

Brody disease is a rare myopathy characterized by exercise-induced muscle stiffness caused by mutations in the ATP2A1 gene. In the largest cohort of Brody patients to date, Molenaar et al. clarify the phenotype and diagnostic possibilities to help improve understanding and recognition of this distinct myopathy.
Keywords: Brody disease, ATP2A1, calcium, phenotype, genotype
Abstract
Brody disease is an autosomal recessive myopathy characterized by exercise-induced muscle stiffness due to mutations in the ATP2A1 gene. Almost 50 years after the initial case presentation, only 18 patients have been reported and many questions regarding the clinical phenotype and results of ancillary investigations remain unanswered, likely leading to incomplete recognition and consequently under-diagnosis. Additionally, little is known about the natural history of the disorder, genotype-phenotype correlations, and the effects of symptomatic treatment. We studied the largest cohort of Brody disease patients to date (n = 40), consisting of 22 new patients (19 novel mutations) and all 18 previously published patients. This observational study shows that the main feature of Brody disease is an exercise-induced muscle stiffness of the limbs, and often of the eyelids. Onset begins in childhood and there was no or only mild progression of symptoms over time. Four patients had episodes resembling malignant hyperthermia. The key finding at physical examination was delayed relaxation after repetitive contractions. Additionally, no atrophy was seen, muscle strength was generally preserved, and some patients had a remarkable athletic build. Symptomatic treatment was mostly ineffective or produced unacceptable side effects. EMG showed silent contractures in approximately half of the patients and no myotonia. Creatine kinase was normal or mildly elevated, and muscle biopsy showed mild myopathic changes with selective type II atrophy. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) activity was reduced and western blot analysis showed decreased or absent SERCA1 protein. Based on this cohort, we conclude that Brody disease should be considered in cases of exercise-induced muscle stiffness. When physical examination shows delayed relaxation, and there are no myotonic discharges at electromyography, we recommend direct sequencing of the ATP2A1 gene or next generation sequencing with a myopathy panel. Aside from clinical features, SERCA activity measurement and SERCA1 western blot can assist in proving the pathogenicity of novel ATP2A1 mutations. Finally, patients with Brody disease may be at risk for malignant hyperthermia-like episodes, and therefore appropriate perioperative measures are recommended. This study will help improve understanding and recognition of Brody disease as a distinct myopathy in the broader field of calcium-related myopathies.
Introduction
Brody disease is a rare, autosomal recessive myopathy with an estimated prevalence of 1 in 10 million (MacLennan, 2000; Voermans et al., 2012). It is caused by mutations in the ATP2A1 gene, encoding the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase type 1 (SERCA1) protein leading to exercise-induced muscle stiffness. Furthermore, myalgia and muscle cramps are described, which may worsen upon exposure to cold temperatures (Odermatt et al., 1996, 2000; Novelli et al., 2004; Guglielmi et al., 2013). EMG can reveal silent contractures defined as prolonged involuntary muscle contractions (following voluntary phasic contractions) without electrical activity detected on EMG (Odermatt et al., 2000; Novelli et al., 2004; Vattemi et al., 2010; Mussini et al., 2015). Various specific and non-specific findings have been described upon examination of muscle biopsies, but there is no general consensus on how often these features are present (Karpati et al., 1986; Benders et al., 1994; Odermatt et al., 2000; Novelli et al., 2004; Vattemi et al., 2010; Sambuughin et al., 2014; Mussini et al., 2015). The current phenotypic characterization of Brody disease is based on 10 case studies with ambiguous results. Moreover, while the focus on Ca2+ metabolism in muscle has grown, and an increasing number of myopathies caused by Ca2+ handling abnormalities are recognized, reports of Brody disease remain scarce (Treves et al., 2017).
Brody described the first case of pronounced exercise-induced muscle stiffness in the absence of myotonic discharges in 1969 (Brody, 1969). In 1986, Karpati discovered that this phenotype arises from SERCA1 protein deficiency in skeletal muscle (Karpati et al., 1986). Ten years later, the causative ATP2A1 gene was identified in two separate families (Odermatt et al., 1996). The disorder is considered to be extremely rare, which is reflected by the limited number of reported cases, with only 18 genetically confirmed patients described in the literature over the past 32 years (Supplementary Table 1).
The SERCA protein plays a key role in regulating Ca2+ homeostasis in skeletal muscle fibres. Following depolarization of the sarcolemma, the dihydropyridine receptor activates the ryanodine receptor type 1 (RYR1). Ca2+ is then released from the sarcoplasmic reticulum into the cytosol where it binds troponin C, effectively initiating the cascade leading to muscle contraction through myosin and actin interaction (cross-bridge cycling). Finally, SERCA lowers the cytoplasmic Ca2+ concentration by actively transporting it back into the sarcoplasmic reticulum causing muscle relaxation (for a review see Lee, 2010; Treves et al., 2017; Allard, 2018). SERCA1, the isoform affected in Brody disease, is uniquely expressed in type II (fast-twitch) skeletal muscle fibres (Rossi and Dirksen, 2006; Lee, 2010). Therefore, exercise-induced muscle stiffness in Brody disease is mainly induced by phasic exercise (rapid, intense contractions performed by fast-twitch fibres) and not by tonic activity (performed by slow-twitch fibres) as required to maintain posture (Karpati et al., 1986).
Unfortunately, almost 50 years after the initial report by Brody, many questions remain unanswered. The prevalence of specific symptoms, signs and findings on ancillary investigations are unknown. Additionally, little is known about the natural history of the disorder, the effects of symptomatic treatment, and genotype-phenotype correlations. This is likely to result in incomplete recognition and difficulties in counselling. Furthermore, with the use of diagnostic whole exome sequencing, mutations in ATP2A1 are likely to be encountered more frequently.
In this study we present a comprehensive review of the clinical features and natural course of the 22 new patients (19 novel ATP2A1 mutations), as well as all 18 previously described patients. We aim to improve the understanding and awareness of Brody disease and provide better means to recognize and diagnose this rare myopathy.
Materials and methods
Patient selection
Literature patients
We reviewed all English publications on Brody disease in PubMed to select the ‘literature cases’ (previously reported patients). We included all genetically confirmed patients reported after the first case report in 1969 until 2018, with either homozygous or compound heterozygous ATP2A1 mutations. We numbered them chronologically from L1 to L18.
Newly identified patients
We identified all patients that were clinically and genetically diagnosed with Brody disease by the Assistance Publique des Hôpitaux de Paris, U.F. de Cardiogénétique et Myogénétique in France (11 patients from nine families) and the genetics department of the Radboud University Medical Center in the Netherlands (three patients from two families). The patients that had not previously been described in literature were labelled as ‘new patients’. After contacting authors of previously published Brody disease literature we included two more new patients diagnosed by the Groupe de recherche interdisciplinaire sur les maladies neuromusculaires (GRIMN) in Quebec, Canada. Additionally, one patient was referred by the Queens Square Centre for Neuromuscular Diseases in London, UK; one patient was referred by the Salford Royal NHS Foundation Trust hospital in Manchester, UK; two patients from the Neuromuskuläres Zentrum in Frankfurt am Main, Germany; and finally, two more patients were referred from the Galdakao-Usansolo hospital and University Hospital Donostia, both in Spain. This resulted in 22 new Brody disease patients that we numbered N1 to N22.
Clinical features
A data sheet was sent to corresponding authors of the literature cases and to clinicians of the new patients, including history, physical examination, results of ancillary investigations, effects of medication and progression of symptoms (Supplementary Table 2).
Ancillary investigations
Results of ancillary investigations were ascertained from case reports in the literature, the completed data sheets, and from reviewing available medical files (i.e. laboratory testing, EMG, muscle biopsy, and genetic analysis). Stored muscle tissue from previously performed biopsies was used to perform additional immunohistochemical testing, SERCA activity measurements, and SERCA1 western blot analysis.
SERCA immunohistochemistry
Immunohistochemical analysis was performed on samples from three new patients. Fragments of biopsies were frozen in liquid nitrogen-precooled isopentane. Serial 7-μm thick cryosections were saturated with 3% bovine serum albumin in phosphate-buffered saline (PBS) for 1 h and stained with monoclonal antibodies against SERCA1 or SERCA2 (1/500; Affinity Bioreagents) for 1 h. After washing with PBS, cryosections were stained with goat anti-mouse 647 IgG antibody for 45 min (Thermo Fisher) and mounted in Mowiol® (Dabco).
SERCA activity measurement
SERCA activity was previously measured in muscle samples from four literature patients (Patients L1, L5, L6 and L13). Additionally, we measured compound SERCA activity in samples from three new patients (Patients N1, N10 and N12) and one literature patient (Patient L11). These measurements were performed by the Radboud Center for Mitochondrial Medicine on remaining muscle tissue from previously performed muscle biopsies. Because SERCA1 is the predominant isoform expressed in quadriceps and deltoid muscle, compound SERCA activity is a reliable estimate of SERCA1 activity (Yang and Yoo, 1986; Benders et al., 1994; Pauw-Gommans et al., 2006; Mandroukas et al., 2010). The muscle tissue was snap-frozen immediately after obtaining the sample and stored at −80°C. The compound SERCA activity was measured in muscle homogenates using procedures based on previously described methods (Benders et al., 1994; Vattemi et al., 2010).
