

Abstract
Whenever new substances are proposed for use as sources of nutrients in food supplements, foods for the general population or foods for specific groups, EFSA is requested by the European Commission to perform an assessment of their safety and of the bioavailability of the nutrient from the proposed source. This guidance describes the scientific data required to allow an evaluation of the safety of the source within the established framework for risk assessment of food additives and novel food ingredients and the bioavailability of the nutrient from this source. This document is arranged in five main sections: one on technical data aimed at characterising the proposed source and at identifying potential hazards resulting from its manufacture and stability in food; one on existing authorisations and evaluation, providing an overview of previous assessments on the proposed source and their conclusions; one on proposed uses and exposure assessment section, allowing an estimate of the dietary exposure to the source and the nutrient based on the proposed uses and use levels; one on toxicological data, describing approaches which can be used to identify (in conjunction with data on manufacture and composition) and to characterise hazards of the source and any relevant breakdown products; the final section on bioavailability focuses on determining the extent to which the nutrient from the proposed source is available for use by the body in comparison with one or more forms of the same nutrient that are already permitted for use on the positive lists. This guidance document should replace the previous guidance issued by the Scientific Committee for Food and published in 2001.
Keywords: EFSA Panel guidance, nutrient sources, nutrients, vitamins, minerals, bioavailability, applications
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1439/full
Summary
The Panel on Food Additives and Nutrient Sources added to Food (ANS Panel) requested the European Food Safety Authority (EFSA) to update existing scientific guidance on the data needed for the assessment of sources of nutrients proposed for use in the manufacture of foods in the light of the experience accrued over the years with this type of assessment and to reflect the current thinking in risk assessment.
This guidance describes the scientific data required to allow an evaluation of the safety of the source within the established framework for risk assessment and the bioavailability of the nutrient from this source.
The use of chemical substances as ‘sources’ of vitamins and minerals in food is regulated in the European Union (EU) by the establishment of positive lists of substances, annexed to the relevant sectorial legislation, i.e.:
Directive 2002/46/EC for food supplements;
Regulation (EC) No 1925/2006 for ‘fortified’ foods;
Regulation (EU) No 609/2013, covering infant formula and follow‐on formula (IF and FOF); processed cereal‐based food and baby food (PCBF); food for special medical purposes (FSMP); and total diet replacement for weight control (TDR).
For the purpose of this guidance, vitamins and minerals listed in the annexes of the above mentioned legislation are referred to as ‘nutrients’.
The positive list of Regulation (EU) No 609/2013 also contains other additional substances (i.e. amino acids, carnitine, taurine, nucleotides, choline and inositol), which are also covered by the term ‘nutrient’ used in this guidance.
According to the Directive and the two Regulations above, the chemical substances used as sources which may be added to food, including food supplements and foods for specific groups, should be safe and also bioavailable, a property which is described, in the relevant legislation, as ‘available to be used by the body’.
Whenever new substances are proposed for inclusion in any of the above positive lists of sources, EFSA is requested by the European Commission to perform an assessment of their safety and bioavailability.
The nutritional, physiological function or safety aspects of nutrients as such are outside the scope of the evaluations carried out by the ANS Panel. Hence, these aspects of the nutrient are not to be considered for this updated guidance on evaluation of sources of nutrients. In addition, the term bioavailability as used in this guidance addresses the comparative bioavailability, namely the bioavailability of the nutrient from the proposed source compared with the bioavailability of the nutrient in forms that are already permitted for use on the positive lists. Hence, the term bioavailability is confined to the difference in bioavailability of the nutrient released from two different sources.
This guidance presents a common format for the organisation of the information to be presented in order to assist the applicant in the preparation of a well‐structured application to demonstrate the safety of a proposed source and the extent to which the nutrient is available to be used by the body. Adherence to this format will facilitate access to information and scientific data in applications which will help EFSA to carry out its evaluation and to deliver its scientific opinion in an effective and consistent way.
For the safety assessment of the source, data requirements which should be covered in all applications relate to the description of the source, manufacturing process (including possible residuals or contaminants), technical specifications, proposed uses and use levels, and anticipated intake of the source and the corresponding intake of the nutrient. Information on existing authorisation and evaluations should also be provided.
The first consideration to be made in assessing the safety of a proposed source of nutrients is whether and to what extent the proposed source dissociates in the lumen of the human gastrointestinal (GI) tract. This document provides guidance both on the type of experimental data to be generated to establish dissociation of the source in the human GI tract lumen and on the toxicological data required, based on the expected dissociation of the source.
The toxicological data required follows the same tiered approach applied to the evaluations of new food additives and novel food ingredients, integrating the core areas of kinetics, genotoxicity, repeated dose toxicity testing, and reproductive and developmental toxicity. For sources intended to be used in infants below 16 weeks of age, the toxicity testing required shall be aligned to the latest recommendations issued by the EFSA Scientific Committee with respect to the risk assessment of substances present in food intended for this age group.
In principle, for sources which extensively and readily dissociate in the GI tract into constituents found in the diet and/or human body or have already been assessed (e.g. as food additives), existing data will be the basis for the assessment. Tier 1 toxicity testing would be required for sources which are absorbed unchanged from the GI tract lumen and/or when there are no existing data. The need for additional testing of sources should be determined by the non‐nutrient component generated following dissociation. The need for higher level of testing is determined by results from Tier 1 testing for sources which are absorbed unchanged. However there is no absorption trigger for Tier 2 testing for sources which are absorbed intact but dissociate into components of the diet and/or the human body by first pass metabolism (either in the GI wall or liver before reaching the systemic circulation). For sources which are absorbed intact and either do not dissociate before reaching the systemic circulation or dissociate into non‐nutrient components which are not natural components of the diet and/or the human body require Tier 2 testing.
It is acknowledged that it is not always possible to determine directly whether the nutrient from the proposed source is available to be used by the body, and therefore, a range of surrogate tests are proposed as examples that will generate data to be used in assessing the bioavailability of the nutrient from the proposed source. These data should allow a comparison between the behaviour of the proposed source and one or more sources of the same nutrient, already permitted for use in foods. Provided the dissociation between the two (or more) substances is similar, the nutrient from the proposed source may be assumed to be as ‘available to be used by the body’ as the nutrient released from the comparator source(s) and no further testing for bioavailability would be required. In the event that the dissociation behaviour between the new proposed source and one of the reference sources is not comparable, then additional testing would be required. Such tests may include more complex dissociation test (e.g. with a simulated gastrointestinal digestion), in vitro studies (e.g. Caco‐2 cell models) and in vivo studies. Selection of the tests is to be determined on a case‐by‐case basis.
The guidance document was subject to consultation between the ANS Panel and the NDA Panel and to public consultation (from 15‐12‐2017 to 25‐2‐2018) before finalisation.
1. Introduction
This guidance document refers to the data needed for the assessment of sources of nutrients proposed for use in the manufacture of foods. It describes the scientific data required to allow an evaluation of the safety of the source within the established framework for risk assessment and the bioavailability of the nutrient from this source.
1.1. Background and Terms of Reference as provided by the ANS Panel
1.1.1. Background
The use of chemical substances as ‘sources’ of vitamins and minerals in food is regulated in the European Union (EU) by the establishment of positive lists of substances, annexed to the relevant sectorial legislation, i.e.:
Directive 2002/46/EC for food supplements1;
Regulation (EC) No 1925/2006 for ‘fortified’ foods2
Regulation (EU) No 609/2013, covering infant formula and follow‐on formula (IF and FOF); processed cereal‐based food and baby food (PCBF); food for special medical purposes (FSMP); and total diet replacement for weight control (TDR)3
For the purpose of this guidance, vitamins and minerals listed in the annexes of the above mentioned legislation are referred to as ‘nutrients’.
The positive list of Regulation (EU) No 609/2013 also contains additional substances (i.e. amino acids, carnitine, taurine, nucleotides, choline and inositol), which are also covered by the term ‘nutrient’ in this guidance.
According to the Directive and the two Regulations above, the chemical substances used as sources of nutrients which may be added to food, including food supplements and foods for specific groups, should be safe and also bioavailable, a property which is described as ‘available to be used by the body’.
Whenever new substances are proposed for inclusion in any of the above positive lists of sources, the European Food Safety Authority (EFSA) is requested by the European Commission to perform an assessment of their safety and bioavailability.
An opinion expressed by the Scientific Committee on Food (SCF) in 2001 provides guidance to the applicants on the nature and extent of the information that should be submitted to establish bioavailability of the nutrient or other ingredient and safety of the source (SCF, 2001a). In this guidance document, reference is made to another opinion expressed by the SCF in 2001, namely the ‘Guidance on submissions for Food Additive Evaluations by the Scientific Committee on Food’, for the toxicological data needed in support of these applications (SCF, 2001b).
Between 2005 and 2009, the AFC Panel and the ANS Panel evaluated approximately 200 dossiers of chemical substances proposed for use as sources. These assessments were used as the scientific basis for the European Commission to draw up positive lists of substances to be included in the relevant legislation.
In July 2012, with the adoption by the ANS Panel of the Scientific Opinion on ‘Guidance on submission for food additive evaluations’ (EFSA ANS Panel, 2012), the latter guidance document from the SCF has become obsolete.
At its 52nd meeting the ANS Panel indicated that the scientific principles that have been incorporated into the 2012 guidance document for food additive evaluations should equally be applied to the evaluation of sources of nutrients and therefore an update of the respective guidance document was warranted.
Moreover, recently there have been cases in which the evaluation of the safety of a source has been combined with an assessment of the substance as a novel food ingredient. The updated guidance on sources should make provisions for the data requirement needed under these circumstances, e.g. by making reference to the relevant food sector legislation and/or existing guidance.
The nutritional, physiological function or safety aspects of nutrients as such are outside the scope of the evaluations carried out by the ANS Panel and as such are not to be considered for this updated guidance on evaluation of sources of nutrients.
Finally, the ANS Panel noted that with the adoption of Commission Regulation (EU) No 2017/228*, the evaluation of nutrient sources and other substances with physiological effects added to foods will be transferred to the NDA Panel on 1 July 2018. Therefore the NDA Panel was involved in the Working Group and consulted prior to the endorsement of this Guidance for public consultation and its finalisation.
1.1.2. Terms of Reference
In accordance with Article 29(1) of Regulation (EC) No 178/20024, the European Food Safety Authority asks its scientific Panel on Food Additives and Nutrient Sources added to Food (ANS) to provide a scientific opinion on guidance on submissions for evaluations of sources of nutrients proposed for use in the manufacture of food, including food supplements and foods for specific groups of the population.
1.1.3. Interpretation of Terms of Reference
For the purpose of this guidance, and in line with the definition given in the relevant Legislation, the Panel noted that bioavailability of a nutrient from a source is described as it being ‘available to be used by the body’. Based on the terms of reference for individual assessments, the Panel has traditionally interpreted this as the bioavailability of the nutrient from the source. However, considerations on systemic availability of the source have always been taken into account in determining the toxicological data requirements for the safety assessment of the source itself.
The evaluation of safety of nutrient sources does not include evaluation of the nutritional, physiological function or safety of nutrient as such, and therefore, these aspects have not been considered in this updated guidance on evaluation of sources of nutrients.
As it is difficult to measure ‘availability to be used by the body’ for most nutrients, the Panel has proposed a range of approaches that can be applied in order to evaluate bioavailability of the nutrient. These approaches are comparative studies which take into consideration the bioavailability of the chemical forms of the nutrient which are already on the positive lists.
Current Legislation treats the substances that are included in the positive lists in isolation. However these substances are often incorporated into different matrices, such as formulated products (tablets, capsules, etc.) or foods, and the particular formulation or product influences the bioavailability of the nutrient. However, the Panel noted that the influence of other components is not part of the evaluation of a source.
Regulation (EC) No 1925/2006 not only contains a positive list of substances that can be added to food, but also, in its Annex III, Part C, lists those substances that are placed under Union Scrutiny as a consequence of an assessment conducted in the context of Article 8 of the above mentioned Regulation (i.e. substances for which the possibility of harmful effects on health is identified but scientific uncertainty persists). In those cases, the applicable implementing rules5 allows a file to be submitted by a food business operator or any other interested party to EFSA for a safety assessment in line with relevant guidance documents adopted or endorsed by EFSA. This guidance however is not specifically intended to provide advice for the preparation of such a file, despite the applicability of certain parts of it to general safety assessments of food ingredients.
