

Georgakis et al. report that genetic predisposition to elevated high-density lipoprotein cholesterol (HDL-C) is associated with reduced risk of small vessel stroke, and lower white matter hyperintensity volume. HDL-C-raising strategies could be considered for the prevention of ischaemic small vessel disease.
Keywords: lipids, small vessel disease, Mendelian randomization, high-density lipoprotein, lacunar stroke
Abstract
Blood lipids are causally involved in the pathogenesis of atherosclerosis, but their role in cerebral small vessel disease remains largely elusive. Here, we explored associations of genetic determinants of blood lipid levels, lipoprotein particle components, and targets for lipid-modifying drugs with small vessel disease phenotypes. We selected genetic instruments for blood levels of high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triglycerides, for cholesterol and triglycerides components of size-defined lipoprotein particles, and for lipid-modifying drug targets based on published genome-wide association studies (up to 617 303 individuals). Applying two-sample Mendelian randomization approaches we investigated associations with ischaemic and haemorrhagic manifestations of small vessel disease [small vessel stroke: 11 710 cases, 287 067 controls; white matter hyperintensities (WMH): 10 597 individuals; intracerebral haemorrhage: 1545 cases, 1481 controls]. We applied the inverse-variance weighted method and multivariable Mendelian randomization as our main analytical approaches. Genetic predisposition to higher HDL-C levels was associated with lower risk of small vessel stroke [odds ratio (OR) per standard deviation = 0.85, 95% confidence interval (CI) = 0.78–0.92] and lower WMH volume (β = –0.07, 95% CI = −0.12 to −0.02), which in multivariable Mendelian randomization remained stable after adjustments for LDL-C and triglycerides. In analyses of lipoprotein particle components by size, we found these effects to be specific for cholesterol concentration in medium-sized high-density lipoprotein, and not large or extra-large high-density lipoprotein particles. Association estimates for intracerebral haemorrhage were negatively correlated with those for small vessel stroke and WMH volume across all lipid traits and lipoprotein particle components. HDL-C raising genetic variants in the gene locus of the target of CETP inhibitors were associated with lower risk of small vessel stroke (OR: 0.82, 95% CI = 0.75–0.89) and lower WMH volume (β = −0.08, 95% CI = −0.13 to −0.02), but a higher risk of intracerebral haemorrhage (OR: 1.64, 95% CI = 1.26–2.13). Genetic predisposition to higher HDL-C, specifically to cholesterol in medium-sized high-density lipoprotein particles, is associated with both a lower risk of small vessel stroke and lower WMH volume. These analyses indicate that HDL-C raising strategies could be considered for the prevention of ischaemic small vessel disease but the net benefit of such an approach would need to be tested in a randomized controlled trial.
Introduction
Cerebral small vessel disease (SVD) accounts for ∼20% of all ischaemic strokes (Sudlow and Warlow, 1997) and most cases of intracerebral haemorrhage (ICH) (Qureshi et al., 2001, 2009). SVD is the leading cause of vascular dementia (O'Brien and Thomas, 2015; Iadecola et al., 2019) and an independent predictor of mortality (Debette et al., 2019; Georgakis et al., 2019). Manifestations of SVD on MRI are highly prevalent in the ageing population with figures reaching 90% for white matter hyperintensities (WMH) in patients aged 65 years and above (de Leeuw et al., 2001; Pantoni, 2010; Wardlaw et al., 2019). However, the mechanisms underlying SVD are poorly understood, thus impeding the development of effective strategies for prevention.
Blood lipids are a well-established risk factor for large artery atherosclerosis (Collins et al., 2016) and lipid-modifying therapies have shown benefits in reducing risk of both coronary artery disease and stroke [Cholesterol Treatment Trialists’ (CTT) Collaboration et al., 2010; Chou et al., 2016]. Yet, their role in SVD remains largely elusive. Current guidelines for secondary stroke prevention recommend treatment with statins after ischaemic stroke or transient ischaemic attack [European Stroke Organisation (ESO) Executive Committee and ESO Writing Committee, 2008; Kernan et al., 2014; Intercollegiate Stroke Working Party, 2016; Stroke Foundation, 2017) referring to clinical trials data and meta-analyses (Amarenco et al., 2006; Amarenco and Labreuche, 2009; Manktelow and Potter, 2009). However, most trials provided no sub-analyses for ischaemic stroke subtypes. The J-STARS trial, the only study providing sub-analyses, found statins to reduce recurrence of large artery stroke but not small vessel stroke (SVS) (Hosomi et al., 2015). Results from the SPARCL trial suggest that statins may increase the risk of ICH in patients with stroke or transient ischaemic attack (Amarenco et al., 2006), especially in patients with SVS as an entry event (Goldstein et al., 2008).
Mendelian randomization makes use of genetic variants that are associated with an exposure or risk factor as instruments, and investigates their associations with disease outcomes thus overcoming some of the key limitations of observational studies such as confounding and reverse causation (Hopewell and Clarke, 2016; Holmes et al., 2017). Hence, Mendelian randomization analyses can assess the causal relevance of a risk factor for disease and facilitate prioritization of interventions to be tested in clinical trials (Holmes et al., 2017; O'Donnell and Sabatine, 2018) as has specifically been demonstrated for lipid-modifying drugs (Khera and Kathiresan, 2017; Ference et al., 2018). In fact, there are several examples where Mendelian randomization studies have predicted the success or failure of clinical trials (Ference et al., 2015, 2016, 2017b, 2019b; Gill et al., 2019; Ray et al., 2019). The availability of large scale genome-wide association studies (GWAS) for an expanding range of phenotypes and the development of two-sample Mendelian randomization approaches enable the exploration of associations for which there is a paucity of evidence from clinical trials, as is the case for lipids and cerebral SVD.
Here, we leveraged data from the largest GWAS currently available on blood lipid levels (617 303 individuals) (Willer et al., 2013; Klarin et al., 2018) and on both ischaemic (SVS, WMH volume) and haemorrhagic (ICH) manifestations of cerebral SVD (Woo et al., 2014; Malik et al., 2018a; Rutten-Jacobs et al., 2018) with the aim to: (i) examine the effects of genetic determinants of blood levels of high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triglycerides on SVD manifestations; (ii) explore associations between genetic determinants of size-defined lipoprotein particle fractions with these phenotypes; and (iii) determine the effects of genetic predisposition to HDL-C raising, LDL-C lowering, and triglyceride lowering through variants in genes encoding targets of lipid-modifying drugs on SVD manifestations.
Materials and methods
This study follows the guidelines for strengthening the reporting of Mendelian randomization studies (STROBE-MR) (Davey Smith et al., 2019). We applied two-sample Mendelian randomization analyses, which allow selection of genetic variants as instruments for a risk factor (blood lipid traits) in one sample and explore associations of theses variants with outcomes (manifestations of SVD) in another sample (Davey Smith and Hemani, 2014; Burgess et al., 2015). By overcoming the requirement for assessing the exposure and outcome in the same dataset, this approach enables the exploration of associations in publicly available summary statistics from large GWASs with a corresponding increase in power. Also, two-sample Mendelian randomization is less prone to the winner’s curse bias than one-sample Mendelian randomization (Davey Smith and Hemani, 2014; Taylor et al., 2014).