Western blot of SERCA1 protein
Western blot analysis of SERCA1 protein was previously performed in muscle samples from five literature cases. Additionally, we assessed SERCA1 protein content in muscle homogenates from four new patients and one literature patient. Muscle homogenates (2% w/v) were prepared by homogenization using a Potter/Elvehjem tissue homogenizer in 250 mM sucrose, 2 mM EDTA, and 10 mM Tris-HCl (pH 7.6). Protein concentration was determined using the Lowry method. Approximately 20 µg of total protein was loaded per lane on a 10% polyacrylamide gel. Electrophoresis and western blotting were performed following standard laboratory methods, after which SERCA1 protein was detected using anti-SERCA1 monoclonal antibody (ab2819, Abcam, ITK Diagnostics) and a goat-anti-mouse peroxidase-conjugated secondary antibody (P044701-2; Agilent-Dako), followed by chemiluminescent detection. To check for equal loading, blots were stained for GAPDH using anti-GAPDH monoclonal antibody (ab8245, Abcam, ITK Diagnostics).
Genetic analysis of ATP2A1
Mutation analysis of the ATP2A1 gene of the new patients was performed by the U.F. Cardiogénétique et Myogénétique (11 patients), by the genetics department of the Radboud University Nijmegen Medical Centre (four patients), by the Jonquière Hospital (two patients), by the Salford Royal National Foundation Trust (one patient), by the Neuromuskuläres Zentrum – Sozialpädistrisches Zentrum Frankfurt Mitte (two patients), and by the Institute of Genetic Medicine at Newcastle University (two patients). For patients diagnosed in France, genomic DNA was isolated from blood samples by using QIAsymphony® DSP DNA Midi kit (Qiagen) on a QIAsymphony® nucleic acid extraction machine (Qiagen). The 23 exons of the ATP2A1 gene (NM_004320.4) were investigated by Sanger sequencing in eight patients (primers, PCR and Sanger sequencing conditions available on request). Three patients were analysed using targeted high-throughput sequencing. DNA was captured on an in-house SeqCap® EZ library probe (Roche NimbleGen), representing the exonic regions and their flanking regions, for 41 genes involved in skeletal muscle excitability and neuromuscular transmission, including the ATP2A1 gene. Then the capture product was amplified and sequenced on a Miseq sequencer (Illumina). The mean number of reads for targeted regions was higher than 500, thus allowing a reliable detection of copy number variations as well as point mutations. Genetic analysis in the other centres was performed using similar methods.
DNA analysis of the literature patients has been previously described (Odermatt et al., 1996, 2000; Novelli et al., 2004; Vattemi et al., 2010; Sambuughin et al., 2014; Mussini et al., 2015).
Ethical considerations
This study was performed according to the guidelines of the local research ethics committees.
Statistical analysis
Descriptive statistics were used to assess the prevalence of specific symptoms, signs, and results of ancillary investigations (dichotomous outcome: absent or present). For the analysis of the prevalence, we included only those patients in whom data were available of that specific item.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Results
Clinical features
We received completed questionnaires from clinicians of all 22 newly diagnosed patients and from 11 of 18 previously described patients. Four literature patients could not be traced by clinicians from the original centre and we did not receive a reply from the authors of three other literature patients. For these seven patients, only the information from the original articles was used.
Symptoms
Overall, we obtained clinical information on 40 patients with Brody disease from 28 families around the world. Some of the clinical data of the 18 literature patients have been previously summarized (Voermans et al., 2012).
Almost all patients reported onset of symptoms in childhood (38/40). Often symptoms were first noticed during gym classes in primary school or when playing outside with peers, especially during fast alternating movements (e.g. skipping rope or running). The mean age of presentation was 19.2 ± 15.0 years old, and the mean age at diagnosis was 27.3 ± 14.6 years old, resulting in an average doctor delay of 9.9 ± 13.7 years.
Exercise-induced muscle stiffness was reported in all patients, 63% of patients reported stiffness at the start of exercise. The muscle groups predominantly affected were lower limbs (38/38), upper limbs (33/38), and eyelids (24/38). Patients variably reported muscle cramps (defined as involuntary and painful sustained muscle contractures) at rest or during exercise (18/34). Myalgia was commonly reported (20/34) and usually had a generalized distribution. The majority of patients mentioned an increase of symptoms upon exposure to cold temperatures (25/36). Weakness was reported by 31% of patients (11/35).
Physicians reported their patient’s disability with a score of 1 (no significant disability and able to carry out all activities and duties; 10/23), or 2 (slight disability but able to look after own affairs; 13/23) on the Modified Rankin Scale (MRS). Impact on daily life was mostly due to the inability to run, difficulties when practising sports and struggling to climb multiple stairs quickly. Heterozygous family members of patients (carriers) did not exhibit the Brody disease phenotype (this information was not systematically gathered but often encountered as family history in medical files).
Rhabdomyolysis and malignant hyperthermia
Two patients suffered from (an) episode(s) of suspected malignant hyperthermia (MH) following administration of general anaesthetics (Patients L15 and L16). Clinical description of the episodes of these patients has been published in detail previously (Novelli et al., 2004; Guglielmi et al., 2013; Sambuughin et al., 2014). In short, Patient L15 suffered from an episode of generalized muscle contracture associated with fever, rhabdomyolysis (creatine kinase up to 53 000 U/l) and myoglobinuria. He also experienced two episodes of rhabdomyolysis following exercise at ages 19 and 22 with creatine kinase levels up to 221 600 IU/l and 100 000 IU/l, respectively. Patient L16 developed skeletal muscle rigidity with slightly elevated creatine kinase after general anaesthesia. One other patient (Patient N15) experienced an episode of exertional rhabdomyolysis. Patient L11 experienced several episodes of unexplained hyperthermia. The RYR1 gene, which is associated with malignant hyperthermia susceptibility (MHS), was analysed in Patients L15–L17, and showed no mutations (Patient L15) and one non-pathogenic variant (Patients L16 and L17; siblings). Eight other patients have undergone general anaesthesia without any adverse reactions.
Physical examination
Delayed relaxation following repetitive contractions or sustained contraction was observed in 22/33 and 27/34 patients, respectively. Delayed relaxation specifically of the eyelids was seen after repetitive eyelid-closure in 19/31 patients and after sustained forceful eyelid-closure in 17/29 patients. Physical examination revealed no muscle atrophy in any of the patients (0/33) and mild proximal muscle weakness in 3/35 patients (Patients L1, L10 and N15; reported as ‘mild proximal weakness’ or MRC score of ≥4 of proximal lower limbs). Several patients were described as having an athletic appearance (20/30) and/or as having hypertrophic muscles (10/34). Percussion myotonia was reported in 4/34 patients and tendon reflexes were normal except for Patients N16 and N18 with decreased tendon reflexes, and Patient N17 with increased reflexes. A more detailed overview of all clinical features can be found in Table 2.
Table 2.
Ancillary investigations
| Lab | EMG | Muscle biopsy | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | CK (IU/l) | EMG performed | Silent cont. | Myotonic discharges | Muscle biopsy performed | Biopsied muscle | Atrophy type I fibres | Atrophy type II fibres | Marked fibre size variability | ↑Internal nuclei | ↓SERCA staining IHC | ↓SERCA immunoblot | ↓SERCA activity |
| Literature patients | |||||||||||||
| L1 | 50 | + | − | + | Biceps | − | + | + | + | + | Low | ||
| L2 | 62 | + | − | − | + | Biceps | − | + | + | ||||
| L3 | 246 | + | + | − | + | Quadriceps | − | + | + | + | + | ||
| L4 | |||||||||||||
| L5 | + | − | 50% | ||||||||||
| L6 | + | Quadriceps | 50% | ||||||||||
| L7 | |||||||||||||
| L8 | |||||||||||||
| L9 | + | − | − | ||||||||||
| L10 | + | + | − | ||||||||||
| L11 | Slightly elevated | + | + | − | + | Peroneus | + | + | − | + | 67% | ||
| L12 | Slightly elevated | + | + | − | |||||||||
| L13 | 256 | + | + | − | + | Quadriceps | − | + | + | + | − | + | 80% |
| L14 | 260–450 | + | + | − | |||||||||
| L15 | 700–1300 | + | + | − | + | Quadriceps | − | + | + | + | − | + | |
| L16 | 183 | + | Quadriceps | − | + | + | + | + | + | ||||
| L17 | Normal | ||||||||||||
| L18 | 733 | + | + | − | +a | Biceps/deltoid | − | + | + | + | + | ||
| New patients | |||||||||||||
| N1 | 219 | − | + | Quadriceps | + | + | + | 59% | |||||
| N2 | + | + | − | + | Quadriceps | + | − | ||||||
| N3 | + | + | − | + | |||||||||
| N4 | + | + | − | − | |||||||||
| N5 | + | − | − | − | |||||||||
| N6 | 1.5× normal | + | + | − | + | Deltoid | − | + | + | + | + | + | |
| N7 | + | + | − | ||||||||||
| N8 | + | + | − | + | Deltoid | ||||||||
| N9 | + | + | − | − | |||||||||
| N10 | + | + | − | + | Deltoid | − | + | + | + | − | + | 59% | |
| N11 | 111 | + | + | − | − | ||||||||
| N12 | 93 | + | − | − | + | Quadriceps | − | − | +/− | − | + | 68% | |
| N13 | + | − | − | − | |||||||||
| N14 | 99 | + | − | − | − | ||||||||
| N15 | 72 | + | − | − | + | Quadriceps | − | − | − | ||||
| N16 | 713 | + | + | − | − | ||||||||
| N17 | 854 | + | − | − | + | Quadriceps | + | + | + | + | |||
| N18 | 500 | + | − | − | + | Tibialis ant. | + | + | + | + | + | ||
| N19 | 131 | − | − | ||||||||||
| N20 | 103 | − | − | ||||||||||
| N21 | 2–6× normal | + | − | − | + | Biceps | + | − | + | + | |||
| N22 | 300–600 | + | − | − | + | Quadriceps | − | − | − | − | |||
| Total + | 30 | 18 | 0 | 22 | 3 | 13 | 11 | 14 | 6 | 10 | 8 | ||
| Total − | 4 | 10 | 30 | 9 | 12 | 4 | 1 | 3 | 6 | 0 | 0 | ||
| Total known | 34 | 28 | 30 | 31 | 15 | 17 | 12 | 17 | 12 | 10 | 8 | ||
| Percentage + and +/− | 88% | 64% | 0% | 71% | 20% | 77% | 92% | 82% | 50% | 100% | 100% | ||
↑ = increased; ↓ = decreased; += present/performed; − = not present/not performed; CK = creatine kinase; Cont. = contractures; IHC = immunohistochemistry.