In addition, pursuant to Regulation (EU) No 609/2013, substances included in the positive list for the foods for specific groups, should not only be safe and bioavailable but also have a nutritional or physiological effect and be suitable for the persons for whom the food is intended. The Panel noted that bioavailability of a nutrient from different sources currently included in the positive lists may be different and some sources may not be suitable for the intended uses (i.e., infant formula and follow‐on formula, processed cereal‐based food and baby food, food for special medical purposes and total diet replacement for weight control). In assessing such products, there is a need for a holistic assessment which in addition to the assessment of the safety of the source and the bioavailability of the nutrient also takes into consideration the intended use(s). The principles for the assessment of the suitability of the proposed sources to the persons for whom the food is intended are outside the scope of this guidance.
1.2. General principles
This document should be read in conjunction with the latest available version of the document ‘Administrative guidance on submissions for safety evaluation of substances added for specific nutritional purposes in the manufacture of foods’ prepared by the Food information and composition, food waste Unit of the Health and Food Safety Directorate‐General of the European Commission (European Commission, online).
The assessment of the safety of the nutrient itself and the data required for these assessments do not fall within the remit of this guidance. The scientific assessment will therefore deal with the safety of a particular source and the bioavailability of a given nutrient from that source. The evaluation of the nutrient itself, in relation to establishing dietary reference values (DRVs), is outside the scope of the assessment. However, if the proposed uses and use levels of the source are likely to reach the tolerable upper level (UL) for that nutrient, this will be taken into account in the safety assessment.
With respect to the safety of the source and the relevant dissociation products or products naturally occurring in biological milieux, the principle of the assessment is not different from the safety assessment of food additives, for which updated guidance was adopted by the ANS Panel in 2012 (EFSA ANS Panel, 2012). This introduced the principles of a tiered approach to toxicological testing to reflect 3Rs principles (replacement, reduction and refinement).
Recently, the European Commission has requested the assessment of the safety of sources that would also fall under the definition of ‘novel food ingredients’ (NFI) according to the applicable Regulation (EU) No 2015/2283†. Also in this case, the guidance has been updated to make reference to the latest available guidance issued by the EFSA Panel on Dietetic Products, Nutrition and Allergies (EFSA NDA Panel, 2016).
Similarly, in the event of sources consisting of, containing, or produced from genetically modified microorganisms, the guidance has also been updated in order to refer to the latest currently applicable guidance (EFSA FEEDAP Panel, 2018).
In assessing the safety of a proposed source, a critical consideration is the behaviour or degree of dissociation expected to occur in the gastrointestinal tract. When a source would be assumed to be extensively dissociated in the gastrointestinal (GI) tract, the risk assessment can usually be based on existing toxicological information of the resulting compounds. However, there might be cases where toxicological information of resulting components is not available or not adequate for risk assessment which would require producing new toxicological data.
In the context of this guidance, the term ‘dissociation’ intends to cover breakdown in whatever form (e.g. but not limited to: dissociation of salts, complexes, chelates; ester hydrolysis; etc.).
Previous versions of this guidance required data on the bioavailability of the nutrient from the source but provided limited information on the purpose of these data or of study designs best able to generate such data. A more detailed description of various approaches that can be used for assessing the bioavailability of the nutrient from the source is included in this guidance, but these are not intended to be exhaustive or prescriptive. Applicants are advised to select and design the actual tests on a case‐by‐case basis taking into account physicochemical data, and any other relevant information on the compound.
The guidance is arranged in the following five main sections describing the data required for assessment:
A general overview
-
Data requirements for dossiers
The Technical data section characterises the proposed source and identifies potential hazards resulting from its manufacture (e.g. impurities, residuals), and stability in food (e.g. degradation products).
The Existing authorisations and evaluation section provides an overview of previous assessments on the proposed source and their conclusions.
The Proposed uses and exposure assessment section allows an estimate of the dietary exposure to the source and the nutrient based on the proposed uses and use levels for different EU Member States and various groups in the population.
The Toxicological data section describes approaches which can be used to identify (in conjunction with data on manufacture and composition) and characterise hazards of the source and any relevant breakdown products.
The Bioavailability data section seeks to determine the extent to which the nutrient from the proposed source is ‘available for use by the body’ by comparing it with one or more forms of the same nutrient that are on the positive lists.
With respect to the data required for hazard identification of the proposed source, this guidance adopts the tiered approach described in the ‘Guidance on submission for food additive evaluations’ (EFSA ANS Panel, 2012) which balances data requirements against the risk. The tiered approach initially uses less complex tests to obtain hazard data; these are then evaluated to determine if they are sufficient for risk assessment or, if not, to plan studies at higher tiers. The intention is that in developing their dossier, applicants will be able to more readily identify relevant data needs which will allow adequate assessment of risks to humans from the intended use whilst strengthening the scientific basis for the assessment. In addition, this approach takes into consideration animal welfare by adopting animal testing strategies in line with the 3Rs (replacement, refinement and reduction).
Where there is evidence that a source would dissociate in the human GI tract into well characterised components, the toxicological assessment should be based on the existing databases for these components and, depending on the time course of the dissociation, additional testing on the source itself may not be necessary.
1.2.1. Sources that fall under the definition of Novel Food Ingredients (NFI) according to Regulation (EU) No 2015/2283
As specified in Regulation (EU) No 2015/2283, vitamins, minerals and other substances used in accordance with Directive 2002/46/EC, Regulation (EC) No 1925/2006 or Regulation (EU) No 609/2013 are considered to fall within the definition of novel food where:
a production process not used for food production within the Union before 15 May 1997, which gives rise to significant changes in the composition or structure of a food, affecting its nutritional value, metabolism or level of undesirable substances has been applied;
they contain or consist of engineered nanomaterials.
Engineered nanomaterials are defined in Regulation (EU) No 2015/2283 as any intentionally produced material that has one or more dimensions of the order of 100 nm or less or that is composed of discrete functional parts, either internally or at the surface, many of which have one or more dimensions of the order of 100 nm or less, including structures, agglomerates or aggregates, which may have a size above the order of 100 nm but retain properties that are characteristic of the nanoscale.
Properties that are characteristic of the nanoscale include:
those related to the large specific surface area of the materials considered; and/or
specific physico‐chemical properties that are different from those of the non‐nanoform of the same material.
In the case of sources that fall under the definition of NFI, the information to be provided must be in accordance with the latest applicable guidance (EFSA NDA Panel, 2016).
For new proposed sources that are also novel foods and which are being assessed for both uses simultaneously, the requirements of both sets of guidance are generally applicable. If differences exist in the requirements, application of the more stringent test is sufficient to meet all requirements.
1.2.2. Assessing the safety of a proposed new source
The first consideration is to determine to what extent the source dissociates in the lumen of the human GI tract based on data from the expected dissociation of the source in the human GI tract (key elements of the design of the study and reporting as well as decision criteria are described in more detail in Appendix D).
The outcome of the dissociation test will inform the toxicological data required for the assessment.
-
If data from the dissociation test demonstrate that the source is extensively and readily dissociated in the GI tract, the safety assessment will be based on toxicological information on the resulting compounds.
If the products of dissociation and/or natural separation that occurs in biological milieus already have established health‐based guidance values (HBGV) and/or other DRVs these are used as the basis for risk assessment and no further toxicological data are needed. For products covered by Commission Delegated Regulation (EU) 2016/20176, the defined minimum and maximums levels can be used;
If the non‐nutrient components do not have established HBGVs, toxicological data requirements, in line with the tiered approach described in the 2012 ANS Panel Guidance on Food Additives evaluations and the 2016 NDA Panel Guidance on Novel Foods are needed (EFSA ANS Panel, 2012; EFSA NDA Panel, 2016);
If data from the dissociation test demonstrate that the source does not extensively and readily dissociate (see Appendix D) in the lumen of the GI tract, then the source is likely to be absorbed at least partly unchanged from the GI tract, and the tiered approach to toxicological testing as described in the 2012 ANS Panel Guidance on Food Additives evaluations and 2016 NDA Guidance on Novel Foods would apply (EFSA ANS Panel, 2012; EFSA NDA Panel, 2016).
It is anticipated that Tier 1 testing on the nutrient source would generally be sufficient if it can be proven that after absorption the nutrient source is undergoing presystemic metabolism in the intestinal wall or in the liver. Decisions on further testing should be made on a case‐by‐case basis, following the 2012 ANS Panel Guidance on Food Additives evaluations.
In the case where a source is absorbed unchanged and not fully metabolised in the intestinal wall or presystemically, the 2012 ANS Panel Guidance on Food Additives evaluations would trigger Tier 2 testing. However, for components which are naturally present in food or the body, then the requirements for Tier 1 testing of the source would generally be sufficient.
Outcome of the safety assessment
The Panel would not normally define an acceptable daily intake (ADI) for a source, but would rather base its conclusions on a comparison between the estimated exposure to the proposed source and a reference point derived from the toxicological dataset provided.
However, if an ADI or a HBGV has been defined for other uses of the same substance, the Panel may consider this comparison of estimated exposure with this value is appropriate (e.g. source already authorised as a food additive).
For sources that are not genotoxic and not carcinogenic, when using margin of exposure (MOE) approaches, the Panel would generally consider a margin of at least 100 between a reference point and the anticipated exposure sufficient to account for the uncertainty for extrapolating between animal species and human, and among human individuals. However, the Panel considers each MOE on a case‐by‐case basis to determine whether the magnitude of the MOE between the anticipated exposure from the proposed uses and use levels and the point of departure are sufficient to conclude that there would be no safety concern. In the consideration, the Panel also takes into account the uncertainties identified in the database and a potential higher sensitivity of vulnerable population groups.
Unavoidable genotoxic and carcinogenic residuals
Substances assessed for their proposed use as sources cannot have genotoxic or genotoxic and carcinogenic activity.
This paragraph therefore concerns exclusively unavoidable genotoxic and carcinogenic residuals or contaminants, for which the Panel considered that it would be possible to use a MOE approach. The Scientific Committee described that for contaminants a MOE of 10,000 or higher, if it is based on the BMDL10 from an animal study, and taking into account overall uncertainties in the interpretation, would be of low concern from a public health point of view and might be reasonably considered as a low priority for risk management actions (EFSA, 2005). However, the Panel considered that for unavoidable residuals, the MOE should be at least 10,000 and preferably as large as possible, and that this should be reflected in the specifications. Whenever possible, it would be prudent to establish levels of this type of residuals in the specifications as low as reasonably achievable. The Panel noted that for the assessment of unavoidable genotoxic residuals, the Threshold of Toxicological Concern (TTC) approach could be applied (EFSA Scientific Committee, 2012).
1.2.3. Assessing the bioavailability of the nutrient from the proposed source
As described previously in Section 1.1.3, it is generally difficult to measure the ‘availability of a nutrient to be used by the body’; therefore, this guidance recommends a range of surrogate tests to generate data that could be used to assess bioavailability. For these approaches, the bioavailability of the nutrient from the proposed source should be compared using the in vitro tests or in vivo studies in humans described below (see Section 2.5), with the bioavailability of the same nutrient from one or more sources already on the positive lists.
Choice of the source used as a comparator must be described and justified by the applicant and results obtained with the novel source should be discussed in the context of what is known about the bioavailability of the nutrient under examination, including considerations on the bioavailability of the nutrient from the source chosen as a comparator.
A range of possible approaches are described in more detail in Section 2.5 and Appendix D.
1.2.4. Assessing the exposure to the source and resulting intake of the nutrient
The assessment of the exposure to the source is the qualitative and/or quantitative evaluation of its likely intake by the European population. In addition to an estimation of the exposure to the source, it is of particular relevance to estimate also the intake of the nutrient resulting from the proposed use(s) and use level(s) of the source, also taking into account the dietary intake all sources, such as natural occurrence in other foods.
In case of sources added to food (e.g. food fortification), the exposure estimates of the source are determined based on consumption data for the food in which the source is intended to be added and by summing the contribution of each food in which the source is intended to be added. The resulting intake of the nutrient is subsequently calculated.