Study design and data sources
The data sources used for this study are detailed in Supplementary Table 1. In Mendelian randomization analyses, we examined associations of blood lipid levels, size-defined lipoprotein particle fractions, and lipid-modifying drug targets, with ischaemic and haemorrhagic SVD phenotypes. We selected genetic instruments from the GWAS summary statistics of the Million Veteran Program (MVP) (Klarin et al., 2018), the Global Lipids Genetics Consortium (GLGC) (Willer et al., 2013), and from a GWAS on nuclear magnetic resonance (NMR) measured circulating metabolites (Kettunen et al., 2016). We then examined associations of the selected instruments with SVS in the GWAS summary statistics of the MEGASTROKE Consortium (Malik et al., 2018a), with WMH volume in a GWAS analysis that we undertook in the UK Biobank neuroimaging dataset (Alfaro-Almagro et al., 2018), and with ICH in the International Stroke Genetics Consortium (ISGC) GWAS meta-analysis (Woo et al., 2014).
Genetic instrument selection
Blood lipid levels
We selected genetic instruments for the blood levels of HDL-C, LDL-C, and triglycerides, based on the results of the GWAS multi-ethnic meta-analysis of the MVP and the GLGC samples (617 303 individuals) (Klarin et al., 2018). Specifically, we used independent genetic variants that reached genome-wide level of significance (P < 5 × 10−8) for their associations with HDL-C, LDL-C and triglycerides, in the conditional GWAS meta-analyses as instruments. We identified 312 instruments for HDL-C, 219 for LDL-C, and 253 for triglycerides (Supplementary Table 2). In our primary analyses, we weighted the instruments based on the joint regression coefficients from the conditional GWAS meta-analysis of MVP and GLGC. As the GLGC further excluded participants on lipid-lowering treatment (Willer et al., 2013), to exclude sources of biases related to treatment-mediated effects on blood lipids in the MVP dataset, we performed sensitivity analyses weighting the instruments using the GLGC effect sizes only. Both MVP and GLGC were imputed to the 1000 Genomes Project (Phase 3 and Phase 1, respectively) (1000 Genomes Project Consortium et al., 2012) and included adjustments for age, age2, sex, and population structure.
In a secondary approach, we restricted our selection of instruments to HDL-C-, LDL-C-, and triglyceride-specific variants. In particular, we used the GLGC dataset (188 577 individuals), for which we had access to the full GWAS summary statistics (Willer et al., 2013), and identified those independent genetic variants associated with HDL-C, LDL-C, or triglycerides at genome-wide significance (P < 5 × 10−8), but showed associations of P > 0.01 with the other two traits. We found 19 HLD-C-specific, 25 LDL-C-specific, and four triglyceride-specific variants (Supplementary Table 3) and performed sensitivity analyses using them as instruments.
Size-defined lipoprotein particle fractions
We then selected genetic instruments for cholesterol and triglyceride concentrations in size-defined lipoprotein particles available from a GWAS for NMR-measured circulating metabolites on 24 925 European individuals (Kettunen et al., 2016). The GWAS analyses were imputed to the 1000 Genomes Project (Phase 1) and adjusted for age, sex, time from last meal, and population structure (Kettunen et al., 2016). Based on summary statistics for each trait, we extracted variants after clumping for linkage disequilibrium (LD) at r2 < 0.1 that reached genome-wide significance (P < 5 × 10−8). The identified instruments for each metabolite are available in Supplementary Table 4.
Variants in genes encoding known lipid-modifying drug targets
Next, we selected variants clumped for linkage disequilibrium at r2 < 0.1 within a region of 100 kb upstream or downstream from genes encoding known drug targets that were associated with the respective lipid trait at a genome-wide significant level (P < 5 × 10−8) in the GLGC dataset (Willer et al., 2013). Specifically, we searched for genetic variants in the CETP locus (encoding the target of CETP inhibitors) associated with HDL-C levels; variants in the loci of HMGCR (target of statins), NPC1L1 (target of ezetimibe), PCSK9 (target of PCSK9 inhibitors), ABCG5 and ABCG8 (targets of bile acid resins), and LDLR (therapeutic target of the LDL receptor) associated with LDL-C levels; and variants in the PPARA locus (target for fibrates) associated with triglyceride levels, in accordance with similar approaches applied by other studies (Ference et al., 2012, 2015, 2017b, 2019a; Anderson et al., 2016; Harrison et al., 2018; Nowak and Arnlov, 2018). We identified 24 HDL-C raising variants in CETP, and for LDL-C lowering targets, four variants in HMGCR, three in NPC1L1, 11 in PCSK9, six in ABCG5/G8, and eight in LDLR (Supplementary Table 5). No triglyceride-lowering variants were identified in the PPARA locus based on our selection criteria for instruments.
For each genetic instrument, we estimated the proportion of variance explained for the respective phenotype and measured instrument strength with F-statistics (Supplementary Tables 2–5). F was >10 for all selected instruments, indicating a low probability for weak instrument bias (Palmer et al., 2012). Furthermore, we performed power calculations (Burgess, 2014) to identify the range of association estimates that we had >80% power (1 − β) to detect at α = 0.05 (Supplementary Table 6).
Associations with outcomes
The outcomes examined in this study were ischaemic and haemorrhagic manifestations of SVD including SVS, WMH volume, and ICH. Genetic association estimates for SVS—defined according to the TOAST (Trial of Org 10172 in Acute Stroke Treatment) criteria (Adams et al., 1993)—were obtained from the MEGASTROKE multi-ethnic GWAS meta-analysis (Malik et al., 2018a, b) on 11 710 cases and 287 067 controls. For WMH volume, we performed a GWAS analysis in the UK Biobank Imaging dataset (10 597 individuals of White-British ancestry), based on the measurements of WMH volume in T1 and T2 FLAIR MRI sequences, as previously described, following adjustments for age, sex, and the first 10 principal components (Rutten-Jacobs et al., 2018). We further examined ICH, as well as ICH subtypes defined according to haemorrhage location (deep and lobar). We used summary statistics from the ISGC GWAS meta-analysis including 1545 cases of spontaneous ICH defined by acute neurological onset and compatible neuroimaging showing intraparenchymal haemorrhage (664 lobar, 881 deep) and 1481 controls of European ancestry (Woo et al., 2014).
Statistical analysis
Main analyses
We applied two-sample Mendelian randomization analyses based on association estimates derived from the abovementioned sources. Following extraction of the association estimates between the instruments and the outcomes and harmonization of the direction of estimates by effect alleles, we computed Mendelian randomization estimates for each instrument with the Wald estimator and standard errors with the Delta method. All Mendelian randomization estimates were scaled to 1 − SD (standard deviation) increment in the lipid levels or the lipoprotein particle fractions. We then pooled individual Mendelian randomization estimates using random-effects inverse-variance weighted (IVW) meta-analyses. IVW is the most widely used main method for Mendelian randomization analysis because it provides robust causal estimates under absence of directional pleiotropy (Burgess et al., 2013).
