones were collected for medical examinations shortly after the accident and the residual serum (1–3 mL) was stored frozen. In 1988, the analytical developments allowed the determination of TCDD in such small serum volumes (Landi et al., 1997, 1998; Needham et al., 1997/1998). Other PCDD/Fs and DL‐PCBs were not analysed in these samples. In later years, blood samples were collected from the exposed subjects and analysed for TCDD and also other PCDD/Fs and DL‐PCBs. Baccarelli et al. (2008) used such data to investigate associations of levels of TCDD but also PCDD/Fs and DL‐PCBs in blood from exposed mothers and TSH levels in blood from newborns. The blood samples of these mothers were collected 16.5–22 years after the incident.
In addition, in order to investigate mainly reproductive effects in women exposed during the incident, the Seveso Women's Health Study (SWHS) was started in 1996 (Eskenazi et al., 2000). The inclusion criteria were the following: (i) women who were 0–30 years old (Phase I) or 31–40 years old (Phase II) in 1976, (ii) who had adequate stored sera collected between 1976 and 1981, and (iii) who resided in the most heavily exposed zones A (n = 234) or B (n = 1,039) at the time of the incident. A wide range of reproductive and other endpoints were studied, including endometriosis, menstrual cycle characteristics, birth outcomes, thyroid hormones and bone density and structure (Eskenazi et al., 2002a,b, 2003, 2014; Chevrier et al., 2014; Wesselink et al., 2014). Levels of TCDD in blood were those determined in samples collected in 1976/1981 and in 1996/1998. In 2014, the Seveso Second Generation Health Study was initiated to follow‐up the children born to women exposed to TCDD during the Seveso incident in 1976.
Yusho Cohort
In 1968, an accidental food poisoning involving around 2,000 people occurred in Japan due to the ingestion of rice oil contaminated with a commercial PCB mixture (Kanechlor‐400), which included high levels of PCDFs (Kuratsune et al., 1971, 1972). The incident also affected a large number of chickens due to the use of a by‐product of the rice oil purification in feed. It took about 9 months before the NDL‐PCBs were detected in the oil (about 2 mg/g). Later studies showed the presence of PCDD/Fs and DL‐PCBs in the oil, reported to contribute 487 and 120 ng WHO1998‐TEQ/g (Yao et al., 2002). This incident is known as Yusho or ‘Yusho oil disease’, and led to a number of studies in the population exposed. A series of diagnostic criteria have been set for the inclusion of subjects as Yusho patients (over 1,900 persons), such as proven history of ingestion of the contaminated oil, prominent dermatological, ophthalmological and mucosal signs, several non‐specific symptoms and signs, and levels and pattern of PCBs (and polychlorinated quarterphenyls (PCQs)) in blood (Mitoma et al., 2015). The congener 2,3,4,7,8‐PeCDF was identified as the most relevant compound for causing the symptoms and, since 2004, blood levels of this congener were adopted as an additional criterion for being considered a Yusho patient (Mitoma et al., 2015).
A number of studies have been performed in Yusho patients and various adverse effects were reported, such as skin lesions, effects on bone, immunological and reproductive effects, including risk of fetal Yusho disease (FYD) and altered sex ratio. Also mortality is followed up (e.g. Mitoma et al., 2015). However, in this assessment, only those studies in which the levels of the target compounds were described in blood or tissues were considered (see Section 2.2). PCBs in blood samples of Yusho patients have been measured in annual medical checks since 1973, and the first analysis of PCDD/Fs and DL‐PCBs in blood samples from Yusho patients started in 2001/2002 (Todaka et al., 2007, 2013; Kajiwara et al., 2011; Mitoma et al., 2015). It has been shown that PCDFs contribute to around 65% to the total TEQ in the blood of Yusho patients (Todaka et al., 2007). The mean total WHO1998‐TEQ concentrations (PCDD/Fs and DL‐PCBs) were reported to be 161.4 pg/g fat (range: 8.2–1,325) in Yusho patients in 2002 (n = 279). These values were around 3.5‐fold higher than the mean in control subjects. The difference in total TEQ was mainly due to PCDFs with a 10‐fold difference between means in cases and controls. Male Yusho patients showed lower mean blood concentrations than females.
Yucheng Cohort
Some 10 years later, in 1979, an incident similar to Yusho occurred in Taiwan, again caused by the ingestion of rice oil used for cooking contaminated with PCBs and their heat‐degradation products, mainly PCDFs as well as PCQs. This incident is known as Yucheng (‘oil syndrome’) and it was estimated that around 2,000 persons ingested the contaminated oil. It took 6 months to detect the presence of PCBs in the rice oil (Hsu et al., 1985). A PCB24 level of 176 μg/g oil was reported by Soong and Ling (1997), and a PCDD/F level around 24 ng TEQ25 /g (corresponding to 22 ng WHO2005‐TEQ/g). The concentration of non‐ortho PCBs (PCB‐77, ‐126, ‐169) in the oil was relatively low when applying WHO2005‐TEFs, being 0.38 ng WHO2005‐TEQ/g. Dioxin‐like mono‐ortho PCBs were not reported.
The first clinical observations in the Yucheng subjects comprised small study groups with poorly defined controls (Hsu et al., 1994). From 1985, epidemiological studies were designed, focused on subjects prenatally exposed, i.e. children born to Yucheng mothers or/and fathers, and adequate control groups. In these subjects (born between 1978 and 1992), a range of effects have been investigated, such as reproductive development, mental and cognitive development, skin and teeth lesions, immunological effects, and mortality (Wang et al., 2003; Guo et al., 2004; Li et al., 2013a,b).
In this assessment, only those studies were considered in which the levels of the target compounds in blood or tissues were described (see Section 2.2). The first analysis of blood samples from Yucheng subjects started in 1979 and continued over the following years including exposed adult subjects (Kashimoto et al., 1981; Chen et al., 1985; Lundgren et al., 1988; Guo et al., 1997; Hsu et al., 2005), as well as levels in children born to Yucheng parents (Ryan et al., 1994). From the samples analysed between 1979 and 1981, the average serum level of 2,3,4,7,8‐PeCDF in Yucheng mothers around the time of delivery was 6,940 pg/g fat (corresponding to 2,082 pg WHO2005‐TEQ/g fat). In 1992, the average value was reported to be 1,090 pg/g fat (327 pg WHO2005‐TEQ/g fat). In children, the median value of 2,3,4,7,8‐PeCDF in samples collected in 1991 was 89 pg/g fat (27 pg WHO2005‐TEQ/g fat). In all cases, these levels were reported to be higher than those found in matched control populations sampled in 1991 (level of 2,3,4,7,8‐PeCDF of 19 pg/g fat in a pooled serum sample of matched control children) (Guo et al., 2004).
3.1.4.1.2. Occupational exposure cohorts
Studies in Vietnam veterans
A number of epidemiological studies have been conducted among Vietnam veterans who were exposed to the herbicides sprayed during the Vietnam War. Most of these studies belong to two main studies, the Air Force Health Study (AFHS) and the Korean Veterans Health Study (KVHS) that considered, respectively, American and Korean military personnel serving in Vietnam.
The AFHS is a prospective study of veterans (all males) of Operation Ranch Hand that was the unit responsible of aerial spraying of herbicides in Vietnam from 1962 to 1971. The study aims to determine whether exposure to the herbicides or their contaminants adversely affected the health and survival of these veterans. From 1962 through 1965 the main herbicides used in Vietnam by the Operation Ranch Hand were small quantities of Agent Purple (2,4‐D26; 2,4,5‐T27), Blue (Cacodylic acid), Pink (2,4,5‐T), and Green (2,4,5‐T). From 1965 through 1970 more than 11 million gallons of Agent Orange (2,4‐D; 2,4,5‐T) were sprayed, and smaller quantities of White (2,4‐D; picloram) and Blue. From 1970 through 1971, only Agents White and Blue were used for defoliation. The herbicide Agent Orange was a 1:1 mixture of 2,4‐D and 2,4,5‐T contaminated with TCDD (from < 0.05 to almost 50 ng/kg). TCDD was unintentionally formed in the production of 2,4,5‐T. The veterans were exposed to herbicides during flight operations and maintenance of the aircraft and herbicide spray equipment (Michalek et al., 1998; Landgren et al., 2015). The comparison group were Air Force veterans who also served in Southeast Asia during the same period but that were not occupationally exposed to the herbicides.
The veterans underwent medical examinations in 1982, 1985, 1987, 1992, 1997 and 2002. When analytical techniques allowed the determination of TCDD in blood samples, levels of TCDD were determined in blood samples collected in the different examination years. Comparison veterans had TCDD levels < 10 pg/g fat (considered to be the threshold for background TCDD exposure), while the Ranch Hand veterans had higher levels, with a median of 12.8, 12.5, 11.6, 11.43 pg/g fat at the 1987, 1992, 1997 and 2002 follow‐up exams, respectively.
The initial TCDD level at the end of duty in Vietnam in the Ranch Hand veterans was estimated for those subjects with TCDD levels above background (10 pg/g fat). The estimation was done using a first‐order kinetic model with a constant half‐life of 8.7 years or in some cases 7.6 years (Michalek et al., 1996; Michalek and Pavuk, 2008). Veterans were assigned to four exposure categories: ‘Comparison’, ‘Background’, ‘Low’ and ‘High’. The median initial dose among all veterans having TCDD > 10 pg/g fat was 94 pg/g fat and this value was selected as cut‐off to separate the ‘Low’ and ‘High’ categories. So veterans were allocated to the exposure categories as follows: ‘Ranch Hand Background’ (≤ 10 pg/g fat), ‘Ranch Hand Low’ (> 10 but ≤ 94 pg/g fat), ‘Ranch Hand High’ (> 94 pg/g fat), and the ‘Comparison’ veterans category (≤ 10 pg/g fat).
In the AFHS, exposure to TCDD has been studied in relation to a number of effects, such as cancer, effects on thyroid function, diabetes, effects on the immune and nervous systems, birth defects, as well as general health status and post‐service mortality (Michalek and Pavuk, 2008; Buffler et al., 2011).
The KVHS is a large‐scale study in Korean veterans that served in Vietnam War between 1964 and 1973. Exposure was estimated through an Agent Orange exposure index based on the proximity of the military unit to the area that was sprayed with Agent Orange, and in 2007, blood samples of 102 Korean veterans were analysed for TCDD (concentration range 0.2–11.4 pg/g fat) (Yi et al., 2013).
3.1.4.1.3. Cohorts with background exposure
Background levels mainly result from exposure via the diet by the general population and may vary in time and geography. Over the past decades, there was a decline in the exposure in industrialised countries resulting in reduced levels in blood and human milk (see Section 3.2.4). A number of studies investigated potential effects associated with background levels in blood or human milk. The different studies are described in chronological order.
Duisburg Cohort
Originally, the Duisburg birth‐cohort comprised of 232 pregnant women and 234 newborns, born between 2000 and 2002, and living in the highly industrialised city Duisburg (Germany). The city is located at the Rhine River and belongs to the Ruhr District, an important agglomeration area in Germany for coal mining, steel production and other heavy industries. (Wilhelm et al., 2008). The city has about 500,000 inhabitants and elevated concentrations of PCDD/Fs in deposited dust in the southern part of the city have been reported. Inclusion criteria were: infants from German or Turkish speaking families, born at term with an Apgar score of at least eight and parity 1–3, without serious complications or illness during pregnancy or parturition and congenital anomalies. Due to self‐selection of the pregnant mothers, it was not possible to calculate a participation rate.
Blood samples were taken from the pregnant women around gestational week 32 and human milk was collected by the mothers within the first three weeks after delivery. In addition, cord blood was collected after parturition. The 17 target PCDD/Fs and 12 DL‐PCBs were measured in the samples, with a median value of 19.33 pg WHO2005‐TEQ/g fat for the sum of PCDD/Fs and DL‐PCBs.
The children have been followed since birth for many different outcomes, such as neurodevelopmental effects, effects on gonadal hormones and on thyroid function (Wilhelm et al., 2008; Winneke et al., 2014).
Russian Children's Study
The Russian Children's Study is a longitudinal cohort study of peripubertal boys and their mothers, residents of Chapaevsk (Russia), to study the impact of environmental contaminants (i.e. organochlorine pesticides, PCDD/Fs, PCBs and lead) on growth and sexual maturation. Chapaevsk is a city located around 800 km South‐East of Moscow, in the region of Samara, with a previous extensive manufacturing of chemical warfare agents (before 1949) and chlorine‐containing industrial and agricultural chemicals ((e.g. hexachlorocyclohexane from 1967 to 1987), hexachlorobenzene, PCP, methyl chloroform, vinyl chloride). The manufacturing process and waste incineration resulted also in widespread contamination by PCDD/Fs and metals, including lead (Revich et al., 2001; Burns et al., 2009; Williams et al., 2010).
The boys were identified from the town's health insurance information system, and were enrolled between 2003 and 2005 at 8–9 years of age. The boys were not eligible if their address was unavailable, they were likely to move during the study, or they had severe cerebral palsy. Of the 572 eligible boys, 516 agreed to participate (90%). Of these, 10 boys living in orphanages were excluded because of missing birth or family history.
Subjects underwent a physical examination and both boys and mothers provided blood samples in which the levels of PCDD/Fs, PCBs and lead were analysed. In a subsample (70% of the participants) also other organochlorines (hexachlorobenzene (HCB), β‐hexachlorocyclohexane (β‐HCH) and dichlorodiphenyldichloroethylene (DDE)) were analysed in mothers (Humblet et al., 2010) and boys (Lam et al., 2015). In 350 boys, the median HCB, β‐HCH and DDE was 159, 168 and 287 ng/g fat, respectively. According to the authors, the concentrations of HCB were much higher than in populations in the US (the 25th percentile being approximately eightfold higher than the median level in the US), whereas DDE levels were in a similar range. The median blood lead concentration in the boys at age 8–9 was 3 μg/dL (Hauser et al., 2008).
A health, lifestyle and diet questionnaire was developed and filled in by the mothers or guardian, as well as a semiquantitative Food Frequency Questionnaire (FFQ) to inform on the child's typical dietary intake in the previous years. Long duration of breastfeeding, age, consumption of local food and having a mother employed at the plant were factors associated with higher serum concentrations of PCDD/Fs and PCBs (both DL‐PCBs and NDL‐PCBs) at age 8–9, whereas higher BMI and having parents with long education was associated with lower concentrations (Burns et al., 2009). Median total WHO2005‐TEQ in 482 boys was 21.1 pg/g fat. PCDDs contributed most to the total TEQ (38%), followed by non‐ortho PCBs (29%), PCDFs (25%) and mono‐ortho PCBs (8%) (Burns et al., 2009). Similar determinants of blood concentrations were found in the mothers, although with opposite direction for the association with breastfeeding of their children (Humblet et al., 2010).
PCDD/F and PCB levels have been studied in relation to reproductive effects such as semen parameters, pubertal timing and growth (Korrick et al., 2011; Burns et al., 2016; Mínguez‐Alarcón et al., 2017). Lead levels as well as organochlorine levels have been studied in relation to onset of puberty (Hauser et al., 2008; Williams et al., 2010; Lam et al., 2015). These endpoints are the same as those investigated for PCDD/Fs and DL‐PCBs, and the implications of this co‐exposure are discussed under each endpoint where results from the Russian Children's Study are presented.
Hokkaido Study on Environment and Children's Health
In Japan, the Hokkaido Study on Environment and Children's Health was established in 2002 as a hospital‐based prospective birth cohort study to investigate the possible adverse effects of PCBs, PCDD/Fs, PFASs and other environmental contaminants on fetal growth, risk of infections and allergy, and neurodevelopment (Nakajima et al., 2006; Konishi et al., 2009; Miyashita et al., 2011).
Between 2002 and 2005, a total of 514 (30% of those approached) native Japanese pregnant women living in Sapporo and the surrounding industrialised areas who were between the 23–35 weeks of gestation and no serious illness or any other medical complications, were enrolled. A self‐administered questionnaire survey provided information on potential confounding variables in relation to the past medical history of the mothers and their partners and other, such as smoking habits, dietary intake during pregnancy, and caffeine and alcohol intake. PCDD/Fs and DL‐PCBs were determined in 426 of the maternal blood samples collected according to the weeks of gestation: 23–31 weeks, 32–34 weeks, 35–41 weeks, and within a week after delivery. The mean (range) total PCDD‐TEQ, PCDF‐TEQ, non‐ortho PCB‐TEQ, mono‐ortho PCB‐TEQ and total TEQ were 7.4 (1.7–29.3), 2.6 (0.6–7.8), 4.6 (0.7–23.2), 0.4 (0.1–1.5) and 14.9 (3.2–43.4) pg WHO2005‐TEQ/g fat, respectively (as reported for 398 maternal blood samples by Konishi et al., 2009).
Flemish Environment and Health Study (FLEHS)
In Belgium, the cross‐sectional FLEHS was initiated in 2002. A total of three cycles have been performed: FLEHS I (2002–2006), FLEHS II (2007–2011) and FLEHS III (2012–2015). The studies included a total of 5,825 participants, who had been living in Flanders for at least 10 years. In FLEHS II and III, additional participants were recruited from hot spot areas. The following age groups were considered: mothers and their newborns, adolescents (14–15 years old), adults (20–40 years old), and older adults (50–65 years old). A questionnaire provided information regarding personal background, lifestyle factors, and food intake. Maternal blood and cord blood, and adolescent and adult blood were collected from the participants in which a number of compounds were analysed, including PBDEs, PFOS/PFOA and metals (e.g. As, Cd, Pb). PCDD/Fs and DL‐PCBs were measured by CALUX in the FLEHS II cycle, with geometric means (95% CI) of 108 (101, 114) pg BEQ/g fat for PCDD/Fs, and 32.1 (30.1, 34.2) pg BEQ/g fat for DL‐PCBs (Schoeters et al., 2012; Croes et al., 2014). Most analyses from the FLEHS are cross‐sectional but a few have cohort design. The study investigated possible adverse effects on male and female pubertal development, semen quality, postnatal growth and anthropometric parameters among others (Dhooge et al., 2011; Croes et al., 2014; Delvaux et al., 2014; Iszatt et al., 2016).
Norwegian Mother and Child Cohort (MoBa)
The MoBa is a prospective population‐based pregnancy cohort that includes 95,000 pregnant women and their 114,000 children born between 1999 and 2008 (Magnus et al., 2006, 2016). The women filled in questionnaires throughout pregnancy and donated blood and urine in the second trimester. Furthermore, parents replied to questionnaires at regular time points while children were growing up (e.g. 6 months, 18 months and 3 years). Maternal exposure to PCDD/Fs and DL‐PCBs (year 2002–2009, n = 83,524) has been assessed by a semiquantitative FFQ and a database on concentrations of PCDD/Fs and DL‐PCBs in food in Norway (Caspersen et al., 2013). Maternal median dietary intake was 3.92 (interquartile range (IQR) 2.59) pg WHO2005‐TEQ/kg bw per week (LB). Only 2.3% of the participants exceeded an intake of 14 pg TEQ/kg bw per week. The FFQ is a valid tool for ranking pregnant women according to high and low intakes of energy, nutrients and food (Brantsæter et al., 2008). Furthermore, in a non‐pregnant adult population, the blood concentrations (total TEQ) predicted by dietary intakes assessed by the same tools and demographic variables were highly correlated with measured levels in blood (Kvalem et al., 2012). Associations between maternal dietary exposure to PCDD/Fs and DL‐PCBs and different outcomes in the entire cohort were studied (Papadopoulou et al., 2013; Caspersen et al., 2016a).
The MoBa subcohorts BraMat (Stolevik et al., 2011, 2013) and the ADHD Study (Caspersen et al., 2016b) have been included in the present opinion.
Amsterdam/Zaandam mother‐child Cohort
Since 1987, a longitudinal cohort study on the effects of background levels of PCDD/Fs and DL‐PCBs was initiated in the Netherlands, in the region of Amsterdam/Zaandam. This mother‐children study included women with optimal pregnancy, gestational age between 37 and 41 weeks, birth weight above 2,500 g, and all children breastfed. The children have been studied during their neonatal (n = 60), toddler (n = 60) and prepubertal period (currently 14–19 years old) (n = 41) (Pluim et al., 1994; Leijs et al., 2017).
Levels of the 17 PCDD/Fs were determined in human milk 3–4 weeks after birth as a measure of prenatal exposure with a mean (range) of 32.6 (9.05–88.8) pg I‐TEQ/g fat. Lactational intake at 11 weeks was estimated as a measure of the postnatal exposure considering the levels in breast milk multiplied by the human milk intake. In addition, levels in blood serum of the 17 PCDD/Fs and 3 DL‐PCBs (PCB‐77, ‐126 and ‐169) were measured in the prepuberty period with mean (range) values of 2.2 (0.4–6.4) and 2.2 (0.04–7.8) pg WHO2005‐TEQ/g fat, respectively (Leijs et al., 2012, 2017).
Subjects in this cohort have been examined during their neonatal period for haematological profile and plasma enzymatic activity (e.g. AST) (Pluim et al., 1994). Neurodevelopment has been studied when the subjects were toddlers (Ilsen et al., 1996) and during prepuberty (Ten Tusscher et al., 2014). During prepuberty, subjects have been examined also for pubertal development (Leijs et al., 2008), effects on immunology and haematology (Leijs et al., 2009), on thyroid hormones levels (Leijs et al., 2012) and energy metabolism (Leijs et al., 2017).
Rotterdam and Groningen Cohorts
Two other cohorts were set up in the Netherlands in the early nineties to study potential adverse effects of pre‐ and postnatal exposure to PCBs and PCDD/Fs on children's health. The first cohort in the Rotterdam area contained 207 children of which 105 were breastfed for at least 6 weeks and 102 were exclusively bottle‐fed with infant formula. The second cohort was established around Groningen and contained 211 women, of which 104 breastfed their infants as compared to 107 that formula‐fed their infants. Pregnant women were recruited between June 1990 and June 1992. Prenatal exposure to PCBs was estimated by analysing PCB‐118, ‐138, ‐153 and ‐180 in maternal and cord blood, and later also in blood from 42‐month‐old children, using GC‐ECD (Koopman‐Esseboom et al., 1994a). Postnatal exposure was assessed by analysing the 17 PCDD/Fs and 3 DL‐PCBs (PCB‐77, ‐126 and ‐169) by GC–HRMS. In addition, 23 non‐planar PCBs were analysed by GC‐ECD, including the mono‐ortho DL‐PCB‐105, ‐118, ‐156, as well as the di‐ortho PCB‐170 and ‐180 which at that time were considered dioxin‐like congeners. Initially, I‐TEFs (NATO) and WHO‐PCB‐TEFs (Ahlborg et al., 1994) were applied. When applying WHO2005‐TEFs on the levels reported by Koopman‐Esseboom et al. (1994a), median milk levels of PCDD/Fs, non‐ortho‐ and mono‐ortho‐PCBs would be 29, 16 and 2 pg WHO2005‐TEQ/g fat. The infant formula did not contain detectable amounts of PCDD/Fs or PCBs. At ages 2 weeks, and 18 and 42 months, all children were examined for neurological effects, using various tests (Huisman et al., 1995a,b; Koopman‐Esseboom et al., 1996; Lanting et al., 1998; Patandin et al., 1999).
The 105 mother–children pairs from the breastfed group from Rotterdam were also examined for the relation between blood levels of PCDD/Fs and PCBs and those of thyroid hormones (maternal total T3 and total T4, infant total T3, total T4, free T4 and TSH) (Koopman‐Esseboom et al., 1994b). At 42 months of age, children from this cohort were examined for immunological effects using a questionnaire and analysing the blood for antibodies against measles, mumps and rubella virus, for which they were vaccinated at the age of 14 months (Weisglas‐Kuperus et al., 2000). In a subgroup of 85 children, monocytes, granulocytes, lymphocytes and white blood cells were counted. In this study, the TEQ levels were recalculated with WHO1998‐TEFs, showing median levels for PCDD/F‐TEQ, non‐ortho PCB‐TEQ and mono‐ortho PCB‐TEQ of 36, 15 and 14 pg WHO1998‐TEQ/g fat. Children from this cohort were also examined for birth weight and growth rate and sex‐specific behaviour (Patandin et al., 1998).
3.1.4.2. Chloracne and other dermal effects
Chloracne is a persistent cystic and hyperkeratotic skin malady and was the first recorded unequivocal toxic effect of PCDD/Fs probably in manufacturing workers during the 19th century in Germany (Panteleyev and Bickers, 2006). The most important features are non‐inflammatory alterations of keratinisation of the pilosebaceous glands resulting in initial erythema followed by comedones, cysts, pustules and abscesses that lead to scarring in the long term. Hair follicles can disappear and the process probably induces metaplasia of the sebaceous gland. In most patients, the face and neck are the most common areas of the skin affected by the lesions but chloracne is also observed on areas such as back, groin, genitalia and thighs. Chloracne has similarities, but is not identical, to common acne vulgaris including the lack of bacterial infection in the early stages although eventually secondary infection may occur. It may persist as long as PCDD/Fs are circulating in blood but indications are that levels have to be of a sufficiently high enough level long term to trigger or continue chloracnegenic symptoms. Chloracne has also been reported in occupational exposure to polyhalogenated naphthalenes and 3,4,3’,4’‐tetrachloroazoxy‐ and –azobenzenes (Taylor, 1979). Chloracne may not develop for weeks following exposure, but once established it can persist for years.
Attempts to estimate critical blood levels or body burdens of PCDD/Fs to cause chloracne have been difficult particularly because of a lag time between clinical observations and analytical studies. Average adipose levels of TCDD and hexaCDD at the time of occupational exposure can be back‐calculated as approximately 18,000 pg TEQ/g of adipose lipid (Beck et al., 1989). Average body burden of PCDD/Fs in members of a family who developed chloracne after consuming contaminated olive oil was estimated as 130,000–310,000 pg TEQ/kg bw (Rodriguez‐Pichardo et al., 1991). For chloracne caused in the Yucheng episode, a body burden of 2,000,000 pg TEQ/kg bw was estimated (Ryan et al., 1990). However, these estimations contain large degrees of uncertainties and assumptions. Two patients who had severe and mild chloracne due to TCDD poisoning had blood levels of 144,000 and 26,000 pg/g blood lipid, respectively, (Geusau et al., 2001). The Ukrainian case report of severe chloracne following ingestion by a male adult of pure TCDD is a well‐documented example showing levels of 108,000 pg/g blood lipid measured within weeks of poisoning and an estimated initial body burden of 20,000,000 pg/kg bw (Sorg et al., 2009; Saurat et al., 2012). In contrast, chloracne occurrence was not reported in a Ranch Hand study in which blood levels of TCDD at time of estimation were only modestly raised above background (Burton et al., 1998). However, it is clear that across individuals blood tissue levels of PCDD/Fs, although very high, do not correlate with development and persistence of chloracne, but probably also depend on factors such as the type of dioxin‐like chemical involved, levels in sebum and probably unknown susceptibility factors including age (Beck et al., 1989; Mocarelli et al., 1990; Baccarelli et al., 2005; Panteleyev and Bickers, 2006). There is insufficient information to determine critical levels of DL‐PCBs likely to cause chloracne independent of co‐exposure to PCDFs.
Although a skin disorder, and frequently observed historically in occupational circumstances, chloracne can occur after ingestion of PCDD/Fs and episodes of severe poisoning via food are well described (Bertazzi, 1991; Rodriguez‐Pichardo et al., 1991; Guo et al., 1999b; Geusau et al., 2001; Sorg et al., 2009). Development of chloracne does not seem to be predictive of any other documented disorders in the patients (Baccarelli et al., 2005). In the severe Yusho and Yucheng poisoning episodes, due to rice oils contaminated with PCBs and PCDFs (see Section 3.1.4.1.1), pigmentation of the skin and ocular problems, including Meibomian gland abnormalities were also observed (Nakamura et al., 2005) but direct comparison with TCDD‐like effects is difficult due to the initial complex mixtures ingested (Masuda, 1985; Guo et al., 1999b; Mitoma et al., 2015).
In conclusion, chloracne caused by PCDD/Fs is now considered to be the most reliable and specific indicator of toxicity in humans but its occurrence only in accidental, deliberate or occupational incidents at levels of PCDD/Fs far higher than most chronic exposures, means that incidences of this are not really pertinent to the risk assessment of much lower exposures especially those of the general population.
3.1.4.3. Reproductive effects (including organs)
In the studies summarised below, some have measured changes in sex hormone concentrations in serum in addition to other endpoints. As changes in serum sex hormones can contribute to mechanistic explanations for adverse reproductive endpoints, they are reported in conjunction with the studies. However, in this opinion, changes in serum sex hormone levels in adults or children were not considered by the CONTAM Panel to be an adverse effect by themselves. A table with an overview of all studies that reported changes in serum levels of sex hormones, including studies captured by the literature search that were not addressing other endpoints considered adverse by themselves, is included in Annex A.10. Overall, the results considering the changes in serum sex hormone levels were mainly studies in males and most often reported no association.
3.1.4.3.1. Male reproductive effects
The main adverse effects identified were effects on semen quality, cryptorchidism and pubertal development. The studies are discussed below and more details are shown in Annex A.8.1 (Table 65 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2).
Effects on semen quality
In the Seveso study, potential implications of the incident on male fertility were examined in men of three different age groups at the time of the incident (Mocarelli et al., 2008). Men aged 1–9 years (mean age 6.5, n = 71) at that time, had some 20 years later reduced mean sperm concentration (48.6 vs 67.1 million/mL; p = 0.025), reduced per cent progressive motility (32.4% vs 40.0%; p < 0.001) and reduced total motile sperm count (41.8 vs 68.4 × 106; p = 0.018), and reduced oestradiol (76.2 vs 95.9 pmol/L; p = 0.001) relative to a comparison group which comprised of healthy volunteer blood donors with similar age (n = 82) (Mocarelli et al., 2008) (see Annex A.8.1, Table 65 therein). The median serum TCDD concentration among these Seveso men was 210 pg/g fat in 1976–1977, whereas the assumed concentration for the comparison group was ≤15 pg/g fat in 1976–1977 (assumed level based on LOQs of the method). Residents in Seveso who were 10–17 years of age at the time of the incident (n = 44, median serum TCDD = 164 pg/g fat) had increased total sperm count (302.8 vs 206.5 × 106; p = 0.042) and increased total motile sperm count (121.9 vs 72.2 × 106; p = 0.036), but reduced oestradiol (74.4 vs 92.9 pmol/L; p < 0.001). No sperm related differences were observed between Seveso men who were 18–26 years of age at the time of the incident (n = 20, median serum TCDD = 123 pg/g fat) and a comparison group with similar age. The participation rate differed between the Seveso men and the comparison group (33% vs 49%, respectively). The CONTAM Panel noted that 16 men with serum TCDD above 2,000 pg/g fat were excluded without particular reason given in the manuscript (10 men in age group 1–9 years and 6 men in age group 10–17 years at explosion in 1976). In addition, the comparison group comprised of blood donors, which might not be representative of the general Seveso male population. The relatively low numbers of men included in the study make it difficult to conduct exposure–response analysis with reasonable statistical power. Nevertheless, the data were split into four quartiles based on serum TCDD levels, showing median levels of 68, 142, 345, and 733 pg/g fat, and sperm concentrations of, respectively, 55, 55, 57 and 48 million/mL, as compared to 73 million/mL for the controls with a TCDD concentration < 15 pg/g fat (sperm concentrations estimated from Figure 3a in Mocarelli et al. (2008)). The CONTAM Panel noted there was a difference between exposed and the comparison group for sperm concentrations and progressive motility but there was no dose–response association among the exposed.
In a second study among the Seveso residents, Mocarelli et al. (2011) concluded that ‘in utero and lactational exposure of children to relatively low dioxin doses can permanently reduce sperm quality’. The study included 39 boys born between 1977 and 1984 to mothers exposed to TCDD during the incident, and 58 comparison boys born to mothers exposed only to background levels of PCDD/Fs and DL‐PCBs. For estimating the TCDD levels at the actual time of conception, the original levels in 1976–1977 were extrapolated by applying a half‐life of 4 years based on Kreuzer et al. (1997). The estimated median TCDD concentrations in blood among the exposed mothers at conception was 26.0 pg/g fat (11.8, 232.3; 5th and 95th percentiles). In the group of exposed women who breastfed the participating boys, the median level was 19 pg/g fat (range: 11.8–117.3; 5th and 95th percentiles). In the comparison mothers, it was assumed to be 10 pg/g fat based on levels reported by Eskenazi et al. (2004) in 4 pooled samples (20–21 women per pool) from women aged 20 to 40 from the non‐ABR zone. The results showed that sons of exposed mothers that were breastfed (n = 21) had lower sperm concentrations (36.3 vs 86.3 million/mL, p = 0.002), lower total sperm count (116.9 vs 231.1 × 106, p = 0.02), lower progressive motility (35.8 vs 44.2%, p = 0.03) and lower total motile count (38.7 vs 98 million, p = 0.01) than the 36 background exposed breastfed comparisons (see Annex A.8.1, Table 65 therein). When splitting up the exposed group into a low and high TCDD level, there was a further decrease in sperm concentration and motility in those in the high TCDD group. The breastfed boys from the higher exposed mothers (n = 9, median TCDD 60 pg/g fat) showed a lower sperm concentration (median 23.8 million/mL) than the 36 breastfed controls (82.5 million/mL, p < 0.0003). Also, the breastfed boys of mothers with lower exposure (n = 12, median TCDD 13 pg/g fat), showed a significantly lower total sperm concentration (47.3 million/mL, p < 0.05) than the breastfed controls. In comparison with both the controls and the formula‐fed boys from Seveso, the breastfed Seveso boys showed significantly higher follicle‐stimulating hormone (FSH) and lower Inhibin‐B concentrations in the blood, both considered markers for impaired spermatogenesis. There were no differences between the 18 formula‐fed exposed and the 22 formula‐fed and 36 breastfed comparisons.
A study among boys/men from the Russian Children's Study, reported that high serum TCDD concentrations at age 8–9 years were associated with impaired semen quality later in life (Mínguez‐Alarcón et al., 2017). The study also showed significant associations for PCDD‐TEQs, but not for PCDF‐TEQs, DL‐PCB‐TEQs or total TEQs (based on WHO2005‐TEFs). The study population comprised 516 boys enrolled at age 8–9 years during the period 2003–2005 that were followed for up to 10 years. At the age of 18–19 years, 133 young men provided 256 semen samples. The median serum TCDD concentration in these participants at enrolment was 2.9 (range: 0.4–12.1) pg/g fat, and the median PCDD‐TEQ concentration was 8.7 (range: 1.0–36.0) pg WHO2005‐TEQ/g fat. Compared to the lowest peripubertal TCDD exposure quartile, the men in the highest quartile had 40% (95% CI, 18, 66%) lower sperm concentration, 29% (3, 64%) lower total sperm count, and 30% (2, 70%) lower total motile sperm count (see Table 10 and Annex A.8.1, Table 65 therein). The corresponding figures (all statistically significant) for the highest PCDD‐TEQ exposure quartile were 39%, 36% and 40%, respectively. Statistically significant trends (i.e. p‐values < 0.05) were observed only for TCDD and PCDD‐TEQs. The authors provided the information that higher sum of serum PCDD/F‐TEQs were associated with lower sperm parameters, but the trend was statistically significant only for sperm concentration. Men in the highest vs. the lowest exposure quartile had 36% decrease in sperm concentration (p for trend = 0.04), 32% decrease in total sperm count (p for trend = 0.09) and 36% decrease in total motile sperm count (p for trend = 0.12) (L. Mínguez‐Alarcón, J Burns, R. Hauser, 2017–2018, documentation submitted to EFSA). The boys were also highly exposed to organochlorine pesticides and these were correlated with total TEQ (Spearman correlation coefficients 0.72 for β‐HCH, 0.51 for DDE, 0.53 for HCB, n = 350). These organochlorine pesticides were previously shown to affect pubertal development (see pubertal development later in this Section). The study authors performed further analyses on a subgroup of 138 participants for which analyses of both PCDD/Fs and organochlorine pesticides were available. Serum levels of HCB, β‐HCH or DDE were not associated with the sperm parameters with the exception that increasing β‐HCH or DDE exposure was associated with lower sperm volume (adjusted p for trend β‐HCH = 0.04, DDE = 0.09). The results showed that the associations of sperm parameters with TCDD were not affected by further adjustment for HCB, β‐HCH and DDE. The associations between PCDD‐TEQs and semen volume, total sperm count and total motile sperm count became stronger after adjusting for HCB, β‐HCH and DDE, but for sperm concentration the associations became somewhat weaker. The boys in the study also had high exposure to NDL‐PCBs28 (median 235 ng/g fat) that showed high correlation (Spearman correlation coefficient 0.82) with total TEQ from PCDD/Fs and DL‐PCBs. The Spearman correlation between NDL‐PCBs and PCDD‐TEQs (r = 0.65, p < 0.0001) was somewhat lower than similar correlations for PCDF‐TEQs (r = 0.70, p < 0,0001) and DL‐PCB‐TEQ (r = 0.73, p < 0,0001) (L. Mínguez‐Alarcón, J Burns, R. Hauser, 2017–2018, see documentation submitted to EFSA). The study authors kindly provided EFSA with mutually adjusted associations between TCDD and NDL‐PCBs and semen parameters (the further adjustments come in addition to adjustments for BMI, smoking status, alcohol drinker, season and abstinence time that were already included in the original model). There was no significant association between NDL‐PCBs and semen parameters. The associations between TCDD and semen parameters became slightly stronger after adjustment for NDL‐PCBs. After this adjustment, men in the highest quartile of TCDD had 49% lower sperm concentration (p for trend = 0.0005 vs. 0.005 before adjustment), 40% lower sperm count (p for trend = 0.01 vs. 0.05 before adjustment) and 43% lower total motile sperm count (p for trend = 0.01 vs. 0.05 before adjustment). The associations with total TEQ also slightly increased after adjustment for NDL‐PCBs, but did not reach statistical significance. Lead in blood was not correlated with total TCDD (Spearman correlation coefficient 0.04) and adjustment for lead in blood did not change the associations between TCDD and sperm parameters (L. Mínguez‐Alarcón, J Burns, R. Hauser, 2017–2018, documentation submitted to EFSA).
Table 10.
Multivariable adjusted mean semen parameters by quartiles (Q) of serum PCDDs, PCDFs and PCBs among 133 young men in the Russian Children's Study contributing 256 semen samples. The table is based on data in Mínguez‐Alarcón et al. (2017) and data submitted to EFSA by the study authors (in italics) (see Documentation provided to EFSA)
| Median (pg/g fat) | Volume (mL) | Sperm concentration (mill/mL) | Total sperm count (mill) | Motile sperm (%) | Total motile sperm count (mill) | |
|---|---|---|---|---|---|---|
| TCDD a (pg/g fat) | ||||||
| Q1 [0.35−1.70] | 0.77 | 2.7 (2.2, 3.2) | 57.0 (45.0, 72.1) | 128 (95.6, 173) | 61.6 (58.6, 64.7) | 78.0 (56.0, 109) |
| Q2 [1.77−2.90] | 2.45 | 2.9 (2.5, 3.4) | 51.8 (42.4, 63.3) | 136 (105.0, 175) | 65.4 (63.4, 67.4) | 87.9 (67.1, 115) |
| Q3 [3.00−4.30] | 3.40 | 2.6 (2.1, 2.9) | 38.6 (28.2, 52.9)f | 85.8 (60.4, 122) | 59.5 (56.0, 62.9) | 50.1 (33.5, 74.8) |
| Q4 [4.40−12.1] | 5.80 | 3.1 (2.5, 3.7) | 34.5 (25.0, 47.7)f | 91.6 (63.5, 132) | 60.1 (56.6, 63.7) | 54.1 (36.0, 81.4) |
| p, trend | 0.55 | 0.005 | 0.05 | 0.17 | 0.05 | |
| TCDD a , b (pg/g fat) | ||||||
| Q1 [0.35−1.70] | 0.77 | 2.69 (2.20, 3.19) | 60.7 (47.4, 77.7) | 136 (99.8, 186) | 61.9 (58.7, 65.2) | 83.8 (58.9, 119) |
| Q2 [1.77−2.90] | 2.45 | 2.89 (2.45, 3.31) | 53.7 (43.3, 66.6) | 138 (105, 179) | 65.4 (63.3, 67.5) | 88.6 (67.0, 117) |
| Q3 [3.00−4.30] | 3.40 | 2.59 (2.13, 3.05) | 36.7 (26.8, 50.3) f | 81.5 (57.7, 115) f | 59.3 (55.6, 62.9) | 47.4 (31.8, 70.6) f |
| Q4 [4.40−12.1] | 5.80 | 3.11 (2.47, 3.75) | 31.2 (22.7, 42.8) f | 81.8 (56.1, 119) f | 59.4 (66.7, 63.0) | 47.6 (31.3, 72.6) f |
| p, trend | 0.48 | 0.0005 | 0.01 | 0.11 | 0.01 | |
| PCDD‐TEQ a (pg/g fat) | ||||||
| Q1 [0.95–5.62] | 3.53 | 3.2 (2.7, 3.6) | 64.7 (53.5, 78.2) | 172 (136.0, 217) | 63.4 (60.7, 66.1) | 108.0 (82.5, 141) |
| Q2 [5.69–8.42] | 7.10 | 2.6 (2.1, 3.1) | 37.3 (27.6, 50.4)f | 85.0 (58.9, 123)f | 59.4 (56.1, 62.8) | 49.2 (32.6, 73.5)f |
| Q3 [8.68–13.3] | 10.5 | 2.4 (2.1, 2.8)f | 41.9 (32.2, 54.8)f | 87.7 (63.0, 122)f | 63.3 (60.5, 66.1) | 54.9 (38.1, 79.1)f |
| Q4 [13.7–36.0] | 20.1 | 3.2 (2.6, 3.8) | 39.4 (28.9, 53.6)f | 109 (78.7, 150)f | 60.7 (57.2, 64.3) | 65.1 (45.1, 93.8)f |
| p, trend | 0.89 | 0.02 | 0.04 | 0.55 | 0.05 | |
| PCDF‐TEQ a (pg/g fat) | ||||||
| Q1 [0.55–3.20] | 2.76 | 2.9 (2.6, 3.4) | 49.3 (36.4, 66.7) | 128 (93.2, 176) | 63.4 (60.8, 65.9) | 80.3 (56.6, 114) |
| Q2 [3.29–4.66] | 3.86 | 2.3 (1.9, 2.8) | 43.3 (32.3, 58.0) | 83.1 (57.2, 121) | 59.3 (55.7, 62.9) | 48.1 (31.8, 72.6) |
| Q3 [4.76–6.87] | 5.52 | 3.1 (2.5, 3.6) | 39.1 (30.9, 49.6) | 103 (76.7, 140) | 61.1 (58.2, 63.9) | 62.3 (44.6, 87.2) |
| Q4 [7.10–50.6] | 9.87 | 3.0 (2.5, 3.6) | 47.8 (36.2, 63.1) | 126 (94.5, 168) | 63.0 (59.6, 66.5) | 78.2 (56.5, 108) |
| p, trend | 0.48 | 0.78 | 0.82 | 0.90 | 0.82 | |
| PCDF‐TEQ a , b (pg/g fat) | ||||||
| Q1 [0.55–3.20] | 2.76 | 2.9 (2.5, 3.3) | 52.3 (37.8, 72.3) | 132 (94.4, 185) | 63.2 (60.4, 66.1) | 82.5 (57.0, 119) |
| Q2 [3.29–4.66] | 3.86 | 2.2 (1.8, 2.6) | 42.8 (31.7, 57.9) | 77.7 (53.1, 114) | 58.8 (54.9, 62.7) | 44.5 (29.2, 67.9) |
| Q3 [4.76–6.87] | 5.52 | 3.0 (2.5, 3.6) | 39.3 (31.0, 49.9) | 104 (76.8, 139) | 61.0 (58.2, 63.9) | 62.3 (44.5, 87.0) |
| Q4 [7.10–50.6] | 9.87 | 3.1 (2.5, 3.8) | 43.3 (31.9, 58.7) | 118 (85.2, 163) | 63.0 (59.3, 66.8) | 73.4 (50.6, 107) |
| p, trend | 0.29 | 0.33 | 0.79 | 0.99 | 0.82 | |
| PCDD/F‐TEQ a (pg/g fat) | ||||||
| Q1 [1.95–9.13] | 7.0 | 3.0 (2.6, 3.4) | 63.6 (52.0, 77.8) | 165 (133, 203) | 64.0 (61.7, 66.4) | 105 (82.9, 133) |
| Q2 [9.16–13.8] | 10.9 | 2.7 (2.1, 3.20 | 37.4 (27.7, 50.5) f | 82.8 (56.7, 121) f | 58.0 (54.5, 61.4) f | 46.7 (30.6, 71.3) f |
| Q3 [13.9–20.4] | 15.9 | 2.5 (2.2, 2.9) | 40.9 (31.2, 53.8) f | 91.7 (65.8, 128) f | 63.4 (60.6, 66.2) | 57.5 (39.8, 83.1) f |
| Q4 [20.5–62.4] | 32.8 | 3.2 (2.6, 3.8) | 41.0 (30.2, 55.6) f | 112 (81.5, 154) f | 61.4 (57.9, 65.0) | 67.6 (41.2, 96.9) f |
| p, trend | 0.83 | 0.04 | 0.09 | 0.67 | 0.12 | |
| Co‐PCB TEQ a , c (pg/g fat) | ||||||
| Q1 [0.52–4.63] | 3.78 | 2.8 (2.3, 3.4) | 56.5 (44.0, 72.6) | 131 (97.6, 175) | 63.1 (60.3, 66.0) | 81.9 (59.6, 112) |
| Q2 [4.66–6.87] | 5.97 | 2.9 (2.5, 3.3) | 36.9 (26.2, 51.8) | 95.6 (64.1, 142) | 60.8 (57.8, 63.7) | 57.0 (36.5, 89.0) |
| Q3 [6.88–9.97] | 8.19 | 2.8 (2.2, 3.3) | 37.4 (27.9, 50.2) | 88.4 (62.2, 125) | 62.1 (58.6, 65.6) | 53.7 (36.0, 80.1) |
| Q4 [10.1–67.2] | 13.8 | 2.9 (2.4, 3.4) | 51.4 (40.1, 65.9) | 127 (95.3, 168) | 60.9 (57.3, 64.6) | 76.0 (54.7, 106) |
| p, trend | 0.89 | 0.73 | 0.88 | 0.47 | 0.77 | |
| Co‐PCB TEQ a , b , c (pg/g fat) | ||||||
| Q1 [0.52–4.63] | 3.78 | 2.8 (2.2, 3.4) | 59.2 (45.3, 77.3) | 136 (101, 187) | 63.7 (60.6, 66.8) | 86.5 (61.9, 121) |
| Q2 [4.66–6.87] | 5.97 | 2.9 (2.5, 3.3) | 36.7 (26.0, 51.9) | 95.3 (63.1, 144) | 61.0 (57.8, 64.3) | 57.2 (36.1, 90.7) |
| Q3 [6.88–9.97] | 8.19 | 2.8 (2.2, 3.3) | 37.7 (28.0, 50.8) | 89.3 (62.9, 127) | 62.2 (58.6, 65.7) | 54.5 (36.5, 81.4) |
| Q4 [10.1–67.2] | 13.8 | 2.8 (2.2, 3.4) | 46.8 (35.5, 61.7) | 110 (77.2, 158) | 59.3 (54.7, 63.9) | 64.3 (42.5, 97.2) |
| p, trend | 0.93 | 0.18 | 0.28 | 0.22 | 0.23 | |
| Total TEQ a (pg/g fat) | ||||||
| Q1 [4.88–16.8] | 12.4 | 3.0 (2.5, 3.5) | 51.9 (38.3, 70.4) | 131 (94.4, 181) | 61.8 (58.7, 64.9) | 80.4 (55.5, 116) |
| Q2 [17.0–21.4] | 19.5 | 2.6 (2.2, 3.1) | 38.9 (28.7, 52.6) | 85.9 (57.9, 128) | 61.4 (58.4, 64.3) | 51.8 (33.8, 79.4) |
| Q3 [21.7–32.5] | 25.9 | 2.9 (2.4, 3.5) | 42.1 (33.9, 52.2) | 102 (78.2, 132) | 61.2 (58.1, 64.4) | 60.8 (45.2, 82.0) |
| Q4 [33.3–107] | 47.8 | 2.8 (2.3, 3.3) | 44.8 (33.4, 60.2) | 112 (82.4, 151) | 61.9 (58.1, 65.6) | 67.7 (47.8, 95.9) |
| p, trend | 0.84 | 0.61 | 0.68 | 0.99 | 0.68 | |
| Total TEQ a , b (pg/g fat) | ||||||
| Q1 [4.88–16.8] | 12.4 | 2.9 (2.4, 3.4) | 57.5 (40.7, 81.3) | 141 (97.2, 203) | 61.7 (58.1, 62.3) | 86.3 (57.0, 131) |
| Q2 [17.0–21.4] | 19.5 | 2.6 (2.1, 3.00 | 41.5 (30.3, 57.0) | 90.0 (60.0, 136) | 61.3 (58.2, 64.4) | 54.2 (34.9, 84.4) |
| Q3 [21.7–32.5] | 25.9 | 2.9 (2.3, 3.4) | 43.1 (34.6, 53.8) | 103 (79.4, 134) | 61.2 (58.1, 64.4) | 61.9 (45.8, 83.6) |
| Q4 [33.3–107] | 47.8 | 2.9 (2.3, 3.6) | 37.2 (25.4, 54.5) | 97.9 (63.6, 151) | 62.1 (57.1, 67.0) | 59.5 (36.3, 97.4) |
| p, trend | 0.83 | 0.15 | 0.26 | 0.99 | 0.19 | |
| NDL‐PCBs a , d (ng/g fat) | ||||||
| Q1 [75.9–195] | 151 | 3.0 (2.5, 3.5) | 52.1 (39.3, 69.0) | 132 (96.8, 179) | 62.7 (59.6, 65.7) | 81.7 (58.0, 115) |
| Q2 [197–284] | 228 | 2.6 (2.2, 3.1) | 45.2 (33.3, 61.4) | 99.0 (66.6, 147) | 62.2 (59.7, 64.8) | 61.2 (39.8, 94.2) |
| Q3 [288–434] | 334 | 2.7 (2.3, 3.1) | 36.1 (27.3, 47.6) | 84.6 (61.1, 117) | 61.7 (58.1, 65.3) | 50.9 (34.9, 74.0) |
| Q4 [435–1,726] | 693 | 3.0 (2.4, 3.6) | 45.3 (34.2, 60.0) | 118 (88.7, 156) | 59.6 (55.9, 63.3) | 68.6 (49.7, 94.6) |
| p, trend | 0.91 | 0.35 | 0.50 | 0.21 | 0.39 | |
| NDL‐PCBs a , d , e (ng/g fat) | ||||||
| Q1 [75.9–195] | 151 | 3.0 (2.5, 3.6) | 47.6 (35.7, 63.6) | 123 (89.8, 169) | 62.8 (59.7, 65.9) | 76.4 (53.8, 109) |
| Q2 [197–284] | 228 | 2.7 (2.2, 3.2) | 42.3 (31.1, 57.6) | 93.9 (62.3, 142) | 62.3 (59.5, 65.1) | 58.2 (37.2, 91.2) |
| Q3 [288–434] | 334 | 2.7 (2.2, 3.1) | 36.8 (28.0, 48.3) | 85.9 (62.1, 119) | 61.7 (58.1, 65.2) | 51.6 (35.6, 74.9) |
| Q4 [435‐1,726] | 693 | 2.9 (2.3, 3.5) | 51.8 (37.9, 70.9) | 131 (91.7, 185) | 59.5 (55.3, 63.6) | 75.9 (50.7, 114) |
| p, trend | 0.62 | 0.97 | 0.93 | 0.25 | 0.77 | |
Data are presented as predicted estimates (95% CI) adjusted for BMI, smoking status, alcohol drinker, season, and abstinence time at the mean level of continuous covariates and adjusted for frequency of categorical measures. Motile sperm and total motile sperm count models were further adjusted by time to start semen analysis.
Further adjustment for NDL‐PCB concentrations.
PCB‐77, ‐81, ‐126 and ‐169.
NDL‐PCBs including mono‐ortho PCBs. Sum of PCB‐18, ‐28, ‐52, ‐49, ‐44, ‐74, ‐66, ‐101, ‐99,‐ 87, ‐110, ‐118, ‐105, ‐151, ‐149, ‐146, ‐153, ‐138/158, ‐128, ‐167, ‐156, ‐157, ‐178, ‐187, ‐183, ‐177, ‐172, ‐180, ‐170, ‐189, ‐201, ‐196/203, ‐195, ‐194 and ‐206, ng/g fat.
Further adjustment for TCDD concentrations (pg/g fat).
p ≤ 0.05.
A cross‐sectional study within the FLEHS in Belgium including 101 men 20–40 years of age, studied the association between dioxin‐like biological activity in blood (as measured by CALUX) and male reproductive parameters (Dhooge et al., 2006). The mean BEQ level was 14.6 pg/g fat (IQR 7.1–17.6), or 90 pg/L blood (IQR 43–110). When focussing on serum levels above 16 pg/L (cut‐off due to sensitivity assay; n = 90), increased BEQ levels were associated with a decrease in semen volume (16%, p = 0.03) and free and total testosterone levels, but a tendency to an increase in sperm concentration (25.2%, p = 0.07). There were no associations with total sperm count (p = 0.53), sperm morphology (p = 0.85). The authors stressed that the bioassay‐based BEQs should only be considered as relative measures of exposure within the population and that further inter‐laboratory validation is needed.
A cross‐sectional study on semen quality among 319 men from Greenland, Warsaw (Poland), Kharkiv (Ukraine) and Sweden did not find consistent associations between BEQ and the outcomes investigated (Toft et al., 2007).
A cross‐sectional study in Turkey compared the PCDD/F and DL‐PCB concentrations in adipose tissue among 23 fertile and 22 infertile men. With the exception of TCDF (p = 0.0029) and OCDF (p = 0.01), no differences were obtained between the fertile and infertile men regarding other PCDD/Fs or the 12 DL‐PCBs (Cok et al., 2008).
A cross‐sectional case (n = 40) control (n = 8) study on men recruited in a fertility clinics in Belgium did not find significant difference between cases and controls for PCDD/Fs and DL‐PCB‐BEQs (Den Hond et al., 2015).
In summary, the strongest associations were between the exposure to TCDD during infancy/prepuberty and impaired semen quality were observed in the Seveso population (Mocarelli et al., 2008, 2011), as well as in the Russian Children's Study (Mínguez‐Alarcón et al., 2017). The Russian Children's Study observed an association at much lower TCDD levels than the Seveso Cohort, but contrary to Seveso, other PCDD/Fs and DL‐PCBs were shown to contribute significantly to the TEQ level. The Russian Children's Study showed significant associations for PCDD‐TEQs, and for the sum of PCDD/F‐TEQs, but not for PCDF‐TEQs, DL‐PCB‐TEQs or total TEQs (based on WHO2005‐TEFs). The association between TCDD and semen parameters became slightly stronger after adjustment for NDL‐PCBs, but was not changed by adjustment for exposure to organochlorine pesticides determined in a subgroup. It should also be noted that the Seveso boys included in Mocarelli et al. (2008) experienced a rapid increase in TCDD levels shortly after the incident, and that this occurred solely postnatally. Also, the study by Mocarelli et al. (2011) indicates that post‐natal exposure by breastfeeding may be of higher importance for the associations with sperm parameters than in utero exposure, since no differences were observed in boys that were formula‐fed.
The evidence from the Seveso studies suggests that there may be a postnatal period of sensitivity that might expand into puberty.
Cryptorchidism
The hypothesised association between POPs and cryptorchidism, i.e. failure of the testicular descent to the bottom of the scrotum, was investigated in two Danish‐Finnish collaborations by the same research team (Virtanen et al., 2012; Koskenniemi et al., 2015) (see Annex A.8.1, Table 65 therein). Both studies were nested case–control studies and the first, which included 95 cases with measured levels of PCDD/Fs or DL‐PCBs in placenta (reflecting fetal exposure), but did not observe any associations (Virtanen et al., 2012). The second study only included 44 cases and measured levels of PCBs, PCDD/Fs and PBDEs in subcutaneous adipose tissue biopsies collected at the operation (Koskenniemi et al., 2015). The mean age at the operation was 2.3 years among the cases and 2.9 years among the controls. Although not significant in the unadjusted analysis, there were significant associations in the adjusted analyses between the sum of PCDD/Fs and cryptorchidism (OR = 3.69, 95% CI 1.45–10.9, p = 0.01) as well as between the total WHO1998‐TEQs and cryptorchidism (OR = 3.21, 95% CI 1.29–9.09, p = 0.02). The reasons for the huge difference between the unadjusted and the adjusted estimates (the ORs increased from 1.41 to 3.69 and from 1.17 to 3.21, respectively) were not explained by the authors. The study was small and the control group included boys with inguinal hernia which has been associated with cryptorchidism. However, the authors stated that there are no data which suggest that boys with inguinal hernia would be less exposed than healthy children. In addition, the boys were recruited at different ages which may have had an influence on the representativeness of the case group. It was not possible to adjust for lipid content in the samples, information about duration of breastfeeding and obstetric data were obtained almost 10 years after the operation for parts of the participants, and data on body size of the children were lacking. With the exception of a positive correlation between PCB‐WHO1998‐TEQ and serum levels of luteinising hormone (LH) at the age of 3 months, no other statistically significant associations were observed between exposures and reproductive hormones (Virtanen et al., 2012).
Taken together, the two studies do not provide sufficient evidence for an association between PCDD/Fs or DL‐PCBs and cryptorchidism.
Pubertal development
Three papers from the Russian Children's Study (for details about the cohort, see Section 3.1.4.1.3) report on associations between exposure to PCDD/Fs and DL‐PCBs and pubertal development. The boys were examined at enrolment at age 8–9 years and then yearly until the age 17 or 18 years (see Annex A.8.1, Table 65 therein).
There was a dose‐related association between serum TCDD and PCDD‐TEQs in the boys (n = 489, median sum of PCDD/F‐ and non‐ortho‐PCB‐TEQs: 21.1 pg WHO2005‐TEQ/g fat, median TCDD: 2.8 pg/g fat) at age 8–9 years and later pubertal onset measured by testicular volume when boys were at age 11–12 years (Korrick et al., 2011). This was observed across quartiles of TCDD and for PCDD‐TEQs, but not for PCDF‐TEQs, non‐ortho‐PCB‐TEQs (PCB‐77, ‐81, ‐126, ‐169) or for sum of PCDD/Fs and non‐ortho‐PCB‐TEQs. Serum TEQ concentrations of PCDD/Fs and non‐ortho PCBs were not associated with pubertal development reported as Tanner stage two or higher for genitalia. Adjusting the analyses for serum levels of sum PCBs28 at age 8–9 years strengthened the associations between PCDD‐TEQs and testicular volume, which is in accordance with a (non‐significant) tendency for earlier onset of puberty with increasing serum sum PCBs28 in a model adjusted for TEQ at age 8–9 years in the same study. A limitation of the study is that at study enrolment, 12% of boys at 8 years and 18% of boys at 9 years had already entered puberty as measured by testicular volume.
The CONTAM Panel noted that reduced growth, which is also associated with exposure to both PCDD/Fs and NDL‐PCBs in this cohort (see Section 3.1.4.5), may have implications for later pubertal onset. Lead in blood, which did not correlate with TCDD (described in ‘Effects on semen quality’ earlier in this Section) was also inversely associated with height, weight and pubertal onset in this cohort (Hauser et al., 2008).
Humblet et al. (2011a) found that total WHO2005‐TEQ in serum (median 25 pg TEQ/g fat) collected from 444 mothers from the Russian Children's Study when the boys were 8 or 9 years old was not associated with pubertal onset assessed at age 12–13 in their offspring by genital staging 2 or higher, testicular volume and pubic hair staging in unadjusted or adjusted models. There was an association between maternal sum PCBs at offspring age 8–9 and earlier onset of genitalia stage 2 in offspring at age 11–12. In sensitivity analyses, introduction of total TEQ in sons’ blood at enrolment did not affect the association. The correlation between total TEQ in the mothers and sons was 0.36. In secondary analyses, in the subgroup of boys that was breastfed for > 6 months (40%), there was a dose‐related delay in pubertal onset assessed by genital staging 2 and increasing quartiles (Q) of maternal total TEQ. Testicular volume and pubic hair staging was not associated.
At follow‐up, when the boys were 17–18 years (n = 473), 98% were sexually mature measured as genital staging 5 and 97% were sexually mature measured by testicular volume of 20 mL or more (Burns et al., 2016). There was a significant dose‐related association between both later pubertal onset and later sexual maturity (measured by testicular volume and genitalia staging), and higher total WHO2005‐TEQ at age 8–9. The associations were stronger in models adjusted for the serum sum of NDL‐PCBs28 in addition to other covariates. Significant association with earlier pubertal onset and earlier sexual maturity was found for NDL‐PCBs only when adjusted for total TEQ. Pubic hair staging was not associated with serum total TEQ or NDL‐PCBs.
High concentrations of organochlorine pesticides (HCB, β‐HCH and DDE) in a subset (n = 355) of the boys in the Russian Children's Study indicate high co‐exposure (see Section 3.1.4.1.3 for a general description of the cohort). These pesticides were reported to be associated with later onset of puberty (HCB) and later sexual maturity (HCB, β‐HCH) in the same cohort of Russian boys (Lam et al., 2014, 2015). The authors clarified that the correlation (Spearman rank) between organochlorine pesticides and total TEQ was high (0.72, β‐HCH) to moderate (0.51, DDE, and 0.53, HCB), and the correlations between serum PCBs and organochlorine pesticides were in similar range (0.71–0.49) (L. Mínguez‐Alarcón, J Burns, R. Hauser, 2017–2018, documentation submitted to EFSA). This made it difficult to create categories of high vs. low co‐exposure to multiple organochlorine compounds, and the authors could not provide further adjusted analyses.
A small underpowered study (n = 14) on boys born between 1986 and 1991 in the Netherlands reported a negative trend with current exposure to PCDD/Fs and DL‐PCBs for age of first ejaculation (self‐reported) (Leis et al., 2008). No association was seen with pubic hair growth, axillary hair, genital stage or testicular volume. The study was too small to draw any conclusions.
A study with only 23 boys from Taiwan reported no association with sex characteristics (Tanner stage) and exposure to PCDD/Fs and PCBs in utero (Su et al., 2012).
A cross‐sectional study on 80 Belgian 17‐year‐olds observed no association between PCDD/Fs and DL‐PCBs reported as CALUX‐BEQs and genital development, pubic hair growth and testicular volume or sex hormones (Den Hond et al., 2002). Another cross‐sectional study on 324 Belgian boys aged 13.6–17 years found no significant association between genital or pubic hair development or sex hormones and CALUX‐BEQs (Croes et al., 2014).
In summary, longitudinal analyses from one cohort, the Russian Children's Study, that followed the boys for 9 years, show dose‐related association between exposure to TCDD and total WHO2005‐TEQ measured in the boys’ blood at age 8–9 years and delayed pubertal onset as well as delayed sexual maturity. These associations were strengthened by adjustment for exposure to the sum of the 23 measured mono‐ and di‐ortho PCBs28, which on the other hand was associated with earlier pubertal onset. The delay in pubertal onset with increasing maternal blood concentrations in the subgroup of Russian boys breastfed > 6 months may indicate a sensitive period postnatally. However, the length of time between pregnancy and sampling of maternal blood precludes this conclusion. First, there are clear indications of decreasing TEQ trends in blood from women in the region from 2000–2009 (average decrease in total TEQ 30%), a time span encompassing the inclusion period in the Russian Children's Study (Humblet et al., 2011b). Second, it is likely that the levels in children breastfed > 6 months at age 8–9 are better correlated with maternal blood levels (and thus prenatal exposure) than the levels in formula fed children are, because of the additional exposure during breastfeeding.
The moderate to high correlation between TEQ from the sum of PCDD/Fs and DL‐PCBs and organochlorine pesticides, together with the high blood concentration of organochlorine pesticides which also affects pubertal development in this population complicates interpretation of possible causal relationship between exposure to PCDD/Fs and DL‐PCBs and timing of puberty.
3.1.4.3.2. Female reproductive effects
The main endpoints studied were endometriosis, pubertal development and other effects on female reproduction. The studies are discussed below and more details are shown in Annex A.8.2 (Table 66 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Endometriosis
Twelve studies addressed endometriosis, of which 11 had cross‐sectional case–control design and one was a prospective cohort study (see Annex A.8.2, Table 66 therein). The cohort study was from Seveso (Eskenazi et al., 2002a) and reported a non‐significant twofold increased risk of endometriosis in the group of women with serum TCDD levels above 100 pg/g fat vs. those with less than 20 pg/g fat in blood sampled after the incident in 1976. No dose response was observed.
Exposure in the case–control studies was much lower than in Seveso:
Six studies reported no difference in blood levels of PCDD/Fs and DL‐PCBs between cases and controls (Pauwels et al., 2001; Fierens et al., 2003; De Felip et al., 2004; Niskar et al., 2009; Porpora et al., 2009; Cai et al., 2011)
One reported lower levels in cases than in controls (Tsukino et al., 2005)
Four reported higher levels in endometriosis cases than in controls (Heilier et al., 2005; Simsa et al., 2010; Martínez‐Zamora et al., 2015; Ploteau et al., 2017).
Several of the case–control studies had low number of cases. Furthermore, in some studies important confounders were not considered.
Since the only prospective study did not observe a dose response, and since the cross‐sectional case–control studies indicating associations had some limitations (e.g. low number of cases and lack of confounder adjustment), the CONTAM Panel concluded that the available studies on endometriosis were insufficient to be used as a basis for the risk assessment.
Pubertal development
In the Seveso Women's Health Study, no associations were observed between TCDD and age at menarche (Warner et al., 2004). Two cross‐sectional studies from Belgium found an association between CALUX‐BEQs and earlier breast development (Den Hond et al., 2002; Croes et al., 2014). No correlations were observed between CALUX‐BEQs and serum hormone levels. Moreover, no association was found for pubic hair development (Den Hond et al., 2002).
A small study from Taiwan including 33 girls at eight years of age did not show any differences in fundus and uterus length between girls exposed to high vs. low total TEQ (Su et al., 2012). With the exception of oestradiol the hormone levels were generally unaffected.
Other effects on female reproduction
Seven studies addressed other effects on female reproduction, including menstrual cycle characteristics, ovarian function, time to pregnancy, uterine leiomyoma, and age at menopause (see Annex A.8.2, Table 66 therein). Five of the studies were investigated in the SWHS.
Different menstrual cycle characteristics were investigated in three studies (see Annex A.8.2, Table 66 therein). Among women from Taiwan, placental PCDD/F and DL‐PCB TEQ level was higher in women with irregular menstrual cycles at the age of 18 years than among women with regular menstrual cycles at the age of 18 years (p = 0.032) (Chao et al., 2007). In addition, placental PCB‐TEQ level was higher in women with menstrual cycles longer than 33 days vs less than 33 days (p = 0.006). Among the premenarcheal women in the Seveso study, a 10‐fold increase in serum TCDD level was associated with borderline statistically significant lengthening of the menstrual cycle by 0.93 days (95% CI −0.01, 1.86) and a reduction of the odds of scanty menstrual flow (OR = 0.33, 95% CI 0.10, 1.06) (Eskenazi et al., 2002b). No such tendencies were observed among the post‐menarcheal women. In both menarche groups, TCDD levels were associated with decreased odds of having irregular cycles (OR = 0.46, 95% CI 0.23, 0.95) but not with days of flow (adjusted β = 0.16, 95% CI −0.08, 0.41). A third study performed in Flanders (Belgium) did not find any associations between exposure and menstrual cycle characteristics (Den Hond et al., 2002). In summary, the studies do not give any consistent support for an association between TCDD exposure levels and irregular menstrual cycles.
Time to pregnancy was investigated in one study. Based on exposures near the time of explosion among the SWHS, every 10‐fold increase in serum TCDD resulted in a 25% increase in time to pregnancy and about a doubling in odds of infertility (Eskenazi et al., 2010). The results were similar for the extrapolated TCDD levels and for the sensitivity analysis. The median age at explosion was 17.2 years and the median age at pregnancy attempt was 25.7 years.
The SWHS is the only study which has investigated the hypothesised association between TCDD and ovarian function (Warner et al., 2007), leiomyomas (fibroids) (Eskenazi et al., 2007) and age at menopause (Eskenazi et al., 2005). The most pronounced association was observed for uterine fibroids where women with the lowest exposure had the highest risk (Eskenazi et al., 2007). Among the women who had ovulated, there were no associations between TCDD and serum progesterone or oestradiol (Warner et al., 2007). Since there was only one study per outcome the evidence was considered insufficient.
3.1.4.3.3. Birth outcomes
The main effects studied have been sex ratio, birth weight, and other birth outcomes. The studies are discussed below and more details are shown in Annex A.8.3 (Table 67 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Sex ratio
A study from Seveso (for details on the description of the cohort see Section 3.1.4.1.1) reported that exposed men fathered relatively lower numbers of boys than background‐exposed men (Mocarelli et al., 2000) (see Annex A.8.3, Table 67 therein). Participants in the Seveso study that later became parents (239 men, 296 women) were sampled in 1976/77 and the children (346 girls and 328 boys) were born between 1977 and 1996. Sex ratio (number of newborn males divided by total number of newborns) was lower than the value of 0.514 in the control population (living outside the A/B/R zones or with TCDD < 15 pg/g fat). In fathers having TCDD blood levels > 15 pg/g fat at sampling, there was a significant inverse trend with increasing exposure (p for trend 0.008). The sex ratio (in children) was lower when fathers were younger than 19 years at exposure. This may point towards higher sensitivity when fathers are exposed at low age, but is also consistent with boys (later becoming young fathers) having higher TCDD blood levels than adults (older fathers) after the incident (Mocarelli et al., 2008). Sex ratio was not associated with TCDD blood levels of mothers. The CONTAM Panel noted that it is unknown if boys below 19 years of age (for physiological reasons) could be more sensitive than older men to changes in sex ratio in their children. The CONTAM Panel also noted that a lower sex ratio in the Seveso region with highest exposure (Zone A) was already observed in the period 1973–1976, i.e. before the incident (Mocarelli et al., 2000). The authors suggested that this might be due to an increased exposure from increased release of TCDD into the environment after installation of a small incinerator for waste from the TCP‐production, which started in 1971.
A study on sex ratio in offspring combined two cohorts of highly exposed workers from a herbicide production plant in Ufa, Russia, which produced 2,4,5‐TCP, hexachlorophene and 2,4,5‐trichlorophenoxy acetic acid (2,4,5‐T) (Ryan et al., 2002). Samples from more than 60 workers and children taken in 1992, 25 years after cessation of 2,4,5‐T production, showed higher exposure to TCDD and PeCDD than expected from background exposure. Similar exposure was indicated in samples from a cohort of TCP workers. Resampling was done for 20 of the previous 2,4,5‐T workers in 1997–2000 and in addition 23 samples were taken from the cohort of TCP workers. The study included all children born at any time after 9 months from the date from which the workers were employed, and most were born in 1965–1972 and fewer up to 1991. Data on birth year and children's sex were taken from company archive records and confirmed by interview. For the general population of Ufa, the birth data were from the State Region Statistical Department. Birth data for in total 227 children from workers showed that the sex ratio of the children from 150 male workers was 0.31 and from 48 female workers 0.51, showing a significantly lower ratio of boys being born from male workers than the normal 0.512 in Ufa. TCDD exposure was measured only in small subsamples of the cohorts and shown to be high. However, for most participants the individual exposures are largely unknown.
In a study on sex ratio in children of men who worked in two plants producing Agent Orange, serum TCDD concentration at conception was calculated based on a pharmacokinetic model and TCDD levels in blood samples from 1987 (Schnorr et al., 2001). Men with no self‐reported occupational TCDD exposure served as matched reference population based on residence, age and race. Data on offspring were obtained by interview with wives or ex‐wives of study men (220 referent wives, 200 worker's wives, 1,339 pregnancies). Workers and referents had similar sex ratio in their offspring and there was no change in sex ratio across quartiles of estimated paternal TCDD level at conception.
‘t Mannetje et al. (2017) reported a lower adjusted OR for male birth outcome in offspring from phenoxy herbicide workers in New Zealand with current serum TCDD levels > 4 pg/g fat in 2007–2008 or with serum TCDD levels > 20 pg/g fat when back‐calculated to the time of child birth by use of a one‐compartment model and a constant TCDD half‐life of 7.6 years. The overall sex ratio in offspring from fathers was 0.55, and thus, higher than the expected 0.51, but for fathers with TCDD ≥ 20 pg/g fat at time of birth, the sex ratio was 0.47. In a low number of female workers, there was no association between TCDD exposure and altered sex ratio in offspring.
In summary, three studies from three different cohorts (Italy, Russia and New Zealand) show that high TCDD exposure in men is associated with a lower ratio of boys to girls in offspring. The exact exposure in Ufa was not known as it was measured only in a subgroup of the 198 men and 25 years after exposure. One study from the US with similar or higher paternal TCDD exposure found no change in sex ratio, but the exposure assessment was uncertain due to calculation of the level at conception based on levels in 1987, long after cessation of Agent Orange production. The exposure in Seveso was measured in individual participants, but not assessed in relation to exposure at conception. It is unknown whether exposure at conception is of higher relevance than exposure previous in life. Also in the cohort from New Zealand the exposure at child birth is associated with high uncertainty due to back‐calculation 24 to 39 years after the end of exposure.
Despite high uncertainty in the back‐calculations, there appears to be a relationship between high TCDD exposure in fathers and lower sex ratio in offspring, as this has been consistently observed across three different cohorts. In order to include possible supportive evidence, the CONTAM Panel investigated reports on sex ratio in populations with accidental exposure to PCDD/Fs and DL‐PCBs, without measured exposure levels in those affected, and such studies which are summarised below.
No significant alteration in sex ratio in offspring of mothers previously accidentally exposed to contaminated rice oil in the Yucheng incident in Taiwan was reported (Rogan et al., 1999). Later, a decreased sex ratio in offspring from exposed fathers was reported (Del Río Gómez et al., 2002). In particular, a lower sex ratio in offspring from fathers exposed before age 20 was reported. The lack of change in sex ratio in children from exposed mothers was also confirmed in this study.
From the Yusho incident in Japan, no significant alteration in sex ratio in offspring was reported in offspring from certified patients (Yoshimura et al., 2001). The exception was an observation of lower sex ratio in (grand)children (F2) from daughters (F1) of exposed (grand)mothers (F0) in the Yusho incident. However, no change in sex ratio was seen in the F1 generation, neither from exposed fathers nor from exposed mothers in the same study (Tsukimori et al., 2012). An assessment of sex ratio in two regions particularly affected (Tamanoura and Naru) by the rice oil incident in comparison with a reference region (Nagasaki Prefecture) indicated a statistical insignificant increased risk of lower sex ratio (RR = 0.95, 95% CI: 0.89 to 1.01) the first decade after the incident in 1968. In the same period, the rate of stillbirths was significantly increased in the affected areas (Yorifuji et al., 2013).
These studies partly support the reduced sex ratio in offspring of exposed fathers observed in occupational cohorts and in the Seveso incident.
Birth weight and other birth outcomes
The birth outcome most frequently investigated was birth weight (see Annex A.8.3, Table 67 therein). Some studies showed significant negative associations between maternal PCDD/Fs and DL‐PCBs exposure and birth weight (Vartiainen et al., 1998; Tajimi et al., 2005; Tsukimori et al., 2012; Konishi et al., 2009; Papadopoulou et al., 2013, 2014), whereas others did not observe any statistically significant associations (Eskenazi et al., 2003; Halldorsson et al., 2009; Vafeiadi et al., 2014; Wesselink et al., 2014; Govarts et al., 2016). The strengths and the consistency of the associations varied and in general the studies have some major weaknesses such as exposure measured up to 40 years after the birth (Tsukimori et al., 2012) and doubts regarding confounders exemplified by a study where smoking was not associated with birth weight (Konishi et al., 2009). In addition, some studies indicated that the associations were mainly observed among male infants (Tsukimori et al., 2012; Konishi et al., 2009) or was not observed when the analysis was restricted to primiparae (Vartiainen et al., 1998). Moreover, the study by Tajimi et al. (2005) compared more than 40 PCDD/Fs and PCB congeners or combination of congeners and the only congener that show statistically significant association with birth weight after being adjusted for potential confounders was OCDD (Tajimi et al., 2005). Regarding small for gestational age (SGA) or intrauterine growth restrictions (IUGR), no or less consistent associations with exposures were observed (Papadopoulou et al., 2014; Eskenazi et al., 2003). In general, paternal exposure was not associated with birth outcomes such as low birth weight or preterm birth (Lawson et al., 2004; Michalek et al., 1998; Schnorr et al., 2001) although an association with infant death was observed in one study (Michalek et al., 1998). As for the studies where maternal exposure was investigated, also the studies regarding paternal exposures have some serious methodological drawbacks such as exposure measured many years after birth and self‐reported outcome data.
One study among Danish children indicated an association between dioxin‐like chemicals and accelerated early childhood growth, so called rapid catch‐up growth (Wohlfahrt‐Veje et al., 2014).
One small study from Japan (Yusho) investigated the association between exposure to PCDDs, PCDFs, and coplanar PCBs and the so called FYD (delivered descendants with low birth weights and hyperpigmented skin and mucosa) (Tsukimori et al., 2013). Although some strong negative associations were observed, the study did only include 10 cases and the blood samples were collected up to 40 years after the delivery.
One study showed significant associations between PCDD/Fs and DL‐PCBs and preterm delivery (Tsukimori et al., 2008). However, the women in the study were interviewed many years after the exposure and had to recall events that took place years before the interview.
Finally, one study showed an inverse association between PCDD/Fs and DL‐PCBs and anogenital distance (AGD, anus to upper penis), whereas no associations were observed for the corresponding measures among the girls (ACD, anus to clitoris; AFD, anus to fourchette) (Vafeiadi et al., 2013).
In summary, the CONTAM Panel concluded that the available studies on birth weight and other birth outcomes were inconclusive and could not be used as a basis for the risk assessment.
3.1.4.4. Thyroid disease and thyroid hormones
Studies related to the exposure to PCDD/Fs and DL‐PCBs and thyroid disease and thyroid hormones are discussed below and more details are shown in Annex A.8.4 (Table 68 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
The thyroid diseases relevant for this opinion are hypothyroidism, associated with decreased serum levels of thyroid hormones (T4 and T3) from the thyroid gland, and hyperthyroidism, associated with increased serum levels of thyroid hormones. Sometimes these conditions are also accompanied by goitre (enlargement of the thyroid gland). Secretion of thyroid hormones is regulated by the TSH from the pituitary gland. Most T3 and T4 in serum are bound to proteins, but minor parts are present as free hormones. T4 is deiodinated to T3, which is the active hormone. The most important biomarkers at assessment of thyroid function are serum/plasma concentrations of TSH, free T4 and free T3. Changes in free hormones are a result of changes in thyroid function, governed by pituitary‐derived TSH, while levels of total T4 and T3 also are affected by levels of thyroid binding proteins, and may therefore change without clinical implications for thyroid function. At normal pituitary function, TSH is the most sensitive marker of hypothyroidism and increases before lower levels of T4 or T3 are detected. Primary hyperthyroidism is accompanied by high levels of free and total T3 and T4, and also by an abnormally low TSH.
Studies in adults
Details of the studies are shown in Annex A.8.4 (Table 68 therein).
Calvert et al. (1999) performed a cross‐sectional study of associations between TCDD and thyroid hormones in workers (n = 282) who had been occupationally exposed to TCDD in the period 1951–1972. The workers were examined together with 260 age‐ and sex‐matched referents from the same geographical area. For TSH and total T4, there was no difference between workers and referents, or between quartiles within workers. The free T4 index was higher among workers and higher in the upper quartile than in the other workers, but free T4 index is not a good measure of thyroid function.
Johnson et al. (2001) examined associations between serum TCDD levels (current and back‐extrapolated) and thyroid hormones in 32 men who had been spraying herbicides (2,4,5‐T, and 2,4‐D) in Australia, mainly in the 1960s and 1970s. The odds ratios for abnormally high TSH varied between 1.4 and 1.9 in the five examination years, none of them significantly different from 1. There was no increase in thyroid disease among Ranch Hands compared to referents, or in the ‘high’ exposure group; the OR was 1.3 in the latter group, based on only 12 cases, so the power was limited.
Bloom et al. (2006) performed a cross‐sectional study of associations between serum levels of the 17 PCDD/Fs and 4 DL‐PCBs and thyroid hormones in 37 New York anglers. Those recruited were selected among 300 anglers who consumed sportfish and had ‘high’ PCB levels (n = 22) plus 15 non‐consumers. Thyroid hormones were determined in biobanked samples from 1993 to 1994. T3 and T4 results were considered invalid due to limited storage stability (many below reference range). An inverse association was found between sum of concentrations of PCDD/Fs and DL‐PCBs, and free T4 (but not with TSH), but there were no associations with TEQs (sum of PCDD/Fs and DL‐PCB‐TEQ). The study had limited power due to the small sample size.
Turyk et al. (2006) did a similar study on associations between TEQs of PCDD/Fs and 4 coplanar PCBs and thyroid hormones in 29 male Great Lakes anglers and 27 referents. After various adjustments (including total PCBs), no significant associations remained. The study had limited power due to the small sample size.
Darnerud et al. (2010) studied associations between levels of PCDD/F‐TEQs and PCB‐TEQs (but not PCB‐126) in breast milk (3 weeks after delivery) and thyroid hormones (TSH, Total T3, free T4) in mothers (late pregnancy, N = 281) and children (3 weeks, N = 150, and 3 months of age, N = 115). The mean PCDD/F TEQ in breast milk was 9 pg WHO1998‐TEQ/g fat (N = 80). After adjustment for potential confounders, no significant associations were found between. As PCB‐126 was not analysed, the study did not meet the eligibility criteria, but is nevertheless mentioned since thyroid hormones in infants are a potential critical effect.
The most informative studies are described in more detail below.
Individuals in the Ranch Hand Cohort (n = 1,009) were compared regarding thyroid hormones (TSH, total T4, T3 uptake, free thyroxine index) and history of thyroid disease (Pavuk et al., 2003) with an internal reference group (n = 1,429) engaged in the area in the same calendar period (1962–1971) but not in herbicide spraying. Serum samples were collected on five occasions (1982, 1985, 1987, 1992 and 1997). The mean serum TCDD concentrations (pg/g fat, only measured in samples from 1987) were 4.6 pg/g in the reference group and 5.8, 16, and 69 in three groups of Ranch Hand Cohort (‘background’, n = 409, ‘low’, n = 273, and ‘high’, n = 275). The ‘low’ and ‘high’ groups had back‐extrapolated levels ≤ 94 and > 94 pg/g (means 55 and 303 pg/g). The mean TSH was significantly higher (5–10% higher) in the ‘high’ exposure group (n = 287) compared with the referents, while there was no difference for serum T4 or T3 uptake (as an indirect measure of T3 level). The odds ratios for abnormally high TSH varied between 1.4 and 1.9 in the five examination years, none of them significantly different from 1. Age, race and military occupation (e.g. pilots or ground crew), and thyroid medications were adjusted for. There was no increase in thyroid disease among Ranch Hand individuals as compared to referents, or in the ‘high’ exposure group; the OR 1.3 in the latter group, was based on only 12 cases, so the power was limited.
Chevrier et al. (2014) performed a longitudinal study of the association between exposure to TCDD and thyroid hormones in the Seveso Women's Health Study (for details on the cohort see Section 3.1.4.1). Serum TCDD was measured in samples from 1976 (n = 981) and in a subgroup (n = 260) also in 1996. Median TCDD levels in 1976 and 1996 were 60 and 7.0 pg/g fat, respectively. In the 1996 subgroup, also WHO2005‐TEQs (17 PCDD/Fs and 12 DL‐PCBs) were calculated. Thyroid hormones (TSH, total and free T4, free T3) were analysed in samples collected in 1996 and 2008. Potential confounders were adjusted for. Some associations were found between 1976 logTCDD levels and total T4 in 1996, but no significant associations with free T4, free T3 or TSH. In general, there were no associations between 1996 TCDD levels and thyroid hormones, but with one of the models there was an inverse association with total T4, and a slight inverse association with free T3. There were no significant associations between TCDD levels and hypo‐ or hyperthyroidism.
Studies in newborn or children
Details of the studies are shown in Annex A.8.4 (Table 68 therein).
Eight studies examined associations between the 17 PCDD/Fs and/or 12 DL‐PCBs and/or CALUX‐BEQs and effects on the thyroid in newborns (prenatal exposure) or children. Several studies of pre‐ or perinatal exposure (Nagayama et al., 1998a; Leijs et al., 2012, n = 29) had too low statistical power, or number and calculations were unclear (Han et al., 2011). Taken together, the studies (see below) by Matsuura et al. (2001), Wang et al. (2005), Wilhelm et al. (2008), and Maervoet et al. (2007) do not suggest any effect of low‐moderate exposure to dioxins and adverse effects on the thyroid in children, for details see below. The study by Baccarelli et al. (2008), see details below, in more highly exposed children from Seveso provides relatively strong support for a causal association between predominantly prenatal exposure to TCDD and increased TSH, indicating possible subclinical hypothyroidism.
Matsuura et al. (2001) examined associations between PCDD/F‐ and DL‐PCB‐WHO1998‐TEQs exposure via human milk and thyroid hormones in 337 Japanese newborns (TSH) and at the age of 1 year (T4, free T4, T3, and TSH). Thyroid hormones were also analysed in 53 bottle‐fed children. Median PCDD/F and DL‐PCB WHO1998‐TEQ levels were 20–25 pg/g (only shown in diagram). There was no difference in thyroid hormones (TSH in newborns or thyroid hormones: TSH, T3, T4, free T4 at 1 year of age) between breastfed or bottle‐fed children. Nor was there any association between total TEQs in human milk and thyroid hormones in the 337 breastfed children.
Wang et al. (2005) performed a cross‐sectional study of PCDD/Fs and DL‐PCBs and thyroid hormones (TSH, total T4 and T3, free T4 and thyroxine‐binding globulin (TBG) in cord blood serum in 188 newborns in Taiwan. The mean WHO1998‐TEQs (PCDD/Fs and DL‐PCBs) was 16 pg/g fat. In unadjusted analyses, where TEQ levels were categorised into ‘high’ and ‘low’ (cut‐off: median) and stratified by sex, TSH tended to be lower in the ‘high’ TEQ groups, and in female newborns total T3 and TBG was significantly higher in this group. There were some positive rank correlations in female infants, adjusted for maternal age, between PCDF‐TEQs and total T3 and TBG, but not with free T4. There were significant inverse rank correlations between PCDD‐TEQs and mono‐ortho PCB‐TEQs and TSH in females. In analyses adjusted for age and other PCDD/F/PCB congeners, there was an inverse association between non‐ortho PCB‐TEQs with free T4 and the product free T4 and TSH. The authors interpret this as a deficient feedback of low free T4 on the pituitary. In summary, this study finds no clear significant associations between total TEQs and the most important thyroid biomarkers free T4 and TSH.
In mother–children pairs from Duisburg (see description of the cohort in Section 3.1.4.1), Wilhelm et al. (2008) studied associations between prenatal PCDD/Fs and DL‐PCBs exposure (TEQs in maternal blood at about 32 weeks, and human milk – during the first 3 weeks) and thyroid hormones (in cord serum, n = 127). Thyroid hormones were also analysed in the maternal serum samples. The median WHO2005‐TEQ (PCDD/Fs and DL‐PCBs) level was 19 pg WHO‐TEQ/g in maternal blood (n = 182) and 20 pg WHO‐TEQ/g in human milk (n = 149). A number of potential confounders were adjusted for. There were only about 80 pairs of maternal blood total TEQ‐cord blood thyroid hormones and 60 pairs of human milk total TEQ‐cord blood thyroid hormones (TSH, T3, T4, free T3, free T4), with substantial overlap of these pairs. No significant associations were found between maternal TEQs and cord blood thyroid hormones. The point estimates for cord serum TSH (median level 7.8) even indicated a slight inverse association with human milk TEQs.
Maervoet et al. (2007) studied associations between prenatal exposure (as measured by CALUX assay in cord blood) and thyroid hormones (also in cord blood) in about 138 neonates (born 2002–2004) in Belgium. The median CALUX‐BEQ was 26 pg/g. A number of potential confounders were adjusted for. There were significant inverse associations between CALUX‐BEQs and ln free T3 and ln free T4, but no association with cord blood TSH (median level 6.6 mU/L).
Nagayama et al. (2007a) examined breast milk levels of PCDD/Fs and DL‐PCBs in 19 mother who gave birth to children with congenital hypothyroidism and in 38 mothers, matched for age and parity who gave birth to children with normal thyroid function. A case–control analysis was performed and when the breast milk level (collected within 4 weeks after delivery) of PCDD/Fs or DL‐PCBs was ≥ the median level in a control group (from which the 38 control mothers were selected) the level was considered high. The ORs of giving birth to a child with congenital hypothyroidism were significantly increased when having breast milk PCDDs, PCDFs, DL‐PCBs, or dioxin‐like TEQs (expressed per g milk) were all significantly increased (ORs 6, 10, 19, and 10, respectively). ORs were also significantly increased for OC pesticides (DDT, HCH, chlordane, and HCB). While this study provides some support for an association between PCDD/Fs and DL‐PCBs in breast milk and risk of congenital hypothyroidism in newborns, the data cannot be used for quantitative risk assessment.
In women living in Seveso at the time of the incident, Baccarelli et al. (2008) performed a study of the association between maternal exposure to TCDD and blood TSH in neonates born 1994–2005. The main study was only based on mothers’ residence area in the contaminated area 1976–1979 (56 children born to mothers from zone A, 425 from zone B and 533 from a non‐contaminated reference area). In another study, mean TCDD levels in 1993–1995 were found to be 61 pg/g fat in women from zone A, 18 pg/g fat in zone B, and 6 pg/g fat in the referent area. The median TCDD levels in plasma measured 1–2 years after the incident were 447 pg/g fat (zone A) and 94 pg/g fat (zone B). In a small subgroup, maternal serum PCDD/Fs and DL‐PCBs (WHO2005‐TEQs) collected in 1992–1998 (chloracne study) were compared with neonatal TSH in 51 children born 1994–2005. Associations were adjusted for a number of potential confounders. In the area‐based study, blood TSH was significantly associated with residence area of mothers (zone A: 1.6 μU/mL, zone B: 1.4 μU/mL, and reference population: 1.0 μU/mL). The difference in average blood TSH between zones was present through the whole observation period (M. Bonzini, 2016–2017, see documentation submitted to EFSA). The OR for blood TSH > 5.0 μU/mL was 6.6 (95% CI 2.4–19; 9/56 children) in mothers from zone A and 1.9 (0.94–3.9; 21/425 children) in mothers from zone B. In children of mothers from the reference area, 2.8% (15/533) had blood TSH > 5.0 μU/mL. A study of blood TSH in siblings showed that TSH decreased with order of siblings in zone A and B, but not in the reference area. In the subgroup with 51 children, neonatal blood TSH was significantly associated with plasma TCDD and with TEQs based on PCDD/F and DL‐PCBs. Blood sample for routine TSH analysis was usually taken about 3 days after birth. This was also the case in the smaller subgroup, but for this group the timing of blood TSH was missing for several of the children whose mothers had high plasma TCDD levels (M. Bonzini, 2016–2017, see documentation submitted to EFSA). Timing of blood TSH sampling is important since TSH may decrease with a factor of 5–10 during the first 2–3 days after birth (Alexander, 2017; Steinmaus et al., 2010). It is unlikely that timing has confounded the large group‐based study, but in the small substudy with individual TCDD levels, missing data on timing of blood TSH makes the quantitative association between TCDD levels and TSH uncertain in this subgroup. The larger, area‐based study provides relatively strong support for an association for prenatal/perinatal exposure to TCDD and increased blood TSH in neonates, but it lacks individual dioxins biomonitoring data.
Croes et al. (2014) examined cross‐sectional associations between PCDD/Fs and DL‐PCBs (CALUX assay) and thyroid hormones (TSH, free T3 and free T4) in 606 children aged 14–15 years in Flanders, Belgium. Children were recruited from two industrial areas (‘hot spots’) and from other parts of Flanders (‘reference group’). Geometric mean levels for the CALUX assay (in BEQs) was 108 pg/g fat in the reference group and much lower (48 and 70 pg/g fat) in the two hot spots. The authors discuss that this difference could be due to differences in diet and socioeconomic status in these areas. In analyses adjusted for some confounders, a positive association was found (in the total group) between BEQs and free T4 and an inverse association between BEQs and TSH. It is difficult to interpret these results without knowing if associations between BEQs and thyroid hormones were due to differences between areas or within area.
Xu et al. (2014) studied cross‐sectional associations between PCDD/Fs and DL‐PCBs and thyroid hormones in 8‐year old children, 21 from an e‐waste recycling area and 24 from a control area. Median PCDD/F‐TEQs were 155 pg/g fat in the e‐waste area and 139 pg/g fat in the control area. Median DL‐PCB‐TEQs were 16 and 6 ng/g fat. No associations were found between PCDD/F or DL‐PCB levels and thyroid hormones. Since the groups were very small, this study is not useful in the risk assessment.
Su et al. (2015) compared levels of thyroid hormones (TSH, T4, free T4, T3) in 56 eight‐year old children with PCDD/F‐PCB‐TEQs above or below the median, which was 14.8 pg/g fat. No adjustment for potential confounders was applied and no significant differences were found.
Since the potential association between perinatal exposure to PCDD/Fs and DL‐PCBs and thyroid hormones in infants is a key issue (Baccarelli et al., 2008), the CONTAM Panel also reviewed an early study (Koopman‐Esseboom et al., 1994b), published before the cut‐off of the literature review. In a cross‐sectional study of 78 mother–infant pairs the authors examined associations between PCDD/Fs and DL‐PCBs in human milk (total mean 75 pg TEQ29 /g fat, 2 weeks after delivery) and thyroid hormones (total and free T4, total T3, and TSH) in maternal plasma (end of pregnancy), cord plasma and infant plasma at 2 weeks and 3 months age. Associations were found between PCDD/F‐TEQs in milk and maternal total T4 and T3, but not free T4 or TSH. No association was found with cord blood thyroid hormones. Significant positive associations (Spearman rank correlations) were found between human milk PCDD/F‐TEQs and TSH, both at 2 weeks and at 3 months. No adjustment for potential confounders was performed. The authors note that since human milk PCDD/F‐TEQs are correlated with maternal plasma PCDD/F‐TEQs, the study could not disentangle prenatal and postnatal exposure. The CONTAM Panel notes that TSH levels determined a couple of weeks/months after birth are much more stable than TSH levels in the first couple of days after birth. But since this is the only study of associations between PCDD/F‐TEQs and TSH levels at this age, and it was not adjusted for potential confounders, it is insufficient to use for risk assessment.
In conclusion, from studies reporting high exposure (resulting from accidental exposure or incidents) to TCDD or PCDD/F and DL‐PCB‐TEQs there is insufficient evidence for an association with thyroid function/disease in adults. The study by Baccarelli et al. (2008) in highly exposed children from Seveso provides relatively strong support for a causal association between prenatal exposure to TCDD and increased neonatal blood TSH concentration, indicating possible subclinical hypothyroidism. However, the association has only been demonstrated at high exposure since most studies of low‐moderate exposure to PCDD/Fs and DL‐PCBs (resulting from background exposure) in newborns or children do not suggest any adverse effects on thyroid function in children.
3.1.4.5. Type 2 diabetes and obesity
Studies related to the exposure to PCDD/Fs and DL‐PCBs and type 2 diabetes and obesity are discussed below and more details are shown in Annex A.8.5 (Table 69 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
There are a number of studies that have investigated the hypothesis between PCDD/Fs and/or DL‐PCBs and diabetes and obesity, respectively. Since both outcomes are part of the so called metabolic syndrome (a cluster of conditions that also are risk factors for cardiovascular disease: diabetes, abdominal obesity, high cholesterol and high blood pressure) it is reasonable to present them together.
Overall, the studies show mixed results and no clear pattern can be observed (see Annex A.8.5, Table 69 therein). There are however methodological weaknesses with the studies. Statistically significant associations have been observed with diabetes or obesity in cross‐sectional studies (Longnecker and Michalek, 2000; Fierens et al., 2003; Kang et al., 2006; Uemura et al., 2008, 2009; Nakamoto et al., 2013). However, in cross‐sectional studies reversed causality cannot be excluded, especially regarding these outcomes. There are also studies with either no associations, very few participants (n < 15), unclear study population, very few cases (n < 10) or poor quality of the outcomes as well as studies which indicating associations only for specific subgroups (Cranmer et al., 2000; Johnson et al., 2001; Baccarelli et al., 2005; Chen et al., 2008; Warner et al., 2013; Steenland et al., 1999, 2001; Leijs et al., 2017; Pelcova et al., 2009; Yi et al., 2013; Delvaux et al., 2014; Iszatt et al., 2016). These studies are difficult to draw any firm conclusions from. The most convincing associations are from the Ranch Hand veteran studies regarding diabetes, especially when the exposure characterisation was refined by for instance addressing the number of days of actual herbicide spraying and calendar period of service (Michalek et al., 1999a; Michalek and Pavuk, 2008). In addition, a small study (n = 29 matched pairs) showed a statistically significant but small associations between TCDD and SI (insulin sensitivity index) (Kern et al., 2004).
A study on the Russian Children's Study (for details about the cohort see Section 3.1.4.1) reported associations of higher serum total TEQ at age 8–9 years old with lower BMI z scores during the three years follow‐up period. The study suggested greater effects of sum PCBs30 on in particular linear growth than PCDD/Fs and mono‐ortho PCBs, leading the authors to suggest the effects on growth not to be primarily AHR mediated (Burns et al., 2011).
In summary, the CONTAM Panel concluded that the available studies on diabetes and obesity were inconclusive and could not be used as a basis for the risk assessment.
3.1.4.6. Cardiovascular effects
Studies related to the exposure to PCDD/Fs and DL‐PCBs and cardiovascular effects are discussed below and more details are shown in Annex A.8.6 (Table 70 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
The strongest support for increased cardiovascular risk comes from the US study by Steenland et al. (1999, 2001) with very high occupational exposure (serum TCDD > 1,000 pg/g fat). In three cohorts with somewhat lower exposure levels (mean serum TCDD levels of 100–1,000 pg/g) the results in the ground crew in the US Ranch Hand study by Ketchum and Michalek (2005) suggest increased cardiovascular risk, while results in a Dutch occupational study (Boers et al., 2010) and the long term follow‐up outcome in Seveso cohort (Pesatori et al., 1998; Bertazzi et al., 2001) did not (see Annex A.8.6, Table 70 therein). These studies are described more in detail below.
Based on the Seveso study, Pesatori et al. (1998) compared cardiovascular mortality from 1976 through 1991 in inhabitants in zone A (n~800), B (n~6,000) and R (n~38,000) with the whole population in the region of Lombardy as reference. The ecological analysis used age‐ and sex‐stratified data but there were no data on potential confounders on the individual level. Education, employment and housing were considered similar to that in the province of Milan. Relative risks (RR, 95% CI) (all cardiovascular disease) in men were 1.6 (1.1–2.5, based on 21 deaths) in zone A, 0.9 (0.7–1.1) in zone B, and 1.1 (1.0–1.2) in zone R. In women, the RRs in the three zones were 1.0, 1.0 and 1.1, respectively. For subgroups of cardiovascular disease, findings in men and women were not consistent. The authors discussed the possibility of TCDD exposure, or psychosocial stress as possible causes for increased cardiovascular mortality.
Bertazzi et al. (2001) reported results from 5 more years of follow‐up (until 1996) of the same study populations, also using the same reference group. The RR (95% CI) of cardiovascular mortality was 1.1 (0.8–1.5, 37 deaths) in zone A (both sexes combined), and 0.9 (0.8–1.1, 228 deaths) in zone B. Numbers for subgroups in zone A were still small. Since an increased cardiovascular mortality was found only in men in the early period after the Seveso incident, the authors discussed psychosocial stress as a likely cause.
Mortality through 1999 in US military staff spraying herbicides (Ranch Hand, n = 1,262) was compared with mortality in an internal reference group (n = 19,078) of other Air Force military staff engaged in the area in the same calendar period (1962–1971) but not in herbicide spraying (Ketchum and Michalek, 2005). Only age and military occupation (e.g. pilots or ground crew) was adjusted for in the total cohort; information on smoking was not available. RRs were estimated using Cox regression. The overall RR (95% CI) for cardiovascular mortality was 1.3 (1.0–1.6). For the ground crew (n = 587), the RR was 1.7 (1.2–2.4) based on 40 deaths, reference group non‐exposed ground crew. Serum TCDD concentrations (pg/g fat) were determined in 2,452 persons who attended any physical examination between 1987–1997, including 457 of the Ranch Hand group. Half of them had ‘High’ estimated (back‐extrapolated) TCDD levels (> 118 pg/g fat, median 245 pg/g fat), one forth ‘Low’ (32–117 pg/g fat, median 65 pg/g fat) and the remaining had ‘background’ levels (< 32 pg/g fat). The levels actually measured (around 1990) before back‐extrapolation were about 5 times lower. The point estimates for RR of cardiovascular mortality in this subgroup (only 29 deaths) were 1.3 (all), 1.5 (‘High’), 1.8 (‘Low’) and 0.8 (‘Background’) with a trend test p‐value of 0.07, adjusted for birth year, smoking and heredity (some individual data on confounders were available for the subgroup that had attended physical examinations).
A cohort study of 3,538 male workers from 12 US plants producing TCP and other chemicals contaminated with TCDD in the period 1942–1984 was reported by Steenland et al. (1999). A job‐exposure matrix was developed for assigning an exposure score to each worker, which was the basis for grouping workers in septiles. There was no information on smoking or other potential confounders. Cox‐regression showed an increasing trend for mortality in ischaemic heart disease with the following point estimate HRs in septiles 2–7: 1.23, 1.34, 1.30, 1.39, 1.57 (95% CI 0.96–2.6) and 1.75 (95% CI 1.1–2.9). However, also lung cancer risk tended to increase with the TCDD exposure score, making the lack of smoking data problematic. In a subsequent paper (Steenland et al., 2001), the TCDD exposure for the exposure scores was estimated (in a risk assessment of cancer). A cumulative exposure score of 10,000 would correspond to 19,000 pg/g‐years (lipid adjusted). This means that a cumulative exposure score of 20,000, which was the cut‐off between septiles 6 and 7 in Steenland et al. (1999) would correspond to an estimated cumulative exposure of about 38,000 pg/g‐years (lipid adjusted), e.g. a mean serum TCDD level of about 2,000 pg/g fat over a 20‐year period. In a later publication by Cheng et al. (2006) the same 3,538 workers were studied, but exposure was modelled in a different way, using the CADM model. Cheng et al. estimated the TCDD exposure to have been on average 4‐5 times higher than that estimated by Steenland et al. (2001).
Boers et al. (2010) examined mortality in a Dutch cohort of 2,106 workers exposed to TCDD in the production of chlorophenoxy herbicides and other chemicals in the period 1955–1985 (factory A, producing mainly 2,4,5‐T) or 1965–1986 (factory B, producing mainly MCPA and MCPP, but also 2,4‐D). Mortality was followed up through 2006. About half of the workers in each factory (539 in factory A and 411 in factory B) were considered ‘exposed’ (to chlorophenoxy herbicides or chlorophenols or their contaminants). Out of the exposed workers in factory A, 260 had been more exposed (main production: mean back‐extrapolated serum TCDD in 1993: 600 pg/g fat, or clean‐up after an explosion: mean back‐extrapolated TCDD: 1,800 pg/g). These levels were, based on 31 exposed and 16 non‐exposed (mean TCDD 8 pg/g) individuals and back‐extrapolated levels were sensitive to estimated half‐time (Heederik et al., 1998). Hazard ratios (HR, 95% CI) calculated for cardiovascular mortality were 1.07 (0.7–1.6, 77 cases) in factory A and 1.06 (0.7–1.7, 31 cases) in factory B with non‐exposed workers as referents. HRs (95% CI) for ischaemic heart disease mortality were 1.15 (0.7–2.0) and 1.56 (0.8–3.1) in factories A and B, respectively. No information about smoking or other potential confounders was available. Lung cancer mortality was similar in exposed and non‐exposed workers. A previous analysis (Hooiveld et al., 1998) of mortality in factory A showed a higher risk of cardiovascular disease.
The following studies were considered less useful for risk assessment, but are described briefly.
Kang et al. (2006) examined the prevalence of ‘heart conditions’ by telephone interviews in 1999–2000 in survivors from the US army staff (Army Chemical Corps, ACC) spraying herbicides (n = 1,499) in Vietnam. The comparison group was other personnel from the US Army with similar type and period of service but not in Vietnam (n = 1,428). Serum TCDD was determined (in 1999–2000) in 795 Vietnam veterans and 102 in the comparison group. The mean TCDD levels were 4.3 ng/g fat in 357 men who had sprayed herbicides and 2.7 ng/g fat in 413 men who had not. The difference in self‐reported heart conditions between the groups (adjusted OR 1.1, 95% CI 0.9–1.4 for Vietnam yes/no). Within the Vietnam veterans the OR was 1.4 (95% CI 1.1–1.9) for having spraying (yes/no). A serious limitation is the imprecise cardiovascular outcome.
Survivors from the ACC were also interviewed in 2013, as reported by Cypel et al. (2016). Associations between self‐reported physician‐diagnosed hypertension and having sprayed herbicides in Vietnam were studied by comparing 1,477 Vietnam veterans with 1,609 Non‐Vietnam ACC staff (about 75% of those approached). Self‐reports were validated against medical records, and in some cases blood pressure measurements. About 60% of the Vietnam veterans had sprayed herbicides vs. about 30% of the non‐Vietnam veterans. The OR (adjusted for a number of potential confounders) for hypertension was 1.8 (95% CI 1.4–2.3) for Vietnam sprayers vs. Vietnam non‐sprayers, 1.7 (95% CI 1.3–2.3) for non‐Vietnam sprayers vs. non‐Vietnam non‐sprayers, and 1.3 (95% CI 0.95–1.7) for Vietnam sprayers vs. non‐Vietnam sprayers. Mean serum TCDD levels in 1999–2000 were 3.5 and 2.5 pg/g fat in the Vietnam staff and 2.4 and 2.2 pg/g fat in the non‐Vietnam staff. It is unclear why the OR for non‐Vietnam sprayers was high in spite of relatively low TCDD levels.
Calvert et al. (1998) examined the history and prevalence of cardiovascular disease in workers who had been occupationally exposed to TCDD at two US plants producing TCP in 1951–1972. Out of 586 eligible workers 400 were alive and could be located, and 70% (n = 281) agreed to participate. They were compared with 260 age‐ and sex‐matched referents from the same geographical area (28% of those invited). The mean serum TCDD in the workers was 220 pg/g fat (back‐extrapolated estimate 1,900 pg/g fat), and they were grouped as ‘High’ (≥ 238 pg/g fat) or ‘Low’ (< 238 pg/g fat). The adjusted odds ratios for previous myocardial infarction (n = 31 in the workers), angina pectoris (n = 24), arrhythmia (n = 34) and hypertension (n = 131) were not significantly increased in the total group or in the ‘High’ exposure group. The power was limited due to few cases in the subgroups, and the participation rate was low in the referents.
Manuwald et al. (2012) examined mortality from 1952 to 2007 in 1,145 men and 389 women working in a German chemical plant between 1952 and 1984. The plant produced not only 2,4,5‐T but also other herbicides and insecticides. Standardised mortality ratios (SMRs) were calculated with the population of Hamburg as reference. Exposure to TCDD was estimated from yearly work histories, and serum (or in some cases fat) analyses of TCDD in 275 workers (Flesch‐Janys et al., 1998). There was also exposure to other PCDD/Fs (Flesch‐Janys et al., 1998). The overall SMR for cardiovascular mortality was 1.16 (95% CI 1.02–1.3, 251 deaths) in men and 0.74 (95% CI 0.6–0.9, 58 deaths) in women. The median estimated cumulative TCDD exposure was 77 pg/g‐years (lipid adjusted) in men and 20 in women. No information was available on potential confounders, but smoking prevalence in a subgroup of workers did not differ from that of the general population.
Two studies published data on cardiovascular disease and risk factors in workers exposed to TCDD at a Czech plant producing 2,4,5‐trichlorophenoxyacetic acid (TCPA) in the 1960s. Pelclova et al. (2007) examined microvascular function in 15 men and a control group of 14 unexposed men. The exposed men had a median TCDD of 110 pg/g fat in 2004. Back‐calculated levels were 120–9,000 pg/g. In the exposed group (mean age 59 and mean BMI 29), 8/15 had a diagnosis of ischemic heart disease (IHD) (4/8 myocardial infarction), 9/15 had hypertension and 13/15 had hyperlipidemia. The control group (mean age 54 and mean BMI 27) was healthy. Microvascular function was significantly worse in the exposed group compared with the control group. The same group published a case report (Pelclova et al., 2009) describing various findings in 11 men from the same plant. Many of them were treated for hyperlipidemia, had hypertension, and increased intima media thickness. The study groups were too small to be useful in risk assessment.
Lin et al. (2012) studied mortality using Cox regression in a cohort (n = 2,361) of participants in NHANES 1999–2004. Their median exposure level (based in 17 PCDD/Fs and 9 DL‐PCBs) was 19 WHO2005‐TEQ pg/g fat. There were 75 cardiovascular deaths until 2006. The multivariable‐adjusted hazard ratio in the upper 25% (> 28 pg/g fat) was 1.7 (95% CI 0.6–4.5) compared to the lowest 25% (< 13 pg/g fat). Thus the power of the study was limited.
Thus, the strongest support for increased cardiovascular risk comes from a large US study (Steenland et al., 1999) with very high occupational exposure (serum TCDD > 1,000 pg/g fat). At somewhat lower exposure levels (mean serum TCDD levels of 100–1,000 pg/g), the results are inconsistent; the study in the ground crew in the US Ranch Hand study by Ketchum and Michalek (2005) suggests increased cardiovascular risk, while results in a Dutch occupational study (Boers et al., 2010) and the long‐term follow‐up outcome in Seveso cohort (Bertazzi et al., 2001) do not. The NHANES study (Lin et al., 2012) provides insufficient support for increased cardiovascular risk at low‐level exposure in the general population. In conclusion, increased cardiovascular risk from exposure to TCDD has only been demonstrated at very high exposure, much higher than blood concentrations resulting from exposure at the present TWI of 14 pg TEQ/kg bw per week.
3.1.4.7. Hepatic disorders and digestive effects
Studies related to the exposure to PCDD/Fs and DL‐PCBs and hepatic disorders and digestive effects are discussed below and more details are shown in Annex A.8.7 (Table 71 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Previously considered epidemiological studies of occupational or accidental high exposure to PCDD/Fs have been suggestive of possible hepatic injury but specific diagnoses have not been consistent. Five studies were found comparing blood PCDD/F levels of occupational or accidental exposed cohorts and potential non‐cancer hepatic and digestive disorders or abnormal function (see Annex A.8.7, Table 71 therein).
Although in Seveso subjects after 20 years higher TCDD levels were associated with previously diagnosed chloracne cases, there was no association with increased prevalence of liver disease, such as jaundice or gastrointestinal diseases either in themselves or their children (Baccarelli et al., 2005). Studies of the US Ranch Hand Veterans have reported increased incidences of unspecified ‘other liver disorders’, particularly associated with increased plasma transaminase or lactate dehydrogenase (LDH) levels, and elevated, non‐statistically significant hepatomegaly associated with blood TCDD levels but this may have been due to confounding factors (Michalek et al., 2001a). Ketchum and Michalek (2005) found no evidence of significant increased mortality of US Ranch Hand Veterans associated with all digestive diseases although this was based on small group numbers (10; 0.8 vs. 0.5% for controls). A further retrospective mortality study after >3 decades of Dutch cohorts of chlorophenoxy herbicide manufacturing workers presumed to be exposed to PCDD/Fs showed no greater incidences of diseases of the digestive system from reference groups (Boers et al., 2010).
Neuberger et al. (1999) examined a cohort of Austrian workers who > 20 years previously were exposed to high levels of TCDD in a chemical factory producing 2,4,5‐T and had developed chloracne. Comparison was made with both non‐exposed controls from the same occupational health centre and with non‐chemical plant industrial workers at another site. Significant association was found between blood TCDD levels and elevated levels of plasma transaminases and urinary coproporphyrin I/III ratios indicative of persistent disturbed liver metabolism. Seven patients who had a history of liver disorders had a mean TCDD level of 801 pg/g fat compared to a mean of 407 pg/g fat in those that did not.
In summary, based on the few existing studies, the CONTAM Panel concluded that there is no evidence for an association of hepatic or digestive diseases with prolonged accidental or occupational exposure to PCDD/Fs.
3.1.4.8. Effects on the immune system
Studies related to the exposure to PCDD/Fs and DL‐PCBs and effects on the immune system are discussed below and more details are shown in Annex A.8.8 (Table 72 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Studies in adults
Details of the studies are shown in Annex A.8.8 (Table 72 therein).
Jung et al. (1998) studied in a retrospective case–control approach association of blood levels of PCDD/Fs with frequency of infectious diseases, levels of tetanus antibodies and autoantibodies in formerly pesticide factory workers. No such associations were observed. In a subgroup of individuals at higher exposure levels, and compared these to case controls, effects on proliferation of lymphocytes were tested, in particular resistance to chromate. Here, reduced resistance was found to be associated with exposure, and this was interpreted as an effect on functionality. This measure is not well accepted or widely used. In the study, an important flaw was comprised by the absence of information on co‐exposure to other occupational exposures. The ultimate study was small and concomitant exposure to other chemicals was not accounted for.
Halperin et al. (1998) performed a study in workers from two factories. The test subjects were divided in four TCDD exposure groups. They also compared the subjects to matched referents from the same neighbourhood as the test subjects. Lower numbers of activated T cells were found associated with TCDD exposure, while in the highest exposure group increased mitogen responsiveness in lymphocytes was observed. These data suggest an association between immunological changes in workers and high serum concentrations of TCDD, but the clinical relevance is unknown, while ‘chance findings’ because of assessing multiple immunological variables cannot be excluded.
Michalek et al. (1999b) investigated Vietnam veterans exposed to TCDD from 1962 to 1971. In 1987 and 1992, blood samples were taken to measure TCDD levels, and these were extrapolated to the time in service to put the subjects into four exposure categories. No consistent association with immunoglobulin levels, autoantibody levels, lymphocyte subpopulations, or responsiveness to recall antigens were observed.
Kitamura et al. (2000) recruited 92 municipal waste operators (88 males, 4 females) aged 48 ± 16 years of age. Their actual activities at the workplace were quite different. The only significant correlation with PCDDs, PCDFs and total TEQ exposure found in this cross‐sectional study, after adjustment for age, smoking, and alcohol status, was suppressed NK activity and suppressed phytohaemagglutinin (PHA)‐induced stimulation of proliferation.
Nagayama et al. (2001) reported on effects of PCDD/Fs and DL‐PCBs on immune parameters in Yusho patients. No exposure‐related effects were noted on immune parameters, except an increment of rheumatoid factor, at the higher concentration (TEQ > 200 pg/g fat). Except for the fact that the subjects were Yusho patients, there is no other information on these patients than the age range. For this reason it is not possible to draw conclusions from this study.
Baccarelli et al. (2002, 2004) performed two small studies from the Seveso area (see Section 3.1.4.1.1 for the description of the cohort), and correlated total immunoglobulin levels (IgM, IgG, IgA) and complement factor C3 and C4. Plasma IgG was inversely associated with TCDD plasma concentration (r = −0.35; P = 0.0002). The measures were done approximately 20 years after the incident occurred. After adjusting for age, sex, smoking, and consumption of domestic livestock, the inverse association between plasma TCDD and IgG remained highly significant. IgM, IgA and complement component C3 and C4 plasma concentrations did not exhibit a consistent association with TCDD plasma levels.
Baccarelli et al. (2005) studied individuals that lived at the Seveso area at the time of the incident, and controls in the area less exposed. Twenty years after the incident they studied the association of TCDD exposure and chloracne. In addition, they studied the general health condition, including allergic rhinitis, asthma, eczema, urticaria, in addition to herpes and hepatitis. Whereas TCDD levels were still elevated in individuals suffering from chloracne, no differences in prevalence of any of these diseases was seen between chloracne patients and controls, hence no suggestion of immune effects could be seen.
Nakamoto et al. (2013) investigated the relationship of PCDD/F‐ and DL‐PCB‐TEQs levels in blood and allergic and other diseases. The authors claimed a reduced occurrence of allergies (especially atopic dermatitis) with higher levels of total exposure. However, while atopic dermatitis develops mainly during infancy, blood levels of PCDD/Fs and DL‐PCBs were measured during adulthood, and it is difficult to draw a conclusion on the causality. For PCDDs and PCDFs, an inverse relation with rhinitis was also observed. Moreover, allergy was self‐reported while no independent verification was evident. In addition, information on the family history of allergies was not presented, whereas this could potentially be an important confounder.
Saberi Hosnijeh et al. (2011, 2012, 2013) investigated immune parameters in occupational cohorts of workers exposed from 1953 to 1969, or from 1955 to 1986, with accidental high exposure in 1963. The studies were based on a small numbers of subjects on the same or overlapping cohorts. Exposure was assessed in serum in 2007–2008, while earlier exposure was assessed based on a kinetic model with and estimated half‐life of 7.1 years. No associations were noted between exposure and parameters of humoral immunity (total immunoglobulin levels, specific IgE titres, complement). A relationship was noted of current exposure with eczema, but not with maximal exposure. This suggests that this is a chance finding. Also no effects on lymphocyte subpopulations were noted nor significant associations with plasma levels of IL1RA, sCD27, sCD30. Only if subjects with chronic diseases were excluded, ILRA1 reached significance. The CONTAM Panel noted that no correction for multiple testing was carried out. Other exposures potentially influencing were not considered. In addition, residual confounding was not taken into account.
One study performed by Croes et al. (2014) was a cross‐sectional investigation on 407 adolescents in Flanders. They correlated serum concentrations of DL‐PCBs and PCDD/Fs to animal allergy and asthma, respectively, and found positive associations. Several main cofounders, such as age, (passive) smoking, asthma and allergy in family, breastfeeding, food consumption were taken into account. This finding is in contrast to studies where no relationships or inverse relationships were noted.
Dinse et al. (2016) examined cross‐sectional serum concentrations of a large number of environmental chemicals, including PCDDs, PCDFs, DL‐PCBs, to autoimmunity, measured as the presence of antinuclear antibodies in a cross‐sectional analysis of 4,340 participants in the NHANES study. No such associations were observed.
In summary, these studies do not lend strong support to effects on the immune system of exposure at adolescence or adulthood to PCDD/Fs and DL‐PCBs. Studies are retrospective, and show shortcomings, such as the limitations in the exposure assessment and possible confounding, while the outcomes analysed often differed. It should be mentioned that measures to gauge potential effects on the immune system are not very sensitive. Whereas some studies show effects on total immunoglobulin levels (notably IgG), or on lymphocyte subpopulations other studies did not show that. Endpoints such as allergic conditions have been shown to be associated or inversely associated with exposure, hence the picture is not consistent.
Studies on exposure during development
Details of the studies are shown in Annex A.8.8 (Table 72 therein).
Nagayama et al. (1998b) investigated the association of lymphocyte subpopulations in a cohort of 36 children (1–year old) with PCDD/Fs and DL‐PCBs. They observed enhanced CD4+ and decreased CD8+ levels, hence an increase tendency to increase CD4/CD8 ratios, associated with higher post‐natal exposure expressed as TEQ values in human milk. There were no overt signs of serious diseases, but other information on the subjects or possible confounders were not provided.
In 1999, Van den Heuvell et al. (2002) performed a study on 207 adolescents, born between 1980 and 1983. A decreased prevalence of allergic responses were seen with increasing exposure, which may be corroborated by the findings with specific IgE, and suggest that exposure influenced the immune status of the adolescents. However, since this was a cross‐sectional study and the information is of limited value.
In a study by Nagayama et al. (2007b), lymphocyte subsets were assessed in peripheral blood of 92 children at about 10 months after birth, and correlated to concentration of organochlorine compounds in their mothers’ breast milk. The authors found an association of exposure to PCDD/Fs and DL‐PCBs and an increased ratio of CD4/CD8 cells, as well as an increased percentage of CD3 cells, but the clinical relevance of this finding is unclear.
Leijs et al. (2009) analysed immunological and haematological parameters in a cohort of children that had been followed since 1987 until adolescent. Immunological parameters comprised leucocyte and differential cell counting. A statistical significant decrement in numbers of polymorphonuclear leucocytes was found in the higher concentration group. In isolation, this effect may not indicate immunosuppression. In addition, the number of subjects in this study was low, so that no conclusions can be drawn
Miyashita et al. (2011) correlated prenatal exposure to dioxin‐like compounds with allergies and infections during infancy. The prospective birth cohort study included mothers visiting the Sapporo children's hospital. From the 1,796 mothers who were asked to participate, 514 agreed. Of these, 23 mothers were excluded after delivery for legitimate reasons. Exposure was well characterised and outcomes were reliable. Higher levels of exposure in males were reported to be associated with an increased incidence of otitis media, whereas a marginally significant association with allergies during infanthood was observed. The CONTAM Panel considered that due to the extended number of outcome measured, chance findings are likely.
Stolevik et al. (2011, 2013) reported results of a prospective cohort study, in which they found an association of maternal dietary exposure at background levels to PCBs and TCDD during pregnancy, and the occurrence wheeze as an indicator for respiratory infection in young children. In addition, they reported an inverse association with antibody titres to measles vaccination. Weaknesses of this study include the inherent uncertainty in the dietary exposure estimates for exposure, in addition to the relative small fraction of the Bramat Cohort that eventually entered the study. They also observed an inverse relation with eczema, although this was not statistically significant when corrected for the body weight of the mothers.
Weisglas‐Kuperus et al. (2000) investigated a number of immunological and clinical parameters in children. Primary vaccinations against mumps, measles, and rubella were given to 206 of the cohort of 207 children at approximately 14 months of age as part of the National Immunization Program. Associations with antibody levels were observed for the NDL‐PCBs in blood. PCDD/Fs and DL‐PCBs were not examined. PCDD/Fs and DL‐PCBs were measured in milk but do not show a relation with the levels of antibodies or illnesses investigated in this study.
In summary, whereas some studies may suggest adverse effects on the immune system at exposure at background levels during development, the available studies do not provide sufficient evidence for an association between PCDD/Fs or DL‐PCBs and effects on the immune system.
3.1.4.9. Effects on the nervous system
Studies related to the exposure to PCDD/Fs and DL‐PCBs and effects on the nervous system are discussed below and more details are shown in Annex A.8.9 (Table 73 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Neurodevelopment in children
Details of the studies are shown in Annex A.8.9 (Table 73 therein).
Neurodevelopmental effects of pre‐ and postnatal exposure to PCDD/Fs and DL‐PCBs were addressed in several prospective cohorts with general background exposure. The different neurodevelopmental evaluations performed in the various cohorts examined a variety of endpoints, and results from more than one cohort were available only for few endpoints. The number of participants in each study was in general low (n < 100–200), with the exception of one study using dietary exposure instead of measured levels to assess the exposure. Studies from the same cohort are presented together below.
Results from the Dutch cohort with children from Rotterdam and Groningen (see general description of the cohort in Section 3.1.4.1.1) have partly been published before 1998. The results of these earlier studies are summarised under each endpoint in this section, but are not included in Annex A.8.9 (Table 73 therein).
Motor development in the children in the Dutch cohort was evaluated in relation to PCDD/Fs and DL‐PCB exposure on the second week after birth (Huisman et al., 1995a), at 18 months (Huisman et al., 1995b) and at 3.5 years (Lanting et al., 1998). Higher levels of PCDD/Fs and DL‐PCBs in human milk were related to lower neonatal neurological optimality score in the second week after birth (Huisman et al., 1995a). At 18 months, there were no associations with neurological optimality score. Also, at the follow‐up at age 3.5 years, there were no associations between lactational exposure and neurological optimality (Lanting et al., 1998).
The association between exposure to PCDD/Fs and DL‐PCBs in maternal milk and motor and/or cognitive development in children in the Dutch cohort was assessed at 3, 7 and 18 months (Koopman‐Esseboom et al., 1996), at 42 months (Patandin et al., 1999) and again at 6.5 years (Vreugdenhil et al., 2002). By use of the Bayley Scale of Infant Development borderline significant negative influence of total TEQ in milk on psychomotor development was seen at 3 months, however, at seven months long breastfeeding duration was associated with increased score for both psychomotor and mental development. At 18 months follow‐up, these associations were no longer observed (Koopman‐Esseboom et al., 1996). Cognitive abilities in breastfed children (measured with K‐ABC) and verbal comprehension (measured with Reynell Language Developmental Scales (RLDS)) was not associated with pre‐ and postnatal exposure to PCDD/Fs and DL‐PCBs at 42 months (Patandin et al., 1999). When the children were 6.5 years of age (Vreugdenhil et al., 2002), total TEQ in milk was not significantly related to neurodevelopmental outcomes measured by the McCarthy Scales of Children Abilities subscales. However, milk total TEQ was associated with lower motor scores in children born to parents with lower verbal IQ scores. This may indicate an adverse association in children with less optimal parental and home characteristics. At school age (7.5 years), play behaviour in the children from the Rotterdam branch of the Dutch cohort showed that higher PCDD/F‐TEQ in maternal milk was associated with more feminised play behaviour in both sexes (Vreugdenhil et al., 2002).
In 6‐month‐old children in Sapporo, no association between maternal total TEQ and scores on Bayley Scale of Infant Development was seen (Nakajima et al., 2006). The mean exposure levels in maternal blood fat were more than threefold lower than the mean concentration in maternal milk fat in the Dutch cohort where the same instrument was used. This conclusion of no association was confirmed with a larger sample size from the same cohort at 6 and 18 months (Nakajima et al., 2017). No associations with cognitive development were found at 42 months as well (Ikeno et al., 2018).
In a study on 11‐year‐old children from Hong Kong with low level prenatal exposure, no association between exposure and neurocognitive function was found (Hui et al., 2016).
Significant associations between one developmental milestone (‘cannot crawl’) out of six milestones was associated with blood fat BEQs from Danish mothers (Halldorsson et al., 2009). In addition, the four scores on motor development combined were associated with BEQs. The exposure was associated with high consumption of fat and meat and the authors stated that the bioassay results may be confounded by polycyclic aromatic hydrocarbons (PAHs).
Language and communication skills were investigated in a large group of three year old children in Norway in relation to maternal dietary intake of PCDD/Fs and DL‐PCB during pregnancy (Caspersen et al., 2016a) (see Section 3.1.4.1.3 for the description of the MoBa Cohort). The weekly dietary intake was lower than the current TWI (14 pg WHO2005‐TEQ/kg bw) in 98% of the participants. Children of mothers with high dietary intake (> 14 pg WHO2005‐TEQ/kg bw per week) had a small increase in odds (OR = 1.1) of having ‘incomplete grammar’. Girls had increased odds for moderate or severe language delay and lower communication skills.
In the ADHD study, which is a subcohort in the Norwegian MoBa Cohort, maternal dietary intake of PCDD/Fs and DL‐PCBs was not associated with ADHD symptoms, verbal/non‐verbal IQ, or executive functions including working memory in 3.5‐year‐old children. The maternal exposure during pregnancy was associated with poorer expressive language skills in girls, although the sex‐specific associations were not significantly different (Caspersen et al., 2016b).
In the Duisburg birth cohort study (see Section 3.1.4.1.3 for the description of the Cohort), several neurodevelopmental endpoints have been investigated. In the study assessing neurodevelopment at 2 weeks, 12, 18 and 24 months (Wilhelm et al., 2008) by use of similar methods as previously used in the Dutch Cohort (BSID and NOS), no associations between maternal total WHO2005‐TEQ from PCDD/Fs and DL‐PCBs during pregnancy and the outcomes were observed.
When the children from the Duisburg Cohort were 6–8 years, associations between exposure and masculine and feminine behaviour was assessed (Winneke et al., 2014). There were significant interactions between sex and exposure. In multiple regressions, association with increased feminine behaviour was observed in boys, whereas in girls decreased feminine behaviour was seen. Less pronounced associations were seen for masculine score, but was significantly decreased in girls with increased sum of PCDD/Fs and DL‐PCB‐WHO2005‐TEQ in maternal milk.
Empathising, systemising and autistic traits were investigated at the age of 10 years in the Duisburg Cohort (Nowack et al., 2015). Increasing PCDD/Fs and DL‐PCBs in maternal blood during pregnancy was inversely associated with deficits in social response in the total group. There was significant interaction between sex and exposure and the negative associations between maternal blood PCDD/Fs and DL‐PCBs and social responsive scales were more pronounced in girls than in boys. No association between exposure and empathising and systemising behaviour was observed, contrary to what could be expected keeping the results at 6–8 years in mind (Winneke et al., 2014).
Attention‐related behaviour at age 8–9 year has also been assessed in relation to PCDD/F and DL‐PCB exposure in the Duisburg Cohort (Neugebauer et al., 2015). Association between maternal concentration of PCDD/Fs and DL‐PCBs with more omission in one subtest was observed.