A different paradigm applies to the sources used in food supplements and in food for specific groups, since in these two cases the exposure estimates are not applicable to the whole European population but only to those who are likely to use these products for particular nutritional purposes. In those cases, it is anticipated that incorporation of the proposed source in food supplements and/or foods for specific groups will be in such a way that a pre‐defined daily intake of the nutrient is achieved.
The estimates of human exposure to the source and the corresponding intake of the nutrient are compared to the relevant health based guidance values, for example established tolerable upper levels (ULs) for a nutrient.
2. Data requirements for dossiers
2.1. Technical data
A synopsis of categories applicable to substances proposed as new sources is presented in Table 1. The data required for the characterisation of the aforesaid substances added to food are not different from those already outlined for the evaluation of food additives (EFSA ANS Panel, 2012) and novel food ingredients (EFSA NDA Panel, 2016), and depends on the nature and origin of the substance(s) under assessment. Depending on the nature of the source, additional data requirements may be described in other relevant guidance documents, as presented in Table 1.
Table 1.
Categories of sources and relevant EFSA Guidance documents outlining specific data requirements for their characterisation
| Category | Examples from past EFSA opinions | Reference | |
|---|---|---|---|
| 1 | Single substances, chemically characterised; sources consisting of, isolated from or produced from material of inorganic mineral origin | Vanadium citrate, stannic chloride, chromium picolinate, copper aspartate, orotic acid salts, inorganic mineral constituents isolated from rocks and utilised as inorganic or organic salts or chelates | EFSA ANS Panel (2012), EFSA NDA Panel (2016) |
| 2 | Mixtures of single substances, chemically characterised | Selenium amino acid chelate; iron(II)‐humic acid/fulvic acid chelate | EFSA ANS Panel (2012) |
| 3 | Complex mixtures not derived from botanical sources, possibly not fully chemically characterised (chemical characterisation extent depending on the proposed use and use levels) | EFSA ANS Panel (2012) | |
| 4 | Sources consisting of, isolated from or produced from animals or parts thereof | Blood peptonates | EFSA ANS Panel (2012) |
| 5 | Sources of botanical origin | Iodised ethyl esters of poppy seed oil | EFSA Scientific Committee (2009, 2014) |
| 6 | Sources consisting of, isolated from, or produced from cell culture or tissue culture derived from animals, plants, fungi or algae | EFSA NDA Panel (2016) | |
| 7 | Natural, derivatised and synthetic polymers | EFSA ANS Panel (2012) | |
| 8 | Sources containing microorganisms or derived from microorganisms, fungi or algae |
Chromium‐enriched yeast Also concerning sources consisting of, isolated from or produced from GMMs |
EFSA (2007b) and subsequent updates, EFSA BIOHAZ Panel (2018), EFSA GMO Panel (2011), EFSA FEEDAP Panel (2018) |
| 9 | Nano‐sized materials, engineered and unintentionally produced | EFSA NDA Panel (2016), EFSA Scientific Committee (2018) |
The chemistry and specifications of a substance (or mixture of substances), in terms of chemical structure(s) and physicochemical properties, is critical information required for risk assessment and subsequent risk management. The purity of a single substance needs to be defined by specifications, and adequate chemical characterisation of simple mixtures needs to be performed. It may not always be possible to fully characterise more complex mixtures, but as much information as possible is required to understand the extent to which variability in composition is controlled during manufacture. Information on the manufacturing process is used in risk assessment to identify impurities, residuals, reaction intermediates, precursors, and reagents that could have an influence in the toxicological evaluation.
Hazardous substances that might need to be controlled in the material of commerce need to be identified and specified (e.g. genotoxic compounds, heavy metals). Information requirements for analytical methods to detect and measure a source in food and during storage and over time, when used in different food types, are critical. The identification of degradation products might trigger toxicological evaluation of one or more degradation products to characterise any additional hazards and risks. Where laboratory test methods are used, validation criteria for the analytical techniques and/or methods should be provided to demonstrate their sensitivity, specificity and associated uncertainty.
2.1.1. Identity of the substance
The information required with respect to the identity of a source will depend on the category to which the substance belongs and is set out in detail in Appendix A. Where requested information is not applicable or is not submitted on any of the points set out below, reasons and a scientific justification should be given.
In complex mixtures (e.g. extracts, protein hydrolysates), all constituents cannot in general be fully chemically characterised and/or identified: a qualitative and quantitative characterisation of the main constituents should be performed, at least via sum parameters.
The experience with previous assessments has shown that the characterisation of coordination complexes or chelates is a relevant issue for the assessment of proposed sources, particularly for minerals. A chemical complex may exhibit a reversible association of molecules, atoms, or ions through weak chemical bonds; however, coordination complexes are known that are quite stable in that they are bound together by considerably strong bonds. The characterisation of coordination complexes is carried out on the basis of their physical, spectral and analytical data: results of elemental analysis and spectral data of the uncomplexed ligands and their metal complexes must be found to be in good agreement with their structures, this eventually proving the purity of all the substances. Specifically, there is a requirement for scientific evidence that demonstrates the existence of the proposed complex as the sole entity, with no relevant presence of residual unbound material and other impurities.
Information should also be provided on the identity and the quantity of impurities or by‐products, residues and contaminants. The type and the spectrum of potential target analytes should be considered in the light of the sources and the production process. For example, for substances produced via microbial fermentation, the presence of undesirable microbial metabolites, such as mycotoxins, has to be investigated. For substances isolated via extraction, residues of the solvent(s) used should be provided.
2.1.2. Proposed specifications
The proposed chemical and microbiological specifications of the source should be submitted in a format modelled on recent EU or other internationally accepted specifications. An example is provided in Appendix B.
Similar to what is required for the evaluation of new food additives (EFSA ANS Panel, 2012), in order to ensure that specifications are representative of the actual material of commerce, the analytical data supporting the specifications should also be obtained on several batches (preferably at least 5) of the source that have been independently produced for a given method of a manufacture. A rationale for the proposed specifications should be provided.
2.1.3. Manufacturing process
As is the case in the evaluation of new food additives (EFSA ANS Panel, 2012) and novel food ingredients (EFSA NDA Panel, 2016), the information provided for nutrient sources is used in the risk assessment to identify potential impurities, reaction intermediates, by‐products or contaminants that could present a hazard. Where hazards are identified (e.g. genotoxic compounds, heavy metals, nanoparticles (non‐engineered nanomaterials)), they might need to be controlled in the material of commerce. The same level of detail as that required for food additives and NFI is therefore expected also for the proposed sources, allowing conclusions to be drawn regarding the impact of the process on the safety of the source and bio‐availability of the nutrient from the source. In all cases a detailed description of the manufacturing process should be provided covering the following: method of manufacture including information on: (i) raw materials, starting substances, and other reagents and solvents used; (ii) operational limits and key parameters of the production process; (iii) measures implemented for production control and quality assurance (e.g. HACCP, GMP, ISO); (iv) a production flow‐chart, including quality control checks.
For substances from chemical syntheses: (i) factors such as reaction sequence, side reactions, purification steps, and preparation of the product to be commercialised, which may assist in determining likely impurities (including nanoparticles) and their influence on the toxicological evaluation; (ii) information on substances entering the manufacturing process, e.g. identity of extraction solvent(s), reagents, reaction conditions (e.g. temperature, duration of reaction, catalyst), special precautions (e.g. light, temperature), chemical or physical decontamination/purification methods (e.g. solvent extraction, crystallisation);
For substances derived from botanical sources: (i) information on the method(s) of manufacture, including the process by which the raw material is converted into a preparation, such as extraction or other procedure(s), and plant‐to‐extract ratio; (ii) information on substances entering the manufacturing process, e.g. identity of the extraction solvent, reagents, special precautions (light, temperature); (iii) standardisation criteria (e.g. see European Pharmacopoeia) (EFSA Scientific Committee, 2009). For sources derived from plant, animal or microbiological sources, the applicant should describe in detail the process by which the raw material is converted into a preparation, e.g. extraction or other procedure(s), as well as standardisation procedures. Information should also be provided on the handling of the sources, for example the growth and harvesting conditions for plants and fungi (e.g. wild or cultivated, cultivation practices including the use of pesticides, and time of harvest in relation to both season and stage of the plant growth); the breeding, rearing, feeding and farming conditions for farmed animals or the hunting, catching or collecting and killing of wild living animals; the culture conditions for microbes and microalgae.
For sources derived from plants, but especially when intended for use as an ingredient for supplements, specific considerations and complementary information is provided in the EFSA Guidance on safety assessment of botanicals and botanical preparations (EFSA Scientific Committee, 2009) and its subsequent extension, the Scientific Opinion on a Qualified Presumption of Safety (QPS) approach for the safety assessment of botanicals and botanical preparations (EFSA Scientific Committee, 2014).
For sources that would fall under the definition of NFI, the information reported in the EFSA NDA Panel, 2016 guidance is to be followed.
If the applicant requests that the detailed description of the manufacturing process is treated confidentially, a non‐confidential description of the manufacturing process should also be provided, alongside a justification for the confidentiality claims made.
2.1.4. Methods of analysis in food
By analogy to what is required for the evaluation of food additives (EFSA ANS Panel, 2012), a minimum of a single laboratory validated analytical method should be provided for the determination of the source and its degradation and reaction products in the foods to which the substance is intended to be added. The method(s) provided should be specific and fit‐for‐purpose. They should be applicable to all the food categories to which the substance may be added. Method(s) should be given in full except where the analytical methods used are well established and may be given by reference only.
2.1.5. Mode of incorporation, reaction and fate in food(s) to which the source is added
Where the source is intended to be used in fortified foods, details on how the source is to be incorporated into the food should be provided.
The stability of the source during storage, as produced and in food, should be described in order to identify and characterise potential hazards which might arise from degradation products. In particular data should be provided on:
the chemical/physicochemical stability of the source in its preparation and under the conditions of storage and effect of storage temperature, environment (light, oxygen, moisture, relative humidity (water activity)) or any other factor that might influence the stability of the source preparation.
The chemical/physicochemical stability of the source during storage of the processed food: e.g. effect of the nature of the food to which the substance is added, processing temperature, pH, water activity or any other factor. The nature and reactivity of any degradation products and nature of interaction/reaction of degradation products with food components.
The duration of the stability testing may depend on the type of the source and its proposed uses and should cover at least the end of the shelf‐life. Accelerated conditions (usually at higher temperature) may be used as an alternative to stability testing under normal conditions. If the source is used as an ingredient added to other foods, its stability in the processed foods should be investigated in real foods or in relevant model systems (e.g. effect of processing temperature, pH and other constituents in the processed foods).
2.2. Information on existing authorisations and evaluations
Information on existing authorisations and evaluations should be provided for the source and the nutrient. This should include details of the following:
the body which carried out the evaluation;
when the evaluation was undertaken;
details of the evaluation identifying the critical studies and the reference points (e.g. no‐observed‐adverse‐effect‐levels (NOAELs)/lowest‐observed‐adverse‐effect levels (LOAELs) and BMDL values) which are used to derive HBGV (e.g. ULs, ADIs) and the uncertainty factors used in this evaluation;
any uncertainties described.
2.3. Proposed uses and exposure assessment
Estimation of the intakes of the proposed source by the European population are needed for the risk characterisation. Intakes should be estimated based on the proposed use levels of the source and data on actual food consumption. Furthermore, a rationale for the target population, precautions and restrictions of use should be provided, with cross‐referencing to relevant safety data. Based on the estimated intakes of the source, the resulting intakes of the nutrient should be calculated. Where potential health hazards have been identified on the basis of the composition, toxicological or other data, they should be discussed and adequately addressed in the proposed conditions of use to ensure that the consumption of the source is safe for the target population. Information provided in this section should be as precise and complete as possible.
2.3.1. Target population
The applicant should specify the intended target population, e.g. adults, the general population or certain defined population subgroups. Similarly, it should be clearly specified if certain subgroups of the population are excluded from the intended uses (e.g. pregnant and lactating women, infants, etc.).
2.3.2. Proposed uses and use levels
This section should provide a justification for the use of the proposed source (not just a general justification for the nutrient), accompanied by information on the types of products in which the source is intended to be added/used. The information provided in this section will form the basis for the exposure assessment.