Patient had two biopsies (Mussini et al., 2015).
Table 1.
Clinical characteristics
| Patient | History taking | Physical examination | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient: family | Sex/ AAD | Decade of onset | Pattern of involvement | Muscle stiffness Ex | Muscle stiffness at SOE | Muscle cramps | Myalgia | Increase in cold | Muscle | Athletic app. | DR of upper limb | DR of eyelids | |||||
| weakness | atrophy | hypertrophy | after rep. con. | after sus. con. | after rep. con. | after sus. con. | |||||||||||
| Literature patients | |||||||||||||||||
| L1: 1.1 | M/42 | 1st | UL+LL | + | + | Ex | − | +a | − | + | |||||||
| L2: 1.2 | M | Child | UL+LL | + | + | + | |||||||||||
| L3: 2.1 | M | Child | LL | + | − | + | + | + | + | ||||||||
| L4: 2.2 | M | + | |||||||||||||||
| L5: 3.1 | M/41 | Child | G | + | − | Ex | Ex+P | + | − | − | + | + | + | + | + | + | |
| L6: 3.2 | M/42 | Child | G | + | − | Ex | − | + | − | − | + | + | + | + | + | + | |
| L7: 3.3 | M/47 | Child | G | + | − | Ex | Ex+Pa | + | − | − | + | + | + | + | + | − | |
| L8: 4.1 | M/38 | 1st | G | + | + | Ex | − | + | − | − | − | − | + | + | + | + | |
| L9: 4.2 | M/28 | Child | G | + | + | Ex | Ex | + | − | − | − | + | + | + | + | + | |
| L10: 5.1 | F | 1st | G | + | + | +a | +/− | + | + | ||||||||
| L11: 6.1 | M/56 | 1st | G | + | + | − | + | − | − | +/− | + | − | + | − | + | ||
| L12: 6.2 | M/52 | 1st | G | + | + | − | + | + | − | − | +/− | + | − | + | − | + | |
| L13: 7.1 | M/42 | Child | G | + | + | − | − | − | − | − | − | − | + | + | + | + | |
| L14: 7.2 | F | 2nd | G | + | + | − | − | − | − | − | − | − | + | + | + | + | |
| L15: 8.1 | M | 1st | G | + | − | R | P | + | − | − | |||||||
| L16: 9.1 | M | Child | LL | R+P | + | − | − | + | − | − | + | − | |||||
| L17: 9.2 | F | Child | + | ||||||||||||||
| L18: 10.1 | M/50 | 1st | G | + | Ra | − | + | − | − | − | − | + | + | − | − | ||
| New patients | |||||||||||||||||
| N1: 11.1 | M/32 | 1st | G | + | − | − | − | + | − | − | − | + | + | + | + | + | |
| N2: 12.1 | F | 1st | G | + | + | − | − | − | |||||||||
| N3: 13.1 | M/36 | 1st | G | + | + | − | − | + | − | − | + | + | + | + | + | + | |
| N4: 13.2 | F/39 | 1st | UL+LL+E | + | + | − | − | − | − | − | − | + | + | − | + | ||
| N5: 14.1 | F/22 | Child | G | + | + | − | − | + | − | − | + | + | − | + | − | + | |
| N6: 15.1 | M/43 | Child | G | + | + | − | Ex+P | + | − | − | + | + | + | − | + | − | |
| N7: 16.1 | F/20 | Child | G | + | − | Ex | P | + | + | + | |||||||
| N8: 17.1 | M/53 | Child | G | + | − | Ex | − | +/− | − | − | + | + | + | − | + | − | |
| N9: 18.1 | F/17 | Child | UL+LL | + | − | − | − | + | − | − | + | + | + | + | + | ||
| N10: 19.1 | F/26 | 1st | UL+LL | + | + | − | − | + | − | − | − | − | + | + | − | − | |
| N11: 19.2 | F/33 | Child | UL+LL | + | + | − | − | + | − | − | − | − | + | + | − | − | |
| N12: 20.1 | M/8 | 1st | UL+LL+E | + | − | − | Ex+P | + | − | − | − | + | − | + | + | + | |
| N13: 20.2 | M/6 | 1st | UL+LL | + | − | − | Ex+P | + | − | − | − | + | + | + | + | + | |
| N14: 21.1 | F/42 | 1st | UL+LL | + | + | Ex | Ex | − | − | − | − | − | + | + | − | ||
| N15: 22.1 | M/27 | 1st | UL+LL | + | + | Ex | Ex+P | − | +a | − | +b | − | − | − | − | − | |
| N16: 23.1 | M/25 | 1st | UL+LL+E | + | + | Ex | Ex | + | − | − | − | + | + | + | + | ||
| N17: 24.1 | M/28 | 1st | UL+LL+E | + | − | Ex | Ex+P+R | − | − | − | − | + | − | + | + | − | |
| N18: 25.1 | M/56 | 1st | UL+LL | + | − | P | + | − | − | + | + | + | + | − | + | ||
| N19: 26.1 | F/8 | 1st | LL | + | + | Ex | Ex | − | − | − | − | − | − | − | − | ||
| N20: 26.2 | M/5 | 1st | LL | + | + | − | − | − | − | − | − | − | − | − | |||
| N21: 27.1 | F/30 | 1st | LL+E | + | + | Ex+R | Ex+P | − | − | − | − | − | − | − | − | − | |
| N22: 28.2 | M/51 | 3rd | UL+LL | + | + | Ex | Ex+P | + | − | − | − | + | − | + | − | ||
| Total + and +/− | 38 | 19 | 18 | 20 | 26 | 3 | 0 | 13 | 20 | 22 | 27 | 19 | 17 | ||||
| Total − | 0 | 11 | 16 | 14 | 10 | 32 | 33 | 21 | 10 | 11 | 7 | 12 | 12 | ||||
| Total known | 38 | 30 | 34 | 34 | 36 | 35 | 33 | 34 | 30 | 33 | 34 | 31 | 28 | ||||
| Percentage + and +/− | 100% | 63% | 52% | 59% | 72% | 9% | 0% | 38% | 67% | 67% | 79% | 61% | 59% | ||||
App. = appearance; AAD = age at diagnosis; Child = childhood; con. = contraction; DR = delayed relaxation; E = eyelids; Ex = during exercise; F = facial; G = generalized (UL+LL+F + E); LL = lower limbs; P = post-exercise; R = in rest; rep. = repetitive; SOE = start of exercise; sus. = sustained; UL = upper limbs.
Rare, mild or minor.
Only in calf muscles.
Progression of symptoms
We gathered follow-up data of reported symptoms and physical examination (mean duration of 9.3 years, range 1–29 years) in 27 patients. In another three patients only limited information on progressiveness of symptoms was available (Patients L1, N5 and N10). Of these 30 patients, seven patients reported that symptoms had increased (23%). Of them, four reported increased muscle stiffness, four reported an increase in myalgia, three reported an increase in muscle weakness (self-reported), and three mentioned increased muscle cramps. No development of weakness or atrophy was seen by physicians during follow-up physical examination.
Treatment
Different medications were used as symptomatic treatment for muscle stiffness, cramps, and myalgia; dantrolene (n = 5), verapamil (n = 9), mexiletine (n = 2), carbamazepine (n = 2), ibuprofen (n = 2), nifedipine (n = 2), acetazolamide (n = 1), or unknown muscle relaxants (n = 2). Verapamil improved symptoms in three patients but was stopped in two of them because of side effects. Mexiletine improved symptoms in one patient but was similarly stopped because of side effects. The other medications showed no effect and only 1 of 18 treated patients received long-term successful treatment (Patient N15; verapamil). Approximately half of patients (13/31) did not ask for treatment and two patients refused treatment.