In summary, various neurodevelopmental outcomes at different age have been investigated, but few outcomes have been assessed in several cohorts and/or at similar age. The median exposure in the mothers was highest in the Dutch cohort, followed by the mothers in the Duisburg and Hokkaido (Sapporo) Cohorts, with approximately threefold lower median exposure. The Bayley Scale of Infant Development was applied in these three cohorts. Borderline significant adverse associations with exposure were seen only in the Dutch cohort at 3 months. In the Dutch cohort, some adverse associations between PCDD/F and DL‐PCB exposure and also other outcomes observed during the first months of life appeared to be transient and no longer present at later follow‐up. Play behaviour showed however a significant higher feminine score at age 7.5 years in the Dutch cohort. Also, in the Duisburg Cohort, sex‐specific alteration in behaviour in relation to exposure was present at age 6‐8 years. In contrast to the Dutch study, the directions in feminised behaviour were opposite in boys (increased) and girls (decreased) in the Duisburg Cohort. At 10 years, the associations with play behaviour were no longer seen. Attention‐related behaviour was investigated in the Duisburg Cohort and the MoBa Cohort, but strong or consistent associations were not seen. Other endpoints have only been investigated in single cohorts. There are some indications of sex‐specific associations for some outcomes, but these are not consistent or confirmed in several cohorts. The available information is therefore not sufficient to form a basis for the risk assessment.
Occupational studies
Details of the studies are shown in Annex A.8.9 (Table 73 therein).
A study on Ranch Hand veterans (Michalek et al., 2001b) indicated associations between exposure to TCDD from Agent Orange spraying assessed in blood samples taken 15–26 years after end of exposure and peripheral neuropathy 30–40 years after end of the exposure. This suggests an association with high TCDD exposure and peripheral neuropathy later in life. The authors stated that the results need to be interpreted with care due to possible residual confounding by diabetes; out of the 14 veterans with probable peripheral neuropathy, 13 had diabetes or findings suggestive for prediabetes. Cognitive functioning among Ranch Hand veterans was assessed 11 years after the end of the Vietnam War in relation to blood TCDD measured 15–26 years after the end of exposure. Some measures of memory functioning were somewhat decreased in those with highest back‐calculated TCDD exposure, but the findings were of unknown clinical significance according to the authors (Barrett et al., 2001).
A cross‐sectional follow‐up examination in 1996 of 13 workers from the Czech Republic that had been exposed to high levels of TCDD as a by‐product of herbicide production approximately 30 years earlier showed that the TCDD levels in blood in 1996 was significantly correlated with several neuropsychological variables (Pelclova et al., 2001). The frequency of polyneuropathy electromyographic (EMG) results was significantly decreased from examination in the 1970s (38%) to 1996 (23%). At 35 years follow‐up, no substantial changes in neuropsychological endpoints compared to those in 1996 were observed (Pelclova et al., 2002). However, the associations with TCDD blood levels in 1996 were no more significant, possibly because not all workers completed the full spectrum of tests. Similar observations were reported also at 37–39 years follow‐up in 13–15 patients and in 8–11 participants at 40 years follow‐up (Urban et al., 2007; Pelclova et al., 2009).
In summary, the results from cohorts occupationally exposed to high levels of TCDD indicate persistent associations between the exposure and different neurophysiological and neuropsychological outcomes. However, back‐calculations to original exposures based on levels in blood sampled decades after the end of exposures are highly uncertain in these studies. Furthermore, the studies from workers in the former Czech Republic suffered from low numbers of participants.
3.1.4.10. Effects on teeth and bone
Studies related to the exposure to PCDD/Fs and DL‐PCBs and effects on teeth and on bone are discussed below and more details are shown in Annex A.8.10 (Table 74 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Development of teeth
Details of the studies are shown in Annex A.8.10 (Table 74 therein).
Hard dental tissues are not remodelled after formation, and formation occurs at defined time intervals for different types of teeth, starting from fourth week in utero (first primary tooth) and ending at approximately 20 years of age (roots of the wisdom teeth).
Twenty‐five years after the Seveso incident, dental effects in study participants who were ≤ 9.5 years of age at time of the incident and living in the A, B and R zones in Seveso (n = 48) were compared with subjects of similar age from the non‐ABR zone (n = 65) (Alaluusua et al., 2004). Regardless of zone, tooth enamel defects were primarily seen in those who had been under 5 years of age at the time of the incident (25 out of 27 subjects with enamel effects). The prevalence of tooth enamel defects was higher (42%) in zone ABR subjects than in non‐ABR subjects (26%), and increased with serum TCDD levels in the ABR zone subjects. The OR (95% CI) for enamel defects in the two higher TCDD tertiles (non‐ABR subjects as referents) was 2.4 (1.3–4.5). Furthermore, lateral incisors or second premolars were missing in a higher proportion of ABR zone subjects (12.5%) than in non‐ABR subjects (4.6%). The frequency of hypodontia increased with serum TCDD level.
Similar associations between exposure to PCDD/Fs and DL‐PCBs as those reported from Seveso have previously (before 1998, the inclusion year cut‐off for the present opinion) been reported in breastfed children from Finland (Alaluusua et al., 1996, 1999). Concentration of PCDD/Fs in breast milk from women collected four weeks after giving birth in the Helsinki area in 1987 was determined (mean 19.8 pg I‐TEQ/g fat, range: 3.8–99.4), and the exposure of their children was calculated using interview‐reported breastfeeding duration (mean 10.5 months, range 1–36). Enamel hypomineralisation in the first permanent molars (mineralised during the first two years of life) was seen in 17 of 102 children at age 6–7 years and in total, 35 first molars were affected. The frequency and severity of the lesions correlated with the total exposure from breast milk expressed in I‐TEQ, taking milk level, duration of breastfeeding and decrease in breast milk level during lactation into account and calculated by the authors as area under the curve (‘low exposure’ < 8, ‘moderate exposure’ 8–16, ‘high exposure’ > 16 (pg × year/g milk fat)). At ‘low’, ‘moderate’ and ‘high’ exposure 1/23 (4%), 8/49 (16%) and 8/30 (27%) had mild to severe effects, whereas 0/23 (0%), 3/49 (6%) and 6/30 (20%) had moderate or severe effects. Of note, neither the duration of breastfeeding alone nor the total I‐TEQs in the milk alone was significantly associated with the occurrence of mineralisation changes. In a later review, the authors described results from an unpublished study on Finnish children that were born in the period 1995–1999 (Alaluusua and Lukinmaa, 2006). In this study, no statistically significant correlation was found between child exposure via milk/placenta and molar‐incisor hypomineralisation. The authors stated that the total exposure levels were clearly lower due to lower concentrations in milk/placenta and shorter breastfeeding durations.
In children of women from Taiwan exposed during the Yucheng incident, abnormalities in teeth were reported (Rogan et al., 1988). This was confirmed in a study on Yucheng children where levels of 2,3,4,7,8‐PeCDF and 1,2,3,4,7,8‐HxCDF in the children were analysed (Wang et al., 2003). In this study, the prevalence of children having teeth in the neonatal period, missing teeth or having rotated teeth and having teeth with developmental defects (fusion, microdontia, pigmentation, enamel hypoplasia and impaction) was higher in children of exposed mothers than in the control group.
In Finnish children born in 1997–2000, the prevalence of natal and neonatal teeth was low (1:1,188 and 1:1,013, respectively). No difference in milk PCDD/F and DL‐PCB concentration between cases (n = 12) and controls (n = 11) was found (Alaluusua et al., 2002).
In summary, in three different population groups (Seveso, Helsinki, Yucheng) childhood exposure to TCDD and/or other PCDD/Fs was dose‐relatedly associated with tooth enamel hypomineralisation or enamel defects. Hypomineralisation has mainly been shown in permanent teeth, and is likely to be a postnatal effect. Hypomineralisation weakens the enamel and is adverse as it increases the risk of caries and impaired tooth health later in life.
Bone
In contrast to teeth, bone has a continuous turnover. Bone mineral density, size and strength were determined through DXA scans in participants of the SWHS (Eskenazi et al., 2014) (see Annex A.8.10, Table 74 therein. A 10‐fold increase in TCDD was positively associated with measures of bone strength and size but differed somewhat by whether exposure occurred before or after peak bone mass. In perimenopausal/menopausal women, the association between higher TCDD and bone strength was positive when exposure occurred after peak bone mass and negative when exposure occurred before peak bone mass. In premenopausal women who were exposed after peak bone mass, there was no clear association. However, in premenopausal women exposed before peak bone mass, higher TCDD was associated with indices of increased bone strength and size. This was stronger when peak exposure was before the age of 5 years.
The level of 2,3,4,7,8‐PeCDF, a congener that was elevated in serum sampled 36–39 years after the Yucheng incident in Taiwan was not significantly associated with bone mineral density measured 42 years after the incident (Fukushi et al., 2016).
In summary, the CONTAM Panel noted that limited evidence from one cohort indicated some changes in bone parameters and that observations at a later age might be more sensitive for assessing possible associations between early life TCDD exposure and measures such as bone strength.
3.1.4.11. Cancer
In 2012, IARC evaluated TCDD and concluded there was sufficient evidence in humans for carcinogenicity (Group 1 carcinogen) based on data from experimental animals, epidemiological studies, and from the common mechanism of action (IARC, 2012). In a previous report (IARC, 1997), it was concluded that there was limited evidence.
Studies related to the exposure to PCDD/Fs and DL‐PCBs and cancer are discussed below and more details are shown in Annex A.8.11 (Table 75 therein), including number of participants, exposure levels, measured outcomes, quantification of associations and p‐values, as well as considerations of the internal validity or risk of bias (see Section 2.2 for further details).
Industrial accidents or contamination incidents
There are several studies using the Seveso Cohort (see Section 3.1.4.1.3 for a general description of the Cohort) that determine the outcome of exposure to TCDD at 15 to > 30 years after the incident. These studies have looked at cancer incidence and mortality. The results of these studies are reported briefly below.
Bertazzi et al. (2001) undertook a 20‐year follow‐up mortality study and, over the entire observation period, found no increase in either all‐cause or all‐cancer mortality in either zone A or B. Latency of effects was also investigated but few consistent trends were observed. In men after 15 years there was an increase in mortality from all cancers in zones A and B. The risk of Hodgkin's lymphoma was increased in the first 10 years (RR = 4.9, 95% CI 1.5–16.4). Non‐Hodgkin's lymphoma (NHL) increased after 15 years (RR = 2.8, 95% CI 1.1–7.0) as did myeloid leukaemia (RR = 3.8, 95% CI 1.2–12.5). There were significant increases in Hodgkin's disease, multiple myeloma and myeloid leukaemia in zone B (too few cases in zone A to conclude). After 15 years, lung cancer showed a significant increase (RR = 1.3, 95% CI 1.0–1.7) as did rectal cancer (RR = 2.4, 95% CI 1.2–4.6). The most consistent increase was for lymphohaematopoietic neoplasia in both genders across zones A and B with a latency pattern observed for women.
In a 25‐year follow‐up reported on mortality looking only at TCDD there was an excess of lymphatic and haematopoietic neoplasia detected in Zones A and B, respectively (RR = 2.23, 95% CI 1.00–4.97; RR = 1.59, 95% CI 1.09–2.33). The number of cases was again small: Zone A ‐ 6 deaths; Zone B ‐ 28 deaths (Consonni et al., 2008).
Pesatori et al. (2008) reported no significant increase in the incidence of pituitary tumours (1976–1996) although there was a trend towards a higher risk in zones A and B. The incidence of pituitary tumours in the least contaminated zone (R) was not different from the reference population. In 2009, Pesatori et al. reported the all cancer incidence covering the 20‐year period from 1977 to 1996 with the aim of identifying late occurring health effects as a result of exposure to TCDD. An excess of lymphatic and haematopoietic neoplasia was detected in zones A and B, respectively (RR = 1.39, 95% CI 0.52–3.71; RR = 1.56, 95% CI 1.07–2.27) but the number of cases was low (zone A ‐ 4 cases; zone B ‐ 29 cases). A slight but non‐significant increase in risk of breast cancer was observed in zone A after 15 years based on 5 cases. The all cancer incidence was not different from that expected in any of the contaminated zones (A, B and R). There was no clear pattern of cancer cases in terms of time since the incident or the zone of exposure due to the small number of cases.
Warner et al. (2002) using data from the Seveso Women's Health Study showed that individual serum TCDD levels correlated with breast cancer incidence at 20 years after the incident. The hazard ratio (HR) for breast cancer increased to 2.1 and was associated with a 10‐fold rise in serum TCDD. The average time from exposure to breast cancer development was 15.2 years after the explosion. The sample size in this study was small (15 cases) and the average age of the population studied was only 40 years, which is relatively young for development of breast cancer. Furthermore, this study was conducted only 20 years after the incident so longer follow‐up was deemed appropriate. In a later study, the same group (Warner et al., 2011) found the HR adjusted for all cancers was increased significantly after > 30 years follow‐up but that the increase in HR in breast cancer noted after 10–15 years was no longer significant. The sample size was low with 66 cancer cases of which 50% were breast cancer. It was noted that the majority of the exposed individuals had still not reached menopause when breast cancer incidence would be expected to rise. The second most common cancer was thyroid (n = 7).
Occupational exposure
Details of the studies are shown in Annex A.8.11 (Table 75 therein).
Air Force Health Study
The Air Force Health Study is a 20‐year longitudinal study examining health, mortality rates and reproductive outcomes of US Air Force veterans involved in Operation Ranch Hand. This unit was responsible for aerial spraying of herbicides from 1962 to 1971 and included TCDD contaminated Agent Orange (see Section 3.1.4.1.3 for a general description of the cohort). For all of these studies, there was mixed exposure to a range of chemicals.
In Ketchum et al. (1999) reported that there was no association between any cancer (all sites and any type) and TCDD category (background, low or high) within 20 years of service. Due to the lack of a dose–response relationship and variable results, the authors concluded that also the prevalence of skin cancer was not related to TCDD exposure.
Akhtar et al. (2004) reported that overall there was no significant increase in cancer incidence at any site compared to the national rates. However, an increased incidence of prostate cancer and melanoma was found in white Ranch Hand veterans compared to national cancer rates. Subdividing this further to those who had spent at most two years in Southeast Asia there was an increased risk of cancer at any site, in the prostate and of melanoma in those exposed to the highest concentrations of TCDD.
Ketchum and Michalek (2005) reported on 20‐year follow‐up post‐service mortality of the Air Force veterans exposed to herbicides, including TCDD. The RR for all‐cause mortality was not increased significantly (RR = 1.0, 95% CI 1.0–1.3) nor the risk of death from cancer (RR = 1.0).
Kang et al. (2006) reported on health effects on Army Chemical Corps (ACC) personnel who served in Vietnam and were involved in spraying from helicopters. The ACC were thought to be similar in exposure to the Operation Ranch Hand personnel who sprayed from fixed wing aircraft. In this study, samples from 795 of the veterans and 102 non‐veterans were analysed for TCDD. The medical history was taken by telephone and checked by Army records. There was a significant increase in the prevalence of all cancer excluding melanoma skin cancer (adjusted OR = 1.46, 95% CI 1.02–2.10). However, there was a non‐significant difference between those who sprayed Agent Orange and those who did not.
In a separate study, analyses of cancer incidence and serum TCDD concentration showed that all sites cancer relative risk increased with TCDD concentration (RR = 1.6, 95% CI 1.2–2.2; Pavuk et al., 2005). The major sites of cancer risk were digestive and respiratory tract and melanoma. The risk of prostate cancer increased with years of service in Southeast Asia but not with increasing TCDD levels. However, there was an interaction between years of service and TCDD with the greatest risk being in those individuals with the highest TCDD concentrations (3rd and 4th quartiles) and the longest years of service in Southeast Asia (3rd and 4th quartiles).
As prostate cancer is common in US males, the link between prostate cancer and exposure to herbicide by spraying was investigated further. Ranch Hand veterans were assigned to a ‘Low’ or ‘High’ exposure category based on a 20‐years median cumulative TCDD level. Pavuk et al. (2006) found no overall increase in the risk of prostate cancer and Ranch Hand veterans vs. the comparisons (other Air Force veterans who server in Southeast Asia but were not involved in spraying). However, if stratified by high TCDD concentration and service in Southeast Asia before 1969, then a positive association was found with prostate cancer (n = 15). The confounding factor in this study was that time spent in Southeast Asia alone increased the risk of prostate cancer in the comparison group. In a later study, Li et al. (2013a,b) investigated whether exposure to Agent Orange and high level of CALUX‐determined BEQ was associated with biochemical recurrence (normally measured as prostate‐specific antigen (PSA) level) of prostate cancer after radical prostatectomy. Men exposed to Agent Orange showed a median level of 22 vs. 15 in non‐exposed individuals. Exposed and non‐exposed were combined and divided over a high and low group, showing median BEQ levels of, respectively, 25 vs 14 pg BEQ/g fat. Neither exposure to Agent Orange or high BEQ was associated with recurrence.
In 2008, Michalek and Pavuk investigated the incidence of diabetes and cancer in the veterans. Without stratification there was no difference in the cancer risk overall or in the risk of all‐site SEER (Surveillance Epidemiology and End Results) cancer. Taking into account calendar dates of service (before or during 1968), number of days spraying (30 days pre 1967 ≥) and the years of service (≤ 2 years) there was a significant increase in cancer risk in the high TCDD group (RR = 2.2, 95% CI 11–4.4).
Landgren et al. (2015) investigated monoclonal gammopathy of undetermined significance (MGUS) in the Operation Ranch Hand cohort. MGUS is believed to be a precursor state for multiple myeloma. The Ranch Hand Operation veterans had increased risk of MGUS and had higher blood TCDD levels compared to veterans who had not been involved in spraying.
Dutch herbicide cohort
Boers et al. (2010) reported there was no association between TCDD exposure and mortality of all cancers. There was a suggestion that predicted TCDD levels were associated with NHL (HR = 1.36, 95% CI 1.06–1.74) but this did not hold up when factory B workers (non‐exposed) were included in the analyses. In a later study, Boers et al. (2012) did not confirm previously reported increased risks of respiratory cancer and NHL. Increased risk was observed for all cancer in both factory A and B (HR = 1.31, 95% CI 0.86–2.01 and HR = 1.54, 95% CI 1.0–2.37, respectively). Increased risks of urinary and genital cancer were observed for factory A but these could not be linked to a specific exposure.
Hooiveld et al. (1998) reported increased risks of cancer mortality, respiratory cancer and NHL in workers exposed to phenoxy herbicides, chlorophenols and contaminants. The exposure‐related increases in risk were greatest with the higher TCDD exposure.
Heederik et al. (1998) showed increased risk of lung cancer in subjects exposed to medium and high TCDD (RR = 6.4, 95% CI 0.8–53.1 and RR = 6.8, 95% CI 0.9–54.4) based on 6 and 8 cases, respectively.
Other studies
Details of the studies are shown in Annex A.8.11 (Table 75 therein).
De Roos et al. (2005) examined organochlorine chemicals in plasma of individuals in a population‐based case–control study in the USA. Increased risk of NHL was associated with PCDFs and DL‐PCB‐156, but also with the NDL‐PCBs 180 and 194. The study showed a 35% increased risk of NHL per 10 pg WHO1998‐TEQ/g fat (95% CI 1.02–1.79).
In a study carried out in 1992 of German chemical factory workers exposed to TCDD between 1952 and 1984, Becher et al. (1988) found an increased SMR for total cancer mortality (SMR = 1.41, 95% CI 1.17–1.68). TCDD and TEQ exposures were positively linked to cancer mortality although the authors expressed less confidence in the TEQ. No latency effects were observed. An increase in SMR for lung cancer was also observed (SMR = 1.51, 95% CI 1.07–2.08), however the degree of uncertainty was too high to show a dose–response function. .
A study in Germany investigated mortality of workers from an herbicide plant exposed to TCDD in 23 years follow‐up after closure of the plant (Manuwald et al., 2012). Exposure was associated with increased risk of all cancers (SMR = 1.37, 95% CI 1.21–1.56). In terms of death, respiratory cancers accounted for 10.9% and digestive cancers for 8.3% of total. Mortality was increased for breast, bladder and kidney cancer in women with the most robust estimate being for breast cancer (SMR = 1.86, 95% CI 1.12–2.91). In men, respiratory, oesophageal, bladder, kidney and rectal cancer were increased. However, there was no clear exposure–response relationship with estimated TCDD levels. Flesch‐Janys et al. (1998) also investigated a cohort of former herbicide and insecticide workers from Germany exposed to PCDD/Fs. The all cancer SMR was 1.41 (95% CI 1.17–1.68) with TCDD showing a significant trend in SMR with increasing cumulative PCDD/F exposure.
Steenland et al. (1999) undertook a cohort mortality study on chemical workers who had been exposed to TCDD in the USA. For all cancers combined, the SMR was 1.13 (95% CI 1.02–1.35). No marked specificity for any one cancer type was noted. Positive trends were observed for all cancers combined and for lung cancer. Regression analyses showed a significant trend for cancer with a 15‐year lag time. The excess cancer was limited to the highest exposure groups who were likely exposed to 100–1,000 times the concentrations of the general population.
In a further study of samples from one of the eight plants used in the earlier study, Steenland et al. (2001) showed a positive trend for all cancer risk with increasing cumulative exposure to TCDD. The authors calculated the excess lifetime risk (lagged 15 years) of exposure to 1 pg/kg bw per day over 75 years as 0.05–0.9% against a background lifetime risk of 12%.
Cheng et al. (2006) reanalysed the exposure to TCDD and the association with cancer in about 3,500 of the chemical workers producing TCP and other chemicals contaminated with TCDD as reported by Steenland et al. (1999, 2001). Cheng et al. used an age‐ and concentration‐dependent model to estimate previous exposure to TCDD in the workers, while Steenland et al. (2001) had used back‐calculation with a fixed half‐life of TCDD. Cheng et al. estimated a cumulative exposure, which was 4‐5 times higher than the estimate used by Steenland et al. (2001), and by EPA. Therefore, while Cheng et al. (2006) could confirm a significant association between TCDD and cancer in when 5% of the workers with the highest exposure were excluded, their dose–response analyses showed 4‐5 times lower risk per year with a certain estimated serum TCDD concentration.
Collins et al. (2009a) reported on a study of 1,615 workers that had been exposed to TCDD during production of TCP. This study had the advantage of a long follow‐up of 62 years with the average being 36 years. There was no increase in SMR for all cancers (SMR = 1.0, 95% CI 0.8–1.1) or for lung cancer (SMR = 0.7, 95% CI 0.5–0.9). There was however greater number of deaths from leukaemia (SMR = 1.9, 95% CI 1–3.2) and NHL (M+SMR = 1.3, 95% CI 0.6–2.5) than expected. There were four deaths from soft tissue sarcoma (SMR = 4.1, 95% CI 1.1–10.5). In a separate study, the mortality of workers manufacturing pentachlorophenol in the USA and exposed to TCDD as a contaminant, Collins et al. (2009b) found deaths from all causes combined and all cancers combined at the expected levels. There was an increase in the SMR 2.4 (95% CI 1.0–4.8) for NHL; however, this was based on only eight deaths.
In a study from New Zealand where workers manufactured trichlorophenol, estimates of exposure to TCDD and the risk of cancer development was assessed (McBride et al., 2009). No increase in total cancer was detected (SMR = 1.1, 95% CI: 0.9–1.4) but there fewer deaths from lung cancer than expected (SMR = 0.8, 95% CI: 0.4–1.5).
In an attempt to examine the effect of chronic exposure to PCDD/Fs and DL‐PCBs in the general population, Lin et al. (2012) examined 242 deaths as part of the NHANES study and found 72 cancer deaths. There was an increase in all cause death for individuals over 40 years of age associated with logarithmically expressed total TEQ. While there was a graded dose–response trend for cancer mortality, this did not reach statistical significance.
In summary, several studies showed a positive association with all cancers combined. However, there was no clear link to any specific site. Due to the lack of clear dose–response relationship and multiple co‐exposures, the CONTAM Panel does not consider these studies suitable for the risk assessment.
3.1.4.12. Other effects
The literature search and the selection for relevance as described in Section 2.2 identified studies related to the exposure to PCDD/Fs and DL‐PCBs and effects other than the ones described in previous sections. Two of the studies met the eligibility criteria (Michalek et al., 2001c; Gupta et al., 2006). These studies covered several outcomes but as no robust conclusions can be drawn from single studies no risk assessment could be performed on the basis of their findings. In addition, three studies (Wang et al., 2006; Cao et al., 2008; Rennert et al., 2012) examined associations between PCDD/Fs and DL‐PCBs and serum sex hormone levels or related proteins/peptides without other endpoints being measured. Results from these three studies are presented in Annex A.10.
3.1.5. Effects in farm and companion animals
The literature search performed as described in Section 2.2 identified a number of studies in which the effects of PCDD/Fs and/or DL‐PCBs exposure on farm and companion animals have been examined. Some of these described the effects observed following accidental exposure to the target compounds (field studies), which lack information on the actual levels of exposure. Others described the effects after exposure to the target compounds under controlled conditions. The CONTAM Panel decided that the controlled studies should be the basis of the evaluation and possible derivation of a reference dose, while field studies are used to complement and to fill specific data gaps, as appropriate.
3.1.5.1. Ruminants
3.1.5.1.1. Cows
Nine studies describing different effects in cows were identified (see Annex A.11.1, Table 88 therein). A number of these describe cases in which increased levels in milk were observed and compared with various parameters to those in cows with lower levels in milk. Lloyd et al. (1991) describe two case studies with farms with substantial numbers of diseased and dead cows related to the presence of chemical and municipal waste incinerators, although without data on potential levels of PCDD/Fs and DL‐PCBs in the animals. Spagnuolo et al. (2011) studied buffalo cows from the Campania region in Italy near sites with open burning of waste, with total TEQ levels in milk around 22 pg/g fat (method not provided). They observed higher levels of products linked to oxidative damage and reduced levels of antioxidants in blood of the animals. There was also an increase in chromosome aberrations but no significant effect on SCEs (Genualdo et al., 2012). Cows from two farms near a steel plant in the Susa valley, with milk levels of 8.6 and 18.6 pg TEQ/g fat (primarily DL‐PCBs), also showed a clear increase in chromosome aberrations and slight increase in SCEs in circulating lymphocytes (Iannuzzi et al., 2009; Di Meo et al., 2011). A follow‐up study including a control group with a milk level of 1.8 pg TEQ/g fat, showed that levels of antioxidants and total antioxidant capacity in plasma were lower in the two groups of cows with the higher TEQ levels in milk, whereas a higher extent of oxidative damage in plasma proteins and lipids was observed in these cows (Spagnuolo et al., 2012).
Cigliano et al. (2016) investigated levels of oxidative stress markers in blood of young cows contaminated with DL‐ and NDL‐PCBs due to a nearby decontamination plant. Upon transfer to a clean area, levels of DL‐ and NDL‐PCBs in pericaudal fat decreased during the 6 months observation which at least partly was due to growth. The stress markers showed an improved situation, with levels comparable to control animals (being of a different breed). Also, a correlation between PCB levels in individual cows and levels of tumour necrosis factor‐α (TNFα) (positive) and haptoglobin (negative) markers were observed.
The CONTAM Panel noted that in these field studies the animals might also have been exposed to other contaminants, like metals and PAHs, which may have contributed to the observed effects. In addition, the chromosomal damage observed in cows might be the consequence of an indirect genotoxic effect due to oxidative stress‐related damage (see Section 3.1.3).
The CONTAM Panel was of the opinion that the decrement in antioxidant levels and the increase in oxidative damage in plasma proteins are not adverse per se.
Therefore, the CONTAM Panel concluded that these studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in cows.
3.1.5.1.2. Sheep
Eight papers on potential effects of PCDD/Fs and DL‐PCBs in sheep were identified (Annex A.11.1, Table 88 therein). Three of these concerned the same study with sheep treated with PCB‐118 (Gutleb et al., 2011; Zimmer et al., 2013; Krogenaes et al., 2014). There was apparently a cross‐contamination with PCB‐153 and standards were not checked for the presence of more potent DL‐PCBs and PCDD/Fs. As such these studies were not considered suitable for risk assessment.
Perucatti et al. (2006) investigated two sheep flocks in contaminated areas in the South of Italy, showing milk levels for PCDD/Fs of 51 and 40 pg TEQ/g fat (TEFs scheme not reported). Soil and grass levels were 4.9 and 1.3 pg WHO2005‐TEQ/g, respectively. These levels correspond to background levels for grass in other countries, in particular in winter. One of the flocks showed relatively high frequencies of abortions and abnormal fetuses compared to a sheep from a control farm. Analysis of circulating lymphocytes showed a slight increase in SCEs and chromosome abnormalities. Milk TEQ levels in the control farm were not reported. A possible exposure to other contaminants cannot be excluded. Similar effects were reported by Iannuzzi et al. (2004) for two other flocks with 10‐fold lower TEQ levels in milk (around 5 pg TEQ/g fat).
Based on similar considerations as discussed above for cows, the CONTAM Panel concluded that these studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in sheep.
3.1.5.1.3. Goats
Eight studies were identified that described effects on goats (Annex A.11.1, Table 88 therein). Six of these were based on the same animal experiment with pregnant does that received three times per week an oral dose of PCB‐126 in corn oil, starting on GD60 till delivery (about GD150). On average, the dose was 49 ng/kg bw per day, of which 20% was estimated to be present in the does, 6 weeks after delivery and 1% in the kids at 9 months of age (Lyche et al., 2004a). The kids were lactated during the first 6 weeks and were followed‐up for a period of 9 months. Various plasma samples as well as tissues sampled from does and kids were analysed. There was clear sequestration of PCB‐126 in the liver of does and kids with lipid‐based levels of, respectively, 50 and 3 ng/g, being seven‐ and threefold higher than in adipose tissue (Lyche et al., 2004a). Various endpoints were studied. There were no effects on body or tissue weights. Levels of monocytes were decreased in the kids, but there was no effect on lymphocyte proliferation after stimulation with various mitogens (Lyche et al., 2004b). Studies on immunological effects indicated increased titres against tetanus toxoid and decreased titres against reovirus in the kids (Lyche et al., 2006). Whereas these data may suggest some influence on the immune system, the results seem not consistent and were only observed at one dose of PCB‐126. Parameters like bone size, mineral content and density, and bone strength were not affected (Lundberg et al., 2006). Cortisol levels in the kids were decreased during a part of the 9 month period and showed a stronger increase after blood drawing in males (Zimmer et al., 2009). There was no clear effect on levels of progesterone, oestradiol and LH in female kids (Lyche et al., 2004c). In male kids, there was a slight reduction in plasma testosterone levels but no effects on sperm parameters or testes histology (Oskam et al., 2005).
The CONTAM Panel concluded that the studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in goats.
3.1.5.2. Pigs
Two studies on potential effects of PCDD/Fs and DL‐PCBs were identified in pigs (Annex A.11.2, Table 89 therein). A study by Ryan (1983) described mortality in young pigs raised on a PCP treated wooden floor. The route of exposure was unclear, but it seemed unlikely to be via the sows’ milk. The level of hexa‐, hepta‐ and octachlorinated PCDD congeners reported for one skin sample would correspond to a WHO2005‐TEQ level of 14 pg TEQ/g fat. The tetra congeners were below the detection limit and other PCDD/Fs were not reported. The authors argued that PCP could not have caused the mortality. A study by Lavrusenko et al. (1996) on immune effects was poorly described and does not allow derivation of a NOAEL.
The CONTAM Panel concluded that these two studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in pigs.
3.1.5.3. Rabbits
Three studies were identified where rabbits had been exposed to TCDD (Annex A.11.3, Table 90 therein). In the study by Schwetz et al. (1973), a single exposure to TCDD (31.6, 63, 126, 252, 500 μg/kg bw) was given by oral, i.p. or dermal route. The LD50 for oral dosing in rabbits was calculated to be 115 μg/kg bw, as compared to 275 μg/kg bw after dermal exposure. For i.p. dosing, an LD50 could not be determined but a NOAEL of 32 μg/kg bw was observed. For oral and dermal exposure, no NOAELs could be determined.
Accidental exposure was associated with the Seveso incident where high mortality rates were observed (up to 100%) in farmed rabbits, especially in zone A, the most contaminated area (Fanelli et al., 1980). To investigate whether animals had died from the incident, TCDD levels were determined in livers from dead rabbits from zones A, B, R and the surrounding area, showing mean levels of, respectively, 85, 90, 26 and 13 ng/g tissue. The authors concluded that it was not possible to estimate the doses killing the animals.
Kimbrough et al. (1977) observed hyperkeratosis in the ear lobe and necrotic changes were found in the liver of TCDD exposed rabbits. These animals were exposed through the spraying of contaminated oil on an arena used by horses. In this case, the level and route of exposure could not be ascertained.
The CONTAM Panel concluded that these studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in rabbits.
3.1.5.4. Horses
The literature search identified three studies in which horses were accidentally exposed to TCDD or individual PCDD/Fs (Annex A.11.4, Table 91 therein). Two studies describe an incident with intoxications, including 57 deaths of horses residing on a riding arena that was treated with contaminated waste oil for dust control (Carter et al., 1975; Kimbrough et al., 1977). The soil contained trichlorophenol but also TCDD at levels around 30 mg/kg. However, the route of exposure (oral and/or dermal) and actual intake of the animals is unclear, and tissue samples were not analysed. There was also a mixed exposure. Another paper (Kerkvliet et al., 1992) describes an incident with similar effects in horses and dead foals. The source of the contamination was shavings prepared from PCP‐treated wood. The exposure route and intake is unclear, but liver tissue samples from two euthanised horses were shown to contain levels corresponding to 272 and 248 pg WHO2005‐TEQ/g tissue (based in the 10 congeners reported). Fat from the first horse contained a level equivalent to 313 pg WHO2005‐TEQ/g tissue. Higher chlorinated congeners, in particular 1,2,3,6,7,8‐HxCDD contributed most to the TEQ levels.
The CONTAM Panel concluded that the studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in horses.
3.1.5.5. Poultry
The literature search carried out as described in Section 2.2 identified a number of studies on the adverse effects of PCDD/Fs and/or DL‐PCBs in poultry (chicken, quail, duck, pheasant and turkey). Although exposure in ovo by transfer from exposed adult birds is an important aspect for assessing adverse effect in the developing embryo and survival of hatchlings, most of the studies have been conducted on newly hatched chicks or by dosing of egg.
For the risk assessment, the CONTAM Panel focused on studies in which more than one dose group had been administered to the adult birds or in ovo (Annex A.11.5, Tables 92 and 93 therein, respectively). The toxicity of individual PCDDs, PCDFs and DL‐PCBs varies between mammals and birds and between species of birds. As discussed in Section 1.3.1, the CONTAM Panel decided to use the TEFs established for mammals in 2005 (van den Berg et al., 2006), and not the previously established TEFs for birds.
3.1.5.5.1. Chicken
Adverse effects in chickens led to the discovery of several feed contamination incidents associated with PCDD/Fs and PCBs (see Section 1.3.1). The reports of an accumulation of fluid in the pericardium affecting millions of USA farmed birds in the 1950s and 1960s led to the isolation of a chick oedema factor in feeds arising from added fats that had been extracted from hides treated with chlorophenols. The active contaminants were identified as PCDDs and this finding was an important driver to understand the mechanism of toxicity and safety assessment of these chemicals (Metcalfe, 1972; Flick et al., 1972). A single dose of TCDD can be toxic to older growing birds (Greig et al., 1973), but the contamination of feed for newly hatched chicks or their exposure in ovo from contaminated laying hens is of the most interest for farmed birds. Contamination of poultry feed by PCDD/Fs and DL‐PCBs in Belgium in 1999 caused a drop in egg production. A few weeks later this was followed by a marked reduction in hatchability, reduced weight gain and increased mortality of chicks that showed subcutaneous oedema, ataxia, degenerative changes of skeletal and cardiac muscles known as chick oedema disease (Bernard et al., 1999). Levels in the feed were shown to be in the low μg TEQ/kg range but the feed also contained high levels of NDL‐PCBs (Bernard et al., 2002).
Effect data from these incidents are less suitable for deriving a reference point for chickens and a number of studies have been performed under more controlled conditions.
Studies in young and adult birds
Three‐day‐old White Leghorn chicks were treated by oral gavage daily with 0.01, 0.1, 1 and 10 μg/kg bw of TCDD, or 0.1, 1, 10 or 100 μg/kg bw of a mixture of HxCDDs daily for up to 21 days (Schwetz et al., 1973). Birds given TCDD at 1 and 10 μg/kg bw or the HxCDDs at 10 and 100 μg/kg bw had a high incidence of mortality by 21 days (no survival at the highest doses), exhibiting pericardial, peritoneal, subcutaneous and pulmonary oedema as well as liver hypertrophy.
One‐day‐old White Leghorn chicks administered daily by gavage 1 or 5 μg/kg bw of TCDF had 16% and 100% mortality, respectively, by 21 days. There was marked incidence of subcutaneous and pericardial oedema, depletion of splenic lymphocytes and thymic atrophy in both groups. These effects were not associated with hepatic histopathology changes which were only mild at the higher dose (McKinney et al., 1976; Goldstein et al., 1976). In comparison, PCB‐169 administered at 400 mg/kg diet also caused subcutaneous and pericardial oedema, depletion of splenic lymphocytes, thymic and bursa atrophy and mild liver necrosis and fatty infiltration with 100% mortality. In contrast, non‐DL‐hexachlorobiphenyls showed mild incidences of these adverse effects but moderate hepatic pathological changes.
Two‐week‐old White Leghorn cockerels were given an i.p. dose of 0.1, 1.0, 10, 100 and 1,000 μg TCDD/kg bw on days 1, 2 and 3. All survived and were culled on day 5. A dose‐related decrease in the size of the Bursa of Fabricius (a site of B‐lymphocyte development) relative to body weight was observed with a NOAEL of 1 μg/kg bw per day (Sawyer et al., 1986).
In a study to model exposures of birds from contaminated soil after and before remediation, White Leghorn male and female chickens were dosed with TCDD by i.m. injection twice weekly for up to 6 weeks resulting in a dose of on average 8.6 or 1,700 ng per day per bird which approximated to 5.6 and 1,099 ng/kg bw per day. The low‐dose hens were reported to have received a total of 360 ng TCDD approximately over six weeks. All egg production from hens of the high doses mated with control males ceased after 12 days of treatment (Alonso et al., 1998; Peden‐Adams et al., 1998). Although changes in B‐cell and T‐cell peripheral mitogen‐induced lymphocyte proliferation were observed, these were inconclusive as to being related with treatment and dose.
Studies in ovo
Few studies have been conducted on development in ovo or hatchlings of eggs from exposed chickens. More studies on the effects of TCDD or DL‐PCBs on chicken embryo development have been conducted by direct injection of fertilised eggs with single or multi doses, expressed in weight or molar weight/egg or gram of egg. Sites of injection of the eggs have been varied, mostly either into the air cell or yolk sacs; time of injection after fertilisation and diffusion of different PCDD/Fs/DL‐PCBs are factors not fully standardised. Particular emphasis has been placed on hatching, cardiovascular, neurological, immunological or hormone endpoints. Doses were standardised to TEQs using WHO2005‐TEFs (Annex A.11.5, Table 93 therein). The CONTAM Panel selected studies in which adverse effects were seen at low exposures in multiple dose investigations.
F1 generation 14‐day old chicks and F1 adults from eggs of hens exposed to a low i.m. dose of TCDD (8.6 ng per day, total 360 ng) described in the previous section, showed decreases in blood B‐cell proliferative response compared to control but these were not statistically significant (Peden‐Adams et al., 1998). On the F1 hens reaching sexual maturity, onset of egg production was delayed by up to 2 weeks compared to controls (Alonso et al., 1998). In parallel studies, hatchlings from eggs injected into the yolk sac with TCDD in olive oil did not survive beyond 14‐days at doses > 200 ppt (> 200 pg/g egg). The LD50 for survival from these egg injection studies was estimated to be 20 pg TCDD/g egg.
Early cardiovascular lesions, linked to chick oedema disease seen in early accidental poisoning of chickens or produced experimentally (Flick et al., 1972), have been observed in embryogenesis after dosing eggs with TCDD or DL‐PCBs. Dose responses in cardiotoxicity in the chicken embryo comparing TCDD, PeCDD, TCDF, 2,3,4,7,8‐PeCDF and PCB‐77 demonstrated the correlation with their ability to activate the AHR signalling pathway (Heid et al., 2001). In a previous study, TCDD and PCB‐126 injected into the yolk sac were shown to induce a dose‐related increase in heart weight after day 10 in embryos of White‐Leghorn‐Babcock and the more sensitive strain Plymouth‐Rock‐Barred embryos (NOAELs < 160 and 35 pg/g egg, respectively), while the NDL‐PCB‐153 had no effect (Walker and Catron, 2000). Morphometric analysis of the more sensitive Plymouth Rock‐Barred strain showed that 0.06–0.45 pmol TCDD/g egg (19–144 pg/g) induced a dose‐related increase in left and right ventricle cavity areas without wall hypertrophy, consistent with cardiomyopathy. At 0.24 pmol TCDD/g, significant increases were also observed in cardiac atrial natriuretic factor mRNA (an early marker of heart failure) and decreased responsiveness to isoproterenol‐induced positive chronotropy consistent with dilated hypertrophy and eventually subcutaneous and peritoneal oedema. There was a decrease in myocyte proliferation and increased apoptosis but the affected cell‐type could not be identified (Ivnitski et al., 2001). Anomalies of the cardiac outflow septa and the coronary arteries, including retroaortic coronary, double right coronary and incomplete lumens, were detected in embryo development after injection of eggs in the air sac with 1 or 3 pmol of TCDD/g (320 or 960 pg/g egg) after preincubation for 4.5 days (Wikenheiser et al., 2013). These anomalies, increased mortality and haemorrhaging were much more frequent with the higher dose.
In a comparison of the influence of the route of injection, newly laid White Leghorn chicken eggs were injected with 0, 10, 30, 60, 100, 300, or 1,000 pg TCDD/g egg and allowed to develop to hatching (Henshel et al., 1997a). Injection into the yolk sac was more toxic than into the air sac (LD50 122 pg/g and 297 pg/g, respectively). Common gross teratogenic changes observed in the dead embryos and fetuses injected with either injection method included pericardial and abdominal oedema, deformed heart, red embryos, deformed or missing eyes, abnormal limbs, short beak, and abnormally shaped head and body.
A dose–response study of chicken eggs injected in the air sac with TCDD (10–1,000 pg/g egg) concluded that brain asymmetry occurred during development of embryos especially affecting the forebrain even at the lower doses (Henshel et al., 1997b). However, in an assessment of PCB‐77 and PCB‐126 after injection into the yolk sac, the usefulness of this type of endpoint has been questioned (Lipsitz et al., 1997). Instrinsic structural asymmetry and during development, as well as vehicle can act as confounding factors.
Exposure of chick embryos to TCDD has been shown to modulate gonadal steroid production depending on time, sex and dose (50–200 pg/g egg) (Sechman et al., 2011). TCCD decreased hatchability at 400 pg/g egg and delayed growth, accompanied by increases in T3/T4 ratio in resulting chickens raised to 6 weeks dosed at ED4 (Bruggeman et al., 2003, 2005).
After injection into the air sac of eggs with 51, 130, 320 or 800 pg/g of PCB‐126, developing chicks were examined 1 day before estimated hatching (day 20). There was dose dependent mortality with a NOAEL of 51 pg/g and an estimated LD50 of 1,010 pg/g. The decrease in mass of both thymus and bursa (sites of T‐ and B‐lymphocyte maturation, respectively) were observed, for the latter organ even at the lowest dose (Fox and Grasman, 1999). Viable lymphoid cells were particularly sensitive with full rather than partial incubation time before dosing. With the same dose range of PCB‐126, total numbers of thymocytes with surface marker TCRαβ+ fell in a dose‐dependent manner (Grasman and Whitacre, 2001). Further studies suggest that thymic atrophy in this chicken model is associated with elevated apoptotic thymocytes on embryo day 20 (Goff et al., 2005).
Cohen‐Barnhouse et al. (2011a,b) compared the susceptibility of avian domestic species including White Leghorn chicken, to toxic endpoints after injection of eggs into the air sac with TCDD, 2,3,4,7,8‐PeCDF, or TCDF at doses ranging from 0.044 to 37 pmol/g egg (15‐12,000 pg/g) depending on the species and PCDD/F. The site of injection was chosen for ease and speed of delivery of chemical. Embryo mortality in the chicken was dose‐related for all three chemicals. The order of potency was TCDF > TCDD = PeCDF with LD50 values of 100 and 210 pg/g egg for TCDF and TCDD, respectively. Chickens could be regarded as more sensitive to TCDD (NOAEL of 61 pg/g egg) and TCDF for hatchability than pheasants, which in turn were greater than quail. However, the sensitivity of chicken compared to the other species could not be predicted from more detailed examinations of deformities, body and organ weights and histopathology (Cohen‐Barnhouse et al., 2011b).
In summary, among the few studies in chicken, a NOAEL of three i.p. doses of 1 μg/kg TCDD to young birds has been shown with respect to decrease in the weight of the Bursa of Fabricius, a site of B‐cell development. An i.m. regimen dose of 360 ng/kg of TCDD to adult birds over 6 weeks had no effect on egg production. However, egg production of the F1 hens was delayed by 2 weeks.
Comparative studies by injection of eggs into the yolk had an LD50 estimated as 20 pg/g egg. However, other injection studies had higher NOAEL and LD50 values. Conclusions from injection into the egg are susceptible to confounders of timing, dose route and exact endpoints examined. The CONTAM Panel concluded that the in ovo studies could not be used for the risk assessment. Direct comparison of PCDD/F levels in dosed hens and maternally exposed eggs, together with outcomes, were not available.
3.1.5.5.2. Quail
Studies in adult birds
Only studies in which quail had been administered single doses of PCB‐77 and/or ‐126 were identified. These studies were not considered useful to derive a reference point.
Studies in ovo
There have been limited studies of maternal exposure of eggs. Injection of eggs was into the air sac or the yolk.
Quail received three i.p. injections of PCB‐77 (5 mg/kg) and showed no overt toxicity (Boily et al., 2003a,b). Eggs were collected but there was no apparent embryo toxicity. When fertile eggs were injected into the yolk with 0.2, 1.0 and 2.0 μg/g egg of PCB‐77 or 2, 10 and 20 μg/g of PCB‐105 and incubated for 6 days, significant embryo toxicity was observed with 1.0 and 2.0 μg/g of PCB‐77 but not with 10 and 20 μg/g of PCB‐105. Studies showed that endpoints based on retinoid metabolism may be highly pertinent to toxicity.
Quail were more resistant than chicken or pheasant to embryotoxicity caused by TCDD, 2,3,4,7,8‐PeCDF, or TCDF after injection of eggs into the air sac in a range of 0.22–37 pmol/g egg with for TCDD‐induced mortality a NOAEL of 1,824 pg/g egg and an LD50 of 9,700 pg/g. In contrast to chicken, the order of chemical potency was PeCDF > TCDF > TCDD (Cohen‐Barnhouse et al., 2011a). However, with other chronic toxicity endpoints, such as deformities, body and organ weights and histopathology, the comparisons were not as clear as with embryo mortality and could not be used to derive a dose response (Cohen‐Barnhouse et al., 2011b).
3.1.5.5.3. Ducks
Studies in adult birds
No studies were identified in farmed ducks.
Studies in ovo
No studies were identified that were helpful for risk assessment purposes (see Annex A.11.5).
3.1.5.5.4. Pheasant
Studies in adult birds
Seven‐month‐old pheasant hens were given orally PCB‐105 (low TEFWHO 2005 of 0.00003) for 10 weeks to achieve cumulative doses of 0.6, 6 and 60 mg/kg bw (equivalent to 18, 180 and 1,800 ng WHO2005‐TEQ/kg bw) (Hornung et al., 1998). Hens were then bred with untreated roosters for 8 weeks. Fertilised egg production, embryo and chick mortality, and body and heart weight of chicks at 1 day, and after 21 days were not significantly affected by treatment of the hens. No malformations or pericardial oedema were detected.
Studies in ovo
No studies with TCDD or PCDD/Fs with equivalent TEFs were identified in which eggs were exposed maternally. Air sac, yolk sac and albumen sites of injection have been employed to study embryo toxicity.
Toxicity following injection of TCDD into the yolk or albumin of ring‐necked pheasant eggs was assessed at 1 day and 28 days post‐hatching. Both sites of injection showed a dose response for mortality of embryos over 21 days with a NOAEL of 100 pg/g egg and LD50 values of 2,182 and 1,354 pg/g egg, respectively. There was no evidence of other more sensitive organ, histological or systems endpoints such as body weight, bursa weight, cardiac morphometrics and signs of oedema (Nosek et al., 1993).
The pheasant may be less sensitive to injection into the air sac of TCDD, 2,3,4,7,8‐PeCDF or TCDF with respect to embryo toxicity compared to chicken, but more sensitive than the quail, with a NOAEL for TCDD of 99 pg/g egg and an LD50 of 1,200 pg/g. Chemical potency was PeCDF > TCDF > TCDD. Detailed changes in embryo development such as deformities, changes in organ weights, and histopathology were not consistently related to dose within species and/or chemical and could not be used to evaluate relative response (Cohen‐Barnhouse et al., 2011a,b).
3.1.5.5.5. Turkey
Studies in adult birds
Only studies in which turkeys had been administered fat contaminated with PCDD/Fs were identified. These studies were not considered useful to derive a reference point.
Studies in ovo
The sensitivity of turkey eggs during incubation to PCB‐77 injected in to the yolk was markedly less than the chicken with a NOAEL of 200 ng/g egg for mortality and no signs in survivors of effects on the thymus or oedema. The difference in sensitivity appeared to correlate with binding to AHR (Brunström and Lund, 1988). A lowest‐observed‐effect level (LOEL) of 20 ng/g egg was observed for PCB‐126 (Brunström, 1989).
3.1.5.6. Fish
The literature search carried out as described in Section 2.2 identified a number of studies on the adverse effects of PCDD/Fs and/or DL‐PCBs in different farmed species of fish in Europe (trout, salmon, sea bream, sea bass, sole, carp and tilapia) (see Annex A.11.6, Tables 94 to 102 therein), as well as studies performed in some of these species in ovo (see Annex A.11.6, Tables 103 and 104 therein). The studies have been divided into trout, salmon and other fish species.
For this evaluation, the CONTAM Panel decided to focus on studies in which more than one dose group had been administered, and when the exposure was either oral (gavage or via feed) or i.p. Waterborne exposure studies, field studies, studies with one dose group only were thus not considered relevant for the purpose of the risk assessment.
3.1.5.6.1. Studies in trout
Some relevant characteristics of the studies in trout are shown in Annex A.11.6 (Table 94 therein).
Rainbow trout were fed nominal doses of TCDD of 0.0023, 2.3 or 2,300 μg/kg diet every six days for 105 days (stated to correspond to doses of 0.0000096, 0.0108 or 6.3 μg TCDD/kg fish per day; Hawkes and Norris, 1977). Fish in the highest treatment group had 88% mortality following 71 days of exposure. There were no effects of exposure to TCDD on food consumption, growth, fin erosion or survival in the two lower exposure groups. The results in this study indicate that the NOAEL for TCDD in rainbow trout was 0.0108 μg TCDD/kg fish per day.
Rainbow trout were exposed by single i.p. injection to TCDD doses of 0 (vehicle), 1, 5, 25 or 125 μg TCDD/kg bw (Spitsbergen et al., 1988a). After 80 days, there was no mortality in the control or the lowest dose group, and 20%, 90% and 95% mortality in the 5, 25 and 125 μg TCDD/kg bw groups, respectively. The 80‐day LD50 was determined to be 10 μg/kg bw. Another group of rainbow trout were injected (i.p.) with 0, 0.1, 1 or 10 μg TCDD/kg bw. Gross and microscopic lesions were present in fish at the highest dose, but not in the other groups. Haematological effects included reduced numbers of leucocytes and thrombocytes in fish treated with 1 or 10 μg TCDD/kg bw compared to the control group; no haematological data were presented for the trout exposed to 0.1 μg TCDD/kg bw. It was not possible to establish a NOAEL for this study due to the limited statistical analysis and variable number of fish tested for different endpoints.
Spitsbergen et al. (1988b) administered TCDD by single i.p. injection at doses of 0, 0.01, 0.1, 1 or 10 μg/kg bw to rainbow trout to assess the effects on growth, mortality, and morphological lesions. No mortality was observed five weeks after treatment; however fish exposed to the highest dose had reduced feed intake and activity, and fin necrosis (which was not observed in any of the other groups). Two weeks after TCDD exposure, control fish and fish from the 0.01, 0.1 and 1 μg TCDD/kg groups were challenged with infectious haematopoietic necrosis virus (IHNV). Mortality in TCDD‐exposed fish was not significantly different from that in the vehicle control group, though there appeared to be more severe and diffuse lesions of IHNV disease in fish in the exposed groups compared to the controls. The NOAEL in this study was 0.1 μg TCDD/kg bw per day based on growth and fin necrosis.
Kleeman et al. (1988) treated rainbow trout with graded doses of TCDD (1, 5, 25, 125 μg/kg bw) by single i.p. injection. The 80‐day LD50 was 10 μg/kg bw. Fin necrosis was observed in rainbow trout treated with 25 and 125 μg TCDD/kg bw. However, the number of fish affected in each treatment group was not given. After 80 days, rainbow trout exposed to 5 μg TCDD/kg bw had significantly lower body weight than the fish treated with 1 μg TCDD/kg and the control group. The NOAEL based on growth was 1 μg TCDD/kg bw.
Van der Weiden et al. (1992) administered TCDD to juvenile rainbow trout by a single i.p. injection of 0.006, 0.03, 0.06, 0.3, 0.6 or 3.06 μg TCDD/kg bw. Growth was reduced in fish in the highest treatment group six weeks after exposure, and there was 20% mortality in this group after 12 weeks. Haemorrhages were present on fins and skin of fish in the 0.30, 0.60 and 3.06 μg TCDD/kg bw groups after six weeks, however the number of fish affected in each treatment group was not given. The NOAEL based on growth was 0.6 μg TCDD/kg bw.
Congener toxicity varies considerably in mammals and fish, one of the major differences being the insensitivity of the latter to DL‐PCBs (Van den Berg et al., 1998). For example, the TEF for PCB‐126 for humans and mammals is 0.1, and 0.005 for fish. The TEFs for fish established in 1998 were not re‐evaluated in 2005, only those for humans and mammals (van den Berg et al., 2006).
Exposure to 14C‐labelled PCB‐126 (0, 12.4 or 126 μg/kg wet weight) for 30 days had no effects on growth, survival and liver or thyroid histology in juvenile rainbow trout (Brown et al., 2002).
Rainbow trout were given single i.p. injections with either a low (100 μg/kg bw) or high (400 μg/kg bw) dose of PCB‐126, and their swimming performance and metabolic rates were compared with control fish from one to nine days post‐injection (Bellehumeur et al., 2016). Swimming speed was highest in the fish exposed to high PCB‐126 treatment. However, the initial condition factor was also highest in this group mainly due to their greater body mass. Trout in both PCB‐126 exposure groups had impaired recovery following intense exercise compared with control fish. PCB‐treated fish had reduced spleen somatic indices, and muscle glucose and glycogen contents compared with control fish, whereas plasma cortisol and glucose levels were increased, indicative of higher metabolic costs during recovery and muscle restoration. It was not possible to establish a NOAEL for this study due to differences in fish size among the groups at the start of the study.
Rainbow trout were fed 0, 0.5 or 50 μg PCB‐126/kg bw per day for five days followed by three weeks starvation (Quabius et al., 2000). Exposure to PCB‐126 did not affect basal hormone or glucose levels in fish directly following treatment. After starvation, plasma cortisol was higher in all groups. Adrenocorticotropic hormone levels increased with increasing PCB‐126 dose, and trout fed the high‐PCB‐126 diet had impaired hyperglycaemia after starvation.
3.1.5.6.2. Studies in salmon
No studies in salmon were eligible according to the criteria described in Section 2.2 (see also Annex A.11.6, Tables 95 and 96 therein). However, the CONTAM Panel found that controlled feeding studies primarily designed to study transfer (see Section 3.1.1.4) also provided some evidence regarding toxicity, since parameters indicative of fish health were measured. The studies assessed in this section had only one dose group, and salmon were exposed to mixtures of POPs (including PCDD/Fs and DL‐PCB) typically present in fish oil. However, due to the large volume of Atlantic salmon produced in Europe, these studies have been evaluated with regard to effects of chronic exposure despite having been designed to evaluate the effect of using clean fish oil in feeds on POP levels in farmed salmon.
Exposure to feed containing 1.5 ng PCDD/F‐WHO2005‐TEQ/kg feed and 2.7 ng sum of PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg diet for 18 months had no effect on growth, feed conversion or production‐related diseases (fin/skin erosion, bone deformity, cataract) compared to Atlantic salmon fed a diet containing 0.3 ng PCDD/F‐WHO1998‐TEQ/kg feed and 0.5 ng sum PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg diet for 18 months (Berntssen et al., 2010b; Lock et al., 2011).
Olli et al. (2010) raised Atlantic salmon on feed containing either a mean level of 0.2 ng PCDD/F‐WHO2005‐TEQ/kg diet (0.5 ng sum PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg feed) or 1.4 ng PCDD/F‐WHO2005‐TEQ/kg feed (2.8 ng sum PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg diet) for an entire production cycle (approximately 17 months). There were no significant differences in nutrient digestibility, thermal growth co‐efficient or condition factor between the fish given the different diets. The feeding regime in this study was 10% overfeeding. In order to convert the exposure to a daily dose of PCDD/Fs and DL‐PCBs the feeding rate used in the study above by Berntssen et al. (2010a) was applied by the CONTAM Panel. No adverse effects were seen in Atlantic salmon in either study at the highest dose tested which was 0.02 ng PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg bw per day (in both studies). In another study on salmon exposed to a fish oil‐based diet or a vegetable oil‐based diet, no adverse effects were seen on fish growth, condition factor, nutrient digestibility or nutrient utilisation after 12 months exposure to a fishmeal‐ and fish oil‐based diet containing 8.1 ng sum PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg diet (Torstensen et al., 2008; Berntssen et al., 2010b), equivalent to a dose of 0.06 ng sum PCDD/F+DL‐PCB‐WHO2005‐TEQ/kg bw based on the feeding regime in the study by Berntssen et al. (2010a).
3.1.5.6.3. Studies in other fish
Few studies were identified on the adverse effects of PCDD/Fs and/or DL‐PCBs in fish species other than trout and salmon with controlled exposure and more than one dose group. Most of the studies identified were field studies, or mixed exposure or studies in which only one dose group had been administered.
Yellow perch (Perca flavescens), carp (Cyprinus carpio), largemouth bass (Micropterus salmonides) and bullhead catfish (Ictalurus melas) were exposed to 1, 5, 25 or 125 μg TCDD/kg bw by single i.p. injection (Kleeman et al., 1988). TCDD toxicity tended to be greater in yellow perch, carp, and bullhead than in the other three species. In the six species tested in this study, the LD50 of TCDD ranged between 3 and 16 μg/kg bw and was significantly lower in yellow perch and carp than it was in largemouth bass. Growth reduction caused by TCDD was both species‐ and dose dependent. Fin necrosis was observed in all fish species at 25 and 125 μg/kg bw. Cutaneous haemorrhages were present in TCDD‐treated perch and carp, and cutaneous hyperpigmentation was only seen in TCDD‐treated carp and largemouth bass. The NOAEL was 1 μg TCDD/kg bw in yellow perch, and 5 μg TCDD/kg bw in the other species (see Annex A.11.6, Table 97 to 102 therein, for details).
Van der Weiden et al. (1994) administered TCDD to juvenile carp (Cyprinus carpio) by single i.p. injection of 0.01, 0.03, 0.05, 0.27, 0.57 and 2.93 μg/kg bw. Growth was reduced in fish in the highest treatment group six weeks after exposure, and there was 60% mortality in this group after 12 weeks. Histopathological effects were observed in the spleen at doses of 0.05 μg/kg and higher, and in the liver at doses of 0.57 and 2.93 μg TCDD/kg bw, however these data were not statistically analysed. Significant decreases were found in haemoglobin and haematocrit at the highest dose, and significant increases in spleen and kidney weight at this dose after 12 weeks. The NOAEL in this study was 0.57 μg TCDD/kg bw, based on growth, organ weight and haematological parameters.
Hart et al. (1999) administered TCDD to tilapia (Oreochromis niloticus) by i.p. injection of 1 or 5 μg/kg bw per day for five consecutive days. Cellularity and morphology of the spleen and pronephros were evaluated 2 days following dosing, and total cell counts were significantly reduced in both the spleen and pronephros at the highest dose. Histological examination revealed cellular depletion within lymphoid regions of the spleen and pronephros, and increased numbers of apoptotic cells in the 5 μg/kg bw treated fish. The NOAEL in this study was 1 μg TCDD/kg bw, based on histological parameters.
Quabius et al. (2000) fed tilapia (Oreochromis mossambicus) diets with 0, 0.5 or 50 μg PCB‐126/kg bw per day for five days followed by three weeks starvation. Exposure to PCB‐126 did not affect resting plasma adrenocorticotropic hormone, cortisol or glucose levels. Adrenocorticotropic hormone levels increased with increasing PCB‐126 dose and tilapia fed the high‐PCB‐126 diet had impaired hyperglycaemia after starvation.
Summary of the studies in trout, salmon and other species
In summary, toxicological responses to PCDD/Fs and DL‐PCBs exposure include fin necrosis, haemorrhages, reduced growth and mortality in fish. Sensitivity was species‐ and dose dependent. The lowest LOAEL in rainbow trout was 1 μg TCDD/kg bw with a NOAEL of 0.1 μg TCDD/kg bw. A NOAEL of 1 μg TCDD/kg bw was identified for yellow perch and tilapia, and 0.57 μg TCDD/kg bw for carp. A feeding study with Atlantic salmon showed no effects after prolonged exposure to PCDD/Fs and DL‐PCBs at 0.02 ng WHO2005‐TEQ/kg bw per day (the highest dose tested).
3.1.5.6.4. Studies in fish eggs
The literature search carried out as described in Section 2.2 identified several in ovo studies on the adverse effects of PCDD/Fs and/or DL‐PCBs in fish (see Annex A.11.6, Table 103 therein). For this evaluation, the CONTAM Panel focussed on studies in which more than one dose group had been administered, and when the exposure was by injection (not waterborne).
Dose‐dependent effects of TCDD in developing fish include oedema around the yolk sac and heart, craniofacial malformations, vascular dysfunction, decreased growth and increased mortality (Spitsbergen et al., 1991; Johnson et al., 1998; Wright and Tillitt, 1999). Other symptoms of TCDD exposure in developing fish include spinal and tail deformities and haemorrhages (Tillitt et al., 2017).
Morphometric effects were examined in rainbow trout eggs (from four individual females) injected with 0, 38, 113, 200, 300, 500 or 1,000 pg TCDD/g (Carvalho et al., 2004). The LD50 for eggs pooled from four different females, was 325 pg TCDD/g. Dose‐dependent decreases were found in cranial‐ and total length in swim‐up fry approximately 65 days post‐fertilisation (and 32 days post‐hatch). The sensitivity to dioxin exposure varied slightly among fry from different females, with a NOAEL of 38 pg TCDD/g for effect on body length in fry from one female, and 113 pg TCDD/g in fry from the other three females. Biochemical, histological and behavioural parameters were also examined in this study in fry which had been injected with 0, 38, 113 or 300 pg/g egg (Carvalho and Tillitt, 2004). Retinal ganglion cell densities were significantly decreased in fish exposed to 113 or 300 pg TCDD/g, compared to controls. Dose‐dependent visual‐ and motor function deficiencies were detected in three different tests on fry (28–32 dph), with a NOAEL of 38 pg TCDD/g for all endpoints.
In summary, the lowest NOAEL was 38 pg TCDD/g egg based on effects of visual acuity, motor detection ability, light sensitivity and retinal ganglion cell densities in trout fry. The CONTAM Panel did not identify evidence suitable for converting the egg concentrations to fish exposure, and the results from fish eggs where thus not considered further.
3.1.5.7. Companion animals (cats and dogs)
Several studies were found in the literature search that mentioned effects in cats or dogs. Five of these were not further considered as they were not reporting adverse effects rather the papers reported enzyme induction or levels of contamination by mixed exposure to organochlorine pesticides and other PCBs (Korytko et al., 1999; Kunisue et al., 2005, 2007; Sonne et al., 2008; Storelli et al., 2009).
The study by Schwetz et al. (1973) (Annex A.11.7, Table 105 therein) showed lethality in dogs by oral administration of TCDD (3 mg/kg). However, no further details were provided. Microscopic damage to liver, kidneys and spleen was observed in two cats that were exposed via contaminated arenas (Kimbrough et al., 1977). In this study, the route and extent of exposure could not be determined.
The CONTAM Panel considered that these studies are not suitable for deriving a reference point for PCDD/Fs and DL‐PCBs in cats and dogs.
3.1.5.8. Fur animals
The literature search carried out as described in Section 2.2 identified a number of studies on the effects of PCDD/Fs and DL‐PCBs in mink. Based on the exposure type, the mink studies fall into two separate categories:
Studies in which mink were fed on laboratory animal feed supplemented with predesigned concentrations of PCDD/Fs and/or DL‐PCBs, and
Studies in which mink were fed on feed supplemented with fish contaminated with PCDD/Fs, DL‐PCBs and other pollutants.
Some details about these studies are described in Annex A.11.8 (Tables 106 and 107 therein, respectively).
The CONTAM Panel considered the studies with predesigned concentrations of PCDD/Fs and/or DL‐PCBs best‐suited for risk assessment in mink. However, the studies in which mink were fed contaminated fish were also assessed and regarded as supplementary evidence because the exposure type resembles that of humans in real life. In most of the mink studies, exposure was given as feed concentration of the examined compounds. To enable comparison with the studies on laboratory animals, feed concentrations have here been converted to doses per kg bw. In some cases, the authors had measured feed intake and provided the daily doses based on these data in the paper; in those instances, the study‐specific doses have been applied. In all other cases, the conversion has been made using a factor for daily feed intake of mink, 100 g/kg bw, derived from three independent studies (Bleavins and Aulerich, 1981; Moore et al., 2012; Bursian et al., 2013a,b).
Two early studies conducted in the 1980s addressed the toxicity of PCB‐169. In the first of them, diets containing 0.1 or 0.5 mg/kg PCB‐169 were fed to adult female mink from one month prior to breeding through parturition (Aulerich et al., 1985). All mink in the higher dosage group died within 60 days, while those fed 0.1 mg/kg (300 ng WHO2005‐TEQ/kg bw per day) showed 50% mortality after 3 months exposure (control: 0%). In the second study, 10‐fold lower concentrations of PCB‐169 were fed to adult female mink for 135 days (Aulerich et al., 1987). In the 0.05 mg/kg group, 50% of the animals died in the course of the study. The lower dose, 0.01 mg/kg (corresponding to 30 ng WHO2005‐TEQ/kg bw per day), was the LOAEL due to, e.g. changes in organ weights and thyroid hormone levels (T3 and free T3 reduced). Two further early studies also failed to establish a NOAEL. Hochstein et al. (1988) administered TCDD to adult male mink as a single oral dose (0, 2.5, 5.0, or 7.5 μg/kg bw) and observed the mink for 28 days. The body weight of the mink decreased significantly vs. control already at the lowest dose. Thus, 2.5 μg TCDD/kg bw was the LOAEL. In the study by Aulerich et al. (1988), newborn mink kits were administered daily 0.1 or 1.0 μg TCDD/kg bw by i.p. injection for the first 12 days post‐partum. The higher dose of TCDD caused mortality (> 50%), while the lower one reduced body weight gain. The LOAEL was therefore 100 ng TCDD/kg bw per day.
The NOAEL was, however, obtained in some studies carried out at a later date. Beckett et al. (2008) exposed adult female mink to PCB‐126 at concentrations equivalent to 0, 24, 240 and 2,400 ng WHO2005‐TEQ/kg feed in a standard ranch diet. The mink were bred with non‐exposed males, and the exposure lasted for up to 133 days (3 weeks prior to breeding, 6 weeks after delivery). The study was designed to explore the effect of PCB‐126 on reproductive performance and kit survival and growth in mink. None of the successfully mated female mink at 240 and 2,400 ng WHO2005‐TEQ/kg feed whelped, indicating embryonal or fetal death. The NOAEL was 24 ng WHO2005‐TEQ/g feed, but control kit survival to weaning was only 56% (at the lowest dose, 57%) precluding any assessment of the effect of PCB‐126 on this variable at the NOAEL level.
In another controlled feeding study (Bursian et al., 2012), adult female mink were treated with vehicle or four increasing doses (as ng WHO2005‐TEQ/kg bw per day) of TCDD (2.1, 4.6, 6.0, 8.4), 2,3,4,7,8‐PeCDF (4.0, 7.6, 9.0, 15), or TCDF (5.2, 12, 21, 25), which were added in their feed. After 2–3 months on the diets, the females were mated with untreated male mink and euthanised when kits were 6 weeks old. Some of the kits from all dosage groups were maintained on their respective diets until they reached the age of 27 weeks. Tissues were subjected to histological analysis. In juvenile mink at the age of 27 weeks, maxillary and mandibular squamous epithelial proliferation increased dose dependently for all three compounds. The change originated from gingiva, led to cyst formation adjacent to teeth, and caused osteoporosis in the jaw bones. The lowest LOAEL was 4.6 ng TEQ/kg bw per day with a NOAEL of 2.1 ng TEQ/kg bw per day, obtained from the TCDD‐exposed group. Dose‐related effects on other tissues were few (see Annex A.11.8, Table 106 therein) and reported in a separate paper (Moore et al., 2012). None of the doses tested for TCDD or 2,3,4,7,8‐PeCDF caused any adverse effects. The NOAEL for TCDF was 21 ng WHO2005‐TEQ/kg bw per day based on histopathological changes in the highest TCDF dosage group.
Hochstein et al. (1998) fed adult female mink on diets supplemented with 0, 0.001, 0.01, 0.1, 1, 10, or 100 μg TCDD/kg for up to 125 days. The original study design included a breeding programme in which the female mink were intended to be mated with untreated males so that the effects of dietary TCDD exposure on female reproductive performance and fetal development could be determined. However, the containment room proved unsuitable for mink breeding with only few mink becoming pregnant, and this aspect of the study was abandoned. Mink mortality was dose‐ and time dependent. By 125 days, all animals in the two highest dosage groups and 5/8 mink of the 1 μg/kg group had succumbed. In the surviving mink, the proportion of band (immature) neutrophils of leucocytes was increased in all treatment groups, suggesting a low‐grade inflammation, but this was not dose‐related. Moreover, no change was recorded in mature (segmented) or total neutrophils. Therefore, the CONTAM Panel did not use this change in the derivation of a NOAEL. Based on mortality, the NOAEL was 0.1 μg/kg feed, which corresponds to 5.5 ng TCDD/kg bw per day, but it should be noted that the sensitive reproductive endpoints could not be analysed.
In another reproductive study by Hochstein et al. (2001), mature female standard dark mink were fed 16, 53, 180 or 1,400 ng/kg TCDD mixed in their diet for 131‐132 days (control diet contained 6 ng/kg TCDD). Between days 35 and 64, the females were mated with unexposed males. However, breeding success was low while not dose‐related. No females whelped at the lowest and highest doses, whereas 5/12, 3/12 and 8/12 mink whelped in the control, 53 and 180 ng/kg groups, respectively. Serum iron was reduced and total carbon dioxide (a measure of serum bicarbonate) was elevated already at the 16 ng/kg dosage level. Terminal body weight showed a dose‐dependent downward tendency, but the change attained statistical significance only at the highest dose. This study was not considered further due to poor fertility rate in controls.
The second group of mink studies consisted of those in which exposure was through fish contaminated with PCDD/Fs and DL‐PCBs, which constituted a variable portion of the diet (see Annex A.11.8, Table 107 therein). Heaton et al. (1995a,b) fed contaminated carp to adult female mink for about two months before and throughout the reproductive period. The exposure levels were 1.0, 19.4, 40.0, and 80.8 ng BEQ/kg diet, the levels being based on a bioassay with H4IIE rat hepatoma cells. Kit survival and growth were adversely affected already at the lowest dose, with the LOAEL being 3.6 ng BEQ/kg bw per day. In a multigenerational feeding study (Restum et al., 1998) even the lowest exposure level (equivalent to 0.32 ng WHO2005‐TEQ/kg bw per day; LOAEL) reduced vulvar swelling in the P1 generation and decreased F1‐1 kits’ body weight at 3 and 6 weeks. It also increased spleen weight in F1‐1 animals and caused hepatocellular lipidosis in P1 mink. In two reproductive studies, Bursian et al. (2006a,b) reported reduced kit survival between 3 and 6 weeks of age and lower body weight of the kits at 3 weeks. However, the most sensitive adverse effect was mandibular and maxillary squamous cell proliferation, for which the NOAEL proved to be 0.5 ng WHO1998‐TEQ/kg bw per day.
In two further fish‐feeding studies (Bursian et al., 2006c; Martin et al., 2006), the NOAEL for mandibular and maxillary squamous cell proliferation was found to be 2.5 ng WHO1998‐TEQ/kg bw per day. This dosage level (and higher ones) led to reductions in plasma retinyl palmitate and total esters (in juvenile females) and in kidney total retinyl esters (in kits and juvenile mink). However, the CONTAM Panel was of the opinion that changes in retinoid concentrations alone cannot be used for deriving a reference point. Finally, in two other studies conducted by Bursian et al. (2013a,b), the NOAEL for kit survival was 0.4 ng WHO2005‐TEQ/kg bw per day, but there was a dose‐dependent increase in the occurrence of mandibular and maxillary squamous cell hyperplastic foci, starting from this dosage level. Therefore, it was the LOAEL for the two studies.
The CONTAM Panel noted that the identities, concentrations and influences of all the other contaminants in the fish fed to mink cannot be reliably assessed. Therefore, the Panel decided to base the assessment on those studies in which mink were fed on laboratory animal feed supplemented with predesigned concentrations of PCDD/Fs and/or DL‐PCBs. However, the fact that lower TEQ levels were reported to cause the typical gingival lesion in studies with exposure to a contaminant mixture vs. studies with exposure to a single PCDD/F or DL‐PCB, suggest a modulatory action by other contaminants. This is in keeping with the findings that e.g. heavy metals can influence, by either enhancing or inhibiting, AHR activation by ligands such as TCDD (Anwar‐Mohamed and El‐Kadi et al., 2012).
In summary, in the studies with controlled exposure the lowest LOAEL was 4.6 ng TEQ/kg bw per day with a NOAEL of 2.1 ng TCDD/kg bw per day for the characteristic morphological change caused by PCDD/Fs and DL‐PCBs in mink, i.e. proliferation of the squamous gingival epithelium in mouth in juvenile mink, from the two‐generation study of Bursian et al. (2012).
3.1.6. Mode of action
Studies to inform the mode of action section were retrieved and selected as described in Section 2.2. The role of AHR in TCDD‐toxicity with the aim to support the health risk assessment process is currently being developed on the AOP‐wiki ( https://aopwiki.org/), but this work could not yet be used for the purpose of this risk assessment.
The current consensus states that the molecular initiating event of toxicity in vertebrates of the prototype dioxin, TCDD, is the binding to AHR and its consequent activation. AHR is a highly conserved, over 600 million‐year‐old protein (Hahn et al., 2017) that has proven to have important physiological functions throughout life, including early development of organs such as the immune, hepatic, cardiovascular and reproductive systems. At the cellular level, AHR is involved in the control of cell proliferation and differentiation, while at the molecular level AHR regulates transcription of a large number of physiologically important genes and may have effects on processes involving epigenetic mechanisms (Mulero‐Navarro and Fernández‐Salguero, 2016). The current view is that ensuing TCDD toxicity results from inappropriate (in terms of timing, location and/or degree) and sustained activation of AHR (Bock and Kohle, 2006; Denison et al., 2011; Mulero‐Navarro and Fernández‐Salguero, 2016). Compared to most other PCDD/F and DL‐PCB congeners, TCDD has a higher binding affinity to the AHR and exhibits a greater AHR activation potency.
3.1.6.1. Discovery of the AHR: CYP1A1 induction
It was initially shown that the PAH compounds 3‐methylcholanthrene and benzo[a]pyrene can induce aryl hydrocarbon hydroxylase (AHH) activity in rat liver (Conney et al., 1957). There were marked differences between mice strains in their AHH inducibility by 3‐methylcholanthrene (Nebert and Gelboin, 1969). Genetic crossing studies revealed that AHH inducibility is primarily inherited as an autosomal dominant trait in mouse (Gielen et al., 1972; Nebert et al., 1972), and the genetic locus responsible for induction was the Ah (aryl hydrocarbon) locus. The mediator protein of AHH induction proved to be the AHR (Poland et al., 1976).
Studies in mutant Hepa‐1 mouse hepatoma cells showed that to induce the cytochrome P‐450 subtype responsible for AHH activity (now termed CYP1A1), another protein was required. This protein was named the AH receptor nuclear translocator (ARNT) (Hoffman et al., 1991). Furthermore, a specific nucleotide sequence, ‘dioxin‐responsive element’ or DRE was identified upstream of the Cyp1a1 gene (Jones et al., 1986; Denison et al., 1988). AHR and ARNT were shown to form a heterodimer that binds to this DRE and thereby regulates the expression of Cyp1a1 (Reyes et al., 1992).
3.1.6.2. AHR signalling pathways
Canonical pathway
Cyp1a1 induction by AHR agonists, foremost TCDD, has served as a key model in more recent studies on molecular mechanisms of AHR function. AHR acts as ligand‐activated transcription factor. In Figure 7, the canonical AHR signalling pathway is depicted with solid black arrows. In its inactive state, AHR resides in the cytosol in a protein complex comprising a dimer of HSP90 and the co‐chaperones AIP (also known as XAP2) and p23 (Murray and Perdew et al., 2012). TCDD penetrates the cell membrane by diffusion. Binding of TCDD to AHR (1. in Figure 7) causes a transformation of its PAS‐A domain conformation (see Section 3.1.6.4, Henry and Gasiewicz, 2003; Soshilov and Denison, 2008). This conformational change results in enhanced nuclear translocation of the AHR (Richter et al., 2001). Once inside the nucleus, the AHR sheds its cytoplasmic partner proteins (2. in Figure 7) and dimerises with ARNT to bind to the DNA at DREs (3. in Figure 7) in the promoter regions of the genes regulated by the AHR (5. to 7. in Figure 7; Jackson et al., 2015).
Figure 7.
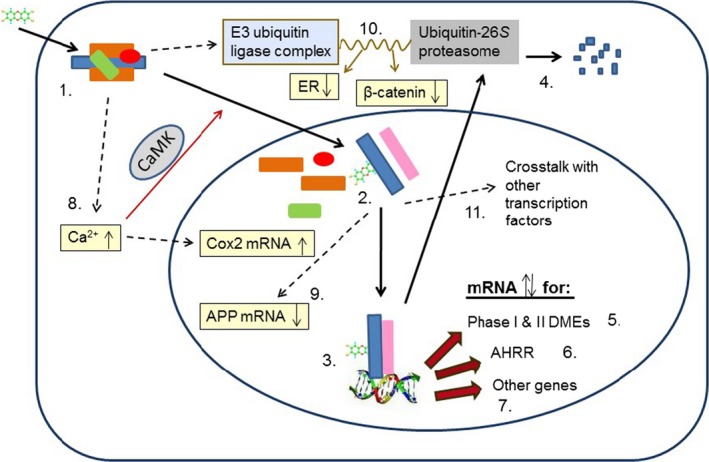
A schematic and simplified diagram of some salient features of AHR signalling pathways
- Black solid arrows depict the canonical pathway, dashed arrows alternative pathways, and the red solid arrow an example of their intersection. The blue bar represents AHR, pink bar ARNT, green bar AIP, brown bars a dimer of HSP90, and the red dot p23 (Modified from Lindén et al., 2010).ER: oestrogen receptor; CaMK: Ca2+/calmodulin‐dependent kinase; Cox2: cyclooxygenase 2; APP: acute‐phase protein; DME: drug‐metabolising enzyme. See text for details
AHR activity is terminated by nuclear export of the receptor (Davarinos and Pollenz, 1999; Ikuta et al., 1998), and by its ubiquitin‐mediated degradation by the 26S proteasome (4. in Figure 7; Davarinos and Pollenz, 1999; Roberts and Whitelaw, 1999; Ma and Baldwin, 2000).
In addition to Cyp1a1, a number of other Phase I and Phase II metabolism genes are consistently upregulated by the AHR (5. in Figure 7), constituting the bulk of the so‐called AHR battery of genes. In mammals, this includes Cyp1a2, Cyp1b1, Cyp2s1, Cyp2a5, Aldh3a1, Gsta1, Ugt1a6 (in humans, UGT1A8, UGT1A9 and UGT1A10) and Nqo1 (Arpiainen et al., 2005; Yeager et al., 2009; Deb and Bandiera, 2010; Kalthoff et al., 2010; Ma, 2012; Li et al., 2015). It is well known that the expression of an additional wide variety of genes involved in general metabolism and endocrine regulation can be modified by TCDD‐activated AHR (Fletcher et al., 2005; Boutros et al., 2008; Franc et al., 2008; Sato et al., 2008). Many of these genes, which are referred to as the ‘other genes’ in Figure 7 (7.), are evolutionarily conserved and/or play key roles in cell/organ function and homeostasis over the life course.
It is also well‐known that the concentration of a large number of endogenous molecules, whose normal concentration ranges are controlled by phase I and II enzymes (Figure 7 (5.)), as well as other more substrate‐specific enzymes, and/or carrier proteins, can be modulated by sustained activation of AHR. This modulation may affect levels of endogenous signalling molecules such as steroid (Annex A.10) and thyroid hormones (see Sections 3.1.4.4 and 3.1.6.10.5), which upon binding to their nuclear receptors, can regulate additional gene cascades involved in fetal development, cell and organ function, as well as homeostasis over the life course. Adverse outcomes may emerge when physiological hormone levels or dynamics can no longer be maintained.
The spectra of ‘other genes’ (Figure 7 (7.)) affected by TCDD in cell studies seem to be species‐ and strain‐dependent with very little overlap (Boverhof et al., 2006; Boutros et al., 2008; Franc et al., 2008; see also Section 3.1.6.4). The observed variation in gene expression could be related to the differences in sensitivity to TCDD‐exposure among strains and species.
Induction of Cyp1a1/2 and Cyp1b1 may accelerate the metabolic activation of xenobiotics, like heterocyclic aromatic amines and PAHs, and thereby indirectly predispose to cancer (Kim and Guengerich, 2005; Hodek et al., 2013; Shimada and Fujii‐Kuriyama, 2004). It may also result in transformation of oestradiol to carcinogenic catechol oestrogens (He and Feng, 2015). However, findings in animal models suggest that the induction of CYP1A1/2 is predominantly protective to the organism by enhancing the detoxification of xenobiotics (Nebert et al., 2004; Uno et al., 2004). For information on the relationship between mono‐oxygenase induction and TCDD toxicity, see Section 3.1.6.4.
Although the induction of Cyp1a1 is believed to provide a good general model of gene regulation by the AHR, the outcome can also be repression of gene activity. The molecular mechanism(s) for transcriptional gene repression by the AHR are still poorly defined (Riddick et al., 2004). Among the genes induced by TCDD are at least two, whose products act as suppressors of AHR activity, thus forming a feedback loop: AHR repressor (Ahrr; 6. in Figure 10) and TCDD‐inducible poly(ADP‐ribose) polymerase (Tiparp).
Figure 10.
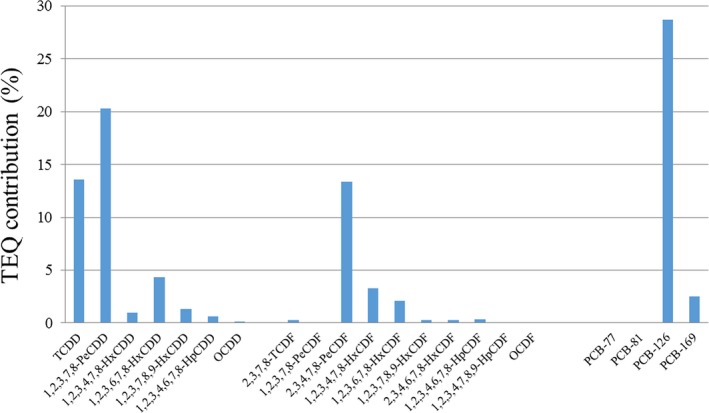
Relative contribution of PCDD/Fs and DL‐PCBs to the WHO 2005‐TEQ level in boys from the Russian Children's Study, for the median (P50) level (based on Burns et al., 2009)
Alternative pathways
In vitro, TCDD causes some responses which are too fast (10–15 min) to occur through the canonical pathway and may not require the presence of ARNT (Matsumura, 2012). These responses depend on the cell line used but generally include increases in cellular Ca2+ concentration (8. in Figure 7) and activation of cytosolic phospholipase A2, c‐SRC kinase and cyclo‐oxygenase 2 (COX‐2), followed by inflammatory changes (reviewed in Matsumura, 2009, 2012). Elevated cellular Ca2+ levels activate Ca2+/calmodulin‐dependent kinases (CaMK), which, in turn, may modulate nuclear translocation of the AHR (red arrow in Figure 7) (Monteiro et al., 2008; Gilot et al., 2011). The in vivo relevance of these findings has been shown for body weight loss and developmental hydronephrosis in mice (Yoshioka et al., 2014; Fujisawa et al., 2018). Other examples of alternative pathways include the AHR‐mediated repressed expression of acute‐phase proteins such as serum amyloid A 3 in mouse hepatocytes (9. in Figure 7) (Patel et al., 2009) and functioning of the AHR as a nuclear E3 ubiquitin ligase. Thereby, it directs oestrogen (ER) and androgen receptors (AR) as well as β‐catenin to proteasomal degradation (10. in Figure 7) (Ohtake et al., 2007; Ohtake and Kato et al., 2011).
Cross‐talk
AHR has extensive cross‐talk with other cellular signalling systems including the oestrogen receptor (ER), as well as the nuclear receptors for thyroid hormone (THR) and retinoic acid (RAR and RXR) (11. in Figure 7). Of these, the most diverse interactions of dioxin‐activated AHR have been described with the ER and include transcriptional repression of ER (Tian et al., 1998a,b; Safe et al., 2000), recruitment of ER to AHR‐regulated genes thus diverting it from its own target genes (Matthews et al., 2005), competition with ER for ARNT as a partner or coactivator (Rüegg et al., 2008), and targeting of ER to proteasomal destruction as described above (10. in Figure 7). Clearly, the ER‐AHR interactions may depend on cell type, promoter context, oestrogen level and receptor expression patterns (Kuo et al., 2013). In the case of retinoid receptors, the shown interaction of Silencing mediator of retinoic acid and thyroid hormone receptor (SMRT), a co‐regulator of many nuclear receptors including RAR, with AHR (Nguyen et al., 1999) can form the basis for AHR‐retinoid cross‐talk (Fallone et al., 2004; Widerak et al., 2006).
Epigenetic mechanisms
Recent studies have reported that TCDD exposure of rodent dams in F0 generation can possibly affect untreated progeny of subsequent generations in an adverse manner (Bruner‐Tran and Osteen, 2011; Manikkam et al., 2012a; Sanabria et al., 2016). There is evidence for epigenetic modifications in AHR signalling involving histones, DNA and micro‐RNAs, which may underlie the reported transgenerational effects of TCDD in rats (Manikkam et al., 2012a,b) and zebrafish (Baker et al., 2014). As to other effects, epigenetic mechanisms appear to play a prominent part in the induction of xenobiotic‐metabolising enzymes by the AHR (Shen and Whitlock, 1989; Beedanagari et al., 2010a,b; Ko and Puga et al., 2012; Kurita et al., 2014), and also in transcriptional regulation of the Ahr gene (Garrison and Denison, 2000; Garrison et al., 2000). For carcinogenicity, it is noteworthy that TCDD‐activated AHR may repress the transcription of some tumour suppressor proteins including p53 by promoter methylation (Ray and Swanson, 2004) and micro‐RNA induction (Gordon et al., 2015). Furthermore, micro‐RNA seems to mediate some immunological effects of TCDD (Singh et al., 2012; Hanieh and Alzahrani, 2013).
3.1.6.3. Role of AHR and its signalling pathways in the toxicity of dioxins
The indispensability of the AHR for the toxicity of TCDD and related compounds was initially suggested by the finding that the toxicity of PCDD/F congeners correlates with their binding affinity to the AHR (Poland and Knutson, 1982; Safe, 1986). Definitive evidence became available with the generation of AHR knockout (AHRKO) mice and rats. Three independent laboratories produced their own AHRKO mouse lines, and these all consistently showed that the AHR is required for all major toxicities of TCDD examined so far, including acute lethality, thymic atrophy, the principal features of the liver lesion, teratogenicity, developmental toxicity to male reproductive organs, immune toxicity, porphyria, disrupted retinoic acid homeostasis and reduced plasma thyroxine levels (Fernandez‐Salguero et al., 1996; Mimura et al., 1997; Vorderstrasse et al., 2001; Lin et al., 2002; Nishimura et al., 2005; Davies et al., 2008). Likewise, AHRKO rats have proven to be resistant to TCDD‐induced inhibition of body weight gain, thymic atrophy, hepatomegaly and histological alterations in the liver, as well as haematological and serum biochemical changes (Harrill et al., 2013, 2016). Although AHR appears to be a prerequisite for TCDD toxicity, it is still possible that TCDD can cause some minor biochemical effects in its absence, as suggested by a small set of genes (32) whose expression levels were altered in AHRKO mouse livers vs. vehicle controls in response to TCDD treatment. However, this occurred at a high dose of TCDD (1,000 μg/kg) (Tijet et al., 2006).
While the studies in AHRKO animals confirm the essentiality of the AHR in the major toxicities of TCDD, they do not demonstrate whether these arise via the canonical or alternative signalling pathways. This issue has been addressed by additional mouse models, in which: (1) the AHR has been unable to translocate into the nucleus upon ligand binding; (2) the AHR has been unable to bind to the DRE; (3) ARNT protein expression has been rendered very low, only about 10% of normal (ARNT hypomorphs); or (4) ARNT has been deleted in hepatocytes alone. These models have unequivocally shown that the toxic outcomes of TCDD so far analysed (hepatotoxicity, thymic atrophy and teratogenicity) are mediated by the canonical signalling pathway (Bunger et al., 2003, 2008; Walisser et al., 2004; Nukaya et al., 2010).
Nevertheless, as described above (see ‘Alternative pathways’), hydronephrosis induced neonatally with TCDD in mice might occur via an alternative pathway with cytosolic phospholipase A2α playing a predominant role in its pathogenesis (Yoshioka et al., 2014), and the two pathways may have distinct or even opposite effects on the regulation of Th17 and Treg cell balance in the immune system (Mohinta et al., 2015).
3.1.6.4. Relationship between structure and function of the AHR
The AHR protein has a modular structure being composed of functional domains (see Figure 8) (Burbach et al., 1992; Whitelaw et al., 1993; Jain et al., 1994; Coumailleau et al., 1995; Fukunaga et al., 1995; Lindebro et al., 1995; Sogawa et al., 1995; Bacsi and Hankinson, 1996; Ikuta et al., 1998; Pongratz et al., 1998; Jones and Whitlock, 2001; Kumar et al., 2001).
Figure 8.
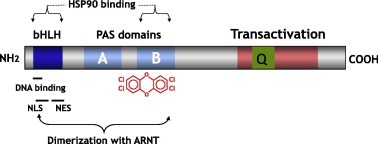
Major functional domains of AHR
- The position of the TCDD molecule shows the approximate location of the ligand‐binding domain (PAS B and flanking amino acids). In the transactivation domain, there are at least three interacting subunits (acidic, glutamine‐rich and proline/serine/threonine‐rich), of which only the glutamine‐rich (Q) subdomain is depicted.NLS: nuclear localisation signal; NES: nuclear export signal (from Lindén et al., 2010).
Genetic variation in some of the functional domains of the AHR is associated with differences in sensitivity to TCDD toxicity. The higher sensitivity of C57BL/6 vs DBA/2 mouse strains (and C57BL/6Jb‐1/b‐1 vs. the subsequently generated congenic C57BL/6Jd/d mice differing only at the Ahr locus), is mainly based on a single amino‐acid difference (alanine vs. valine) in the ligand‐binding domain of the AHR (Poland et al., 1994). This variation results in about a 10‐fold difference in binding affinity, which is reflected in a difference of a similar magnitude in sensitivity to a variety of TCDD toxicities, including acute lethality, liver lesions, thymic atrophy, porphyria, teratogenicity, myelotoxicity and immune toxicity (Poland and Glover, 1980; Chapman and Schiller, 1985; Hong et al., 1987; Birnbaum et al., 1990; Kerkvliet et al., 1990a,b; Harper et al., 1993; Davies et al., 2008). In birds, AHR binding affinity also correlates with susceptibility to TCDD and DL‐PCBs, and two amino acids (one of which is equivalent to the critical amino acid in mice) are major determinants in this respect (Head et al., 2008; Manning et al., 2012; Farmahin et al., 2013).
Although the primary structure of the human AHR is closely similar to that of the most TCDD‐sensitive species, guinea pig (Korkalainen et al., 2001), functionally more important appears to be the fact that in the ligand‐binding domain of the human AHR, there is valine at the same position as in the TCDD‐insensitive mouse AHRd isoform (for polymorphisms in human AHR and implications to risk assessment, see Section 3.1.6.8). The human AHR also displays lower TCDD binding affinity compared with the product of the TCDD‐sensitive mouse b‐1 allele (Manchester et al., 1987; Ramadoss and Perdew, 2004). As described below (see Section 3.1.6.6), this feature cannot be generalised to all AHR agonists, because the human receptor displays a higher relative ligand binding affinity than its mouse b‐1 counterpart for some ligands, such as indirubin. Moreover, these two receptors appear to regulate distinct batteries of genes, even when expressed in the same host. In primary hepatocytes isolated from C57BL/6J mice (with the b‐1 allele) or from transgenic mice expressing the human AHR specifically in their hepatocytes, treatment with TCDD showed that only 18% of the genes induced by either receptor were also similarly altered by the other, foremost genes of the AHR battery. The corresponding figure for gene repression was 49% (Flaveny et al., 2010). Similarly, while TCDD altered the expression levels of 1,547 and 475 genes in rat and human primary hepatocytes, respectively, only 158 differentially expressed orthologues were common between rat and human hepatocytes, these included xenobiotic‐metabolising enzymes of the AHR battery. Enrichment analysis of the differentially expressed orthologues revealed 49 and 34 enriched human and rat pathways, respectively. Only 12 of these were shared between the two species, including AHR signalling, CAR‐ and PXR‐mediated regulation of xenobiotic metabolism, and oestradiol metabolism (Black et al., 2012). In further support to species‐specific gene regulation, in a large dose–response and time‐course study covering a total of seven time‐points between 1 and 48 h, exposure to 10 nM TCDD resulted in 495, 2,305 and 711 differentially expressed orthologous genes in human, mouse, and rat primary hepatocytes, respectively. However, only 16 orthologues were differentially expressed across hepatocytes from all three species. Comparable EC50 values were obtained for AHR battery genes including Cyp1a1 and Tiparp. TCDD‐mediated differential gene expression mapped to 41 human, 153 mouse, and 142 rat canonical pathways, of which 10 were common to hepatocytes from all three species and included AHR and Nrf2 signalling, as well as lipid transport, processing, and metabolism (Forgacs et al., 2013). Finally, a study using primary B lymphocytes from three species identified TCDD‐elicited differential expression of 515 human, 2,371 mouse and 712 rat orthologous genes over a 24‐h period (Kovalova et al., 2017). Again, merely 28 orthologues were differentially expressed in response to TCDD in lymphocytes from all three species, but the ten most upregulated genes included genes of the AHR battery in all of them. TCDD‐induced differential gene expression mapped to 34 mouse, 25 rat and 27 human KEGG canonical pathways, among which 5 were in common to the three species and included extracellular matrix (ECM)‐receptor interaction, focal adhesion and regulation of actin cytoskeleton.
With respect to the transcriptomic studies in cells with TCDD, the CONTAM Panel noted that these show clear differences in the degree of up‐ or downregulation of the various genes, but also in the dose levels at which such gene regulation occurs. Despite the apparently species‐dependent patterns of gene expression responses of hepatocytes in general, the genes of the AHR battery largely respond in the same manner across hepatocytes from different species, including human. Except for AHR signalling, there seem to be few if any common pathways consistently enriched across the cell studies.
In contrast to the alanine/valine polymorphism in the ligand‐binding domain of the AHR, which seems to uniformly determine sensitivity to TCDD′s toxic effects, certain changes in AHR transactivation domain appear to be capable of exerting a selective influence on TCDD toxicities. In a rat substrain, Han/Wistar (Kuopio; Han Wistar), a single‐nucleotide mutation at an intron/exon junction of the Ahr gene gives rise to splice variants which produce two proteins, both of them carrying a modified transactivation domain (Pohjanvirta et al., 1998). This mutation is the major reason for the exceptional resistance (over 1,000‐fold higher than in the most sensitive rats) of Han Wistar rats to the acute lethality of TCDD (Unkila et al., 1994; Tuomisto et al., 1999). However, the resistance is endpoint‐dependent, and for a number of biochemical and toxic effects of TCDD, including induction of Phase I and II enzymes, thymus atrophy, fetolethality and derailed vitamin A status, Han Wistar rats exhibit sensitivity equal or similar to that of other rat strains (Pohjanvirta et al., 2011). Thereby, TCDD‐triggered responses in rats can be divided in two categories: those robust and insensitive to the mutation in the AHR transactivation domain structure (type I; such as induction of Phase I and Phase II xenobiotic‐metabolising enzymes, thymus atrophy, fetolethality, derailed vitamin A status, hypercholesterolemia, reduced serum thyroxine and melatonin levels, incisor tooth lesion, and avoidance of novel foods) and those sensitive to it (type II; such as acute lethality, the wasting syndrome, liver toxicity, tumour promotion, hepatic lipid peroxidation and biliverdin accumulation, hyperbilirubinaemia, hypoglycaemia, increased serum amino acid and free fatty acid levels) (Viluksela et al., 1999; Pohjanvirta et al., 2011). Surprisingly, the resistance of Han Wistar rats is highest for TCDD and then decreases with reducing AHR affinity of PCDD/F congeners in such a way that both 1,2,3,4,7,8‐hexa‐ and 1,2,3,4,6,7,8‐heptaCDD are actually more toxic to these rats than TCDD, and their sensitivity to the latter congener is comparable to that of Sprague–Dawley rats (Pohjanvirta et al., 1995).
It was recently shown that interference of the transactivation function of the human AHR with short peptides also resulted in selective suppression of gene regulation by the AHR (Ren et al., 2016). The transactivation domain of the AHR is further restructured (at the glutamine‐rich subdomain) in another highly TCDD‐resistant laboratory animal, the hamster (Korkalainen et al., 2000), which (similar to the Han Wistar rat) is still normally responsive to cytochrome P‐450 induction (Gasiewicz et al., 1986). Overall, a high linear correlation across mammalian species has been demonstrated between sensitivity to the acute lethality of TCDD and AHR transactivation domain structure (number of glutamine residues in the glutamine‐rich subdomain) combined with binding free energy between AHR and TCDD (Wang et al., 2013). With regard to risk assessment, it is important to note that the most often used indicator of AHR activation, induction of drug‐metabolising enzymes, does not correlate with TCDD resistance in the animal models of altered AHR transactivation domain (Han Wistar rat, hamster). Conversely, Cyp1a1 induction is a highly sensitive but non‐specific (Hu et al., 2007) marker of AHR activation and thus indicates the exposure range where the first biochemical alterations may emerge.
The wide inter‐ and intraspecies sensitivity differences that exist in acute toxicity of TCDD and other dioxins taper off in developmental toxicity, which occurs in a similar dose range across species (Couture et al., 1990). This is also true for the Long‐Evans (Turku/AB) and Han Wistar rat strains, but the abnormalities induced by TCDD vary strain dependently (Huuskonen et al., 1994). The sensitivities to toxicity other than those mentioned above, generally seem to follow the patterns of acute toxicity, based on the somewhat scant data available. For example, TCDD carcinogenicity requires ≥ 1,000‐fold higher doses in hamsters than in Sprague–Dawley rats or sensitive mouse strains (Knerr and Schrenk, 2006). Similarly, Han Wistar rats are approximately 100‐fold more resistant to hepatic tumour promotion by TCDD than Long‐Evans (Turku/AB) rats (Viluksela et al., 2000).
3.1.6.5. Timing and duration of AHR activation
An adequate level of AHR activity, in the right place and at the right time, is beneficial and even essential to the normal development and functioning of the vertebrate organism. For example, studies in AHR‐deficient mouse models have demonstrated that AHR activity is indispensable for the normal development of intestinal lymphoid follicles and a crucial regulator in maintaining intraepithelial lymphocyte numbers in both the intestine and the skin (Kiss et al., 2011; Li et al., 2011). It is further involved in various other physiological processes including liver and vascular development, male and female reproduction, cell cycle and diurnal rhythms and UV radiation‐induced skin tanning (Jux et al., 2011; Dietrich, 2012; Ichihara, 2012; Karman et al., 2012a; Tischkau, 2012). In contrast, sustained activity can be harmful, as evidenced by a mouse model with constitutively active AHR. Based on comparisons of CYP1A1 induction in thymus and liver, the transcriptional activity of the AHR in these mice corresponds to that observed in wild‐type animals one day after a single oral treatment with 3 (thymus) or 0.3 (liver) μg TCDD per kg bw, i.e. fairly low doses (Andersson et al., 2002). The mice showed hepatomegaly and thymus atrophy, two typical TCDD toxicities, and additionally uterus weight was lowered, epididymal sperm count was reduced by 45%, bone stiffness was decreased (only in female mice) and the sizes of B‐lymphocyte subpopulations were altered (Andersson et al., 2003, 2012; Brunnberg et al., 2006; Wejheden et al., 2010). Moreover, these mice develop invasive tumours (hamartomas) of the glandular part of the stomach (close to and with the limiting ridge) at an early age, correlating with increased mortality (Andersson et al., 2002, 2005).
Inappropriate timing of AHR activation can also result in unwanted effects. When exposed to TCDD prenatally (on GD15 or earlier), rat pups showed reduced serum LH and FSH levels due to diminished synthesis of the mRNA for their β‐subunits in the pituitary (Mutoh et al., 2006; Takeda et al., 2009). The lowered pituitary gonadotropin gene expression is specific for the fetus and does not occur in adult rats after either oral or intracerebroventricular injection of TCDD. Furthermore, it imprints persistent defects in sexual behaviour (discernible at adulthood) as well as in the maturation of gonadal tissues (Takeda et al., 2009). Its primary reasons appear to be perpetually declined gonadotropin‐releasing hormone (GnRH) synthesis in the hypothalamus and induction of histone acetylases in the pituitary (Takeda et al., 2012, 2014a). Prenatal exposure to TCDD lowers pituitary gonadotropin mRNA expression also in mice, and this occurs AHR‐dependently in the C57BL/6 and DBA/2 strains (Takeda et al., 2014b) (see also Section 3.1.6.10.3 ).
3.1.6.6. Natural AHR ligands
In addition to xenobiotics such as PCDD/Fs and PAHs (and the microbial metabolites mentioned above), a wide variety of other factors can activate the AHR. However, no single substance has so far stood out as a primary endogenous ligand for AHR.
One of the most potent ligands for the receptor is FICZ (6‐formylindolo[3,2‐b]carbazole), which is readily generated from tryptophan by UV light but also enzymatically (Smirnova et al., 2016; Wei et al., 1999). Also, other tryptophan metabolites, such as kynurenine and kynurenic acid, bind (though weakly) to the AHR, thereby activating it (DiNatale et al., 2010; Nuti et al., 2014). Furthermore, a number of other endogenous and dietary compounds can act as AHR agonists, antagonists or partial agonists (Phelan et al., 1998; Schaldach et al., 1999; Adachi et al., 2001; Savouret et al., 2001; Song et al., 2002; Safe et al., 2012). Although many of these exogenously and endogenously generated ligands were first hypothesised or identified from in vitro studies, increasing strong evidence supports their role as AHR regulators in vivo, especially related to immunological aspects in the skin, lung and gut (Stockinger et al., 2014). However, at present, it is unclear how prominent a role these compounds play in the regulation of AHR signalling elsewhere in the body, as dietary AHR ligands appear to be metabolised rapidly in the intestine and/or the liver during their first pass (Pohjanvirta et al., 2002; de Waard et al., 2008). It should also be noted that chemicals may not always need to be ligands for the AHR to activate its signalling (Gradelet et al., 1997; Anwar‐Mohamed and El‐Kadi et al., 2012). The inhibition of CYP1A1 catalytic activity with a consequent increase in endogenous FICZ levels might be one indirect mechanism of AHR activation (Wincent et al., 2012).
3.1.6.7. Species‐, ligand‐ and gender‐specific gene regulation
As described in the preceding sections, there are functionally critical differences in AHR structure both among and within species. These differences involve either the ligand‐binding or transactivation domain of the AHR and result in, respectively, non‐selective or selective divergencies in sensitivity to the manifestations of TCDD toxicity. Moreover, at the gene expression level in cell studies, the changes brought about by TCDD seem to be largely species‐dependent.
Evidence is further mounting to show that PCDD/Fs and DL‐PCBs do not cause a uniform pattern of effects on gene regulation, either. For example, at TEF‐adjusted equipotent hepatic levels in mice, 2,3,7,8‐TCDF and PCB‐126 were reported to alter the expression levels of only subsets of the genes affected by TCDD (Kopec et al., 2010; Nault et al., 2013). This was also the case for 2,3,7,8‐TCDF and 2,3,4,7,8‐PeCDF in rat primary hepatocytes (Rowlands et al., 2011a). It should be noted that the genes of the AHR battery belong to the ones usually induced by all potent AHR agonists, although even their induction profiles may be distinct (Kopec et al., 2010; Rowlands et al., 2011a; Mahiout et al., 2017).
This ligand dependency extends to other AHR agonists as well. While the human AHR displays some 10‐fold lower affinity to TCDD than its C57BL/6 mouse counterpart, it binds certain compounds such as indirubin with higher affinity than the mouse AHR (Flaveny et al., 2009). PAHs cause AHR‐dependent oocyte apoptosis in mouse ovaries, but TCDD does not (Matikainen et al., 2001). Although 3‐methylcholanthrene induces Cyp1a1 maximally to the same degree as TCDD, it does not produce the same signs of toxicity even after a daily administration of as much as 100 mg/kg bw for 20 days to rats (Poland and Glover, 1974; Neal et al., 1979). Likewise, even as closely related compounds as kynurenine and kynurenic acid elicit different effects (Maaetoft‐Udsen et al., 2012).
In fish, DL‐PCBs have relatively low potency compared with humans, and the relative potencies of PCDF and PCB congeners may widely deviate from the mammalian pattern among bird species (Walker and Peterson, 1991; Kennedy et al., 1996; Abnet et al., 1999; Herve et al., 2010; Hahn and Karchner et al., 2012). The reasons for these differences have not been fully elucidated, but some key factors appear to be ligand stability and affinity, primary structure of the ligand‐binding domain, differential binding of ligands within the ligand‐binding pocket, ligand‐induced unique AHR conformations, and ligand‐specific coregulator recruitment (Riddick et al., 1994; Henry and Gasiewicz, 2008; Zhang et al., 2008; Petkov et al., 2010; Powis et al., 2011; DeGroot et al., 2012; Nuti et al., 2014). On the other hand, nucleotide specificity of DNA binding at DREs seems to be constant for all AHR agonists (DeGroot and Denison, 2014). AHR functional differences may also largely account for the conspicuous diversity in, e.g. histopathological changes caused by TCDD across animal species (Pohjanvirta and Tuomisto, 1994).
TCDD toxicity has also been shown to depend on gender, but without a constant pattern. In TCDD‐sensitive rat strains, males are approximately twofold more resistant than females to liver tumour formation, acute lethality and induction of hepatic microsomal enzymes (Lucier et al., 1973; Kociba et al., 1978; Pohjanvirta et al., 1993). In mice, male animals are 5‐ to 13‐fold more susceptible to the acute lethality of TCDD than females (Pohjanvirta et al., 2012). Males are also the more sensitive gender to TCDD toxicity in guinea pigs (Enan et al., 1998a,b).
3.1.6.8. AHR in relation to human dioxin risk assessment
The studies described above have indicated that AHR binding and activation is the molecular initiating event of TCDD toxicity. They further advocated for the key role of the canonical signalling pathway. At present, it is not clear what the exact natural role of the AHR‐pathway is and what the natural ligand is that activates the pathway. As such, it is difficult to understand how compounds activating this pathway in a non‐physiological way may affect the normal functioning of this pathway. This applies especially for compounds that are not easily degraded and as such may cause a continuous rather than transient stimulation of this pathway.
An important question is if there are differences in sensitivity between humans and the species used for toxicological studies. Human AHR resembles that in the DBA/2 mouse strain in having the same amino acid (V) at the critical site in the ligand‐binding domain that is responsible for the lower TCDD affinity of the AHR in this strain compared with the TCDD‐responsive C57BL/6 mouse AHR, which has A in this position. This renders the TCDD binding affinity of the human AHR also about 10‐fold lower than that of C57BL/6 AHR (Manchester et al., 1987; Ramadoss and Perdew, 2004; Connor and Aylward, 2006). However, simultaneously there exists a 12‐fold variation range in the human population in TCDD binding affinity to the AHR (Nebert et al., 2004; Okey, 2007). Single‐nucleotide polymorphisms (SNPs) are known to exist in human AHR, primarily localising in the transactivation domain, but it is not known whether any of these are associated with altered susceptibility to PCDD/Fs and DL‐PCBs (Okey, 2007; Celius and Matthews, 2010; Rowlands et al., 2010). Recently, it was reported that an SNP at the human AHR promoter appears to influence AHR expression levels and may therefore modify sensitivity to PCDD/Fs and DL‐PCBs (Liu et al., 2015). As to the functional partner proteins of the AHR signalling pathway, none of the established SNPs in the structures of human ARNT, HSP90 and AIP are predicted to markedly increase this sensitivity (Rowlands et al., 2011b; Urban et al., 2011, 2012). Of greater functional consequence, however, might be an alternative exon of ARNT, which has been shown to contribute to malignancy in human lymphoid cell lines (Gardella et al., 2016), and a variant of AHRR, which can result in enhanced inducibility of CYP1A2 (and thereby increase the risk of some cancers), and which may be common in certain populations (Cavaco et al., 2013; Hung et al., 2013). AHRR mutation has further been reported to correlate with advanced endometriosis in humans (Wu et al., 2012).
The CONTAM Panel decided to apply the current TEF scheme established by experts under coordination of WHO in 2005 (Van den Berg et al., 2006). Nevertheless, it is important to be aware of potential differences in relative potencies and these TEFs, especially when interpreting the outcomes of epidemiological studies. The current TEF values for PCDD/Fs and DL‐PCBs are primarily based on in vivo and in vitro rodent toxicity assays. Furthermore, most studies do not include critical endpoints like sperm production and pubertal onset following exposure of the mother. Only one study analysed the consistency of the TEQ concept using a rat developmental toxicity study design and a mixture of six PCDD/Fs and three DL‐PCBs (Hamm et al., 2003). It was concluded that effects of the mixture were observed at a TEQ dose within a factor 2 compared to TCDD, the lower response being caused by lower accumulation of certain congeners in tissues. Combined data from various in vitro assays on human cells with in silico models showed that the human cell models responded only weakly to DL‐PCBs, based on endpoints like EROD induction and mRNA levels of CYPs 1A1 and 1B1, and the AHRR (Larsson et al., 2015). As shown in studies using human lymphocytes, primary keratinocytes, primary hepatocytes and HepG2 hepatoma cells (Silkworth et al., 2005; van Ede et al., 2014; Larsson et al., 2015; van Ede et al., 2016), PCB‐126 proved to be ≥ 30–fold less potent than suggested by its WHO2005‐TEF value. In addition, these studies showed that several PCDD/Fs (2,3,4,7,8‐PeCDF, 1,2,3,4,7,8‐HxCDF, 1,2,3,4,6,7,8‐HpCDD, 1,2,3,4,7,8,9‐HpCDF) could be more than 10‐fold more potent than implied by their TEFs. It should be stressed that differences in kinetics were not included in these studies, since the main aim was to evaluate the toxic potential of levels measured in blood and tissues. This may be especially relevant for the two HpCDD/Fs that show a lower absorption and different distribution in the body compared with lower chlorinated congeners (see Section 3.1.1.3).
The CONTAM Panel noted that these studies are only based on a few endpoints, which are suitable to be determined in human cells but seem not directly related to the critical effects. Furthermore, mice also appear to be much less sensitive for PCB‐126 for these endpoints (induction of EROD activity and CYP1A mRNA in liver), whereas for effects on the immune system, the potency of PCB‐126 is according to the TEF value or even higher (Haws et al., 2006). The question arises whether this also applies to other critical adverse effects like those on sperm production, and whether similar might be the case for humans. Wimmerová et al. (2016) studied correlations between relative potencies (REPs) of PCDD/Fs and DL‐PCBs for thyroid impacts (thyroid volume and free T4) and those for CYP1A1 and CYP1B1 gene expression in human peripheral blood mononuclear cells. The authors concluded that while CYP1A1 seems well‐suited for toxicity evaluation of PCDD/F mixtures, CYP1B1 is more apt for that of PCDF/PCB mixtures.
3.1.6.9. AHR in relation to farm and companion animals risk assessment
With regard to farm and companion animals, AHR structural variations play a notable role in the risk assessment for PCDD/Fs and DL‐PCBs in the case of birds and some fish species. While most mammals have only a single AHR, birds possess at least two (AHR1 and AHR2), of which AHR1 is probably the dominant subtype (Yasui et al., 2007). Differences in the ligand‐binding domain structure similar to those in mouse strains described above (see Section 3.1.6.4) may broadly explain why chickens are more sensitive to dioxin toxicity than turkeys, which in turn are more sensitive than Japanese quail or ducks (Head et al., 2008). In addition, this order of sensitivities is also reflected in CYP1A1 induction in primary hepatocyte cultures (Kennedy et al., 1996). Fish are endowed with at least five classes of AHR‐related genes (Hahn et al., 2017). In the common model fish species, zebrafish and killifish, AHR2 is the predominant AHR form regulating the response to TCDD and DL‐PCBs in embryos (Hahn and Karchner et al., 2012). However, in white sturgeon, both AHRs (AHR1 and AHR2) appear to be highly responsive to dioxin‐like compounds, exhibiting in in vitro reporter gene assays lower EC50 concentrations for TCDD than any other AHRs of vertebrates tested so far. Furthermore, because of species‐specific differences in relative potencies of PCDD/Fs and DL‐PCBs, the current TEFs for fish may underestimate by 10‐fold the toxicity, based on TEQ levels, in white sturgeons (Doering et al., 2014). Similar limitations of the salmonid‐based TEFs have been reported for other fish species (Rigaud et al., 2014).
3.1.6.10. Mode of action for specific toxicity outcomes
In previous sections, the involvement of the AHR in the initial molecular mechanisms of action of PCDD/Fs and DL‐PCBs is described. However, the precise complex sequences of key events which lead on to different toxic endpoints and pathological outcomes in vivo are still unclear. Understanding these is of high importance for consideration of relevance of observed effects in animals to humans and for the overall risk assessment.
The CONTAM Panel decided to describe only the modes of action for the most relevant endpoints based on the data available, like reproductive toxicity, effects on the thyroid and tooth development. It was decided to also describe the current knowledge on chloracne, carcinogenicity and immunotoxic effects, although these effects were not considered for the risk assessment on humans.
3.1.6.10.1. Chloracne
Although there have been many reports of chemicals causing chloracne and the clinical aspects are clearly documented, the mechanisms are still to be completely understood, but seem to implicate activation of skin stem cells and shifts in the differentiation of progeny (Panteleyev and Bickers, 2006; Bock, 2016). In a patient with severe chloracne, focal dermal CYP1A1 protein was highly induced with sustained atrophy of the sebaceous glands and marked changes in gene expression associated with the AHR signalling pathway (see Section 3.1.6.2), lipid metabolism, morphogenesis and inflammation (Saurat et al., 2012). LIGR1 expressing multipotent progenitor cells, believed to be involved in the turnover of sebaceous gland cells, appear to be the target of TCDD in a mouse model (Fontao et al., 2017).
3.1.6.10.2. Carcinogenicity
PCDD/Fs and DL‐PCBs cause tumours in experimental animals at multiple sites (Knerr and Schrenk, 2006). They have also been associated with increased incidence of cancer in humans exposed either occupationally, accidentally or even to environmental levels at higher ranges but this can be contentious (IARC, 1997; Boffetta et al., 2011) (see Section 3.1.4.11).
Experimentally, the evidence is robust that the development of cancer caused by PCDD/Fs and DL‐PCBs in rodents is not due to direct genotoxicity (see Section 3.1.3). Instead, the working hypothesis is that PCDD/Fs and DL‐PCBs should be considered as non‐genotoxic promoters of carcinogenesis probably by a receptor mediated mechanism. Rodent studies using a two‐stage initiation/promotion approach showed TCDD to be a potent promoter of skin, ovarian and liver cancer following initiation by classic genotoxic agents. For instance, TCDD is a promoter of dermal papilloma formation initiated by administration of N‐Methyl‐N′‐nitro‐N‐nitrosoguanidine to hairless mice (Poland et al., 1982). Many studies have demonstrated quantitatively the ability of PCDD/Fs and DL‐PCBs to promote rat liver carcinogenesis initiated by diethylnitrosamine (DEN) both by the quantitation of tumour numbers and markers of preneoplastic foci (Pitot et al., 1980; Dragan and Schrenk, 2000).
As with other toxicity endpoints, the MOA of PCDD/Fs and DL‐PCBs causing tumours invokes binding and activation of the AHR which is followed by translocation to the nucleus, binding to DREs and modulation of gene transcription. Mice with a low affinity AHR for PCDD/Fs show resistance to DEN‐initiated hepatocarcinogenesis promoted by TCDD (Kennedy et al., 2014) but comparative carcinogenesis studies on Ahr‐null rats and mice have not been reported. It is envisaged that for rodent liver tumours sustained near‐maximal activation of AHR with a persistent range of detrimental gene expression (for a major proportion of the life span) is the pivotal molecular initiating event or first key event (Budinsky et al., 2014; Becker et al., 2015). Subsequent key events have been considered as shifts in altered focal cell proliferation and apoptosis, followed by histological changes leading to hyperplasia albeit susceptible to other modulating factors. PCDD/Fs and DL‐PCBs may induce a variety of responses associated with deregulation of cell–cell communication invoking downregulation of proteins of gap junctions, adherens junctions and desmosomes such as connexin 43, E‐cadherin, plakoglobin and β‐catenin (Vondráček and Machala, 2016). There is evidence that these latter Key Events are quantifiable and dose‐dependent but are probably subject to thresholds at lower doses (Becker et al., 2015). The effect of TCDD on apoptosis seems to be p53 dependent (Becker et al., 2015). Cell proliferation induced by PCDDs can be dose‐ and time dependent. In the liver, histological changes may be linked to regeneration following hepatotoxicity.
Despite key events being defined for rodent liver carcinogenesis, the pertinent underpinning molecular events associated with gene transcription after AHR activation leading to early stages of cancer are still to be elucidated (Becker et al., 2015). Many biochemical processes have been explored, including the role of oxidative stress and oestrogens but no clear pathway has been demonstrated. It should be noted that many of the applied in vivo models for studying cancer include initiation by a genotoxic agent. AHR‐dependent promotion by TCDD of DEN‐initiated hepatocarcinogenesis in mice has been shown to be dependent upon IL‐1‐like cytokine receptor signalling pathways. This supports a link between AHR activation and acute inflammatory liver toxicity and tumour promotion by TCDD (Kennedy et al., 2014).
3.1.6.10.3. Immunotoxicity
TCDD and also various PCDD/Fs and DL‐PCBs have shown to cause immunotoxic effects in laboratory animals. For instance, a single dose of TCDD in the low μg/kg bw range suppresses both antibody‐ and cell‐mediated immune responses to a wide variety of model antigens and foreign proteins in mice, indicative of increasing susceptibility to a number of infectious agents (Kerkvliet, 2012).
Cells that are components of the immune system express various levels of the AHR (reviewed by Esser and Rannug, 2015). The AHR has proven to be a critical regulator of the immune system (mainly studied in mice). AHR signalling dampens inflammation, and AHR‐deficient mice are hypersensitive to endotoxin (Sekine et al., 2009; Bessede et al., 2014). AHR activity has also been shown to be indispensable for the normal development of intestinal lymphoid follicles and a crucial regulator in maintaining intraepithelial lymphocyte numbers in both the intestine and the skin, critical barrier tissues (Kiss et al., 2011; Li et al., 2011). In the skin, the AHR is activated by metabolites produced by Malassezia yeasts (Gaitanis et al., 2008; Magiatis et al., 2013), which occur on the skin of all humans, and by UV radiation (Navid et al., 2013). In the intestine, AHR signalling is a key to sustaining a balance between protection against infection and tolerance to harmless antigens. AHR activation by dietary AHR agonists triggers intestinal innate lymphoid cells to secrete IL‐22, which stimulates mucosal epithelial cells to generate antimicrobial peptides (Lee et al., 2012). These peptides inhibit colonisation of e.g. C. albicans but not that of lactobacilli (Zelante et al., 2013). Deficiency of dietary AHR agonists or their precursors such as tryptophan reduces AHR activity and the production of these peptides, resulting in aggravated gut irritability and colitis (Hashimoto et al., 2012). In support of this, AHR‐null mice showed exacerbated experimental colitis, whereas AHR agonists, including TCDD, attenuated it in AHR wild‐type mice (Benson and Shepherd, 2011; Furumatsu et al., 2011; Singh et al., 2011). The relationship between microbiota and AHR is bi‐directional: Microbes produce AHR activating metabolites (Cheng et al., 2015; Sridharan et al., 2014) while AHR activity helps maintain commensal microbial composition in the lumen (Asselin and Gendron, 2014).
Among the Th‐cell subsets, especially Th17 cells express AHR at high levels, and AHR signalling seems to fine‐tune the balance between Treg and Th17 cells, thereby regulating autoimmune reactions (Esser, 2012). AHR agonists can shift the balance between inflammatory, autoimmune‐prone Th17 responses and immunosuppressive responses driven by Tregs (Esser and Rannug, 2015). The activation of the AHR with TCDD led to the differentiation of murine naïve CD4+ T cells into FoxP3+ Treg, while also enhanced IL‐17 and IL‐22 expression was observed in murine naïve CD4+ T cells cultured under Th17 conditions (reviewed by Van Voorhis et al., 2013). IL‐22, a member of the IL‐10 cytokine family, is important in microbial immunity. Sherr and Monti (2013) showed AHR‐dependent control of B cell differentiation from the haematopoietic stem cell through the pro‐B cell, mature B cell and antibody‐secreting plasma cell stages, explaining eventual suppression of humoral immune responses by TCDD (Sulentic and Kaminski, 2011).
Earlier work pointed to effects of TCDD and compounds such as DL‐PCBs at the thymic level, including effects on thymus epithelium (De Heer et al., 1994) and inhibited thymocyte maturation and reduced expression of thymic major histocompatibility complex molecules (Holladay, 1999), possibly caused by AHR‐dependent nuclear translocation of NF‐κB and expression of Fas ligand in thymic stromal cells, resulting in apoptosis in T cells (Camacho et al., 2005).
Also effects on bone marrow and haemopoiesis were indicated (reviewed by Kerkvliet, 2002). The AHR normally curbs excessive proliferation of haematopoietic stem cells; inappropriate stimulation of AHR activity may compromise the ability of these cells to balance quiescence, proliferation, migration, and differentiation (Gasiewicz et al., 2014). Also in the periphery, information suggests the inappropriate activation of cells, leading to anergy or death, and the consequent premature termination of the immune response. Enhanced activation of B cells, dendritic cells, and CD4+ T cells by TCDD has been described as well as the earlier disappearance of the latter two populations from peripheral lymphoid organs (reviewed by Kerkvliet, 2002).
Apart from the role of the AHR on various cells that are components of the immune system, the specific underlying biochemical mechanisms by which TCDD and related compounds disrupt immunological functions remain unclear. Zordoky and El‐Kadi (2009) found that in cells of the immune system, AHR agonists repress or transactivate NF‐κB response genes, while on the other hand NF‐κB activators such as the inflammatory cytokines IL‐1b, IL‐6, TNFα and lipopolysaccharide suppress constitutive and AHR‐induced CYP1A1, suggesting a link between inflammation, oxidative stress and CYP expression.
Taken together, the current information suggests that TCDD and other AHR agonists such as PCDD/Fs and DL‐PCBs, after interaction with the AHR, involve NF‐κB‐dependent pathways that may lead to dysregulation of components of the immune system, and eventually lead to adverse effects such as repressed resistance to infections or effects on autoimmune phenomena.
3.1.6.10.4. Reproductive toxicity
The normal functioning of both male and female reproductive systems is critically dependent on the tightly regulated feedback system conducted by the hypothalamus‐pituitary‐gonad axis. The AHR has been detected at all levels of this axis providing thus multiple ways for dioxins to interfere with and disrupt reproduction physiology (Karman et al., 2012b). PCDD/Fs and DL‐PCBs also alter thyroid hormone balance, which can indirectly lead to the same outcome.
The effects of PCDD/Fs and DL‐PCBs on reproductive organs and their regulatory systems depend on the timing of exposure, whether it occurs during development or at adult age. In neonatal rat brain, the Ahr gene is expressed in virtually all GABA/glutamatergic neurons also containing oestrogen receptors in the anteroventral periventricular nucleus of the hypothalamus (Hays et al., 2002). These neurons are there important for regulation of the gonadotropin‐releasing hormone neurons and thus for puberty onset in both genders as well as for ovulation in the female (Clarkson and Herbison, 2006).
Gestational TCDD exposure (1 μg/kg) abolishes the gender difference in anteroventral periventricular nucleus expression of glutamic acid decarboxylase 67, a key enzyme in GABA synthesis (Hays et al., 2002). This probably bears on the findings that female rats exposed in utero to TCDD have alterations in their reproductive functions at adult age including early puberty, constant oestrus, and premature reproductive senescence (Gray and Ostby, 1995). Male offspring, in turn, exhibit delayed puberty and a feminine pattern of gonadotropin release consisting of an LH surge in response to oestradiol and progesterone in castrated adult rats. In addition, their sexual behaviour is feminised and daily sperm production may be reduced (Mably et al., 1992a,b; Bjerke and Peterson, 1994). The demasculinisation of male pups can be due to the prevention by TCDD of perinatal LH and testosterone surges (Mably et al., 1992c; Mutoh et al., 2006) (see Section 3.1.6.5). This impact of TCDD involves depressed expression of the gene for the β‐subunit of LH in the pituitary (Takeda et al., 2012). Subsequently, the suppression of perinatal LH surge results in permanently reduced GnRH gene expression in the hypothalamus (Takeda et al., 2014a). Similar effects may be linked to delayed puberty and decreased sperm production and motility observed in humans (Korrick et al., 2011; Mocarelli et al., 2008, 2011; Burns et al., 2016; Mínguez‐Alarcón et al., 2017) (see Section 3.1.4.3)
A lowered boy‐to‐girl ratio has been reported in the progeny of men exposed to TCDD and to a lesser extent also in Yucheng victims (see Section 3.1.4.3.3). Hsu et al. (2016) observed that 8% of a group of male Yucheng patients showed an X/Y sperm ratio higher than 1.4, which was not observed in controls. Repeated treatment of Holtzman rat dams (F0 generation) with TCDD (loading dose 400 ng/kg bw prior to mating, followed by weekly maintenance doses of 80 ng/kg during mating, pregnancy and the lactation period) led to a reduced ventral prostate weight in F1 rats and a markedly decreased male/female ratio in their F2 offspring (Ikeda et al., 2005). A dose‐dependently reduced male/female ratio was recorded in ICR mice when male mice were weekly treated with TCDD prior to mating (Ishihara et al., 2007). In mice, TCDD failed to affect the Y‐bearing/X‐bearing sperm ratio. However, at two‐cell embryo stage, the proportion of male embryos was significantly lower in the TCDD group than in the controls, suggesting a diminished ability of Y‐bearing sperm to conceive the ova (Ishihara et al., 2010).
In adult male Wistar rats and marmoset monkeys, a fairly low single dose of TCDD (3 μg/kg bw) has been reported to decrease the intercellular contact between Sertoli cells or between Sertoli and neighbouring germ cells, along with sloughing off of immature spermatids into the tubular lumen (Rune et al., 1991a,b; Chahoud et al., 1992). This could be due to TCDD‐induced oxidative stress (Wong and Cheng, 2011). Of note, in mice harbouring constitutively active AHR, epididymal sperm count is reduced by 45% (see Section 3.1.6.5). In marmosets, early cessation of maturation in spermiogenesis was also recorded. As to Leydig cells, histochemical evidence of impaired steroid synthesis was obtained in both species already at 1 μg/kg bw. In another study on Sprague–Dawley rats, a reduction in Leydig cell volume was found 4 weeks after a single dose of 12.5 μg/kg bw TCDD (Johnson et al., 1992). TCDD may also suppress testosterone synthesis in adult male rats by inhibiting the LH‐stimulated mobilisation of cholesterol to cytochrome P450scc, which converts cholesterol to pregnenolone (Moore et al., 1991). Evidence of effects of PCDD/Fs on semen quality has also been reported in epidemiological studies (see Section 3.1.4.3.1).