Sources intended for use in food supplements (Directive 2002/46/EC)
In the case of sources that are intended for use in food supplements, the anticipated daily intake of the source should be provided, alongside the corresponding anticipated daily intake of the nutrient:
e.g. the source is intended to be used in food supplements at a typical/maximum intake level of X mg source per day which corresponds to a recommended typical/maximum of Y mg of nutrient per day.
If different use levels are anticipated for different population subgroups, these should be specified in detail.
Sources intended for use in foods for specific groups (Regulation (EU) No 609/2013
In the case of sources that are intended for use in foods for specific subgroups of the population (i.e. infant and follow‐on formula, processed cereal‐based baby food and baby food, food for special medical purposes, total diet replacement for weight control), the anticipated daily intake of the source should be provided, alongside the corresponding anticipated daily intake of the nutrient:
e.g. the source is intended to be added to foods [specify food] product for specific groups [specify subgroup] at a typical/maximum level of X mg source per kg or L (of the food), which corresponds to a typical/maximum of Y mg of nutrient per kg or L (of the food).
The applicant should provide an indication of the proposed daily intake of the food product.
For nutrients added to foods for which regulatory limits define the maximum concentration of the nutrient:
e.g. the source in intended to be added to foods [specify food] at a typical/maximum level which corresponds to a maximum level not to exceed the regulatory limits established for these products as defined by Commission Delegated Regulation (EU) 2016/127.
Sources intended for use in foods (Regulation 1925/2006)
In the case of sources that are intended for use in foods (e.g. for fortification purposes), the applicant should specify in which type of products the source is intended to be incorporated, and the quantities added to these products. If applicable, typical and maximum use levels should be indicated.
e.g. the source is intended to be used in [specify food product] at a typical/maximum level of [X] mg source per kg or L, corresponding to typical/maximum level of [Y] mg nutrient per kg or L.
In order to support the calculation of the most refined possible exposure estimations, each food product or food category in which the source is intended to be used should be defined at the highest level of detail possible, using the FoodEx classification system (used for the EFSA comprehensive database) (EFSA, 2011).
2.3.3. Anticipated intake of the source and corresponding intake of the nutrient
On the basis of the information provided in Sections 2.31 and 2.3.2, estimations of anticipated daily intakes of the source are required (per kg body weight and in absolute amounts). Estimations of mean and high (at least 95th percentile) anticipated daily intakes of the source are needed for each target population group (including, if relevant, vulnerable groups such as children, pregnant and lactating women). The concurrent consumption of all food categories in which a source is proposed to be used should be addressed in the estimations, possibly considering different consumption scenarios. The highest estimated daily intake (i.e. at least the 95th percentile) among the population groups from a representative database (e.g. EFSA Comprehensive European Food Consumption Database) is recommended to be used as the starting point for the safety evaluation of the source. For the intake assessment on the basis of ‘per kg body weight’, the EFSA guidance on default values and rounding should be taken into account (EFSA Scientific Committee, 2012). Chronic intake estimates should be provided by default. In case the available data from toxicological or human data raise concerns regarding an acute effect, acute intake estimates should also be considered. The application should document the methodological aspects of the intake assessment; in particular:
the sources of data used (sources of food consumption data);
the scientific principles and methods applied;
the assumptions made and their rationale; in particular with respect to the assignment of a food to a particular food category or with respect to the model used for the calculation of high intake levels.
The Panel proposes a tiered approach where the first step makes use of the summary statistics of the EFSA Comprehensive Food consumption Database. Summary statistics of food consumption are available on the EFSA website in the form of spreadsheets, both for chronic and acute consumption. Detailed information on the database and guidance on its use have been published (EFSA, 2011). Anticipated daily intakes for mean and high‐percentile consumers can be calculated through the combination of the intended use level in each food category with mean and high chronic consumption values from the database, respectively.
The use of the EFSA Food Additive Intake Model (FAIM) tool (which is also based on summary statistics of the EFSA Comprehensive Food Consumption Database) may serve as an appropriate alternative for the first exposure estimate. The FAIM tool was developed to support the calculation of chronic exposure to food additives in the regulatory framework of food additives Regulation (EU) 1333/2008. Exposure assessment of food additives and intake assessment of sources added to food for the general population share common principles. Thus, the FAIM tool may be used by applicants for the intake assessment of sources incorporate to food for the general population, where the food categories to which the source is intended to be added, reasonably match with the food categories covered in the FAIM tool. It allows the applicant to estimate the mean and high‐level exposure to the sources for different population groups throughout several European countries by means of pre‐defined exposure calculation worksheets. For the calculation of high percentiles of daily intake, the model assumes that an individual might be a high‐level consumer of one food category only and would be an average consumer of all the remaining food groups. Thus, the FAIM tool adds the highest of the high‐levels of intake from one food category (calculated for consumers only) to the mean intake values for the remaining categories (calculated for the total population).
Summary statistics from the EFSA Comprehensive European Food Consumption Database (incl. FAIM tool) provide valuable estimates of intake. In some cases, such estimates provide sufficient information, if high intake estimates are below HBGV (e.g. acceptable or tolerable daily intake). In other cases, where more refined estimates are needed, the applicant should consider more detailed assessments, such as intake calculations based on individual data from national food consumption surveys (Tier 2).
The applicant should consider and discuss the uncertainties related to the assessment; in particular, sources of under‐ or over‐estimations. To this end, the guidance from the EFSA Scientific Committee related to uncertainties in dietary exposure assessment should be considered (EFSA, 2007a).
Once the intake has been estimated for the source, the resulting intake of the nutrient should be calculated. An example is provided in Appendix C.
For nutrients added to foods where regulatory limits define the maximum concentration of the nutrient, a reference to these regulatory limits can be provided as an estimated intake of the nutrient, with an indication that the addition of the source will not exceed these regulatory maximums for any nutrient.
2.3.4. Information on background exposure to the nutrient from food
The applicant should provide estimates of intake of the nutrient from the diet in relevant population groups (e.g. from published literature).
To this end it should be noted that EFSA NDA Panel opinions on DRVs contain estimates of the dietary intake of nutrients obtained from food consumption data available through the EFSA Comprehensive Food Consumption Database (EFSA, 2011).
2.4. Toxicological data
Toxicity studies should be carried out with the source meeting the specifications proposed in Appendix B and manufactured according to the production process described in Section 2.1.3. If the test material used in the toxicity studies does not meet the specifications proposed in Appendix B or is not manufactured according to the production process described in Section 2.1.3, a rationale should be provided to justify why the results can be used to assess the safety of the source.
It is generally considered that the tiered toxicity testing approach proposed for food additives and novel foods should be considered an appropriate approach also for new proposed sources. It integrates the core areas of kinetics, genotoxicity, repeated dose toxicity testing (subchronic, chronic toxicity and carcinogenicity) and reproductive and developmental toxicity (EFSA ANS Panel, 2012; EFSA NDA Panel, 2016). Additional studies may be needed to examine specific biological processes which may not be fully considered in the core areas for evaluation. Other studies that may be relevant include, e.g. immunotoxicity and food intolerance, studies on neurotoxicity, endocrine activity and mode of action.
In the case of sources, for example functional endpoints related to the purported function of the nutrient may be considered as supportive evidence of bioavailability (see Section 2.5).
Deviations from the tiered approach applicable to food additives and novel foods and/or its non‐applicability should be reasoned with sound scientific arguments.
For sources intended to be used in infants below 16 weeks of age, the toxicity testing required shall be aligned to the requirements set up in the EFSA Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age (EFSA Scientific Committee, 2017).
2.4.1. Tiered approach for toxicity testing
A decision tree to decide on the approach for toxicological testing of sources is presented in Figure 1.
Figure 1.
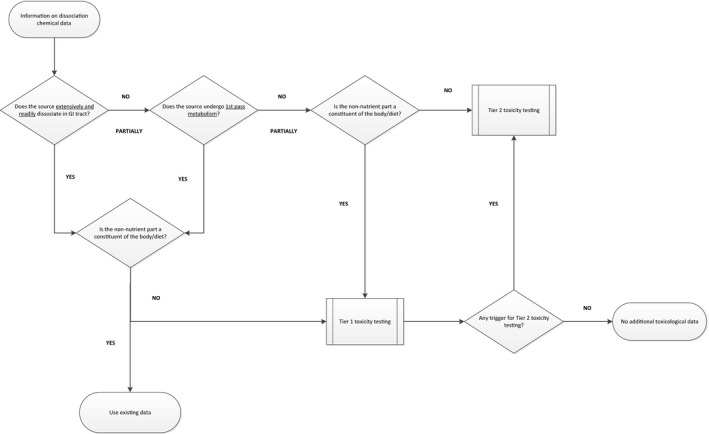
Decision tree for the toxicological testing of sources
If data from the dissociation tests demonstrate that the source is extensively and readily dissociated in the GI tract lumen, the safety assessment will take into consideration existing toxicological information on the resulting compounds.
If the non‐nutrient component(s) part of the source are not constituents of the diet and/or human body, the toxicological data requirements would be in line with the tiered approach devised for safety evaluations of food additives and novel foods (EFSA ANS Panel, 2012; EFSA NDA Panel, 2016). From these data, a toxicological reference point (RP) is established from which either a HBGV could be derived for comparison with the exposure estimate or a comparison could be performed of the exposure estimate with the RP (margin of exposure approach).
If data from the dissociation test demonstrate that the source is not extensively and readily dissociated in the GI tract lumen but is likely to be absorbed unchanged from the GI tract lumen, then the tiered approach to toxicological testing (EFSA ANS Panel, 2012; EFSA NDA Panel, 2016) would apply.
Hence, the minimum dataset to be provided for evaluation should comprise:
-
Basic test battery for genotoxicity testing:
-
–
A bacterial reverse mutation assay (OECD TG 471), and
-
–
An in vitro mammalian cell micronucleus test (OECD TG 487).
-
–
A modified 90‐day toxicity test (OECD TG 408 with extended parameters from the OECD TG 407).
The Panel noted that the revised TG 408 with extended parameters from the OECD TG 407 is in the process of being updated to add endocrine disruptor relevant endpoints.7
Tier 1 toxicity testing on the source would generally be required also in cases in which it can be proven that after absorption the source is undergoing presystemic metabolism in the GI walls or in the liver leading to substances that are normal constituent of the body and/or the diet.
If a source is absorbed unchanged and is not fully metabolised in the GI walls or presystemically, the ANS Panel Guidance on Food Additives evaluations would trigger Tier 2 toxicity testing.
Hence, in addition to the tests above:
A chronic toxicity (12 months) and carcinogenicity in a single species, generally the rat (either two separate studies according to OECD TGs 452 and 451 or the combined study according to OECD TG 453).
Reproductive and developmental toxicity testing comprising a prenatal developmental toxicity study (OECD TG 414) in the rabbit and an Extended One‐Generation Reproduction Toxicity Study (EORGTS) (OECD TG 443).
However, the need for this higher level of testing should be determined, in the case of sources, by the non‐nutrient component(s) generated. For components generated by first pass metabolism, which are normal constituents of either the diet or the body it is anticipated that Tier 1 testing of the source would generally be sufficient.
2.4.2. Additional studies
Decisions on further testing should be made on a case‐by‐case basis, following the 2012 ANS Panel Guidance on Food Additives evaluations (EFSA ANS Panel, 2012).
2.5. Bioavailability data
Sources of a nutrient are usually incorporated into products (tablet, capsule, etc.) or foods, and the matrix may affect the bioavailability of the nutrient. The Panel also acknowledged that there are a number of other modulating factors (e.g. nutritional status) that may influence bioavailability in the individual subject. In the context of this Guidance, the individual variability of bioavailability is not the focus. The Guidance aims to consider the relative bioavailability of the nutrient from the source compared under identical experimental conditions with the bioavailability of the nutrient in forms that are already permitted for use, i.e. are on the positive lists.
There is wide variation in the bioavailability of different nutrients, and also between different forms of the same nutrient. Therefore, the assessment of the bioavailability of a nutrient from its source should be performed on a case‐by‐case basis. Various approaches can be used for assessing the bioavailability of a nutrient (Figure 2).
Figure 2.