Ancillary investigations
Laboratory testing
Serum resting creatine kinase levels (n = 24) ranged between 50 IU/l and 1300 IU/l. Creatine kinase was considered normal in 12/24 patients and slightly to moderately elevated in 12/24 patients (Table 2).
EMG
None of the patients had myotonic discharges on EMG (n = 30). Silent contractures were present in 18/28 patients (Table 2). EMG showed possible myopathic motor unit potentials in only one patient (Patient N15). Results from nerve conduction velocity testing (n = 8) and the McMannis long exercise test (n = 2) were normal.
Histology
There were morphological data available for 22 patients (10 of 18 literature patients and 12 of 22 new patients). The results are summarized in Table 2.
Atrophy of type II fibres was seen in 76% (13/17) of patients, of which two also demonstrated type I atrophy. Only one biopsy (Patient N21) showed exclusive atrophy of type I fibres. In four biopsies further specification of type II atrophy was documented, with specific type IIA atrophy in two and specific type IIB atrophy in the other two samples (Sambuughin et al., 2014; Mussini et al., 2015). Type II atrophy was seen in both normal and athletic build patients to the same extent.
Variability of muscle fibre size was markedly increased in 92% (11/12) of samples.
The percentage of internal nuclei was increased in 82% (14/17) of samples (Table 2). Patient L18 underwent two muscle biopsies (at ages 24 and 42), showing a significant increase in the percentage of internal nuclei in the second biopsy (Mussini et al., 2015). Nuclear centralization and nuclear clumping was sporadically seen in the second biopsy, not to the extent as can be observed in centronuclear myopathies.
In nine biopsies the distribution of type I and type II muscle fibres was quantified. In quadriceps muscle biopsies (n = 7), the observed percentages of type II fibres ranged from 58 to 85%; mean 75% (Karpati et al., 1986; Vattemi et al., 2010; Guglielmi et al., 2013) [normal percentage of type II fibres in healthy quadriceps is 50–65% (Pauw-Gommans et al., 2006)]. Furthermore, the percentage of type II fibres in the m. biceps brachii biopsy of Patient L1 (71%) was also increased [normal value: 59 ± 8% (Dahmane et al., 2001)]. However, in three patients (Patients N2, N10 and N18) the percentage of type II fibres was reduced. In two biopsies (Patients L18 and N17), a specific decrease in the amount of type IIB fibres was found (Mussini et al., 2015) (Table 2).
Discrete necrosis was found in only one biopsy (Patient L18). The same biopsy also showed sparse regeneration, and two other biopsies showed degeneration (Patients N12 and N22) (Mussini et al., 2015).
In two patients, nuclear abnormalities were described. In the muscle biopsy of Patient L16, multiple nuclei were shown to be abnormally elongated (Sambuughin et al., 2014). For Patient L18, a similar elongation of nuclei was described. Furthermore, the two biopsies of Patient L18 showed several other nuclear abnormalities at the ultrastructural level (Mussini et al., 2015).
In vitro contracture test
The in vitro contracture test (IVCT), used to assess MHS, was performed in three patients (Patients L1, L15 and L16) (Halsall and Ellis, 1979; Hopkins et al., 2015). In Patients L15 and L16 (both suffered from episodes of suspected malignant hyperthermia), and in Patient L1 (not suspected of MHS) the IVCT demonstrated a positive result for caffeine and halothane, indicating MHS (Karpati et al., 1986; Novelli et al., 2004; Guglielmi et al., 2013; Sambuughin et al., 2014).
SERCA activity
SERCA activity was previously measured in four literature patients. A variety of techniques were used making it difficult to compare the results on a quantitative level. Nonetheless, all evaluations showed a decreased activity. For Patient L1, SERCA activity was measured as ATP-dependent Ca2+ transport in membrane fractions and was shown to have a consistently low rate of transport (Karpati et al., 1986). In Patients L5 and L6, compound SERCA activity was reduced by 50% (Benders et al., 1994). In Patient L13, SERCA activity was determined as Ca2+ dependent ATP transport in whole muscle homogenates and shown to be reduced by ∼80% (Vattemi et al., 2010).
Odermatt et al. (2000) transfected HEK-293 cells with wild-type and the mutant ATP2A1 of Patient L11. They showed that the mutant SERCA1 had an altered affinity for Ca2+, so that Ca2+ transport would be nearly non-existent at physiological Ca2+ levels (Odermatt et al., 2000). We performed SERCA activity analysis in this patient and found SERCA activity to be reduced by 67%. Additionally, we determined SERCA activity in three new patients and found similar results with a reduction of SERCA activity by 59% (Patients N1 and N10), and 68% (Patient N12) (Table 2).
SERCA expression
Immunohistochemistry was performed in eight literature cases and three new patients (Fig. 1). Compound SERCA staining in type II muscle fibres mainly results in visualization of SERCA1 since this is the predominant isoform in type II fibres (>90%) (Lee, 2010). A severe reduction of compound SERCA was found in type II fibres in Patients L1 and L3. In both patients, staining of SERCA in type I fibres was normal (Karpati et al., 1986). SERCA1 staining revealed a severe reduction in six patients (6/12) and was normal in the other patients (6/12) (Karpati et al., 1986; Benders et al., 1994; Odermatt et al., 2000; Vattemi et al., 2010; Guglielmi et al., 2013; Sambuughin et al., 2014; Mussini et al., 2015). The presence of the SERCA2 isoform was determined by immunohistochemistry in eight biopsies, with normal expression in seven patients and increased expression in one patient (Odermatt et al., 2000; Guglielmi et al., 2013; Sambuughin et al., 2014; Mussini et al., 2015).
Figure 1.
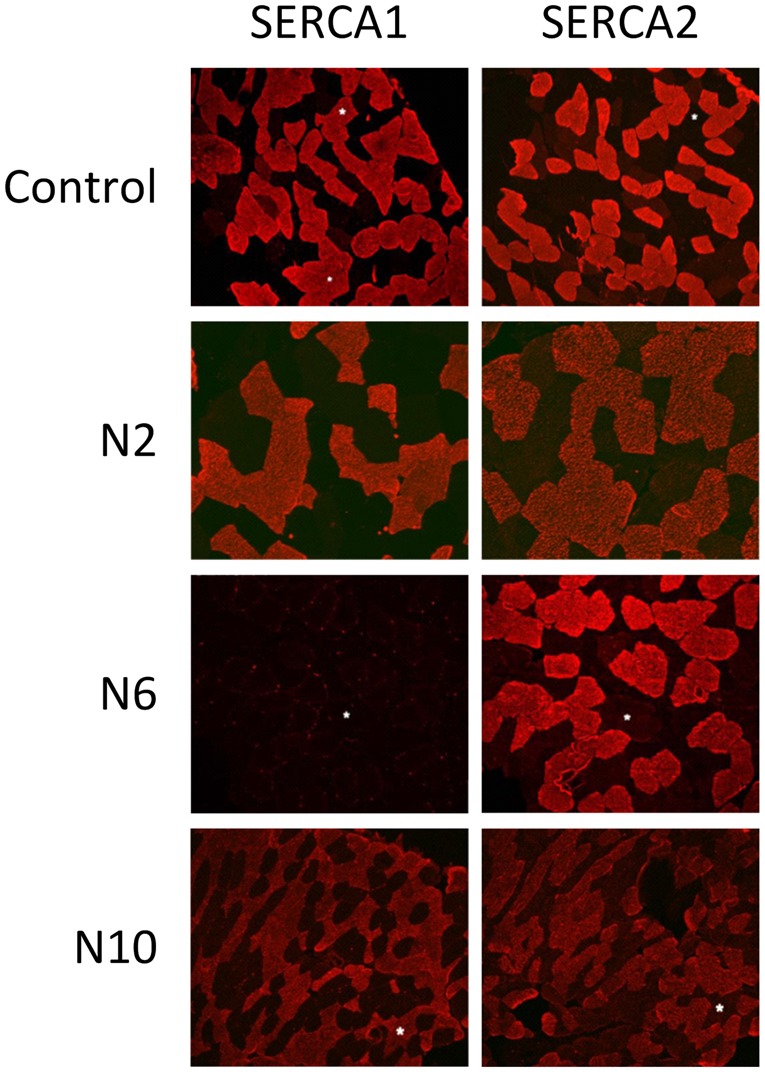
Immunohistochemistry of SERCA1 and SERCA2 in controls and Brody disease. Staining of SERCA1 was reduced in Patient N6 (deltoid muscle), and normal in Patients N2 (quadriceps muscle) and N10 (deltoid muscle). SERCA2 staining was normal in all samples. Interestingly, Patients N6 and N10 carry the same mutation consisting of a large e9 deletion. In the biopsy of Patient N6 (homozygous e9 deletion) SERCA1 staining was reduced, and in the biopsy of Patient N10 (compound heterozygous; e9 deletion and e15 missense mutation) staining was present. This discrepancy is most likely due to the second mutation of Patient N10 having less of an effect on protein expression resulting in normal staining.