In adult female mice, the AHR influences ovarian follicle growth, e.g. by promoting proliferation of granulosa cells and regulating their aromatase expression (the enzyme that converts testosterone to oestradiol); it further modifies ovulation (Karman et al., 2012a). These findings are in keeping with the reported irregular oestrous cycles and reduced ovulatory rate in adult rats treated with TCDD, which are characterised by prolonged dioestrus (Li et al., 1995) or by persistent oestrus or dioestrus (Franczak et al., 2006). Moreover, TCDD dose dependently downregulates uterine oestrogen and progesterone receptors (both cytosolic and nuclear) in female rats (Romkes and Safe, 1988). Chronic exposure to TCDD also brings about premature reproductive senescence in female rats due to endocrine disruption, foremost diminished circulating oestradiol levels (Franczak et al., 2006; Valdez et al., 2007). This may emanate from repressed ovarian expression of Cyp17a1, whose product is an important enzyme in the oestrogen biosynthesis pathway (Valdez et al., 2009), and from induction of xenobiotic‐metabolising enzymes (Martucci and Fishman, 1993; see Section 3.1.6.2). In human luteinising granulosa cells in vitro, TCDD reduced oestradiol secretion and decreased aromatase mRNA expression at extremely low concentrations (down to fM) (Baldridge et al., 2015). This may be involved in dose‐related increases in time‐to‐pregnancy and infertility associated with individual serum TCDD levels in women from Seveso as reported by Eskenazi et al. (2010).
3.1.6.10.5. Effects on thyroid hormones and the thyroid gland
TCDD decreases serum T4 concentrations within 24 h in rats (Jones et al., 1987b). Total and free T4 usually exhibit parallel changes (Potter et al., 1983, 1986; Gorski and Rozman, 1987). In short‐term studies, both the duration and magnitude of T4 reduction are partly dependent on the dose, with lethal doses causing an irreversible decline of 50–70% prior to death compared with controls (Potter et al., 1986; Gorski and Rozman, 1987; Pohjanvirta et al., 1989). The effect of TCDD on serum T3 is much less consistent with increased (Potter et al., 1986), decreased (Pazdernik and Rozman, 1985; Rozman et al., 1985), and unaltered levels (Potter et al., 1983; Rozman and Greim, 1986; Gorski and Rozman, 1987; Jones et al., 1987b) having been reported in rats. Changes in free T3 appear to be negligible (Gorski et al., 1988). In other laboratory animal species, such a drastic drop in T4 is usually not seen after TCDD exposure. In hamsters, T4 concentration is persistently elevated by TCDD (Henry and Gasiewicz, 1987), and may also be increased at high doses and at late phases of acute toxicity in mice (Birnbaum et al., 1990) and guinea pigs (McKinney et al., 1985). After in utero exposure, however, serum T4 is lowered in TCDD‐exposed mice, and in an AHR‐dependent manner (Nishimura et al., 2005). As to TSH levels in TCDD‐exposed animals, they are either elevated (Potter et al., 1986; Pohjanvirta et al., 1989) or unchanged (Gorski and Rozman, 1987; Henry and Gasiewicz, 1987; Gorski et al., 1988) in rats. Of note, they are also elevated in hamsters (Henry and Gasiewicz, 1987).
A major mechanism by which TCDD brings about the decrease of T4 in rats is its accelerated hepatic clearance through biliary excretion due to induction of UGT (especially UGT1A) activity (McKinney et al., 1985; Rozman et al., 1985; Pohjanvirta et al., 1989, 1990b; Martin et al., 2012). UGT is also induced in hamsters despite their opposite response in serum T4, but the absolute UGT activity towards T4 is 3‐4 times lower than in rats (Henry and Gasiewicz, 1987). On the other hand, 5’‐deiodinases I and II, which are responsible for conversion of T4 to T3 in a tissue‐specific manner (Larsen et al., 1981), do not appear to be involved in TCDD's effects on thyroid hormone levels (Viluksela et al., 2004). As to the influence of TCDD on thyroid hormone receptors (THRs), the data are conflicting. While TCDD was reported to competitively and dose‐dependently inhibit the binding of T3 to THRs (Jung et al., 2007), another study found TCDD to augment THR‐mediated responses to T3 (Yamada‐Okabe et al., 2004). Overall though, as assessed by a variety of endpoints, TCDD‐treated rats appear to be functionally euthyroid (Potter et al., 1986; Kelling et al., 1987; Roth et al., 1988).
The thyroid gland itself may be morphologically affected by TCDD. A sublethal dose of TCDD has been reported to diminish follicular colloid in the rat thyroid gland and produce papillary projections into the follicular lumen, suggestive of high tissue activity (Gupta et al., 1973). Follicular cell hypertrophy without adenomas or carcinomas of these cells has also been found in rats after chronic exposure to TCDD (Yoshizawa et al., 2010). On the other hand, an acutely lethal dose may distend thyroid follicles and flatten follicular epithelium, characteristic of dampened functional activity (Rozman et al., 1986). Moreover, wide interindividual variation in thyroid architecture, regardless of TCDD dose, has been observed in rats (Potter et al., 1986), which probably contributes to the negative histopathological findings in other studies (Potter et al., 1986; Pohjanvirta et al., 1989),
While DL‐PCBs by and large modify thyroid hormone balance the same way as TCDD (decrease serum T4 by inducing hepatic UGT1A activity), they also appear to have some specific effects. Foremost, their hydroxylated (and, possibly, sulfated) metabolites may compete with T4 for binding to transthyretin (which transports thyroxine and retinol) in serum, providing an alternative or contributing mechanism of action for T4 reduction in rats (Kato et al., 2004; Grimm et al., 2013). As adult rats are virtually deficient of thyroxine‐binding globulin (Rouaze‐Romet et al., 1992), transthyretin is their major T4 carrier, and thyroid hormone balance is much more vulnerable in them than humans in which transthyretin plays only a minor role (~ 10%) as T4 transporter. Moreover, basal T4 glucuronidation rate appears to be some 10‐fold higher in rats than humans (Richardson et al., 2014).
PCB‐118 was shown to cause inflammation and stimulate the production of Il‐6 and TNF‐α in rat thyroid gland, at least partially through the c‐Jun N‐terminal kinase (JNK) pathway (Xu et al., 2016). In Fischer rat thyroid cell line‐5 (FRTL‐5) cells, PCB‐118 decreased cell viability and the sodium‐iodide symporter (NIS; catalyses the active accumulation of iodide in thyroid epithelial cells) protein abundance through the protein kinase B (Akt)/forkhead box protein O3a (FoxO3a) signalling pathway (Yang et al., 2015). In cultured human thyroid epithelial cells, concentrations of PCB‐118 that did not affect cell viability reduced thyroglobulin and T4 concentrations and increased Akt mRNA and protein levels while decreasing NIS mRNA and protein levels (Guo et al., 2015a). Thus, human thyroid might be affected by dl‐PCBs through the same pathway as rat thyroid. NIS mRNA expression was also diminished by TCDD and PCB‐126 in a primary porcine thyrocyte culture. These compounds further diminished the activity of cathepsin B, which is responsible for partial proteolysis of thyroglobulin in thyrocytes (Pocar et al., 2006).
Evidence has been provided that in rats, induced CYP1A1 can generate such hydroxylated metabolites of the mono‐ortho PCB‐105 and PCB‐118 that are directly capable of activating the THR (Gauger et al., 2007; Giera et al., 2011). A mixture of non‐ortho‐ and mono‐ortho PCBs may thereby potentiate the effects of its individual congeners in tissues where CYP1A1 is inducible. On the other hand, hydroxylated metabolites of mono‐ortho PCBs have also been shown to be capable of suppressing THR‐mediated transcription in vitro (Miyazaki et al., 2008). These interactions might account for the unexpected finding that whereas PCB‐126 and PCB‐118 individually increased or had no effect, respectively, on serum T3 at an interim evaluation in a chronic exposure study in rats, their co‐exposure led to a significant decrease in serum T3 (Yoshizawa et al., 2010).
3.1.6.10.6. Effects on teeth development
Tooth defects have proven to be among the most sensitive responses to TCDD in vivo (see Section 3.1.4.10). In utero and lactational exposure of rat pups reduced the size or prevented the development of their third lower molars (line‐ and dose‐dependently) at single doses of 0.03 or 0.1 μg TCDD/kg bw and higher when the dams were treated on GD15 (Kattainen et al., 2001). The defective development of molar teeth is associated with their increased susceptibility to caries. After a single dose of TCDD on GD15, cariogenic lesions were detectable in the enamel of pup molars already at the lowest maternal dose tested, 0.03 μg/kg (Miettinen et al., 2006).
In particular, TCDD interferes with mineralisation of the dental matrices in developing teeth. The outcome is dependent on species, strain, dose and timing. The most critical window of sensitivity appears to be during the early morphogenesis, from tooth initiation to the early bud stage, after which the sensitivity substantially decreases. In rats and mice, the third molar tooth develops last (in rats, the first morphological signs of the first, second, and third molars appear on GD13, GD14–15, and GD20, respectively (Shellis and Berkovitz et al., 1981)) and is probably therefore more vulnerable than the continuously erupting incisors (see below) for TCDD's impacts. For example, in rats, a single dose of 1 μg/kg bw to the dam resulted in the absence of third molars in the pups exposed both in utero and via lactation, and the frequency of missing molars was the greater the earlier the dam was exposed: 100%, 88% and 50% of the offspring exposed on GD11, GD13 and GD19, respectively. All third molars developed in offspring of dams exposed on PND0, PND2 or PND4 (Miettinen et al., 2002).
At a higher dose of TCDD, the development of third molars can be prevented even postnatally in rats. After a single dose of 50 or 1,000 μg TCDD/kg bw to lactating dams of Han Wistar rats on day 1 after delivery, about half the pups lacked one or more third molars by day 22. The root tips of the more advanced first and second molars were prematurely closed and root formation was arrested, but eruption was not affected (the crowns appeared normal). Moreover, there was total lack of mineralisation in the third molar cusps (Lukinmaa et al., 2001).
A further analysis of these study data disclosed that the first and second molars of the exposed pups, but not controls, exhibited retention of enamel matrix and abnormally thick predentin. Immunostaining for AHR and CYP1A1 in ameloblasts and odontoblasts (which produce enamel and dentin, respectively) was reduced, suggesting that TCDD affects dentin mineralisation through the AHR (Gao et al., 2004). Both AHR and ARNT are expressed during early tooth development with the expression becoming intense in secretory odontoblasts and ameloblasts (Sahlberg et al., 2002). However, AHR may not be the only determinant in this respect. Inbred mouse strains, all harbouring high‐affinity Ahr alleles, exhibited differential responses in third molars to TCDD after in utero exposure, suggesting that there is a genetic component, beyond the Ahr gene, mediating the effects of TCDD on molar development (Keller et al., 2007, 2008).
In addition to molars, TCDD has been reported to afflict the continuously erupting incisor teeth in rats. A single extremely high dose of 1,000 μg/kg bw to adult TCDD‐resistant Han Wistar rats and a 20‐week repeated exposure to total doses of 17 or 170 μg/kg bw in Han Wistar and TCDD‐sensitive Long‐Evans (Turku/AB) rats led to odontoblastic and pulpal cell death and the consequent arrest of dentin formation. The pulp was often lingually exposed. Furthermore, the post‐secretory enamel organ underwent precocious squamous metaplasia with pronounced proliferation. In the latter study, Han Wistar and Long‐Evans (Turku/AB) rats were equally affected, which established this effect to be of type I (i.e. independent of the structural variation in AHR transactivation domain in these model strains) (see Section 3.1.6.4) (Alaluusua et al., 1993; Kiukkonen et al., 2002).
In vitro studies on mouse embryonic teeth have revealed that TCDD blocks mouse molar tooth development by enhancing apoptosis in the dental epithelium if the exposure starts at the initiation stage, while exposure at later stages results in a smaller tooth size and deformation of cusps (Partanen et al., 2004). Both stem cells and EGFR signalling appear to be involved in the tooth effects of TCDD. Twice‐a‐week administration of TCDD (1 or 5 μg/kg bw) for 5 weeks to Sprague–Dawley rats (starting on PND6) inhibited the proliferation and odontogenic differentiation of stem cells of the apical papilla. Concomitantly, TCDD reduced molar tooth root length (Guo et al., 2015b). When molar teeth of mouse embryos were exposed to TCDD in vitro, depolarisation of odontoblasts and ameloblasts and the consequent failure of dentin matrix mineralisation, enamel matrix deposition and cusp formation was found in wild‐type and heterozygous mouse teeth, whereas in EGFR‐deficient mice only the cuspal contour was slightly modified. Moreover, supplementation of the culture medium with EGF prevented most of the adverse effects of TCDD in teeth expressing EGFR (Partanen et al., 1998). In another in vitro study on mouse embryonic mandibular molars, the high concentration of TCDD used (1 μM) specifically reduced or prevented the expression of the dentin sialophosphoprotein gene (whose product is required for normal mineralisation of teeth) in secretory odontoblasts and decreased it in presecretory ameloblasts, providing a conceivable explanation for the retardation of dentin mineralisation by TCDD in vivo (Kiukkonen et al., 2006).
In addition to rats and mice, developmental tooth abnormalities after TCDD exposure have also been reported in rhesus monkeys (Yasuda et al., 2005) and humans (see Sections 3.1.2.2 and 3.1.4.10). As to other congeners, the rank order of relative potencies of 1,2,3,7,8‐PeCDD, 1,2,3,4,7,8‐HxCDD and 1,2,3,4,6,7,8‐HpCDD in producing pulpal perforations in rat incisor teeth in vivo were found to be in agreement with their TEF values (Simanainen et al., 2002). Studies on DL‐PCBs and PCDFs in teeth were not identified.
3.1.7. Consideration of critical effects and dose–response analysis for the human risk assessment
In order to identify critical studies for the new risk assessment, the CONTAM Panel decided to use the previous assessment by the SCF as a starting point. The current TWI of 14 pg TEQ/kg bw per week (SCF, 2001) was based on studies in rats showing decreased sperm counts and decreased anogenital distance, both in male offspring of dams treated with TCDD. These effects had been observed in a number of other studies, in addition to a series of other effects like endometriosis and cognitive effects in monkeys and effects on the immune system of offspring of treated rats. For the study on reduced sperm count (Faqi et al., 1998), the body burden corresponding to the LOAEL was estimated to be 40 ng/kg bw, for the study showing decreased anogenital distance (Ohsako et al., 2001), a NOAEL body burden of 20 ng/kg bw was estimated. Using a one‐compartment model (half‐life of 7.6 years and absorption fraction of 0.5), the estimated human daily intake resulting in a body burden of 40 ng/kg bw was calculated to be 20 pg/kg bw per day. Applying a total UF of 9.6 (factor 3.2 for toxicokinetic differences between humans, and 3 to extrapolate the LOAEL to a NOAEL), this was translated into a TDI of 2 pg/kg bw per day, and extended to a TWI of 14 pg/kg bw per week. The Ohsako et al. (2001) study, showing a NOAEL body burden of 20 ng/kg bw and requiring only a UF of 3.2, would have resulted in a 1.5‐fold higher TWI.
Since that assessment by the SCF, a large number of valuable animal studies have been performed with various critical endpoints but also many intermediate endpoints that help understanding the mechanism behind the adverse effects observed in animals and humans. In addition, new results from epidemiological studies have been published that may be used as the basis of the risk assessment. The CONTAM Panel decided to base its assessment on human studies but to also evaluate the relevant studies in experimental animals in support of the epidemiological studies, including the derivation of a possible HBGV based on these studies. In this way also the effect of applying kinetic instead of one‐compartment modelling will be demonstrated.
3.1.7.1. Critical effects in epidemiological studies
Based on a thorough review of the literature, the CONTAM Panel noted that since the assessment by the SCF (2001) many studies have been performed to study the effects of PCDD/Fs and DL‐PCBs in humans using a number of different endpoints (see Section 3.1.4). In some of these studies, the incidence (or prevalence) in the exposed group was compared to those in a control group, in others exposure‐response relations were studied within the same study population, e.g. by comparing quantiles of exposure.
A number of effects were associated with exposure to PCDD/Fs and DL‐PCBs. Associations with exposure were reported for developmental male reproductive endpoints such as impaired semen quality and later onset of puberty in boys, and other developmental outcomes as lower sex ratio (boys to girls) after exposure of boys/men. Furthermore, increased TSH levels in newborns after in utero exposure were observed, as well as impaired teeth development in boys and girls after perinatal exposure. Increased exposure has also been associated with all cancers combined but not specific tumours. These endpoints are further discussed in Sections 3.1.7.1.1 to 3.1.7.1.5 below.
Chloracne caused by PCDD/Fs is considered a specific adverse effect in humans, observed in various incidents. However, it occurs at levels of PCDD/Fs far higher than background exposure (see Section 3.1.4.2) and is not further discussed.
3.1.7.1.1. Developmental male reproductive endpoints
The developing male reproductive organs are sensitive targets for TCDD both in experimental animals (see Section 3.1.2.1) and in humans. Although the sequence of developmental events and maturation of the male reproductive system is similar in mammals, there are important differences between rodents and humans in terms of control and timing of morphological and hormonal events (reviewed by Picut et al., 2018).
After birth, the influence of oestrogens from the placenta disappears, releasing the negative feedback on the hypothalamic–pituitary–gonadal axis. The hormonal activity of the testes and the hypothalamic–pituitary axis is high during the first few months after birth (until approximately 6 months in boys), a period often referred to as the mini‐puberty. In boys, the levels of FSH and LH are high during the first three months of life. The Leydig cells activated by LH proliferate and produce testosterone, with a peak between one and three months of life. Induced by FSH, the Sertoli cells proliferate and the Inhibin‐B production increases. To some extent, the gonocytes also proliferate in this period. After the mini‐puberty, the hypothalamic–pituitary–gonadal axis is quiescent until the onset of puberty, by a poorly understood mechanism. The prenatal and early postnatal period as well as the puberty are sensitive to endocrine disrupting chemicals such as TCDD.
Associations between the exposure to TCDD during infancy/prepuberty and impaired semen quality were observed in the Seveso population (Mocarelli et al., 2008, 2011), as well as in the Russian Children's Study (Mínguez‐Alarcón et al., 2017). There appear to be some distinct differences between the exposures in these two cohorts, in terms of timing of exposure and in terms of congener composition as well as the exposure levels. At least, part of the Seveso boys included in Mocarelli et al. (2008) experienced on top of the existing background exposure to PCDD/Fs and DL‐PCBs a rapid increase in TCDD levels shortly after the incident, and this TCDD increase occurred solely postnatally. The studied boys were at different ages at the time of the incident, but impaired sperm quality was only seen in boys exposed before puberty. A second study from Seveso, with lower number of participants, involved sons of mothers that were exposed during the incident and a reference group of male blood donors, having mothers not exposed during the incident (Mocarelli et al., 2011). In these groups, respectively, 54% and 62% of the boys were breastfed. The results of this study indicate that postnatal exposure by breastfeeding may be of higher importance for impaired sperm quality than in utero exposure, since impaired semen quality was observed in breastfed sons of exposed mothers but not in formula‐fed sons of exposed mothers. The lower sperm concentrations in breastfed sons of exposed mothers were accompanied by lower serum Inhibin‐B and higher serum FSH concentrations at adult age, whereas oestradiol, testosterone and LH levels were unaffected. It could not be deduced whether hormonal effects were cause or consequence of the affected sperm parameters, and no changes in Inhibin‐B and FSH were observed in other studies (these parameters were not reported in the Russian Children's Study). Overall, the CONTAM Panel concluded that the evidence from both Seveso studies suggests a postnatal period of sensitivity that might expand into puberty. A potential problem in these studies was that exposure to other PCDD/Fs and DL‐PCBs was not taken into account.
In the Russian Children's Study, the study‐design does not allow discrimination between pre‐and postnatal exposure. Contrary to the Seveso studies, also other PCDD/Fs and DL‐PCBs were analysed and shown to contribute significantly to the TEQ level in addition to TCDD, based on WHO2005‐TEFs. Associations were most significant for serum levels of TCDD and PCDD‐TEQ, a bit less for PCDD/F‐TEQ, but not significant for PCDF‐TEQ, DL‐PCB‐TEQ or total‐TEQ. All associations, including those with PCDFs and DL‐PCBs, were somewhat stronger when adjusting for NDL‐PCBs, although NDL‐PCBs alone were not significantly associated with sperm parameters. In addition, the boys in the Russian Children's Study had high exposure to organochlorine pesticides, and in particular HCB. However, serum levels of HCB, β‐HCH or DDE were not associated with sperm concentrations, and adjustment did not influence the associations between PCDD/F levels at age 8–9 and the sperm quality after sexual maturation (see Section 3.1.4.3.1).
Based on the evidence from experimental animal studies and the human cohort studies that observed associations with the serum levels at young age, the CONTAM Panel concluded that impaired semen quality is likely to be a causal effect of exposure to TCDD, other PCDDs and possibly PCDFs. Regarding DL‐PCBs, and contrary to experimental animals (e.g. Wakui et al., 2010, see Annex A.5), there is no conclusive evidence based on humans studies. The CONTAM Panel noted the uncertainty in relation to the association with PCDF exposure, but other studies on semen quality in relation to high PCDF exposure, e.g. from the rice oil incidents, were not available. The dose–response relationships for semen quality from the Russian Children's Study and for the studies from Seveso are further elaborated in Section 3.1.7.4 below.
The CONTAM Panel acknowledged that cryptorchidism causes reduced fertility, but this endpoint was not studied in the Seveso cohort or in the Russian Children's Study. The two available human studies on cryptorchidism did not provide sufficient evidence for an association with PCDD/F or DL‐PCB exposure.
It is not known whether pubertal timing in boys is related to semen quality later in life, but changes in pubertal timing may be an indication of disturbance of the hormonal homeostasis, which may again have implications for sperm production. The poorly understood trigger of pubertal onset in humans appears to involve altered sensitivity of the hypothalamus, pituitary and reproductive glands to androgens and adrenal production of dehydroepiandrosterone, supposed to promote maturation of the hypothalamic nerve cells producing and releasing GnRH. The resulting increase in LH from the pituitary stimulates testosterone production in Leydig cells, and, together with adrenal dehydroepiandrosterone, stimulation of hair growth around the genitals and armpits. The pituitary increase in FSH production following hypothalamic GnRH production stimulates Sertoli cell proliferation, testicular growth and subsequently sperm development (as reviewed by Picut et al., 2018).
Delayed puberty was observed in rats after TCDD exposure (indicated by delayed balanopreputial separation, see Section 3.1.2.1.3). In humans, associations between PCDD/F‐ and DL‐PCB‐TEQ levels and delayed male pubertal development were seen in the Russian Children's Study. Growth, which is also connected to pubertal onset, was also delayed with increasing exposure in this cohort. The other available cohorts and cross‐sectional studies, with lower number of participants and lower exposure level, did not provide any significant associations with pubertal development. Studies on changes in serum sex hormone levels most often reported no association, but whether these samples were taken in the correct time period for such possible changes, and if the exposure levels were sufficiently high in the cohorts not showing significant effects for other endpoints, is unclear (see Section 3.1.4.3 and Annex A.10). In the Russian Children's Study, the serum level of HCB was also associated with delayed puberty, whereas exposure to NDL‐PCBs was associated with earlier puberty. This complicates interpretation of a possible relationship between exposure to PCDD/Fs and DL‐PCBs, and timing of puberty. The CONTAM Panel concluded that there was insufficient information to conclude on causal associations.
3.1.7.1.2. Lower sex ratio
Lower sex ratio (lower proportion of boys) in offspring was seen consistently following high TCDD exposure of fathers in the Seveso incident and following high occupational exposure of men (see Section 3.1.4.3.3). In Seveso, the sex ratio was already decreased prior to the incident, possibly because of increased TCDD exposure starting a few years before the time of the incident. In the occupational studies and in the Seveso Cohort the serum TCDD concentrations in fathers were in the range 100–250 pg/g fat. The exact exposure is uncertain, in particular in the occupational studies, because TCDD was quantified in samples taken many years after the exposure, and extrapolated to exposure levels at the time of conception based on several assumptions. The observed lower sex ratio was also supported by observations after high accidental exposure to PCDFs and DL‐PCBs due to consumption of contaminated rice oil. The individual exposures were not quantified in these population studies. Furthermore, a dose‐dependent lower sex ratio was seen after TCDD treatment of male mice prior to mating (Ishihara et al., 2007).
Therefore, the CONTAM Panel concluded that a lower sex ratio in offspring after exposure to PCDD/Fs of the fathers to be is likely a causal effect. For DL‐PCBs, no studies were identified. The CONTAM Panel noted that the mode of action of reduced sex ratio as a result of such PCDD/F exposure is not well characterised (see Section 3.1.6.10.4), but it has been suggested that lower sex ratio may result from reduced fertility (Jacobsen et al., 2000). Given the uncertainties in the exposure assessment associated with studies on sex ratio, this was not further considered for dose–response assessment.
3.1.7.1.3. Increased TSH level in newborns
Newborn children (born in 1994–2005) of mothers who lived in the most affected Zones A or B during the Seveso incident, had higher levels of TSH in blood than children born in the same period from mothers who lived in less contaminated areas (Baccarelli et al., 2008, see Section 3.1.4.4). The median TCDD level in serum collected 0–2 years after the incident in Zone A and B was reported to be 447 and 94 pg/g fat, respectively. The total exposure in TEQs at the time of the incident is unclear (see Section below). TCDD levels in serum of mothers when the children were born about 20 years later were 3 to 5 times lower. The positive association between living in TCDD‐contaminated areas and TSH in newborns is likely to be causal, since the groups examined and the differences in TSH were relatively large, and no obvious confounders were identified. However, this area‐based study lacks individual TCDD data, and is therefore not suitable for quantitative risk assessment. The results from the substudy (Baccarelli et al., 2008) with individual TCDD and TEQ data from a subgroup of the women are considered uncertain due to small numbers and unclear timing of collection of samples from the newborns with the highest TSH levels.
The CONTAM panel concluded that a causal association between relatively high TCDD exposure and increased TSH in newborns in Seveso is likely, but the results are not suitable for risk assessment. Most studies of low‐moderate exposure to PCDD/Fs and DL‐PCBs (resulting from background exposure) in newborns do not suggest any adverse effects on thyroid function.
3.1.7.1.4. Impaired development of teeth
Association between exposure and tooth enamel hypomineralisation or enamel defects was reported in three different population groups (see Section 3.1.6.10). Teeth formation occurs at defined time intervals for different types of teeth. Development of the first primary human teeth starts from the fourth week in utero, whereas enamel of permanent teeth is formed postnatally. Most studies on the association between exposure to PCDD/Fs and DL‐PCBs and development of teeth were on permanent molars, and thus considered a postnatal effect in humans. Hypomineralisation weakens the enamel and is adverse as it increases the risk of caries and impaired tooth health later in life. Also in rats, mice and rhesus monkeys the teeth development, hypomineralisation and susceptibility to caries was shown to be affected by exposure to TCDD and other PCDDs (Section 3.1.6.10). The reported associations between exposure and developmental effects on teeth are likely to be causal.
In the participants in the two highest tertiles of exposure in the Seveso study (with higher odds of enamel defects than the lower tertile), blood TCDD concentrations were 238 to 26,000 pg/g fat (Alaluusua et al., 2004). As noted by the authors, the incidence in the non‐ABR zone, which served as controls in this study, was also relatively high. This can potentially be due to the high TCDD levels in children from this zone (see Section 3.1.7.2.2). The reported significant association between exposure to total PCDD/Fs via breastmilk (expressed in I‐TEQ) and enamel hypomineralisation in teeth in children born in 1987 in Finland, increased in frequency and severity with the total exposure (Alaluusua et al., 1996, 1999). The exposure (area under the curve (AUC)) was expressed as ‘low exposure’ (< 8 pg × year/g milk fat), ‘moderate exposure’ (8–16 pg × year/g milk fat), and ‘high exposure’ (> 16 pg × year/g milk fat). Mean levels in breast milk were 19.6 and 24.6 pg I‐TEQ/g fat, in rural and urban regions in Finland, respectively (Vartiainen et al., 1997). The CONTAM Panel noted that since neither the duration of breastfeeding alone nor the total I‐TEQs in the milk alone was significantly associated with the occurrence of mineralisation changes, a direct comparison of the exposure levels with serum concentrations in the Seveso study participants is not appropriate. In a study conducted with Finnish children approximately 10 years later, such effects were no more observed. However, the total exposure of children was lower due to lower PCDD/F and DL‐PCB concentrations in the mothers and their milk and also the breastfeeding duration was shorter (Alaluusua and Lukinmaa, 2006). The CONTAM Panel concluded that exposure to PCDD/Fs via breast milk may lead to increased enamel defects in teeth in the children. However, these data were less suitable for dose–response assessment, because the association was seen only for the accumulated exposure from breast milk, and the parameters were not reported separately. However, a calculated exposure31 of 8 pg × year/g milk fat (the upper range of the low exposure category in Alaluusua et al., 1996), would correspond to an initial milk level at the start of the breastfeeding of 10.4 pg/g fat, based on the reported average duration of breastfeeding of 10.5 months. When extending this period to 12 months, an initial milk level of 9.2 pg/g fat would result in the exposure of 8 pg × year/g milk fat. These calculations take into account a 25% decrease in the milk level of a one‐year period.
3.1.7.1.5. Cancer
IARC has concluded that there is sufficient evidence in humans for carcinogenicity of TCDD, 2,3,4,7,8‐PeCDF and PCB‐126 (Group 1 carcinogen) based on data from experimental animals, epidemiological studies, and from the common mechanism of action (IARC, 2012).
The CONTAM Panel identified several studies showing a positive association with all cancers combined. However, there was no clear link to any specific site and no evidence of direct genotoxicity (see Section 3.1.3). Due to the lack of a clear dose–response relationship and multiple co‐exposures, the CONTAM Panel does not consider these studies suitable for the risk assessment. The decision not to focus on these carcinogenic effects in the risk assessment is in line with the previous assessments by the WHO, the SCF and JECFA. However, it should be noted that in some studies follow‐up of highly exposed groups may still be too short to reveal an increased incidence of certain tumours.
3.1.7.2. Dose–response relationships in epidemiological studies
Contrary to previous assessments, the CONTAM Panel was asked to consider not only TCDD but also other PCDD/Fs and DL‐PCBs. Furthermore, the existing TEF‐values, established under the remit of the WHO in 2005 were requested to be the basis. This provided a number of additional challenges.
A first issue is whether the current TEFs are actually suitable when evaluating the associations between serum levels and certain effects in human studies. The outcome of the EU SYSTEQ project suggested that the potencies of a number of congeners may seriously deviate from WHO2005‐TEFs (see Section 3.1.7.2.1).
A second important issue is that in some studies only TCDD was analysed, whereas other PCDD/Fs and DL‐PCBs are likely to have contributed to the exposure and the observed effects. Even in Seveso, where the explosion released in particular TCDD, the background exposure to PCDD/Fs and DL‐PCBs was probably at its historical peak (Hagenmaier and Walczok, 1996), causing substantially higher background levels in the population than the present day levels in Europe. There are, however, few studies that did analyse samples from that period of time. Nevertheless, the CONTAM Panel evaluated the potential contribution of other PCDD/Fs and DL‐PCBs to the serum TEQ levels in these studies (see Section 3.1.7.2.1).
3.1.7.2.1. TEF values
The relative potencies of the various PCDD/Fs and DL‐PCBs are of importance when evaluating studies on adverse effects in animals and humans. Over the past decades, insight into these relative potencies has changed, as reflected by changes in the TEF values. In 1988, this resulted in a set of so‐called I‐TEFs for PCDD/Fs and an additional set of TEFs for DL‐PCBs. In 1998 and again in 2005, these TEFs were updated in meetings organised by WHO (Van den Berg et al., 1998, 2006) (see Section 1.3.1). TEFs are weighted values based on animal studies and supported by in vitro studies. In practice, the so‐called REPs determined for each congener show a large range of values, due to factors like animal species/strain, measured endpoint and duration of exposure. Furthermore, the latest WHO2005‐TEF values are rounded based on a log scale, and as such present more an order of magnitude of different potencies (so values of 1, 0.3, 0.1, 0.03, 0.01, 0.0003, 0.0001 and 0.00003). Different PCDD/F congeners with similar numbers of chlorines were assigned the same TEF value, so the hexachlorinated all have a TEF of 0.1, the heptachlorinated all have a TEF of 0.01, and the octachlorinated have a TEF of 0.0003. All mono‐ortho PCBs were assigned the same TEF value of 0.00003.
This approach implies that TEFs are not a precise estimate of the toxic potency of a congener, which may affect the interpretation of both human and animal studies. For this reason, the CONTAM Panel decided to consider only animal studies performed with TCDD, since studies with other congeners, potentially showing a lower point of departure, would merely question the current TEF rather than forming the basis of an HBGV. It should be stressed that TEFs are weighted factors based on a range of relative potencies from various studies and endpoints, and determined to discrete points on a log scale.
In the case of human studies, selecting those with only TCDD would probably discriminate their use, especially when focusing on the lower end of the exposure distributions where other PCDD/Fs and DL‐PCBs could contribute significantly. It should also be considered that only for congeners that contribute to the TEQ levels, the uncertainty in the TEFs may be relevant. For this reason, it was decided to use these studies but to estimate the relative contribution of the different congeners to the TEQ when interpreting the results of some of the studies discussed below.
It is also important to realise that REPs from animal studies are based on treatment levels (external dose), and not levels measured in tissues (internal dose). This may, e.g. be relevant when dealing with levels in human serum or tissues. In relation to this issue of the external dose, the outcome of the EU SYSTEQ project is of interest (Larsson et al., 2015). This project dealt with the issue that once present in the body, relative potencies of congeners may change. Higher potencies (so‐called consensus toxicity factors) than the WHO2005‐TEFs were e.g. observed for 2,3,4,7,8‐PeCDF and 1,2,3,4,7,8‐HxCDF, both congeners that may contribute much to TEQ levels. In addition, also species differences in relative potencies are important. For example, it was shown that various types of human cells, both primary cells and cell‐lines, are much less sensitive to PCB‐126 than suggested by the TEF of 0.1. The CONTAM Panel noted that this is based on endpoints like induction of CYP 1A1, 1A2, 1B1, AHRR and EROD activity. However, according to the database with REPs used in the last TEF evaluation, a similar situation seems to apply to mice, with lower potency of PCB‐126 when focusing on CYPs and related activities (Haws et al., 2006). On the other hand, when considering immunotoxic effects, like plaque forming cells or IgG production, the reported relative potencies of PCB‐126 are much larger, including studies in which EROD activity showed a lower potency of this congener (Harper et al., 1993). The question is if this also applies to some of the identified critical endpoints like reproductive effects, and whether this may be the case for humans. Since such effects seem difficult to measure in vitro and no incidents with PCB‐126 as the major contributor to the TEQ are known, it seems difficult to obtain more certainty on this issue. Nevertheless, since this DL‐PCB is by far the most important contributor, not only to DL‐PCB‐TEQ, but also to total‐TEQ levels in blood and human milk, it seems important to consider this potentially lower potency when evaluating epidemiological studies.
Another important issue is the differences in kinetics between congeners. Budinsky et al. (2006), evaluated data on TCDD and 2,3,4,7,8‐PeCDF from an NTP study. For the latter compound, REPs for EROD induction measured after 14, 31 and 53 weeks were 0.10, 0.18 and 0.44 when based on administered dose. For liver tumour incidence, the REP was 0.27 for administered dose. However, when basing it on the liver concentrations, the REP for this congener would decrease to 0.014. This difference is due to the higher liver sequestration of 2,3,4,7,8‐PeCDF. Whether the bound fraction is active, can be disputed but according to Budinsky et al. (2006), there are indications that it is.
Although the CONTAM Panel decided to base the assessment on the existing TEFs, i.e. WHO2005‐TEFs, these issues above are of importance when evaluating epidemiological studies and selecting the point of departure, as discussed below.
3.1.7.2.2. Background exposure in the Seveso Cohort
In the studies by Mocarelli et al. (2008, 2011), blood samples in exposed boys or mothers were taken within several weeks up to one year after the incident. At a later stage, these were analysed for levels of TCDD, but not for other PCDD/Fs and DL‐PCBs. Since the Seveso incident occurred in a time period when the environmental exposure to PCDD/Fs and DL‐PCBs was around its historical peak, the contribution of other congeners than TCDD to the TEQ levels in both the exposed and the reference groups is an important issue. Unfortunately, analysis of PCDD/Fs and DL‐PCBs in human samples started at a much later stage and there are few studies that can be used to estimate the background levels in 1976. To investigate the existing levels at the time of the explosion, Eskenazi et al. (2004) determined serum levels of PCDD/Fs and DL‐PCBs in a number of pooled samples from women of various age classes from the non‐ABR zone, collected just after the incident. Pools were based on 20–21 people per sample and data were initially expressed in WHO1998‐TEFs (see Table 11).
Table 11.
Levels of TCDD, other PCDD/Fs and DL‐PCBs and total TEQ in pooled blood samples from girls and women from zone non‐ABR (n = 20–21 per pool) (based on Eskenazi et al., 2004 and Warner et al., 2014)
| Age group | TCDD | Other PCDD/Fsand DL‐PCBs | Total TEQ | TCDD contribution |
|---|---|---|---|---|
| (years) | (pg/g fat) | (pg TEQ/g fat)a | (pg TEQ/gfat)a | (%) |
| 0–12 | 48 | 72 | 120 | 40 |
| 0–12 | 33 | 80 | 114 | 29 |
| Mean (0–12) (WHO1998‐TEFs) | 41 | 76 | 117 | 35 |
| 12–20 | 17 | 59 | 76 | 22 |
| 12–20 | 22 | 53 | 75 | 29 |
| 12–20 | 20 | 79 | 99 | 20 |
| Mean (12–20) (WHO1998‐TEFs) | 20 | 64 | 83 | 24 |
| 20–40 | 10 | 92 | 102 | 10 |
| 20–40 | 9 | 117 | 126 | 7 |
| 20–40 | 12 | 81 | 92 | 13 |
| 20–40 | 11 | 89 | 100 | 11 |
| Mean (20–40) (WHO1998‐TEFs) | 10 | 95 | 105 | 10 |
| Median (all ages) (WHO1998‐TEFs)b | 17 | 82 | 99 | 17 |
| Median (all ages) (WHO2005‐TEFs)b | 17 | 55 | 72 | 24 |
| Median (all ages) (SYSTEFs)c | 17 | 95 | 112 | 15 |
DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TCDD: 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin; TEQ: toxic equivalents; TEF: toxic equivalency factor.
Based on WHO1998‐TEFs.
Based on median congener levels as presented by Warner et al. (2014).
Converted using consensus toxicity factors as proposed by Larsson et al. (2015).
Warner et al. (2014) compared the TEQ levels in these nine pools, based on WHO1998‐ and WHO2005‐TEFs and also presented the median levels and range for individual PCDD/F and DL‐PCB congeners. Based on WHO1998‐TEFs, the median was 17 pg/g fat for TCDD, 29 pg/g fat for the TEQ for other PCDD/Fs, 53 pg/g fat for DL‐PCB‐TEQ and 99 pg/g fat for total‐TEQ. When applying WHO2005‐TEFs, the median values for PCDD/Fs w/o TCDD decreased from 29 to 24 pg/g fat, and for DL‐PCBs from 53 to 31 pg TEQ/g fat. This means a reduction of 17% for the TEQ of PCDD/Fs w/o TCDD, of 41% for DL‐PCB‐TEQ, and of 33% for the sum‐TEQ of these PCDD/Fs and DL‐PCBs. The range (min–max) presented in the same paper for the different congeners shows a similar decrease for the sum‐TEQ w/o TCDD. The CONTAM Panel was not able to obtain the data on individual congeners for each pool, meaning that only an overall assessment could be made on the effect of using WHO2005‐TEFs on the background for specific age groups (see below). Figure 9A shows the congener pattern based on the median levels, and reveals that in addition to TCDD, PCB‐126, 2,3,4,7,8‐PeCDF and PeCDD contribute most to the WHO2005‐TEQ level.
Figure 9.
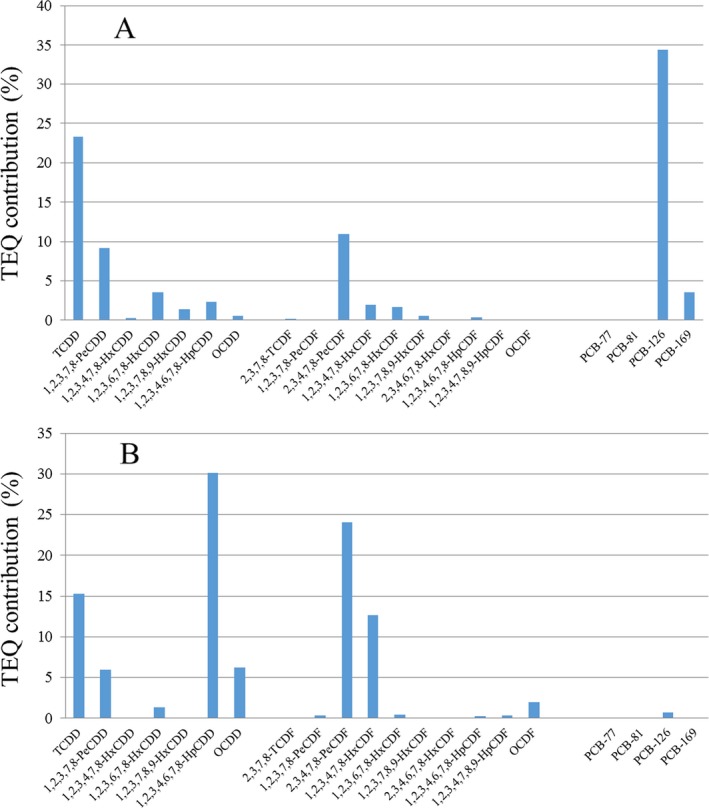
Congener pattern for TEQ contribution of PCDD/Fs and non‐ortho DL‐PCBs based on the median levels for the pooled samples and applying (A) WHO 2005‐TEFs or (B) consensus toxicity factors (SYSTEQ)
Regarding the effect of using internal rather than external TEFs, the last row in Table 11 shows the levels when applying the consensus toxicity factors (SYSTEQ), as proposed by Larsson et al. (2015). The congener pattern based on these values is shown in Figure 9B. The total TEQ and the contribution of the dioxin‐like compounds other than TCDD would be much higher than that obtained with the WHO2005‐TEFs, despite the fact that the contribution of the DL‐PCBs would be reduced from around 60% to 1%. This is caused by the fact that consensus toxicity factors for several other PCDD/Fs are higher than the WHO2005‐TEFs.
Table 11 also shows that the samples taken from the younger age groups in the ‘non‐ABR zone’, within the first year after the incident, show a relatively high contribution of TCDD to the total TEQ. The ‘non‐ABR Zone’ is the area surrounding the exposure zones A, B and R. Part of the subjects belonging to the ‘non‐ABR Zone’ may have been living close to the factory (but not in the direction of the wind on the day of the incident). Mocarelli et al. (2000) actually mentioned that the pollution with TCDD already started some years before the incident following the installation of a waste incinerator at the plant. They argued that this may explain some of the high levels in blood of men from Zone A who were actually not in the area during the incident.
3.1.7.2.3. Dose–response assessment in pivotal studies
Dose–response analysis of decreased sperm concentrations in boys exposed at 1–9 years in the Seveso Cohort (Mocarelli et al., 2008)
This study observed decreased sperm concentrations in boys exposed at an age between 1 and 9 years (mean ± SD: 6.2 ± 2.5 years) as compared to controls (healthy blood donors) selected from an area further away from Seveso. The exposed boys showed an average TCDD serum level of 210 pg/g fat. Dividing the boys into quartiles with median levels of 68, 142, 345 and 733 pg/g fat did not show a clear dose–response curve, meaning that sperm concentrations were lower in all quartiles as compared to the control group, although not statistical significant.
Concerning the contribution from the background, as shown in Table 11, for the age group 0–12 years, the analysis of the two pooled samples from girls of the non‐ABR zone showed mean WHO1998‐TEQ levels of 117 pg/g fat of which TCDD contributed 41 pg TEQ/g fat. Therefore, 76 pg TEQ/g fat came from other PCDD/Fs and DL‐PCBs. When applying on this WHO1998‐TEQ level of 76 pg/g fat the 33% reduction due to the change of some of the TEFs in 2005 (see above), the contribution of the other PCDD/Fs and DL‐PCBs to the total TEQ level would be on average 51 pg WHO2005‐TEQ/g fat. As mentioned above, the relatively high TCDD levels in these girls suggest increased exposure also in the non‐ABR Zone, but the men in the control group of the study on sperm effects were from areas not affected by the explosion, and as such are unlikely to have been affected by this increased TCDD exposure. Blood from these men taken at the time of the incident was not available and Mocarelli et al. (2008) assumed the TCDD level in this group to be ≤ 15 pg/g fat, being the LOQ of the analytical method.
Assuming similar exposure to these other PCDD/Fs and the DL‐PCBs in the various zones and no difference between boys and girls at this age, the background level of 51 pg WHO2005‐TEQ/g fat could then be added to the TCDD levels measured in the boys exposed at young age (1–9 years) (Table 12). The average TCDD level in this group was 210 pg/g fat, and thus the average TEQ level in the exposed boys would be 261 pg WHO2005‐TEQ/g fat (210 for TCDD + 51 for others). The group was split in quartiles with median TCDD levels of 68, 142, 345 and 733 pg TCDD/g, the TCDD level in the controls was assumed to be 15 pg/g fat or lower. Including other congeners, the levels in the controls would increase to maximal 66 pg TEQ/g fat (≤ 15 + 51). This is lower than the average 117 pg TEQ/g fat measured in the pooled samples from girls (Table 11), due to lower TCDD level and application of other TEFs. In the case of the lowest quartile, with a level of 68 pg TCDD/g fat, adding the average background from other PCDD/Fs and DL‐PCBs of 51 pg TEQ/g fat would result in a total TEQ level of 119 pg TEQ/g fat, i.e. at least 1.8‐fold the level in the controls. When focusing on PCDD/Fs only (44% of the background TEQ of 51, or 22 pg TEQ/g fat), the PCDD/F‐TEQ level in the whole group of exposed boys would be 232 (210 + 22), in the lowest quartile 90 (68 + 22) and in the controls maximum 37 (≤ 15 + 22) pg TEQ/g fat (i.e. 6 and 2.5‐fold higher than control boys for the average and lowest quartile, respectively). Ratios would increase slightly if the actual TCDD levels in controls would be lower than 15 pg/g fat.
Table 12.
Effect of adding average background exposure to other PCDD/Fs and DL‐PCBs in the studies of Mocarelli et al. (2008, 2011), based on pooled samples from girls and women of the non‐ABR zone
| TCDD | Other PCDD/Fs and DL‐PCBs | Total TEQ | DL‐PCBsb | PCDD/Fs | |
|---|---|---|---|---|---|
| (pg/g fat) | (pg TEQ/g fat) | (pg TEQ/g fat) | (pg TEQ/g fat) | (pg TEQ/g fat) | |
| Mocarelli et al. ( 2008 ) | |||||
| Girls | |||||
| 0–12 Avg (WHO1998‐TEFs) | 41 | 76 | 117 | ||
| 0–12 Avg (WHO2005‐TEFs)a | 41 | 51 | 92 | 29 | 63 |
| Boys 1–9 all (WHO2005‐TEFs) | 210 | 51 | 261 | 29 | 232 |
| Boys 1–9 Q1 (WHO2005‐TEFs) | 68 | 51 | 119 | 29 | 90 |
| Controls 1–9 (WHO2005‐TEFs) | ≤ 15c | 51 | 51–66 | 29 | 22–37 |
| Mocarelli et al. ( 2011 ) | |||||
| Women | |||||
| 20–40 Avg(WHO1998‐TEFs) | 11 | 94 | 105 | 36 | 39 |
| 20–40 Avg (WHO2005‐TEFs)a | 11 | 64 | 75 | ||
|
Mothers of boys breastfed all (WHO2005‐TEFs) boys breastfed low (WHO2005‐TEFs) boys breastfed high (WHO2005‐TEFs) controls 1‐9 (WHO2005‐TEFs) |
19 13 59 10 |
64 64 64 64 |
83 77 123 74 |
36 36 36 36 |
47 41 87 38 |
TCDD: 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents; TEF: toxic equivalency factor.
Reduction in TEQ of PCDD/F and DL‐PCB congeners other than TCDD by 33% due to application of WHO2005‐TEFs
Based on 56% contribution of DL‐PCBs to WHO2005‐TEQ.
Not measured but assumed to be below the LOQ of 15 pg/g fat.
This evaluation suggests that there would still be a clear difference between the serum levels of PCDD/Fs and DL‐PCBs in the exposed boys, even in the lowest quartile, compared to the controls. The study showed a significant difference in sperm concentrations between the whole group of men exposed as young boys and the controls. However, the CONTAM Panel noted that when the exposed group was split in quartiles, no dose response is observed and as such the data are not suitable for BMD modelling.
Decreased sperm concentrations in boys exposed in utero and via breastfeeding in the Seveso Cohort (Mocarelli et al., 2011)
The study included 39 men born between 1977 and 1984 to mothers exposed to TCDD (i.e. 1–8 years after the 1976 incident), and 58 comparison men born to mothers exposed only to background levels of PCDD/Fs and DL‐PCBs. Semen samples were collected from exposed men with an average age of 22.5 years as compared to 24.6 years for the comparison group. In this study, the effect on sperm production was only observed in men that were breastfed. Formula‐fed boys of exposed mothers showed a slight but non‐significant decrease when compared to controls.
The median TCDD concentrations in blood among all the exposed mothers at conception, estimated from previous measurements, was 26 pg/g fat (5th and 95th percentiles 12 and 232). When focusing on the women who breastfed, the median level at the time of conception was estimated to be 19 pg/g fat (5th and 95th percentiles 12 and 117). The breastfed boys were split in two groups showing a larger effect on sperm parameters for the boys born to mothers with higher serum levels. The median TCDD levels for these two groups were 13.1 (n = 12) and 58.9 (n = 9) pg/g fat. All estimated levels were based on TCDD levels in the blood taken within the first year after the incident which were subsequently extrapolated to the time of conception (on average 5.7 years after the incident), using a half‐life of 4 years based on Kreuzer et al. (1997).
In the comparison mothers, TCDD levels were assumed to be 10 pg/g fat, based on the levels reported by Eskenazi et al. (2004) in four pooled samples from women aged 20–40 from the non‐ABR zone (see Table 11). On average, the total WHO1998‐TEQ level in these pools was 105 pg WHO1998‐TEQ/g fat. Considering WHO2005‐TEFs, when applying the reduction of 33% for the other PCDD/Fs and the DL‐PCBs as explained above, this would result in a non‐TCDD background of 64 pg WHO2005‐TEQ/g fat (see Table 12). Total WHO2005‐TEQ levels for the control and exposed mothers who breastfed would be, respectively, 74 and 83 pg/g fat. The P5–P95 range for the latter group would be 76–181 pg TEQ/g fat, a 1‐ to 2.5‐fold difference from controls. For the two groups of exposed mothers who breastfed, the serum TEQ levels would be 77 and 123 pg WHO2005‐TEQ/g fat, a 1‐ and 1.7‐fold difference from the controls.
When focusing on PCDD/Fs only (44% contribution to background), the background would be 28 pg WHO2005‐TEQ/g fat, meaning TEQ levels of 38 and 47 (range 40–145) for control and exposed mothers. This implies a one‐ to fourfold difference.
The above‐mentioned background levels of PCDD/Fs and DL‐PCBs are based on levels in pooled samples collected in 1976/1977. To estimate the TCDD levels at the actual time of conception (up to 7 years later), the original levels in 1976‐1977 were extrapolated using a half‐life of 4 years. Worldwide, the exposure to PCDD/Fs and DL‐PCBs started to decline in the 1970s. Thus it seems likely that the background levels in this study also decreased during the period between the incident and the birth of the boys. However, this decline may be less than that of TCDD due to different half‐lives and continued exposure via food. The CONTAM Panel was unable to estimate the actual decrease in these background levels.
The data show an increased effect for breastfed boys which becomes stronger at the higher level when split in two groups (mothers with low and high TCDD levels). The group with the lower serum TCDD levels showed a significant difference from the controls, suggesting a LOAEL for TCDD only of 13 pg/g fat. The CONTAM Panel noted that for this group, the difference from the existing background levels of other PCDD/Fs and DL‐PCBs could have been very small. Furthermore, the data were considered not suitable for BMD‐modelling. Therefore, it was decided to not use this study for deriving the HBGV but merely as supportive evidence.
Decreased sperm concentrations in boys in the Russian Children's Study (Mínguez‐Alarcón et al., 2017)
Boys from the city of Chapaevsk (Russia) were studied for potential effects of PCDD/Fs, PCBs, organochlorine pesticides and lead on a number of endpoints, including delayed onset of puberty and semen parameters. Blood was collected at the age of 8 to 9 years and analysed for the various contaminants. At the age of 18–19, sperm was collected from 133 participants.
Total TEQ levels in serum varied between 1.9 and 107 pg WHO2005‐TEQ/g fat, with a median of 22 (Burns et al., 2009). In addition, the boys had relatively high levels of PCBs (sum of more than 30 PCB congeners), with a median of 235 ng/g fat (range 58–1,500). Figure 10 shows the relative contribution of the various congeners to the WHO2005‐TEQ level. The highest contribution comes from PCB‐126, followed by PeCDD, 2,3,4,7,8‐PeCDF and TCDD.
EFSA was asked to derive an HBGV for the sum of PCDD/Fs and DL‐PCBs. When focusing on this study, a significant association was found for TCDD, PCDD‐TEQ and PCDD/F‐TEQ, but not for total‐TEQ, PCDF‐TEQ and DL‐PCB‐TEQ. A possible explanation is that PCB‐126, which contributes most to the DL‐PCB‐TEQ level, may be less potent in humans than indicated by the TEF‐value of 0.1. There are actually strong indications from in vitro studies that this is the case (see Section 3.1.7.2.1). To the contrary, there are no data implying that this is also the case for PCDFs. No studies were performed on victims of, e.g. the rice‐oil incidents, where exposure came primarily from PCDFs and DL‐PCBs. Another possible explanation is that PCDFs and DL‐PCBs showed a stronger association with NDL‐PCBs than PCDDs. Additional information showed that the associations for PCDF‐TEQ, DL‐PCB‐TEQ and total‐TEQ became stronger when adjusted for NDL‐PCBs although the trend did not become significant (see Table 10 in Section 3.1.4.3.1). Similar results were found for TCDD and PCDD/F‐TEQ.
The CONTAM Panel noted that since the current exposure is based on PCDD/Fs or total‐TEQ, focusing on TCDD or PCDD‐TEQ only could be misleading, and not adhering to the TEQ principle. At the same time there are strong indications that the DL‐PCB‐TEQ level may be seriously overestimated based on the current TEF of PCB‐126. Therefore, it was decided to base the assessment on the inverse association between PCDD/F‐TEQ levels and sperm concentrations.
A decreased sperm concentration was already observed in the second quartile, with a median PCDD/F‐TEQ level of 10.9 pg WHO2005‐TEQ/g fat (range: 9.2–13.8) as compared to a median of 7.0 (range: 2.0–9.1) in the first quartile. No further decrease was observed at higher levels.
In the absence of individual data and a clear dose response, it was decided not to perform BMD‐modelling but to use the median serum level in the lowest quartile, i.e. 7.0 pg WHO2005‐TEQ/g fat, as the starting point for estimating the associated daily exposure.
3.1.7.3. Critical effects in studies in experimental animals
Concerning the animal studies, it was decided to select only those published since the previous assessment and showing effects considered as adverse at dose levels that correspond to a body burden lower than the ones forming the basis of the current TWI. A two‐step selection process was applied to identify these studies from the ones that were initially retrieved from the literature search (see Section 3.1.2.1.1). This initially resulted in the selection of 28 studies on rats, four on mice and six on monkeys, but these numbers were further reduced (see Annex A.6).
These studies were then further assessed for critical effects on the basis of toxicological endpoints, rather than intermediate changes in homeostasis or gene expression. Furthermore, studies were selected for critical effects that were observed with only TCDD rather than with other PCDD/Fs, DL‐PCBs or mixtures to avoid uncertainties arising from the setting of the TEFs. This resulted in eight studies in rats and one in mice which were considered to show relevant adverse endpoints at doses leading to body burdens in the range at, or below, those used for the current TWI, and might be suitable for deriving an HBGV. Adverse outcomes were either general toxicity or developmental and reproductive effects, mostly those previously recognised at higher exposure levels (Table 9). The CONTAM Panel considered that none of the primate studies were suitable due to the few animals involved, their unknown selection and survival rates (see Section 3.1.2.2).
3.1.7.3.1. General toxicity
General aspects of TCDD toxicity seen in many experimental investigations in rodents concern histopathological, clinical chemistry and haematological endpoints. Female Sprague–Dawley rats, administered TCDD by gavage 4 or 5 days a week for 4 weeks with doses from 3 to 1,000 ng/kg bw per day, showed, e.g. liver hyperplasia and thymus atrophy (Harrill et al., 2016). A NOAEL of 100 ng/kg bw per day was established, corresponding to a NOAEL body burden of 789 ng/kg bw. In a study of longer duration with female Long‐Evans (Turku/AB) rats, a dosing regimen of a single loading dose followed by weekly doses over 20 weeks was used (Viluksela et al., 2000). This rat strain is relatively sensitive to TCDD compared to Han Wistar rats, ascribed to differences in the transactivation domain of the AHR (see Section 3.1.6.4). A NOAEL body burden of 26 ng/kg bw was estimated based on body weight loss, blood AST levels and histopathological findings in the liver. In comparison, the more resistant Han Wistar rats were approximately 10‐fold less sensitive, based on these outcomes and thymic atrophy. These are classical endpoints of TCDD toxicity in rats and many other experimental animals, reflecting the fundamental and wide ranging repercussions of disturbing AHR function.
Outcome of these types of studies with TCDD are also clearly dependent on time, in addition to the body burden. In an NTP carcinogenicity study with female Sprague–Dawley rats, groups of animals were exposed to TCDD at 3 to 100 ng/kg bw, 5 days per week for up to 105 weeks (NTP, 2006a, 2006b, 2006c, 2006d–e). Besides hepatocellular adenomas and cholangiocarcinomas observed at the highest dose at the end of the study, a number of non‐neoplastic changes were noted especially related to hepatic toxicity such as multinucleated hepatocytes, necrosis, bile duct hyperplasia and portal fibrosis. A NOAEL for these adverse outcomes after this time was established at 2.1 ng/kg bw per day and estimated body burden of 85 ng/kg bw, significantly lower than that recorded for similar adverse endpoints noted after a shorter exposure in this study. The importance of the temporal factor was exemplified by interim culls where NOAELs for the same hepatic toxicity outcome declined with time, i.e. at 14 weeks 71 ng/kg bw per day, at 31 weeks 33 ng/kg bw per day, and at 53 weeks 16 ng/kg bw per day, yet the body burden remained almost constant.
3.1.7.3.2. Effects on bone
An adverse outcome less commonly reported and identified in further studies of the same experimental animals as used by Viluksela et al. (2000), was the effect on bone strength and morphology in rats with low body burdens of TCDD over the 20 weeks exposure. Breaking forces of tibia and other bone parameters were reduced by TCDD with a calculated NOAEL body burden of 28 ng/kg bw, based on the dry weight liver levels reported in this paper (Jämsä et al., 2001). Again, Han Wistar rats seemed to be less sensitive.
3.1.7.3.3. Immunotoxic effects
In the same study as Harrill et al. (2016), blood lymphocyte populations were identified as a sensitive target based on marked suppression of the percentage of LPS‐induced IgM+ cells (Phadnis‐Moghe et al., 2016). Yet, an increment of proliferation was observed (ex vivo LPS‐induced), indicating a dysregulation of the humoral immune response with a NOAEL of 22 ng/kg bw per day corresponding to a body burden of 250 ng/kg bw.
3.1.7.3.4. Reproductive and developmental effects
In establishing the TWI set by the SCF in 2001, reproductive and developmental outcomes were considered critical endpoints. Since then, a number of pertinent studies have been reported, but the CONTAM Panel concluded that few met the strict criteria for use in the present risk assessment. Two studies were conducted according to OECD/GLP guidelines, in which TCDD was either administered orally as a single dose to dams on GD15, or chronically for 12 weeks to females during premating and continued through mating and gestation, and the consequences studied in F1 males (Bell et al., 2007b,c).
Following the single gavage dose of TCDD to mated female Wistar (Han) rats at GD15 of 50, 200 or 1,000 ng/kg bw, the mean litter size of the highest dose group was decreased during lactation and pup weights were decreased. Balanopreputial separation in these F1 males was delayed (on average by 2.8 days). There was no evidence of decreased sperm counts and the increase in abnormal sperm on PND70 was considered associated with the delayed puberty. Testes weights were slightly but significantly less than controls at PND70 and PND120 although fertility was not affected. A NOAEL based on the decreased pup weight at PND1 to PND7 of 50 ng/kg bw was established, corresponding to a maternal body burden of 19 and 20 ng/kg bw on GD16 and GD21, respectively (Table 9). Correction for other tissues and for acute to chronic exposure (additional factor of 2.6) resulted in body burdens of 56 and 59 ng/kg bw.
In the second study, an increased TCDD body burden was established prior to pregnancy by providing a diet containing TCDD which resulted in doses of 2.4, 8 or 40 ng/kg bw per day over 12 weeks of a premating period, then mating and through gestation. After parturition mothers were returned to control diet during lactation of litters. At the higher exposures, there was an increase in litter loss and offspring had decreased weights. As in the acute study, balanopreputial separation was significantly delayed but at all three exposures levels, not just the highest, with a LOAEL of 2.4 ng/kg bw per day (body burden of 42 and 50 ng/kg bw on GD16 and GD21, respectively). Sperm parameters of the F1 males at PND70 and PND120 showed no significant effects, with the exception of the proportion of abnormal sperm at the highest dose and ~ 10% decrease in testes weight at PND70. Again these were ascribed to the delay in developmental puberty and had no effect on fertility of these male F1 rats.
These studies suggested that in rats low maternal exposures to TCDD during pregnancy may influence early rate of development of offspring but effects on sperm counts and quality or the fertility of adult F1 offspring could not be used as critical endpoints.
In the study by Faqi et al. (1998) that was also described by the SCF in their risk assessment (see Section 1.3.3), an elevated maternal body burden was established by s.c. injection of Wistar rats with TCDD and maintained by a weekly s.c. 5‐fold lower dose. The daily sperm production on PND70 and 170 was affected with a LOAEL at a measured body burden of 25 ng/kg bw (Table 9). Also, sperm number in the epididymis was affected. Only a small additional reduction in semen parameters was observed at higher exposure. The CONTAM Panel noted that in this study it appeared that delay in pubertal development could not explain the decreased sperm production at PND70, as a delay in balano preputial separation was only observed at higher dose (BB of 137 ng/kg bw).
Embryo toxicity was explored with pregnant NIH mice after exposure to 0, 2, 50, or 100 ng TCDD/kg bw per day by gavage during days 1‐8 of gestation (Li et al., 2006). Embryonic loss occurred with the mid and high doses, particularly at pre‐implantation stage, but not with 2 ng/kg bw per day (Table 9), corresponding to a NOAEL body burden of 9 ng/kg bw.
Based on these results and estimated body burdens, the CONTAM Panel selected the following studies on general toxicity endpoint for further dose–response considerations: Faqi et al. (1998), Bell et al. (2007b,c), NTP (2006a), Jämsä et al. (2001), Li et al. (2006) and Viluksela et al. (2000) (see Section 3.1.7.4).
3.1.7.4. Dose–response relationships in experimental animal studies
The CONTAM Panel performed Benchmark dose (BMD) modelling on the results obtained in the selected rat and mice studies (see Section 3.1.7.3). The EFSA guidance on the use of BMD in risk assessment (EFSA Scientific Committee, 2017) was used. The full details of the BMD analysis are reported in Appendix C for the different studies. For some studies, the applied dose was used as input. In the case of Faqi et al. (1998) and Jämsä et al. (2001), the dosing regimen (loading dose plus weekly maintenance dose, applied s.c.) did not allow this and the modelling was performed with estimated levels in the body (body burden). The analyses resulted in the BMDL values and corresponding body burdens reported in Table 13. For details on the BMD modelling and body burden calculations, see Appendix C).
Table 13.
BMD modelling and corresponding body burdens (BB) for critical effects from the selected rat and mice studies
| Critical endpoint | Reference | BMDL | BMDU | BB at the BMDL (ng/kg bw) |
|---|---|---|---|---|
| Studies in rats | ||||
| Decreased sperm production and sperm counts in F1 male Wistar rats | Faqi et al., 1998 | Based on BB in ng/kg bw | ||
| At PND70 | BMDL10 = 0.15 | 11 | 0.15 | |
| At PND170 | BMDL10 = 0.0014 | 14 | 0.0014 | |
| Decreased pup weight from PND1 to PND7 in male F1 Wistar (Han) rats | Bell et al. (2007b) | Single dose ng/kg bw | ||
| Individual data set using litter variability | BMDL5 = 68 | 792 | 78 (GD16) | |
| Delay in BPS in male F1 Wistar (Han) rats | Bell et al. (2007b) | Based on ng/kg bw per day | 51 (GD16) | |
| BMDL5 = 3.5 | 21 | 61 (GD21) | ||
| Hepatopathy in female Sprague–Dawley rats | NTP (2006a) | Based on ng/kg bw per day BMDL10 = | ||
| Multinucleated hepatocytes | 3.8 | 7.7 | 97 | |
| Fatty change | 4.3 | 9.5 | Not calculated | |
| Necrosis | 8.0 | 49 | Not calculated | |
| Oval cell hyperplasia | 8.2 | 15 | Not calculated | |
| Bile duct hyperplasia | 8.1 | 15 | Not calculated | |
| Hepatopathy | 4.9 | 9.6 | Not calculated | |
| Increase in liver weight and AST and increased incidence of histopathological findings in the liver of Long‐Evans (Turku/AB) rats | Viluksela et al. (2000) | Not modelleda | – | – |
| Decreased tibia length, tibia geometry parameters, tibia ash weight, and increased plasma ALP activity in Long‐Evans (Turku/AB) rats | Jämsä et al. (2001) | Based on BB in ng/kg bw | ||
| Tibial length | BMDL5 = 372 | 1,280 | 372 | |
| Tibial pericircumferences | BMDL5 = 104 | 505 | 104 | |
| Tibial endo circumferences | BMDL5 = 47.8 | 290 | 48 | |
| Tibial CSA | BMDL5 = 13.8 | 455 | 14 | |
| Tibia ash weight | BMDL5 = 15.9 | 210 | 16 | |
| Plasma ALP activity b | – | – | – | |
| Studies in mice | ||||
| Embryonic loss in female NIH mice day 1–3 | Li et al. (2006) | Based on ng/kg bw per day BMDL10 = 11.4 | 52 | 53 |
BMD: benchmark dose; BMDL: benchmark dose lower confidence limit; BMDU: benchmark dose upper confidence limit; BPS: balanopreputial separation.
No BMD analysis performed for Viluksela et al. (2000) for the following reasons: (i) The histopathological effects in the liver were observed only in the mid‐ and high‐dose groups with a steep increase from the low‐dose and control groups (100% response). These type of data sets are not ideal for the dose–response analysis, in particular in studies with low number of animals. (ii) Changes in enzymatic activities and liver weights are not tabled in the publication but are reported only in diagrams of low graphic quality, making the digitisation very uncertain.
The results of the modelling show that this is not the most sensitive parameter of the study and therefore no additional actions were taken. The modelling of ALP was not further considered in the opinion.
The data show the lowest BMDLs for the study by Faqi et al. (1998) on reduced sperm production. It was noted that, although there is a clear monotonic dose–response trend in daily sperm production, the lowest tested dose induced a mean reduction of about 19% and 40% over the control groups at 70 and 170 PND, respectively. The dose selection therefore resulted in a poor characterisation of the dose–response relationship around the effect size selected as BMR, in particular in the latter time‐point. As a consequence, wide BMDL–BMDU confidence intervals were calculated at PND70 (BMDU/BMDL ratio: 70) and PND170 (BMDU/BMDL ratio: 20,000), resulting in a high uncertainty on the RP selection when using the BMD modelling output. Although the Panel noted uncertainties connected to the possible blinding of the spermatid counting in coulter chambers (See Section 3.1.2.1.3), it was decided to use the lowest dose as the LOAEL. The CONTAM Panel decided to recalculate the body burden originally calculated by the SCF (2001) in the dams for the study by Faqi et al. (1998), because it was noted that the dosing regimen with an s.c. loading dose and maintenance dose should not be considered as an acute dose and as such no further correction was applied. The LOAEL body burden in dams for decreased sperm production in male offspring, as observed at all doses at PND70 and PND170, was estimated to be 25 ng/kg bw (see Appendix B for calculations). This was the lowest body burden associated with adverse effects in experimental animals.
3.1.8. Derivation of a human HBGV
PCDD/Fs and DL‐PCBs are carcinogenic but likely not via a direct genotoxic mechanism (see Section 3.1.3). Therefore, the CONTAM Panel considered it appropriate to set a health‐based guidance value.
There is a large body of evidence attesting to the binding of PCDD/Fs and DL‐PCBs to the AHR as a prerequisite for their toxic manifestations. AHR binding and its consequent activation can thus be considered the molecular initiating event. Various adverse effects may result from this initial event although the modes of action have not been clarified. Overall, among the endpoints assessed, effects on sperm quality were found consistently and this endpoint provided the lowest reference point. It may be linked to changes in onset of puberty and possibly also lower sex ratio.
3.1.8.1. Selection of the reference point
As detailed in Section 3.1.4.3.1, three studies in humans (Mocarelli et al., 2008, 2011; Mínguez‐Alarcón et al., 2017) examined associations between TCDD, PCDD/F‐ or total TEQ and sperm quality. All three studies suggest associations between contaminant levels and decreased sperm concentration. In the exposed groups in Seveso (Mocarelli et al., 2008, 2011), sperm concentrations were decreased by about 30% and 60%. In the boys from the Russian Children's Study (Mínguez‐Alarcón et al., 2017), the reduction was about 40%. However, in this study, the decrease in sperm concentration occurred already at a PCDD/F‐TEQ level of 11 pg/g fat (LOAEL), with no further decrease at higher levels (Table 10). In the Seveso studies, sperm concentrations differed between exposed and control groups with much higher estimated TCDD, PCDD/F‐TEQ, and total TEQ levels with an apparent NOAEL level higher than the LOAEL level in the Russian Children's Study (Table 14). Therefore, the dose–response relations in the Seveso and in the Russian Children's Study may appear inconsistent since the levels at the second quartile (showing an effect) in the Russian Children's Study are lower than the estimated levels in the controls from the Seveso Cohort. However, the TEQs in Seveso had to be estimated from other studies as the actual levels in the controls from Seveso were not determined. Also the TEF values add uncertainty to this comparison. In this context, it is relevant to further note that the timing of exposure in Seveso (Mocarelli et al., 2008) and the boys from the Russian Children's Study differed, and other co‐exposures are likely to have differed as well. This makes direct comparison between these studies difficult.
Table 14.
Measured and estimated median serum levels of TCDD and PCDD/F‐TEQ in affected and unaffected boys in the Seveso and Russian Children's Study
| Studya | TCDD (pg/g fat) | PCDD/F‐TEQ (pg/g fat) | ||
|---|---|---|---|---|
| Group | ‘Unaffected’b | ‘Affected’ | ‘Unaffected’b | ‘Affected’ |
|
Seveso Boys 1–9 years (all) Boys (1–9 years) (Q1)c |
≤ 15d ≤ 15d |
210 68 |
22–37d 22–37d |
232d 90d |
|
Seveso Mothers of boys (all) Mothers of boys (Q1) |
10d 10d |
19 13 |
38d 38d |
47d 41d |
|
Russian Children's Study Boys (9 years) |
2.5 | 3.4 | 7.0 | 10.9 |
TCDD: 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Respective studies are Mocarelli et al. (2008), Mocarelli et al. (2011) and Mínguez‐Alarcón et al. (2017).
In young boys, Seveso meaning healthy blood donors, for mothers Seveso meaning those that formula‐fed the boys, in the Russian Children's Study the boys in the lowest quartile.
Difference with controls in sperm concentrations not statistically significant.
Estimated based on mean of pooled samples, not measured within the study.
The CONTAM Panel noted that these studies were comparable in size. In contrast to the two Seveso studies, the Russian Children's Study included two semen samples for most participants. The three studies measured sperm concentration in a similar way, and they adjusted for potential confounders. The Russian Children's Study had the advantage of a very narrow age range (18–19 years), while the Seveso studies had a broader age range, and adjusted for age. One drawback with the first study from Seveso (Mocarelli et al., 2008) was that the reference group (healthy blood donors) may in some aspects not be directly comparable with the men from Seveso. In the Seveso studies, semen was collected at home, while in the Russian Children's Study semen was collected in the laboratory, which is another advantage. Analysis of semen was performed within an hour at both sites. A major difference was the measurement of not only TCDD but also other PCDD/Fs and DL‐PCBs in the Russian Children's Study. This study showed effects at the lowest serum levels, with a NOAEL of 7.0 pg WHO2005‐TEQ/g fat for the sum of PCDD/F TEQ, which was the median in the lowest quartile. No significant association was observed when including also the Co‐PCB‐TEQ.32 The CONTAM Panel considered that this may be related to a much lower potency of PCB‐126 in humans than expressed by the current WHO2005‐TEF. An additional possibility is effects of co‐exposure to NDL‐PCBs,28 since the associations between TCDD or total TEQ and semen parameters became slightly stronger after adjustment for these, although there were no significant association between NDL‐PCBs and semen parameters. Therefore, the CONTAM Panel only evaluated the association with PCDD/F‐TEQ levels. For these levels, the median values in quartiles 2–4 were 10.9, 15.9 and 32.8 pg WHO2005‐TEQ/g fat, respectively. The mean sperm concentration in the lowest quartile of PCDD/F‐TEQ was 64 million/mL (Table 10) and the mean levels in quartile 2–4 were about 40 million/mL. This difference was considered biologically relevant.
The CONTAM Panel therefore decided to use the NOAEL of the Russian Children's Study (median serum level of 7.0 pg WHO2005‐TEQ/g fat for the sum of PCDD/F‐TEQ in the lowest quartile) as reference point for the HBGV and for derivation of the human exposure associated with this serum concentration at the age of 9 years.
For experimental animals, as described in Section 3.1.7.3, the lowest dose causing adverse effects corresponded to a LOAEL body burden of 25 ng/kg bw for reduced sperm production in rats born to treated dams (Faqi et al., 1998). Also, in this study, an almost maximal response (decreases in daily sperm production to 60, 54 and 51% of that of controls at PND170) was observed at this dose level. For effects on bone, the body burden at the lowest BMDL05 was estimated to be 14 ng/kg bw (see Table 13), based on the study by Jämsä et al. (2001). The CONTAM Panel noted that the observed effects on bone parameters in rats could be relevant in relation to the mineralisation defects observed on human teeth. With the application of UFs and toxicokinetic modelling, estimated daily intakes would be in a similar range as the one derived from the critical study in humans (see Appendix D for details).
3.1.8.2. Modelling of human dietary exposure associated with the reference point for the HBGV
In order to estimate the intake leading to the critical serum levels or body burden, the CONTAM Panel considered several options. Previously, the SCF used a one‐compartment model to calculate the EDI leading to the critical body burden in women of child‐bearing age, in practice being the steady‐state level. However, several physiological models have been developed that take into account not only accumulation in body fat, but also induction of liver CYP enzymes, liver sequestration and growth. Induction of liver enzymes results in increased clearance and reduced half‐life at higher body burdens. Growth will result in the ‘dilution’ of the existing body burden and thus an apparent shorter half‐life in children. Since the most critical effect was observed in boys exposed before the age of 10 years, i.e. reduced sperm concentrations, a model including growth is more suitable. Furthermore, levels in milk and the duration of breastfeeding influence the serum level and body burden in children, which needs to be taken into account when estimating the daily human intake leading to the critical serum concentration. In addition, infants are already exposed in utero and will have a starting level at birth depending on the body burden of the mother.
For the modelling, it was decided to use an age of 35 years for mothers, in order to cover a common age for having the first child. However, the actual increase in serum levels at child‐bearing age is rather minor (see Figure 13 below).
Figure 13.
Serum levels (Ca, pg/g fat) in a woman breastfed for 12 months in infancy, with milk containing 5.9 pg/g fat, and then exposed to 0.25 pg/kg bw per day for 34 years
Emond model
As a first option, the model for TCDD developed by Emond et al. (2005) was evaluated by transferring the ACSLX codes into Berkeley‐Madonna, and subsequently into R. The adaptations for including a breastfeeding period (Emond et al., 2016) were also evaluated but this model seems to require further investigation.
Using the model without breastfeeding, it was estimated that for a woman of 35 years, a daily intake of 2 pg/kg bw would result in a serum level of 51 pg/g fat, and adipose tissue and liver levels of 19 and 919 pg/g fat. This means a ratio of 2.7 between lipid‐based levels in serum and adipose tissue. When converted to wet weight, the liver and adipose tissue levels would be 62 and 15 pg/g ww (based on lipid levels of, respectively, 6.7 and 80%), implying a ratio of 4.1 between the two. The CONTAM Panel noted several potential discrepancies between these calculations and the data reported on human levels (see Section 3.2.4):
The liver to adipose ratio on wet weight basis for people with low exposure is reported to be around 0.1 for TCDD (see Section 3.2.4), whereas the model arrives at ratios of 4.1, 1.7, 0.8 and 0.6 at exposures of 2, 0.2, 0.02, 0.002 pg/kg bw per day. The higher ratio, even at low exposure implies that a higher fraction of the TCDD body burden is present in the liver. This fraction in the liver can be estimated to be 28, 14, 7 and 5% at intake levels of, respectively, 2, 0.02, 0.2 and 0.002 pg/kg bw per day for a person of 70 kg, based on fractions for adipose tissue and liver of 27 and 2.6% of the body weight, respectively, and assuming no contributions of other tissues. This apparent overestimation by the model of the liver content at low exposure might be due to a too high binding to CYP1A2 or too high liver concentration of CYP1A2, thought to be responsible for the sequestration. However, regarding the relatively low fraction in the liver at low exposure levels, the consequence for the estimated levels in serum and resulting EDI might be low.
It was also noted that in this model, sequestration increases at relatively low body burdens, with a half‐maximal increase in the ratio liver to adipose tissue at a body burden around 50 ng/kg ww. This is lower than the value of 100 ng/kg bw estimated by Carrier et al. (1995), based on rat data for TCDD from Abraham et al. (1988) and human data for 2,3,4,7,8‐PeCDF (300 but 150 ng/kg bw when applying a TEF of 0.5). However, since sequestration occurs at relatively high levels, also this fact may not have a large consequence for estimating the EDI leading to the NOAEL blood level in boys of 7 pg/g fat.
More critical seems the ratio obtained with the Emond model etween the lipid‐based level in serum and adipose tissue, being 2.7, independent of the dosing. Various studies have shown that this ratio is normally around 1 (see Section 3.2.4). When evaluating the total balance in the calculations (intake vs body burden), it appears that the estimated serum levels are too high, rather than that the adipose tissue level would be too low. As a result, the EDI used for establishing the HBGV would be different by a factor of 2.7 when basing it either on serum levels or the level in adipose tissue. When focusing on the NOAEL in boys from the Russian Children's Study of around 7 pg WHO2005‐TEQ/g fat at 9 years of age, the EDI would be either 0.3 when based on the serum level, or 1 pg WHO2005‐TEQ/kg bw per day based on the adipose tissue level, assuming that the level would be similar to that in serum. As already mentioned, this does not take into account the effect of breastfeeding during the first 6 to 12 months in life, since this could not be included in the Emond model.
Concentration‐ and Age‐Dependent Model (CADM)
The CONTAM Panel decided to also evaluate a second so‐called CADM model, being the one developed by Carrier et al. (1995) and optimised by Aylward et al. (2005). The original model takes into account liver sequestration, but was optimised by including the loss of TCDD ‘due to simple lipid partitioning from the circulation across the intestinal lumen into fecal contents’, based on the studies by Moser and McLachlan (1999).
This model was further adapted by Ruiz et al. (2014) to include a growth curve and a breastfeeding period. These model codes for Berkeley Madonna were implemented and evaluated. A number of issues were noted and the model was modified accordingly by the CONTAM Panel (see Appendix E for model codes):
First of all, it turned out that the growth curve started at a body weight around 8 kg for a newborn baby. The CONTAM Panel considered this was unrealistic, and therefore implemented the growth curves applied by Emond et al. (2017) for men (in case of boys) and women (in case of mothers), based on Pelekis et al. (2003).
Second, the exposure after the breastfeeding period was assumed to be constant for the rest of life (pg/day) rather than expressed on a pg per kg bw per day. This was changed to a constant intake per kg bw. However, the about double exposure (per kg bw) during childhood was also evaluated.
The breastfeeding was based on data by Kerger et al. (2007) and could not be varied in terms of concentration in milk and lactation duration. This was overcome by the CONTAM Panel by assuming a constant milk intake of 800 mL during breastfeeding, containing 3.5% milk fat and concentration in milk (in pg/g fat) and duration that can be varied.
Furthermore, the original model assumed that for infants the absorption fraction is rather low, as compared to 97% for adults, but the absorption rate calculated for 10‐month old children in the paper by Abraham et al. (1996) is a clear underestimation (see Section 3.1.1.3). Therefore, absorption was kept constant at 97%.
The relatively short half‐life in infants, expressed in a higher elimination rate by the liver (ke), seems to be primarily based on dilution by growth, which is already taken into account in the model. Therefore, it was assumed by the CONTAM Panel to be similar to adults.
The body burden at birth (termed ‘cb init’) is dependent on that of the mother. Cord blood is often used as an indicator of the body burden in the infant. For TCDD, lipid‐based levels in cord blood and blood of mothers are similar, although fat content of cord blood is lower. Newborns also have a relatively low fat content of around 10%, whereas the CADM assumes 25% throughout life. Such fat content is reached after 6 months and this change in fat percentage over time was not built into the model. However, for the initial body burden expressed per kg bw, the 10% fat content at birth was taken into account by assuming that the TEQ/g fat in the newborn is equal to that in the breast milk.
The CADM model was developed for TCDD and estimates the levels in the fat compartment (lipid based), liver (wet weight) and total body (wet weight), the latter based on relative fractions of 25% and 3% of the body weight for the fat compartment and the liver. Blood levels are not predicted by the model but can be assumed to be similar to those in adipose tissue when adjusted for lipid. Once absorbed, TCDD distributes between liver and fat taking into account induction of CYP1A2 enzyme. As a result, the fraction accumulating in the liver will increase with the dose. Figure 11 shows the sequestration effect for this model, which shifts to higher body burdens and lower maximum storage in liver, compared to the model by Emond. It also shows a lower ratio at low body burdens. Induction of CYP1A2 also results in a shorter half‐life at higher dose levels.
Figure 11.
Fraction of the body burden present in the liver, assuming that all TCDD is present in body fat and liver. Calculations were performed for a 35 year old woman, using the models developed by Emond et al. (2005) and CADM developed by Carrier et al. (1995) and optimised by Aylward et al. (2005). EC 50s for the fraction in the liver are around body burdens of, respectively, 50 and 100 ng/kg bw, corresponding to body fat levels of 95 and 260 ng/kg fat
Using this modified model, a constant intake of 2 pg/kg bw per day by a woman for 35 years would lead to a level in adipose tissue of 31.6 pg/g fat, and levels in liver and whole body of 17.9 and 8.4 pg/g ww. Serum levels are not predicted by the model but assumed to be similar to those in adipose tissue. The TCDD level of 31.6 pg/g fat can be compared with the 51 and 19 pg/g fat for serum and adipose tissue, respectively, observed in the Emond model. Including breastfeeding of these women, when they were infants, for 6 or 12 months with milk containing 32 pg/g fat, would only slightly increase the TCDD level in body fat when these women are 35 years of age (respectively 32.1 and 32.5 pg/g fat). Intake for 35 years of 1, 0.5 and 0.25 pg/kg bw per day is predicted to lead to adipose tissue levels of 19.2, 10.9 and 5.9 pg/g fat (when including breastfeeding for 12 months).
Using CADM, an intake level of 0.5 pg/kg bw per day would result in milk levels between 9 and 11 pg/g fat for women aged 20 and 35 years, respectively. As shown in Section 3.3.1, current median exposure of adults to PCDD/Fs and DL‐PCBs is estimated to be 0.57 (LB) and 0.72 (UB) pg TEQ/kg bw per day. Levels in pooled human milk samples collected between 2009 and 2014 from various European Countries were on average 7.4 pg TEQ/g fat (see Section 3.2.4). This implies that the levels estimated by the CADM are in the correct range although a bit higher than those measured. It should be pointed out that the model was developed for TCDD, which contributes only 3.4% to the current exposure, implying considerable uncertainty with respect to other congeners. The calculations also do not take into account historical exposure which may have been higher, but this would have caused higher levels than those predicted. However, there are indications that the levels in food and also exposure did stabilise during the last decade (Adamse et al., 2017).
Using this model for exposure of boys during the first 9 years in life, without breastfeeding, would lead to levels in body fat of 24, 12.5, 6.5, 3.3 and 1.7 pg/g fat for intakes of 4, 2, 1, 0.5 and 0.25 pg/kg bw per day, respectively (Table 15). This does take into account the level at birth based on a fat fraction of 10% at birth and body fat levels of mothers after similar exposure for 35 years.
Table 15.
Expected serum levels of boys at age 9 years for different durations of breastfeeding (0, 6 and 12 months), and subsequent dietary exposure up to 9 years of age being equal to, or double that, of the mothers
| Intake of mothers (pg/kg bw per day) | Human milk level (pg/g fat)a | Breast feeding duration (months) | Exposure of boys after weaning similar to mothers | Exposure boys after weaning double of mothers | ||
|---|---|---|---|---|---|---|
| Intake by boys (pg/kg bw per day) | Serum level in boys at 9 years (pg/g fat) | Intake by boys (pg/kg bw per day) | Serum level in boys at 9 years (pg/g fat) | |||
| 0.13 | 3.1 |
0 6 12 |
0.13 |
0.8 2.0 3.0 |
0.25 |
1.6 2.7 3.8 |
| 0.25 | 5.9 |
0 6 12 |
0.25 |
1.7 3.7 5.6 |
0.5 |
3.2 5.2 7.0 |
| 0.5 | 10.9 |
0 6 12 |
0.5 |
3.3 6.8 9.9 |
1 |
6.3 9.7 12.6 |
| 1 | 19.2 |
0 6 12 |
1 |
6.5 12.0 16.6 |
2 |
12.3 17.4 21.7 |
| 2 | 32.5 |
0 6 12 |
2 |
12.5 20.4 26.5 |
4 |
23.3 30.2 35.7 |
Ignoring higher intake during mother's childhood.
The breastfeeding has an important effect on the levels in children since exposure expressed per kg bw is rather high. To account for this, first a breastfeeding period of 12 months was modelled. Levels in milk were assumed to be equal to those in the body fat of mothers, resulting from a similar long‐term intake as used for the boys after breastfeeding. Studies have shown that levels in adipose tissue and breast milk are similar (see Section 3.2.4). As an example, an intake by mothers of 2 pg/kg bw per day results in a body fat level of 32.5 pg/g fat. Figure 12 shows that in boys, breastfeeding with milk containing this level results in a peak level in body fat at 12 months of around 130 pg/g fat, which then decreases to a level at 9 years of 26.5 pg/g fat. Taking into account that after breastfeeding the intake of children is twice as high as that of adults, because of a higher energy requirement (i.e. 4 pg/kg bw per day in this scenario), this would result in a body fat level of 35.7 pg/g fat at age 9 years. This twofold higher exposure of children was confirmed for those consumption surveys that included the various age groups (see Annex B). Decreasing the breastfeeding period to 6 months would decrease this level to 30.2 pg/g fat, as compared to 20.4 pg/g fat when assuming a similar intake by the boys as the mothers, i.e. 2 pg/kg bw per day. Table 15 shows serum levels for these and other scenarios.
Figure 12.
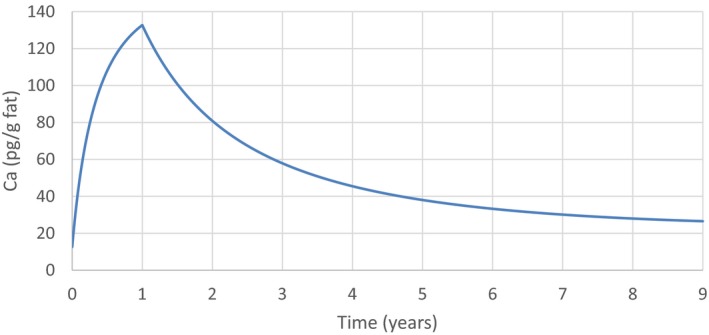
Levels in adipose tissue (Ca, pg/g fat) calculated for boys exposed for 9 years to 2 pg/kg bw per day after a breastfeeding period of 12 months with levels in milk of 32.5 pg/g fat (i.e. the level resulting from exposure of mothers for 35 years to 2 pg/kg bw per day)
The results in Table 15 are based on the assumption that lipid‐based serum levels in the infants at birth are similar to those in the mother, and thus in human milk. In practice, several studies show that for TCDD this may be the case but not for PCDD/F‐TEQ and DL‐PCB‐TEQ. For PCDD/F‐TEQ, the ratio was reported to be around 0.6, for DL‐PCB‐TEQ even lower. However, when reducing the initial serum level of the infant by a factor 2, this had only a minor effect on the serum level at 9 years of age.
The data show that breastfeeding contributes considerably to the serum levels at 9 years of age, at lower levels at least doubling the serum level. The milk levels are not linear with the exposure of mothers, due to sequestration and higher elimination at higher exposure. As a result the impact of breastfeeding decreases at higher exposure of mothers.
3.1.8.3. Setting of the HBGV
The CONTAM Panel decided that the data from epidemiological studies were adequate for deriving the HBGV, and to use the data obtained with experimental animals only as supportive evidence.
Similar to the SCF in 2001, and in line with what is done for other persistent compounds, the CONTAM Panel decided to set the HBGV on a weekly basis because the risk is derived from the long‐term accumulation of these compounds in the body rather than from an occasional elevated exposure. Setting the HBGV on a weekly base was judged to be acceptable, but extending it to a month may be interpreted as accepting a high temporary increase in serum levels, and as such, an increased exposure of sensitive tissues during a critical window (see Appendix F for details).
It was decided to base the HBGV on the NOAEL of 7.0 pg PCDD/F‐WHO2005‐TEQ/g serum fat, being the median serum level in the lowest quartile, for effects on sperm concentration in the Russian Children's Study (Mínguez‐Alarcón et al., 2017). The CADM simulations indicate that following breastfeeding for 12 months, and a similar intake of sons after breastfeeding as for mothers, the intake should be below 0.3 pg TEQ/kg bw per day in order not to reach a serum concentration of 7.0 pg PCDD/F‐WHO2005‐TEQ/g fat at 9 years of age.
When taking into account 12 months breastfeeding followed by twofold higher intake by boys than by adults, the intake by the mothers should be below 0.25 pg PCDD/F‐WHO2005‐TEQ/kg bw per day.
Figure 13 below shows a scenario on the serum levels in a woman breastfed for 12 months in infancy, and then exposed to 0.25 pg/kg bw per day for 34 years. The concentration in the milk was 5.9 pg/g fat, and assumed to be constant. This milk concentration is based on the predicted level of the body fat of her mother at age 35, having the same exposure scenario. The body burden at birth was set at 0.59 pg/g ww, based on 10% body fat in the infant and the concentration in the body fat of the mother of 5.9 pg/g fat.
The next graph (Figure 14) shows the serum level in boys breastfed for 12 months with this milk with 5.9 pg/g fat (800 mL per day, 3.5% fat), followed by a twofold higher intake of 0.5 pg/kg bw per day for an additional 8 years, thus resulting in a serum level of 7 pg/g fat.
Figure 14.
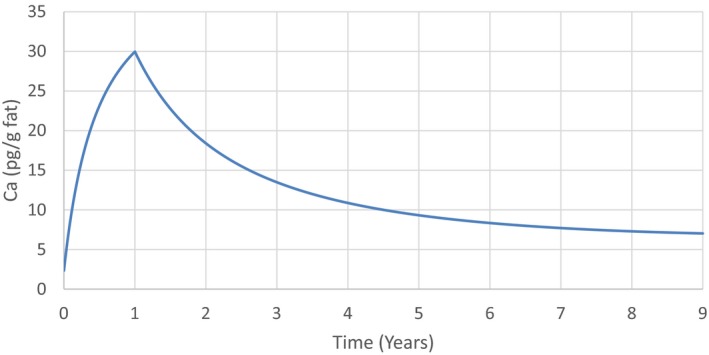
Serum level (Ca, pg/g fat) in boys breastfed for 12 months with milk with 5.9 pg/g fat (800 mL per day, 3.5% fat), followed by an intake of 0.5 pg/kg bw per day for an additional 8 years, thus resulting in a serum level of 7 pg/g fat
Since this HBGV is based on a NOAEL obtained in a study with a relatively large number of boys (n = 133) and repeated semen sampling, it was decided not to apply additional UFs.
Considering the discussion above, the data suggest that the long‐term intake should remain below 0.25 pg WHO2005‐TEQ/kg bw per day or 1.75 pg WHO2005‐TEQ/kg bw per week to ensure that serum levels in boys remain below the NOAEL for effects on sperm concentrations of 7.0 pg WHO2005‐TEQ/g fat, also when breastfed for 12 months. The value of 1.75 was rounded to 2 considering the uncertainty in the estimation of the critical serum level and corresponding daily intake (see Section 3.5).
Therefore, a TWI was established of 2 pg WHO2005‐TEQ/kg bw per week.
Although this TWI is based on findings on PCDD/F‐TEQ only, the CONTAM Panel concluded that the TWI should apply to the sum of PCDD/Fs and DL‐PCBs. However, the studies indicate that the current TEFs require re‐evaluation. In particular, PCB‐126, which contributes most to the DL‐PCB‐TEQ level, may be less potent in humans than indicated by the TEF‐value of 0.1 (see Section 3.1.7.2.1).
The CONTAM Panel noted that the TWI is based on serum levels sampled from boys at the age of 8–9 years. The critical window for the effects on sperm may actually be at younger age or during puberty. The TWI is protective for the general population and prevents women from reaching a concentration in the blood that could lead to harmful pre‐ and postnatal effects. The modelling of concentrations in serum takes into account the much higher exposure during infancy from both breast milk and food. The CONTAM Panel considered the modelling sufficiently accurate because there are no indications that the serum levels in the boys from the Russian Children's Study during the first 9 years would have followed a different pattern than predicted by the model.
Based on the available data, this TWI should be protective towards all endpoints. These include lower sex ratio, higher TSH levels in newborns and developmental enamel defects on teeth, the latter appearing to occur at only slightly higher exposure than the developmental effects on semen quality (see Section 3.1.7.1.4).
3.1.9. Critical effects and reference points in farm and companion animal studies
For most farm and companion animals, no studies were identified that could be used for deriving a NOAEL or LOAEL. For chickens, a NOAEL of 5.6 ng/kg bw per day and LOAEL of 1,099 ng/kg bw per day was identified, showing that egg production had ceased after 12 days of treatment with a high dose of TCDD.
Many studies have been performed on different species of farmed fish, but only few of these were suitable to derive a NOAEL/LOAEL. The lowest NOAEL in rainbow trout was 11 ng TCDD/kg bw (and a LOAEL of 6.3 μg TCDD/kg bw), where other studies indicated NOAELs of 0.1 μg TCDD/kg bw or higher. For yellow perch and tilapia, a NOAEL of 1 μg TCDD/kg bw was identified and for carp 0.57 μg TCDD/kg bw. A feeding study with Atlantic salmon showed no effects after prolonged exposure to PCDD/Fs and DL‐PCBs at 20 pg WHO2005‐TEQ/kg bw per day, but this was the highest dose tested.
Several studies with mink were identified. A two‐generation study with mink orally exposed to TCDD, showed the lowest NOAEL of 2.1 ng/kg bw per day for the characteristic morphological change caused by TCDD and other PCDD/Fs and DL‐PCBs, i.e. proliferation of the squamous gingival epithelium in the mouth of juveniles.
3.2. Occurrence data
3.2.1. Occurrence data submitted to EFSA
A total of 1,006,148 results (individual congeners) from 46,982 samples on PCDD/Fs and DL‐PCBs on food and feed were available in the EFSA database. The CONTAM Panel decided to only consider the samples taken after year 2010. Thus, 176,351 result from preceding years (2002–2009) were excluded from the original data set. The resulting data set included a total of 824,905 analytical results corresponding to 35,252 samples submitted to EFSA between 2010 and December 2016.
Data providers were contacted to clarify a number of possible inconsistencies which were identified during the data check. The following modifications were made to the initial data set based on the feedback received:
All results were converted to the unit of measure (pg/g) laid down in Commission Regulation (EC) No 1881/200633 as subsequently amended.34
The product description of a number of records allowed a more accurate FoodEx classification. In these cases, the samples were reclassified to a more specific, lower level.
Samples reported as ‘suspect sampling’ (32,334 results corresponding to 1,328 samples) or prescreened with bioanalytical methods (e.g. CALUX bioassay) and found positive before their confirmatory analysis (7,360 results corresponding to 311 samples) were not taken into account in the current assessment.
Only results of samples reported to be analysed by GC–HRMS or GC–MS/MS (reported by the data providers also as HRGC–HRMS, GC–HRMS (magnetic sector) or GC–QqQ‐MS/MS) were considered sufficiently reliable to be included in the assessment. Results obtained with other analytical methods or without information on the analytical method were excluded from the assessment (49,519 results corresponding to 4,438 samples).
18 samples with extremely high values or duplicate submission of congeners were eliminated after confirmation of a possible error by the data provider.
2,159 analytical results corresponding to 142 samples were found to be duplicate submission and were disregarded.
1,652 results corresponding to 588 samples were confirmed as originating from Total Diet Studies (TDS). These were disregarded as it would cause a bias to mix data on processed food with the results of analyses on unprocessed food (which is the case for the vast majority of the data). Results of TDS studies published in the literature are reported in Sections 3.3.2 and 3.3.1.
Expression of results
According to Commission Regulation (EC) No 1881/2006 as subsequently amended,34 levels of foodstuffs of terrestrial animal origin except offals, marine oils and vegetable fats and oils are to be given on a fat weight basis. Products of aquatic origin except marine oil, offals of terrestrial animals and products of plant origin except vegetable fats and oils are expressed on a whole weight basis; feedstuffs have to be expressed on 88% dry matter basis.
Samples which should have been reported on fat weight basis according to legislation but were submitted to EFSA on whole weight basis, and for which transformation was not possible because their fat content was missing, were excluded from the current assessment (n = 3,324 results corresponding to 1,186 samples). Results for feed samples that were not expressed on 88% dry matter (30%) were recalculated with an assumed moisture content of the matrix, since excluding those samples would have led to the loss of a substantial number of feed samples. For the assumption of moisture content of different feedstuffs, Feedipedia,35 an open access information system on animal feed resources, developed by a joint project of FAO and three other organisations, was used.
Missing values for individual congeners
A total of 5,838 analytical results corresponding to 1,946 samples in which at least one congener out of the 17 PCDD/Fs or 12 DL‐PCBs group was missing were excluded from the data set (see Annex B, Table B.1).
3.2.1.1. Occurrence data on food
Application of performance criteria
The data set for the current human exposure assessment contained 24,987 food samples, in which at least either all of the 17 PCDD/Fs, or all of the 12 DL‐PCBs were analysed.
These samples were checked for compliance with analytical performance criteria based on Commission Regulation (EU) No 2017/644 and taking into account the existing Maximum Levels (MLs) as laid down in Commission Regulation (EC) No 1881/2006 as subsequently amended,34 and Action Levels (ALs) as given in Commission Recommendation 2014/663/EU. These were expressed in pg WHO2005‐TEQ/g.
Some of the legislation categories of the MLs and ALs were not perfectly in line with the ones defined by the FoodEx system for the occurrence data. In case no other information was available for further specifying the given FoodEx category, the following assumptions were made for the application of the performance criterion on the occurrence data:
The legislation distinguishes and sets different MLs for wild caught and farmed eels. Where the information was not available on the production method of the eel, it was assumed to be farmed.
In addition, in case of meat products, sausages, as no information was available on their source animal, all meat products were assumed to be made of pig meat and the corresponding ML was applied on them.
In 2012, in the last DATA report on the monitoring of PCDD/Fs and DL‐PCBs in food and feed (EFSA, 2012), two criteria for analytical performance were applied to reduce the uncertainty in the exposure assessment. First of all, samples with an LOQ higher than one fifth of the corresponding MLs for the sum of PCDD/Fs and DL‐PCBs were excluded from the assessment. In addition, LB and UB estimates were compared as a second criterion. Samples were excluded when the percentage difference between the UB and LB estimates of the TEQ‐based sum of all congeners, taking into account UB as a reference, was greater than a predefined threshold value. The cut‐off percentage varied depending on the level in the sample, meaning that at lower levels a larger difference was accepted. The first criterion was also applied in the current assessment. However, the second criterion was not applied as the CONTAM Panel considered that its application would result in an overestimation on the exposure, because it was shown that in particular data in the lower end of the distribution were excluded. See some examples in Table 16.
Table 16.
Effect of application of the second criterion (i.e. based on the difference between LB and UB estimates of the TEQ‐based sum of all congeners) on the occurrence values and on the number of samples presented on example categories
| Occurrence values and number of samples without application of the second criterion: | ||||||||
|---|---|---|---|---|---|---|---|---|
| FOODEX L1 | FOODEX L2 | FOODEX L3 | Expression of results | N | Mean LB | Mean UB | P95 LB | P95 UB |
| pg WHO2005‐TEQ/g | ||||||||
| Animal and vegetable fats and oils | Animal fat | Pork lard | Fat weight | 489 | 0.10 | 0.21 | 0.32 | 0.40 |
| Animal and vegetable fats and oils | Animal fat | Tallow | Fat weight | 547 | 0.62 | 0.73 | 1.73 | 1.79 |
| Eggs and egg products | Eggs, fresh | Eggs, fresh | Fat weight | 134 | 0.74 | 0.88 | 3.21 | 3.21 |
| Eggs and egg products | Eggs, fresh | Whole egg, chicken | Fat weight | 2,312 | 1.18 | 1.31 | 4.32 | 4.32 |
| Milk and dairy products | Liquid milk | Cow milk | Fat weight | 935 | 0.75 | 0.92 | 1.79 | 2.01 |
| Milk and dairy products | Liquid milk | Goat milk | Fat weight | 102 | 1.21 | 1.40 | 3.71 | 3.73 |
| Fish and other seafood | Fish meat | Fish meat | Whole weight | 565 | 0.80 | 0.82 | 3.19 | 3.19 |
| Fish and other seafood | Fish meat | Salmon and trout | Whole weight | 857 | 0.87 | 0.94 | 5.82 | 5.82 |
| Animal and vegetable fats and oils | Animal fat | Pork lard | Fat weight | 427 | 0.10 | 0.20 | 0.35 | 0.44 |
| Animal and vegetable fats and oils | Animal fat | Tallow | Fat weight | 391 | 0.79 | 0.86 | 2.10 | 2.16 |
| Eggs and egg products | Eggs, fresh | Eggs, fresh | Fat weight | 86 | 1.17 | 1.22 | 3.98 | 3.99 |
| Eggs and egg products | Eggs, fresh | Whole egg, chicken | Fat weight | 1,617 | 1.72 | 1.77 | 6.16 | 6.22 |
| Milk and dairy products | Liquid milk | Cow milk | Fat weight | 660 | 0.89 | 0.94 | 2.05 | 2.15 |
| Milk and dairy products | Liquid milk | Goat milk | Fat weight | 70 | 1.57 | 1.68 | 4.17 | 4.19 |
| Fish and other seafood | Fish meat | Fish meat | Whole weight | 550 | 0.82 | 0.83 | 3.75 | 3.75 |
| Fish and other seafood | Fish meat | Salmon and trout | Whole weight | 777 | 1.00 | 1.04 | 6.36 | 6.39 |
LB: lower bound; P95: 95th percentile; UB: upper bound.
In this Scientific Opinion, samples with an LOQ higher than one‐fifth of the corresponding ML for the sum of PCDD/Fs or higher than one third of the corresponding AL (action level) for the sum of DL‐PCBs were excluded as follows:
For the sum of PCDD/Fs,
For samples based on whole weight and on fat weight with more than 2% fat, where the full set of 17 PCDD/Fs were reported, those for which the LOQ for the PCDD/F‐TEQ was higher than one fifth of the corresponding ML for the 17 PCDD/Fs, were excluded.
The ML level expressed on a fat basis is not applicable for specific foods containing < 2% fat. For these foods the ML is applicable on a product basis (whole weight basis), calculated from the ML established on a fat basis multiplied by 0.02. If the total LOQ was higher than one‐fifth of the ML × 0.02, the sample was excluded. After this step the samples were transformed to fat weight.
A total of 822 food samples did not comply with the criterion and were therefore excluded.
For the sum of DL‐PCBs,
For samples on whole weight and on fat weight with more than 2% fat, where the full set of 12 DL‐PCBs were reported, those for which the LOQ for DL‐PCB‐TEQ was higher than one third of the AL for the 12 DL‐PCBs were excluded. For samples with less than 2% fat, this criterion was not applied, as indicated in Commission Recommendation 2014/663/EU.
A total of 46 food samples did not comply with the criterion and were therefore excluded.
The criterion was not applied separately on the sum of PCDD/Fs and DL‐PCBs with their corresponding MLs, but the samples fulfilling the above two criteria have been included in the final data set.
Overall, the final food data set that contained samples fulfilling the above criterion and all the conditions listed in Section 3.2.1 were:
19,965 food samples with all 29 congeners determined (17 PCDD/Fs and 12 DL‐PCBs);
20,273 food samples with all 17 PCDD/F congeners determined (including samples with the 29 congeners);
22,974 food samples with all 12 DL‐PCB congeners determined (including samples with the 29 congeners).
The food samples were submitted by 23 European countries (Figure 15) between the years 2010 and 2016 (Figure 16). The Mean and P95 levels for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs in a number of food categories are shown in Table 17. More data on summary statistics of occurrence values are presented in Annex B (Table 2a,b,c, respectively).
Figure 15.
Number of food samples in the final data set encoded by European countries (n = 20,273 food samples with results for the 17 PCDD/Fs: from these 19,965 also had the 12 DL‐PCBs reported)
Figure 16.
Number of food samples in the final data set reported by year (n = 20,273 food samples with results for the 17 PCDD/Fs: from these 19,965 also had the 12 DL‐PCBs reported)
Table 17.
Mean and P95 levels for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs in selected food groups (see Annex B for additional food items)
| Foodstuff | Unit | Sum of PCDD/Fs (WHO2005‐PCDD/F‐TEQ) | Sum of PCDD/Fs and DL‐PCBs (WHO2005‐PCDD/F‐PCB‐TEQ) | ||||
|---|---|---|---|---|---|---|---|
| n | Mean (LB/UB) | P95a (LB/UB) | n | Mean (LB/UB) | P95a (LB/UB) | ||
| Meat and meat products (excluding edible offal) | pg/g fat | ||||||
| Beef | 869 | 0.53/0.61 | 1.68/1.68 | 866 | 2.14/2.23 | 6.08/6.08 | |
| Veal | 152 | 0.31/0.45 | 1.09/1.18 | 152 | 1.32/1.47 | 4.57/4.69 | |
| Sheep | 241 | 0.50/0.57 | 1.43/1.43 | 240 | 0.95/1.05 | 2.55/2.56 | |
| Chicken | 573 | 0.14/0.26 | 0.45/0.58 | 565 | 0.30/0.43 | 0.93/1.09 | |
| Duck | 97 | 0.25/0.32 | 0.56/0.61 | 96 | 0.40/0.48 | 1.17/1.29 | |
| Turkey | 145 | 0.12/0.25 | 0.51/0.60 | 145 | 0.26/0.42 | 1.02/1.36 | |
| Pigs | 459 | 0.08/0.16 | 0.33/0.36 | 454 | 0.14/0.24 | 0.45/0.52 | |
| Horse, asses, mules or hinnies | pg/g fat | 80 | 2.65/2.68 | 7.10/7.19 | 80 | 6.23/6.26 | 15.9/16.0 |
| Boar meat | pg/g fat | 207 | 2.61/2.78 | 10.5/10.6 | 207 | 5.28/5.45 | 24.6/24.6 |
| Venison meat | 148 | 0.89/1.00 | 2.53/2.64 | 148 | 3.51/3.63 | 9.32/9.32 | |
| Liver of terrestrial animals referred to in 5.1 with the exception of sheep and derived products thereof | pg/g ww | ||||||
| Beef | 183 | 0.06/0.07 | 0.19/0.19 | 181 | 0.15/0.15 | 0.41/0.41 | |
| Veal | 74 | 0.04/0.05 | 0.12/0.12 | 74 | 0.10/0.11 | 0.30/0.30 | |
| Chicken | 18 | 0.02/0.03 | – | 18 | 0.02/0/04 | – | |
| Pigs | 55 | 0.12/0.13 | – | 55 | 0.13/0.14 | – | |
| Liver of sheep and derived products thereof | pg/g ww | 282 | 0.49/0.51 | 1.77/1.81 | 282 | 0.71/0.74 | 2.29/2.29 |
| Liver of game animals | pg/g ww | 75 | 1.72/1.73 | 7.28/7.28 | 75 | 2.40/2.40 | 13.5/13.5 |
| Muscle meat of fish and fishery products | pg/g ww | ||||||
| Salmon and trout | 907 | 0.27/0.33 | 1.93/1.95 | 857 | 0.88/0.94 | 5.82/5.82 | |
| Herring | 401 | 1.22/1.25 | 3.37/3.37 | 399 | 2.34/2.39 | 6.36/6.36 | |
| Mackerel | 322 | 0.37/0.43 | 1.23/1.24 | 317 | 1.36/1.44 | 4.72/4.78 | |
| Eels | 258 | 0.97/1.00 | 3.28/3.28 | 258 | 9.17/9.21 | 32.2/32.2 | |
| Sprat | 91 | 1.57/1.58 | 2.86/2.87 | 91 | 3.43/3.47 | 6.39/6.40 | |
| Sardine/pilchard | 177 | 0.29/0.29 | 0.89/0.89 | 177 | 1.65/1.66 | 3.96/3.96 | |
| Bream | 106 | 0.61/0.62 | 2.41/2.41 | 100 | 2.92/2.94 | 12.9/13.0 | |
| Carp | 88 | 0.09/0.13 | 0.16/0.19 | 87 | 0.32/0.42 | 0.58/0.62 | |
| Cod and whiting | 384 | 0.04/0.08 | 0.09/0.21 | 375 | 0.17/0.28 | 0.48/0.88 | |
| Halibut | 466 | 0.31/0.35 | 0.92/0.94 | 466 | 1.12/1.16 | 3.32/3.36 | |
| Tuna | 117 | 0.01/0.04 | 0.05/0.16 | 101 | 0.13/0.17 | 0.45/0.45 | |
| Crab | 275 | 0.62/0.63 | 2.28/2.28 | 274 | 1.26/1.27 | 4.18/4.18 | |
| Mussel | 325 | 0.18/0.21 | 0.57/0.59 | 320 | 0.54/0.57 | 1.68/1.68 | |
| Oyster | 235 | 0.41/0.41 | 0.93/0.93 | 235 | 0.89/0.89 | 2.11/2.11 | |
| Fish meat unspecified | 589 | 0.14/0.16 | 0.62/0.62 | 655 | 0.80/0.82 | 3.19/3.19 | |
| Fish offal | pg/g ww | 911 | 4.33/4.89 | 12.7/13.1 | 911 | 21.7/22.0 | 60.3/60.5 |
| Marine oils (for human consumption) | pg/g fat | ||||||
| Fish oil | 21 | 0.13/0.24 | – | 21 | 1.22/1.34 | – | |
| Cod liver oil | 7 | 0.55/0.63 | – | 7 | 3.01/3.09 | – | |
| Raw milk and dairy products, including butter fat | pg/g fat | 1,801 | 0.28/0.44 | 0.92/1.06 | 1,786 | 0.73/0.88 | 1.92/2.04 |
| Hen eggs and egg products | pg/g fat | ||||||
| Chicken | 2,338 | 0.47/0.58 | 1.79/1.79 | 2,312 | 1.18/1.31 | 4.32/4.32 | |
| Duck | 67 | 0.76/0.79 | 2.51/2.51 | 67 | 1.05/1.08 | 3.91/3.91 | |
| Fat of the following animals: | pg/g fat | ||||||
| Bovine animals and sheep (tallow) | 550 | 0.23/0.34 | 0.82/0.85 | 547 | 0.62/0.73 | 1.73/1.79 | |
| Chicken | 109 | 0.31/0.42 | 0.58/0.62 | 107 | 0.14/0.26 | 0.53/0.70 | |
| Pigs | 494 | 0.07/0.17 | 0.23/0.31 | 489 | 0.10/0.21 | 0.32/0.40 | |
| Vegetable oils | pg/g fat | 206 | 0.06/0.12 | 0.32/0.40 | 205 | 0.08/0.16 | 0.35/0.43 |
| Foods for infants and young children(g) | pg/g ww | 500 | 0.00/0.01 | 0.02/0.04 | 472 | 0.01/0.02 | 0.04/0.07 |
| Vegetables and vegetable products (including fungi) | pg/g ww | 164 | 0.02/0.05 | 0.12/0.21 | 136 | 0.05/0.08 | 0.26/0.28 |
LB: lower bound; UB: upper bound; P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
The mean and P95 LB/UB levels of the sum of the 17 PCDD/Fs and 12 DL‐PCBs (29 congeners) in ‘Livestock meat including offal’ were, respectively, 1.43/1.54 and 5.06/5.12 pg WHO2005‐TEQ/g fat weight. In various species within ‘Livestock meat’, the mean levels ranged from 0.12/0.20 to 6.23/6.26 pg WHO2005‐TEQ/g fat weight.
In ‘Milk and milk products’, the mean and P95 LB/UB levels of the sum of the 29 congeners were, respectively, 0.73/0.88 and 1.92/2.04 pg WHO2005‐TEQ/g fat weight, in eggs and egg products 1.17/1.30 and 4.38/4.39 pg WHO2005‐TEQ/g fat weight, in ‘Animal and vegetable fat’ 0.42/0.53 and 1.59/1.65 pg WHO2005‐TEQ/g fat weight, in Vegetables 0.05/0.08 and 0.26/0.28 pg WHO2005‐TEQ/g whole weight, and in ‘Fish and seafood’ 4.35/4.45 and 21.0/21.6 pg WHO2005‐TEQ/g whole weight. For various fish species, the mean LB/UB levels ranged from 0.10/0.10 to 9.17/9.21 pg WHO2005‐TEQ/g whole weight.
For the 17 PCDD/Fs, the mean and P95 LB/UB levels in ‘Livestock meat including offal’ were, respectively, 0.50/0.60 and 1.54/1.61 pg WHO2005‐TEQ/g fat weight. The levels varied between different species of ‘Livestock meat’, showing mean LB/UB levels from 0.08/0.16 to 2.65/2.68 pg WHO2005‐TEQ/g fat weight. In ‘Milk and milk products’, the mean and P95 LB/UB levels were, respectively, 0.28/0.43 and 0.92/1.06 pg WHO2005‐TEQ/g fat weight, in ‘Eggs and egg products’ 0.51/0.62 and 2.02/2.02 pg WHO2005‐TEQ/g fat weight, in ‘animal and vegetable fat’ 0.20/0.29 and 0.66/0.70 pg WHO2005‐TEQ/g fat weight, in ‘Vegetables’ 0.02/0.05 and 0.12/0.21 pg WHO2005‐TEQ/g whole weight, and in ‘Fish and seafood’ 0.95/1.05 and 4.30/4.66 pg WHO2005‐TEQ/g whole weight. The levels varied between various fish species, showing mean LB/UB levels from 0.01/0.04 to 2.66/2.67 pg WHO2005‐TEQ/g whole weight.
The highest mean LB/UB concentrations for the sum of PCDD/Fs and DL‐PCBs (29 congeners) were found in some rarely consumed foods such as certain game birds (Mallard meat’ and ‘Pheasant meat’ with 39.8/39.8 and 8.29/8.55 pg WHO2005‐TEQ/g fat weight, respectively‘), ‘Fish liver’ (22.1/22.6 pg WHO2005‐TEQ/g whole weight), and ‘Brown meat of crabs’ (6.10/6.17 pg WHO2005‐TEQ/g whole weight). High mean LB/UB concentrations of the 17 PCDD/F congeners were found in the same categories: game birds (Mallard meat’ and ‘Pheasant meat’ with 2.16/2.19 and 1.76/2.02 pg WHO2005‐TEQ/g fat weight, respectively‘), ‘Fish liver’ (4.41/4.95 pg WHO2005‐TEQ/g whole weight), and ‘Brown meat of crabs’ (3.22/3.29 pg WHO2005‐TEQ/g whole weight).
Special food groups
Special food groups were identified in the occurrence data for which a more specific categorisation was possible than the FoodEx1. As distinguishing these groups was not possible in the consumption data (Comprehensive Database), only the occurrence data was presented in this form.
Chicken Eggs
Standard Sample Description (SSD) system of EFSA allows adding specific information about the production method of the sample. Submitting this information is not compulsory, but may be added by the data provider. Definitions of possible production methods according to the SSD system is shown in Table 18.
Table 18.
Definitions of production methods according to the Standard Sample Description system
| Production method | Definition |
|---|---|
| Battery productiona | Production of animals in cages (applies to poultry, rabbits) |
| Free range productionb | Animals have continuous daytime access to open air enclosures covered with vegetation |
| Traditional production | Production of food using traditional / artisan methods |
| Organic production | A method of production which places the highest emphasis on environmental protection and, with regard to livestock production, animal welfare considerations. It avoids or largely reduces the use of synthetic chemical inputs such as fertilisers, pesticides, additives and medicinal products. ( http://www.organic-europe.net/europe_eu/statistics-eurostat.asp) |
| Non‐organic production | Products produced without use of organic production methods |
| Farmed domestic or cultivated | Animals produced in captivity (applies also to game and fish), plants produced by cultivation |
| Outdoor/Open‐air growing condition | Cultivation of plants and rearing of animals without the use of climate‐controlled or protective structures |
| Production method unknown | |
| Wild or gathered or hunted | Animal or plant products harvested from their natural environment |
Battery production of eggs is prohibited since 1 January 2012 (Council Directive 1999/74/EC).
The category possibly includes eggs where the chickens were non‐caged, but they were not in outdoor conditions. Note that interpretation of categories could be different among the data providers.
Differences between chicken eggs from battery, free range and other types of production were investigated and the occurrence values for the different categories are shown in Tables 19, 20 and 21 for the sum of PCDD/Fs and DL‐PCBs, for the 17 PCDD/Fs and for the 12 DL‐PCBs, respectively. In general, eggs from hens with outdoor access showed higher levels of both PCDD/Fs and DL‐PCBs than those kept indoors, assuming that the latter also applies to most of the free range samples.
Table 19.
Sum of PCDD/F and DL‐PCB levels expressed in pg WHO2005‐TEQ/g fat weight in chicken eggs among different production methods
| Production method of chicken eggs | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Outdoor/Open‐air growing condition | 412 | 1.58 | 0.36 | 5.12 | 1.69 | 0.56 | 5.13 |
| Organic production | 419 | 1.18 | 0.62 | 3.68 | 1.28 | 0.71 | 3.79 |
| Non‐organic production | 125 | 1.35 | 0.37 | 5.25 | 1.47 | 0.61 | 5.25 |
| Farmed Domestic or cultivated | 21 | 0.18 | 0.10 | – | 0.53 | 0.49 | – |
| Battery production | 102 | 0.20 | 0.12 | 0.50 | 0.36 | 0.26 | 0.65 |
| Free range production | 524 | 0.58 | 0.16 | 2.41 | 0.78 | 0.43 | 2.47 |
| Traditional production | 37 | 0.57 | 0.18 | – | 0.64 | 0.25 | – |
| Production method unknown | 983 | 1.39 | 0.30 | 4.60 | 1.49 | 0.43 | 4.61 |
P95: 95th percentile; DL‐PCB: dioxin‐like polychlorinated biphenyl; PCDD/F: polychlorinated dibenzo‐p‐dioxin and dibenzofuran; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 20.
Sum of PCDD/F levels expressed in pg WHO2005‐TEQ/g fat weight in chicken eggs among different production methods
| Production method of eggs | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Outdoor/Open‐air growing condition | 414 | 0.46 | 0.15 | 1.95 | 0.57 | 0.34 | 1.95 |
| Organic production | 420 | 0.52 | 0.27 | 1.70 | 0.61 | 0.39 | 1.75 |
| Non‐organic production | 125 | 0.92 | 0.19 | 3.60 | 1.04 | 0.42 | 3.62 |
| Farmed Domestic or cultivated | 21 | 0.13 | 0.10 | – | 0.36 | 0.34 | – |
| Battery production | 103 | 0.10 | 0.05 | 0.26 | 0.24 | 0.17 | 0.48 |
| Free range production | 530 | 0.37 | 0.07 | 1.54 | 0.53 | 0.31 | 1.56 |
| Intensive Industrial production | 1 | – | 0.09 | – | 0.19 | – | |
| Traditional production | 37 | 0.20 | 0.09 | – | 0.26 | 0.14 | – |
| Production method unknown | 1,000 | 0.56 | 0.16 | 1.87 | 0.64 | 0.28 | 1.87 |
P95: 95th percentile; PCDD/Fs: polychlorinated dibenzo‐p‐dioxin and dibenzofuran; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 21.
Sum of DL‐PCB levels expressed in pg WHO2005‐TEQ/g fat weight in chicken eggs among different production methods
| Production method of eggs | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Outdoor/Open‐air growing condition | 443 | 1.09 | 0.20 | 3.13 | 1.10 | 0.20 | 3.13 |
| Organic production | 449 | 0.65 | 0.27 | 1.43 | 0.66 | 0.28 | 1.43 |
| Non‐organic production | 134 | 0.42 | 0.18 | 1.54 | 0.42 | 0.18 | 1.54 |
| Farmed Domestic or cultivated | 21 | 0.05 | 0.00 | – | 0.16 | 0.14 | – |
| Battery production | 136 | 0.12 | 0.06 | 0.50 | 0.14 | 0.07 | 0.51 |
| Free range production | 556 | 0.21 | 0.07 | 0.75 | 0.24 | 0.13 | 0.75 |
| Traditional production | 37 | 0.37 | 0.09 | – | 0.38 | 0.11 | – |
| Production method unknown | 1,060 | 0.87 | 0.14 | 2.89 | 0.88 | 0.15 | 2.89 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Eel
In Tables 22, 23 and 24, the occurrence values for the sum of PCDD/Fs and DL‐PCBs, the 17 PCDD/Fs or the 12 DL‐PCBs, respectively, in eel according to different production methods as reported by the data providers are shown. Comparing the differences between farmed and wild caught eel, it was concluded that levels in farmed eel were substantially lower, but the number of samples was low. DL‐PCBs contribute much more to the total TEQ levels than PCDD/Fs.
Table 22.
Sum of PCDD/Fs and DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in eel among different production methods
| Production method of eel | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Non‐organic production | 12 | 3.31 | 2.89 | – | 3.31 | 2.90 | – |
| Wild or gathered or hunted | 126 | 10.72 | 8.75 | 23.23 | 10.73 | 8.75 | 23.23 |
| Farmed Domestic or cultivated | 19 | 2.17 | 1.62 | – | 2.20 | 1.62 | – |
| Production method unknown | 101 | 9.24 | 3.66 | 37.35 | 9.32 | 3.88 | 37.35 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 23.
Sum of PCDD/Fs expressed in pg WHO2005‐TEQ/g whole weight in eel among different production methods
| Production method of eel | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Non‐organic production | 12 | 1.18 | 0.78 | – | 1.19 | 0.79 | – |
| Wild or gathered or hunted | 126 | 0.94 | 0.71 | 2.32 | 0.95 | 0.73 | 2.32 |
| Farmed Domestic or cultivated | 19 | 0.38 | 0.36 | – | 0.41 | 0.40 | – |
| Production method unknown | 101 | 1.08 | 0.51 | 3.93 | 1.16 | 0.59 | 3.93 |
P95: 95th percentile; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 24.
Sum of DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in eel among different production methods
| Production method of eel | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Non‐organic production | 12 | 2.12 | 1.69 | – | 2.12 | 1.69 | – |
| Wild or gathered or hunted | 126 | 9.78 | 8.22 | 22.23 | 9.79 | 8.22 | 22.23 |
| Farmed Domestic or cultivated | 19 | 1.79 | 1.23 | – | 1.79 | 1.23 | – |
| Production method unknown | 119 | 8.92 | 3.67 | 38.12 | 8.92 | 3.69 | 38.12 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
It is known that in several European countries the catching and/or selling of wild eel from contaminated areas is discouraged or in some countries even prohibited.
Salmon and trout
Tables 25, 26 and 27 show the occurrence values for the sum of PCDD/Fs and DL‐PCBs, the 17 PCDD/Fs or the 12 DL‐PCBs, respectively, in salmon and trout according to different production methods as reported by the data providers.
Table 25.
Levels of the sum of PCDD/Fs and DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in salmon and trout among different production methods
| Production method of salmon and trout | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Organic production | 18 | 1.05 | 1.18 | – | 1.05 | 1.18 | – |
| Non‐organic production | 66 | 0.41 | 0.26 | 0.92 | 0.46 | 0.31 | 0.98 |
| Wild or gathered or hunted | 83 | 3.90 | 3.82 | 8.79 | 3.98 | 3.85 | 8.82 |
| Farmed Domestic or cultivated | 168 | 0.43 | 0.31 | 0.94 | 0.48 | 0.38 | 0.94 |
| Traditional production | 38 | 0.47 | 0.46 | – | 0.47 | 0.46 | – |
| Production method unknown | 483 | 0.60 | 0.34 | 1.15 | 0.67 | 0.45 | 1.20 |
| Other production method | 1 | – | 0.07 | – | – | 0.22 | – |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 26.
Levels of the sum of PCDD/Fs expressed in pg WHO2005‐TEQ/g whole weight in salmon and trout among different production methods
| Production method of salmon and trout | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Organic production | 23 | 0.32 | 0.31 | – | 0.32 | 0.31 | – |
| Non‐organic production | 66 | 0.10 | 0.05 | 0.26 | 0.16 | 0.11 | 0.27 |
| Wild or gathered or hunted | 83 | 1.38 | 1.42 | 3.18 | 1.41 | 1.45 | 3.20 |
| Farmed Domestic or cultivated | 168 | 0.12 | 0.07 | 0.29 | 0.16 | 0.14 | 0.34 |
| Traditional production | 38 | 0.15 | 0.15 | – | 0.15 | 0.15 | – |
| Production method unknown | 528 | 0.18 | 0.08 | 0.36 | 0.24 | 0.17 | 0.47 |
| Other production method | 1 | – | 0.01 | – | – | 0.16 | – |
P95: 95th percentile; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 27.
Levels of the sum of DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in salmon and trout among different production methods
| Production method of salmon and trout | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95a | Mean | Median | P95a | ||
| Organic production | 18 | 0.71 | 0.75 | – | 0.71 | 0.75 | – |
| Non‐organic production | 67 | 0.30 | 0.22 | 0.72 | 0.30 | 0.22 | 0.72 |
| Wild or gathered or hunted | 83 | 2.52 | 2.57 | 5.94 | 2.57 | 2.57 | 5.94 |
| Farmed Domestic or cultivated | 168 | 0.31 | 0.25 | 0.67 | 0.32 | 0.25 | 0.67 |
| Traditional production | 38 | 0.32 | 0.32 | – | 0.32 | 0.32 | – |
| Production method unknown | 522 | 0.50 | 0.25 | 1.43 | 0.50 | 0.25 | 1.43 |
| Other production method | 1 | – | 0.06 | – | – | 0.06 | – |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Salmon and trout samples reported as ‘wild or gathered or hunted’ had higher levels of all congener groups than those reported with other production method categories. In this respect, it must be noted that the majority of wild or gathered salmon/trout originated from countries with coastline to the Baltic Sea (see also Tables 28, 29 and 30).
Table 28.