Given the correlation between HDL-C, LDL-C, and triglyceride levels, and between cholesterol and triglyceride concentrations in specific size-defined lipoprotein particles, we further performed multivariable Mendelian randomization to disentangle their independent associations with SVD phenotypes (Burgess and Thompson, 2015). For HDL-C, LDL-C, and triglyceride blood levels, we used the respective instruments and adjusted for their effects on the other two traits from the GLGC dataset. For cholesterol concentration in HDL particles, we combined all unique variants associated with either total HDL-C levels or size-defined HDL cholesterol concentration and adjusted for their effects on blood LDL-C and triglyceride levels. Similarly, for cholesterol concentration in LDL and larger particles, we combined all variants associated with either total LDL-C levels or size-defined LDL and larger particle cholesterol concentrations and adjusted for their effects on HDL-C and triglyceride levels. Finally, we combined instruments for either total circulating triglyceride levels or for particle-specific triglyceride concentrations and adjusted for their effects on HDL-C and LDL-C.
For all analyses, we corrected for multiple comparisons with the false discovery rate (FDR) approach and set statistical significance at a q-value < 0.05. Associations not reaching this threshold, but showing a P < 0.05, were considered suggestive of an association.
Assessment of pleiotropy and sensitivity analyses
The IVW method was our primary Mendelian randomization analysis approach, but the derived estimates might be biased in case of directional pleiotropy. As a measure of pleiotropy, we assessed heterogeneity across the Mendelian randomization estimates for each instrument in the IVW Mendelian randomization analyses with the Cochran’s Q statistic (Bowden et al., 2018). Under presence of nominal heterogeneity (P from Cochran’s Q < 0.10) we further applied alternative Mendelian randomization methods, which are more robust to the use of pleiotropic instruments. These were the weighted median estimator and the Mendelian randomization (MR)-Egger regression. The weighted median estimator allows the use of invalid instruments under the assumption that at least half of the instruments used in the Mendelian randomization analysis are valid (Hartwig et al., 2017). The MR-Egger regression allows for the estimation of an intercept term, which can be used as an indicator of unbalanced directional pleiotropy (Bowden et al., 2015). MR-Egger provides less precise estimates and relies on the assumption that the strengths of potential pleiotropic instruments are independent of their direct associations with the outcome (Bowden et al., 2015). The intercept obtained from MR-Egger regression was used as a measure of directional pleiotropy (P < 0.05 indicated statistical significance) (Bowden et al., 2015).
In case of evidence of directional pleiotropy (as assessed by both the Cochran’s Q statistic and the intercept in the MR-Egger regression) and inconsistent results between the different approaches, we further applied the generalized summary data-based Mendelian randomization (GSMR) approach. This method uses all variants reaching genome-wide significance as instruments by accounting for linkage disequilibrium correlation between them and further identifies and eliminates outliers that exert apparent pleiotropic effects on both the risk factor and the outcome using the HEIDI-outlier method (Zhu et al., 2018). GSMR further provides a measure of remaining global heterogeneity following exclusion of outliers that also takes into account the low linkage disequilibrium across the used instruments.
All analyses were performed in R (v3.5.0; The R Foundation for Statistical Computing) using the MendelianRandomization and the gsmr packages.
Data availability
The data used for the current study are publicly available and may also become available from the corresponding author on reasonable request.
Results
Genetic determinants of blood lipid levels and ischaemic small vessel disease
The primary results of the IVW Mendelian randomization analyses for the associations between genetic determinants of blood lipid levels and SVS and WMH volume are presented in Fig. 1. Genetic predisposition to elevated HDL-C levels were associated with both a lower risk of SVS [odds ratio (OR): 0.85, 95% CI: 0.78–0.92, P = 5 × 10−4] and lower WMH volume (β: −0.07, 95% CI: −0.12 to −0.02, P = 0.004). We further found genetic predisposition to higher triglyceride levels to be associated with higher risk of SVS and a suggestive association between genetic predisposition to higher LDL-C levels and SVS risk. In multivariable Mendelian randomization, the associations between genetic determinants of HDL-C levels and SVS and WMH volume remained stable and statistically significant (Fig. 1). In contrast, the association between genetic determinants of triglyceride levels and SVS was attenuated when adjusting for HDL-C and LDL-C.
Figure 1.

Mendelian randomization associations of genetic determinants of blood lipid levels (HDL-C, LDL-C, triglycerides) with risk of small vessel stroke and WMH volume. Shown are the results derived from random-effects IVW (inverse-variance weighted) Mendelian randomization and multivariable Mendelian randomization (MVMR) analyses adjusting for the effects of the genetic variants on all the three blood lipid traits. SD = standard deviation; TG = triglycerides.
The Mendelian randomization results were stable when weighting the genetic instruments for the three lipid traits based on their association estimates in the GLGC dataset, which excluded individuals on lipid-modifying treatment (Supplementary Figs 1 and 2). In Mendelian randomization analyses restricted to the instruments specifically associated with HDL-C, LDL-C, or triglycerides, the association estimates of genetic determinants of HDL-C for both risk of SVS (OR: 0.78, 95% CI: 0.62–0.98) and WMH volume (β: −0.27, 95% CI: −0.45 to −0.08) were even stronger (Supplementary Fig. 3). GSMR-HEIDI, which identifies and excludes pleiotropic outlier variants, also showed significant associations between genetic predisposition to higher HDL-C and both lower SVS risk and lower WMH volume (Supplementary Figs 1 and 2).
Genetic determinants of size-defined lipoprotein particle fractions and ischaemic small vessel disease
To obtain a deeper understanding of the observed associations, we next selected genetic instruments for cholesterol and triglyceride concentrations in size-defined lipoprotein particles and examined their associations with SVS and WMH volume (Fig. 2 and Supplementary Table 7). We found genetic predisposition to higher cholesterol concentration in the medium-sized, but not in the large- or extra-large sized HDL particles, to be associated with both lower SVS risk (OR: 0.84, 95% CI: 0.73–0.96, P = 0.007) and lower WMH volume (β: −0.09, 95% CI: −0.16 to −0.02, P = 0.009). There was no heterogeneity and the associations remained significant when adjusting for the effects of the instruments on circulating LDL-C and triglyceride levels (Fig. 2 and Supplementary Table 8).
Figure 2.

Mendelian randomization associations of genetic determinants of cholesterol (C) and triglyceride (TG) concentrations in size-defined lipoprotein particles with risk of small vessel stroke and WMH volume. Shown are the results derived from random-effects inverse-variance weighted (IVW) Mendelian randomization and multivariable Mendelian randomization (MVMR) analyses. MVMR for cholesterol in HDL particles adjusted for LDL-C and triglycerides; for cholesterol in LDL and larger particles adjusted for HDL-C and triglycerides; and for triglycerides in any particles adjusted for HDL-C and LDL-C. IDL = intermediate density lipoprotein; L = large; M = medium; S = small; VLDL = very low density lipoprotein; XL = extra-large.
Because of evidence for heterogeneity (Cochran’s Q P < 0.10) and inconsistent results for the associations of genetic determinants of total HDL-C with SVS risk and WMH volume across sensitivity analyses (weighted median and MR-Egger) (Fig. 3 and Supplementary Figs 1 and 2), we next restricted the set of instruments for total HDL-C to those associated with medium-sized HDL-C (P < 5 × 10−8). These analyses revealed stronger association estimates between genetic predisposition to higher HDL-C and both lower risk of SVS (OR: 0.69, 95% CI: 0.56–0.84, P = 4 × 10−4) and lower WMH volume (β: −0.23, 95% CI: −0.35 to −0.10, P = 2 × 10−4) (Fig. 3). Moreover, the estimates were highly consistent in alternative Mendelian randomization approaches with no evidence for heterogeneity, thus suggesting that heterogeneity in the overall analyses was driven by non-medium sized HDL-C increasing variants.