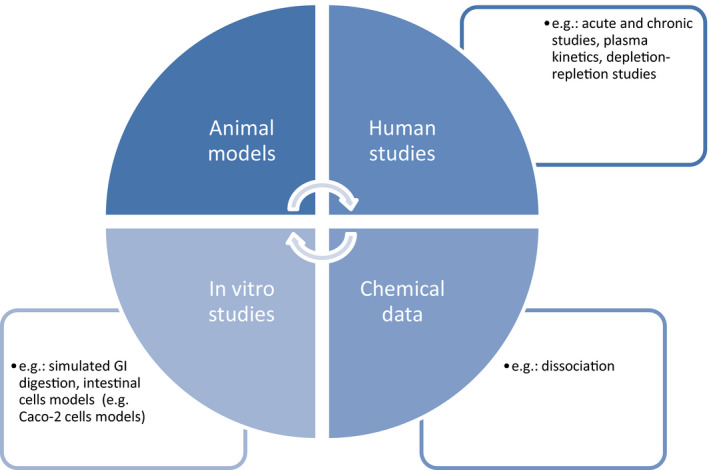
Illustrative approaches for assessing bioavailability of a nutrient
The Panel did not consider that these approaches should be followed according a predefined hierarchy of the evidence, rather the choice should be made by applicants after careful consideration and discussion of what is already known about the bioavailability of the nutrient under examination.
The testing strategy should be justified by the applicant based on elements such as the properties of the source (e.g. water soluble or lipophilic compounds; nutrients released in the GI tract or nutrient source absorbed intact) and the target population (particularly in the case of vulnerable population groups).
The applicant should give a rationale for the selection of data. There are, however, specific circumstances (e.g. when the proposed source would be the only source of the nutrient for the intended population) under which human data in the relevant population would be considered essential for the assessment.
Data from chemical dissociation tests may be considered sufficient, and no further testing for bioavailability required, when the nutrient from the proposed source can be assumed to be ‘available to be used by the body’ on the basis of such tests. Alternative testing strategies include more complex dissociation test (e.g. with a simulated intestinal digestion), in vitro absorption studies (e.g. intestinal cells models such as Caco‐2 cells models, or brush border membrane models) and in vivo studies. Results from toxicological testing (e.g. Tier 1 toxicity testing, see Section 2.4.1), if they include functional parameters related to the purported role of the nutrient, would not be sufficient for a quantitative assessment of bioavailability per se but can be considered as supportive evidence for assessing bioavailability.
The sections below provide an overview of experimental studies that could be used to generate data on bioavailability and criteria for assessing the results.
Experimental studies need to be designed on a case‐by‐case basis and, when relevant, may need to provide data comparing the proposed source with one or more sources of the same nutrient from the positive lists. In designing such studies, the applicant should provide a rationale for their design choices, e.g. sampling times, culture conditions, and should define the criteria for assessing ‘availability to be used by the body’. Bioavailability studies should be carried out with the source meeting the specifications proposed in Appendix B and manufactured according to the production process described in Section 2.1.3. If the test material used in the bioavailability study does not meet the specifications proposed in Appendix B or is not manufactured according to the production process described in Section 2.1.3, a rationale should be provided to justify why the results of the bioavailability study can be used to assess the bioavailability of the nutrient from the source.
2.5.1. Test methods and models
Chemical data
The aim of this approach is to generate data which can predict the fate of the source in the human body once it is ingested. This testing phase is, however, focussed on the initial phase of the digestive process and the tests should therefore be conducted under conditions which could mimic the process of human digestion, e.g. at a temperature of 37°C, using different buffers to simulate the different environments of the GI tract (preferably pH 2, 4 and 6.8). The time course for the dissociation of the source could be measured for a better understanding of the rate and the extent of the dissociation, and this could also be compared to an already established source of the same nutrient.
Although dissociation tests have been performed for decades and reported in publications and submissions, no validated, standardised methods are available which can be recommended to investigate dissociation under GI conditions. Certain elements contained in OECD Guidelines for the Testing of Chemicals, (Section 1 Physical–Chemical properties Test No. 111: Hydrolysis as a Function of pH) and in the European Pharmacopoeia (9th edition, 5.17.1 Recommendations on Dissolution testing) may be considered relevant also for the conduct of dissociation tests with a proposed source.
Sampling times should be determined on a case‐by‐case basis, but the rationale for this must be provided. Replicate measurements for each of the sampling time should be performed.
In vitro studies
Several in vitro systems simulating human GI digestion are described in the literature, which can assist in providing experimental evidence as whether, and to what extent, and over what time, the nutrient component can be released from a source. The simulated gastrointestinal digestion was described by Schricker et al. (1981) to investigate relative iron absorption from meals. Later, this method has been used by other authors to investigate the release of other components of nutritional interest from different food matrixes (Gil‐Izquierdo et al., 2002; Minekus et al., 2014). The most detailed description of the method is provided by Versantvoort et al. (2004, online).
This method can be expanded to incorporate a further step in which the in vitro dialysability is tested (Sandberg, 2005). The tests are also used to study interactions of meals with the nutrient.
Until now, however, there is no standardised method available (Marze, 2017).
In vitro systems with biological systems can be used to assess whether the nutrient source and/or the nutrient is entering the cells, imitating the cells lining the small intestine (Etcheverry et al., 2012). The Caco2‐cell system is a well‐established model for water soluble chemicals (Bessems et al., 2014). Under special culture conditions Caco‐2 cell monolayers are capable to express intestinal CYP isozymes, phase‐II enzymes and transporters. Details of the methods are described in the literature (Ferruzza et al., 2012a,b; Natoli et al., 2012; Brück et al., 2017). Validation of the method and a standard protocol are also described in the literature (Marino et al., 2005; Hubatsch et al., 2007). Other systems have been proposed, e.g. brush border membrane vesicles (Moghimipour et al., 2016), or human small intestinal epithelial cells (Takenaka et al., 2014).). The latter two systems, however, are not validated to a similar extent and standard protocols are not available.
The Caco‐2 cell‐system has some advantages over other in vitro systems as it is more convenient and has a high throughput and it is more accurate than isolated brush border membrane vesicles. Rates of absorption can be determined as apparent permeability coefficients (see for example: Hubatsch et al., 2007).
In vitro models may assist in the evaluation of the stability of the compound in the GI tract and can provide quantitative information which relates to the absorption of the nutrient from the source and can measure some components of bioavailability. Reviews of the available methods, their applicability to the investigation of nutrient bioavailability and their predictive values are available in the literature (Etcheverry et al., 2012).
New developments to improve the in vitro system mimicking in wider array of parameters the human gut have used stem cells and produced gastrointestinal organoids (Schweinlin et al., 2016) This system has been used to study the interaction of the gut wall with the microbiome and also the interaction of the gut wall with infectious agents (Hill and Spence, 2016; In et al., 2016). It is possible that the system might also be used as a tool to investigate gastrointestinal absorption.
Animal models
Although animal models are known to have a limited capability to predict bioavailability in humans, they can still provide useful insight for some nutrients (García and Díaz‐Castro, 2013; Musther et al., 2014). In a comparative approach, results obtained in experimental animals can provide useful data on the bioavailability of a new proposed source with respect to already established ones.
In the case of new toxicological data that are being generated in accordance with the tiered approach presented in this guidance, ideally the studies could be expanded to cover also toxicokinetics investigations.
Human studies
Human bioavailability studies have a long tradition and comparative designs can be used applying different protocols and techniques. In a comparative design, studies in humans have the highest predictive value, even if performed in a limited number of healthy subjects (EMA, 2010). Other study designs can also be considered such as studies with chronic dosing and depletion/repletion studies.
In mass balance studies, the net dietary balance, often referred to as retention, of the nutrient is determined from simultaneous measurements of intake and excretion (e.g. urinary and faecal) of a radio‐ or stable isotope of the nutrient.
Measurement of the concentration–time profile of the nutrient in plasma may also be used as a basis to assess bioavailability of a nutrient following single or repeated oral administration of a new proposed source. More details are provided in Appendix D.
For the assessment of nutrient bioavailability the selection of approach should be undertaken on a case by case basis.
3. Concluding remarks
In compiling the data in support of the safety of a source and bioavailability of the nutrient from the source, applicants should also seek to interpret the data and draw conclusions.
The significant findings of each toxicity study (both unpublished and published) should be highlighted, together with the method for the identification of the reference point, (BMDL values or the NOAEL), and any other relevant information. The reasons for disregarding any findings should be carefully explained. Where necessary, the conclusions should include an interpretation of the importance of the findings in terms of possible mechanisms underlying any effects observed, a discussion of whether these are relevant to humans and, if so, the possible importance of the extrapolation of such findings to humans.
In terms of demonstrating that the nutrient is ‘available to be used by the body’ from the proposed source, the applicant should seek to draw conclusions comparing the results obtained with the proposed source and a reference source. The conclusions should allow determination whether the bioavailability of the nutrient from the proposed source is equivalent, higher or lower than from a reference source. The implications of this classification for the safety of the source at the proposed uses and use levels, and with respect to relevant health‐based guidance values (e.g. ULs) should be clearly stated.
Glossary
- Dietary Reference Value
The complete set of reference values for nutrient intake comprising Population Reference Intakes (PRI), Average Requirements (AR), Adequate Intakes (AI), Lower Threshold Intakes (LTI) and Reference Intakes (RI). DRVs are typically used as a basis for reference values in food labelling and for establishing food‐based dietary guidelines
- Nutrient
For the purpose of this document, the term nutrient covers the substances listed in the ‘positive lists’ of the relevant legislation and any subsequent update. The status as of 13/3/2017 is shown in the table below:
| Nutrient | Directive 2002/46/EC(a) | Regulation (EC) No 1925/2006(a) | Regulation (EU) No 609/2013(a) | |||
|---|---|---|---|---|---|---|
| FS | FF | IF | PCBF | FSMP | TDR | |
| Vitamins | ||||||
| Vitamin A | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin D | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin E | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin K | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin B1 (thiamine) | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin B2 (riboflavin) | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Niacin | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Pantothenic acid | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin B6 | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Folic acid (folate) | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin B12 | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Biotin | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Vitamin C | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Minerals | ||||||
| Calcium | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Magnesium | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Iron | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Copper | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Iodine | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Zinc | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Manganese | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Sodium | ☑ | ☑ | ☑ | ☑ | ☑ | |
| Potassium | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| Selenium | ☑ | ☑ | ☑ | ☑ | ☑ | |
| Chromium | ☑ | ☑ | ☑ | ☑ | ||
| Molybdenum | ☑ | ☑ | ☑ | ☑ | ||
| Fluoride | ☑ | ☑ | ☑ | ☑ | ||
| Chloride | ☑ | ☑ | ||||
| Phosphorus | ☑ | ☑ | ||||
| Boron | ☑ | ☑ | ☑ | ☑ | ||
| Silicon | ☑ | |||||
| Other substances | ||||||
| Amino acids | ☑ | ☑ | ☑ | ☑ | ||
| Carnitine and taurine | ☑ | ☑ | ☑ | ☑ | ||
| Nucleotides | ☑ | ☑ | ☑ | |||
| Choline | ☑ | ☑ | ☑ | ☑ | ||
| Inositol | ☑ | ☑ | ☑ | ☑ | ||
FS: food supplements; FF: fortified foods; IF: infant formula and follow‐on formula; PCBF: processed cereal‐based food and baby food; FSMP: food for special medical purposes; TDR: total diet replacement for weight control.
☑ included in the Annexes of the legislation cited above.
(a): As amended.