Levels of the sum of PCDD/Fs and DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in salmon samples from possible Baltic and non‐Baltic origin
| N | Lower bound | Upper bound | Lower bound | Upper bound | |
|---|---|---|---|---|---|
| Mean | Mean | P95 | P95 | ||
| All salmon/trout | 857 | 0.87 | 0.94 | 5.82 | 5.82 |
| Baltic salmon/trouta | 275 | 1.90 | 1.93 | 8.30 | 8.33 |
| Non‐Baltic salmon/trout | 582 | 0.39 | 0.47 | 0.92 | 0.94 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Samples from Germany, Denmark, Finland, Poland, Sweden were available and were included in the table.
Table 29.
Levels of the sum of PCDD/Fs expressed in pg WHO2005‐TEQ/g whole weight in salmon samples from possible Baltic and non‐Baltic origin
| N | Lower bound | Upper bound | Lower bound | Upper bound | |
|---|---|---|---|---|---|
| Mean | Mean | P95 | P95 | ||
| All salmon | 907 | 0.27 | 0.33 | 1.93 | 1.95 |
| Baltic salmona | 296 | 0.62 | 0.65 | 2.89 | 2.91 |
| Non‐Baltic salmon | 611 | 0.10 | 0.17 | 0.30 | 0.36 |
P95: 95th percentile; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Samples from Germany, Denmark, Finland, Poland, Sweden were available and were included in the table.
Table 30.
Levels of the sum of DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in salmon samples from possible Baltic and non‐Baltic origin
| N | Lower Bound | Upper Bound | Lower Bound | Upper Bound | |
|---|---|---|---|---|---|
| Mean | Mean | P95 | P95 | ||
| All salmon | 897 | 0.63 | 0.64 | 3.92 | 3.92 |
| Baltic salmona | 290 | 1.33 | 1.34 | 5.94 | 5.94 |
| Non‐Baltic salmon | 607 | 0.30 | 0.30 | 0.67 | 0.67 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
Samples from Germany, Denmark, Finland, Poland, Sweden were available and were included in the table.
Salmon and trout samples from countries with coastline to the Baltic Sea were also investigated and compared with samples from other regions. Tables 28, 29 and 30 show the LB and UB mean and P95 levels for the sum of PCDD/Fs and DL‐PCBs, the 17 PCDD/Fs or the 12 DL‐PCBs, respectively, of all salmon and trout samples and the same samples distinguished based on possible Baltic and non‐Baltic origin. Higher levels of PCDD/Fs and DL‐PCBs were found in samples of possible Baltic origin than in those of non‐Baltic origin.
Herring
Herring samples possibly originating from the Baltic Sea were also investigated and compared with samples from other regions. Tables 31, 32 and 33 show the LB and UB mean and P95 contamination levels for the sum of PCDD/Fs and DL‐PCBs, the 17 PCDD/Fs or the 12 DL‐PCBs, respectively, of all herring samples and the same samples distinguished from possible Baltic (originating from countries with coastline to the Baltic Sea) and non‐Baltic origin. Higher levels of PCDD/Fs and DL PCBs were found in samples of possible Baltic origin.
Table 31.
Levels of the sum of PCDD/Fs and DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in herring from possible Baltic or non‐Baltic origin
| N | Lower Bound | Upper Bound | Lower Bound | Upper Bound | |
|---|---|---|---|---|---|
| Mean | Mean | P95 | P95 | ||
| All herring | 399 | 2.34 | 2.39 | 6.36 | 6.36 |
| Baltic herringa | 197 | 3.01 | 3.05 | 7.26 | 7.26 |
| Non‐Baltic herring | 202 | 1.70 | 1.74 | 5.64 | 5.64 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Samples from Germany, Denmark, Lithuania, Poland, Sweden were available and were included in the table.
Table 32.
Levels of the sum of PCDD/Fs expressed in pg WHO2005‐TEQ/g whole weight in herring from possible Baltic or non‐Baltic origin
| N | Lower bound | Upper bound | Lower bound | Upper bound | |
|---|---|---|---|---|---|
| Mean | Mean | P95 | P95 | ||
| All herring | 401 | 1.22 | 1.25 | 3.37 | 3.37 |
| Baltic herringa | 199 | 1.59 | 1.61 | 4.10 | 4.10 |
| Non‐Baltic herring | 202 | 0.85 | 0.89 | 2.95 | 2.95 |
P95: 95th percentile; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Samples from Germany, Denmark, Lithuania, Poland, Sweden were available and were included in the table.
Table 33.
Levels of the sum of DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in herring from possible Baltic or non‐Baltic origin
| N | Lower bound | Upper bound | Lower bound | Upper bound | |
|---|---|---|---|---|---|
| Mean | Mean | P95 | P95 | ||
| All herring | 404 | 1.12 | 1.13 | 2.93 | 2.93 |
| Baltic herringa | 199 | 1.40 | 1.43 | 3.31 | 3.31 |
| Non‐Baltic herring | 205 | 0.84 | 0.84 | 2.64 | 2.64 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
Samples from Germany, Denmark, Lithuania, Poland, Sweden were available and were included in the table.
Fatty fish other than eel and salmon/trout
In Tables 34, 35 and 36, the occurrence values for the sum of PCDD/Fs and DL‐PCBs, the 17 PCDD/Fs or the 12 DL‐PCBs, respectively, in other fatty fish according to different production methods as reported by the data providers are shown. For most of the fatty fish samples other than eel and salmon/trout, the production method was not reported. Nevertheless, the data clearly indicate that levels of both PCDD/Fs and DL‐PCBs in farmed fatty fish are lower than those in wild caught fatty fish.
Table 34.
Levels of the sum of PCDD/Fs and DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in fatty fish other than eel and salmon/trout among different production methods
| Production method of fatty fisha | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95b | Mean | Median | P95b | ||
| Organic production | 1 | – | 0.81 | – | – | 0.81 | – |
| Non‐organic production | 130 | 3.05 | 2.712 | 7.75 | 3.06 | 2.72 | 7.75 |
| Wild or gathered or hunted | 100 | 2.15 | 1.94 | 4.69 | 2.26 | 2.06 | 4.79 |
| Farmed Domestic or cultivated | 65 | 0.24 | 0.20 | 0.68 | 0.36 | 0.32 | 0.88 |
| Production method unknown | 1,329 | 1.50 | 0.88 | 5.06 | 1.54 | 0.93 | 5.06 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Fatty fish other than eel and salmon/trout contains data on: Carp (Cyprinus), Char (Salvelinus), Halibut (Hippoglossus spp.), Herring (Clupea), Mackeral (Scomber), Sardine and pilchard (Sardinia), Smelt (Osmerus), Sprat (Sprattus sprattus) and Swordfish (Xiphiidae spp.).
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 35.
Levels of the sum of PCDD/Fs expressed in pg WHO2005‐TEQ/g whole weight in fatty fish other than eel and salmon/trout among different production methods
| Production method of fatty fisha | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95b | Mean | Median | P95b | ||
| Organic production | 1 | – | 0.22 | – | – | 0.22 | – |
| Non‐organic production | 130 | 1.55 | 1.26 | 4.17 | 1.57 | 1.26 | 4.17 |
| Wild or gathered or hunted | 100 | 0.84 | 0.67 | 2.47 | 0.87 | 0.69 | 2.49 |
| Farmed Domestic or cultivated | 65 | 0.07 | 0.06 | 0.15 | 0.13 | 0.13 | 0.21 |
| Production method unknown | 1,337 | 0.51 | 0.25 | 2.15 | 0.55 | 0.30 | 2.16 |
P95: 95th percentile; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Fatty fish other than eel and salmon/trout contains data on: Carp (Cyprinus), Char (Salvelinus), Halibut (Hippoglossus spp.), Herring (Clupea), Mackeral (Scomber), Sardine and pilchard (Sardinia), Smelt (Osmerus), Sprat (Sprattus sprattus) and Swordfish (Xiphiidae spp.).
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Table 36.
Levels of the sum of DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in fatty fish other than eel and salmon/trout among different production methods
| Production method of fatty fisha | N | Lower bound estimate | Upper bound estimate | ||||
|---|---|---|---|---|---|---|---|
| Mean | Median | P95b | Mean | Median | P95b | ||
| Organic production | 1 | – | 0.59 | – | – | 0.59 | – |
| Non‐organic production | 130 | 1.45 | 1.29 | 3.45 | 1.50 | 1.29 | 3.45 |
| Wild or gathered or hunted | 100 | 1.31 | 1.17 | 3.01 | 1.40 | 1.27 | 3.02 |
| Farmed Domestic or cultivated | 65 | 0.17 | 0.12 | 0.60 | 0.24 | 0.17 | 0.67 |
| Production method unknown | 1,387 | 1.07 | 0.61 | 3.06 | 1.07 | 0.61 | 3.06 |
P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents
Fatty fish other than eel and salmon/trout contains data on: Carp (Cyprinus), Char (Salvelinus), Halibut (Hippoglossus spp.), Herring (Clupea), Mackeral (Scomber), Sardine and pilchard (Sardinia), Smelt (Osmerus), Sprat (Sprattus sprattus) and Swordfish (Xiphiidae spp.).
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Crabs
The CONTAM Panel noted that crab meat samples reported as brown crab meat (n = 14) contained substantially higher levels than the two reported as white crab meat or that did not report the type of crab meat (n = 258). This is consistent with studies in brown crabs (Oehme et al., 1990) and Chinese mitten crabs (Hoogenboom et al., 2015d), that show much higher levels in brown meat than in white meat.
Since brown crab meat or mixed brown‐ and white crab meat is consumed in some European countries, and Foodex did not include data on the type of crab meat consumed, all samples from crab were included in the mean occurrence in crab meat (n = 274) (see Tables 37, 38 and 39).
Table 37.
Levels of the sum of PCDD/Fs and DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in crabs among different parts analysed
| Crabs ‐ part analysed | N | Lower bound Mean | Upper bound Mean |
|---|---|---|---|
| Brown meat | 14 | 6.10 | 6.17 |
| White meat | 2 | 0.38 | 0.42 |
| Not reported | 258 | 1.01 | 1.01 |
DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Table 38.
Levels of the sum of PCDD/Fs expressed in pg WHO2005‐TEQ/g whole weight in crabs among different parts analysed
| Crabs ‐ part analysed | N | Lower bound Mean | Upper bound Mean |
|---|---|---|---|
| Brown meat | 14 | 3.22 | 3.29 |
| White meat | 2 | 0.26 | 0.29 |
| Not reported | 259 | 0.48 | 0.49 |
PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Table 39.
Levels of the sum of DL‐PCBs expressed in pg WHO2005‐TEQ/g whole weight in crabs among different parts analysed
| Crabs ‐ part analysed | N | Lower bound Mean | Upper bound Mean |
|---|---|---|---|
| Brown meat | 14 | 2.88 | 2.88 |
| White meat | 2 | 0.13 | 0.13 |
| Not reported | 270 | 0.51 | 0.51 |
DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
Grouping of food categories for the exposure assessment
In view of the exposure assessment, food data were grouped at different FoodEx levels, and other merged categories used as supplements to FoodEx for the present assessment, taking into consideration several factors including the similarities between food categories, the number of samples, and the concentrations observed.
At the most detailed level (Foodex level 3), the food was retained if more than 6 samples were available in the category. If less than 6 samples were available, the levels were compared with similar foods belonging to other categories:
if the levels were similar, the samples were either grouped together with the similar food, resulting in a new category, or the food was taken into account at a higher (parent) level (FoodEx level 2) if this broader category was well‐represented by the available categories at FoodEx level 3.
-
if the levels reported were very different, the category was excluded as it was considered insufficiently covered. Two exceptions were made in order to avoid excluding potentially relevant categories:
-
–
In the case of ‘Pasta with eggs’, levels derived from the contamination levels of ‘Chicken eggs’ were taken into account, assuming an average 15% egg content in these products.
-
–
In the case of ‘Fish roe’, levels of category ‘Fish products’ were used, as the levels analysed for its parent category ‘Fish Offal’ would result in an overestimation of exposure.
-
–
Samples in the categories of the least detailed FoodEx classification (FoodEx level 1) were excluded in case no information was available. Altogether, 218 samples were excluded from the assessment from the 19,965 samples with PCDD/F and DL‐PCB data and also from the 20,273 samples with PCDD/F data as they did not fulfil the above conditions. In total, 135 categories were created for the linking between food occurrence and food consumption data (see Annex B, Table 3A and B for the 29 congeners and the 17 congeners, respectively).
Supplementary information on limit of quantification values and left censored data
By applying the performance criteria presented in Section 3.2.1.1, paragraph ‘Application of performance criteria’ and disregarding samples analysed by inappropriate analytical methods or without information on the applied analytical method (see Section 3.2.1), the retained samples are deemed to represent a reliable quality. Thus, the CONTAM Panel did not consider it relevant to show the ranges of LOQ values per category.
Moreover, the percentage of left‐censored data among the food categories is not considered informative, as the final values considered in the assessment are the sums of the individual analytical results of the 29 PCDD/F and DL‐PCB congeners, or the 17 PCDD/F congeners.
3.2.1.2. Occurrence data in feed
Application of performance criteria
The data set for the current farm and companion animal exposure assessment contained 2,442 feed samples, in which at least either all of the 17 PCDD/Fs, or all the 12 DL‐PCBs were analysed.
Similar to the food samples (see Section 3.2.1.1), these samples were checked for compliance with analytical performance criteria based on Commission Regulation (EU) No 2017/771 and taking into account the existing action thresholds (see Section 1.3.4 Legislation). These were expressed in ng WHO2005‐TEQ/kg, based on 88% dry matter.
For samples where the full set of 17 PCDD/Fs were analysed, those for which the total LOQ was higher than one‐fifth of the corresponding ML for the sum‐TEQ of 17 PCDD/Fs were excluded. A total of 319 feed samples did not comply with the criterion and were therefore excluded.
For samples where the full set of 12 DL‐PCBs were analysed, those for which the total LOQ was higher than one‐third of the AT for the sum‐TEQ of the 12 DL‐PCBs were excluded. A total of 33 feed samples did not comply with the criterion and were therefore excluded.
Overall, the final feed data set that contained samples fulfilling the above criterion, and all the conditions listed in Section 3.2.1 were:
1,830 feed samples with all 29 congeners determined (17 PCDD/Fs and 12 DL‐PCBs);
1,844 feed samples with all 17 PCDD/F congeners determined (including samples with the 29 congeners);
2,131 feed samples with all 12 DL‐PCB congeners determined (including samples with the 29 congeners).
The feed samples were submitted by 12 European countries (Figure 17) between the years 2010 to 2016 (Figure 18). The mean and P95 levels for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs in a number of food categories are shown in Table 40. More data on summary statistics of occurrence values reported in feed for the 29, 17 and 12 congeners data are presented in Annex B (Tables 2D, E and F, respectively).
Figure 17.
Number of feed samples in the final data set encoded by European countries (n = 1,844 feed samples with results for 17 PCDD/Fs reported; from these 1,830 also had the 12 DL‐PCBs)
Figure 18.
Number of feed samples in the final data set reported by year (n = 1,844 feed samples with results for the 17 PCDD/Fs reported; from these 1,830 also had the 12 DL‐PCBs)
Table 40.
Mean and P95 levels for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs in selected feed groups in ng/kg expressed in 88% dry matter (see Annex B for additional feed items)
| Feedstuff | Sum of PCDD/Fs (WHO2005‐PCDD/F‐TEQ) | Sum of PCDD/Fs and DL‐PCBs (WHO2005‐PCDD/F‐PCB‐TEQ) | ||||
|---|---|---|---|---|---|---|
| n | Mean (LB/UB) | P95a (LB/UB) | n | Mean (LB/UB) | P95a (LB/UB) | |
| Animal fat (including milk fat and egg fat) | 96 | 0.10/0.17 | 0.40/0.57 | 96 | 0.33/0.42 | 1.36/1.53 |
| Compound feed for food producing animals (except fish) | 532 | 0.02/0.03 | 0.08/0.09 | 527 | 0.03/0.05 | 0.13/0.15 |
| Compound feed for fish | 224 | 0.13/0.17 | 0.36/0.42 | 224 | 0.54/0.58 | 1.18/1.19 |
| Compound feed for pet animals | 19 | 0.08/0.09 | – | 19 | 0.11/0.14 | – |
| Feed materials of mineral origin | 176 | 0.02/0.13 | 0.13/0.18 | 173 | 0.03/0.15 | 0.15/0.23 |
| Feed materials of plant origin except roughages, vegetable oils and their by‐products | 230 | 0.02/0.03 | 0.06/0.09 | 226 | 0.03/0.04 | 0.15/0.17 |
| Forages and roughage, and products derived thereof | 82 | 0.05/0.07 | 0.20/0.20 | 82 | 0.10/0.12 | 0.27/0.28 |
| Fish and Fish meal with the exception of fish protein, hydrolysed | 106 | 0.22/0.28 | 0.68/0.68 | 106 | 0.63/0.69 | 1.55/1.56 |
| Fish oil | 82 | 0.80/0.90 | 2.49/2.49 | 82 | 3.33/3.43 | 8.33/8.35 |
| Land animal products other than animal fat | 46 | 0.02/0.06 | – | 45 | 0.04/0.08 | – |
| Vegetable oils and their by‐products | 137 | 0.12/0.21 | 0.34/0.42 | 137 | 0.17/0.27 | 0.38/0.48 |
| Miscellaneous(b) | 95 | 0.08/0.14 | 0.36/0.36 | 95 | 0.10/0.18 | 0.36/0.50 |
LB: lower bound; UB: upper bound; P95: 95th percentile; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
The 95th percentile estimates obtained with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Defined by Commission Regulation (EU) 68/2013 as amended by Commission Regulation (EU) 2017/1017. Category includes glucose molasses, products from the bakery and pasta industry, other unspecified miscellaneous feedstuffs.
3.2.2. Previously reported occurrence data in the open literature
A number of studies providing results for PCDD/Fs and DL‐PCBs in different food and feed commodities have been published in the literature. The paragraphs below, which do not claim to be complete, give an overview of some of the occurrence values reported.
Food
Several international bodies have reported the occurrence of PCDD/Fs and DL‐PCBs in food when estimating the dietary exposure to these compounds for a risk assessment (see Section 1.3.3).
The SCF (2000, 2001) based its exposure assessment on the occurrence data compiled in the Scientific Cooperation (EU SCOOP) Task 3.2.5. It included data from 10 European countries, taken from rural and industrial sites covering the period 1982–1999. Products of vegetable origin showed concentrations of 0.02–0.03 pg I‐TEQ/g whole food basis, eggs of 1 pg I‐TEQ/g fat, while wild and farmed freshwater fish exhibited higher mean levels of around 10 pg I‐TEQ/g fat for PCDD/Fs and 30 pg PCB‐TEQ36/g fat for DL‐PCBs. Meat (i.e. poultry, beef, veal, pork and mixed meat) showed lower levels of 0.4–0.7 pg I‐TEQ/g fat (PCDD/Fs) and 0.3–1.5 pg PCB‐TEQ/g (DL‐PCBs). Concentrations in milk and dairy products ranged from 0.6 to 1.0 pg I‐TEQ/g fat (PCDD/Fs) and from 0.6 to 1.3 pg PCB‐TEQ/g fat (DL‐PCBs).
JECFA estimated the dietary intake based on occurrence data of PCDD/Fs and DL‐PCBs in food submitted by industrialised countries from (Western) Europe, North America, Asia and Oceania collected after 1995 (FAO/WHO, 2002). The data from Europe were submitted by 10 European countries. Data were aggregated into six major food categories. For Western Europe, weighted concentrations were reported to be 0.07, 0.16, 0.47, 0.08 and 0.04 pg/g whole food basis for dairy, eggs, fish, meat and vegetable products, respectively.
EFSA published two technical reports on the levels of PCDD/Fs and DL‐PCBs in food and feed (EFSA, 2010a,b, 2012). In its latest report, data for 10,468 food samples were included, collected between 1995 and 2010 (with the majority of samples between 2003 and 2010), from 26 European countries (EFSA, 2012). The food groups showing the highest values (expressed in WHO2005‐TEQs, mean UB values) were ‘Fish liver and derived products’ (28.28 pg/g ww), ‘liver of terrestrial animals’ (10.98 pg/g fat) and ‘Muscle meat eel’ (9.76 pg/g ww). Regarding the contribution of the different contaminant groups to the total WHO2005‐TEQ, non‐ortho PCBs were the main contributors (representing 21.0–74.5%) followed by PCDD/Fs which represented 12.4–73.2% of the total TEQ level. A total of 9.7% of the food samples exceeded the permitted ML for PCDD/Fs and DL‐PCBs. A time trend analysis was performed in three food groups which had the highest number of years covered, number of countries and number of data available for each year. These food groups were ‘Raw milk and dairy products’, ‘Hen eggs and egg products’ and ‘Muscle meat from fishes other than eels’). An overall decreasing tendency was observed, which was statistically significant in ‘Raw milk and dairy products’ (56% reduction in 10 years), ‘Muscle meat from fishes other than eels’ (98% reduction in 10 years). It should, however, be noted that when restricting the analysis to the years with the most robust estimates, generally between 2002/2003 and 2010, the decrease was no longer statistically significant. The reasons for the change observed cannot solely be attributed to risk management measures. There were also improvements throughout the years in both analytical methods and/or sampling designs of the monitoring programmes could influence the levels.
Fürst and Bernsmann (2011) and Fürst (personal communication, 2017) analysed raw milk from all dairies in North Rhine‐Westphalia/Germany for PCDD/Fs and PCBs. In order to investigate whether seasonal trends exist, each dairy was sampled four times per year in March, May, July and September. All samples were taken from the collection tanks in the dairies directly before processing. Between 1990 and 2014, this monitoring programme was repeated every 4 years following the same sampling and analytical strategy. In total, 654 samples were analysed over the years. Between 1990 and 2014 the median PCDD/F levels decreased from 1.27 to 0.35 (mean: 1.35 to 0.36; 95th percentile: 2.04 to 0.49) pg I‐TEQ/g milk fat. These results indicate a median decrease for PCDD/Fs in raw milk of around 75% between 1990 and 2014. However, the results obtained in the 2010 and 2014 surveys indicate a stagnation in the decline of PCDD/F levels in milk. While the initial two sampling periods only focused on PCDD/Fs, since 1998 also DL‐PCBs were included and show a mean decrease from 1.85 to 0.58 pg WHO1998‐TEQ/g milk fat indicating a decline of around 70%. Calculated with WHO1998‐TEFs, the corresponding PCDD/F levels decreased from 0.73 to 0.43 pg TEQ/g fat. While the mean share of DL‐PCBs to total WHO1998‐TEQs amounted to 72% in 1998, this was only 58% in 2014.
Since 2012, several authors and organisations have reported the occurrence levels in different food commodities in Europe. For example, in France, for its second TDS, PCDD/F and DL‐PCBs were analysed in samples purchased between 2007 and 2009 (ANSES, 2011). In total, 19,830 products were purchased and prepared as consumed, then grouped into 1,319 composite samples that were analysed. The highest mean concentrations of PCDD/Fs and DL‐PCBs were found in ‘Fish’ and ‘Molluscs and Crustaceans’ (0.65 and 0.48 pg WHO1998‐TEQ/g fresh weight, respectively). In 2012, the UK TDS (FERA, 2012) collected 986 individual food samples collected across the UK and prepared as normal for consumption. Individual samples were split into 20 food groups. The highest levels (LB and UB levels) were reported (in pg WHO2005‐TEQ/g fat) for Fish (LB: 3.498, UB: 3.499) and Offal (LB: 1.923, UB: 1.925) while Meat products (LB: 0.201, UB: 0.203), Potatoes (LB: 0.156, UB: 0.186), Poultry (LB: 0.136, UB: 0.148), Cereals (LB: 0.053, UB: 0.134), Fats and oils (LB: 0.096, UB: 0.124) and Nuts (LB: < 0.013, UB: 0.045) showed lower values.
Adamse et al. (2017) published levels of PCDD/Fs and DL‐PCBs in 2,475 food samples of animal origin produced in the Netherlands for the period 2001–2011, including beef, veal, lamb, chicken, pork, liver, deer, milk and eggs. The median levels were similar to those reported by EFSA (2012) but P95 levels were in general much lower. The fraction of non‐compliant samples was 1% or less for most products, except for liver and sheep meat/fat. The authors also concluded that there was no clear decreasing trend in the mean levels of PCDD/Fs and DL‐PCBs for the products and period studied, and attributed this to a plateauing of current background levels. For some products, levels were consistently below the LOQ, not allowing trend analysis. It was shown that improving the sensitivity of the analytical method results in an increase in LB and decrease of UB levels, and as such results in wrong interpretation of the trend.
Lundebye et al. (2017) determined inter alia PCDD/F and PCBs in wild and farmed Atlantic salmon. In wild Atlantic salmon (n = 87), the mean PCDD/F and DL‐PCB levels were 0.43 ± 0.18 and 0.57 ± 0.15 pg WHO2005‐TEQ/g wet weight, respectively. The contamination in farmed salmon (n = 130) was somewhat lower with respective mean levels reported as 0.23 ± 0.07 and 0.29 ± 0.09 pg WHO2005‐TEQ/g wet weight. In contrast, the total lipid content was significantly higher in farmed (14 ± 3 mg/g) than wild Atlantic salmon (8 ± 3 mg/g). In escaped farmed salmon (n = 12), mean PCDD/F and DL‐PCB levels were determined as 0.46 ± 0.40 and 0.46 ± 0.17 pg WHO2005‐TEQ/g wet weight, respectively. The total lipid content in these escaped farmed salmon was reported as 9 ± 3 mg/g.
Feed
The presence of high levels of PCDD/Fs and/or DL‐PCBs in feed or materials used for feed production has been the source of a number of contamination incidents in the feed and food chain (see Section 1.3.1). The European Commission's former SCAN (2000) evaluated the contribution of PCDD/Fs and PCBs in feedingstuffs to the contamination of food of animal origin. Only levels of PCDD/Fs were reported as the database for DL‐PCBs was scarce. Feed groups considered were vegetable feed materials (including, e.g. roughage, roots and tubers and cereals), feed materials of animal origin (including, e.g. fishmeal, fish oil, meat and bone meal, animal fat), other feed material (including trace elements, macrominerals, binders and anticaking agents) and soil. Fish oil and fishmeal were the most contaminated feed materials (mean PCDD/Fs values ranging from 0.14 to 4.8 ng WHO‐TEQ/kg dry matter), followed by animal fat (mean 1 ng WHO‐TEQ/kg dry matter). Other feed material of plant and animal origin were reported to contain mean values of ≤ 0.2 ng WHO‐TEQ/kg dry matter. Soil was reported to have a mean PCDD/F concentration of 5 ng WHO‐TEQ/kg dry matter.
The two EFSA reports mentioned above also compiled the occurrence data generated by European countries on feed (EFSA, 2010a,b, 2012). In the 2012 report, a total of 3,329 feed samples were included, mostly covering the period 2003–2010. The feed groups best represented were ‘Feed materials of plant origin, oils excluded’ and ‘Compound feed, excluding feed for fur animals, pets and fish’ which together represented 56.2% of the data. On the other hand, ‘Additives binders and anti‐caking agents’ and ‘Animal fat’ were poorly represented. The feed groups with the highest mean UB levels of PCDD/Fs and DL‐PCBs (expressed in WHO2005‐TEQs) were ‘Fish oil’, ‘Feed for fur animals, pets and fish’ and ‘Fish, other aquatic animals, and product derived therof’ with 8.61, 1.24 and 1.00 ng/kg 88% dry matter, respectively. As for food, the non‐ortho PCBs were the major contributors to the total WHO2005‐TEQs, except in the groups ‘Additives binders and anti‐caking agents’ where PCDDs represented more than 80% of the total TEQ, and in the groups ‘Additives compounds of trace element’, ‘Other feed additives’ and ‘Feed materials of plant origin, oils excluded’ in which PCDFs were the major contributor (29.0–92.4% of the total TEQ). The percentage of samples exceeding the EU legal limits was lower than for food, with about 2.3% for the sum of PCDD/Fs and DL‐PCBs. The highest percentages of results above the MLs were reported for ‘Fish oil’ and ‘Vegetable oils and their by‐products’.
Since then, several authors have reported the levels of PCDD/Fs and/or DL‐PCBs in feed samples collected in European countries. For example, Sissener et al. (2013) analysed in commercial Norwegian fish feed (for aquaculture fish species in Norway) a number of compounds including PCDD/Fs, DL‐PCBs, pesticide residues, PBDEs and metals. The samples were collected from 2000 to 2010. The PCDD/F and DL‐PCB levels in the fish feeds ranged from 0.28 to 6.17 ng WHO1998‐TEQ/kg, and the authors reported a decrease over time for the years studied, especially for the period 2001–2004. The levels correlated significantly with the percentage inclusion of fish oil in Norwegian fish feeds from 2002 to 2010.
Godliauskiene et al. (2017) analysed PCDD/Fs and DL‐PCBs in 114 samples of feed and feed ingredients in Lithuania covering the years 2007–2014. The samples included sugar beet pulp, grass meal, mixed feed, various premixes, mineral supplements, fishmeal and others. Fishmeal had the highest levels of sum of PCDD/Fs and DL‐PCBs (0.44 ng WHO‐TEQ37/kg), while feed premixes and feed of plant origin had the lowest levels (0.06 ng WHO‐TEQ/kg). PCDD/Fs were the major contributors to the total TEQ in compound feed, mineral feed and vegetable oils (60%), while DL‐PCBs were the major contributors in fishmeal (70%). Three samples exceeded the EU maximum levels (fishmeal and zinc oxide).
Regarding companion animals, several studies have reported the concentrations of PCDD/Fs and/or DL‐PCBs in commercial feed for cats or dogs. Kunisue et al. (2005) analysed the levels of the 17 PCDD/Fs and 12 DL‐PCBs in dry or canned feed for dogs (n = 3) and cats (n = 8). The LB mean (SD) concentration in dog feed was 0.27 (0.072) pg WHO1998‐TEQ/g fat, while in cat feed it was 0.68 (0.76) pg WHO1998‐TEQ/g fat, which, considering average lipid contents of 10 and 13%, corresponds to 0.03 and 0.09 ng/kg feed, respectively. Mono‐ortho PCBs contributed most to the total TEQ in both cat‐ and dog feed.
Adamse et al. (2015) evaluated Dutch monitoring data for the period 2001 to 2011. For most matrices less than 1% of the samples exceeded to ML set in the EU, with the exception of fishmeal (4.1%), clay minerals (3.4%) and vegetable oil (1.7%). Non‐compliance rates, but for many products also the P95, were lower than reported by EFSA (2010a,b, 2012).
In conclusion, given the different years and place of sampling and analysis, the increase in analytical sensitivity, the different TEF schemes applied and the basis of the reported occurrence data (lipid/fresh weight, dry weight, 88% dry matter), the results of the previously reported occurrence data in the open literature are in good concordance with the occurrence data submitted to EFSA by data providers used for the exposure estimation in the present risk assessment. The occurrence data indicate a PCDD/F and PCB decline in the past 30 years; however, this decrease seems to have levelled off in the last 10 years as also reported for levels in humans (see Section 3.2.4 ).
3.2.3. Food and feed processing
As PCDD/Fs and PCBs are lipophilic compounds that accumulate in fatty tissues, all processing steps which alter the lipid content of a food commodity will have an impact on their levels in the processed sample. For example, trimming of visible fatty tissue from a food sample already leads to a substantial reduction of the PCDD/Fs and PCB content of the respective product.
Domingo (2011) reviewed the literature on the influence of different cooking processes on the concentrations of PCDD/Fs and PCBs, and other contaminants, in various foodstuffs. The studies indicate that in general, cooking procedures that release or remove fat from the product tend to reduce the PCDD/F and PCB concentrations in the cooked food. Moreover, it seems that the influence of cooking depends not only on the particular cooking process, but also on the specific food commodity. Stachiw et al. (1988) studied the potential of reducing TCDD levels during various cooking practices on restructured (deboned, washed, dewatered and chopped) carp fillets. Charbroiling resulted in greater TCDD reduction than roasting uncovered. Increasing internal temperature from 60 to 80°C increased the TCDD loss from the carp fillets. The loss was also apparent when increasing the diameter of the fillets from 7.5 to 10 cm. Rawn et al. (2013) determined PCDD/Fs and PCBs in composites of 18 different fish products which were prepared as baked, boiled and fried. PCB and PCDD/F concentrations were generally reduced during cooking, although there was little difference between the levels in some raw and processed products. On average, PCDD/F losses were observed in both the finfish, such as catfish, grouper, mackerel, red snapper and monkfish (3.6%) and non‐finfish products, including octopus, scallops, sea squirts and squids (25%). The results refer to the sum of the 17 toxic PCDD/F congeners on a wet weight basis.
Schecter et al. (1998a) investigated the effect of broiling on the levels of PCDD/Fs and non‐ortho PCBs in ground beef (hamburgers), bacon and cat fish. On a sample or serving basis, the total amount of pg TEQ for PCDD/Fs and coplanar PCBs decreased on average by approximately 50% after broiling. However, due to weight loss, the mean concentration (mean of 3 individual samples, pg TEQ/kg, wet weight) remained the same in hamburgers, increased by 83% in bacon and decreased by 34% in catfish. The CONTAM Panel noted that the levels measured in the individual samples were in the pg/kg (ppq) range and varied substantially.
Schecter et al. (1998a) also reported a marked increase in PCDD/F‐TEQ levels in French fries fried in lard (pork fat) when compared with uncooked fries.
Reductions in PCDD/F and PCB levels were also reported by Hori et al. (2005) who investigated the effects of grilling, boiling and ‘tsumire’ (a typical Japanese cooking practice) on the changes in PCDD/F and PCB levels in mackerel and beef. Grilling sliced mackerel resulted in a 31% reduction of PCDD/Fs. Boiling and ‘tsumire’ reduced the PCDD/F levels in sliced mackerel by 14 and 21%, respectively. For beef, the authors report reductions in PCDD/F levels between 42 and 44%.
When reviewing the literature, Domingo (2011) concluded that the reduction of PCB levels, especially in fish may be explained by the loss of lipids, and the evaporation of water and PCBs from the fillets during the processing step. The influence of the surface area and the thickness of the fillets on the contaminant reduction following various processing techniques were also demonstrated. Moreover, the initial lipid concentration in the fish may have an impact on the PCB loss during processing (Sherer and Price, 1993). The authors report reductions of 20 to 30% of the PCBs in fish after baking, broiling, microwave cooking, poaching and roasting. Higher reductions of more than 50% of the PCB levels in fish were found after frying. Planche et al. (2017) investigated the effects of pan cooking on PCBs, PCDD/Fs and other contaminants in meat. The average PCB losses after cooking were 18±5 % for rare, 30±3 % for medium, and 48±2% for well‐done meat. In contrast, PCDD/Fs losses were not significant.
The surveys on the processing effects on meat and meat derived products were mostly performed on samples with relatively low environmental levels which introduce some uncertainty in the interpretation of the results; hence small changes in concentration during processing may not have been apparent. Petroske et al. (1998) and Rose et al. (2001) investigated the effects of different processing techniques on tissues from cattle which had been dosed with several PCDD/F congeners. Petroske et al. (1998) studied the effects of pan‐frying on hamburger patties derived from tissues which were obtained from steers exposed to pentachlorophenol‐treated wood and dosed with 12 PCDD/F congeners. The 110 g hamburger patties (fat content 20%) were placed in a stainless steel frying pan on a hot plate and heated to an internal temperature of 74°C. The final hot plate temperature was approximately 210°C. Fats and juices were also collected from the pan and subjected to PCDD/F analysis. Assuming that the fats and juices released from the hamburger patties during the frying process are discarded, pan frying ground beef patties reduced the amount of PCDD/Fs actually consumed by 40–50%. The loss of lipids correlated well with the loss of PCDD/Fs with a mean of 47.6%. The recovery (determined as difference between PCDD/F congener levels in unprocessed patties and sum of cooked patties, fats and juices) amounted to 82‐99%. Thus, the authors assumed that there is probably no significant degradation or conversion of any PCDD/F congener to a differently substituted congener during the cooking process.
Rose et al. (2001) studied the changes in concentration of five PCDD/F congeners after different cooking practices of beef from treated cattle. The preparation and cooking methods included burgers (fried, grilled and barbecued), roasts (cooked using conventional and microwave ovens) and stews (open pan and pressure cooked). For some cooking methods, the authors report concentration changes on a whole weight basis between the raw and cooked product, which could be attributed to changes arising from the loss of water and elimination of PCDD/Fs with released fat. The data show that those cooking processes that release or remove fat from the product tend to reduce the total PCDD/Fs amount in the prepared food. As reported by Petroske et al. (1998), also this survey showed that none of the cooking procedures studied which resemble typical home‐cooking practices had any substantial effect on the total PCDD/Fs amount present, i.e. PCDD/Fs were neither formed from potential precursors nor degraded as a result of the applied cooking process.
Wu et al. (2011) performed cooking experiments to investigate the generation of PCDD/Fs during cooking at high temperatures with sucralose and 1,3‐dichloro‐2‐propanol as chlorine containing flavourings. Their results indicate that PCDD/Fs can be generated during cooking processes, such as burning or when cooking with reactive organochlorine compounds. However, the PCDD/Fs are more likely to be present in the smoke (gas phase) than in the edible portion. The results indicate that good ventilation should be maintained during cooking to avoid PCDD/F intake from the air.
Data on the influence of processing practices on the PCDD/F and PCB concentration in foodstuffs of plant origin are scarce. While Hori et al. (2001) investigated the effects of cooking processes on the PCDD/F and PCB levels in komatsuna, a popular green leafy vegetable in Japan, Tsutsumi et al. (2002) studied the respective effects on two types of spinach. Both groups found a substantial reduction in PCDD/Fs and PCB levels after washing and a further decrease after boiling of up to 80%. These results indicate that washing vegetables before cooking may already have a considerable impact on the PCDD/Fs and PCB levels in the food processed for consumption.
There has been considerable focus on the possible reduction of PCDD/Fs and DL‐PCBs in feed ingredients. Fish oils and fish meals can be decontaminated by various processing techniques involving the use of activated carbon and hexane extraction, respectively. Such processing leads to significant reduction in the levels of these contaminants; reduction of PCDD/Fs and DL‐PCBs in fish oil by 82–95% and 26–55%, respectively, and reduction of PCDD/Fs and DL‐PCBs in fish meal by 70–97% and 60–93% respectively (EFSA CONTAM Panel et al., 2017a,b, 2018a,b). Fish oils consumed directly as food supplements are also refined in order to reduce their contamination levels.
In summary, typical household cooking practices neither lead to degradation nor to generation of appreciable amounts of PCDD/Fs and PCBs. At high temperatures, some release of lower chlorinated congeners may occur due to their greater volatility compared to the higher chlorinated congeners. Several studies have shown that application of various cooking practices may lead to substantial PCDD/F and PCB losses of even more than 50% in the processed products as consumed compared to the raw commodity. Exposure to PCDD/Fs and PCBs can therefore be lowered by discarding the fat which is released from food during processing. On the other hand, the PCDD/F and PCB levels in processed foods may exceed the levels in the corresponding raw materials if cooking oils are used which contain appreciable amounts of PCDD/F. The data indicate the importance of taking food processing into account when reliably estimating human exposure to PCDD/Fs and PCBs, as the consideration of only raw foodstuffs may lead to an over‐ or underestimation of the exposure.
3.2.4. Levels in humans
Analytical methods
As PCDD/Fs and PCBs are lipophilic compounds that accumulate in humans, lipid rich matrices, such as adipose tissue, human milk and blood are the tissues of choice when estimating human exposure. Initially the determination of PCDD/Fs and PCBs in humans was restricted to cases where a sufficient sample amount for analysis was available, such as adipose tissue or to samples with substantial concentrations far above background exposure, like human milk from contaminated areas, such as Vietnam (Baughman, 1974). The increasing knowledge on human exposure during the past four decades goes in parallel with the improvement of the analytical instrumental capabilities, in particular concerning the substantial lowering of the analytical limits of detection.
In principle, the modern analytical methods follow the same approach as methods applied for feed and food analyses. The analytical methods are generally based on application of bioassays, such as CALUX and/or GC–HRMS and GC–MS/MS (see Sections 1.3.2 and 3.5). Modern procedures based on GC–HRMS and GC–MS/MS allow a congener‐specific determination of PCDD/Fs and DL‐PCBs in biological samples at low concentrations even with sample weights of less than 20 g human milk or blood.
Irrespective of the analytical method applied, generally two approaches are followed; (i) initial extraction and gravimetric determination of lipids with a subsequent PCDD/F/DL‐PCB analysis from an aliquot, or (ii) extraction of the analytes from the original sample with a separate determination of lipids by enzymatic analysis. The type of approach may be crucial for lipid‐adjusted results of liver and blood samples, as different methods may cover diverging blood fractions. This is not only true for the choice of solvents for lipid extraction but also for the selection of blood fractions included for enzymatic determination of lipids. The latter may cover besides triglycerides also free and/or total cholesterol, phospholipids and other blood fractions as the term ‘total lipids’ is not defined (Bernert et al., 2007). Thus, dependent on the polarity of the solvents which are generally adapted to the extraction of the analytes of interest, specific blood fractions that are included in the enzymatic analysis may not be covered by the solvent extraction.
In summary, the variability in the exact composition of the lipid fraction and in the per cent lipid determined by different methods should be considered when comparing PCDD/F and DL‐PCBs levels in biological samples, such as blood or human milk from different laboratories (Patterson et al., 1989a,b).
Levels in human milk and time trends
The use of human milk samples to estimate human body burden with PCDD/Fs and PCBs has the advantage that (i) sample collection is non‐invasive, (ii) it can give information on the body burden of the mother as well as on the exposure of the child, and (iii) it has a relatively high lipid content. The disadvantage is that those measurements are restricted to lactating women in a relatively narrow age window.
The first results from comprehensive human milk studies on PCDD/F were reported by Fürst et al. (1986), van den Berg et al. (1986), Rappe et al. (1986) and Ende (1986). These studies showed similar results. All congeners identified were 2,3,7,8‐chlorine substituted. While on a concentration basis OCDD generally represented more than 50% of the total PCDD amount, the levels of the other PCDD congeners declined with decreasing grade of chlorination. In all samples the levels of the three HxCDD congeners with 2,3,7,8‐chlorine substitution were nearly of the same relative proportion with 1,2,3,6,7,8‐HxCDD dominating. The PCDF levels were generally considerably lower than those of the PCDDs. The share of OCDF did by no means contribute to the sum of PCDFs at such a ratio as OCDD amounted to the total PCDD level. Similar to HxCDDs, the isomer distribution of the HxCDF congeners was quite constant in the human milk samples analysed in the above studies. In contrast to 1,2,3,4,7,8‐, 1,2,3,6,7,8‐, and 2,3,4,6,7,8‐HxCDF, 1,2,3,7,8,9‐HxCDF was generally not identified above the limit of detection. The predominant PCDF congener detected in the human milk samples was generally 2,3,4,7,8‐PeCDF.
In the following years, human milk became an important matrix to globally survey the PCDD/F and PCB human exposure by numerous research groups and national institutions. Lakind et al. (2001), Lakind (2007), Srogi (2008), Ulaszewska et al. (2011) and Fång et al. (2013) give comprehensive overviews on PCDD/Fs and PCBs in human milk of mothers from different geographical areas. Irrespective of the origin, the patterns found in human milk resemble to a great extent those that were analysed in the surveys already performed in the 1980s. However, substantial differences were found in the concentrations of PCDD/Fs and PCBs in the samples. Generally, the levels were higher in human milk samples from industrialised countries compared to respective samples from developing countries.
Results of WHO coordinated human milk studies
Even in developed countries, analytical capabilities are not always available to adequately analyse PCDD/Fs and PCBs in biological samples. Therefore, since 1987, WHO (partly in cooperation with the United Nations Environment Programme (UNEP)) conducted several coordinated surveys on the occurrence of POPs, including PCDD/Fs and PCBs in human milk. These surveys were not primarily intended to compare levels of POPs among countries, but rather to examine levels within countries over time. Therefore, strict protocols had to be followed with respect inter alia to selection of donors, location and time of sampling, storage and pooling of samples, and shipping of the samples to the laboratory (WHO, 2007). Six rounds were performed between 1987 and 2015. To ensure consistency in the analytical measurements, all samples of the third to sixth round were analysed for PCDD/Fs and PCBs by one laboratory, the European Reference Laboratory for dioxins and PCBs in Feed and Food in Freiburg/Germany.
The latest results of these surveys are given in Table 41 as UB values calculated with the WHO2005‐TEFs (Malisch and Schächtele, 2018, see documentation submitted to EFSA). Due to the low limits of quantification of the method applied, there were virtually no differences between the LB and UB levels, and thus only the UB levels are given. All pools contain the same number of individual samples collected based on the above strict protocol. In case more than one pool was analysed in a specific round from a country, the median of the respective concentrations is shown in the table. Unfortunately, a number of countries did not participate in all rounds. In this case, the results of the latest round(s) are given. The data from the last round including samples collected in 2014/2015 show similar results that are in general much lower than respective samples collected in the earlier rounds.
Table 41.
PCDD/Fs, DL‐PCBs and total PCDD/F/DL‐PCBs in human milk pools from various European countries as analysed in the frame of WHO coordinated studies. All levels are given as pg WHO2005‐TEQ/g fat (upper bound). The last column shows the relative proportion of PCDD/F‐TEQ in relation to total TEQs
| Country | Year | PCDD/Fs | DL‐PCBs | PCDD/F/DL‐PCBs | Ratio PCDD/F‐TEQ : total‐TEQ |
|---|---|---|---|---|---|
| (pg WHO2005‐TEQ/g fat) | (%) | ||||
| Belgium | 2015 | 4.03 | 2.47 | 6.50 | 62 |
| 2010 | 6.95 | 3.74 | 10.7 | 65 | |
| 2006 | 8.49 | 4.38 | 12.9 | 66 | |
| Bulgaria | 2014 | 4.18 | 1.96 | 6.14 | 68 |
| 2001 | 5.07 | 3.21 | 8.28 | 61 | |
| Croatia | 2014 | 2.40 | 2.42 | 4.82 | 50 |
| 2001 | 5.42 | 4.92 | 10.3 | 52 | |
| Cyprus | 2006 | 3.85 | 1.85 | 5.70 | 68 |
| The Czech Republic | 2014 | 3.82 | 3.53 | 7.35 | 52 |
| 2006 | 6.28 | 6.96 | 13.2 | 47 | |
| 2001 | 6.04 | 7.37 | 13.4 | 45 | |
| Finland | 2007 | 4.83 | 2.10 | 6.93 | 70 |
| 2001 | 8.05 | 3.68 | 11.7 | 69 | |
| Georgia | 2014 | 3.12 | 3.44 | 6.56 | 48 |
| 2009 | 3.95 | 4.85 | 8.80 | 45 | |
| Germany | 2002 | 10.6 | 8.47 | 19.1 | 56 |
| Hungary | 2006 | 4.25 | 1.52 | 5.77 | 74 |
| 2001 | 5.99 | 2.19 | 8.18 | 73 | |
| Ireland | 2010 | 4.58 | 2.04 | 6.62 | 69 |
| 2002 | 6.82 | 3.10 | 9.92 | 69 | |
| Italy | 2001 | 10.6 | 9.54 | 20.1 | 53 |
| Lithuania | 2015 | 4.15 | 3.15 | 7.30 | 57 |
| 2009 | 4.87 | 4.26 | 9.13 | 53 | |
| Luxembourg | 2006 | 8.92 | 6.75 | 15.7 | 57 |
| 2002 | 12.6 | 9.10 | 21.7 | 58 | |
| Moldavia | 2015 | 4.37 | 4.72 | 9.09 | 48 |
| 2009 | 7.54 | 7.47 | 15.0 | 50 | |
| Norway | 2006 | 4.62 | 3.18 | 7.80 | 59 |
| 2001 | 6.26 | 4.67 | 10.9 | 57 | |
| Romania | 2014 | 5.71 | 3.92 | 9.63 | 59 |
| 2001 | 7.29 | 5.88 | 13.2 | 55 | |
| Slovak Republic | 2006 | 5.10 | 4.85 | 9.95 | 51 |
| 2001 | 7.39 | 6.21 | 13.6 | 54 | |
| Spain | 2002 | 10.0 | 5.66 | 15.7 | 64 |
| Sweden | 2007 | 5.03 | 4.18 | 9.21 | 55 |
| 2001 | 8.32 | 5.96 | 14.3 | 58 | |
| Switzerland | 2009 | 5.04 | 4.84 | 9.88 | 51 |
| The Netherlands | 2014 | 4.48 | 2.68 | 7.16 | 63 |
| 2001 | 15.9 | 7.59 | 23.5 | 68 | |
| Ukraine | 2001 | 8.54 | 10.7 | 19.2 | 44 |
PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; DL‐PCBs: dioxin‐like polychlorinated biphenyls; TEQ: toxic equivalents.
It is noteworthy that the relative TEQ contribution of PCDD/Fs to total PCDD/Fs/DL‐PCBs‐TEQs between countries shows some variation as it ranges between 44% and 74%. In contrast, within a country the ratio is quite consistent over time.
Results of European individual human milk studies performed after 2010
Individual human milk studies comprising samples from Europe collected after 2010 are scarce. Within their assessment of temporal trends of PCDD/Fs and PCBs in human milk from Stockholm/Sweden, Fång et al. (2013) analysed two pools each collected in 2010 and 2011, respectively. While the PCDD/F levels in the two pools from 2010 amounted to 2.7 and 3.6 pg WHO2005‐TEQ/g fat, the corresponding levels in the two pools from 2011 were 3.1 and 2.7 pg WHO2005‐TEQ/g fat. The DL‐PCBs were found to be 5.2 and 6.2 pg WHO2005‐TEQ/g fat in the 2010 pools and 5.5 and 4.6 pg WHO2005‐TEQ/g fat in the pools collected in 2011.
Schuhmacher et al. (2013) analysed human milk samples for PCDD/Fs and DL‐PCBs periodically between 1998 and 2012 from primiparae mothers, living in the vicinity of a hazardous waste incinerator in Catalonia/Spain. The study was part of a surveillance programme that focused on determining a potential impact on human health derived from emissions from the incinerator which began its regular operation in 1999. Twenty milk samples were collected in 2012 from women aged between 26 and 44 years. The samples showed levels of 4.79 ± 2.76 (range: 1.06–12.3) and 2.48 ± 1.15 (range: 0.71–5.28) pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs, respectively. Compared to the results of human milk samples from the same area analysed in 1998, the mean PCDD/F and DL‐PCB levels in 2012 were significantly lower with decreases of 61% and 86%, respectively. The authors concluded that the incinerator does not pose a significant impact on the population living in the neighbourhood and interpreted the decline of the human milk levels as a result of the general reduction of PCDD/F and PCB emissions and the continued decrease of dietary exposure to these contaminants.
Parera et al. (2013) monitored PCDD/Fs in human milk in 2008 and 2012 in women living nearby a modern solid waste incinerator in Spain. Samples from a total of 46 women aged between 18 and 40 years old were collected. Women were stratified in four groups according to age (≤ 30 years old or > 30 years old) and previous child (none or one). For women ≤ 30 years old, the total PCDD/Fs concentration (pg WHO2005‐TEQ/g fat) in 2008 was 3.4 (first child) and 3.2 (second child), while in 2012 it was reported as 3.8 and 2.9, respectively. For women > 30 years old the PCDD/Fs concentrations in 2008 were 6.6 (first child) and 4.8 (second child), while in 2012 they were 3.4 (first child) and 2.3 (second child) pg WHO2005‐TEQ/g fat.
Kamińska et al. (2014) collected 40 human milk samples between 2008 and 2010 from mothers living in Central Poland. While 20 women aged 23–34 years resided in an urban area, the other 20 specimens were from women aged 22–38 years living in a rural area. In the samples from the urban area, mean levels of 5.58 ± 3.11 (range: 0.31–10.6) and 1.85 ± 1.58 (range: 0.02–4.5) pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs, respectively, were found. The respective mean levels in the rural area were 4.52 ± 2.86 (range: 0.26–11.0) and 1.92 ± 1.34 (range: 0.02–4.8) pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs.
Antignac et al. (2016) analysed 96 human milk samples collected between 2011 and 2014 from French women (age 30.3 ± 4.7 years) and found median levels of 6.07 and 4.31 pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs, respectively.
Global temporal trends on PCDD/F and DL‐PCB levels in human milk
In a comprehensive review, Lakind (2007) evaluated global data on PCDD/Fs in human milk and showed that the levels decreased globally between 1975 and 2005. A few studies reported results on annual sampling and analysis of human milk from European countries over a number of years. Based on the analyses of more than 1,000 individual samples, Fürst (2006) reported a decline of the median PCDD/F levels in human milk collected from mothers living in North‐Rhine‐Westphalia/Germany from 33.9 to 9.8 pg I‐TEQ/g fat between 1989 and 2003. BfR (2011) compiled data on PCDD/F in human milk from all over Germany where multiple year sampling and analyses of human milk is performed by several institutions since the middle of the 1980s and reported a decrease of WHO1998‐PCDD/F‐TEQs from 35.7 to 6.3 pg/g fat for the time period 1986‐2009.
Lignell et al. (2009) analysed human milk samples collected annually between 1996 and 2006 from randomly recruited primiparae living in Uppsala County/Sweden. Results were adjusted for life‐style factors that are associated with POP body burdens, such as age, body mass index (BMI), and weight changes during and after pregnancy. Applying a multiple linear regression analysis, annual declines of 6.7, 4.6 and 6.5% for PCDDs, PCDFs and mono‐ortho‐PCBs, each calculated with the WHO2005‐TEFs were calculated, indicating a faster decrease for PCDDs and mono‐ortho PCBs compared to PCDFs.
Fång et al. (2013) assessed temporal trends for PCDD/Fs and DL‐PCBs in archived pooled human milk samples collected annually between 1972 and 2011 from women aged 27‐31 years living in Stockholm/Sweden. Each pool consisted of 50 g milk. The number of donating mothers (not exclusively from primiparae) in each pool ranged from 9 to 140. While in 1972 the levels for PCDD/Fs and DL‐PCBs were found to be 26 and 30 pg WHO2005‐TEQs/g fat, respectively, the corresponding levels in 2011 only amounted to 2.7 and 1.9 pg WHO2005‐TEQs/g fat. Concentrations for PCDD/F‐ and DL‐PCB‐TEQs show a statistically significant relative annual decline between 1972 and 2011 of 6.1% and 6.9%, respectively. While the contribution of PCDD/F‐WHO2005‐TEQ to total TEQ in the pools from the 1970s was around 50%, the data from 2010/2011 point more to 55–60%. However, these latter results should be considered with caution because of the variable number of donating mothers in the different pools with in some cases the number of donors being small.
In summary, the occurrence data from the past few years as reported by individual studies as well as by the WHO field studies, indicate that the current median PCDD/F and DL‐PCB levels in human milk from European mothers are generally below 10 pg WHO‐TEQ/g fat. Pooled human milk samples collected as part of the WHO field studies across European countries in 2014/2015 revealed levels of 2.4‐5.7 pg WHO2005‐TEQ/g fat and 4.8–9.6 pg WHO2005‐TEQ/g fat for PCDD/Fs and for the sum of PCDD/Fs and DL‐PCBs, respectively. The data indicate a substantial global decline of PCDD/F and DL‐PCB levels in human milk since the first measured samples collected in the early 1980s. This may be an indication that the measures to decrease the environmental release were effective where applied (UNEP, 2013). However, available results from the last decade are quite similar. Whether this is an indication that the concentrations of PCDD/Fs and DL‐PCBs in human milk are levelling off can only be answered if data from future years of monitoring are available.
Parameters that influence PCDD/F and DL‐PCB levels in human milk
In order to gain knowledge on the parameters that influence the levels of PCDD/Fs and PCBs in human milk, a number of studies include questionnaires which have to be filled out by the mothers. These questionnaires ask for information inter alia concerning personal characteristics (e.g. age, height, weight, number of breastfed children, length of breastfeeding period(s)), living conditions (e.g. rural/urban, potential emission sources in the vicinity, type of heating system), food consumption and smoking habits.
Number of breastfed children and length of nursing periods
It has been demonstrated that the previously mentioned parameters may have a substantial impact on the PCDD/F and DL‐PCB levels in human milk. This is especially true for the number of breastfed infants and the length of the nursing periods. PCDD/F and PCB levels in human milk from primipara mothers were found to be higher than those from multipara mothers (Fürst et al., 1992a; Beck et al., 1994; Yang et al., 2002; Sasamoto et al., 2006; Uechara et al., 2006). Moreover, it was found that the PCDD/F levels declined with the length of the nursing period(s) with some differences depending on the congener (Fürst et al., 1992a; Beck et al., 1994; Vigh et al., 2013). Vigh et al. (2013) collected human milk at days 5, 12 and 84 post‐partum from 22 primipara mothers (age: 27.7 ± 4.4 years) who delivered their infants during 2007. At the three time‐points, the total PCDD/F levels were 2.13 ± 1.36, 1.85 ± 1.19 and 1.65 ± 1.02 pg WHO2005‐TEQ/g fat. The respective levels found for DL‐PCBs were 1.04 ± 0.86, 0.85 ± 0.83 and 0.75 ± 0.82 pg WHO2005‐TEQ/g fat. The main decline was found between days 5 and 12, and the decline in elimination of the higher chlorinated PCDDs was faster than that of the lower chlorinated ones. These results are in good agreement with the data by Beck et al. (1994) who analysed human milk samples at weeks 1, 6 and 12 post‐partum from primipara mothers. In contrast, the levels increased with the age of the mother (Fürst et al., 1992a; Beck et al., 1994; Lignell et al., 2009, 2011; Ulaszewska et al., 2011).
Food
There is general consensus that food, especially of animal origin, contributes more than 90% to human PCDD/F and PCB body burdens (Fürst et al., 1992b) for those who are not occupationally exposed. Thus, it becomes apparent that certain foods, particularly if highly contaminated, may have an important impact on the PCDD/F and DL‐PCB levels in human milk (Li et al., 2009; Lignell et al., 2011). Lignell et al. (2011) showed that women with the highest consumption of fatty Baltic fish the year before pregnancy had 11–16% higher levels of mono‐ortho PCB‐TEQ, PCDF‐TEQ and BDE‐153 in their milk than women with no consumption of such fish. Beck et al. (1994) reported that the PCDD/F levels in six human milk samples from vegans and vegetarians were slightly lower than those in 60 samples from mothers with a common mixed diet.
Weight change
The investigations into the influence of weight change during breastfeeding on the body burden of persistent lipophilic contaminants of the mothers show variable results, as reviewed by Lakind (2007) and Lignell et al. (2011, 2016). However, it has to be stated that most of the studies were performed with organic pesticides, such as DDT/DDE, HCB, HCH and NDL‐PCBs. In contrast, respective studies on PCDD/Fs and DL‐PCBs are scarce. Lignell et al. (2011) reported that based on a multiple regression model, levels of PCDD/Fs and DL‐PCBs were positively associated with weight loss after delivery and inversely associated with prepregnancy BMI and weight gain during pregnancy. However, prepregnancy BMI, weight gain during pregnancy and weight loss after delivery explained a smaller part of the variation in PCB‐ and PCDD/F‐levels than age and sampling year. In a further study, Lignell et al. (2016) examined associations between weight loss and concentrations of chlorinated POPs (including PCB‐153) in human milk in 32 lactating women participating in a weight loss study. Most women lost weight during the study (0.45 ± 0.30 kg per week). Among these women, the concentration of PCB‐153 in human milk was significantly (p = 0.04) higher at follow‐up than at baseline. Weight loss was significant and positively associated with an increase of PCB 153 of 2.0% per percent weight loss. Despite the fact that PCB‐153 is an NDL‐PCB, the result supports the conclusion of the Lignell et al. (2011) study because of the lipophilic nature of this compound which resembles the PCDD/Fs.
Area of domicile
The area of domicile, whether urban or rural, had no effect on the body burden with these contaminants (Lindström et al., 1988; Beck et al., 1994). This latter finding was not confirmed in studies from other geographical areas. In their review, Ulaszewska et al. (2011) summarised results published between 2000 and 2009 from human milk studies that analysed PCDD/Fs and PCBs. Besides the levels in human milk from different geographical regions, the review focussed on studies that reported on factors that potentially influence the body burden, such as the effect of living in an urban or industrialised area, living near an incinerator or dumping sites and the effects of smoking. The findings in these studies are sometimes conflicting, especially with regard to the potential impact of the area of living (urban vs. rural areas) or specific diets on the body burden. The impact of these factors is obviously highly dependent on the specific situation and the existence of a potential hotspot.
Smoking
A possible effect of smoking on the PCDD/F levels in human milk is still debated. Fürst et al. (1992a) reported that mothers who are active or passive smokers contain on average significantly lower PCDD/F levels than non‐smoking women. While this finding was confirmed by several other studies, the contrary result was observed in other investigations (Ulaszewska et al., 2011).
In summary, the above studies show that certain parameters can influence the concentrations of PCDD/Fs and DL‐PCBs in human milk, and therefore it is crucial to take the year of sampling, personal characteristics and the lifestyle of the mothers into consideration to avoid wrong conclusions when comparing the results of the studies.
Levels in human blood and other biological tissues
To assess the body burden of lipophilic contaminants in children, non‐lactating women and men, the matrices of choice are adipose tissue and/or preferably blood.
Due to the limited analytical sensitivity in the 1980s, the assessment of body burden of potentially exposed persons was performed by analysing adipose tissue obtained by biopsy or by liposuction (Rappe et al., 1987; Schecter and Ryan, 1988; Beck et al., 1989). The suitability of blood serum as a matrix for body burden measurements was investigated by Patterson et al. (1988). He analysed paired human serum and adipose tissue samples from 50 persons for TCDD. After adjusting the adipose tissue and serum TCDD levels each for total lipid content, the mean of the partitioning ratio of adipose tissue to serum TCDD was 1.09. Moreover, the TCDD levels in adipose tissue and serum were highly correlated (r = 0.98). Schecter et al. (1990) analysed paired adipose tissue and plasma from 20 patients for PCDD/Fs and found also ratios for the congeners close to 1 with some exceptions for the higher chlorinated congeners. These findings indicated that serum is a suitable matrix for measurements of human PCDD/Fs and PCBs body burden and broadened the possibilities for large scale investigations in cohorts as drawing of blood is a much less invasive sampling procedure than collecting adipose tissues. In addition, because of the considerable improvement of the analytical sensitivity which allows an analysis of blood for PCDD/Fs and PCBs with less than 20 mL, even small children could be included in the surveys.
Consonni et al. (2012) performed a comprehensive worldwide literature review on blood levels of PCDD/Fs and PCBs in the general (non‐exposed) adult population. The review comprised 187 studies published between 1989 and 2010 from 26 countries. The year of blood collection ranged from 1985 to 2008. Data for DL‐PCBs in these studies were first reported in 1992. PCDD/F and/or DL‐PCB congener levels were reported in 161 studies. Based on 13,370 blood samples, for PCDD/Fs a range of 2.3–63.4 (mean: 12.4; median: 10.8) pg WHO2005‐TEQ/g fat was calculated. For DL‐PCBs, the range based on 8,390 blood samples was 1.8–42.4 (mean: 3.7; median: 1.9) pg WHO2005‐TEQ/g fat. As the statistics include both individual and pooled samples, the results were weighted by number of subjects. The number of subjects included in the evaluation was 26,110 and 20,152 for PCDD/Fs and DL‐PCBs, respectively. The authors reported that blood samples from European countries showed higher levels of most PCDD/F, DL‐PCB congeners and TEQs compared to specimens from non‐European countries. A positive association of subjects’ age with most congeners and with TEQ values (1–3% increase per year) was found, confirming previous findings. Significant decreases (6–7% decrease per year) over time (1985–2008) were documented for PCDD/Fs and TEQs. No significant decrease was found for non‐ortho‐PCBs, notably PCB‐126.
As already demonstrated for human milk, also the PCDD/F and PCB levels in human blood may be substantially affected by consumption of highly contaminated food. This was demonstrated by Kiviranta et al. (2000) who analysed blood plasma from 47 fishermen in Finland. Those 26 fishermen who ate fatty fish from the Baltic Sea at least twice a week, had substantially higher levels (mean: 180; median 170 pg I‐TEQ/g fat) compared to controls who ate fish less than once a week and showed mean and median levels of only 33 and 32 pg I‐TEQ/g fat, respectively. Moreover, the authors reported that individual congener patterns in the investigated human blood samples were similar to congener patterns in the fish species consumed. In a follow‐up publication, Kiviranta et al. (2002) presented the results as WHO1998‐TEQ and also reported DL‐PCB levels analysed in blood from those 26 fishermen who were considered as highly exposed. The mean and median PCDD/F levels were reported as 220 and 210 (range: 58–500) pg WHO1998‐TEQ/g fat, respectively, and the mean and median DL‐PCB levels were given as 110 and 96 (range: 22–400) pg WHO1998‐TEQ/g fat, respectively. Unfortunately, no further data were reported for the controls.
As part of the Norwegian Fish and Game Study, Kvalem et al. (2009) related dietary patterns to intake and blood concentrations of PCDD/Fs, DL‐PCBs and six NDL‐PCBs from ‘representative’ and ‘high’ consumers. A statistically significant correlation was observed between the estimated dietary intake of PCDD/Fs and PCBs and their respective serum levels. Fatty fish was found to be the major source of DL‐PCBs and NDL‐PCBs in all consumers, contributing to approximately 70% of total TEQ intake. Four dietary patterns were identified by principal component analysis. Two were related to high intakes, one was dominated by oily fish, and the fourth one by fish liver and seagull eggs. Only the latter was closely associated with higher blood concentrations of PCDD/Fs and PCBs.
Blood samples collected after 2010
Analyses for PCDD/Fs and DL‐PCBs in blood samples collected after 2010 are scarce. Fromme et al. (2015) collected blood samples in 2013 from 70 subjects (37 females and 33 males) between 4 and 76 years old. The participants were recruited in the neighbourhood of a reclamation plant in a rural area in Southern Germany. Median levels of 4.5 and 2.6 pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs, respectively, were found. The 95th percentiles were reported as 17.9 and 13.2 pg WHO2005‐TEQ/g fat for PCDD/Fs and DL‐PCBs, respectively. A significant correlation of PCDD/F‐TEQ with DL‐PCB‐TEQ (r: 0.862, p < 0.001) was found. Higher concentrations for PCDD/F and DL‐PCBs were found with increasing age. No significant differences between females and males were observed.
In a further study, Fromme et al. (2016) analysed 42 blood samples collected in 2013 from randomly selected healthy subjects (age: 20–68 years) living in Munich and the surrounding areas for a number of persistent organic contaminants, including PCDD/Fs and DL‐PCBs. Mean levels of 7.2 and 4.5 pg WHO2005‐TEQ/g fat were found for PCDD/Fs and DL‐PCBs, respectively. While the median and maximum levels for PCDD/Fs were 6.2 and 20.6 pg WHO2005‐TEQ/g fat, the corresponding DL‐PCB levels amounted to 4.1 and 11.0 pg WHO2005‐TEQ/g fat. Compared with the data reported by Wittsiepe et al. (2008), the mean levels have declined by 53 and 28% for PCDD/Fs and DL‐PCBs. As the concentrations measured in the current samples resemble the respective levels in human milk, they seem to indicate the background contamination of the general population, at least, in Germany.
Parera et al. (2013) also monitored PCDD/Fs in blood samples between 1995 and 2012 in the general population living nearby a modern solid waste incinerator in Spain. The study group was established just after the plant was put into service and included subjects living < 1 km from the incinerator plant (considered ‘exposed’), subjects living > 3 km from the incinerator plant (considered ‘non‐exposed) and subjects living in a nearby town (also considered ‘non‐exposed’). In addition, a group of workers of the plant was included. Blood pools were prepared by mixing together individual samples according to age, sex and exposure groups. The PCDD/F levels determined in samples collected in 2012 from subjects considered ‘exposed’ (n = 68), ‘non‐exposed living > 3 km from the incinerator plant’ (n = 94), ‘non‐exposed living in a nearby town’ (n = 86) and workers (n = 10) were 13.1, 13.8, 13.1 and 13.2 pg WHO2005‐TEQ/g fat, respectively. Values of PCDD/Fs for the total exposed and total of unexposed groups were calculated as weighted geometrical means.
Comparison of PCDD/F and PCB levels in various matrices
The use of different matrices for body burden assessments poses the question of comparability of the results. Wittsiepe et al. (2007b) analysed blood and milk samples from 169 lactating women belonging to the Duisburg cohort for PCDD/F and DL‐PCBs. For PCDD/Fs, the WHO1998‐TEQ were in the range of 2.7–55.1 pg/g fat (median: 15.3, arithmetic mean: 16.8) for blood, and 1.8–34.7 pg/g fat (median: 13.3, arithmetic mean: 13.8) for milk, respectively. For DL‐PCBs, the WHO1998‐TEQ were in the range of 1.4–42.2 pg/g fat (median: 10.8, arithmetic mean: 11.6) for blood, and 1.2–50.1 pg/g fat (median: 13.0, arithmetic mean: 13.4) for milk, respectively. Within each matrix, high correlations were found between PCDD/F and DL‐PCB TEQ levels (r = 0.86 (blood) or 0.83 (milk); both significant at p < 0.05). For the various congeners, different distributions in the two body compartments were reported. In blood, higher chlorinated substances were found in higher concentrations when compared to milk (e.g. OCDD 3.3‐fold), whereas lower chlorinated congeners such as pentachlorinated biphenyls are predominantly enriched in milk in concentrations about 1.5‐fold higher than those found in blood. The authors concluded that the different lipophilicity of the substances, the molecule diameter and molecular weight influencing the membrane permeability might be responsible for this observation. Overall, good correlations between lipid‐adjusted PCDD/F‐TEQs (r = 0.83) and DL‐PCB‐TEQs (r = 0.91) in blood and milk were found.
Congener‐specific differences between milk and blood lipids were also shown by Nakano et al. (2005), with several of the PCDD/F congeners at higher concentrations in blood as compared to milk.
‘t Mannetje et al. (2012) performed a literature review to assess the relationship between PCDD/F and PCB congener levels in human blood and human milk, expressed as serum/milk ratio. The results from the different studies revealed that the mean reported serum/milk ratio (lipid adjusted) was greater than 1 for all individual PCDD/F congeners. For the lower chlorinated PCDDs (up to six chlorines), the mean serum/milk ratio was around 1.3; increasing to approximately 3 for hepta‐CDD and 6 for OCDD. For PCDFs, this pattern was less consistent, but nevertheless had similarities to the pattern seen for PCDDs.
Besides blood and human milk, Schecter et al. (1998b) also included adipose tissue, placenta and cord blood when they investigated the partitioning of PCDD/F and three DL‐PCB (PCB‐77, ‐126 and ‐169) in these matrices from five women. Despite some distinct differences, especially in the measured higher chlorinated PCDD congeners, the mean concentrations for the sum of PCDD/Fs and the three DL‐PCBs calculated as TEQs38 (lipid adjusted) were similar with values of 11.6 pg TEQ/g for adipose tissue, 12.1 pg TEQ/g for predelivery blood, 10.5 pg TEQ/g for placenta, 9.98 pg TEQ/g for post‐partum blood, and 10.2 pg TEQ/g for human milk. The exception was cord blood which showed only a TEQ of 5.81 pg/g, (all data lipid adjusted). Thus, the analysis of maternal blood, placenta and cord blood may give a valuable indication of the prenatal PCDD/F and DL‐PCB exposure of infants.
PCDD/F and co‐planar‐PCBs in blood, adipose tissue, human milk, cord blood and placenta collected from 44 pregnant Japanese women were analysed by Nakano et al. (2005). Correlations were observed between PCDD/F‐TEQ (fresh weight) in blood and in adipose tissue (r = 0.913, p < 0.0001), human milk (r = 0.695, p = 0.0007), and cord blood (r=0.759, p < 0.0001). Total concentrations of dioxins and total TEQ in cord blood were approximately 56% and 39% lower than corresponding values in maternal blood.
Aylward et al. (2014) reviewed reported data from more than 100 studies on the ratios of cord/maternal blood concentrations for a range of lipophilic chemicals, including PCDD/Fs and PCBs. Six studies had data on partitioning of PCDD/Fs between cord blood and maternal blood, and the authors reported wet weight ratios generally in the range of 0.1 to 0.4, with lipid‐adjusted ratios near 1 for most congeners which is consistent with the difference in cord vs. maternal blood lipid content. Cord/maternal blood ratios appear to be generally higher for the PCDF congeners compared to PCDD congeners. For PCBs, central estimates of ratios of cord/maternal blood levels generally ranged between 0.1 and 0.4 on a wet weight basis. On a lipid‐adjusted basis, the reported central estimates of cord/maternal blood ratios were typically below or close to 1 for all PCB congeners.
Thoma et al. (1990) analysed adipose tissue and liver samples from 28 people from the Munich area for 2,3,7,8‐chlorine substituted PCDD/Fs. The data show that for up to 6 chlorine substituents, the lipid adjusted PCDD liver/adipose tissue ratio is about 2. With higher degree of chlorination the ratio increases to 12 for OCDD. In contrast to PCDDs, the ratios of the PCDF congeners increase continuously from 2.2 for TCDF to 15.4 for HpCDF and drops to 7.4 for OCDF. The ratio greater than 1 suggests binding to specific proteins in the liver (sequestration, see Section 3.1.1.3).
Beck et al. (1990) reported on the analysis of three paired adipose tissue and liver samples from infants who died suddenly and unexpectedly (SIDS) for PCDD/Fs. The liver/adipose tissue ratio (lipid adjusted) of PCDDs were similar to the ones of the before mentioned Thoma et al. (1990) study. Both studies showed that the liver/adipose tissue fat ratios for the higher chlorinated congeners HxCDF, HpCDF, HpCDD, OCDF and OCDD are markedly higher than those for the lower chlorinated congeners, suggesting differences in the degree of sequestration between congeners.
Bajanowski et al. (2002) analysed PCDD/Fs in liver, kidney, subcutaneous fatty tissue and spleen from 27 SIDS cases. The cases were subdivided into 2 groups consisting of 15 infants who died in 1991/1992 and in 12 infants who died in 1996/1997. The main findings can be summarised as follows: (i) there was a substantial decrease in the PCDD/F concentration in infant tissues from 1991/1992 to 1996/1997, (ii) the birth order was inversely and the duration of breastfeeding directly proportional to the PCDD/F concentrations, (iii) the levels in the biological samples decreased from exclusively to partially and non‐breast fed infants. Moreover, some distinct differences were found for the ratio of various congeners between liver and adipose tissue. While the liver/adipose tissue ratio (lipid adjusted) for OCDD and 1,2,3,4,6,7,8‐HpCDD in the two highest groups was calculated as 11.2 and 7.0, the respective ratios of the other PCDDs with 2,3,7,8‐chlorine substitution amounted only to 0.8–1.7. For PCDFs, the highest ratio in the most exposed groups was found for OCDF at 16.9. The ratios for the other PCDF congeners with 2,3,7,8‐chlorine substitution were determined as 1.4‐9.9. Similar ratios were found in the samples originating from non‐breast fed infants. When transformed into I‐TEQs, the ratio between liver and adipose tissue ranged between 2.3 and 2.8.
Abraham et al. (1996) studied intake, faecal excretion and body burden of PCDD/F in two breastfed and two formula‐fed infants during the first year of life. The PCDD/F intake was calculated up to 50 times higher in the breastfed infants. PCDD/F concentrations were measured in blood fat of the mothers and of all infants at the age of 11 months. PCDD/F concentrations in the formula‐fed infants (2.4, 2.6 pg I‐TEQ/g fat) were less than 20% of maternal values (16.9, 13.8 pg I‐TEQ/g fat) and more than 10 times lower than in the infants breastfed for 6‐7 months (29.2, 37.5 pg I‐TEQ/g fat). The PCDD/F levels in blood of the two breastfeeding mothers were found to be 12.3 and 10.5 pg I‐TEQ/g fat. The PCB‐126 levels in the blood of the two breastfeeding mothers were determined as 105 and 86 pg/g fat. The corresponding levels in the two breastfed infants were 287 and 374 pg/g fat. While in the two mothers of the formula‐fed infants, the PCB‐126 levels in blood amounted to 193 and 52 pg/g fat, the levels in the infants were only 24 and 18 pg/g fat.
In summary, the PCDD/F and DL‐PCB levels (lipid adjusted) in different human matrices from background populations within European countries are generally similar. However, on a congener‐specific basis, several congeners, in particular higher chlorinated PCDD/Fs showed a substantial enrichment in the liver due to sequestration. This must be taken into account when occurrence data from different matrices are used to estimate human body burden or to assess potential health effects.
3.3. Dietary exposure assessment
3.3.1. Dietary exposure assessment for humans
Mean and high dietary exposure
Chronic dietary exposure was estimated across Europe following the methodology described in Section 2.3. A total of 35 dietary surveys, carried out in 19 different Member States, were selected for this assessment. These dietary surveys and the number of subjects available per age class are described in Annex B (Table 4).
Two exposure assessments were carried out:
Assessment group A (29 congeners): taking into account the occurrence values of all the 29 congeners (17 PCDD/Fs and 12 DL‐PCBs).
Assessment group B (17 congeners): taking into account only the occurrence data of the 17 PCDD/F congeners.
Assessment group A
Table 42 summarises the chronic dietary exposure estimates for the 29 PCDD/Fs and DL‐PCBs across the 35 dietary surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in Annex B (Table 5A, Assessment group A).
Table 42.
Summary statistics of chronic dietary exposure assessment to the 29 PCDD/Fs and DL‐PCBs across European dietary surveys (Assessment group A)
| Age classa | N | Mean dietary exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 6 | 0.44 | 0.66 | 0.65 | 0.93 | 1.16 | 1.42 |
| Toddlers | 10 | 0.68 | 0.88 | 1.25 | 1.53 | 2.12 | 2.57 |
| Other children | 18 | 0.56 | 0.71 | 1.15 | 1.39 | 2.01 | 2.45 |
| Adolescents | 17 | 0.30 | 0.39 | 0.66 | 0.79 | 1.27 | 1.50 |
| Adults | 17 | 0.42 | 0.49 | 0.64 | 0.76 | 1.11 | 1.30 |
| Elderly | 14 | 0.39 | 0.52 | 0.67 | 0.77 | 1.27 | 1.37 |
| Very elderly | 12 | 0.43 | 0.57 | 0.64 | 0.73 | 1.21 | 1.32 |
| Age classa | N |
95th percentile dietary exposure (pg WHO2005‐TEQ/kg bw per day) |
|||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 5 | 1.38 | 1.89 | 1.96 | 2.45 | 2.81 | 3.28 |
| Toddlers | 7 | 2.02 | 2.38 | 2.75 | 3.19 | 5.05 | 5.94 |
| Other children | 18 | 1.52 | 1.73 | 3.10 | 3.47 | 6.02 | 6.63 |
| Adolescents | 17 | 0.91 | 1.07 | 1.93 | 2.08 | 4.06 | 4.34 |
| Adults | 17 | 0.94 | 1.18 | 1.94 | 2.08 | 2.87 | 3.11 |
| Elderly | 14 | 0.76 | 0.93 | 2.13 | 2.38 | 3.61 | 3.82 |
| Very elderly | 9 | 0.84 | 1.04 | 1.90 | 2.13 | 2.55 | 2.78 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Section 2.4 describes the age range within each age class.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Not including intake from breast milk.
The mean LB exposure across age groups and surveys ranged from 0.30 to 2.12 pg WHO2005‐TEQ/kg bw per day and the mean UB exposure range was 0.39–2.57 pg WHO2005‐TEQ/kg bw per day. At the 95 percentile exposure, the LB range across age groups and surveys was from 0.76 to 6.02 pg WHO2005‐TEQ/kg bw per day and at the UB from 0.93 to 6.63 pg WHO2005‐TEQ/kg bw per day. The highest exposures were calculated for the age classes Toddlers and Other children.
Median of the mean exposure ratios of PCDD/Fs and DL‐PCBs between either Toddlers and Adults, or Other Children and Adults, calculated for those surveys that included these age classes, were, respectively, 2.2 and 1.9.
Dietary exposure in specific groups of the population, namely pregnant and lactating women (each represented with one survey), were within the range of exposure estimates for the adult population. The LB mean exposure was 0.70 and 0.99 pg WHO2005‐TEQ/kg bw per day, while UB mean exposure was 0.82 and 1.12 pg WHO2005‐TEQ/kg bw per day for pregnant women and lactating women, respectively. Regarding the P95 exposure, LB estimate was 1.99 and 3.43 pg WHO2005‐TEQ/kg bw per day, while the UB was 2.20 and 3.64 pg WHO2005‐TEQ/kg bw per day for them, pregnant women and lactating women, respectively.
Assessment group B
Table 43 summarises the chronic dietary exposure estimate for the 17 PCDD/Fs across the 35 dietary surveys. Detailed summary statistics on the exposure estimates calculated for each dietary survey are presented in Annex B (Table 5B, Assessment group B).
Table 43.
Summary statistics of chronic dietary exposure assessments to the 17 PCDD/Fs across European dietary surveys (Assessment group B)
| Age classa | N | Mean dietary exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 6 | 0.18 | 0.35 | 0.28 | 0.47 | 0.47 | 0.70 |
| Toddlers | 10 | 0.28 | 0.45 | 0.51 | 0.75 | 0.92 | 1.28 |
| Other children | 18 | 0.21 | 0.34 | 0.40 | 0.59 | 0.89 | 1.25 |
| Adolescents | 17 | 0.11 | 0.17 | 0.24 | 0.34 | 0.48 | 0.68 |
| Adults | 17 | 0.14 | 0.20 | 0.25 | 0.34 | 0.36 | 0.51 |
| Elderly | 14 | 0.19 | 0.26 | 0.23 | 0.31 | 0.42 | 0.52 |
| Very elderly | 12 | 0.20 | 0.27 | 0.23 | 0.31 | 0.39 | 0.50 |
| Age classa | N | 95th percentile dietary exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 5 | 0.57 | 0.95 | 0.70 | 1.04 | 1.06 | 1.44 |
| Toddlers | 7 | 0.67 | 1.02 | 0.89 | 1.20 | 1.76 | 2.42 |
| Other children | 18 | 0.47 | 0.72 | 0.99 | 1.31 | 1.75 | 2.27 |
| Adolescents | 17 | 0.30 | 0.43 | 0.58 | 0.80 | 1.30 | 1.62 |
| Adults | 17 | 0.41 | 0.50 | 0.57 | 0.72 | 0.97 | 1.12 |
| Elderly | 14 | 0.35 | 0.52 | 0.69 | 0.85 | 1.29 | 1.37 |
| Very elderly | 9 | 0.38 | 0.55 | 0.69 | 0.83 | 0.87 | 1.03 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Section 2.4 describes the age range within each age class.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Not including intake from breast milk.
The mean LB exposure across age groups and surveys ranged from 0.11 to 0.92 pg WHO2005‐TEQ/kg bw per day and the mean UB exposure range was 0.17–1.28 pg WHO2005‐TEQ/kg bw per day. At the 95 percentile exposure, the LB range across age groups and surveys was from 0.30 to 1.76 pg WHO2005‐TEQ/kg bw per day and at the UB from 0.43 to 2.42 pg WHO2005‐TEQ/kg bw per day. The highest exposures were calculated for the age classes Toddlers and Other Children.
Similarly, as for Assessment group A, dietary exposure in specific groups of the population, namely pregnant and lactating women (each represented with one survey), were within the range of exposure estimates for the adult population. The LB mean exposure was 0.28 and 0.29 pg WHO2005‐TEQ/kg bw per day for pregnant women and lactating women, respectively, while UB mean exposure was 0.39 pg WHO2005‐TEQ/kg bw per day in both surveys. Regarding the P95 exposure, LB estimate was 0.86 and 0.99 pg WHO2005‐TEQ/kg bw per day, while the UB was 0.99 and 1.08 pg WHO2005‐TEQ/kg bw per day for them, pregnant women and lactating women, respectively.
The difference between LB and UB estimates was generally found to be small for both assessment groups, indicating that the uncertainty due to left‐censored data is minor.
Contribution of the individual congeners and congener families to the total dietary exposure
Average contribution of individual congeners and congener families to the exposure was investigated.
The percentages of contribution to the mean LB TEQ exposure of the 29 PCDD/Fs and DL‐PCBs and their corresponding families (in Assessment group A) are shown in Figure 19, while contributions to the mean LB TEQ exposure of the 17 PCDD/Fs and their corresponding families (in Assessment group B) are presented on Figure 20. Contributions to the corresponding UB exposures are presented in Annex B (Table 7).
Figure 19.
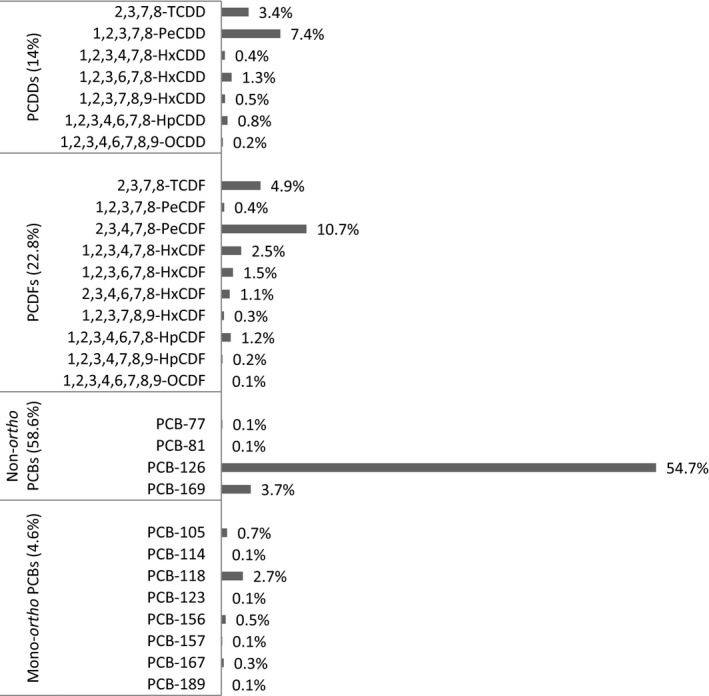
Percentage contribution of each congener (weighted with TEF) and their corresponding families to the overall LB mean exposure of the 29 PCDD/Fs and DL‐PCBs (Assessment group A)
Figure 20.
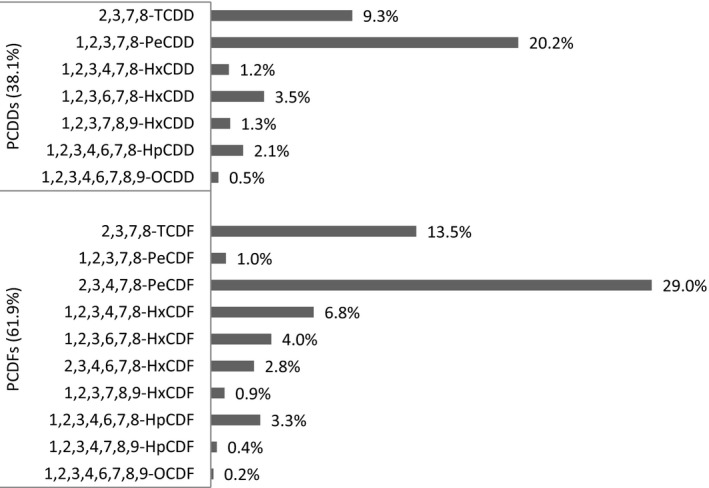
Percentage contribution of each congener (weighted with TEF) and their corresponding families to the overall LB mean exposure of the 17 PCDD/Fs (Assessment group B)
The figures show that with the current WHO2005‐TEFs, PCB‐126 contributes most to the exposure, followed by 2,3,4,7,8‐PeCDF, PeCDD, TCDF, PCB‐169 and TCDD. The HxCDDs and HxCDFs contribute, respectively, 2.2% and 5.4% to the exposure, whereas the heptas and octas contribute very little, similar to most other DL‐PCBs. Consequently, considering the congener families, PCDD/Fs constituted 37% and DL‐PCBs 63% of the mean total intake. The non‐ortho PCBs had the biggest contribution (59%) to the overall LB exposure, followed by the PCDFs with a contribution of 23%. Taking into consideration only the families of the 17 congeners of PCDDs and PCDFs (Assessment group B), in the LB exposure scenario, 62% of the exposure is coming from the PCDFs.
Contribution of different food groups to the total dietary exposure
Figures 21 (29 PCDD/Fs and DL‐PCBs) and 22 (17 PCDD/Fs) summarise the proportions of contributors of exposure (LB) in seven broad food groups together with the magnitude of exposure for each the surveys in all age groups (Toddlers, Other Children, Adolescents, Adults, Elderly and Very elderly). As shown in the figures, food of animal origin are main contributors across all surveys.
Figure 21.
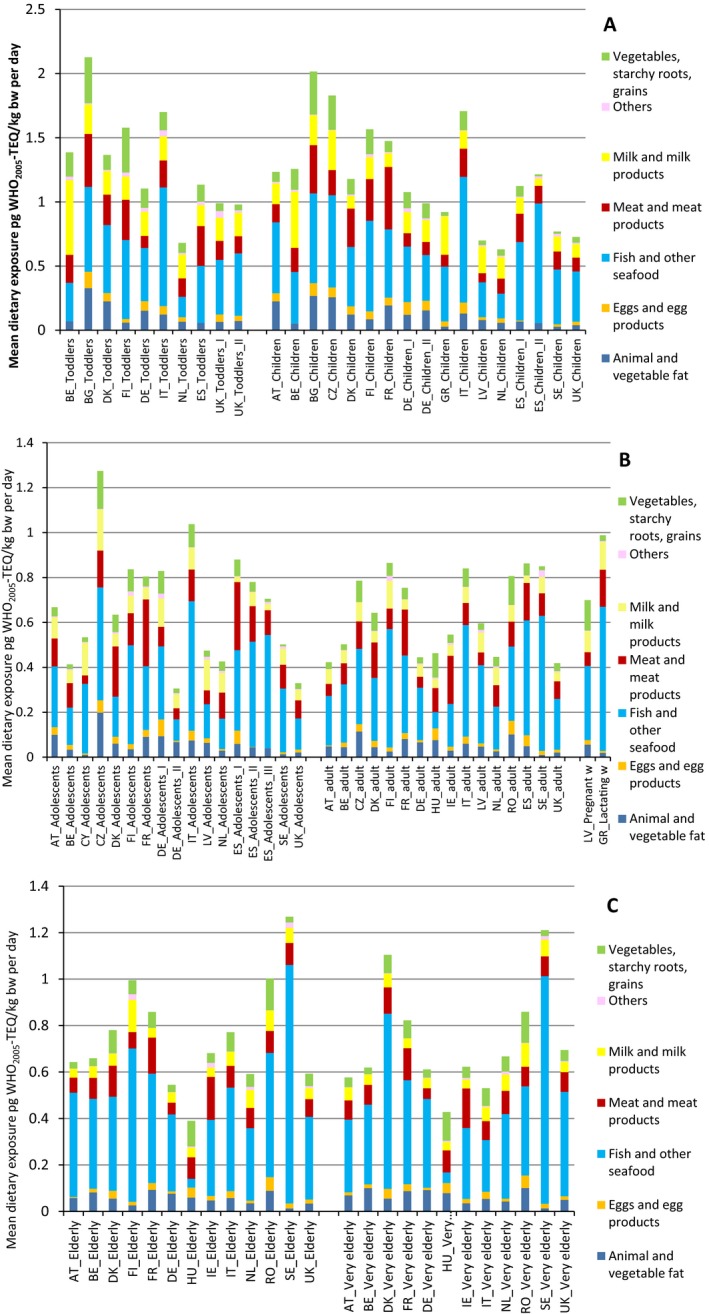
Contributors of exposure of the PCDD/Fs and DL‐PCBs LB exposure (Assessment group A) among different countries/surveys in: (A) Toddlers and Other Children, (B) Adolescents and Adults, including the survey of pregnant women from Latvia and lactating women from Greece, and (C) Elderly and Very Elderly age groups
The contribution to the exposure of the food groups which have more than 5% contribution to the LB exposure estimates in at least one dietary survey is summarised by age groups for Assessment group A (29 PCDD/Fs and DL‐PCBs) in Annex B (Table 6A), and for Assessment group B (17 PCDD/Fs) in Annex B (Table 6B).
In both Assessment groups, for Infants the main contributors were ‘Butter and butter oil’ (contributing from 6.1% to 19.6% in four dietary surveys), ‘Fatty fish’ (contributing from 5.8% to 26.3% in four dietary surveys) and ‘Potatoes and potato products’ (contributing from 5% to 29.6% in five dietary surveys). The fact that the latter food category appears as one of the major contributors is probably not because of its high contamination levels, but due its high consumption among surveys where the overall exposure is low, thus it can represent more than 5% of the total exposure.
For Toddlers, the categories ‘Fatty fish’ (contributing from 5.9% to 13.9% in nine dietary surveys), ‘Cheese’ (contributing from 5.9% to 21.8% in eight dietary surveys) and ‘Livestock meat’ (contributing from 7.7% to 16.2% in eight dietary surveys) were found to be the main sources of exposure, but also the categories ‘Fish products’ (contributing from 5.3% to 31.4% in seven dietary surveys) and ‘Unspecified fish meat’ (contributing from 7.6% to 22.3% in four dietary surveys) have a high contribution.
Similarly, for the age groups of Other Children, Adolescents, Adults and Elderly the main contributors were ‘Fatty fish’ (up to 56% contribution), ‘Unspecified fish meat’ (up to 53.4% contribution), ‘Cheese’ (up to 21.8% contribution) and ‘Livestock meat’ (up to 33.8% contribution).
The most highly contaminated foods are not appearing as main contributors, as their consumption is not frequent among the general population. However, frequent consumption of generally highly contaminated specimens, such as seagull eggs, fish liver and offals (See Annex B, Table 2A and B) could result in elevated exposure.
Figure 22.
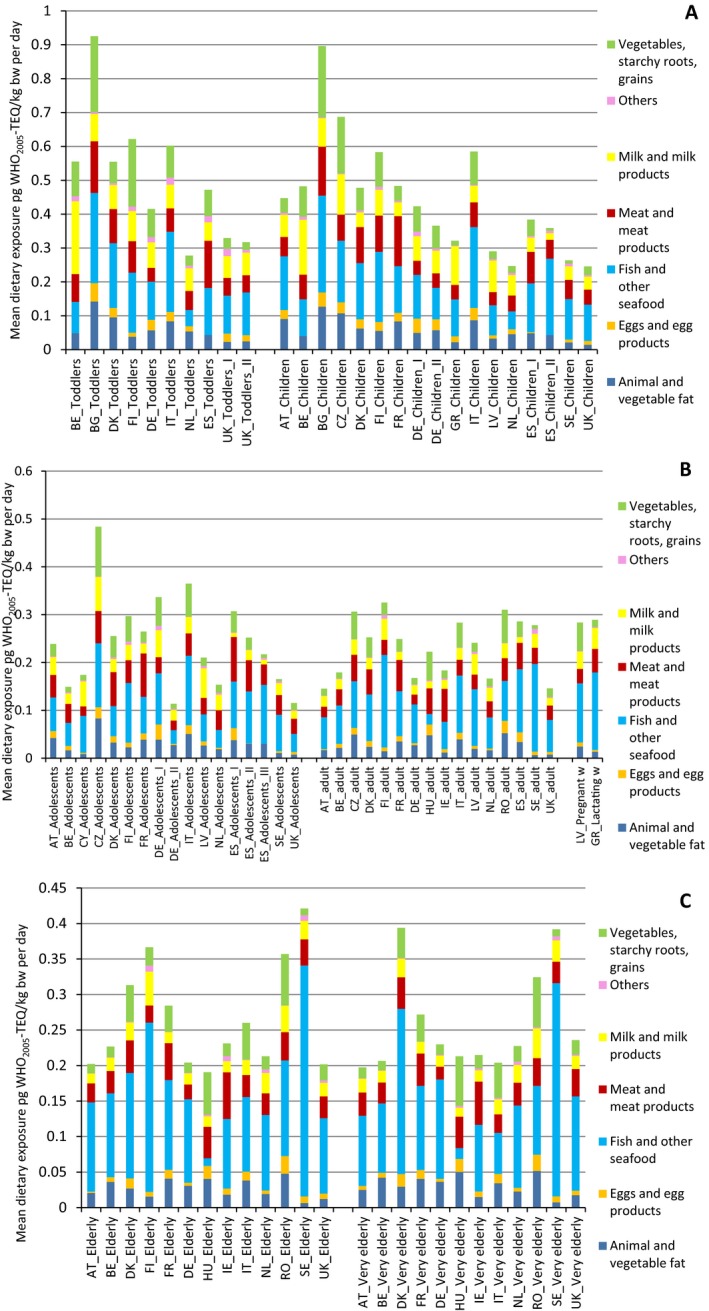
Contributors of exposure of the PCDD/Fs LB exposure (Assessment group B) among different countries/surveys in (A) Toddlers and Other Children, (B) Adolescents and Adults, including the survey of pregnant women from Latvia and lactating women from Greece, and (C) Elderly and Very Elderly age groups
Exposure of infants through breastfeeding
For the exposure assessment of breastfed infants below 6 months of age, a median age of three months was selected, equivalent to a body weight of about 6.1 kg, with an estimated average daily milk consumption of about 800 mL and a high consumption of 1,200 mL. The median occurrence levels were taken from the milk pools collected in European countries and analysed within the WHO‐field‐study 2014/2015 (see Table 41).
For breastfed infants with average human milk consumption, the daily exposure to PCDD/Fs, DL‐PCBs and sum of PCDD/Fs and DL‐PCBs was estimated to be 19.1, 14.5 and 32.9 pg WHO2005‐TEQ/kg bw, respectively.
For breastfed infants with high human milk consumption, the daily exposure to PCDD/Fs, DL‐PCBs and sum of PCDD/Fs and DL‐PCBs was estimated to be 28.6, 21.7 and 49.3 pg WHO2005‐TEQ/kg bw, respectively.
Previously reported dietary exposure assessments
Several international bodies have estimated the dietary exposure to PCDD/Fs and DL‐PCBs while performing their risk assessments related to the presence of these compounds in food (see Section 1.3.3). The WHO (1998) estimated a daily intake of PCDD/Fs in the order of 1–3 pg I‐TEQ/kg bw per day for a 60 kg adult based on the available information derived from food surveys from industrialised countries. It was indicated in this report that this value would be greater by a factor of 2–3 if DL‐PCBs were to be included in the estimations.
JECFA assessed the dietary intake of PCDD/Fs using the GEMS/Food regional diets, and national food consumption data. For PCDD/Fs, the median estimates ranged from 33 to 42 pg WHO1998‐TEQ/kg bw per month, while for DL‐PCBs it ranged from 9 to 47 pg WHO1998‐TEQ/kg bw per month (FAO/WHO, 2002).
The EFSA estimated the dietary intake of these compounds in its two technical reports on the monitoring of PCDD/Fs and DL‐PCBs in food and feed (EFSA, 2010a,b, 2012) (see Section 3.2.2). In its latest report, the EFSA estimated the chronic dietary intake based on dietary surveys for 68 population groups across 17 European Member States, covering seven age classes. Exposure to the sum of PCDD/Fs and DL‐PCBs was estimated considering occurrence data for the period 2008–2010. The UB average exposure (calculated with WHO2005‐TEFs, UB and LB was similar) ranged from 0.57 to 2.54 pg TEQ/kg bw per day across dietary surveys, while the 95th percentile was between 1.2 and 9.9 pg TEQ/kg bw per day. Toddlers and Other children were the age groups with the highest exposure (average estimates ranging from 1.08 to 2.54 pg TEQ/kg bw per day). Infants were less exposed than Toddlers and Other Children, with an estimated dietary intake of around 1.1 pg TEQ/kg bw per day, noting that human milk consumption was not taken into account in the estimation. With these estimates, the percentage of individuals exposed above the SCF TWI of 14 pg TEQ/kg bw was between 1.0 and 52.9%.
‘Fish and seafood’ was the food category which contributed most (30–75%) to the total UB exposure in Adolescents, Adults, Elderly and Very Elderly population groups, followed by ‘Meat and meat products’ (9–34%) and/or ‘Milk and dairy products’ (7–25%). In Infants and Toddlers, the major contributor was the category ‘Milk and dairy products’ (28–50%), followed in Infants by ‘Foods for infants and young children’ (22–31%) and in Toddlers by ‘Fish and seafood products’ (11–36%) or ‘Meat and meat products’ (10–34%). In Other Children, the exposure profile varied, with ‘Milk and dairy products’, ‘Meat and meat products’ and ‘Fish and other seafood products’ as major contributors depending on the country surveyed (EFSA, 2012).
Comparing the estimates from the periods 2002–2004 and 2008–2010, a statistically significant decrease of between 16.6% and 79.3% was observed across the different population group. This decrease may be attributable to the effects of the European risk management measures to reduce the exposure of the European population, but could also in part be due to improvements in the analytical methods and sampling designs of the monitoring programmes over the years (EFSA, 2012).
Since then, studies on dietary exposure to PCDD/Fs and DL‐PCBs in several European countries have been published. For example, Bramwell et al. (2017) reported on the 2012 UK TDS (see Section 3.2.2) and estimated the dietary exposure by combining the occurrence data with the total consumption for each food group derived from 4‐day food diaries kept by approximately 500 adult participants for the period 2011–2012. The average UB dietary exposure (in WHO2005‐TEQs) was estimated to be 0.52 pg TEQ/kg bw per day. The same authors also carried out a Duplicate Diet Study in which 24‐h duplicate diet samples were collected from 10 adult volunteers living in the UK in the period 2011–2012 (Bramwell et al., 2017). The average UB exposure estimate was in this case 0.27 pg TEQ/kg bw per day. The food groups with the greatest contribution to the exposure were ‘Fish and seafood’, ‘Meat’ and ‘Milk and dairy’.
In France, for its second Total Diet Study that analysed samples purchased between 2007 and 2009 (ANSES, 2011), the occurrence data were combined with the consumption data from the French Individual and National Food Consumption Survey (INCA 2). The daily mean (range) exposure (in WHO1998‐TEQs) was estimated at 0.47 (0.39–0.51) pg TEQ/kg bw per day for adults and 0.76 (0.70–0.85) pg TEQ/kg bw per day for children (3–6 years old). In both age categories, the main contributors to the dietary exposure were butter and fish (both around 20%) followed by cheese (around 14%) and meat (around 10%). Mean exposure among women of childbearing age was 0.46 pg TEQ/kg bw per day (ANSES, 2011).
In conclusion, the mean UB exposure range for the different age groups in the present risk assessment (0.39–2.57 pg WHO2005‐TEQ/kg bw per day) is in good concordance with the chronic human PCDD/F and DL‐PCB dietary intake estimated by EFSA in its 2012 technical report (0.57–2.54 pg WHO2005‐TEQ/kg bw per day) based on occurrence data from the sampling years 2008–2010. Compared to 1.2–9.9 pg WHO2005‐TEQ/kg bw per day estimated in 2012 (EFSA, 2012), the 95th percentile UB dietary PCDD/F and DL‐PCB exposure in the present opinion is slightly lower with 1.04–6.63 pg WHO2005‐TEQ/kg bw per day.
The data indicate that the present mean human dietary PCDD/F and DL‐PCB exposure has not substantially changed compared to estimates based on food samples collected between 2008 and 2010.
Non‐dietary exposure assessment
Human body burden with environmental contaminants can result from either accidental, occupational or environmental (background) exposure. Generally, accidental and occupational exposure is limited to small subgroups of the general population. Some examples of accidental and occupational exposure episodes are given in Section 3.1.4, together with the outcome of epidemiological evaluations.
Environmental (background) contamination due to diffuse emission sources affects the entire general population which may be exposed by several routes:
Inhalation of air including particulates in the air
Ingestion of contaminated soil, e.g. adhered to fruit or vegetables
Dermal absorption
Consumption of food
Other sources, e.g. clay supplements.
Fürst et al. (1992b) performed a pathway analysis taking into account known routes of human exposure to PCDD/Fs based on the then available occurrence and consumption data. Inhalation of air and ingestion of particulates from air is dependent on the levels in outdoor and indoor air, and thus a reliable estimation of the PCDD/F intake should be based on the different length of stay and the fact that the breathing frequency is reduced during the sleeping period. As occurence data of indoor air were scarce, the authors estimated the contribution of air to daily human PCDD/F intake using an average annual ambient air concentration of 0.2 pg I‐TEQ/m3 and an average inhalation rate of 20 m3 per day which resulted in a potential daily intake of 4 pg I‐TEQ/person. This intake might be considerably higher in individual cases, in particular of elevated levels in indoor air or due to PCDD/F emitting sources in the vicinity of the residential neighbourhood.
An additional PCDD/F intake via inhalation can result from smoking. Based on the analysis of the ten best selling brands in Germany which revealed levels between 0.08 and 0.15 pg I‐TEQ/cigarette in the mainstream smoke (Ball et al., 1990), smoking of 20 cigarettes per day would result in an additional daily intake of 1.6–3.0 pg I‐TEQ/person. Higher levels were reported by Löfroth and Zeführ (1992) who analysed PCDD/Fs in mainstream and sidestream smoke from one common Swedish commercial brand. While the mainstream smoke contained 0.9 pg I‐TEQ/cigarette, around 2 pg I‐TEQ/cigarette were determined in the sidestream smoke. Smith et al. (2004) determined PCDD/Fs in the mainstream smoke of seven brands covering a range of tar deliveries from 1–14 mg. Mainstream smoke of ultra‐lights and lights products contained around 0.01 pg WHO1998‐TEQ/cigarette, and a full‐flavour brand produced 0.03 pg WHO1998‐TEQ/cigarette.
Exposure to soil can either occur by direct ingestion, which may be the case especially for small children, or by particles, e.g. adhered to fruits or vegetables. The contribution of soil to human intake is highly dependent on the surrounding, whether rural or urban and the potential presence of PCDD/F emission sources. Assuming soil concentrations between 5 and 50 pg I‐TEQ/g and a daily ingestion of 0.1 g, the contribution from soil would be 0.5–5.0 pg I‐TEQ/person/day (Fürst et al., 1992b).
Theelen (1991) investigated the dermal absorption of soil to human PCDD/F exposure. Using an average of 7 pg I‐TEQ/g soil, and assuming a dermal application of 1 mg soil/cm2, an unprotected area of 2,000 cm2 and a dermal absorption rate of 1%, they calculated a potential daily uptake of 0.15 pg I‐TEQ/person.
Compared to the daily dietary intake of 70–200 pg I‐TEQ/person estimated with the occurence and consumption data then available, it was concluded that the non‐dietary intake generally contributed less than 10% to human daily PCDD/F intake. It has also to be taken into account that the estimation dealt only with potential exposure which is presumably higher than the actual uptake due to different absorption rates for the various exposure routes (Fürst et al., 1992b).
The consumption of clay supplements may be another source of exposure, since some of these clays have been shown to contain very high levels of PCDDs. In the EU, such clays are, e.g. used by ethnic minorities during pregnancy.
Recent representative European data on PCDD/F and DL‐PCB in ambient air and soil could not be identified. With decreasing exposure via food, the relative contribution from other sources might have changed. However, taking even the levels in air and soil from the earlier pathway analysis as a conservative approach and the exposure via food from this assessment into account, it can be concluded that dietary intake is by far the predominant pathway of human PCDD/F and DL‐PCB exposure.
3.3.2. Dietary exposure assessment for farm and companion animals
Mean and high dietary exposure assessment
Two assessments have been undertaken in estimating exposure for farm and companion animals:
Assessment group A (29 congeners): taking into account the occurrence values of all the 29 congeners (17 PCDD/Fs and 12 DL‐PCBs).
Assessment group B (17 congeners): taking into account only the occurrence data of the 17 PCDD/F congeners.
For all species, mean exposures have been estimated based on the 95th percentile and the mean LB and UB concentrations, respectively. According to EFSA (2010c), caution is needed when calculating exposure (95th percentile) where data on less than 60 samples are available, since the results may not be statistically robust. Therefore, in this Opinion, no exposure estimations have been made where data on < 60 samples are available. Furthermore, EFSA (2010c) has indicated that estimates of mean exposure based on data for < 10 samples may also be unreliable, and therefore where data on less than 10 samples have been provided these have not been used to estimate the mean LB and UB exposures.
For many livestock in Europe, feeds are supplied in the form of commercially produced species‐specific blends or compound feeds, and where these data were available, mean exposures have been calculated using the concentrations reported and assumed intakes given in Annex A.3.
For those livestock categories for which insufficient data on species‐specific compound feeds were provided, the CONTAM Panel identified example diets and feed inclusion rates (see Annex A.3 for details), and used concentrations of PCDD/Fs and DL‐PCBs in individual feed materials to estimate P95 and mean exposure.
As reported in Annex A.3, a wide range of feeds and feeding systems are used for livestock in Europe. It must be stressed that the feed intakes or diet compositions used in estimating exposures in this Scientific Opinion are not ‘average’ diets, nor are they an attempt to describe ‘worst‐case’ scenarios. Rather, they are intended to provide an indication of likely exposure to PCDD/Fs and DL‐PCBs across a range of feeding systems in Europe.
In the tables below, the dietary concentrations originally expressed as 88% DM, have been converted to – and are reported on ‐ a DM basis (as pg WHO2005‐TEQ/kg DM).
Ruminants and horses
For ruminants, insufficient data on species‐specific compound feeds were available, and therefore exposures have been estimated using example rations and concentrations in individual feed materials (see Annex A.3 for details). For horses, sufficient data were available to allow exposure to be made from species‐specific compound feeds.
For ruminants and horses, forages are essential components of their diets, and exposure has been estimated using levels of PCDD/Fs and DL‐PCBs reported for the feed category ‘Forages and roughages’. In addition, levels in maize silage have been reported, and therefore exposure estimates have been made for dairy cows and beef cattle fed maize silage‐based diets.
Estimated P95 and mean exposures are given below for ruminants and horses to the sum of PCDD/Fs and DL‐PCBs (29 congeners) (Table 44) and the sum of PCDD/Fs (17 congeners) (Table 45).
Table 44.
Estimated mean and P95 exposures to the sum of PCDD/Fs and DL‐PCBs (29 congeners) for ruminants and horses derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | ||||||
|---|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | |||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | ||||||||
| Horsesa | LB | 84 | – | 760 | – | 1.7 | – | |
| UB | 100 | – | 905 | – | 2.0 | – | ||
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | ||||||||
| Dairy cows: forage‐based diet | LB | 82 | 249 | 1,706 | 5,163 | 2.6 | 7.9 | |
| UB | 100 | 257 | 2,066 | 5,310 | 3.2 | 8.2 | ||
| Dairy cows: maize silage‐based diet | LB | 47 | 140 | 1,284 | 3,827 | 2.0 | 5.9 | |
| UB | 65 | 165 | 1,767 | 4,509 | 2.7 | 6.9 | ||
| Beef cattle: cereal‐based diet | LB | 30 | 88 | 299 | 876 | 0.75 | 2.2 | |
| UB | 43 | 96 | 427 | 961 | 1.1 | 2.4 | ||
| Beef cattle: maize silage‐based diet | LB | 46 | 110 | 302 | 726 | 1.0 | 2.4 | |
| UB | 64 | 117 | 420 | 775 | 1.4 | 2.6 | ||
| Lactating sheep | LB | 70 | 218 | 197 | 610 | 2.5 | 7.6 | |
| UB | 87 | 226 | 242 | 634 | 3.0 | 7.9 | ||
| Lactating goats | LB | 38 | 124 | 130 | 422 | 2.2 | 7.0 | |
| UB | 50 | 126 | 168 | 428 | 2.8 | 7.1 | ||
| Fattening goats | LB | 80 | 264 | 120 | 396 | 3.0 | 9.9 | |
| UB | 96 | 267 | 143 | 400 | 3.6 | 10 | ||
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples available to estimate P95 exposure.
Table 45.
Estimated mean and P95 exposure to the sum of PCDD/Fs (17 congeners) for ruminants and horses derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Horsesa | LB | 41 | – | 371 | – | 0.82 | – |
| UB | 56 | – | 507 | – | 1.1 | – | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Dairy cows: forage‐based diet | LB | 41 | 149 | 856 | 3,087 | 1.3 | 4.8 |
| UB | 58 | 166 | 1,194 | 3,431 | 1.8 | 5.3 | |
| Dairy cows: maize silage‐based diet | LB | 29 | 93 | 789 | 2,551 | 1.2 | 3.9 |
| UB | 45 | 120 | 1,224 | 3,272 | 1.9 | 5.0 | |
| Beef cattle: cereal‐based diet | LB | 13 | 44 | 131 | 440 | 0.33 | 1.1 |
| UB | 25 | 60 | 247 | 603 | 0.62 | 1.5 | |
| Beef cattle: maize silage‐based diet | LB | 25 | 80 | 166 | 530 | 0.55 | 1.8 |
| UB | 42 | 94 | 275 | 623 | 0.92 | 2.1 | |
| Lactating sheep | LB | 35 | 128 | 98 | 359 | 1.2 | 4.5 |
| UB | 50 | 145 | 140 | 407 | 1.8 | 5.1 | |
| Lactating goats | LB | 19 | 80 | 65 | 271 | 1.1 | 4.5 |
| UB | 30 | 86 | 101 | 291 | 1.7 | 4.9 | |
| Fattening goats | LB | 41 | 164 | 61 | 246 | 1.5 | 6.2 |
| UB | 56 | 173 | 83 | 260 | 2.1 | 6.5 | |
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples were available to estimate P95 exposure.
Pigs and poultry
Estimates of P95 and mean exposures by pigs and poultry to PCDD/Fs and DL‐PCBs are given in Tables 46 (sum of 29 congeners) and 47 (sum of 17 congeners), respectively.
Table 46.
Estimates of mean and P95 exposure to the sum of PCDD/Fs and DL‐PCBs (29 congeners) for pigs and poultry derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Pigs: growing and fattening | LB | 27 | 143 | 82 | 429 | 0.82 | 4.3 |
| UB | 49 | 168 | 148 | 504 | 1.5 | 5.1 | |
| Lactating sowa | LB | 25 | – | 151 | – | 0.76 | – |
| UB | 42 | – | 250 | – | 1.3 | – | |
| Poultry (starters) | LB | 17 | 33 | 1.3 | 2.5 | 1.9 | 3.5 |
| UB | 32 | 68 | 2.4 | 5.1 | 3.4 | 7.3 | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Pigs: starter | LB | 27 | 172 | 26 | 172 | 1.3 | 8.6 |
| UB | 45 | 206 | 45 | 206 | 2.2 | 10 | |
| Fattening chickens | LB | 32 | 192 | 3.9 | 23 | 1.9 | 11 |
| UB | 52 | 223 | 6.2 | 27 | 3.1 | 13 | |
| Laying hens | LB | 30 | 182 | 3.6 | 22 | 1.8 | 10 |
| UB | 47 | 208 | 5.7 | 25 | 2.8 | 12 | |
| Fattening turkeys | LB | 21 | 122 | 8.4 | 49 | 0.70 | 4.1 |
| UB | 36 | 145 | 14 | 58 | 1.2 | 4.9 | |
| Fattening ducks | LB | 26 | 175 | 3.7 | 25 | 1.2 | 8.2 |
| UB | 44 | 207 | 6.1 | 29 | 2.0 | 9.7 | |
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples were available to estimate P95 exposure.
Table 47.
Estimates exposure to the sum of PCDD/Fs (17 congeners) for pigs and poultry derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Pigs: growing and fattening | LB | 16 | 61 | 48 | 183 | 0.48 | 1.8 |
| UB | 31 | 66 | 94 | 198 | 0.94 | 1.9 | |
| Lactating sowa | LB | 21 | – | 123 | – | 0.62 | – |
| UB | 31 | – | 186 | – | 0.93 | – | |
| Poultry (starters) | LB | 6.8 | 21 | 0.51 | 1.5 | 0.73 | 2.2 |
| UB | 20 | 40 | 1.46 | 3.03 | 2.09 | 4.34 | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Pigs: starter | LB | 9.2 | 80 | 9.2 | 80 | 0.46 | 4.0 |
| UB | 26 | 131 | 26 | 131 | 1.3 | 6.5 | |
| Fattening chickens | LB | 17 | 95 | 2.1 | 11 | 1.0 | 5.7 |
| UB | 34 | 139 | 4.1 | 17 | 2.1 | 8.3 | |
| Laying hens | LB | 16 | 98 | 2.0 | 12 | 0.98 | 5.9 |
| UB | 32 | 134 | 3.9 | 16 | 1.9 | 8.0 | |
| Fattening turkeys | LB | 7.0 | 56 | 2.9 | 22 | 0.24 | 1.9 |
| UB | 21 | 92 | 8.3 | 37 | 0.69 | 3.1 | |
| Fattening ducks | LB | 9.5 | 87 | 1.3 | 12 | 0.44 | 4.1 |
| UB | 26 | 134 | 3.6 | 19 | 1.2 | 6.3 | |
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples were available to estimate P95 exposure.
For growing/fattening pigs and lactating sows, these were derived from data for species‐specific compound feeds, while for pig (starters) exposures have been estimated using example rations and concentration in individual feed materials (see Annex A.3 for details). In the case of lactating sows, insufficient data were available to predict P95 LB and UB exposures reliably.
For poultry (starters) data on species‐specific compound feeds were available, while for fattening chickens (broilers), laying hens, fattening turkeys and ducks insufficient data on species‐specific compound feeds were available, and therefore exposures have been estimated using example rations and concentrations in individual feed materials (see Appendix C for details of rations used).
Farmed fish (salmonids, carp)
For farmed fish, the data on the levels of PCDD/Fs and DL‐PCBs in complete feedingstuffs (n = 224) were not species specific, although a number of species of fish are farmed in the EU, each requiring different feed formulations. Since the Atlantic salmon is the leading species in European aquaculture, the data on compound feed for fish have been used to estimate exposure by this species. For carp, no species‐specific data on concentrations of PCDD/Fs and DL‐PCBs in compound feeds for fish could be identified. However, since carp are able to metabolise carbohydrates more effectively than salmon, their feeds are more cereal‐based, and therefore exposures by carp have been estimated using example rations and concentrations in individual feed materials (see Annex A.3 for details of rations used) and are reported in Tables 48 (sum of 29 congeners) and 49 (sum of 17 congeners).
Table 48.
Estimated mean and P95 exposure to the sum of PCDD/Fs and DL‐PCBs (29 congeners) for farmed fish derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Salmonids | LB | 609 | 1,345 | 24 | 54 | 12 | 27 |
| UB | 660 | 1,349 | 26 | 54 | 13 | 27 | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Carp | LB | 203 | 608 | 4.5 | 13 | 4.5 | 13 |
| UB | 227 | 630 | 5.0 | 14 | 5.0 | 14 | |
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Table 49.
Estimated mean and P95 exposure to the sum of PCDD/Fs (17 congeners) for farmed fish derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Salmonids | LB | 145 | 411 | 5.8 | 16 | 2.9 | 8.2 |
| UB | 194 | 476 | 7.8 | 19 | 3.9 | 9.5 | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Carp | LB | 35 | 135 | 0.78 | 3.0 | 0.78 | 3.0 |
| UB | 54 | 169 | 1.20 | 3.7 | 1.20 | 3.7 | |
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Rabbits and mink
For rabbits, there was sufficient data on species‐specific compound feeds to allow mean (but not P95) estimates of exposure to be made, while for mink estimates of mean and P95 exposures were made by using example rations and concentrations in individual feed materials (see Tables 50 and 51) (see Annex A.3 for details of rations used).
Table 50.
Estimated mean and P95 exposure to the sum of PCDD/Fs and DL‐PCBs (29 congeners) for rabbits and farmed mink derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Rabbitsa | LB | 46 | – | 7.0 | – | 3.5 | – |
| UB | 60 | – | 9.1 | – | 4.5 | – | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Mink | LB | 73 | 205 | 5.5 | 15 | 2.7 | 7.4 |
| UB | 84 | 212 | 6.3 | 16 | 3.1 | 7.7 | |
bw: body weight; LB: lower bound; UB: upper bound. DM: dry matter.
Insufficient samples were available to estimate P95 exposure
Table 51.
Estimated mean and P95 exposure to the sum of PCDD/Fs (17 congeners) for rabbits and farmed mink derived from LB and UB concentrations in species‐specific compound feeds or feed materials
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in species‐specific compound feeds | |||||||
| Rabbitsa | LB | 26 | – | 3.9 | – | 1.9 | – |
| UB | 37 | – | 5.5 | – | 2.8 | – | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Mink | LB | 40 | 83 | 3.0 | 6.3 | 1.4 | 3.0 |
| UB | 51 | 96 | 3.8 | 7.2 | 1.9 | 3.5 | |
bw: body weight; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples were available to estimate P95 exposure.
Companion animals (dogs and cats)
Data on specific manufactured feed for dogs were available to estimate mean (but not P95), exposures. No data on levels of PCDD/Fs and DL‐PCBs in specific manufactured feed for cats were available and therefore exposures were estimated using example rations (see Annex A.3 for details) and concentrations in individual feed materials. The exposures are reported in Tables 52 (sum of 29 congeners) and 53 (sum of 17 congeners), respectively.
Table 52.
Estimated P95 and mean exposure to the sum of PCDD/Fs and DL‐PCBs (29 congeners) by companion animals (dogs and cats)
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in specific manufactured compound feeds | |||||||
| Dogsa | LB | 141 | – | 51 | – | 2.0 | – |
| UB | 173 | – | 62 | – | 2.5 | – | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Cats | LB | 49 | 159 | 2.9 | 9.5 | 0.70 | 2.4 |
| UB | 59 | 167 | 3.5 | 10 | 0.88 | 2.5 | |
bw: body weigh; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples were available to estimate P95 exposure.
Table 53.
Estimated P95 and mean exposure to the sum of PCDD/Fs (17 congeners) by companion animals (dogs and cats)
| Diet concentration (pg WHO2005‐TEQ/kg DM) | Exposure (pg WHO2005‐TEQ/day) | Exposure (pg WHO2005‐TEQ/kg bw per day) | |||||
|---|---|---|---|---|---|---|---|
| Mean | P95 | Mean | P95 | Mean | P95 | ||
| Estimates derived from LB and UB concentrations in specific manufactured compound feeds | |||||||
| Dogsa | LB | 129 | – | 46 | – | 1.9 | – |
| UB | 135 | – | 49 | – | 2.0 | – | |
| Estimates derived from LB and UB concentrations in feeds and their relative proportions in diets | |||||||
| Cats | LB | 26 | 71 | 1.6 | 4.23 | 0.39 | 1.1 |
| UB | 36 | 85 | 2.2 | 5.12 | 0.54 | 1.3 | |
bw: body weigh; LB: lower bound; UB: upper bound; DM: dry matter.
Insufficient samples were available to estimate P95 exposure.
Concluding remarks
For all farm and companion animals combined, the mean LB and UB exposures to the sum of PCDD/Fs and DL‐PCBs (29 congeners) were 2.4/3.0 pg WHO2005‐TEQ/kg bw per day, and ranged from 0.68 (minimum LB) to 13 (maximum UB) pg WHO2005‐TEQ/kg bw per day. The combined mean LB and UB for the 95th percentile for all animals were 8.0/8.8, respectively, with ranges of 2.1 (minimum LB) and 27 (maximum UB) pg WHO2005‐TEQ/kg bw per day. For exposure to the sum of PCDD/Fs (17 congeners) the mean LB and UB exposures were 1.0/1.6, pg WHO2005‐TEQ/kg bw per day, and ranged from 0.24 (minimum LB) to 3.9 (maximum UB) pg WHO2005‐TEQ/kg bw per day, while the mean P95 LB and UB were 3.8/4.8, respectively, and ranged from of 1.1 to (LB) and 9.5 (UB) pg WHO2005‐TEQ/kg bw per day.
The highest estimated exposure was for salmonids (for 29 congeners mean LB and UB = 12/13 pg WHO2005‐TEQ/kg bw per day; P95 LB and UB = 27/27 pg WHO2005‐TEQ/kg bw per day). However, there was considerable variation in estimates of exposure. For ruminants, the highest LB and UB P95 exposures were for fattening goats, and these were approximately three to four times higher than the lowest estimated exposure (for beef cattle on a cereal‐based diet). For pigs and poultry, the highest exposure was for fattening chickens, while estimates derived from data on species‐specific compound feeds (for growing and fattening pigs, lactating sows and starter poultry) tended to be lower than those based on diet formulations.
The CONTAM Panel noted the marked differences in diet concentrations and exposures between cats and dogs. For dogs, limited data on levels of PCDD/Fs and DL‐PCBs were provided to EFSA to allow mean exposures to be calculated. However, similar compound feed data were not available for cats and therefore estimates of exposure were based on example diets provided by the Pet Food Manufacturers Association (PFMA). Although compound feeds for companion animals contain a range of feeds, including cereals and by‐products of food manufacture, their diets may also include products of animal origin, including fish meal and fish oil. However, the formulations for cats provided by the PFMA did not include feeds of fish or animal origin, and this may explain the differences in intake and exposure between cats and dogs.
It is widely recognised that soil ingestion by farmed livestock and horses can be considerable. Estimates of up to 30% of the daily dry matter intake have been reported by grazing sheep and 18% by grazing cattle (Thornton and Abrahams, 1983), while higher levels of PCDD/Fs and DL‐PCBs reported in eggs from free‐range poultry have been attributed, in part at least, to soil intake (Schoeters and Hoogenboom, 2006; Kijlstra et al., 2007). For chickens, no clear estimate for soil consumption is available, but a few grams per day seems feasible. Even low levels in soil (few ng TEQ/kg DM) can thus result in exposure considerably higher than from feed. Rose et al. (2012) have reported that TEQ values in meat samples from outdoor pigs tended to be slightly higher than those from comparable ages reared indoors, which the authors attributed to additional intake from soil. No attempt has been made in this Opinion to account for the intakes, by grazing livestock, of PCDD/Fs and DL‐PCBs present in soil, and therefore the estimates of exposure given above for these species must be considered to be underestimates.
Previously reported exposure assessments for farm animals
In 2000, the European Commission's former SCAN published a risk assessment on PCDD/F levels in feedingstuffs and their contribution to the contamination of food of animal origin (SCAN, 2000). The risk assessment was based on all then available occurrence data on PCDD/Fs in feedingstuffs and a list of typical diets for the main animal species and categories representative of farming practices in the EU. DL‐PCBs could not be included in the assessment because of lack of occurrence data. Special attention was paid to soil as a potential source of PCDD/F contamination. The SCAN calculated the total PCDD/F concentration for each diet using the low, mean and high occurrence values for the PCDD/F content determined for each ingredient or group of ingredients to identify the main sources of contamination. The evaluation indicated that greatest concerns arose from the use of fishmeal and fish oil of European origin, in particular when used in diets for farmed fish and where fishmeal is incorporated into diets of other food producing animals.
Overall, the SCAN concluded that no adverse effects from PCDD/Fs would be expected in mammals, birds and fish exposed to the levels of background pollution measured at the time of the assessment. However, toxic consequences would be expected in animals challenged by severe accidental contamination with PCDD/Fs or PCBs. Moreover, the SCAN stated that consumption of contaminated soil during grazing may drastically increase the exposure of PCDD/Fs in free‐ranging animals. As soil is deposited onto the aerial surfaces of all plants it may also be consumed by other animals post‐harvest. However, the bioavailability of dioxins adsorbed on mineral or organic soil particles was considered limited (SCAN, 2000).
The importance of soil ingestion and its contribution to livestock exposure, especially in sheep is illustrated in EFSA's ‘Scientific Opinion on the risk to public health related to the presence of high levels of dioxins and dioxin‐like PCBs in liver from sheep and deer’ (EFSA CONTAM Panel, 2011).
In the past 20 years, a number of incidents were identified where, due to a criminal or grossly negligent action of individuals, massive feed contamination occured causing seizure and destruction of tens of thousands tonnes of feed and food and slaughter of thousands of heavily exposed food producing animals. Section 1.3.1 gives further information on the most important contamination incidents.
3.4. Risk characterisation
3.4.1. Human risk characterisation
The CONTAM Panel evaluated the current exposure using mean levels for PCDD/Fs or the sum of PCDD/Fs and DL‐PCBs in various food groups, expressed in WHO2005‐TEQs. This was performed using the different food consumption surveys from European countries, taking into account different age classes. The exposure was subsequently compared with the newly established TWI of 2 pg TEQ/kg bw per week. Since the exposure was estimated on a daily basis, the values were first extrapolated to a weekly basis, simply by multiplication with a factor 7 (Tables 54 and 55).
Table 54.