Figure 3.

Mendelian randomization (MR) associations of genetic determinants of HDL-C with risk of small vessel stroke and WMH volume. Shown are the results from random-effects inverse-variance weighted (IVW), weighted median and MR-Egger analyses when (A) using the full set of genetic instruments and (B) restricting the analyses to instruments also associated with cholesterol concentration in medium-sized HDL.
To explore whether the observed associations were specific to genetic predisposition to higher cholesterol concentration in the medium-sized HDL, we next expanded our analyses to other components of the HDL particles (Supplementary Fig. 4). In this post hoc analysis, we found similar association estimates between genetic determinants of the concentration of any of the medium-sized HDL particle components (total cholesterol, cholesterol-esters, free cholesterol, total lipids, and phospholipids) and SVS risk as well as WMH volume suggesting that the associations are driven by the medium-sized HDL particles as a whole.
Regarding other lipoprotein particle components, we further found genetic predisposition to higher concentration of triglycerides in the small-sized HDL particles to be associated with higher risk of SVS (Fig. 2 and Supplementary Tables 7–9).
Genetic variants in loci of lipid-modifying drug targets and ischaemic small vessel disease
We next selected genetic variants in genes encoding known HDL-C-raising or LDL-C-lowering drug targets and examined their associations with ischaemic SVD phenotypes. HDL-C-raising variants in the CETP locus were associated with lower risk of SVS (OR: 0.82, 95% CI = 0.75–0.89, P = 9 × 10−6) and lower WMH volume (β = −0.08, 95% CI = −0.13 to −0.02, P = 0.008) (Figs 4, 5A and B). While there was heterogeneity in the association between CETP variants and SVS (P = 0.03), the results remained significant in the weighted median and MR-Egger approaches (Supplementary Table 10). As previous analyses from the REVEAL trial (HPS3/TIMI55–REVEAL Collaborative Group et al., 2017) had shown the beneficial effects of cholesteryl-ester transfer protein (CETP) inhibitors on vascular disease to be mainly driven by their LDL-C lowering and not their HDL-C raising capacity, we further explored the associations between genetic predisposition to LDL-C lowering through CETP variants and ischaemic SVD manifestations. While genetic predisposition to LDL-C lowering was associated with lower risk of SVS and lower WMH volume in univariable IVW Mendelian randomization analyses, these effects were entirely reversed after adjusting for the HDL-C raising effects of the variants in multivariable Mendelian randomization (Supplementary Table 11). Analyses for genetic variants in LDL-C lowering drug target loci showed no statistically significant results (Fig. 4).
Figure 4.

Mendelian randomization associations of HDL-C-raising and LDL-C-lowering genetic variants in the loci of known lipid-modifying drug targets with risk of SVS and WMH volume. Shown are the results derived from random-effects inverse-variance weighted (IVW) Mendelian randomization analyses. The results are scaled per 1 SD increment in circulating HDL-C levels (HDL-C-raising drug targets) and per 1 SD increment in circulating LDL-C levels (LDL-C-lowering drug targets).
Genetic associations of lipid traits with intracerebral haemorrhage
IVW-Mendelian randomization analyses showed no significant associations of genetic determinants of HDL-C, LDL-C, and triglycerides with risk of ICH (Fig. 6). When examining lipoprotein particle fractions, we found associations of the opposite direction, as compared to both SVS and WMH volume (Fig. 7). However, confidence intervals were wide, likely due to lack of statistical power (Supplementary Fig. 5 and Supplementary Tables 6 and 12). Across drug target loci (Supplementary Fig. 6) we found HDL-C raising variants in the CETP locus to be associated with a higher risk of ICH (OR: 1.64, 95% CI: 1.26–2.13, P = 2.6 × 10−4) (Fig. 5C). This effect was significant for both deep (OR: 2.01, 95% CI: 1.27–3.18, P = 0.003) and lobar ICH (OR: 1.78, 95% CI: 1.06–2.89, P = 0.028) (Supplementary Fig. 7).
Figure 6.
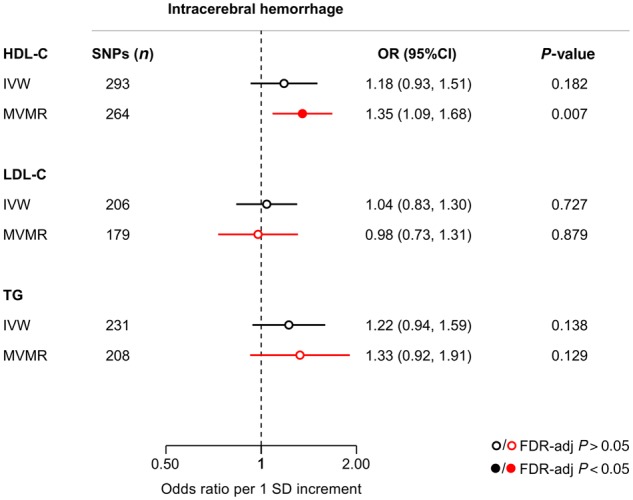
Mendelian randomization associations of genetic determinants of blood lipid levels (HDL-C, LDL-C, triglycerides) with risk of intracerebral haemorrhage. Shown are the results derived from random-effects inverse-variance weighted (IVW) Mendelian randomization and multivariable Mendelian randomization (MVMR) analyses. MVMR for cholesterol in HDL particles adjusted for LDL-C and triglycerides; for cholesterol in LDL and larger particles adjusted for HDL-C and triglycerides; and for triglycerides in any particles adjusted for HDL-C and LDL-C. TG = triglycerides.
Figure 7.

Comparisons of association estimates for genetic determinants of lipid traits (blood lipid levels and concentrations of lipoprotein particle components) between SVS, WMH volume, and ICH. Comparisons of the Mendelian randomization association estimates between genetic determinants of lipid traits and risk of SVS with the Mendelian randomization association estimates for WMH volume and risk of ICH. Shown are the meta-regression slopes for the comparisons of these association estimates for: (A) risk of SVS and WMH volume, (B) risk of SVS and risk of ICH, and (C) WMH volume and risk of ICH. Estimates are scaled per 1 SD increment.
Figure 5.

Mendelian randomization associations between HDL-C raising genetic variants in the CETP locus and (A) risk of SVS, (B) WMH volume, and (C) risk of ICH. Shown are the results from the random-effects inverse-variance weighted (IVW) Mendelian randomization approach. The results are scaled per 1 SD increment in circulating HDL-C levels.
Discussion
The main findings from this study can be summarized as follows: (i) we found significant associations between genetic predisposition to higher HDL-C levels and both lower risk of SVS and lower WMH volume; (ii) associations were specific for cholesterol concentrations in the medium and not large or extra-large sized HDL particles; (iii) exploring genetic variants at loci for targets of lipid-modifying drugs, we found HDL-C raising variants in CETP to be associated with a lower SVS risk and lower WMH volume; and (iv) we found these HDL-C raising variants in CETP to be associated with a higher risk of ICH, with consistent results for both lobar and deep ICH.