- Source
For the purpose of this guidance, the terms ‘source’ identifies the chemical substances used as sources of the nutrients listed above
- Tolerable upper intake level
The maximum intake of substances in food, such as nutrients or contaminants, that can be consumed daily over a lifetime without adverse health effects
Abbreviations
- ADI
acceptable daily intake
- ANS
EFSA Panel on Food Additives and Nutrient Sources added to Food
- AUC
area under the curve
- BMDL10
benchmark dose level
- CI
confidence interval
- DRVs
Dietary Reference Values
- EORGTS
Extended One‐Generation Reproduction Toxicity Study
- FAIM
Food Additive Intake Model
- FEEDAP
EFSA Panel on Additives and Products or Substances used in Animal Feed
- FOF
follow‐on formula
- FSMP
Food for special medical purposes
- GI
gastrointestinal
- HBGV
health‐based guidance values
- IF
infant feed
- LOAEL
lowest‐observed‐adverse‐effect level
- MOE
margin of exposure
- NDA
EFSA Panel on Dietetic Products, Nutrition and Allergies
- NFI
novel food ingredients
- NOAEL
no‐observed‐adverse‐effect‐level
- PCBF
processed cereal‐based food and baby food
- QPS
Qualified Presumption of Safety
- RP
Reference point
- SCF
Scientific Committee on Food
- TDR
total diet replacement
- TTC
Threshold of Toxicological Concern
- UL
tolerable upper level
Appendix A – Information on identity of a substance
1.
| Category 1 | Single substances, chemically characterised; sources consisting of, isolated from or produced from material of inorganic mineral origin |
| 1.1 | Chemical name (i.e. unequivocal trivial name), and chemical name according to IUPAC nomenclature rules |
| 1.2 | CAS number, E Number (where appropriate), EC (or EINECS) number, and other identification numbers, as available from established scientific sources. |
| 1.3 | Synonyms, trade names, abbreviations |
| 1.4 | Molecular and structural formulae |
| 1.5 | Molecular weight (or atomic weight for elements) (g/mol, Da) |
| 1.6 | Particle size, shape and distribution, if applicable |
| 1.7 | Spectroscopic data (printout) such as IR, UV‐VIS, NMR or MS spectra or other data |
| 1.8 | Description of physical and chemical properties: appearance, melting point, boiling point, specific gravity, stereochemistry (if any) |
| 1.9 | Solubility (reference e.g. JECFA general method for solubility (JECFA, 2006)) in water and other common solvents |
| 1.10 | Influence of pH on solubility; ionisation constant(s) |
| 1.11 | Octanol‐to‐water partition ratio (KOW) |
| 1.12 | Other data that the applicant considers may be useful to support the identity of the substance |
| Category 2 | Mixtures of simple substances, chemically characterised |
| 2.1 | Chemical name, when appropriate, according to IUPAC nomenclature rules |
| 2.2 | Chemical composition: identity of the components of the mixture, as required for item 1 |
| 2.3 | CAS number, E Number (where appropriate), EC (or EINECS) number, and other identification numbers, as available from established scientific sources |
| 2.4 | Synonyms, trade names, abbreviations |
| 2.5 | Proportion of each component in the mixture |
| 2.6 | Molecular and structural formulae of each component in the mixture |
| 2.7 | Molecular weight (or atomic weight for elements) (g/mol, Da) of each component in the mixture |
| 2.8 | Spectroscopic and chromatographic data (printout of spectra/chromatogram) which allow the identification of the components of the mixture |
| 2.9 | Description of physical and chemical properties: appearance, stereochemistry, of each component (unless not applicable) |
| 2.10 | Solubility (reference e.g. JECFA general method for solubility (JECFA, 2006)) in water and other common solvents |
| 2.11 | Particle size, shape and distribution, if applicable |
| 2.12 | Other data that the applicant considers may be useful to support the identity of the substance |
| Category 3 | Complex mixtures not derived from botanical sources, possibly not fully chemically characterised (chemical characterisation extent depending on the proposed use and use levels) |
| 3.1 | Starting materials or source materials |
| 3.2 | Species, in case of animal origin |
| 3.3 | Chemical name, when appropriate, according to IUPAC nomenclature rules of each component |
| 3.4 | CAS number, E Number (where appropriate), EC (or EINECS) number, and other identification numbers, as available from established scientific sources of each component. A mixture should also be identified with appropriate identification number(s), if any available, from established scientific sources |
| 3.5 | Synonyms, trade names, abbreviations |
| 3.6 | Chemical description, the level of principal components in so far as these are known and level of unidentified components |
| 3.7 | Description of physical and chemical properties |
| 3.8 | Solubility (reference e.g. JECFA general method for solubility (JECFA, 2006)) in water and other common solvents |
| 3.9 | Particle size, shape and distribution, if applicable |
| 3.10 | Other data that the applicant considers may be useful to support the identity of the substance |
| Category 4 | Sources consisting of, isolated from, or produced from animals and parts thereof |
| 4.1 | Scientific (Latin) name (zoological family, genus, species, subspecies, breed, if applicable) |
| 4.2 | Synonyms that may be used interchangeably with the scientific name |
| 4.3 | Common names (if a trivial or common name is used extensively, it should be linked to the scientific name and part used) |
| 4.4 | Part used |
| 4.5 | Geographical origin (continent, country, region) |
| Category 5 | Sources of botanical origin |
| In addition to information listed in 1–3 and 5 | |
| 5.1 | Scientific (Latin) name (botanical family, genus, species, subspecies, variety with author's name, chemotype, if applicable) |
| 5.2 | Synonyms (botanical name) that may be used interchangeably with the preferred scientific name |
| 5.3 | Common names (if a trivial or a common name is used extensively in the monograph, it should be firmly linked to the scientific name and part used) |
| 5.4 | Part used (e.g. root, leaf, seed) |
| 5.5 | Geographical origin (continent, country, region) |
| 5.6 | Growth and harvesting conditions (wild or cultivated; cultivation practices, time of harvest in relation to both season and stage of the plant growth) |
| Furthermore, data on the chemical composition of the plant‐derived proposed source should be provided with emphasis on the concentrations of relevant constituents of relevance; this includes the concentrations of the following: | |
| 5.7 | Compounds classified according to their chemical structure (e.g. flavonoids, terpenoids, alkaloids) |
| 5.8 | Constituents being characteristic for the food additive (chemical fingerprint, markers) |
| 5.9 | Constituents that provide reasons for concern due to their chemical, pharmacological or toxicological properties |
| 5.10 | Information on maximum levels for microorganisms and possible contaminants, including heavy metals, mycotoxins, pesticide residues, and polycyclic aromatic hydrocarbons (PAHs), should be provided (EFSA Scientific Committee, 2009) |
| Category 6 | Sources consisting of, isolated from, or produced from cell culture or tissue culture derived from animals, plants, fungi or algae |
| 6.1 | Biological source (taxonomic information on family, genus, species, subspecies, variety) |
| 6.2 | Organ and tissue or part of the organism sourced |
| 6.3 | Laboratory or culture collection sourced |
| 6.4 | Information on the identity of cells |
| 6.5 | Cells or tissue substrate used as a Novel Food |
| 6.6 | Type of culture |
| Category 7 | Natural, derivatised and synthetic polymers |
| 7.1 | Chemical name (i.e. unequivocal trivial name) and chemical name according to IUPAC |
| 7.2 | CAS number, E Number (where appropriate), EC (or EINECS) number, and other identification numbers, as available from established scientific sources |
| 7.3 | Synonyms, trade names, abbreviations |
| 7.4 | Chemical and structural formula |
| 7.5 | Molecular weight (or atomic weight for elements) (g/mol, Da) or number average molecular weight and weight average molecular weight (if feasible) |
| 7.6 | Structural formulae of monomers and starting materials, other agents involved in the polymerisation |
| 7.7 | Degree of substitution, percentages of substituted groups (where appropriate) |
| 7.8 | Description of physical and chemical properties |
| 7.9 | Solubility (reference e.g. JECFA general method for solubility (JECFA, 2006)) in water and other common solvents |
| 7.10 | Particle size, shape and distribution, if applicable |
| 7.11 | Other data that the applicant considers may be useful to identify the mixture and its components |
| Category 8 | Sources containing microorganisms or derived from microorganism, fungi or algae |
| The following information is required for substances of microbial origin: | |
| 8.1 | The microbial origin of food additives produced by fermentation or cultivation, including:
|
| 8.2 | Whether the microorganism fulfils the requirements for a Qualified Presumption of Safety (QPS) (EFSA, 2007b and subsequent updates, e.g. EFSA BIOHAZ Panel, 2018). In such cases, no further data on the microorganism itself are required |
| 8.3 | Information on residual levels of toxins |
| 8.4 | Information on the production process |
| 8.5 | Information on the identity of residual intermediates or microbial metabolites in the final product |
| 8.6 | Other relevant information as recommended by applicable guidance documents (EFSA GMO Panel, 2011; EFSA FEEDAP Panel, 2018) |
| Category 9 | Nano‐sized materials, engineered and unintentionally produced |
| In addition to the information listed in 1–3 and 7, the information reported in Table 1 of the EFSA Guidance on Engineered Nanomaterials (ENMs) is required (EFSA Scientific Committee et al., 2018 ): | |
| 9.1 | Chemical composition, identity: information on chemical composition of the ENM including purity, nature of any impurities, coatings or surface moieties, encapsulating materials, processing chemicals, dispersing agents and/or other formulants (e.g. stabilisers) |
| 9.2 | Particle size (primary/secondary): information on primary particle size, size range, and number size distribution (indicating batch‐to‐batch variation, if any). The same information needed for secondary particles (e.g. agglomerates and aggregates), if present. Two methods to be used, one being electron microscopy |
| 9.3 | Physical form and morphology: the information should indicate whether the ENM is present in a particle‐, tube‐, rod‐shape, crystal or amorphous form, and whether it is in free particulate form or in an agglomerated/aggregated state as well as whether the preparation is in the form of a powder, solution, suspension or dispersion |
| 9.4 | Particle and mass concentration: information on concentration in terms of particle number, and particle mass per volume when in dispersion and per mass when as dry powder |
| 9.5 | Specific surface area: information on specific surface area of the ENM, essential for dry powders |
| 9.6 | Surface chemistry: information on ENM surface, including any chemical/biochemical modifications that could modify the surface reactivity, or add a new functionality |
| 9.7 | Surface charge: information on zeta potential of the ENM |
| 9.8 | Redox potential: conditions under which redox potential was measured need to be documented |
| 9.9 | Solubility and partition properties: information on solubility of the ENM in relevant solvents and their partitioning between aqueous and organic phase (e.g. as log KOW if appropriate) |
| 9.10 | pH: essential for liquid dispersions (e.g. aqueous suspensions) |
| 9.11 | Viscosity: information on viscosity of liquid dispersions |
| 9.12 | Density and pour density: information on density/porosity of unformulated ENM and pour density (essential for granular materials) |
| 9.13 | Dustiness: information on dustiness of powder products, such as spices, creamers, and soup powders |
| 9.14 | Chemical reactivity, catalytic activity: information on relevant chemical reactivity or catalytic activity of the ENM and of any surface coating of the ENM |
| 9.15 | Photocatalytic activity: information on photocatalytic activity of relevant materials used in food packaging, coatings and printing inks, and internal reactions |
Appendix B – Example of proposed specifications for sources
1.
| Name of the source | |
| Synonyms | |
| Definition | |
| EINECS | |
| Colour Index No | |
| Chemical names | |
| Chemical formula | |
| Molecular/Atomic weight/Weight average molecular weight | |
| Particle size of powder | |
| Assay | |
| Description | |
| Appearance of a solution | |
| Identification | |
| Spectrophotometry, spectrometry, chromatography, Infrared, X‐ray diffraction | |
| Density/specific gravity | XX (20°C) (25/25°C) |
| Refractive index | |
| Specific rotation | |
| pH | XX–XX (XX% aqueous solution) |
| Degree of hydrolysis/decomposition/properties during burning | |
| Precipitation reaction | |
| Colour reaction | |
| Melting range or point | XX–XX°C |
| Viscosity | |
| Solubility | |
| Boiling point | |
| Specific identification tests and parameters | |
| Congealing range | |
| Distillation range | |
| Drop point | |
| Isoelectric point | |
| Solidification point | |
| Sublimation point | |
| Vapour pressure | |
| Microscopic observation/examination | |
| Purity | |
| Loss on drying | |
| Loss on ignition | |
| Water or HCl insoluble matter | |
| Water content | |
| Conductivity | |
| Acid/Hydroxyl value | |
| Acidity/alkalinity | |
| Oil content | |
| Fat | |
| Protein | |
| Total sugars | |
| Starch | |
| Sodium chloride | |
| Ash | Not more than XX% (XXX°C) |
| Viscosity | Not less/more than XXX mPa.s |
| Wax | |
| Residual Solvents | Not more than XX mg/Kg |
| Residue on ignition | |
| Non‐volatile residue | |
| Organic volatile impurities | |
| Aldehydes | |
| Unsaponifiable matter | |
| Saponification value | |
| Ester value | |
| Iodine value | |
| Peroxide value/peroxides | |
| Oxidising/reducing substances | |
| Readily carbonisable substances | |
| Specific parameters for impurities | |
| Other specific parameters indicating the degree of purity | |
| Chlorinated compounds | |
| 3‐Monochloropropane‐1,2‐diol (3‐MCPD) | |
| Polycyclic aromatic hydrocarbons | |
| Organic compounds other than colouring matters | |
| Pentachlorophenol | |
| Epoxides | |
| Mercury | Not more than XX μg/Kg |
| Cadmium | Not more than XX μg/Kg |
| Arsenic | Not more than XX μg/Kg |
| Lead | Not more than XX μg/Kg |
| Aluminium/aluminium oxides | Not more than XX μg/Kg (expressed as Al) |
| Copper | |
| Nickel | |
| Antimony | |
| Chromium | |
| Selenium | |
| Fluorides | |
| Microbiological criteria | |
| Salmonella spp. | |
| Escherichia coli (coliforms) Staphylococcus aureus | |
| Yeasts and moulds | |
| Total bacterial count | |
| Total plate count | |
| Other safety or purity related microbiological criteria | |
Appendix C – Examples of exposure estimates to a nutrient from a source
1.