Weekly intake of the sum of PCDD/Fs and DL‐PCBs (29 congeners)
| Age classa | N | Mean dietary exposure (pg WHO2005‐TEQ/kg bw per week) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 6 | 3.1 | 4.6 | 4.6 | 6.5 | 8.1 | 9.9 |
| Toddlers | 10 | 4.8 | 6.2 | 8.8 | 10.7 | 14.8 | 18.0 |
| Other children | 18 | 3.9 | 5.0 | 8.1 | 9.7 | 14.1 | 17.2 |
| Adolescents | 17 | 2.1 | 2.7 | 4.6 | 5.5 | 8.9 | 10.5 |
| Adults | 17 | 2.9 | 3.4 | 4.5 | 5.3 | 7.8 | 9.1 |
| Elderly | 14 | 2.7 | 3.6 | 4.7 | 5.4 | 8.9 | 9.6 |
| Very elderly | 12 | 3.0 | 4.0 | 4.5 | 5.1 | 8.5 | 9.2 |
| Age classa | N | 95th percentile dietary exposure (pg WHO2005‐TEQ/kg bw per week) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 5 | 9.7 | 13.2 | 13.7 | 17.2 | 19.7 | 23.0 |
| Toddlers | 7 | 14.1 | 16.7 | 19.3 | 22.3 | 35.4 | 41.6 |
| Other children | 18 | 10.6 | 12.1 | 21.7 | 24.3 | 42.1 | 46.4 |
| Adolescents | 17 | 6.4 | 7.5 | 13.5 | 14.6 | 28.4 | 30.4 |
| Adults | 17 | 6.6 | 8.3 | 13.6 | 14.6 | 20.1 | 21.8 |
| Elderly | 14 | 5.3 | 6.5 | 14.9 | 16.7 | 25.3 | 26.7 |
| Very elderly | 9 | 5.9 | 7.3 | 13.3 | 14.9 | 17.9 | 19.5 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound; DL‐PCBs: dioxin‐like polychlorinated biphenyls; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Section 2.4 describes the age range within each age class.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Not including intake from breast milk.
Table 55.
Weekly intake of PCDD/Fs (17 congeners)
| Age classa | N | Mean dietary exposure (pg WHO2005‐TEQ/kg bw per week) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 6 | 1.3 | 2.5 | 2.0 | 3.3 | 3.3 | 4.9 |
| Toddlers | 10 | 2.0 | 3.2 | 3.6 | 5.3 | 6.4 | 9.0 |
| Other children | 18 | 1.5 | 2.4 | 2.8 | 4.1 | 6.2 | 8.8 |
| Adolescents | 17 | 0.8 | 1.2 | 1.7 | 2.4 | 3.4 | 4.8 |
| Adults | 17 | 1.0 | 1.4 | 1.8 | 2.4 | 2.5 | 3.6 |
| Elderly | 14 | 1.3 | 1.8 | 1.6 | 2.2 | 2.9 | 3.6 |
| Very elderly | 12 | 1.4 | 1.9 | 1.6 | 2.2 | 2.7 | 3.5 |
| Age classa | N | 95th percentile dietary exposure (pg WHO2005‐TEQ/kg bw per week) | |||||
|---|---|---|---|---|---|---|---|
| Minimumb | Medianb | Maximumb | |||||
| LB | UB | LB | UB | LB | UB | ||
| Infants c | 5 | 4.0 | 6.7 | 4.9 | 7.3 | 7.4 | 10.1 |
| Toddlers | 7 | 4.7 | 7.1 | 6.2 | 8.4 | 12.3 | 16.9 |
| Other children | 18 | 3.3 | 5.0 | 6.9 | 9.2 | 12.3 | 15.9 |
| Adolescents | 17 | 2.1 | 3.0 | 4.1 | 5.6 | 9.1 | 11.3 |
| Adults | 17 | 2.9 | 3.5 | 4.0 | 5.0 | 6.8 | 7.8 |
| Elderly | 14 | 2.5 | 3.6 | 4.8 | 6.0 | 9.0 | 9.6 |
| Very elderly | 9 | 2.7 | 3.9 | 4.8 | 5.8 | 6.1 | 7.2 |
bw: body weight; LB: lower bound; N: number of surveys; UB: upper bound; PCDD/Fs: polychlorinated dibenzo‐p‐dioxins and dibenzofurans; TEQ: toxic equivalents.
Section 2.4 describes the age range within each age class.
The 95th percentile estimates obtained on dietary surveys/age classes with less than 60 observations may not be statistically robust (EFSA, 2011a). Those estimates were not included in this table.
Not including intake from breast milk.
Breastfed infants are known to have a higher exposure than Toddlers and Other Children (see Section 3.3.1). The exposure of breastfed infants should not be compared to the TWI. The reason is that the TWI was set to prevent a level in breast milk that would result in serum levels in children that have been associated with adverse effects. The factor of 2 higher exposure in Toddlers and Other Children, that was accounted for in the toxicokinetic modelling (see Section 3.1.8.2), needs to be considered when comparing their exposure to the TWI.
Another important issue is that the TWI was based on the association between reduced sperm concentrations and serum PCDD/F‐TEQ levels. When taking into account the DL‐PCB‐TEQ levels, the association was no longer significant. The CONTAM Panel took into account results from studies showing that the relative potency of the most relevant DL‐PCB, i.e. PCB‐126, may be much lower in humans than suggested by its current TEF of 0.1. Therefore it is relevant to compare not only the exposure of the total TEQ with the TWI, but also that of the PCDD/F‐TEQ only. It should be mentioned that this uncertainty on the relative potency of PCB‐126 was an important reason to round the TWI to 2 pg/kg bw per week.
Concerning the sum of PCDD/Fs and DL‐PCBs (29 congeners), for Adults the mean LB and UB exposure varied from 2.9 and 9.1 pg TEQ/kg bw per week, which exceeds the TWI by a factor 1.5 to 4.6. Similar is observed for Adolescents, Elderly and Very Elderly (LB–UB range 2.1–10.5 pg TEQ/kg bw per week).
For Toddlers and Other Children, the LB–UB exposure ranged from 3.9 to 18 pg TEQ/kg bw per week, being 2.0‐ to 9‐fold higher than the TWI.
Infants showed an intake of 3.1–9.9 pg TEQ/kg bw per week from sources other than breast milk.
When focussing on the higher end of the exposure (P95), the intake of the sum of PCDD/Fs and DL‐PCBs by Adolescents, Adults, Elderly and Very Elderly ranged from 5.3 to 30.4 pg TEQ/kg bw per week, being 2.7‐ to 15.2‐fold the TWI. Again, Toddlers and Other Children showed a higher intake, being 10.6 to 46.4 pg TEQ/kg bw per week (5.3‐ to 23.2‐fold the TWI). Infants showed an intake of 9.7–23.0 pg TEQ/kg bw per week from sources other than breast milk.
In comparison to the sum of PCDD/Fs and DL‐ PCBs, the exposure to PCDD/F‐TEQ only (17 congeners) was, in general, a factor 2.4 lower for the mean and a factor 2.7 lower for the P95 exposure.
For Adolescents, Adults, Elderly and Very Elderly, the mean LB and UB exposure varied from 0.8 to 4.8 pg TEQ/kg bw per week, being, respectively, a factor 0.4 and 2.4 of the TWI.
For Toddlers and Other Children, LB–UB exposure ranged from 1.5 to 9.0 pg TEQ/kg bw per week, being 0.8‐ and 4.5‐fold the TWI. Infants showed an intake of 1.3–4.9 pg TEQ/kg bw per week from sources other than breast milk.
When focussing on the higher end of the exposure (P95), the intake of the PCDD/F‐TEQ by Adolescents, Adults, Elderly and Very Elderly ranged from 2.1 to 11.3 pg TEQ/kg bw per week, being 1.1‐ to 5.7‐fold the TWI. Again, Toddlers and Other Children showed a higher intake, being 3.3 to 16.9 pg TEQ/kg bw per week (1.6‐ to 8.5‐fold the TWI). Infants showed an intake of 4.0–10.1 pg TEQ/kg bw per week from sources other than breast milk.
It can be concluded that the intake of PCDD/Fs and DL‐PCBs by Adolescents, Adults, Elderly and Very Elderly for all mean and P95 estimates exceeds the TWI of 2 pg TEQ/kg bw per week, by up to a factor 15.
For Toddlers and Other Children, the exceedances are approximately a factor of 2 higher than in the older age groups. But since higher exposure at young age was taken into account when deriving the TWI, the exceedances are in a similar range to the older age groups.
The intake of PCDD/F‐TEQs is more than twofold lower than the intake of total TEQs (sum of PCDD/Fs and DL‐PCBs). As a result, only part of the mean exposure exceeds the TWI, but all estimated P95 intakes are higher than the TWI, by up to a factor 5.7 when focussing on Adolescents, Adults, Elderly and Very Elderly. In general, the difference between LB and UB estimates is rather small and the exceedance is not due to a high fraction of left‐censored data and too high LOQs.
The CONTAM Panel concluded that the current exposure to PCDD/Fs and DL‐PCBs is of concern.
3.4.2. Farm and companion animal risk characterisation
The CONTAM Panel reviewed the available studies in order to derive a possible NOAEL or LOAEL for farm and companion animals. Although some studies describe effects following incidents or from field studies, none of the studies on ruminants, pigs or horses were suitable to derive an exposure level that could be compared with the current mean and P95 intake from feed. It was the same for rabbits, and cats and dogs.
Several feed‐related incidents revealed that chickens are sensitive to the effects of PCDD/Fs and DL‐PCBs, showing chicken oedema disease, decreased hatching of eggs and mortality. Only few controlled studies were identified. One study indicated a LOAEL of 1,099 ng/kg bw per day and NOAEL of 5.6 ng/kg bw per day. All egg production from hens of the high doses ceased after 12 days of treatment. When compared with the mean and P95 UB intakes of 2.8 and 12 pg WHO2005‐TEQ/kg bw per day estimated for laying hens, a large margin is observed.
No suitable studies were derived for ducks and turkeys. There were some studies on quails and pheasants, which implied that these species are more resistant than chicken to embryotoxicity caused by TCDD, 2,3,4,7,8‐PeCDF, or TCDF after injection of eggs into the air sac. No specific data on exposure of these species were obtained but when farmed, it might be assumed that feed levels and also exposure might be similar to laying hens.
Studies with salmon, trout and other fish, showed that toxicological responses to PCDD/Fs and DL‐PCBs include fin necrosis, haemorrhages, reduced growth and mortality. Sensitivity was species‐ and dose dependent. Many studies have been performed on different species of farmed fish, however few studies were suitable to derive an exposure level that could be compared with the current mean and P95 intake from feed. In rainbow trout, the lowest NOAEL was 11 ng TCDD/kg bw (and a LOAEL of 6.3 μg TCDD/kg bw), where other studies indicated NOAELs of 0.1 μg TCDD/kg bw or higher. A NOAEL of 1 μg TCDD/kg bw was identified for yellow perch and tilapia, and 0.57 μg TCDD/kg bw for carp. A feeding study with Atlantic salmon showed no effects after prolonged exposure to PCDD/Fs and DL‐PCBs at 20 pg WHO2005‐TEQ/kg bw per day (the highest dose tested). The mean and P95 UB exposure of salmonids, based on data reported to EFSA, was estimated to be 13 and 27 pg WHO2005‐TEQ/kg bw/d. When compared with the dose of 20 pg WHO2005‐TEQ/kg bw per day reported to not cause any effects in salmon, it appears that the P95 exposure exceeds this level. However, no higher doses were tested and when compared to NOAELs and LOAELs reported for other fish species, including trout, the margin seems to be much larger. For carp, the estimated mean and P95 UB intake was 5 and 14 pg WHO2005‐TEQ /kg bw per day. Again, when compared to reported NOAELs and LOAELs for various fish species, this seems not to imply a risk.
A fairly large number of studies with mink were identified, both with feed prepared with pure standards and feed prepared with contaminated fish. The studies with controlled exposure of mink showed a lowest NOAEL of 2.1 ng TCDD/kg bw per day in a two‐generation study for the characteristic morphological change caused by TCDD and other PCDD/Fs and DL‐PCBs, i.e. proliferation of the squamous gingival epithelium in the mouth of juvenile mink. Based on reported levels in feed materials, for mink a mean and P95 UB exposure of 3.1 and 7.7 pg WHO2005‐TEQ/kg bw per day was estimated. This implies a 677‐ and 273‐fold margin with the NOAEL of 2.1 ng TCDD/kg bw per day.
The CONTAM Panel concluded that information of levels causing effects in farm and companion animals is rather limited but that the estimated exposure of various species, based on current levels, does not imply a risk. Exposure from contaminated soil was not included in the calculations.
3.5. Uncertainty analysis
The evaluation of the inherent uncertainties in the assessment of exposure to PCDD/Fs and DL‐PCBs has been performed following the guidance of the Opinion of the Scientific Committee related to Uncertainties in Dietary Exposure Assessment (EFSA, 2007). In addition, the report on ‘Characterizing and Communicating Uncertainty in Exposure Assessment’ has been considered (WHO/IPCS, 2008).
3.5.1. Uncertainty in exposure estimates
3.5.1.1. Food
Occurrence data
The number of occurrence data submitted differed considerably depending on food products, with most of the samples related to foods of animal origin, and on the reporting data provider, with most of the food samples (~ 77%) collected in only four European countries (namely Germany, France, Norway and Denmark). Country‐based differences in the levels of PCDD/Fs and DL‐PCBs in food commodities might not be evenly represented in the available data, introducing an uncertainty on the representativeness of the overall statistics. The lower the number of samples, the higher is the uncertainty related to the levels of PCDD/Fs and DL‐PCBs in food commodities such as, for example most of the food categories of plant origin.
Differences in levels in food across European countries may result in over‐ or underestimation of exposure for certain food consumption surveys. This may be in particular relevant for fish, regarding its contribution to exposure. Certain fish species (e.g. salmon and herring) from the Baltic area were shown to contain much higher levels than those reported by countries close to other fishing areas (see Section 3.2.1). It was decided not to remove these data from the database or selectively use them for certain consumption surveys, since at least part of this fish can be exported to other European countries, provided that they are compliant with the legal limits. Furthermore, some food surveys from countries surrounding the Baltic Sea indicated frequent consumption of herring, which may be locally caught. It is also not clear what fraction of the fish in countries around the Baltic Sea is imported from other areas. It is also possible that the consumption of other commodities produced and consumed locally in high contaminated areas (e.g. eggs, livestock meat, etc.) could result in higher exposure levels.
According to Eurostat, the large majority of salmon/trout on the EU market is farmed. To assess the influence of wild caught salmon/trout in the occurrence data, the exposure was calculated both including and excluding salmon and trout reported as wild caught.
The LB and UB mean levels of the sum of PCDD/Fs and DL‐PCBs in salmon/trout decreased from 0.88 to 0.55 pg WHO2005‐TEQ/kg and from 0.94 to 0.61 pg WHO2005‐TEQ/kg fresh weight by excluding wild‐caught salmon and trout. The calculated decrease in the mean exposure ranged from 0% to 10% (similar for LB and UB). At the 95th‐percentile exposure, the decrease in the LB exposure ranged from 3% to 23% (at the UB from 3% to 13%), with the highest impact in surveys reporting higher contribution from salmon/trout to the total intake (see Annex B, Table 8). The CONTAM Panel considered this uncertainty small for the exposure assessment based on mean levels in all salmon/trout in the database. However, on an individual basis, the CONTAM Panel noted that the impact on exposure can be large when fatty fish, e.g. from the Baltic Sea is regularly consumed.
It has to be considered that almost all occurrence data come from official food and feed control and thus were mainly generated to check for compliance with legal limits. Therefore, the sensitivity of the applied analytical methods are generally fitted to check compliance to the legal limits and not necessarily to background levels. This is to some extent covered by the performance criteria for analytical methods applied in official control and also used for data selection. Since the differences between LB and UB levels were not substantial, this should have made a minor contribution to the uncertainty.
Consumption data
Uncertainties and limitations related to the use of the EFSA Comprehensive Food Consumption Database have already been described by EFSA (EFSA, 2011a) and are not further detailed in this Scientific Opinion. These relate to the use of different dietary survey methodologies, standard portion sizes, representativeness of samples included in surveys, or to the inclusion of consumption surveys covering only few days to estimate high percentiles of chronic exposure. This last point would lead to an overestimation of the P95 exposure.
Dietary exposure assessment
Important sources of uncertainty are related to the different assumptions done, mainly for the linkage between the occurrence and consumption data. In particular:
Sampled foods were assumed to represent consumed foods, which could lead to both over‐ and underestimation of the exposure.
Occurrence samples not sufficiently described (e.g. classified only at the first level of FoodEx) were excluded, which could lead to both over‐ and underestimation of the exposure.
A number of foodstuffs were grouped mainly at the second level of the FoodEx system, assuming homogeneity of the contamination levels, which could lead to both over‐ and underestimation of the exposure.
In case of two widely consumed heterogeneous food categories presenting less than six samples, the contamination levels were estimated from the contamination levels of related products (namely fish roe from fish products, and pasta with eggs from eggs), which could lead to both over‐ and underestimation of the exposure.
In case of occurrence data expressed on a fat weight basis, the contamination level was combined with the fat per cent of the consumed foods, as reported in the national consumption surveys in the Comprehensive Database. Where the fat content was missing, the random hot‐deck imputation method (see Section 2.3.1.4) was used to input a value. This could lead to both over‐ and underestimation of the exposure.
For the exposure assessment, the CONTAM Panel applied criteria to reduce the uncertainty in the levels and inherent exposure assessment. The main criterion is that the LOQ should be fivefold lower than the ML for PCDD/F‐TEQ, or threefold lower than the AL for DL‐PCB‐TEQ. Another criterion for the acceptable difference between LB and UB, previously applied by EFSA, was omitted because it resulted in the possible removal of levels in the lower part of the distribution. Nevertheless, the difference in the estimated UB and LB exposure was small (on average 17%). This implies that the uncertainty in the exposure assessment is rather small. The uncertainty was further reduced by applying fat percentages reported in the consumption surveys, rather than those reported by providers of data on PCDD/F and DL‐PCB levels. This was the case for samples of animal origin (except offals and fish) and for vegetable fats and oils, where the analytical results are expressed in fat weight. In practice, laboratories may focus on samples with higher fat levels or unprocessed foods, like milk from dairy farms. As a result of this approach, the estimated exposure may be lower than in previous assessments, especially for surveys where such products contribute most to the exposure.
Effect of cooking/processing was not taken into account. The human exposure estimations in this opinion are based on consumption data and occurrence levels in raw food commodities. It is known that typical household cooking practices neither lead to degradation nor to generation of appreciable amounts of PCDD/Fs and DL‐PCBs. However, changes in the fat content of food commodities during cooking/processing practices may lead to changes of the lipophilic contaminants in the processed food compared to the raw food commodity. Due to lack of representative data on these possible changes the effect of cooking and processing could not be taken into account. This may have added to the uncertainty in the exposure estimation to some extent.
3.5.1.2. Feed
Occurrence data
In case of feed, to estimate the chronic dietary exposure of farm and companion animals to PCDD/Fs and DL‐PCBs, 1,844 feed samples had the 17 PCDD/Fs reported, and from them 1,830 the 12 DL‐PCBs as well. 72% of the data on feed samples were reported by two Member States. Country‐based differences in the levels of PCDD/Fs and DL‐PCBs in food commodities might not be evenly represented in the available data, introducing an uncertainty on the representativeness of the EU of the pool statistics.
Feed composition
The representativeness of feeds analysed is limited, and there is a wide discrepancy in the geographical spread of samples reported (possible over‐/underestimation).
Data on species‐specific compound feeds have been provided for some, but not all species. In the absence of data on occurrence in compound or complementary feeds for individual species, typical dietary compositions have been assumed, which may not be typical of all farm and companion animal diets (possible over‐/underestimation).
There were limited or no data available on some key ingredients, e.g. oilseed meals. The formulations therefore assume no exposure from these feeds (possible underestimation).
For ruminants and horses, forages are a major constituent of their diets. Although data on samples of forages were reported in the category ‘Forages and roughages’, these were not sufficiently characterised (e.g. as fresh, ensiled or dried grass, maize silage or legumes) to allow them to be used to assess exposure (possible underestimation).
Single diet formulations have been assumed for each species, although there are large differences in feeding systems and diet formulations for farm and companion animals in the EU (possible over‐/underestimation).
Feed intakes
A single level of feed intake has been assumed for each livestock species/companion animal, but in practice this will vary for a given live weight or level of activity/productivity (possible over‐/underestimation).
Single levels of production or activity have been assumed, but these can vary markedly resulting in greater or lesser amounts of feed required or consumed (possible over‐/underestimation).
Exposure
In estimating exposure to PCDD/Fs and DL‐PCBs, various assumptions have been made, particularly in respect of the types and amounts of feed consumed by farm and companion animals, and this will contribute to the uncertainty associated with the estimates of exposure. The main areas of uncertainty/concern relate to the extent to which the feeds reported are representative of feeds used for farm and companion animals in the EU, the composition of the diets assumed for each of the livestock species/companion animals, and the estimates of feed consumed (possible over‐/underestimation).
In some situations, ingestion of soil by livestock, particularly ruminants, horses, and free‐range poultry and pigs, can be considerable. Although it is widely accepted that PCDD/Fs and DL‐PCBs are present in soils, no attempt has been made to account for exposure of animals from soil (possible underestimation).
3.5.2. Uncertainties in hazard identification and characterisation
There are compounds other than PCDD/Fs and DL‐PCBs that bind to the AHR, are persistent and thus accumulate in the food chain. Examples are polychlorinated naphthalenes (PCNs), but also brominated and mixed halogenated PXDD/Fs and DL‐PXBs. Although these compounds are not included in the TEQ principle, their presence in food may result in an underestimation of the exposure to persistent AHR‐agonists. There is limited information on the occurrence of these compounds and contribution to the exposure to dioxin‐like compounds. On the other hand, it is unknown whether such compounds could also have contributed to effects observed in epidemiological studies.
TEFs have been set for oral exposure and are rounded figures based on a wide range of relative potencies in animal and cell based studies. As such they may not reflect the real potency in humans, and not accurately reflect the potency of measured levels in human tissues. A particular case is PCB‐126 that showed much lower potency in various human cell‐lines. The poor association between effects on sperm quality and DL‐PCBs, but also total‐TEQ, observed in the Russian Children's Study seems to confirm these observations. For this reason, the derivation of the TWI was based on PCDD/F‐TEQ only. This is especially relevant since PCB‐126 is the most important congener in terms of contribution to the TEQ levels in human tissues, but also food and feed.
Recent studies suggest that there is little overlap in the gene expression profiles of PCDD/F and DL‐PCBs contributing most to TEQ levels. This suggests that the TEQ principle may not apply for various types of effects. However, this does not relate to the well‐known genes, encoding for several biotransformation enzymes, but merely to a large set of other genes, expressed at higher levels and to a much lower extent.
3.5.2.1. Experimental animal studies
Body burden calculations were based on a small number of studies with radiolabelled TCDD, showing a specific distribution between body fat, liver and other tissues. It was assumed that a similar distribution would occur in other studies with rodents and body burden calculations were based on these distributions. This could have caused some uncertainties in the estimated body burdens for animals. However, these studies were used as supportive evidence and not for derivation of the HBGV.
There are wide differences in sensitivity to the toxicity of TCDD, the prototype compound of PCDD/Fs and DL‐PCBs, both among and within species. These differences can stay fairly constant across all toxic effects as in the case of inbred mouse strains with high‐ or low‐affinity AHR. On the other hand, they may also critically depend on response as in the case of the rat substrain model Han Wistar vs. Long‐Evans (Turku/AB) rat, and guinea pig vs. hamster, both of which exhibit a broad divergence to some toxicities (exemplified by acute lethality), but much less so with others effects, namely induction of xenobiotic‐metabolising enzymes and fetotoxicity.
Because of the inclusion criteria used in the present opinion, the uncertainty in the studies on experimental animals is considered low. However, the CONTAM Panel noted higher uncertainty in the results from the study showing effects at the lowest body burden, being the one by Faqi et al. (1998) on semen parameters, since it was not described in the paper whether the sperm analyses were performed blinded. However, since the HBGV is derived from human studies, this study was only used as supportive evidence.
3.5.2.2. Epidemiological studies in humans
The evaluation and use of epidemiological studies in this assessment is subject to several uncertainties. First, given the amount of blood needed to quantify PCDD/Fs and DL‐PCBs and the relatively high analytical cost (compared to quantifying NDL‐PCBs, for example), relatively few studies were available and, for certain outcomes many of the studies evaluated were modest in size (n < 100). The accompanying uncertainty is that many studies had relatively low statistical power to detect associations with the outcome explored. The lack of replication by other studies means that chance findings could sometimes not be excluded. This is especially true if the magnitude of the hypothesised effect is small.
Another uncertainty frequently encountered in this opinion is the suboptimal design of many studies, particularly those examining the potential health consequences of accidental or occupational exposures to PCDD/Fs and DL‐PCBs. This was, for example, the case in some of the studies from the Ranch Hand Veterans, Yusho, Yucheng and Seveso cohorts. As these cohorts were initiated after exposure had occurred, exposure was in many cases quantified using back‐calculation from blood samples drawn many years later. In addition, information on important confounders had to be retrospectively assessed. These limitations substantially increase the risk of bias and confounding. The large studies on workers from chemical plants producing TCP often had to rely on job tasks for exposure assessment, with blood analyses of TCDD only in subgroups. This usually causes non‐differential misclassification (attenuating a true association) but in some cases could cause confounding, if job tasks are associated with relevant outcomes for other reasons than exposure to PCDD/Fs and DL‐PCBs.
Confounding is common in epidemiological studies and occurs when other factors can affect serum contaminant levels as well as the outcomes examined. Examples of factors which may be associated with PCDD/F and DL‐PCB levels are time period (decreasing levels over time), age (usually increasing with age), sex (sometimes), BMI (due to amount of body fat), diet and breastfeeding. For the critical outcomes identified in this opinion, it was concluded that most relevant confounders were accounted for in statistical analyses. However, there is always the possibility that some identified factor (biological or lifestyle related) that is associated with both outcome and exposure may have acted as a confounder. However, in the absence of any clear hypothesis on how such confounding may occur, and replication of the findings in different populations where sources of exposure are likely to differ (such as for the semen quality studies in Seveso and the Russian Children's Study), associations presented cannot be assumed to be caused by such residual confounding. There is also strong support from experimental animal studies that developmental male reproductive effects, including sperm production is sensitive to TCDD exposure.
Co‐exposure to other chemical compounds, correlated with PCDD/F and DL‐PCB levels, also constitutes confounding, provided that there are similar effects on the outcome from exposure to such compounds. If the correlation between serum levels of PCDD/Fs and DL‐PCBs and such compounds is high, it is difficult to disentangle effects of the respective compounds.
If study participants have special characteristics (e.g. pregnant women, groups with specialised diet, or certain diseases), they may not be representative for the general population. Nevertheless it is important that health‐based guidance values are also protective for sensitive subgroups of the population.
Many epidemiological studies of exposure to PCDD/Fs and DL‐PCBs use biomarkers, which are indirectly linked to an adverse effect (usually a disease). Such examples are serum levels of sex hormones, biomarkers of glucose homeostasis, or various markers of semen quality. As discussed in the EFSA Guidelines on Biological relevance, there should be prior knowledge of the relation between changes in biomarkers and organ damage or disease states if such biomarkers are used as potential critical effects (EFSA Scientific Committee, 2017b). Sometimes there is uncertainty whether changes in biomarkers are only adaptive or are clearly related to an adverse effect.
Effects on thyroid hormones
Associations between serum levels of PCDD/Fs and DL‐PCBs, and thyroid disease and/or thyroid hormones were analysed in several studies. As mentioned in Section 3.1.4.4, the most important biomarkers at assessment of thyroid function are serum/plasma concentrations of TSH, free T4 and free T3. Some studies examined only total T3 and T4, making conclusions highly uncertain. A potentially critical study for the present opinion was the study by Baccarelli et al. (2008) on TSH in newborn children. In a subgroup, individual levels of TCDD, PCDD/F and DL‐PCB TEQs in mothers were associated with TSH in their newborn. But since the timing of TSH sampling (which is very important for TSH levels in the first 2–3 days after birth) was missing for several cases with high TCDD exposure and high TSH, the CONTAM Panel could not use that subgroup for dose–response analysis, also considering that the small number of observations was driving the relationship. If it could have been confirmed that the samples from these newborns in fact were taken after 2–3 days, when the initial peak of TSH had decreased, this outcome could have been used in the derivation of the HBGV. This conclusion, not to use increased TSH in newborns as critical outcome, is uncertain in particular when taking into account that the association was supported by comparison of TSH levels in infants born to mothers with residence in different zones. However, for these mother/infant pairs, no individual serum levels on PCDD/Fs and DL‐PCBs were available.
Effects on semen parameters
In the cohort from Seveso (Mocarelli et al., 2008) and in the Russian Children's Study (Mínguez‐Alarcón et al., 2017), the sperm count was reduced in the first quartile in comparison with the controls, or in the second quartile of the whole group, respectively, and no clear further reduction was observed with higher exposure. This hampers a proper exposure‐response assessment and introduces uncertainty. Similar dose–response relationship, with a decrease in sperm production at lowest dose and then no further decrease at higher levels, was also seen in male rats following treatment of mother dams (Faqi et al., 1998). In these rats, a 50% reduction in sperm production did not result in reduced fertility. Because humans have lower sperm production than rats, even a modest decrease could affect fertility in men with an already reduced sperm production.
The studies from Seveso on reduced sperm concentrations (Mocarelli et al., 2008, 2011) focussed on TCDD only, being the only congener released in the explosion at the factory. The unexposed group in the study published in 2008 constituted healthy blood donors, and it is not known whether their overall health status was better than among Seveso cohort participants, and this introduces uncertainty. Another question in the Seveso studies is the background exposure to other PCDD/Fs and DL‐PCBs. An additional study on women from the non‐ABR zone around Seveso allowed an estimation of the contribution of other PCDD/Fs and DL‐PCBs to the exposure, but the original data did not allow an accurate assessment of the other congeners in the Seveso cohort. The unanswered questions around background exposure become even more important in the study published in 2011, involving sons of exposed mothers and adds uncertainty. However, the effects observed in boys exposed in their prepubertal period in Mocarelli et al. (2008) and in the breastfed boys in the second generation (Mocarelli et al., 2011), together, suggest a sensitive window in young boys.
In the Russian Children's Study, there was high exposure also to other organochlorines (HCB, β‐HCH and DDE). However, since adjustment for these compounds was possible (see Section 3.1.4.3.1), and these compounds were not strongly associated with semen quality, uncertainty due to such co‐exposure is considered low, but if present it would lead to overestimation of the risk.
Overall, only three studies reported the association between increased serum levels of TCDD or PCDD/Fs and decreased sperm concentrations. The levels in the Russian Children's Study are lower than those from Seveso.
3.5.2.3. Farm and companion animal studies
The CONTAM Panel decided to apply the mammalian WHO2005‐TEFs also for fish and birds, as the WHO1998‐TEFs for fish and birds which were mainly derived from tissue concentrations in fish and egg injections studies, studies with cultured avian hepatocytes, and studies with cultured thymus cells in birds were not re‐evaluated by international bodies after 1998. Furthermore, the primary focus in the present risk assessment was on oral exposure. This may have added some uncertainty in the case of fish due to the absent or very low response to mono‐ortho PCBs compared to mammals and birds.
A major adverse effect of PCDD/Fs in chickens is cardiotoxicity, but the dose–response relationship between effects observed in study with eggs and exposure via maternal exposure of the hens is not clear. In addition, the applicability to other bird species requires clarification as does the relevance of the mode of action to non‐avian species.
No suitable studies with farm or companion animals were identified that could be used to derive a LOAEL or NOAEL. Field studies with sheep and cows indicated effects on biomarkers at relatively low exposure but the relevance of these endpoints requires further studies and co‐exposure to other contaminants could not be excluded.
3.5.3. Uncertainty in dose–response assessment and HBGV derivation
It was decided to base the HBGV on a weekly basis to account for the fact that an occasional exceedance of the TDI may not have a large impact on the levels in the blood. Worst‐case calculations showed that extension to a monthly basis may not prevent such an increase in blood levels and as such an elevated exposure of the fetus or sensitive tissues during a critical window (see Appendix F). No studies or toxicokinetic models were identified dealing with this specific aspect.
3.5.3.1. BMD modelling
Various studies with experimental animals were selected based on the applied dose and relevance of the endpoint. The study with the lowest dosing was the study by Faqi et al. (1998) but this study showed no clear dose response, resulting in a large BMDL‐BMDU interval. It was therefore decided to select the lowest dose as a LOAEL and this is associated with uncertainty, as explained in Section 3.5.3.
3.5.3.2. Toxicokinetic modelling
Lactation results in the loss of PCDD/Fs and DL‐PCBs by the mother and hence a gradual decrease in the milk levels. This also will result in a decreased exposure of infants. Existing kinetic models do not take into account variations in levels of PCDD/Fs and DL‐PCBs during pregnancy and lactation to an extent that would fit the purpose of the modelling by the CONTAM Panel. Since not taken into account this may result in an overestimation of child exposure and hence a more conservative HBGV. In addition, body fat content was kept constant for infants and children, where it is known that infants will have a high body fat content after 6 months and thereafter the % fat in the body decreases. This implies that the initial serum level at birth may be higher than shown by the graphs but peak serum levels and subsequent decrease may be less affected. It may have an effect of the relative amount stored initially in the liver. Also variations in the body weight and fraction of adipose tissue in the mother may affect the transfer to infants, which was not taken into account by the modelling. It is unclear how this could have affected the levels in children.
The models used for estimating the daily intake that would lead to exceedance of the critical serum levels were developed for TCDD, meaning that they may be less accurate for other relevant PCDD/Fs and DL‐PCBs. As shown in the exposure assessment, PeCDD and 2,3,4,7,8‐PeCDF are the most relevant PCDD/Fs. The half‐lives for these compounds are longer than the one for TCDD. This implies that the models are likely to underestimate the serum levels for these compounds. It was however decided not to apply another UF. Future studies should include the modelling of at least the most relevant PCDD/Fs, based on contribution to TEQ levels. Similar may apply for PCB‐126, but regarding the uncertainty in the potency, this was not further evaluated.
3.5.4. Uncertainty in the risk characterisation
In the study by Mínguez‐Alarcón et al. (2017), the association with reduced sperm concentrations was most significant for TCDD or PCDD‐TEQ, but still existing for PCDD/F‐TEQ. There was no association with DL‐PCB‐TEQ or sum‐TEQ. As already mentioned above, this implies that DL‐PCBs and in particular PCB‐126 may be less potent in humans than in rats. As shown in the exposure assessment, DL‐PCBs contribute more than half to human exposure, based on WHO2005‐TEFs. As a result, the exceedance of the new TWI may be smaller than calculated using the WHO2005‐TEFs. Although this introduced uncertainty, the CONTAM Panel decided that the TWI should not be adapted to compensate for this potential lower potency of PCB‐126. Rather, it indicates that the current TEF value of PCB‐126 and possibly also other PCDD/Fs and DL‐PCBs should be re‐evaluated. The opinion of the CONTAM Panel is that uncertainty would have been much larger if DL‐PCB TEQs had been included.
3.5.5. Summary of uncertainties
In Table 56, a summary of the uncertainty evaluation is presented, highlighting the main sources of uncertainty and indicating an estimate of whether the respective source of uncertainty might have led to an over‐ or underestimation of the exposure or the resulting risk.
Table 56.
Summary of qualitative evaluation of the impact of uncertainties on the risk assessment of exposure of PCDD/Fs and DL‐PCBs in food and feed
| Sources of uncertainty | Direction |
|---|---|
| Exposure | |
| Extrapolation of the occurrence data to the whole of Europe | +/− |
| Consumption data: different methodologies/representativeness/underreporting/misreporting/no portion size standard | +/− |
| Use of data from food consumption surveys covering only a few days to estimate high percentiles (95th) long‐term (chronic) exposure | + |
| Occurrence samples not sufficiently described (e.g. classified only at the first level of FoodEx) were excluded | +/− |
| Imputation of missing fat percentages of certain foods in the Comprehensive Database | +/− |
| Effect of cooking/processing not taken into account | +/− |
| Contribution of other persistent AHR agonists | − |
| Extrapolation of the occurrence data on feed mainly submitted by two countries to the whole of Europe | +/− |
| Soil and clay ingestion not taken into account | − |
| Hazard characterisation | |
| Uncertainty in the relative potency of PCB‐126 | + |
| The use of WHO2005‐TEFs for all species | +/− |
| Uncertainties in WHO2005‐TEFs being rounded figures based on a wide range of relative potencies in animal and cell based studies | +/− |
| Epidemiological studies | |
| Uncertainty about systemic TEFs | +/− |
| Lack of measurements on PCDD/Fs and DL‐PCBs other than TCDD | + |
| Non‐differential misclassification of exposure | − |
| True exposure being higher or lower than the estimate of exposure | +/− |
| True outcome is more or less prevalent than the estimate of the outcome | +/− |
| Confounding by other factors | +/− |
| Low number of epidemiological studies on the critical endpoint at low exposure | +/− |
| Critical study | |
| Co‐exposure to other compounds which may impair semen quality | + |
| Uncertainty regarding critical window for effect on semen quality outcome | +/− |
(a): + = uncertainty with potential to cause over‐estimation of exposure/risk; − = uncertainty with potential to cause under‐estimation of exposure/risk.
The CONTAM Panel considered that the impact of the uncertainties on the risk assessment of PCDD/Fs in food is moderate. For the sum of PCDD/F and DL‐PCBs, due to the uncertainty in the relative potency of PCB‐126 in humans, the impact of the uncertainties on the risk assessment is high. Overall, the assessment is likely to be conservative.
The CONTAM Panel considered that the impact of the uncertainties on the risk assessment of PCDD/Fs and DL‐PCBs for farm and companion animals is high and is incomplete due to lack of data.
4. Conclusions
PCDD/Fs are two groups of tricyclic planar compounds. Dependent on the number of chlorine atoms and their positions at the rings 75 PCDDs and 135 PCDFs, termed ‘congeners’, can occur. PCDD/Fs have never been produced on an industrial scale and have no technological use. They are formed unintentionally in a number of industrial and thermal processes. In contrast to PCDD/Fs, PCBs had widespread use in open and closed systems, generally in the form of complex technical mixtures. They were produced with an estimated total world production of 1.2–1.5 million tonnes between 1929 and the end of the 1970s, when their production was abandoned in the majority of countries. A subgroup of 12 PCB congeners that are non‐ortho or mono‐ortho chlorine substituted and contain at least four chlorine substituents can easily adopt a coplanar structure and show toxicological properties similar to TCDD. This subgroup is termed DL‐PCBs. Due to their lipophilic properties and poor degradation, PCDD/Fs and DL‐PCBs accumulate in the food chain.
4.1. Occurrence and exposure
4.1.1.
4.1.1.1.
Food
Most of the samples were derived from animal origin matrices.
The mean and P95 LB/UB levels of the sum of the 17 PCDD/Fs and 12 DL‐PCBs (29 congeners) in ‘Livestock meat including offal’ were, respectively, 1.43/1.54 and 5.06/5.12 pg WHO2005‐TEQ/g fat weight. The levels varied between the various species within this category, showing mean levels from 0.12/0.20 to 6.23/6.26 pg WHO2005‐TEQ/g fat weight.
In ‘Milk and milk products’, the mean and P95 LB/UB levels of the sum of PCDD/Fs and DL‐PCBs were, respectively, 0.73/0.88 and 1.92/2.04 pg WHO2005‐TEQ/g fat weight, in eggs and egg products 1.17/1.30 and 4.38/4.39 pg WHO2005‐TEQ/g fat weight, in ‘Animal and vegetable fat’ 0.42/0.53 and 1.59/1.65 pg WHO2005‐TEQ/g fat weight, and in ‘Vegetables’ 0.05/0.08 and 0.26/0.28 pg WHO2005‐TEQ/g whole weight,
In ‘Fish and seafood’, the mean and P95 LB/UB levels of the sum of the 17 PCDD/Fs and 12 DL‐PCBs were 4.35/4.45 and 21.0/21.6 pg WHO2005‐TEQ/g whole weight. The levels varied between different fish species, showing mean LB/UB levels between 0.10/0.10 and 9.17/9.21 pg WHO2005‐TEQ/g whole weight.
For the 17 PCDD/Fs, the mean and P95 LB/UB levels in ‘Livestock meat including offal’ were, respectively, 0.50/0.60 and 1.54/1.61 pg WHO2005‐TEQ/g fat weight. In meat from various species, the mean LB/UB levels ranged from 0.08/0.16 to 2.65/2.68 pg WHO2005‐TEQ/g fat weight.
In ‘Milk and milk products’, the mean and P95 LB/UB levels for PCDD/Fs were, respectively, 0.28/0.43 and 0.92/1.06 pg WHO2005‐TEQ/g fat weight, in ‘Eggs and egg products’ 0.51/0.62 and 2.02/2.02 pg WHO2005‐TEQ/g fat weight, in ‘Animal and vegetable fat’ 0.20/0.29 and 0.66/0.70 pg WHO2005‐TEQ/g fat weight, in ‘Vegetables’ 0.02/0.05 and 0.12/0.21 pg WHO2005‐TEQ/g whole weight.
In ‘Fish and seafood’, the mean and P95 LB/UB levels for PCDD/Fs were 0.95/1.05 and 4.30/4.66 pg WHO2005‐TEQ/g whole weight. In various fish species, the mean LB/UB levels ranged from 0.01/0.04 to 2.66/2.67 pg WHO2005‐TEQ/g whole weight.
Highest mean LB/UB concentrations for the sum of PCDD/Fs and DL‐PCBs (29 congeners) were found in some rarely consumed foods such as certain game birds (Mallard meat’ and ‘Pheasant meat’ with 39.8/39.8 and 8.29/8.55 pg WHO2005‐TEQ/g fat weight, respectively‘), ‘Fish liver’ (22.1/22.6 pg WHO2005‐TEQ/g whole weight), and ‘Brown meat of crabs’ (6.10/6.17 pg WHO2005‐TEQ/g whole weight).
High mean LB/UB concentrations of the PCDD/Fs (17 congeners) were found in the same categories: game birds (Mallard meat’ and ‘Pheasant meat’ with 2.16/2.19 and 1.76/2.02 pg WHO2005‐TEQ/g fat weight, respectively‘), ‘Fish liver’ (4.41/4.95 pg WHO2005‐TEQ/g whole weight), and ‘Brown meat of crabs’ (3.22/3.29 pg WHO2005‐TEQ/g whole weight).
-
To estimate the human chronic dietary intake, two exposure assessments were carried out:
-
–
taking into account the occurrence values of 19,675 samples with all the 29 PCDD/F and DL‐PCB congeners,
-
–
taking into account the occurrence values of 20,273 samples with the 17 PCDD/F congeners (including samples with all 29 congeners analysed).
-
–
The difference between the LB and UB estimations across all age classes was small for both exposure assessments.
For the sum of PCDD/Fs and DL‐PCBs (29 congeners), the mean UB exposure ranged from 0.4 to 2.6 pg WHO2005‐TEQ/kg bw per day. At the 95th percentile exposure, the UB estimates ranged from 0.9 to 6.6 pg WHO2005‐TEQ/kg bw per day.
For the sum of PCDD/Fs (17 congeners), the mean UB exposure ranged from 0.2 to 1.3 pg WHO2005‐TEQ/kg bw per day. At the 95th percentile exposure, the UB estimates ranged from 0.4 to 2.4 pg WHO2005‐TEQ/kg bw per day.
The highest exposures to the sum of PCDD/Fs and DL‐PCBs (29 congeners) and to the sum of PCDD/Fs (17 congeners) were estimated for the age classes Toddlers and Other Children, and was about twofold higher than in Adolescents and Adults.
For average contribution of individual congeners to the overall mean LB WHO2005‐TEQ exposure (29 congeners), PCB‐126 contributes most to the exposure, followed by 2,3,4,7,8‐PeCDF, 1,2,3,7,8‐PeCDD, 2,3,7,8‐TCDF, PCB‐169 and 2,3,7,8‐TCDD. As a group, the non‐ortho PCBs showed the highest contribution (59%), followed by the PCDFs (23%), PCDDs (14%) and mono‐ortho PCBs (5%). Considering only the sum of PCDDs and PCDFs (17 congeners), the PCDFs contributed 62%.
The main contributors to the mean dietary exposure for the age group Infants were ‘Butter and butter oil’ (contributing from 6.1% to 19.6%) and ‘Fatty fish’ (contributing from 5.8% to 26.3%).
For Toddlers, the categories ‘Fatty fish’ (contributing from 5.9% to 13.9%), ‘Cheese’ (contributing from 5.9% to 21.8%) and ‘Livestock meat’ (contributing from 7.7% to 16.2%) were found to be the main sources of exposure.
Similarly, for the age groups of Other Children, Adolescents, Adults and Elderly the main contributors were ‘Fatty fish’ (up to 56% contribution), ‘Unspecified fish meat’ (up to 53.4% contribution), ‘Cheese’ (up to 21.8% contribution) and ‘Livestock meat’ (up to 33.8% contribution).
Feed
The LB/UB mean levels of the sum of PCDD/Fs and DL‐PCBs (29 congeners) in ‘Fish oil’ were 3.33/3.38 ng WHO2005‐TEQ/kg, in ‘Fish meal’ 0.60/0.62 ng WHO2005‐TEQ/kg, while in complete feed for fish they were 0.54/0.56 ng WHO2005‐TEQ/kg. In ‘Animal fat (for feed)’, LB/UB mean levels were 0.33/0.37 ng WHO2005‐TEQ/kg and in ‘Vegetable fat and oil’ 0.17/0.22 ng WHO2005‐TEQ/kg (all expressed in 88% dry matter).
The LB/UB mean levels of the PCDD/Fs (17 congeners) in these categories were as follows: ‘Fish oil’ 0.80/0.85 ng WHO2005‐TEQ/kg, ‘Fish meal’ 0.21/0.24 ng WHO2005‐TEQ/kg, and complete feeds for fish 0.13/0.15 ng WHO2005‐TEQ/kg. The levels in ‘Animal fat (for feed)’ and ‘Vegetable fat and oil’ were 0.10/0.13 and 0.10/0.16 ng WHO2005‐TEQ/kg, respectively (all expressed in 88% dry matter).
To estimate the chronic dietary exposure of farm and companion animals, 1,830 feed samples had all 29 congeners determined (17 PCDD/Fs and 12 DL‐PCBs), and 1,844 feed samples had 17 PCDD/F congeners determined (including samples with all 29 congeners analysed).
The highest estimated exposure for the sum of PCDD/Fs and DL‐PCBs (29 congeners) was for ‘Salmonids’ (mean LB/UB = 12/13 pg WHO2005‐TEQ/kg bw per day; P95 LB/UB = 27/27 pg WHO2005‐TEQ/kg bw per day). ‘Carp’ had a lower estimated exposure (mean LB/UB = 4.5/5.0 pg WHO2005‐TEQ/kg bw per day; P95 LB/UB = 13/14 pg WHO2005‐TEQ/kg bw per day).
For ruminants, the highest mean and P95 exposures to the sum of PCDD/Fs and DL‐PCBs (in pg WHO2005‐TEQ/kg bw per day) were for ‘Fattening goats’ (mean LB/UB = 3.0/3.6; P95 LB/UB = 9.9/10), and these were approximately three to four times higher than the lowest estimated exposures, that were estimated for ‘Beef cattle on a cereal‐based diet’ (mean UB/LB = 0.75/1.1, P95 LB/UB = 2.2/2.4).
For pigs, the highest exposure for the 29 congeners (in pg WHO2005‐TEQ/kg bw per day) was for ‘Pigs: starters’ (mean LB/UB = 1.3/2.2, P95 LB/UB = 8.6/10), followed by that in ‘Pigs: growing and fattening’ (mean LB/UB = 0.82/1.5, P95 LB/UB = 4.3/5.1) and in ‘Lactating sow’ (mean LB/UB = 0.76/1.3).
For poultry, the highest exposure for the 29 congeners (in pg WHO2005‐TEQ/kg bw per day) was for ‘Fattening chickens’ (mean UB/LB = 1.9/3.1, P95 LB/UB = 11/13), followed by ‘Laying hens’ (mean LB/UB = 1.8/2.8, P95 LB/UB = 10/12) and ‘Starter poultry’ (mean LB/UB = 1.9/3.4, P95 LB/UB = 3.5/7.3). The estimated exposure for ‘Fattening turkeys’ and ‘Fattening ducks’ was lower (mean LB/UB = 0.70/1.2, P95 LB/UB = 4.1/4.9, and mean LB/UB = 1.2/2.0, P95 LB/UB = 8.2/9.7, respectively).
For rabbits, the mean LB/UB exposure was 3.5/4.5 pg WHO2005‐TEQ/kg bw per day, while for mink the values estimated were lower (mean LB/UB = 2.7/3.1; P95 LB/UB = 7.4/7.7). Insufficient data on species‐specific compound feeds for rabbits and mink were available to reliably predict P95 exposures.
For companion animals, the CONTAM Panel noted the marked differences in estimated diet concentrations and exposures between cats and dogs. For dogs, the mean LB/UB exposure was 2.0/2.5 pg WHO2005‐TEQ/kg bw per day, based on data on compound feeds for dogs. In contrast, data on compound feed data were not available for cats but based on individual feed ingredients the mean exposure was estimated to be 0.70/0.88 pg WHO2005‐TEQ/kg bw per day, with P95 LB/UB exposures of 2.4/2.5 pg WHO2005‐TEQ/kg bw per day, respectively.
As for the 29 congeners, the highest exposure to the sum of PCDD/Fs (17 congeners), in pg WHO2005‐TEQ/kg bw per day, was for ‘Salmonids’ (mean LB/UB = 2.9/3.9; P95 LB/UB = 8.2/9.5), and was higher than that of ‘Carp’ (mean LB/UB = 0.78/1.20; P95 LB/UB = 3.0/3.7).
For ruminants, the highest exposures (in pg WHO2005‐TEQ/kg bw per day) were again estimated for ‘Fattening goats’ (mean LB/UB = 1.5/2.1; P95 LB/UB = 6.2/6.5), while the lowest was observed for ‘Beef cattle on a cereal‐based diet’ (mean UB/LB = 0.33/0.62, P95 LB/UB = 1.1/1.5).
For pigs, the highest exposure to the 17 PCDD/Fs (in pg WHO2005‐TEQ/kg bw per day) was for ‘Pigs: starters’ (mean LB/UB = 0.46/1.3; P95 LB/UB = 4.0/6.5) and the lowest for ‘Pigs: growing and fattening’ (mean LB/UB = 0.48/0.94; P95 LB/UB = 1.8/1.9) and ‘Lactating sows’ (mean LB/UB = 0.62/0.93).
For poultry, the highest exposure (in pg WHO2005‐TEQ/kg bw per day) was for ‘Fattening chickens’ (mean UB/LB = 1.0/2.1, P95 LB/UB = 5.7/8.3), while the lowest was estimated for ‘Fattening turkeys’ (mean LB/UB = 0.24/0.69; P95 LB/UB = 1.9/3.1).
For rabbits, the mean LB/UB exposure (based on data for compounds feeds) was 1.9/2.8 pg WHO2005‐TEQ/kg bw per day. In the absence of similar data for mink, exposures were estimated using data for individual feeds, and this resulted in lower estimates of exposure (mean LB/UB = 1.4/1.9; P95 LB/UB = 3.0/3.5).
For companion animals, again the exposure estimated for dogs (mean LB/UB = 1.9/2.0 pg WHO2005‐TEQ/kg bw per day) was higher than that of cats (mean LB/UB = 0.39/0.54; P95 LB/UB = 1.1/1.3).
4.2. Hazard identification and characterisation
4.2.1. Toxicokinetics
Laboratory animals
In rodents, PCDD/Fs and DL‐PCBs are well absorbed and distributed to various tissues, and transferred to the fetus. The major accumulation is in adipose tissue and in liver, with a liver/adipose tissue ratio that increases with the applied dose. At least in mice this is shown to be due to binding to CYP1A2 in the liver.
In all species of laboratory animals, the biotransformation of TCDD, being slow, mainly consists of hydroxylation at a lateral or peri‐position. It is a detoxification process.
There can be some differences in the rate and products of TCDD biotransformation, but these do not seem to account for the strain‐ or species‐specific sensitivities to TCDD toxicity.
In rats, 2,3,7,8‐TCDF and 1,2,3,7,8‐PeCDF are effectively metabolised, but higher chlorinated PCDFs are metabolised to a much lower degree. Except for PCB‐77, most of the DL‐PCBs are not readily metabolised.
In rats and mice, faecal excretion dominates over excretion via urine. Metabolites are excreted rapidly in bile and urine. At least in rats, the higher chlorinated PCDDs seem to exist predominantly unmetabolised in faeces.
Half‐lives are in the order of several weeks and short when compared to humans.
Humans
PCDD/Fs and DL‐PCBs are well absorbed and subsequently distributed to liver and body lipids. Levels of the more relevant congeners in blood are in equilibrium with those in adipose tissue. At high exposure, PCDD/Fs and DL‐PCBs can show higher lipid‐based levels in liver than in adipose tissue.
Most PCDD/Fs and DL‐PCBs are poorly metabolised but some hydroxylated metabolites have been identified.
Compared to laboratory animals, most PCDD/Fs and DL‐PCBs show long half‐lives (several years) which vary between congeners and depending on the levels, age, BMI and sex.
Farm and companion animals
PCDD/Fs, with the exception of the higher chlorinated congeners, and DL‐PCBs are effectively absorbed. Most are poorly degraded but some metabolites of TCDD and some DL‐PCBs have been identified. The parent compounds are accumulated in body fat and liver in a congener specific manner. They are also transferred to milk and eggs.
Transfer in food‐producing animals
The transfer in dairy cows has been studied in a number of controlled experiments and follow‐up studies of incidents. Although to a lesser extent, this also applies for laying hens, growing pigs and sheep. These studies show congener and species specific differences in the excretion and accumulation in meat, body fat and liver.
Long time periods are required to decrease levels after termination of the exposure. Elimination via milk and eggs is a major factor in the decrease of body burdens in lactating ruminants and laying hens, respectively. For meat producing animals, growth of animals contributes to the reduction of the levels.
TRs and BCFs were derived for several species, describing the relation between intake and levels in milk and eggs, or accumulation in tissues. At prolonged exposure (steady state), the daily TEQ amount in milk or eggs may be more than 1/3 of the daily ingested dose.
For dairy cows, laying hens and fattening pigs, toxicokinetic models have been developed that can be used to describe levels in edible products based on levels in feed and duration of exposure and post‐exposure decrease.
PCDD/Fs and DL‐PCBs are accumulated to a greater extent in fillet of farmed oily fish (such as salmon and trout) than in leaner fish such as carp and seabream. BCFs were derived for several fish species, describing the relation between intake and accumulation in fillet.
PCDD/Fs and DL‐PCBs accumulate to a greater degree in the liver than in fillet of lean fish such as cod.
Toxicokinetic models have been developed for salmon enabling the prediction of fillet concentrations of PCDD/F and DL‐PCBs from known feed concentrations.
4.2.2. Toxicity in experimental animals
The CONTAM Panel selected studies not evaluated by the SCF in 2000 and 2001, that could potentially show effects at lower body burdens than the ones used as basis for the TWI set by the SCF in their assessment in 2001. The CONTAM Panel decided to focus on studies in which only TCDD had been dosed to the animals.
The studies on rodents confirmed that developmental effects were seen at body burdens in a similar range as those that were the basis for the previous risk assessment by the SCF.
In rats the adverse effects at such low body burdens were reduced sperm production (LOAEL body burden 25 ng/kg bw), delayed puberty development (LOAEL body burden 42–50 ng/kg bw), altered bone parameters (NOAEL body burden 28 ng/kg bw) and hepatopathy (NOAEL body burden 26 ng/kg bw). In mice, the lowest extrapolated body burden at the NOAEL was 9 ng/kg bw, based on embryo loss.
Studies in primates treated during gestation and lactation showed dental effects and effects on sperm concentration at high dose.
4.2.3. Observations in humans
The CONTAM Panel selected studies in humans which analysed in tissues (e.g. blood, human milk, adipose tissue) of the subjects under study for either (i) TCDD or any other congener dominating the TEQ, e.g. due to a contamination incident, (ii) the 17 PCDD/Fs and 12 DL‐PCBs, (iii) the 17 PCDD/Fs and 4 non‐ortho DL‐PCBs, (iv) the 17 PCDD/Fs and 3 non‐ortho DL‐PCBs (including PCB‐126), or (v) the total TEQs (or BEQs analysed by, e.g. CALUX). Studies assessing dietary exposure with validated methods in relation to outcomes were also included.
The epidemiological studies have been conducted in subjects/cohorts exposed to PCDD/Fs and DL‐PCBs at different life stages under different exposure conditions, e.g. from industrial accidents or contamination incidents, from occupational exposure or from background levels mainly via the diet in the general population.
Chloracne
Chloracne is the most unequivocal toxicity outcome observed occurring in accidental, occupational and unresolved poisoning cases with PCDD/Fs and DL‐PCBs, children appearing to be particularly sensitive. However, chloracne only occurs after high exposures (resulting in serum levels > 20,000 pg/g fat) and is not relevant for deriving a health based guidance value for the general population. There is insufficient information with respect to DL‐PCBs, since even in the rice oil incidents with PCB‐oil, 2,3,4,7,8‐PeCDF contributed most to the TEQ level.
Male reproductive effects
Associations between exposure to TCDD during infancy/prepuberty and impaired semen quality were observed in three prospective studies (two after the Seveso incident and one from the Russian Children's Study). Based on weight of evidence, including also experimental animal studies, the associations were considered causal.
Impaired semen quality was observed in men in Seveso but only in those that were prepubertal at the time of the incident. Even in the lowest quartile the serum levels of TCDD were high compared to present‐day levels in Europe. In another study on adult men born to mothers that were exposed during the Seveso incident, impaired semen quality was observed only in those who had been breastfed. Together, this evidence indicates that there may be a postnatal period of sensitivity that might expand into puberty.
In the Russian Children's Study, which included boys exposed to high environmental background levels, associations of serum TCDD with impaired semen quality were observed. Significant associations were observed also for the sum of PCDD‐TEQ and PCDFs‐TEQ, but not for DL‐PCB‐TEQ or total TEQ. The association between TCDD and semen parameters became slightly stronger after adjustment for NDL‐PCBs, but were not changed by adjustment for exposure to organochlorine pesticides.
There is insufficient evidence for an association between PCDD/Fs or DL‐PCBs and cryptorchidism.
For changes in time of pubertal onset and sexual maturity, observed in one cohort only (the Russian Children's Study), there was insufficient information to conclude on causal associations.
Female reproductive effects
For endometriosis, the only available prospective study did not observe a dose response, and since the available case–control studies indicating associations had limitations, the available evidence was insufficient to be used as a basis for the risk assessment.
The few available studies indicated no association between exposure and pubertal development.
The evidence was insufficient for other female reproductive effects (menstrual cycle characteristics, ovarian function, time to pregnancy, uterine leiomyoma, and age at menopause).
Birth outcomes
A relationship between high TCDD exposure in fathers and lower sex ratio in offspring (lower number of boys relative to girls), has been consistently observed across three different cohorts, and is likely to be causal.
The studies on other birth outcomes (birth weight, preterm birth, fetal Yusho disease and anogenital distance) were inconclusive and could not be used as a basis for the risk assessment.
Thyroid disease and thyroid hormones
In adults, epidemiological studies provide insufficient support for an association between TCDD, other PCDDs, PCDFs or DL‐PCBs and thyroid disease or thyroid function.
A study in children born to mothers highly exposed to TCDD in Seveso indicates a causal association between TCDD and increased neonatal TSH. Studies with low‐moderate exposure to TCDD, other PCDDs, PCDFs or DL‐PCBs do not suggest any adverse effects on the thyroid.
Type 2 diabetes and obesity
The studies were inconclusive and could not be used as a basis for the risk assessment.
Cardiovascular effects
An epidemiological study of very high occupational exposure to TCDD (serum TCDD > 1,000 pg/g fat) indicates increased risk of cardiovascular mortality.
At lower exposures to TCDD, other PCDDs, PCDFs or DL‐PCBs, epidemiological studies provide insufficient support for an association with cardiovascular risk.
Hepatic disorders and digestive effects
Following accidental or occupational exposure, evidence for a causal association with hepatic or digestive diseases is insufficient.
Effects on the immune system
Some studies suggest adverse effects on the immune system at background exposure during development, but the available studies do not provide sufficient evidence for an association between PCDD/Fs or DL‐PCBs and the functionality of the immune system.
Effects on the nervous system
Various neurodevelopmental outcomes at different ages have been investigated in children, but few outcomes have been assessed in several cohorts and/or at similar age. The available information is not sufficient to form a basis for the risk assessment.
There is insufficient information to draw conclusions on effects on the nervous system after exposure in adult life.
Effects on teeth and bone
In three different population groups, childhood exposure to TCDD and/or other PCDD/Fs was dose‐relatedly associated with tooth enamel hypomineralisation or enamel defects. Hypomineralisation of permanent teeth is likely to be causally related to exposure and is likely to be a postnatal effect.
Limited evidence from one cohort indicates associations between PCDD/F and DL‐PCB exposure and some changes in bone parameters.
Cancer
While several studies (many with multiple co‐exposures) showed a positive association with all cancers combined there was no clear link to any specific cancer site.
There was no clear dose–response relationship between exposure and cancer development.
4.2.4. Adverse effects in farm and companion animals
Ruminants
No studies were identified that could be used for the risk assessment.
Pigs
No studies were identified that could be used for the risk assessment.
Rabbits
It was not possible to determine a NOAEL from the studies with rabbits exposed to TCDD.
Horses
In the three studies identified in horses, there was mixed exposure to contaminants and no NOAEL could be identified.
Poultry
Chicks treated with PCDD/Fs or DL‐PCBs by gavage had high incidences of mortality during development, which was associated with pericardial, peritoneal and pulmonary oedema as well as atrophy of the thymus and Bursa of Fabricius, depletion of splenic lymphocytes and delayed egg production when mature. After i.p. dosing with TCDD, young chickens showed a decrease in the Bursa of Fabricius after 5 days with a NOAEL of 1 μg/kg bw per day.
Studies on eggs following in ovo injection, showed poor hatchability and associated cardiomyopathy and teratogenicity, associated with effects like thymic atrophy and changes in thyroid hormone levels. However, the CONTAM Panel concluded that the in ovo studies could not be used for the risk assessment, since they are confounded by timing and route of administration.
Studies of ducks, turkeys, pheasants and quails and their eggs were not useful for risk assessment but illustrated that these species were less susceptible than chicken to PCDD/Fs and DL‐PCBs for some adverse outcomes.
Fish
Fin necrosis, haemorrhages, reduced growth and mortality were the toxicological responses to PCDD/Fs and DL‐PCBs exposure observed in fish.
The lowest LOAEL in rainbow trout was 1 μg TCDD/kg bw, with a NOAEL of 0.1 μg TCDD/kg bw.
A NOAEL of 1 μg TCDD/kg bw was identified for yellow perch and tilapia, and 0.57 μg TCDD/kg bw for carp.
Companion animals (cats and dogs)
Several studies in cats and dogs reported non‐adverse effects, e.g. enzyme induction. Lethality was observed in dogs at high dose.
Microscopic changes were observed in the liver, kidney and spleen of cats but the route and extent of exposure could not be determined.
Fur animals
Mink are sensitive to the toxicity of PCDD/Fs and DL‐PCBs, and the most sensitive response in mink (NOAEL of 2.1 ng TCDD/kg bw per day in a two‐generation feeding study) proved to be proliferation of the squamous gingival epithelium in mouth. This may lead to cyst formation adjacent to teeth and cause osteoporosis in jaw bones.
Co‐exposure of mink to a mixture of toxicants (by feeding on contaminated fish) appeared to augment the toxicity of PCDD/Fs and DL‐PCBs, with a LOAEL of 0.4 ng WHO2005‐TEQ/kg bw per day being obtained for mandibular and maxillary squamous cell hyperplastic foci.
4.2.5. Mode of action
Binding to the AHR is the molecular initiating event of the toxicities of PCDD/Fs and DL‐PCBs. Toxicity is due to inappropriate (in terms of timing, location and/or degree) and sustained activation of the AHR.
AHR signalling can proceed via canonical or alternative pathways. The major toxicities of PCDD/Fs and DL‐PCBs appear to be primarily mediated by the canonical pathway, in which the AHR acts as a ligand‐activated transcription factor.
In animal models, structural variations in the ligand‐binding or transactivation domain of the AHR are associated with non‐selective or selective differences, respectively, in sensitivity to the manifestations of TCDD toxicity.
The human AHR has a lower binding affinity to TCDD when compared to rats and most mouse strains. This may differ for other PCDD/Fs and DL‐PCB congeners.
PCDD/Fs and DL‐PCBs affect the expression of a large number of genes and these seem to be species‐ and congener‐dependent, indicating additional modes of action.
MOA for carcinogenicity
There is no robust evidence that the development of cancer caused by TCDD and other PCDD/Fs in experimental animals is associated with direct genotoxicity.
Rodent studies demonstrate that TCDD is a potent promoter of skin, ovary and liver cancer following initiation with genotoxic agents such as diethylnitrosamine (DEN) and N‐methyl‐N’‐nitrosoguanidine.
Hepatic neoplastic changes may be linked to liver regeneration in response to toxicity.
MOA for reproductive toxicity
In rats, gestational TCDD exposure abrogates the gender difference in anteroventral periventricular nucleus expression of glutamic acid decarboxylase 67, a key enzyme in GABA synthesis, and prevents perinatal LH and testosterone surges in male pups. These changes may underlie the alterations in reproductive functions discernible at adult age, including early puberty, constant oestrus, and premature reproductive senescence in females, and delayed puberty, feminised sexual behaviour and (possibly) reduced daily sperm production in males.
A decreased male‐to‐female ratio has been reported in rat F2 generation after treatment of F0 dams with TCDD, and in the offspring of mouse or human males exposed to TCDD. In mice, suggestive evidence was found of a diminished ability of Y‐bearing sperm to conceive the ova.
In adult male rats and marmosets, TCDD impaired testosterone synthesis in Leydig cells and adversely affected spermiogenesis. Similarly, in mice with a constitutively active AHR, epididymal sperm count was reduced by 45%.
Exposure of adult female rodents to TCDD has been found to lead to irregular oestrous cycles and reduced ovulatory rate, possibly due to repressed ovarian expression of Cyp17a1, induction of xenobiotic‐metabolising enzymes, and inhibition of oestrogen receptor function by the activated AHR.
MOA for effects on thyroid hormones
In rats, TCDD decreases dose‐dependently circulating total and free T4 concentrations, accompanied by an inconsistent impact on serum T3 levels. Functionally, TCDD‐treated rats appear to be euthyroid.
The decrease in T4 in rats is primarily due to accelerated hepatic clearance of T4 through biliary excretion as a result of induction of UDP‐glucuronosyltransferase (UGT) (especially UGT1A) activity.
In addition to inducing hepatic UGT activity, DL‐PCBs may decrease serum T4 levels via competition of their hydroxylated metabolites with T4 for binding to transthyretin.
There is no consistent pattern of thyroid histopathological effects of TCDD. In in vitro studies, TCDD and DL‐PCBs have been shown to reduce the protein or mRNA expression of the sodium‐iodide symporter in animal and human thyroid cells.
MOA for tooth effects
In rats, a highly sensitive response to in utero exposure to TCDD is a reduction in size or total missing of third molar teeth in pups. This is associated with an increased susceptibility of their molar teeth to caries. At higher doses in rats, TCDD may also affect the continuously erupting incisor teeth.
TCDD especially interferes with mineralisation of the dental matrices in developing teeth, with the most critical window of sensitivity being during the early morphogenesis of teeth.
In vitro studies have revealed that at the initiation stage, TCDD blocks mouse molar tooth development by enhancing apoptosis in the dental epithelium and inhibiting the proliferation and differentiation of stem cells of the apical papilla.
EGFR signalling and the dentin sialophosphoprotein gene appear to be involved in the mineralisation defects caused by TCDD.
4.3. Critical effects, dose–response assessment and derivation of a health based guidance value in humans
The association between serum levels and the decreased sperm concentrations observed in the Russian Children's Study and in the Seveso studies was selected as the critical effect.
In the Russian Children's Study, an association between decreased sperm concentrations and increasing serum levels of TCDD, PCDD‐TEQ and PCDD/F‐TEQ was observed. A NOAEL serum level for PCDD/Fs of 7.0 pg WHO2005‐TEQ/g fat at age 9 years was selected, based on the median level in the lowest quartile.
A toxicokinetic model was used to estimate the daily intake leading to a serum level of 7.0 pg WHO2005‐TEQ/g fat at the age of 9 years in boys, taking into account breastfeeding for 12 months by mothers with similar exposure. In the calculations, the twofold higher dietary exposure of Toddlers and Other Children was taken into account. The model includes the concentration‐dependent distribution between liver and body fat, the degradation in the liver and the direct loss via lipids in the faeces.
It was estimated that a level in human milk of 5.9 pg TEQ/g fat, resulting from the constant exposure of mothers to 0.25 pg TEQ/kg bw per day, and subsequent exposure via food to 0.5 pg TEQ/kg bw/d, would result in the NOAEL serum level of 7.0 pg WHO2005‐TEQ/g fat at the age of 9 years.
Taking the uncertainties into account, a TWI of 2 pg WHO2005‐TEQ/kg bw per week was established. The CONTAM Panel decided to base the HBGV on a weekly basis since this is not expected to result in a critical increase in levels in serum. This could not be assumed for extension to a longer, e.g. monthly intake, in the absence of studies and toxicokinetic models that can exclude that a single high dose with e.g. half of the tolerable monthly intake could result in a peak in the serum level.
The CONTAM Panel noted that in the Russian Children's Study, no association was observed for DL‐PCB‐TEQ or the sum‐TEQ of PCDD/Fs and DL‐PCBs. This might be explained by observations from in vitro studies with human cells, showing that PCB‐126 is much less potent in humans than suggested by the WHO2005‐TEF of 0.1. PCB‐126 is the DL‐PCB contributing most to the current intake of PCDD/Fs and DL‐PCBs, but also in the serum of boys from the Russian Children's Study.
4.4. Risk characterisation in humans
When comparing the mean current exposure to PCDD/Fs and DL‐PCBs of Adolescents, Adults, Elderly and Very Elderly, an up to fivefold exceedance of the TWI was observed (highest UB). At the P95, this ranged from 3 to 15. Toddlers and Other Children showed a factor of 2 higher exceedance than older age groups. When calculating the intake leading to the critical serum level of 7.0 pg WHO2005‐TEQ/g fat at the age of 9 years this factor was taken into account.
Regarding the potentially lower potency of PCB‐126, the CONTAM Panel also evaluated the current exposure to PCDD/Fs only. The mean exposure of Adolescents and adult age groups were up to twofold higher than the TWI (highest UB). At the P95 this was up to sixfold higher.
Breastfed infants are known to have a higher exposure than Toddlers and Other Children. The exposure of breastfed infants should not be compared to the TWI. The reason is that the TWI was set to prevent a level in breast milk that would result in serum levels in children that have been associated with adverse effects.
4.5. Critical effects and reference points in farm and companion animals
No studies were identified that could be used to derive a NOAEL or LOAEL for ruminants, pigs, horses, rabbits, ducks, turkeys, quails, pheasants, cats and dogs.
For laying hens, a NOAEL of 5.6 ng/kg bw per day and corresponding LOAEL of 1.1 μg/kg bw per day was identified, showing that egg production had ceased after 12 days of treatment with a high dose of TCDD. In chicks, a NOAEL of 0.1 μg TCDD/kg bw was observed, a 10‐fold higher dose showing mortality.
In rainbow trout, the lowest LOAEL was 1 μg TCDD/kg bw, with a NOAEL of 0.1 μg TCDD/kg bw, based on growth, fin erosion and survival.
In Atlantic salmon no effects were observed after prolonged exposure to PCDD/Fs and DL‐PCBs at 20 pg WHO2005‐TEQ/kg bw per day via the feed (the highest dose tested).
For yellow perch and tilapia, a NOAEL of 1 μg TCDD/kg bw was identified based on growth, fin necrosis and cutaneous haemorrhages.
For carp, a NOAEL of 0.57 μg TCDD/kg bw was identified based on growth, organ weight and haematological parameters.
For mink, the lowest LOAEL of 4.6 ng TEQ/kg bw per day with corresponding NOAEL of 2.1 ng/kg bw per day was observed in a two‐generation study following oral exposure to TCDD, showing proliferation of the squamous gingival epithelium in the mouth of juveniles.
4.6. Risk characterisation in farm and companion animals
None of the studies on ruminants, pigs, horses, rabbits, cats or dogs were suitable to derive a critical exposure level (NOAEL/LOAEL) that could be compared with the current mean and P95 intake from feed. Similar applies to ducks, turkeys, quails, and pheasants.
For laying hens, a large margin is observed between the estimated mean and P95 UB intakes of 2.8 and 12 pg WHO2005‐TEQ/kg bw per day and the NOAEL for reduced egg production of 5.6 ng/kg bw per day. This also applied for young chicks, with similar exposure and a higher NOAEL.
For farmed fish, when the mean and P95 UB exposure of salmonids of, respectively, 13 and 27 pg WHO2005‐TEQ/kg bw per day was compared with the dose of 20 pg WHO2005‐TEQ/kg bw per day reported not to cause any effects in salmon, it appears that the P95 exposure exceeds this level. However, no higher doses were tested and when compared to NOAELs and LOAELs reported for other fish species, including trout (NOAEL of 11 ng TCDD/kg bw), the margin is much larger.
For carp, comparison of the estimated mean and P95 UB intake of, respectively, 5 and 14 pg WHO2005‐TEQ/kg bw per day with the reported NOAEL of 0.57 μg TCDD/kg bw does not imply a risk.
For mink, comparison of the estimated mean and P95 UB exposure of, respectively, 3.1 and 7.7 pg WHO2005‐TEQ/kg bw per day with the lowest NOAEL identified of 2.1 ng TCDD/kg bw per day does not imply a risk.
The CONTAM Panel concluded that information on levels causing effects in farm and companion animals is limited but that the estimated exposure of various species, based on current levels, does not imply a risk. Exposure from contaminated soil was not included in the calculations.
5. Recommendations
To reduce the uncertainty in the risk assessment, the CONTAM Panel recommends that,
The current WHO2005‐TEFs should be re‐evaluated in order to take into account new in vivo and in vitro data. In particular, more insight into the relative potency of PCB‐126 in humans is required.
There is a specific need to derive systemic TEFs for PCDD/Fs and DL‐PCBs for use in epidemiological studies, also taking into account the results from human cells.
There should be an evaluation of the relative exposure contribution of other persistent chemicals, acting as agonists on the AHR, taking into account their toxic potencies.
To evaluate the applicability of the TEQ principle, more research and understanding is needed on reported congener‐specific effects of PCDD/Fs and DL‐PCBs, including their relevance at low doses.
Further improvement of toxicokinetic models is needed, including parameters dealing with pregnancy, breastfeeding and occasional exposure to high levels. Inclusion of PCDD/Fs, other than TCDD, and DL‐PCBs is required. The use of in vitro models for further refinement should be considered.
Data from both experimental animal and epidemiological studies should be reported in a way that allows a better dose–response evaluation in order to improve the risk assessment. There is a need to develop a consensus methodology for data sharing between individual researchers and public health authorities.
There is a need for prospective developmental epidemiological studies on PCDD/Fs and DL‐PCBs at low to moderate doses on, in particular, male reproductive outcomes and effects on the thyroid system. Follow‐up studies on existing and previous cohorts with good information on pre‐ and postnatal exposure should be considered.
Validated and cost‐effective methods are needed to assess exposure in small amounts/volumes of biological samples of animals and humans.
Studies on adverse effects at low doses in farm and companion animals are needed.
To better understand the adverse effects of PCDD/Fs and DL‐PCBs, more insight is needed into the mode of action, especially in relation to observed critical effects.
Mechanistic studies on transgenerational (third generation) effects are needed.
To improve human exposure estimation, more occurrence data are needed on food of plant origin, especially where individual results of certain foods indicate potential higher contamination.
More data are needed on feed, provided by a greater number of European countries.
There is a need for an updated risk‐benefit assessment of fish consumption that takes exposure to PCDD/Fs and DL‐PCBs into account.
It should be considered whether specific TEFs for farm and companion animal species should be updated or derived.
Documentation provided to EFSA
Bonzini M, 2016–2017. Data provided by Matteo Bonzini about the study Baccarelli et al. (2008) from the Seveso Cohort and used in Section 3.1.4 on Observations in humans.
Mínguez‐Alarcón L, Burns J and Hauser R, 2017–2018. Data provided by Lidia Mínguez‐Alarcón, Jane Burns and Russ Hauser about the Russian Children's Study and the study by Mínguez‐Alarcón et al. (2017), and used in Section 3.1.4 on Observations in humans.
Malisch R and Schächtele A, 2018. Data provided in February 2018 by Rainer Malisch and Alexander Schächtele on the PCDD/F‐ and PCB‐related results of the WHO/UNEP‐coordinated exposure studies 2000–2015 performed in European countries, and used in Section 3.2.4 on Levels in humans
Glossary
- Body burden
The total amount of a chemical in the body
- Canonical pathway of the AHR
The pathway of AHR‐mediated signal transduction elucidated originally for Cyp1a1 induction. Key steps along it are ligand binding‐triggered transformation of the AHR, its translocation into the nucleus, heterodimerisation with ARNT, and binding to DREs in promoter regions of AHR‐regulated genes, followed by alterations in the expression levels of these genes
- HR, RR, OR
Relative risk (RR), odds ratio (OR) and hazard ratio (HR) are statistics frequently used to quantify risk in epidemiological studies. The choice of statistics depends on the prevalence of the outcome under consideration and whether the outcome is time dependent or not. A relative risk (RR) is the ratio of the probability (p) of a certain event occurring in one group divided by the probability of the same event occurring in another. In the context of this opinion an event would be some sort of medical condition such a disease or abnormal levels of certain biomarker (such as hypothyroidism) and the exposure would be quantified concentrations of dioxins (or individual congeners). Formally, relative risk would then be defined as RR = event among exposed/event among comparisons. The word odds refer to the ratio of the probability of an event occurring in a group of subjects divided by the probability of the event not occurring. That is if the probability of hypothyroidism is p = 0.25 then the odds are p/(1 – p) = 0.25/(1 – 0.25) = 1/3 as the probability of hypothyroidism is, in this ‘unrealistic example’, one third of the probability of not having this condition. An odds ratio (OR) is therefore simply the ratio of the odds of the same event occurring in two different group of subjects. More formally, if the probability of an event occurring in one group is p1 and the probability of the same event occurring in another groups is p2 then the OR = (P1/(1 – P1))/(P2/(1 – P2)). Note: If the probability of an event is very low (p ≪ 1), such as for cancer, then OR and RR will converge. However, when the probability of an event is relatively prevalent such as for disease conditions such as asthma or dyslipidaemia (in middle aged subjects) the OR tend to be inflated compared to RR. In simplified terms, a hazard ratio (HR) is like RR but is used when the probability of an event occurring is not constant with respect to time
Abbreviations
- β‐HCH
β‐hexachlorocyclohexane
- ACC
Army Chemical Corps
- ACD
anus to clitoris distance
- ADME
absorption, distribution, metabolism and excretion
- AFD
anus to fourchette distance
- AFHS
Air Force Health Study
- AGD
anogenital distance
- AHH
aryl hydrocarbon hydroxylase
- AHR
aryl hydrocarbon receptor
- AHRKO
AHR knockout
- AIC
Akaike information criterion
- AL
action Level
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- ALT
alanine transaminase
- AM
algal meat
- ARNT
AH receptor nuclear translocator
- AST
aspartate aminotransferase
- ATSDR
Agency for Toxic Substances and Disease Registry
- BB
body burden
- BCF
bioconcentration factor
- BEQ
bioanalytical equivalents
- BMD
benchmark dose
- BMDL
benchmark dose lower confidence limit
- BMDU
benchmark dose upper confidence limit
- BMI
body mass index
- BMR
benchmark response
- BPS
balanopreputial separation
- BTF
biotransfer factor
- bw
body weight
- CA
chromosomal aberrations
- CADM
concentration‐ and age‐dependent model
- CALUX
Chemical Activated LUciferase gene eXpression assay
- CAR
constitutive activated/androstane receptor
- CI
confidence interval
- CONTAM
EFSA Panel on Contaminants in the Food Chain
- COR
carry‐over rate
- COT
UK Committee on Toxicity
- CYP
cytochrome P450
- DATA Unit
EFSA Evidence Management Unit
- DDE
dichlorodiphenyldichloroethylene
- DEN
diethylnitrosamine
- deNFO
decontaminated northern fish oil
- DHA
docosahexaenoic acid
- DL‐PCBs
dioxin‐like PCBs
- DM
dry matter
- DME
drug‐metabolising enzyme
- DRE
dioxin‐responsive element
- dw
dry weight
- ECD
electron capture detector
- ECEH
European Centre for Environment and Health
- ECM
extracellular matrix
- EDI
estimated daily intake
- EGFR
epidermal growth factor receptor
- EHDI
estimated human daily intake
- ELS
Extensive Literature Search
- EMA
electromyography
- ER
oestrogen receptor
- EROD
ethoxyresorufin‐O‐deethylase
- EU‐RL
European Reference Laboratory
- FAO/WHO
Food and Agriculture Organization of the United Nations/World Health Organization
- FFQ
Food Frequency Questionnaire
- FRTL‐5
(6‐formylindolo[3,2‐b]carbazole
- FLEHS
Flemish Environment and Health Study
- FO
fish oil
- FoxO3a
forkhead box protein O3a
- FSH
follicle‐stimulating hormone
- FYD
fetal Yusho disease
- GC
gas chromatography
- GD
gestation day
- GGT
gamma‐glutamyl transferase
- GI
gastrointestinal
- GLP
Good laboratory practice
- GM
Geometric mean
- GnRH
Gonadotropin‐releasing hormone
- GST
glutathione S‐transferase
- HBGV
health‐based guidance value
- HCB
hexachlorobenzene
- HR
hazard ratio
- HRMS
high‐resolution mass spectrometry
- IARC
International Agency for Research on Cancer
- IgG
immunoglobulin G
- IgM
immunoglobulin M
- IHD
ischemic heart disease
- IHNV
infectious haematopoietic necrosis virus
- IL
interleukin
- i.m.
intramuscular
- i.p.
intraperitoneal
- IPCS
International Programme on Chemical Safety
- IQR
interquartile range
- IUGR
intrauterine growth restriction
- i.v.
intravenous
- JECFA
Joint FAO/WHO Committee on Food Additives
- JNK
c‐Jun N‐terminal kinase
- KO
knockout
- KVHS
Korean Veterans Health Study
- LB
lower bound
- LD50
lethal dose, median
- LDH
lactate dehydrogenase
- LH
luteinising hormone
- LOAEL
lowest‐observed‐adverse‐effect level
- LOEL
LOWEST‐observed‐effect level
- LOD
limit of detection
- LOQ
limit of quantification
- LPS
lipopolysaccharide
- MB
middle bound
- MCPA
2‐methyl‐4‐chlorophenoxyacetic acid
- MCPP
methylchlorophenoxypropionic acid
- MGUS
monoclonal gammopathy of undetermined significance
- ML
maximum level
- MoA
mode of action
- MoBa
Norwegian Mother and Child Cohort
- MOE
margin of exposure
- MRM
multiple reaction monitoring
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- MWI
municipal waste incinerator
- m/z
mass‐to‐charge ratio
- NDL‐PCBs
non‐dioxin‐like PCBs
- NES
nuclear export signal
- NFO
northern fish oil
- NHANES
National Health and Nutrition Examination Survey
- NHL
non‐Hodgkin's lymphoma
- NLS
nuclear localisation signal
- NOAEL
no‐observed‐adverse‐effect level
- NOEL
no‐observed‐effect level
- NRL
National Reference Laboratories
- NTP
US National Toxicology Program
- OECD
Organisation for Economic Co‐operation and Development
- OR
odds ratio
- PAH
polycyclic aromatic hydrocarbon
- PBDEs
polybrominated diphenyl ethers
- PBPK
physiologically based pharmacokinetic (model)
- PCBs
polychlorinated biphenyls
- PCDDs
polychlorinated dibenzo‐p‐dioxins
- PCDFs
polychlorinated dibenzofurans
- PCN
polychlorinated naphthalene
- PCP
pentachlorophenol
- PCQs
polychlorinated quarterphenyls
- PFMA
Pet Food Manufacturers Association
- PFOS
perfluorooctanesulfonic acid
- PFOA
perfluorooctanoic acid
- PHA
phytohaemagglutinin
- PK
pharmacokinetic
- PMTDI
provisional maximum tolerable daily intake
- PND
postnatal day
- p.o.
per dose
- POP
persistent organic pollutant
- PSA
prostate‐specific antigen
- RAR
retinoic acid receptor
- RfD
reference dose
- RLDS
Reynell Language Developmental Scales
- RO
rapeseed oil
- ROS
reactive oxygen species
- RR
relative risk
- RXR
retinoic X receptor
- s.c.
subcutaneous
- SCAN
Scientific Committee on Animal Nutrition
- SCE
Sister chromatid exchange
- SCF
Scientific Committee on Food
- SD
Standard deviation
- SFO
southern fish oil
- SGA
small for gestational age
- SGOT
serum glutamic oxaloacetic transaminase
- SIM
selected ion monitoring
- SMR
Standardised mortality ratio
- SMRT
Silencing mediator of retinoic acid and thyroid hormone receptor
- SNP
single‐nucleotide polymorphism
- SO
soya bean oil
- SOD
superoxide dismutase
- SSD
Standard Sample Description
- SWHS
Seveso Women's Health Study
- T3
triiodothyronine
- T4
thyroxine
- TBG
thyroxine‐binding globulin
- TCDD
2,3,7,8‐tetrachlorodibenzo‐p‐dioxin
- TCPA
2,4,5‐trichlorophenoxyacetic acid
- TDI
tolerable daily intake
- TDS
Total Diet Study
- TEF
toxic equivalency factor
- TEQ
toxic equivalents
- THR
thyroid hormone receptor
- TNF
tumour necrosis factor
- TR
transfer rate
- TSH
thyroid‐stimulating hormone
- TWI
tolerable weekly intake
- UB
upper bound
- UF
uncertainty factor
- UGT
UDP‐glucuronosyltransferase
- UNEP
United Nations Environment Programme
- US‐EPA
United States Environmental Protection Agency
- VO
vegetable oil
- WG
Working Group
- WHO
World Health Organization
- ww
wet weight
Appendix A – Maximum and action levels as laid down in the European legislation
1.
Appendix B – Body burden estimation in rodent studies
B.1. Principle
For the selection of studies that could show effects at lower body burdens as those forming the basis of the risk assessment by SCF (2001), it was important to develop criteria for calculating the BB from either an external dose (feed level or injected dose), or measured levels in the liver, adipose tissue or both.
In mammals, the body burden of TCDD, i.e. the total amount in the body divided by body weight, predominantly rests in the liver and the adipose tissue. At low dose, the adipose tissue dominates, whereas at increasing dose liver deposition increases by a process referred to as ‘hepatic sequestration’. This may result in the body burden being dominated by liver deposition. Based on studies by Diliberto et al. (1995, 2001) in mice and Hurst et al. (2000a,b) in rats, Figure B.1 shows the distribution of TCDD in the liver and adipose tissue compared to the whole body. Given empirical relationships on the deposition of TCDD in the liver, the adipose tissue or both, the body burden can be estimated. The formulas describing the fitted curves were used for the calculations of body burdens based on measured levels in the liver and/or adipose tissue.
Figure B.1.
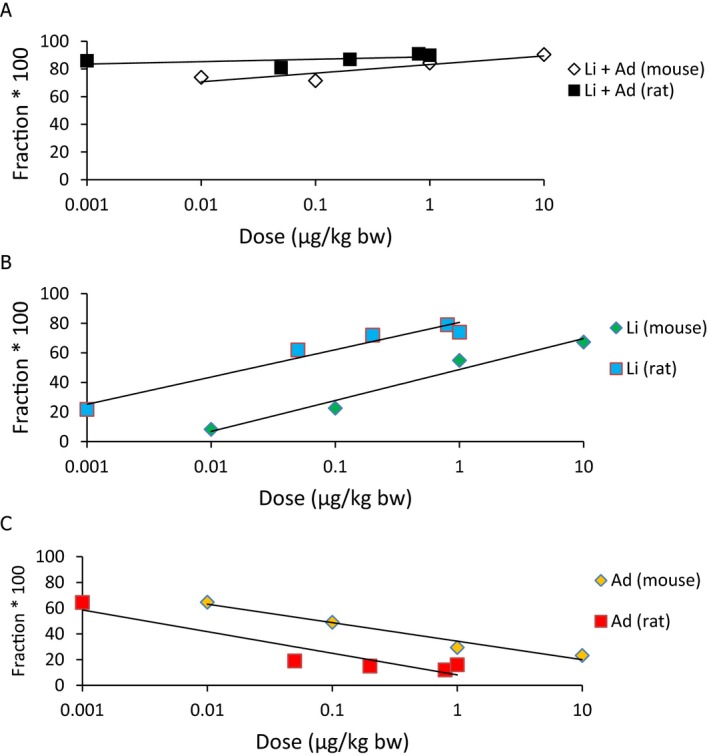
The percentage of the amount of [3H]‐TCDD in the body residing in the liver (Li) and/or adipose tissue (Ad) after single p.o. gavage exposure of rat (Hurst et al., 2000b) or mouse (Diliberto et al., 1995)
X‐axis: Administered dose (μg/kg bw); Y‐axis: Percentage of the body burden.
Mouse: single p.o. gavage doses of 0.01, 0.1, 1, 10 μg/kg bw. Vehicle: corn oil (Diliberto et al., 1995, 2001, measurements: 7 days after dose administration).
Rat: single p.o. gavage dose of 0,001; 0.05, 0.20, 0.80, or 1.0 μg/kg bw to pregnant dams at GD 15; Vehicle: corn oil (Hurst et al., 2000b, measurements: GD 16).
Modelling
A. Sum of liver and adipose tissue
Rat y = 0.7548ln(x) + 88.77 (square)
Mouse y = 2.7013ln(x) + 83.21 (diamond)
B. Liver
Rat y = 8.022ln(x) + 80.59 (square)
Mouse y= 9.0898l(x) + 48.74 (diamond)
C. Adipose tissue
Rat y = −7.296ln(x) + 8.15 (square)
Mouse y = −6.245ln(x) + 34.36 (diamond)
Figure B.2.
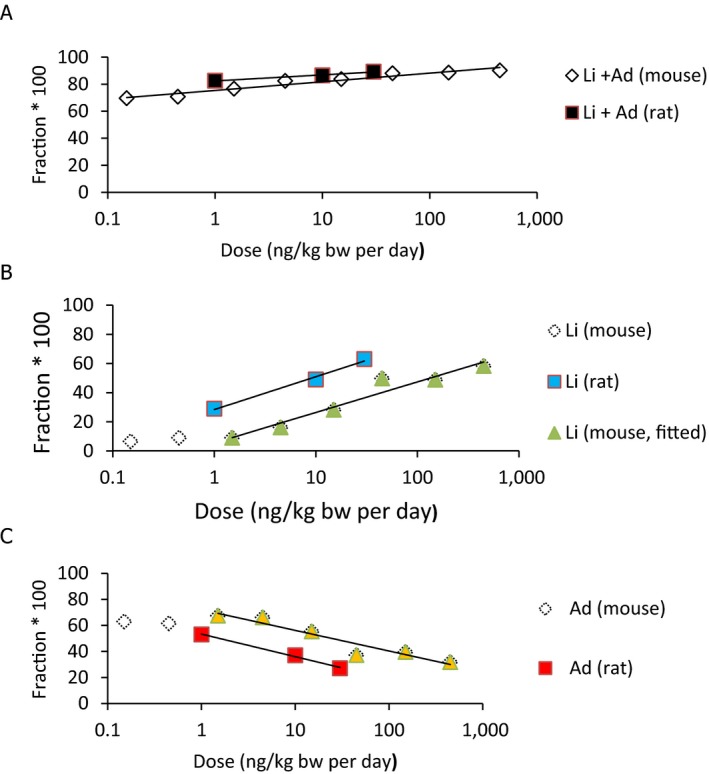
The percentage of the amount of [3H]‐2378‐TCDD in the body residing in the liver (Li) and/or adipose tissue (Ad) after repeated gavage exposure of rat (Hurst et al., 2000a) or mouse (Diliberto et al., 2001)
X‐axis: Administered dose (ng/kg bw per day); Y‐axis: Percentage of the body burden.
Modelling
A. Sum of liver and adipose tissue
Rat y = 1.934ln(x) + 82.39 (square)
Mouse y = 2.777ln(x) + 75.35 (diamond)
B. Liver
Rat y = 9.796ln(x) + 28.38 (square)
Mouse y = 9.099ln(x) + 5.459 (diamond)
C. Adipose tissue
Rat y = −7.538ln(x) + 53.33 (square)
Mouse y = −6.884ln(x) + 72.04 (diamond)
B.2. Body burden calculations for NOAELs and LOAELs in selected studies
Faqi et al. ( 1998 )
In the Faqi study, dams were treated subcutaneously before mating and throughout mating, pregnancy and lactation. The animals received an initial loading dose of 25, 60 or 300 ng TCDD/kg bw 2 weeks prior to mating, followed by weekly maintenance doses of 5, 12 or 60 ng/kg bw. Effects on male reproduction were studied on postnatal days (PND) 70 and 170. For this study, no NOAEL was identified and the lowest dose was the LOAEL (i.e. loading dose of 25 ng/kg bw, and weekly maintenance dose of 5 ng/kg bw). For this dose scheme, the TCDD concentration determined in maternal liver and adipose tissue at GD21 amounted to respectively 80 and 150 ng/kg tissue (Figure 1 of the manuscript). Assuming the female rat body at GD21 to consist of 12% adipose tissue and 4.5% liver tissue, the body burden was calculated to be 150 × 0.12 + 80 × 0.045 = 21.6 ng/kg bw. The fraction of the body burden residing in the liver + adipose tissue after the initial loading dose of 25 ng/kg bw is estimated to be 0.7548 × ln(0.025) + 88.77 = 86%. Likewise, at its equivalent repeated dose of 25/7 = 3.6 ng/kg bw per day this fraction would be 1.934 × ln(3.6) + 82.39 = 85%. It was decided not to apply an additional factor to correct for the single maintenance dose applied each week, also because an s.c. dose does not behave similar as a single gavage dose, and is likely to distribute more slowly to other tissues.
The chronic maternal GD21 body burden at the 25/5 ng/kg exposure protocol thus is estimated to be 21.6/0.86 = 25.1 ng/kg bw.
The other two dose groups showed levels in liver of respectively 140 and 640 ng/kg, and in adipose tissue of 240 and 780 ng/kg. The fraction of TCDD residing in the liver was calculated to be 87% and 88%, meaning that body burdens amount to 41 and 137 ng/kg bw for the medium and high dose.
In Bell et al. (2007b), pregnant rat dams were given a single oral gavage dose of 0, 50, 200 or 1,000 ng TCDD/kg bw at GD15 (Bell et al., 2007a).
For the NOAEL dose of 50 ng/kg bw, the GD16 and GD21 body burdens were experimentally determined at 18.8 resp. 19.6 ng/kg bw, based on the summation of liver, adipose tissue and (negligible) fetal deposition (Bell et al., 2007a; 5). Correcting these body burdens for the fraction of the body burden residing in the liver and adipose tissue ((0.87, as calculated from y = 0.7548ln0.05) + 88.77), then gives a GD16 body burden of 18.8/0.87 = 21.6, resp. GD21 body burden of 19.6/0.87 = 22.5 ng TCDD/kg. Extrapolating the acute exposure to chronic exposure, applying a factor 2.6 (SCF, 2001), then results in an estimated chronic GD16 body burden of 21.6 × 2.6 = 56.2 ng/kg bw, and GD21 body burden of 22.5 × 2.6 = 58.5 ng/kg bw.
Bell (2007a,c)
In Bell et al. (2007c), female rats were subchronically exposed via the diet to 0, 2.4, 8, 46 ng TCDD/kg bw per day during 12 weeks prior to mating, and continued during mating and pregnancy. For the LOAEL of 2.4 ng TCDD/kg bw per day, GD16 and GD21 body burdens were experimentally determined to be 35.4 resp. 42.4, based on the summation of liver, adipose tissue and (negligible) fetal deposition (Bell et al., 2007a; Table 5). Correcting these body burdens for the fraction of the body burden residing in the liver and adipose tissue (0.84, as calculated with y = 1.934ln(2.4) + 82.39), then gives a subchronic GD16 body burden of 35.4/0.84 = 42.1 ng/kg bw, and GD21 body burden of 42.4/0.84 = 50.4 ng/kg bw.
Viluksela et al. ( 2000 ) and Jämsä et al. ( 2001 )
In the Viluksela et al. study, 10‐week‐old female Long‐Evans and Han Wistar rats were exposed for 20 weeks with TCDD. To rapidly achieve the kinetic steady state, the first dose was a loading dose, which was five times as high as the 19 consecutive maintenance doses (see Table 1 of the manuscript). Over the 20‐week period, the dosing, on average, corresponded with daily doses of 0, 1, 10, 100 and 1,000 ng/kg bw per day. At the end of the exposure the concentration in the liver of Long‐Evans rats dosed with 1 and 10 ng/kg bw per day was measured to be 0.16 and 2.1 ng/g ww, respectively. Concentrations in livers of Han Wistar rats, treated with 100 and 1,000 ng/kg bw per day were 20.7 and 166 ng/g wet weight, respectively.
For the Long‐Evans rats, the NOAEL was 1 ng/kg bw per day. At this dose level the liver concentration in Long‐Evans rats amounted to 0.16 ng/g ww, i.e. 160 ng/kg liver. Assuming the liver to account for 4.5% of body weight, this gives the estimate for the body burden, based on liver only, of 160 × 0.045 = 7.2 ng/kg bw. At a dose level of 1 ng/kg bw per day, the fraction residing in the liver after repeated exposure in the rat is given by: fractional body burden = 9.796ln(1) + 28.38, which gives 28.38%. The estimated total body burden corresponding to the NOAEL of 1 ng/kg bw per day is therefore 7.2/0.28 ≈ 26 ng/kg bw.
The Jämsä et al. (2001) study can be considered as co‐studies to the Viluksela et al. (2000) study, i.e. based on the same animal study. Hence, the reported liver concentrations were slightly different, i.e. 0.67, 7.37 and 56.13 ng/g dw for the low, medium and high dose. Using a factor of 3.8, based on Viluksela et al. (2000), these correspond to wet weight based levels of 0.18, 1.94 and 14.77 ng/g. Based on the average daily doses of 1, 10 and 100 ng/kg bw, it can be calculated that 28.4, 50.9 and 73.5% of the body burden resides in the liver. Based again on a relative liver weight of 4.5%, the body burdens would be respectively 28, 171 and 904 ng/kg bw for the three dose groups.
As for the Viluksela et al. study, a NOAEL of 1 ng TCDD/kg bw per day was found for the Jämsä study. Consequently, the body burden corresponding with the NOAEL would be 28 ng/kg bw.
Harrill et al. ( 2016 ), Phadnis‐Moghe et al. ( 2016 )
The Harrill et al. study is a subacute, oral gavage study in which rats were exposed for 4 weeks/5 days per week. The NOAEL in this study is 100 ng/kg bw per day, corresponding to a daily exposure of 100 × 5/7 = 71 ng/kg bw per day.
At the end of the exposure period, for this dosing, the concentration in adipose tissue and liver amounted to around 3.2 and 10.2 ng/g tissue (Estimated from Figure 8 of the manuscript). Assuming the liver and adipose tissue to account for 8 and 4.5% of the body weight then gives an estimate for the body burden of 3.2 × 0.08 + 10.2 × 0.045 = 0.72 ng/g, or 720 ng/kg bw. At a dose level of 71 ng/kg bw, the fraction residing in the liver + adipose tissue after repeated exposure in the rat is given by: fractional body burden = 1.934 × ln(71) + 82.39, which gives 90.6%.
The estimate of the body burden corresponding at the 4‐week NOAEL of 71 ng/kg bw per day therefore is 720/0.91 = 789 ng/kg bw.
The study by Phadnis‐Moghe et al. (2016) is based on the same animal study as Harrill et al. (2016), but focussing on immunological effects. The NOAEL in this study is 22 ng/kg bw per day, corresponding to a daily exposure of 22 × 5/7 = 15.7 ng/kg bw per day.
According to Harrill et al. (2016), this dose results in concentrations in adipose tissue and liver to around 1.5 and 2.2 ng/g tissue (estimated from Figure 8 of the manuscript). Assuming the liver and adipose tissue to account for 8% and 4.5% of the body weight then gives an estimate for the body burden of 1.5 × 0.08 + 2.2 × 0.045 = 0.22 ng/g, or 220 ng/kg bw. At a dose level of 15.7 ng/kg bw, the fraction residing in the liver + adipose tissue after repeated exposure in the rat is given by: fractional body burden = 1.934 × ln(15.7) + 82.39, which gives 87.7%.
The estimate of the body burden corresponding at the 4‐week NOAEL of 15.7 ng/kg bw per day therefore is 220/0.88 ≈ 250 ng/kg bw.
NTP ( 2006a )
In this study, the administered dose levels were 3, 10, 22, 46 and 100 ng/kg bw per day, 5 days a week, corresponding to 2.1, 7.1, 15.7, 32.9 and 71.4 ng/kg bw per day (Walker et al., 2006). A dose level of 2.1 ng/kg bw per day was identified as NOAEL, given an exposure duration of 105 weeks. After 105 weeks exposure, measured concentrations of TCDD in the liver and in the adipose tissue were, respectively, 681 and 505 pg/g tissue (NTP, 2006a, 2006b, 2006c, 2006d–e, Table 13). Assuming the liver to account for 4.5% of body weight and the adipose tissue for 8%, this gives an estimate for the body burden of 681 × 0.045 + 505 × 0.08 = 71 ng/kg bw.
At a dose level of 2.1 ng/kg bw, the fraction residing in the liver + adipose tissue after repeated exposure in the rat is given by: fractional body burden = 1.934 × ln(2.1) + 82.39, which gives 83.8%.
The estimate of the body burden corresponding at the 105 week NOAEL of 2.1 ng/kg bw per day therefore is 71/0.84 = 85 ng/kg bw.
Li et al. ( 2006 )
The Li et al. study refers to an acute reproductive toxicity study in which pregnant mice were orally exposed to 0, 2, 50 or 100 ng TCDD/kg bw, throughout GD1–8, GD1–3 or GD4–8. A NOAEL of 2 ng/kg bw was identified for each of the applied dosing regimen. Though this study presents tissue dosimetry (see Tables 5 and 6 of the manuscript), the provided data were based on a biological assay and not on the basis of chemical measurement. Therefore, these data were discarded as basis for a body burden calculation.
Assuming linear kinetics at short exposure duration, it was assumed that the body burden increased linearly with the exposure duration, i.e. 3 days for the GD1–3 exposure period and 8 days for the GD1–8 exposure period. In concordance with SCF (2001), an absorption fraction of 0.6 was assumed. At short exposure a repeated exposure of a bioaccumulating compound like TCDD (half‐life in the rat ≈ 20 days) behaves approximately linear. So, in this case, a scaling factor of 3 was used for the 3‐day GD1–3 dosing and 8 for the 8‐day GD1–8 dosing. Furthermore, a factor of 2.6 was used to extrapolate a single acute dose to equivalent chronic fetal exposure. Hence, the corresponding chronic body burden ranged from 2 × 3 × 0.6 × 2.6 = 9.4 ng/kg bw to 2 × 8 × 0.6 × 2.6 = 25.0 ng/kg bw at the end of the GD1–3 and GD1–8 exposure period.
The factor 0.6 stands for the fraction absorbed of the administered dose. This is in concordance of the fraction absorbed for a single p.o. dose during pregnancy applied in the SCF, 2001 evaluation (Ohsako study). The factor 2.6 stems from the single dose → chronic exposure scaling for fetal exposure during pregnancy. Its use is explained in the Opinion.
B.3. Body burden calculations for BMDLs in selected studies
In Bell et al. (2007b), pregnant rat dams were given a single oral gavage dose of 0, 50, 200, 1,000 ng TCDD/kg bw at GD15 (Bell et al., 2007b) with a BMDL5 = 68 ng/kg bw.
For the 50 and 200 ng/kg bw doses, Bell et al. (2007a) mentions the measured GD16/GD21 body burden based on the summation of liver, adipose tissue and (negligible) fetal deposition (see Table, third column). Correcting the measured body burdens for the fraction of the whole body burden residing in the liver and the adipose tissue (see Table, fourth column) then gives the whole body burden for acute exposure (see Table, fifth column).
| Dose (ng/kg bw) | GD | Measured body burden (ng/kg bw) | Fraction of whole body burden in liver + adipose tissuea | Extrapolated whole body burden for acute exposure (ng/kg bw) |
|---|---|---|---|---|
| 50 | 16 | 18.8 | 0.87 | 21.6 |
| 21 | 19.6 | 0.87 | 22.5 | |
| 200 | 16 | 80 | 0.88 | 90.9 |
| 21 | 66 | 0.88 | 75.3 |
GD: gestation day; bw: body weight.
y = 0.7548 × ln(0.05) + 88.77 = 86.5 resp. y = 0.7548 × ln(0.2) + 88.77 = 87.5.
Assuming the body burden to increase linearly in the dose range 50–200, the acute GD16/GD21 body burdens corresponding with the BMDL5 of 68 ng/kg bw then are 30 and 29 ng/kg bw, for GD16 and GD21, respectively. Extrapolation to the chronic body burden by applying the correction factor of 2.6 for higher exposure of the fetus, results in BBs of 78 and 75 ng/kg bw.
This study concerns repeated oral dosing of rats (0, 2.4, 8, 46 ng/kg bw per day during 12 weeks prior to mating and continued during mating and pregnancy), with a BMDL5 = 3.5 ng/kg bw per day.
For the 2.4 and 8 ng/kg bw per day doses Bell et al. (2007a) mentions the measured GD16/GD21 body burden based on the summation of liver, adipose tissue and (negligible) fetal deposition (see Table, third column). Correcting the measured body burdens for the fraction of the whole body burden residing in the liver and adipose tissue (see Table, fourth column), this gives the whole body burden for chronic exposure (see Table, fifth column).
| Dose (ng/kg bw per day) | GD | Measured body burden (ng/kg bw) | Fraction of whole body burden in liver + adipose tissuea | Extrapolated whole body burden for chronic exposure (ng/kg bw) |
|---|---|---|---|---|
| 2.4 | 16 | 35.4 | 0.84 | 42 |
| 21 | 42.4 | 0.84 | 50 | |
| 8 | 16 | 74.9 | 0.86 | 87 |
| 21 | 91.2 | 0.86 | 106 |
GD: gestation day; bw: body weight.
y = 1.934 × ln(2.4) + 82.39 = 84 resp. y = 1.934 × ln(8) + 82.39 = 86.
Assuming the body burden to increase linearly at low dose, the chronic GD16/GD21 body burdens corresponding with the BMDL of 3.5 ng/kg bw per day then are 50.9 and 61.3 ng/kg bw, for GD16 and GD21, respectively.
Jämsä et al. ( 2001 )
Female rats were treated for 20 weeks, first with a single loading dose and subsequently with weekly maintenance doses, corresponding to an average daily dose of 0, 1, 10 and 100 ng/kg bw per day. Based on tissue levels, body burdens were calculated of 0, 28, 171 and 904 ng/kg bw. A number of effects on bones were reported, for which the CONTAM Panel calculated a number of BMDL5 levels based on body burdens, varying from 13.8 to 372 ng/kg bw.
NTP ( 2006a )
In this study, rats were chronically exposed to on average 2.1, 7.1, 15.7, 32.9 and 71.4 ng/kg bw per day with a lowest BMDL10 of 3.8 ng/kg bw per day (5 days/week), corresponding with a chronic daily dose of 2.7 ng/kg bw per day.
Based on 105‐week liver + adipose tissue disposition, the chronic body burden corresponding with the 2.1 ng/kg bw per day dose was calculated to be 681 × 0.045 + 505 × 0.0 8 = 71 ng/kg bw (based on 4.5% and 8% relative weights of the liver and adipose tissue). Similarly, the body burden corresponding with the 7.1 ng/kg bw per day dose was calculated to be 2,213 × 0.045 + 752 × 0.08 = 160 ng/kg bw.
The extrapolated body burden at the dose level of 2.7 ng/kg bw per day then is 82 ng/kg bw.
At a dose level of 2.7 ng/kg bw per day, the fraction of the body burden residing in the liver + adipose tissue after repeated exposure in the rat is:
The estimate of the chronic body burden corresponding with the BMDL10 of 2.7 ng/kg bw per day then is 82/0.84 = 97 ng/kg bw.
Li et al. ( 2006 )
This study refers to an acute reproductive toxicity study in which mice were orally exposed to 0, 2, 50 or 100 ng/kg bw per day, throughout GD1–8, GD 1–3 or GD4–8.
Given a GD1–3 BMDL10 of 11.4 ng/kg bw per day, the corresponding chronic body burden is 11.4 × 3 × 0.6 × 2.6 = 53.4 ng/kg bw (in this case, applying the factor of 2.6 to correct for the higher transfer to the fetus after an acute dose).
Faqi et al. ( 1998 )
In the Faqi et al. study, dams were treated subcutaneously before mating and throughout mating, pregnancy and lactation. The animals received an initial loading bolus dose of 25, 60 or 300 ng TCDD/kg bw, 2 weeks prior to mating, followed by weekly maintenance bolus doses of, respectively, 5, 12 or 60 ng/kg bw. Effects on male reproduction were studied on PND70 and PND170. The lowest dose resulted in an almost maximal response without a clear dose response at higher levels. Therefore, BMD modelling was not considered informative, as also shown by the large difference between BMDL and BMDU (see Table 13). Similar to the SCF (2001), the lowest dose was considered to be a LOAEL. As shown in Appendix B.2 the corresponding body burden is 25.1 ng/kg bw.
Appendix C – BMD modelling of rodent studies
C.1. Introduction
The appendix contains the details of the BMD modelling performed on experimental animal data. In this introduction a general description of the approach followed in the modelling is given.
Selection of the BMR
The benchmark dose (BMD) is defined as the dose that corresponds with an estimated change in response compared with the background response. The benchmark response (BMR) is the estimated response corresponding with the BMD of interest.
In most of the modelling performed in this opinion, the CONTAM Panel applied default BMR of 5% and 10% for continuous and quantal data, respectively, indicated in the EFSA guidance on BMD in risk assessment (EFSA Scientific Committee, 2017a). Deviations from the default BMR were considered on a case by case basis and are justified in the specific modelling reports in this appendix.
A 90% confidence interval around the BMD will be estimated, the lower bound is reported by BMDL and the upper bound by BMDU.
Software used
Results are obtained using the EFSA web‐tool for BMD analysis
Fitting benchmark dose models is based on the R‐package http://www.rivm.nl/en/Documents_and_publications/Scientific/Models/PROAST, version 65.7.
Averaging results from multiple fitted benchmark dose models (used only for modelling of quantal data) is based on the methodology in Wheeler and Bailer (2008).
Specification of deviations from default assumptions
No deviations from general assumption were introduced.
The CONTAM Panel selected the following default models:
Default set of fitted models for continuous data:
| Model | Number of parameters | Formula | |
|---|---|---|---|
| Null | 1 |
|
|
| Full | No. of groups |
|
|
| Exp model 3 | 3 |
|
|
| Exp model 5 | 4 |
|
|
| Hill model 3 | 3 |
|
|
| Hill model 5 | 4 |
|
Default set of fitted models for quantal data:
| Model | Number of parameters | Formula | |
|---|---|---|---|
| Null | 1 |
|
|
| Full | No. of groups | y = group mean | |
| Logistic | 2 |
|
|
| Probit | 2 |
|
|
| Log‐logistic | 3 |
|
|
| Log‐probit | 3 |
|
|
| Weibull | 3 |
|
|
| Gamma | 3 |
|
|
| Two‐stage | 3 |
|
|
| LVM – Exponential | Depend on underlying model | See continuous models | |
| LVM – Hill |
Procedure for selection of BMDL
BMDL was selected applying the following flowchart given in the EFSA Scientific Committee (2017) guidance: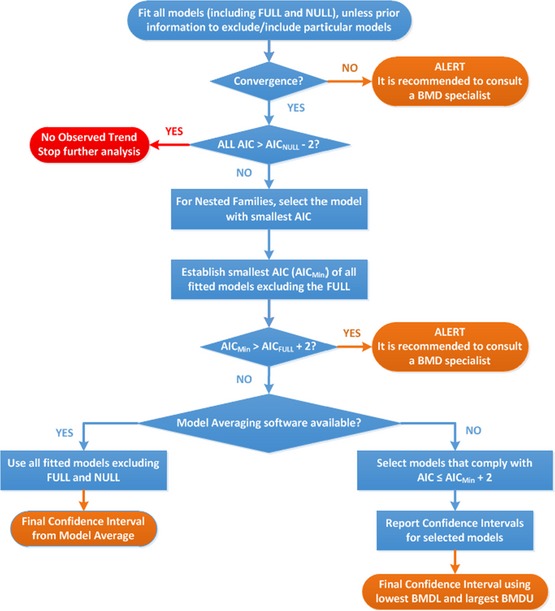
Flowchart for selection of BMDL
C.2. Faqi et al. (1998) – daily sperm production at PND70
Data description
The endpoint to be analysed is: mean daily sperm production at PND70.
Subset of the data is taken for time, retaining value(s) 1.
Data used for analysis:
| Dose body burden (ng/kg bw) | Mean daily sperm production at PND70 (×106) | SD | N |
|---|---|---|---|
| 0.0 | 34.4 | 4.3 | 20 |
| 25.1 | 28.0 | 5.7 | 20 |
| 41.0 | 25.2 | 5.6 | 20 |
| 137.0 | 23.1 | 4.9 | 20 |
bw: body weight; PND: postnatal day; SD: standard deviation.
Selection of the BMR
A BMR of 10% was selected deviating from the default BMR of 5% recommended for continuous data (EFSA Scientific Committee, 2017a,b). The selection of the 10% BMR was justified by the nature of the endpoint and variability observed in the controls and treatment group. The BMR of 10% corresponds approximately to the SD of the control group.
Results
Response variable: mean daily sperm production
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 20.32 | 5 | −30.64 |
| null‐ | Yes | −0.37 | 2 | 4.74 |
| Expon. m3‐ | Yes | 19.64 | 4 | −31.28 |
| Expon. m5‐ | Yes | 20.32 | 5 | −30.64 |
| Hill m3‐ | Yes | 19.66 | 4 | −31.32 |
| Hill m5‐ | Yes | 20.32 | 5 | −30.64 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.03583
estimate for a‐ : 34.17
estimate for CED‐ : 1.484
estimate for d‐ : 0.3061
HILL
estimate for var‐ : 0.03581
estimate for a‐ : 34.17
estimate for CED‐ : 1.892
estimate for d‐ : 0.3622
Final BMD Values (ng/kg bw)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 0.26 | 10.1 | 1.48 |
| Hill m3‐ | 0.15 | 10.9 | 1.89 |
Lowest BMDL and highest BMDU Values (ng/kg bw)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 0.148 | 10.9 |
Visualisation
C.3. Faqi et al. (1998) – daily sperm production at PND170
Data description
The endpoint to be analysed is: mean daily sperm production at PND170.
Subset of the data is taken for time, retaining value(s) 2.
Data used for analysis:
| Dose body burden (ng/kg bw) | Mean daily sperm production at PND170 (×106) | SD | N |
|---|---|---|---|
| 0.0 | 45.6 | 6.2 | 20 |
| 25.1 | 27.5 | 7.2 | 20 |
| 41.0 | 24.8 | 5.9 | 20 |
| 137.0 | 23.4 | 5.6 | 20 |
bw: body weight; PND: postnatal day; SD: standard deviation.
Selection of the BMR
A BMR of 10% was selected deviating from the default BMR of 5% recommended for continuous data (EFSA Scientific Committee, 2017a,b). The selection of the 10% BMR was justified by the nature of the endpoint and variability observed in the controls and treatment group. The BMR of 10% corresponds approximately to the SD of the control group.
Results
Response variable: mean daily sperm production
Fitted Models
| Model | Converged | Loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 9.30 | 5 | −8.60 |
| null‐ | Yes | −28.80 | 2 | 61.60 |
| Expon. m3‐ | Yes | 7.20 | 4 | −6.40 |
| Expon. m5‐ | Yes | 9.30 | 5 | −8.60 |
| Hill m3‐ | Yes | 8.52 | 4 | −9.04 |
| Hill m5‐ | Yes | 9.30 | 5 | −8.60 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.0464
estimate for a‐ : 45.18
estimate for CED‐ : 2.8
estimate for c‐ : 0.5036
estimate for d‐ : 0.9386
HILL
estimate for var‐ : 0.04731
estimate for a‐ : 44.88
estimate for CED‐ : 0.01714
estimate for d‐ : 0.25
Final BMD Values (ng/kg bw)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m5‐ | 0.00 | 14.30 | 2.80 |
| Hill m3‐ | 0.01 | 0.17 | 0.02 |
Lowest BMDL and highest BMDU Values (ng/kg bw)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 0.00135 | 14.3 |
Visualisation
C.4. Bell et al. (2007b) – Decreased pup weight from PND1 to PND7 in male F1 Wistar (Han) rats
Data description
The endpoint to be analysed is: pup weight from PND1 to PND7 in male F1 rats, following a single dosing of the dams.
Data used for analysis:
| Dose (ng/kg bw) | Pup weight (g) | Dam |
|---|---|---|
| 0 | 14.600000 | 53 |
| 0 | 14.500000 | 54 |
| 0 | 16.250000 | 56 |
| 0 | 14.133333 | 57 |
| 0 | 14.475000 | 58 |
| 0 | 14.550000 | 59 |
| 0 | 8.725000 | 60 |
| 0 | 14.500000 | 61 |
| 0 | 11.425000 | 62 |
| 0 | 14.320000 | 63 |
| 0 | 14.500000 | 64 |
| 0 | 13.175000 | 65 |
| 0 | 13.833333 | 66 |
| 0 | 12.412500 | 67 |
| 0 | 14.250000 | 68 |
| 0 | 12.680000 | 69 |
| 0 | 12.940000 | 70 |
| 0 | 15.275000 | 71 |
| 0 | 17.275000 | 72 |
| 0 | 14.875000 | 73 |
| 0 | 14.975000 | 74 |
| 0 | 15.516667 | 75 |
| 50 | 14.040000 | 109 |
| 50 | 10.100000 | 111 |
| 50 | 14.150000 | 112 |
| 50 | 14.666667 | 113 |
| 50 | 13.485714 | 114 |
| 50 | 11.300000 | 115 |
| 50 | 16.100000 | 116 |
| 50 | 13.200000 | 117 |
| 50 | 14.971429 | 118 |
| 50 | 12.266667 | 119 |
| 50 | 14.750000 | 120 |
| 50 | 17.600000 | 121 |
| 50 | 12.533333 | 122 |
| 50 | 14.025000 | 123 |
| 50 | 13.833333 | 124 |
| 50 | 13.700000 | 125 |
| 50 | 16.650000 | 126 |
| 50 | 16.100000 | 128 |
| 50 | 13.950000 | 129 |
| 50 | 15.733333 | 130 |
| 200 | 13.200000 | 163 |
| 200 | 13.483333 | 164 |
| 200 | 14.525000 | 165 |
| 200 | 12.820000 | 166 |
| 200 | 14.950000 | 167 |
| 200 | 12.575000 | 168 |
| 200 | 14.240000 | 169 |
| 200 | 13.883333 | 170 |
| 200 | 12.050000 | 171 |
| 200 | 10.800000 | 172 |
| 200 | 12.550000 | 173 |
| 200 | 12.466667 | 174 |
| 200 | 14.000000 | 175 |
| 200 | 13.900000 | 176 |
| 200 | 16.675000 | 179 |
| 200 | 13.920000 | 180 |
| 200 | 12.875000 | 181 |
| 200 | 14.200000 | 182 |
| 200 | 15.950000 | 183 |
| 200 | 13.180000 | 184 |
| 200 | 14.200000 | 185 |
| 1,000 | 9.600000 | 221 |
| 1,000 | 13.180000 | 222 |
| 1,000 | 10.242857 | 224 |
| 1,000 | 10.600000 | 225 |
| 1,000 | 11.440000 | 226 |
| 1,000 | 9.375000 | 227 |
| 1,000 | 12.700000 | 229 |
| 1,000 | 11.225000 | 230 |
| 1,000 | 8.866667 | 231 |
| 1,000 | 13.800000 | 232 |
| 1,000 | 11.380000 | 234 |
| 1,000 | 13.760000 | 237 |
| 1,000 | 14.600000 | 238 |
| 1,000 | 13.800000 | 239 |
| 1,000 | 13.820000 | 240 |
bw: body weight.
The following outliers were identified for Hill m3‐:
| Dose | pup.wgt | Dam | |
|---|---|---|---|
| 23 | 0 | 8.1 | 60 |
| 26 | 0 | 7.5 | 60 |
Results
Response variable: pup weight
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 189.29 | 5 | −368.58 |
| null‐ | Yes | 149.88 | 2 | −295.76 |
| Expon. m3‐ | Yes | 189.04 | 4 | −370.08 |
| Expon. m5‐ | Yes | 189.18 | 5 | −368.36 |
| Hill m3‐ | Yes | 189.05 | 4 | −370.10 |
| Hill m5‐ | Yes | 189.18 | 5 | −368.36 |
Confidence intervals for the BMD are based on 200 generated data sets.
Estimated Model Parameters
EXP
estimate for var‐ : 0.01875
estimate for a‐ : 13.87
estimate for CED‐ : 358.3
estimate for d‐ : 1.26
HILL
estimate for var‐ : 0.01875
estimate for a‐ : 13.87
estimate for CED‐ : 352.7
estimate for d‐ : 1.307
Final BMD Values (ng/kg bw per day)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 67.6 | 792 | 358.3 |
| Hill m3‐ | 68.5 | 784 | 352.7 |
Lowest BMDL and highest BMDU Values (ng/kg bw; single dose)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 67.55 | 792.4 |
Visualisation
Advanced Plots
C.5. Bell et al. (2007c) – delay in balanopreputial separation (BPS) in male F1 Wistar (Han) rats
Data description
The endpoint to be analysed is: mean day of BPS in male F1 rat males.
Data used for analysis:
| Dose (ng/kg bw per day) | Mean PND for BPS (day) | SD | N |
|---|---|---|---|
| 0.0 | 45.4 | 2.2 | 99 |
| 1.8 | 47.2 | 2.3 | 98 |
| 9.3 | 47.3 | 2.6 | 93 |
| 53.0 | 49.8 | 4.9 | 67 |
bw: body weight; PND: postnatal day; BPS: balanopreputial separation; SD: standard deviation.
Results
Response variable: mean day of BPS
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 485.98 | 5 | −961.96 |
| null‐ | Yes | 448.91 | 2 | −893.82 |
| Expon. m3‐ | Yes | 483.91 | 4 | −959.82 |
| Expon. m5‐ | No | 483.87 | 5 | −957.74 |
| Hill m3‐ | Yes | 483.95 | 4 | −959.90 |
| Hill m5‐ | Yes | 483.87 | 5 | −957.74 |
ATTENTION: The Panel noted that the criterion for accepting the modelling (AICmin < AICfull +2) is formally not respected. However, the difference between AICmin (−959.90) and AICfull (−961.96) is very close to 2; therefore, it was considered appropriate to accept the modelling.
Estimated Model Parameters
EXP
estimate for var‐ : 0.003892
estimate for a‐ : 45.4
estimate for CED‐ : 9.063
estimate for d‐ : 0.306
HILL
estimate for var‐ : 0.003891
estimate for a‐ : 45.4
estimate for CED‐ : 9.113
estimate for d‐ : 0.298
Final BMD Values (ng/kg bw per day)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 3.5 | 20.6 | 9.06 |
| Hill m3‐ | 3.7 | 20.6 | 9.11 |
Lowest BMDL and highest BMDU Values (ng/kg bw per day)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 3.5 | 20.6 |
Visualisation
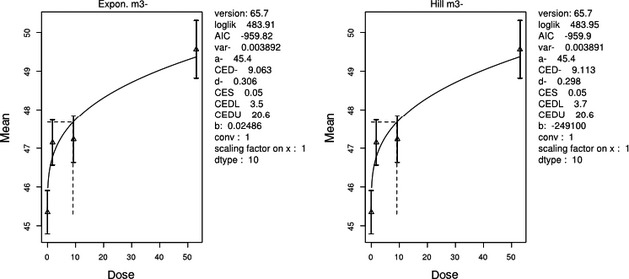
C.6. NTP (2006a) – incidence of multinucleated hepatocytes
Data description
The endpoint to be analysed is: incidence of multinucleated hepatocytes
Data used for analysis:
| Dose (ng/kg bw per day) | Incidence of multinucleated hepatocytes | N |
|---|---|---|
| 0 | 0 | 53 |
| 3 | 0 | 54 |
| 10 | 16 | 53 |
| 22 | 26 | 53 |
| 46 | 36 | 53 |
| 100 | 51 | 53 |
bw: body weight.
Results
Response variable: incidence of multinucleated hepatocytes
Fitted Models
| Model | No. of par | loglik | AIC | Accepted | BMDL | BMDU | BMD | Conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −215.25 | 432.50 | NA | NA | NA | NA | |
| full | 6 | −110.96 | 233.92 | NA | NA | NA | NA | |
| two.stage | 3 | −116.38 | 238.76 | No | NA | NA | 4.09 | Yes |
| log.logist | 3 | −115.91 | 237.82 | No | NA | NA | 5.81 | Yes |
| Weibull | 3 | −116.13 | 238.26 | No | NA | NA | 4.68 | Yes |
| log.prob | 3 | −114.80 | 235.60 | Yes | 4.36 | 7.80 | 6.01 | Yes |
| gamma | 3 | −115.97 | 237.94 | No | NA | NA | 5.00 | Yes |
| logistic | 2 | −130.02 | 264.04 | No | NA | NA | 11.10 | Yes |
| probit | 2 | −130.70 | 265.40 | No | NA | NA | 10.90 | Yes |
| LVM: Expon. m3‐ | 3 | −115.87 | 237.74 | No | NA | NA | 5.25 | Yes |
| LVM: Hill m3‐ | 3 | −115.09 | 236.18 | Yes | 4.04 | 7.65 | 5.79 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 1e‐06
estimate for BMD‐ : 4.089
estimate for c : 0.08755
log.logist
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.815
estimate for c : 1.639
Weibull
estimate for a‐ : 1e‐06
estimate for BMD‐ : 4.68
estimate for c : 1.111
log.prob
estimate for a‐ : 1e‐06
estimate for BMD‐ : 6.012
estimate for c : 0.9814
gamma
estimate for a‐ : 1e‐06
estimate for BMD‐ : 4.997
estimate for cc : 1.222
logistic
estimate for a‐ : ‐2.204
estimate for BMD‐ : 11.05
probit
estimate for a‐ : ‐1.283
estimate for BMD‐ : 10.85
EXP
estimate for a‐ : 2.718
estimate for CED‐ : 5.248
estimate for d‐ : 0.25
estimate for th : 0
estimate for sigma : 0.25
HILL
estimate for a‐ : 4.482
estimate for CED‐ : 5.792
estimate for d‐ : 0.3172
estimate for th : 0
estimate for sigma : 0.25
Weights for Model Averaging (ng/kg bw per day)
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.06 | 0.1 | 0.08 | 0.31 | 0.1 | 0 | 0 | 0.11 | 0.23 |
Final BMD Values (ng/kg bw per day)
| BMDL | BMDU |
|---|---|
| 3.8 | 7.74 |
Confidence intervals for the BMD are based on 1,000 generated data sets.
Visualisation
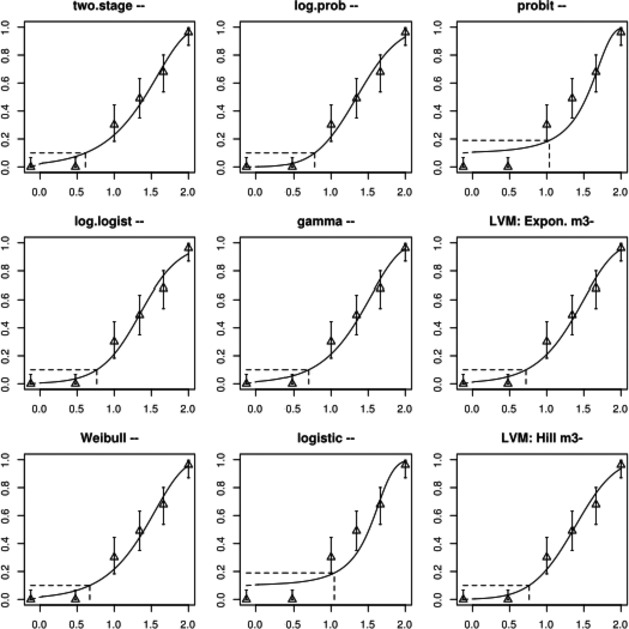
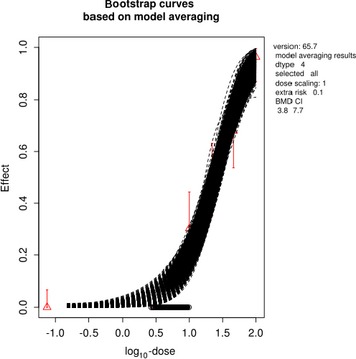
C.7. NTP (2006a) – incidence of diffuse fatty change
Data description
The endpoint to be analysed is: incidence of diffuse fatty change
Data used for analysis:
| Dose (ng/kg bw per day) | Incidence of diffuse fatty change | N |
|---|---|---|
| 0 | 0 | 53 |
| 3 | 2 | 54 |
| 10 | 12 | 53 |
| 22 | 17 | 53 |
| 46 | 30 | 53 |
| 100 | 48 | 53 |
bw: body weight.