Immunoblot analysis was previously performed in five literature patients, either of specific SERCA1 content (Patients L13, L15 and L16) or compound SERCA content (Patients L1 and L3). All immunoblot analyses demonstrated absent or severely decreased amounts of SERCA protein. Additionally, we performed specific SERCA1 western blot analysis in five patients with also marked decreased/absent SERCA1 content in all (Fig. 2).
Figure 2.

Western blot analysis of SERCA1 in Brody disease and control. All Brody disease samples demonstrated severely reduced or absent SERCA1 protein content. See Supplementary material for full gels.
Genetic analysis
All ATP2A1 mutations are listed in Table 3. The location of null mutations (stop mutations, frameshift mutations, splice mutations) within the ATP2A1 gene is shown in Fig. 3A. The location of mutated or deleted amino acids (missense mutations or in-frame deletion of one codon) within the SERCA1 protein are shown in Fig. 3B. Missense mutations and in-frame deletions of one codon affected highly conserved amino acids in different domains of the protein (Fig. 3B and C). Some mutations were recurrent: p.Leu67del (Families 7 and 14), c.2464dup (Families 14, 16 and 27), deletion of exon 9 (Families 15 and 19), p.Arg560Cys (Families 21 and 22). Among the 33 different mutations, 11 were missense, three were in-frame deletions of one single codon, seven were frameshift mutations (four out-of-frame small deletions, three out-of-frame small insertions or duplications), six were stop mutations, four were splice site mutations (three affecting canonical donor GT sites at the +1 position of introns 3, 19 and 20, one affecting canonical acceptor AG site at −1 position in intron 15), and two were large rearrangements (deletion of exon 9, deletion of the whole gene).
Table 3.
Genetic characteristics
| Genotype | Mutation 1 | Location | Mutation type | Mutation 2 | Location | Mutation type | |
|---|---|---|---|---|---|---|---|
| Literature patients | |||||||
| L1 | Homozygous | c.440del p.Pro147Leufs*34 | e 5 | fs 1-bp del | - | - | - |
| L2 | Homozygous | c.440del p.Pro147Leufs*34 | e 5 | fs 1-bp del | - | - | - |
| L3 | Compound heterozygous | c.2025C>A p.Cys675* | e 15 | Nonsense | c.219+1G>C | i 3 (ds) | Splice |
| L4 | Compound heterozygous | c.2025C>A p.Cys675* | e 15 | Nonsense | c.219+1G>C | i 3 (ds) | Splice |
| L5 | Homozygous | c.592C>T p.Arg198* | e 7 | Nonsense | - | - | - |
| L6 | Homozygous | c.592C>T p.Arg198* | e 7 | Nonsense | - | - | - |
| L7 | Homozygous | c.592C>T p.Arg198* | e 7 | Nonsense | - | - | - |
| L8 | Homozygous | c.162_163insAA p.Glu55Lysfs*43 | e 3 | fs 2-bp ins | - | - | - |
| L9 | Homozygous | c.162_163insAA p.Glu55Lysfs*43 | e 3 | fs 2-bp ins | - | - | - |
| L10 | Homozygous | c.100G>T p.Glu34* | e 1 | Stop | c.1167C>A p.Tyr389* | e 10 | Nonsense |
| L11 | Homozygous | c.2366C>T p.Pro789Leu | e 17 | Missense | - | - | |
| L12 | Homozygous | c.2366C>T p.Pro789Leu | e 17 | Missense | - | - | |
| L13 | Compound heterozygous | c.198_200del p.Leu67del | e 3 | In-frame 3-bp del | c.1817_1819del p.Glu606del | e 15 | In-frame 3-bp del |
| L14 | Compound heterozygous | c.198_200del p.Leu67del | e 3 | in-frame 3-bp del | c.1817_1819del p.Glu606del | e 15 | In-frame 3-bp del |
| L15 | Compound heterozygous | c.178del p.Leu60Serfs*37 | e 3 | fs 1-bp del | c.200T>G p.Leu67Arg | e 3 | Missense |
| L16 | Compound heterozygous | c.704T>A p.Ile235Asn | e 8 | Missense | c.2944G>A p.Glu982Lys | e 21 | Missense |
| L17 | Compound heterozygous | c.704T>A p.Ile235Asn | e 8 | Missense | c.2944G>A p.Glu982Lys | e 21 | Missense |
| L18 | Compound heterozygous | c.1742_1743del p.Ser581Cysfs*7 | e 14 | fs 2-bp del | c.2744+1G>T | i 19 (ds) | Splice |
| New patients | |||||||
| N1 | Homozygous | c.490C>T p.Arg164* | e 6 | Nonsense | - | - | - |
| N2 | Homozygous | c.547G>A p.Glu183Lys | e 7 | Missense | - | - | - |
| N3 | Homozygous | c.2245G>A p.Glu749Lys | e 16 | Missense | - | - | - |
| N4 | Homozygous | c.2245G>A p.Glu749Lys | e 16 | Missense | - | - | |
| N5 | Compound heterozygous | c.198_200del p.Leu67del / | e 3 | In-frame 3-bp del | c.2464dup p.Arg822Profs*39 | e 17 | fs 1-bp dup |
| N6 | Homozygous | c.(928+1_929-1)_(1094+1_1095-1)del | e 9 and sr | large del e 9 | - | - | - |
| N7 | Compound heterozygous | c.1271_1273del p.Ser424del | e 11 | In-frame 3-bp del | c.2464dup p.Arg822Profs*39 | e 17 | fs 1-bp dup |
| N8 | Homozygous | c.2765T>C p.Leu922Pro | e 20 | Missense | - | - | - |
| N9 | Homozygous | c.2101-1G>T | i 15 (as) | splice | - | - | - |
| N10 | Compound heterozygous | c.(928+1_929-1)_(1094+1_1095-1)del | e 9 and sr | Large del e 9 | c.1811G>A p.Arg604His | e 15 | Missense |
| N11 | Compound heterozygous | c.(928+1_929-1)_(1094+1_1095-1)del | e 9 and sr | Large del e 9 | c.1811G>A p.Arg604His | e 15 | Missense |
| N12 | Homozygous | c.100G>T p.Glu34* | e 1 | Stop | - | - | - |
| N13 | Homozygous | c.100G>T p.Glu34* | e 1 | Stop | - | - | - |
| N14 | Homozygous | c.1678C>T p.Arg560Cys | e 14 | Missense | - | - | - |
| N15 | Compound heterozygous | c.1678C>T p.Arg560Cys | e 14 | Missense | c.2441T>C p.Leu814Pro | e 17 | Missense |
| N16 | Homozygous | c.1966C>T p.Arg656* | e 15 | Nonsense | - | - | - |
| N17 | Compound heterozygous | c.(?_-235)_(*417_?)del | all e and i | Large del all e and i | c.2464dup p.Arg822Profs*39 | e 17 | fs 1-bp dup |
| N18 | Compound heterozygous | c.(?_-235)_(*417_?)del | all e and i | Large del all e and i | c.2464dup p.Arg822Profs*39 | e 17 | fs 1-bp dup |
| N19 | Homozygous | c.2862+1G>A | i 20 (ds) | Splice | - | - | - |
| N20 | Homozygous | c.2862+1G>A | i 20 (ds) | Splice | - | - | - |
| N21 | Compound heterozygous | c.361dupG p.Glu121Glyfs*3 | e5 | fs 1-bp dup | c.2464dup p.Arg822Profs*39 | e 17 | In-frame 1-bp dup |
| N22 | Compound heterozygous | c.428G>A p.Arg143Gln | e 5 | Missense | c.1317_1318del p.Glu439Aspfs*80 | e 12 | fs 2-bp del |
Mutations are numbered according to transcript NM_004320.4.
as = acceptor site; ds = donor site; del = deletion; dup = duplication; e = exon(s); fs = frameshift; ins = insertion; i = intron(s); sr = surrounding regions.
Figure 3.

Diversity of ATP2A1 mutations in patients with Brody disease. (A) Localization of stop mutations (asterisk), frameshift mutations (dollar sign), splice site mutations (downward arrow), and large scale rearrangements (horizontal lines with dashed line ends) are shown on a schematic gene architecture that encompasses the 23 exons of transcript NM_4320.4. (B) Localization of missense mutations and in-frame deletions of one single codon. The corresponding mutated or deleted amino acids are displayed in a schematic representation of the SERCA1 protein according to crystallographic data. The protein comprises 10 alpha helices, one actuator domain (A), one nucleotide binding domain (N) and one phosphorylation domain (P). Mutated amino acids are located in different regions of the protein. (C) The SERCA1 protein is highly conserved, as shown by the conservation of amino acids that are affected in our patients with Brody disease. Missense mutations are shown in the single letter amino acid code, and deletion of an amino acid is shown by a delta symbol.
Discussion
This study reports the features of the largest cohort of Brody disease patients to date, consisting of 22 new patients carrying 19 novel mutations plus the 18 previously published patients. The findings classify Brody disease as a distinct myopathy in the broader field of calcium-related myopathies.