The following one is a theoretical example based on a source intended to be used in food (food fortification).
Exposure of the source from its proposed use levels (mg/kg food or L food) in the different food categories for which it is intended can be estimated based on consumption data from the EFSA Comprehensive European Food Consumption Database.
The terms ‘children’ and ‘the elderly’ correspond, respectively, to ‘other children’ and the merging of the categories ‘elderly’ and ‘very elderly’ in the EFSA guidance ‘Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment’ (EFSA, 2011).
The range (min–max) is reported. In brackets, the number of dietary surveys available in the database.
| Estimated exposure (mg/day) | Infants | Toddlers | Children | Adolescents | Adults | The elderly |
|---|---|---|---|---|---|---|
| (12 weeks–11 months) | (12–35 months) | (3–9 years) | (10–17 years) | (18–64 years) | (≥ 65 years) | |
| Exposure to source at the proposed use levels | ||||||
| Mean | 50–400 (6) | 200–3,000 (10) | 10–3,000 (18) | 10–5,000 (17) | 10–3,000 (17) | 50–2,000 (14) |
| High level consumers | 100‐2,000 (5) | 1,000–8,000 (8) | 20–9,000 (18) | 20–20,000 (17) | 300–15,000 (16) | 100–10,000 (14) |
The corresponding intake of the nutrient from the proposed source can be estimated based on the following information:
Because Source is comprised of [example: 10% (w/w)] nutrient, the corresponding estimated exposure to nutrient (expressed as mg/day) can be calculated.
The range (min–max) is reported. In brackets, the number of dietary surveys available in the database.
| Estimated exposure (mg/day) | Infants | Toddlers | Children | Adolescents | Adults | The elderly |
|---|---|---|---|---|---|---|
| (12 weeks–11 months) | (12–35 months) | (3–9 years) | (10–17 years) | (18–64 years) | (≥ 65 years) | |
| Corresponding nutrient intake from the proposed use of source in foods (from EFSA database) | ||||||
| Mean | 5–40 (6) | 20–300 (10) | 1–300 (18) | 1–500 (17) | 1–300 (17) | 5–200 (14) |
| High level consumers | 10–200 (5) | 100–800 (8) | 2–900 (18) | 2–2,000 (17) | 30–1,500 (16) | 10–1,000 (14) |
Appendix D – Minimum criteria for acceptability of data and interpretation of results from studies assessing bioavailability
General considerations
All the tests should be carried out with the material complying with the proposed specifications for the source.
An established source of the tested nutrient (i.e. one of the substances included in the positive lists of the relevant sectorial Legislation) should be used as a comparator. If that is the case, a justification should be provided for the choice of the comparator substance.
The analytical methods used must be well characterised, fully validated and documented. Performance data have to be provided to demonstrate the quality of the method including accuracy, precision, specificity, limit of detection, limit of quantitation, linearity and range. Within study, validation should be performed using quality control samples in each analytical run.
Chemical tests
For interpretation of the results, the following parameters will be considered:
| Expression of dissociation | Quantity (% tested material) | Time |
|---|---|---|
| Extensively and readily | At least 80% | Within 15 min |
| Extensively | At least 80% | Within 45 min |
| Readily | 50–80% | Within 15 min |
| Poorly | < 50% | Within 45 time |
The test should be performed in replicates. The results from the dissolution tests of the two sources should be compared. The following parameter will be considered to establish similarity between the two dissociation profiles:
If the two‐sided 90th confidence interval (CI) is fully contained in the range of 0.8–1.25, the test release of the nutrient from the new sources is considered equivalent to the reference source.
If the lower CI limit is below 0.8, the release of the nutrient from the new source is lower than the release of the nutrient from the reference source.
If the upper CI limit is higher than 1.25, the release of the nutrient from the new source is higher than the release of the nutrient from the reference source.
In vitro studies
The test should be performed in replicates. The resulting apparent permeability coefficients of the two sources should be compared.
The following parameter will be considered to establish similarity between the two permeability coefficients:
If the 90th two‐sided CI is fully contained in the range of 0.8–1.25, the rate of absorption of the nutrient from the new sources is considered equivalent to that of the reference source.
If the lower CI limit is below 0.8, the rate of absorption of the nutrient from the new source is lower than that of the nutrient from the reference source.
If the upper CI limit is higher than 1.25, the rate of absorption of the nutrient from the new source is higher than that of the nutrient from the reference source.
Animal models
No specific advice is given for animal studies.
Human studies
A study protocol is to be established beforehand. The study is to be performed respecting ethical principles for medical research involving humans (Declaration of Helsinki in the current version (2013)) and all regulatory requirements have to be followed. All previous relevant literature should be considered when designing the study.
Primary endpoint:
The primary endpoint is to be selected depending upon the nutrient under assessment and study designed accordingly.
If demonstration of equivalent bioavailability is based on the comparison of the plasma/blood concentration–time profiles of the concentration of the nutrient from the source and from the comparator, then the primary endpoint is the area under the curve (AUC 0‐t) covering at least 80% of AUC(0‐∞).
Selection of subjects: In order to reduce variability, it is advisable to perform the studies in healthy volunteers. Subjects should preferably be non‐smokers and without a history of alcohol or drug abuse. Testing conditions should be standardised for the new proposed source and the already established source used as a comparator. Therefore, temporal variability in biological parameters should be considered.
Study design: As a standard design the randomised, two‐period, two‐sequence single dose crossover design is recommended.
It might be necessary to perform the study after introducing steady‐state conditions. In this case, a parallel study design can be an appropriate option. In this case, the treatment groups should be comparable in all known variables important for the handling of the substance by the body (e.g. age, body weight, sex).
Number of subjects: The number of subjects in the study should be based on an appropriate sample size calculation.
Sampling (blood/plasma): The number of samples should be sufficient to adequately describe the concentration‐time profile. The sampling schedule should be of a duration long enough to provide AUC(from time 0 to the last measured sample) covering at least 80% of AUC (from time 0 to time ∞).
For endogenous substances, the sampling schedule should allow characterisation of the endogenous baseline profile for each subject in each period. Often, a baseline is determined from two to three samples taken before administration. In other cases, sampling at regular intervals throughout day(s) or weeks prior to administration may be necessary in order to account for fluctuations in the endogenous baseline due to circadian rhythms.
Interpretation of the results: The following decision rule will be considered to establish similarity of bioavailability. The assessment of similarity is based upon 90% confidence intervals for the ratio of the population geometric means (test/reference) for the parameters under consideration.
If the two‐sided 90th CI is fully contained in the range of 0.8–1.25, the test release of the nutrient from the new sources is considered similar to the reference source.
If the lower CI limit is below 0.8, the bioavailability of the nutrient from the new source is lower than that of the reference source.
If the upper CI limit is higher than 1.25, the bioavailability of the nutrient from the new source is higher than that of the reference source.
References
EDQM (European Directorate for the Quality of Medicines & HealthCare), 2016. European Pharmacopoeia 8.0. Chapter 5: General texts: 5.17.1 Recommendations on dissolution testing.
EMA (European Medicines Agency), 2010. Committee for Medicinal Products for Human Use (CHMP) Guideline on the investigation of bioequivalence. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
EMA (European Medicines Agency), 2011. Committee for Medicinal Products for Human Use (CHMP). Appendix IV of the Guideline on the Investigation on Bioequivalence (CPMP/EWP/QWP/1401/98 Rev.1): Presentation of Biopharmaceutical and Bioanalytical Data in Module 2.7.1. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/11/WC500117887.pdf
Suggested citation: EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS) , Younes M, Aggett P, Aguilar F, Crebelli R, Dusemund B, Filipicč M, Frutos MJ, Galtier P, Gundert‐Remy U, Kuhnle GG, Lambré C, Leblanc J‐C, Lillegaard IT, Moldeus P, Mortensen A, Oskarsson A, Stankovic I, Waalkens‐Berendsen I, Woutersen RA, Wright M, Di Domenico A, Fairweather‐Tait S, McArdle H, Smeraldi C and Gott D, 2018. Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources. EFSA Journal 2018;16(6):5294, 35 pp. 10.2903/j.efsa.2018.5294
Requestor: Panel on Food Additives and Nutrient Sources added to Food (ANS Panel)
Question number: EFSA‐Q‐2016‐00150
Panel members: Peter Aggett, Fernando Aguilar, Riccardo Crebelli, Birgit Dusemund, Metka Filipicč, Maria Jose Frutos, Pierre Galtier, David Gott, Ursula Gundert‐Remy, Gunter Georg Kuhnle, Claude Lambré, Jean‐Charles Leblanc, Inger Therese Lillegaard, Peter Moldeus, Alicja Mortensen, Agneta Oskarsson, Ivan Stankovic, Ine Waalkens‐Berendsen, Rudolf Antonius Woutersen, Matthew Wright and Maged Younes.
Acknowledgements: The Panel wishes to thank EFSA staff members: Agnes De Sesmaisons‐Lecarre and Andrea Germini and the NDA Panel for the support provided to this scientific output.
Adopted: 16 May 2018
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2018.EN-1439/full
This guidance document is applicable for applications submitted until 26 March 2021.
Notes
Directive 2002/46/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements OJ L 183, 12.7.2002, p. 51–57.
Regulation (EC) No 1925/2006 of the European Parliament and of the Council of 20 December 2006 on the addition of vitamins and minerals and ovf certain other substances to foods OJ L 404, 30.12.2006, p. 26–38.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009 OJ L 181, 29.6.2013, p. 35–56.
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. OJ L 31, 1.2.2002, p. 1–24.
Commission Implementing Regulation (EU) No 307/2012 of 11 April 2012 establishing implementing rules for the application of Article 8 of Regulation (EC) No 1925/2006 of the European Parliament and of the Council on the addition of vitamins and minerals and of certain other substances to foods. OJ L 102, 12.4.2012, p. 2–4.
Commission Delegated Regulation (EU) 2016/127 of 25 September 2015 supplementing Regulation (EU) No 609/2013 of the European Parliament and of the Council as regards the specific compositional and information requirements for infant formula and follow‐on formula and as regards requirements on information relating to infant and young child feeding. OJ L 25, 2.2.2016, p. 1–29.