Results
Response variable: incidence of diffuse fatty change
Fitted Models
| Model | No. of par | loglik | AIC | Accepted | BMDL | BMDU | BMD | Conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −204.85 | 411.70 | NA | NA | NA | NA | |
| full | 6 | −122.99 | 257.98 | NA | NA | NA | NA | |
| two.stage | 3 | −123.96 | 253.92 | Yes | 4.49 | 8.02 | 5.84 | Yes |
| log.logist | 3 | −125.53 | 257.06 | No | NA | NA | 6.58 | Yes |
| Weibull | 3 | −124.10 | 254.20 | Yes | 3.80 | 8.48 | 5.96 | Yes |
| log.prob | 3 | −125.62 | 257.24 | No | NA | NA | 6.42 | Yes |
| gamma | 3 | −124.14 | 254.28 | Yes | 3.61 | 8.51 | 5.92 | Yes |
| logistic | 2 | −132.92 | 269.84 | No | NA | NA | 15.30 | Yes |
| probit | 2 | −132.73 | 269.46 | No | NA | NA | 14.70 | Yes |
| LVM: Expon. m3‐ | 3 | −124.29 | 254.58 | Yes | 3.75 | 8.69 | 6.05 | Yes |
| LVM: Hill m3‐ | 3 | −124.58 | 255.16 | Yes | 4.07 | 9.02 | 6.34 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.842
estimate for c : 0.1528
log.logist
estimate for a‐ : 1e‐06
estimate for BMD‐ : 6.582
estimate for c : 1.414
Weibull
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.957
estimate for c : 1.072
log.prob
estimate for a‐ : 1e‐06
estimate for BMD‐ : 6.419
estimate for c : 0.8239
gamma
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.919
estimate for cc : 1.096
logistic
estimate for a‐ : ‐2.298
estimate for BMD‐ : 15.28
probit
estimate for a‐ : ‐1.36
estimate for BMD‐ : 14.73
EXP
estimate for a‐ : 2.199
estimate for CED‐ : 6.046
estimate for d‐ : 0.2991
estimate for th : 0
estimate for sigma : 0.25
HILL
estimate for a‐ : 2.086
estimate for CED‐ : 6.336
estimate for d‐ : 0.4467
estimate for th : 0
estimate for sigma : 0.25
Weights for Model Averaging (ng/kg bw per day)
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.23 | 0.05 | 0.2 | 0.04 | 0.19 | 0 | 0 | 0.16 | 0.12 |
Final BMD Values (ng/kg bw per day)
| BMDL | BMDU |
|---|---|
| 4.28 | 9.48 |
Confidence intervals for the BMD are based on 1,000 generated data sets.
Visualisation
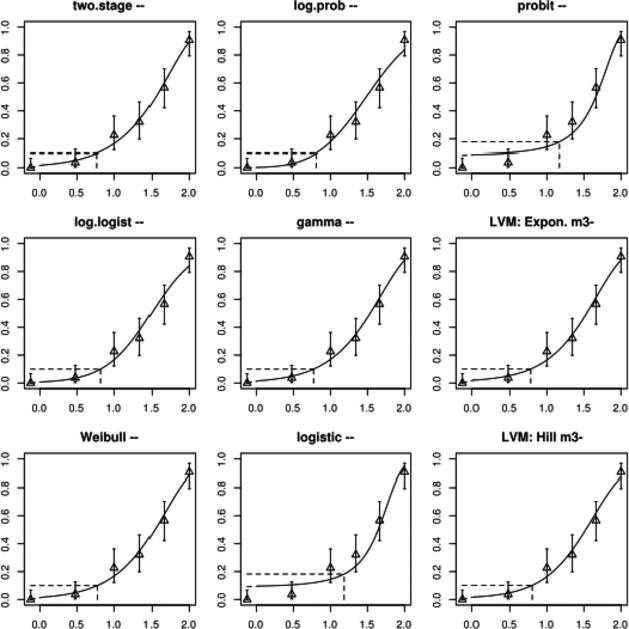
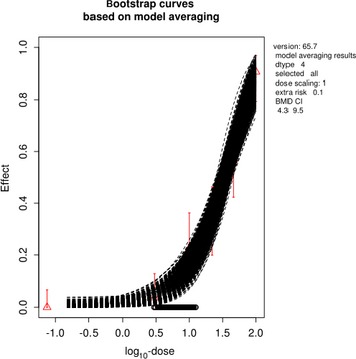
C.8. NTP (2006a) – incidence of liver necrosis
Data description
The endpoint to be analysed is: incidence of liver necrosis.
Data used for analysis:
| Dose (ng/kg bw per day) | Incidence of liver necrosis | N |
|---|---|---|
| 0 | 1 | 53 |
| 3 | 4 | 54 |
| 10 | 4 | 53 |
| 22 | 8 | 53 |
| 46 | 10 | 53 |
| 100 | 17 | 53 |
bw: body weight.
Results
Response variable: incidence of liver necrosis
Fitted Models
| Model | No. of par | loglik | AIC | Accepted | BMDL | BMDU | BMD | Conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −127.98 | 257.96 | NA | NA | NA | NA | |
| full | 6 | −114.81 | 241.62 | NA | NA | NA | NA | |
| two.stage | 3 | −115.79 | 237.58 | Yes | 19.90 | 45.1 | 28.3 | Yes |
| log.logist | 3 | −115.24 | 236.48 | Yes | 5.03 | 41.1 | 15.6 | Yes |
| Weibull | 3 | −115.20 | 236.40 | Yes | 4.96 | 41.1 | 15.6 | Yes |
| log.prob | 3 | −115.37 | 236.74 | Yes | 4.89 | 41.0 | 14.9 | Yes |
| gamma | 3 | −115.17 | 236.34 | Yes | 4.87 | 40.6 | 15.6 | Yes |
| logistic | 2 | −117.16 | 238.32 | No | NA | NA | 48.8 | Yes |
| probit | 2 | −116.95 | 237.90 | Yes | 37.00 | 61.9 | 45.7 | Yes |
| LVM: Expon. m3‐ | 3 | −115.10 | 236.20 | Yes | 7.47 | 41.7 | 16.3 | Yes |
| LVM: Hill m3‐ | 3 | −115.12 | 236.24 | Yes | 6.30 | 41.5 | 16.2 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 0.04276
estimate for BMD‐ : 28.27
estimate for c : 1e‐06
log.logist
estimate for a‐ : 0.02203
estimate for BMD‐ : 15.57
estimate for c : 0.6953
Weibull
estimate for a‐ : 0.02148
estimate for BMD‐ : 15.64
estimate for c : 0.6398
log.prob
estimate for a‐ : 0.02283
estimate for BMD‐ : 14.86
estimate for c : 0.3672
gamma
estimate for a‐ : 0.02102
estimate for BMD‐ : 15.6
estimate for cc : 0.599
logistic
estimate for a‐ : ‐2.6
estimate for BMD‐ : 48.79
probit
estimate for a‐ : ‐1.508
estimate for BMD‐ : 45.73
EXP
estimate for a‐ : 1.667
estimate for CED‐ : 16.3
estimate for d‐ : 0.3274
estimate for th : 0
estimate for sigma : 0.25
HILL
estimate for a‐ : 1.663
estimate for CED‐ : 16.23
estimate for d‐ : 0.3758
estimate for th : 0
estimate for sigma : 0.25
Weights for Model Averaging (ng/kg bw per day)
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.07 | 0.13 | 0.13 | 0.11 | 0.14 | 0.05 | 0.06 | 0.15 | 0.15 |
Final BMD Values (ng/kg bw per day)
| BMDL | BMDU |
|---|---|
| 8.01 | 49.4 |
Confidence intervals for the BMD are based on 1,000 generated data sets.
Visualisation
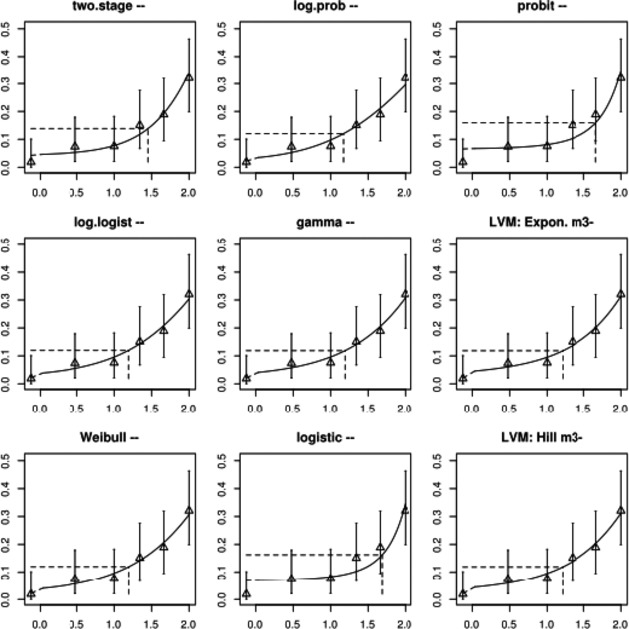
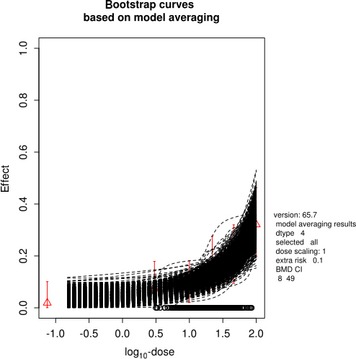
C.9. NTP (2006a) – incidence of oval cell hyperplasia
Data description
The endpoint to be analysed is: incidence of oval cell hyperplasia.
Data used for analysis:
| Dose (ng/kg bw per day) | Incidence of oval cell hyperplasia | N |
|---|---|---|
| 0 | 0 | 53 |
| 3 | 4 | 54 |
| 10 | 3 | 53 |
| 22 | 20 | 53 |
| 46 | 38 | 53 |
| 100 | 53 | 53 |
bw: body weight.
Results
Response variable: incidence of oval cell hyperplasia
Fitted Models
| Model | No. of par | loglik | AIC | Accepted | BMDL | BMDU | BMD | Conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −210.19 | 422.38 | NA | NA | NA | NA | |
| full | 6 | −92.49 | 196.98 | NA | NA | NA | NA | |
| two.stage | 3 | −95.66 | 197.32 | Yes | 5.67 | 13.4 | 8.11 | Yes |
| log.logist | 3 | −97.98 | 201.96 | No | NA | NA | 13.60 | Yes |
| Weibull | 3 | −96.33 | 198.66 | Yes | 6.58 | 15.3 | 10.80 | Yes |
| log.prob | 3 | −97.19 | 200.38 | No | NA | NA | 14.00 | Yes |
| gamma | 3 | −96.71 | 199.42 | No | NA | NA | 12.60 | Yes |
| logistic | 2 | −97.91 | 199.82 | No | NA | NA | 13.70 | Yes |
| probit | 2 | −97.06 | 198.12 | Yes | 10.80 | 15.0 | 12.70 | Yes |
| LVM: Expon. m3‐ | 3 | −95.72 | 197.44 | Yes | 6.62 | 13.3 | 9.51 | Yes |
| LVM: Hill m3‐ | 3 | −96.18 | 198.36 | Yes | 7.38 | 14.5 | 10.50 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 1e‐06
estimate for BMD‐ : 8.113
estimate for c : 4.718
log.logist
estimate for a‐ : 0.03326
estimate for BMD‐ : 13.61
estimate for c : 2.835
Weibull
estimate for a‐ : 0.02128
estimate for BMD‐ : 10.81
estimate for c : 1.766
log.prob
estimate for a‐ : 0.03601
estimate for BMD‐ : 13.97
estimate for c : 1.701
gamma
estimate for a‐ : 0.03086
estimate for BMD‐ : 12.58
estimate for cc : 2.925
logistic
estimate for a‐ : ‐3.12
estimate for BMD‐ : 13.71
probit
estimate for a‐ : ‐1.823
estimate for BMD‐ : 12.74
EXP
estimate for a‐ : 1.788
estimate for CED‐ : 9.512
estimate for d‐ : 0.6389
estimate for th : 0
estimate for sigma : 0.25
HILL
estimate for a‐ : 1.707
estimate for CED‐ : 10.51
estimate for d‐ : 0.9222
estimate for th : 0
estimate for sigma : 0.25
Weights for Model Averaging (ng/kg bw per day)
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.21 | 0.02 | 0.11 | 0.05 | 0.07 | 0.06 | 0.14 | 0.2 | 0.13 |
Final BMD Values (ng/kg bw per day)
| BMDL | BMDU |
|---|---|
| 8.21 | 15.2 |
Confidence intervals for the BMD are based on 1,000 generated data sets.
Visualisation
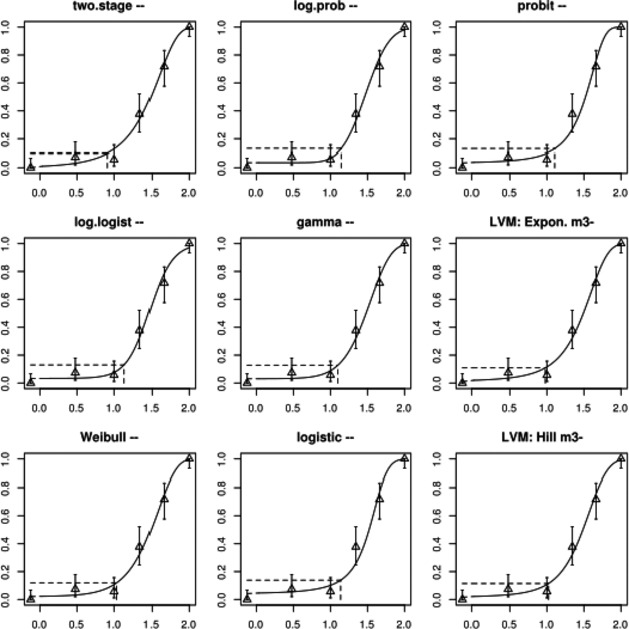
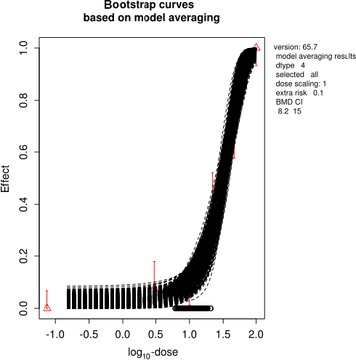
C.10. NTP (2006a) – incidence of bile duct hyperplasia
Data description
The endpoint to be analysed is: Incidence of Bile duct hyperplasia.
Data used for analysis:
| Dose (ng/kg bw per day) | Incidence of bile duct hyperplasia | N |
|---|---|---|
| 0 | 5 | 53 |
| 3 | 4 | 54 |
| 10 | 7 | 53 |
| 22 | 22 | 53 |
| 46 | 40 | 53 |
| 100 | 53 | 53 |
bw: body weight.
Results
Response variable: incidence of bile duct hyperplasia
Fitted Models
| Model | No. of par | loglik | AIC | Accepted | BMDL | BMDU | BMD | Conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −215.99 | 433.98 | NA | NA | NA | NA | |
| full | 6 | −117.00 | 246.00 | NA | NA | NA | NA | |
| two.stage | 3 | −117.88 | 241.76 | Yes | 6.42 | 14.0 | 11.10 | Yes |
| log.logist | 3 | −119.11 | 244.22 | No | NA | NA | 13.10 | Yes |
| Weibull | 3 | −117.78 | 241.56 | Yes | 7.38 | 15.5 | 11.00 | Yes |
| log.prob | 3 | −118.46 | 242.92 | No | NA | NA | 13.20 | Yes |
| gamma | 3 | −117.89 | 241.78 | Yes | 8.02 | 16.4 | 11.90 | Yes |
| logistic | 2 | −118.43 | 240.86 | Yes | 9.05 | 12.6 | 10.70 | Yes |
| probit | 2 | −118.16 | 240.32 | Yes | 8.50 | 11.6 | 9.94 | Yes |
| LVM: Expon. m3‐ | 3 | −118.15 | 242.30 | Yes | 6.04 | 14.7 | 9.58 | Yes |
| LVM: Hill m3‐ | 3 | −118.07 | 242.14 | Yes | 6.85 | 15.2 | 10.40 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 0.0781
estimate for BMD‐ : 11.08
estimate for c : 95.26
log.logist
estimate for a‐ : 0.08561
estimate for BMD‐ : 13.07
estimate for c : 2.81
Weibull
estimate for a‐ : 0.07807
estimate for BMD‐ : 10.97
estimate for c : 1.823
log.prob
estimate for a‐ : 0.0875
estimate for BMD‐ : 13.2
estimate for c : 1.658
gamma
estimate for a‐ : 0.08182
estimate for BMD‐ : 11.9
estimate for cc : 2.833
logistic
estimate for a‐ : ‐2.449
estimate for BMD‐ : 10.7
probit
estimate for a‐ : ‐1.44
estimate for BMD‐ : 9.941
EXP
estimate for a‐ : 1.441
estimate for CED‐ : 9.58
estimate for d‐ : 0.9671
estimate for th : 0
estimate for sigma : 0.25
HILL
estimate for a‐ : 1.433
estimate for CED‐ : 10.39
estimate for d‐ : 1.191
estimate for th : 0
estimate for sigma : 0.25
Weights for Model Averaging (ng/kg bw per day)
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.11 | 0.03 | 0.12 | 0.06 | 0.11 | 0.17 | 0.22 | 0.08 | 0.09 |
Final BMD Values (ng/kg bw per day)
| BMDL | BMDU |
|---|---|
| 8.08 | 15.4 |
Confidence intervals for the BMD are based on 1,000 generated data sets.
Visualisation
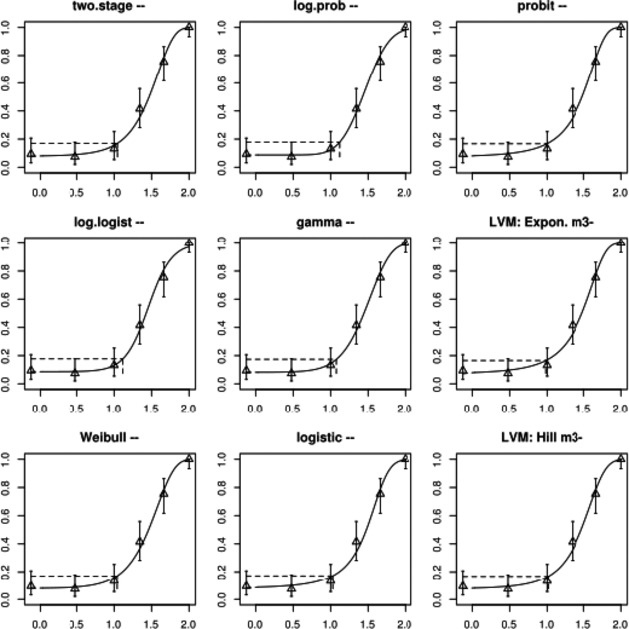
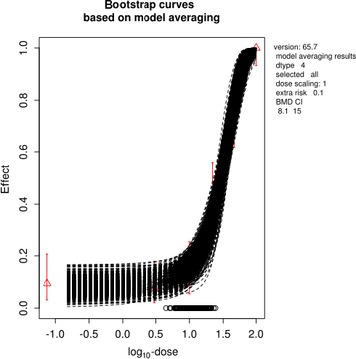
C.11. NTP (2006a) – incidence of toxic hepatopathy
Data description
The endpoint to be analysed is: incidence of toxic hepatopathy.
Data used for analysis:
| Dose (ng/kg bw per day) | Incidence of toxic hepatopathy | N |
|---|---|---|
| 0 | 0 | 53 |
| 3 | 2 | 54 |
| 10 | 8 | 53 |
| 22 | 30 | 53 |
| 46 | 45 | 53 |
| 100 | 53 | 53 |
bw: body weight.
Results
Response variable: incidence of toxic hepatopathy
Fitted Models
| Model | No. of par | loglik | AIC | Accepted | BMDL | BMDU | BMD | conv |
|---|---|---|---|---|---|---|---|---|
| null | 1 | −218.21 | 438.42 | NA | NA | NA | NA | |
| full | 6 | −89.81 | 191.62 | NA | NA | NA | NA | |
| two.stage | 3 | −91.24 | 188.48 | Yes | 3.85 | 8.41 | 5.84 | Yes |
| log.logist | 3 | −92.02 | 190.04 | No | NA | NA | 7.96 | Yes |
| Weibull | 3 | −90.82 | 187.64 | Yes | 4.43 | 8.28 | 6.25 | Yes |
| log.prob | 3 | −91.92 | 189.84 | No | NA | NA | 8.50 | Yes |
| gamma | 3 | −90.81 | 187.62 | Yes | 4.65 | 8.50 | 6.54 | Yes |
| logistic | 2 | −97.18 | 198.36 | No | NA | NA | 9.90 | Yes |
| probit | 2 | −96.53 | 197.06 | No | NA | NA | 9.50 | Yes |
| LVM: Expon. m3‐ | 3 | −90.75 | 187.50 | Yes | 4.60 | 8.63 | 6.58 | Yes |
| LVM: Hill m3‐ | 3 | −90.83 | 187.66 | Yes | 4.97 | 8.90 | 6.86 | Yes |
Estimated Model Parameters
two.stage
estimate for a‐ : 1e‐06
estimate for BMD‐ : 5.844
estimate for c : 3.576
log.logist
estimate for a‐ : 0.007129
estimate for BMD‐ : 7.958
estimate for c : 2.37
Weibull
estimate for a‐ : 1e‐06
estimate for BMD‐ : 6.245
estimate for c : 1.492
log.prob
estimate for a‐ : 0.01565
estimate for BMD‐ : 8.496
estimate for c : 1.444
gamma
estimate for a‐ : 1e‐06
estimate for BMD‐ : 6.544
estimate for cc : 1.926
logistic
estimate for a‐ : ‐2.929
estimate for BMD‐ : 9.902
probit
estimate for a‐ : ‐1.726
estimate for BMD‐ : 9.501
EXP
estimate for a‐ : 2.515
estimate for CED‐ : 6.582
estimate for d‐ : 0.3594
estimate for th : 0
estimate for sigma : 0.25
HILL
estimate for a‐ : 2.153
estimate for CED‐ : 6.861
estimate for d‐ : 0.6314
estimate for th : 0
estimate for sigma : 0.25
Weights for Model Averaging (ng/kg bw per day)
| two.stage | log.logist | Weibull | log.prob | gamma | logistic | probit | EXP | HILL |
|---|---|---|---|---|---|---|---|---|
| 0.12 | 0.06 | 0.19 | 0.06 | 0.19 | 0 | 0 | 0.2 | 0.18 |
Final BMD Values (ng/kg bw per day)
| BMDL | BMDU |
|---|---|
| 4.94 | 9.58 |
Confidence intervals for the BMD are based on 1000 generated data sets.
Visualisation
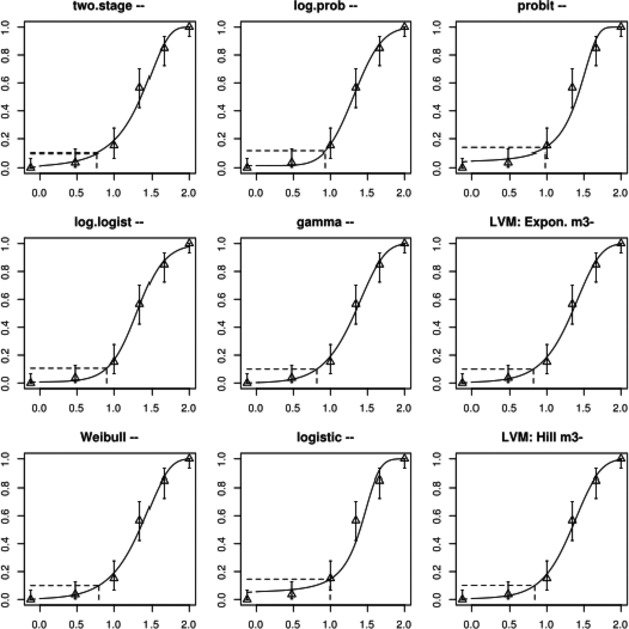
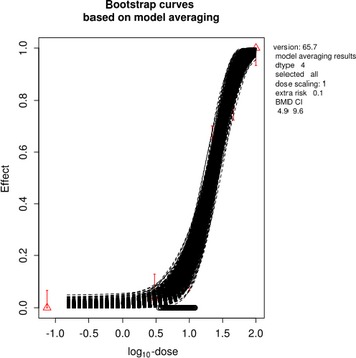
C.12. Jämsä et al. (2001) – tibial length in Long‐Evans rats
Data description
The endpoint to be analysed is: tibial length.
Data used for analysis:
| Dose body burden (ng/kg bw) | Tibial length (mm) | SD | N |
|---|---|---|---|
| 0 | 37.1 | 0.4 | 5 |
| 28 | 36.8 | 0.5 | 5 |
| 171 | 36.0 | 0.5 | 5 |
| 904 | 35.1 | 0.6 | 5 |
bw: body weight.
Results
Response variable: tibial length
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 59.20 | 5 | −108.40 |
| null‐ | Yes | 45.46 | 2 | −86.92 |
| Expon. m3‐ | Yes | 58.81 | 4 | −109.62 |
| Expon. m5‐ | Yes | 59.19 | 5 | −108.38 |
| Hill m3‐ | Yes | 58.82 | 4 | −109.64 |
| Hill m5‐ | Yes | 59.20 | 5 | −108.40 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.0001635
estimate for a‐ : 37.14
estimate for CED‐ : 698.9
estimate for d‐ : 0.4345
HILL
estimate for var‐ : 0.0001633
estimate for a‐ : 37.14
estimate for CED‐ : 697.5
estimate for d‐ : 0.4424
Final BMD Values (ng/kg bw BB)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 374 | 1270 | 699 |
| Hill m3‐ | 372 | 1280 | 698 |
Lowest BMDL and highest BMDU Values (ng/kg bw BB)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 372 | 1,280 |
Visualisation
C.13. Jämsä et al. (2001) – tibial pericircumferences in Long‐Evans rats
Data description
The endpoint to be analysed is: tibial pericircumferences
Data used for analysis:
| Dose body burden (ng/kg bw) | Tibial pericircumferences (mm) | SD | N |
|---|---|---|---|
| 0 | 9.48 | 0.15 | 5 |
| 28 | 9.39 | 0.14 | 5 |
| 171 | 9.09 | 0.18 | 5 |
| 904 | 8.58 | 0.32 | 5 |
bw: body weight.
Results
Response variable: tibial pericircumferences
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 48.66 | 5 | −87.32 |
| null‐ | Yes | 33.82 | 2 | −63.64 |
| Expon. m3‐ | Yes | 48.52 | 4 | −89.04 |
| Expon. m5‐ | Yes | 48.66 | 5 | −87.32 |
| Hill m3‐ | Yes | 48.53 | 4 | −89.06 |
| Hill m5‐ | Yes | 48.66 | 5 | −87.32 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.0004575
estimate for a‐ : 9.493
estimate for CED‐ : 260
estimate for d‐ : 0.555
HILL
estimate for var‐ : 0.0004569
estimate for a‐ : 9.493
estimate for CED‐ : 258
estimate for d‐ : 0.5719
Final BMD Values (ng/kg bw BB)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 104 | 505 | 260 |
| Hill m3‐ | 104 | 501 | 258 |
Lowest BMDL and highest BMDU Values (ng/kg bw BB)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 104 | 505 |
Visualisation
 Plot for response ‘peri’: exphill
Plot for response ‘peri’: exphill
Advanced Plots
 Advanced plots for response ‘peri’
Advanced plots for response ‘peri’
C.14. Jämsä et al. (2001) – tibial endocircumferences in Long‐Evans rats
Data description
The endpoint to be analysed is: tibial endocircumferences
Data used for analysis:
| Dose body burden (ng/kg bw) | Tibial endocircumferences (mm) | SD | N |
|---|---|---|---|
| 0 | 5.66 | 0.11 | 5 |
| 28 | 5.62 | 0.07 | 5 |
| 171 | 5.32 | 0.19 | 5 |
| 904 | 4.76 | 0.25 | 5 |
bw: body weight.
Results
Response variable: tibial endocircumferences
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 41.62 | 5 | −73.24 |
| null‐ | Yes | 23.15 | 2 | −42.30 |
| Expon. m3‐ | Yes | 41.32 | 4 | −74.64 |
| Expon. m5‐ | Yes | 41.62 | 5 | −73.24 |
| Hill m3‐ | Yes | 41.36 | 4 | −74.72 |
| Hill m5‐ | Yes | 41.62 | 5 | −73.24 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.0009396
estimate for a‐ : 5.68
estimate for CED‐ : 131
estimate for d‐ : 0.6473
HILL
estimate for var‐ : 0.0009364
estimate for a‐ : 5.68
estimate for CED‐ : 130.8
estimate for d‐ : 0.6801
Final BMD Values (ng/kg bw BB)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 47.8 | 290 | 131.0 |
| Hill m3‐ | 49.1 | 285 | 130.8 |
Lowest BMDL and highest BMDU Values (ng/kg bw BB)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 47.8 | 290 |
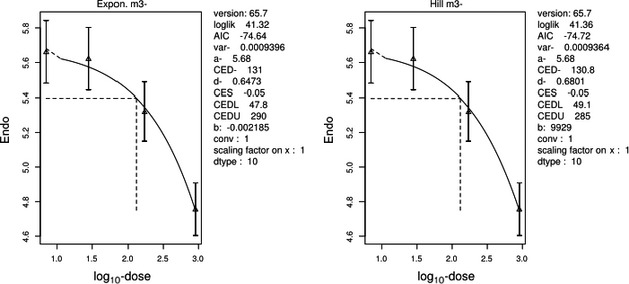
C.15. Jämsä et al. (2001) – tibial cross‐sectional area of cortex (CSA) in Long‐Evans rats
Data description
The endpoint to be analysed is: CSA.
Data used for analysis:
| Dose body burden (ng/kg bw) | Tibial CSA (mm2) | SD | N |
|---|---|---|---|
| 0 | 4.60 | 0.17 | 5 |
| 28 | 4.50 | 0.16 | 5 |
| 171 | 4.32 | 0.15 | 5 |
| 904 | 4.00 | 0.30 | 5 |
bw: body weight.
Results
Response variable: tibial cross‐sectional area of cortex (CSA)
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 34.34 | 5 | −58.68 |
| null‐ | Yes | 24.98 | 2 | −45.96 |
| Expon. m3‐ | Yes | 34.33 | 4 | −60.66 |
| Expon. m5‐ | Yes | 34.34 | 5 | −58.68 |
| Hill m3‐ | Yes | 34.33 | 4 | −60.66 |
| Hill m5‐ | Yes | 34.34 | 5 | −58.68 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.001891
estimate for a‐ : 4.6
estimate for CED‐ : 118.6
estimate for d‐ : 0.5048
HILL
estimate for var‐ : 0.00189
estimate for a‐ : 4.6
estimate for CED‐ : 118.5
estimate for d‐ : 0.527
Final BMD Values (ng/kg bw BB)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 13.8 | 455 | 118.6 |
| Hill m3‐ | 14.2 | 451 | 118.5 |
Lowest BMDL and highest BMDU Values (ng/kg bw BB)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 13.8 | 455 |
Visualisation
C.16. Jämsä et al. (2001) – Tibial ash weight
Data description
The endpoint to be analysed is: tibial ash weight.
Data used for analysis:
| Dose body burden (ng/kg bw) | Ash weight (mg) | SD | N |
|---|---|---|---|
| 0 | 232 | 8 | 5 |
| 28 | 226 | 3 | 5 |
| 171 | 213 | 9 | 5 |
| 904 | 189 | 13 | 5 |
bw: body weight.
Results
Response variable: tibial ash weight
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 36.16 | 5 | −62.32 |
| null‐ | Yes | 19.99 | 2 | −35.98 |
| Expon. m3‐ | Yes | 36.12 | 4 | −64.24 |
| Expon. m5‐ | Yes | 36.16 | 5 | −62.32 |
| Hill m3‐ | Yes | 36.13 | 4 | −64.26 |
| Hill m5‐ | Yes | 36.16 | 5 | −62.32 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.001581
estimate for a‐ : 232.2
estimate for CED‐ : 69.07
estimate for d‐ : 0.5462
HILL
estimate for var‐ : 0.001579
estimate for a‐ : 232.2
estimate for CED‐ : 70.32
estimate for d‐ : 0.5809
Final BMD Values (ng/kg bw BB)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 15.9 | 210 | 69.07 |
| Hill m3‐ | 16.9 | 209 | 70.32 |
Lowest BMDL and highest BMDU Values (ng/kg bw BB)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 15.9 | 210 |
Visualisation
C.17. Jämsä et al. (2001) – Plasma ALP activity in Long‐Evans rats
Data description
The endpoint to be analysed is: ALP.
Data used for analysis:
| Dose body burden (ng/kg bw) | ALP | SD | N |
|---|---|---|---|
| 0 | 151 | 13 | 5 |
| 28 | 132 | 8 | 5 |
| 171 | 190 | 23 | 5 |
| 904 | 305 | 17 | 5 |
bw: body weight; ALP: alkaline phosphatase.
Selection of the BMR
A BMR of 10% was selected deviating from the default BMR of 5% recommended for continuous data (EFSA Scientific Committee, 2017a,b). The selection of the 10% BMR was justified by the nature of the endpoint and variability observed in the controls and treatment group. The BMR of 10% corresponds approximately to the SD of the control group.
Results
Response variable: ALP
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | 23.22 | 5 | −36.44 |
| null‐ | Yes | −6.06 | 2 | 16.12 |
| Expon. m3‐ | Yes | 15.95 | 4 | −23.90 |
| Expon. m5‐ | Yes | 19.97 | 5 | −29.94 |
| Hill m3‐ | Yes | 15.29 | 4 | −22.58 |
| Hill m5‐ | Yes | 19.97 | 5 | −29.94 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.007948
estimate for a‐ : 140.8
estimate for CED‐ : 122.1
estimate for c‐ : 2.163
estimate for d‐ : 4
HILL
estimate for var‐ : 0.00795
estimate for a‐ : 140.8
estimate for CED‐ : 118.2
estimate for c‐ : 2.167
estimate for d‐ : 4
Final BMD Values (ng/kg bw BB)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m5‐ | 63.2 | 136 | 122.1 |
| Hill m5‐ | 61.9 | 134 | 118.2 |
Lowest BMDL and highest BMDU Values (ng/kg bw BB)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 61.9 | 136 |
Visualisation
Advanced Plots
REMARK: The AIC of the best model (minimum AIC) is more than two units larger than that of the full model. The results of the modelling show that this is not the most sensitive parameter of the study and therefore no additional actions were taken. The modelling of ALP was not further considered in the opinion.
C.18. Li et al. (2006) – Embryo loss day 1–3
Data description
The endpoint to be analysed is: mean embryo loss day 1–3
Data used for analysis:
| Dose (ng/kg bw per day) | Mean embryo loss (implantation sites) | SE | N |
|---|---|---|---|
| 0 | 10.8 | 1.4 | 10 |
| 2 | 11.4 | 2.2 | 10 |
| 50 | 8.2 | 2.0 | 10 |
| 100 | 2.0 | 1.3 | 10 |
bw: body weight.
Selection of the BMR
BMR (benchmark response) of 10% was selected deviating from the default BMR of 5% recommended for continuous data (EFSA Scientific Committee, 2017a,b). The selection of the 10% BMR was justified by the nature of the endpoint and variability observed in the controls and treatment group. The Panel noted that the SD in the control group is around 40% of the mean value. In view of the nature of the endpoint it was decided not to select the control group SD as BMR.
Results
Response variable: Mean embryo loss
Fitted Models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| full | Yes | −45.97 | 5 | 101.94 |
| null‐ | Yes | −66.03 | 2 | 136.06 |
| Expon. m3‐ | Yes | −45.98 | 4 | 99.96 |
| Expon. m5‐ | Yes | −45.98 | 5 | 101.96 |
| Hill m3‐ | Yes | −46.02 | 4 | 100.04 |
| Hill m5‐ | Yes | −46.02 | 5 | 102.04 |
Estimated Model Parameters
EXP
estimate for var‐ : 0.5833
estimate for a‐ : 9.862
estimate for CED‐ : 29.03
estimate for d‐ : 2.535
HILL
estimate for var‐ : 0.5845
estimate for a‐ : 10.03
estimate for CED‐ : 32.32
estimate for d‐ : 4
Final BMD Values (ng/kg bw per day)
| Model | BMDL | BMDU | BMD |
|---|---|---|---|
| Expon. m3‐ | 11.4 | 51.5 | 29.03 |
| Hill m3‐ | 20.3 | 37.1 | 32.32 |
Lowest BMDL and highest BMDU Values (ng/kg bw per day)
| Subgroup | bmdl.lowest | bmdu.highest |
|---|---|---|
| All | 11.4 | 51.5 |
Visualisation
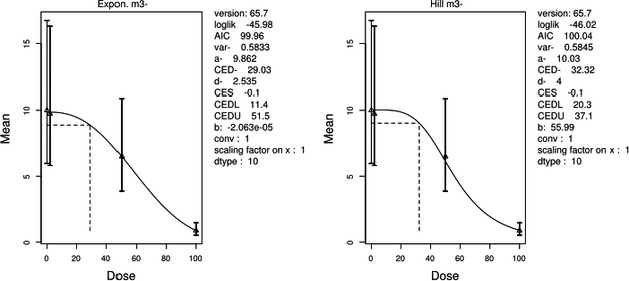
Appendix D – Estimated daily intake based on LOAEL/NOAEL body burdens observed in experimental animal studies
1.
As the serum concentrations in children in the human epidemiological studies cannot be directly compared with the body burden in dams from the rodent studies, the CONTAM Panel undertook an exercise to estimate the human daily intake corresponding to the observed critical reference points (expressed as body burdens) derived from animal studies after applying similar assessment factors as in the risk assessment carried out by the SCF (2001).
The lowest body burden in dams associated with adverse effects (LOAEL) was estimated to be 25 ng/kg bw observed in the study by Faqi et al. (1998) for effects on sperm concentrations in male offspring (see Section 3.1.7.3). Conducting a BMD analysis of the body burdens of the dams in this study did not result in a reliable BMDL (see Section 3.1.7.4). Applying an UF of 3 for extrapolating a LOAEL to a NOAEL would result in an estimated NOAEL body burden in the dams of 8.4 ng/kg bw.
As a conservative approach, the default UF of 3.2 for interindividual toxicokinetic differences is applied on the NOAEL body burden, rather than the corresponding EDI leading to this body burden. This results in a body burden of 2.6 ng/kg bw. CADM was used to estimate the EDI that would lead to this body burden in a pregnant woman after 35 years of exposure. This corresponded to a daily intake of 0.46 pg/kg bw per day,39 after being breastfed for 12 months with milk containing a level of 10 pg/g fat (which would be similar to the concentration in the adipose tissue after 35 years). The twofold higher exposure of a woman during childhood was not taken into account in these calculations, but this was shown to have a minor effect.40 The EDI of 0.46 pg/kg bw per day could then be extrapolated to an estimated weekly intake of about 3 pg/kg bw per week.
For effects on bone, the body burden at the lowest BMDL05 was estimated to be 14 ng/kg bw (see Table 13), based on the study by Jämsä et al. (2001). Applying a UF of 3.2 to account for interindividual differences in toxicokinetics amongst humans would give a body burden of 4.3 for this second most sensitive endpoint, which is 1.7‐fold higher than the 2.6 ng/kg bw derived for effects on semen quality in rats.
Appendix E – Berkeley Madonna modelling codes for the CADM model
1.
The model was developed by Carrier et al. (1995), optimised by Aylward et al. (2005), and further adapted by Ruiz et al. (2014). It was modified by the CONTAM Panel as indicated in Section 3.1.8.2).


Appendix F – Choice of weekly or monthly intake
1.
HBGVs are routinely set on a daily basis. However, for compounds where the accumulation over time results in levels that present the risk, EFSA decided to base them on a weekly basis. A monthly intake has been considered in previous risk assessments (JECFA, FAO/WHO, 2002). In this way, the occasional consumption of a higher contaminated food product can easily lead to exceedance of a tolerable daily but, e.g. not a weekly intake. The question is to what extent the period can be extended to a week, month or even longer without increasing the risk on adverse effects. In the case of PCDD/Fs and DL‐PCBs, a major problem is that it is unclear when, and how long, the critical windows for certain effects in humans occur. In rats, e.g. it has been established that exposure via the dams around GD15.5–17.5 leads to the effects on sperm parameters at a later stage in life. For boys, the timing and duration of the critical period for effects of PCDD/Fs on sperm is not known. In Mínguez‐Alarcón et al. (2017), a NOAEL of 7 pg WHO2005‐TEQ/g fat for PCDD/Fs was established based on serum levels in 9‐year‐old boys.
As shown by the modelling (Figure 14 of the opinion), serum levels increase during breastfeeding and then decline during the first 9 years of age. The issue is which single dose could increase the existing blood levels and as such lead to increased levels in sensitive tissues. Unfortunately, the currently available models were not designed to model this short‐term effect on the blood levels. In a worst‐case situation, it could be assumed that within hours after a meal, all the absorbed contaminants are concentrated in the blood, followed by gradual redistribution to the liver and body fat. As such, a single high intake could lead to a peak in the blood and it may take some time before the levels return to those before the meal. The daily intake can be compared with the total TEQ‐amount in the blood. Assuming a blood volume of 8% of the body weight, 55% serum and 5 g lipids per litre serum, the absolute amount in the blood before the meal was calculated. This was compared with the total ingested dose in one meal, assuming that 50% of the whole tolerable intake for 1 day, week or month would be present in a single meal. Serum levels, calculated for boys (age 1–9 years) breastfed for 12 months and then having an intake twofold higher than in adults were used. As an example, using TCDD:
At age 9, a body weight of 39 kg and thus an amount of blood of 3.1 L, results in a total of 3.1 L × 55% × 5 g/L = 8.6 g fat in the blood.
The level at age 9 years resulting from breastfeeding with milk containing 5.9 pg/g fat and daily intake of 0.5 pg/kg bw per day was estimated to be 7.0 pg/g fat (using CADM), corresponding to an overall amount in blood of 8.6 × 7 = 60 pg.
An intake of 0.5 pg/kg bw per day means 15 pg/kg bw per month, and 50% of this amount in one meal corresponds to 39 × 15 × 50% = 296 pg.
The total amount in blood could raise from 60 to maximal 356 pg, meaning a 356/60 = 5.9‐fold increase.
As shown in Figure F.1, the intake of 0.5 pg/kg bw via one meal would lead to an absolute amount at the peak of maximal 1.2‐fold of the amount present in the blood. Assuming that half of the whole tolerable weekly intake is present in one meal could lead to an intake of 0.5 × 39 × 7 × 50% = 68 pg or maximal 2.1‐fold the absolute amount in blood, i.e. a potential doubling of the serum level. For the monthly situation, this would be 5.9‐fold as shown by the example. The largest fold‐increase is calculated for age 9 and is caused by the decrease in the serum level over this period. Nevertheless, for the monthly scenario, already at the age of 2 years the amount ingested is about twofold higher than the amount in the blood.
Figure F.1.
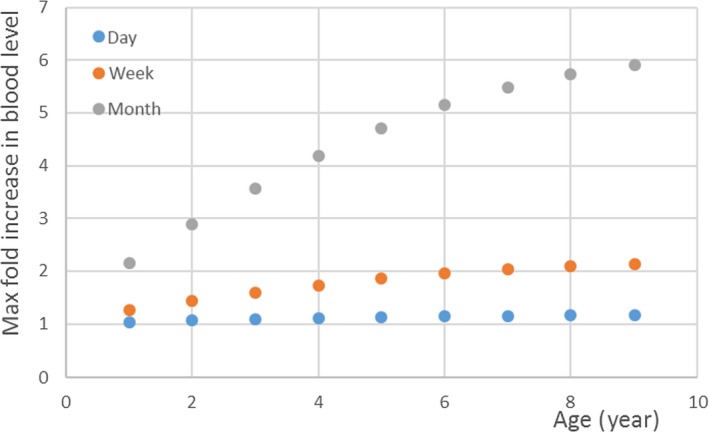
Comparison of peak exposure (absolute amount) for boys with existing absolute amount of TEQs, assuming that 50% of the whole daily, weekly or monthly dose is ingested in one occasion
The CONTAM Panel is aware that this probably presents a worst‐case situation since the distribution to the liver and body fat will probably dampen the effect. Therefore, setting the HBGV on a weekly base was judged to be acceptable, but extending it to a month may be interpreted as accepting a high temporary increases of serum levels, and as such, an increased exposure of sensitive tissues during a critical window. Further studies and improvement of PBK models are required to get more insight in the effect of a single high dose on blood levels and the potential risk of extending the period over which the HBGV could apply to a month or longer.
Annex A – Additional information for the risk assessment for human and animal health related to the presence of dioxins and DL‐PCBs in food and feed
1.
Annex A can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2018.5333
Description: The annex is a word file which presents information for the risk assessment for human and animal health related to the presence of dioxins and DL‐PCBs in food and feed.
Annex B – Occurrence data in food and feed submitted to EFSA and dietary exposure assessment for humans
1.
Annex B can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2018.5333
Description: The annex is an excel file which presents tables on PCDD/Fs and DL‐PCBs on occurrence data in food and feed and dietary exposure assessment for humans.
Supporting information
Additional information for the risk assessment for human and animal health related to the presence of dioxins and DL‐PCBs in food and feed
Occurrence data in food and feed submitted to EFSA and dietary exposure assessment for humans
Suggested citation: EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) , Knutsen HK, Alexander J, Barregård L, Bignami M, Brüschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hogstrand C, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Fürst P, Håkansson H, Halldorsson T, Lundebye A‐K, Pohjanvirta R, Rylander L, Smith A, van Loveren H, Waalkens‐Berendsen I, Zeilmaker M, Binaglia M, Gómez Ruiz JÁ, Horváth Z, Christoph E, Ciccolallo L, Ramos Bordajandi L, Steinkellner H and Hoogenboom LR, 2018. Scientific Opinion on the risk for animal and human health related to the presence of dioxins and dioxin‐like PCBs in feed and food. EFSA Journal 2018;16(11):5333, 331 pp. 10.2903/j.efsa.2018.5333
Requestor: European Commission
Question number: EFSA‐Q‐2015‐00028
Panel members: Jan Alexander, Lars Barregård, Margherita Bignami, Beat Brüschweiler, Sandra Ceccatelli, Bruce Cottrill, Lutz Edler, Michael Dinovi, Bettina Grasl‐Kraupp, Christer Hogstrand, Laurentius (Ron) Hoogenboom, Helle Katrine Knutsen, Carlo Stefano Nebbia, Isabelle P Oswald, Annette Petersen, Martin Rose, Alain‐Claude Roudot, Tanja Schwerdtle, Christiane Vleminckx, Günter Vollmer and Heather Wallace.
Acknowledgements: EFSA wishes to thank the Working Group members: Manolis Kogevinas (until 14 September 2016), George Loizou (until 23 January 2017), and the hearing experts: Matteo Bonzini, Jane Burns, Claude Emond, Aleksander Giwercman, Russ Hauser, Lidia Mínguez‐Alarcón and Paolo Mocarelli, for the support provided to this scientific output. The CONTAM Panel acknowledges all European competent institutions and other stakeholders that provided occurrence data on PCDD/Fs and DL‐PCBs in food and feed, and supported the data collection for the Comprehensive European Food Consumption Database.
Amendment: An additional sheet has been inserted in Annex B so as to present the congener specific occurrence values for the 29 congeners in the samples reported in Table 2A. This addition does not materially affect the contents or outcome of this scientific output. To avoid confusion, the older version has been removed from the EFSA Journal.
Adopted: 14 June 2018
Reproduction of the images listed below is prohibited and permission must be sought directly from the copyright holder:
Figure 3: © Taylor and Francis; © Elsevier Ltd; Figures 7 and 8: © Elsevier Inc.
This publication is linked to the following EFSA Supporting Publications articles: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1136/full, http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1137/full, http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1374/full
Amended: 18 February 2019
Notes
Opinion of the Scientific Committee on Food on the risk assessment of dioxins and dioxin‐like PCBs in food. Update based in new scientific information available since the adoption of the SCF opinion of 22nd November 2000 (adopted on 30 May 2001). http://ec.europa.eu/food/fs/sc/scf/out90_en.pdf
WHO Technical Report series, 909. Evaluation of certain food additives and contaminants, p. 121–146. Available at http://whqlibdoc.who.int/trs/WHO_TRS_909.pdf
Available at: http://www.efsa.europa.eu/en/efsajournal/doc/911.pdf
OJ L 243, 24.9.1996, p. 31–35.
Commission Regulation (EU) 2017/644 of 5 April 2017 laying down methods of sampling and analysis for the control of levels of dioxins, dioxin‐like PCBs and non‐dioxin‐like PCBs in certain foodstuffs and repealing Regulation (EU) No 589/2014. OJ L 92, 6.4.2017, p. 9–34.
Commission Regulation (EU) 2017/771 of 3 May 2017 amending Regulation (EC) No 152/2009 as regards the methods for the determination of the levels of dioxins and polychlorinated biphenyls. OJ L 115, 4.5.2017, p. 22–42.
In literature, both terms ‘lipid’ and ‘fat’ are cited, generally without stating which fractions or constituents were actually measured. Therefore, in this opinion. both terms are considered synonymous, unless otherwise stated.
The total amount of a chemical in the body.
OJ L 37, 13.2.1993, p. 1–3.
OJ L 364, 20.12.2006, p. 5–24.
Commission Recommendation (EU) 2016/688 of 2 May 2016 on the monitoring and management of the presence of dioxins and PCBs in fish and fishery products from the Baltic región. OJ L 118, 4.5.2016, p. 16–23.
Commission Recommendation 2014/663/EU of 11 September 2014 amending the Annex to Recommendation 2013/711/EU on the reduction of the presence of dioxins, furans and PCBs in feed and food. OJ L 272, 13.9.2014, p. 17–18.
OJ L 140, 30.5.2002, p. 10.
PubMed, Entrez Global Query Cross‐Database Search System, National Center for Biotechnology Information (NCBI), National Library of Medicine (NLM), Department of the National Institutes of Health (NIH), United States Department of Health and Human Services. Available online: http://www.ncbi.nlm.nih.gov/pubmed/
Commission Regulation (EU) No 2017/1017 of 15 June 2017 amending Regulation (EU) 68/2013 on the Catalogue of feed materials. OJ L 159, p. 25–65.
Facts and figures in the common fisheries policy. Basic statistical data. 2018 Edition. European Union 2018.
Assumptions: VRL = 0.04; VRF = 0.07. Calculated: fraction of body burden residing in liver and adipose tissue = 0.88.
For plasma, in some studies, this applied for total lipid levels, based on total cholesterol, triglycerides and phospholipids (Patterson et al., 1988); other studies applied a correction factor when only total cholesterol and triglycerides were determined or based it on gravimetrically determined levels after hexane extraction (Iida et al., 1999).
Additonal information about details of these studies was obtained from Schramm et al., 2009.
Population in the Zone at the time of explosion (Needham et al., 1997/1998) and ha from Mocarelli (2001).
Di‐ to octa‐chlorinated PCBs determined by GC/MS. Individual congeners not specified.
TEF scheme: NATO/CCMS, 1988 and TEFs for no‐PCBs from Safe (1994); levels converted with WHO2005‐TEFs.
2,4‐dichloro‐phenoxyacetic acid.
2,4,5‐trichlorophenoxyacetic acid.
Sum of PCB‐18, ‐28, ‐52, ‐49, ‐44, ‐74, ‐66, ‐101, ‐99,‐ 87, ‐110, ‐118, ‐105, ‐151, ‐149, ‐146, ‐153, ‐138/158, ‐128, ‐167, ‐156, ‐157, ‐178, ‐187, ‐183, ‐177, ‐172, ‐180, ‐170, ‐189, ‐201, ‐196/203, ‐195, ‐194 and ‐206.
TEQs for PCDD/Fs were calculated with I‐TEFs, and TEQs for DL‐PCBs with the TEFs proposed by Ahlborg et al. (1994).
Sum of PCB‐18, ‐28, ‐52, ‐49, ‐44, ‐74, ‐66, ‐101, ‐99,‐ 87, ‐110, ‐118, ‐105, ‐151, ‐149, ‐146, ‐153, ‐138/158, ‐128, ‐167, ‐156, ‐157, ‐178, ‐187, ‐183, ‐177, ‐172, ‐180, ‐170, ‐189, ‐201, ‐196/203, ‐195, ‐194 and ‐206.
AUC = C × (1−e−ke × t) / ke, where C = Concentration in milk (pg/g fat), ke = 0.2877/year, t = duration of breastfeeding (years) (Alaluusua et al., 1996).
PCB‐77, ‐81, ‐126 and ‐169.
Commission Regulation of 2 December 2011 amending Regulation (EC) No 1881/2006 as regards maximum levels for dioxins, dioxin‐like PCBs and non‐dioxin‐like PCBs in foodstuffs. OJ L 320, 3.12.2011, p. 18–23.
As of 1 May 2018.
PCB‐TEQs calculated with the PCB‐TEFs by Ahlborg et al. (1994).
The authors stated that TEQ values were estimated using WHO1998‐TEFs in the period 2007–2012 and WHO2005‐TEFs for the period 2012–2014.
TEQs for PCDD/Fs were calculated with I‐TEFs and for the three DL‐PCBs with the TEFs proposed by Ahlborg et al. (1994).
Using CADM, a body burden of 25 ng/kg bw in humans (i.e. the LOAEL body burden in dams from the study by Faqi et al., 1998) would result from an EDI of 8.8 pg/kg bw per day (after being breastfed with milk containing 85.2 pg/g fat). When applying the UFs of 3 and 3.2, this would be translated to 0.92 pg/kg bw per day. When using the estimated NOAEL body burden of 8.4 ng/kg bw, the corresponding EDI would be 1.92 pg/kg bw per day (milk level of 32 pg/g fat) and after applying the UF of 3.2 translates to 0.6 pg/kg bw per day. These higher estimates that result when applying the UFs at a later stage are caused by the longer half‐life with lower body burdens.
In the model, it is possible to add an additional dose during a certain period and this can be used to double the exposure during childhood. However, in practice there will be a more gradual change in the exposure per kg bw, so not changing by 50% at the start of adolescence. This could be an important future improvement of the model.
References
- ‘t Mannetje A, Coakley J, Mueller JF, Harden F, Toms LM and Douwes J, 2012. Partitioning of persistent organic pollutants (POPs) between human serum and breast milk: a literature review. Chemosphere, 89, 911–918. [DOI] [PubMed] [Google Scholar]
- ‘t Mannetje A, Eng A, Walls C, Dryson E, Kogevinas M, Brooks C, McLean D, Cheng S, Smith AH and Pearce N, 2017. Sex ratio of the offspring of New Zealand phenoxy herbicide producers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Occupational and Environmental Medicine, 74, 24–29. [DOI] [PubMed] [Google Scholar]
- Ábalos M, Parera J, Estevez A, Sole M, Fabregat MC, Pina B, Navarro A and Abad E, 2011. Decontamination trends in the aquacultured fish gilthead seabream (Sparus aurata) after feeding long‐term a PCDD/F spiked feed. Chemosphere, 82, 64–71. [DOI] [PubMed] [Google Scholar]
- Abnet CC, Tanguay RL, Heideman W and Peterson RE, 1999. Transactivation activity of human, zebrafish, and rainbow trout aryl hydrocarbon receptors expressed in COS‐7 cells: greater insight into species differences in toxic potency of polychlorinated dibenzo‐p‐dioxin, dibenzofuran, and biphenyl congeners. Toxicology and Applied Pharmacology, 159, 41–51. [DOI] [PubMed] [Google Scholar]
- Abraham K, Krowke R and Neubert D, 1988. Pharmacokinetics and biological activity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. 1. Dose‐dependent tissue distribution and induction of hepatic ethoxyresorufin O‐deethylase in rats following a single injection. Archives of Toxicology, 62, 359–368. [DOI] [PubMed] [Google Scholar]
- Abraham K, Wiesmüller T, Brunner H, Krowke R, Hagenmaier H and Neubert D, 1989. Elimination of various polychlorinated dibenzo‐p‐dioxins and dibenzofurans (PCDDs and PCDFs) in rat faeces. Archives Toxicology, 63, 75–78. [DOI] [PubMed] [Google Scholar]
- Abraham K, Knoll A, Ende M, Papke O and Helge H, 1996. Intake, fecal excretion, and body burden of polychlorinated dibenzo‐p‐dioxins and dibenzofurans in breast‐fed and formula‐fed infants. Pediatric Research, 40, 671–679. [DOI] [PubMed] [Google Scholar]
- Adachi J, Mori Y, Matsui S, Takigami H, Fujino J, Kitagawa H, Miller CA III, Kato T, Saeki K and Matsuda T, 2001. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. The Journal of Biological Chemistry, 276, 31475–31478. [DOI] [PubMed] [Google Scholar]
- Adamse P, Van der Fels‐Klerx HJ, Schoss S, de Jong J and Hoogenboom RLAP, 2015. Concentrations of dioxins and dioxin‐like PCBs in feed materials in the Netherlands, 2001‐11. Food Additives and Contaminants Part a‐Chemistry Analysis Control Exposure and Risk Assessment, 32, 1301–1311. [DOI] [PubMed] [Google Scholar]
- Adamse P, Schoss S, Theelen RMC and Hoogenboom RLAP, 2017. Levels of dioxins and dioxin‐like PCBs in food of animal origin in the Netherlands during the period 2001‐2011. Food Additives and Contaminants Part a‐Chemistry Analysis Control Exposure & Risk Assessment, 34, 78–92. [DOI] [PubMed] [Google Scholar]
- Adolphs J, Kleinjung F, Numata J, Mielke H, Abraham K, Schafft H, Müller‐Graf C and Greiner M, 2013. A probabilistic model for the carry‐over of PCDD/Fs from feed to growing pigs. Chemosphere, 93, 474–479. [DOI] [PubMed] [Google Scholar]
- Ahlborg UG, 1988. Nordisk dioxinriskbedömning (Nordic risk assessment of dioxins). Stockholm: National Institute of Environmental Medicine (with English summary) Available online: https://books.google.it/books?id=AXuHM1E6JIoC%26pg=PA1%26hl=it%26source=gbs_selected_pages%26cad=2#v=onepage%26q%26f=false [Google Scholar]
- Ahlborg UG, Becking GC, Birnbaum LS, Brouwer A, Derks H, Feeley M, Golor G, Hanberg A, Larsen JC, Liem AKD, Safe SH, Schlatter C, Waern F, Younes M and Yrjanheikki E, 1994. Toxic equivalency factors for dioxin‐like PCBs. Report on a WHO‐ECEH and IPCS Consultation, December 1993. Chemosphere, 28, 1049–1067. [Google Scholar]
- Ahlborg UG, Alexander J, Darnerud PO, Dybing E, Hanberg A, Johansson N, Madsen C, Rappe C, Utne Skåre J, Tuomisto J and Wicklund‐Glynn ???, 1995. Proceedings from the Nordic Meeting on USEPA Dioxin reevaluation. Available online: http://norden.diva-portal.org/smash/get/diva2:702032/FULLTEXT01.pdf
- Akhtar FZ, Garabrant DH, Ketchum NS and Michalek JE, 2004. Cancer in US Air Force veterans of the Vietnam War. Journal of Occupational and Environmental Medicine, 46, 123–136. [DOI] [PubMed] [Google Scholar]
- Alaluusua S and Lukinmaa PL, 2006. Developmental dental toxicity of dioxin and related compounds ‐ a review. International Dental Journal, 56, 323–331. [DOI] [PubMed] [Google Scholar]
- Alaluusua S, Lukinmaa PL, Pohjanvirta R, Unkila M and Tuomisto J, 1993. Exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin leads to defective dentin formation and pulpal perforation in rat incisor tooth. Toxicology, 81, 1–13. [DOI] [PubMed] [Google Scholar]
- Alaluusua S, Lukinmaa PL, Vartiainen T, Partanen M, Torppa J and Tuomisto J, 1996. Polychlorinated dibenzo‐p‐dioxins and dibenzofurans via mother's milk may cause developmental defects in the child's teeth. Environmental Toxicology and Pharmacology, 1, 193–197. [DOI] [PubMed] [Google Scholar]
- Alaluusua S, Lukinmaa PL, Torppa J, Tuomisto J and Vartiainen T, 1999. Developing teeth as biomarker of dioxin exposure. Lancet, 353, 206–206. [DOI] [PubMed] [Google Scholar]
- Alaluusua S, Kiviranta H, Leppaniemi A, Holtta P, Lukinmaa PL, Lope L, Jarvenpaa AL, Renlund M, Toppari J, Virtanen H, Kaleva M and Vartiainen T, 2002. Natal and neonatal teeth in relation to environmental toxicants. Pediatric Research, 52, 652–655. [DOI] [PubMed] [Google Scholar]
- Alaluusua S, Calderara P, Gerthoux PM, Lukinmaa PL, Kovero O, Needham L, Patterson DG, Tuomisto J and Mocareili P, 2004. Developmental dental aberrations after the dioxin accident in Seveso. Environmental Health Perspectives, 112, 1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander EK, 2017. 2017 Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and the postpartum (vol 27, pg 315, 2017). Thyroid, 27, 1212–1212. [DOI] [PubMed] [Google Scholar]
- Alonso KR, Peden‐Adams MM, Liu JY, Charbonneau C, Henshel D and Dickerson RL, 1998. Effects of in ovo exposure to 2,3,7,8‐TCDD on F1 generation adult chickens (Gallus gallus). Chemosphere, 37, 1873–1883. [DOI] [PubMed] [Google Scholar]
- Andersen ME, Mills JJ, Gargas ML, Kedderis L, Birnbaum LS, Neubert D and Greenlee WF, 1993. Modeling receptor‐mediated processes with dioxin: implications for pharmocokinetics and risk assessment. Risk Analysis, 13, 25–36. [DOI] [PubMed] [Google Scholar]
- Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A and Poellinger L, 2002. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proceedings of the National Academy of Sciences of the United States of America, 99, 9990–9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson P, Ridderstad A, McGuire J, Pettersson S, Poellinger L and Hanberg A, 2003. A constitutively active aryl hydrocarbon receptor causes loss of peritoneal B1 cells. Biochemical and Biophysical Research Communications, 302, 336–341. [DOI] [PubMed] [Google Scholar]
- Andersson P, Rubio C, Poellinger L and Hanberg A, 2005. Gastric hamartomatous tumours in a transgenic mouse model expressing an activated dioxin/Ah receptor. Anticancer Research, 25, 903–911. [PubMed] [Google Scholar]
- Andersson P, Brunnberg S, Wejheden C, Poellinger L and Hanberg A, 2012. Transgenic mice with a constitutively active AHR: a model for human exposure to dioxin and other AHR ligands In: Pohjanvirta R. (ed.). The AH receptor in biology and toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 373–385. [Google Scholar]
- ANSES (French Agency for Food, Environmental and Occupational Health and Safety), 2011. Second French Total Diet Study (TDS 2) Report 1 Inorganic contaminants, minerals, persistent organic pollutants, mycotoxins and phytoestrogens. ANSES Opinion.
- Antignac JP, Main KM, Virtanen HE, Boquien CY, Marchand P, Venisseau A, Guiffard I, Bichon E, Wohlfahrt‐Veje C, Legrand A, Boscher C, Skakkebæk NE, Toppari J and Le Bizec B, 2016. Country‐specific chemical signatures of persistent organic pollutants (POPs) in breast milk of French, Danish and Finnish women. Environmental Pollution, 218, 728–738. [DOI] [PubMed] [Google Scholar]
- Anwar‐Mohamed A and El‐Kadi AOS, 2012. Modulation of AHR function by heavy metals and disease states In: Pohjanvirta R. (ed.). The AH receptor in biology and toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 343–372. [Google Scholar]
- Aragon AC, Kopf PG, Campen MJ, Huwe JK and Walker MK, 2008. In utero and lactational 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin exposure: effects on fetal and adult cardiac gene expression and adult cardiac and renal morphology. Toxicological Sciences, 101, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima A, Kato H, Ooshima Y, Tateishi T, Inoue A, Muneoka A, Ihara T, Kamimura S, Fukusato T, Kubota S, Sumida H and Yasuda M, 2009. In utero and lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) induces a reduction in epididymal and ejaculated sperm number in rhesus monkeys. Reproductive Toxicology, 28, 495–502. [DOI] [PubMed] [Google Scholar]
- Arima A, Kato H, Ise R, Ooshima Y, Inoue A, Muneoka A, Kamimura S, Fukusato T, Kubota S, Sumida H and Yasuda M, 2010. In utero and lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) induces disruption of glands of the prostate and fibrosis in rhesus monkeys. Reproductive Toxicology, 29, 317–322. [DOI] [PubMed] [Google Scholar]
- Arpiainen S, Raffalli‐Mathieu F, Lang MA, Pelkonen O and Hakkola J, 2005. Regulation of the Cyp2a5 gene involves an aryl hydrocarbon receptor‐dependent pathway. Molecular Pharmacology, 67, 1325–1333. [DOI] [PubMed] [Google Scholar]
- Asselin C and Gendron FP, 2014. Shuttling of information between the mucosal and luminal environment drives intestinal homeostasis. FEBS Letters, 588, 4148–4157. [DOI] [PubMed] [Google Scholar]
- ATSDR (Agency for Toxic Substances and Disease Registry), 1994. Toxicological profile for chlorinated dibenzofurans (CDFs). Agency for Toxic Substances and Disease Registry, Atlanta, GA. May 1994.
- ATSDR (Agency for Toxic Substances and Disease Registry), 1998. Toxicological profile for chlorinated dibenzo‐para‐dioxins. U.S. Department of Health and Human Services. Public Health Service. Agency for Toxic Substances and Disease Registry. December 1998.
- ATSDR (Agency for Toxic Substances and Disease Registry), 2012. Addendum to the toxicological profile for chlorinated dibenzo‐p‐dioxins (CDDs). Agency for Toxic Substances and Disease Registry, Atlanta, GA. November 2012.
- Aulerich RJ, Bursian SJ, Breslin WJ, Olson BA and Ringer RK, 1985. Toxicological manifestations of 2,4,5‐hexachlorobiphenyl, 2’,4’,5’‐hexachlorobiphenyl, 2,3,6,2’,3’,6’‐hexachlorobiphenyl, and 3,4,5,3’,4’,5’‐hexachlorobiphenyl and Aroclor 1254 in mink. Journal of Toxicology and Environmental Health, 15, 63–79. [DOI] [PubMed] [Google Scholar]
- Aulerich RJ, Bursian SJ, Evans MG, Hochstein JR, Koudele KA, Olson BA and Napolitano AC, 1987. Toxicity of 3,4,5,3’,4’,5’‐hexachlorobiphenyl to mink. Archives of Environmental Contamination and Toxicology, 16, 53–60. [DOI] [PubMed] [Google Scholar]
- Aulerich RJ, Bursian SJ and Napolitano AC, 1988. Biological effects of epidermal growth‐factor and 2,3,7,8‐tetrachlorodibenzo‐para‐dioxin on developmental parameters of neonatal mink. Archives of Environmental Contamination and Toxicology, 17, 27–31. [DOI] [PubMed] [Google Scholar]
- Aylward LL and Hays SM, 2002. Temporal trends in human TCDD body burden: decreases over three decades and implications for exposure levels. Journal of Exposure Analysis and Environmental Epidemiology, 12, 319–328. [DOI] [PubMed] [Google Scholar]
- Aylward LL, Brunet RC, Carrier G, Hays SM, Cushing CA, Needham LL, Patterson DG, Gerthoux PM, Brambilla P and Mocarelli P, 2005. Concentration‐dependent TCDD elimination kinetics in humans: toxicokinetic modeling for moderately to highly exposed adults from Seveso, Italy, and Vienna, Austria, and impact on dose estimates for the NIOSH cohort. Journal of Exposure Analysis and Environmental Epidemiology, 15, 51–65. [DOI] [PubMed] [Google Scholar]
- Aylward LL, Collins JJ, Bodner KM, Wilken M and Bodnar CM, 2013. Elimination rates of dioxin congeners in former chlorophenol workers from midland, Michigan. Environmental Health Perspectives, 121, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward LL, Hays SM, Kirman CR, Marchitti SA, Kenneke JF, English C, Mattison DR and Becker RA, 2014. Relationships of chemical concentrations in maternal and cord blood: a review of available data. Journal of Toxicology and Environmental Health, Part B, 17, 175–203. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Mocarelli P, Patterson DG, Bonzini M, Pesatori AC, Caporaso N and Landi MT, 2002. Immunologic effects of dioxin: new results from Seveso and comparison with other studies. Environmental Health Perspectives, 110, 1169–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A, Pesatori AC, Masten SA, Patterson DG, Needham LL, Mocarelli P, Caporaso NE, Consonni D, Grassman JA, Bertazzi PA and Landi MT, 2004. Aryl‐hydrocarbon receptor‐dependent pathway and toxic effects of TCDD in humans: a population‐based study in Seveso, Italy. Toxicology Letters, 149, 287–293. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Pesatori AC, Consonni D, Mocarelli P, Patterson DG, Caporaso NE, Bertazzi PA and Landi MT, 2005. Health status and plasma dioxin levels in chloracne cases 20 years after the Seveso, Italy accident. British Journal of Dermatology, 152, 459–465. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Giacomini SM, Corbetta C, Landi MT, Bonzini M, Consonni D, Grillo P, Patterson DG Jr, Pesatori AC and Bertazzi PA, 2008. Neonatal thyroid function in Seveso 25 years after maternal exposure to dioxin. PLoS Med, 5, e161 10.1371/journal.pmed.0050161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacsi SG and Hankinson O, 1996. Functional characterization of DNA‐binding domains of the subunits of the heterodimeric aryl hydrocarbon receptor complex imputing novel and canonical basic helix‐loop‐helix protein‐DNA interactions. The Journal of Biological Chemistry, 271, 8843–8850. [DOI] [PubMed] [Google Scholar]
- Bajanowski T, Fürst P, Wilmers K, Beike J, Köhler H and Brinkmann B, 2002. Dioxin in infants – an environmental hazard? International Journal of Legal Medicine, 116, 27–32. [DOI] [PubMed] [Google Scholar]
- Baker TR, King‐Heiden TC, Peterson RE and Heideman W, 2014. Dioxin induction of transgenerational inheritance of disease in zebrafish. Molecular and Cellular Endocrinology, 398, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MG, Marks GT, Rawlins RG and Hutz RJ, 2015. Very low‐dose (femtomolar) 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) disrupts steroidogenic enzyme mRNAs and steroid secretion by human luteinizing granulosa cells. Reproductive Toxicology, 52, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M, Päpke O and Lis A, 1990. Polychlordibenzodioxine und Polychlordibenzofurane in Cigarettenrauch. Beitrage zur Tabakforschung International, 14, 393–402. [Google Scholar]
- Barrett DH, Morris RD, Akhtar FZ and Michalek JE, 2001. Serum dioxin and cognitive functioning among veterans of Operation Ranch Hand. Neurotoxicology, 22, 491–502. [DOI] [PubMed] [Google Scholar]
- Baughman RW, 1974. Tetrachloro‐p‐dioxins in the environment: high resolution mass spectrometry at the picogram level. PhD Thesis, Harvard University. [Google Scholar]
- Beatty PW, Vaughn WK and Neal RA, 1978. Effect of alteration of rat hepatic mixed‐function oxidase (MFO) activity on toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Toxicology and Applied Pharmacology, 45, 513–519. [DOI] [PubMed] [Google Scholar]
- Becher H, Steindorf K and Flesch‐Janys D, 1988. Quantitative cancer risk assessment for dioxins using an occupational cohort. Environmental Health Perspectives, 106, 663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H and Mathar W, 1985. Analysenverfahren zur Bestimmung von ausgewählten PCB‐Einzelkomponenten in Lebensmitteln. Bundesgesundhbl, 28, 1–12. [Google Scholar]
- Beck H, Eckart K, Mathar W and Wittkowski R, 1989. Levels of PCDDs and PCDFs in adipose‐tissue of occupationally exposed workers. Chemosphere, 18, 507–516. [Google Scholar]
- Beck H, Dross A, Kleemann WJ and Mathar W, 1990. PCDD and PCDF concentrations in different organs from infants. Chemosphere, 20, 903–910. [Google Scholar]
- Beck H, Dross A and Mathar W, 1994. PCDD and PCDF exposure and levels in humans in Germany. Environmental Health Perspectives, 102(Suppl 1), 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker RA, Patlewicz G, Simon TW, Rowlands JC and Budinsky RA, 2015. The adverse outcome pathway for rodent liver tumor promotion by sustained activation of the aryl hydrocarbon receptor. Regulatory Toxicology and Pharmacology, 73, 172–190. [DOI] [PubMed] [Google Scholar]
- Beckett KJ, Yamini B and Bursian SJ, 2008. The effects of 3,3’,4,4’,5‐pentachlorobiphenyl (PCB 126) on mink (Mustela vison) reproduction and kit survivability and growth. Archives of Environmental Contamination and Toxicology, 54, 123–129. [DOI] [PubMed] [Google Scholar]
- Beedanagari SR, Taylor RT, Bui P, Wang F, Nickerson DW and Hankinson O, 2010a. Role of epigenetic mechanisms in differential regulation of the dioxin‐inducible human CYP1A1 and CYP1B1 genes. Molecular Pharmacology, 78, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beedanagari SR, Taylor RT and Hankinson O, 2010b. Differential regulation of the dioxin‐induced Cyp1a1 and Cyp1b1 genes in mouse hepatoma and fibroblast cell lines. Toxicology Letters, 194, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JG, McGhee F, Dick JR and Tocher DR, 2005. Dioxin and dixon‐like polychlorinated biphenyls (PCBS) in Scottish farmed salmon (Salmo salar): effects of replacement of dietary marine fish oil vegetable oils. Aquaculture, 243, 305–314. [Google Scholar]
- Bell DR, Clode S, Fan MQ, Fernandes A, Foster PMD, Jiang T, Loizou G, MacNicoll A, Miller BG, Rose M, Tran L and White S, 2007a. Relationships between tissue levels of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD), mRNAs, and toxicity in the developing male Wistar(Han) rat. Toxicological Sciences, 99, 591–604. [DOI] [PubMed] [Google Scholar]
- Bell DR, Clode S, Fan MQ, Fernandes A, Foster PMD, Jiang T, Loizou G, MacNicoll A, Miller BG, Rose M, Tran L and White S, 2007b. Toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the developing male Wistar(Han) rat. I: no decrease in epididymal sperm count after a single acute dose. Toxicological Sciences, 99, 214–223. [DOI] [PubMed] [Google Scholar]
- Bell DR, Clode S, Fan MQ, Fernandes A, Foster PMD, Jiang T, Loizou G, MacNicoll A, Miller BG, Rose M, Tran L and White S, 2007c. Toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the developing male Wistar(Han) rat. II: chronic dosing causes developmental delay. Toxicological Sciences, 99, 224–233. [DOI] [PubMed] [Google Scholar]
- Bell JG, Dick JR, Strachan F, Guy DR, Berntssen MHG and Sprague M, 2012. Complete replacement of fish oil with a blend of vegetable oils affects dioxin, dioxin‐like polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs) in 3 Atlantic salmon (Salmo salar) families differing in flesh adiposity. Aquaculture, 324, 118–126. [Google Scholar]
- Bellehumeur K, Lapointe D, Cooke SJ and Moon TW, 2016. Exposure to sublethal levels of PCB‐126 impacts fuel metabolism and swimming performance in rainbow trout. Comparative Biochemistry and Physiology B‐Biochemistry and Molecular Biology, 199, 97–104. [DOI] [PubMed] [Google Scholar]
- Benedetto A, Brizio P, Guaraldo P, Stella C, Cappa C, Baioni E, Spalenza V, Nebbia C and Abete MC, 2016. Dioxins, DL‐PCB and NDL‐PCB accumulation profiles in livers from sheep and cattle reared in North‐western Italy. Chemosphere, 152, 92–98. [DOI] [PubMed] [Google Scholar]
- Benson JM and Shepherd DM, 2011. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn's disease. Toxicological Sciences, 120, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg V, Lyche JL, Gutleb AC, Lie E, Skaare JU, Aleksandersen M and Ropstad E, 2010. Distribution of PCB 118 and PCB 153 and hydroxylated PCB metabolites (OH‐CBs) in maternal, fetal and lamb tissues of sheep exposed during gestation and lactation. Chemosphere, 80, 1144–1150. [DOI] [PubMed] [Google Scholar]
- Berge JA, Hylland K, Schlabach M and Ruus A, 2011. Accumulation of polychlorinated dibenzo‐p‐dioxins and furans in Atlantic Cod (Gadus morhua) cage experiments in a Norwegian Fjord. Journal of Toxicology and Environmental Health‐Part a‐Current Issues, 74, 455–465. [DOI] [PubMed] [Google Scholar]
- Bernard A, Hermans C, Broeckaert F, de Poorter G, de Cock A and Hoins G, 1999. Food contamination by PCBs and dioxins; an isolated episode in Belgium is unlikely to have affected public health. Nature, 401, 231–232. [DOI] [PubMed] [Google Scholar]
- Bernard A, Broeckaert F, De Poorter G, De Cock A, Hermans C, Saegerman C and Houins G, 2002. The Belgian PCB/dioxin incident: analysis of the food chain contamination and health risk evaluation. Environmental Reserach, 88, 1–18. [DOI] [PubMed] [Google Scholar]
- Bernert JT, Turner WE, Patterson DG and Needham LL, 2007. Calculation of serum “total lipid” concentrations for the adjustment of persistent organohalogen toxicant measurements in human samples. Chemosphere, 68, 824–831. [DOI] [PubMed] [Google Scholar]
- Berntssen MH, Giskegjerde TA, Rosenlund G, Torstensen BE and Lundebye AK, 2007. Predicting World Health Organization toxic equivalency factor dioxin and dioxin‐like polychlorinated biphenyl levels in farmed Atlantic salmon (Salmo salar) based on known levels in feed. Environmental Toxicology and Chemistry, 26, 13–23. [DOI] [PubMed] [Google Scholar]
- Berntssen MHG, Julshamn K and Lundebye A‐K, 2010a. Chemical contaminants in aquafeeds and Atlantic salmon (Salmo salar) following the use of traditional‐ versus alternative feed ingredients. Chemosphere, 78, 637–646. [DOI] [PubMed] [Google Scholar]
- Berntssen MHG, Olsvik PA, Torstensen BE, Julshamn K, Midtun T, Goksoyr A, Johansen J, Sigholt T, Joerum N, Jakobsen JV, Lundebye AK and Lock EJ, 2010b. Reducing persistent organic pollutants while maintaining long chain omega‐3 fatty acid in farmed Atlantic salmon using decontaminated fish oils for an entire production cycle. Chemosphere, 81, 242–252. [DOI] [PubMed] [Google Scholar]
- Berntssen MH, Maage A, Julshamn K, Oeye BE and Lundebye AK, 2011. Carry‐over of dietary organochlorine pesticides, PCDD/Fs, PCBs, and brominated flame retardants to Atlantic salmon (Salmo salar L.) fillets. Chemosphere, 83, 95–103. [DOI] [PubMed] [Google Scholar]
- Berntssen MH, Sanden M, Hove H and Lie Ø, 2016. Modelling scenarios on feed‐to‐fillet transfer of dioxins and dioxin‐like PCBs in future feeds to farmed Atlantic salmon (Salmo salar). Chemosphere, 163, 413–421. [DOI] [PubMed] [Google Scholar]
- Bertazzi PA, 1991. Long‐term effects of chemical disasters‐ lessons and results from Seveso. Science of the Total Environment, 106, 5–20. [DOI] [PubMed] [Google Scholar]
- Bertazzi PA, Bernucci I, Brambilla G, Consonni D and Pesatori AC, 1998. The Seveso studies on early and long‐term effects of dioxin exposure: a review. Environmental Health Perspectives, 106, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertazzi PA, Consonni D, Bachetti S, Rubagotti M, Baccarelli A, Zocchetti C and Pesatori AC, 2001. Health effects of dioxin exposure: a 20‐year mortality study. American Journal of Epidemiology, 153, 1031–1044. [DOI] [PubMed] [Google Scholar]
- Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EM, Macchiarulo A, Vacca C, Iannitti R, Tissi L, Volpi C, Belladonna ML, Orabona C, Bianchi R, Lanz TV, Platten M, Della Fazia MA, Piobbico D, Zelante T, Funakoshi H, Nakamura T, Gilot D, Denison MS, Guillemin GJ, DuHadaway JB, Prendergast GC, Metz R, Geffard M, Boon L, Pirro M, Iorio A, Veyret B, Romani L, Grohmann U, Fallarino F and Puccetti P, 2014. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature, 511, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BfR (Bundesinstitut für Risikobewertung), 2011. Frauenmilch: dioxingehalte sinken kontinuierlich ‐ Information Nr. 011/2011 des BfR vom 23.03.2011. Available online: http://www.bfr.bund.de/cm/343/frauenmilch_dioxingehalte_sinken_kontinuierlich.pdf
- Bhavsar SP, Gewurtz SB, Helm PA, Labencki TL, Marvin CH, Fletcher R, Hayton A, Reiner EJ and Boyd D, 2010. Estimating sediment quality thresholds to prevent restrictions on fish consumption: application to polychlorinated biphenyls and dioxins‐furans in the Canadian great lakes. Integrated Environmental Assessment and Management, 6, 641–652. [DOI] [PubMed] [Google Scholar]
- Binh ND, Oanh NTK and Parkpian P, 2014. Photodegradation of dioxin in contaminated soil in the presence of solvents and nanoscale TiO 2 particles. Environmental Technology, 35, 1121–1132. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, Decad GM and Matthews HB, 1980. Disposition and excretion of 2378‐tertachlorodibenzofuran in the rat. Toxicology and Applied Pharmacology, 55, 342–352. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, Decad GM, Matthews HB and McConnell EE, 1981. Fate of 2,3,7,8‐tetrachlorodibenzofuran in the monkey. Toxicology and Applied Pharmacology, 57, 189–196. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS and Couture LA, 1988. Disposition of octachlorodibenzo‐p‐dioxin (OCDD) in male rats. Toxicology and Applied Pharmacology, 93, 22–30. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, McDonald MM, Blair PC, Clark AM and Harris MW, 1990. Differential toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in C57BL/6J mice congenic at the Ah Locus. Fundamental and Applied Toxicology: Official Journal of the Society of Toxicology, 15, 186–200. [DOI] [PubMed] [Google Scholar]
- Bjerke DL and Peterson RE, 1994. Reproductive toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in male rats: different effects of in utero versus lactational exposure. Toxicology and Applied Pharmacology, 127, 241–249. [DOI] [PubMed] [Google Scholar]
- Black MB, Budinsky RA, Dombkowski A, Cukovic D, LeCluyse EL, Ferguson SS, Thomas RS and Rowlands JC, 2012. Cross‐species comparisons of transcriptomic alterations in human and rat primary hepatocytes exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicological Sciences, 127, 199–215. [DOI] [PubMed] [Google Scholar]
- Blanco SL, Sobrado C, Quintela C, Cabaleiro S, Gonzalez JC and Vieites JM, 2007. Dietary uptake of dioxins (PCDD/PCDFs) and dioxin‐like PCBs in Spanish aquacultured turbot (Psetta maxima). Food Additives and Contaminants, 24, 421–428. [DOI] [PubMed] [Google Scholar]
- Bleavins MR and Aulerich RJ, 1981. Feed consumption and food passage time in mink (Mustela vison) and European ferrets (Mustela putorius furo). Laboratory Animal Science, 31, 268–269. [PubMed] [Google Scholar]
- Bloom M, Vena J, Olson J and Moysich K, 2006. Chronic exposure to dioxin‐like compounds and thyroid function among New York anglers. Environmental Toxicology and Pharmacology, 21, 260–267. [DOI] [PubMed] [Google Scholar]
- BMUB (German Federal Ministry for the Environment), 2005. Waste Incineration — A Potential Danger? Federal Ministry for the Environment, Nature Conservation and Nuclear Safety. Available online: http://www.bmub.bund.de/fileadmin/bmu-import/files/english/pdf/application/pdf/muellverbrennung_dioxin_en.pdf
- Bock KW and Kohle C, 2006. Ah receptor: dioxin‐mediated toxic responses as hints to deregulated physiologic functions. Biochemical Pharmacology, 72, 393–404. [DOI] [PubMed] [Google Scholar]
- Bock KW, 2016. Toward elucidation of dioxin‐mediated chloracne and Ah receptor functions. Biochemical Pharmacology, 112, 1–5. [DOI] [PubMed] [Google Scholar]
- Boers D, Portengen L, Bueno‐de‐Mesquita HB, Heederik D and Vermeulen R, 2010. Cause‐specific mortality of Dutch chlorophenoxy herbicide manufacturing workers. Occupational and Environmental Medicine, 67, 24–31. [DOI] [PubMed] [Google Scholar]
- Boers D, Portengen L, Turner WE, Bueno‐de‐Mesquita HB, Heederik D and Vermeulen R, 2012. Plasma dioxin levels and cause‐specific mortality in an occupational cohort of workers exposed to chlorophenoxy herbicides, chlorophenols and contaminants. Occupational and Environmental Medicine, 69, 113–118. [DOI] [PubMed] [Google Scholar]
- Boffetta P, Mundt KA, Adami HO, Cole P and Mandel JS, 2011. TCDD and cancer: a critical review of epidemiologic studies. Critical Reviews in Toxicology, 41, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boily MH, Ndayibagira A and Spear PA, 2003a. Retinoids, LRAT and REH activities in eggs of Japanese quail following maternal and in ovo exposures to 3,3’,4,4’‐tetrachlorobiphenyl. Ecotoxicology, 12, 9–21. [DOI] [PubMed] [Google Scholar]
- Boily MH, Ndayibagira A and Spear PA, 2003b. Retinoid metabolism (LRAT, REH) in the yolk‐sac membrane of Japanese quail eggs and effects of mono‐ortho‐PCBs. Comparative Biochemistry and Physiology C‐Toxicology and Pharmacology, 134, 11–23. [DOI] [PubMed] [Google Scholar]
- Boutros PC, Yan R, Moffat ID, Pohjanvirta R and Okey AB, 2008. Transcriptomic responses to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in liver: comparison of rat and mouse. BMC Genomics, 9, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boverhof DR, Burgoon LD, Tashiro C, Sharratt B, Chittim B, Harkema JR, Mendrick DL and Zacharewski TR, 2006. Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicological Sciences: An Official Journal of the Society of Toxicology, 94, 398–416. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Dellatte E, Fochi I, Iacovella N, Miniero R and di Domenico A, 2007. Depletion of selected polychlorinated biphenyl, dibenzodioxin, and dibenzofuran congeners in farmed rainbow trout (Oncorhynchus mykiss): a hint for safer fish farming. Chemosphere, 66, 1019–1030. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Fochi I, Falce M, De Filippis SP, Ubaldi A and di Domenico A, 2008. PCDD and PCDF depletion in milk from dairy cows according to the herd metabolic scenario. Chemosphere, 73, S216–S219. [DOI] [PubMed] [Google Scholar]
- Brambilla G, Fochi I, De Filippis SP, Iacovella N and di Domenico A, 2009. Pentachlorophenol, polychlorodibenzodioxin and polychlorodibenzofuran in eggs from hens exposed to contaminated wood shavings. Food Additives and Contaminants, 26, 258–264. [DOI] [PubMed] [Google Scholar]
- Brambilla G, De Filippis SP, Iamiceli AL, Iacovella N, Abate V, Aronica V, Di Marco V and di Domenico A, 2011. Bioaccumulation of dioxin‐like substances and selected brominated flame retardant congeners in the fat and livers of black pigs farmed within the Nebrodi Regional Park of Sicily. Journal of Food Protection, 74, 261–269. [DOI] [PubMed] [Google Scholar]
- Bramwell L, Mortimer D, Rose M, Fernandes A, Harrad S and Pless‐Mulloli T, 2017. UK dietary exposure to PCDD/Fs, PCBs, PBDD/Fs, PBBs and PBDEs: comparison of results from 24‐h duplicate diets and total diet studies. Food Additives and Contaminants Part a‐Chemistry Analysis Control Exposure and Risk Assessment, 34, 65–77. [DOI] [PubMed] [Google Scholar]
- Branson DR, Takahashi IT, Parker WM and Blau GE, 1985. Bioconcentration kinetics of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in rainbow‐trout. Environmental Toxicology and Chemistry, 4, 779–788. [Google Scholar]
- Brantsæter AL, Haugen M, Alexander J and Meltzer HM, 2008. Validity of a new food frequency questionnaire for pregnant women in the Norwegian Mother and Child Cohort Study (MoBa). Maternal and Child Nutrition, 4, 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster DW and Birnbaum LS, 1987. Disposition and excretion of 2,3,4,7,8‐pentachlorodibenzofuran in the rat. Toxicology and Applied Pharmacology, 90, 243–252. [DOI] [PubMed] [Google Scholar]
- Brewster DW and Birnbaum LS, 1988. Disposition of 1,2,3,7,8‐pentachlorodibenzofuran in the rat. Toxicology and Applied Pharmacology, 95, 490–498. [DOI] [PubMed] [Google Scholar]
- Brouwer A, Klasson‐Wehler E, Bokdam M, Morse DC and Traag WA, 1990. Competitive inhibition of thyroxin binding to transthyretin by monohydroxy metabolites of 3,4,3’,4’‐tetrachlorobiphenyl. Chemosphere, 20, 1257–62. TK [Google Scholar]
- Brown SB, Fisk AT, Brown M, Villella M, Muir DCG, Evans RE, Lockhart WL, Metner DA and Cooley HM, 2002. Dietary accumulation and biochemical responses of juvenile rainbow trout (Oncorhynchus mykiss) to 3,3’,4,4’,5‐pentachlorobiphenyl (PCB 126). Aquatic Toxicology, 59, 139–152. [DOI] [PubMed] [Google Scholar]
- Bruggeman V, Swennen Q, De Ketelaere B, Onagbesan O, Tona K and Decuypere E, 2003. Embryonic exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in chickens: effects of dose and embryonic stage on hatchability and growth. Comparative Biochemistry and Physiology C‐Toxicology and Pharmacology, 136, 17–28. [DOI] [PubMed] [Google Scholar]
- Bruggeman B, Onagbesan O, Dumez L, De Ketelaere B and Decuypere E, 2005. Effects of early prenatal exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) on postnatal reproduction in the laying hen (Gallus gallus). Comparative Biochemistry and Physiology C‐Toxicology and Pharmacology, 141, 349–355. [DOI] [PubMed] [Google Scholar]
- Bruner‐Tran KL and Osteen ???, 2011. Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reproductive Toxicology, 31, 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunnberg S, Andersson P, Lindstam M, Paulson I, Poellinger L and Hanberg A, 2006. The constitutively active Ah receptor (CA‐Ahr) mouse as a potential model for dioxin exposure–effects in vital organs. Toxicology, 224, 191–201. [DOI] [PubMed] [Google Scholar]
- Brunström B and Lund J, 1988. Differences between chick and turkey embryos in sensitivity to 3,3’,4,4’‐tetrachloro‐biphenyl and in concentration/affinity of the hepatic receptor for 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Comparative Biochemistry and Physiology C, 91, 507–512. [DOI] [PubMed] [Google Scholar]
- Brunström B, 1989. Toxicity of coplanar polychlorinated‐biphenyls in avian embryos. Chemosphere, 19, 765–768. [Google Scholar]
- Bruns‐Weller E, Knoll A and Heberer T, 2010. High levels of polychlorinated dibenzodioxins/furans and dioxin like‐PCBs found in monitoring investigations of sheep liver samples from Lower Saxony, Germany. Chemosphere, 78, 653–658. [DOI] [PubMed] [Google Scholar]
- Budinsky RA, Paustenbach D, Fontaine D, Landenberger B and Starr TB, 2006. Recommended relative potency factors for 2,3,4,7,8‐pentachlorodibenzofuran: the impact of different dose metrics. Toxicological Sciences, 91, 275–285. [DOI] [PubMed] [Google Scholar]
- Budinsky RA, Schrenk D, Simon T, Van den Berg M, Reichard JF, Silkworth JB, Aylward LL, Brix A, Gasiewicz T, Kaminski N, Perdew G, Starr TB, Walker NJ and Rowlands JC, 2014. Mode of action and dose‐response framework analysis for receptor‐mediated toxicity: the aryl hydrocarbon receptor as a case study. Critical Reviews in Toxicology, 44, 83–119. [DOI] [PubMed] [Google Scholar]
- Buffler PA, Ginevan ME, Mandel JS and Watkins DK, 2011. The air force health study: an epidemiologic retrospective. Annals of Epidemiology, 21, 673–687. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC and Bradfield CA, 2003. Resistance to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. The Journal of Biological Chemistry, 278, 17767–17774. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Glover E, Moran SM, Walisser JA, Lahvis GP, Hsu EL and Bradfield CA, 2008. Abnormal liver development and resistance to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin toxicity in mice carrying a mutation in the DNA‐binding domain of the aryl hydrocarbon receptor. Toxicological Sciences, 106, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach KM, Poland A and Bradfield CA, 1992. Cloning of the Ah‐receptor cDNA reveals a distinctive ligand‐activated transcription factor. Proceedings of the National Academy of Sciences of the United States of America, 89, 8185–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burka LT, McGown SR and Tomer KB, 1991. Identification of the biliary metabolites of 2,3,7,8‐tetrachlorodibenzofuran in the rat. Chemosphere, 21, 1231–1242. [Google Scholar]
- Burns JS, Williams PL, Sergeyev O, Korrick S, Lee MM, Revich B, Altshul L, Patterson DG Jr, Turner WE, Needham LL, Saharov I and Hauser R, 2009. Predictors of serum dioxins and PCBs among peripubertal Russian Boys. Environmental Health Perspectives, 117, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JS, Williams PL, Sergeyev O, Korrick S, Lee MM, Revich B, Altshul L, Del Prato JT, Humblet O, Patterson DG Jr, Turner WE, Needham LL, Starovoytov M and Hauser R, 2011. Serum dioxins and polychlorinated biphenyls are associated with growth among Russian boys. Pediatrics, 127, E59–E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JS, Lee MM, Williams PL, Korrick SA, Sergeyev O, Lam T, Revich B and Hauser R, 2016. Associations of peripubertal serum dioxin and polychlorinated biphenyl concentrations with pubertal timing among Russian boys. Environmental Health Perspectives, 124, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursian SJ, Beckett KJ, Yamini B, Martin PA, Kannan K, Shields KL and Mohr FC, 2006a. Assessment of effects in mink caused by consumption of carp collected from the Saginaw River, Michigan, USA. Archives of Environmental Contamination and Toxicology, 50, 614–623. [DOI] [PubMed] [Google Scholar]
- Bursian SJ, Sharma C, Aulerich RJ, Yamini B, Mitchell RR, Beckett KJ, Orazio CE, Moore D, Svirsky S and Tillitt DE, 2006b. Dietary exposure of mink (Mustela vison) to fish from the Housatonic River, Berkshire County, Massachusetts, USA: effects on organ weights and histology and hepatic concentrations of polychlorinated biphenyls and 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin toxic equivalence. Environmental Toxicology and Chemistry, 25, 1541–1550. [DOI] [PubMed] [Google Scholar]
- Bursian SJ, Sharma C, Aulerich RJ, Yamini B, Mitchell RR, Orazio CE, Moore DRJ, Svirsky S and Tillitt DE, 2006c. Dietary exposure of mink (Mustela vison) to fish from the Housatonic River, Berkshire County, Massachusetts, USA: effects on reproduction, kit growth, and survival. Environmental Toxicology and Chemistry, 25, 1533–1540. [DOI] [PubMed] [Google Scholar]
- Bursian SJ, Moore J, Newsted JL, Link JE, Fitzgerald SD, Bello N, Bhat VS, Kay D, Zhang XW, Wiseman S, Budinsky RA, Giesy JP and Zwiernik MJ, 2012. Incidence of jaw lesions and activity and gene expression of hepatic P4501A enzymes in mink (Mustela vison) exposed to dietary 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, 2,3,7,8‐tetrachlorodibenzofuran, and 2,3,4,7,8‐pentachlorodibenzofuran. Environmental Toxicology and Chemistry, 31, 2545–255. [DOI] [PubMed] [Google Scholar]
- Bursian SJ, Kern J, Remington RE, Link JE and Fitzgerald SD, 2013a. Dietary exposure of mink (Mustela vison) to fish from the upper Hudson River, New York, USA: effects on reproduction and offspring growth and mortality. Environmental Toxicology and Chemistry, 32, 780–793. [DOI] [PubMed] [Google Scholar]
- Bursian SJ, Kern J, Remington RE, Link JE and Fitzgerald SD, 2013b. Dietary exposure of mink (Mustela vison) to fish from the upper Hudson River, New York, USA: effects on organ mass and pathology. Environmental Toxicology and Chemistry, 32, 794–801. [DOI] [PubMed] [Google Scholar]
- Burton JE, Michalek JE and Rahe AJ, 1998. Serum dioxin, chloracne, and acne in veterans of operation ranch hand. Archives of Environmental Health, 53, 199–204. [DOI] [PubMed] [Google Scholar]
- Cai LY, Izumi S, Suzuki T, Goya K, Nakamura E, Sugiyama T and Kobayashi H, 2011. Dioxins in ascites and serum of women with endometriosis: a pilot study. Human Reproduction, 26, 117–126. [DOI] [PubMed] [Google Scholar]
- Calvert GM, Wall DK, Sweeney MH and Fingerhut MA, 1998. Evaluation of cardiovascular outcomes among US workers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Environmental Health Perspectives, 106, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert GM, Sweeney MH, Deddens J and Wall DK, 1999. Evaluation of diabetes mellitus, serum glucose, and thyroid function among United States workers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Occupational and Environmental Medicine, 56, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho IA, Singh N, Hegde VL, Nagarkatti M and Nagarkatti PS, 2005. Treatment of mice with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin leads to aryl hydrocarbon receptor‐dependent nuclear translocation of NF‐kappa B and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells. Journal of Immunology, 175, 90–103. [DOI] [PubMed] [Google Scholar]
- Cao Y, Winneke G, Wilhelm M, Wittsiepe J, Lemm F, Fuerst P, Ranft U, Imoehl M, Kraftg M, Oesch‐Bartlomowicz B and Kraemer U, 2008. Environmental exposure to dioxins and polychlorinated biphenyls reduce levels of gonadal hormones in newborns: results from the Duisburg cohort study. International Journal of Hygiene and Environmental Health, 211, 30–39. [DOI] [PubMed] [Google Scholar]
- Carabano R and Piquer J, 1998. The digestive system of the rabbit In: de Blas C. and Wiseman J. (eds.). The Nutrition of the Rabbit. CABI Publishing, Oxfordshire: pp. 1–16. [Google Scholar]
- Carrier G, Brunet RC and Brodeus J, 1995. Modelling of the toxicokinetics of polychlorinated dibenzo‐p‐dioxins and dibenzofurans in mammalians, including humans .1. Nonlinear distribution of PCDD/PCDF body burden between liver and adipose tissues. [DOI] [PubMed]
- Carter CD, Kimbrough RD, Liddle JA, Cline RE, Zack MM Jr, Barthel WF, Koehler RE and Phillips PE, 1975. Tetrachlorodibenzodioxin: an accidental poisoning episode in horse arenas. Science, 188, 738–740. [DOI] [PubMed] [Google Scholar]
- Carvalho PSM and Tillitt DE, 2004. 2,3,7,8‐TCDD effects on visual structure and function in swim‐up rainbow trout. Environmental Science and Technology, 38, 6300–6306. [DOI] [PubMed] [Google Scholar]
- Carvalho PSM, Noltie DB and Tillitt DE, 2004. Intra‐strain dioxin sensitivity and morphometric effects in swim‐up rainbow trout (Oncorhynchus mykiss). Comparative Biochemistry and Physiology C‐Toxicology and Pharmacology, 137, 133–142. [DOI] [PubMed] [Google Scholar]
- Caspersen IH, Knutsen HK, Brantsaeter AL, Haugen M, Alexander J, Meltzer HM and Kvalem HE, 2013. Dietary exposure to dioxins and PCBs in a large cohort of pregnant women: results from the Norwegian Mother and Child Cohort Study (MoBa). Environmental International, 59, 398–407. [DOI] [PubMed] [Google Scholar]
- Caspersen IH, Haugen M, Schjolberg S, Vejrup K, Knutsen HK, Brantster AL, Meltzer HM, Alexander J, Magnus P and Kvalem HE, 2016a. Maternal dietary exposure to dioxins and polychlorinated biphenyls (PCBs) is associated with language delay in 3 year old Norwegian children. Environment International, 91, 180–187. [DOI] [PubMed] [Google Scholar]
- Caspersen IH, Aase H, Biele G, Brantsæter AL, Haugen M, Kvalem HE, Skogan AH, Zeiner P, Alexander J, Meltzer HM and Knutsen HK, 2016b. The influence of maternal dietary exposure to dioxins and PCBs during pregnancy on ADHD symptoms and cognitive functions in Norwegian preschool children. Environment International, 94, 649–660. [DOI] [PubMed] [Google Scholar]
- Cavaco I, Hombhanje FW, Gil JP and Kaneko A, 2013. Frequency of the functionally relevant aryl hydrocarbon receptor repressor (AhRR) Pro185Ala SNP in Papua New Guinea. Drug Metabolism and Pharmacokinetics, 28, 519–521. [DOI] [PubMed] [Google Scholar]
- Celius T and Matthews J, 2010. Functional analysis of six human aryl hydrocarbon receptor variants in human breast cancer and mouse hepatoma cell lines. Toxicology, 277, 59–65. [DOI] [PubMed] [Google Scholar]
- Chahoud I, Hartmann J, Rune GM and Neubert D, 1992. Reproductive toxicity and toxicokinetics of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. 3. Effects of single doses on the testis of male rats. Archives of Toxicology, 66, 567–72. [DOI] [PubMed] [Google Scholar]
- Chao HR, Wang SL, Lin LY, Lee WJ and Papke O, 2007. Placental transfer of polychlorinated dibenzo‐p‐dioxins, dibenzofurans, and biphenyls in Taiwanese mothers in relation, to menstrual cycle characteristics. Food and Chemical Toxicology, 45, 259–265. [DOI] [PubMed] [Google Scholar]
- Chapman DE and Schiller CM, 1985. Dose‐related effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in C57BL/6J and DBA/2J mice. Toxicology and Applied Pharmacology, 78, 147–157. [DOI] [PubMed] [Google Scholar]
- Chen PH, Wong CK, Rappe C and Nygren M, 1985. Polychlorinated‐biphenyls, dibenzofurans and quaterphenyls in toxic rice‐bran oil and in the blood and tissues of patients with PCB poisoning (Yu‐Cheng) in Taiwan. Environmental Health Perspectives, 59, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J‐W, Wang S‐L, Liao P‐C, Chen HY, Ko Y‐C and Lee C‐C, 2008. Relationship between insulin sensitivity and exposure to dioxins and polychlorinated biphenyls in pregnant women. Environmental Research, 107, 245–253. [DOI] [PubMed] [Google Scholar]
- Cheng H, Aylward L, Beall C, Starr TB, Brunet RC, Carrier G and Delzell E, 2006. TCDD exposure‐response analysis and risk assessment. Risk Analysis, 26, 1059–1071. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Jin UH, Allred CD, Jayaraman A, Chapkin RS and Safe S, 2015. Aryl hydrocarbon receptor activity of tryptophan metabolites in young adult mouse colonocytes. Drug Metabolism and Disposition, 43, 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier J, Warner M, Gunier RB, Brambilla P, Eskenazi B and Mocarelli P, 2014. Serum dioxin concentrations and thyroid hormone levels in the Seveso women's health study. American Journal of Epidemiology, 180, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigliano L, Nebbia C, Rychen G, Feidt C, Girolami F, Rossetti C and Spagnuolo MS, 2016. Evaluation of serum markers of blood redox homeostasis and inflammation in PCB naturally contaminated heifers undergoing decontamination. Science of the Total Environment, 542, 653–664. [DOI] [PubMed] [Google Scholar]
- Clarkson J and Herbison AE, 2006. Development of GABA and glutamate signaling at the GnRH neuron in relation to puberty. Molecular and Cellular Endocrinology, 254–255, 32–8. [DOI] [PubMed] [Google Scholar]
- Cohen‐Barnhouse AM, Zwiernik MJ, Link JE, Fitzgerald SD, Kennedy SW, Herve JC, Giesy JP, Wiseman S, Yang Y, Jones PD, Wan Y, Collins B, Newsted JL, Kay D and Bursian SJ, 2011a. Sensitivity of Japanese Quail (Coturnix japonica), Common Pheasant (Phasianus colchicus), and White Leghorn Chicken (Gallus gallus domesticus) Embryos to In Ovo Exposure to TCDD, PeCDF, and TCDF. Toxicological Sciences, 119, 93–103. [DOI] [PubMed] [Google Scholar]
- Cohen‐Barnhouse AM, Zwiernik MJ, Link JE, Fitzgerald SD, Kennedy SW, Giesy JP, Wiseman S, Jones PD, Newsted JL, Kay D and Bursian SJ, 2011b. Developmental and posthatch effects of in ovo exposure to 2,3,7,8‐TCDD, 2,3,4,7,8‐PECDF, and 2,3,7,8‐TCDF in Japanese quail (Coturnix japonica), common pheasant (Phasianus colchicus), and white leghorn chicken (Gallus gallus domesticus) embryos. Environmental Toxicology and Chemistry, 30, 1659–1668. [DOI] [PubMed] [Google Scholar]
- Cok I, Donmez MK, Satiroglu MH, Aydinuraz B, Henkelmann B, Shen H, Kotalik J and Schramm KW, 2008. Concentrations of polychlorinated dibenzo‐p‐dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), and dioxin‐like PCBs in adipose tissue of infertile men. Archives of Environmental Contamination and Toxicology, 55, 143–152. [DOI] [PubMed] [Google Scholar]
- Collins JJ, Bodner K, Aylward LL, Wilken M, Swaen G, Budinsky R, Rowlands C and Bodnar CM, 2009a. Mortality rates among workers exposed to dioxins in the manufacture of pentachlorophenol. Journal of Occupational and Environmental Medicine, 51, 1212–1219. [DOI] [PubMed] [Google Scholar]
- Collins JJ, Bodner K, Aylward LL, Wilken M and Bodnar CM, 2009b. Mortality rates among trichlorophenol workers with exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. American Journal of Epidemiology, 170, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conney AH, Miller EC and Miller JA, 1957. Substrate‐induced synthesis and other properties of benzpyrene hydroxylase in rat liver. The Journal of Biological Chemistry, 228, 753–766. [PubMed] [Google Scholar]
- Connor KT and Aylward LL, 2006. Human response to dioxin: aryl hydrocarbon receptor (AhR) molecular structure, function, and dose‐response data for enzyme induction indicate an impaired human AhR. Journal of Toxicology and Environmental Health Part B, Critical Reviews, 9, 147–171. [DOI] [PubMed] [Google Scholar]
- Consonni D, Pesatori AC, Zocchetti C, Sindaco R, D'Oro LC, Rubagotti M and Bertazzi PA, 2008. Mortality in a population exposed to dioxin after the Seveso, Italy, accident in 1976: 25 years of follow‐up. American Journal of Epidemiology, 167, 847–858. [DOI] [PubMed] [Google Scholar]
- Consonni D, Sindaco R and Bertazzi PA, 2012. Blood levels of dioxins, furans, dioxin‐like PCBs, and TEQs in general populations: a review, 1989–2010. Environment International, 44, 151–162. [DOI] [PubMed] [Google Scholar]
- Coristine S, Haffner GD, Ciborowski JJH, Lazar R, Nanni ME and Metcalfe CD, 1996. Elimination rates of selected di‐ortho, mono‐ortho, and non‐ortho substituted polychlorinated biphenyls in rainbow trout (Oncorhynchus mykiss). Environmental Toxicology and Chemistry, 15, 1382–1387. [Google Scholar]
- Costera A, Feidt C, Marchand P, Le Bizec B and Rychen G, 2006. PCDD/F and PCB transfer to milk in goats exposed to a long‐term intake of contaminated hay. Chemosphere, 64, 650–657. [DOI] [PubMed] [Google Scholar]
- COT (Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment), 2001. Statement on the tolerable daily intake for dioxins and dioxin‐like polychlorinated biphenyls. COT/2001/07.
- Coumailleau P, Poellinger L, Gustafsson JA and Whitelaw ML, 1995. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. The Journal of Biological Chemistry, 270, 25291–25300. [DOI] [PubMed] [Google Scholar]
- Couture LA, Elwell MR and Birnbaum LS, 1988. Dioxin‐like effects observed in male rats following exposure to octachlorodibenzo‐p‐dioxin during a 13 week study. Toxicology and Applied Pharmacology, 93, 31–46. [DOI] [PubMed] [Google Scholar]
- Couture LA, Abbott BD and Birnbaum LS, 1990. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin: recent advances toward understanding the mechanism. Teratology, 42, 619–627. [DOI] [PubMed] [Google Scholar]
- Cranmer M, Louie S, Kennedy RH, Kern PA and Fonseca VA, 2000. Exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) is associated with hyperinsulinemia and insulin resistance. Toxicological Sciences, 56, 431–436. [DOI] [PubMed] [Google Scholar]
- Croes K, Den Hond E, Bruckers L, Loots I, Morrens B, Nelen V, Colles A, Schoeters G, Sioen I, Covaci A, Vandermarken T, Van Larebeke N and Baeyens W, 2014. Monitoring chlorinated persistent organic pollutants in adolescents in Flanders (Belgium): concentrations, trends and dose‐effect relationships (FLEHS II). Environment International, 71, 20–28. [DOI] [PubMed] [Google Scholar]
- Curtis LR, Kerkvliet NI, Baecher‐Steppan L and Carpenter HM, 1990. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin pretreatment of female mice altered tissue distribution but not hepatic metabolism of a subsequent dose. Fundamental and Applied Toxicology, 14, 523–531. [DOI] [PubMed] [Google Scholar]
- Cypel YS, Kress AM, Eber SM, Schneiderman AI, Davey VJ, 2016. Herbicide exposure, Vietnam service, and hypertension risk in army chemical corps veterans. Journal of Occupational and Environmental Medicine, 58, 1127–1136. [DOI] [PubMed] [Google Scholar]
- Dahl P, Lindström G, Wiberg K and Rappe C, 1995. Absorption of polychlorinated biphenyls, dibenzo‐p‐dioxins and dibenzofurans by breast fed children. Chemosphere, 30, 2297–2306. [DOI] [PubMed] [Google Scholar]
- Darnerud PO, Lignell S, Glynn A, Aune M, Tornkvist A and Stridsberg M, 2010. POP levels in breast milk and maternal serum and thyroid hormone levels in mother‐child pairs from Uppsala, Sweden. Environment International, 36, 180–187. [DOI] [PubMed] [Google Scholar]
- Davarinos NA and Pollenz RS, 1999. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. The Journal of Biological Chemistry, 274, 28708–28715. [DOI] [PubMed] [Google Scholar]
- Davies R, Clothier B, Robinson SW, Edwards RE, Greaves P, Luo J, Gant TW, Chernova T and Smith AG, 2008. Essential role of the AH receptor in the dysfunction of heme metabolism induced by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Chemical Research in Toxicology, 21, 330–340. [DOI] [PubMed] [Google Scholar]
- De Felip E, di Domenico A, Falleni M, Ferri F, Iacovella N, Menale G, Tafani P, Tommasino G and Turrio‐Baldassarri L, 1994. Polychlorodibenzodioxin and polychlorodibenzofuran levels in dielectric fluids containing polychlorobiphenyls. Toxicological and Environmental Chemistry, 46, 239–260. [Google Scholar]
- De Felip E, Porpora MG, di Domenico A, Ingelido AM, Cardelli M, Cosmi EV and Donnez J, 2004. Dioxin‐like compounds and endometriosis: a study on Italian and Belgian women of reproductive age. Toxicology Letters, 150, 203–209. [DOI] [PubMed] [Google Scholar]
- De Filippis SP, Chirollo C, Brambilla G, Anastasio A, Sarnelli P, De Felip E, di Domenico A, Iamiceli AL and Cortesi ???, 2013. Polychlorodibenzodioxin and ‐furan and dioxin‐like polychlorobiphenyl distribution in tissues and dairy products of dairy buffaloes. Journal of Agricultural and Food Chemistry, 61, 6552–6561. [DOI] [PubMed] [Google Scholar]
- De Heer C, Dewaal EJ, Schuurman HJ, Vos JG and Van Loveren H, 1994. The intrathymic target‐cell for the thymotoxic action of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Experimental and Clinical Immunogenetics, 11, 86–93. [DOI] [PubMed] [Google Scholar]
- De Roos AJ, Hartge P, Lubin JH, Colt JS, Davis S, Cerhan JR, Severson RK, Cozen W, Patterson DG Jr, Needham LL and Rothman N, 2005. Persistent organochlorine chemicals in plasma and risk of non‐Hodgkin's lymphoma. Cancer Reserach, 65, 11214–11226. [DOI] [PubMed] [Google Scholar]
- de Waard PW, Peijnenburg AA, Baykus H, Aarts JM, Hoogenboom RL, van Schooten FJ and de Kok TM, 2008. A human intervention study with foods containing natural Ah‐receptor agonists does not significantly show AhR‐mediated effects as measured in blood cells and urine. Chemico‐Biological Interactions, 176, 19–29. [DOI] [PubMed] [Google Scholar]
- Deb S and Bandiera SM, 2010. Characterization of a new cytochrome P450 enzyme, CYP2S1, in rats: its regulation by aryl hydrocarbon receptor agonists. Toxicology, 267, 91–98. [DOI] [PubMed] [Google Scholar]
- Decad GM, Birnbaum LS and Matthews HB, 1981a. Distribution and excretion of 2,3,7,8‐tetrachlorodibenzofuran in C57BL/6J and DBA/2J mice. Toxicology and Applied Pharmacology, 59, 564–73. [DOI] [PubMed] [Google Scholar]
- Decad GM, Birnbaum LS and Matthews HB, 1981b. 2,3,7,8‐Tetrachlorodibenzofuran tissue distribution and excretion in guinea pig. Toxicology and Applied Pharmacology, 57, 231–40. [DOI] [PubMed] [Google Scholar]
- DeCaprio AP, McMartin DM, O'Keefe PW, Rej R, Silkworth JB and Kaminsky LS, 1986. Subchronic oral toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the guinea pig: comparisons with a PCB‐containing transformer fluid pyrolysate. Fundamental and Applied Toxicology, 6, 454–463. As cited in ATSDR (1994). [DOI] [PubMed] [Google Scholar]
- DeGroot D, He G, Fraccalvieri D, Bonati L, Pandini A and Denison MS, 2012. AHR ligands: promiscuity in binding and diversity in response In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 63–79. [Google Scholar]
- DeGroot DE and Denison MS, 2014. Nucleotide specificity of DNA binding of the aryl hydrocarbon receptor: ARNT complex is unaffected by ligand structure. Toxicological Sciences, 137, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKrey GK, Teagarden RE, Lenberg JL and Titus RG, 2013. 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin slows the progression of experimental cutaneous leishmaniasis in susceptible BALB/c and SCID mice. PLOS ONE, 8, e76259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Río Gómez I, Marshall T, Tsai P, Shao T‐S and Guo YL, 2002. Number of boys born to men exposed to polychlorinated biphenyls. The Lancet ‐ Research Letters, 360, 143–144. [DOI] [PubMed] [Google Scholar]
- Delvaux I, Van Cauwenberghe J, Den Hond E, Schoeters G, Govarts E, Nelen V, Baeyens W, Van Larebeke N and Sioen I, 2014. Prenatal exposure to environmental contaminants and body composition at age 7‐9 years. Environmental Research, 132, 24–32. [DOI] [PubMed] [Google Scholar]
- Den Hond E, Roels HA, Hoppenbrouwers K, Nawrot T, Thijs L, Vandermeulen C, Winneke G, Vanderschueren D and Staessen JA, 2002. Sexual maturation in relation to polychlorinated aromatic hydrocarbons: Sharpe and Skakkebaek's hypothesis revisited. Environmental Health Perspectives, 110, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Hond E, Tournay H, De Sutter P, Ombelet W, Baeyens W, Covaci A, Cox B, Nawrot TS, Van Larebeke N and D'Hooghe T, 2015. Human exposure to endocrine disrupting chemicals and fertility: a case–control study in male subfertility patients. Environment International, 84, 154–160. [DOI] [PubMed] [Google Scholar]
- Denison MS, Fisher JM and Whitlock JP Jr, 1988. The DNA recognition site for the dioxin‐Ah receptor complex. Nucleotide sequence and functional analysis. The Journal of Biological Chemistry, 263, 17221–17224. [PubMed] [Google Scholar]
- Denison MS, Soshilov AA, He G, DeGroot DE and Zhao B, 2011. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicological Sciences, 124, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks HJGM, Berende PLM, Olling M, Everts H, Liem AKD and de Jong APJM, 1994. Pharmacokinetic modelling of polychlorinated dibenzo‐p‐dioxins (PCDDs) and furans (PCDFs) in cows. Chemosphere, 28, 711–715. [Google Scholar]
- Dhooge W, van Larebeke N, Koppen G, Nelen V, Schoeters G, Vlietinck R, Kaufman JM and Comhaire F, 2006. Serum dioxin‐like activity is associated with reproductive parameters in young men from the general Flemish population. Environmental Health Perspectives, 114, 1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhooge W, Den Hond E, Koppen G, Bruckers L, Nelen V, van de Mieroop E, Bilau M, Croes K, Baeyens W, Schoeters G and van Larebeke N, 2011. Internal exposure to pollutants and sex hormone levels in Flemish male adolescents in a cross‐sectional study: associations and dose‐response relationships. Journal of Exposure Science and Environmental Epidemiology, 21, 106–113. [DOI] [PubMed] [Google Scholar]
- Di Meo GP, Perucatti A, Genualdo V, Caputi‐Jambrenghi A, Rasero R, Nebbia C and Iannuzzi L, 2011. Chromosome fragility in dairy cows exposed to dioxins and dioxin‐like PCBs. Mutagenesis, 26, 269–272. [DOI] [PubMed] [Google Scholar]
- Dietrich C, 2012. The AHR in the control of cell cycle and apoptosis In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 467–484. [Google Scholar]
- Diletti G, Ceci R, Conte A, De Benedictis A, Migliorati G and Scortichini G, 2008. Milk contamination from dioxins in Italy: source identification and intervention strategies In: Mehmetli E and Koumanova B (eds.). The fate of persistent organic pollutants in the environment. Springer, Netherlands: pp. 301–314. [Google Scholar]
- Diliberto JJ, Akubue PI, Luebke RW and Birnbaum LS, 1995. Dose‐response relationships of tissue distribution and induction of CYP1A1 and CYP1A2 enzymatic activities following acute exposure to 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) in mice. Toxicology and Applied Pharmacology, 130, 197–208. [DOI] [PubMed] [Google Scholar]
- Diliberto JJ, Jackson JA and Birnbaum LS, 1996. Comparison of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) disposition following pulmonary, oral, dermal, and parenteral exposures to rats. Toxicology and Applied Pharmacology, 138, 158–168. [DOI] [PubMed] [Google Scholar]
- Diliberto JJ, Burgin DE and Birnbaum LS, 1999. Effects of CYP1A2 on disposition of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, 2,3,4,7,8‐pentachlorodibenzofuran, and 2,2’,4,4’,5,5’‐hexachlorobiphenyl in CYP1A2 knockout and parental (C57BL/6N and 129/Sv) strains of mice. Toxicology and Applied Pharmacology, 159, 52–64. [DOI] [PubMed] [Google Scholar]
- Diliberto JJ, DeVito MJ, Ross DG and Birnbaum LS, 2001. Subchronic exposure of [3H]‐2378‐Tetrachlorodibenzo‐p‐dioxin (TCDD) in female B6C3F1 mice: relationship of steady‐state levels to disposition and metabolism. Toxicological Sciences, 61, 241–255. [DOI] [PubMed] [Google Scholar]
- DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, Omiecinski CJ and Perdew GH, 2010. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin‐6 in the presence of inflammatory signaling. Toxicological Sciences, 115, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinse GE, Jusko TA, Whitt IZ, Co CA, Parks CG, Satoh M, Chan EKL, Rose KM, Walker NJ, Birnbaum LS, Zeldin DC, Weinberg CR and Miller FW, 2016. Associations between selected xenobiotics and antinuclear antibodies in the national health and nutrition examination survey, 1999–2004. Environmental Health Perspectives, 124, 426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering JA, Farmahin R, Wiseman S, Kennedy SW, Giesy JP and Hecker M, 2014. Functionality of aryl hydrocarbon receptors (AhR1 and AhR2) of white sturgeon (Acipenser transmontanus) and implications for the risk assessment of dioxin‐like compounds. Environmental Science and Technology, 48, 8219–8226. [DOI] [PubMed] [Google Scholar]
- Domingo JL, 2011. Influence of cooking processes on the concentrations of toxic metals and various organic environmental pollutants in food: a review of the published literature. Critical Reviews in Food Science and Nutrition, 51, 29–37. [DOI] [PubMed] [Google Scholar]
- Dragan YP and Schrenk D, 2000. Animal studies addressing the carcinogenicity of TCDD (or related compounds) with an emphasis on tumour promotion. Food Additives and Contaminants, 17, 289–302. [DOI] [PubMed] [Google Scholar]
- Dragin N, Dalton TP, Miller ML, Shertzer HG and Nebert DW, 2006. For dioxin‐induced birth defects, mouse or human CYP1A2 in maternal liver protects whereas mouse CYP1A1 and CYP1B1 are inconsequential. Journal of Biological Chemistry, 281, 18591–18600. [DOI] [PubMed] [Google Scholar]
- Drew MD, Ogunkoya AE, Janz DM and Van Kessel AG, 2007. Dietary influence of replacing fish meal and oil with canola protein concentrate and vegetable oils on growth performance, fatty acid composition and organochlorine residues in rainbow trout (Oncorhynchus mykiss). Aquaculture, 267, 260–268. [Google Scholar]
- Van Ede KI, van Duursen MBM and van den Berg M, 2016. Evaluation of relative effect potencies (REPs) for dioxin‐like compounds to derive systemic or human‐specific TEFs to improve human risk assessment. [DOI] [PMC free article] [PubMed]
- EFSA (European Food Safety Authority), 2005. Opinion of the Scientific Panel on Contaminants in the Food Chain on a request from the Commission related to the presence of non dioxin‐like polychlorinated biphenyls (PCB) in feed and food. (Question No EFSA‐Q‐2003‐114). Adopted on 8 November 2005. EFSA Journal 2005;3(11):284, 137 pp. 10.2903/j.efsa.2005.284 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2007. Guidance of the Scientific Committee on a request from EFSA related to Uncertainties in Dietary Exposure Assessment. EFSA Journal 2007;5(1):438, 54 pp. 10.2903/j.efsa.2007.438 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010a. Results of the monitoring of dioxin levels in food and feed. EFSA Journal 2010;8(3):1385, 36 pp. 10.2903/j.efsa.2010.1385. Available online: http://www.efsa.europa.eu [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010b. Standard sample description for food and feed. EFSA Journal 2010;8(1):1457, 54 pp. 10.2903/j.efsa.2010.1457. Available online: http://www.efsa.europa.eu [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2010c. Management of left‐censored data in dietary exposure assessment of chemical substances. EFSA Journal 2010;8(3):1557, 96 pp. 10.2903/j.efsa.2010.1557. Available online: http://www.efsa.europa.eu [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011a. Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097. Available online: http://www.efsa.europa.eu/efsajournal.htm [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011b. The food classification and description system FoodEx 2 (draft‐revision 1). EFSA Supporting Publications 2011;8(12):215, 438 pp. 10.2903/sp.efsa.2011.EN-215. Available online: http://www.efsa.europa.eu [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2012. Update of the monitoring of dioxins and PCBs levels in food and feed. EFSA Journal 2012;10(7):2832, 82 pp. 10.2903/j.efsa.2012.2832. Available online: http://www.efsa.europa.eu/efsajournal [DOI] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2012. Guidance for the preparation of dossiers for sensory additives. EFSA Journal 2012;10(1):2534, 26 pp. https://doi.org/10. 2903/j.efsa.2012.2534 [Google Scholar]
- EFSA (European Food Safety Authority), 2015. Scientific statement on the health‐based guidance value for dioxins and dioxin‐like PCBs. EFSA Journal 2015;13(5):4124, 14 pp. 10.2903/j.efsa.2015.4124 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2008. Statement of EFSA on the risks for public health due to the presence of dioxins in pork from Ireland. EFSA Journal 2008;6(12):911, 15 pp. 10.2903/j.efsa.2008.911 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2011. Scientific Opinion on the risk to public health related to the presence of high levels of dioxins and dioxin‐like PCBs in liver from sheep and deer. EFSA Journal 2011;9(7):2297, 71 pp. 10.2903/j.efsa.2011.2297. Available online: http://www.efsa.europa.eu/efsajournal [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), 2012. Scientific Opinion on the presence of dioxins (PCDD/Fs) and dioxin‐like PCBs (DL‐PCBs) in commercially available foods for infants and young children. EFSA Journal 2012;10(12):2983, 29 pp. 10.2903/j.efsa.2012.2983. Available online: http://www.efsa.europa.eu/efsajournal [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Knutsen HK, Alexander J, Barregard L, Bignami M, Bruschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hoogenboom LR, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Lundebye A‐K, Metzler M, Colombo P and Hogstrand C, 2017a. Scientific opinion on the assessment of a decontamination process for dioxins and dioxin‐like PCBs in fish oil by physical filtration with activated carbon. EFSA Journal 2017;15(7):4961, 10 pp. 10.2903/j.efsa.2017.4961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Knutsen HK, Alexander J, Barregard L, Bignami M, Bruschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hoogenboom LR, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Lundebye A‐K, Metzler M, Colombo P and Hogstrand C, 2017b. Scientific Opinion on the assessment of decontamination processes for dioxins and dioxin‐like PCBs in fish oil by physical filtration with activated carbon. EFSA Journal 2017;15(12):5081, 13 pp. 10.2903/j.efsa.2017.5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Knutsen HK, Alexander J, Barregard L, Bignami M, Bruschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hoogenboom LR, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Lundebye A‐K, Metzler M, Colombo P and Hogstrand C, 2018a. Scientific Opinion on the assessment of a decontamination process for dioxins and PCBs from fishmeal by replacement of fish oil. EFSA Journal 2018;16(2):5174, 10 pp. 10.2903/j.efsa. 2018.5174 [DOI] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain), Knutsen HK, Alexander J, Barregard L, Bignami M, Bruschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl‐Kraupp B, Hoogenboom LR, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A‐C, Schwerdtle T, Vleminckx C, Vollmer G, Wallace H, Lundebye A‐K, Metzler M, Colombo P and Hogstrand C, 2018b. Scientific Opinion on the assessment of a decontamination process for dioxins and PCBs from fish meal by hexane extraction and replacement of fish oil. EFSA Journal 2018;16(2):5173, 9 pp. https://doi.org/doi. org/10.2903/j.efsa.2018.5173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen KH, More S, Mortensen A, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Silano V, Solecki R, Turck D, Aerts M, Bodin L, Davis A, Edler L, Gundert‐Remy U, Sand S, Slob W, Bottex B, Abrahantes JC, Marques DC, Kass G and Schlatter JR, 2017a. Update: Guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen HK, More S, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Schlatter JR, Silano V, Solecki R, Turck D, Younes M, Bresson J‐L, Griffin J, Hougaard Benekou S, vanLoveren H , Luttik R, Messean A, Penninks A, Ru G, Stegeman JA, dervan Werf W , Westendorf J, Woutersen RA, Barizzone F, Bottex B, Lanzoni A, Georgiadis N and Alexander J, 2017b. Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA Journal 2017;15(8):4970, 73 pp. 10.2903/j.efsa.2017.4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emond C, Birnbaum LS and DeVito MJ, 2004. Physiologically based pharmacokinetic model for developmental exposures to TCDD in the rat. Toxicological Sciences, 80, 115–133. [DOI] [PubMed] [Google Scholar]
- Emond C, Michalek JE, Birnbaum LS and DeVito MJ, 2005. Comparison of the use of a physiologically based pharmacokinetic model for dioxin exposure assessments. Environmental Health Perspectives, 113, 1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emond C, Birnbaum LS and DeVito MJ, 2006. Use of a physiologically based pharmacokinetic model for rats to study the influence of body fat mass and induction of CYP1A2 on the pharmacokinetics of TCDD. Environmental Health Perspectives, 114, 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emond C, DeVito M, Warner M, Eskenazi B, Mocarelli P and Birnbaum LS, 2016. An assessment of dioxin exposure across gestation and lactation using a PBPK model and new data from Seveso. Environmental International, 92–93, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emond C, Ruiz P and Mumtaz M, 2017. Physiologically based pharmacokinetic toolkit to evaluate environmental exposures: applications of the dioxin model to study real life exposures. Toxicology and Applied Pharmacology, 315, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enan E, El‐Sabeawy F, Scott M, Overstreet J and Lasley B, 1998a. Alterations in the growth factor signal transduction pathways and modulators of the cell cycle in endocervical cells from macaques exposed to TCDD. Toxicology and Applied Pharmacology, 151, 283–293. [DOI] [PubMed] [Google Scholar]
- Enan E, El‐Sabeawy F, Overstreet J, Matsumura F and Lasley B, 1998b. Mechanisms of gender‐specific TCDD‐induced toxicity in guinea pig adipose tissue. Reproduction and Toxicology, 12, 357–369. [DOI] [PubMed] [Google Scholar]
- Ende M, 1986. Bericht über das Muttermilchuntersuchungsprogramm Niedersachsen 1986 Niedersächsisches Ärzteblatt 13/1986, 18‐19.