Clinical features
Brody disease typically manifests in the first decade of life, but patients usually do not present to a physician until their third decade. All patients suffered from exercise-induced muscle stiffness, resulting in an inability to run and mild-to-moderate difficulties in daily life in most. Delayed muscle relaxation occurred especially after explosive, short and repetitive contractions, which is explained by the selective type II muscle fibre involvement. Symptoms were usually present in upper and lower limbs and eyelids. Eyelid involvement is most likely the result of the high percentage of type II muscle fibres in orbicularis oculi muscle (∼90%) (Hwang et al., 2011).
One-third of patients reported muscle weakness, while physical examination revealed only mild muscle weakness in three patients. Patients might acknowledge stiffness as weakness or rate their disability as weakness. Physical examination revealed no atrophy; in contrast, an athletic build was observed in approximately two-thirds of patients. Symptoms were progressive in 23% of patients but did not become debilitating (MRS score of 1 or 2). However, with a relatively young age at last evaluation in our cohort (mean 34, range 6–56 years old) it is possible that progression of symptoms occurs in late adulthood. This would be supported by the progression of myopathic changes in the second muscle biopsy in Patient L18 at the age of 42 years (18 years after the first muscle biopsy). Nevertheless, pronounced muscle weakness and atrophy excludes Brody disease as a diagnosis. This might be related to the selective type II involvement that allows for compensation with normally functioning type I muscle fibres throughout life.
Ancillary investigations
EMG
EMG is characterized by the presence of silent contractures and absence of myotonic discharges. Therefore, when EMG shows no myotonic discharges in patients clinically suspected of having a myotonic disorder, provocation of possible silent contractures is recommended by performing rapid contractions during EMG. Furthermore, we recommend using ‘silent contractures’ instead of ‘silent cramps’, as cramps have a strict electromyographical definition that does not apply to patients with Brody disease (i.e. action potentials that gradually increase and later decrease in number and frequency) (Stern and Bernick, 1990; Parisi et al., 2003).
Malignant hyperthermia and in vitro contracture test
The MH-like episodes in Brody disease were most likely anaesthesia- or exercise-induced rhabdomyolysis without signs of hypermetabolism, as can be seen in other myopathies (Klingler et al., 2005). The association between Brody disease and MH is likely related to the accumulation of Ca2+ ions in the cytoplasm due to decreased reuptake of Ca2+ in the sarcoplasmic reticulum (Sambuughin et al., 2014; Rosenberg et al., 2015). In MH caused by RYR1/CACNA1S mutations the hypermetabolic state is due to a rapid continuous cycle of Ca2+ efflux (via RYR1) and influx (via SERCA) from/into the sarcoplasmic reticulum, consuming large amounts of ATP; whereas in Brody disease, activity of the RYR1 channel is normal but the dysfunctional SERCA1 protein causes the prolonged contraction. Furthermore, RYR1 channels are located in both types of skeletal muscle fibres whereas SERCA1 is only expressed in type II muscle fibres. Therefore, the overall accumulation of Ca2+ ions would be less pronounced in Brody disease leading to a higher threshold for developing MH-like events compared to classical MH. The finding that all four IVCTs in patients with Brody disease were positive is likely due to the increased cytoplasmic Ca2+ concentration, which does not necessarily indicate an increased risk of classic malignant hyperthermia (Sambuughin et al., 2014).
One of the patients (Patient L15), who was clinically suspected of MHS and had a positive IVCT, also carried an autosomal dominant chromosomal translocation [(2;7)(p11.2;p12.1)] (Novelli et al., 2004; Guglielmi et al., 2013). The genes in the breakpoints and adjacent regions of this translocation are not known to be associated with MHS (such as RYR1, CACNA1S, STAC3, CASQ1, or JSRP1) (Stowell, 2014; Rosenberg et al., 2015). His father also carried this translocation and had a positive IVCT but did not suffer from MH episodes (Novelli et al., 2004; Guglielmi et al., 2013). As the positive IVCT in this family co-segregated with the chromosomal translocation, the ATP2A1 mutations may not be the only cause of the episode of MH in this patient.
Nonetheless, as these rhabdomyolysis or MH-like events are potentially life-threatening conditions, we consider Brody disease patients at risk for these reactions and advise taking necessary perioperative precautions: avoid administration of succinylcholine and inhalational agents, and monitor vital functions including core temperature (Glahn et al., 2010; Rosenberg et al., 2015).
Histology
Systematic re-evaluation of the biopsy reports showed fibre type II atrophy, increased fibre size variability, and an increased number of internal nuclei. Hence, Brody disease is very unlikely in the case of a normal biopsy. The atrophy of type II fibres most likely results from selective involvement of these muscle fibres. Given the athletic appearance in many patients, the common finding of atrophic muscle fibres was somewhat unexpected. Pseudohypertrophy is unlikely to explain the athletic build as no excessive fatty infiltration or fibrosis was seen in the biopsies. In one patient (Patient L16), type I fibres were hypertrophic, which could serve as a compensatory mechanism and explain the athletic build (Sambuughin et al., 2014). However, this has not been demonstrated in other patients and is therefore unlikely to fully explain this phenomenon. Further investigation, e.g. muscle ultrasound and/or MRI could help to unravel the discrepancy between the muscular build and histological findings.
SERCA expression
In Brody disease, immunohistochemical staining of SERCA1 is reduced or normal and therefore not a reliable tool to assess SERCA1 expression (Benders et al., 1994, 1996; Guglielmi et al., 2013). In contrast, the immunoblot analysis showed reduced or absent SERCA1 protein in all tested patients.
Although immunohistochemistry and immunoblot analysis both assess protein expression, in some cases the results do not fully correlate. This is most likely to be because of the lower sensitivity of immunohistochemistry, where a moderately decreased SERCA1 content could still lead to full colour saturation due to the abundant natural expression of SERCA1 in skeletal muscle. Furthermore, the antibodies used for immunohistochemistry might attach to cross reactive epitopes resulting in false positive results. Guglielmi et al. (2013) demonstrated that the decreased SERCA1 content detected in patients with Brody disease by western blot was probably not due to a decreased solubility of the mutated protein compared to wild-type SERCA1.
All performed western blot analyses were qualitatively assessed as reduced or absent protein expression, resulting in no specific correlation with genotype. We cannot exclude that with the use of quantitative western blot techniques such a correlation could be demonstrated. Similarly, no correlation was found between genotype and immunohistochemistry results, with both normal and abnormal staining seen in all different mutation types (i.e. null mutations, missense mutations, in-frame deletions, and large scale rearrangements).
SERCA activity
SERCA activity was determined in different muscles and using different techniques across patients, all showing severely reduced residual activity. If Ca2+ removal from the cytosol in type II muscle fibres was only regulated by SERCA1, this would be predicted to result in a severe phenotype. The mild phenotype suggests that different compensatory mechanisms preserve the ability of patients to relax their muscles at a reasonable speed after contraction (Karpati et al., 1986; Guglielmi et al., 2013). Ectopic expression of SERCA2 in type II muscle fibres could be one of these mechanisms (Sambuughin et al., 2014). The first evidence for the upregulation of SERCA2 in patients with Brody disease was an increased immunohistochemical staining of SERCA2, in type I as well as in type II fibres (Sambuughin et al., 2014). However, ectopic expression of SERCA2 was deemed unlikely by Guglielmi et al. (2013) who did not detect increased expression of SERCA2 in type II fibres by immunohistochemical analysis. Other possible compensatory mechanisms are downregulation of myoregulin (SERCA1 inhibitor), or increasing the mitochondrial calcium-uptake mechanisms and Ca2+ transportation from the cytoplasm to the extracellular space (PMCA, Na+/Ca2+-antiporter) (MacLennan, 2000). In normal conditions there is little contribution of the Na+/Ca2+-antiporter in total Ca2+ efflux from the cytosol (Melzer et al., 1995). However, it is possible that in Brody disease this antiporter and other alternative Ca2+ efflux pathways are upregulated to compensate for the decreased SERCA1 activity. Interestingly, this theory has been tested in cattle with congenital pseudomyotonia, an animal model of Brody disease caused by recessive ATP2A1 mutations with a similar phenotype (Drogemuller et al., 2008; Bianchini et al., 2014; Dorotea et al., 2015; Mascarello and Sacchetto, 2016). In these cattle it has been confirmed that the loss of SERCA1 was not compensated by increased expression of SERCA2. Conversely, immunoblot analysis revealed an increased expression of PMCA in pathological muscle (Dorotea et al., 2015). Furthermore, it was demonstrated that proteasomal inhibition can functionally rescue mutated SERCA1 in muscle fibres from pseudomyotonia cattle by avoiding the premature disposal of mutated SERCA1 (Bianchini et al., 2014). Future research would be needed to test if similar compensatory mechanisms can be identified in patients with Brody disease.