Our Mendelian randomization results provide evidence for a protective role of HDL-C on ischaemic SVD. This agrees with findings from two small observational studies. In the Women’s Healthy Ageing Project, midlife HDL-C levels among 135 females were inversely associated with WMH volume after 20 years, independently of other vascular risk factors (Aljondi et al., 2018). Similarly, in a cross-sectional study of 817 participants aged ≥50 years, higher HDL-C levels were associated with lower volumes of both deep and periventricular WMH after adjusting for vascular risk factors (Yin et al., 2018). The mechanisms underlying the observed inverse association between HDL-C levels and ischaemic SVD are unknown but may involve protective effects on the vascular endothelium (Sorrentino et al., 2010; Prosser et al., 2012; Tran-Dinh et al., 2013; Monette et al., 2016). Endothelial cells, including those of the brain microvasculature (Lapergue et al., 2010; Fung et al., 2017), express receptors, which upon HDL binding, induce intracellular signalling eventually leading to vasodilatory (Yuhanna et al., 2001; Spieker et al., 2002; Nofer et al., 2004), anti-inflammatory (Cockerill et al., 1995; Nicholls et al., 2005; Murphy et al., 2008), antioxidative (Garner et al., 1998; Lee et al., 2005; Terasaka et al., 2007), and anti-thrombotic effects (Viswambharan et al., 2004; Calkin et al., 2009).
Our findings contrast with Mendelian randomization analyses on atherosclerotic phenotypes supporting no association of genetic determinants of HDL-C levels with coronary artery disease (Voight et al., 2012; Holmes et al., 2015; White et al., 2016) and large artery stroke (Hindy et al., 2018) thus suggesting differential effects of HDL-C on cerebral SVD and large artery atherosclerosis. A disparity in the effect of lipid levels between small and large vessel pathologies has also been reported for LDL-C: previous Mendelian randomization studies found strong effects of genetic predisposition to higher LDL-C on the risk of coronary artery disease, large artery stroke, and peripheral artery disease (Holmes et al., 2015; Hindy et al., 2018; Valdes-Marquez et al., 2019; Emanuelsson et al., 2019), but no effect on risk of retinopathy and neuropathy, which are typically related to small vessel pathology (Emanuelsson et al., 2019). Future studies should explore potentially distinct mechanisms through which blood lipids influence the risk of small versus large vessel disease.
Analysing size-defined lipoprotein particle subfractions we found that the protective effects of HDL-C on ischaemic SVD are specific for medium-sized, and not larger HDL particles. In additional analyses, this effect seemed to be not specific to a particular component of the HDL particles but rather uniform across the different components, thus suggesting that medium-sized HDL particles as a whole could underlie this observation. HDL comprises a heterogeneous pool of lipoprotein particles (Kontush and Chapman, 2010) and the few observational studies that have performed analyses stratified by particle size indeed found differential effects on vascular outcomes (Martin et al., 2015; Wurtz et al., 2015; Joshi et al., 2016; Holmes et al., 2018). There are technical challenges related to different methods of HDL subfractioning (Superko et al., 2012), making it challenging to compare our results with those from previous studies. Still, our results agree with the general notion that the favourable effects observed for HDL are predominantly exerted by the smaller and denser HDL particles (Yu et al., 2003; Williams, 2012; Martin et al., 2014). Of note, previous Mendelian randomization studies on blood lipids that showed no significant associations between HDL-C levels and atherosclerotic phenotypes did not consider particle subfractions (Holmes et al., 2015; White et al., 2016; Hindy et al., 2018). Conceivably, disregarding subfractions might result in masking causal effects of potential biological relevance. As such, our findings highlight the importance of sub-analyses stratifying by lipoprotein particle size, but the complexity of the potential underlying mechanisms necessitates further study of our observations.
Importantly, we found HDL-C raising genetic variants in the CETP locus to also associate with lower SVS risk and WMH volume. Pharmacological CETP inhibition leads to an increase in the circulating pool of HDL particles (Armitage et al., 2019). While initial randomized trials investigating CETP inhibitors on top of statins found no benefit of CETP inhibition on vascular risk (Barter et al., 2007; Schwartz et al., 2012; Lincoff et al., 2017), the most recent REVEAL trial showed a reduced risk for major coronary events (HPS3/TIMI55–REVEAL Collaborative Group et al., 2017). In light of the relatively small effect (relative risk reduction in REVEAL: 9%) it seems unlikely that CETP inhibitors will achieve approval for prevention of cardiovascular disease (Hegele, 2017; Badimon, 2018). However, none of these trials explicitly reported effects on risk of SVS or other SVD manifestations. Our Mendelian randomization results suggest that post hoc analyses should consider stratifying for stroke subtypes, and that HDL-C raising approaches might show promise as a strategy for lowering the burden of ischaemic SVD.
The exact mechanism by which CETP inhibition might reduce risk of SVS and WMH volume is poorly understood. In the REVEAL trial, the reduction in vascular risk by CETP inhibition was mediated by a reduction in LDL-C rather than an increase in HDL-C (HPS3/TIMI55–REVEAL Collaborative Group et al., 2017). In our analyses, most of the HDL-C raising CETP variants also showed strong associations with lower LDL-C levels. Yet, in multivariable Mendelian randomization analyses adjusting for the effects of the variants on both HDL-C and LDL-C, we found only the effects of genetic predisposition to higher HDL-C through these CETP variants to remain consistent in terms of magnitude and directionality. Thus, although we were not sufficiently powered to entirely disentangle the effects of the two traits, our results suggest that in contrast with the REVEAL trial results, the effects of CETP variants on SVD manifestations might be primarily exerted by HDL-C raising. Administration of CETP inhibitors increases HDL particle size (Brousseau et al., 2004) and genetic predisposition to higher CETP concentration is associated with increased concentrations of medium- and large-sized, but not smaller HDL particles (Blauw et al., 2019). However, whether the expected effects of CETP inhibition on SVS and WMH volume are mediated through increases in the pool of specific HDL subparticles would need to be explored in future studies.
Previous observational and genetic studies found high HDL-C and low LDL-C levels to be associated with a higher risk of ICH (Wang et al., 2013; Anderson et al., 2016; Sun et al., 2019). Also, clinical trials have shown that LDL-C lowering with statins might increase risk for ICH (Amarenco et al., 2006; Goldstein et al., 2008). We found HDL-C raising variants in the CETP locus to be associated with a higher risk of both deep and lobar ICH, which relate to different vascular pathologies. Specifically, deep ICH has been associated with hypertensive SVD, whereas lobar ICH is typically related to cerebral amyloid angiopathy (Martini et al., 2012). While speculative, low serum LDL-C and high HDL-C levels may be associated with a fragile vascular endothelium, eventually leading to vessel permeability and a higher susceptibility to rupture (Konishi et al., 1993).
The main analytical approaches used in the current study, IVW and multivariable Mendelian randomization, are sensitive to directional pleiotropy (Lawlor et al., 2008; Burgess and Thompson, 2015). Specifically, if the single nucleotide polymorphisms used as genetic instruments for blood lipid levels associate with manifestations of SVD through pathways independent of blood lipid levels, the results could be biased. To ameliorate this risk, we performed a series of sensitivity analyses, which are based on statistical models that are more robust to pleiotropy, are focused on a subset of genetic instruments that are more specifically associated with the blood lipid traits under study, or excluded outlier single nucleotide polymorphisms with out of average effects on SVD manifestations, which are more likely to exert pleiotropic effects. Importantly, our results for an association between genetic determinants of HDL-C levels with risk of SVS and WMH volume were robust across these sensitivity analyses, thus supporting the results of the main analyses.