- Erickson MD, 1989. PCDFs and related compounds produced from PCB fires A review. Chemosphere, 19, 161–165. [Google Scholar]
- Eskenazi B, Mocarelli P, Warner M, Samuels S, Vercellini P, Olive D, Needham L, Patterson D and Brambilla P, 2000. Seveso Women's Health Study: a study of the effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on reproductive health. Chemosphere, 40, 1247–1253. [DOI] [PubMed] [Google Scholar]
- Eskenazi B, Mocarelli P, Warner M, Samuels S, Vercellini P, Olive D, Needham LL, Patterson DG, Brambilla P, Gavoni N, Casalini S, Panazza S, Turner W and Gerthoux PM, 2002a. Serum dioxin concentrations and endometriosis: a cohort study in Seveso, Italy. Environmental Health Perspectives, 110, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B, Warner M, Mocarelli P, Samuels S, Needham LL, Patterson DG, Lippman S, Vercellini P, Gerthoux PM, Brambilla P and Olive D, 2002b. Serum dioxin concentrations and menstrual cycle characteristics. American Journal of Epidemiology, 156, 383–392. [DOI] [PubMed] [Google Scholar]
- Eskenazi B, Mocarelli P, Warner M, Chee WY, Gerthoux PM, Samuels S, Needham LL and Patterson DG, 2003. Maternal serum dioxin levels and birth outcomes in Women of Seveso, Italy. Environmental Health Perspectives, 111, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B, Mocarelli P, Warner M, Needham L, Patterson DG, Samuels S, Turner W, Gerthoux PM and Brambilla P, 2004. Relationship of serum TCDD concentrations and age at exposure of female residents of Seveso, Italy. Environmental Health Perspectives, 112, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B, Warner M, Marks AR, Samuels S, Gerthoux PM, Vercellini P, Olive DL, Needham L, Patterson DG and Mocarelli P, 2005. Serum dioxin concentrations and age at menopause. Environmental Health Perspectives, 113, 858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B, Warner M, Samuels S, Young J, Gerthoux PM, Needham L, Patterson D, Olive D, Gavoni N, Vercellini P and Mocarelli P, 2007. Serum dioxin concentrations and risk of uterine leiomyoma in the Seveso Women's Health Study. American Journal of Epidemiology, 166, 79–87. [DOI] [PubMed] [Google Scholar]
- Eskenazi B, Warner M, Marks AR, Samuels S, Needham L, Brambilla P and Mocarelli P, 2010. Serum dioxin concentrations and time to pregnancy. Epidemiology, 21, 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskenazi B, Warner M, Sirtori M, Fuerst T, Rauch SA, Brambilla P, Mocarelli P and Rubinacci A, 2014. Serum dioxin concentrations and bone density and structure in the Seveso Women's Health Study. Environmental Health Perspectives, 122, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C, 2012. The physiological role of AHR in the mouse immune system In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 499–510. [Google Scholar]
- Esser C and Rannug A, 2015. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacological Reviews, 67, 259–279. [DOI] [PubMed] [Google Scholar]
- European Commission , 2001. Integrated Pollution Prevention and Control (IPPC) ‐ Reference Document on Best Available Techniques in the Non Ferrous Metals Industries ‐ December 2001. Available online: http://eippcb.jrc.ec.europa.eu/reference/BREF/nfm_bref_1201.pdf
- Fader KA, Nault R, Ammendolia DA, Harkema JR, Williams KJ, Crawford RB, Kaminski NE, Potter D, Sharratt B and Zacharewski TR, 2015. 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin alters lipid metabolism and depletes immune cell populations in the Jejunum of C57BL/6 Mice. Toxicological Sciences, 148, 567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallone F, Villard PH, Seree E, Rimet O, Nguyen QB, Bourgarel‐Rey W, Fouchier F, Barra Y, Durand A and Lacarelle B, 2004. Retinoids repress Ah receptor CYP1A1 induction pathway through the SMRT corepressor. Biochemical and Biophysical Research Communications, 322, 551–556. [DOI] [PubMed] [Google Scholar]
- Fanelli R, Bertoni MP, Castelli MG, Chiabrando C, Martelli GP, Noseda A, Garattini S, Binaghi C, Marazza V and Pezza F, 1980. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin toxic effects and tissue levels in animals from the contaminated area of Seveso, Italy. Archives of Environmental Contamination and Toxicology, 9, 569–577. [DOI] [PubMed] [Google Scholar]
- Fång J, Nyberg E, Bignert A and Bergman A, 2013. Temporal trends of polychlorinated dibenzo‐p‐dioxins and dibenzofurans and dioxin‐like polychlorinated biphenyls in mothers’ milk from Sweden, 1972‐2011. Environment International, 60, 224–231. [DOI] [PubMed] [Google Scholar]
- Fängström B, Hovander L, Bignert A, Athanassiadis I, Linderholm L, Grandjean P, Weihe P and Bergman A, 2005. Concentrations of polybrominated diphenyl ethers, polychlorinated biphenyls, and polychlorobiphenylols in serum from pregnant Faroese women and their children 7 years later. Environmental Science and Technology, 39, 9457–63. [DOI] [PubMed] [Google Scholar]
- FAO/WHO (Food and Agricultural Organization/World Health Organization), 2002. Evaluation of certain food additives and contaminants. Fifty‐seventh report of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). WHO Technical Report Series 909. Polychlorinated dibenzodioxins, polychlorinated dibenzofurans and coplanar polychlorinated biphenyls.
- Faqi AS, Dalsenter PR, Merker HJ and Chahoud I, 1998. Reproductive toxicity and tissue concentrations of low doses of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in male offspring rats exposed throughout pregnancy and lactation. Toxicology and Applied Pharmacology, 150, 383–392. [DOI] [PubMed] [Google Scholar]
- Farmahin R, Manning GE, Crump D, Wu D, Mundy LJ, Jones SP, Hahn ME, Karchner SI, Giesy JP, Bursian SJ, Zwiernik MJ, Fredricks TB and Kennedy SW, 2013. Amino acid sequence of the ligand‐binding domain of the aryl hydrocarbon receptor 1 predicts sensitivity of wild birds to effects of dioxin‐like compounds. Toxicological Sciences, 131, 139–152. [DOI] [PubMed] [Google Scholar]
- Feil VJ, Huwe JK, Zaylskie RG, Davison KL, Anderson VL, Marchello M and Tiernan TO, 2000. Chlorinated dibenzo‐p‐dioxin and dibenzofuran concentrations in beef animals from a feeding study. Journal of Agricultural and Food Chemistry, 48, 6163–6173. [DOI] [PubMed] [Google Scholar]
- FERA (The Food and Environment Research Agency), 2012. Organic Environmental Contaminants in the 2012 Total Diet Study Samples. Report to the Food Standards Agency. Report Number: FD 12/04. The Food and Environment Research Agency. Sand Hutton. York. YO41 1LZ. UK.
- Fernandes AR, Foxall C, Lovett A, Rose M and Dowding A, 2011. The assimilation of dioxins and PCBs in conventionally reared farm animals: occurrence and biotransfer factors. Chemosphere, 83, 815–822. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Salguero PM, Hilbert DM, Rudikoff S, Ward JM and Gonzalez FJ, 1996. Aryl‐hydrocarbon receptor‐deficient mice are resistant to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐induced toxicity. Toxicology and Applied Pharmacology, 140, 173–179. [DOI] [PubMed] [Google Scholar]
- Ferrario J, Byrne C, Winters D, Boone T, Vigo C and Dupuy A, 2003. Chlorinated dioxins and furans from kelp and copper sulfate: initial investigations of dioxin formation in mineral feed supplements. Organohalogen Compounds, 63, 183–186. [Google Scholar]
- Fiedler H, 2001. Global and local disposition of PCBs In: Robertson LW. and Hansen LG. (eds). PCBs: Recent Advances in Environmental Toxicology and Health Effects. The University Press of Kentucky, Lexington: pp. 11–15. [Google Scholar]
- Field JA and Sierra‐Alvarez R, 2008. Microbial transformation and degradation of polychlorinated biphenyls. Environmental Pollution, 155, 1–12. [DOI] [PubMed] [Google Scholar]
- Fierens S, Mairesse H, Heilier JF, De Burbure C, Focant JF, Eppe G, De Pauw E and Bernard A, 2003. Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: findings in a population‐based study in Belgium. Biomarkers, 8, 529–534. [DOI] [PubMed] [Google Scholar]
- Firestone D, Clower M, Borsetti AP, Teske RH and Long PE, 1979. Polychlorodibenzo‐p‐dioxin and pentachlorophenol residues in milk and blood of cows fed technical pentachlorophenol. Journal of Agricultural and Food Chemistry, 27, 1171–1177. [DOI] [PubMed] [Google Scholar]
- Fisk AT, Yarechewski AL, Metner DA, Evans RE, Lockhart WL and Muir DCG, 1997. Accumulation, depuration and hepatic mixed‐function oxidase enzyme induction in juvenile rainbow trout and lake whitefish exposed to dietary 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Aquatic Toxicology, 37, 201–220. [Google Scholar]
- Flaveny CA, Murray IA, Chiaro CR and Perdew GH, 2009. Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Molecular Pharmacology, 75, 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaveny CA, Murray IA and Perdew GH, 2010. Differential gene regulation by the human and mouse aryl hydrocarbon receptor. Toxicological Sciences, 114, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesch‐Janys D, Becher H, Gurn P, Jung D, Konietzko J, Manz A and Papke O, 1996. Elimination of polychlorinated dibenzo‐p‐dioxins and dibenzofurans in occupationally exposed persons. Journal of Toxicology and Environmental Health, 47, 363–378. [DOI] [PubMed] [Google Scholar]
- Flesch‐Janys D, Steindorf K, Gurn P and Becher H, 1998. Estimation of the cumulated exposure to polychlorinated dibenzo‐p‐dioxins/furans and standardized mortality ratio analysis of cancer mortality by dose in an occupationally exposed cohort. Environmental Health Perspectives, 106, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher N, Hanberg A and Håkansson H, 2001. Hepatic vitamin A depletion is a sensitive marker of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) exposure in four rodent species. Toxicological Sciences, 62, 166–75. [DOI] [PubMed] [Google Scholar]
- Fletcher N, Wahlstrom D, Lundberg R, Nilsson CB, Nilsson KC, Stockling K, Hellmold H and Hakansson H, 2005. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) alters the mRNA expression of critical genes associated with cholesterol metabolism, bile acid biosynthesis, and bile transport in rat liver: a microarray study. Toxicology and Applied Pharmacology, 207, 1–24. [DOI] [PubMed] [Google Scholar]
- Flick DF, Firestone D and Higginbotham GR, 1972. Studies of the chick edema disease part 9 response of chicks fed or singly administered synthetic edema producing compounds. Poultry Science, 51, 2026–2034. [DOI] [PubMed] [Google Scholar]
- Fontao F, Barnes L, Kaya G, Saurat JH and Sorg O, 2017. High susceptibility of Lrig1 sebaceous stem cells to TCDD in mice. Toxicological Sciences, 160, 230–243. [DOI] [PubMed] [Google Scholar]
- Forgacs AL, Dere E, Angrish MM and Zacharewski TR, 2013. Comparative analysis of temporal and dose‐dependent TCDD‐elicited gene expression in human, mouse, and rat primary hepatocytes. Toxicological Sciences, 133, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier A, Rychen G, Marchand P, Toussaint H, Le Bizec B and Feidt C, 2013. Polychlorinated biphenyl (PCB) decontamination kinetics in lactating goats (Capra hircus) following a contaminated corn silage exposure. Journal of Agricultural and Food Chemistry, 61, 7156–7164. [DOI] [PubMed] [Google Scholar]
- Fox LL and Grasman KA, 1999. Effects of PCB 126 on primary immune organ development in chicken embryos. Journal of Toxicology and Environmental Health‐Part A, 58, 233–244. [DOI] [PubMed] [Google Scholar]
- Frakes RA, Zeeman CQT and Mower B, 1993. BIOACCUMULATION OF 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) by fish downstream of pulp and paper‐mills in Maine. Ecotoxicology and Environmental Safety, 25, 244–252. [DOI] [PubMed] [Google Scholar]
- Franc MA, Moffat ID, Boutros PC, Tuomisto JT, Tuomisto J, Pohjanvirta R and Okey AB, 2008. Patterns of dioxin‐altered mRNA expression in livers of dioxin‐sensitive versus dioxin‐resistant rats. Archives of Toxicology, 82, 809–830. [DOI] [PubMed] [Google Scholar]
- Franczak A, Nynca A, Valdez KE, Mizinga KM and Petroff BK, 2006. Effects of acute and chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on the transition to reproductive senescence in female Sprague‐Dawley rats. Biology of Reproduction, 74, 125–30. [DOI] [PubMed] [Google Scholar]
- Fries GF, Paustenbach DJ, Mather DB and Luksemburg WJ, 1999. A congener specific evaluation of transfer of chlorinated dibenzo‐p‐dioxins and dibenzofurans to milk of cows following ingestion of pentachlorophenol‐treated wood. Environmental Science and Technology, 33, 1165–1170. [Google Scholar]
- Fries GF, Paustenbach DJ and Luksemburg WJ, 2002. Complete mass balance of dietary polychlorinated dibenzo‐p‐dioxins and dibenzofurans in dairy cattle and characterization of the apparent synthesis of hepta‐ and octachlorodioxins. Journal of Agricultural and Food Chemistry, 50, 4226–4231. [DOI] [PubMed] [Google Scholar]
- Friesen EN, Skura BJ, Ikonomou MG, Oterhals A and Higgs DA, 2015. Influence of terrestrial lipid and protein sources and activated carbon‐treated fish oil on levels of persistent organic pollutants and fatty acids in the flesh of Atlantic salmon. Aquaculture Research, 46, 358–381. [Google Scholar]
- Fromme H, Albrecht M, Appel M, Hilger B, Völkel W, Liebl B and Roscher E, 2015. PCBs, PCDD/Fs, and PBDEs in blood samples of a rural population in South Germany. International Journal of Hygiene and Environmental Health, 218, 41–46. [DOI] [PubMed] [Google Scholar]
- Fromme H, Hilger B, Albrecht M, Gries W, Leng G and Völkel W, 2016. Occurrence of chlorinated and brominated dioxins/furans, PCBs, and brominated flame retardants in blood of German adults. International Journal of Hygiene and Environmental Health, ???, 380–388. [DOI] [PubMed] [Google Scholar]
- FSAI (Food Safety Authority of Ireland), 2009. Review of the 2008 Irish Dioxin Incident. Available online: https://www.fsai.ie/uploadedFiles/News_Centre/Events/archive/Rhodri%20Evans%20FSAI.pdf
- Fujisawa N, Yoshioka W, Yanagisawa H and Tohyama C, 2018. Roles of cytosolic phospholipase A(2)alpha in reproductive and systemic toxicities in 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐exposed mice. Archives of Toxicology, 92, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga BN, Probst MR, Reisz‐Porszasz S and Hankinson O, 1995. Identification of functional domains of the aryl hydrocarbon receptor. The Journal of Biological Chemistry, 270, 29270–29278. [DOI] [PubMed] [Google Scholar]
- Fukushi J, Tokunaga S, Nakashima Y, Motomura G, Mitoma C, Uchi H, Furue M and Iwamoto Y, 2016. Effects of dioxin‐related compounds on bone mineral density in patients affected by the Yusho incident. Chemosphere, 145, 25–33. [DOI] [PubMed] [Google Scholar]
- Fürst P, Meemken HA and Groebel W, 1986. Determination of polychlorinated dibenzodioxins and dibenzofurans in human milk. Chemosphere, 15, 1977–1980. [DOI] [PubMed] [Google Scholar]
- Fürst P, Fürst C and Wilmers K, 1992a. PCDDs and PCDFs in human milk – statistical evaluation of a 6‐years survey. Chemosphere, 25, 1029–1038. [Google Scholar]
- Fürst P, Beck H and Theelen R, 1992b. Assessment of human intake of PCDDs and PCDFs from different environmental sources. Toxic Substances Journal, 12, 133–150. [Google Scholar]
- Fürst P, 2006. Dioxins, polychlorinated biphenyls and other organohalogen compounds in human milk ‐ Levels, correlations, trends and exposure through breastfeeding. Molecular Nutrition and Food Research, 50, 922–933. [DOI] [PubMed] [Google Scholar]
- Fürst P and Bernsmann T, 2011. Decline of PCDD/PCDF and PCB in dairy products 1990–2010. Organohalogen Compounds, 73, 360–363. [Google Scholar]
- Furumatsu K, Nishiumi S, Kawano Y, Ooi M, Yoshie T, Shiomi Y, Kutsumi H, Ashida H, Fujii‐Kuriyama Y, Azuma T and Yoshida M, 2011. A role of the aryl hydrocarbon receptor in attenuation of colitis. Digestive Diseases and Sciences, 56, 2532–2544. [DOI] [PubMed] [Google Scholar]
- Gaitanis G, Magiatis P, Stathopoulou K, Bassukas ID, Alexopoulos EC, Velegraki A and Skaltsounis AL, 2008. AhR ligands, malassezin, and indolo[3,2‐b]carbazole are selectively produced by Malassezia furfur strains isolated from seborrheic dermatitis. The Journal of Investigative Dermatology, 128, 1620–1625. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sahlberg C, Kiukkonen A, Alaluusua S, Pohjanvirta R, Tuomisto J and Lukinmaa PL, 2004. Lactational exposure of Han/Wistar rats to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin interferes with enamel maturation and retards dentin mineralization. Journal of Dental Research, 83, 139–144. [DOI] [PubMed] [Google Scholar]
- Gardella KA, Muro I, Fang G, Sarkar K, Mendez O and Wright CW, 2016. Aryl hydrocarbon receptor nuclear translocator (ARNT) isoforms control lymphoid cancer cell proliferation through differentially regulating tumor suppressor p53 activity. Oncotarget, 7, 10710–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison PM and Denison MS, 2000. Analysis of the murine AhR gene promoter. Journal of Biochemical and Molecular Toxicology, 14, 1–10. [DOI] [PubMed] [Google Scholar]
- Garrison PM, Rogers JM, Brackney WR and Denison MS, 2000. Effects of histone deacetylase inhibitors on the Ah receptor gene promoter. Archives of Biochemistry and Biophysics, 374, 161–171. [DOI] [PubMed] [Google Scholar]
- Gasiewicz TA, Olson JR, Geiger LE and Neal RA, 1983a. Absorption, distribution and metabolism of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in experimental animals. Environmental Science Research, 26, 495–525. [Google Scholar]
- Gasiewicz TA, Geiger LE, Rucci G, Neal RA, 1983b. Distribution, excretion, and metabolism of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metabolism and Disposition, 11, 397–403. [PubMed]
- Gasiewicz TA, Rucci G, Henry EC and Baggs RB, 1986. Changes in hamster hepatic cytochrome P‐450, ethoxycoumarin O‐deethylase, and reduced NAD(P): menadione oxidoreductase following treatment with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Partial dissociation of temporal and dose‐response relationships from elicited toxicity. Biochemical Pharmacology, 35, 2737–2742. [DOI] [PubMed] [Google Scholar]
- Gasiewicz TA, Singh KP and Bennet JA, 2014. The Ah receptor in stem cell cycling, regulation and quiescence. Annals of the New York Academy of Sciences, 1310, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger KJ, Giera S, Sharlin DS, Bansal R, Iannacone E and Zoeller RT, 2007. Polychlorinated biphenyls 105 and 118 form thyroid hormone receptor agonists after cytochrome P4501A1 activation in rat pituitary GH3 cells. Environmental Health and Perspectives, 115, 1623–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrs BC, Riddle MM, Williams WC and Smialowicz RJ, 1997. Alterations in the developing immune system of the F344 rat after perinatal exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. II. Effects on the pup and the adult. Toxicology, 122, 229–240. [DOI] [PubMed] [Google Scholar]
- Gehrs BC and Smialowicz RJ, 1999. Persistent suppression of delayed‐type hypersensitivity in adult F344 rats after perinatal exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology, 134, 79–88. [DOI] [PubMed] [Google Scholar]
- Genualdo V, Perucatti A, Iannuzzi A, Di Meo GP, Spagnuolo SM, Caputi‐Jambrenghi A, Coletta A, Vonghia G and Iannuzzi L, 2012. Chromosome fragility in river buffalo cows exposed to dioxins. Journal of Applied Genetics, 53, 221–226. [DOI] [PubMed] [Google Scholar]
- Geusau A, Abraham K, Geissler K, Sator MO, Stingl G and Tschachler E, 2001. Severe 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) intoxication: clinical and laboratory effects. Environmental Health Perspectives, 109, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen JE, Goujon FM and Nebert DW, 1972. Genetic regulation of aryl hydrocarbon hydroxylase induction. II. Simple Mendelian expression in mouse tissues in vivo . The Journal of Biological Chemistry, 247, 1125–1137. [PubMed] [Google Scholar]
- Giera S, Bansal R, Ortiz‐Toro TM, Taub DG and Zoeller RT, 2011. Individual polychlorinated biphenyl (PCB) congeners produce tissue‐ and gene‐specific effects on thyroid hormone signaling during development. Endocrinology, 152, 2909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilot D, Le Meur N, Giudicelli F, Le Vee M, Lagadic‐Gossmann D, Theret N and Fardel O, 2011. RNAi‐based screening identifies kinases interfering with dioxin‐mediated up‐regulation of CYP1A1 activity. PloS One, 6, e18261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri AK, 1986. Mutagenic and genotoxic effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin – a review. Mutation Research, 168, 241–248. [DOI] [PubMed] [Google Scholar]
- Girolami F, Spalenza V, Benedetto A, Manzini L, Badino P, Abete MC and Nebbia C, 2016. Comparative liver accumulation of dioxin‐like compounds in sheep and cattle: possible role of AhR‐mediated xenobiotic metabolizing enzymes. Science of the Total Environment, 571, 1222–1229. [DOI] [PubMed] [Google Scholar]
- Godliauskiene R, Tamosiunas V and Naujalis E, 2017. Polychlorinated dibenzo‐p‐dioxins/furans and dioxin‐like polychlorinated biphenyls in food and feed in the Lithuanian market. Toxicological and Environmental Chemistry, 99, 65–77. [Google Scholar]
- Goff KF, Hull BE and Grasman KA, 2005. Effects of PCB 126 on primary immune organs and thymocyte apoptosis in chicken embryos. Journal of Toxicology and Environmental Health‐Part a‐Current Issues, 68, 485–500. [DOI] [PubMed] [Google Scholar]
- Goldstein JA, McKinney JD, Lucier GW, Hickman P, Bergman H and Moore JA, 1976. Toxicological assessment of hexachlorobiphenyl isomers and 2,3,7,8,‐tetrachlorodibenzofuran in chicks. II. Effects on drug metabolism and porphyrin accumulation. Toxicology and Applied Pharmacology, 36, 81–92. [DOI] [PubMed] [Google Scholar]
- Gordon MW, Yan F, Zhong X, Mazumder PB, Xu‐Monette ZY, Zou D, Young KH, Ramos KS and Li Y, 2015. Regulation of p53‐targeting microRNAs by polycyclic aromatic hydrocarbons: implications in the etiology of multiple myeloma. Molecular Carcinogenesis, 54, 1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JR and Rozman K, 1987. Dose‐response and time course of hypothyroxinemia and hypoinsulinemia and characterization of insulin hypersensitivity in 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD)‐treated rats. Toxicology, 44, 297–307. [DOI] [PubMed] [Google Scholar]
- Gorski JR, Muzi G, Weber LW, Pereira DW, Arceo RJ, Iatropoulos MJ and Rozman K, 1988. Some endocrine and morphological aspects of the acute toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Toxicologic Pathology, 16, 313–20. [DOI] [PubMed] [Google Scholar]
- Govarts E, Remy S, Bruckers L, Den Hond E, Sioen I, Nelen V, Baeyens W, Nawrot TS, Loots I, Van Larebeke N and Schoeters G, 2016. Combined effects of prenatal exposures to environmental chemicals on birth weight. International Journal of Environmental Research and Public Health, 13, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradelet S, Astorg P, Pineau T, Canivenc MC, Siess MH, Leclerc J and Lesca P, 1997. Ah receptor‐dependent CYP1A induction by two carotenoids, canthaxanthin and beta‐apo‐8’‐carotenal, with no affinity for the TCDD binding site. Biochemical Pharmacology, 54, 307–315. [DOI] [PubMed] [Google Scholar]
- Grasman KA and Whitacre LL, 2001. Effects of PCB 126 on thymocyte syurface marker expression and immune organ development in chicken embryos. Journal of Toxicology and Environmental Health, Part A, 62, 191–206. [DOI] [PubMed] [Google Scholar]
- Gray LE Jr and Ostby JS, 1995. In utero 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) alters reproductive morphology and function in female rat offspring. Toxicology and Applied Pharmacology, 133, 285–94. [DOI] [PubMed] [Google Scholar]
- Gray LE, Ostby JS and Kelce WR, 1997a. A dose‐response analysis of the reproductive effects of a single gestational dose of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in male Long Evans hooded rat offspring. Toxicology and Applied Pharmacology, 146, 11–20. [DOI] [PubMed] [Google Scholar]
- Gray LE, Wolf C, Mann P and Ostby JS, 1997b. In utero exposure to low doses of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) alters reproductive development of female Long Evans hooded rat offspring. Toxicology and Applied Pharmacology, 146, 237–244. [DOI] [PubMed] [Google Scholar]
- Greig JB, Jones G, Butler WH and Barnes JM, 1973. Toxic effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Food and Cosmetics Toxicology, 11, 585–595. [DOI] [PubMed] [Google Scholar]
- Grimm FA, Lehmler HJ, He X, Robertson LW and Duffel MW, 2013. Sulfated metabolites of polychlorinated biphenyls are high‐affinity ligands for the thyroid hormone transport protein transthyretin. Environmental Health Perspectives, 121, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm FA, Hu D, Kania‐Korwel I, Lehmler H‐J, Ludewig G, Hornbuckle KC, Duffel MW, Bergman A and Robertson LW, 2015. Metabolism and metabolites of polychlorinated biphenyls. Critical Reviews in Toxicology, 45, 245–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grova N, Feidt C, Laurent C and Rychen G, 2002. C‐14 Milk, urine and faeces excretion kinetics in lactating goats after an oral administration of C‐14 polycyclic aromatic hydrocarbons. International Dairy Journal, 12, 1025–1031. [Google Scholar]
- Guo YL, Ryan JJ, Lau BPY, Yu ML and Hsu CC, 1997. Serum levels of PCB/PCDF congeners 14 years after accidental exposure to contaminated rice oil. Archives of Environmental Contamination and Toxicology, 33, 104–108. [DOI] [PubMed] [Google Scholar]
- Guo Y, Hendrickx AG, Overstreet JW, Dieter J, Stewart D, Tarantal AF, Laughlin L and Lasley BL, 1999a. Endocrine biomarkers of early fetal loss in cynomolgus macaques (Macaca fascicularis) following exposure to dioxin. Biology of Reproduction, 60, 707–713. [DOI] [PubMed] [Google Scholar]
- Guo YLL, Yu ML, Hsu CC and Rogan WJ, 1999b. Chloracne, goiter, arthritis, and anemia after polychlorinated biphenyl poisoning: 14‐year follow‐up of the Taiwan Yucheng cohort. Environmental Health Perspectives, 107, 715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YM, Wang SY, Wang XR and Lasley B, 2000. Effect of TCDD on maternal toxicity and chorionic gonadotropin ‐ bioactivity in the immediate post‐implantation period of macaque. Biomedical and Environmental Sciences, 13, 26–31. [PubMed] [Google Scholar]
- Guo YLL, Lambert GH, Hsu CC and Hsu MML, 2004. Yucheng: health effects of prenatal exposure to polychlorinated biphenyls and dibenzofurans. International Archives of Occupational and Environmental Health, 77, 153–158. [DOI] [PubMed] [Google Scholar]
- Guo H, Yang H, Chen H, Li W, Tang J, Cheng P, Xie Y, Liu Y, Ding G, Cui D, Zheng X and Duan Y, 2015a. Molecular mechanisms of human thyrocyte dysfunction induced by low concentrations of polychlorinated biphenyl 118 through the Akt/FoxO3a/NIS pathway. Journal of Applied Toxicology, 35, 992–8. [DOI] [PubMed] [Google Scholar]
- Guo H, Zhang L, Wei K, Zhao J, Wang Y, Jin F and Xuan K, 2015b. Exposure to a continuous low dose of tetrachlorodibenzo‐p‐dioxin impairs the development of the tooth root in lactational rats and alters the function of apical papilla‐derived stem cells. Archives of Oral Biology, 60, 199–207. [DOI] [PubMed] [Google Scholar]
- Gupta BN, Vos JG, Moore JA, Zinkl JG and Bullock BC, 1973. Pathologic effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in laboratory animals. Environmental Health Perspectives, 5, 125–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Ketchum N, Roehrborn CG, Schecter A, Aragaki CC and Michalek JE, 2006. Serum dioxin, testosterone, and subsequent risk of benign prostatic hyperplasia: a prospective cohort study of air force veterans. Environmental Health Perspectives, 114, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutleb AC, Lilienthal H, Erhard HW, Zimmer KE, Skaare JU and Ropstad E, 2011. Effects of pre‐ and postnatal polychlorinated biphenyl exposure on emotional reactivity observed in lambs before weaning. Ecotoxicology and Environmental Safety, 74, 1396–1401. [DOI] [PubMed] [Google Scholar]
- Hagenmaier H and Walczok M, 1996. Time trends in levels, patterns and profiles for PCDD/PCDF in sediment cores of Lake Constance. Organohalogen Compounds, 28, 101–104. [Google Scholar]
- Hahn ME and Karchner SI, 2012. Structural and functional diversification of AHRs during metazoan evolution In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 389–403. [Google Scholar]
- Hahn ME, Karchner SI and Merson RR, 2017. Diversity as opportunity: insights from 600 million years of AHR evolution. Current Opinion in Toxicology, 2, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakk H, Larsen GL and Feil VJ, 2001. Metabolism of C‐14 1,2,7,8‐tetrachlorodibenzo‐p‐dioxin in a ruminating Holstein calf. Xenobiotica, 31, 443–455. [DOI] [PubMed] [Google Scholar]
- Hakk H, Diliberto JJ and Birnbaum LS, 2009. The effect of dose on 2,3,7,8‐TCDD tissue distribution, metabolism and elimination in CYP1A2 (‐/‐) knockout and C57BL/6N parental strains of mice. Toxicology and Applied Pharmacology, 241, 119–126. [DOI] [PubMed] [Google Scholar]
- Halldorsson TI, Thorsdottir I, Meltzer HM, Strom M and Olsen SF, 2009. Dioxin‐like activity in plasma among Danish pregnant women: dietary predictors, birth weight and infant development. Environmental Reserach, 109, 22–28. [DOI] [PubMed] [Google Scholar]
- Halperin W, Vogt R, Sweeney MH, Shopp G, Fingerhut M and Petersen M, 1998. Immunological markers among workers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Occupational and Environmental Medicine, 55, 742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm JT, Chen CY and Birnbaum LS, 2003. A mixture of dioxins, furans, and non‐ortho PCBs based upon consensus toxic equivalency factors produces dioxin‐like reproductive effects. Toxicological Sciences, 74, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G, Ding G, Lou X, Wang X, Han J, Shen H, Zhou Y and Du L, 2011. Correlations of PCBs, DIOXIN, and PBDE with TSH in Children's blood in areas of computer E‐waste recycling. Biomedical and Environmental Sciences, 24, 112–116. [DOI] [PubMed] [Google Scholar]
- Hanieh H and Alzahrani A, 2013. MicroRNA‐132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti‐inflammation: a new Ahr‐based exploration. European Journal of Immunology, 43, 2771–2782. [DOI] [PubMed] [Google Scholar]
- Hansson M, Barregård L, Sällsten G, Svensson BG and Rappe C, 1997. Polychlorinated dibenzo‐p‐dioxin and dibenzofuran levels and patterns in polyvinylchloride and chloralkali industry workers. International Archives of Occupational and Environmental Health, 70, 51–56. [DOI] [PubMed] [Google Scholar]
- Harnly ME, Petreas MX, Flattery J and Goldman LR, 2000. Polychlorinated dibenzo‐p‐dioxin and polychlorinated dibenzofuran contamination in soil and home‐produced chicken eggs near pentachlorophenol sources. Environmental Science and Technology, 34, 1143–1149. [Google Scholar]
- Harper N, Connor K and Safe S, 1993. Immunotoxic potencies of polychlorinated biphenyl (PCB), dibenzofuran (PCDF) and dibenzo‐p‐dioxin (PCDD) congeners in C57BL/6 and DBA/2 mice. Toxicology, 80, 217–227. [DOI] [PubMed] [Google Scholar]
- Harrill JA, Hukkanen RR, Lawson M, Martin G, Gilger B, Soldatow V, Lecluyse EL, Budinsky RA, Rowlands JC and Thomas RS, 2013. Knockout of the aryl hydrocarbon receptor results in distinct hepatic and renal phenotypes in rats and mice. Toxicology and Applied Pharmacology, 272, 503–518. [DOI] [PubMed] [Google Scholar]
- Harrill JA, Layko D, Nyska A, Hukkanen RR, Manno RA, Grassetti A, Lawson M, Martin G, Budinsky RA, Rowlands JC and Thomas RS, 2016. Aryl hydrocarbon receptor knockout rats are insensitive to the pathological effects of repeated oral exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Journal of Applied Toxicology, 36, 802–814. [DOI] [PubMed] [Google Scholar]
- Hart LJ, Gogal RM, Smith SA, Smith BJ, Robertson J and Holladay SD, 1999. Leukocyte hypocellularity in the spleen and pronephros of tilapia (Oreochromis niloticus) exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) may result from antiproliferative effects and enhanced apoptosis. Toxic Substance Mechanisms, 18, 21–38. [Google Scholar]
- Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, Wild B, Camargo SM, Singer D, Richter A, Kuba K, Fukamizu A, Schreiber S, Clevers H, Verrey F, Rosenstiel P and Penninger JM, 2012. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature, 487, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser R, Sergeyev O, Korrick S, Lee MM, Revich B, Gitin E, Burns JS and Williams PL, 2008. Association of blood lead levels with onset of puberty in Russian boys. Environmental Health Perspectives, 116, 976–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CL and Norris LA, 1977. Chronic oral toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) to rainbow trout. Transactions of the American Fisheries Society, 106, 641–645. [Google Scholar]
- Haws LC, Su SH, Harris M, DeVito MJ, Walker NJ, Farland WH, Finley B and Birnbaum LS, 2006. Development of a refined database of mammalian relative potency estimates for dioxin‐like compounds. Toxicological Sciences, 89, 4–30. [DOI] [PubMed] [Google Scholar]
- Hays LE, Carpenter CD and Petersen SL, 2002. Evidence that GABAergic neurons in the preoptic area of the rat brain are targets of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin during development. Environmental Health Perspectives, 110(Suppl 3), 369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward DG, Nortrup D, Gardner A and Clower M, 1999. Elevated TCDD in chicken eggs and farm‐raised catfish fed a diet with ball clay from a Southern United States mine. Environmental Research Section A, 81, 248–256. [DOI] [PubMed] [Google Scholar]
- He X and Feng S, 2015. Role of metabolic enzymes P450 (CYP) on activating procarcinogen and their polymorphisms on the risk of cancers. Current Drug Metabolism, 16, 850–863. [DOI] [PubMed] [Google Scholar]
- Head JA, Hahn ME and Kennedy SW, 2008. Key amino acids in the aryl hydrocarbon receptor predict dioxin sensitivity in avian species. Environmental Science and Technology, 42, 7535–7541. [DOI] [PubMed] [Google Scholar]
- Heaton SN, Bursian SJ, Giesy JP, Tillitt DE, Render JA, Jones PD, Verbrugge DA, Kubiak TJ and Aulerich RJ, 1995a. Dietary exposure of mink to carp from Saginaw Bay, Michigan. 1. Effects on reproduction and survival, and the potential risks to wild mink populations. Archives of Environmental Contamination and Toxicology, 28, 334–343. [DOI] [PubMed] [Google Scholar]
- Heaton SN, Bursian SJ, Giesy JP, Tillitt DE, Render JA, Jones PD, Verbrugge DA, Kubiak TJ and Aulerich RJ, 1995b. Dietary exposure of mink to carp from Saginaw bay, Michigan. 2. Hematology and liver pathology. Archives of Environmental Contamination and Toxicology, 29, 411–417. [DOI] [PubMed] [Google Scholar]
- Heederik D, Hooiveld M and Bueno‐de‐Mesquita HB, 1998. Modelling of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin levels in a cohort of workers with exposure to phenoxy herbicides and chlorophenols. Chemosphere, 37, 1743–1754. [DOI] [PubMed] [Google Scholar]
- Heid SE, Walker MK and Swanson HI, 2001. Correlation of cardiotoxicity mediated by halogenated aromatic hydrocarbons to aryl hydrocarbon receptor activation. Toxicological Sciences, 61, 187–196. [DOI] [PubMed] [Google Scholar]
- Heilier JF, Nackers F, Verougstraete V, Tonglet R, Lison D and Donnez J, 2005. Increased dioxin‐like compounds in the serum of women with peritoneal endometriosis and deep endometriotic (adenomyotic) nodules. Fertility and Sterility, 84, 305–312. [DOI] [PubMed] [Google Scholar]
- Hellou J, Mackay D and Banoub J, 1999. Levels, persistence and bioavailability of organic contaminants present in marine harbor sediments impacted by raw sewage. Chemosphere, 38, 457–473. [DOI] [PubMed] [Google Scholar]
- Henry EC and Gasiewicz TA, 1987. Changes in thyroid hormones and thyroxine glucuronidation in hamsters compared with rats following treatment with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology and Applied Pharmacology, 89, 165–74. [DOI] [PubMed] [Google Scholar]
- Henry EC and Gasiewicz TA, 2003. Agonist but not antagonist ligands induce conformational change in the mouse aryl hydrocarbon receptor as detected by partial proteolysis. Molecular Pharmacology, 63, 392–400. [DOI] [PubMed] [Google Scholar]
- Henry EC and Gasiewicz TA, 2008. Molecular determinants of species‐specific agonist and antagonist activity of a substituted flavone towards the aryl hydrocarbon receptor. Archives of Biochemistry and Biophysics, 472, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshel DS, Hehn B, Wagey R, Vo M and Steeves JD, 1997a. The relative sensitivity of chicken embryos to yolk‐ or air‐cell‐injected 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Environmental Toxicology and Chemistry, 16, 725–732. [Google Scholar]
- Henshel DS, Martin JW and Dewitt JC, 1997b. Brain asymmetry as a potential biomarker for developmental TCDD intoxication: a dose‐response study. Environmental Health Perspectives, 105, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heres L, Hoogenboom R, Herbes R, Traag W and Urlings B, 2010. Tracing and analytical results of the dioxin contamination incident in 2008 originating from the Republic of Ireland. Food Additives and Contaminants, Part A, 27, 1733–1744. [DOI] [PubMed] [Google Scholar]
- Hermsen SAB, Larsson S, Arima A, Muneoka A, Ihara T, Sumida H, Fukusato T, Kubota S, Yasuda M and Lind PM, 2008. In utero and lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) affects bone tissue in rhesus monkeys. Toxicology, 253, 147–152. [DOI] [PubMed] [Google Scholar]
- Herve JC, Crump DL, McLaren KK, Giesy JP, Zwiernik MJ, Bursian SJ and Kennedy SW, 2010. 2,3,4,7,8‐pentachlorodibenzofuran is a more potent cytochrome P4501A inducer than 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in herring gull hepatocyte cultures. Environmental Toxicology and Chemistry/SETAC, 29, 2088–2095. [DOI] [PubMed] [Google Scholar]
- Higginbotham GR, Huang A, Firestone D, Verrett J, Ress J and Campbell AD, 1968. Chemical and toxicological evaluations of isolated and synthetic chloro derivatives of dibenzo‐p‐dioxin. Nature, 220, 702–703. [DOI] [PubMed] [Google Scholar]
- Higgins JPT and Green S (eds), 2011. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available online: http://www.cochrane-handbook.org
- Hochstein JR, Aulerich RJ and Bursian SJ, 1988. Acute toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin to mink. Archives of Environmental Contamination and Toxicology, 17, 33–37. [DOI] [PubMed] [Google Scholar]
- Hochstein JR, Bursian SJ and Aulerich RJ, 1998. Effects of dietary exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in adult female mink (Mustela vison). Archives of Environmental Contamination and Toxicology, 35, 348–353. [DOI] [PubMed] [Google Scholar]
- Hochstein JR, Render JA, Bursian SJ and Aulerich RJ, 2001. Chronic toxicity of dietary 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin to mink. Veterinary and Human Toxicology, 43, 134–139. [PubMed] [Google Scholar]
- Hodek P, Koblihová J, Kizek R, Frei E, Arlt VM and Stiborová M, 2013. The relationship between DNA adduct formation by benzo[a]pyrene and expression of its activation enzyme cytochrome P450 1A1 in rat. Environmental Toxicology and Pharmacology, 36, 989–96. [DOI] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA and Hankinson O, 1991. Cloning of a factor required for activity of the AH (dioxin) receptor. Science (New York, NY), 252, 954–958. [DOI] [PubMed] [Google Scholar]
- Holladay SD, 1999. Prenatal immunotoxicant exposure and postnatal autoimmune disease. Environmental Health Perspectives, 107, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrand H, Gadomski D, Mandalakis M, Tysklind M, Irvine R, Andersson P and Gustafsson O, 2006. Origin of PCDDs in ball clay assessed with compound‐specific chlorine isotope analysis and radiocarbon dating. Environmental Science and Technology, 40, 3730–3735. [DOI] [PubMed] [Google Scholar]
- Holoubek I, 2001. Polychlorinated biphenyl (PCB) contaminated sites worldwide In: PCBs: Recent Advances in Environmental Toxicology and Health Effects. Eds Robertson LW, Hansen LG, The University Press of Kentucky, Lexington, pp. 17–26. [Google Scholar]
- Hong LH, McKinney JD and Luster MI, 1987. Modulation of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD)‐mediated myelotoxicity by thyroid hormones. Biochemical Pharmacology, 36, 1361–1365. [DOI] [PubMed] [Google Scholar]
- Hoogenboom LAP, Bovee TFH, Portier L, Bor G, van der Weg G, Onstenk C and Traag WA, 2004a. The German bakery waste incident; use of a combined approach of screening and confirmation for dioxins in feed and food. Talanta, 63, 1249–1253. [DOI] [PubMed] [Google Scholar]
- Hoogenboom LAP, Kan CA, Bovee TFH, van der Weg G, Onstenk C and Traag WA, 2004b. Residues of dioxins and PCBs in fat of growing pigs and broilers fed contaminated feed. Chemosphere, 57, 35–42. [DOI] [PubMed] [Google Scholar]
- Hoogenboom LAP, Kan CA, Zeilmaker MJ, van Eijkeren JCH and Traag WA, 2006. Carry‐over of dioxins and PCBs from feed and soil to eggs at low contamination levels. Food Additives and Contaminants, 23, 518–527. [DOI] [PubMed] [Google Scholar]
- Hoogenboom LAP, van Eijkeren JCH, Zeilmaker MJ, Mengelers MJB, Herbes R, Immerzeel J and Traag WA, 2007. A novel source for dioxins present in recycled fat from gelatin production. Chemosphere, 68, 814–823. [DOI] [PubMed] [Google Scholar]
- Hoogenboom R, Zeilmaker M, van Eijkeren J, Kan K, Mengelers M, Luykx D and Traag W, 2010. Kaolinic clay derived dioxins in the feed chain from a sorting process for potatoes. Chemosphere, 78, 99–105. [DOI] [PubMed] [Google Scholar]
- Hoogenboom L, Hoffer S, Mennen M, Morgenstern P and Traag W, 2012. Dioxins formed during fires; a threat to the food chain? Organohalogen Compounds, 74, 1600–1603. [Google Scholar]
- Hoogenboom R, ten Dam G, Immerzeel J and Traag W, 2014. Building related sources of PCBs in eggs from free‐range hens. Organohalogen Compounds, 76, 1700–1703. [Google Scholar]
- Hoogenboom R, Traag W, Fernandes A and Rose M, 2015a. European developments following incidents with dioxins and PCBs in the food and feed chain. Food Control, 50, 670–683. [Google Scholar]
- Hoogenboom R, Stark M‐L, Spolders M, Zeilmaker MJ, Traag WA, ten Dam G and Schafft HA, 2015b. Accumulation of polychlorinated dibenzo‐p‐dioxins, dibenzofurans, and biphenyls in livers of young sheep. Chemosphere, 122, 137–144. [DOI] [PubMed] [Google Scholar]
- Hoogenboom R, Klop A, Herbes R, Eijkeren JCH, Zeilmaker MJ, van Vuuren AM and Traag WA, 2015c. Carry‐over of polychlorinated dibenzo‐p‐dioxins and dibenzofurans (PCDD/Fs) and polychlorinated biphenyls (PCBs) in dairy cows fed smoke contaminated maize silage or sugar beet pulp. Chemosphere, 137, 214–220. [DOI] [PubMed] [Google Scholar]
- Hoogenboom RL, Kotterman MJ, Hoek‐van Nieuwenhuizen M, van der Lee MK, Mennes WC, Jeurissen SM and van Leeuwen SP, 2015d. Dioxins, PCBs and heavy metals in Chinese mitten crabs from Dutch rivers and lakes. Chemosphere, 123, 1–8. [DOI] [PubMed] [Google Scholar]
- Hooiveld M, Heederik DJ, Kogevinas M, Boffetta P, Needham LL, Patterson DG Jr and Bueno‐de‐Mesquita HB, 1998. Second follow‐up of a Dutch cohort occupationally exposed to phenoxy herbicides, chlorophenols, and contaminants. American Journal of Epidemiology, 147, 891–901. [DOI] [PubMed] [Google Scholar]
- Hori T, Nakagawa R, Tobiishi K, Iida T, Tsutsumi T, Sasaki K and Toyoda M, 2001. Effects of cooking on concentrations of polychlorinated dibenzo‐p‐dioxins and related compounds in green leafy vegetable ‘komatsuna’. Shokuhin Eiseigaku Zasshi, 42, 339–342. [DOI] [PubMed] [Google Scholar]
- Hori T, Nakagawa R, Tobiishi K, Iida T, Tsutsumi T, Sasaki K and Toyoda M, 2005. Effects of cooking on concentrations of polychlorinated dibenzo‐p‐dioxins and related compounds in fish and meat. Journal of Agricultural and Food Chemistry, 53, 8820–8828. [DOI] [PubMed] [Google Scholar]
- Horii Y, van Bavel B, Kannan K, Petrick G, Nachtigall K and Yamashita N, 2008. Novel evidence for natural formation of dioxins in ball clay. Chemosphere, 70, 1280–1289. [DOI] [PubMed] [Google Scholar]
- Hornung MW, Miller L, Goodman B, Melancon MJ and Peterson RE, 1998. Lack of developmental and reproductive toxicity of 2,3,3’,4,4’‐pentachlorobiphenyl (PCB 105) in ring‐necked pheasants. Archives of Environmental Contamination and Toxicology, 35, 646–653. [DOI] [PubMed] [Google Scholar]
- Hsu ST, Ma CI, Hsu SK, Wu SS, Hsu NH and Yeh C, 1985. Discovery and epidemiology of PCB poisoning in Taiwan: a four‐year follow‐up. Environmental Health Perspectives, 59, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C‐C, Yu M‐Lm, Chen T‐CJ, Guo Y‐LL and Rogan WJ, 1994. The Yu‐cheng rice oil poisoning incident In: Schecter A. (ed.). Dioxin and Health. [Google Scholar]
- Hsu JF, Guo YL, Yang SY and Liao PC, 2005. Congener profiles of PCBs and PCDD/Fs in Yucheng victims fifteen years after exposure to toxic rice‐bran oils and their implications for epidemiologic studies. Chemosphere, 61, 1231–1243. [DOI] [PubMed] [Google Scholar]
- Hsu P‐C, Li M‐C, Lee Y‐C, Kuo P‐L and Guo YL, 2016. Polychlorinated biphenyls and dibenzofurans increased abnormal sperm morphology without alterations in aneuploidy: the Yucheng study. Chemosphere, 165, 294–297. [DOI] [PubMed] [Google Scholar]
- Hu W, Sorrentino C, Denison MS, Kolaja K and Fielden MR, 2007. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Molecular Pharmacology, 71, 1475–1486. [DOI] [PubMed] [Google Scholar]
- Hui LL, Lam HS, Lau EYY, Nelson EAS, Wong TW and Fielding R, 2016. Prenatal dioxin exposure and neurocognitive development in Hong Kong 11‐year‐old children. Environmental Research, 150, 205–212. [DOI] [PubMed] [Google Scholar]
- Huisman M, Koopmanesseboom C, Fidler V, Haddersalgra M, Vanderpaauw CG, Tuinstra L, Weisglaskuperus N, Sauer PJJ, Touwen BCL and Boersma ER, 1995a. Perinatal exposure to polychlorinated‐biphenyls and dioxins and its effect on neonatal neurological development. Early Human Development, 41, 111–127. [DOI] [PubMed] [Google Scholar]
- Huisman M, KoopmanEsseboom C, Lanting CI, van der Paauw CG, Tuinstra L, Fidler V, WeisglasKuperus N, Sauer PJJ, Boersma ER and Touwen BCL, 1995b. Neurological condition in 18‐month‐old children perinatally exposed to polychlorinated biphenyls and dioxins. Early Human Development, 43, 165–176. [DOI] [PubMed] [Google Scholar]
- Humblet O, Williams PL, Korrick SA, Sergeyev O, Emond C, Birnbaum LS, Burns JS, Altshul L, Patterson DG Jr, Turner WE, Lee MM, Revich B and Hauser R, 2010. Predictors of serum dioxin, furan, and PCB concentrations among women from chapaevsk, Russia. Environmental Science and Technology, 44, 5633–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humblet O, Williams PL, Korrick SA, Sergeyev O, Emond C, Birnbaum LS, Burns JS, Altshul L, Patterson DG, Turner WE, Lee MM, Revich B and Hauser R, 2011a. Dioxin and polychlorinated biphenyl concentrations in Mother's Serum and the timing of pubertal onset in sons. Epidemiology, 22, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humblet O, Sergeyev O, Altshul L, Korrick SA, Williams PL, Emond C, Birnbaum L, Burns JS, Lee MM, Revich B, Shelepchikov A, Feshin D and Hauser R, 2011b. Termporal trends in serum concentrations of polychlorinated dioxins, furans, and PCBs among adult women living in Chapaevsk, Russia: a longitudinal study from 2000 to 2009. Environmental Health, 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung WT, Lambert GH, Huang PW, Patterson DG Jr and Guo YL, 2013. Genetic susceptibility to dioxin‐like chemicals’ induction of cytochrome P4501A2 in the human adult linked to specific AhRR polymorphism. Chemosphere, 90, 2358–2364. [DOI] [PubMed] [Google Scholar]
- Hurst CH, DeVito MJ and Birnbaum LS, 2000a. Tissue disposition of 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) in maternal and developing Long‐Evans rats following subchronic exposure. Toxicological Sciences, 57, 275–283. [DOI] [PubMed] [Google Scholar]
- Hurst CH, DeVito MJ, Woodrow‐Setzer R and Birnbaum LS, 2000b. Acute administration of 2,3,7,8‐tetrachloro‐p‐dibenzodioxin (TCDD) in pregnant long Evans rats: association of measured tissue concentration with developmental effects. Toxicological Scienes, 57, 275–283. [DOI] [PubMed] [Google Scholar]
- Hutzinger O, Safe S and Zitko V, 1974. Commercial PCB preparations: properties and composition In: The Chemistry of PCB's. CRC Press. [Google Scholar]
- Huuskonen H, Unkila M, Pohjanvirta R and Tuomisto J, 1994. Developmental toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the most TCDD‐resistant and ‐susceptible rat strains. Toxicology and Applied Pharmacology, 124, 174–180. [DOI] [PubMed] [Google Scholar]
- Huuskonen S, LindstromSeppa P, Koponen K and Roy S, 1996. Effects of non‐ortho‐substituted polychlorinated biphenyls (Congeners 77 and 126) on cytochrome P4501A and conjugation activities in rainbow trout (Oncorhynchus mykiss). Comparative Biochemistry and Physiology C‐Toxicology and Pharmacology, 113, 205–213. [Google Scholar]
- Huwe J, Zaylskie R and Feil V, 2001. Distribution of dioxins into lipid stores; another look at levels in typical sampling matrices and retail meat cuts. Organohalogen Compounds, 51, 310–313. [Google Scholar]
- Huwe J and Smith DJ, 2005. Laboratory and on‐farm studies on the bioaccumulation and elimination of dioxins from a contaminated mineral supplement fed to dairy cows. Journal of Agricultural and Food Chemistry, 53, 2362–2370. [DOI] [PubMed] [Google Scholar]
- Huybrechts I, Sioen I, Boon PE, Ruprich J, Lafay L, Turrini A, Amiano P, Hirvonen T, De Neve M, Arcella D, Moschandreas J, Westerlund A, Ribas‐Barba L, Hilbig A, Papoutsou S, Christensen T, Oltarzewski M, Virtanen S, Rehurkova I, Azpiri M, Sette S, Kersting M, Walkiewicz A, Serra‐Majem L, Volatier JL, Trolle E, Tornaritis M, Busk L, Kafatos A, Fabiansson S, De Henauw S and Van Klaveren J, 2011. Dietary Exposure Assessments for Children in Europe (the EXPOCHI project): rationale, methods and design. Archives of Public Health, 69, 12 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannuzzi L, Perucatti A, Di Meo GP, Polimeno F, Ciotola F, Incarnato D, Peretti V, Caputi‐Jambrenghi A, Pecoraro A, Manniti F, D'Alessandro A and Vonghia G, 2004. Chromosome fragility in two sheep flocks exposed to dioxins during pasturage. Mutagenesis, 19, 355–359. [DOI] [PubMed] [Google Scholar]
- Iannuzzi L, Perucatti A, Genualdo V, Incarnato D, Peretti V, Rasero R, Nebbia C and Di Meo GP, 2009. Chromosome analyses in dairy cows exposed to dioxins and dioxin‐like PCBs using the SCE test. Italian Journal of Animal Science, 8, 93–95. [Google Scholar]
- IARC (International Agency for Research on Cancer), 1997. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 69. Polychlorinated dibenzo‐para‐dioxins and polychlorinated dibenzofurans, IARC, Lyon, France. [PMC free article] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer), 2012. IARC monographs on the evaluation of carcinogenic risks to humans. Chemical Agents and related occupations. Volume 100F. A review of human carcinogens. [PMC free article] [PubMed]
- Iben C, Bohm J, Tausch H, Leibetseder J and Luf W, 2003. Dioxin residues in the edible tissue of broiler chicken. Journal of Animal Physiology and Animal Nutrition, 87, 142–148. [DOI] [PubMed] [Google Scholar]
- Ichihara S, 2012. Role of AHR in the development of the liver and blood vessels In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 413–421. [Google Scholar]
- Iida T, Hirakawa H, Matsueda T, Nagayama J and Nagata T, 1999. Polychlorinated dibenzo‐p‐dioxins and related compounds: correlations of levels in human tissues and in blood. Chemosphere, 38, 2767–2774. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Tamura M, Yamashita J, Suzuki C and Tomita T, 2005. Repeated in utero and lactational 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin exposure affects male gonads in offspring, leading to sex ratio changes in F2 progeny. Toxicology and Applied Pharmacology, 206, 351–355. [DOI] [PubMed] [Google Scholar]
- Ikeno T, Miyashita C, Nakajima S, Kobayashi S, Yamazaki K, Saijo Y, Kita T, Sasaki S, Konishi K, Kajiwara J, Hori T and Kishi R, 2018. Effects of low‐level prenatal exposure to dioxins on cognitive development in Japanese children at 42 months. Science of the Total Environment, 618, 1423–1430. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Eguchi H, Tachibana T, Yoneda Y and Kawajiri K, 1998. Nuclear localization and export signals of the human aryl hydrocarbon receptor. The Journal of Biological Chemistry, 273, 2895–2904. [DOI] [PubMed] [Google Scholar]
- Ilsen A, Briet JM, Koppe JG, Pluim HJ and Oosting J, 1996. Signs of enhanced neuromotor maturation in children due to perinatal load with background levels of dioxins ‐ Follow‐up until age 2 years and 7 months. Chemosphere, 33, 1317–1326. [DOI] [PubMed] [Google Scholar]
- Inomata T, Sekiguchi M, Hirayama S, Akahori F, Shirai M, Kashiwazaki N, Ito J, Hisamatsu S, Sakita K and Ninomiya H, 2009. An assessment of mutagenic effect of 3,3’,4,4’,5 Pentachlorobiphenyl (PCB126) in Muta Mouse foetuses. Journal of Veterinary Medical Science, 71, 529–533. [DOI] [PubMed] [Google Scholar]
- Inui H, Itoh T, Yamamoto K, Ikushiro S‐I and Sakaki T, 2014. Mammalian Cytochrome P450‐dependent metabolism of polychlorinated dibenzo‐p‐dioxins and coplanar polychlorinated biphenyls. International Journal of Molecular Sciences, 15, 14044–14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Warita K, Tanida T, Sugawara T, Kitagawa H and Hoshi N, 2007. Does paternal exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) affect the sex ratio of offspring? The Journal of Veterinary Medical Science, 69, 347–52. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Ohsako S, Tasaka K, Harayama H, Miyake M, Warita K, Tanida T, Mitsuhashi T, Nanmori T, Tabuchi Y, Yokoyama T, Kitagawa H and Hoshi N, 2010. When does the sex ratio of offspring of the paternal 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) exposure decrease: in the spermatozoa stage or at fertilization? Reproduction and Toxicology, 29, 68–73. [DOI] [PubMed] [Google Scholar]
- Ishimura R, Ohsako S, Miyabara Y, Sakaue M, Kawakami T, Aoki Y, Yonemoto J and Tohyama C, 2002. Increased glycogen content and glucose transporter 3 mRNA level in the placenta of Holtzman rats after exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology and Applied Pharmacology, 178, 161–171. [DOI] [PubMed] [Google Scholar]
- Isosaari P, Vartiainen T, Hallikainen A and Ruohonen K, 2002. Feeding trial on rainbow trout: comparison of dry fish feed and Baltic herring as a source of PCDD/Fs and PCBs. Chemosphere, 48, 795–804. [DOI] [PubMed] [Google Scholar]
- Isosaari P, Kiviranta H, Lie O, Lundebye AK, Ritchie G and Vartiainen T, 2004. Accumulation and distribution of polychlorinated dibenzo‐p‐dioxin, dibenzofuran, and polychlorinated biphenyl congeners in Atlantic salmon (Salmo salar). Environmental Toxicology and Chemistry, 23, 1672–1679. [DOI] [PubMed] [Google Scholar]
- Iszatt N, Stigum H, Govarts E, Murinova LP, Schoeters G, Trnovec T, Legler J, Thomsen C, Koppen G and Eggesbo M, 2016. Perinatal exposure to dioxins and dioxin‐like compounds and infant growth and body mass index at seven years: a pooled analysis of three European birth cohorts. Environment International, 94, 399–407. [DOI] [PubMed] [Google Scholar]
- Ivnitski I, Elmaoued R and Walker MK, 2001. 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) inhibition of coronary development is preceded by a decrease in myocyte proliferation and an increase in cardiac apoptosis. Teratology, 64, 201–212. [DOI] [PubMed] [Google Scholar]
- Jackson DP, Joshi AD and Elferink CJ, 2015. Ah receptor pathway intricacies; signaling through diverse protein partners and DNA‐motifs. Toxicology Research, 4, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen R, Bostofte E, Engholm G, Hansen J, Skakkebaek NE and Moller H, 2000. Fertility and offspring sex ratio of men who develop testicular cancer: a record linkage study. Human Reproduction, 15, 1958–1961. [DOI] [PubMed] [Google Scholar]
- Jain S, Dolwick KM, Schmidt JV and Bradfield CA, 1994. Potent transactivation domains of the Ah receptor and the Ah receptor nuclear translocator map to their carboxyl termini. The Journal of Biological Chemistry, 269, 31518–31524. [PubMed] [Google Scholar]
- Jämsä T, Viluksela M, Tuomisto JT, Tuomisto J and Tuukkanen J, 2001. Effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on bone in two rat strains with different aryl hydrocarbon receptor structures. Journal of Bone and Mineral Research, 16, 1812–1820. [DOI] [PubMed] [Google Scholar]
- JEA (Japanese Environmental Agency), 1999. Report on Tolerable Daily Intake (TDI) of Dioxins and Related Compounds (Japan). Available online: https://www.env.go.jp/en/chemi/dioxins/tdi_report.pdf
- Jensen DJ and Hummel RA, 1982. Secretion of TCDD in milk and cream following the feeding of TCDD to lactating dairy cows. Bulletin of Environmental Contamination and Toxicology, 29, 440–446. [DOI] [PubMed] [Google Scholar]
- Jobst H and Aldag R, 2000. Dioxine in Lagerstätten‐Tonen. Zeitschrift für Umweltchemie und Ökotoxikologie, 12, 2–4. [Google Scholar]
- Johansson N and Hanberg A, 2000. Report from a Nordic meeting on the 1998 WHO consultation on assessment odf the health risks of dioxins; re‐evaluation of the tolerable daily intake (TDI. Organohalogen Compounds, 48, 252–255. [Google Scholar]
- Johnson L, Dickerson R, Safe SH, Nyberg CL, Lewis RP and Welsh TH Jr, 1992. Reduced Leydig cell volume and function in adult rats exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin without a significant effect on spermatogenesis. Toxicology, 76, 103–18. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Tietge JE, Jensen KM, Fernandez JD, Linnum AL, Lothenbach DB, Holcombe GW, Cook PM, Christ SA, Lattier DL and Gordon DA, 1998. Toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin to early life stage brook trout (Salvelinus fontinalis) following parental dietary exposure. Environmental Toxicology and Chemistry, 17, 2408–2421. [Google Scholar]
- Johnson ES, Shorter C, Bestervelt LL, Patterson DG, Needham LL, Piper WN, Lucier G and Nolan CJ, 2001. Serum hormone levels in humans with low serum concentrations of 2,3,7,8‐TCDD. Toxicology and Industrial Health, 17, 105–112. [DOI] [PubMed] [Google Scholar]
- Jones PB, Durrin LK, Galeazzi DR and Whitlock JP Jr, 1986. Control of cytochrome P1‐450 gene expression: analysis of a dioxin‐responsive enhancer system. Proceedings of the National Academy of Sciences of the United States of America, 83, 2802–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D, Safe S, Morcom E, Holcomb M, Coppock C and Ivie W, 1987a. Bioavailability of tritiated 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) administered to Holstein dairy cows. Chemosphere, 16, 1743–1748. [Google Scholar]
- Jones MK, Weisenburger WP, Sipes IG and Russell DH, 1987b. Circadian alterations in prolactin, corticosterone, and thyroid hormone levels and down‐regulation of prolactin receptor activity by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology and Applied Pharmacology, 87, 337–50. [DOI] [PubMed] [Google Scholar]
- Jones D, Safe S, Morcom E, Holcomb M, Coppock C and Ivie W, 1989. Bioavailability of grain and soil‐borne tritiated 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) administered to lactating Holstein cows. Chemosphere, 18, 1257–1263. [Google Scholar]
- Jones PD, Kannan K, Newsted JL, Tillitt DE, Williams LL and Giesy JP, 2001. Accumulation of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin by rainbow trout (Onchorhynchus mykiss) at environmentally relevant dietary concentrations. Environmental Toxicology and Chemistry, 20, 344–350. [PubMed] [Google Scholar]
- Jones LC and Whitlock JP Jr, 2001. Dioxin‐inducible transactivation in a chromosomal setting. Analysis of the acidic domain of the Ah receptor. The Journal of Biological Chemistry, 276, 25037–25042. [DOI] [PubMed] [Google Scholar]
- Julshamn K, Duinker A, Berntssen M, Nilsen BM, Frantzen S, Nedreaas K and Maage A, 2013. A baseline study on levels of polychlorinated dibenzo‐p‐dioxins, polychlorinated dibenzofurans, non‐ortho and mono‐ortho PCBs, non‐dioxin‐like PCBs and polybrominated diphenyl ethers in Northeast Arctic cod (Gadus morhua) from different parts of the Barents Sea. Marine Pollution Bulletin, 75, 250–258. [DOI] [PubMed] [Google Scholar]
- Jung D, Berg PA, Edler L, Ehrenthal W, Fenner D, Flesch‐Janys D, Huber C, Klein R, Koitka C, Lucier G, Manz A, Muttray A, Needham L, Papke O, Pietsch M, Portier C, Patterson D, Prellwitz W, Rose DM, Thews A and Konietzko J, 1998. Immunologic findings in workers formerly exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin and its congeners. Environmental Health Perspectives, 106, 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KK, Kim SY, Kim TG, Kang JH, Kang SY, Cho JY and Kim SH, 2007. Differential regulation of thyroid hormone receptor‐mediated function by endocrine disruptors. Archives of Pharmcal Reserach, 30, 616–23. [DOI] [PubMed] [Google Scholar]
- Jux B, Kadow S, Luecke S, Rannug A, Krutmann J and Esser C, 2011. The aryl hydrocarbon receptor mediates UVB radiation‐induced skin tanning. Journal of Investigative Dermatology, 131, 203–10. [DOI] [PubMed] [Google Scholar]
- Kajiwara J, Todaka T, Hirakawa H, Hori T, Onozuka D, Takao Y, Hirata T, Iida T, Uchi H and Furue M, 2011. The difference between male and female dioxin concentrations in the blood of yusho patients. Fukuoka Acta Medica, 102, 140–144. [PubMed] [Google Scholar]
- Kakeyama M, Sone H, Miyabara Y and Tohyama C, 2003. Perinatal exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin alters activity‐dependent expression of BDNF mRNA in the neocortex and male rat sexual behavior in adulthood. Neurotoxicology, 24, 207–217. [DOI] [PubMed] [Google Scholar]
- Kalthoff S, Ehmer U, Freiberg N, Manns MP and Strassburg CP, 2010. Interaction between oxidative stress sensor Nrf2 and xenobiotic‐activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP‐glucuronosyltransferase 1A10. The Journal of Biological Chemistry, 285, 5993–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamińska J, Ligocka D, Zieliński M, Czerska M and Jakubowski M, 2014. The use of PowerPrep and HRGC/HRMS for biological monitoring of exposure toPCDD, PCDF and dl‐PCB in Poland. International Journal of Hygiene and Environmental Health, 217, 11–16. [DOI] [PubMed] [Google Scholar]
- Kang HK, Dalager NA, Needham LL, Patterson DG Jr, Lees PS, Yates K and Matanoski GM, 2006. Health status of Army Chemical Corps Vietnam veterans who sprayed defoliant in Vietnam. American Journal of Industrial Medicine, 49, 875–884. [DOI] [PubMed] [Google Scholar]
- Kanimura H, Koga N, Oguri K, Yoshimura H, Honda Y and Nakano M, 1998. Enhanced faecal excretion of 23478‐pentachlorodibenzofuran in rats by a long‐terms treatment with activated charcoal beads. Xenobiotica, 18, 585–574. [DOI] [PubMed] [Google Scholar]
- Karl H and Lahrssen‐Wiederholt M, 2009. Dioxin and dioxin‐like PCB levels in cod liver and muscle from different fishing grounds of the North and Baltic Sea and the North Atlantic. Journal für Verbraucherschutz und Lebensmittelsicherheit, 4, 247e255. [Google Scholar]
- Karl H, Kammann U, Aust M‐O, Manthey‐Karl M, Lüth A and Kanisch G, 2016. Large scale distribution of dioxins, PCBs, heavy metals, PAH metabolite and radionuclides in cod (Gadus morhua) from the North Atlantic and its adjacent seas. Chemosphere, 149, 294–303. [DOI] [PubMed] [Google Scholar]
- Karman BN, Basavarajappa MS, Craig ZR and Flaws JA, 2012a. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin activates the aryl hydrocarbon receptor and alters sex steroid hormone secretion without affecting growth of mouse antral follicles in vitro . Toxicology and Applied Pharmacology, 261, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman BN, Hernández‐Ochoa I, Ziv‐Gal A and Flaws JA, 2012b. Involvement of the AHR in development and functioning of the female and male reproductive systems In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 437–466. [Google Scholar]
- Kashimoto T, Miyata H, Kunita S, Tung TC, Hsu ST, Chang KJ, Tang SY, Ohi G, Nakagawa J and Yamamoto SI, 1981. Role of polychlorinated dibenzofuran in Yusho (PCB poisoning). Archives of Environmental Health, 36, 321–326. [DOI] [PubMed] [Google Scholar]
- Kashimoto T and Miyata H, 1987. Differences between Yusho and other kinds of poisoning involving only PCBs In: Waid JS, (ed.). PCBs and the environment, Vol. III. CRC Press, Baton Raton: pp 1–26. [Google Scholar]
- Katic J, Cemeli E, Baumgartner A, Laubenthal J, Bassano I, Stolevik SB, Granum B, Namork E, Nygaard UC, Lovik M, van Leeuwen D, Loock KV, Anderson D, Fucic A and Decordier I, 2010. Evaluation of the genotoxicity of 10 selected dietary/environmental compounds with the in vitro micronucleus cytokinesis‐block assay in an interlaboratory comparison. Food and Chemical Toxicology, 48, 2612–2623. [DOI] [PubMed] [Google Scholar]
- Kato Y, Ikushiro S, Haraguchi K, Yamazaki T, Ito Y, Suzuki H, Kimura R, Yamada S, Inoue T and Degawa M, 2004. A possible mechanism for decrease in serum thyroxine level by polychlorinated biphenyls in Wistar and Gunn rats. Toxicological Sciences, 81, 309–315. [DOI] [PubMed] [Google Scholar]
- Kattainen H, Tuukkanen J, Simanainen U, Tuomisto JT, Kovero O, Lukinmaa PL, Alaluusua S, Tuomisto J and Viluksela M, 2001. In utero/lactational 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin exposure impairs molar tooth development in rats. Toxicology and Applied Pharmacology, 174, 216–24. [DOI] [PubMed] [Google Scholar]
- Kedderis LB, Diliberto JJ, Linko P, Goldstein JA and Birnbaum LS, 1991. Disposition of 2,3,7,8‐tetrabromodibenzo‐p‐dioxin and 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the rat ‐ biliary‐excretion and induction of cytochromes CYP1A1 and CYP1A2. Toxicology and Applied Pharmacology, 111, 163–172. [DOI] [PubMed] [Google Scholar]
- Keller JM, Huet‐Hudson YM and Leamy LJ, 2007. Qualitative effects of dioxin on molars vary among inbred mouse strains. Archives of Oral Biology, 52, 450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JM, Huet‐Hudson Y and Leamy LJ, 2008. Effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on molar development among non‐resistant inbred strains of mice: a geometric morphometric analysis. Growth Development and Aging, 71, 3–16. [PubMed] [Google Scholar]
- Kelling CK, Menahan LA and Peterson RE, 1987. Hepatic indices of thyroid status in rats treated with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Biochemical Pharmacology, 36, 283–91. [DOI] [PubMed] [Google Scholar]
- Kennedy SW, Lorenzen A, James CA and Collins BT, 1993. Ethoxyresorufin‐O‐deethylase and porphyrin analysis in chicken embryo hepatocyte cultures with a fluorescence multiwell plate reader. Analytical Biochemistry, 211, 102–112. [DOI] [PubMed] [Google Scholar]
- Kennedy SW, Lorenzen A, Jones SP, Hahn ME and Stegeman JJ, 1996. Cytochrome P4501A induction in avian hepatocyte cultures: a promising approach for predicting the sensitivity of avian species to toxic effects of halogenated aromatic hydrocarbons. Toxicology and Applied Pharmacology, 141, 214–230. [DOI] [PubMed] [Google Scholar]
- Kennedy GD, Nukaya M, Moran SM, Glover E, Weinberg S, Balbo S, Hecht SS, Pitot HC, Drinkwater NR and Bradfield CA, 2014. Liver tumor promotion by 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin is dependent on the Aryl hydrocarbon receptor and TNF/IL‐1 receptors. Toxicological Sciences, 140, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerger BD, Leung H‐W, Scott PK and Paustenbach DJ, 2007. An adaptable internal dose model for risk assessment of dietary and soil dioxin exposures in young children. Toxicological Sciences, 100, 224–237. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI, Baecher‐Steppan L, Smith BB, Youngberg JA, Henderson MC and Buhler DR, 1990a. Role of the Ah locus in suppression of cytotoxic T lymphocyte activity by halogenated aromatic hydrocarbons (PCBs and TCDD): structure‐activity relationships and effects in C57Bl/6 mice congenic at the Ah locus. Fundamental and Applied Toxicology: Official Journal of the Society of Toxicology, 14, 532–541. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI, Steppan LB, Brauner JA, Deyo JA, Henderson MC, Tomar RS and Buhler DR, 1990b. Influence of the Ah locus on the humoral immunotoxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin: evidence for Ah‐receptor‐dependent and Ah‐receptor‐independent mechanisms of immunosuppression. Toxicology and Applied Pharmacology, 105, 26–36. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI, Wagner SL, Schmotzer WB, Hackett M, Schrader K and Hultgren B, 1992. Dioxin intoxication from chronic exposure of horses to pentachlorophenol‐contaminated wood shavings. Journal of the American Veterinary Medical Association, 201, 296–302. [PubMed] [Google Scholar]
- Kerkvliet NI, 2002. Recent advances in understanding the mechanisms of TCDD immunotoxicity. International Immunopharmacology, 2, 277–291. [DOI] [PubMed] [Google Scholar]
- Kerkvliet NI, 2012. TCDD: an environmental immunotoxicant reveals a novel pathway of immunoregulation. A 30‐year odyssey. Toxicologic Pathology, 40, 138–142. [DOI] [PubMed] [Google Scholar]
- Kern PA, Said S, Jackson WG Jr and Michalek JE, 2004. Insulin sensitivity following agent orange exposure in Vietnam veterans with high blood levels of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. The Journal of Clinical Endocrinology and Metabolism, 89, 4665–4672. [DOI] [PubMed] [Google Scholar]
- Ketchum NS and Michalek JE, 2005. Postservice mortality of Air Force veterans occupationally exposed to herbicides during the Vietnam War: 20‐year follow‐up results. Military Medicine, 170, 406–413. [DOI] [PubMed] [Google Scholar]
- Ketchum NS, Michalek JE and Burton JE, 1999. Serum dioxin and cancer in veterans of Operation Ranch Hand. American Journal of Epidemiology, 149, 630–639. [DOI] [PubMed] [Google Scholar]
- Kijlstra A, Traag WA and Hoogenboom LAP, 2007. Effect of flock size on dioxin levels in eggs from chickens kept outside. Poultry Science, 86, 2042–2048. [DOI] [PubMed] [Google Scholar]
- Kim D and Guengerich FP, 2005. Cytochrome P450 activation of arylamines and heterocyclic amines. Annual Review of Pharmacology and Toxicology, 45, 27–49. [DOI] [PubMed] [Google Scholar]
- Kim M, Kim D‐G, Choi S‐W, Guerrero P, Norambuena J and Chung GS, 2011. Formation of polychlorinated dibenzo‐p‐dioxins/dibenzofurans, PCDD/Fs. from a refinery process for zinc oxide used in feed additives: a source of dioxin contamination in Chilean pork. Chemosphere, 82, 1225–1229. [DOI] [PubMed] [Google Scholar]
- Kimbrough RD, Carter CD, Liddle JA and Cline RE, 1977. Epidemiology and pathology of a tetrachlorodibenzodioxin poisoning episode. Archives of Environmental Health, 32, 77–86. [DOI] [PubMed] [Google Scholar]
- Kimura E, Kubo K, Matsuyoshi C, Benner S, Hosokawa M, Endo T, Ling W, Kohda M, Yokoyama K, Nakajima K, Kakeyama M and Tohyama C, 2015. Developmental origin of abnormal dendritic growth in the mouse brain induced by in utero disruption of aryl hydrocarbon receptor signaling. Neurotoxicology and Teratology, 52, 42–50. [DOI] [PubMed] [Google Scholar]
- King FG, Dedrick RL, Collins JM, Matthews HB and Birnbaum LS, 1983. A physiologically model for the pharmacokinetics of 2,3,7,8‐tetrachlorodibenzofuran in several species. Toxicology and Applied Pharmacology, 67, 390–400. [DOI] [PubMed] [Google Scholar]
- Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C and Diefenbach A, 2011. Natural Aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science, 334, 1561–156. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Kikuchi Y, Watanabe S, Waechter G, Sakurai H and Takada T, 2000. Health effects of chronic exposure to polychlorinated dibenzo‐p‐dioxins (PCDD), dibenzofurans (PCDF) and coplanar PCB (Co‐PCB) of municipal waste incinerator workers. Journal of Epidemiology, 10, 262–270. [DOI] [PubMed] [Google Scholar]
- Kiukkonen A, Viluksela M, Sahlberg C, Alaluusua S, Tuomisto JT, Tuomisto J and Lukinmaa PL, 2002. Response of the incisor tooth to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in a dioxin‐resistant and a dioxin‐sensitive rat strain. Toxicological Sciences, 69, 482–9. [DOI] [PubMed] [Google Scholar]
- Kiukkonen A, Sahlberg C, Lukinmaa PL, Alaluusua S, Peltonen E and Partanen AM, 2006. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin specifically reduces mRNA for the mineralization‐related dentin sialophosphoprotein in cultured mouse embryonic molar teeth. Toxicology and Applied Pharmacology, 216, 399–406. [DOI] [PubMed] [Google Scholar]
- Kiviranta H, Vartiainen T, Verta M, Tuomisto JT and Tuomisto J, 2000. High fish‐specific dioxin concentrations in Finland. Lancet, 355, 1883–1885. [DOI] [PubMed] [Google Scholar]
- Kiviranta H, Vartiainen T and Tuomisto J, 2002. Polychlorinated dibenzo‐p‐dioxins, dibenzofurans, and biphenyls in fishermen in Finland. Environmental Health Perspectives, 110, 355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeman JM, Olson JR, Chen SM and Peterson RE, 1986. Metabolism and disposition of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in rainbow trout. Toxicology and Applied Pharmacology, 83, 391–401. [DOI] [PubMed] [Google Scholar]
- Kleeman JM, Olson JR and Peterson RE, 1988. Species‐differences in 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin toxicity and biotransformation in fish. Fundamental and Applied Toxicology, 10, 206–213. [DOI] [PubMed] [Google Scholar]
- Knerr S and Schrenk D, 2006. Carcinogenicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in experimental models. Molecular Nutrition and Food Research, 50, 897–907. [DOI] [PubMed] [Google Scholar]
- Ko CI and Puga A, 2012. Epigenetic mechanisms in AHR function In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 157–178. [Google Scholar]
- Kociba RJ, Keyes DG, Beyer JE, Carreon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Barnard SD, Hummel RA and Humiston CG, 1978. Results of a two‐year chronic toxicity and oncogenicity study of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in rats. Toxicology and Applied Pharmacology, 46, 279–303. [DOI] [PubMed] [Google Scholar]
- Koga N, Beppu M and Yoshimura H, 1990. Metabolism in vivo of 3,4,5,3’,4’‐pentachlorobiphenyl and toxicological assessment of the metabolite in rats. Journal of pharmacobio‐dynamics, 13, 497–506. [DOI] [PubMed] [Google Scholar]
- Konishi K, Sasaki S, Kato S, Ban S, Washino N, Kajiwara J, Todaka T, Hirakawa H, Hori T, Yasutake D and Kishi R, 2009. Prenatal exposure to PCDDs/PCDFs and dioxin‐like PCBs in relation to birth weight. Environmental Research, 109, 906–913. [DOI] [PubMed] [Google Scholar]
- Koopman‐Esseboom C, Huisman M, Weisglas‐Kuperus N, Boersma ER, de Ridder MA, Van der Paauw CG, Tuinstra LG and Sauer PJ, 1994a. Dioxin and PCB levels in blood and human milk in relation to living areas in The Netherlands. Chemosphere, 29, 2327–2338. [DOI] [PubMed] [Google Scholar]
- Koopman‐Esseboom C, Morse DC, Weisglaskuperus N, Lutkeschipholt IJ, Vanderpaauw CG, Tuinstra L, Brouwer A and Sauer PJJ, 1994b. Effects of dioxins and polychlorinated‐biphenyls on thyroid‐hormone status of pregnant‐women and their infants. Pediatric Research, 36, 468–473. [DOI] [PubMed] [Google Scholar]
- Koopman‐Esseboom C, Weisglas‐Kuperus N, deRidder MA , derVan Paauw CG , Tuinstra LG and Sauer PJ, 1996. Effects of polychlorinated biphenyl/dioxin exposure and feeding type on infants’ mental and psychomotor development. Pediatrics, 97, 700–706. [PubMed] [Google Scholar]
- Kopec AK, Burgoon LD, Ibrahim‐Aibo D, Burg AR, Lee AW, Tashiro C, Potter D, Sharratt B, Harkema JR, Rowlands JC, Budinsky RA and Zacharewski TR, 2010. Automated dose‐response analysis and comparative toxicogenomic evaluation of the hepatic effects elicited by TCDD, TCDF, and PCB126 in C57BL/6 mice. Toxicological Sciences: an Official Journal of the Society of Toxicology, 118, 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen K, Kukkonen JVK and Lindstrom‐Seppa P, 1998. Chemical accumulation and biotransformation enzyme activities of rainbow trout embryos in waterborne exposure to PCB‐77. Marine Environmental Research, 46, 475–478. [Google Scholar]
- Koponen K, Lindstrom‐Seppa P and Kukkonen JVK, 2000. Accumulation pattern and biotransformation enzyme induction in rainbow trout embryos exposed to sublethal aqueous concentrations of 3,3’,4,4 ‘‐tetrachlorobiphenyl. Chemosphere, 40, 245–253. [DOI] [PubMed] [Google Scholar]
- Korenaga T, Fukusato T, Ohta M, Asaoka K, Murata N, Arima A and Kubota S, 2007. Long‐term effects of subcutaneously injected 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on the liver of rhesus monkeys. Chemosphere, 67, S399–S404. [DOI] [PubMed] [Google Scholar]
- Korkalainen M, Tuomisto J and Pohjanvirta R, 2000. Restructured transactivation domain in hamster AH receptor. Biochemical and Biophysical Research Communications, 273, 272–281. [DOI] [PubMed] [Google Scholar]
- Korkalainen M, Tuomisto J and Pohjanvirta R, 2001. The AH receptor of the most dioxin‐sensitive species, guinea pig, is highly homologous to the human AH receptor. Biochemical and Biophysical Research Communications, 285, 1121–1129. [DOI] [PubMed] [Google Scholar]
- Korrick SA, Lee MM, Williams PL, Sergeyev O, Burns JS, Patterson DG, Turner WE, Needham LL, Altshul L, Revich B and Hauser R, 2011. Dioxin exposure and age of pubertal onset among Russian boys. Environmental Health Perspectives, 119, 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korytko PJ, Casey AC, Bush B and Quimby FW, 1999. Induction of hepatic cytochromes P450 in dogs exposed to a chronic low dose of polychlorinated biphenyls. Toxicological Sciences, 47, 52–61. [DOI] [PubMed] [Google Scholar]
- Koshakji RP, Harbison RD and Bush MT, 1984. Studies on the metabolic fate of [14C]2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the mouse. Toxicology and Applied Pharmacology, 73, 69–77. [DOI] [PubMed] [Google Scholar]
- Koskenniemi JJ, Virtanen HE, Kiviranta H, Damgaard IN, Matomaki J, Thorup JM, Hurme T, Skakkebaek NE, Main KM and Toppari J, 2015. Association between levels of persistent organic pollutants in adipose tissue and cryptorchidism in early childhood: a case‐control study. Environmental Health, 14, 78–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalova N, Nault R, Crawford R, Zacharewski TR and Kaminski NE, 2017. Comparative analysis of TCDD‐induced AhR‐mediated gene expression in human, mouse and rat primary B cells. Toxicology and Applied Pharmacology, 316, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kransler KM, McGarrigle BP and Olson JR, 2007a. Comparative developmental toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the hamster, rat and guinea pig. Toxicology, 229, 214–225. [DOI] [PubMed] [Google Scholar]
- Kransler KM, Tonucci DA, McGarrigle BP, Napoli JL and Olson JR, 2007b. Gestational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin alters retinoid homeostasis in maternal and perinatal tissues of the Holtzman rat. Toxicology and Applied Pharmacology, 224, 29–38. [DOI] [PubMed] [Google Scholar]
- Kransler KM, McGarrigle BP, Russell RJ and Olson JR, 2008. Effects of Helicobacter infection on developmental toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in Holtzman rats. Lab Animal, 37, 171–175. [DOI] [PubMed] [Google Scholar]
- Kransler KM, McGarrigle BP, Swartz DD and Olson JR, 2009. Lung development in the Holtzman Rat is adversely affected by gestational exposure to 2,3,7,8‐Tetrachlorodibenzo‐p‐Dioxin. Toxicological Sciences, 107, 498–511. [DOI] [PubMed] [Google Scholar]
- Kreuzer PE, Csanády GA, Baur C, Kessler W, Päpke O, Greim H and Filser JG, 1997. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) and congeners in infants. A toxicokinetic model of human lifetime body burden by TCDD with special emphasis on its uptake by nutrition. Archives of Toxicology, 71, 383–400. [DOI] [PubMed] [Google Scholar]
- Krogenaes AK, Ropstad E, Gutleb AC, Hardnes N, Berg V, Dahl E and Fowler PA, 2014. In utero exposure to environmentally relevant concentrations of PCB 153 and PCB 118 disrupts fetal testis development in sheep. Journal of Toxicology and Environmental Health‐Part a‐Current Issues, 77, 628–649. [DOI] [PubMed] [Google Scholar]
- Kuehl DW, Cook PM, Batterman AR, Lothenbach DB, Butterworth BC and Johnson DL, 1985. Bioavailability of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin from municipal incinerator fly‐ash to fresh‐water fish. Chemosphere, 14, 427–437. [Google Scholar]
- Kuehl DW, Cook PM, Batterman AR, Lothenbach D and Butterworth BC, 1987. Bioavailability of polychlorinated dibenzo‐p‐dioxins and dibenzofurans from contaminated Wisconsin River sediment to carp. Chemosphere, 16, 667–679. [Google Scholar]
- Kumar MB, Ramadoss P, Reen RK, Vanden Heuvel JP and Perdew GH, 2001. The Q‐rich subdomain of the human Ah receptor transactivation domain is required for dioxin‐mediated transcriptional activity. The Journal of Biological Chemistry, 276, 42302–42310. [DOI] [PubMed] [Google Scholar]
- Kunisue T, Nakanishi S, Watanabe M, Abe T, Nakatsu S, Kawauchi S, Sano A, Horii A, Kano Y and Tanabe S, 2005. Contamination status and accumulation features of persistent organochlorines in pet dogs and cats from Japan. Environmental Pollution, 136, 465–476. [DOI] [PubMed] [Google Scholar]
- Kunisue T, Takayanagi N, Tsubota T and Tanabe S, 2007. Persistent organochlorines in raccoon dogs (Nyctereutes procyonoides) from Japan: Hepatic sequestration of oxy‐chlordane. Chemosphere, 66, 203–211. [DOI] [PubMed] [Google Scholar]
- Kuo LC, Cheng LC, Lin CJ and Li LA, 2013. Dioxin and estrogen signaling in lung adenocarcinoma cells with different aryl hydrocarbon receptor/estrogen receptor alpha phenotypes. American Journal of Respiratory Cell and Molecular Biology, 49, 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuratsune M, Yoshimura T, Matsuzaka J and Yamaguchi A, 1971. Yusho, a poisoning caused by rice oil contaminated with polychlorinated biphenyls. HSMHA Health Reports, 86, 1083–1091. [PMC free article] [PubMed] [Google Scholar]
- Kuratsune M, Yoshimura T, Matsuzaka J and Yamaguchi A, 1972. Epidemiologic study on Yusho, a poisoning caused by ingestion of rice oil contaminated with a commercial brand of polychlorinated biphenyls. Environmental Health Perspectives, 1, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita H, Schnekenburger M, Ovesen JL, Xia Y and Puga A, 2014. The Ah receptor recruits IKKalpha to its target binding motifs to phosphorylate serine‐10 in histone H3 required for transcriptional activation. Toxicological Sciences, 139, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvalem HE, Knutsen HK, Thomsen C, Haugen M, Stigum H, Brantsæter AL, Frøshaug M, Lohman N, Päpke O, Becher G, Alexander J and Meltzer HM, 2009. Role of dietary patterns for dioxin and PCB exposure. Molecular Nutrition and Food Research, 53, 1538–1451. [DOI] [PubMed] [Google Scholar]
- Kvalem HE, Brantsaeter AL, Meltzer HM, Stigum H, Thomsen C, Haugen M, Alexander J and Knutsen HK, 2012. Development and validation of prediction models for blood concentrations of dioxins and PCBs using dietary intakes. Environment International, 50, 15–21. [DOI] [PubMed] [Google Scholar]
- LaKind JS, Berlin CM and Naiman DQ, 2001. Infant exposure to chemicals in breast milk in the United States: what we need to learn from a breast milk monitoring program. Environmental Health Perspectives, 109, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakind JS, 2007. Recent global trends and physiologic origins of dioxins and furans in human milk. Journal of Exposure Science and Environmental Epidemiology, 17, 510–524. [DOI] [PubMed] [Google Scholar]
- Lam T, Williams PL, Lee MM, Korrick SA, Birnbaum LS, Burns JS, Sergeyev O, Revich B, Altshul LM, Patterson DG Jr, Turner WE and Hauser R, 2014. Prepubertal organochlorine pesticide concentrations and age of pubertal onset among Russian boys. Environment International, 73, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T, Williams PL, Lee MM, Korrick SA, Birnbaum LS, Burns JS, Sergeyev O, Revich B, Altshul LM, Patterson DG Jr and Hauser R, 2015. Prepubertal serum concentrations of organochlorine pesticides and age at sexual maturity in Russian boys. Environmental Health Perspectives, 123, 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgren O, Shim YK, Michalek J, Costello R, Burton D, Ketchum N, Calvo KR, Caporaso N, Raveche E, Middleton D, Marti G and Vogt RF Jr, 2015. Agent orange exposure and monoclonal gammopathy of undetermined significance: an operation ranch hand veteran cohort study. JAMA Oncology, 1, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi MT, Needham LL, Lucier G, Mocarelli P, Bertazzi PA and Caporaso N, 1997. Concentrations of dioxin 20 years after Seveso. Lancet, 349, 1811–1811. [DOI] [PubMed] [Google Scholar]
- Landi MR, Consonni D, OPatterson DG, Needham LL, Lucier G, Brambilla P, Cazzaniga MA, Mocarelli P, Pesatori AC, Bertzzi PA and Caporaso NE, 1998. 2,3,7,8‐Tetrachlorodibenzo‐p‐Dioxin Plasma Levels in Seveso 20 Years after the Accident. Environmental Health Perspectives, 106, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanting CI, Patandin S, Fidler V, Weisglas‐Kuperus N, Sauer PJJ, Boersma ER and Touwen BCL, 1998. Neurological condition in 42‐month‐old children in relation to pre‐ and postnatal exposure to polychlorinated biphenyls and dioxins. Early Human Development, 50, 283–292. [DOI] [PubMed] [Google Scholar]
- Larsen PR, Silva JE and Kaplan MM, 1981. Relationships between circulating and intracellular thyroid hormones: physiological and clinical implications. Endocrine Reviews, 2, 87–102. [DOI] [PubMed] [Google Scholar]
- Larsson M, van den Berg M, Brenerova P, van Duursen MB, van Ede KI, Lohr C, Luecke‐Johansson S, Machala M, Neser S, Pencikova K, Poellinger L, Schrenk D, Strapacova S, Vondracek J and Andersson PL, 2015. Consensus toxicity factors for polychlorinated dibenzo‐p‐dioxins, dibenzofurans, and biphenyls combining in silico models and extensive in vitro screening of AhR‐mediated effects in human and rodent cells. Chemical Research in Toxicology, 28, 641–650. [DOI] [PubMed] [Google Scholar]
- Laurent C, Feidt C, Grova N, Mpassi D, Lichtfouse E, Laurent F and Rychen G, 2002. Portal absorption of 14C after ingestion of spiked milk with 14C‐benzo[a]pyrene or 14C‐TCDD in growing pigs. Chemosphere, 48, 843–848. [DOI] [PubMed] [Google Scholar]
- Lavrusenko GP, Zheltov VA, Ponomarev VN, Lavrov AL, Volkov VN, Yerokhina LM, Malogolovkin SA, Novikov BV, Dmitrenko VV and Shadymova NG, 1996. Effect of dioxine on the immune system of farm animals. Russian Agricultural Sciences, ???, 8–14. [Google Scholar]
- Lawson CC, Schnorr TM, Whelan EA, Deddens JA, Dankovic DA, Piacitelli LA, Sweeney MH and Connally LB, 2004. Paternal occupational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin and birth outcomes of offspring: birth weight, preterm delivery, and birth defects. Environmental Health Perspectives, 112, 1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD and Colonna M, 2012. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature Immunology, 13, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson S and Summers JD, 2008. Commercial Poultry Nutrition, 3rd Edition Nottingham University Press, Guelph, Ontario, Canada: 412 pp. [Google Scholar]
- Leijs MM, Koppe JG, Olie K, van Aalderen WM, Voogt P, Vulsma T, Westra M and ten Tusscher GW, 2008. Delayed initiation of breast development in girls with higher prenatal dioxin exposure; a longitudinal cohort study. Chemosphere, 73, 999–1004. [DOI] [PubMed] [Google Scholar]
- Leijs MM, Koppe JG, Olie K, van Aalderen WMC, de Voogt P and ten Tusscher GW, 2009. Effects of dioxins, PCBs, and PBDEs on immunology and hematology in adolescents. Environmental Science and Technology, 43, 7946–7951. [DOI] [PubMed] [Google Scholar]
- Leijs MM, ten Tusscher GW, Olie K, van Teunenbroek T, van Aalderen WM, de Voogt P, Vulsma T, Bartonova A, Krayer von Krauss M, Mosoiu C, Riojas‐Rodriguez H, Calamandrei G and Koppe JG, 2012. Thyroid hormone metabolism and environmental chemical exposure. Environmental Health, 11(Suppl 1), S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijs MM, Koppe JG, Vulsma T, Olie K, vanAalderen WMC , deVoogt P , Legler J and ten Tusscher GW, 2017. Alterations in the programming of energy metabolism in adolescents with background exposure to dioxins, dl‐PCBs and PBDEs. Plos One, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung HW, Ku RH, Paustenbach DJ and Andersen ME, 1988. A physiological‐based pharmacokinetic model for 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in C57BL/6J and DBA/2J mice. Toxicology Letters, 42, 15–28. [DOI] [PubMed] [Google Scholar]
- Leung HW, Poland A, Paustenbach DJ, Murray FJ and Andersen ME, 1990a. Pharmacokinetics of [125I]‐2‐iodo‐3,7,8‐trichlorodibenzo‐p‐dioxin in mice: analysis with a physiological modeling approach. Toxicology and Applied Pharmacology, 103, 411–419. [DOI] [PubMed] [Google Scholar]
- Leung HW, Paustenbach DJ, Murray FJ and Andersen ME, 1990b. A physiological pharmacokinetic description of the tissue distribution and enzyme‐inducing properties of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the rat. Toxicology and Applied Pharmacology, 103, 399–410. [DOI] [PubMed] [Google Scholar]
- Leung H‐W, Kerger BD and Paustenbach DJ, 2006. Elimination half‐lives of selected polychlorinated dibenzodioxins and dibensofurans in breast‐fed human infants. Journal of Toxicology and Environmental Health, Part A, 69, 437–443. [DOI] [PubMed] [Google Scholar]
- Li X, Johnson DC and Rozman KK, 1995. Effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) on estrous cyclicity and ovulation in female Sprague‐Dawley rats. Toxicology Letters, 78, 219–22. [DOI] [PubMed] [Google Scholar]
- Li B, Liu HY, Dai LJ, Lu JC, Yang ZM and Huang L, 2006. The early embryo loss caused by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin may be related to the accumulation of this compound in the uterus. Reproductive Toxicology, 21, 301–306. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang L, Wu Y, Liu Y, Zhou P, Wen S, Liu J, Zhao Y and Li X, 2009. A national survey of polychlorinated dioxins, furans (PCDD/Fs) and dioxin‐like polychlorinated biphenyls (dl‐PCBs) in human milk in China. Chemosphere, 75, 1236–1242. [DOI] [PubMed] [Google Scholar]
- Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C and Veldhoen M, 2011. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell, 147, 629–640. [DOI] [PubMed] [Google Scholar]
- Li M‐C, Tsai P‐C, Chen P‐C, Hsieh C‐J, Guo Y‐LL and Rogan WJ, 2013a. Mortality after exposure to polychlorinated biphenyls and dibenzofurans: 30 years after the “Yucheng Accident”. Environmental Research, 120, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lan L, Klaassen Z, Shah SR, Moses KA and Terris MK, 2013b. High level of dioxin‐TEQ in tissue is associated with Agent Orange exposure but not with biochemical recurrence after radical prostatectomy. Prostate Cancer and Prostatic Diseases, 16, 376–381. [DOI] [PubMed] [Google Scholar]
- Li CY, Renaud HJ, Klaassen CD and Cui JY, 2015. Age‐specific regulation of drug‐processing genes in mouse liver by ligands of xenobiotic‐sensing transcription factors. Drug Metabolism and Disposition, 44, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem AKD, Hoogerbrugge R, Kootstra PR, van der Velde EG and de Jong APJM, 1991. Occurrence of dioxins in cow's milk in the vicinity of municipal waste incinerators and a metal reclamation plant in the Netherlands. Chemosphere, 23, 1975–1984. [Google Scholar]
- Lignell S, Aune M, Darnerud PO, Cnattingius S and Glynn A, 2009. Persistent organochlorine and organobromine compounds in mother's milk from Sweden 1996‐2006: Compound‐specific temporal trends. Environmental Research, 109, 760–767. [DOI] [PubMed] [Google Scholar]
- Lignell S, Aune M, Darnerud PO, Soeria‐Atmadja D, Hanberg A, Larsson S and Glynn A, 2011. Large variation in breast milk levels of organohalogenated compounds is dependent on mother's age, changes in body composition and exposures early in life. Journal of Environmental Monitoring, 13, 1607–1616. [DOI] [PubMed] [Google Scholar]
- Lignell S, Winkvist A, Bertz F, Rasmussen KM, Glynn A, Aune M and Brekke HK, 2016. Environmental organic pollutants in human milk before and after weight loss. Chemosphere, 159, 96–102. [DOI] [PubMed] [Google Scholar]
- Lin TM, Ko K, Moore RW, Simanainen U, Oberley TD and Peterson RE, 2002. Effects of aryl hydrocarbon receptor null mutation and in utero and lactational 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin exposure on prostate and seminal vesicle development in C57BL/6 mice. Toxicological Sciences, 68, 479–487. [DOI] [PubMed] [Google Scholar]
- Lin YS, Caffrey JL, Hsu PC, Chang MH, Faramawi MF and Lin JW, 2012. Environmental exposure to dioxin‐like compounds and the mortality risk in the U.S. population. International Journal of Hygiene and Environmental Health, 215, 541–546. [DOI] [PubMed] [Google Scholar]
- Lindebro MC, Poellinger L and Whitelaw ML, 1995. Protein‐protein interaction via PAS domains: role of the PAS domain in positive and negative regulation of the bHLH/PAS dioxin receptor‐Arnt transcription factor complex. The EMBO Journal, 14, 3528–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindén J, Lensu S, Tuomisto J and Pohjanvirta R, 2010. Dioxins, the aryl hydrocarbon receptor and the central control of body energy balance. Frontiers in Neuroendocrinology, 31, 452–478. [DOI] [PubMed] [Google Scholar]
- Lindström GUM, Sjostrom M, Swanson SE, Furst P, Kruger C, Meemken HA and Groebel W, 1988. Multivariate statistical approach to a data set of dioxin and furan contaminations in human‐milk. Bulletin of Environmental Contamination and Toxicology, 40, 641–646. [DOI] [PubMed] [Google Scholar]
- Lipsitz L, Powell D, Bursian S and Tanaka D, 1997. Assessment of cerebral hemispheric symmetry in hatchling chickens exposed in ovo to polychlorinated biphenyl congeners. Archives of Environmental Contamination and Toxicology, 32, 399–406. [DOI] [PubMed] [Google Scholar]
- Liu G, Asanoma K, Takao T, Tsukimori K, Uchi H, Furue M, Kato K and Wake N, 2015. Aryl hydrocarbon receptor SNP‐130 C/T associates with dioxins susceptibility through regulating its receptor activity and downstream effectors including interleukin 24. Toxicology letters, 232, 384–392. [DOI] [PubMed] [Google Scholar]
- Lloyd OL, Lloyd MM, Williams FLR, McKenzie A and Hay A, 1991. Toxicity from ragwort and fat cow syndrome, or from industrial chemicals: the value of epidemiologic analysis for interpreting clinicopathological findings. Science of the Total Environment, 106, 83–96. [DOI] [PubMed] [Google Scholar]
- Lock EJ, Fjelldal PG, Torstensen BE, Bjornevik M, Breck O, Johansen J, Reynolds P, Sigholt T, Joerum N, Jakobsen JV, Ruohonen K, Waagbo R and Berntssen MHG, 2011. Dietary decontaminated fish oil has no negative impact on fish performance, flesh quality or production‐related diseases in Atlantic salmon (Salmo salar). Aquaculture Nutrition, 17, 760–772. [Google Scholar]
- Löfroth G and Zeführ Y, 1992. Polychlorinated dibenzo‐p‐dioxins (PCDDs) and dibenzofurans (PCDFs) in mainstream and sidestream cigarette smoke. Bulletin of Environmental Contamination and Toxicology, 48, 789–794. [DOI] [PubMed] [Google Scholar]
- Longnecker MP and Michalek JE, 2000. Serum dioxin level in relation to diabetes mellitus among Air Force veterans with background levels of exposure. Epidemiology, 11, 44–48. [DOI] [PubMed] [Google Scholar]
- Loonen H, Tonkes M, Parsons JR and Govers HAJ, 1994. Bioconcentration of polychlorinated dibenzo‐p‐dioxins and polychlorinated dibenzofurans in guppies after aqueous exposure to a complex PCDD PCDF mixture ‐ Relationship with molecular‐structure. Aquatic Toxicology, 30, 153–169. [Google Scholar]
- Lovett AA, Foxall CD, Ball DJ and Creaser CS, 1998. The Panteg monitoring project: comparing PCB and dioxin concentrations in the vicinity of industrial facilities. Journal of Hazardous Materials, 61, 175–185. [Google Scholar]
- Lucier GW, McDaniel OS, Hook GE, Fowler BA, Sonawane BR and Faeder E, 1973. TCDD‐induced changes in rat liver microsomal enzymes. Environmental Health Perspectives, 5, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinmaa PL, Sahlberg C, Leppäniemi A, Partanen AM, Kovero O, Pohjanvirta R, Tuomisto J and Alaluusua S, 2001. Arrest of rat molar tooth development by lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology and Applied Pharmacology, 173, 38–47. [DOI] [PubMed] [Google Scholar]
- Lundberg R, Lyche JL, Ropstad E, Aleksandersen M, Ronn M, Skaare JU, Larsson S, Orberg J and Lind PM, 2006. Perinatal exposure to PCB 153, but not PCB 126, alters bone tissue composition in female goat offspring. Toxicology, 228, 33–40. [DOI] [PubMed] [Google Scholar]
- Lundebye AK, Berntssen MHG, Lie O, Ritchie G, Isosaari P, Kiviranta H and Vartiainen T, 2004. Dietary uptake of dioxins (PCDD/PCDFs) and dioxin‐like PCBs in Atlantic salmon (Salmo salar). Aquaculture Nutrition, 10, 199–207. [Google Scholar]
- Lundebye AK, Lock Erik‐Jan, Rasinger JD, Nøstbakken OJ, Hannisdal R, Karlsbakk E, Wennevik V, Madhun AS, Madsen L, Graff IE and Ørnsrud R, 2017. Lower levels of Persistent Organic Pollutants, metals and the marine omega 3‐fatty acid DHA in farmed compared to wild Atlantic salmon (Salmo salar). Environmental Research, 155, 49–59. [DOI] [PubMed] [Google Scholar]
- Lundgren K, Collman GW, Wangwuu S, Tiernan T, Taylor M, Thompson CL and Lucier GW, 1988. Cytogenetic and chemical‐detection of human exposure to polyhalogenated aromatic‐hydrocarbons. Environmental and Molecular Mutagenesis, 11, 1–11. [DOI] [PubMed] [Google Scholar]
- Lyche JL, Skaare JU, Larsen HJS and Ropstad E, 2004a. Levels of PCB 126 and PCB 153 in plasma and tissues in goats exposed during gestation and lactation. Chemosphere, 55, 621–629. [DOI] [PubMed] [Google Scholar]
- Lyche JL, Larsen HJS, Skaare JU, Tverdal A, Dahl E, Johansen GM and Ropstad E, 2004b. Effects of perinatal exposure to low doses of PCB 153 and PCB 126 on lymphocyte proliferation and hematology in goat kids. Journal of Toxicology and Environmental Health‐Part a‐Current Issues, 67, 889–904. [DOI] [PubMed] [Google Scholar]
- Lyche JL, Oskam IC, Skaare JU, Reksen O, Sweeney T, Dahl E, Farstad W and Ropstad E, 2004c. Effects of gestational and lactational exposure to low doses of PCBs 126 and 153 on anterior pituitary and gonadal hormones and on puberty in female goats. Reproductive Toxicology, 19, 87–95. [DOI] [PubMed] [Google Scholar]
- Lyche JL, Larsen HJ, Skaare JU, Tverdal A, Johansen GM and Ropstad E, 2006. Perinatal exposure to low doses of PCB 153 and PCB 126 affects maternal and neonatal immunity in goat kids. Journal of Toxicology and Environmental Health, Part A, 69, 139–158. [DOI] [PubMed] [Google Scholar]
- Ma Q and Baldwin KT, 2000. 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin‐proteasome pathway. Role of the transcription activaton and DNA binding of AhR. The Journal of Biological Chemistry, 275, 8432–8438. [DOI] [PubMed] [Google Scholar]
- Ma Q, 2012. Overview of AHR functional domains and the classical AHR signaling pathway: induction of drug metabolizing enzymes In: Pohjanvirta R. (ed). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 35–45. [Google Scholar]
- Maaetoft‐Udsen K, Shimoda LM, Frokiaer H and Turner H, 2012. Aryl hydrocarbon receptor ligand effects in RBL2H3 cells. Journal of Immunotoxicology, 9, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mably TA, Bjerke DL, Moore RW, Gendron‐Fitzpatrick A and Peterson RE, 1992a. In utero and lactational exposure of male rats to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. III Effects on spermatogenesis and reproductive capability. Fundamental and Applied Toxicology, 114, 108–116. [DOI] [PubMed] [Google Scholar]
- Mably TA, Moore RW, Goy RW and Peterson RE, 1992b. In utero and lactational exposure of male rats to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. 2. Effects on sexual behavior and the regulation of luteinizing hormone secretion in adulthood. Toxicology and Applied Pharmacology, 114, 108–17. [DOI] [PubMed] [Google Scholar]
- Mably TA, Moore RW and Peterson RE, 1992c. In utero and lactational exposure of male rats to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. 1. Effects on androgenic status. Toxicology and Applied Pharmacology, 114, 97–107. [DOI] [PubMed] [Google Scholar]
- Maervoet J, Vermeir G, Covaci A, Van Larebeke N, Koppen G, Schoeters G, Nelen V, Baeyens W, Schepens P and Viaene MK, 2007. Association of thyroid hormone concentrations with levels of organochlorine compounds in cord blood of neonates. Environmental Health Perspectives, 115, 1780–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiatis P, Pappas P, Gaitanis G, Mexia N, Melliou E, Galanou M, Vlachos C, Stathopoulou K, Skaltsounis AL, Marselos M, Velegraki A, Denison MS and Bassukas ID, 2013. Malassezia yeasts produce a collection of exceptionally potent activators of the Ah (dioxin) receptor detected in diseased human skin. The Journal of Investigative Dermatology, 133, 2023–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus P, Irgens LM, Haug K, Nystad W, Skjaerven R and Stoltenberg C and Moba Study G , 2006. Cohort profile: The Norwegian Mother and Child Cohort Study (MoBa). International Journal of Epidemiology, 35, 1146–1150. [DOI] [PubMed] [Google Scholar]
- Magnus P, Birke C, Vejrup K, Haugan A, Alsaker E, Daltveit AK, Handal M, Haugen M, Hoiseth G, Knudsen GP, Paltiel L, Schreuder P, Tambs K, Vold L and Stoltenberg C, 2016. Cohort profile update: The Norwegian Mother and Child Cohort Study (MoBa). International Journal of Epidemiology, 45, 382–388. [DOI] [PubMed] [Google Scholar]
- Mahiout S, Lindén J, Esteban J, Sánchez‐Pérez I, Sankari S, Pettersson L, Håkansson H and Pohjanvirta R, 2017. Toxicological characterisation of two novel selective aryl hydrocarbon receptor modulators in Sprague‐Dawley rats. Toxicology and Applied Pharmacology, 326, 54–65. [DOI] [PubMed] [Google Scholar]
- Malisch R, 2000. Increase of the PCDD/F‐contamination of milk, butter and meat samples by use of contaminated citrus pulp. Chemosphere, 40, 1041–1053. [DOI] [PubMed] [Google Scholar]
- Malisch R and Kotz A, 2014. Dioxins and PCBs in feed and food‐Review from European perspective. Science of the Total Environment, 491, 2–10. [DOI] [PubMed] [Google Scholar]
- Manchester DK, Gordon SK, Golas CL, Roberts EA and Okey AB, 1987. Ah receptor in human placenta: stabilization by molybdate and characterization of binding of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, 3‐methylcholanthrene, and benzo(a)pyrene. Cancer Research, 47, 4861–4868. [PubMed] [Google Scholar]
- Manikkam M, Guerrero‐Bosagna C, Tracey R, Haque MM and Skinner MK, 2012a. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One, 7, e31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M, Tracey R, Guerrero‐Bosagna C and Skinner MK, 2012b. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS One, 7, e46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning GE, Farmahin R, Crump D, Jones SP, Klein J, Konstantinov A, Potter D and Kennedy SW, 2012. A luciferase reporter gene assay and aryl hydrocarbon receptor 1 genotype predict the LD50 of polychlorinated biphenyls in avian species. Toxicology and Applied Pharmacology, 263, 390–401. [DOI] [PubMed] [Google Scholar]
- Manuwald U, Velasco Garrido M, Berger J, Manz A and Baur X, 2012. Mortality study of chemical workers exposed to dioxins: follow‐up 23 years after chemical plant closure. Occupational and Environmental Medicine, 69, 636–642. [DOI] [PubMed] [Google Scholar]
- Martin PA, Mayne GJ, Bursian S, Palace V and Kannan K, 2006. Changes in thyroid and vitamin A status in mink fed polyhalogenated‐aromatic‐hydrocarbon‐contaminated carp from the Saginaw River, Michigan, USA. Environmental Research, 101, 53–67. [DOI] [PubMed] [Google Scholar]
- Martin LA, Wilson DT, Reuhl KR, Gallo MA and Klaassen CD, 2012. Polychlorinated biphenyl congeners that increase the glucuronidation and biliary excretion of thyroxine are distinct from the congeners that enhance the serum disappearance of thyroxine. Drug Metabolism and Disposition, 40, 588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Zamora MA, Mattioli L, Parera J, Abad E, Coloma JL, van Babel B, Galceran MT, Balasch J and Carmona F, 2015. Increased levels of dioxin‐like substances in adipose tissue in patients with deep infiltrating endometriosis. Human Reproduction, 30, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Martucci CP and Fishman J, 1993. P450 enzymes of estrogen metabolism. Pharmacology and Therapeutics, 57, 237–257. [DOI] [PubMed] [Google Scholar]
- Mason G and Safe S, 1986. Synthesis, biologic and toxic effects of the major 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin metabolites in the rat. Toxicology, 41, 153–9. [DOI] [PubMed] [Google Scholar]
- Masuda Y, 1985. Health‐status of Japanese and Taiwanese after exposure to contaminated rice oil. Environmental Health Perspectives, 60, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH and Tilly JL, 2001. Aromatic hydrocarbon receptor‐driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nature Genetics, 28, 355–360. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Akahane M, Kanagawa Y, Koike S, Yoshimura T, Mitoma C, Shibata S, Uchi H, Furue M and Imamura T, 2009. Variation in half‐life of penta‐chlorodibenzofuran (PeCDF) blood level among Yusho patients. Chemosphere, 77, 658–662. [DOI] [PubMed] [Google Scholar]
- Matsumura F, 2009. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin‐activated Ah receptor to cause toxic effects. Biochemical Pharmacology, 77, 608–626. [DOI] [PubMed] [Google Scholar]
- Matsumura F, 2012. Nongenomic route of action of TCDD: identity, characteristics, and toxicological significance In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 197–215. [Google Scholar]
- Matsuura N, Uchiyama T, Tada H, Nakamura Y, Kondo N, Morita M and Fukushi M, 2001. Effects of dioxins and polychlorinated biphenyls (PCBs) on thyroid function in infants born in Japan – the second report from research on environmental health. Chemosphere, 45, 1167–1171. [DOI] [PubMed] [Google Scholar]
- Matthews J, Wihlen B, Thomsen J and Gustafsson JA, 2005. Aryl hydrocarbon receptor‐mediated transcription: ligand‐dependent recruitment of estrogen receptor alpha to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐responsive promoters. Molecular and Cellular Biology, 25, 5317–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride DI, Collins JJ, Humphry NF, Herbison P, Bodner KM, Aylward LL, Burns CJ and Wilken M, 2009. Mortality in Workers Exposed to 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin at a Trichlorophenol Plant in New Zealand. Journal of Occupational and Environmental Medicine, 51, 1049–1056. [DOI] [PubMed] [Google Scholar]
- McDonald P, Greenhalgh JFD, Morgan CA, Edwards R, Sinclair L and Wilkinson R, 2011. Animal Nutrition. 7th Edition, Benjamin Cummings. 692 pp. [Google Scholar]
- McKinney JD, Chae K, Gupta BN, Moore JA and Goldstein HA, 1976. Toxicological assessment of hexachlorobiphenyl isomers and 2,3,7,8 tetrachlorodibenzofuran in chicks. I. Relationship of chemical parameters. Toxicology and Applied Pharmacology, 36, 65–80. [DOI] [PubMed] [Google Scholar]
- McKinney JD, Fawkes J, Jordan S, Chae K, Oatley S, Coleman RE and Briner W, 1985. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) as a potent and persistent thyroxine agonist: a mechanistic model for toxicity based on molecular reactivity. Environmental Health Perspectives, 61, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan MS, Thoma H, Reissinger M and Hutzinger O, 1990. PCDD/F in an agricultural food chain; part 1: PCDD/F mass balance of a lactating cow. Chemosphere, 20, 1013–1020. [Google Scholar]
- McLachlan MS, 1993. Digestive tract absorption of polychlorinated dibenzo‐p‐dioxins, dibenzofurans, and biphenyls in a nursing infant. Toxicology and Applied Pharmacology, 123, 68–72. [DOI] [PubMed] [Google Scholar]
- McLachlan MS and Richter W, 1998. Uptake and transfer of PCDD/Fs by cattle fed naturally contaminated feedstuffs and feed contaminated as a result of sewage sludge application. 1 Lactating cows. Journal of Agricultural and Food Chemistry, 46, 1166–1172. [DOI] [PubMed] [Google Scholar]
- Meerts IA, Assink Y, Cenijn PH, van den Berg JH, Weijers BM, Bergman Å, Koeman JH and Brouwer A, 2002. Placental transfer of a hydroxylated polychlorinated biphenyl and effects on fetal and maternal thyroid hormone homeostasis in the rat. Toxicological Sciences, 68, 361–71. [DOI] [PubMed] [Google Scholar]
- Merten C, Ferrari P, Bakker M, Boss A, Hearty A, Leclercq C, Lindtner O, Tlustos C, Verger P, Volatier JL and Arcella D, 2011. Methodological characteristics of the national dietary surveys carried out in the European Union as included in the European Food Safety Authority (EFSA) Comprehensive European Food Consumption Database. Food Additives and Contaminants: Part A, 28, 975–995. [DOI] [PubMed] [Google Scholar]
- Metcalfe LD, 1972. Proposed source of chick edema factor. Journal ‐ Association of Official Analytical Chemists, 55, 542–546. [PubMed] [Google Scholar]
- Michalek JE, Tripathi RC, Caudill SP and Pirkle JL, 1992. Investigation of TCDD half‐life heterogeneity in veterans of Operation Ranch Hand. Journal of Toxicology and Environmental Health, 35, 29–38. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Pirkle JL, Caudill SP, Tripathi RC, Patterson DC and Needham LL, 1996. Pharmacokinetics of TCDD in veterans of operation ranch hand: 10‐year follow‐up. Journal of Toxicology and Environmental Health, 47, 209–220. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Rahe AJ and Boyle CA, 1998. Paternal dioxin, preterm birth, intrauterine growth retardation, and infant death. Epidemiology, 9, 161–167. [PubMed] [Google Scholar]
- Michalek JE, Akhtar FZ and Kiel JL, 1999a. Serum dioxin, insulin, fasting glucose, and sex hormone‐binding globulin in veterans of operation ranch hand. Journal of Clinical Endocrinology and Metabolism, 84, 1540–1543. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Ketchum NS and Check IJ, 1999b. Serum dioxin and immunologic response in veterans of Operation Ranch Hand. American Journal of Epidemiology, 149, 1038–1046. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Ketchum NS and Longnecker MP, 2001a. Serum dioxin and hepatic abnormalities in veterans of Operation Ranch Hand. Annals of Epidemiology, 11, 304–311. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Akhtar FZ, Arezzo JC, Garabrant DH and Albers JW, 2001b. Serum dioxin and peripheral neuropathy in veterans of Operation Ranch Hand. Neurotoxicology, 22, 479–490. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Akhtar FZ, Longnecker MP and Burton JE, 2001c. Relation of serum 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) level to hematological examination results in veterans of Operation Ranch Hand. Archives of Environmental Health, 56, 396–405. [DOI] [PubMed] [Google Scholar]
- Michalek JE, Pirkle JL, Needham LL, Patterson DG, Caudill SP, Tripathi RC and Mocarelli P, 2002. Pharmacokinetics of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in Seveso adults and veterans of operation Ranch Hand. Journal of Exposure Analysis and Environmental Epidemiology, 12, 44–53. [DOI] [PubMed] [Google Scholar]
- Michalek JE and Pavuk M, 2008. Diabetes and cancer in veterans of Operation Ranch Hand after adjustment for calendar period, days of spraying, and time spent in Southeast Asia. Journal of Occupational and Environmental Medicine, 50, 330–340. [DOI] [PubMed] [Google Scholar]
- Miettinen HM, Alaluusua S, Tuomisto J and Viluksela M, 2002. Effect of in utero and lactational 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin exposure on rat molar development: the role of exposure time. Toxicology and Applied Pharmacology, 184, 57–66. [PubMed] [Google Scholar]
- Miettinen HM, Sorvari R, Alaluusua S, Murtomaa M, Tuukkanen J and Viluksela M, 2006. The effect of perinatal TCDD exposure on caries susceptibility in rats. Toxicological Sciences, 91, 568–75. [DOI] [PubMed] [Google Scholar]
- Milbrath MOG, Wenger Y, Chang C‐W, Emond C, Garabrant D, Gillespie BW and Jolliet O, 2009. Apparent half‐lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast‐feeding. Environmental Health Perspectives, 117, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M and Fujii‐Kuriyama Y, 1997. Loss of teratogenic response to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes to Cells: Devoted to Molecular and Cellular Mechanisms, 2, 645–654. [DOI] [PubMed] [Google Scholar]
- Mínguez‐Alarcón L, Sergeyev O, Burns JS, Williams PL, Lee MM, Korrick SA, Smigulina L, Revich B and Hauser R, 2017. A longitudinal study of peripubertal serum organochlorine concentrations and semen parameters in young men: the Russian Children's Study. Environmental Health Perspectives, 125, 460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitoma C, Mine Y, Utani A, Imafuku S, Muto M, Akimoto T, Kanekura T, Furue M and Uchi H, 2015. Current skin symptoms of Yusho patients exposed to high levels of 2,3,4,7,8‐pentachlorinated dibenzofuran and polychlorinated biphenyls in 1968. Chemosphere, 137, 45–51. [DOI] [PubMed] [Google Scholar]
- Miyashita C, Sasaki S, Saijo Y, Washino N, Okada E, Kobayashi S, Konishi K, Kajiwara J, Todaka T and Kishi R, 2011. Effects of prenatal exposure to dioxin‐like compounds on allergies and infections during infancy. Environmental Research, 111, 551–558. [DOI] [PubMed] [Google Scholar]
- Miyazaki W, Iwasaki T, Takeshita A, Tohyama C and Koibuchi N, 2008. Identification of the functional domain of thyroid hormone receptor responsible for polychlorinated biphenyl‐mediated suppression of its action in vitro . Environmental Health Perspectives, 116, 1231–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukawa H, Nomiyama K, Kunisue T, Watanabe MX, Subramanian A, Iwata H, Ishizuka M and Tanabe S, 2015. Organohalogens and their hydroxylated metabolites in the blood of pigs from an open waste dumping site in south India: association with hepatic cytochrome P450. Environmental Research, 138, 255–263. [DOI] [PubMed] [Google Scholar]
- Mocarelli P, 2001. Seveso: a teaching story. Chemosphere, 43, 391–402. [DOI] [PubMed] [Google Scholar]
- Mocarelli P, Patterson SG, Marocchi A and Needham LL, 1990. Pilot study (Phase II) for determining PCDD and PCDF levels in serum of Seveso, Italy, residents collected at the tiem of exposure: future plans. Chemosphere, 20, 967–974. [Google Scholar]
- Mocarelli P, Gerthoux PM, Ferrari E, Patterson DG, Kieszak SM, Brambilla P, Vincoli N, Signorini S, Tramacere P, Carreri V, Sampson EJ, Turner WE and Needham LL, 2000. Paternal concentrations of dioxin and sex ratio of offspring. Lancet, 355, 1858–1863. [DOI] [PubMed] [Google Scholar]
- Mocarelli P, Gerthoux PM, Patterson DG Jr, Milani S, Limonta G, Bertona M, Signorini S, Tramacere P, Colombo L, Crespi C, Brambilla P, Sarto C, Carreri V, Sampson EJ, Turner WE and Needham LL, 2008. Dioxin exposure, from infancy through puberty, produces endocrine disruption and affects human semen quality. Environmental Health Perspectives, 116, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarelli P, Gerthoux PM, Needham LL, Patterson DG, Limonta G, Falbo R, Signorini S, Bertona M, Crespi C, Sarto C, Scott PK, Turner WE and Brambilla P, 2011. Perinatal exposure to low doses of dioxin can permanently impair human semen quality. Environmental Health Perspectives, 119, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohinta S, Kannan AK, Gowda K, Amin SG, Perdew GH and August A, 2015. Differential regulation of Th17 and T regulatory cell differentiation by aryl hydrocarbon receptor dependent xenobiotic response element dependent and independent pathways. Toxicological Sciences, 145, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molcan T, Swigonska S, Orlowska K, Myszczynski K, Nynca A, Sadowska A, Ruszkowska M, Jastrzebski JP and Ciereszko RE, 2017. Structural‐functional adaptations of porcine CYP1A1 to metabolize polychlorinated dibenzo‐p‐dioxins. Chemosphere, 168, 205–216. [DOI] [PubMed] [Google Scholar]
- Monteiro P, Gilot D, Le Ferrec E, Rauch C, Lagadic‐Gossmann D and Fardel O, 2008. Dioxin‐mediated up‐regulation of aryl hydrocarbon receptor target genes is dependent on the calcium/calmodulin/CaMKIalpha pathway. Molecular Pharmacology, 73, 769–777. [DOI] [PubMed] [Google Scholar]
- Moore JA, McConnell EE, Dalgard DW and Harris MW, 1979. Comparative toxicity of three halogenated dibenzofurans in guinea pigs, mice and rhesus monkeys. Annals of the New York Academy of Sciences, 75, 151–163. As cited in ATSDR (1994). [DOI] [PubMed] [Google Scholar]
- Moore RW, Jefcoate CR and Peterson RE, 1991. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin inhibits steroidogenesis in the rat testis by inhibiting the mobilization of cholesterol to cytochrome P450scc. Toxicology and Applied Pharmacology, 109, 85–97. [DOI] [PubMed] [Google Scholar]
- Moore JN, Zwiernik MJ, Newsted JL, Fitzgerald SD, Link JE, Bradley PW, Kay D, Budinsky R, Giesy JP and Bursian SJ, 2012. Effects of dietary exposure of mink (Mustela vison) to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, 2,3,4,7,8‐pentachlorodibenzofuran, and 2,3,7,8‐tetrachlorodibenzofuran on reproduction and offspring viability and growth. Environmental Toxicology and Chemistry, 31, 360–369. [DOI] [PubMed] [Google Scholar]
- Moran FM, Tarara R, Chen J, Santos S, Cheney A, Overstreet JW and Lasley BL, 2001. Effect of dioxin on ovarian function in the cynomolgus macaque (M. fascicularis). Reproductive Toxicology, 15, 377–383. [DOI] [PubMed] [Google Scholar]
- Morris DL, Jeong HG, Jordan SD, Kaminski NE and Holsapple MP, 1998. Characterization of the effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in B6C3F1 and DBA/2 mice following single and repeated exposures. Archives of Toxicology, 72, 157–168. [DOI] [PubMed] [Google Scholar]
- Moser GA and McLachlan MS, 1999. A non‐absorbable dietary fat substitute enhances elimination of persistent lipophilic contaminants in humans. Chemosphere, 39, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Moser GA and McLachlan MS, 2001. The influence of dietary concentration on the absorption and excretion of persistent lipophilic organic pollutants in the human intestinal tract. Chemosphere, 45, 201–211. [DOI] [PubMed] [Google Scholar]
- Moser GA and McLachlan MS, 2002. Modeling digestive tract absorption and desorption of lipophilic organic contaminants in humans. Environmental Science and Technology, 36, 3318–3325. [DOI] [PubMed] [Google Scholar]
- Mulero‐Navarro S and Fernández‐Salguero PM, 2016. New trends in aryl hydrocarbon receptor biology. Frontiers in Cell and Developmental Biology, 4, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA and Perdew G, 2012. Role of chaperone proteins in AHR function In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 47–61. [Google Scholar]
- Mutoh J, Taketoh J, Okamura K, Kagawa T, Ishida T, Ishii Y and Yamada H, 2006. Fetal pituitary gonadotropin as an initial target of dioxin in its impairment of cholesterol transportation and steroidogenesis in rats. Endocrinology, 147, 927–36. [DOI] [PubMed] [Google Scholar]
- Nagayama J, Okamura K, Iida T, Hirakawa H, Matsueda T, Tsuji H, Hasegawa M, Sato K, Ma HY, Yanagawa T, Igarashi H, Fukushige J and Watanabe T, 1998a. Postnatal exposure to chlorinated dioxins and related chemicals on thyroid hormone status in Japanese breast‐fed infants. Chemosphere, 37, 1789–1793. [DOI] [PubMed] [Google Scholar]
- Nagayama J, Tsuji H, Iida T, Hirakawa H, Matsueda T, Okamura K, Hasegawa M, Sato K, Ma HY, Yanagawa T, Igarashi H, Fukushige J and Watanabe T, 1998b. Postnatal exposure to chlorinated dioxins and related chemicals on lymphocyte subsets in Japanese breast‐fed infants. Chemosphere, 37, 1781–1787. [DOI] [PubMed] [Google Scholar]
- Nagayama J, Tsuji H, Iida T, Hirakawa H, Matsueda T and Ohki M, 2001. Effects of contamination level of dioxins and related chemicals on thyroid hormone and immune response systems in patients with “Yusho”. Chemosphere, 43, 1005–1010. [DOI] [PubMed] [Google Scholar]
- Nagayama J, Kohno H, Kunisue T, Kataoka K, Shimomura H, Tanabe S and Konishi S, 2007a. Concentrations of organochlorine pollutants in mothers who gave birth to neonates with congenital hypothyroidism. Chemosphere, 68, 972–976. [DOI] [PubMed] [Google Scholar]
- Nagayama J, Tsuji H, Iida T, Nakagawa R, Matsueda T, Hirakawa H, Yanagawa T, Ji Fukushige and Watanabe T, 2007b. Immunologic effects of perinatal exposure to dioxins, PCBs and organochlorine pesticides in Japanese infants. Chemosphere, 67, S393–S398. [DOI] [PubMed] [Google Scholar]
- Nakajima S, Saijo Y, Kato S, Sasaki S, Uno K, Kanagami N, Hirakawa H, Hori T, Tobiishi K, Todaka T, Nakamura Y, Yanagiya S, Sengoku Y, Iida T, Sata F and Kishi R, 2006. Effects of prenatal exposure to polychlorinated biphenyls and dioxins on mental and motor development in Japanese children at 6 months of age. Environmental Health Perspectives, 114, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Saijo Y, Miyashita C, Ikeno T, Sasaki S, Kajiwara J and Kishi R, 2017. Sex‐specific differences in effect of prenatal exposure to dioxin‐like compounds on neurodevelopment in Japanese children: Sapporo cohort study. Environmental Research, 159, 222–231. [DOI] [PubMed] [Google Scholar]
- Nakamoto M, Arisawa K, Uemura H, Katsuura S, Takami H, Sawachika F, Yamaguchi M, Juta T, Sakai T, Toda E, Mori K, Hasegawa M, Tanto M, Shima M, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M and Satoh H, 2013. Association between blood levels of PCDDs/PCDFs/dioxin‐like PCBs and history of allergic and other diseases in the Japanese population. International Archives of Occupational and Environmental Health, 86, 849–859. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Miyazaki W, Ohnishi Y and Ishibashi T and Study Group Y , 2005. Ophthalmic findings in Yusho. Journal of dermatological science, S57–S63. [Google Scholar]
- Nakano S, Noguchi T, Takekoshi H, Suzuki G and Nakano M, 2005. Maternal‐fetal distribution and transfer of dioxins in pregnant women in Japan, and attempts to reduce maternal transfer with Chlorella (Chlorella pyrenoidosa) supplements. Chemosphere, 61, 1244–1255. [DOI] [PubMed] [Google Scholar]
- NATO/CCMS (North Atlantic Treaty Organization, Committee on the Challenges of Modem Society), 1988. International toxicity equivalency factor (I‐TEF) method of risk assessment for complex mixtures of dioxins and related compounds. Report No. 176. Brussels: North Atlantic Treaty Organization. [Google Scholar]
- Nault R, Forgacs AL, Dere E and Zacharewski TR, 2013. Comparisons of differential gene expression elicited by TCDD, PCB126, betaNF, or ICZ in mouse hepatoma Hepa1c1c7 cells and C57BL/6 mouse liver. Toxicology Letters, 223, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R, Fader KA, Ammendolia DA, Dornbos P, Potter D, Sharratt B, Kumagai K, Harkema JR, Lunt SY, Matthews J and Zacharewski T, 2016. Dose‐dependent metabolic reprogramming and differential gene expression in TCDD‐elicited hepatic fibrosis. Toxicological Sciences, 154, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navid F, Bruhs A, Schuller W, Fritsche E, Krutmann J, Schwarz T and Schwarz A, 2013. The Aryl hydrocarbon receptor is involved in UVR‐induced immunosuppression. The Journal of Investigative Dermatology, 133, 2763–2770. [DOI] [PubMed] [Google Scholar]
- Neal RA, Beatty PW and Gasiewicz TA, 1979. Studies of the mechanisms of toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Annals of the New York Academy of Sciences, 320, 204–213. [DOI] [PubMed] [Google Scholar]
- Neal RA, Olson JR, Gasiewicz TA and Geiger LE, 1982. The toxicokinetics of 2, 3, 7, 8‐tetrachlorodibenzo‐p‐dioxin in mammalian systems. Drug Metabolism Reviews, 13, 355–85. [DOI] [PubMed] [Google Scholar]
- Neal R, Gasiewicz T, Geiger L, Olson J and Sawahata T, 1984. Metabolism of 2 3 7 8 tetrachlorodibenzo‐p‐dioxin in mammalian systems. In: Poland A and Kimbrough RD. pp. 49–60.