References
- Bessems JG, Loizou G, Krishnan K, Clewell HJ III, Bernasconi C, Bois F, Coecke S, Collnot EM, Diembeck W, Farcal LR, Geraets L, Gundert‐Remy U, Kramer N, Küsters G, Leite SB, Pelkonen OR, der Schrö K, Testai E, Wilk‐Zasadna I and Zaldívar‐Comenges JM, 2014. PBTK modelling platforms and parameter estimation tools to enable animal‐free risk assessment: recommendations from a joint EPAA–EURL ECVAM ADME workshop. Regulatory Toxicology and Pharmacology, 68, 119–139. [DOI] [PubMed] [Google Scholar]
- Brück S, Strohmeier J, Busch D, Drozdzik M and Oswald S, 2017. Caco‐2 cells – expression, regulation and function of drug transporters compared with human jejunal tissue. Biopharmaceutics and Drug Disposition, 38, 115–126. 10.1002/bdd.2025 [DOI] [PubMed] [Google Scholar]
- EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards), Ricci A, Allende A, Bolton D, Chemaly M, Davies R, Girones R, Koutsoumanis K, Lindqvist R, Nørrung B, Robertson L, Ru G, Fernández Escámez PS, Sanaa M, Simmons M, Skandamis P, Snary E, Speybroeck N, Ter Kuile B, Threlfall J, Wahlström H, Cocconcelli PS, Peixe L, Maradona MP, Querol A, Suarez JE, Sundh I, Vlak J, Barizzone F, Correia S and Herman L, 2018. Statement on the update of the list of QPS‐recommended biological agents intentionally added to food or feed as notified to EFSA 7: suitability of taxonomic units notified to EFSA until September 2017. EFSA Journal 2018;16(1):5131, 43 pp. 10.2903/j.efsa.2018.5131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2005. Opinion of the Scientific Committee on a request from EFSA related to A Harmonised Approach for Risk Assessment of Substances Which are both Genotoxic and Carcinogenic. EFSA Journal 2005;3(10):282, 33 pp. 10.2903/j.efsa.2005.282 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2007a. Opinion of the Scientific Committee related to uncertainties in dietary exposure assessment. EFSA Journal 2007;5(1):438, 54 pp. 10.2903/j.efsa.2007.438 [DOI] [Google Scholar]
- EFSA (European Food safety Authority), 2007b. Opinion of the Scientific Committee on a request from EFSA on the introduction of a Qualified Presumption of Safety (QPS) approach for assessment of selected microorganisms referred to EFSA. EFSA Journal 2007;5(12):587, 16 pp. 10.2903/j.efsa.2007.587 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2011. Use of the EFSA Comprehensive European Food Consumption Database in Exposure Assessment. EFSA Journal 2011;9(3):2097, 34 pp. 10.2903/j.efsa.2011.2097 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), 2012. Guidance for submission for food additive evaluations. EFSA Journal 2012;10(7):2760, 60 pp. 10.2903/j.efsa.2012.2760 [DOI] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), Rychen G, Aquilina G, Azimonti G, Bampidis V, Bastos ML, Bories G, Chesson A, Cocconcelli PS, Flachowsky G, Gropp J, Kolar B, Kouba M, López‐Alonso M, López Puente S, Mantovani A, Mayo B, Ramos F, Saarela M, Villa RE, Wallace RJ, Wester P, Glandorf B, Herman L, Kärenlampi S, Aguilera J, Anguita M, Brozzi R and Galobart J, 2018. Guidance on the characterisation of microorganisms used as feed additives or as production organisms. EFSA Journal 2018;16(3):5206, 24 pp. 10.2903/j.efsa.2018.5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms), 2011. Scientific Opinion on Guidance on the risk assessment of genetically modified microorganisms and their products intended for food and feed use. EFSA Journal 2011;9(6):2193, 54 pp. 10.2903/j.efsa.2011.2193 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), Turck D, Bresson J‐L, Burlingame B, Dean T, Fairweather‐Tait S, Heinonen M, Hirsch‐Ernst KI, Mangelsdorf I, McArdle H, Naska A, Neuhäuser‐Berthold M, Nowicka G, Pentieva K, Sanz Y, Siani A, Sjödin A, Stern M, Tomé D, Vinceti M, Willatts P, Engel K‐H, Marchelli R, Pöting A, Poulsen M, Salminen S, Schlatter J, Arcella D, Gelbmann W, de Sesmaisons‐Lecarré A, Verhagen H and van Loveren H, 2016. Guidance on the preparation and presentation of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283. EFSA Journal 2016;14(11):4594, 24 pp. 10.2903/j.efsa.2016.4594 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2009. Guidance on safety assessment of botanicals and botanical preparations intended for use as ingredients in food supplements, on request of EFSA. EFSA Journal 2009;7(9):1249, 19 pp. 10.2093/j.efsa.2009.1249 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Scientific Opinion on the applicability of the Margin of Exposure approach for the safety assessment of impurities which are both genotoxic and carcinogenic in substances added to food/feed. EFSA Journal 2012;10(3):2578, 5 pp. 10.2903/j.efsa.2012.2578 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2014. Scientific Opinion on a Qualified Presumption of Safety (QPS) approach for the safety assessment of botanicals and botanical preparations. EFSA Journal 2014;12(3):3593, 38 pp. 10.2903/j.efsa.2014.3593 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen HK, More S, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Schlatter JR, Silano V, Solecki R, Turck D, Bresson J‐L, Dusemund B, Gundert‐Remy U, Kersting M, Lambré C, Penninks A, Tritscher A, Waalkens‐Berendsen I, Woutersen R, Arcella D, Court Marques D, Dorne J‐L, Kass GEN and Mortensen A, 2017. Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age. EFSA Journal 2017;15(5):4849, 58 pp. 10.2903/j.efsa.2017.4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen HK, More S, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Schlatter JR, Silano V, Solecki R, Turck D, Younes M, Chaudhry Q, Cubadda F, Gott D, Oomen A, Weigel S, Karamitrou M, Schoonjans A and Mortensen A, 2018. Guidance on risk assessment of the application of nanoscience and nanotechnologies in the food and feed chain: part 1, human and animal health. EFSA Journal 2018;16(7):5327, 114 pp. 10.5327/j.efsa.2018.5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMA (European Medicines Agency, formerly EMEA), 2010. Guideline on the investigation of bioequivalence. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
- Etcheverry P, Grusak MA and Fleige LE, 2012. Application of in vitro bioaccessibility and bioavailability methods for calcium, carotenoids, folate, iron, magnesium, polyphenols, zinc, and vitamins B6, B12, D, and E. Frontiers in Physiology, 3, 317. 10.3389/fphys.2012.00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Commission , online. ‘Administrative guidance on submissions for safety evaluation of substances added for specific nutritional purposes in the manufacture of foods’. Available online: http://ec.europa.eu/food/safety/docs/labelling_nutrition-vitamins_minerals-adm_guidance_safety_substances_en.pdf
- Ferruzza S, Rossi C, Scarino ML and Sambuy Y, 2012a. A protocol for differentiation of human intestinal Caco‐2 cells in asymmetric serum‐containing medium. Toxicology in Vitro 26, 1252–1255. 10.1016/j.tiv.2012.01.008 [DOI] [PubMed] [Google Scholar]
- Ferruzza S, Rossi C, Scarino ML and Sambuy Y, 2012b. A protocol for in situ enzyme assays to assess the differentiation of human intestinal Caco‐2 cells. Toxicology in Vitro 26, 1247–1251. 10.1016/j.tiv.2011.11.007 [DOI] [PubMed] [Google Scholar]
- García Y and Díaz‐Castro J, 2013. Advantages and disadvantages of the animal models v. in vitro studies in iron metabolism: a review. Animal, 7, 1651–1658. [DOI] [PubMed] [Google Scholar]
- Gil‐Izquierdo A, Pilar Zafrilla P and Tomás‐Barberán FA, 2002. An in vitro method to simulate phenolic compound release from the food matrix in the gastrointestinal tract. European Food Research and Technology 214, 155–159. [Google Scholar]
- Hill DR and Spence JR, 2016. Gastrointestinal organoids: understanding the molecular basis of the host‐microbe interface. Cell Mol Gastroenterol Hepatol, 3, 138–149. 10.1016/j.jcmgh.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubatsch I, Ragnarsson EG and Artursson P, 2007. Determination of drug permeability and prediction of drug absorption in Caco‐2 monolayers. Nature Protocols, 2, 2111–2119. [DOI] [PubMed] [Google Scholar]
- In JG, Foulke‐Abel J, Estes MK, Zachos NC, Kovbasnjuk O and Donowitz M, 2016. Human mini‐guts: new insights into intestinal physiology and host‐pathogen interactions. Nature Reviews Gastroenterology and Hepatology, 13, 633–642. 10.1038/nrgastro.2016.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2006. Combined Compendium of food additive specifications. Volume 4 of the Joint FAO/WHO Expert Committee on Food Additives. World Health Organization, Geneva. Available online: ftp://ftp.fao.org/docrep/fao/009/a0691e/a0691e00a.pdf
- Marino AM, Yarde M, Patel H, Chong S and Balimane PV, 2005. Validation of the 96 well Caco‐2 cell culture model for high throughput permeability assessment of discovery compounds. International Journal of Pharmaceutics 297, 235–241. [DOI] [PubMed] [Google Scholar]
- Marze S, 2017. Bioavailability of nutrients and micronutrients: advances in modeling and in vitro approaches. Annual Review of Food Science and Technology, 8, 35–55 (Volume publication date February 2017). 10.1146/annurev-food-030216-030055 [DOI] [PubMed] [Google Scholar]
- Minekus M, Alminger M, Alvito P, Ballance S, Bohn T, Bourlieu C, Carrière F, Boutrou R, Corredig M, Dupont D, Dufour C, Egger L, Golding M, Karakaya S, Kirkhus B, Le Feunteun S, Lesmes U, Macierzanka A, Mackie A, Marze S, McClements DJ, Ménard O, Recio I, Santos CN, Singh RP, Vegarud GE, Wickham MS, Weitschies W and Brodkorb A, 2014. A standardised static in vitro digestion method suitable for food ‐ an international consensus. Food Function 5, 1113–1124. 10.1039/c3fo60702j [DOI] [PubMed] [Google Scholar]
- Moghimipour E, Tabassi SAS, Ramezani M, Handali S and Löbenberg R, 2016. Brush border membrane vesicle and Caco‐2 cell line: two experimental models for evaluation of absorption enhancing effects of saponins, bile salts, and some synthetic surfactants. Journal of Advanced Pharmaceutical Technology and Reserach, 7, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musther H, Olivares‐Morales A, Hatley OJ, Liu B and Rostami Hodjegan A, 2014. Animal versus human oral drug bioavailability: do they correlate? European Journal of Pharmaceutical Sciences 57, 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli M, Leoni BD, D'Agnano I, Zucco F and Felsani A, 2012. Good Caco‐2 cell culture practices. Toxicology in Vitro, 26, 1243–1246. 10.1016/j.tiv.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Sandberg A‐S, 2005. Methods and options for in vitro dialyzability; benefits and limitations. International Journal for Vitamin and Nutrition Research, 75, 395–404. [DOI] [PubMed] [Google Scholar]
- SCF (Scientific Committee on Food), 2001a. Guidance on submissions for safety evaluation of sources of nutrients or of other ingredients proposed for use in the manufacture of foods. Opinion expressed on July 2001. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out100_en.pdf
- SCF (Scientific Committee on Food), 2001b. Guidance on submissions for food additives evaluations. Opinion expressed on July 2001. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out98_en.pdf
- Schricker BR, Miller DD, Rasmussen RR and Van Campen D, 1981. A comparison of in vivo and in vitro methods for determining availability of iron from meals. American Journal of Clinical Nutrition 34, 2257–2263. [DOI] [PubMed] [Google Scholar]
- Schweinlin M, Wilhelm S, Schwedhelm I, Hansmann J, Rietscher R, Jurowich C, Walles H1 and Metzger M, 2016. Development of an advanced primary human in vitro model of the small intestine. Tissue Engineering Part C Methods, 22, 873–883. 10.1089/ten.tec.2016.0101 [DOI] [PubMed] [Google Scholar]
- Takenaka T, Harada N, Kuze J, Chiba M, Iwao T and Matsunaga T, 2014. Human small intestinal epithelial cells differentiated from adult intestinal stem cells as a novel system for predicting oral drug absorption in humans. Drug Metabolism and Disposition, 42, 1947–1954. [DOI] [PubMed] [Google Scholar]
- Versantvoort CHM, Rompelberg CJM and Sips AJAM, 2004. Online report 630030 001. Available online: http://www.rivm.nl/dsresource?objectid=b25873b8-d1ae-4639-ac51-12eec0c5f543&type=org&disposition=inline