Our study has several strengths. The use of large genetic datasets enabled us to explore associations with a range of phenotypes, covering key manifestations of cerebral SVD. Also, the use of GWAS data for NMR-derived measurements enabled analyses stratified for lipoprotein particle subfractions. We further performed multiple tests for the detection of unbalanced pleiotropy and used multiple sensitivity analyses including advanced approaches such as GSMR-HEIDI. These analyses showed consistent results, thus minimizing the possibility of bias in the Mendelian randomization analyses. Finally, we explored the effects of HDL-C raising or LDL-C lowering genetic variants in genes encoding known lipid-modifying drug targets; this approach has previously been validated with the Mendelian randomization effects being comparable to those derived from randomized controlled trials.
Our study also has limitations. First, Mendelian randomization examines the lifetime effect of genetic determinants of blood lipid levels, which might differ from the effects of clinical lipid-modifying interventions. Second, we were not sufficiently powered to identify significant associations for ICH, and especially for ICH subtypes. Similarly, the non-significant, but still suggestive associations between LDL-C levels and SVS risk should be tested in larger datasets offering greater statistical power. Third, we had no access to the full summary statistics from the meta-analysis of the MVP and the GLGC studies. Hence, some analyses were restricted to the smaller GLGC dataset. Fourth, we are not aware of any sufficiently powered GWAS on cerebral microbleeds that would more accurately capture the spectrum of haemorrhagic SVD pathology than the currently used phenotype of ICH. While SVD is an important cause of ICH as a severe clinical manifestation, SVD more frequently manifests with subclinical cerebral microbleeds. Future GWAS on cerebral microbleeds will facilitate Mendelian randomization analyses on the relationship with blood lipids. Finally, we could not identify valid triglyceride-lowering variants in the locus of the target for fibrates. Hence, we could not explore their associations with SVD phenotypes. Future studies leveraging even larger GWAS datasets on blood lipid levels might identify genetic instruments for the full range of lipid-modifying drug classes.
In conclusion, our results suggest causal associations between higher HDL-C levels and both a lower risk of SVS and lower WMH volume, which were driven by cholesterol concentrations in medium-sized, and not larger HDL particles. HDL-C raising strategies might be of benefit for the prevention of ischaemic SVD. Considering the predicted increase in risk of ICH, the net benefit of such an approach would need to be tested in a randomized controlled trial.
Supplementary Material
Acknowledgements
This research has been conducted using the UK Biobank Resource (UK Biobank application 2532). We acknowledge the contributions by the MEGASTROKE Consortium, the ISGC, the GLGC, the MVP, as well as by Kettunen et al. (2016) for performing the original GWASs and for making summary statistics data publicly available.
Funding
M.K.G. has been supported by scholarships from the Onassis Foundation and the German Academic Exchange Service (DAAD). C.D.A. received grants from the National Institutes of Health and the National Institute of Neurological Disorders and Stroke (Grant numbers: R01NS103924, K23NS086873). This project has received funding from the European Union’s Horizon 2020 research and innovation programme (No 666881), Small vessel diseases in a mechanistic perspective: Targets for Intervention (to M.D.) and No 667375, Common mechanisms and pathways in stroke and Alzheimer's disease (to M.D.); the German Research Foundation (Deutsche Forschungsgemeinschaft) as part of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy ID 390857198) and the Collaborative Research Center 1123 (B3) (to M.D.); the Corona Foundation (to M.D.); the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain) (to M.D.); the e: Med program (Systems medicine of myocardial infarction and stroke) (to M.D.) and the FP7/2007-2103 European Union project Exploitation of Genomic Variants Affecting Coronary Artery Disease and Stroke Risk for Therapeutic Intervention (grant agreement number Health-F2-2013-601456) (to M.D.).
Competing interests
C.D.A. receives sponsored research support from the National Institutes of Health of the United States, the American Heart Association, Massachusetts General Hospital, and Bayer AG, and has consulted for ApoPharma, Inc. J.C.H. receives personal fellowship support from the British Heart Foundation [FS/14/55/30806]. J.C.H. works in the Clinical Trial Service Unit & Epidemiological Studies Unit of the Nuffield Department of Population Health at the University of Oxford, which has received research grants from Abbott, AstraZeneca, Bayer, Boehringer Ingelheim, GlaxoSmithKline, The Medicines Company, Merck, Mylan, Novartis, Pfizer, Roche, Schering, and Solvay, which are governed by University of Oxford contracts that protect their independence. In line with the Clinical Trial Service Unit & Epidemiological Studies Unit staff policy, J.C.H. does not take any personal payments directly or indirectly from industry (with reimbursement sought only for the costs of travel and accommodation to attend scientific meetings) for clinical trial involvement. M.K.G., R.M., K.G.P., and M.D. have no competing interests to declare.
Glossary
Abbreviations
- GLGC
Global Lipids Genetics Consortium
- GWAS
genome-wide association study
- HDL-C
high-density lipoprotein cholesterol
- ICH
intracerebral haemorrhage
- LDL-C
low-density lipoprotein cholesterol
- SVD
small vessel disease
- SVS
small vessel stroke
- WMH
white matter hyperintensities
References
- 1000 Genomes Project ConsortiumAbecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams HP Jr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in acute stroke treatment. Stroke 1993; 24: 35–41. [DOI] [PubMed] [Google Scholar]
- Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JLR, Griffanti L, Douaud G, et al. Image processing and quality control for the first 10,000 brain imaging datasets from UK Biobank. Neuroimage 2018; 166: 400–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljondi R, Szoeke C, Steward C, Gorelik A, Desmond P. The effect of midlife cardiovascular risk factors on white matter hyperintensity volume and cognition two decades later in normal ageing women. Brain Imaging Behav 2018. Advance Access published on September 26, 2018. doi: 10.1007/s11682-018-9970-5. [DOI] [PubMed] [Google Scholar]
- Amarenco P, Bogousslavsky J, Callahan A 3rd, Goldstein LB, Hennerici M, Rudolph AE, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 2006; 355: 549–59. [DOI] [PubMed] [Google Scholar]
- Amarenco P, Labreuche J. Lipid management in the prevention of stroke: review and updated meta-analysis of statins for stroke prevention. Lancet Neurol 2009; 8: 453–63. [DOI] [PubMed] [Google Scholar]
- Anderson CD, Falcone GJ, Phuah CL, Radmanesh F, Brouwers HB, Battey TW, et al. Genetic variants in CETP increase risk of intracerebral hemorrhage. Ann Neurol 2016; 80: 730–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage J, Holmes MV, Preiss D. Cholesteryl ester transfer protein inhibition for preventing cardiovascular events: jACC review topic of the week. J Am Coll Cardiol 2019; 73: 477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon L. CETP inhibition and HDL: what is the trial REVEALing? Cardiovasc Res 2018; 114: e15–16. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357: 2109–22. [DOI] [PubMed] [Google Scholar]