- Nebert DW and Gelboin HV, 1969. The in vivo and in vitro induction of aryl hydrocarbon hydroxylase in mammalian cells of different species, tissues, strains, and developmental and hormonal states. Archives of Biochemistry and Biophysics, 134, 76–89. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Goujon FM and Gielen JE, 1972. Aryl hydrocarbon hydroxylase induction by polycyclic hydrocarbons: simple autosomal dominant trait in the mouse. Nature: New Biology, 236, 107–110. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP, Okey AB and Gonzalez FJ, 2004. Role of aryl hydrocarbon receptor‐mediated induction of the CYP1 enzymes in environmental toxicity and cancer. Journal of Biological Chemistry, 279, 23847–23850. [DOI] [PubMed] [Google Scholar]
- Needham LL, Gerthoux PM, Patterson DG, Brambilla P, Turner WE, Beretta C, Pirkle JL, Colombo L, Sampson EJ, Tramacere PL, Signorini S, Meazza L, Carreri V, Jackson RJ and Mocarelli P, 1997/1998. Serum dioxin levels in Seveso, Italy, population in 1976. Teratogenesis Carcinogenesis and Mutagenesis, 17, 225–240. [PubMed] [Google Scholar]
- Negishi T, Shimomura H, Koyama T, Kawasaki K, Ishii Y, Kyuwa S, Yasuda M, Kuroda Y and Yoshikawa Y, 2006. Gestational and lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin affects social behaviors between developing rhesus monkeys (Macaca mulatta). Toxicology Letters, 160, 233–244. [DOI] [PubMed] [Google Scholar]
- Neuberger M, Rappe C, Bergek S, Cai H, Hansson M, Jager R, Kundi M, Lim CK, Wingfors H and Smith AG, 1999. Persistent health effects of dioxin contamination in herbicide production. Environmental Research, 81, 206–214. [DOI] [PubMed] [Google Scholar]
- Neugebauer J, Wittsiepe J, Kasper‐Sonnenberg M, Schoneck N, Scholmerich A and Wilhelm M, 2015. The influence of low level pre‐ and perinatal exposure to PCDD/Fs, PCBs, and lead on attention performance and attention‐related behavior among German school‐aged children: results from the Duisburg Birth Cohort Study. International Journal of Hygiene and Environmental Health, 218, 153–162. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, Hoivik D, Lee JE and Safe S, 1999. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Archives of Biochemistry and Biophysics, 367, 250–257. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Miyabara Y, Suzuki JS, Sato M, Aoki Y, Satoh M, Yonemoto J and Tohyama C, 2001. Induction of metallothionein in the livers of female Sprague‐Dawley rats treated with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Life Sciences, 69, 1291–1303. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Miyabara Y, Sato M, Yonemoto J and Tohyama C, 2002. Immunohistochemical localization of thyroid stimulating hormone induced by a low oral dose of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in female Sprague‐Dawley rats. Toxicology, 171, 73–82. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Yonemoto J, Miyabara Y, Sato M and Tohyama C, 2003. Rat thyroid hyperplasia induced by gestational and lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Endocrinology, 144, 2075–2083. [DOI] [PubMed] [Google Scholar]
- Nishimura N, Yonemoto J, Miyabara Y, Fujii‐Kuriyama Y and Tohyama C, 2005. Altered thyroxin and retinoid metabolic response to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in aryl hydrocarbon receptor‐null mice. Archives of Toxicology, 79, 260–267. [DOI] [PubMed] [Google Scholar]
- Niskar AS, Needham LL, Rubin C, Turner WE, Martin CA, Patterson DG Jr, Hasty L, Wong L‐Y and Marcus M, 2009. Serum dioxins, polychlorinated biphenyls, and endometriosis: a case‐control study in Atlanta. Chemosphere, 74, 944–949. [DOI] [PubMed] [Google Scholar]
- Nohara K, Fujimaki H, Tsukumo S, Ushio H, Miyabara Y, Kijima M, Tohyama C and Yonemoto J, 2000. The effects of perinatal exposure to low doses of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on immune organs in rats. Toxicology, 154, 123–133. [DOI] [PubMed] [Google Scholar]
- Nordic Ministry Council Report , 1988. In Swedish: Nordisk Dioxinriskbedömning, Nordiska dioxin arbetsgruppen. Rapport från en Nordisk Expertgrupp. Nordisk Ministerråd Miljörapport 1988:7. Document prepared by Ahlborg UG, Håkansson H, Waern F, Hanberg A, Institute of Environmental Medicine, Karolinska Institutet.
- Norén K, Weistrand C and Karpe F, 1999. Distribution of PCB congeners, DDE, hexachlorobenzene, and methylsulfonyl metabolites of PCB and DDE among various fractions of human blood plasma. Archives of Environmental Contamination and Toxicology, 37, 408–414. [DOI] [PubMed] [Google Scholar]
- Nosek JA, Sullivan JR, Craven SR, Gendronfitzpatrick A and Peterson RE, 1993. Embryotoxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the ring‐necked pheasant. Environmental Toxicology and Chemistry, 12, 1215–1222. [Google Scholar]
- Nottebrock C, Riecke K, Kruse M, Shakibaei M and Stahlmann R, 2006. Effects of 2,3,7,8‐tetrachloro‐dibenzo‐p‐dioxin on the extracellular matrix of the thymus in juvenile marmosets (Callithrix jacchus). Toxicology, 226, 197–207. [DOI] [PubMed] [Google Scholar]
- Nowack N, Wittsiepe J, Kasper‐Sonnenberg M, Wilhelm M and Scholmerich A, 2015. Influence of low‐level prenatal exposure to PCDD/Fs and PCBs on empathizing, systemizing and autistic traits: results from the Duisburg Birth Cohort Study. Plos One, 10, e0129906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC (National Research Council), 2007a. Nutrient requirements of small ruminants: sheep, goats, cervids and new world camelids. National Academies Press, Washington, DC. [Google Scholar]
- NRC (National Research Council), 2007b. Nutrient requirement of horses. 6th Revised Edition National Academies Press, Washington, DC. [Google Scholar]
- NTP (National Toxicology Programe), 2006a. Technical report on the toxicology and carcinogenesis studies of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) (CAS NO. 1746‐01‐6) in female Harlan Sprague‐Dawley rats (gavage studies). NTP TR 521. NIH Publication No. 06‐4468. National Institutes of Health. Public Health Service. U.S. Department of Health and Human Services. [PubMed]
- NTP (National Toxicology Programe), 2006b. Technical report on the toxicology and carcinogenesis studies of 3,3’,4,4’,5‐Pentachlorobiphenyl (PCB 126) (CAS NO. 57465‐28‐8) in female Harlan Sprague‐Dawley rats (gavage studies). NTP TR 520. NIH Publication No. 06‐4454. National Institutes of Health. Public Health Service. U.S. Department of Health and Human Services. [PubMed]
- NTP (National Toxicology Programe), 2006c. Technical report on the toxicology and carcinogenesis studies of 2,3,4,7,8‐pentachlorodibenzofuran (PeCDF) (CAS NO. 57117‐31‐4) in female Harlan Sprague‐Dawley rats (gavage studies). NTP TR 525. NIH Publication No. 06‐4461. National Institutes of Health. Public Health Service. U.S. Department of Health and Human Services. [PubMed]
- NTP (National Toxicology Programe), 2006d. Technical report on the toxicology and carcinogenesis studies of a mixture of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) (CAS NO. 1746‐01‐6), 2,3,4,7,8‐pentachlorodibenzofuran (PeCDF) (CAS NO. 57117‐31‐4), and 3,3’,4,4’,5‐pentachlorobiphenyl (PCB 126) (CAS NO. 57465‐28‐8) in female Harlan Sprague‐Dawley rats (gavage studies).. NTP TR 526. NIH Publication No. 06‐4462. National Institutes of Health. Public Health Service. U.S. Department of Health and Human Services.
- NTP (National Toxicology Programe), 2006e. Technical report on the toxicology and carcinogenesis studies of a binary mixture of 3,3’,4,4’,5‐pentachlorobiphenyl (PCB 126) (CAS NO. 57465‐28‐8) and 2,3’,4,4’,5‐pentachlorobiphenyl (PCB 118) (CAS NO. 31508‐00‐6) in female Harlan Sprague‐Dawley rats (gavage studies). NTP TR 531. NIH Publication No. 06‐4467. National Institutes of Health. Public Health Service. U.S. Department of Health and Human Services.
- NTP (National Toxicology Programe), 2010. Technical report on the toxicology and carcinogenesis studies of 2,3’,4,4’,5‐pentachlorobiphenyl (PCB 118) (CAS NO. 31508‐00‐6) in female Harlan Sprague‐Dawley rats (gavage studies). NTP TR 559. NIH Publication No. 11‐5900. National Institutes of Health Public Health Service. U.S. Department of Health and Human Services. [PubMed]
- Nukaya M, Walisser JA, Moran SM, Kennedy GD and Bradfield CA, 2010. Aryl hydrocarbon receptor nuclear translocator in hepatocytes is required for aryl hydrocarbon receptor‐mediated adaptive and toxic responses in liver. Toxicological Sciences, 118, 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuti R, Gargaro M, Matino D, Dolciami D, Grohmann U, Puccetti P, Fallarino F and Macchiarulo A, 2014. Ligand binding and functional selectivity of l‐tryptophan metabolites at the mouse aryl hydrocarbon receptor (mAhR). Journal of Chemical Information and Modeling, 54, 3373–3383. [DOI] [PubMed] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development), 2013. Guidance Document on Residues in Livestock. Series on Pesticides No. 73. OECD, Paris, France. 77 pp.
- Oehme M, Bartonova A and Knutzen J, 1990. Estimation of polychlorinated dibenzofuran and dibenzo‐p‐dioxin contamination of a coastal region using isomer profiles in crabs. Environmental Science and Technology, 24, 1836–184. [Google Scholar]
- Ogura I, 2004. Half‐life of each dioxin and PCB congener in the human body. Organohalogen Compounds, 66, 3376–3384. [Google Scholar]
- Ohsako S, Miyabara Y, Nishimura N, Kurosawa S, Sakaue M, Ishimura R, Sato M, Takeda K, Aoki Y, Sone H, Tohyama C and Yonemoto J, 2001. Maternal exposure to a low dose of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) suppressed the development of reproductive organs of male rats: dose‐dependent increase of mRNA levels of 5alpha‐reductase type 2 in contrast to decrease of androgen receptor in the pubertal ventral prostate. Toxicological Sciences, 60, 132–143. [DOI] [PubMed] [Google Scholar]
- Ohtake F and Kato S, 2011. The E3 ubiquitin ligase activity of transcription factor AHR permits nongenomic regulation of biological pathways In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 143–156. [Google Scholar]
- Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii‐Kuriyama Y and Kato S, 2007. Dioxin receptor is a ligand‐dependent E3 ubiquitin ligase. Nature, 446, 562–566. [DOI] [PubMed] [Google Scholar]
- Okey AB, 2007. An aryl hydrocarbon receptor odyssey to the shores of toxicology: the Deichmann Lecture, International Congress of Toxicology‐XI. Toxicological Sciences, 98, 5–38. [DOI] [PubMed] [Google Scholar]
- Olie K, Vermeulen PL and Hutzinger O, 1977. Chlorodibenzo‐p‐dioxins and chlorodibenzofurans are trace components of fly ash and flue gas of some municipal incinerators in the Netherlands. Chemosphere, 6, 455–459. [Google Scholar]
- Olli J, Breivik H, Morkore T, Ruyter B, Johansen J, Reynolds P, Thorstad O and Berge G, 2010. Removal of persistent organic pollutants from Atlantic salmon (Salmo salar L.) diets: influence on growth, feed utilization efficiency and product quality. Aquaculture, 310, 145–155. [Google Scholar]
- Olling M, Derks HJGM, Berende PLM, Liem ADK and de Jong APJM, 1991. Toxicokinetics of eight 13C‐labelled polychlorinated dibenzo‐p‐dioxins and ‐furans in lactating cows. Chemosphere, 23, 1377–1385. [Google Scholar]
- Olson JR, Gasiewicz TA and Neal RA, 1980. Tissue distribution, excretion, and metabolism of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the Golden Syrian hamster. Toxicology and Applied Pharmacology, 56, 78–85. [DOI] [PubMed] [Google Scholar]
- Olson JR, 1986. Metabolism and disposition of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in guinea‐pigs. Toxicology and Applied Pharmacology, 85, 263–273. [DOI] [PubMed] [Google Scholar]
- Oskam IC, Lyche JL, Krogenaes A, Thomassen R, Skaare JU, Wiger R, Dahl E, Sweeney T, Stien A and Ropstad E, 2005. Effects of long‐term maternal exposure to low doses of PCB126 and PCB153 on the reproductive system and related hormones of young male goats. Reproduction, 130, 731–742. [DOI] [PubMed] [Google Scholar]
- Ounnas F, Feidt C, Toussaint H, Marchand P, Bizec BL, Rychen G and Jurjanz S, 2010. Polychlorinated biphenyl and low polybrominated diphenyl ether transfer to milk in lactating goats chronically exposed to contaminated soil. Environmental Science and Technology, 44, 2682–2688. [DOI] [PubMed] [Google Scholar]
- Panteleyev AA and Bickers DR, 2006. Dioxin‐induced chloracne – reconstructing the cellular and molecular mechanisms of a classic environmental disease. Experimental Dermatology, 15, 705–730. [DOI] [PubMed] [Google Scholar]
- Papadopoulou E, Caspersen IH, Kvalem HE, Knutsen HK, Duarte‐Salles T, Alexander J, Meltzer HM, Kogevinas M, Brantsaeter AL and Haugen M, 2013. Maternal dietary intake of dioxins and polychlorinated biphenyls and birth size in the Norwegian Mother and Child Cohort Study (MoBa). Environment International, 60, 209–216. [DOI] [PubMed] [Google Scholar]
- Papadopoulou E, Kogevinas M, Botsivali M, Pedersen M, Besselink H, Mendez MA, Fleming S, Hardie LJ, Knudsen LE, Wright J, Agramunt S, Sunyer J, Granum B, Gutzkow KB, Brunborg G, Alexander J, Meltzer HM, Brantsaeter AL, Sarri K, Chatzi L, Merlo DF, Kleinjans JC and Haugen M, 2014. Maternal diet, prenatal exposure to dioxin‐like compounds and birth outcomes in a European prospective mother‐child study (NewGeneris). Science of the Total Environment, 484, 121–128. [DOI] [PubMed] [Google Scholar]
- Parera J, Abalos M, Perez‐Vendrell AM, Brufau J, de Juan F, Escribano F, Abad E and Rivera J, 2008. Occurrence and bioaccumulation study of PCDD and PCDF from mineral feed additives. Chemosphere, 73, S252–260. [DOI] [PubMed] [Google Scholar]
- Parera J, Serra‐Prat M, Palomera E, Mattioli L, Abalos M, Rivera J and Abad E, 2013. Biological monitoring of PCDD/Fs and PCBs in the City of Mataro. A population‐based cohort study (1995‐2012). Science of the Total Environment, 461, 612–617. [DOI] [PubMed] [Google Scholar]
- Partanen AM, Alaluusua S, Miettinen PJ, Thesleff I, Tuomisto J, Pohjanvirta R and Lukinmaa PL, 1998. Epidermal growth factor receptor as a mediator of developmental toxicity of dioxin in mouse embryonic teeth. Laboratory Investigation, 78, 1473–81. [PubMed] [Google Scholar]
- Partanen AM, Kiukkonen A, Sahlberg C, Alaluusua S, Thesleff I, Pohjanvirta R and Lukinmaa PL, 2004. Developmental toxicity of dioxin to mouse embryonic teeth in vitro: arrest of tooth morphogenesis involves stimulation of apoptotic program in the dental epithelium. Toxicology and Applied Pharmacology, 194, 24–33. [DOI] [PubMed] [Google Scholar]
- Patandin S, Koopman‐Esseboom C, De Ridder MAJ, Weisglas‐Kuperus N and Sauer PJJ, 1998. Effects of environmental exposure to polychlorinated biphenyls and dioxins on birth size and growth in Dutch children. Pediatric Research, 44, 538–545. [DOI] [PubMed] [Google Scholar]
- Patandin S, Lanting CI, Mulder PGH, Boersma ER, Sauer PJJ and Weisglas‐Kuperus N, 1999. Effects of environmental exposure to polychlorinated biphenyls and dioxins on cognitive abilities in Dutch children at 42 months of age. Journal of Pediatrics, 134, 33–41. [DOI] [PubMed] [Google Scholar]
- Patel RD, Murray IA, Flaveny CA, Kusnadi A and Perdew GH, 2009. Ah receptor represses acute‐phase response gene expression without binding to its cognate response element. Laboratory I, 89, 695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson DG, Needham LL, Pirkle JL, Roberts DW, Bagby J, Garrett WA, Andrews JS, Falk H, Bernert JT, Sampson EJ and Houk VN, 1988. Correlation between serum and adipose‐tissue levels of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in 50 persons from Missouri. Archives of Environmental Contamination and Toxicology, 17, 139–143. [DOI] [PubMed] [Google Scholar]
- Patterson DG, Fürst P, Alexander LR, Isaacs SG, Turner WE and Needham ????, 1989a. Analysis of human serum for PCDDs/PCDFs: a comparison of three extraction procedures. Chemosphere, 19, 89–96. [Google Scholar]
- Patterson DG, Furst P, Henderson LO, Isaacs SG, Alexander LR, Turner WE, Needham LL and Hannon H, 1989b. Partitioning of invivo bound PCDDs/PCDFs among various compartments in whole‐blood. Chemosphere, 19, 135–142. [Google Scholar]
- Patterson RM, Stachlewitz R and Germolec D, 2003. Induction of apoptosis by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin following endotoxin exposure. Toxicology and Applied Pharmacology, 190, 120–134. [DOI] [PubMed] [Google Scholar]
- Pauwels A, Schepens PJC, D'Hooghe T, Delbeke L, Dhont M, Brouwer A and Weyler J, 2001. The risk of endometriosis and exposure to dioxins and polychlorinated biphenyls: a case‐control study of infertile women. Human Reproduction, 16, 2050–2055. [DOI] [PubMed] [Google Scholar]
- Pavuk M, Schecter AJ, Akhtar FZ and Michalek JE, 2003. Serum 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) levels and thyroid function in Air Force veterans of the Vietnam War. Annals of Epidemiology, 13, 335–343. [DOI] [PubMed] [Google Scholar]
- Pavuk M, Michalek JE, Schecter A, Ketchum NS, Akhtar FZ and Fox KA, 2005. Did TCDD exposure or service in Southeast Asia increase the risk of cancer in air force Vietnam veterans who did not spray agent orange? Journal of Occupational and Environmental Medicine, 47, 335–342. [DOI] [PubMed] [Google Scholar]
- Pavuk M, Michalek JE and Ketchum NS, 2006. Prostate cancer in US Air Force veterans of the Vietnam war. Journal of Exposure Science and Environmental Epidemiology, 16, 184–190. [DOI] [PubMed] [Google Scholar]
- Pazdernik TL and Rozman KK, 1985. Effect of thyroidectomy and thyroxine on 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐induced immunotoxicity. Life Sciences, 36, 695–703. [DOI] [PubMed] [Google Scholar]
- Peden‐Adams M, Alonso K, Godard C, Skipper S, Mashburn W, Hoover J, Charbonneau C, Henshel D and Dickerson R, 1998. Effects of environmentally relevant concentrations of 2,3,7,8‐TCDD on domestic chicken immune function and CYP450 activity: F1 generation and egg injection studies. Chemosphere, 37, 1923–1939. [DOI] [PubMed] [Google Scholar]
- Pelclova D, Fenclova Z, Dlaskova Z, Urban P, Lukas E, Prochazka B, Rappe C, Preiss J, Kocan A and Vejlupkova J, 2001. Biochemical, neuropsychological, and neurological abnormalities following 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) exposure. Archives of Environmental Health, 56, 493–500. [DOI] [PubMed] [Google Scholar]
- Pelclova D, Fenclova Z, Preiss J, Prochazka B, Spacil J, Dubska Z, Okrouhlik B, Lukas E and Urban P, 2002. Lipid metabolism and neuropsychological follow‐up study of workers exposed to 2,3,7,8‐tetrachlordibenzo‐p‐dioxin. International Archives of Occupational and Environmental Health, 75(Suppl), S60–66. [DOI] [PubMed] [Google Scholar]
- Pelclova D, Prazny M, Skrha J, Fenclova Z, Kalousova M, Urban P, Navratil T, Senholdova Z and Smerhovsky Z, 2007. 2,3,7,8‐TCDD exposure, endothelial dysfunction and impaired microvascular reactivity. Human and Experimental Toxicology, 26, 705–713. [DOI] [PubMed] [Google Scholar]
- Pelclova D, Fenclova Z, Urban P, Ridzon P, Preiss J, Kupka K, Malik J, Dubska Z and Navratil T, 2009. Chronic health impairment due to 2,3,7,8‐tetrachloro‐dibenzo‐p‐dioxin exposure. Neuroendocrinology Letters, 30, 219–224. [PubMed] [Google Scholar]
- Pelekis M, Nicolich MJ and Gauthier JS, 2003. Probabilistic framework for the estimation of the adult and child toxicokinetic intraspecies uncertainty factors. Risk Analysis, 23, 1239–1255. [DOI] [PubMed] [Google Scholar]
- Perucatti A, Di Meo GP, Albarella S, Ciotola F, Incarnato D, Jambrenghi AC, Peretti V, Vonghia G and Iannuzzi L, 2006. Increased frequencies of both chromosome abnormalities and SCEs in two sheep flocks exposed to high dioxin levels during pasturage. Mutagenesis, 21, 67–75. [DOI] [PubMed] [Google Scholar]
- Pesatori AC, Zocchetti C, Guercilena S, Consonni D, Turrini D and Bertazzi PA, 1998. Dioxin exposure and non‐malignant health effects: a mortality study. Occupational and Environmental Medicine, 55, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesatori AC, Baccarelli A, Consonni D, Lania A, Beck‐Peccoz P, Bertazzi PA and Spada A, 2008. Aryl hydrocarbon receptor‐interacting protein and pituitary adenomas: a population‐based study on subjects exposed to dioxin after the Seveso, Italy, accident. European Journal of Endocrinology, 159, 699–703. [DOI] [PubMed] [Google Scholar]
- Petkov PI, Rowlands JC, Budinsky R, Zhao B, Denison MS and Mekenyan O, 2010. Mechanism‐based common reactivity pattern (COREPA) modelling of aryl hydrocarbon receptor binding affinity. SAR and QSAR in Environmental Research, 21, 187–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroske E, Zaylskie RG and Feil VJ, 1998. Reduction in polychlorinated dibenzodioxin and dibenzofuran residues in hamburger meat during cooking. Journal of Agricultural and Food Chemistry, 46, 3280–3284. [Google Scholar]
- Phadnis‐Moghe AS, Chen W, Li J, Crawford RB, Bach A, D'Ingillo S, Kovalova N, Suarez‐Martinez JE, Kaplan BLF, Harrill JA, Budinsky R, Rowlands JC, Thomas RS and Kaminski NE, 2016. Immunological characterization of the aryl hydrocarbon receptor (AHR) knockout rat in the presence and absence of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Toxicology, 368, 172–182. [DOI] [PubMed] [Google Scholar]
- Phelan D, Winter GM, Rogers WJ, Lam JC and Denison MS, 1998. Activation of the Ah receptor signal transduction pathway by bilirubin and biliverdin. Archives of Biochemistry and Biophysics, 357, 155–163. [DOI] [PubMed] [Google Scholar]
- Picut CA, Ziejewski MK and Stanislaus D, 2018. Comparative aspects of pre‐ and postnatal development of the male reproductive system. Birth Defects Research, 110, 190–227. [DOI] [PubMed] [Google Scholar]
- Pirard C and De Pauw E, 2006. Toxicokinetic study of dioxins and furans in laying chickens. Environment International, 32, 466–469. [DOI] [PubMed] [Google Scholar]
- Pitot HC, Goldsworthy T, Campbell HA and Poland A, 1980. Quantitative‐evaluation of the promotion by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Research, 40, 3616–3620. [PubMed] [Google Scholar]
- Planche C, Ratel J, Blinet P, Mercier F, Angenieux M, Chafey C, Zinck J, Marchond N, Chevolleau S, Marchand P, Dervilly‐Pinel G, Guerin T, Debrauwer L and Engel E, 2017. Effects of pan cooking on micropollutants in meat. Food Chemistry, 232, 395–404. [DOI] [PubMed] [Google Scholar]
- Ploteau S, Cano‐Sancho G, Volteau C, Legrand A, Venisseau A, Vacher V, Marchand P, Le Bizec B and Antignac J‐P, 2017. Associations between internal exposure levels of persistent organic pollutants in adipose tissue and deep infiltrating endometriosis with or without concurrent ovarian endometrioma. Environment International, 108, 195–203. [DOI] [PubMed] [Google Scholar]
- Pluess N, Poiger H, Schlatter C and Buser HR, 1987. The metabolism of some pentachlorodibenzofurans in the rat. Xenobiotica, 17, 209–216. [DOI] [PubMed] [Google Scholar]
- Pluess N, Poiger H, Hohbach C and Schlatter C, 1988. Subchronic toxicity of some chlorinated dibenzofurans PCDFs and a mixture of PCDFs and chlorinated dibenzodioxins PCDDs in rats. Chemosphere, 17, 973–984. As cited in ATSDR (1994). [Google Scholar]
- Pluim HJ, Koppe JG, Olie K, Vanderslikke JW, Slot PC and Vanboxtel CJ, 1994. Clinical laboratory manifestations of exposure to background levels of dioxins in the perinatal‐period. Acta Paediatrica, 83, 583–587. [DOI] [PubMed] [Google Scholar]
- Pocar P, Klonisch T, Brandsch C, Eder K, Fröhlich C, Hoang‐Vu C and Hombach‐Klonisch S, 2006. AhR‐agonist‐induced transcriptional changes of genes involved in thyroid function in primary porcine thyrocytes. Toxicological Sciences, 89, 408–14. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R and Tuomisto J, 1994. Short‐term toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in laboratory animals: effects, mechanisms, and animal models. Pharmacological Reviews, 46, 483–549. [PubMed] [Google Scholar]
- Pohjanvirta R, Kulju T, Morselt AFW, Tuominen R, Juvonen R, Rozman K, Männistö P, Collan Y, Sainio E‐L and Tuomisto J, 1989. Target tissue morphology and serum biochemistry following 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) exposure in a TCDD‐susceptible and a TCDD‐resistant rat strain. Fundamental and Applied Toxicology, 12, 698–712. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Vartiainen T, Uusi‐Rauva A, Mönkkönen J and Tuomisto J, 1990a. Tissue distribution, metabolism, and excretion of 14C‐TCDD in a TCDD‐susceptible and a TCDD‐resistant rat strain. Pharmacology and Toxicology, 66, 93–100. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Håkansson H, Juvonen R and Tuomisto J, 1990b. Effects of TCDD on vitamin A status and liver microsomal enzyme activities in a TCDD‐susceptible and a TCDD‐resistant rat strain. Food and Chemical Toxicology, 28, 197–203. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Unkila M and Tuomisto J, 1993. Comparative acute lethality of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD), 1,2,3,7,8‐pentachlorodibenzo‐p‐dioxin and 1,2,3,4,7,8‐hexachlorodibenzo‐p‐dioxin in the most TCDD‐susceptible and the most TCDD‐resistant rat strain. Pharmacology and Toxicology, 73, 52–56. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Unkila M, Lindèn J, Tuomisto JT and Tuomisto J, 1995. Toxic equivalency factors do not predict the acute toxicities of dioxins in rats. Eur. J. Pharmacol. Environmental Toxicology and Pharmacology, 293, 341–353. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Wong JM, Li W, Harper PA, Tuomisto J and Okey AB, 1998. Point mutation in intron sequence causes altered carboxyl‐terminal structure in the aryl hydrocarbon receptor of the most 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐resistant rat strain. Molecular Pharmacology, 54, 86–93. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Korkalainen M, McGuire J, Simanainen U, Juvonen R, Tuomisto JT, Unkila M, Viluksela M, Bergman J, Poellinger L and Tuomisto J, 2002. Comparison of acute toxicities of indolo[3,2‐b]carbazole (ICZ) and 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in TCDD‐sensitive rats. Food and Chemical Toxicology, 40, 1023–1032. [DOI] [PubMed] [Google Scholar]
- Pohjanvirta R, Korkalainen M, Moffat ID, Boutros PC and Okey AB, 2011. Role of the AHR and its structure in TCDD toxicity In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 181–196. [Google Scholar]
- Pohjanvirta R, Miettinen H, Sankari S, Hegde N and Lindén J, 2012. Unexpected gender difference in sensitivity to the acute toxicity of dioxin in mice. Toxicology and Applied Pharmacology, 262, 167–176. [DOI] [PubMed] [Google Scholar]
- Poiger H and Schlatter C, 1979. Biological degradation of TCDD in rats. Nature, 281, 706–7. [DOI] [PubMed] [Google Scholar]
- Poiger H, Buser HR, Weber H, Zweifel U and Schlatter C, 1982. Structure elucidation of mammalian TCDD‐metabolites. Experientia, 38, 484–6. [DOI] [PubMed] [Google Scholar]
- Poiger H and Buser HR, 1984. The metabolism of TCDD in the dog and rat In: Poland A. and Kimbrough R. (eds.). Biological Mechanisms of Dioxin Action, Banbury Report 18, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, pp. 39–47. [Google Scholar]
- Poiger H and Schlatter C, 1985. Influence of phenobarbital and TCDD on the hepatic metabolism of TCDD in the dog. Experientia, 41, 376–8. [DOI] [PubMed] [Google Scholar]
- Poiger H and Schlatter C, 1986. Pharmacokinetics of 2,3,7,8‐TCDD in man. Chemosphere, 15, 1489–1494. [Google Scholar]
- Poiger H, Pluess N and Schlatter C, 1989a. Subchronic toxicity of some chlorinated dibenzofurans in rats. Chemosphere, 18, 265–275. As cited in ATSDR (1994). [Google Scholar]
- Poiger H, Pluess N and Buser HR, 1989b. The metabolism of selected PCDFs in the rat. Chemosphere, 18, 259–264. [Google Scholar]
- Poland A and Glover E, 1974. Comparison of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, a potent inducer of aryl hydrocarbon hydroxylase, with 3‐methylcholanthrene. Molecular Pharmacology, 10, 349–359. [PubMed] [Google Scholar]
- Poland A and Glover E, 1980. 2,3,7,8,‐Tetrachlorodibenzo‐p‐dioxin: segregation of toxicity with the Ah locus. Molecular Pharmacology, 17, 86–94. [PubMed] [Google Scholar]
- Poland A and Knutson JC, 1982. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annual Review of Pharmacology and Toxicology, 22, 517–554. [DOI] [PubMed] [Google Scholar]
- Poland A, Glover E and Kende AS, 1976. Stereospecific, high affinity binding of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. The Journal of Biological Chemistry, 251, 4936–4946. [PubMed] [Google Scholar]
- Poland AP, Panel D and Glover E, 1982. Tumor promotion by TCDD in skin of HRS/J hairless mice. Nature, 3000, 271–273. [DOI] [PubMed] [Google Scholar]
- Poland A, Teitelbaum P and Glover E, 1989a. [125I]2‐iodo‐3,7,8‐trichlorodibenzo‐p‐dioxin‐binding species in mouse liver induced by agonists for the Ah receptor: characterization and identification. Molecular Pharmacology, 36, 113–120. [PubMed] [Google Scholar]
- Poland A, Teitelbaum P, Glover E and Kende A, 1989b. Stimulation of in vivo hepatic uptake and in vitro hepatic binding of [125I]2‐lodo‐3,7,8‐trichlorodibenzo‐p‐dioxin by the administration of agonist for the Ah receptor. Molecular Pharmacology, 36, 121–127. [PubMed] [Google Scholar]
- Poland A, Palen D and Glover E, 1994. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Molecular Pharmacology, 46, 915–921. [PubMed] [Google Scholar]
- Pongratz I, Antonsson C, Whitelaw ML and Poellinger L, 1998. Role of the PAS domain in regulation of dimerization and DNA binding specificity of the dioxin receptor. Molecular and Cellular Biology, 18, 4079–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porpora MG, Medda E, Abballe A, Bolli S, De Angelis I, di Domenico A, Ferro A, Ingelido AM, Maggi A, Panici PB and De Felip E, 2009. Endometriosis and organochlorinated environmental pollutants: a case‐control study on Italian women of reproductive age. Environmental Health Perspectives, 117, 1070–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CL, Sipes IG and Russell DH, 1983. Hypothyroxinemia and hypothermia in rats in response to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin administration. Toxicology and Applied Pharmacology, 69, 89–95. [DOI] [PubMed] [Google Scholar]
- Potter CL, Moore RW, Inhorn SL, Hagen TC and Peterson RE, 1986. Thyroid status and thermogenesis in rats treated with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology and Applied Pharmacology, 84, 45–55. [DOI] [PubMed] [Google Scholar]
- Powis M, Celius T and Matthews J, 2011. Differential ligand‐dependent activation and a role for Y322 in aryl hydrocarbon receptor‐mediated regulation of gene expression. Biochemical and Biophysical Research Communications, 410, 859–865. [DOI] [PubMed] [Google Scholar]
- Quabius ES, Nolan DT, Allin CJ and Bonga SEW, 2000. Influence of dietary exposure to polychlorinated biphenyl 126 and nutritional state on stress response in tilapia (Oreochromis mossambicus) and rainbow trout (Oncorhynchus mykiss). Environmental Toxicology and Chemistry, 19, 2892–2899. [Google Scholar]
- Quaß U, Fermann M and Bröker G, 2004. The European dioxin air emission inventory project—final results. Chemosphere, 54, 1319–1327. [DOI] [PubMed] [Google Scholar]
- Ramadoss P and Perdew GH, 2004. Use of 2‐azido‐3‐[125I]iodo‐7,8‐dibromodibenzo‐p‐dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Molecular Pharmacology, 66, 129–136. [DOI] [PubMed] [Google Scholar]
- Ramsey JC, Hefner JG, Karbowski RJ, Braun WH and Gehring PJ, 1982. The in vivo biotransformation of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the rat. Toxicology and Applied Pharmacology, 65, 180–184. [DOI] [PubMed] [Google Scholar]
- Rappe C, Nygren M, Lindström G and Hansson M, 1986. Dioxins and dibenzofurans in biological samples of European origin. Chemosphere, 15, 1635–1639. [Google Scholar]
- Rappe C, Andersson R, Bergqvist PA, Brohede C, Hansson M, Kjeller LO, Lindström G, Marklund S, Nygren M, Swanson SE, Tysklind M and Wiberg K, 1987. Overview on environmental fate of chlorinated dioxins and dibenzofurans. Sources, levels and isomeric pattern in various matrices. Chemosphere, 16, 1603–1618. [Google Scholar]
- Rawn DFK, Breakell K, Verigin V, Tittlemier SA, Del Gobbo L, Diamond M, Vanderlinden L and Sit D, 2013. Impacts of cooking technique on polychlorinated biphenyl and polychlorinated dioxins/furan concentrations in fish and fish products with intake estimates. Journal of Agricultural and Food Chemistry, 61, 989–997. [DOI] [PubMed] [Google Scholar]
- Ray SS and Swanson HI, 2004. Dioxin‐induced immortalization of normal human keratinocytes and silencing of p53 and p16INK4a. The Journal of Biological Chemistry, 279, 27187–27193. [DOI] [PubMed] [Google Scholar]
- Rebourcet D, Odet F, Verot A, Combe E, Meugnier E, Pesenti S, Leduque P, Dechaud H, Magre S and Le Magueresse‐Battistoni B, 2010. The effects of an in utero exposure to 2,3,7,8‐tetrachloro‐dibenzo‐p‐dioxin on male reproductive function: identification of Ccl5 as a potential marker. International Journal of Andrology, 33, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeuwijk NM, Talidda A, Malisch R, Kotz A, Tritscher A, Fiedler H, Zeilmaker MJ, Kooijman M, Wienk KJH, Traag WA and Hoogenboom RLAP, 2013. Dioxins (polychlorinated dibenzo‐p‐dioxins and polychlorinated dibenzofurans) in traditional clay products used during pregnancy. Chemosphere, 90, 1678–1685. [DOI] [PubMed] [Google Scholar]
- Ren L, Thompson JD, Cheung M, Ngo K, Sung S, Leong S and Chan WK, 2016. Selective suppression of the human aryl hydrocarbon receptor function can be mediated through binding interference at the C‐terminal half of the receptor. Biochemical Pharmacology, 107, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert A, Wittsiepe J, Kasper‐Sonnenberg M, Binder G, Fuerst P, Cramer C, Kraemer U and Wilhelm M, 2012. Prenatal and early life exposure to polychlorinated dibenzo‐p‐dioxins, dibenzofurans and biphenyls may influence dehydroepiandrosterone sulfate levels at prepubertal age: results from the Duisburg Birth Cohort Study. Journal of Toxicology and Environmental Health Part A, 75, 1232–1240. [DOI] [PubMed] [Google Scholar]
- Restum JC, Bursian SJ, Giesy JP, Render JA, Helferich WC, Shipp EB, Verbrugge DA and Aulerich RJ, 1998. Multigenerational study of the effects of consumption of PCB‐contaminated carp from Saginaw Bay, Lake Huron, on mink. 1. Effects on mink reproduction, kit growth and survival, and selected biological parameters. Journal of Toxicology and Environmental Health‐Part A, 54, 343–375. [DOI] [PubMed] [Google Scholar]
- Revich B, Aksel E, Ushakova T, Ivanova I, Zhuchenko N, Klyuev N, Brodsky B and Sotskov Y, 2001. Dioxin exposure and public health in Chapaevsk, Russia. Chemosphere, 43, 951–966. [DOI] [PubMed] [Google Scholar]
- Reyes H, Reisz‐Porszasz S and Hankinson O, 1992. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science, 256, 1193–1195. [DOI] [PubMed] [Google Scholar]
- Richardson VM, Ferguson SS, Sey YM and Devito MJ, 2014. In vitro metabolism of thyroxine by rat and human hepatocytes. Xenobiotica, 44, 391–403. [DOI] [PubMed] [Google Scholar]
- Richter CA, Tillitt DE and Hannink M, 2001. Regulation of subcellular localization of the aryl hydrocarbon receptor (AhR). Archives of Biochemistry and Biophysics, 389, 207–217. [DOI] [PubMed] [Google Scholar]
- Riddick DS, Huang Y, Harper PA and Okey AB, 1994. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin versus 3‐methylcholanthrene: comparative studies of Ah receptor binding, transformation, and induction of CYP1A1. The Journal of Biological Chemistry, 269, 12118–12128. [PubMed] [Google Scholar]
- Riddick DS, Lee C, Bhathena A, Timsit YE, Cheng PY, Morgan ET, Prough RA, Ripp SL, Miller KK, Jahan A and Chiang JY, 2004. Transcriptional suppression of cytochrome P450 genes by endogenous and exogenous chemicals. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 32, 367–375. [DOI] [PubMed] [Google Scholar]
- Riecke K, Grimm D, Shakibaei M, Kossmehl P, Schulze‐Tanzil G, Paul M and Stahlmann R, 2002. Low doses of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin increase transforming growth factor beta and cause myocardial fibrosis in marmosets (Callithrix jacchus). Archives of Toxicology, 76, 360–366. [DOI] [PubMed] [Google Scholar]
- Rier SE, Martin DC, Bowman RE, Dmowski WP and Becker JL, 1993. Endometriosis in Rhesus Monkeys (Macaca mulatta) following chronic exposure to 2,3,7,8,‐tetrachlorodibenzo‐p‐dioxin. Fundamental and Applied Toxicology, 21, 433–441. [DOI] [PubMed] [Google Scholar]
- Rier SE, Turner WE, Martin DC, Morris R, Lucier GW and Clark GC, 2001a. Serum levels of TCDD and dioxin‐like chemicals in rhesus monkeys chronically exposed to dioxin: correlation of increased serum PCB levels with endometriosis. Toxicological Sciences, 59, 147–159. [DOI] [PubMed] [Google Scholar]
- Rier SE, Coe CL, Lemieux AM, Martin DC, Morris R, Lucier GW and Clark GC, 2001b. Increased tumor necrosis factor‐alpha production by peripheral blood leukocytes from TCDD‐exposed rhesus monkeys. Toxicological Sciences, 60, 327–337. [DOI] [PubMed] [Google Scholar]
- Rigaud C, Couillard CM, Pellerin J, Legare B and Hodson PV, 2014. Applicability of the TCDD‐TEQ approach to predict sublethal embryotoxicity in Fundulus heteroclitus. Aquatic Toxicology (Amsterdam, Netherlands), 149, 133–144. [DOI] [PubMed] [Google Scholar]
- RIVM and RIKILT Wageningen University & Research , 2018. Implementation and verification of PBPK modelling codes of TCDD in rats and humans into Berkeley Madonna. EFSA supporting publication 2018:EN‐1374, 24 pp. 10.2903/sp.efsa.2018.en-1374 [DOI] [Google Scholar]
- Roberts BJ and Whitelaw ML, 1999. Degradation of the basic helix‐loop‐helix/Per‐ARNT‐Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. The Journal of Biological Chemistry, 274, 36351–36356. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Pichardo A, Camacho F, Rappe C, Hansson M, Smith AG and Greig IB, 1991. Chloracne caused by ingestion of olive oil contaminated with PCDDs and PCDFs. Human and Experimental Toxicology, 10, 311–322. [DOI] [PubMed] [Google Scholar]
- Rogan WJ, Gladen BC, Hung KL, Koong SL, Shih LY, Taylor JS, Wu YC, Yang D, Ragan NB and Hsu CC, 1988. Congenital poisoning by polychlorinated biphenyls and their contaminants in Taiwan. Science, 241, 334–336. [DOI] [PubMed] [Google Scholar]
- Rogan WJ, Gladen BC, Guo YL and Hsu CC, 1999. Sex ratio after exposure to dioxin‐like chemicals in Taiwan. Lancet, 353, 206–207. [DOI] [PubMed] [Google Scholar]
- Rohde S, Moser GA, Papke O and McLachlan MS, 1999. Clearance of PCDD/Fs via the gastrointestinal tract in occupationally exposed persons. Chemosphere, 38, 3397–3410. [DOI] [PubMed] [Google Scholar]
- Romkes M and Safe S, 1988. Comparative activities of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin and progesterone as antiestrogens in the female rat uterus. Toxicology and Applied Pharmacology, 92, 368–380. [DOI] [PubMed] [Google Scholar]
- Rooney AA, Boyles AL, Wolfe MS, Bucher JR and Thayer KA, 2014. Systematic review and evidence integration for literature‐based environmental health science assessments. Environmental Health Perspectives, 122, 711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JQ, Ramsey JC, Wentzler TH, Hummel RA and Gehring PJ, 1976. The fate of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin following single and repeated oral doses to the rat. Toxicology and Applied Pharmacology, 36, 209–26. [DOI] [PubMed] [Google Scholar]
- Rose M, Thorpe S, Kelly M, Harrison N and Startin J, 2001. Changes in concentration of five PCDD/F congeners after cooking beef from treated cattle. Chemosphere, 43, 861–868. [DOI] [PubMed] [Google Scholar]
- Rose M, Fernandes A, Foxall C and Dowding A, 2012. Transfer and uptake of polychlorinated dibenzo‐p‐dioxins and furans (PCDD/Fs) and polychlorinated biphenyls (PCBs) into meat and organs of indoor and outdoor reared pigs. Food Additives and Contaminants A, 29, 431–448. [DOI] [PubMed] [Google Scholar]
- Roth W, Voorman R and Aust SD, 1988. Activity of thyroid hormone‐inducible enzymes following treatment with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicology and Applied Pharmacology, 92, 65–74. [DOI] [PubMed] [Google Scholar]
- Rouaze‐Romet M, Vranckx R, Savu L and Nunez EA, 1992. Structural and functional microheterogeneity of rat thyroxine‐binding globulin during ontogenesis. Biochemical Journal, 286(Pt 1), 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands CJ, Staskal DF, Gollapudi B and Budinsky R, 2010. The human AHR: identification of single nucleotide polymorphisms from six ethnic populations. Pharmacogenetics and Genomics, 20, 283–290. [DOI] [PubMed] [Google Scholar]
- Rowlands JC, Budinsky R, Gollapudi B, Novak R, Abdelmegeed M, Cukovic D and Dombkowski A, 2011a. Transcriptional profiles induced by the Aryl Hydrocarbon Receptor agonists 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, 2,3,7,8‐tetrachlorodibenzofuran and 2,3,4,7,8‐pentachlorodibenzofuran in primary rat hepatocytes. Chemosphere, 85, 232–238. [DOI] [PubMed] [Google Scholar]
- Rowlands JC, Urban JD, Wikoff DS and Budinsky RA, 2011b. An evaluation of single nucleotide polymorphisms in the human aryl hydrocarbon receptor‐interacting protein (AIP) gene. Drug Metabolism and Pharmacokinetics, 26, 431–439. [DOI] [PubMed] [Google Scholar]
- Rozman K, Hazelton GA, Klaassen CD, Arlotto MP and Parkinson A, 1985. Effect of thyroid hormones on liver microsomal enzyme induction in rats exposed to 2,3,7,8,‐tetrachlorodibenzo‐p‐dioxin. Toxicology, 37, 51–63. [DOI] [PubMed] [Google Scholar]
- Rozman K and Greim H, 1986. Toxicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in cold‐adapted rats. Archives of Toxicology, 59, 211–5. [DOI] [PubMed] [Google Scholar]
- Rozman K, Pereira D and Iatropoulos MJ, 1986. Histopathology of interscapular brown adipose‐tissue, thyroid, and pancreas in 2,3,7,8‐tetrachlorodibenzo‐para‐dioxin (TCDD)‐treated rats. Toxicology and Applied Pharmacology, 82, 551–559. 10.1016/0041-008X(86),90290-5 [DOI] [PubMed] [Google Scholar]
- RPA and IEH (Risk & Policy Analysts Limited and IEH Consulting Limited), 2018. Extensive literature search, selection for relevance and data extraction of studies related to the toxicity of PCDD/Fs and DL‐PCBs in humans. EFSA supporting publication 2018:EN‐1136, 57 pp. 10.2903/sp.efsa.2018.en-1136 [DOI] [Google Scholar]
- Rüegg J, Swedenborg E, Wahlstrom D, Escande A, Balaguer P, Pettersson K and Pongratz I, 2008. The transcription factor aryl hydrocarbon receptor nuclear translocator functions as an estrogen receptor beta‐selective coactivator, and its recruitment to alternative pathways mediates antiestrogenic effects of dioxin. Molecular Endocrinology, 22, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz P, Aylward LL and Mumtaz M, 2014. Application of pharmacokinetic modelling for 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin exposure assessment. SAR and QSAR in Environmental Research, 25, 873–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rune GM, Desouza P, Krowke R, Merker HJ and Neubert D, 1991a. Morphological and histochemical effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) on marmoset (Callithrix‐jacchus) testes. Archives of Andrology, 26, 143–154. [DOI] [PubMed] [Google Scholar]
- Rune GM, Desouza P, Krowke R, Merker HJ and Neubert D, 1991b. Morphological and histochemical pattern of response in rat testes after administration of 2,3,7,8‐tetra‐chlorodibenzo‐p‐dioxin (TCDD). Histology and Histopathology, 6, 459–467. [PubMed] [Google Scholar]
- Ruus A, Daae IA and Hylland K, 2012. Accumulation of polychlorinated biphenyls from contaminated sediment by Atlantic cod (Gadus morhua): direct accumulation from resuspended sediment and dietary accumulation via the polychaete Nereis virens . Environmental Toxicology and Chemistry, 31, 2472–2481. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, 1983. Higher chlorinated dioxins implicated in the mortality of young ‐pigs kept on a pentachlorophenol‐treated wooden floor. Canadian Veterinary Journal‐Revue Veterinaire Canadienne, 24, 72–75. [PMC free article] [PubMed] [Google Scholar]
- Ryan JJ, Gasiewicz TA and Brown JF, 1990. Human‐body burden of polychlorinated dibenzofurans associated with toxicity based on the Yusho and Yucheng incidents. Fundamental and Applied Toxicology, 15, 722–731. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Hsu CC, Boyle MJ and Guo YL, 1994. Blood serum levels of PCDFs and PCBs in Yu‐Cheng children perinatally exposed to a toxic rice oil. Chemosphere, 29, 1263–1278. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Amirova Z and Carrier G, 2002. Sex ratios of children of Russian pesticide producers exposed to dioxin. Environmental Health Perspectives, 110, A699–A701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi Hosnijeh F, Boers D, Portengen L, Bueno‐de‐Mesquita HB, Heederik D and Vermeulen R, 2011. Long‐term effects on humoral immunity among workers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Occupational and Environmental Medicine, 68, 419–424. [DOI] [PubMed] [Google Scholar]
- Saberi Hosnijeh F, Lenters V, Boers D, Portengen L, Baeten E, Bueno‐de‐Mesquita HB, Heederik DJ, Bloem AC and Vermeulen R, 2012. Changes in lymphocyte subsets in workers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Occupational and Environmental Medicine, 69, 781–786. [DOI] [PubMed] [Google Scholar]
- Saberi Hosnijeh F, Portengen L, Bueno‐de‐Mesquita HB, Heederik D and Vermeulen R, 2013. Circulating soluble CD27 and CD30 in workers exposed to 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD). Cancer Epidemiology, Biomarkers and Prevention, 22, 2420–2424. [DOI] [PubMed] [Google Scholar]
- Safe SH, 1986. Comparative toxicology and mechanism of action of polychlorinated dibenzo‐p‐dioxins and dibenzofurans. Annual Review of Pharmacology and Toxicology, 26, 371–399. [DOI] [PubMed] [Google Scholar]
- Safe S, 1992. Toxicology, structure‐function relationship, and human and environmental health impacts of polychlorinated biphenyls: progress and problems. Environmental Health Perspectives, 100, 259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S, 1994. Polychlorinated biphenyls (PCBs): environmental impact, biochemical and toxic responses, and implications for risk assessment. Critical Reviews in Toxicology, 24, 87–149. [DOI] [PubMed] [Google Scholar]
- Safe S, Wormke M and Samudio I, 2000. Mechanisms of inhibitory aryl hydrocarbon receptor‐estrogen receptor crosstalk in human breast cancer cells. Journal of Mammary Gland Biology and Neoplasia, 5, 295–306. [DOI] [PubMed] [Google Scholar]
- Safe S, Chadalapaka G and Jutooru I, 2012. AHR‐active compounds in the human diet In: Pohjanvirta R. (ed.). The AH Receptor in Biology and Toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 331–342. [Google Scholar]
- Sahlberg C, Pohjanvirta R, Gao Y, Alaluusua S, Tuomisto J and Lukinmaa PL, 2002. Expression of the mediators of dioxin toxicity, aryl hydrocarbon receptor (AHR) and the AHR nuclear translocator (ARNT), is developmentally regulated in mouse teeth. The International Journal of Developmental Biology, 46, 295–300. [PubMed] [Google Scholar]
- Sanabria M, Cucielo MS, Guerra MT, Borges CdS, Banzato TP, Perobelli JE, Araujo Leite GA, Anselmo‐Franci JA and Kempinas WDG, 2016. Sperm quality and fertility in rats after prenatal exposure to low doses of TCDD: A three‐generation study. Reproductive Toxicology, 65, 29–38. [DOI] [PubMed] [Google Scholar]
- Sandal S, Yilmaz B and Carpenter DO, 2008. Genotoxic effects of PCB 52 and PCB 77 on cultured human peripheral lymphocytes. Mutation Research‐Genetic Toxicology and Environmental Mutagenesis, 654, 88–92. [DOI] [PubMed] [Google Scholar]
- Sanden M, Hemre GI, Måge A, Lunestad BT, Espe M, Lundebye AK, Amlund H and Ørnsrud R, 2016. Program for overvåking av fiskefôr. Årsrapport for prøver innsamlet i 2015, National Institute of Nutrition and Seafood Research (NIFES). pp. 20. Available online: https://www.nifes.no/report/forrapport-2015-3/
- Sanderson JT, Aarts JMMJG, Brouwer A, Froese KL, Denison MS and Giesy JP, 1996. Comparison of Ah‐receptor‐mediated luciferase and ethoxyresorufin O‐deethylase induction in H4IIE cells: implications for their use as bioanalytical tools for the detection of polyhalogenated aromatic compounds. Toxicology and Applied Pharmacology, 137, 316–325. [DOI] [PubMed] [Google Scholar]
- Santostefano MJ, Johnson KL, Whisnant NA, Richardson VM, DeVito MJ, Diliberto JJ and Birnbaum LS, 1996. Subcellular localization of TCDD differs between the liver, lungs, and kidneys after acute and subchronic exposure: species /dose comparisons and possible mechanism. Fundamental and Applied Toxicology, 34, 265–275. [DOI] [PubMed] [Google Scholar]
- Sasamoto T, Horii Sh, Ibe A, Takada N and Shirota K, 2006. Concentration changes of PCDDs, PCDFs, and dioxin‐like PCBs in human breast milk samples as shown by a follow‐up survey. Chemosphere, 64, 642–649. [DOI] [PubMed] [Google Scholar]
- Sato S, Shirakawa H, Tomita S, Ohsaki Y, Haketa K, Tooi O, Santo N, Tohkin M, Furukawa Y, Gonzalez FJ and Komai M, 2008. Low‐dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor‐mediated pathway in mouse liver. Toxicology and Applied Pharmacology, 229, 10–19. [DOI] [PubMed] [Google Scholar]
- Saurat JH, Kaya G, Saxer‐Sekulic N, Pardo B, Becker M, Fontao L, Mottu F, Carraux P, Pham XC, Barde C, Fontao F, Zennegg M, Schmid P, Schaad O, Descombes P and Sorg O, 2012. The Cutaneous lesions of dioxin exposure: lessons from the poisoning of Victor Yushchenko. Toxicological Sciences, 125, 310–317. [DOI] [PubMed] [Google Scholar]
- Savouret JF, Antenos M, Quesne M, Xu J, Milgrom E and Casper RF, 2001. 7‐Ketocholesterol is an endogenous modulator for the arylhydrocarbon receptor. The Journal of Biological Chemistry, 276, 3054–3059. [DOI] [PubMed] [Google Scholar]
- Sawahata T, Olson JR and Neal RA, 1982. Identification of metabolites of 2, 3, 7, 8‐tetrachlorodibenzo‐p‐dioxin (TCDD) formed on incubation with isolated rat hepatocytes. Biochemical and Biophysical Research Communications, 105, 341–6. [DOI] [PubMed] [Google Scholar]
- Sawyer T, Jones D, Rosanoff K, Mason G, Piskorska‐Pliszczynska J and Safe S, 1986. The biologic and toxic effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in chickens. Toxicology, 39, 197–206. [DOI] [PubMed] [Google Scholar]
- SCAN (Scientific Committee on Animal Nutrition), 2000. Dioxin Contamination of feedingstuffs and their contribution to the contamination of food of animal origin. Available at: https://ec.europa.eu/food/sites/food/files/safety/docs/animal-feed-undes-sub-out55_en.pdf
- SCF (Scientific Committee on Food), 2000. Opinion on the risk assessment of dioxins and dioxin‐like PCBs in food. SCF/CS/CNTM/DIOXIN/8 Final. SCF, Health and Consumer Protection Directorate‐General, European Commission. Brussels, 141 pp. Available online: http://ec.europa.eu/food/fs/sc/scf/out78_en.pdf
- SCF (Scientific Committee on Food), 2001. Opinion on the risk assessment of dioxins and dioxinslike PCB in food (update based on the new scientific information available since the adoption of the SCF opinion of 22 November 2000) (adopted by the SCF on 30 May 2001). 29 pp. Available online: http://ec.europa.eu/food/fs/sc/scf/out90_en.pdf
- Schaldach CM, Riby J and Bjeldanes LF, 1999. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry, 38, 7594–7600. [DOI] [PubMed] [Google Scholar]
- Schantz S and Bowman RE, 1989. Learning in monkeys exposed perinatally to 2,3,7,8‐ tetrachlorodibenzo‐p‐dioxin (TCDD). Neurotoxicology and Teratology, 11, 13–19. [DOI] [PubMed] [Google Scholar]
- Schantz SL, Ferguson SA and Bowman RE, 1992. Effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on behaviour of monkey in peer groups. Neurotoxicology and Teratology, 14, 433–446. [DOI] [PubMed] [Google Scholar]
- Schecter A and Ryan JJ, 1988. Polychlorinated dibenzo‐para‐dioxin and dibenzofuran levels in human adipose tissue from workers 32 years after occupational exposure to 2,3,7,8‐TCDD. Chemosphere, 17, 915–920. [Google Scholar]
- Schecter A, Ryan JJ, Constable JD, Baughman R, Bangert J, Furst P, Wilmers K and Oates RP, 1990. Partitioning of 2,3,7,8‐chlorinated dibenzo‐p‐dioxins and dibenzofurans between adipose tissue and plasma lipid of 20 Massachusetts Vietnam veterans. Chemosphere, 20, 951–958. [Google Scholar]
- Schecter A, Papke O, Ball M and Ryan JJ, 1991. Partitioning of dioxins and dibenzofurans – whole‐blood, blood‐plasma and adipose‐tissue. Chemosphere, 23, 1913–1919. [Google Scholar]
- Schecter A, Ryan JJ and Constable JD, 1992. Partitioning of dioxin and dibenzofuran congeners between plasma and cell‐fractions of blood from 10 adult male‐patients. Chemosphere, 25, 2017–2022. [Google Scholar]
- Schecter A, Dellarco M, Papke O and Olson J, 1998a. A comparison of dioxins, dibenzofurans and coplanar PCBs in uncooked and broiled ground beef, catfish and bacon. Chemosphere, 37, 1723–1730. [DOI] [PubMed] [Google Scholar]
- Schecter A, Kassis I and Päpke O, 1998b. Partitioning of dioxins, dibenzofurans, and coplanar PCBs in blood, milk, adipose tissue, placenta and cord blood from five American women. Chemosphere, 37, 1817–1823. [DOI] [PubMed] [Google Scholar]
- Schnorr TM, Lawson CC, Whelan EA, Dankovic DA, Deddens JA, Piacitelli LA, Reefhuis J, Sweeney MH, Connally LB and Fingerhut MA, 2001. Spontaneous abortion, sex ratio, and paternal occupational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Environmental Health Perspectives, 109, 1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeters G and Hoogenboom R, 2006. Contamination of free‐range chicken eggs with dioxins and dioxin‐like polychlorinated biphenyls. Molecular Nutrition and Food Research, 50, 908–914. [DOI] [PubMed] [Google Scholar]
- Schoeters G, Den Honda E, Colles A, Loots I, Morrens B, Keune H, Bruckers L, Nawrot T, Sioen I, De Coster S, Van Larebeke N, Nelen V, Van de Mieroop E, Vrijens J, Croes K, Goeyens K and Baeyens W, 2012. Concept of the Flemish human biomonitoring programme. International Journal of Hygiene and Environmental Health, 215, 102–108. [DOI] [PubMed] [Google Scholar]
- Schramm K‐W, Lenk S, Rambeck WA, Mayer R, Henkelmann B and Wehr U, 2009. Influence of dioxin contaminated feed and its effect upon the content in adipose tissues of pigs. Organohalogen Compound, 71, 000712–000714. [Google Scholar]
- Schuhmacher M, Kiviranta H, Ruokojärvi P, Nadal M and Domingo JL, 2013. Levels of PCDD/Fs, PCBs and PBDEs in breast milk of women living in the vicinity of a hazardous waste incinerator: assessment of the temporal trend. Chemosphere, 93, 1533–1540. [DOI] [PubMed] [Google Scholar]
- Schuler F, Schmid P and Schlatter Ch, 1997. Transfer of airborne polychlorinated dibenzo‐p‐dioxins and dibenzofurans into dairy milk. Journal of Agricultural and Food Chemistry, 45, 4162–4167. [Google Scholar]
- Schwartz M and Appel KE, 2005. Carcinogenic risks of dioxin: mechanistic considerations. Regulatory Toxicology and Pharmacology, 43, 19–34. [DOI] [PubMed] [Google Scholar]
- Schwetz BA, Norris JM, Sparschu GL, Rowe UK, Gehring PJ, Emerson JL and Gerbig CG, 1973. Toxicology of chlorinated dibenzo‐p‐dioxins. Environmental Health Perspectives, 5, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MA, Tarara RP, Hendrickx AG, Benirschke K, Overstreet JW and Lasley BL, 2001. Exposure to the dioxin 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) induces squamous metaplasia in the endocervix of cynomolgus macaques. Journal of Medical Primatology, 30, 156–160. [DOI] [PubMed] [Google Scholar]
- Sechman A, Hrabia A, Lis MW and Niedziolka J, 2011. Effect of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) on steroid concentrations in blood and gonads of chicken embryo. Toxicology Letters, 205, 190–195. [DOI] [PubMed] [Google Scholar]
- Sekine H, Mimura J, Oshima M, Okawa H, Kanno J, Igarashi K, Gonzalez FJ, Ikuta T, Kawajiri K and Fujii‐Kuriyama Y, 2009. Hypersensitivity of aryl hydrocarbon receptor‐deficient mice to lipopolysaccharide‐induced septic shock. Molecular and Cellular Biology, 29, 6391–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servos MR, Muir DCG and Webster GRB, 1989. The effect of dissolved organic‐matter on the bioavailability of polychlorinated dibenzo‐p‐dioxins. Aquatic Toxicology, 14, 169–184. [Google Scholar]
- Sévère S, Marchand P, Guiffard I, Morio F, Venisseau A, Veyrand B, Le Bizec B, Antignac JP and Abadie J, 2015. Pollutants in pet dogs: a model for environmental links to breast cancer. Springerplus, 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shellis P and Berkovitz BWB, 1981. The dentition of laboratory rodents and lagomorphs In: Osborn JW, (ed.). Dental Anatomy and Embryology. Blackwell Sci, Oxford, pp. 432–439. [Google Scholar]
- Shen ES and Olson JR, 1987. Relationship between the murine Ah phenotype and the hepatic uptake and metabolism of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Drug Metabolism and Disposition, 15, 653–60. [PubMed] [Google Scholar]
- Shen ES and Whitlock JP Jr, 1989. The potential role of DNA methylation in the response to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. The Journal of Biological Chemistry, 264, 17754–17758. [PubMed] [Google Scholar]
- Shen H, Henkelmann B, Rambeck WA, Mayer R, Wehr U and Schramm K‐W, 2012a. Physiologically based persistent organic pollutant accumulation in pig tissues and their edible safety differences: an in vivo study. Food Chemistry, 132, 1830–1835. [Google Scholar]
- Shen H, Henkelmann B, Rambeck WA, Mayer R, Wehr U and Schramm KW, 2012b. The predictive power of the elimination of dioxin‐like pollutants from pigs: an in vivo study. Environment International, 38, 73–78. [DOI] [PubMed] [Google Scholar]
- Sherer RA and Price PS, 1993. The effect of cooking processes on PCB levels in edible fish tissue. Quality Assurance (San Diego, Calif.), 2, 396–407. [PubMed] [Google Scholar]
- Sherr DH and Monti S, 2013. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Seminars in Immunopathology, 35, 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SI, Wang IC, Wu KY, Li HW, Wang LC and Chang‐Chien GP, 2009. Uptake of polychlorinated dibenzo‐p‐dioxins and dibenzofurans in laying ducks. Journal of Environmental Science and Health. Part A, Toxic/Hazardous Substances and Environmental Engineering, 44, 799–807. [DOI] [PubMed] [Google Scholar]
- Shimada T and Fujii‐Kuriyama Y, 2004. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Science, 95, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkyo R, Sakaki T, Ohta M and Inouye K, 2003. Metabolic pathways of dioxin by CYP1A1: species difference between rat and human CYP1A subfamily in the metabolism of dioxins. Archives of Biochemistry and Biophysics, 409, 180–7. [DOI] [PubMed] [Google Scholar]
- Shirota M, Kaneko T, Okuyama M, Sakurada Y, Shirota K and Matsuki Y, 2007. Internal dose‐effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in gonadotropin‐primed weanling rat model. Archives of Toxicology, 81, 261–269. [DOI] [PubMed] [Google Scholar]
- Shridhar S, Farley A, Reid RL, Foster WG and Van Vugt DA, 2001. The effect of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin on corticotrophin‐releasing hormone, arginine vasopressin, and pro‐opiomelanocortin mRNA levels in the hypothalamus of the cynomolgus monkey. Toxicological Sciences, 63, 181–188. [DOI] [PubMed] [Google Scholar]
- Sijm D, Yarechewski AL, Muir DCG, Webster GRB, Seinen W and Opperhuizen A, 1990. Biotransformation and tissue distribution of 1,2,3,7‐tetrachlorodibenzo‐p‐dioxin, 1,2,3,4,7‐pentachlorodibenzo‐p‐dioxin and 2,3,4,7,8‐pentachlorodibenzofuran in rainbow trout. Chemosphere, 21, 845–866. [Google Scholar]
- Sijm D, Part P and Opperhuizen A, 1993. The influence of temperature on the uptake rate constants of hydrophobic compounds determined by the isolated perfused gills of rainbow trout (Oncorhynchus mykiss). Aquatic Toxicology, 25, 1–14. [Google Scholar]
- Silkworth JB, Koganti A, Illouz K, Possolo A, Zhao M and Hamilton SB, 2005. Comparison of TCDD and PCB CYP1A induction sensitivities in fresh hepatocytes from human donors, Sprague‐Dawley rats, and rhesus monkeys and HepG2 cells. Toxicological Sciences, 87, 508–519. [DOI] [PubMed] [Google Scholar]
- Simanainen U, Tuomisto JT, Tuomisto J and Viluksela M, 2002. Structure‐activity relationships and dose responses of polychlorinated dibenzo‐p‐dioxins for short‐term effects in 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐resistant and ‐sensitive rat strains. Toxicology and Applied Pharmacology, 181, 38–47. [DOI] [PubMed] [Google Scholar]
- Simsa P, Mihalyi A, Schoeters G, Koppen G, Kyama CM, Den Hond EM, Fueloep V and D'Hooghe TM, 2010. Increased exposure to dioxin‐like compounds is associated with endometriosis in a case‐control study in women. Reproductive Biomedicine Online, 20, 681–688. [DOI] [PubMed] [Google Scholar]
- Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M and Nagarkatti PS, 2011. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL‐17 expression and amelioration of experimental colitis. PloS One, 6, e23522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, Singh UP, Guan H, Nagarkatti P and Nagarkatti M, 2012. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PloS One, 7, e45054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissener NH, Julshamn K, Espe M, Lunestad BT, Hemre GI, Waagbø R and Måge A, 2013. Surveillance of selected nutrients, additives and undesirables in commercial Norwegian fish feeds in the years 2000‐2010. Aquaculture Nutrition, 19, 555–572. [Google Scholar]
- Sjödin A, Tullsten A‐K and Klasson‐Wehler E, 1998. Identification of the parent compounds to selectively retained hydroxylated PCB metabolites in rat blood plasma. Organohalogen Compound, 3, 365–8. [Google Scholar]
- Slezak BP, Hatch GE, Devito MJ, Diliberto JJ, Slade R, Crissman K, Hassoun E and Birnbaum LS, 2000. Oxidative stress in female B6C3F1 mice following acute and subchronic exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). Toxicological Sciences, 54, 390–398. [DOI] [PubMed] [Google Scholar]
- Slob W, Olling M, Derks HJGM and de Jong APJM, 1995. Congener‐specific bioavailability of PCDD/Fs and coplanar PCBs in cows: laboratory and field measurements. Chemosphere, 31, 3827–3838. [DOI] [PubMed] [Google Scholar]
- Smirnova A, Wincent E, Vikstrom Bergander L, Alsberg T, Bergman J, Rannug A and Rannug U, 2016. Evidence for new light‐independent pathways for generation of the endogenous aryl hydrocarbon receptor agonist FICZ. Chemical Research in Toxicology, 29, 75–86. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Sykes DC, Cantrell DW and Moldoveanu SC, 2004. Dioxin levels in mainstream smoke from cigarettes with different TPM deliveries. Beiträge zur Tabakforschung international, 21, 205–209. [Google Scholar]
- Sogawa K, Iwabuchi K, Abe H and Fujii‐Kuriyama Y, 1995. Transcriptional activation domains of the Ah receptor and Ah receptor nuclear translocator. Journal of Cancer Research and Clinical Oncology, 121, 612–620. [DOI] [PubMed] [Google Scholar]
- Song J, Clagett‐Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR and DeLuca HF, 2002. A ligand for the aryl hydrocarbon receptor isolated from lung. Proceedings of the National Academy of Sciences of the United States of America, 99, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonne C, Dietz C, Born EW, Leifsson PS and Andersen S, 2008. Is there a link between hypospadias and organochlorine exposure in East Greenland sledge dogs (Canis familiaris)? Ecotoxicology and Environemntal Safety, 69, 391–395. [DOI] [PubMed] [Google Scholar]
- Soong D‐K and Ling Y‐C, 1997. Reassessment of PCDD/Fs and Co‐PCBs toxicity in contaminated trice‐bran oil responsible for the disease “Yu‐Cheng”. Chemosphere, 34, 5–7. [DOI] [PubMed] [Google Scholar]
- Sorg O, Zennegg M, Schmid P, Fedosyuk R, Valikhnovskyi R, Gaide O, Kniazevych V and Saurat JH, 2009. 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) poisoning in Victor Yushchenko: identification and measurement of TCDD metabolites. Lancet, 374, 1179–1185. [DOI] [PubMed] [Google Scholar]
- Soshilov A and Denison MS, 2008. Role of the Per/Arnt/Sim domains in ligand‐dependent transformation of the aryl hydrocarbon receptor. The Journal of Biological Chemistry, 283, 32995–33005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnuolo MS, Sarubbi F, Rossetti C, Grazioli G, Di Meo GP and Iannuzzi L, 2011. Effect of dioxin exposure on several indices of blood redox status in lactating buffalo cows. Journal of Dairy Research, 78, 154–159. [DOI] [PubMed] [Google Scholar]
- Spagnuolo MS, Cigliano L, Nebbia C, Rossetti C, Grazioli G and Iannuzzi L, 2012. Analysis of plasma indices of redox homeostasis in dairy cows reared in polluted areas of Piedmont (northern Italy). Science of the Total Environment, 433, 450–455. [DOI] [PubMed] [Google Scholar]
- Spitaler M, Iben C and Tausch H, 2005. Dioxin residues in the edible tissue of finisching pigs after dioxin feeding. Journal of Animal Physiology and Animal Nutrition, 89, 65–71. [DOI] [PubMed] [Google Scholar]
- Spitsbergen JM, Kleeman JM and Peterson RE, 1988a. Morphologic lesions and acute toxicity in rainbow‐trout (Salmo gairdneri) treated with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Journal of Toxicology and Environmental Health, 23, 333–358. [DOI] [PubMed] [Google Scholar]
- Spitsbergen JM, Schat KA, Kleeman JM and Peterson RE, 1988b. Effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) or Aroclor 1254 on the resistance of rainbow trout, Salmo gairdneri Richardson, to infectious haematopoietic necrosis virus. Journal of Fish Diseases, 11, 73–83. [Google Scholar]
- Spitsbergen JM, Walker MK, Olson JR and Peterson RE, 1991. Pathological alterations in early life stages of lake trout, Salvelinus namaycush, exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin as fertilized eggs. Aquatic Toxicology, 19, 41–71. [Google Scholar]
- Sprague M, Bendiksen EA, Dick JR, Strachan F, Pratoomyot J, Berntssen MHG, Tocher DR and Bell JG, 2010. Effects of decontaminated fish oil or a fish and vegetable oil blend on persistent organic pollutant and fatty acid compositions in diet and flesh of Atlantic salmon (Salmo solar). British Journal of Nutrition, 103, 1442–1451. [DOI] [PubMed] [Google Scholar]
- Sprague M, Walton J, Campbell PJ, Strachan F, Dick JR and Bell JG, 2015. Replacement of fish oil with a DHA‐rich algal meal derived from Schizochytrium sp on the fatty acid and persistent organic pollutant levels in diets and flesh of Atlantic salmon (Salmo salar, L.) post‐smolts. Food Chemistry, 185, 413–421. [DOI] [PubMed] [Google Scholar]
- Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, Lee K and Jayaraman A, 2014. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nature Communications, 5, 5492. [DOI] [PubMed] [Google Scholar]
- Srogi K, 2008. Levels and congener distributions of PCDDs, PCDFs and dioxin‐like PCBs in environmental and human samples: a review. Environmental Chemistry Letters, 6, 1–28. [Google Scholar]
- Stachiw NC, Zabik ME, Booren AM and Zabik MJ, 1988. Tetrachlorodibenzo‐para‐dioxin residue reduction through cooking processing of restructured carp fillets. Journal of Agricultural and Food Chemistry, 36, 848–852. [Google Scholar]
- Steenland K, Piacitelli L, Deddens J, Fingerhut M and Chang LI, 1999. Cancer, heart disease, and diabetes in workers exposed to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Journal of the National Cancer Institute, 91, 779–786. [DOI] [PubMed] [Google Scholar]
- Steenland K, Calvert G, Ketchum N and Michalek J, 2001. Dioxin and diabetes mellitus: an analysis of the combined NIOSH and Ranch Hand data. Occupational and Environmental Medicine, 58, 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmaus C, Miller MD and Smith AH, 2010. Perchlorate in drinking water during pregnancy and neonatal thyroid hormone levels in California. Journal of Occupational and Environmental Medicine, 52, 1217–1224. [DOI] [PubMed] [Google Scholar]
- Stockinger B, Di Meglio P, Gialitakis M and Duarte JH, 2014. The aryl hydrocarbon receptor: multitasking in the immune system. Annual Review of Immunology, 32, 403–32. [DOI] [PubMed] [Google Scholar]
- Stolevik SB, Nygaard UC, Namork E, Haugen M, Kvalem HE, Meltzer HM, Alexander J, van Delft JHM, van Loveren H, Lovik M and Granum B, 2011. Prenatal exposure to polychlorinated biphenyls and dioxins is associated with increased risk of wheeze and infections in infants. Food and Chemical Toxicology, 49, 1843–1848. [DOI] [PubMed] [Google Scholar]
- Stolevik SB, Nygaard UC, Namork E, Haugen M, Meltzer HM, Alexander J, Knutsen HK, Aaberge I, Vainio K, van Loveren H, Lovik M and Granum B, 2013. Prenatal exposure to polychlorinated biphenyls and dioxins from the maternal diet may be associated with immunosuppressive effects that persist into early childhood. Food and Chemical Toxicology, 51, 165–172. [DOI] [PubMed] [Google Scholar]
- Storelli MM, Storelli A, Barone G and Franchini D, 2009. Accumulation of polychlorinated biphenyls and organochlorine pesticide in pet cats and dogs: assessment of toxicological status. Science of the Total Environment, 408, 64–68. [DOI] [PubMed] [Google Scholar]
- Su PH, Huang PC, Lin CY, Ying TH, Chen JY and Wang SL, 2012. The effect of in utero exposure to dioxins and polychlorinated biphenyls on reproductive development in eight year‐old children. Environment International, 39, 181–187. [DOI] [PubMed] [Google Scholar]
- Su PH, Chen HY, Chen SJ, Chen JY, Liou SH and Wang SL, 2015. Thyroid and growth hormone concentrations in 8‐year‐old children exposed in utero to dioxins and polychlorinated biphenyls. The Jourmnal of Toxicological Sciences, 40, 309–319. [DOI] [PubMed] [Google Scholar]
- Sulentic CEW and Kaminski NE, 2011. The long winding road toward understanding the molecular mechanisms for B‐Cell suppression by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin. Toxicological Sciences, 120, S171–S191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson BG, Barregård L, Sällasten G, Nilsson A, Hansson M and Rappe C, 1993. Exposure to polychlorinated dioxins (PCDD) and dibenzofurans (PCDF) from graphite electrodes in a chloralkali plant. Chemosphere, 27, 259–262. [Google Scholar]
- Tai HL, McReynolds JH, Goldstein JA, Eugster HP, Sengstag C, Alworth WL and Olson JR, 1993. Cytochrome P4501A1 mediates the metabolism of 2,3,7,8‐tetrachlorodibenzofuran in the rat and human. Toxicology and Applied Pharmacology, 123, 34–42. [DOI] [PubMed] [Google Scholar]
- Tajimi M, Uehara R, Watanabe M, Oki I, Ojima T and Nakamura Y, 2005. Relationship of PCDD/F and Co‐PCB concentrations in breast milk with infant birthweights in Tokyo, Japan. Chemosphere, 61, 383–388. [DOI] [PubMed] [Google Scholar]
- Takeda T, Matsumoto Y, Koga T, Mutoh J, Nishimura Y, Shimazoe T, Ishii Y, Ishida T and Yamada H, 2009. Maternal exposure to dioxin disrupts gonadotropin production in fetal rats and imprints defects in sexual behavior. Journal of Pharmacology and Experimental Therapeutics, 329, 1091–9. [DOI] [PubMed] [Google Scholar]
- Takeda T, Fujii M, Taura J, Ishii y and Yamada H, 2012. Dioxin silences gonadotropin expression in perinatal pups by inducing histone deacetylases: a new insight into the mechanism for the imprinting of sexual immaturity by dioxin. Journal of Biological Chemistry, 287, 18440–18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda T, Fujii M, Hattori Y, Yamamoto M, Shimazoe T, Ishii Y, Himeno M and Yamada H, 2014a. Maternal exposure to dioxin imprints sexual immaturity of the pups through fixing the status of the reduced expression of hypothalamic gonadotropin‐releasing hormone. Molecular Pharmacology, 85, 74–82. [DOI] [PubMed] [Google Scholar]
- Takeda T, Taura J, Hattori Y, Ishii Y and Yamada H, 2014b. Dioxin‐induced retardation of development through a reduction in the expression of pituitary hormones and possible involvement of an aryl hydrocarbon receptor in this defect: a comparative study using two strains of mice with different sensitivities to dioxin. Toxicology and Applied Pharmacology, 278, 220–229. [DOI] [PubMed] [Google Scholar]
- Taylor JS, 1979. Environmental chloracne: update and overview. Annals New York Academy of Sciences, 320, 295–307. [PubMed] [Google Scholar]
- ten Tusscher GW, Leijs MM, de Boer LCC, Legler J, Olie K, Spekreijse H, van Dijk BW, Vulsma T, Briet J, Ilsen A and Koppe JG, 2014. Neurodevelopmental retardation, as assessed clinically and with magnetoencephalography and electroencephalography, associated with perinatal dioxin exposure. Science of the Total Environment, 491, 235–239. [DOI] [PubMed] [Google Scholar]
- Theelen RMC, 1991. Modelling of human exposure to TCDD and I‐TEQ in the Netherlands: background and occupational In: Biological basis for risk assessment of dioxins and related compounds. Banbury report 35. Gallo MA, Scheuplein RJ, van Heijden K. (eds). Pp 277–290. New York: Cold Spring Harbour Laboratory. [Google Scholar]
- Thoma H, Mücke W and Kauert G, 1990. Comparison of the polychlorinated dibenzo‐p‐dioxin and dibenzofuran in human tissue and human liver. Chemosphere, 20, 433–442. [Google Scholar]
- Thornton I and Abrahams P, 1983. Soil ingestion – a major pathway of heavy metals into livestock grazing contaminated land. The Science of the Total Environment, 28, 287–294. [DOI] [PubMed] [Google Scholar]
- Thornton AS, Oda Y, Stuart GR, Glickman BW and de Boer JG, 2001. Mutagenicity of TCDD in Big Blue transgenic rats. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 478, 45–50. [DOI] [PubMed] [Google Scholar]
- Thorpe S, Kelly M, Startin J, Harrison N and Rose M, 2001. Concentration changes for 5 PCDD/F congeners after administration in beef cattle. Chemosphere, 43, 869–879. [DOI] [PubMed] [Google Scholar]
- Tian Y, Ke S, Thomas T, Meeker RJ and Gallo MA, 1998a. Regulation of estrogen receptor mRNA by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin as measured by competitive RT‐PCR. Journal of Biochemical and Molecular Toxicology, 12, 71–77. [DOI] [PubMed] [Google Scholar]
- Tian Y, Ke S, Thomas T, Meeker RJ and Gallo MA, 1998b. Transcriptional suppression of estrogen receptor gene expression by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD). The Journal of Steroid Biochemistry and Molecular Biology, 67, 17–24. [DOI] [PubMed] [Google Scholar]
- Tietge JE, Johnson RD, Jensen KM, Cook PM, Elonen GE, Fernandez JD, Holcombe GW, Lothenbach DB and Nichols JW, 1998. Reproductive toxicity and disposition of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in adult brook trout (Salvelinus fontinalis) following a dietary exposure. Environmental Toxicology and Chemistry, 17, 2395–2407. [Google Scholar]
- Tijet N, Boutros PC, Moffat ID, Okey AB, Tuomisto J and Pohjanvirta R, 2006. Aryl hydrocarbon receptor regulates distinct dioxin‐dependent and dioxin‐independent gene batteries. Molecular Pharmacology, 69, 140–153. [DOI] [PubMed] [Google Scholar]
- Tillitt DE, Buckler JA, Nicks DK, Candrl JS, Claunch RA, Gale RW, Puglis HJ, Little EE, Linbo TL and Baker M, 2017. Sensitivity of lake sturgeon (Acipenser fulvescens) early life stages to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin and 3,3’,4,4’,5‐pentachlorobiphenyl. Environmental Toxicology and Chemistry, 36, 988–998. [DOI] [PubMed] [Google Scholar]
- Tischkau SA, 2012. AHR and the circadian clock In: Pohjanvirta R. (ed.). The AH receptor in biology and toxicology. John Wiley & Sons Inc, Hoboken, NJ, USA: pp. 511–522. [Google Scholar]
- Tlustos C, Sheridan M, O'Sullivan D, Anderson W and Flynn A, 2012. The dioxin contamination incident in Ireland, 2008: analytical results and congener patterns. Food Additives and Contaminants, Part A, 29, 128–138. [DOI] [PubMed] [Google Scholar]
- Todaka T, Hirakawa H, Hori T, Tobiishi K, Iida T and Furue M, 2007. Concentrations of polychlorinated dibenzo‐p‐dioxins, polychlorinated dibenzofurans, and non‐ortho and mono‐ortho polychlorinated biphenyls in blood of Yusho patients. Chemosphere, 66, 1983–1989. [DOI] [PubMed] [Google Scholar]
- Todaka T, Uchi H, Hirakawa H, Takao Y, Kajiwara J and Furue M, 2013. The changes in dioxin concentrations in the blood of yusho patients from 2004 to 2010. Fukuoka Acta Medica, 104, 118–127. [PubMed] [Google Scholar]
- Toft G, Long M, Krüger T, Hjelmborg PS, Bonde JP, Rignell‐Hydbom A, Tyrkiel E, Hagmar L, Giwercman A, Spanó M, Bizzaro D, Pedersen HS, Lesovoy V, Ludwicki JK and Bonefeld‐Jørgensen EC, 2007. Semen quality in relation to xenohormone and dioxin‐like serum activity among Inuits and three European populations. Environmental Health Perspectives, 115(suppl 1), 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torstensen BE, Espe M, Sanden M, Stubhaug I, Waagbo R, Hemre GI, Fontanillas R, Nordgarden U, Hevroy EM, Olsvik P and Berntssen MHG, 2008. Novel production of Atlantic salmon (Salmo salar) protein based on combined replacement of fish meal and fish oil with plant meal and vegetable oil blends. Aquaculture, 285, 193–200. [Google Scholar]
- Traag WA, Kan CA, van der Weg G, Onstenk C and Hoogenboom LAP, 2006. Residues of dioxins (PCDD/Fs) and PCBs in eggs, fat and livers of laying hens following consumption of contaminated feed. Chemosphere, 65, 1518–1525. [DOI] [PubMed] [Google Scholar]
- Tsukimori K, Tokunaga S, Shibata S, Uchi H, Nakayama D, Lshimam T, Nakano H, Wake N, Yoshimura T and Furue M, 2008. Long‐term effects of polychlorinated biphenyls and dioxins on pregnancy outcomes in women affected by the Yusho incident. Environmental Health Perspectives, 116, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukimori K, Uchi H, Mitoma C, Yasukawa F, Chiba T, Todaka T, Kajiwara J, Yoshimura T, Hirata T, Fukushima K, Wake N and Furue M, 2012. Maternal exposure to high levels of dioxins in relation to birth weight in women affected by Yusho disease. Environmental International, 38, 79–86. [DOI] [PubMed] [Google Scholar]
- Tsukimori K, Uchi H, Tokunaga S, Yasukawa F, Chiba T, Kajiwara J, Hirata T and Furue M, 2013. Blood levels of PCDDs, PCDFs, and coplanar PCBs in Yusho mothers and their descendants: association with fetal Yusho disease. Chemosphere, 90, 1581–1588. [DOI] [PubMed] [Google Scholar]
- Tsukino H, Hanaoka T, Sasaki H, Motoyama H, Hiroshima M, Tanaka T, Kabuto M, Niskar AS, Rubin C, Patterson DG, Turner W, Needham L and Tsugane S, 2005. Associations between serum levels of selected organochlorine compounds and endometriosis in infertile Japanese women. Environmental Research, 99, 118–125. [DOI] [PubMed] [Google Scholar]
- Tsutsumi T, Iida T, Hori T, Nakagawa P, Tobiishi K, Yanagi T, Kono Y, Uchibe H, Matsuda R, Sasaki K and Toyoda M, 2002. Recent survey and effects of cooking processes on levels of PCDDs, PCDFs and co‐PCBs in leafy vegetables in Japan. Chemosphere, 46, 1443–1449. [DOI] [PubMed] [Google Scholar]
- Tuey DB and Matthews HB, 1980. Use of a physiologically compartmental model for the rat to describe the pharmacokinetics of several chlorinated biphenyls in the mouse. Drug Metabolism and Disposition, 8, 397–403. [PubMed] [Google Scholar]
- Tuinstra LGMTh, Roos AH, Berende PLM, van Rhijn JA, Traag WA and Mengelers MJB, 1992. Excretion of polychlorinated dibenzo‐p‐dioxins and ‐furans in milk of cows fed on dioxins in the dry period. Journal of Agricultural and Food Chemistry, 40, 1772–1776. [Google Scholar]
- Tuomisto JT, Viluksela M, Pohjanvirta R and Tuomisto J, 1999. The AH receptor and a novel gene determine acute toxic responses to TCDD: segregation of the resistant alleles to different rat lines. Toxicology and Applied Pharmacology, 155, 71–81. [DOI] [PubMed] [Google Scholar]
- Tuomisto JT, Viluksela M, Pohjanvirta R and Tuomisto J, 2000. Changes in food intake and food selection in rats after 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) exposure. Pharmacology Biochemistry and Behavior, 65, 381–387. [DOI] [PubMed] [Google Scholar]
- Turrio‐Baldassarri L, Alivernini S, Carasi S, Casella M, Fuselli S, Iacovella N, Iamiceli L, La Rocca C, Scarcella C and Battistelli CL, 2009. PCB, PCDD and PCDF contamination of food of animal origin as the effect of soil pollution and the cause of human exposure in Brescia. Chemosphere, 76, 278–285. [DOI] [PubMed] [Google Scholar]
- Turyk ME, Anderson HA, Freels S, Chatterton R Jr, Needham LL, Patterson DG Jr, Steenport DN, Knobeloch L, Imm P, Persky VW and Great Lakes C, 2006. Associations of organochlorines with endogenous hormones in male Great Lakes fish consumers and nonconsumers. Environmental Research, 102, 299–307. [DOI] [PubMed] [Google Scholar]
- Uechara R, Peng G, Nakamura Y, Matsuura N, Kondo N and Tada H, 2006. Human milk survey for dioxins in the general population in Japan. Chemosphere, 62, 1135–1141. [DOI] [PubMed] [Google Scholar]
- Uemura H, Arisawa K, Hiyoshi M, Satoh H, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M and Suzuki T, 2008. Associations of environmental exposure to dioxins with prevalent diabetes among general inhabitants in Japan. Environmental Research, 108, 63–68. [DOI] [PubMed] [Google Scholar]
- Uemura H, Arisawa K, Hiyoshi M, Kitayama A, Takami H, Sawachika F, Dakeshita S, Nii K, Satoh H, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M and Suzuki T, 2009. Prevalence of metabolic syndrome associated with body burden levels of dioxin and related compounds among Japan's general population. Environmental Health Perspectives, 117, 568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulaszewska MM, Zuccato E and Davoli E, 2011. PCDD/Fs and dioxin‐like PCBs in human milk and estimation of infants’ daily intake: a review. Chemosphere, 83, 774–782. [DOI] [PubMed] [Google Scholar]
- UNEP (United Nations Environmental Programme), 2007. Guidelines on best available techniques and provisional guidance on best environmental practices relevant to Article 5 and Annex C of the Stockholm Convention on Persistent Organic Pollutants. http://chm.pops.int/Implementation/BATandBEP/BATBEPGuidelinesArticle5/tabid/187/Default.aspx
- UNEP (United Nations Environmental Programme), 2013. Results of the global survey on concentrations in human milk of persistent organic pollutants by the United Nations Environment Programme and the World Health Organization. http://www.unep.org/chemicalsandwaste/portals/9/POPs/docs/UNEP-POPS-COP.6-INF-33.English.pdf
- UNEP (United Nations Environmental Programme), 2015. Preliminary assessment of efforts made toward the elimination of polychlorinated biphenyls. UNEP/DTIE Chemicals Branch, January 2015. http://www.unep.org/chemicalsandwaste/Portals/9/POPs/PCB/Preliminary%20Assessment%20of%20Efforts%20Made%20Toward%20the%20Elimination%20of%20PCB_UNEP%20Chemicals%20Branch_2015_Final.pdf
- Unkila M, Pohjanvirta R, MacDonald E, Tuomisto JT and Tuomisto J, 1994. Dose‐response and time course of alterations in tryptophan metabolism by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the most TCDD‐susceptible and the most TCDD‐resistant rat strain: relationship with TCDD lethality. Toxicology and Applied Pharmacology, 128, 280–292. [DOI] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Derkenne S, Curran CP, Miller ML, Shertzer HG and Nebert DW, 2004. Oral exposure to benzo[a]pyrene in the mouse: detoxication by inducible cytochrome P450 is more important than metabolic activation. Molecular Pharmacology, 65, 1225–1237. [DOI] [PubMed] [Google Scholar]
- Urban P, Pelclova D, Lukas E, Kupka K, Preiss J, Fenclova Z and Smerhovsky Z, 2007. Neurological and neurophysiological examinations on workers with chronic poisoning by 2,3,7,8‐TCDD: follow‐up 35 years after exposure. European Journal of Neurology, 14, 213–218. [DOI] [PubMed] [Google Scholar]
- Urban JD, Budinsky RA and Rowlands JC, 2011. Single nucleotide polymorphisms in the human aryl hydrocarbon receptor nuclear translocator (ARNT) gene. Drug Metabolism and Pharmacokinetics, 26, 637–645. [DOI] [PubMed] [Google Scholar]
- Urban JD, Budinsky RA and Rowlands JC, 2012. An evaluation of single nucleotide polymorphisms in the human heat shock protein 90 kDa alpha and beta isoforms. Drug Metabolism and Pharmacokinetics, 27, 268–278. [DOI] [PubMed] [Google Scholar]
- US‐EPA (United States ‐ Environmental Protection Agency), 2012. EPA's Reanalysis of Key Issues Related to Dioxin Toxicity and Response to NAS Comments, Volume 1. Washington, DC: US Environmental Protection Agency. EPA/600/R‐10/038F.
- Vafeiadi M, Agramunt S, Papadopoulou E, Besselink H, Mathianaki K, Karakosta P, Spanaki A, Koutis A, Chatzi L, Vrijheid M and Kogevinas M, 2013. In utero exposure to dioxins and dioxin‐like compounds and anogenital distance in newborns and infants. Environmental Health Perspectives, 121, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafeiadi M, Agramunt S, Pedersen M, Besselink H, Chatzi L, Fthenou E, Fleming S, Hardie LJ, Wright J, Knudsen LE, Nielsen JKS, Sunyer J, Carreras R, Brunborg G, Gutzkow KB, Nygaard UC, Lovik M, Kyrtopoulos SA, Segerback D, Merlo DF, Kleinjans JC, Vrijheid M, Kogevinas M and NewGeneris C, 2014. In utero exposure to compounds with dioxin‐like activity and birth outcomes. Epidemiology, 25, 215–224. [DOI] [PubMed] [Google Scholar]
- Valdez K, Shi Z, Ting A and Petroff B, 2007. Effect of chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8‐tetirachlorodibenzo‐p‐dioxin in female rats on ovarian gene expression. Biology of Reproduction, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez KE, Shi Z, Ting AY and Petroff BK, 2009. Effect of chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in female rats on ovarian gene expression. Reproductive Toxicology, 28, 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg M, van der Wielen FWM, Olie K and van Boxtel CJ, 1986. The presence of PCDDs and PCDFs in human breast milk from the Netherlands. Chemosphere, 15, 693–706. [Google Scholar]
- Van den Berg M, De Jongh J, Poiger H and Olson JR, 1994. The toxicokinetics and metabolism of polychlorinated dibenzo‐p‐dioxins (PCDDs) and dibenzofurans (PCDFs) and their relevance for toxicity. Critical Reviews in Toxicology, 24, 1–74. [DOI] [PubMed] [Google Scholar]
- Van den Berg M, Birnbaum L, Bosveld ATC, Brunström B, Cook P, Feeley M, Giesy J, Hanberg A, Hasegawa R, Kennedy SW, Kubiak T, Larsen JC, van Leeuwen FXR, Liem AKD, Nolt C, Peterson RE, Poellinger L, Safe S, Schrenk D, Tillitt D, Tysklind M, Younes M, Wærn F and Zacharewski T, 1998. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environmental Health Perspectives, 106, 775–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg M, Birnbaum LS, Denison M, de Vito M, Farland W, Feeley M, Fiedler H, Hakansson H, Hanberg A, Haws L, Rose M, Safe S, Schrenk D, Tohyama C, Tritscher A, Tuomisto J, Tysklind Walker N and Peterson RE, 2006. The 2005 World Health Organization re‐evaluation of human and mammalian toxic equivalency factors for dioxins and dioxin‐like compounds. Toxicological Sciences, 93, 223–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Heuvell RL, Koppen G, Staessen JA, Den Hond E, Verheyen G, Nawrot TS, Roels HA, Vlietinck R and Schoeters GER, 2002. Immunologic biomarkers in relation to exposure markers of PCBs and dioxins in Flemish adolescents (Belgium). Environmental Health Perspectives, 110, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Molen GW, Kooijman SALM and Slob W, 1996. A generic toxicokinetic model for persistent lipophilic compounds in humans: an application to TCDD. Fundamental and Applied Toxicology, 31, 83–94. [DOI] [PubMed] [Google Scholar]
- Van der Weiden MEJ, Vanderkolk J, Bleumink R, Seinen W and Vandenberg M, 1992. Concurrence of P450‐1A1 induction and toxic effects after administration of a low‐dose of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the rainbow‐trout (Oncorhynchus‐mykiss). Aquatic Toxicology, 24, 123–142. [Google Scholar]
- Van der Weiden MEJ, Bleumink R, Seinen W and Vandenberg M, 1994. Concurrence of P450 1A induction and toxic effects in the mirror carp (Cyprinus carpio), after administration of a low dose of 2,3,7,8‐tetrachloro‐dibenzo‐p‐dioxin. Aquatic Toxicology, 29, 147–162. [Google Scholar]
- van Ede KI, Aylward L, Andersson P, van den Berg M and Duursen MB, 2013. Tissue distribution of dioxin‐like compounds: potential impacts on systemic relative potency estimates. Toxicology Letters, 220, 294–302. [DOI] [PubMed] [Google Scholar]
- van Ede KI, Gaisch KP, van den Berg M and van Duursen MB, 2014. Differential relative effect potencies of some dioxin‐like compounds in human peripheral blood lymphocytes and murine splenic cells. Toxicology Letters, 226, 43–52. [DOI] [PubMed] [Google Scholar]
- Van Eijkeren JCH, Zeilmaker MJ, Kan CA, Traag WA and Hoogenboom LAP, 2006. A PB‐PK based model for the carry‐over of dioxins and PCBs from feed and soil to eggs. Food Additives and Contaminants, 23, 518–527. [DOI] [PubMed] [Google Scholar]
- Van Esterik JCJ, Verharen HW, Hodemaekers HM, Gremmer ER, Nagarajah B, Kamstra JH, Dolle MET, Legler J and van der Ven LTM, 2015. Compound‐ and sex‐specific effects on programming of energy and immune homeostasis in adult C57BL/6JxWB mice after perinatal TCDD and PCB 153. Toxicology and Applied Pharmacology, 289, 262–275. [DOI] [PubMed] [Google Scholar]
- Van Voorhis M, Fechner JH, Zhang X and Mezrich JD, 2013. The aryl hydrocarbon receptor: a novel target for immunomodulation in organ transplantation. Transplantation, 95, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen T, Saarikoski S, Jaakkola JJ and Tuomisto J, 1997. PCDD, PCDF, and PCB concentrations in human milk from two areas in Finland. Chemosphere, 34, 2571–2583. [DOI] [PubMed] [Google Scholar]
- Vartiainen T, Jaakkola JJ, Saarikoski S and Tuomisto J, 1998. Birth weight and sex of children and the correlation to the body burden of PCDDs/PCDFs and PCBs of the mother. Environmental Health Perspectives, 106, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerkamp W, Wever J and Hutzinger O, 1981. The metabolism of chlorinated dibenzofurans by rats. Chemosphere, 10, 397–403. [Google Scholar]
- Vehlow J, Bergfeldt B and Hunsinger H, 2006. PCDD/F and related compounds in solid residues from municipal solid waste incineration – a literature review. Waste Management and Research, 24, 404–420. [DOI] [PubMed] [Google Scholar]
- Vigh E, Colombo A, Benfenati E, Håkansso H, Berglund M, József Bódis and János Garai, 2013. Individual breast milk consumption and exposure to PCBs and PCDD/Fs in Hungarian infants: a time‐course analysis of the first three months of lactation. Science of the Total Environment, 449, 336–344. [DOI] [PubMed] [Google Scholar]
- Viluksela M, Stahl BU, Birnbaum LS and Rozman KK, 1997. Subchronic/chronic toxicity of 1,2,3,4,6,7,8‐heptachlorodibenzo‐p‐dioxin (HpCDD) in rats.1. Design, general observations, hematology, and liver concentrations. Toxicology and Applied Pharmacology, 146, 207–216. [DOI] [PubMed] [Google Scholar]
- Viluksela M, Unkila M, Pohjanvirta R, Tuomisto JT, Stahl BU, Rozman KK and Tuomisto J, 1999. Effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) on liver phosphoenolpyruvate carboxykinase (PEPCK) activity, glucose homeostasis and plasma amino acid concentrations in the most TCDD‐susceptible and the most TCDD‐resistant rat strains. Archives of Toxicology, 73, 323–36. [DOI] [PubMed] [Google Scholar]
- Viluksela M, Bager Y, Tuomisto JT, Scheu G, Unkila M, Pohjanvirta R, Flodstrom S, Kosma VM, Maki‐Paakkanen J, Vartiainen T, Klimm C, Schramm KW, Warngard L and Tuomisto J, 2000. Liver tumor‐promoting activity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in TCDD‐sensitive and TCDD‐resistant rat strains. Cancer Research, 60, 6911–6920. [PubMed] [Google Scholar]
- Viluksela M, Raasmaja A, Lebofsky M, Stahl BU and Rozman KK, 2004. Tissue‐specific effects of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) on the activity of 5’‐deiodinases I and II in rats. Toxicology Letters, 147, 133–42. [DOI] [PubMed] [Google Scholar]
- Vinopal JH and Casida JE, 1973. Metabolic stability of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in mammalian liver microsomal systems and in living mice. Archives of Environmental Contamination and Toxicology, 1, 122–132. [DOI] [PubMed] [Google Scholar]
- Virtanen HE, Koskenniemi JJ, Sundqvist E, Main KM, Kiviranta H, Tuomisto JT, Tuomisto J, Viluksela M, Vartiainen T, Skakkebaek NE and Toppari J, 2012. Associations between congenital cryptorchidism in newborn boys and levels of dioxins and PCBs in placenta. International Journal of Andrology, 35, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VKM , 2014. Benefit‐risk assessment of fish and fish products in the Norwegian diet – an update. Scientific Opinion of the Scientific Steering Committee. VKM Report 15 [293 pp], ISBN: 978‐82‐8259‐159‐1, Oslo, Norway. Available online: http://www.vkm.no
- Vondráček J and Machala M, 2016. Environmental ligands of the aryl hydrocarbon receptor and their effects in models of adult liver progenitor cells. Stem Cells International, Article ID 4326194, 14 pages 10.1155/2016/4326194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorderstrasse BA, Steppan LB, Silverstone AE and Kerkvliet NI, 2001. Aryl hydrocarbon receptor‐deficient mice generate normal immune responses to model antigens and are resistant to TCDD‐induced immune suppression. Toxicology and Applied Pharmacology, 171, 157–164. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil HJI, Slijper FME, Mulder PGH and Weisglas‐Kuperus N, 2002. Effects of perinatal exposure to PCBs and dioxins on play behavior in Dutch children at school age. Environmental Health Perspectives, 110, A593–A598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker R, Poiger H and Schlatter C, 1986. Pharmacokinetics and metabolism of 1,2,3,7,8‐pentachlorodibenzo‐p‐dioxin in the rat. Chemosphere, 15, 1473–1476. [Google Scholar]
- Wahl K, Kotz A, Hädrich J, , Malisch R, Anastassiades M and Sigalova I, 2008. The guar gum case: contamination with PCP and dioxins and analytical problems. Organohalogen Compounds, 70, Available at: http://wwwdioxin20xxorg/pdfs/2008/08-301pdf [Google Scholar]
- Wakui S, Muto T, Motohashi M, Kobayashi Y, Suzuki Y, Takahashi H and Hano H, 2010. Testicular spermiation failure in rats exposed prenatally to 3,3’,4,4’,5‐pentachlorobiphenyl. Journal of Toxicological Sciences, 35, 757–765. [DOI] [PubMed] [Google Scholar]
- Walisser JA, Bunger MK, Glover E, Harstad EB and Bradfield CA, 2004. Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. The Journal of Biological Chemistry, 279, 16326–16331. [DOI] [PubMed] [Google Scholar]
- Walker MK and Peterson RE, 1991. Potencies of polychlorinated dibenzo‐para‐dioxin, dibenzofuran, and biphenyl congeners, relative to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin, for producing early life stage mortality in rainbow trout (Oncorhynchus mykiss). Aquatic Toxicology, 21, 219–238. [Google Scholar]
- Walker MK and Catron TF, 2000. Characterization of cardiotoxicity induced by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin and related chemicals during early chick embryo development. Toxicology and Applied Pharmacology, 167, 210–221. [DOI] [PubMed] [Google Scholar]
- Walker NJ, Wyde ME, Fischer LJ, Nyska A and Bucher JR, 2006. Comparison of chronic toxicity and carcinogenicity of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in 2‐year bioassays in female Sprague‐Dawley rats. Molecular Nutrition and Food Research, 50, 934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Santastefano MJ, Evans MV, Richardson VM, Diliberto JJ and Birnbaum LS, 1997. Determination of parameters responsible for pharmacokinetic behavior of TCDD in female Sprague‐Dawley rats. Toxicology and Applied Pharmacology, 147, 151–168. [DOI] [PubMed] [Google Scholar]
- Wang X, Santostefano MJ, DeVito MJ and Birnbaum LS, 2000. Extrapolation of a PBPK model for dioxins across dosage regimen, gender, strain and species. Toxicological Sciences, 56, 49–60. [DOI] [PubMed] [Google Scholar]
- Wang SL, Chen TT, Hsu JF, Hsu CC, Chang LW, Ryan JJ, Guo YLL and Lambert GH, 2003. Neonatal and childhood teeth in relation to perinatal exposure to polychlorinated biphenyls and dibenzofurans: observations of the Yucheng children in Taiwan. Environmental Research, 93, 131–137. [DOI] [PubMed] [Google Scholar]
- Wang SL, Su PH, Jong SB, Guo YL, Chou WL and Papke O, 2005. In utero exposure to dioxins and polychlorinated biphenyls and its relations to thyroid function and growth hormone in newborns. Environmental Health Perspectives, 113, 1645–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SL, Chang YC, Chao HR, Li CM, Li LA, Lin LY and Papke O, 2006. Body burdens of polychlorinated dibenzo‐p‐dioxins, dibenzofurans, and biphenyls and their relations to estrogen metabolism in pregnant women. Environmental Health Perspectives, 114, 740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IC and Lee W‐J, 2010. Polychlorinated dibenzo‐p‐dioxin, polychlorinated dibenzofurans and polychlorinated biphenyls in farmed fish, water, sediment, and feed. Journal of Environmental Science and Health Part a‐Toxic/Hazardous Substances and Environmental Engineering, 45, 201–210. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang Q, Wu B, Li Y and Lu G, 2013. Correlation between TCDD acute toxicity and aryl hydrocarbon receptor structure for different mammals. Ecotoxicology and Environmental Safety, 89, 84–88. [DOI] [PubMed] [Google Scholar]
- Warner M, Eskenazi B, Mocarelli P, Gerthoux PM, Samuels S, Needham L, Patterson D and Brambilla P, 2002. Serum dioxin concentrations and breast cancer risk in the Seveso Women's Health Study. Environmental Health Perspectives, 110, 625–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M, Samuels S, Mocarelli P, Gerthoux PM, Needham L, Patterson DG and Eskenazi B, 2004. Serum dioxin concentrations and age at menarche. Environmental Health Perspectives, 112, 1289–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M, Eskenazi B, Olive DL, Samuels S, Quick‐Miles S, Vercellini P, Gerthoux PM, Needham L, Patterson DG and Mocarelli P, 2007. Serum dioxin concentrations and quality of ovarian function in women of Seveso. Environmental Health Perspectives, 115, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M, Mocarelli P, Samuels S, Needham L, Brambilla P and Eskenazi B, 2011. Dioxin exposure and cancer risk in the seveso women's health study. Environmental Health Perspectives, 119, 1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M, Mocarelli P, Brambilla P, Wesselink A, Samuels S, Signorini S and Eskenazi B, 2013. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: the Seveso Women's Health Study. Environmental Health Perspectives, 121, 906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M, Mocarelli P, Brambilla P, Wesselink A, Patterson DG Jr, Turner WE and Eskenazi B, 2014. Serum TCDD and TEQ concentrations among Seveso women, 20 years after the explosion. Journal of Exposure Science and Environmental Epidemiology, 24, 588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe MX, Kunisue T, Tao L, Kannan K, Subramanian A, Tanabe S and Iwata H, 2010. Dioxin‐like and perfluorinated compounds in pigs in an Indian open waste dumping site: toxicokinetics and effects on hepatic cytochrome P450 and blood plasma hormones. Environmental Toxicology and Chemistry, 29, 1551–1560. [DOI] [PubMed] [Google Scholar]
- WCA Environment Ltd , 2018. Extensive literature search, selection for relevance and data extraction of studies related to the toxicity of PCDD/Fs and DL‐PCBs in experimental animals. EFSA supporting publication 2018: EN‐1137, 68 pp. 10.2903/sp.efsa.2018.en-1137 [DOI] [Google Scholar]
- Weber H, Poiger H and Schlatter C, 1982. Acute oral toxicity of TCDD‐metabolites in male guinea pigs. Toxicology Letters, 14, 117–22. [DOI] [PubMed] [Google Scholar]
- Weber LW, Ernst SW, Stahl BU and Rozman K, 1993. Tissue distribution and toxicokinetics of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in rats after intravenous injection. Fundamental and Applied Toxicology, 21, 523–34. [DOI] [PubMed] [Google Scholar]
- Wei YD, Rannug U and Rannug A, 1999. UV‐induced CYP1A1 gene expression in human cells is mediated by tryptophan. Chemico‐Biological Interactions, 118, 127–140. [DOI] [PubMed] [Google Scholar]
- Weisglas‐Kuperus N, Patandin S, Berbers GAM, Sas TCJ, Mulder PGH, Sauer PJJ and Hooijkaas H, 2000. Immunologic effects of background exposure to polychlorinated biphenyls and dioxins in Dutch preschool children. Environmental Health Perspectives, 108, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wejheden C, Brunnberg S, Larsson S, Lind PM, Andersson G and Hanberg A, 2010. Transgenic mice with a constitutively active aryl hydrocarbon receptor display a gender‐specific bone phenotype. Toxicological Sciences: An Official Journal of the Society of Toxicology, 114, 48–58. [DOI] [PubMed] [Google Scholar]
- Wesselink A, Warner M, Samuels S, Parigi A, Brambilla P, Mocarelli P and Eskenazi B, 2014. Maternal dioxin exposure and pregnancy outcomes over 30 years of follow‐up in Seveso. Environment International, 63, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MW and Bailer AJ, 2008. Model averaging software for dichotomous dose response risk estimation. Journal of Statistical Software, 26, 1–15.19777145 [Google Scholar]
- White KL Jr, Lysy HH, McCay JA, Anderson AC, 1986. Modulation of serum complement levels following exposure to polychlorinated dibenzo‐p‐dioxins. Toxicology and Applied Pharmacology, 84, 209–219. [DOI] [PubMed] [Google Scholar]
- Whitelaw ML, Gottlicher M, Gustafsson JA and Poellinger L, 1993. Definition of a novel ligand binding domain of a nuclear bHLH receptor: co‐localization of ligand and hsp90 binding activities within the regulable inactivation domain of the dioxin receptor. The EMBO Journal, 12, 4169–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (World Health Organisation), 1998. Consultation on assessment of the health risk of dioxins; re‐evaluation of the tolerable daily intake (TDI): executive summary. Food Additives and Contaminants, 17, 223–240. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organisation), 2000. Assessment of the health risk of dioxins: re‐evaluation of the Tolerable Daily Intake (TDI) In: van Leeuwen FXR, Younes MM. (eds.). Food Additives and Contaminants, Vol. 17, No. 4. Taylor and Francis, London, UK. [Google Scholar]
- WHO (World Health Organization), 2003. Polychlorinated biphenyls: Human health aspects. Concise International Chemical Assessment Document 55. World Health Organization (Geneva), 64 pp. Available from http://www.who.int/ipcs/publications/cicad/en/cicad55.pdf
- WHO (World Health Organization), 2007. Fourth WHO‐Coordinated Survey of Human Milk for Persistent Organic Pollutants in Cooperation with UNEP ‐ Guidelines for Developing a National Protocol. http://www.who.int/foodsafety/chem/POPprotocol.pdf
- WHO (World Health Organisation), 2016. Safety evaluation of certain food additives and contaminants Supplement 1: Non‐dioxin‐like polychlorinated biphenyls. WHO Food Additives Series: 71‐S1. Prepared by the eightieth meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). Available at: http://apps.who.int/iris/bitstream/10665/246225/1/9789241661713-eng.pdf
- WHO/IPCS (World Health Organization/International Programme on Chemical Safety), 2008. Uncertainty and data quality in exposure assessment. Harmonisation project document No. 6. ISBN 978 92 4 156376 5.U.
- WHO/IPCS (World Health Organization/International Programme on Chemical Safety), 2009. Principles and Methods for the Risk Assessment of Chemicals in Food, International Programme on Chemical Safety, Environmental Health Criteria 240. Chapter 6: Dietary Exposure Assessment of Chemicals in Food. Available online: http://www.who.int/ipcs/food/principles/en/index1.html
- Widerak M, Ghoneim C, Dumontier MF, Quesne M, Corvol MT and Savouret JF, 2006. The aryl hydrocarbon receptor activates the retinoic acid receptor alpha through SMRT antagonism. Biochimie, 88, 387–397. [DOI] [PubMed] [Google Scholar]
- Wikenheiser J, Karunamuni G, Sloter E, Walker MK, Roy D, Wilson DL and Watanabe M, 2013. Altering HIF‐1 alpha through 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) exposure affects coronary vessel development. Cardiovascular Toxicology, 13, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Wittsiepe J, Lemm F, Ranft U, Kramer U, Furst P, Roseler SC, Greshake M, Imohl M, Eberwein G, Rauchfuss K, Kraft M and Winneke G, 2008. The Duisburg birth cohort study: influence of the prenatal exposure to PCDD/Fs and dioxin‐like PCBs on thyroid hormone status in newborns and neurodevelopment of infants until the age of 24 months. Mutation Research‐Reviews in Mutation Research, 659, 83–92. [DOI] [PubMed] [Google Scholar]
- Williams PL, Sergeyev O, Lee MM, Korrick SA, Burns JS, Humblet O, DelPrato J, Revich B and Hauser R, 2010. Blood lead levels and delayed onset of puberty in a longitudinal study of russian boys. Pediatrics, 125, E1088–E1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmerová S, van den Berg M, Chovancová J, Patayová H, Jusko TA, van Duursen MB, Palkovičová Murínová Ľ, Canton RF, van Ede KI and Trnovec T, 2016. Relative effect potency estimates of dioxin‐like activity for dioxins, furans, and dioxin‐like PCBs in adults based on cytochrome P450 1A1 and 1B1 gene expression in blood. Environment International, 96, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincent E, Bengtsson J, Mohammadi Bardbori A, Alsberg T, Luecke S, Rannug U and Rannug A, 2012. Inhibition of cytochrome P4501‐dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor. Proceedings of the National Academy of Sciences of the United States of America, 109, 4479–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winneke G, Ranft U, Wittsiepe J, Kasper‐Sonnenberg M, Furst P, Kramer U, Seitner G and Wilhelm M, 2014. Behavioral sexual dimorphism in school‐age children and early developmental exposure to dioxins and PCBs: a follow‐up study of the Duisburg Cohort. Environmental Health Perspectives, 122, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittsiepe J, Erlenkaemper B, Welge P, Hack A and Wilhelm M, 2007a. Bioavailability of PCDD/F from contaminated soil in young Goettingen minipigs. Chemosphere, 67, S355–S364. [DOI] [PubMed] [Google Scholar]
- Wittsiepe J, Fürst P, Schrey P, Lemm F, Kraft M, Eberwein G, Winneke G and Wilhelm M, 2007b. PCDD/F and dioxin‐like PCB in human blood and milk from German mothers. Chemosphere, 67, S286–S294. [DOI] [PubMed] [Google Scholar]
- Wittsiepe J, Schrey P, Lemm F, Eberwein G and Wilhelm M, 2008. Polychlorinated dibenzo‐p‐dioxins/polychlorinated dibenzofurans (PCDD/Fs), polychlorinated biphenyls (PCBs), and organochlorine pesticides in human blood of pregnant women from Germany. Journal of Toxicology and Environmental Health, Part A, 71, 703–709. [DOI] [PubMed] [Google Scholar]
- Wohlfahrt‐Veje C, Audouze K, Brunak S, Antignac JP, Bizec BI, Juul A, Skakkebaek NE and Main KM, 2014. Polychlorinated dibenzo‐p‐dioxins, furans, and biphenyls (PCDDs/PCDFs and PCBs) in breast milk and early childhood growth and IGF1. Reproduction, 147, 391–399. [DOI] [PubMed] [Google Scholar]
- Wolfe WH, Michalek JE, Miner JC, Pirkle JL, Caudill SP, Patterson DG and Needham LL, 1994. Determinants of TCDD half‐life in veterans of Operation Ranch Hand. Journal of Toxicology and Environmental Health, 41, 481–488. [DOI] [PubMed] [Google Scholar]
- Wong EW and Cheng CY, 2011. Impacts of environmental toxicants on male reproductive dysfunction. Trends in Pharmacological Sciences, 32, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PJ and Tillitt DE, 1999. Embryotoxicity of Great Lakes lake trout extracts to developing rainbow trout. Aquatic Toxicology, 47, 77–92. [Google Scholar]
- Wroblewski VJ and Olson JR, 1985. Hepatic metabolism of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) in the rat and guinea pig. Toxicology and Applied Pharmacology, 81, 231–40. [DOI] [PubMed] [Google Scholar]
- Wroblewski VJ and Olson JR, 1988. Effect of monooxygenase inducers and inhibitors on the hepatic metabolism of 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the rat and hamster. Drug Metabolism and Disposition, 16, 43–51. [PubMed] [Google Scholar]
- Wu WZ, Schramm KW, Xu Y and Kettrup A, 2001. Accumulation and partition of polychlorinated dibenzo‐p‐dioxins and dibenzofurans (PCDD/F) in the muscle and liver of fish. Chemosphere, 43, 633–641. [DOI] [PubMed] [Google Scholar]
- Wu J, Dong S, Liu G, Zhang B and Zheng M, 2011. Cooking process: a new source of unintentionally produced dioxins? Journal of Agricultural and Food Chemistry, 59, 5444–5449. [DOI] [PubMed] [Google Scholar]
- Wu CH, Guo CY, Yang JG, Tsai HD, Chang YJ, Tsai PC, Hsu CC and Kuo PL, 2012. Polymorphisms of dioxin receptor complex components and detoxification‐related genes jointly confer susceptibility to advanced‐stage endometriosis in the Taiwanese han population. American Journal of Reproductive Immunology (New York, NY: 1989), 67, 160–168. [DOI] [PubMed] [Google Scholar]
- Xu P, Lou X, Ding G, Shen H, Wu L, Chen Z, Han J, Han G and Wang X, 2014. Association of PCB, PBDE and PCDD/F body burdens with hormone levels for children in an e‐waste dismantling area of Zhejiang Province, China. Science of the Total Environment, 499, 55–61. [DOI] [PubMed] [Google Scholar]
- Xu B, Yang H, Sun M, Chen H, Jiang L, Zheng X, Ding G, Liu Y, Sheng Y, Cui D and Duan Y, 2016. 2,3’,4,4’,5‐Pentachlorobiphenyl induces inflammatory responses in the thyroid through JNK and aryl hydrocarbon receptor‐mediated pathway. Toxicological Sciences, 149, 300–11. [DOI] [PubMed] [Google Scholar]
- Yamada‐Okabe T, Aono T, Sakai H, Kashima Y and Yamada‐Okabe H, 2004. 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin augments the modulation of gene expression mediated by the thyroid hormone receptor. Toxicology and Applied Pharmacology, 194, 201–10. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Suzuki M, Itoh T, Yamamoto K, Kanemitsu M, Matsumura C, Nakano T, Sakaki T, Fukami Y, Imaishi H and Inui H, 2011. Structural basis of species differences between human and experimental animal CYP1A1s in metabolism of 3,3’,4,4’,5‐pentachlorobiphenyl. The Journal of Biochemistry, 149, 487–94. [DOI] [PubMed] [Google Scholar]
- Yang JZ, Agarwal SK and Foster WG, 2000. Subchronic exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin modulates the pathophysiology of endometriosis in the cynomolgus monkey. Toxicological Sciences, 56, 374–381. [DOI] [PubMed] [Google Scholar]
- Yang J, Shin D, Park S, Chang Y, Kom D and Ikonomou MG, 2002. PCDDs, PCDFs, and PCBs concentrations in breast milk from two areas in Korea: body burden of mothers and implications for feeding infants. Chemosphere, 46, 419–428. [DOI] [PubMed] [Google Scholar]
- Yang H, Chen H, Guo H, Li W, Tang J, Xu B, Sun M, Ding G, Jiang L, Cui D, Zheng X and Duan Y, 2015. Molecular mechanisms of 2, 3’, 4, 4’, 5‐pentachlorobiphenyl‐induced thyroid dysfunction in FRTL‐5 cells. PLoS One, 10, e0120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Takasuga T, Masunaga S and Nakanishi J, 2002. Detailed study on the levels of polychlorinated dibenzo‐p‐dioxins, polychlorinated dibenzofurans and polychlorinated biphenyls in Yusho rice oil. Chemosphere, 46, 1461–1469. [DOI] [PubMed] [Google Scholar]
- Yasuda I, Yasuda M, Sumida H, Tsusaki H, Arima A, Ihara T, Kubota S, Asaoka K, Tsuga K and Akagawa Y, 2005. In utero and lactational exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD) affects tooth development in rhesus monkeys. Reproductive Toxicology, 20, 21–30. [DOI] [PubMed] [Google Scholar]
- Yasui T, Kim EY, Iwata H, Franks DG, Karchner SI, Hahn ME and Tanabe S, 2007. Functional characterization and evolutionary history of two aryl hydrocarbon receptor isoforms (AhR1 and AhR2) from avian species. Toxicological Sciences, 99, 101–117. [DOI] [PubMed] [Google Scholar]
- Yeager RL, Reisman SA, Aleksunes LM and Klaassen CD, 2009. Introducing the “TCDD‐inducible AhR‐Nrf2 gene battery”. Toxicological Sciences, 111, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellon SM, Singh D, Garrett TM, Fagoaga OR and Nehlsen‐Cannarella SL, 2000. Reproductive, neuroendocrine, and immune consequences of acute exposure to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin in the Siberian hamster. Biology of Reproduction, 63, 538–54. [DOI] [PubMed] [Google Scholar]
- Yi SW, Ohrr H, Won JU, Song JS and Hong JS, 2013. Serum 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin levels and their association with age, body mass index, smoking, military record‐based variables, and estimated exposure to Agent Orange in Korean Vietnam veterans. Journal of Preventive Medicine and Public Health, 46, 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji T, Kashima S, Tokinobu A, Kato T and Tsuda T, 2013. Regional impact of exposure to a polychlorinated biphenyl and polychlorinated dibenzofuran mixture from contaminated rice oil on stillbirth rate and secondary sex ratio. Environment International, 59, 12–15. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Kamimura H, Oguri K, Honda Y and Nakano M, 1986. Stimulating effects of activated charcoal beads on fecal excretion of 2,3,4,7,8‐pentachlorodibenzofuran. Chemosphere, 15, 219–225. [Google Scholar]
- Yoshimura H, Yonemoto Y, Yamada H, Koga N, Oguri K and Saeki S, 1987. Metabolism in vivo of 3,4,3’,4’‐tetrachlorobiphenyl and toxicological assessment of the metabolites in rats. Xenobiotica, 17, 897–910. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kaneko S and Hayabuchi H, 2001. Sex ration in offspring of those affected by dioxin and dioxin‐like compounds: the Yusho, Seveso, and Yucheng incidents. Occupational Environmental Medicine, 58, 540–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka W, Kawaguchi T, Fujisawa N, Aida‐Yasuoka K, Shimizu T, Matsumura F and Tohyama C, 2014. Predominant role of cytosolic phospholipase A2alpha in dioxin‐induced neonatal hydronephrosis in mice. Scientific Reports, 4, 4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa K, Walker NJ, Nyska A, Kissling GE, Jokinen MP, Brix AE, Sells DM and Wyde ME, 2010. Thyroid follicular lesions induced by oral treatment for 2 years with 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin and dioxin‐like compounds in female Harlan Sprague‐Dawley rats. Toxicologic Pathology, 38, 1037–50. [DOI] [PubMed] [Google Scholar]
- Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D'Angelo C, Massi‐Benedetti C, Fallarino F, Carvalho A, Puccetti P and Romani L, 2013. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin‐22. Immunity, 39, 372–385. [DOI] [PubMed] [Google Scholar]
- Zhang S, Rowlands C and Safe S, 2008. Ligand‐dependent interactions of the Ah receptor with coactivators in a mammalian two‐hybrid assay. Toxicology and Applied Pharmacology, 227, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wang P, Li Y, Chen Z, Li W, Ssebugere P, Zhang Q and Jiang G, 2015. Bioconcentration and trophic transfer of polychlorinated biphenyls and polychlorinated dibenzo‐p‐dioxins and dibenzofurans in aquatic animals from an e‐waste dismantling area in East China. Environmental Science‐Processes and Impacts, 17, 693–699. [DOI] [PubMed] [Google Scholar]
- Zimmer KE, Gutleb AC, Lyche JL, Dahl E, Oskam IC, Krogenaes A, Skaare JU and Ropstad E, 2009. Altered stress‐induced cortisol levels in goats exposed to polychlorinated biphenyls (PCB 126 and PCB 153) during fetal and postnatal development. Journal of Toxicology and Environmental Health‐Part A‐Current Issues, 72, 164–172. [DOI] [PubMed] [Google Scholar]
- Zimmer KE, Kraugerud M, Aleksandersen M, Gutleb AC, Ostby GC, Dahl E, Berg V, Skaare JU, Olsaker I and Ropstad E, 2013. Fetal adrenal development: comparing effects of combined exposures to PCB 118 and PCB 153 in a Sheep Model. Environmental Toxicology, 28, 164–177. [DOI] [PubMed] [Google Scholar]
- Zober A and Päpke O, 1993. Concentrations of PCDDs and PCDFs in human tissue 36 years after accidental dioxin exposure. Chemosphere, 27, 413–418. [Google Scholar]
- Zordoky BNM and El‐Kadi AOS, 2009. Role of NF‐kappa B in the Regulation of Cytochrome P450 Enzymes. Current Drug Metabolism, 10, 164–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional information for the risk assessment for human and animal health related to the presence of dioxins and DL‐PCBs in food and feed
Occurrence data in food and feed submitted to EFSA and dietary exposure assessment for humans