Genetic analysis
DNA alterations at the ATP2A1 gene locus represent the whole spectrum of mutation types: whole gene deletion, single exon deletion, splice site mutations, small exonic frameshift deletions or insertions, missense substitutions, and in-frame small deletions (Table 3 and Fig. 3). Genetic diagnosis of Brody disease relies on the identification of a homozygous or compound heterozygous mutation. Recessive pathogenicity of mutations is evident for null mutations (stop, frameshift, splice, large rearrangements), but may be discussed for missense mutations or single codon deletions, according to the conservation of affected amino acids throughout species and paralogues. The missense mutations in this cohort were located at highly conserved sites (Fig. 3C) and are likely to impair the conformational cycle of the protein and Ca2+ pumping. Brody disease mutations are loss-of-function mutations, either by preventing the protein from being expressed correctly, or by hampering its function. No gradation in severity could be demonstrated in clinical presentation (e.g. age of onset, muscle weakness, disability) or mutation type leading to no particular phenotype-to-genotype correlations. However, with a mean age of 34 years (range 6–56 years), it is possible that certain phenotype-to-genotype correlations could become apparent with advancing age.
This study significantly increases the number of known ATP2A1 mutations contributing to the often difficult interpretation of genetic variants with next generation genetic testing. Pathogenicity of ATP2A1 variants can be assessed using clinical (delayed muscle relaxation), electrophysiological (silent contractures), and laboratory (SERCA activity and western blot of muscle) testing. In parallel, abnormal activity and/or expression of SERCA should lead to extensive analysis of the ATP2A1 gene. This is demonstrated by the decreased SERCA activity and expression found in Patients N10 and N11 (Family 19), which made the geneticist further analyse the genetic data in which previously only one ATP2A1 mutation was identified. This led to the discovery of an additional large-scale deletion of exon 9, confirming Brody disease as the diagnosis.
Treatment
Verapamil and dantrolene, drugs that limit the Ca2+ release from the sarcoplasmic reticulum by inhibition of the RYR1 channel and the dihydropyridine receptor, have been prescribed but often stopped because of side effects (Benders et al., 1994; Ikemoto et al., 2001).
For future research, drugs that lower the cytosolic Ca2+ concentration by different mechanisms could possibly be tested in Brody disease. For instance, drugs that promote the peptide named dwarf open reading frame (DWORF), which decreases the activity of myoregulin (SERCA1 inhibitor), and phospholamban and sarcolipin (SERCA2 inhibitors) (Nelson et al., 2016). Drugs that would directly inhibit myoregulin or that promote Ca2+ efflux from the cytosol by promoting PMCA or the Na+/Ca2+-antiporter could also potentially alleviate symptoms in Brody disease (Anderson et al., 2015). Moreover, drugs inhibiting proteasomal disposal of mutated SERCA1 might have beneficial effects in Brody patients (Bianchini et al., 2014).
To our knowledge, drugs that alter the function/activity of these proteins do not yet exist but would make very interesting pharmacological targets, both for Brody disease and other (cardio)myopathies with abnormal Ca2+ handling.
Diagnostic pathway and considerations
Non-dystrophic myotonia was the most frequent differential diagnosis, but can easily be distinguished from Brody disease. First, Brody disease only involves fast-twitch muscle fibres leading to selective muscle stiffness with short, repetitive and explosive movements, whereas delayed relaxation in myotonic disorders depends mostly on intensity and repetition of contractions and can demonstrate a typical warm-up phenomenon (Trip et al., 2009). Second, myotonic disorders will typically demonstrate myotonic discharges at myography that are not seen in Brody disease (Kuntzer, 2004; Miller, 2008; Trip et al., 2009; Matthews et al., 2010).
Silent contractures can also be found in McArdle disease (glycogen storage disease type V). However, McArdle disease typically presents with a second wind phenomenon, permanently high creatine kinase levels and episodes of rhabdomyolysis during/after (rigorous) exercise that are not or are rarely seen in Brody disease (Godfrey and Quinlivan, 2016). The histopathological changes seen in Brody disease can also be seen in numerous other neuromuscular disorders (e.g. muscular dystrophies, congenital myasthenic syndromes, glucocorticoid myopathy) (Ciciliot et al., 2013; Eymard et al., 2013; Matsumoto et al., 2019). However, in general, these disorders do not display the distinct phenotype of Brody disease (i.e. delayed muscle relaxation with absence of muscle weakness) making them unlikely differential diagnostic candidates.
In line with current practice in (suspected) non-dystrophic myotonias, we recommend specific sequencing of the ATP2A1 gene as a first step (or performing Next Generation Sequencing including ATP2A1 analysis) when a patient is clinically suspected of Brody disease. If an ATP2A1 mutation of unknown pathogenicity is identified, we recommend a muscle biopsy with measurement of SERCA activity and/or western blot analysis of SERCA1 to confirm the pathogenicity of the mutations and diagnose Brody disease.
We suspect that Brody disease is indeed less rare than previously estimated because of the relatively mild phenotype and the delayed relaxation and silent contractures that can be missed if not specifically looked for. The advances in genetic testing with gene panel screening and whole exome/genome sequencing most likely contributes to the recent increase in diagnosed Brody disease patients.
Calcium-related myopathies
In recent years there is an increased identification of mutations in other (novel) genes encoding proteins involved in Ca2+ homeostasis (Snoeck et al., 2015; Treves et al., 2017; Jungbluth et al., 2018). We suggest that Brody disease should be classified as a calcium-related myopathy, in parallel with other myopathies affecting calcium handling such as RYR1-related myopathy and tubular aggregate myopathy linked to STIM1, ORAI1, and CASQ1 mutations (Bohm and Laporte, 2018). STIM1 and ORAI1 are involved in Ca2+ entry activated by depletion of sarcoplasmic reticulum stores. Calsequestrin (CASQ1) acts as the major Ca2+ buffering protein in the sarcoplasmic reticulum of skeletal muscle and is mainly expressed in fast twitch muscle fibres. There is heterogeneous age of onset and disease severity in tubular aggregate myopathy, with CASQ1 mutations representing the milder end of the spectrum. Both Brody disease and tubular aggregate myopathy are associated with exercise-induced muscle cramps, stiffness and myalgia. However, tubular aggregate myopathy often gives rise to progressive muscle weakness, a feature not seen in Brody myopathy (Funk et al., 2013; Bohm and Laporte, 2018).
In summary, we have clarified the phenotype and diagnostic possibilities to improve understanding and recognition of Brody disease as a distinct myopathy in the broader field of calcium-related myopathies. Future research could further clarify the compensatory mechanisms of cytosolic Ca2+ removal involved in Brody disease, which could ultimately lead to targeted treatment options.
Supplementary Material
Acknowledgements
Karina Horsting-Wethly, Berendien Stoltenborg-Hogenkamp, and Doreen Fialho.
Funding
No funding was provided towards this work.
Competing interests
The authors report no competing interests.