- Blauw LL, Noordam R, Soidinsalo S, Blauw CA, Li-Gao R, de Mutsert R, et al. Mendelian randomization reveals unexpected effects of CETP on the lipoprotein profile. Eur J Hum Genet 2019; 27: 422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015; 44: 512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J, Hemani G, Davey Smith G. Invited commentary: detecting individual and global horizontal pleiotropy in mendelian randomization-a job for the humble heterogeneity statistic? Am J Epidemiol 2018; 187: 2681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brousseau ME, Schaefer EJ, Wolfe ML, Bloedon LT, Digenio AG, Clark RW, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med 2004; 350: 1505–15. [DOI] [PubMed] [Google Scholar]
- Burgess S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol 2014; 43: 922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013; 37: 658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, Consortium E-I. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 2015; 30: 543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 2015; 181: 251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, Drew BG, Ono A, Duffy SJ, Gordon MV, Schoenwaelder SM, et al. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation 2009; 120: 2095–104. [DOI] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376: 1670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults: evidence report and systematic review for the US Preventive Services Task Force. JAMA 2016; 316: 2008–24. [DOI] [PubMed] [Google Scholar]
- Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol 1995; 15: 1987–94. [DOI] [PubMed] [Google Scholar]
- Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016; 388: 2532–61. [DOI] [PubMed] [Google Scholar]
- Davey Smith G, Davies NM, Dimou N, Egger M, Gallo V, Golub R, et al. STROBE-MR: guidelines for strengthening the reporting of Mendelian randomization studies. PeerJ Preprints 2019; 7: e27857v1. [Google Scholar]
- Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014; 23: R89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw FE, de Groot JC, Achten E, Oudkerk M, Ramos LM, Heijboer R, et al. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry 2001; 70: 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debette S, Schilling S, Duperron MG, Larsson SC, Markus HS. Clinical significance of magnetic resonance imaging markers of vascular brain injury: a systematic review and meta-analysis. JAMA Neurol 2019; 76: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson F, Nordestgaard BG, Tybjaerg-Hansen A, Benn M. Impact of LDL cholesterol on microvascular versus macrovascular disease: a Mendelian randomization study. J Am Coll Cardiol 2019; 74: 1465–76. [DOI] [PubMed] [Google Scholar]
- European Stroke Organisation (ESO) Executive Committee; ESO Writing Committee. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc Dis 2008; 25: 457–507. [DOI] [PubMed] [Google Scholar]
- Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017a; 38: 2459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Graham I, Tokgozoglu L, Catapano AL. Impact of lipids on cardiovascular health: jACC health promotion series. J Am Coll Cardiol 2018; 72: 1141–56. [DOI] [PubMed] [Google Scholar]
- Ference BA, Kastelein JJP, Ginsberg HN, Chapman MJ, Nicholls SJ, Ray KK, et al. Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. JAMA 2017b; 318: 947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, et al. Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA 2019a; 321: 364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 x 2 factorial Mendelian randomization study. J Am Coll Cardiol 2015; 65: 1552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Ray KK, Catapano AL, Ference TB, Burgess S, Neff DR, et al. Mendelian randomization study of ACLY and cardiovascular disease. N Engl J Med 2019b; 380: 1033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 2016; 375: 2144–53. [DOI] [PubMed] [Google Scholar]
- Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol 2012; 60: 2631–9. [DOI] [PubMed] [Google Scholar]
- Fung KY, Wang C, Nyegaard S, Heit B, Fairn GD, Lee WL. SR-BI mediated transcytosis of HDL in brain microvascular endothelial cells is independent of caveolin, clathrin, and PDZK1. Front Physiol 2017; 8: 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner B, Waldeck AR, Witting PK, Rye KA, Stocker R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem 1998; 273: 6088–95. [DOI] [PubMed] [Google Scholar]
- Georgakis MK, Duering M, Wardlaw JM, Dichgans M. WMH and long-term outcomes in ischemic stroke: a systematic review and meta-analysis. Neurology 2019; 92: e1298–308. [DOI] [PubMed] [Google Scholar]
- Gill D, Georgakis MK, Koskeridis F, Feng Q, Wei W-Q, Theodoratou E, et al. Use of genetic variants related to antihypertensive drugs to inform on efficacy and side effects. Circulation 2019; 140: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LB, Amarenco P, Szarek M, Callahan A 3rd, Hennerici M, Sillesen H, et al. Hemorrhagic stroke in the stroke prevention by aggressive reduction in cholesterol levels study. Neurology 2008; 70: 2364–70. [DOI] [PubMed] [Google Scholar]
- Harrison SC, Holmes MV, Burgess S, Asselbergs FW, Jones GT, Baas AF, et al. Genetic association of lipids and lipid drug targets with abdominal aortic aneurysm: a meta-analysis. JAMA Cardiol 2018; 3: 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 2017; 46: 1985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele RA. CETP inhibitors-a new inning? N Engl J Med 2017; 377: 1284–5. [DOI] [PubMed] [Google Scholar]
- Hindy G, Engstrom G, Larsson SC, Traylor M, Markus HS, Melander O, et al. Role of blood lipids in the development of ischemic stroke and its subtypes: a Mendelian randomization study. Stroke 2018; 49: 820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol 2017; 14: 577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J 2015; 36: 539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Millwood IY, Kartsonaki C, Hill MR, Bennett DA, Boxall R, et al. Lipids, lipoproteins, and metabolites and risk of myocardial infarction and stroke. J Am Coll Cardiol 2018; 71: 620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopewell JC, Clarke R. Emerging risk factors for stroke: what have we learned from mendelian randomization studies? J Cereb Circ 2016; 47: 1673–8. [DOI] [PubMed] [Google Scholar]
- Hosomi N, Nagai Y, Kohriyama T, Ohtsuki T, Aoki S, Nezu T, et al. The Japan Statin treatment against recurrent stroke (J-STARS): a multicenter, randomized, open-label, parallel-group study. EBioMedicine 2015; 2: 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HPS3/TIMI55–REVEAL Collaborative GroupBowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med 2017; 377: 1217–27. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Duering M, Hachinski V, Joutel A, Pendlebury ST, Schneider J, et al. Vascular cognitive impairment and dementia: JACC scientific expert panel. J Am Coll Cardiol 2019; 73: 3326–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intercollegiate Stroke Working Party. National clinical guideline for stroke. 5th edn. London: Royal College of Physicians; 2016.
- Joshi PH, Toth PP, Lirette ST, Griswold ME, Massaro JM, Martin SS, et al. Association of high-density lipoprotein subclasses and incident coronary heart disease: the Jackson Heart and Framingham Offspring Cohort Studies. Eur J Prev Cardiol 2016; 23: 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014; 45: 2160–236. [DOI] [PubMed] [Google Scholar]
- Kettunen J, Demirkan A, Wurtz P, Draisma HH, Haller T, Rawal R, et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat Commun 2016; 7: 11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet 2017; 18: 331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, et al. Genetics of blood lipids among ∼300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 2018; 50: 1514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi M, Iso H, Komachi Y, Iida M, Shimamoto T, Jacobs DR Jr, et al. Associations of serum total cholesterol, different types of stroke, and stenosis distribution of cerebral arteries. The Akita Pathology Study. Stroke 1993; 24: 954–64. [DOI] [PubMed] [Google Scholar]
- Kontush A, Chapman MJ. Antiatherogenic function of HDL particle subpopulations: focus on antioxidative activities. Curr Opin Lipidol 2010; 21: 312–8. [DOI] [PubMed] [Google Scholar]
- Lapergue B, Moreno JA, Dang BQ, Coutard M, Delbosc S, Raphaeli G, et al. Protective effect of high-density lipoprotein-based therapy in a model of embolic stroke. Stroke 2010; 41: 1536–42. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008; 27: 1133–63. [DOI] [PubMed] [Google Scholar]
- Lee CM, Chien CT, Chang PY, Hsieh MY, Jui HY, Liau CS, et al. High-density lipoprotein antagonizes oxidized low-density lipoprotein by suppressing oxygen free-radical formation and preserving nitric oxide bioactivity. Atherosclerosis 2005; 183: 251–8. [DOI] [PubMed] [Google Scholar]
- Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med 2017; 376: 1933–42. [DOI] [PubMed] [Google Scholar]
- Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 2018a; 50: 524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Rannikmae K, Traylor M, Georgakis MK, Sargurupremraj M, Markus HS, et al. Genome-wide meta-analysis identifies 3 novel loci associated with stroke. Ann Neurol 2018b; 84: 934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manktelow BN, Potter JF. Interventions in the management of serum lipids for preventing stroke recurrence. Cochrane Database Syst Rev 2009; 3: CD002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS, Jones SR, Toth PP. High-density lipoprotein subfractions: current views and clinical practice applications. Trends Endocrinol Metab 2014; 25: 329–36. [DOI] [PubMed] [Google Scholar]
- Martin SS, Khokhar AA, May HT, Kulkarni KR, Blaha MJ, Joshi PH, et al. HDL cholesterol subclasses, myocardial infarction, and mortality in secondary prevention: the Lipoprotein Investigators Collaborative. Eur Heart J 2015; 36: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini SR, Flaherty ML, Brown WM, Haverbusch M, Comeau ME, Sauerbeck LR, et al. Risk factors for intracerebral hemorrhage differ according to hemorrhage location. Neurology 2012; 79: 2275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monette JS, Hutchins PM, Ronsein GE, Wimberger J, Irwin AD, Tang C, et al. Patients with coronary endothelial dysfunction have impaired cholesterol efflux capacity and reduced HDL particle concentration. Circ Res 2016; 119: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, et al. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol 2008; 28: 2071–7. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye KA, et al. Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation 2005; 111: 1543–50. [DOI] [PubMed] [Google Scholar]
- Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck Lipinski K, Baba HA, et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest 2004; 113: 569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak C, Arnlov J. A Mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat Commun 2018; 9: 3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JT, Thomas A. Vascular dementia. Lancet 2015; 386: 1698–706. [DOI] [PubMed] [Google Scholar]
- O'Donnell CJ, Sabatine MS. Opportunities and challenges in Mendelian randomization studies to guide trial design. JAMA Cardiol 2018; 3: 967. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res 2012; 21: 223–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- Prosser HC, Ng MK, Bursill CA. The role of cholesterol efflux in mechanisms of endothelial protection by HDL. Curr Opin Lipidol 2012; 23: 182–9. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet 2009; 373: 1632–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med 2001; 344: 1450–60. [DOI] [PubMed] [Google Scholar]
- Ray KK, Bays HE, Catapano AL, Lalwani ND, Bloedon LT, Sterling LR, et al. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N Engl J Med 2019; 380: 1022–32. [DOI] [PubMed] [Google Scholar]
- Rutten-Jacobs LCA, Tozer DJ, Duering M, Malik R, Dichgans M, Markus HS, et al. Genetic study of white matter integrity in UK biobank (N=8448) and the overlap with stroke, depression, and dementia. Stroke 2018; 49: 1340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 2012; 367: 2089–99. [DOI] [PubMed] [Google Scholar]
- Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010; 121: 110–22. [DOI] [PubMed] [Google Scholar]
- Spieker LE, Sudano I, Hurlimann D, Lerch PG, Lang MG, Binggeli C, et al. High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation 2002; 105: 1399–402. [DOI] [PubMed] [Google Scholar]
- Stroke Foundation. Clinical guidelines for stroke management. Melbourne: Stroke Foundation; 2017.
- Sudlow CL, Warlow CP. Comparable studies of the incidence of stroke and its pathological types: results from an International Collaboration. International Stroke Incidence Collaboration. Stroke 1997; 28: 491–9. [DOI] [PubMed] [Google Scholar]
- Sun L, Clarke R, Bennett D, Guo Y, Walters RG, Hill M, et al. Causal associations of blood lipids with risk of ischemic stroke and intracerebral hemorrhage in Chinese adults. Nat Med 2019; 25: 569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Superko HR, Pendyala L, Williams PT, Momary KM, King SB 3rd, Garrett BC. High-density lipoprotein subclasses and their relationship to cardiovascular disease. J Clin Lipidol 2012; 6: 496–523. [DOI] [PubMed] [Google Scholar]
- Taylor AE, Davies NM, Ware JJ, VanderWeele T, Smith GD, Munafo MR. Mendelian randomization in health research: using appropriate genetic variants and avoiding biased estimates. Econ Hum Biol 2014; 13: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaka N, Wang N, Yvan-Charvet L, Tall AR. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc Natl Acad Sci U S A 2007; 104: 15093–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran-Dinh A, Diallo D, Delbosc S, Varela-Perez LM, Dang QB, Lapergue B, et al. HDL and endothelial protection. Br J Pharmacol 2013; 169: 493–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes-Marquez E, Parish S, Clarke R, Stari T, Worrall BB, Hopewell JC. Relative effects of LDL-C on ischemic stroke and coronary disease: a Mendelian randomization study. Neurology 2019; 92: e1176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswambharan H, Ming XF, Zhu S, Hubsch A, Lerch P, Vergeres G, et al. Reconstituted high-density lipoprotein inhibits thrombin-induced endothelial tissue factor expression through inhibition of RhoA and stimulation of phosphatidylinositol 3-kinase but not Akt/endothelial nitric oxide synthase. Circ Res 2004; 94: 918–25. [DOI] [PubMed] [Google Scholar]
- Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012; 380: 572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Dong Y, Qi X, Huang C, Hou L. Cholesterol levels and risk of hemorrhagic stroke: a systematic review and meta-analysis. Stroke 2013; 44: 1833–9. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol 2019; 18: 684–96. [DOI] [PubMed] [Google Scholar]
- White J, Swerdlow DI, Preiss D, Fairhurst-Hunter Z, Keating BJ, Asselbergs FW, et al. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol 2016; 1: 692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013; 45: 1274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PT. Low high-density lipoprotein 3 reduces the odds of men surviving to age 85 during 53-year follow-up. J Am Geriatr Soc 2012; 60: 430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo D, Falcone GJ, Devan WJ, Brown WM, Biffi A, Howard TD, et al. Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet 2014; 94: 511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtz P, Havulinna AS, Soininen P, Tynkkynen T, Prieto-Merino D, Tillin T, et al. Metabolite profiling and cardiovascular event risk: a prospective study of 3 population-based cohorts. Circulation 2015; 131: 774–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin ZG, Wang QS, Yu K, Wang WW, Lin H, Yang ZH. Sex differences in associations between blood lipids and cerebral small vessel disease. Nutr Metab Cardiovasc Dis 2018; 28: 28–34. [DOI] [PubMed] [Google Scholar]
- Yu S, Yarnell JW, Sweetnam P, Bolton CH. High density lipoprotein subfractions and the risk of coronary heart disease: 9-years follow-up in the Caerphilly Study. Atherosclerosis 2003; 166: 331–8. [DOI] [PubMed] [Google Scholar]
- Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 2001; 7: 853–7. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun 2018; 9: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used for the current study are publicly available and may also become available from the corresponding author on reasonable request.