Glossary
Abbreviations
- IVCT
in vitro contracture test
- MH(S)
malignant hyperthermia (susceptibility)
- PMCA
plasma membrane Ca2+ ATPase
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
References
- Allard B. From excitation to intracellular Ca(2+) movements in skeletal muscle: basic aspects and related clinical disorders. Neuromuscul Disord 2018; 28: 394–401. [DOI] [PubMed] [Google Scholar]
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015; 160: 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benders AA, Veerkamp JH, Oosterhof A, Jongen PJ, Bindels RJ, Smit LM, et al. Ca2+ homeostasis in Brody's disease. A study in skeletal muscle and cultured muscle cells and the effects of dantrolene an verapamil. J Clin Invest 1994; 94: 741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benders AA, Wevers RA, Veerkamp JH. Ion transport in human skeletal muscle cells: disturbances in myotonic dystrophy and Brody's disease. Acta Physiol Scand 1996; 156: 355–67. [DOI] [PubMed] [Google Scholar]
- Bianchini E, Testoni S, Gentile A, Cali T, Ottolini D, Villa A, et al. Inhibition of ubiquitin proteasome system rescues the defective sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1) protein causing Chianina cattle pseudomyotonia. J Biol Chem 2014; 289: 33073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm J, Laporte J. Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 2018; 76: 1–9. [DOI] [PubMed] [Google Scholar]
- Brody IA. Muscle contracture induced by exercise. A syndrome attributable to decreased relaxing factor. N Engl J Med 1969; 281: 187–92. [DOI] [PubMed] [Google Scholar]
- Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 2013; 45: 2191–9. [DOI] [PubMed] [Google Scholar]
- Dahmane R, Valen IV, Knez N, Eren I. Evaluation of the ability to make non-invasive estimation of muscle contractile properties on the basis of the muscle belly response. Med Biol Eng Comput 2001; 39: 51–5. [DOI] [PubMed] [Google Scholar]
- Dorotea T, Grunberg W, Murgiano L, Plattet P, Drogemuller C, Mascarello F, et al. Fast-twitch skeletal muscle fiber adaptation to SERCA1 deficiency in a Dutch Improved Red and White calf pseudomyotonia case. Neuromuscul Disord 2015; 25: 888–97. [DOI] [PubMed] [Google Scholar]
- Drogemuller C, Drogemuller M, Leeb T, Mascarello F, Testoni S, Rossi M, et al. Identification of a missense mutation in the bovine ATP2A1 gene in congenital pseudomyotonia of Chianina cattle: an animal model of human Brody disease. Genomics 2008; 92: 474–7. [DOI] [PubMed] [Google Scholar]
- Eymard B, Stojkovic T, Sternberg D, Richard P, Nicole S, Fournier E, et al. Congenital myasthenic syndromes: difficulties in the diagnosis, course and prognosis, and therapy–The French National Congenital Myasthenic Syndrome Network experience. Rev Neurol 2013; 169: S45–55. [DOI] [PubMed] [Google Scholar]
- Funk F, Ceuterick-de Groote C, Martin JJ, Meinhardt A, Taratuto AL, De Bleecker J, et al. Morphological spectrum and clinical features of myopathies with tubular aggregates. Histol Histopathol 2013; 28: 1041–54. [DOI] [PubMed] [Google Scholar]
- Glahn KP, Ellis FR, Halsall PJ, Muller CR, Snoeck MM, Urwyler A, et al. Recognizing and managing a malignant hyperthermia crisis: guidelines from the European Malignant Hyperthermia Group. Br J Anaesth 2010; 105: 417–20. [DOI] [PubMed] [Google Scholar]
- Godfrey R, Quinlivan R. Skeletal muscle disorders of glycogenolysis and glycolysis. Nat Rev Neurol 2016; 12: 393–402. [DOI] [PubMed] [Google Scholar]
- Guglielmi V, Vattemi G, Gualandi F, Voermans NC, Marini M, Scotton C, et al. SERCA1 protein expression in muscle of patients with Brody disease and Brody syndrome and in cultured human muscle fibers. Mol Genet Metab 2013; 110: 162–9. [DOI] [PubMed] [Google Scholar]
- Halsall PJ, Ellis FR. A screening test for the malignant hyperpyrexia phenotype using suxamethonium-induced contracture of muscle treated with caffeine and its inhibition by dantrolene. Br J Anaesth 1979; 51: 753–6. [DOI] [PubMed] [Google Scholar]
- Hopkins PM, Ruffert H, Snoeck MM, Girard T, Glahn KP, Ellis FR, et al. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth 2015; 115: 531–9. [DOI] [PubMed] [Google Scholar]
- Hwang K, Huan F, Kim DJ. Muscle fiber types of human orbicularis oculi muscle. J Craniofac Surg 2011; 22: 1827–30. [DOI] [PubMed] [Google Scholar]
- Ikemoto T, Hosoya T, Aoyama H, Kihara Y, Suzuki M, Endo M. Effects of dantrolene and its derivatives on Ca(2+) release from the sarcoplasmic reticulum of mouse skeletal muscle fibres. Br J Pharmacol 2001; 134: 729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H, Treves S, Zorzato F, Sarkozy A, Ochala J, Sewry C, et al. Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat Rev Neurol 2018; 14: 151–67. [DOI] [PubMed] [Google Scholar]
- Karpati G, Charuk J, Carpenter S, Jablecki C, Holland P. Myopathy caused by a deficiency of Ca2+-adenosine triphosphatase in sarcoplasmic reticulum (Brody's disease). Ann Neurol 1986; 20: 38–49. [DOI] [PubMed] [Google Scholar]
- Klingler W, Lehmann-Horn F, Jurkat-Rott K. Complications of anaesthesia in neuromuscular disorders. Neuromuscul Disord 2005; 15: 195–206. [DOI] [PubMed] [Google Scholar]
- Kuntzer T. Electrophysiological testing in muscle channelopathies. Rev Neurol 2004; 160: S49–54. [DOI] [PubMed] [Google Scholar]
- Lee EH. Ca2+ channels and skeletal muscle diseases. Prog Biophys Mol Biol 2010; 103: 35–43. [DOI] [PubMed] [Google Scholar]
- MacLennan DH. Ca2+ signalling and muscle disease. Eur J Biochem 2000; 267: 5291–7. [DOI] [PubMed] [Google Scholar]
- Mandroukas A, Heller J, Metaxas TI, Christoulas K, Vamvakoudis E, Stefanidis P, et al. Deltoid muscle characteristics in wrestlers. Int J Sports Med 2010; 31: 148–53. [DOI] [PubMed] [Google Scholar]
- Mascarello F, Sacchetto R. Structural study of skeletal muscle fibres in healthy and pseudomyotonia affected cattle. Ann Anat 2016; 207: 21–6. [DOI] [PubMed] [Google Scholar]
- Matsumoto C, Mori-Yoshimura M, Noguchi S, Endo Y, Oya Y, Murata M, et al. Phenotype of a limb-girdle congenital myasthenic syndrome patient carrying a GFPT1 mutation. Brain Dev 2019; 41: 470–3. [DOI] [PubMed] [Google Scholar]
- Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D, et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 2010; 133: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer W, Herrmann-Frank A, Luttgau HC. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim Biophys Acta 1995; 1241: 59–116. [DOI] [PubMed] [Google Scholar]
- Miller TM. Differential diagnosis of myotonic disorders. Muscle Nerve 2008; 37: 293–9. [DOI] [PubMed] [Google Scholar]
- Mussini JM, Magot A, Hantai D, Sternberg D, Chevessier F, Pereon Y. Atypical nuclear abnormalities in a patient with Brody disease. Neuromuscul Disord 2015; 25: 773–9. [DOI] [PubMed] [Google Scholar]
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016; 351: 271–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli A, Valente EM, Bernardini L, Ceccarini C, Sinibaldi L, Caputo V, et al. Autosomal dominant Brody disease cosegregates with a chromosomal (2;7)(p11.2;p12.1) translocation in an Italian family. Eur J Hum Genet 2004; 12: 579–83. [DOI] [PubMed] [Google Scholar]
- Odermatt A, Barton K, Khanna VK, Mathieu J, Escolar D, Kuntzer T, et al. The mutation of Pro789 to Leu reduces the activity of the fast-twitch skeletal muscle sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA1) and is associated with Brody disease. Hum Genet 2000; 106: 482–91. [DOI] [PubMed] [Google Scholar]
- Odermatt A, Taschner PE, Khanna VK, Busch HF, Karpati G, Jablecki CK, et al. Mutations in the gene-encoding SERCA1, the fast-twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat Genet 1996; 14: 191–4. [DOI] [PubMed] [Google Scholar]
- Parisi L, Pierelli F, Amabile G, Valente G, Calandriello E, Fattapposta F, et al. Muscular cramps: proposals for a new classification. Acta Neurol Scand 2003; 107: 176–86. [DOI] [PubMed] [Google Scholar]
- Pauw-Gommans IM, Gerrits KH, de Haan A, van Engelen BG. Muscle slowness in a family with nemaline myopathy. Neuromuscul Disord 2006; 16: 477–80. [DOI] [PubMed] [Google Scholar]
- Rosenberg H, Pollock N, Schiemann A, Bulger T, Stowell K. Malignant hyperthermia: a review. Orphanet J Rare Dis 2015; 10: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve 2006; 33: 715–31. [DOI] [PubMed] [Google Scholar]
- Sambuughin N, Zvaritch E, Kraeva N, Sizova O, Sivak E, Dickson K, et al. Exome analysis identifies Brody myopathy in a family diagnosed with malignant hyperthermia susceptibility. Mol Genet Genomic Med 2014; 2: 472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeck M, van Engelen BG, Kusters B, Lammens M, Meijer R, Molenaar JP, et al. RYR1-related myopathies: a wide spectrum of phenotypes throughout life. Eur J Neurol 2015; 22: 1094–112. [DOI] [PubMed] [Google Scholar]
- Stern LZ, Bernick C. Muscle cramps In: Walker HK, Hall WD, Hurst JW, editors. Clinical methods: the history, physical, and laboratory examinations. 3rd edn Boston; 1990. [PubMed] [Google Scholar]
- Stowell KM. DNA testing for malignant hyperthermia: the reality and the dream. Anesth Analg 2014; 118: 397–406. [DOI] [PubMed] [Google Scholar]
- Treves S, Jungbluth H, Voermans N, Muntoni F, Zorzato F. Ca(2+) handling abnormalities in early-onset muscle diseases: novel concepts and perspectives. Semin Cell Dev Biol 2017; 64: 201–12. [DOI] [PubMed] [Google Scholar]
- Trip J, Drost G, Ginjaar HB, Nieman FH, van der Kooi AJ, de Visser M, et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry 2009; 80: 647–52. [DOI] [PubMed] [Google Scholar]
- Vattemi G, Gualandi F, Oosterhof A, Marini M, Tonin P, Rimessi P, et al. Brody disease: insights into biochemical features of SERCA1 and identification of a novel mutation. J Neuropathol Exp Neurol 2010; 69: 246–52. [DOI] [PubMed] [Google Scholar]
- Voermans NC, Laan AE, Oosterhof A, van Kuppevelt TH, Drost G, Lammens M, et al. Brody syndrome: a clinically heterogeneous entity distinct from Brody disease: a review of literature and a cross-sectional clinical study in 17 patients. Neuromuscul Disord 2012; 22: 944–54. [DOI] [PubMed] [Google Scholar]
- Yang JC, Yoo YJ. Histochemical muscle fiber types of autopsied human gastrocnemius, soleus, peroneus longus and tibialis anterior muscles. Kor J Pathol 1986; 20: 413–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.